Настоящее изобретение относится к производным хромонов, к способам их получения, к содержащим их фармацевтическим композициям и к их терапевтическим применениям в качестве агонистов, частичных агонистов или антагонистов рецептора дофамина D3 (DRD3) для лечения различных неврологических и психиатрических состояний.
Шизофрения является термином, используемым для описания группы патологий неизвестного происхождения, которые поражают примерно 1% всего населения. Данная патология характеризуется разнообразием симптомов, классифицируемых как позитивные симптомы (галлюцинации, бред, беспорядочные мысли) и негативные симптомы (социальная самоизоляция и аффективная тупость) в пубертатном возрасте начала или в возрасте начала полового созревания, и может персистировать в хронической форме с эпизодами обострения в течение многих лет.
Пациентов, больных шизофренией, можно лечить лекарственными средствами, называемыми нейролептиками, также известными под названием антипсихотические средства. Терапевтический эффект антипсихотических средств общеизвестен как результат блокады рецепторов нейромедиатора дофамина в головном мозге. Существует пять подтипов рецепторов дофамина, называемых D1, D2, D3, D4 и D5 (Sokoloff, P. et al., Novel dopamine receptor subtypes as targets for antipsychotic drugs. Annals New-York Academy of Sciences 1995, 757, 278), и общепринятые антипсихотические средства представляют собой антагонисты рецепторов D2 и D3. Однако антипсихотические средства часто ответственны за нежелательные экстрапирамидальные побочные эффекты (ЭПЭ) и аномальные движения, называемые поздними дискинезиями, которые присущи блокаде рецептора D2 в стриарной области головного мозга. Предположили, что блокада рецептора D3 (DRD3) ответственна за терапевтические эффекты антипсихотических средств (Schwartz J.C. et al., Eur. Neuropsychopharmacol. 2003, 13 fsuppl. 4): S 166). Следовательно, фармакологические агенты, которые селективно модулируют функцию DRD3, считают эффективными антипсихотическими средствами, свободными от неврологических побочных эффектов (международная заявка на патент WO91/15513).
Селективное модулирование рецепторов DRD3 может быть достигнуто с помощью молекул, которые селективно связываются с DRD3 и которые действуют как агонисты, как антагонисты или как частичные агонисты. Антипсихотическая активность в результате модулирования функции DRD3 может быть предсказана на животных путем использования моделей шизофрении на мышах (Leriche L. et al., Neuropharmacology 2003, 45, 174). Кроме того, продемонстрировано, что селективная блокада DRD3, но не одновременная блокада DRD2 и DRD3, повышает внеклеточные уровни дофамина и ацетилхолина, другого нейромедиатора, в префронтальной коре (Lacroix L.P. et al., Neuropsychophamacol. 2003, 28, 839). Дофамин и ацетилхолин в данной области головного мозга являются существенными для когнитивной функции. Следовательно, считают, что селективные антагонисты DRD3 могут улучшить познавательную способность, которая нарушена при шизофрении, а также при нейродегенеративных патологиях, таких как болезнь Альцгеймера.
Антипсихотические средства в целом и арипипразол, кветиапин и оланзапин в частности применяют при лечении острой маниакальной фазы биполярного расстройства. Антагонисты или частичные агонисты DRD3, следовательно, также рассматривают как лекарственные средства для лечения биполярного расстройства.
Генетически модифицированные мыши, несущие мутацию, которая выводит из строя DRD3 (DRD3 "нокаут"), являются менее тревожными в поведенческих тестах, предсказывающих анксиогенную или анксиолитическую активность (Steiner H. et al., 1: Physiol Behav. 1997, 63, 137-41). Следовательно, фармакологическое выведение из строя DRD3, такое как получают путем применения антагониста DRD3, описанного в настоящем изобретении, также является лечением тревоги.
Депрессия представляет собой общую патологию настроения, которая характеризуется ощущениями глубокой тоски, пессимистическими мыслями и занижением самооценки, часто сопровождается утратой энергии, энтузиазма и либидо. Неспособность чувствовать удовольствие от обычно приятных впечатлений, также известную под названием ангедония, также рассматривают как общий симптом при депрессии. Значительную роль в удовольствии и мотивации приписывают дофаминергическим нейронам в области головного мозга, называемой прилежащим ядром (Koob G.F. etal., Sem. Neurosci. 1992, 4, 139; Salamone J.D. et al., Behav. Brain Res. 1994, 61, 117). Следовательно, предполагают, что эти нейроны вовлечены в нейробиологию депрессии, в частности ангедонии, и в терапевтические эффекты некоторых антидепрессивных лекарственных средств (Kapur S. and Mann J. Biol. Psychiatry 1992, 32, 1-17; Willner P., Int. Clin. Psychopharmacol. 1997, 12, S7-S14). Продемонстрировано, что различные антидепрессивные терапии селективно повышают экспрессию DRD3 в прилежащем ядре (Lammers C.H. et al., Mol. Psychiatry 2000, 5, 378), что позволяет предположить, что повышение функции DRD3 могло бы быть новым способом антидепрессивной терапии. Повышение функции рецептора D3 DRD3 может быть достигнуто путем применения агонистов или частичных агонистов DRD3, которые могут, следовательно, быть эффективной терапией для депрессии.
Зависимость от лекарств или веществ, вызывающих привыкание, также известная как наркозависимость, представляет собой хроническую и рецидивирующую патологию, при которой поведение, включающее рискованный прием и поиск веществ, вызывающих привыкание, и навязчивое поведение приема лекарств персистирует несмотря на негативные последствия, получаемые пациентом (Deroche-Gamonet V. Et al., Science 2004, 305, 1014; Vanderschuren L.J. et al., Science 2004, 305, 1017). Феномен отмены, который происходит во время абстиненции от веществ, вызывающих привыкание, может запускаться или обостряться за счет стимулов окружающей среды, которые приобретают мотивационную силу в результате того, что они неоднократно связаны с эффектами лекарства, как у человека (Childress A.R. et al., Am. J. Psychiatry 1999, 156, 11; Robinson Т.Е. et al., Brain Research Reviews 1993, 18, 247), так и у животных (Goldberg S.R. et al., NIDA Res. Monogr. 1981, 37, 241; Arroyo M. Psychopharmacology 1999, 140, 331). У животных высокоселективные агонисты или частичные антагонисты DRD3 специфично снижают ответы на стимулы, связанные с кокаином (Pilla M. Nature, 1999, 400, 371; Le Foil, В. Eur. J. Neurosci. 2002, 15, 2016; Vorel S.R. J. Neurosci. 2002, 22, 9595), с опиатом (Frances H. et al., Neuroreport 2004, 15, 2245) или с никотином (Le FoilB. et al., Mot. Psychiatry 2002, 8, 225), в то же время не влияя на первичные эффекты этих лекарств. Плотность DRD3 является аномально высокой в головном мозге кокаиновых наркоманов (Staley J.K. et al., J. Neurosci. 1996, 16, 6106). Поэтому частичные агонисты или антагонисты DRD3 считают эффективными лекарственными средствами для облегчения абстиненции и снижения риска рецидива.
Болезнь Паркинсона представляет собой патологию, характеризующуюся тремором в покое, ригидностью конечностей и акинезией (затруднениями при начале движений). Это заболевание вызвано дегенерацией дофаминергических нейронов. Лечение болезни Паркинсона основано на замещении дофамина посредством введения L-дигидроксифениламина (L-ДОФА) или прямых агонистов дофамина. Длительное применение L-ДОФА, однако, ассоциировано с очень большим числом случаев появления аномальных движений, называемых дискинезиями. В модели болезни Паркинсона на нечеловекообразных приматах продемонстрировано, что модулирование DRD3 высокоселективными частичным агонистом ослабляет дискинезии (Bezard E. et al., Nat. Med. 2003, 6, 762). Соединения, описанные в настоящем документе, следовательно, рассматривают как аддитивные терапевтические средства при болезни Паркинсона. Однако продемонстрировано, что агонист DRD3 повышает нейрогенез у крыс, поэтому агонисты DRD3 также могут быть лекарственными средствами, которые замедляют прогрессирование этого заболевания.
Мутация в гене DRD3 ассоциирована и сегрегирует совместно с эссенциальным тремором, распространенным и наследуемым неврологическим расстройством, которое характеризуется тремором действия всех частей или части тела в отсутствие какой-либо другой неврологической патологии (Jeanneteau et al., Proc. Natl. Acad. Sci. USA 2006, 103, 10753). Эта мутация повышает функцию DRD3. Нормализация функции DRD3 посредством применения частичных агонистов или антагонистов DRD3 могла бы, следовательно, быть эффективной терапией для эссенциального тремора.
Дофамин контролирует эректильную функцию, и дофаминергические агенты предложены в качестве терапии для эректильной дисфункции (Guiliano F., Ramplin О. Physiol Behav. 2004, 63, 189-201). Более конкретно, проэректильные эффекты дофаминергических агонистов опосредованы рецептором D3 у грызунов (Collins G.T. et al., J. Pharmacol. Exp. Ther., 2009, 329, 210-217), и селективный антагонист рецептора D3 задерживает эякуляцию во время коитуса у крысы (Clement P. et al., J. Sex. Med., 2009, 6, 980-988). Агонисты, частичные агонисты и антагонисты DRD3, такие как описаны в настоящем изобретении, могут, таким образом, быть терапией для различных дисфункций эректильной функции.
В литературе упомянуты фенилпиперазинхромоны для применения при борьбе с малярией в Biochemical and Biophysical Research Communications 2007, 358(3), 686. В Indian J. Chem., section B, 2002, 41 B(4), 817, описаны соединения фенилпиперазинометилхромона. Манниховы основания, использующие метоксихромоны, известны из Farmaco Edizione Scientifica 1977, 32(9), 635. В описании патента US 3410851 описаны флавоны, обладающие противосудорожными, анальгезирующими и бронходилататорными свойствами. Соединения по настоящему изобретению отличаются тем фактом, что они имеют углеродную цепь из 4 метиленов между хромоновой группировкой и фенилпиперазином, которая придает им свойство лигандов дофаминергического рецептора D3.
В заявках на патенты WO 2003028728, WO 2004004729 и WO 2006077487 и в описании патента ЕР 1841752 описаны гетероарилфенилпиперазинбутилкарбоксамиды в качестве лигандов DRD3. В заявке на патент WO 2008009741 упомянуты хромен- и тиохроменкарбоксамиды, демонстрирующие сродство к дофаминергическому рецептору D3, для применения в качестве антипсихотических средств. В заявке на патент WO 2006072608 упомянуты арилпиперазины, обладающие модулирующими свойствами в отношении дофаминергических и серотонинергических рецепторов, для применения при нейропсихиатрических расстройствах, таких как шизофрения. В публикации J. Med. Chem. 2009, 52, 151 также упомянуты те же производные. Все эти продукты, описанные в цитируемых выше описаниях патентов, имеют карбоксамидную цепь в своей структуре. Продукты по настоящему изобретению отличаются от описанных соединений тем, что они не имеют карбоксамидной цепи, но неожиданно являются эффективными лигандами дофаминергического рецептора D3.
Как использовано выше, термин "рецептор дофамина D3", "рецептор D3" или "DRD3" означает подтип рецептора дофамина, главным образом экспрессирующийся в лимбической системе (Sokoloff P et al., Nature, 1990, 347, 146-151). DRD3 описан в международной заявке на патент WO 91/15513.
Как использовано выше, термин "частичный агонист рецептора D3" означает соединение, которое образует комплекс с DRD3 и действует как комбинированный агонист-антагонист, то есть он индуцирует физиологический ответ более низкой интенсивности, чем природный медиатор, дофамин. In vitro в клетке, экспрессирующей DRD3, частичный агонист DRD3 продуцирует активный ответ, максимальная интенсивность которого ниже, чем у ответа, продуцируемого дофамином или полным агонистом, например квинпиролом (транс(-)-4aR-4,4а,5,6,7,8,8а,9-октагидро-5-пропил-1Н(или 2Н)пиразоло [3,4g]хинолином). Частичный агонист DRD3 может также частично предотвращать ответ, продуцируемый дофамином или его полными агонистами. In vivo частичный агонист DRD3 продуцирует дофаминергические ответы, в частности, когда уровень дофамина снижен, как в случае у крыс, имеющих повреждения, вызванные 6-гидроксидофамином, или у обезьян с интоксикацией 1-метил-4-фенил-1,2,3,6-тетрагидропиридином (МФТП). Кроме того, in vivo частичный агонист DRD3 может действовать как антагонист, в частности, когда DRD3 подвергается ограниченной стимуляции дофамином.
"Антагонист DRD3" означает молекулу, которая образует комплекс с DRD3 и способна к предотвращению ответа, запускаемого дофамином или его агонистом в клетке, экспрессирующей DRD3.
Как используют в данной заявке, термин "соли" означает соли присоединения неорганической кислоты или основания соединений по настоящему изобретению. Предпочтительно соли являются фармацевтически приемлемыми, то есть они нетоксичны для пациента, которому их вводят.
Выражение "фармацевтически приемлемый" относится к молекулярным субстанциям и композициям, которые не производят какого-либо вредного аллергического эффекта или другой нежелательной реакции при введении животному или человеку.
При использовании в данной заявке выражение "фармацевтически приемлемый эксципиент" включает любой разбавитель, адъювант или эксципиент, такой как консервант, наполнитель, разрыхлитель, увлажняющий агент, эмульгатор, диспергирующий агент, антибактериальный или противогрибковый агент, либо также агенты, которые дадут возможность замедления всасывания и резорбции в тонком кишечнике и пищеварительной системе. Применение этих сред или векторов хорошо известно в данной области техники. За исключением тех случаев, где агент химически несовместим с производным хромона, рассматривают его применение в фармацевтических композициях, содержащих соединения в соответствии с изобретением.
В контексте изобретения термин "лечение", как используют в данной заявке, означает предупреждение или ингибирование возникновения или прогрессирования состояния, к которому применяют этот термин, или одного или более чем одного симптома этого состояния.
"Терапевтически эффективное количество" означает количество производного хромона, которое эффективно при получении желаемого терапевтического эффекта в соответствии с изобретением. Согласно изобретению термин "пациент" относится к человеку или к млекопитающему, отличному от человека, пораженному или склонного к поражению патологией. Предпочтительно пациент является человеком.
В контексте настоящего изобретения С1-4алкильную группу понимают как нормальную или разветвленную углеводородную цепь, содержащую от 1 до 4 атомов углерода, например метильную группу, этильную группу, пропильную группу или бутильную группу.
В контексте настоящего изобретения С1-4алкокси группу понимают как нормальную или разветвленную углеводородную цепь, содержащую от 1 до 4 атомов углерода и атом кислорода, например метокси группу, этокси группу, пропокси группу или бутокси группу.
В контексте настоящего изобретения С1-4тиоалкокси группу понимают как нормальную или разветвленную углеводородную цепь, содержащую от 1 до 4 атомов углерода, атом кислорода и атом серы, например тиометокси группу, тиоэтокси группу, тиопропокси группу или тиобутокси группу.
В контексте настоящего изобретения С1-4диалкиламиногруппу понимают как амин, двузамещенный нормальными или разветвленными С1-4алкильными группами, например группу диметиламино, группу диэтиламино, группу дипропиламино или группу дибутиламино.
В контексте настоящего изобретения галоген понимают как фтор, хлор или бром.
В контексте настоящего изобретения С1-4алогеноалкильную группу понимают как С1-4алкильную группу, монозамещенную, двузамещенную или тризамещенную галогеном, например группу CF3, группу CHF2, группу CH2F, группу CCl3, группу CHCl2, группу CH2Cl, группу CBr3, группу CHBr2 или группу CH2Br group.
В контексте настоящего изобретения С1-4диалкиламиноалкильную группу понимают как С1-4диалкиламиногруппу, как определено выше, связанную с С1-4алкильной группой атомом углерода, например диметиламинометильную группу, диметиламиноэтильную группу, диэтиламинометильную группу или диэтиламиноэтильную группу. В контексте настоящего изобретения C1-4алкоксиалкильную группу понимают как С1-4алкильную группу, как определено выше, связанную с С1-4алкильной группой атомом углерода, например метоксиметильную группу, этоксиметильную группу, метоксиэтильную группу или этоксиэтильную группу.
В контексте настоящего изобретения С1-4гидроксиалкильную группу понимают как алкильную группу, как определено выше, в которой атом водорода замещен гидроксильной группой, например группу CH2OH, группу C2H4OH, группу С3Н6ОН или группу С4Н8ОН.
В контексте настоящего изобретения С1-4алкилкарбонильную группу понимают как алкильную группу, как определено выше, связанную с карбонильной группой атомом углерода, например группу СОСН3, группу СОС2Н5, группу СОС3Н7 или группу COC4H9.
В контексте настоящего изобретения С1-4алкоксикарбонильную группу понимают как алкокси группу, как определено выше, связанную с карбонильной группой атомом углерода, например группу СООСН3, группу СООС2Н5, группу СООС3Н7 или группу COOC4H9.
В контексте настоящего изобретения С1-4фенилалкильную группу понимают как фенильную группу, связанную атомом углерода с алкильной группой, как определено выше.
Изобретение относится к производным хромонов, к способам их получения и к их применению в качестве лекарственного средства, в качестве лигандов рецептора DRD3, для лечения неврологических или психиатрических заболеваний, состояний или расстройств. Эти соединения соответствуют общей формуле 1:

общая формула 1
где:
R1 представляет собой один или более чем один из идентичных или различных заместителей на бензольном кольце, каждый из которых независимо представляет собой атом водорода или атом галогена, или С1-4алкокси группу, или ОН группу, или С1-4алкильную группу или группу -O(СН2)nO-, в которой n=1 или 2.
- R2 представляет собой атом водорода или С1-4алкильную группу.
- А и В независимо представляют собой либо атом азота, либо атом углерода.
- R3 представляет собой атом водорода или один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из: атома галогена, С1-4алкильной группы, С1-4алкокси или С1-4тиоалкокси группы, группы -O(СН2)nO-, в которой n=1 или 2, группы NO2, группы NHSO2R4, группы NHR5, ОН группы, С1-4алогеноалкильной группы, CN группы, C1-4алкоксикарбонильной группы, С1-4алкилкарбонильной группы, C1-4гидроксиалкильной группы и бензильного или фенильного заместителя, необязательно замещенного С1-4алкокси или С1-4алкильной группой, или атомом галогена,
- либо R3 составляет кольцо, конденсированное с бензольным кольцом, несущим его, выбранное из группы, состоящей из нафталина, индола, бензимидазола, карбостирила, бензоксазолона и бензимидазолона.
- R4 представляет собой С1-4алкильную группу, или C1-4диалкиламиногруппу, или С1-4алкоксиалкильную группу, или C1-4диалкиламиноалкильную группу либо фенильную или фенил-С1-4алкильную группу,
- R5 представляет собой атом водорода, или С1-4алкилкарбонильную группу, или С1-4алкоксикарбонильную группу,
а также их фармацевтически приемлемые соли.
В соответствии с изобретением соединения общей формулы (I) представляют собой соединения, где:
- R1 представляет собой один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из С1-4алкокси группы, ОН группы и группы -O(СН2)nO-, в которой n=1 или 2.
В соответствии с изобретением соединения общей формулы (I) представляют собой соединения, где:
- R2 представляет собой атом водорода.
В соответствии с другим воплощением изобретения соединения общей формулы (I) представляют собой соединения, где R3 представляет собой атом водорода, когда А и/или В представляет собой атом азота.
В соответствии с изобретением соединения общей формулы (I) представляют собой соединения, где:
- А и В одновременно представляют собой атом углерода. В соответствии с изобретением соединения общей формулы (I) представляют собой соединения, где:
- R3 представляет собой один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из: атома галогена, С1-4алкокси группы, группы -O(СН2)nO-, в которой n=1 или 2, группы NHSO2R4, ОН группы и CN группы.
В соответствии с другим воплощением изобретения соединения общей формулы (I) представляют собой соединения, где:
-R3 вместе с бензольным кольцом, несущим его, представляет собой индольную группу, или бензимидазольную группу, или карбостирильную группу.
В соответствии с другим воплощением изобретения соединения общей формулы (I) представляют собой соединения, где:
- R1 представляет собой один или два идентичных или различных заместителя, где каждый независимо представляет собой метокси группу или группу -O(СН2)nO-, в которой n=1, или ОН группу.
- R2 представляет собой атом водорода.
- А представляет собой атом углерода и В представляет собой атом азота или атом углерода.
- когда А и В представляют собой атом углерода:
- R3 представляет собой один или два идентичных или различных заместителя, выбранных из группы, состоящей из: атома водорода, CN группы, атома хлора, атома фтора, ОН группы, группы NO2, группы NHSO2R4, группы NHR5, группы CF3, метокси группы,
- или R3 образует кольцо, конденсированное с бензольным кольцом, несущим его, выбранное из группы, состоящей из: бензимидазола, бензоксазолона, индола, бензимидазолона и карбостирила.
-когда А представляет собой атом углерода и В представляет собой атом азота:
- R3 представляет собой атом водорода
- R4 представляет собой метильную группу, или этильную группу, или диметиламиноэтильную группу, или этоксиметильную группу.
- R5 представляет собой атом водорода, группу СОСН3 или группу СООСН3.
Ниже приведены примеры соединений в соответствии с изобретением:
- 6,7-диметокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
- 3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-бензонитрил
- 3-{4-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
- 3-{4-[4-(3-гидроксифенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
- 6,7-диметокси-3-[4-(4-пиримидин-2-ил-пиперазин-1-ил)-бутил]-хромен-4-он
- 6,7-диметокси-3-[4-(4-пиридин-2-ил-пиперазин-1-ил)-бутил]-хромен-4-он
- 3-{4-[4-(2,3-дифторфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
- 3-{4-[4-(1 Н-бензимидазол-4-ил-)пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
- 3-{4-[4-(1Н-индол-4-ил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
- 5-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1Н-хинолин-2-он
- 6,7-диметокси-3-{4-[4-(3-нитрофенил)-пиперазин-1-ил]-бутил}-хромен-4-он
- 3-{4-[4-(3-аминофенил)-пиперазин-1-ил-]-бутил}-6,7-диметоксихромен-4-он
- N-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид
- N-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фен ил)-ацетамид
- метил-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-карбамат
- 7-{4-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
- 7-{4-[4-(2,3-дифторфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
- 7-{4-[4-(3-нитрофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
- 7-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
- N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил-ацетамид
- N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид
- N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-этансульфонамид
- 2-диметиламиноэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амид
- 2-метоксиэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амид
- 7-{4-[4-(1Н-индол-4-ил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
- 3-{4-[4-(3-трифторметилфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
- 6-метокси-3-[4-(4-фенилпиперазин-1-ил)-бутил]-хромен-4-он
- 6-метокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
- 6-метокси-3-{4-[4-(3-трифторметилфенил)-пиперазин-1-ил]-бутил}-хромен-4-он
- 7-{4-[4-(2,3-дихлорфенил)пиперазин-1-ил-]-бутил}-6-метил-[1,3]диоксоло[4,5-g]хромен-8-он
- 6,7-метокси-7,6-гидрокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
- 7-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-3Н-бензоксазо1-2-он
- 4-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1,3-дигидробензимидазол-2-он.
Изобретение также относится к их фармацевтически приемлемым солям, а также к содержащим их фармацевтическим композициям и к их применению в качестве лекарственных средств, предназначенных для лечения расстройств центральной нервной системы.
Изобретение также относится к способу получения этих соединений.
Соединения общей формулы 1 получают в соответствии со схемой 1.

Схема 1
Реакция Фриделя - Крафтса или реакция Фриса с замещенным ароматическим метокси соединением 2 (Y=Me) или замещенным соединением фенола 3 (Y=Н) дает ароматический кетон 3 (Y= Me, H). В этой реакции используют галогенид омега-галогенированной гексановой кислоты, такой как 6-бромгексаноилхлорид. Конденсация происходит с растворителем или без растворителя в присутствии кислоты Льюиса, такой как AlCl3, в соответствии со способом, аналогичным описанному в Chem. Ber. 1939, 72, 1414, или J. Org. Chem. 1955, 20, 38, с хлор- или бромацетилхлоридом или бромидом. В данной заявке в этой реакции используют бромгексаноилхлорид, который конденсируется в орто-положении фенольной функциональной группы с образованием производного 3. Где используют растворитель, можно использовать хлорированный растворитель, такой как метиленхлорид, для реакции при температуре окружающей среды или при низкой температуре либо для реакции при более высокой температуре можно, например, использовать дихлорэтан или 1,1,2,2-тетрахлорэтан. Используемые фенолы с соответствующими заместителями либо имеются в продаже, либо известны из литературы, и их получают путем деметилирования в присутствии агентов, обычно используемых для деметилирования ароматических метокси соединений, такие как НВг и кислоты Льюиса (AlCl3, BBr3). Реакцию Фриделя - Крафтса можно также проводить на метоксилированном ароматическом кольце, богатом электронами. Стадия деметилирования с получением промежуточного соединения 3 может идти после стадии ацилирования. Фенол 3 (Y=Н), ацилированный таким образом, можно циклизировать ацеталем диметилформамида (=ДМФ) или диметиламина (=ДМА) при нагревании с получением галогенированного хромона 4. Эту реакцию замыкания кольца с образованием хромона можно также осуществлять в ДМФ в присутствии PCl5 и эфирата BF3, а также с этилформиатом в присутствии натрия в соответствии с Bull. Soc. Chim. Fr. 1944, 5, 302. Затем галогенобутил-производное хромона 4 объединяют с замещенными арилпиперазинами или гетероарилпиперазинами формулы 5 стандартным способом в присутствии основания, такого как К2СО3 или карбонат цезия, в ацетонитриле или метилэтилкетоне с получением производных формулы 1. Этот метод используют с пиперазинами формулы 5, где А, В и R3 являются такими, как определено выше. Можно использовать вариант этого способа, который включает введение пиперазиновой группировки перед образованием хромонового кольца: таким образом, конденсация пиперазина формулы 5 с галогенированным фенолом формулы 3 в таких же обычных условиях алкилирования в щелочной среде (K2CO3/CH3CN или метилэтилкетон) дает соединения формулы 6. Затем образование хромонового кольца можно осуществлять путем замыкания кольца с ДМФ или ацеталем ДМФ или ДМА. Используя этот способ, введение пиперазина перед циклизацией до хромона дает возможность получить более чистое циклизированное соединение, чем способом образования хромона, начиная с производного 3 (Y=Н). Действительно, условия нагревания для образования кольца ДМФ при повышенной температуре образует диметиламин, который может взаимодействовать с галогенированным производным 3 с получением вторичного продукта (формула 4, Х=NMe2) и требует дополнительной очистки. Специалист в данной области техники сможет выбрать подходящий способ в соответствии с заместителями, которые несет фенилпиперазин 5. Модификации заместителей пиперазина можно также осуществлять на последних стадиях, как, например, используя пиперазин формулы 5 (А=В=С, R3=3-NO2). Восстановление нитрогруппы в продукте формулы 1 (А=В=С, R3=3-NO2) обычно осуществляют путем каталитического восстановления водородом, используя палладий на углероде или никель Ренея, либо путем обработки металлом, таким как железо, в кислой среде с получением соответствующего анилина (формула 1, А=В=С, R3=3-NH2). Анилиновую группу можно, таким образом, ацилировать в присутствии пиридина или другого основания ацетилхлоридом с получением производного ацетамида, метилхлорформиатом с получением метилкарбамата или метансульфонилхлоридом с получением метилсульфонамида. Взаимодействие хлорэтилсульфонилхлорида можно осуществлять таким же путем, а затем виниловое промежуточное соединение можно объединять с диметиламином или с метоксидом натрия с получением соответственно заместителя диметиламиноэтилсульфонамида или метоксиэтилсульфонамида. В литературе упомянуты гетероциклические арилпиперазины, такие как 4-пиперазин-1-ил-1Н-индол, пиперазин-1-ил-1Н-бензимидазол, 7-пиперазин-1-ил-3Н-бензоксазол-2-он, 4-пиперазин-1-ил-1,3-дигидробензимидазол-2-он, 5-пиперазин-1-ил-1Н-хинолин-2-он. Гетероциклические пиперазины могут быть получены путем взаимодействия соответствующих анилинов с азотистыми ипритами (бисхлорэтиламинами). Эти азотистые иприты могут быть N-замещенными защитной группой бензила, которую можно удалить путем простого гидрогенолиза с Pd/C в атмосфере водорода, когда осуществлена конденсация с пиперазином (Fr2504532; Fr2524884; Bioorg. Med. Chem. Lett. 1998, 8, 2675; Bioorg. Med. Chem. Let. 2001, 11, 2345, J. Med. Chem. 2002, 45, 4128; J. Med. Chem. 2004, 47, 871; Synth. Commun. 2006, 36, 1983; Synthesis 1977, 33; Tet Let. 1970, 5265; Chem. Pharm. Bull. 1981, 29, 651 или 1979, 27, 2627; Tet. 2000, 56, 3245).
Таким образом, изобретение также относится к описанным ниже способам получения:
Способ получения соединений общей формулы 1, характеризующийся тем, что получают возможно замещенный хромон формулы 4 (X=Cl, Br, I), который подвергают взаимодействию с пиперазином формулы 5.

Радикалы R1, R2, R3, А и В имеют значения, приведенные выше. Способ получения соединений общей формулы 1, характеризующийся тем, что получают возможно замещенное производное фенола формулы 6, начиная с соединения формулы 3 (X=Cl, Br), и подвергают взаимодействию с ДМФ (=диметилформамид), или диметилацеталем ДМФ, или ДМА (= диметиламин).

Радикалы R1, R3, А и В имеют значения, приведенные выше, в условиях алкилирования в присутствии основания, такого как К2СО3, Cs2CO3 или NEt3, в растворителе, таком как ацетонитрил или метилэтилкетон.
Изобретение также относится к фармацевтической композиции, содержащей по меньшей мере одно соединение общей формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
С учетом селективного модулирования передач сигналов дофамина, осуществляемых посредством рецептора DRD3 в лимбических областях, которые вовлечены в эмоциональные и когнитивные процессы, соединения по изобретению пригодны при различных терапевтических применениях и не препятствуют дофаминергическим передачам сигналов экстрапирамидальной системы, системы передней доли гипофиза или вегетативной системы (например, самого заднего поля). Соединения по изобретению можно, таким образом, применять для получения фармацевтических композиций и лекарственных средств для лечения неврологических или психиатрических заболеваний, состояний или расстройств, в которые вовлечен рецептор DRD3, таких как психотические состояния.
Кроме того, поскольку эффект антидепрессивных лекарственных средств состоит в повышении экспрессии рецептора DRD3 в областях головного мозга, вовлеченных в мотивацию, соединения по изобретению также способны имитировать действие антидепрессивных лекарственных средств. Соединения по изобретению можно, таким образом, применять для получения фармацевтических композиций и лекарственных средств для лечения депрессии.
С учетом роли рецептора DRD3 в состояниях лекарственной зависимости фармацевтические композиции или лекарственные средства на основе соединений, описанных в настоящем изобретении, можно с пользой вводить при состояниях, ассоциированных с абстиненцией, и/или чтобы способствовать детоксикации индивидуумов, зависимых от кокаина, героина, алкоголя, табака и других веществ, вызывающих привыкание.
Соединения в соответствии с изобретением подобно частичным агонистам рецептора DRD3 в целом можно также применять в качестве дополнительного лечения к лечению болезни Паркинсона L-ДОФА.
Соединения в соответствии с изобретением подобно частичным агонистам и антагонистам рецептора DRD3 в целом можно также применять для лечения эссенциального тремора.
Соответственно, соединения формулы 1, основания или соли можно применять для лечения неврологических или психиатрических состояний, в частности состояний, которые можно лечить агонистами, частичными агонистами или антагонистами рецептора DRD3.
Изобретение также относится к способу лечения неврологических или психиатрических состояний, заболеваний или расстройств, который включает введение соединения формулы 1 в терапевтически эффективном количестве пациенту, которому требуется лечение. Изобретение, кроме того, относится к соединениям формулы 1 для их применения в качестве лекарственных средств.
Изобретение также относится к соединениям формулы 1 для получения лекарственного средства для лечения неврологического или психиатрического заболевания или расстройства, либо эректильной дисфункции, либо зависимости от лекарств или от веществ, вызывающих привыкание.
Изобретение относится к соединениям общей формулы (I) для получения лекарственного средства для лечения болезни Паркинсона, психоза, шизофрении, дискинезий, ассоциированных с болезнью Паркинсона, когнитивной недостаточности, возможно ассоциированной с возрастом или с болезнью Альцгеймера, расстройства настроения, эссенциального тремора, тревоги, депрессии, биполярного расстройства, половой импотенции, преждевременной эякуляции, алкоголизма и никотиновой зависимости.
Соединения формулы 1 в соответствии с изобретением можно вводить пероральным, системным, парентеральным, назальным или ректальным путем. Соединение можно, в частности, вводить пероральным путем в соответствующем препарате. Дозировки соединений формулы 1 в композициях по изобретению можно регулировать до получения количества активного вещества, которое эффективно при получении желаемого терапевтического ответа для композиции, характерной для способа введения. Выбранный уровень дозировки, таким образом, зависит от желаемого терапевтического эффекта, пути введения, желаемой продолжительности лечения и других факторов.
Соединения формулы 1 оценивали in vitro в качестве лигандов DRD3 и модуляторов активности этого рецептора в соответствии с изобретением в клетках, экспрессирующих рекомбинантный рецептор DRD3 человека. Константы ингибирования (Ki) измеряли на основании ингибирования связывания [3H] спиперона, как описано авторами Cussac et al., в Naunyn-Schmiedeberg's Arch. Pharmacol. 2000, 361, 569. Авторы изобретения продемонстрировали, что соединения формулы 1 ведут себя как эффективные лиганды со значениями Ki от 0,1 до 30 наномоль·литр-1. Те же соединения проявляют заметное сродство к рецептору дофамина D2, которое является в 10-200 раз более слабым. Соединения формулы 1. оценивали на их агонистическую, частичную агонистическую или антагонистическую активность путем использования МАР-киназного теста на активность на рекомбинантных рецепторах человека, как описано Cussac D. et al., Mol. Pharmacol. 1999, 56, 1025-1030. Собственные активности соединений формулы 1 составляют от 0 (антагонист) до 0,80 (агонист).
Соединения формулы 1 оценивали in vivo в тесте гиперактивности, индуцированной МК-801, у мыши (Leriche L. et al., Neuropharmacology 2003, 45, 174). Значения ED50 соединений формулы 1 составляют от 0,01 до 6 мг/кг.
Суммарная суточная доза соединений для применения в соответствии с данным изобретением, вводимая в однократной дозе или в дробных дозах, может находиться в количествах, например, от 0,001 до примерно 100 мг/кг массы тела в сутки.
Конкретный уровень дозы для любого конкретного пациента будет зависеть от ряда факторов, включая массу тела, общее состояние здоровья, пол, режим питания, продолжительность и путь введения, уровни всасывания и резорбции в тонком кишечнике и выведения, комбинацию с другими лекарственными средствами и тяжесть конкретного состояния, подлежащего лечению.
Например, в качестве неограничивающего примера получение соединений по изобретению продемонстрировано в приведенных ниже Примерах:
Пример 1: 6,7-Диметокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он

Стадия 1: 6-Бром-1-(2,4,5-триметоксифенил)-гексан-1 -он

6 мл (40 ммоль) 1,2,4-триметоксибензола вносят в 80 мл сухого CH2Cl2, и смесь охлаждают до -10ºС при перемешивании. Затем добавляют по каплям 6-бромгексаноилхлорид (6,2 мл, 40 ммоль), растворенный в 20 мл CH2Cl2·AlCl3 (5,6 г, 42 ммоль) постепенно вносят небольшими порциями в реакционную смесь. Реакционную смесь выдерживают при перемешивании в течение 8 ч с возвращением к температуре окружающей среды. Затем реакционную смесь наливают на лед (200 мл) и подкисляют до рН 1, используя HCl. Смесь перемешивают до возвращения ее к температуре окружающей среды, 1 ч. После выпаривания CH2Cl2 смесь экстрагируют AcOEt и органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают. Остаток подвергают флэш-хроматографии на SiO2 с градиентом от чистого гептана до гептана-AcOEt 50-50. Чистые фракции выпаривают с получением 13,6 г (выход =99%) кристаллов. ТСХ SiO2 (гептан-ACOEt 70-30) Rf=0,5; 1H ЯМР (CDCl3): 7,41 (s, 1H), 6,50(s, 1H), 3,95 (s, 3H), 3,91 (s, 3H), 3,87(s, 3Н), 3,43 (t, 2H, J=6,76 Гц), 2,98 (t, 2H, J=6,32 Гц), 1,91 (m, 2H), 1,71 (m, 2H), 1,51 (m, 2H).
Стадия 2: 6-Бром-1-(2-гидрокси-4,5-диметоксифенил)-гексан-1-он

13,6 г продукта, полученного на вышеописанной стадии, растворяют в 80 мл 48% HBr. Смесь нагревают при 90ºС в течение 5 ч. Затем реакционную смесь наливают на лед (300 мл) и экстрагируют AcOEt. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают с получением зеленого масла, которое подвергают флэш-хроматографии на SiO2 с градиентом от чистого гептана до гептана-AcOEt 85-15. Получают 7,33 г (выход =56%) 6-бром-1-(2-гидрокси-4,5-диметоксифенил)-гексан-1-она, 1H ЯМР (CDCl3): 12,7 (s, 1H), 7,08 (s, 1H), 6,46 (s, 1H), 3,91 (s, 3H), 3,87 (s 3H), 3,44 (t, 2H, J=8 Гц), 2,92 (t, 2H, J=7,2 Гц), 1,93 (m, 2H), 1,78 (m, 2H), 1,55 (m 2H); а также 1,4 г ди-деметилированного соединения, 6-бром-1-(2,4,5-дигидро-5,4-метоксифенил)гексан-1-она, 1H ЯМР (CDCl3): 12,5 (s, 1H), 7,22 (s, 1H), 6,45 (s, 1H) 5,20 (s, 1H), 3,93 (s, 3H), 3,42 (t, 2H, J=6,68 Гц), 2,89 (t, 2H, J=7,32 Гц), 1,91 (m 2H), 1,76 (m, 2H), 1,53 (m, 2H).
Стадия 3: 3-(4-Бромбутил)-6,7-диметоксихромен-4-он

1 способ: Раствор А готовят из 500 мг соединения вышеописанной стадии, 6-бром-1-(2-гидрокси-4,5-диметоксифенил)-гексан-1-она (1,5 мл, растворенных в 0,60 мл (4,5 ммоль) Et2O-BF3), и этот раствор охлаждают до 10ºС. Затем добавляют 2,3 мл ДМФ. Кроме того, готовят раствор В из 4 мл ДМФ и добавляют к нему небольшими порциями при 10ºС 470 мг (2,25 ммоль) PCl5. Раствор В нагревают при 55ºС в течение 20 мин, а затем вводят по каплям в раствор А, описанный вначале, с возвращением к температуре окружающей среды. Смесь становится оранжево-желтой и осаждается. Вводят 50 мл 0,1 н. HCl, и смесь экстрагируют AcOEt, органические фазы промывают насыщенным раствором NaCl, отделяют, высушивают над MgSO4, фильтруют и выпаривают. Остаток подвергают флэш-хроматографии на SiO2 с градиентом от чистого гептана до гептана-AcOEt 70-30. Очищенные фракции кристаллизуются после выпаривания. Получают 300 мг 3-(4-бромбутил)-6,7-диметоксихромен-4-она в форме кристаллов (выход =59%); ТСХ SiO2 гептан-AcOEt 50-50 Rf=0,4.
2 способ: Раствор 500 мг (1,5 ммоль) соединения вышеописанной стадии, 6-бром-1-(2-гидрокси-4,5-диметоксифенил)-гексан-1-она, в 30 мл сухого толуола кипятят с обратным холодильником при перемешивании с 0,6 мл (4,5 ммоль) ДМФ диметилацеталя. Кипячение с обратным холодильником продолжают в течение 5 ч. После концентрирования и очистки флэш-хроматографией с градиентом от чистого гептана до гептана-ACOEt 80-20 получают 270 мг (выход =53%) 3-(4-бромбутил)-6,7-диметоксихромен-4-она после выпаривания в форме белых кристаллов, идентичных полученным 1 способом. ТСХ SiO2 гептан-ACOEt 70-30 Rf=0,3. 1H ЯМР (ДМСО): 8,19 (s, 1H), 7,36 (s, 1H), 7,16 (s, 1H), 3,89 (s, 3H), 3,84 (s, 3H), 3,65 (t, 2H, J=6,3 Гц), 2,38 (t, 2Н, J=7,3 Гц), 1,72 (m, 2H), 1,64 (m, 2H), 1,55 (m, 2H).
Стадия 4: 6,7-Диметокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
Бромированное производное, полученное на вышеописанной стадии 3 (150 мг, 0,44 ммоль) суспендируют в 10 мл метилэтилкетона и добавляют 120 мг (0,62 ммоль) 2-метоксифенилпиперазина и 121 мг (0,87 ммоль) К2СО3, а также 10 мг тетрабутиламмония бромида. Смесь кипятят с обратным холодильником в течение 20 ч, а затем концентрируют. Остаток растворяют в воде и экстрагируют этилацетатом. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают с получением бесцветного масла. Флэш-хроматография на SiO2 с элюированием градиентом от CH2Cl2 до CH2Cl2-MeOH 90-10 дает возможность получить масло, которое кристаллизуется в iPr2O. Получают 128 мг (выход =60%) белых кристаллов. Т.пл.ºС=124-130; МС (ИЭР) m/z=453 (МН+); 1H ЯМР (CDCl3): 7,72 (s, 1Н), 7,55 (s, 1H), 6,92 (m, 5H), 3,97 (s, 3Н), 3,86 (s, 3Н), 3,12 (m, 4H), 2,69 (m, 4H), 2,49 (m, 4H), 1,64 (m, 4H).
Продукты приведенных ниже Примеров получают путем такой же последовательности реакций:
Пример 2: 3-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-бензонитрил

Путем конденсации бромированного производного, 3-(4-бромбутил)-6,7-диметоксихромен-4-она, полученного на стадии 3 Примера 1, с 3-цианофенилпиперазином получают 3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-бензонитрил при выходе 40%. Т.пл.ºС=154-155; аналитическая ВЭЖХ Sym C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 30-70-6,8 г/л, рН 4, r.t.=9,72 мин; МС ИЭР, m/z=448 (МН+); 1H ЯМР (ДМСО): 8,17 (s, 1H), 7,13-7,39 (m, 6H), 3,89 (s, 3Н), 3,84 (s, 3Н), 3,19 (m, 4H), 2,47 (m, 4H), 2,38 (t, 2H, J=6,8 Гц), 2,32 (t, 2H, J=6,8 Гц), 1,50 (m, 4H).
Пример 3: 3-{4-[4-(2,3-Дихлорфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он

Подобно примеру 1, но используя 2,3-дихлорфенилпиперазин, получают 3-{4-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он при выходе 62%. Т.пл.ºС=160-162; аналитическая ВЭЖХ Sym C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 40-60-6,8 г/л, рН 4, r.t.=8,80 мин; МС ИЭР, m/z=491; 1H ЯМР (CDCl3): 7,72 (s, 1Н), 7,55 (s, 1H), 7,14 (m, 2H), 6,95 (m, 1H), 6,83 (s, 1H), 3,97 (s, 6H, ОСН3), 3,07 (m, 4Н), 2,64 (m, 4H), 2,48 (m, 4H), 1,64 (m, 4H).
Получение гидрохлорида: 2,64 г основания, полученного выше, растворяют в смеси 100 мл ацетон-МеОН (50-50). Добавляют раствор изопропанола и 2 Н HCl. Осажденную соль отфильтровывают с получением после высушивания в вакууме 2,02 г гидрохлорида (выход =72%). Т.пл.ºС=252-254.
Пример 4: 3-{4-[4-(3-Гидроксифенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он

Используя то же исходное вещество, 3-(4-бромбутил)-6,7-диметоксихромен-4-он, полученное на стадии 3 Примера 1, но с 3-гидроксифенилпиперазином, и используя сосуд для микроволновой реакции (15 мин, 160ºС, 150 Вт), получают 3-{4-[4-(3-гидроксифенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он подобно примеру 1 при выходе 17%. Т.пл.ºС=177-180; аналитическая ВЭЖХ Sym C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-Н2О, KH2PO4 25-75-6,8 г/л, рН 4, r.t.=9,99 мин; МС ИЭР, m/z=439 (МН+); 1H ЯМР (CDCl3): 7,72 (s, 1H), 7,55 (s, 1H), 7,09 (t, 1H, J=8 Гц), 6,83 (s, 1H), 6,49 (d, 1H, J=8,28 Гц), 6,39 (s, 1H), 6,31 (d, 1H, J=7,84 Гц), 3,97 (s, 6H, ОСН3), 3,18 (m, 4H), 2,58 (m, 4H), 2,49 (m, 2H), 2,43 (m, 2H), 1,62 (m, 4H).
Пример 5: 6,7-Диметокси-3-[4-(4-пиримидин-2-ил-пиперазин-1-ил)-бутил]-хромен-4-он

Подобно примеру 1, но используя 2-пиримидинилпиперазин, получают 6,7-диметокси-3-[4-(4-пиримидин-2-илпиперазин-1-ил)-бутил]-хромен-4-он при выходе 77%. Т,пл.ºС=123-124; аналитическая ВЭЖХ Sym C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 20-80-6,8 г/л, рН 4, r.t.=14,31 мин; МС ИЭР, m/z=425 (МН+); 1H ЯМР (CDCl3): 8,34 (d, 2Н, J=4,64 Гц), 8,17 (s, 1H), 7,36 (s, 1H), 7,15 (s, 1H), 6,60 (t, 1H, J=4,6 Гц), 3,89 (s, 3Н), 3,84 (s, 3H), 3,69 (m, 4H), 2,38 (m, 6H), 2,31 (m, 2Н), 1,51 (m, 4H).
Пример 6: 6,7-Диметокси-3-[4-(4-пиридин-2-ил-пиперазин-1-ил)-бутил]-хромен-4-он

Подобно примеру 1, но используя 2-пиридинилпиперазин, получают 6,7-диметокси-3-[4-(4-пиридин-2-илпиперазин-1-ил)-бутил]-хромен-4-он при выходе 50%. Т.пл.ºС=41-143; аналитическая ВЭЖХ XBridge, 4,6×250 мм, 8,5 мкм, элюент: CH3CN-H2O, KH2PO4 20-80-6,8 г/л, рН 4, r.t.=14,21 мин; МС ИЭР, m/z=424 (МН+); 1H ЯМР (ДМСО): 8.17 (s, 1H), 8.09 (d, 1H, J=4.28 Гц), 7.5 (t, 1H, J=7.6 Гц), 7.36 (s, 1H), 7.15 (s, 1H), 6.79 (d, 1H, J=8.6 Гц), 6.61 (t, 1H, J=5.8 Гц), 3.89 (s, 3Н), 3.84 (s, 3Н), 3.43 (m, 4H), 2.39 (m, 6H), 2.33 (m, 2Н), 1.51 (m, 4H).
Пример 7: 3-{4-[4-(2,3-Дифторфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он

Подобно примеру 1, но используя 2,3-дифторфенилпиперазин, описанный в J. Med. Chem. 2006, 49, 3628, получают 3-{4-[4-(2,3-дифторфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он при выходе 33%. Т.пл.ºС=148-151; Анализ: C25H28N2O4F2=458,51, вычислено С% 65,49, Н% 6,16, N% 6,11, обнаружено С% 65,44, Н% 6,29, N% 6,26; МС ИЭР, m/z=459 (МН+); 1H ЯМР (ДМСО): 8,18 (s, 1H), 7,36 (s, 1H), 7,16 (s, 1H), 7,08 (dd, 1H, J=14,4 Гц, J'=6,8 Гц), 6,96 (dd, 1H, J=17,2 Гц, J'=8 Гц), 6,83 (t, 1 Н, J=7,6 Гц), 3,89 (s, 3H), 3,84 (s, 3H), 3,32 (m, 4H), 3,02 (m, 4H), 2,36 (m, 4H), 1,50 (m, 4H).
Пример 8: 3-{4-[4-(1Н-Бензимидазол-4-ил)пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он

Подобно примеру 1, но используя 4-бензимидазолилпиперазин, описанный в Tet. 2000, 56, 3245, получают 3-{4-[4-(1 Н-бензимидазол-4-ил)пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он при выходе 66%. Т.пл.ºС=175-179; аналитическая ВЭЖХ XBridge, 4,6×250 мм, 8,5 мкм, элюент: CH3CN-H2O, KH2PO4 20-80-6,8 г/л, рН 4, r.t.=9,41 мин; МС ИЭР, m/z=463 (МН+); 1H ЯМР (ДМСО): 12,3 (m, 1Н), 8,19 (s, 1Н), 8,04 (s, 1H), 7,37 (s, 1H), 7,16 (s, 1H), 7,03 (m, 2H), 6,48 (m, 1H), 3,89 (s, 3Н), 3,85 (s, 3Н), 3,45 (m, 4H), 3,32 (m, 4H), 2,57 (m, 4H), 2,40 (m, 4H), 1,54 (m, 4H).
Пример 9: 3-{4-[4-(1Н-Индол-4-ил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он

Подобно примеру 1, но используя 4-индолилпиперазин, описанный в J. Med. Chem. 2002, 45, 4128, получают 3-{4-[4-(1Н-индол-4-ил)пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он при выходе 69%. Т.пл.ºС = 197-199; аналитическая ВЭЖХ XBridge, 4,6×250 мм, 8,5 мкм, элюент: CH3CN-Н2О, KH2PO4 30-70-6,8 г/л, рН 4, r.t.=8,15 мин; МС ИЭР, m/z = 462 (МН+); 1H ЯМР (ДМСО): 11,0 (m, 1H), 8,19 (s, 1H), 7,37 (s, 1H), 7,22 (m, 1H), 6,96 (m, 2H), 6,43 (m, 1H), 6,35 (m, 1H), 3,89 (s, 3Н), 3,85 (s, 3Н), 3,09 (m, 4H), 2,57 (m, 4H), 2,40 (m, 4H), 1,54 (m, 4H).
Гидрохлорид: Т.пл.ºС=244; Анализ C27H31N3O4, HCl=510,43 (+5,88% H2O) вычислено С% 63,26, Н% 6,37, N% 8,20, обнаружено С% 62,95, Н% 6,15, N% 7,98.
Пример 10: 5-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1Н-хинолин-2-он

Стадия 1: 5-Амино-1Н-хинолин-2-он

Раствор 2,1 г (11 ммоль) 5-нитро-1Н-хинолин-2-она (Chem. Pharm. Bull. 1981, 29, 651) в 40 мл АсОН гидрогенизируют с 210 мг 10% Pd/C в присутствии водорода в течение 24 ч при энергичном перемешивании. Катализатор отфильтровывают и смесь выпаривают. Остаток подвергают флэш-хроматографии на SiO2 с градиентом от чистого CH2Cl2 до CH2Cl2-MeOH 99-1. После выпаривания получают 1,67 г (выход 97%) желтых кристаллов. 1H ЯМР (ДМСО): 11,38 (s, 1Н), 8,08 (d, 1Н, J=8 Гц), 7,10 (t, 1Н, J=7,6 Гц), 6,44 (d, 1H, J=8 Гц), 6,33 (d, 1Н, J=8 Гц), 6,26 (d, 1Н, J=10 Гц), 5,85 (s, 2H).
Стадия 2: 5-Пиперазин-1-ил-1Н-хинолин-2-он

800 мг производного вышеописанной стадии (4,96 ммоль) вносят в сосуд для микроволновых реакций с 890 мг (4,96 ммоль) бис-2-хлорэтиламина с 1,25 мл 2-(2-метоксиэтокси)этанола и нагревают при 150ºС в течение 20 ч. После добавления 1 н. раствора гидроксида натрия смесь экстрагируют CH2Cl2. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают. Флэш-хроматография с градиентом от чистого CH2Cl2 до CH2Cl2-MeOH-NH4OH 90-9-1 позволяет выделить после выпаривания и растирания в этиловом эфире 260 мг (выход =23%) желтых кристаллов. МС, ИЭР m/z=230 (МН+); 1H ЯМР (ДМСО): 11,67 (s, 1Н), 7,99 .(d, 1Н, J=10 Гц), 7,39 (t, 1Н, J=8 Гц), 6,98 (d, 1Н, J=8,4 Гц), 6,79 (d, 1Н, J=7,6 Гц), 6,45 (d, 1Н, J=10 Гц), 2,90 (m, 4H), 2,86 (m, 4H).
Стадия 3: 5-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1Н-хинолин-2-он
Пиперазин, полученный на вышеописанной стадии, затем конденсируют так же, как на стадии 4 Примера 1, с бромированным производным 3-(4-бромбутил)-6,7-диметоксихромен-4-оном, полученным на стадии 3 Примера 1, но используя ацетонитрил в качестве растворителя. Получают 300 мг (выход = 54%) бледно-желтых кристаллов. Т.пл.ºС=243-246; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 20-80-6,8 г/л, рН 4, r.t.=12,69 мин; МС ИЭР, m/z=490 (МН+).
Пример 11: 6,7-Диметокси-3-{4-[4-(3-нитрофенил)-пиперазин-1-ил]-бутил}- хромен-4-он

Подобно примеру 1, но используя 3-нитрофенилпиперазин, получают 6,7-диметокси-3-{4-[4-(3-нитрофенил)-пиперазин-1-ил]-бутил}-хромен-4-он при выходе 16%. Т.пл.ºС=149-151; аналитическая ВЭЖХ Sym C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 30-70-6,8 г/л, рН 4, r.t.=12,11 мин; МС APCI, m/z=468; 1H ЯМР (CDCl3): 7,72 (s, 1Н), 7,71 (d, 1H, J=7,8 Гц), 7,64 (d, 1H, J=8,04 Гц), 7,55 (s, 1H), 7,36 (t, 1H, J=8,2 Гц), 7,17 (d, 1H, J=8,16 Гц), 6,83 (s, 1H), 3,97 (s, 6H), 3,29 (m, 4H), 2,61 (m, 4H), 2,47 (m, 4H), 1,63 (m, 4H).
Пример 12: 3-{4-[4-(3-Аминофенил)-пиперазин-1-ил-]-бутил}-6,7-диметоксихромен-4-он

Нитросоединение вышеописанного Примера 11 (910 мг, 1,95 ммоль) гидрогенизируют в смеси 50 мл CH2Cl2 и 50 мл EtOH с 91 мг 10% Pd/C в атмосфере водорода в течение 24 ч при энергичном перемешивании. После удаления катализатора фильтрованием и после выпаривания выделяют 720 мг розовых кристаллов. Флэш-хроматография на SiO2 с элюированием градиентом от чистого CH2Cl2 до CH2Cl2-MeOH 95-5 позволяет выделить 550 мг (выход =64%) бежевых кристаллов путем растирания с iPr2O. Т.пл.ºС=175-176; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 20-80-6,8 г/л, рН 4, r.t.=11,46 мин; МС ИЭР, m/z=438 (МН+).
Пример 13: N-(3-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид

427 мг (0,98 ммоль) соединения 3-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-она, полученного в вышеописанном Примере 12, суспендируют в 10 мл CH2Cl2, добавляют 0,16 мл (1,95 ммоль) пиридина и при 0ºС добавляют по каплям 75 мкл (0,98 ммоль) мезилхлорида, растворенного в 2 мл CH2Cl2. Перемешивание поддерживают при температуре окружающей среды в течение 8 ч. Смесь наливают в воду и экстрагируют CH2Cl2. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают. Остаток подвергают флэш-хроматографии SiO2 и элюируют градиентом от 2 до CH2Cl2-MeOH 90-10. После выпаривания полученное масло кристаллизуют в iPr2O с получением 272 мг бежевых кристаллов (выход =54%). Т.пл.ºС=186-189; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 30-70-6,8 г/л, рН 4, r.t.=6,91 мин; МС ИЭР, m/z=516 (МН+); 1H ЯМР (CDCl3): 7,72 (s, 1Н), 7,55 (s, 1H), 7,19 (t, 1H, J=8,2 Гц), 6,83 (s, 1H), 6,78 (s, 1H), 6,73 (d, 1H, J=8,52 Гц), 6,63 (d, 1H, J=7,4 Гц), 6,29 (m, 1H), 3,97 (s, 6H), 3,19 (m, 4H), 2,99 (s, 3Н), 2,59 (m, 4H), 2,49 (m, 2H), 2,44 (m, 2H), 1,59 (m, 4H).
Пример 14: N-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-ацетамид

Подобно Примеру 13, но используя ацетилхлорид и 3-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он, полученный в Примере 12, получают N-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-ацетамид.
Пример 15; Метил-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-карбамат

Подобно примеру 13, но используя метилхлорформиат и 3-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он, полученный в Примере 12, получают метил-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-карбамат.
Пример 16: 7-{4-[4-(2,3-Дихлорфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-д]хромен-8-он

Стадия 1: Получение 6-бром-1-(6-гидроксибензо[1,3]диоксо-5-ил)-гексан-1-она
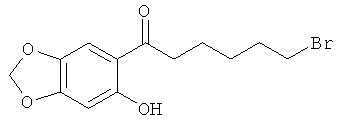
Раствор 1 г (7,2 ммоль) сезамола в 20 мл CH2Cl2 охлаждают до -10ºС при перемешивании. Добавляют 1,1 мл (7,2 ммоль) 6-бромгексаноилхлорида, а затем 1 г (7,6 ммоль) AlCl3 небольшими порциями. Температуре дают подняться до температуры окружающей среды и перемешивание продолжают в течение 18 ч. Гидролиз осуществляют путем добавления льда, и подкисление осуществляют концентрированной HCl (2 мл). Экстракцию осуществляют CH2Cl2, органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают, подвергают флэш-хроматографии на SiO2 с градиентом от чистого гептана до гептана-AcOEt 80-20 с получением 500 мг бледно-желтых кристаллов после выпаривания (выход =22%) МС, ИЭР, m/z=314-316. 1H ЯМР (CDCl3): 7,26 (s, 1Н), 7,07 (s, 1Н), 6,45 (s, 1Н), 5,98 (s, 2H), 3,42 (t, 2H, J=6,8 Гц), 2,87 (t, 2H, J=7,6 Гц), 1,92 (m, 2H), 1,76 (m, 2H), 1,54 (m, 2H).
6-Бром-1-(2-гидрокси-5-метоксифенил)-гексан-1-он получают идентично.
Стадия 2: Получение 6-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-1-(6-гидроксибензо[1,3]диоксол-5-ил)-гексан-1-она

950 мг (3 ммоль) бромированного производного, полученного на вышеописанной стадии, 690 мг (3 ммоль), 2,3-дихлорфенилпиперазина, 1,3 мл (9 ммоль) триэтиламина и 500 мг (3 ммоль) KI добавляют к 10 мл CH3CN. Смесь кипятят с обратным холодильником при перемешивании в течение 20 ч. Добавляют насыщенный раствор NaHCO3 (50 мл) и осуществляют экстракцию AcOEt. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают. Флэш-хроматография на SiO2 с элюированием градиентом от чистого CH2Cl2 до CH2Cl2-MeOH 90-10 позволяет получить после выпаривания и кристаллизации из iPr2O 960 мг (выход =69%) бежевых кристаллов. 1H ЯМР (ДМСО): 7,45 (s, 1Н), 7,30 (m, 2Н), 7,13 (m, 1Н), 6,57 (s, 1H), 6,08 (s, 2H), 2,96 (m, 6H), 2,50 (m, 4H), 2,33 (m, 2H), 1,63 (m, 2H), 1,48 (m, 2H), 1,36 (m, 2H).
Стадия 3: 7-{4-[4-(2,3-Дихлорфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
Раствор 600 мг (1,30 ммоль) соединения, полученного на вышеописанной стадии, в 5 мл диметилформамида диметилацеталя нагревают при 90ºС в течение 5 ч при перемешивании. Добавляют 50 мл воды и осуществляют экстракцию CH2Cl2. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают. Флэш-хроматография на SiO2 с элюированием градиентом от CH2Cl2 до CH2Cl2-MeOH 90-10 позволяет получить после концентрирования и кристаллизации из iPr2O 250 мг (выход =40%) бежевых кристаллов. Т.пл.ºС=140-142; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-Н2О, KH2PO4 40-60-6,8 г/л, рН 4, r.t.=9,51 мин; МС ИЭР, m/z=475-477; Анализ C24H24N2O4Cl2=475,38+0,21 H2O, вычислено С% 60,64, Н% 5,09, N% 5,89, обнаружено С% 60,61, Н% 5,07, N% 6,45; 1H ЯМР (ДМСО): 8,17 (s, 1Н), 7,33 (s, 1Н), 7,29 (m, 2H), 7,22 (s, 1Н), 7,13 (m, 1Н), 6,02 (s, 2H), 2,96 (m, 4H), 2,50 (m, 6H), 2,35 (m, 2H), 1,51 (m, 4H).
Пример 17: 7-{4-[4-(2,3-Дифторфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он

Идентично вышеописанному примеру 16, но используя дифторфенилпиперазин, получают 7-{4-[4-(2,3-дифторфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он. Т.пл.ºС=140-142; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 35-65-6,8 г/л, рН 4, r.t.=9,59 мин; МС ИЭР, m/z=443 (МН+); Анализ C24H24N2O4F2=442,46+0,78 H2O, вычислено С% 65,15, Н% 5,47, N% 6,33, обнаружено С% 65,41, Н% 5,71, N% 6,77; 1H ЯМР (ДМСО): 8,16 (s, 1H), 7,33 (s, 1H), 7,22 (s, 1H), 7,08 (m, 1H), 6,96 (m, 1H), 6,83 (t, 1H, J=8 Гц), 6,20 (s, 2H), 3,02 (m, 4H), 2,50 (m, 4H), 2,34 (m, 4H), 1,51 (m,4H).
Пример 18: 7-{4-[4-(3-Нитрофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он

Идентично вышеописанному примеру 16, но используя 3-нитрофенилпиперазин, получают 6-{4-[4-(3-нитрофенил)-пиперазин-1 -ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он. МС ИЭР, m/z=452 (МН+); 1H ЯМР (ДМСО): 8,17 (s, 1H), 7,62 (s, 1H), 7,57 (d, 1H, J=7,6 Гц), 7,46 (t, 1H, J=8,4 Гц), 7,39 (d, 1H, J=8,4 Гц), 7,33 (s, 1H), 7,22 (s, 1H), 6,20 (s, 2H), 3,24 (m, 4H), 2,50 (m, 4H), 2,35 (m, 4H), 1,50 (m, 4H).
Пример 19: 7-{4-[4-(3-Аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
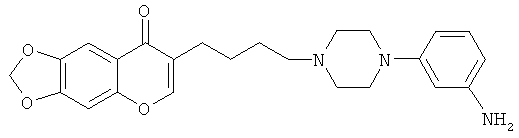
Идентично Примеру 12, но используя соединение, полученное в Примере 18, получают анилин 7-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он. МС ИЭР, m/z=422 (МН+); 1H ЯМР (CDCl3): 7,68 (s, 1H), 7,52 (s, 1H), 7,03 (t, 1H, J=8 Гц), 6,81 (s, 1H), 6,36 (d, 1H, J=8 Гц), 6,25 (d, 1H, J=2 Гц), 6,21 (d, 1H, J=7,6 Гц), 6,08 (s, 2H), 3,16 (m, 4H), 2,57 (m, 2H), 2,47 (t, 2H, J = 6,4 Гц), 2,41 (t, 2H, J=7,6 Гц), 1,59 (m, 4H).
Пример 20: N-(3-{4-[4-(8-оксо-8H-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-илфенилацетамид
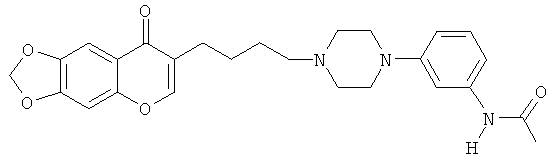
Идентично Примеру 14, но используя 7-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-д]хромен-8-он вместо 3-{4-[4-(3-аминофенил)-пиперазин-1 -ил-]-бутил}-6,7-диметоксихромен-4-она, получают N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-ацетамид.
Пример 21: N-(3-{4-[4-(8-оксо-8H-[1,3]диоксоло[4,5-д]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид

Аналогично Примеру 13, но используя 7-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он, полученный в Примере 19, вместо 3-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-она, получают N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид. Т.пл.ºС=174; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 25-75-6,8 г/л, рН 4, r.t.=13,23 мин; МС, ИЭР, m/z=499 (MH+); 1H ЯМР (ДМСО): 9,51 (s, 1H), 8,16 (s, 1H), 7,33 (s, 1H), 7,22 (s, 1H), 7,13 (t, 1H, J=8,4 Гц), 6,73 (s, 1H), 6,67 (d, 1H, J=8,4 Гц), 6,63 (d, 1H), 6,20 (s, 2H), 3,07 (m, 4H), 2,94 (s, 3H), 2,47 (m, 4H), 2,36 (t, 2H, J=6,4 Гц и 6,8 Гц), 1,49 (m, 4H).
Гидрохлорид: Т.пл.ºС=260, Анализ C25H30ClN3O6S=499,59+0,34% H2O, вычислено С% 56,02, Н% 5,64, N% 7,84, S% 5,98, обнаружено С% 56,37, Н% 5,69, N% 7,65, S% 6,89.
Пример 22: N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-этансульфонамид

Идентично Примеру 13, но используя соответствующие реагенты, получают N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-этансульфонамид. МС, ИЭР, m/z=514 (МН+); 1H ЯМР гидрохлорида (ДМСО): 9,69 (s,1H), 8,22 (s, 1H), 7,34 (s, 1H), 7,25 (s, 1H), 7,19 (t, 1H, J=8,4 Гц), 6,80 (s, 1H), 6,73 (m, 1H), 6,21 (s, 2H), 3,71 (m, 2H), 3,54 (m, 2H), 3,08 (m, 6H), 2,40 (m, 2H), 1,72 (m, 2H), 1,55 (m, 2H).
Пример 23: 2-Диметиламиноэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амид

Стадия 1: Подобно примеру 21, 7-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он конденсируют с 2-хлорэтилсульфонилхлоридом. Получают (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-этенсульфонамид. 1H ЯМР (ДМСО): 9,79 (s, 1Н), 8,16 (s, 1Н), 7,33 (s, 1Н), 7,22 (s, 1H), 7,09 (t, 1H, J=8 Гц), 6,75 (dd, 1H, J=16,4 Гц и 10 Гц), 6,67 (d, 1H), 6,63 (dd, 1H, J=10 Гц и 2 Гц), 6,57 (dd, 1H, J=8 Гц и 1,2 Гц), 6,20 (s, 2H), 6,09 (d, 1H, J=16,4 Гц), 6,01 (d, 1H, J=9,6 Гц), 3,05 (m, 4H), 2,47 (m, 4H), 2,34 (m, 4H), 1,51 (m, 4H).
Стадия 2: Соединение вышеописанной стадии 1 (100 мг, 0,2 ммоль) вносят в герметично закрытую пробирку с 2 мл 2 М раствора диметиламина в МеОН при температуре окружающей среды на 3 ч. Весь объем выпаривают до сухости и остаток растирают с изопропанолом - HCl, гидрохлорид вносят в iPr2O и фильтруют. МС, ИЭР, m/z=557 (МН+); 1H ЯМР (ДМСО) гидрохлорида: 10,10 (s, 1H), 8,23 (s, 1H), 7,22 (m, 2H), 6,78 (m, 3H), 6,21 (s, 2H), 3,77 (m, 2H), 3,68 (m, 2H), 3,55 (m, 2H), 3,45 (m, 2H), 3,13 (m, 6H), 2,76 (s, 6H), 2,40 (m, 2H), 1,74 (m, 2H), 1,55 (m, 2H).
Пример 24: 2-Метоксиэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амид

Подобно примеру 23 раствор метоксида натрия можно использовать с промежуточным соединением стадии 1 Примера 23 с получением 2-метоксиэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амида.
Пример 25: 7-{4-[4-(1Н-Индол-4-ил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он

Путем такой же последовательности реакций, которая указана в Примере 16, но используя 4-индолилпиперазин, получают 7-{4-[4-(1 Н-индол-4-ил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он, Т.пл.ºС=177-179; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 30-70-6,8 г/л, рН 4, r.t.=9,69 мин;
МС, ИЭР, m/z=446 (MH+); 1H ЯМР (ДМСО): 11,0 (s, 1H), 9,51 (s, 1H), 8,18 (s, 1H), 7,33 (s, 1H), 7,23 (m, 2H), 7,00 (d, 1H, J=8 Гц), 6,94 (t, 1H, J=7,2 Гц), 6,42 (d, 1H, J=7,2 Гц), 6,34 (s, 1H), 6,20 (s, 2H), 3,09 (m, 4H), 2,57 (m, 4H), 2,37 (m, 4H), 1,51 (m, 4H).
Гидрохлорид: Анализ C26H27N3O4, HCl=481,98+0,54% H2O, вычислено C% 64,79, H% 5,86, N% 8,72, обнаружено С% 63,78, Н% 5,70, N% 8,46.
Пример 26: 3-{4-[4-(3-Трифторметилфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он

Путем такой же последовательности реакций, как на стадиях 2 и 3 Примера 16, но используя 6-бром-1-(2-гидрокси-4,5-диметоксифенил)-гексан-1-он, полученный на стадии 2 Примера 1, получают 3-{4-[4-(3-трифторметилфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он в форме соли с 1,5 эквивалентами фумаровой кислоты. Т.пл.ºС=220; ТСХ: SiO2 элюирование CHCl3-МеОН 90-10, Rf=0,56; Анализ C26H29F3N2O4, C6H6O6=664,63, вычислено C% 57,82, H% 5,30, N% 4,21, F% 8,57, обнаружено C% 57,71, H% 5,24, N% 4,30, F% 8,80%.
Пример 27: 6-Метокси-3-[4-(4-фенилпиперазин-1-ил)-бутил]-хромен-4-он

Путем такой же последовательности реакций, как на стадиях 2 и 3 Примера 16, но используя 6-бром-1-(2-гидрокси-4-метоксифенил)-гексан-1-он, полученный в соответствии со стадией 1 Примера 1 и используя 1,4-диметоксибензол вместо 1,2,4-триметоксибензола, или используя 4-метоксифенол в качестве исходного вещества в соответствии с таким же методом, как на Стадии 1 Примера 16, получают 6-метокси-3-[4-(4-фенилпиперазин-1-ил)-бутил]-хромен-4-он, полученный в форме белых кристаллов гидрохлорида. Т.пл.ºС=198; ТСХ: SiO2 элюирование CHCl3-МеОН-NH4OH 95-4,5-0,5, Rf=0,45; Анализ C24H29ClN2O3=428,94, вычислено С% 67,20, Н% 6,81, N% 6,53, Cl% 8,26, обнаружено С% 66,78, Н% 6,82, N% 6,47, Cl% 7,95%.
Пример 28: 6-Метокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он

Идентично вышеописанному примеру, но используя соответствующие исходные вещества, получают 6-метокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он в форме белых кристаллов гидрохлорида. Т.пл.ºС=191;
ТСХ: SiO2 элюирование CHCl3-МеОН-NH4OH 95-4,5-0,5, Rf=0,67; Анализ C25H31ClN2O4=458,97, вычислено С% 65,42, Н% 6,81, N% 6,10, Cl% 7,72, обнаружено С% 66,28, Н% 6,88, N% 6,08, Cl% 7,64%.
Пример 29: 6-Метокси-3-{4-[4-(3-трифторметилфенил)-пиперазин-1 -ил]-бутил}-хромен-4-он

Идентично вышеописанному примеру, но используя соответствующие исходные вещества, получают 6-метокси-3-{4-[4-(3-трифторметилфенил)-пиперазин-1-ил]-бутил}-хромен-4-он в форме белых кристаллов гидрохлорида. Т.пл.ºС=180; ТСХ: SiO2 элюирование CHCl3-MeOH-NH4OH 95-4,5-0,5, Rf=0,56;
Анализ C25H28ClF3N2O3=496,45, вычислено С% 60,42, Н% 5,68, N% 5,64, Cl% 7,13, F% 11,48, обнаружено С% 60,23, Н% 5,63, N% 5,63, Cl% 6,97%, F% 11,28.
Пример 30: 7-{4-[4-(2,3-Дихлорфенил)пиперазин-1-ил-]-бутил}-6-метил-[1,3]диоксоло[4,5-g]хромен-8-он

6-[4-(2,3-Дихлорфенил)-пиперазин-1-ил]-1-(6-гидроксибензо[1,3]диоксол-5-ил)-гексан-1-он (200 мг, 0,43 ммоль), полученный на стадии 2 Примера 16, вносят в сосуд для микроволновых реакций с 1 мл диметилацетамида диметилацеталя и нагревают при 160ºС в течение 5 мин. Смесь выбрасывают в воду, а затем экстрагируют AcOEt. Органические фазы отделяют, высушивают над MgSO4, фильтруют и выпаривают. Флэш-хроматография на SiO2 с элюированием градиентом от CH2Cl2 до CH2Cl2-MeOH 90-10 позволяет получить после выпаривания и растирания с iPr2O 30 мг (выход 14%) бежевых кристаллов. Т.пл.ºС=153-155; аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH2CN-H2O, KH2PO4 40-60-6,8 г/л, рН 4, r.t.=11,09 мин; МС, ИЭР, m/z=489-491 (MH+); 1H ЯМР (ДМСО): 7,29 (m, 3H), 7,16 (m, 2H), 6,18 (s, 2H), 2,96 (m, 4H), 2,45 (m, 6H), 2,40 (s, 3H), 2,35 (m, 2H), 1,46 (m, 4H).
Пример 31: 6/7-Метокси-7/6-гидрокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
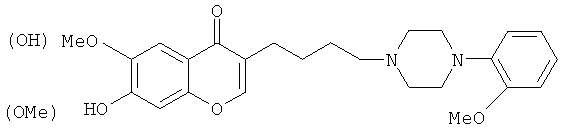
Данное соединение получают путем такой же последовательности реакций, как для Примера 1 на стадиях 3 и 4, но используя в качестве исходного вещества 6-бром-1-(2,4-дигидро-5-метоксифенил)гексан-1-он, полученный в качестве вторичного продукта на стадии 2 деметилирования Примера 1. Аналитическая ВЭЖХ Xbridge C8, 4,6×250 мм, 5 мкм, элюент: CH3CN-H2O, KH2PO4 25-75-6,8 г/л, рН 4, r.t.=11,27 мин; МС, ИЭР, m/z=439 (МН+).
Пример 32: 7-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-3Н-бензоксазол-2-он
Подобно примеру 1, стадии 4, но используя 7-пиперазин-1-ил-3Н-бензоксазол-1-он, описанный в Bioorg. Med. Chem. Let. 2001, 11, 2345, получают 7-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-3Н-бензоксазол-2-он.
Пример 33: 4-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1,3-дигидробензимидазол-2-он

Подобно примеру 1, Стадия 4, но используя производное гидрогенолиза 4-бензилпиперазин-1-ил)-1,3-дигидробензимидазол-2-она, описанного в Bioorg. Med. Chem. Let. 1998, 8, 2675, получают 4-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1,3-дигидробензимидазол-2-он.

Протокол исследований in vivo
Введение
Фенциклидин (РСР, phencyclidine), неконкурентный антагонист в глутаматного рецептора подтипа N-метил-D-аспартат (NMDA, N-methyl-D-aspartate), является анестезирующим агентом, который характеризуется наличием психотомиметических свойств по отношению к человеку (Javitt и Zukin, 1991). РСР вызывает состояние сенсорной изоляции, видимое «опьянение» и галлюцинации, часто с последующим чувством деперсонализации у злоупотребляющих (Snyder, 1980), и симптомы, подобные шизофреническим, у здоровых добровольцев (Jentsch и Roth, 1999;. Luby et al., 1959). РСР также ускоряет развитие психоза у больных шизофренией (Ital et al., 1967). Психотические симптомы, вызванные РСР или фармакологически родственным соединением кетамином, включают в себя и положительные (галлюцинации, бред), и отрицательные симптомы (расстройство формального мышления, социальная самоизоляция), а также когнитивную дисфункцию. Таким образом, эффекты неконкурентного антагониста рецептора NMDA могут быть использованы в качестве убедительной индуцированной лекарствами модели шизофрении, (Javitt, Zukin, 1991; Snyder, 1988). У животных, РСР или МК-801, другой неконкурентный антагонист рецептора NMDA (Wong et al., 1986), вызывает поведенческие отклонения, в том числе гиперактивность, нарушения сенсомоторного стробирования и дефицит социальных взаимодействий, которым противодействуют антипсихотические препараты (Jentsch, Roth, 1999;. Leriche et al., 2003).
Среди систем нейромедиаторов, участвующих в индуцированных МК-801 поведенческих отклонениях, решающую роль играет глутаматная система (Jentsch, Roth, 1999; Moghaddam, Jackson, 2003; Moghaddam, 2003). Это является прямым следствием механизма действия МК-801. Следовательно, многие препараты, которые модулируют глутаматергическую нейротрансмиссию (включая, но не ограничиваясь, соединения, действующие в отношении дофаминергического D3 (Sokoloff, 2012)) как было показано, модулируют МК-801-индуцированные ответы (Jentsch и Roth, 1999; Moghaddam и Jackson, 2003; Moghaddam, 2003, Ieriche et al., 2003, Sokoloff et al., 2012).
Наконец, поскольку все антипсихотические препараты способны ингибировать МК-801-индуцированную гиперактивность, этот простой тест, как обычно считают, имеет ценную прогностическую валидность для исследования возможных антипсихотических характеристик лекарства-кандидата (Jentsch, Roth, 1999; Leriche et al., 2003; Bradford et al., 2010).
Авторы изобретения исследовали влияние различных антипсихотических препаратов и некоторых типичных соединений из текущего патента, оказываемое на МК-801-индуцированную двигательную гиперактивность у мышей, для того чтобы 1) оценить модель с помощью эталонных антипсихотических препаратов и 2) зарегистрировать потенциальный эффект F17779, подобный антипсихотическому.
Материалы и методы
Животные
Животных содержали и испытывали в системе, аккредитированной Ассоциацией по Оценке и Аккредитации ухода за лабораторными животными (AAALAC, Association for the Assessment and Accreditation of Laboratory Animal Care). Их разместили, с ними обращались и заботились о них в соответствии с Руководством по уходу и использованию лабораторных животных (Национальный Исследовательский Совет) и с Европейской директивой N° 86/609, и экспериментальный протокол проводился в строгом соответствии со всеми действующими нормами и с локальными институционными нормами исследований на животных Этического Комитета.
Самцы домовых мышей (20-22 г массы тела по прибытии) были размещены по группам в числе 8 особей на клетку (поликарбонатные клетки Типа III, длина - 375 мм, ширина - 215 мм, высота - 149 мм; площадь поверхности пола 806 см2, покрытие из древесных опилок). Мышей держали в карантине в течение по меньшей мере 4 дней перед использованием в экспериментах. Все мыши были размещены в комнате с кондиционером (при температуре, равной 21±1°С; влажности 55±5% и с включенным светом с 7:00 до 19:00 ч). Мыши имели свободный доступ к стандартному сухому лабораторному корму (А04; SAFE, Augy, France) и воде (отфильтрованной с помощью 0,22 мкм фильтра) на всех этапах размещения и карантина. В день эксперимента домашние клетки с мышами перенесли в комнату с актиметром и помещены в вентилируемые закрытые стеллажи («enceintes»). Условия окружающей среды в экспериментальной комнате были идентичны таковым в комнате для жилья. Все эксперименты проводились с 09:00 до 17:00 часов. Пища и вода не были доступны во время отдельных тестовых сессий внутри актиметра (продолжительность=90 мин). В конце дня, после того, как были сделаны все наблюдения за поведением, животные были умерщвлены при помощи вдыхания 70% CO2.
Лекарственные средства.
В случаях, когда это было возможно, эталонные нейролептики были приобретены у коммерческих поставщиков; остальные были синтезированы на кафедре медицинской химии Исследовательского Института им. Пьера Фабра. (+)-дизоцилпин малеат (МК-801) был получен от компании Sigma. Типичные соединения были синтезированы на кафедре медицинской химии Исследовательского Института им. Пьера Фабра. Каждое соединение растворяли в подходящей среде. Все растворы готовили свежими ежедневно и вводили i.p. (i.p., - внутрибрюшинно, intraperitoneal) в объеме 10 мл/кг. Стерильный 0,9% раствор NaCl (физиологический раствор, Sal (saline)) или подходящую среду использовали в качестве контроля. Дозы выражены в мг/кг свободного основания.
Обработка лекарственными средствами.
Для индукции гиперактивности использовали фиксированную дозу МК-801, равную 0,14 мг/кг i.p., так как на основании результатов, полученных в исследованиях, проведенных для внутренних целей в соответствии с тем же экспериментальным протоколом и в соответствии с литературой (Leriche et al., 2003) эта доза установлена в качестве стандартной нагрузки для индукции гиперактивности.
Наблюдения за поведением
Эксперименты проводили в соответствии с процедурами, описанными Leriche et al. (Neuropharmacology, 2003, 45: 174-81), для оценки эффекта, оказываемого соединениями на гиперактивность, вызванную МК-801.
Мышам делали инъекции i.p., содержащие соединения или физиологический раствор (контрольные группы) и измеряли двигательную активность в течение 30 мин (фаза привыкания фазы, эффект, оказываемый соединением на спонтанную активность). Затем мышам вводили i.p. физиологический раствор (отрицательный контроль) или МК-801 и регистрировали активность в течение дополнительного периода длиной в 1 час для измерения эффекта, оказываемого соединением на индуцированную МК-801 гиперактивность.
Все эксперименты проводились с 09:00 до 17:00 часов. Сначала обслуживающий персонал по уходу за животными переносил мышей в испытательную комнату в 08:00 часов и они привыкали к испытательной комнате в течение не менее чем 1 ч до начала любых манипуляций. Двигательная активность измерялась в актиметре, который состоял из 16 отдельных клеткок для оценки общей активности деятельности (модель 2150, Tecniplast, внутренние размеры: 190×305×190 мм Ш×Д×В, площадь поверхности пола - 580 см2, с опилками на полу) пересеченных инфракрасными лучами (Imetronic, Pessac, France). Баллы за направленную вперед горизонтальную активность увеличиваются каждый раз, когда животное перемещается из одной половины клетки к другой, что соответствует прерыванию двух скрещенных параллельных пучков света в 14 см друг от друга. Вертикальная активность (приподнимание) также была записана.
Статистический анализ
Различия между группами были проанализированы с помощью однофакторного дисперсионного анализа (ANOVA, analysis of variance) и ретроспективного анализа менее значимых различий (LSD, least significance difference). Статистический анализ проводился с использованием программного обеспечения SigmaStat 3.5.
Вычисление ED50
Ингибирование МК-801-индуцированной гиперактивности рассчитывали по формуле:
%inhib.MK=100·[(MSaline+MK - MDrug_dose+MK)/(MSaline+MK - MSaline+Saline)]
Где MSaline+MK - это средняя активность начиная от 30 мин и до 90 мин в группе, предварительно обработанной физиологическим раствором и затем обработанной МК-801, MDrug_dose+MK - это средняя активность начиная от 30 мин и до 90 мин в группе, предварительно обработанной дозой лекарственного средства и затем обработанной МК-801, и MSaline+Saline это средняя активность начиная от 30 мин и до 90 мин в группе, предварительно обработанной физиологическим раствором и затем обработанной физиологическим раствором.
Эффективные дозы, вызывающие 50% ингибирования МК-801 - индуцированной гиперактивности ED50 (МК-801), были рассчитаны с использованием нелинейной регрессии (сигмоидальная кривая доза-эффект с или без переменного наклона с использованием наиболее подходящей модели, данной исправленным критерием информации Экейка (AICC, Akaike′s Information Criterion Corrected)) % ингибирования = f(log[доза]). Эти вычисления были проведены с использованием программного обеспечения GraphPad Prism 4.0.
Результаты в Таблице 2
В следующей ниже Таблице 2 показаны значения ED50 некоторых примеров соединений формулы 1, которые составляют от 0,01 до 6 мг/кг, вместе с другими эталонными антипсихотическими средствами. Эти значения были получены в результате тестирования на гиперактивность, индуцированную МК-801 (Leriche L. et al., Neuropharmacology 2003, 45, 174).

И, как показано на Фигуре 1, МК-801 индуцировал сильную и значимую гиперактивность по сравнению с животными, предварительно обработанными физиологическим раствором и затем обработанными физиологическим раствором. Эти стимулирующие эффекты дозозависимо ингибировались антипсихотическим средством галоперидолом, в зависимости от дозы ингибировали путем антипсихотического средства галоперидола, с ED50, равной 0,05 мг/кг i.p. Пример соединения 25 испытывали в тех же условиях, и он также дозозависимо подавлял индуцированную МК-801 гиперактивность с расчетной EDso, равной 0,12 мг/кг, i.p.
Краткое описание графических материалов
Фигура 1 демонстрирует сравнение между соединением согласно Примеру 25 и галоперидолом (эталонное антипсихотическое средство), в тесте на гиперактивность, вызванную МК-801.
Галоперидол (А) или Пример 25 (В) зависимым от дозы образом ингибировали спонтанную горизонтальную двигательную активность со значительным эффектом при дозах 0,16 и 0,63 мг/кг i.p. для галоперидола или 0,04, 0,63 и 2,5 мг/кг i.p. для Примера 25. Галоперидол (0,01-0,63 мг/кг, i.p.), Пример 25 (0.01-2,5 мг/кг i.p.) или физиологический раствор (Sal) вводили животным и помещали их в актиметр. Тридцать минут спустя вводили МК-801 (0,14 мг/кг, i.p., МК) или Sal и регистрировали двигательную активность в течение последующего периода длительностью 1 час. Результаты представляют собой среднее ± S.E.M. для N=20 животных (А) и N=9-20 животных (В). * Р<0,05, ** Р<0,01 и *** Р<0,001 по сравнению с Sal+Sal (В) и ## Р<0,01 и ### Р<0,005 по сравнению с МК(В); рассчитано с помощью ANOVA c последующим ретроспективным тестом LSD.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ (ПОЛИ)АМИНОАЛКИЛАМИНОАЛКИЛАМИДНЫЕ, АЛКИЛ-МОЧЕВИННЫЕ ИЛИ АЛКИЛ-СУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ ЭПИПОДОФИЛЛОТОКСИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ СРЕДСТВ | 2009 |
|
RU2529676C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ IV, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ НА ИХ ОСНОВЕ | 2003 |
|
RU2297418C9 |
| КОМБИНАЦИОННОЕ ЛЕКАРСТВО | 2003 |
|
RU2328280C2 |
| ПРОИЗВОДНЫЕ НОСКАПИНА (ВАРИАНТЫ), КОМБИНАТОРНАЯ И ФОКУСИРОВАННАЯ БИБЛИОТЕКИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ПРИМЕНЕНИЯ | 2006 |
|
RU2304584C1 |
| ИНДОЛИЛМАЛЕИМИДНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ НАРУШЕНИЙ ИЛИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ Т-ЛИМФОЦИТАМИ И/ИЛИ ПКС | 2003 |
|
RU2340610C2 |
| ПРОИЗВОДНЫЕ 2-ИМИНОПИРРОЛИДИНА | 2002 |
|
RU2270192C2 |
| АЗОЛИЛЬНЫЕ ПРОИЗВОДНЫЕ ХИНОЛИНА И ХИНАЗОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2002 |
|
RU2283841C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |

Изобретение относится к новым производным хромонов общей формулы 1, где: R1 представляет собой один или более чем один из идентичных или различных заместителей на бензольном кольце, каждый из которых независимо представляет собой атом водорода, или атом галогена, или С1-4 алкокси группу, или ОН группу, или группу -O(СН2)nO-, в которой n=1 или 2, R2 представляет собой атом водорода или С1-4 алкильную группу, А и В независимо представляют собой либо атом азота, либо атом углерода, R3 представляет собой атом водорода или один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из: атома галогена, С1-4 алкильной группы, С1-4 алкоксигруппы, группы -O(СН2)nO-, в которой n=1 или 2, группы NO2, группы NHSO2R4, группы NHR5, ОН группы, C1-4 галогеноалкильной группы, CN группы, либо R3 составляет кольцо, конденсированное с бензольным кольцом, несущим его, выбранное из группы, состоящей из индола, бензимидазола, карбостирила, бензоксазолона и бензимидазолона, R4 представляет собой С1-4алкильную группу, или С1-4 диалкиламиногруппу, или С1-4 алкоксиалкильную группу, или С1-4 диалкиламиноалкильную группу, R5 представляет собой атом водорода, или С1-4 алкилкарбонильную группу, или С1-4 алкоксикарбонильную группу, и его фармацевтически приемлемым солям, а также к способам их получения и фармацевтическим композициям на их основе и к их применению в качестве лекарственного средства для расстройств центральной нервной системы, поскольку обладают свойствами дофаминергических лигандов D3. 4 н. и 13 з.п. ф-лы, 1 ил., 2 табл., 33 пр.

1. Соединение общей формулы 1
где:
- R1 представляет собой один или более чем один из идентичных или различных заместителей на бензольном кольце, каждый из которых независимо представляет собой атом водорода или атом галогена, или С1-4 алкокси группу, или ОН группу или группу -O(СН2)nO-, в которой n=1 или 2,
- R2 представляет собой атом водорода или С1-4 алкильную группу,
-А и В независимо представляют собой либо атом азота, либо атом углерода,
- R3 представляет собой атом водорода или один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из: атома галогена, С1-4 алкильной группы, С1-4 алкоксигруппы, группы -O(СН2)nO-, в которой n=1 или 2, группы NO2, группы NHSO2R4, группы NHR5, ОН группы, C1-4 галогеноалкильной группы, CN группы,
- либо R3 составляет кольцо, конденсированное с бензольным кольцом, несущим его, выбранное из группы, состоящей из индола, бензимидазола, карбостирила, бензоксазолона и бензимидазолона,
- R4 представляет собой С1-4алкильную группу, или С1-4 диалкиламиногруппу, или С1-4 алкоксиалкильную группу, или С1-4 диалкиламиноалкильную группу;
- R5 представляет собой атом водорода или С1-4 алкилкарбонильную группу, или С1-4 алкоксикарбонильную группу, а также его фармацевтически приемлемые соли.
2. Соединение по п.1, отличающееся тем, что R1 представляет собой один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из С1-4 алкокси группы, ОН группы и группы -O(СН2)nO-, в которой n=1 или 2.
3. Соединение по п.1, отличающееся тем, что R2 представляет собой атом водорода.
4. Соединение по п.1, отличающееся тем, что R3 представляет собой атом водорода, когда А и/или В представляет собой атом азота.
5. Соединение по п.1, отличающееся тем, что А и В одновременно представляют собой атом углерода.
6. Соединение по п.1, отличающееся тем, что R3 представляет собой один или более чем один из идентичных или различных заместителей, выбранных из группы, состоящей из: атома галогена, С1-4 алкокси группы, группы -O(СН2)nO-, в которой n=1 или 2, группы NHSO2R4, ОН группы и CN группы.
7. Соединение по п.1, отличающееся тем, что R3 вместе с бензольным кольцом, несущим его, представляет собой индольную группу, или бензимидазольную группу, или карбостирильную группу.
8. Соединение по п.1, отличающееся тем, что оно выбрано из приведенной ниже группы соединений:
-6,7-диметокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
-3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-бензонитрил
-3-{4-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
-3-{4-[4-(3-гидроксифенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
-6,7-диметокси-3-[4-(4-пиримидин-2-ил-пиперазин-1-ил)-бутил]-хромен-4-он
-6,7-диметокси-3-[4-(4-пиридин-2-ил-пиперазин-1-ил)-бутил]-хромен-4-он
-3-{4-[4-(2,3-дифторфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
-3-{4-[4-(1Н-бензимидазол-4-ил-)пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
-3-{4-[4-(1Н-индол-4-ил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
-5-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1Н-хинолин-2-он
-6,7-диметокси-3-{4-[4-(3-нитрофенил)-пиперазин-1-ил]-бутил}-хромен-4-он
-3-{4-[4-(3-аминофенил)-пиперазин-1-ил-]-бутил}-6,7-диметоксихромен-4-он
N-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид
-N-(3-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-ацетамид
-метил-(3-{4-[4-(6,7-Диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-фенил)-карбамат
-7-{4-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
-7-{4-[4-(2,3-дифторфенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
-7-{4-[4-(3-нитрофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
-7-{4-[4-(3-аминофенил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
-N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил-ацетамид
-N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-метансульфонамид
-N-(3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-этансульфонамид
-2-диметиламиноэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амид
-2-метоксиэтансульфоновой кислоты (3-{4-[4-(8-оксо-8Н-[1,3]диоксоло[4,5-g]хромен-7-ил)-бутил]-пиперазин-1-ил}-фенил)-амид
-7-{4-[4-(1Н-индол-4-ил)-пиперазин-1-ил]-бутил}-[1,3]диоксоло[4,5-g]хромен-8-он
-3-{4-[4-(3-трифторметилфенил)-пиперазин-1-ил]-бутил}-6,7-диметоксихромен-4-он
-6-метокси-3-[4-(4-фенилпиперазин-1-ил)-бутил]-хромен-4-он
-6-метокси-3-{4-[4-(2-метоксифенил)-пиперазин-1-ил]-бутил}-хромен-4-он
-6-метокси-3-{4-[4-(3-трифторметилфенил)-пиперазин-1-ил]-бутил}-хромен-4-он
-7-{4-[4-(2,3-дихлорфенил)пиперазин-1-ил-]-бутил}-6-метил-[1,3]диоксоло[4,5-g]хромен-8-он
-7-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-3Н-бензоксазол-2-он
4-{4-[4-(6,7-диметокси-4-оксо-4Н-хромен-3-ил)-бутил]-пиперазин-1-ил}-1,3-дигидробензимидазол-2-он.
9. Способ получения соединений общей формулы 1 по любому из пп. 1-8, отличающийся тем, что получают возможно замещенный хромон формулы 4 (X=Cl, Br, I), который подвергают взаимодействию с пиперазином формулы 5,
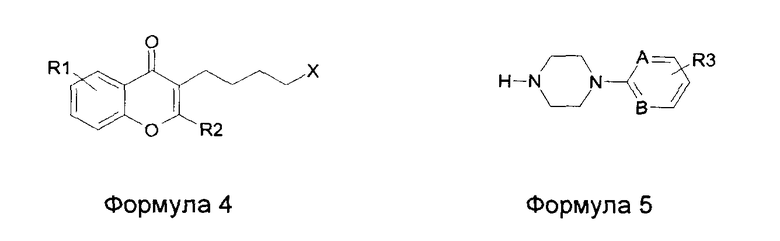
причем радикалы R1, R2, R3, А и В имеют значения, приведенные в п. 1.
10. Способ получения соединений общей формулы 1 по любому из пп.1-8, отличающийся тем, что возможно замещенное производное фенола формулы 6 получают, начиная с соединения формулы 3 (X=Cl, Br),

в условиях алкилирования в присутствии основания в растворителе и соединение формулы 6 затем подвергают взаимодействию с ДМФ (диметилформамидом), или диметилацеталем ДМФ, или ДМА (диметиламина), где значения радикалов R1, R3, А и В такие, как приведены в п. 1.
11. Способ получения соединений общей формулы 1 по п.10, отличающийся тем, что основание выбирают из К2СО3, Cs2CO3 или NEt3.
12. Способ получения соединений общей формулы 1 по п.10, отличающийся тем, что растворитель выбирают из ацетонитрила или метилэтилкетона.
13. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-8 или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, для лечения неврологических или психиатрических состояний.
14. Фармацевтическая композиция по п.13, где неврологическое или психиатрическое состояние выбрано из неврологического или психиатрического заболевания или расстройства, или эректильной дисфункции, или зависимости от лекарств и от веществ, вызывающих привыкание.
15. Соединение по п.1 для применения в качестве лекарственного средства для лечения неврологических или психиатрических состояний, в частности для лечения неврологического или психиатрического заболевания или расстройства, или эректильной дисфункции, или зависимости от лекарств и от веществ, вызывающих привыкание.
16. Соединение по п.15 для получения лекарственного средства для лечения неврологического или психиатрического заболевания или расстройства, или эректильной дисфункции, или зависимости от лекарств и от веществ, вызывающих привыкание.
17. Соединение по п.16, отличающееся тем, что неврологическое или психиатрическое заболевание или расстройство, или эректильная дисфункция, или зависимость от лекарств и от веществ, вызывающих привыкание, выбраны из группы, состоящей из: болезни Паркинсона, психоза, шизофрении, дискинезий, ассоциированных с болезнью Паркинсона, когнитивного дефицита, возможно ассоциированного с возрастом или с болезнью Альцгеймера, расстройства настроения, эссенциального тремора, тревоги, депрессии, биполярного расстройства, половой импотенции, преждевременной эякуляции, алкоголизма и никотиновой зависимости.
| Hackling Anneke et al, J | |||
| Medicinal Chemistry, 2003, v/46,no.18,p.3883-3899 | |||
| WO 2008009741 A1 24.01.2008 | |||
| Воздушная турбина | 1925 |
|
SU1683A1 |
| US 2005197343 A1 08.09.2005 | |||
| Bunini S et al, J.Medicinal Chemistry, 2009,v.52,no.1,p.151-169 | |||
| Micheli f et al, Expert Opinion on Therapeutic Patents, 2008,v.18,no.8,p.821-840 | |||
| RU 2007134584 A 27.03.2009 | |||

