Область техники, к которой относится изобретение
Настоящее изобретение относится к диариловым эфирам, которые являются ингибиторами белка NS5A, кодируемого вирусом гепатита C (HCV), и их применению для лечения или профилактики HCV.
Уровень техники
HCV представляет собой одноцепочечный вирус со смысловой РНК, принадлежащий к семейству флавивирусов рода гепатовирусов. Вирусный геном транслируется в единую открытую рамку считывания, которая кодирует множество структурных и неструктурных белков.
После первичной острой инфекции у большинства инфицированных индивидов развивается хронический гепатит, потому что HCV реплицируется предпочтительно в гепатоцитах, но не является непосредственно цитопатическим. В частности, отсутствие интенсивной ответной реакции T-лимфоцитов и большая предрасположенность вируса к мутированию, по-видимому, способствует высокой частоте хронической инфекции. Хронический гепатит может развиваться до фиброза печени, приводящего к циррозу, заболеванию печени последней стадии, и HCC (злокачественной гепатоме), что делает его основной причиной пересадки печени.
Существует шесть основных генотипов HCV и более чем 50 подтипов, которые разнообразно распределены географически. HCV генотип 1 представляет собой преобладающий генотип в Европе и США. Большая генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, вероятно объясняя сложности разработки вакцины и отсутствие ответной реакции на современную терапию.
Передача HCV может осуществляться через контакт с зараженной кровью или продуктами крови, например, после переливания крови или внутривенного введения лекарственных средств. Введение диагностических тестов, применяемых в отборе крови, привело к тенденции с понижением случаев заражения HCV после переливания. Однако, принимая во внимание медленное развитие до заболевания печени последней стадии, существующие в настоящее время инфекции будут продолжать представлять собой серьезное медицинское и экономическое бремя в течение десятков лет.
Современные методы терапии HCV основаны на (пегилированном) интерфероне-альфа (IFN-α) в комбинации с рибавирином. Данная комбинационная терапия дает устойчивый вирусологический ответ у 40% пациентов, инфицированных HCV генотипа 1, и у приблизительно 80% пациентов, инфицированных генотипами 2 и 3. Наряду с ограниченной эффективностью воздействия на HCV генотипа 1, данная комбинационная терапия обладает значительными побочными эффектами, включая гриппоподобные симптомы, гематологические нарушения и психоневрологические симптомы. Следовательно, существует необходимость в более эффективных, удобных и хорошо переносимых терапевтических методах.
Опыт с лекарственными средствами против ВИЧ, в частности с ингибиторами ВИЧ протеазы, показал, что недостаточно оптимальная фармакокинетика и комплексные режимы дозирования быстро приводят в результате к неумышленным нарушениям соблюдения предписанного режима терапии. Это в свою очередь значит, что 24-часовая минимальная концентрация препарата (минимальная концентрация в плазме) для соответствующих лекарственных средств при ВИЧ режиме часто падает ниже IC90 или ED90 предела в течение значительной части дня. Считают, что 24-часовая минимальная концентрация, по меньшей мере, IC50, и более реалистично, IC90 или ED90, является существенной для снижения скорости развития «ускользнувших от лекарственного средства» мутантов. Достижение необходимых фармакокинетических показателей и метаболизма лекарственного средства для обеспечения данных минимальных концентраций является неотложной проблемой в разработке лекарственных средств.
NS5A белок HCV расположен ближе к 3'-концу от NS4B белка и ближе к 5'-концу от NS5B белка. В результате посттрансляционного расщепления вирусной сериновой протеазой NS3, NS5A созревает до содержащего цинк, трехдоменного фосфопротеина, который существует в виде или гипофосфорилированного (56-кДа, p56), или гиперфосфорилированного типов (58-кДа, p58). NS5A HCV участвует во многих фазах жизненного цикла вируса, включая репликацию вируса и сборку инфекционной частицы, а также модулировании окружения клеток-хозяев. Хотя белку не приписывают ферментативную функцию, сообщают, что он взаимодействует с большим количеством вирусных и клеточных факторов.
Ряд патентов и патентных заявок описывают соединения с NS5A HCV ингибирующей активностью. WO2006/133326 описывает стильбеновые производные, тогда как WO2008/021927 описывает бифенильные производные, обладающие ингибирующей NS5A HCV активностью.
Существует необходимость в ингибиторах HCV, которые могут преодолеть недостатки современной терапии HCV, такие как побочные эффекты, ограниченная эффективность, возникновение резистентности и нарушение соблюдения предписанного режима, а также улучшить длительную ответную реакцию на вирусную нагрузку.
Настоящее изобретение относится к группе диариловых эфиров, ингибирующих HCV, с полезными свойствами, касающимися одного или более из следующих параметров: противовирусная эффективность, подходящий профиль развития устойчивости, отсутствие токсичности и генотоксичности, подходящие фармакокинетические и фармакодинамические параметры и легкость создания рецептур и введения.
Соединения настоящего изобретения могут также быть привлекательными, благодаря тому факту, что они не обладают активностью относительно других вирусов, в частности относительно ВИЧ. Пациенты, инфицированные ВИЧ, часто страдают от коинфицирования, такого как HCV. Лечение данных пациентов HCV ингибитором, который также ингибируют ВИЧ, может приводить к возникновению устойчивых ВИЧ штаммов.
Описание настоящего изобретения
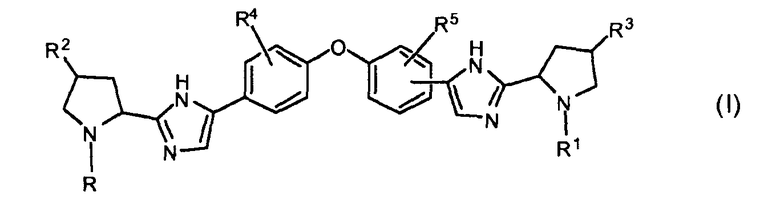
В одном аспекте настоящее изобретение относится к соединениям, которые могут быть представлены формулой I:
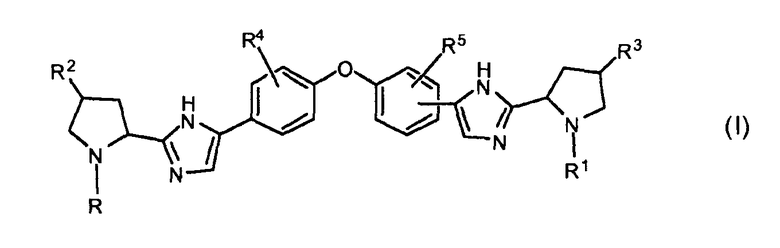
включая их любые возможные стереоизомеры, в которых:
R и R1, независимо друг от друга, представляют собой бензоил, необязательно замещенный одним, двумя или тремя заместителями, каждый из которых независимо выбирают из галогена и C1-C6алкила, или -C(=O)-Het, где Het необязательно замещен одним или двумя заместителями, независимо выбранными из C1-C4алкила, или группу формулы -C(=O)-CH(Rх)-R6, бензилоксикарбонил, C1-C6алкилоксикарбонил, группу формулы H2N-CH(R7)-C(=O)-, группу формулы R8-O-C(=O)-HN-CH(R7)-C(=O)- или -C(=O)-C(=O)-фенил;
R6 представляет собой C1-C4алкил, C3-C6циклоалкил, бензил или фенил, где фенил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из галогена, C1-C6алкила, метокси, трифторметокси, или два заместителя при соседних атомах кольца вместе с фенильным кольцом образуют бензодиоксол, и где C1-C4алкил необязательно замещен моно- или ди-C1-C4алкиламино, фенилсульфонилом, Het, и где бензил необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбирают из галогена, метокси;
Rx выбирают из водорода, гидрокси, C1-C6алкокси, амино, моно- или ди-C1-C6алкиламино, пирролидинила, пиперидинила, морфолинила, имидазолила, C1-C6алкилкарбониламино или C1-C6алкилоксикарбониламино;
Het представляет собой гетероциклическую группу, содержащую один или два гетероатома, выбранные из O и N, и содержащую 4-7 атомов в кольце, причем упомянутое гетероциклическое кольцо соединено с карбонильным атомом углерода через атом углерода кольца, и где по меньшей мере один из упомянутых гетероатомов является смежным с упомянутым атомом углерода кольца,
R2 и R3, независимо друг от друга, представляют собой водород, гидроксил, C1-C4алкил или галоген;
R4 и R5, независимо друг от друга, представляют собой водород, C1-C4алкил, галоген или метокси;
каждый R7 независимо представляет собой водород, фенил или C1-C4алкил, необязательно замещенный метокси или фенилом; и
R8 представляет собой C1-C4алкил или бензил;
или их фармацевтически приемлемой соли и/или сольвату.
В одном варианте осуществления настоящее изобретение относится к соединениям, которые могут быть представлены формулой I:
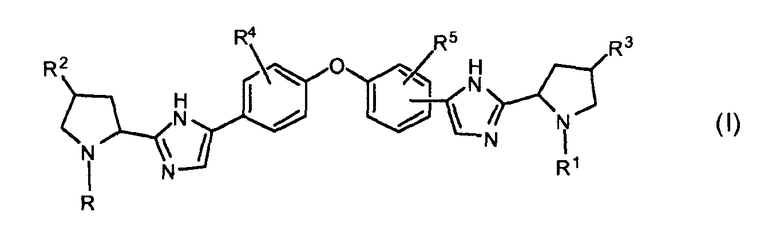
включая их любые возможные стереоизомеры, в которых:
R и R1, независимо друг от друга, представляют собой бензоил, необязательно замещенный одним, двумя или тремя заместителями, каждый из которых независимо выбирают из галогена и C1-C6алкила или -C(=O)-Het, или группу формулы -C(=O)-CH(Rх)-R6, бензилоксикарбонил, C1-C6алкилоксикарбонил, группу формулы H2N-CH(R7)-C(=O)- или группу формулы R8-O-C(=O)-HN-CH(R7)-C(=O)-;
R6 представляет собой C1-C4алкил или фенил, в котором фенил можно быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбирают из галогена и C1-C6алкила;
Rx выбирают из водорода, гидрокси, C1-C6алкокси, амино, моно- или ди-C1-C6алкиламино, пирролидинила, пиперидинила, морфолинила, C1-C6алкилкарбониламино или C1-C6алкилоксикарбониламино;
Het представляет собой гетероциклическую группу, содержащую один или два гетероатома, выбранные из O и N, и содержащую 4-7 атомов в кольце, где упомянутое гетероциклическое кольцо соединено с карбонильным атомом углерода через атом углерода кольца, и где по меньшей мере один из упомянутых гетероатомов является смежным с упомянутым атомом углерода кольца,
R2 и R3, независимо друг от друга, представляют собой водород, гидроксил, C1-C4алкил или галоген;
R4 и R5, независимо друг от друга, представляют собой водород, C1-C4алкил, галоген или метокси;
каждый R7 независимо представляет собой водород, C1-C4алкил или фенил; и
R8 представляет собой C1-C4алкил или бензил; или их фармацевтически приемлемой соли и/или сольвату.
В следующем аспекте настоящее изобретение относится к применению соединений формулы I, как описано в настоящем изобретении, для ингибирования HCV. Альтернативно, настоящее изобретение относится к применению для получения лекарственного средства соединения формулы I, как описано в настоящем изобретении.
Варианты осуществления настоящего изобретения относятся к соединениям формулы (I), или любой их подгруппе, как определено в настоящем изобретении, в которых применяют одно или более из определений для R, R1, R2, R3, R4, R5, R6, R7, R8, Rx и/или Het, как описано в настоящем изобретении.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых R и R1 представляют собой бензилкарбонил или изобутилоксикарбонил, в частности в которых R и R1 представляют собой бензилкарбонил.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых R и R1, независимо друг от друга, могут представлять собой группы формулы -C(=O)-CH(Rх)-R6 или -C(=O)-Het, в частности в которых R и R1, независимо друг от друга, представляют собой группы формулы -C(=O)-CH(Rх)-R6.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых Rx представляет собой гидрокси, C1-C6алкокси, амино, моно- или ди-C1-C6алкиламино, пирролидинил, пиперидинил, морфолинил, C1-C6алкилкарбониламино или C1-C6алкилоксикарбониламино; или в которых Rx представляет собой гидроксил, амино, ди-C1-C4алкиламино или морфолинил; или в которых Rx представляет собой гидрокси, амино или диметиламино.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых R6 представляет собой фенильное кольцо, необязательно замещенное одним галогеном или C1-C6алкилом; или в которых фенильное кольцо не замещают. Альтернативно, R6 выбирают из фенила и изопропила.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых Het, как определено выше, содержит 4, 5 или 6 атомов в кольце; или в которых Het выбирают из 2-пиридинила, 2-пиримидила, 2-пиразинила, 2-имидазолила, 2-тиазолила, 2-тиофенила, 2-пиразолинила, 2-пиперидинила, 2-пирролидинила, 2-пирролила, 2-фуранила, 2-тетрагидрофуранила, 2-оксетанила, 2- или 3-морфолинила и 2-пиперазинила, в частности 2-тетрагидрофуранила.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых R2 и R3 независимо представляют собой водород, гидрокси или фтор, в частности в которых R2 и R3 представляют собой водород.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых R4 и R5 представляют собой водород, метил, метокси или хлор, в частности в которых R4 и R5 представляют собой водород.
Подгруппами соединений формулы I являются подгруппы соединений формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых R8 представляет собой метил.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых группу
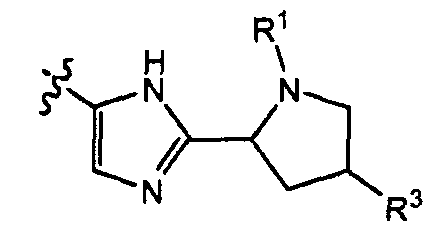
замещают в мета- или пара-положении, относительно кислородного мостика между двумя фенильными группами, в частности, соединения формулы I, как определено в настоящем изобретении, или их подгруппы, в которых упомянутая группа находится в мета-положении.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, в которых группа -HN-CH(R7)-C(=O)- как часть формулы H2N-CH(R7)-C(=O)- или формулы R8-O-C(=O)-HN-CH(R7)-C(=O)-, образует аминокислотный остаток, выбранный из валина (Val), лейцина (Leu), фенилаланина (Phe), MeO-треонина или фенилглицина. Особенно интересными являются L-аминокислотные остатки, такие как L-Val, L-Leu, L-Phe и L-MeO-треонин.
Подгруппами соединений формулы I являются те соединения формулы I, или подгруппы соединений формулы I, как определено в настоящем изобретении, имеющие структурную формулу Y
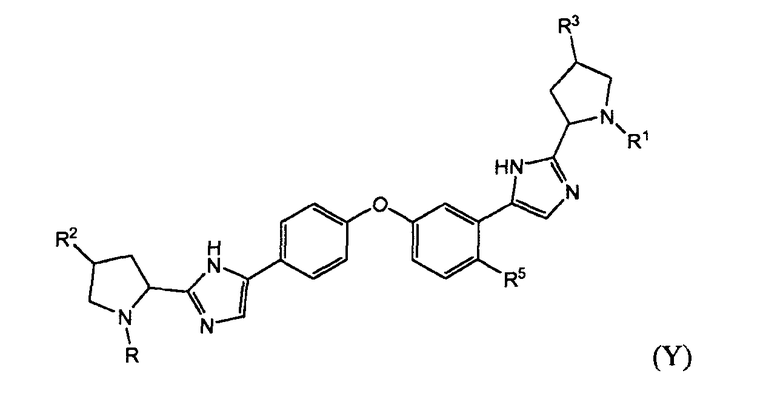
в которой R, R1, R2, R3, R5, R6, R7, R8, Rx и/или Het такие, как описано в настоящем изобретении.
В следующем аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, гидрату или сольвату, для применения для лечения или профилактики (или получения лекарственного средства для лечения или профилактики) HCV инфекции. Примерные HCV генотипы в контексте лечения или профилактики согласно настоящему изобретению включают генотип 1b (преобладающий в Европе) или 1a (преобладающий в Северной Америке). Настоящее изобретение также относится к способу лечения или профилактики HCV инфекции, в частности генотипа 1a или 1b.
Чистые стереоизомерные формы соединений и промежуточных соединений, как упоминается в настоящем изобретении, определяют как изомеры, практически не содержащие другие энантиомерные или диастереомерные формы той же самой основной молекулярной структуры упомянутых соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным соединениям, имеющим стереоизомерный избыток, по меньшей мере, 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров) вплоть до 100% количества стереоизомерного избытка (т.е. 100% одного изомера и отсутствие других), более в частности, соединениям или промежуточным соединениям, имеющим стереоизомерный избыток в количестве от 90% вплоть до 100%, даже более в частности, имеющим стереоизомерный избыток в количестве от 94% вплоть до 100% и самое конкретное, имеющим стереоизомерный избыток в количестве от 97% вплоть до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" следует понимать аналогичным способом, но по отношению к энантиомерам и диастереомерам, соответственно, рассматриваемой смеси.
Чистые стереоизомерные формы или стереоизомеры соединений и промежуточных соединений настоящего изобретения можно получить применением хорошо известных методик. Например, энантиомеры можно отделять друг от друга избирательной кристаллизацией их диастереомерных солей с оптически активными кислотами или основаниями. Их примерами являются винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота и камфорсульфоновая кислота. Альтернативно, энантиомеры можно разделять хроматографическими методами, применяя хиральные неподвижные фазы. Упомянутые стереохимически чистые изомерные формы можно также получить из соответствующих чистых стереоизомерных форм подходящих исходных веществ при условии, что реакция протекает стереоспецифически. Предпочтительно, если требуется конкретный стереоизомер, получать упомянутое соединение стереоспецифическими способами получения. В данных способах предпочтительно применять энантиомерно чистые исходные вещества.
Диастереомерные рацематы соединений формулы I можно получить отдельно общепринятыми способами. Подходящими физическими способами разделения, которые можно преимущественно применять, являются, например, избирательная кристаллизация и хроматография, например, колоночная хроматография или хроматография со сверхкритической подвижной фазой.
Соединения формулы I имеют несколько центров хиральности. Интерес представляют стереогенные центры пирролидинового кольца при 2-атоме углерода. Конфигурация в данном положении может представлять собой конфигурацию, соответствующую L-пролину, т.е.
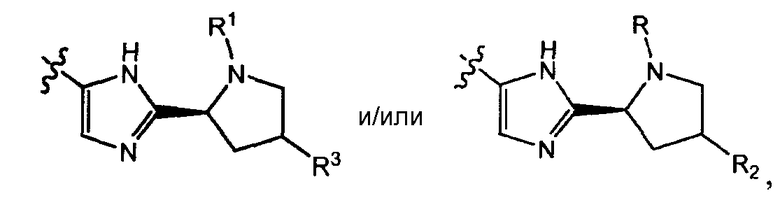
или конфигурацию, соответствующую D-пролину, т.е.
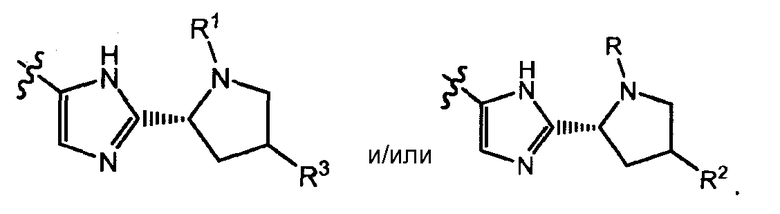
Также интерес представляет стереохимия при атоме углерода, замещенном Rx и R6 в группах формулы -C(=O)-CH(Rх)-R6, и/или аминокислотных остатков, как определено формулой H2N-CH(R7)-C(=O)- или формулой R8-O-C(=O)-HN-CH(R7)-C(=O)-.
Фармацевтически приемлемые соли присоединения включают терапевтически активные нетоксичные солевые формы присоединения кислоты или основания соединений формулы (I) или их подгруппы. Интерес представляют свободные, т.е. несолевые формы соединений формулы I, или любой подгруппы соединений формулы I, упоминаемых в настоящем изобретении.
Фармацевтически приемлемые соли присоединения кислоты можно удобно получить обработкой основной формы подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлороводородная или бромоводородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая (т.е. этандиовая), малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная (т.е. гидроксилбутандиовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. В свою очередь, упомянутые солевые формы можно превратить обработкой подходящим основанием в свободную основную форму.
Соединения формулы (I), содержащие кислый протон, можно также превратить в их соли присоединения основания, в частности солевые формы присоединения металла или амина, обработкой подходящим органическим и неорганическим основанием. Подходящие солевые формы присоединения основания включают, например, аммониевые соли, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и подобные, соли с органическими основаниями, например, бензатином, N-метил-D-глюкамином, гидрабаминовые соли, и соли с аминокислотами, например, аргинином, лизином и подобными.
Термин "сольваты" включает все фармацевтически приемлемые сольваты, которые соединения формулы I, а также их соли, способны образовывать. Данными сольватами являются, например, гидраты, алкоголяты, например этанолаты, пропанолаты и подобные.
Некоторые из соединений формулы I могут также существовать в таутомерных формах. Например, таутомерными формами амидных (-C(=O)-NH-) групп являются иминоспиртовые группы (-C(OH)=N-). Предполагается, что таутомерные формы, хотя явно не показаны в структурных формулах, представленных в настоящем изобретении, включены в объем настоящего изобретения.
Как применяют в настоящем изобретении, "C1-C4алкил", в качестве группы или части группы, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, содержащим 1-4 атома углерода, таким как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил, 2-метил-2-пропил. "C1-C6алкил" включает C1-C4алкильные группы и их высшие гомологи, содержащие 5 или 6 атомов углерода, такие как, например, 1-пентил, 2-пентил, 3-пентил, 1-гексил, 2-гексил, 2-метил-1-бутил, 2-метил-1-пентил, 2-этил-1-бутил, 3-метил-2-пентил, и подобные. Среди C1-C6алкилов представляет интерес C1-C4алкил.
"C1-C6алкокси" или "C1-C6алкилокси" относится к группам формулы -O-C1-C6алкил, в которых C1-C6алкил представляет собой, как определено выше. Примерами C1-C6алкокси являются метокси, этокси, н-пропокси или изопропокси. Среди C1-C6алкокси особый интерес представляют C1-C4алкокси, т.е. группы формулы -O-C1-C4алкил, в которых C1-C4алкил представляет собой как определено выше.
Термин " C3-C6циклоалкил" является общим для циклопропила, циклобутила, циклопентила и циклогексила.
Термин "галоген" является общим для фтора, хлора, брома и йода.
Как применяют в настоящем изобретении, термин "(=O)" или "оксо" образует карбонильную группу при присоединении к атому углерода. Следует отметить, что атом можно замещать только оксогруппой, когда это позволяет валентность атома.
Как применяют в настоящем изобретении с целью определить Het, гетероциклическая группа может содержать один или два гетероатома, выбранные из O и N и содержать 4, 5, 6 или 7 кольцевых атомов.
Когда положение группы в молекуле не указано (например, заместитель при фениле) или представлено «плавающей» связью, данная группа может располагаться при любом атоме данной группы при условии, что полученная в результате структура является химически стабильной. Когда любая переменная присутствует более чем один раз в молекуле, каждое ее значение является независимым.
При применении в настоящем изобретении подразумевается, что термин "соединения формулы I", или "настоящие соединения" или аналогичные термины, включает соединения формулы I, включая возможные стереоизомерные формы и их фармацевтически приемлемые соли и сольваты.
Общие способы получения
Соединения формулы (I), в которых R и R1 имеют одинаковые значения, причем упомянутые соединения представлены формулой (Ia), можно получить реакцией биспирролидинилового производного формулы (II) с промежуточным соединением R-W или R1-W в реакции образования амида и карбамата, как показано на схеме 1. Для реакции образования амида W представляет собой гидрокси или активирующую группу, и для реакции образования карбамата W представляет собой активирующую группу. Активирующими группами являются галогенангидриды, в частности хлорангидриды, смешанные ангидриды или активированные эфиры.
Схема 1
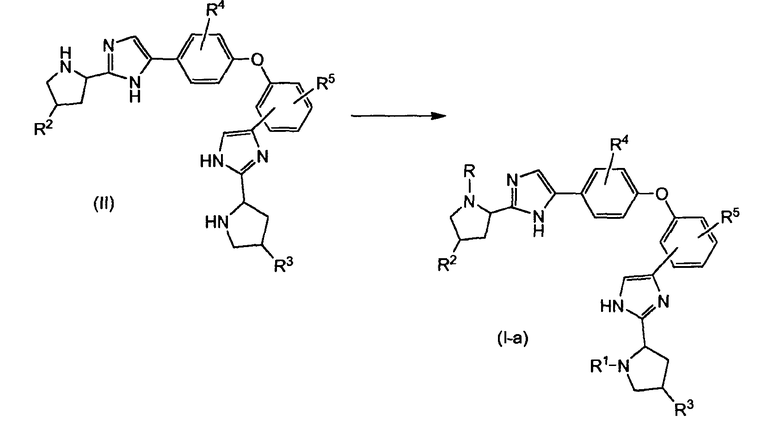
В вышеуказанной и следующих реакционных схемах, α-атом углерода в пирролидиновых кольцах может быть рацемическим или может иметь одну из стереохимических конфигураций (R или S).
Реакция образования амида включает реакцию исходных веществ, т.е. производного карбоновой кислоты и биспирролидинила (II), например, фенилуксусной кислоты, миндальной кислоты, толилуксусной кислоты, с амид-конденсирующим реагентом в реакционно-инертном растворителе, необязательно в присутствии основания. Растворители, которые можно применять, включают углеводороды, такие как дихлорметан (DCM) или хлороформ, простые эфиры, такие как тетрагидрофуран (THF) или 2-метилтетрагидрофуран (MeTHF), углеводородные растворители, такие как толуол или ксилол, диполярные апротонные растворители, такие как DMF, DMA, ацетонитрил, или их смеси. Амид-конденсирующие реагенты включают такие агенты, как N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ), N-изопропоксикарбонил-2-изопропокси-1,2-дигидрохинолин, в частности его хлороводородная соль, (IIDQ), гексафторфосфат N,N,N,N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (HATU), гексафторфосфат бензотриазол-1-илокси-трис-пирролидинофосфония (имеется в продаже под названием PyBOP®), 1,1'-карбонилдиимидазол (CDI), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDI или EDCI), а также его хлороводородная соль, дициклогексилкарбодиимид (DCC) или 1,3-диизопропилкарбодиимид, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония (HBTU) и подобные. Можно добавлять катализатор, например, 1-гидроксибензотриазол (HOBt) или 4-диметиламинопиридин (DMAP). Реакцию обычно проводят в присутствии основания, в частности аминового основания, такого как третичный амин, например, триэтиламин, N-метилморфолин, N,N-диизопропилэтиламин (последний также называют основание Хюнига, DIPEA или DIEA). В одном варианте осуществления реакцию проводят в DMF с HATU в присутствии N,N-диизопропилэтиламина в качестве основания.
Амиды можно также получить реакцией активированной кислоты, такой как галогенангидрид карбоновой кислоты, в частности хлорангидрид, или смешанного ангидрида или активированного эфира с (II). В активированных эфирах W представляет собой арилоксигруппу, такую как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и подобные; или W может представлять собой остаток смешанного ангидрида, т.е. W представляет собой -O-CO-Z или -O-CO-OZ, причем Z в последней группе представляет собой C1-4алкил, такой как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, или Z представляет собой бензил.
Реакции образования карбамата можно проводить, применяя ряд способов, в частности реакцией биспирролидинилового производного формулы (II) с алкилхлорформиатами; хотя и не предпочтительно, реакцией спиртов с карбамоилхлоридами или изоцианатами; с помощью реакций, включающих комплексы металлов или агенты, переносящие ацильную группу. Монооксид углерода и определенные металлические катализаторы можно применять для получения карбаматов. Можно применять в качестве катализаторов металлы, такие как палладий, иридий, уран и платину.
Один подход для получения карбаматов включает применение промежуточных соединений формулы R'-O-CO-Q, в которых R' представляет собой C1-C6алкил или бензил, и Q представляет собой уходящую группу, такую как галоген, в частности хлор и бром, или группы, применяемые в активированных группах для образования амидной связи, такие как те, что упоминались выше. Промежуточные соединения R'-O-CO-Q можно получить из спиртов R'-OH и фосгена, таким образом, получая хлорформиат, или переносом хлора в последнем к другим активным группам.
Реакции с активированными кислотами или реакции образования карбамата можно проводить, применяя аналогичные условия реакции, как описано выше для реакций с амид-конденсирующими реагентами.
Биспирролидиниловые производные формулы (II), в которых одна из пирролидиноимидазолильных групп находится в мета-положении, называемые в настоящем изобретении (II-m), можно получить, как показано на следующей реакционной схеме.
Схема 2
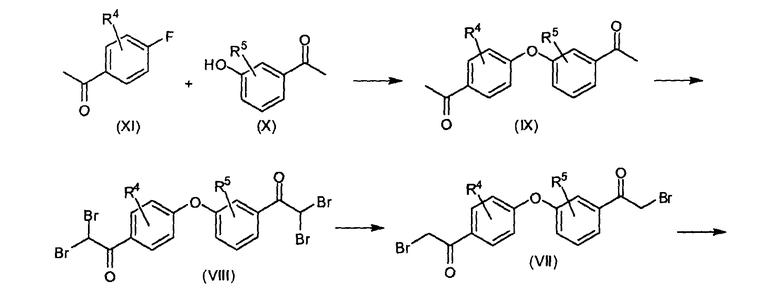
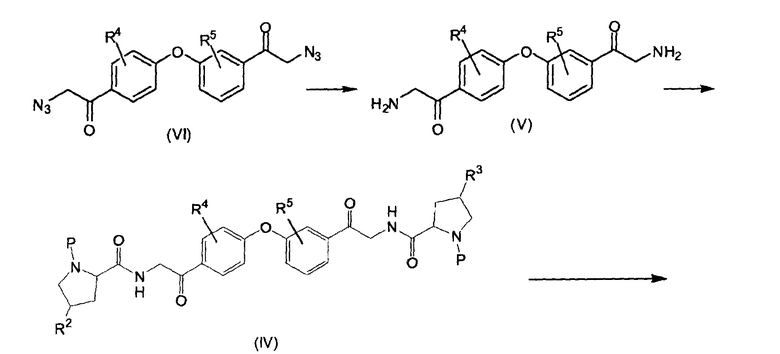
1-(3-(4-ацетилфенокси)фенил)этаноновое производное (IX) можно получить образованием бисфенилэфирной группы реакций ароматического замещения 1-(3-гидроксифенил)этанонового производного (X) на 1-(4-фторфенил)этаноновое производное (XI) в присутствии основания, такого как карбонат калия. Упомянутое соединение (IX) бромируют бромом до 2,2-дибром-1-(3-(4-(2,2-дибромацетил)фенокси)фенил)этанонового производного (VIII). Последнее можно превратить в 2-бром-1-(3-(4-(2-бромацетил)фенокси)фенил)этаноновое производное (VII) реакцией с фосфитом, например, диэтилфосфитом, после чего атомы брома можно заместить азидогруппами, применяя азидную соль, такую как азид натрия. Азидные группы в полученном в результате бисазидном соединении (VI) можно восстановить водородом в присутствии катализатора на основе благородного металла, например, водородом в присутствии Pd, получая 2-амино-1-(3-(4-(2-аминоацетил)фенокси)фенил)этаноновое производное (V). Последнее можно конденсировать с N-защищенным пролиновым производным, получая феноксифенильное производное (IV), в котором P представляет собой амино-защитную группу, например, трет-бутилоксикарбонил (BOC).
На следующей стадии (IV) циклизуют с ацетатом аммония до промежуточного соединения (III), в котором P представляет собой, как определено выше. Удаление P, например, когда P представляет собой BOC, реакцией с кислотой, например, с водной HCl, дает (II-m). Последнее можно применять в качестве исходного вещества для получения различных соединений формулы (I) введением ацильной или карбаматной групп.
Альтернативно, соединение (VII) можно превратить в соединение (III) аналогичными способами, как применяют для превращения соединения (XIV) (схема 3) в соединение (XII), как, например, описано в примере 17.
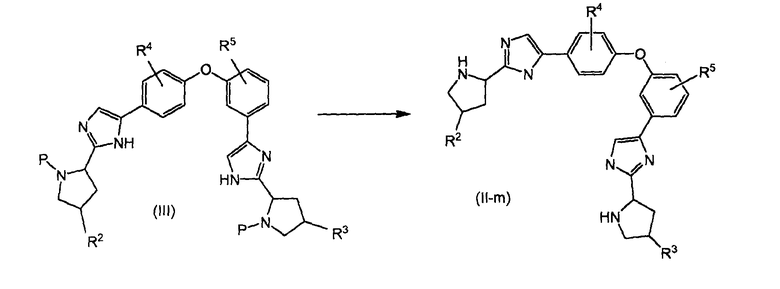
Биспирролидинильные производные формулы (II), в которых обе пирролидиноимидазолильные группы находятся в пара-положении относительно друг друга, называемые в настоящем изобретении (II-p), можно получить, как показано на реакционной схеме 3.
Схема 3

1,1'-(4,4'-оксибис(4,1-фенилен))бис(2-хлорэтаноновое) производное (XIV) можно получить проведением реакции ацилирования Фриделя-Крафтса с диарилэфиром (XV) в присутствии кислоты Льюиса, такой как хлорид алюминия, и хлорангидрида кислоты, возможно применяя дисульфид углерода в качестве растворителя, предпочтительно нагретого до температуры кипения. Ацилирование хлорацетилхлоридом приводит в результате к альфа-галогенкетону (XIV). Полученный в результате галогенированный кетон реагирует с аминокислотой, такой как Boc-L-пролин для получения эфира, применяя растворимое органическое основание, такое как триэтиламин или диизопропилэтиламин. Затем эфирный продукт (XIII) циклизуют, применяя ацетат аммония, до промежуточного соединения (XII). Циклизацию можно осуществлять в плотно закрытой пробирке вместе с источником аммиака в избытке, например, ацетатом аммония, и нагревать стандартным способом или в микроволновой печи для получения промежуточного соединения (XII). Удаление P, например, когда P представляет собой BOC, деблокированием кислотой, например водной HCl, дает (II-p).
Соединения формулы (I), в которой R и R1 имеют различные значения, представленные в настоящем изобретении формулой (I-b), можно получить, как показано на реакционной схеме 4.
Схема 4
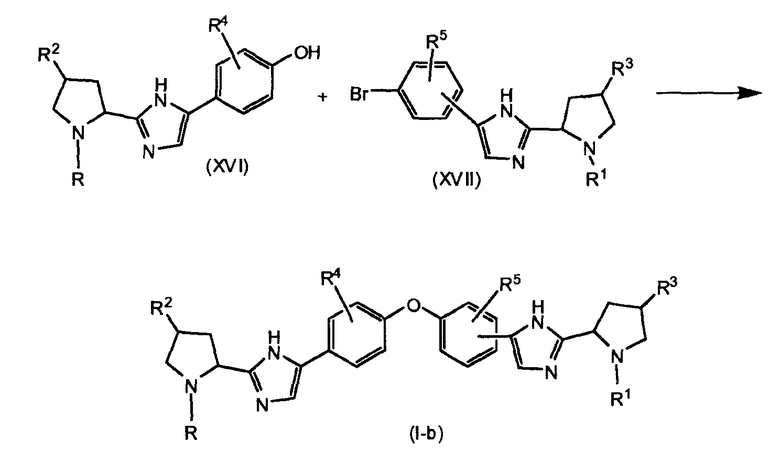
R и R1 в (I-b) представляют собой различные группы, имеющие значения, определенные выше.
Реакцию (XVI) с (XVII) проводят с катализатором на основе переходного металла, в частности Pd° комплекса, такого как тетракистрифенилфосфин палладия, или соли меди(II), такой как трифлат меди(II), или Cu°. Промежуточные соединения формулы (XVI) можно получить из соответствующего защищенного фенола, например, соответствующего метоксианалога, который деметилируют, применяя, например, трибромид бора (BBr3). Соответствующий метоксианалог (XVIa) можно получить, как показано на схеме 5. Соединение формулы (XVIa) можно получить, исходя из соединения формулы (XVIII), применяя тот же тип методик, как описано выше для стадий превращения соединения (IX) в соединение (III).
Схема 5
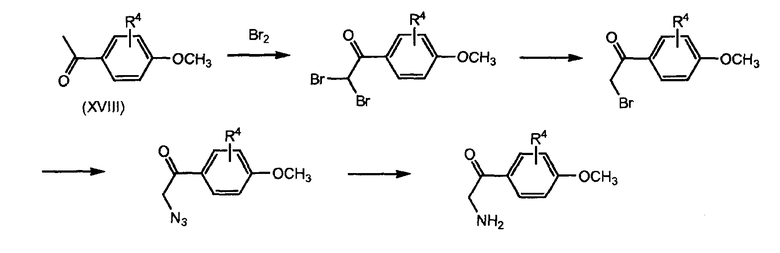
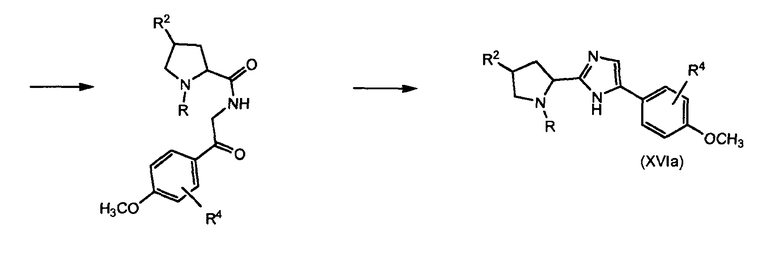
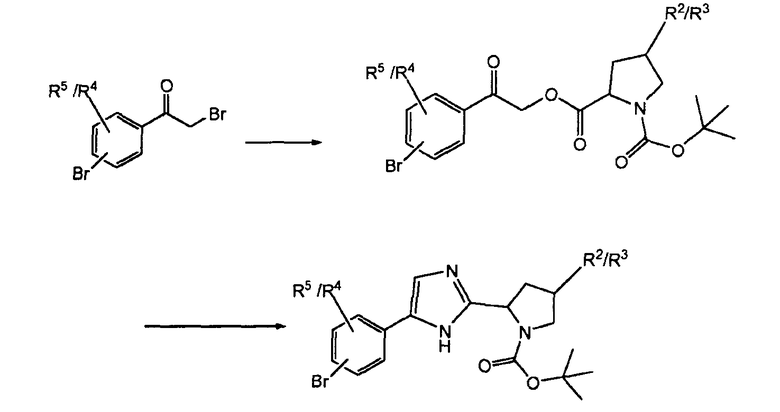
Схема 6 иллюстрирует то, что промежуточные соединения (XVII) можно получить из бром-, бромацетилфенильного производного, применяя аналогичные методики, как описано для превращения соединения (XIV) в соединение (XII).
Схема 6
В следующем аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим терапевтически эффективное количество соединения формулы I, как описано в настоящем изобретении, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в данном контексте представляет собой количество, достаточное для действия профилактическим способом против HCV инфекции, для стабилизации или ослабления HCV инфекции, у инфицированных субъектов или субъектов, которые рискуют быть инфицированными. В еще следующем аспекте настоящее изобретение относится к способу получения фармацевтических композиций, как описано в настоящем изобретении, который включает тщательное смешение фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I, как описано в настоящем изобретении.
Следовательно, соединения настоящего изобретения или любую их подгруппу можно формулировать в виде различных фармацевтических форм для целей введения. В качестве подходящих композиций можно упомянуть все композиции, обычно применяемые для системного введения лекарственных средств. Для получения фармацевтических композиций настоящего изобретения эффективное количество конкретного соединения, необязательно в форме соли присоединения или металлического комплекса, в качестве активного ингредиента, смешивают до тонко перемешанной смеси с фармацевтически приемлемым носителем, где носитель принимает большое разнообразие форм в зависимости от формы препарата, требуемого для введения. Желательно, чтобы данные фармацевтические композиции были в единичной лекарственной форме, подходящей, в частности, для введения перорально, ректально, подкожно или парентеральной инъекцией. Например, при получении композиций в виде пероральной лекарственной формы, можно применять любую из пригодных фармацевтических сред, такую как, например, вода, гликоли, масла, спирты и подобные, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие, разрыхлители и подобные, в случае порошков, пилюль, капсул и таблеток. Благодаря легкости их введения, таблетки и капсулы представляют собой наиболее предпочтительные пероральные стандартные лекарственные формы, и в этом случае, безусловно, применяют твердые фармацевтические носители. Для парентеральных композиций носитель будет обычно содержать стерильную воду, по меньшей мере, в значительной степени, хотя можно включать другие ингредиенты, например, для того чтобы способствовать растворению. Например, можно получить инъецируемые растворы, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Можно также получить инъецируемые суспензии, в которых можно применять подходящие жидкие носители, суспендирующие агенты и подобные. Также включенными являются препараты в твердой форме, предназначенные для превращения, незадолго до применения, в препараты в жидкой форме. В композициях, пригодных для подкожного введения, носитель необязательно содержит агент, улучшающий проникновение, и/или подходящий увлажняющий агент, необязательно смешанный с подходящими добавками любой природы в меньших количествах, где добавки не оказывают значительного вредного воздействия на кожу. Соединения настоящего изобретения можно также вводить пероральной ингаляцией или инсуфляцией в форме раствора, суспензии или сухого порошка, применяя любую хорошо известную систему доставки.
Особенно предпочтительно формулировать вышеупомянутые фармацевтические композиции в стандартных лекарственных формах для простоты введения и однородности дозирования. Стандартная лекарственная форма, как применяют в настоящем изобретении, относится к физически дискретным единицам, подходящим в качестве единичных доз, причем каждая единичная доза содержит предварительно определенное количество активного ингредиента, рассчитанное для получения требуемого терапевтического эффекта, вместе с требуемым фармацевтическим носителем. Примерами данных стандартных лекарственных форм являются таблетки (включая таблетки с рисками или таблетки с покрытием), капсулы, пилюли, суппозитории, пакеты с порошком, капсулы, инъецируемые растворы или суспензии и подобные, и их отдельные соотношения.
Соединения формулы I обладают активностью против HCV, и их можно применять для лечения и профилактики HCV инфекции или заболеваний, связанных с HCV. Последние включают прогрессирующий фиброз печени, воспаление и некроз, ведущие к циррозу, заболеванию печени последней стадии, и HCC. Более того, считают, что ряд соединений настоящего изобретения являются активными против мутировавших штаммов HCV. Кроме того, соединения настоящего изобретения могут иметь подходящий фармакокинетический профиль и привлекательные свойства с точки зрения биодоступности, включая приемлемое время полувыведения, AUC (площадь под кривой) и пиковые величины, и не вызывать неблагоприятные явления, такие как недостаточно быстрое начало действия и задержка в ткани.
Противовирусную активность in vitro против HCV соединений формулы I можно исследовать в клеточной HCV системе репликона согласно Lohmann et al. (1999) Science 285:110-113, с дополнительными изменениями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624 (введенные в настоящее изобретение с помощью ссылки), которые дополнительно иллюстрируются в разделе с примерами. Данная модель, при том что она является неполноценной инфекционной моделью HCV, широко применяется в качестве самой надежной и эффективной модели независимой HCV РНК репликации, доступной в настоящее время. Ясно, что важно отличать соединения, которые специфично препятствуют HCV функциям от соединений, которые оказывают цитотоксический или цитостатический эффекты в HCV модели репликона, и как следствие вызывают снижение количества HCV РНК или связанной с ней концентрации репортерного фермента. Анализы являются известными в данной области техники для оценки клеточной цитотоксичности на основе, например, активности митохондриальных ферментов, применяя флуорогенные красители, такие как резазурин. Кроме того, существуют клеточные обратные скрининги для оценки неселективного ингибирования связанной активности репортерного гена, такой как люцифераза светлячка. Подходящие клеточные типы можно снабдить стабильной трансфекцией люциферазным репортерным геном, чья экспрессия зависит от конститутивно активного генного промотора, и данные клетки можно применять в качестве обратного скрининга для исключения неселективных ингибиторов.
Благодаря их противовирусной активности, в частности их анти-HCV свойствам, соединения формулы I, как описано в настоящем изобретении, являются пригодными для ингибирования HCV репликации, в частности для лечения теплокровных животных, в частности людей, инфицированных HCV, и для профилактики HCV инфекций. Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, в частности человека, инфицированного HCV, или рискующего быть инфицированным HCV, причем упомянутый способ включает введение анти-HCV эффективного количества соединения формулы I, как описано в настоящем изобретении.
Следовательно, соединения формулы I, как описано в настоящем изобретении, можно применять в качестве лекарственного средства, в частности анти-HCV лекарственного средства. Упомянутое применение в качестве лекарственного средства или способ лечения включает системное введение субъектам, инфицированным HCV, или субъектам, подверженным HCV инфекции, количества, эффективного для борьбы с заболеваниями, связанными с HCV инфекцией.
Настоящее изобретение также относится к применению настоящих соединений для получения лекарственного средства для лечения или предотвращения HCV инфекции.
В общем, предполагается, что эффективное ежедневное противовирусное количество будет составлять от приблизительно 0,01 до приблизительно 50 мг/кг, или от приблизительно 0,02 до приблизительно 30 мг/кг веса тела. Может быть уместно вводить требуемую дозу в виде двух, трех, четырех или более поддоз через подходящие интервалы в течение дня. Упомянутые поддозы можно формулировать в виде стандартных лекарственных форм, например, содержащих от приблизительно 1 до приблизительно 500 мг, или от приблизительно 1 до приблизительно 300 мг, или от приблизительно 1 до приблизительно 100 мг, или от приблизительно 2 до приблизительно 50 мг активного ингредиента на стандартную лекарственную форму.
Настоящее изобретение также относится к комбинации соединения формулы I, его фармацевтически приемлемой соли или сольвата, и другого противовирусного соединения, в частности другого анти-HCV соединения. Термин "комбинация" может относиться к продукту, содержащему (a) соединение формулы I, как указано выше, и (b) необязательно другое анти-HCV соединение, в виде комбинированного препарата для одновременного, раздельного или последовательного применения при лечении HCV инфекций.
Следующие примеры предназначены для того, чтобы проиллюстрировать настоящее изобретение, и не следует истолковывать их как ограничивающие его объем.
Примеры получения
Пример 1: 1-(3-(4-ацетилфенокси)фенил)этанон
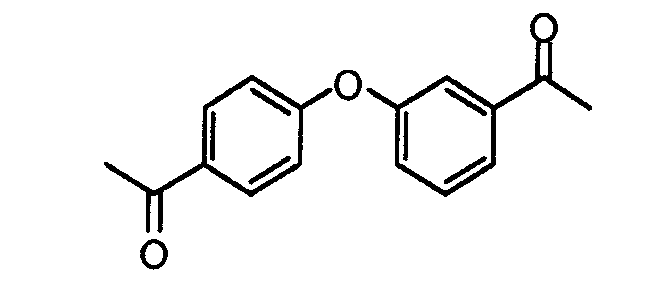
Смесь 1-(3-гидроксифенил)этанона (8 г, 58,7 ммоль), 1-(4-фторфенил)этанона (8,1 г, 58,7 ммоль) и безводного карбоната калия (16,2 г, 117,5 ммоль) в DMSO (150 мл) перемешивали в течение 16 часов при 140°C. Смесь распределяли между этилацетатом и водой. Водный слой подкисляли 1M HCl до pH 6-7 и экстрагировали этилацетатом. Объединенные органические фракции промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Неочищенный продукт хроматографировали на силикагеле (этилацетат/гептан: 1/1), получая в результате 1-(3-(4-ацетилфенокси)фенил)этанон (11 г, 74%) в виде белого твердого остатка.
LC/MS: m/z=256,2 (M+1)+. Точная масса: 255,1.
Пример 2: 2,2-дибром-1-(3-(4-(2,2-дибромацетил)фенокси)фенил)этанон
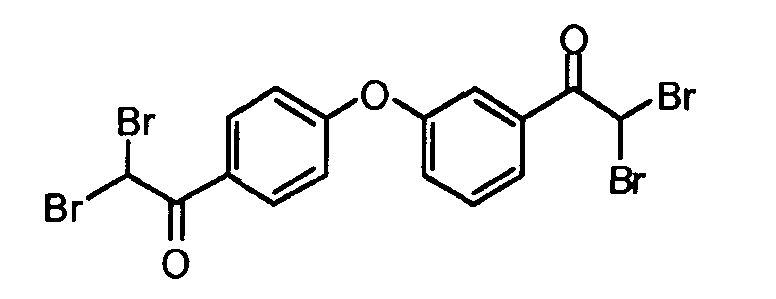
К раствору 1-(3-(4-ацетилфенокси)фенил)этанона (11 г, 43,2 ммоль) в хлороформе (300 мл) добавляли по каплям бром (6,6 мл, 129,7 ммоль). Реакцию перемешивали в течение 4 часов при 80°C, охлаждали до комнатной температуры и концентрировали при пониженном давлении. Смесь распределяли между этилацетатом и водой. Водный слой экстрагировали этилацетатом, и объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученное в результате неочищенное вещество очищали флэш-хроматографией (силикагель: EtOAc/гептан: 3/7) для получения 2,2-дибром-1-(3-(4-(2,2-дибромацетил)фенокси)фенил)этанона в виде твердого остатка желтого цвета (16 г, 65%).
Пример 3: 2-бром-1-(3-(4-(2-бромацетил)фенокси)фенил)этанон
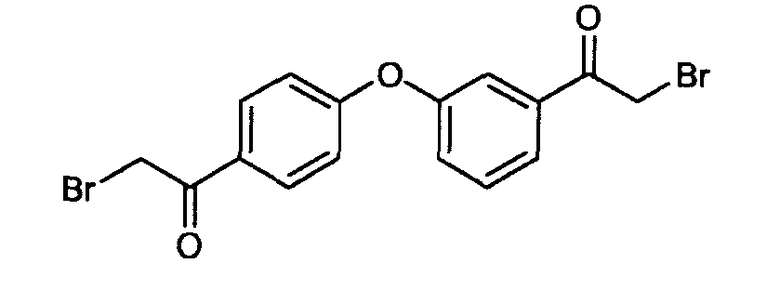
К раствору 2,2-дибром-1-(3-(4-(2,2-дибромацетил)фенокси)фенил)этанона (16 г, 28 ммоль) в тетрагидрофуране (300 мл) при 0°C добавляли триэтиламин (3,6 мл) и диэтилфосфит (10,7 мл, 83,2 ммоль). Реакцию постепенно нагревали до комнатной температуры, и смесь перемешивали в течение 30 минут и концентрировали при пониженном давлении. Смесь распределяли между этилацетатом и водой. Водный слой экстрагировали EtOAc, и объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученное в результате неочищенное вещество очищали флэш-хроматографией (силикагель: EtOAc/гептан: 3/7) для получения 2-бром-1-(3-(4-(2-бромацетил)фенокси)фенил)этанона (10,2 г, 88%) в виде желтого масла.
Пример 3a: 2-бром-1-(4-(3-(2-бромацетил)-4-метоксифенокси)фенил)этанон
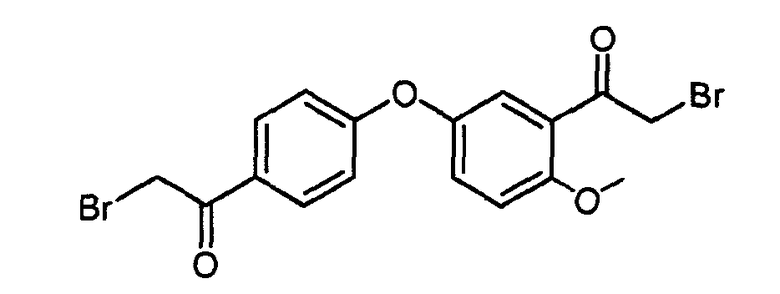
2-бром-1-(4-(3-(2-бромацетил)-4-метоксифенокси)фенил)этанон получали, применяя аналогичную методику, как описано в примере 1-3 с 1-(5-гидрокси-2-метоксифенил)этаноном и 1-(4-фторфенил)этаноном в качестве исходных соединений.
Пример 4: 2-азидо-1-(3-(4-(2-азидоацетил)фенокси)фенил)этанон
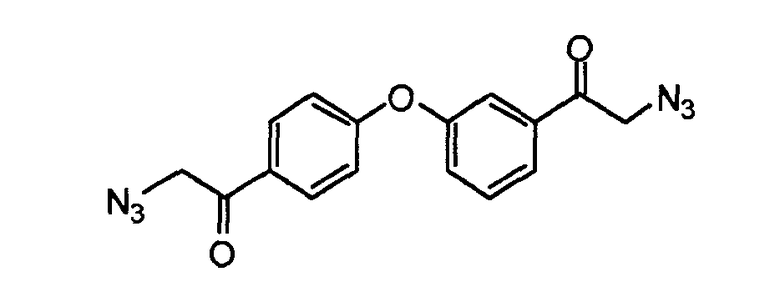
К раствору 2-бром-1-(3-(4-(2-бромацетил)фенокси)фенил)этанона (10,2 г, 24,3 ммоль) в DMSO (200 мл) добавляли азид натрия (3,5 г, 53,4 ммоль). Реакционную смесь перемешивали в течение 60 минут при комнатной температуре и затем концентрировали при пониженном давлении. Неочищенную смесь распределяли между этилацетатом и водой. Водный слой экстрагировали этилацетатом, и объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт применяли как есть на следующей стадии.
Пример 5: дигидрохлорид 2-амино-1-(3-(4-(2-аминоацетил)фенокси)фенил)этанона
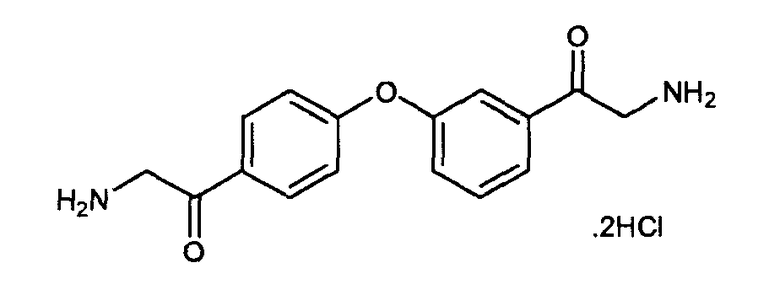
К раствору 2-азидо-1-(3-(4-(2-азидоацетил)фенокси)фенил)этанона (9 г, 26,4 ммоль) в метаноле (400 мл) добавляли HCl (1M в метаноле, 53 мл), и смесь гидрировали, применяя Pd/C (10%) в атмосфере водорода (1 бар), в течение 2 часов. Раствор фильтровали через целит и концентрировали при пониженном давлении. Неочищенный остаток растворяли в дихлорметане и после добавления HCl (6н. в изопропаноле) выпадала гидрохлоридная соль. Фильтрование и сушка в вакууме давали дигидрохлорид 2-амино-1-(3-(4-(2-аминоацетил)фенокси)фенил)этанона.
LC/MS: m/z=285,2 (M+1)+. Точная масса: 284,1
Пример 6: (S)-трет-бутил {2-(2-(3-(4-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбоксамидо)ацетил)фенокси)фенил)-2-оксоэтилкарбамоил)пирролидин-1-карбоксилат
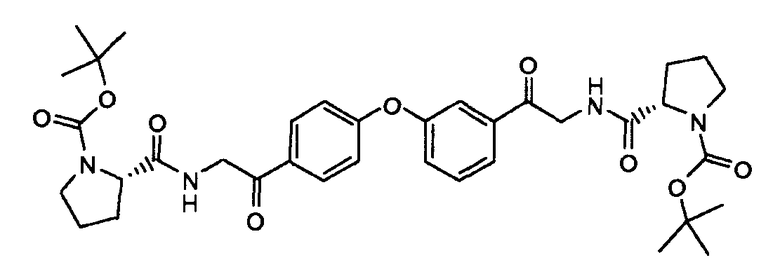
К раствору дигидрохлорида 2-амино-1-(3-(4-(2-аминоацетил)фенокси)фенил)этанона (5,65 г, 15,8 ммоль) в DMF (200 мл) добавляли N,N-диизопропилэтиламин (10,4 мл), HATU (15 г, 39,54 ммоль) и BOC-L-пролин (7,5 г, 34,7 ммоль). Смесь перемешивали при комнатной температуре в течение 60 минут и затем концентрировали при пониженном давлении. Смесь распределяли между EtOAc и водой. Водный слой экстрагировали EtOAc, и объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученное в результате неочищенное вещество очищали флэш-хроматографией (силикагель: дихлорметан/MeOH: 9/1) для получения (S)-трет-бутил 2-(2-(3-(4-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбоксамидо)ацетил)фенокси)фенил)-2-оксоэтилкарбамоил)пирролидин-1-карбоксилата в виде желтого масла (8 г, 74,5%).
LC/MS: m/z=679,3 (M+1)+. Точная масса: 678,3
Пример 7: (S)-трет-бутил 2-(5-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилат
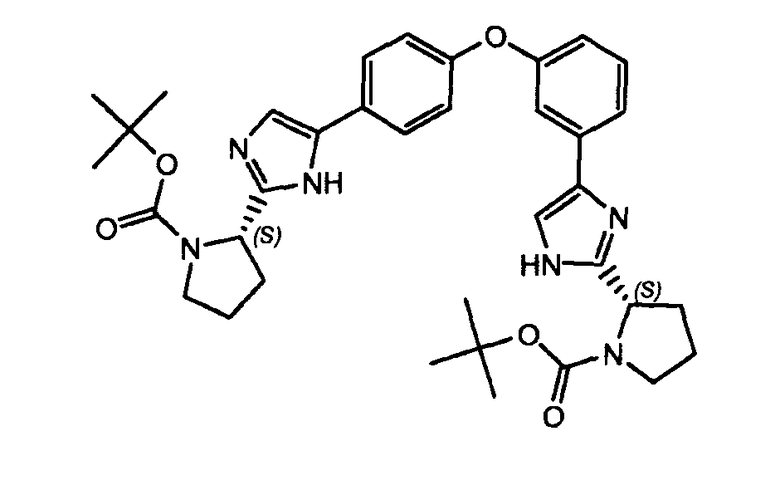
К раствору (S)-трет-бутил 2-(5-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата (1,3 г, 17,6 ммоль) в п-ксилоле (20 мл) добавляли HOAc (0,5 мл) и ацетат аммония (600 мг, 0,88 ммоль) в герметизированной пробирке. Смесь нагревали в микроволновой печи в течение двух часов при 140°C. Смесь распределяли между EtOAc и водой. Водный слой экстрагировали EtOAc, и объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученное в результате неочищенное вещество очищали флэш-хроматографией (силикагель: дихлорметан/MeOH: 9/1) для получения (S)-трет-бутил 2-(5-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата (420 мг, 74%).
LC/MS: m/z=641,2 (M+1)+. Точная масса: 640,3
Пример 8: Гидрохлорид 2-((S)-пирролидин-2-ил)-5-(4-(3-(2-((S)-пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола
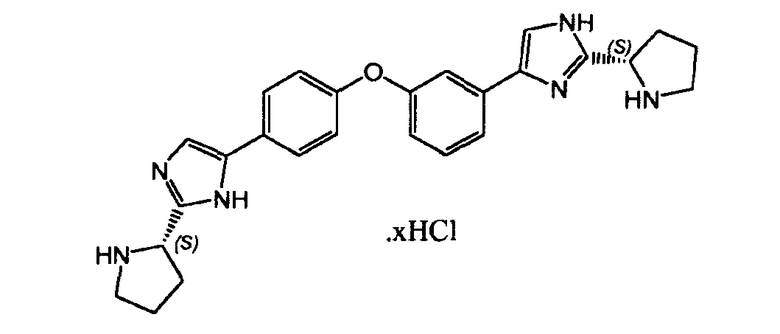
К раствору (S)-трет-бутил 2-(5-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата (500 мг, 0,78 ммоль) в дихлорметане добавляли HCl (1 M, 3 мл), продукт реакции выпадал в осадок после перемешивания при комнатной температуре. Фильтрование и сушка в вакуумной печи давали гидрохлорид 2-((S)-пирролидин-2-ил)-5-(4-(3-(2-((S)-пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола (220 мг).
Пример 9: 2-фенил-1-((S)-2-(5-(4-(3-(2-((S)-1-(2-фенилацетил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-ил)этанон
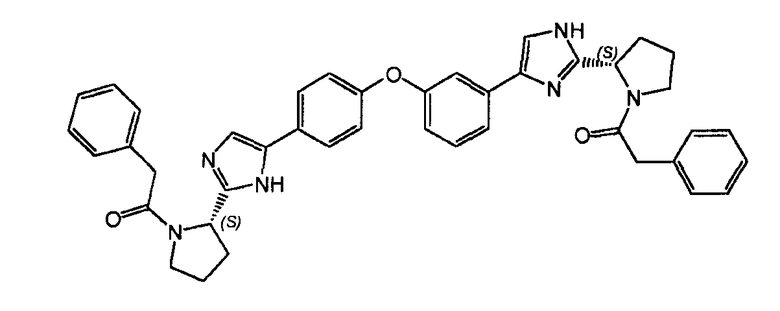
К раствору гидрохлорида 2-((S)-пирролидин-2-ил)-5-(4-(3-(2-((S)-пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола (72 мг) в DMF (20 мл) добавляли N,N-диизопропилэтиламин (0,1 мл), ΗATU (155 мг, 0,4 ммоль) и фенилуксусную кислоту (49 мг, 0,36 ммоль). Реакцию перемешивали в течение 1 часа при комнатной температуре и концентрировали при пониженном давлении. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ для получения 2-фенил-1-((S)-2-(5-(4-(3-(2-((S)-1-(2-фенилацетил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-ил)этанона (6 мг).
LC/MS: m/z=677,6 (M+1)+. Точная масса: 676,3
Пример 10: 2-гидрокси-1-((2S)-2-(5-(4-(3-(2-((2S)-1-(2-гидрокси-2-фенилацетил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-ил)-2-фенилэтанона.
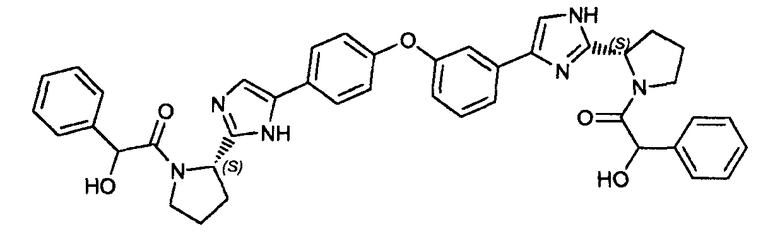
К раствору гидрохлорида 2-((S)-пирролидин-2-ил)-5-(4-(3-(2-((S)-пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола (56 мг) в DMF (15 мл) добавляли N,N-диизопропилэтиламин (0,1 мл), ΗATU (103 мг, 0,27 ммоль) и DL-миндальную кислоту (36 мг, 0,24 ммоль). Реакцию перемешивали в течение 1 часа при комнатной температуре и концентрировали при пониженном давлении. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ для получения 2-гидрокси-1-((2S)-2-(5-(4-(3-(2-((2S)-1-(2-гидрокси-2-фенилацетил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-ил)-2-фенилэтанона (3 мг).
LC/MS: m/z=709,4 (M+1)+. Точная масса: 708,3
Пример 11: 2-м-толил-1-((2S)-2-(5-(4-(3-(2-((S)-1-(2-м-толилацетил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-ил)этанон
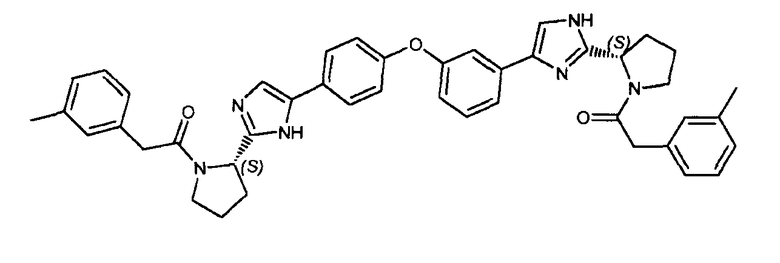
К раствору гидрохлорида 2-((S)-пирролидин-2-ил)-5-(4-(3-(2-((S)-пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола (100 мг, 0,22 ммоль) в DMF (22 мл) добавляли N,N-диизопропилэтиламин (0,2 мл), ΗATU (216 мг, 0,56 ммоль) и 2-м-толилуксусную кислоту (75 мг, 0,5 ммоль). Реакцию перемешивали в течение 1 часа при комнатной температуре и концентрировали при пониженном давлении. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ для получения 2-м-толил-1-((2S)-2-(5-(4-(3-(2-((S)-1-(2-м-толилацетил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-ил)этанона (5 мг).
LC/MS: m/z=705,2 (M+1)+. Точная масса: 704,3
Пример 12: 1,1'-(4,4'-оксибис(4,1-фенилен))бис(2-хлорэтанон)
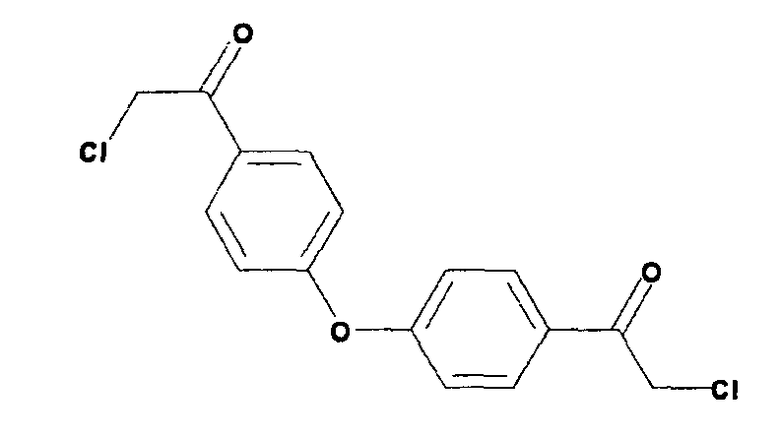
В трехгорлую круглодонную колбу, снабженную обратным холодильником и большой магнитной мешалкой, помещали дифениловый эфир (30 г, 0,176 моль), хлорацетилхлорид (79,6 г, 0,71 моль) и дисульфид углерода (100 мл). Смесь энергично перемешивали при добавлении хлорида алюминия (141 г, 1,06 моль) приблизительно 3 г порциями или до начала кипения реакционной смеси. После завершения добавления реакционную смесь нагревали при 100°C в течение 2,5 часов при перемешивании. Реакцию охлаждали до комнатной температуры и оставляли на один час. Верхний слой (дисульфид углерода) выбрасывали. Нижний слой разделяли поровну на 4 пробирки (1 л каждая), причем каждая содержала лед (600 мл) и концентрированную хлороводородную кислоту (50 мл) при энергичном перемешивании. Смеси объединяли и экстрагировали дихлорметаном (5×400 мл). Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали, и растворители фильтрата удаляли при пониженном давлении для получения коричневого масла. Неочищенную смесь частично очищали флэш-хроматографией, применяя градиент гептана в этилацетате. Лучшие фракции объединяли, и растворитель удаляли при пониженном давлении для получения коричневого масла. Масло перекристаллизовывали из этилацетата для получения заявленного в заголовке соединения (16,25 г). Вторая порция давала дополнительные 25 г.
LC/MS m/z=323 (M+1)+. Точная масса: 322,0
1Н-ЯМР (хлороформ-d, 400 МГц): 8,01 м.д. (д, 4Н), 7,15 м.д. (д, 4Н), 4,69 м.д. (с, 4Н).
Пример 13: (2S,2'S)-1-трет-бутил '2,2-2,2'-(4,4'-оксибис(4,1-фенилен))бис(2-оксоэтан-2,1-диил)дипирролидин-1,2-дикарбоксилат
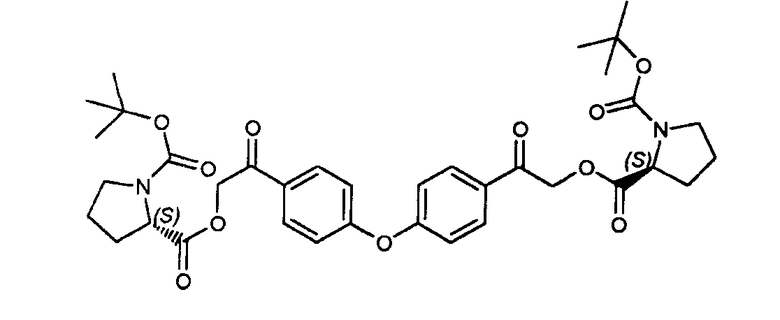
К соединению, полученному в примере 12 (5 г, 15,5 ммоль), в ацетонитриле добавляли Boc-L-пролин (6,99 г, 32,5 ммоль), с последующим добавлением по каплям DIEA (4,4 г, 34,04 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Растворители удаляли при пониженном давлении. Добавляли воду (50 мл), и смесь экстрагировали дихлорметаном (3×150 мл). Органические слои объединяли, сушили (сульфат натрия), фильтровали, и растворители фильтрата удаляли при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией, применяя градиент гептана в этилацетате для получения заявленного в заголовке продукта.
LC/MS: m/z=681 (M+1)+. Точная масса: 680,3
Пример 14: (2S,2'S)-трет-бутил 2,2'-(5,5'-(4,4'-оксибис(4,1-фенилен))бис(1H-имидазол-5,2-диил))дипирролидин-1-карбоксилат
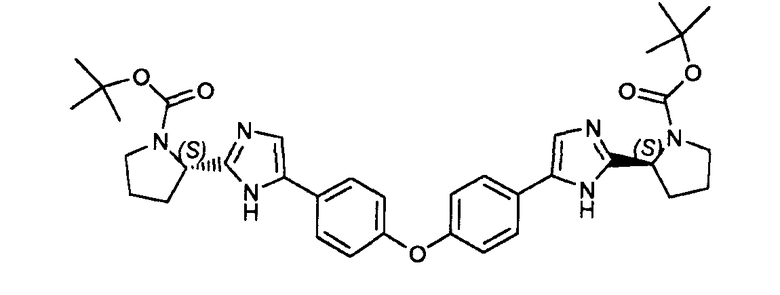
В герметично закрываемую пробирку помещали исходный эфир, полученный в примере 13, ацетат аммония (20 экв.) и ксилол. Смесь перемешивали при кипячении с обратным холодильником в течение нескольких часов для получения заявленного в заголовке продукта. Смесь распределяли между EtOAc и водой. Водный слой экстрагировали EtOAc, и объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученное в результате неочищенное вещество очищали флэш-хроматографией (силикагель: дихлорметан/MeOH: 9/1) для получения заявленного в заголовке продукта.
LC/MS: m/z=641 (M+1)+. Точная масса: 640,3
Пример 15: (S)-5,5'-(4,4'-оксибис(4,1-фенилен))бис(2-((S)-пирролидин-2-ил)-1H-имидазол)
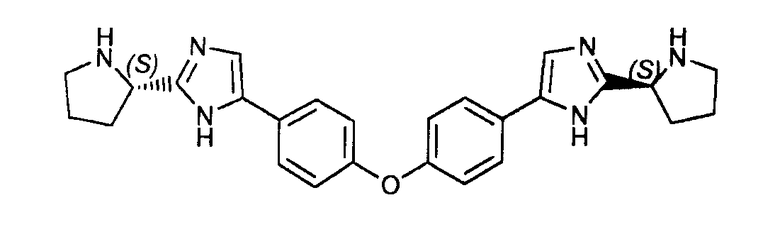
Защитные группы в остатках пролина соединения, полученного в примере 14, удаляли, следуя той же методике, как описано в примере 8.
LC/MS: m/z=441 (M+1)+. Точная масса: 440,2
Кроме того, деблокированные атомы азота остатков пролина ацилировали, применяя ту же методику, как в примере 9.
Пример 16: (S)-трет-бутил 2-(4-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)-4-метоксифенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилат
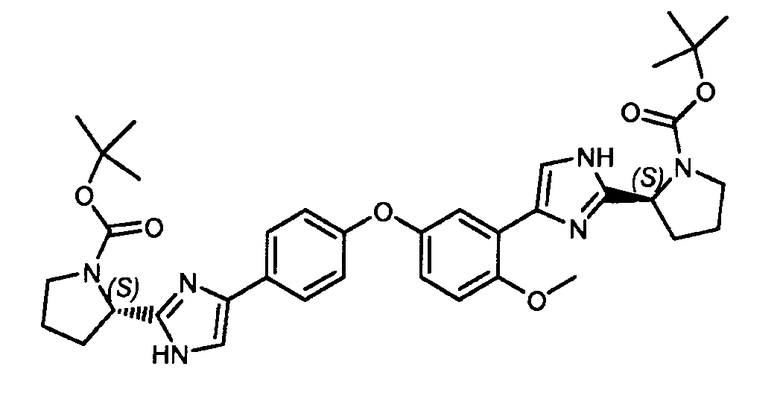
К раствору 2-бром-1-(4-(3-(2-бромацетил)-4-метоксифенокси)фенил)этанона (пример 3a) (10 г, 14 ммоль) в ацетонитриле (150 мл) добавляли N-(трет-бутоксикарбонил)-L-пролин (11,2 г, 28 ммоль) при комнатной температуре. К данному раствору добавляли по каплям основание Хюнига (23 мл, 42 ммоль), после чего смесь перемешивали в течение 6 часов при комнатной температуре. Затем реакционную смесь концентрировали в вакууме, и неочищенный остаток разбавляли дихлорметаном (300 мл). Затем органическую смесь промывали водой (2×300 мл). Объединенные органические фракции сушили над сульфатом магния. После фильтрования и упаривания растворителя смесь очищали флэш-хроматографией (EtOAc/гептан: 3/7) для получения чистого (S)-2-(2-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбонилокси)ацетил)-4-метоксифенокси)фенил)-2-оксоэтил) 1-трет-бутилпирролидин-1,2-дикарбоксилата, который применяли как есть в следующей стадии. Затем (S)-2-(2-(4-(3-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбонилокси)ацетил)-4-метоксифенокси)фенил)-2-оксоэтил) 1-трет-бутилпирролидин-1,2-дикарбоксилат растворяли в ксилоле (150 мл) и добавляли NH4OАc (5 экв.). Реакционную смесь перемешивали в течение 6 часов при 140°C. Затем смесь экстрагировали дихлорметаном, объединенные органические слои промывали водой и сушили над Na2SO4. Смесь фильтровали, растворитель удаляли, и остаток очищали флэш-хроматографией (хлороформ/MeOH: 95/5) для получения заявленного в заголовке соединения (4,1 г, выход: 44%).
1H ЯМР (400 МГц, хлороформ-d) δ м.д. 1,48 (с, 9H) 1,50 (с, 9H) 1,86-2,02 (м, 2H) 2,07-2,23 (м, 4H) 2,91-3,04 (м, 2H) 3,39 (уш.с, 4H) 3,97 (уш.с, 3H) 4,97 (дд, J=7,5, 2,4 Гц, 2H) 6,90 (м, 5H) 6,98 (д, J=8,6 Гц, 2H) 7,14 (с, 2H).
Пример 17: Альтернативное получение (S)-трет-бутил 2-(5-(3-(4-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата
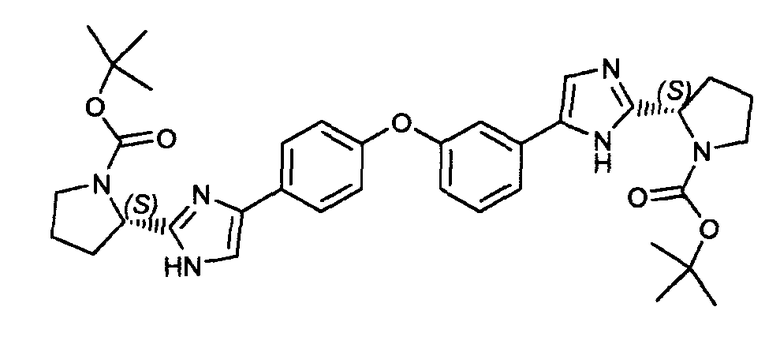
К раствору 2-бром-1-(3-(4-(2-бромацетил)фенокси)фенил)этанона (2,05 г, 5 ммоль) в CH2Cl2 (40 мл) добавляли N-(трет-бутоксикарбонил)-L-пролин (2,15 г, 10 ммоль) при 0°C. Затем осторожно добавляли по каплям триэтиламин (1,0 г, 10 ммоль), и смесь перемешивали при комнатной температуре в течение 6 часов. Затем добавляли CH2Cl2 (150 мл), смесь промывали водой и сушили над Na2SO4. После фильтрования растворитель удаляли, и полученный остаток очищали флэш-хроматографией (н-гексан/этилацетат: 2/1) для получения (S)-2-(2-(3-(4-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбонилокси)ацетил)фенокси)фенил)-2-оксоэтил) 1-трет-бутилпирролидин-1,2-дикарбоксилата с выходом 2,11 г (63%). Смесь (S)-2-(2-(3-(4-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-карбонилокси)ацетил)фенокси)фенил)-2-оксоэтил) 1-трет-бутилпирролидин-1,2-дикарбоксилата (6,80 г, 10 ммоль) и NH4OAc (7,7 г, 100 ммоль) в ксилоле (150 мл) перемешивали в течение 6 часов при 140°C. Затем смесь экстрагировали этилацетатом (300 мл), промывали водой и сушили над Na2SO4. После фильтрования растворитель удаляли, и полученный остаток очищали флэш-хроматографией (хлороформ/MeOH: 95/5) для получения заявленного в заголовке соединения (50%).
1H ЯМР (400 МГц, хлороформ-d) δ м.д. 1,49 (м, 18H) 1,84-2,02 (м, 4H) 2,15 (уш.с, 4H) 3,41 (м, 4H) 4,86-5,03 (м, 2H) 6,87 (д, J=7,0 Гц, 1H) 7,04 (м, 3H) 7,17 (м, 3H) 7,32 (м, 3H).
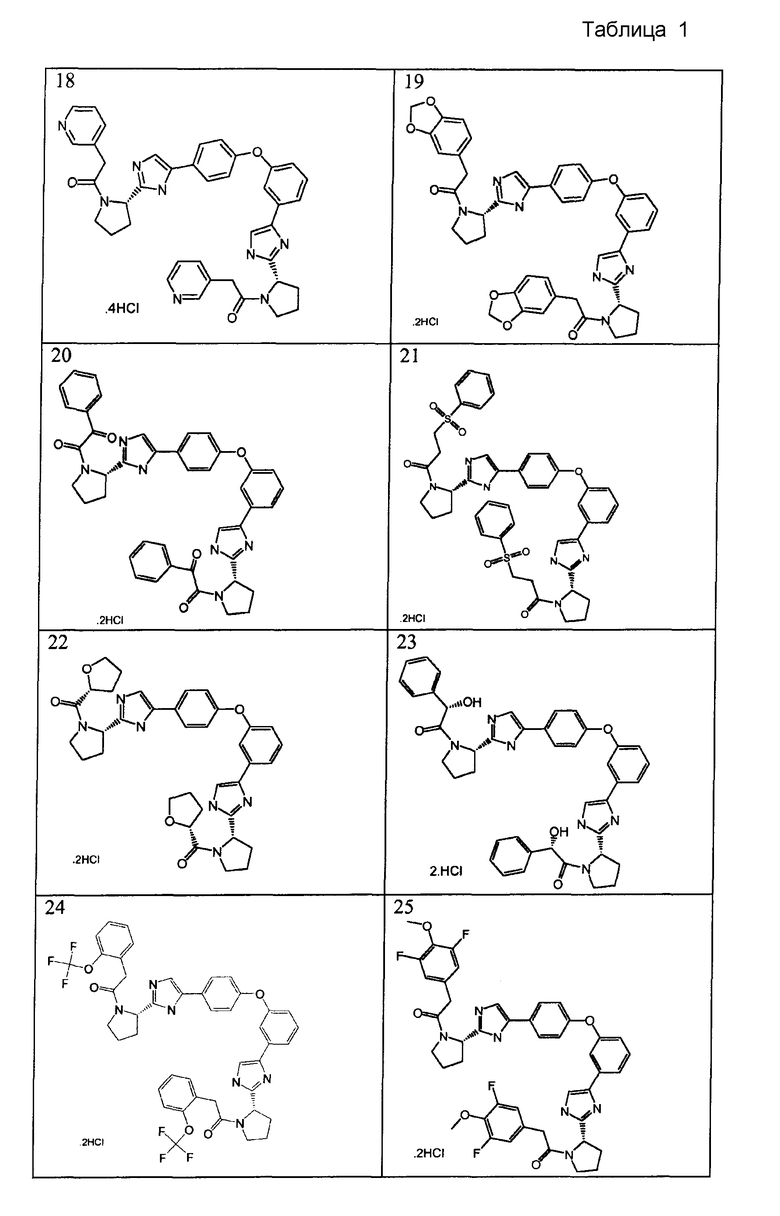
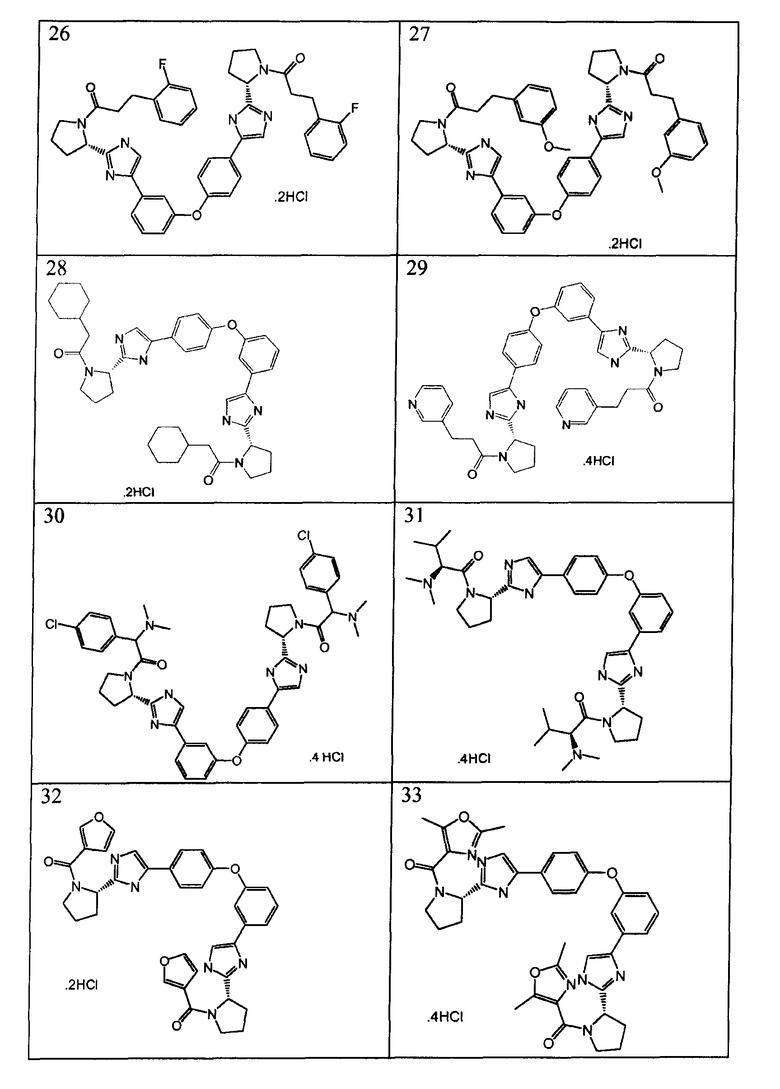
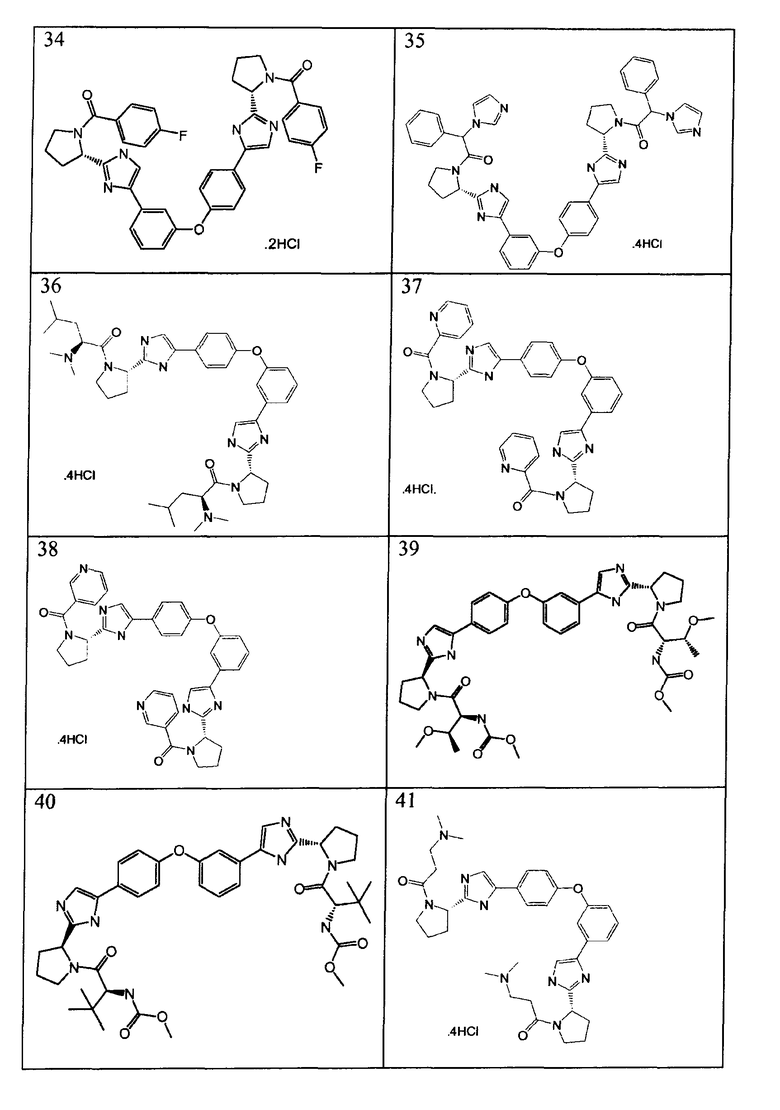
Примеры 18-41
Общая методика:
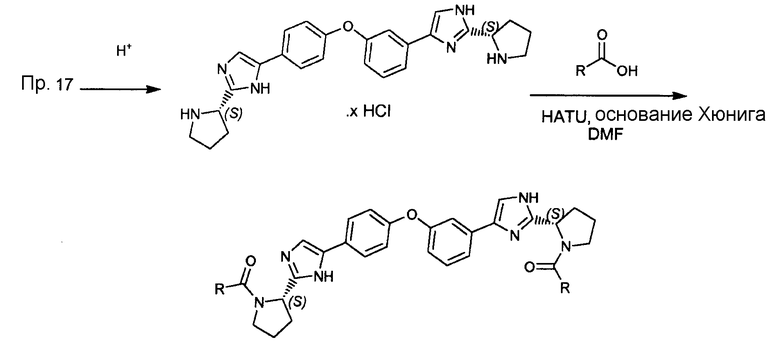
Во-первых, к раствору (S)-трет-бутил 2-(5-(3-(4-(2-((S)-1-(трет-бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата в дихлорметане добавляли по каплям избыток HCl (1M в изопропаноле). Выпавший осадок отфильтровывали и сушили в вакуумной печи для получения гидрохлорида 2-(пирролидин-2-ил)-5-(4-(3-(2-(пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола.
Затем к раствору гидрохлорида 2-(пирролидин-2-ил)-5-(4-(3-(2-(пирролидин-2-ил)-1H-имидазол-4-ил)фенокси)фенил)-1H-имидазола (300 мг) в DMF (7 мл) добавляли основание Хюнига (0,45 мл, 2,72 ммоль), ΗATU (647 мг, 1,70 ммоль) и соответствующую кислоту (1,5 ммоль). Смесь перемешивали в течение 4 часов при комнатной температуре и концентрировали в вакууме. Смесь помещали на Isolute (SCX-3, 15 мл) пробку, и пробку промывали MeOH (4 раза). Затем продукт промывали NΗ3/MeOΗ (2 раза). Затем полученный раствор концентрировали в вакууме, и неочищенный продукт обрабатывали HCl (1 M в воде) и DCM (3/1) до получения твердого остатка. Затем твердый остаток отфильтровывали и промывали водной HCl (1 M). Продукт сушили в вакуумной печи для получения целевого продукта.
Соединения, полученные согласно данной общей методике, перечислены в таблице 1. Как показано в таблице 1, некоторые соединения получали в виде HCl солей, тогда как другие получали в виде свободного основания. Соединения 39, 40, 43 и 44 дополнительно очищали общепринятой хроматографией на силикагеле (MeOH (7н. NΗ3) / DCM: 2/98) или обращенно-фазовой хроматографией.
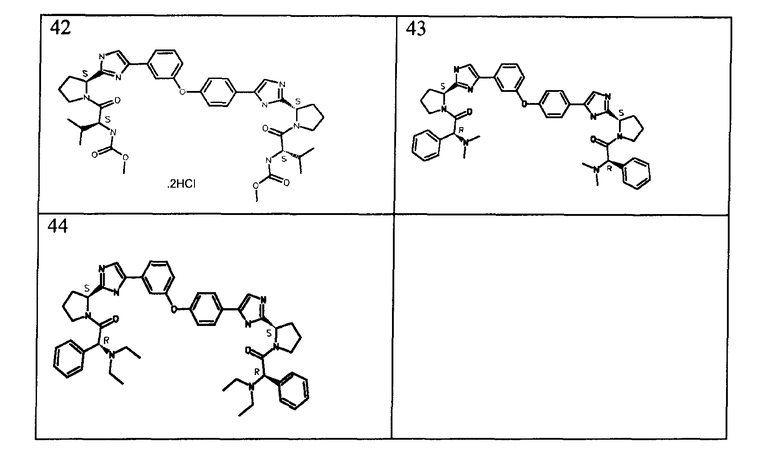
Примеры 45-48
Соединения, перечисленные в таблице 2, получали, применяя ту же общую методику, как для соединений 18-41 выше, но исходя из (S)-трет-бутил 2-(4-(4-(3-(2-((S)-1-(трет- бутоксикарбонил)пирролидин-2-ил)-1H-имидазол-4-ил)-4-метоксифенокси)фенил)-1H-имидазол-2-ил)пирролидин-1-карбоксилата (пример 16). Соединения 45, 46, 47 и 48 дополнительно очищали общепринятой хроматографией на силикагеле (MeOH (7н NΗ3)/DCM: 2/98) или обращенно-фазовой хроматографией.
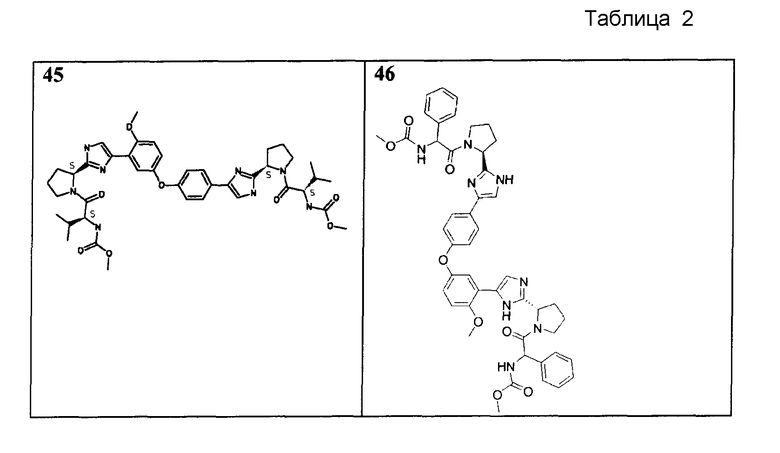
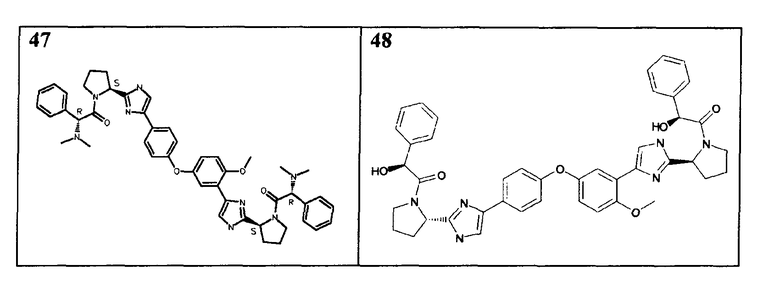
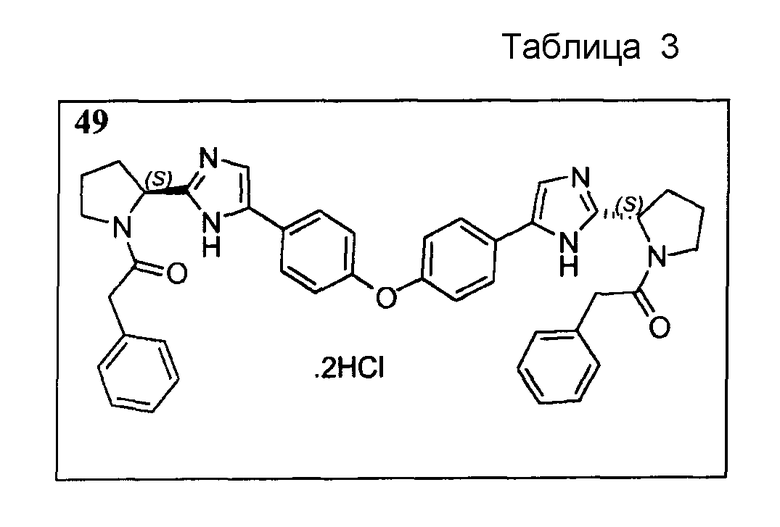
Пример 49
Соединение, представленное в таблице 3, получали, применяя ту же методику, как для соединений 18-41 выше, но исходя из (S)-5,5'-(4,4'-оксибис(4,1-фенилен))-бис(2-((S)-пирролидин-2-ил)-1H-имидазола) (пример 15).
Все соединения охарактеризовывали LC/MS. Время удерживания (Rt) для примерных соединений представлено в таблице 4. В таблице 4 также приведен LC/MS способ, который применяли для определения Rt для каждого из примерных соединений. Применяли следующие LC/MS способы:
Способ A: Waters Acquity UPLC, снабженный PDA детектором (диапазон 210-400 нм) и Waters SQD с двухрежимным источником ионов ES+/-. Применяемой колонкой была Halo C 18, 2,7 мкм, 2,1×50 мм, и она поддерживалась при 50°C. Градиент 95% водной муравьиной кислоты (0,1%)/5% ацетонитрил - 100% ацетонитрил применяли в течение 1,5 минут, выдерживали в течение 0,6 минут, затем возвращали к 100% водной муравьиной кислоте (0,1%) в течение 0,5 минут. Скорость потока была 0,6 мл/мин.
Способ B: жидкостная хроматография: Waters Alliance 2695, УФ-детектор: Waters 996 PDA, диапазон: 210-400 нм; масс-детектор: Waters ZQ, источник ионов: ES+, ES-применяемая колонка: SunFire C18 3,5 мкм 4,6×100 мм. подвижная фаза A: 10 мМ NH4OOCH+ 0,1% HCOOH в H2O; подвижная фаза B: CH3OH; температура колонки: 50°C; скорость потока: 1,5 мл/мин время градиентного элюирования (мин) [%A/%B] 0[65/35] - 7[5/95] - 9,6[5/95] - 9,8[65/35] - 12 [65/35].
Способ C: Waters Acquity UPLC, снабженный PDA детектором (диапазон 210-400 нм) и Waters SQD с двухрежимным источником ионов ES+/-. Применяемой колонкой была XS Strategy 1,7 мкм, 2,1×20 мм, и она поддерживалась при 50°C. Градиент 100% водная муравьиная кислота (0,1%) - 100% ацетонитрил применяли в течение 1,5 минут, выдерживали в течение 0,6 минут, затем возвращали к 100% водной муравьиной кислоте (0,1%) в течение 0,5 минут. Скорость потока была 0,6 мл/мин.
Отобранные соединения дополнительно характеризовали ЯМР. ЯМР спектры регистрировали на Bruker Avance 400 спектрометре, работающем при 400 МГц для 1H и 100 МГц для 13C и с DMSO в качестве растворителя, если не указано особо. В каждом случае тетраметилсилан (TMS) применяли в качестве внутреннего стандарта. Химические сдвиги даны в м.д. и J величины - в Гц.
Мультиплетность показана, применяя следующие сокращения: д - для дублета, т - для триплета, m - для мультиплета и т.д.
Пример 32:
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 2,00-2,16 (м, 6H), 2,42 (м, 2H), 3,71-3,82 (м, 2H), 4,03-4,14 (м, 2H), 5,25-5,41 (м, 1H), 6,79 (м, 2H), 7,07-7,14 (м, 1H), 7,19 (м, 2H), 7,19 (уш.с, 1H), 7,57 (м, 1H), 7,67 (м, 1H), 7,71 (м, 1H), 7,77 (с, 2H), 7,90 (м, 2H), 8,01 (с, 1H), 8,13 (с, 1H), 8,28 (уш.с, 2H).
Пример 39:
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,02-1,05 (м, 6H) 1,88-2,19 (м, 8H) 3,07-3,13 (м, 3H) 3,17-3,20 (м, 3H) 3,37-3,50 (м, 4H) 3,51-3,56 (м, 6H) 3,75-3,86 (м, 2H) 4,20-4,32 (м, 2H) 4,98-5,10 (м, 2H) 6,80 (уш.с, 1H) 6,97 (уш.с, 2H) 7,18-7,26 (м, 2H) 7,29-7,34 (м, 1H) 7,37 (уш.с, 2H) 7,44-7,52 (м, 2H) 7,66-7,75 (м, 2H) 11,63-11,82 (м, 2H).
Пример 40:
1H ЯМР (600 МГц, метанол-d4) δ м.д. 0,84-1,06 (м, 18H) 1,81-2,43 (м, 8H) 3,36-3,44 (м, 2H) 3,61-3,67 (м, 6H) 3,78-3,88 (м, 2H) 3,93-4,03 (м, 2H) 4,26-4,41 (м, 2H) 5,05-5,23 (м, 2H) 6,74-7,08 (м, 4H) 7,15-7,77 (м, 8H).
Пример 45:
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 0,71-0,84 (м, 12H) 1,92-2,22 (м, 8H) 2,28-2,41 (м, 2H) 3,50-3,56 (м, 6H) 3,78-4,00 (м, 4H) 3,94-3,99 (м, 3H) 4,02-4,15 (м, 2H) 5,11-5,23 (м, 2H) 7,04-7,13 (м, 2H) 7,15-7,21 (м, 1H) 7,22-7,30 (м, 2H) 7,68-7,75 (м, 1H) 7,83-7,89 (м, 2H) 7,83-7,89 (м, 2H) 7,89-7,93 (м, 1H) 7,94-7,99 (м, 1H).
Пример 48:
1H ЯМР (600 МГц, ДМСО-d6) δ м.д. 1,87-2,10 (м, 8H) 2,29-2,40 (м, 2H) 3,73-3,89 (м, 4H) 3,96 (уш.с, 3H) 5,13-5,25 (м, 2H) 5,30-5,41 (м, 2H) 7,07-7,13 (м, 2H) 7,16-7,23 (м, 2H) 7,24-7,40 (м, 12H) 7,65-7,98 (м, 5H).
Биологические примеры
Анализ на основе репликона
Соединения формулы (I) исследовали на ингибирующую способность в HCV репликоне. Данный клеточный способ основан на конструкте бицистронной экспрессии, как описано Lohmann et al. (1999) Science vol. 285 pp. 110-113 с изменениями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624, в многоцелевой скрининговой стратегии. По существу, способ был следующим:
В анализе применяли стабильно трансфицированную клеточную линию Huh-7 luc/neo (называемую далее Huh-Luc). Данная клеточная линия содержит РНК, кодирующую конструкт бицистронной экспрессии, содержащий NS3-NS5B области дикого типа HCV типа 1b, транслированные из участка внутренней посадки рибосомы (IRES) из вируса энцефаломиокардита (EMCV) с предшествующей репортерной частью (FfL-люцифераза) и частью селектируемого маркера (neoR, неомицинфосфотрансфераза). Конструкт фланкирован 5' и 3' NTR (нетранслируемыми областями) из HCV типа 1b. Непрерывное культивирование клеток с репликоном в присутствии G418 (neoR) зависит от репликации HCV РНК. Для скрининга противовирусных соединений применяли стабильно трансфицированные клетки с репликоном, которые экспрессируют HCV РНК, которая реплицируется автономно и до больших концентраций, кодирующую, в том числе, люциферазу.
Клетки с репликоном помещали в 384-луночные планшеты в присутствии испытуемых и контрольных соединений, которые добавляли в различных концентрациях. После выдерживания в течение трех дней HCV репликацию измеряли анализом люциферазной активности (применяя стандартные субстраты и реагенты для люциферазного анализа и Perkin Elmer ViewLux™ ultraHTS микропланшетный аппарат для визуализации). Клетки с репликоном в контрольной группе обладали высокой люциферазной экспрессией в отсутствие любого ингибитора. Ингибирующую активность соединения на люциферазную активность отслеживали на Huh-Luc клетках, получая кривую дозовой зависимости для каждого испытуемого соединения. Затем рассчитывали EC50 величины, которые представляют собой количество соединения, требуемое для снижения степени обнаруживаемой люциферазной активности на 50%, или более конкретно, для снижения способности генетически связанной HCV репликоновой РНК реплицироваться.
Результаты
В таблице 4 показаны результаты для репликонов и цитотоксические результаты, полученные для соединений примеров, данных выше.
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2419619C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2441870C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2486189C2 |
| МАКРОЦИКЛИЧЕСКИЕ ФЕНИЛКАРБАМАТЫ, ИНГИБИРУЮЩИЕ HCV | 2008 |
|
RU2490261C2 |
| ПРОИЗВОДНЫЕ БИС-БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2540897C2 |
| ФЕНИЛЭТИНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2538507C2 |
| МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2006 |
|
RU2437886C2 |
| ПИРИМИДИН-ЗАМЕЩЕННЫЕ МАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ HCV | 2008 |
|
RU2481340C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ СРЕДСТВА ПРОТИВ ВИРУСОВ СЕМЕЙСТВА FLAVIVIRIDAE | 2004 |
|
RU2355687C2 |
| МОЛЕКУЛА, ПЕСТИЦИДНАЯ КОМПОЗИЦИЯ НА ЕЕ ОСНОВЕ (ВАРИАНТЫ) И СПОСОБ ПРИМЕНЕНИЯ МОЛЕКУЛЫ (ВАРИАНТЫ) | 2010 |
|
RU2543806C2 |
Изобретение относится к соединению формулы (I), к его возможным стереоизомерам, или к его фармацевтически приемлемым солям, где R и R1, независимо друг от друга, представляют собой бензоил, замещенный одним заместителем, каждый из которых независимо выбирают из галогена или -C(=O)-Het, где Het необязательно замещен двумя заместителями, независимо выбранными из C1-4алкила, или группу формулы -C(=O)-СН(Rx)-R6, C1-6алкилоксикарбонил, группу формулы R8-O-C(=O)-HN-CH(R7)-С(=O)- или -С(=O)-С(=O)-фенил; R6 представляет собой C1-4алкил, C3-6циклоалкил, бензил или фенил, где фенил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбирают из галогена, C1-6алкила, метокси, трифторметокси, или два заместителя при соседних атомах кольца вместе с фенильным кольцом образуют бензодиоксол, и где C1-4алкил замещен диC1-6алкиламино, фенилсульфонилом, Het, и где бензил замещен одним заместителем, каждый из которых независимо выбирают из галогена, метокси; Rx выбирают из водорода, гидрокси, диC1-6алкиламино, имидазолила; Het представляет собой гетероциклическую группу, содержащую один или два гетероатома, выбранных из O и N, и содержащую 5-6 атомов в кольце, где упомянутое гетероциклическое кольцо соединено с карбонильным атомом углерода через атом углерода кольца и где по меньшей мере один из упомянутых гетероатомов является смежным с упомянутым атомом углерода кольца, R2 и R3, независимо друг от друга, представляют собой водород; R4 и R5, независимо друг от друга, представляют собой водород или метокси; каждый R7 независимо представляет собой фенил или C1-4алкил, необязательно замещенный метокси; и R8 представляет собой C1-4алкил. Также изобретение относится к конкретным соединениям и фармацевтической композиции на основе соединения формулы (I). Технический результат: получены новые соединения, полезные в качестве HCV ингибиторов. 3 н. и 8 з.п. ф-лы. 4 табл., 49 пр.
1. Соединение формулы I:
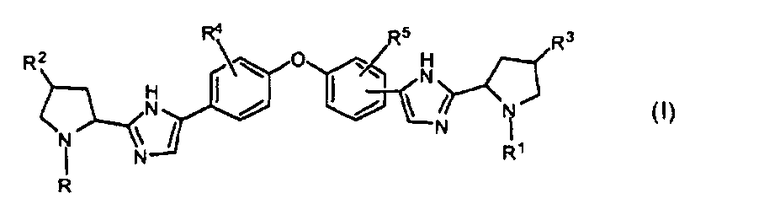
включая его любые возможные стереоизомеры, где:
R и R1, независимо друг от друга, представляют собой бензоил, замещенный одним заместителем, каждый из которых независимо выбирают из галогена или -C(=O)-Het, где Het необязательно замещен двумя заместителями, независимо выбранными из C1-4алкила, или группу формулы -C(=O)-СН(Rx)-R6, C1-6алкилоксикарбонил, группу формулы R8-O-C(=O)-HN-CH(R7)-С(=O)- или -С(=O)-С(=O)-фенил;
R6 представляет собой C1-4алкил, C3-6циклоалкил, бензил или фенил, где фенил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбирают из галогена, C1-6алкила, метокси, трифторметокси, или два заместителя при соседних атомах кольца вместе с фенильным кольцом образуют бензодиоксол, и где C1-4алкил замещен диC1-6алкиламино, фенилсульфонилом, Het, и где бензил замещен одним заместителем, каждый из которых независимо выбирают из галогена, метокси;
Rx выбирают из водорода, гидрокси, диC1-6алкиламино, имидазолила;
Het представляет собой гетероциклическую группу, содержащую один или два гетероатома, выбранных из O и N, и содержащую 5-6 атомов в кольце, где упомянутое гетероциклическое кольцо соединено с карбонильным атомом углерода через атом углерода кольца и где по меньшей мере один из упомянутых гетероатомов является смежным с упомянутым атомом углерода кольца,
R2 и R3, независимо друг от друга, представляют собой водород;
R4 и R5, независимо друг от друга, представляют собой водород или метокси;
каждый R7 независимо представляет собой фенил или C1-4алкил, необязательно замещенный метокси; и
R8 представляет собой C1-4алкил; или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором R2 и R3 представляют собой водород.
3. Соединение по п.1 или 2, в котором R и R1, независимо друг от друга, представляют собой группы формулы -C(=O)-СН(Rx)-R6 или -C(=O)-Het, в частности в которых R и R1, независимо друг от друга, представляют собой группы формулы -С(=O)-СН(Rx)-R6.
4. Соединение по п.1, в котором Rx представляет собой гидрокси, диC1-6алкиламино; или где Rx представляет собой гидроксил, диC1-4алкиламино; или где Rx представляет собой гидрокси.
5. Соединение по п.1, в котором R6 представляет собой фенильное кольцо, необязательно замещенное одним галогеном или C1-6алкилом.
6. Соединение по п.1, в котором R6 выбирают из фенила и изопропила.
7. Соединение по п.1, в котором Het выбирают из 2-пиридинила, 2-пиримидила, 2-пиразинила, 2-имидазолила, 2-пиразолинила, 2-пиперидинила, 2-пирролидинила, 2-пирролила, 2-фуранила, 2-тетрагидрофуранила, 2- или 3-морфолинила и 2-пиперазинила, в частности 2-тетрагидрофуранила.
8. Соединение по п.1, в котором R4 и R5 независимо представляют собой водород или метокси, в частности в котором R4 и R5 представляют собой водород.
9. Соединение по п.1, в котором группа
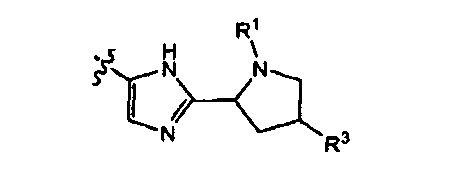
замещена в мета-положении относительно кислородного мостика между двумя фенильными группами.
10. Соединение, выбранное из группы, включающей следующие соединения:
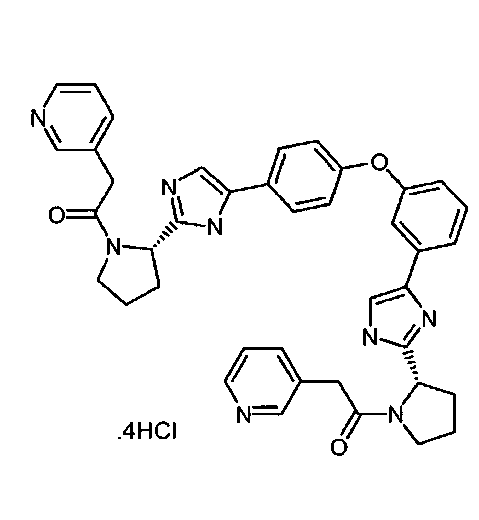 ,
, 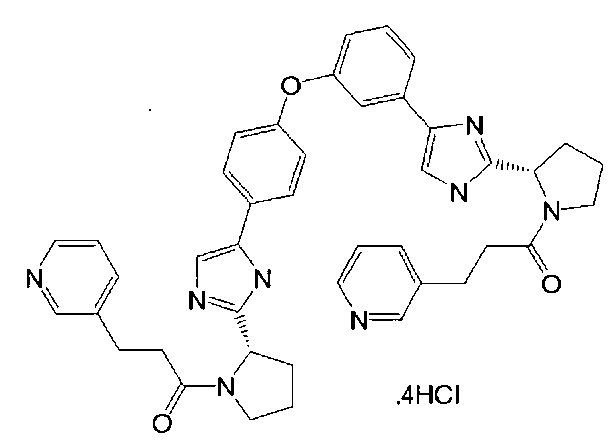 ,
,
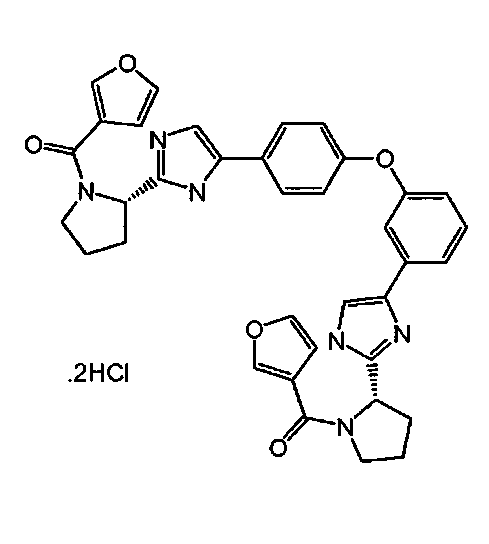 и
и 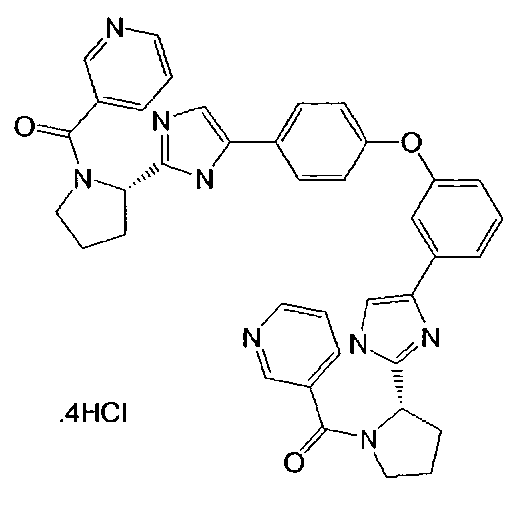 .
.
11. Фармацевтическая композиция для ингибирования репликации HCV, содержащая эффективное количество соединения по любому из пп.1-10 и фармацевтически приемлемый носитель.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| Устройство для защиты рентгеновских установок от высокого напряжения | 1926 |
|
SU7968A1 |
| Электрическое устройство для перевода железнодорожных стрелок с подвижного состава | 1927 |
|
SU8148A1 |

