ОБЛАСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение касается процесса получения правастатина, в частности микробного процесса получения правастатина в промышленных масштабах.
ПРЕДПОСЫЛКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
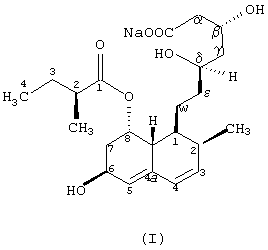
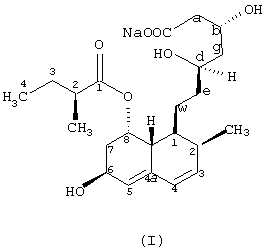
Самым высоким фактором риска атеросклероза, а особенно закупорки коронарных сосудов, является высокий уровень холестерина в плазме. В последние два десятилетия в качестве фермента, определяющего и ограничивающего биосинтез холестерина, интенсивно исследовалась 3-гидрокси-3-метилглутарил-СоА-редуктаза (ЕС. 1.1.1.34). Правастатин, соединение формулы I,
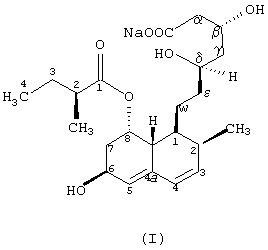
а также другие родственные соединения (компактин, мевинолин, симвастатин) представляют собой конкурентные ингибиторы группы белков с высокой подвижностью-СоА-редуктазы [A. Endo et al., J. Antibiot. 29, 1346-1348 (1976); A. Endo et al., FEES Lett. 72, 323-326 (1976): С.H.Kuo et al., J. Org. Chem. 48,1991 (1983)].
Впервые правастатин был выделен М. Tanaka с соавторами (неопубликованные результаты) из мочи собаки при изучении процесса метаболизма компактина (Arai, М. et al., Sankyo Kenkyusyo Nenpo, 40, 1-38, 1988). В настоящее время правастатин представляет собой агент, снижающий уровень холестерина при наиболее благоприятном механизме действия в терапии. Его наиболее важной чертой является тканевая селективность, а именно ингибирование им синтеза холестерина в двух основных местах его возникновения, таких как печень и тонкий кишечник, в то время как в других органах его ограничивающее внутриклеточное воздействие выявляется с трудом. В то же самое время для большинства органов существенно сдерживающее воздействие на биосинтез холестерина оказывают мевинолин и симвастин (Т. Koga et al., Biochim. Biophys. Acta, 1045, 115-120, 1990).
По химическому строению правастатин существенно отличается от мевинолина и симвастина, обладающих более липофильным характером. Для последних соединений заместитель, присоединенный к С-1 атому углерода гексагидронафталенового скелета, заканчивается 6-членным лактоновым циклом, в то время как в случае правастатина вместо лактонового цикла присутствует биологически активная нециклическая форма вторичной кислой соли натрия. Другим важным структурным отличием является то, что в положении С-6 гексагидронафталинового цикла вместо метальной группы (как в случае мевинолина и симвастина) у правастатина можно обнаружить гидроксильную группу, что приводит к еще более значительному усилению его гидрофильного характера.
В результате указанных выше структурных отличий правастатин способен только в минимальной степени проникать сквозь липофильную мембрану периферических клеток (А.Т.М., Serajuddin et al., J. Pharm. Sci. 80, 830-834. 1991).
Промышленное производство правастатина может быть осуществлено с помощью двух процессов ферментации. В первом процессе на микробиологической стадии получают компактин, а затем при второй ферментации натриевую соль компактиновой кислоты превращают в правастатин микробным гидроксилированием по 6β -позиции.
Согласно опубликованным патентам микробное гидроксилирование компактина может до различной степени осуществляться плеснями, принадлежащими к разным видами, а также нитевидными бактериями, принадлежащими к видам Nocardia, Aciinomadura и Streptomyces (Патентная заявка Бельгии № 895090. Патентная заявка Японии № 5,810,572, патенты США № 4,537,859 и № 4,346,227; а также опубликованная заявка на ЕП № 0605230). Было опубликовано, что биоконверсия субстрата компактина проходит при концентрации 500 мкг/мл с использованием нитевидных плесеней, таких как Mucor hiemalis, Syncephalastrum nigricans, Cunninghamella echinulata и при концентрации в 2000-4000 мкг/мл с использованием штаммов Nocardia, Actinomodura и Streptomyces, принадлежащих к прокариотам.
Основная проблема, возникающая в случае получения правастатина с помощью нитевидных плесеней, состоит в том, что из-за противогрибкового воздействия компактина эти микроорганизмы не способны быть устойчивыми к субстрату компактина, подаваемому в культуру даже при низких концентрациях (Serizawa et al., J. Antibiotics, 36, 887-891, 1983). Токсичность клеток этого субстрата наблюдали также при гидроксилировании Streptomyces carbophilus, которое интенсивно изучали японские исследователи (М. Hosobuchi et al., Biotechnology and Bioengineering, 42, 815-820, 1993).
Японские авторы пытались улучшить способность к гидроксилированию штамма Streptomyces carbophilus, используя метод генной инженерии. Для гидроксилирования компактина необходима система цитохром Р-450 монооксигеназы (Matsuoka et al., Eur. J. Biochem. 184, 707-713, 1989). Однако, по мнению этих авторов, в бактериальной системе цитохром Р-450 монооксигеназы в транспорте электронов участвует не один, а несколько белков, что усложняет использование генной инженерии. Разработка эффективного с точки зрения стоимости способа микробиологического гидроксилирования для получения правастатина является очень трудной и комплексной задачей.
Целью настоящего изобретения является детальная разработка нового микробиологического процесса получения правастатина из компактина в промышленных масштабах, который продуцировал бы правастатин в более предпочтительных условиях, чем известные ранее. В наших исследованиях мы, помимо этого, старались найти штамм микроорганизмов с гидроксилазой, адаптированной для микробных превращений компактина в правастатин в значительных концентрациях.
КРАТКОЕ СОДЕРЖАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение касается микробного процесса получения соединения формулы (I) исходя из соединения субстрат формулы (II),
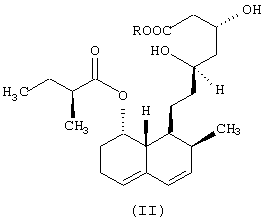
в которой R означает щелочной металл или ион аммония, включающего стадии: (а) культивирования штамма нитевидных плесеней вида Mortierella maculata, способных к 6β -гидроксилированию соединения формулы (II) на питательной среде, содержащей усвояемые источники углерода и азота, а также минеральные соли, (б) подачи указанного субстрата для его превращения в культуру Mortierella maculata, (в) ферментации указанного субстрата вплоть до окончания биоконверсии, (г) отделения соединения формулы (I) от культурального бульона и (д) выделения указанного соединения формулы (I).
Настоящее изобретение касается также биологически чистой культуры штамма Mortierella maculata n. sp. E-97, депонированного Национальной коллекцией сельскохозяйственных и промышленных микроорганизмов Венгрии (National Collection of Agricultural and Industrial Microorganisms, Budapest, Hungary) под инвентарным номером NCAIM(P)F 001266, а также биологически чистой культуры ее мутанта-штамма Mortierella maculata n. sp. Е97/15/13, депонированного Национальной коллекцией сельскохозяйственных и промышленных микроорганизмов Венгрии под инвентарным номером NCAIM(P)F001267.
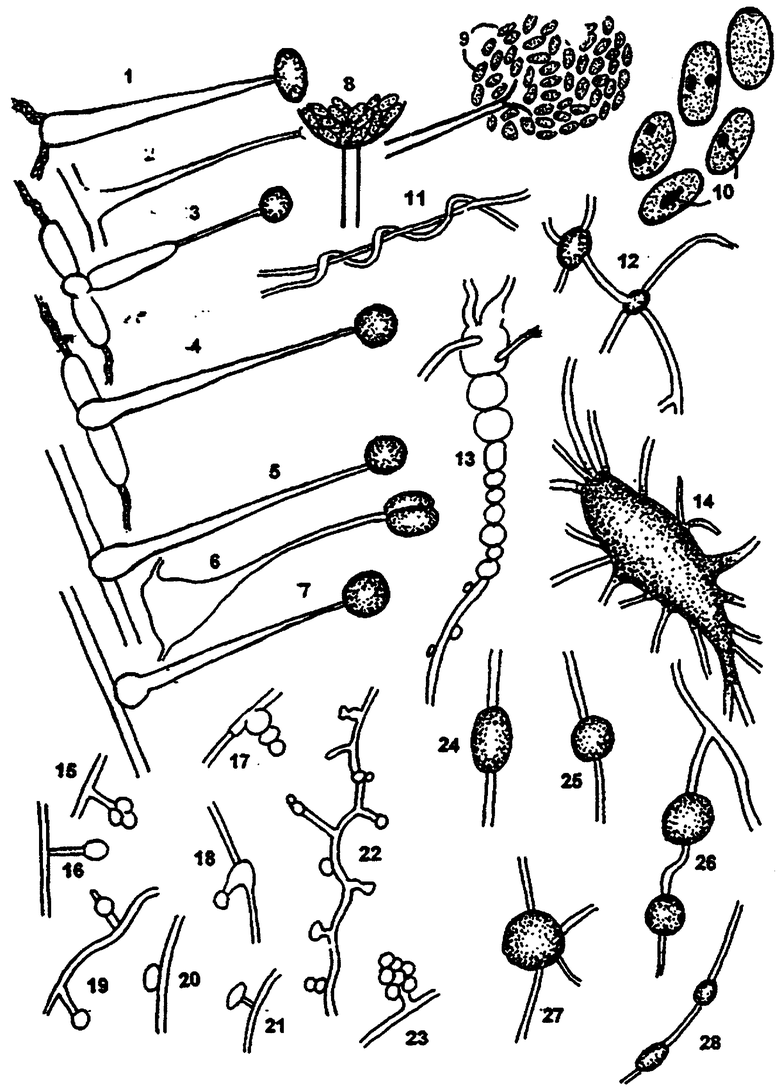
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
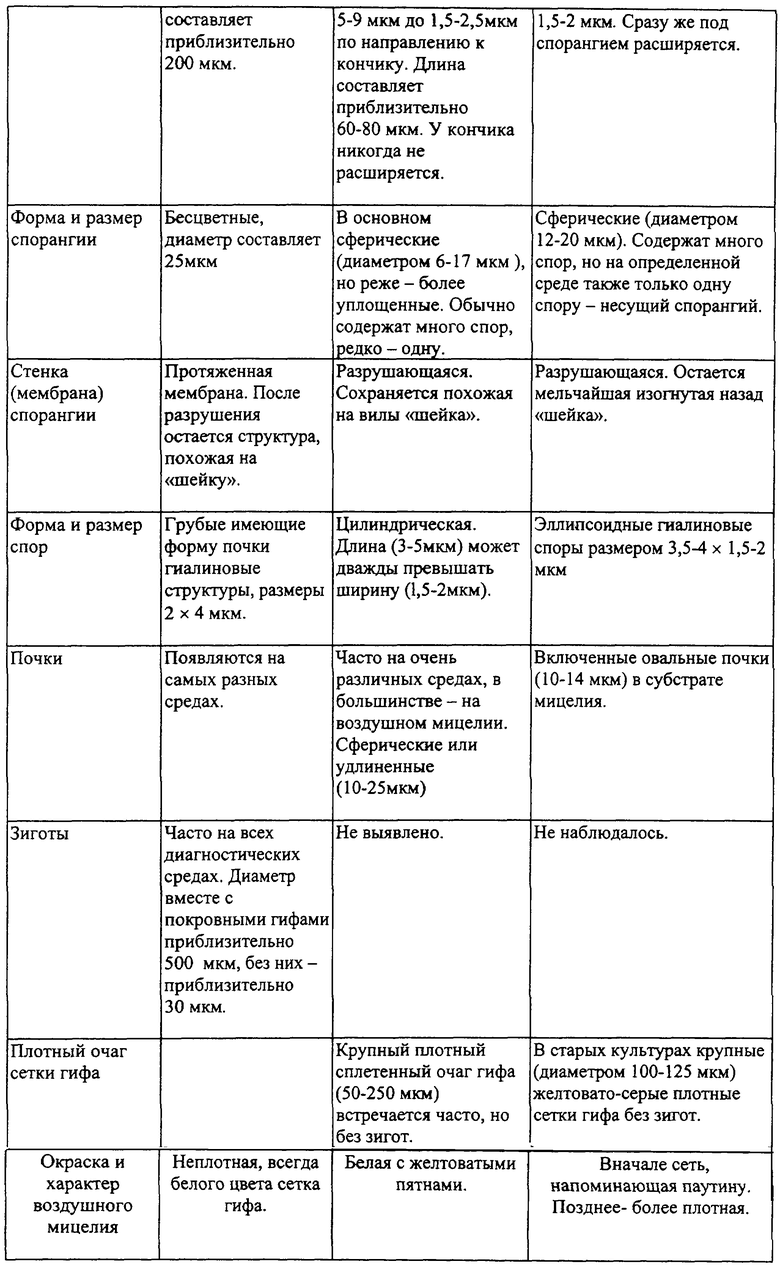
Чертеж представляет иллюстрацию физических характеристик штамма Mortierella maculata n. sp. E-97.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
В ходе нашей предварительной программы, которая охватывала приблизительно 5500 штаммов прокариот и эукариот, было отобрано 23 микроорганизма, способных, напротив, гидроксилировать компактин. Из этих штаммов наиболее пригодными для продуцирования провастатина оказались нитевидные плесени за счет их более высокой устойчивости к компастину по сравнению со штаммами, известными по опубликованным патентам. Согласно таксономическому исследованию этот штамм является новым представителем вида, принадлежащего семейству Mortierella (Mortierella maculata n.sp.). Из отобранных плесеней новый штамм был выделен, с одной стороны, за счет использования методов мутации и отбора, а, с другой стороны, за счет введения гидроксилазы в штамм, который только один был способен гидроксилировать субстрат компастина, превращая его в правастатин в более высоких концентрациях, чем это публиковалось ранее. В качестве мутагенных факторов использовались физические и химические мутагены (УФ-облучение, метилметансульфонат, N-метил-N’-нитpo-N-нитрозогуанидин). После этих мутагенных обработок в целях получения гаплоидных клеток суспензию спор распыляли на чашки Петри с агаром, содержащим беномил. Затем для того, чтобы ввести гидроксилазу, разросшиеся колонии инокулировали на чашки Петри, содержащие 100 мкг/мл 8-де-(2-метилбутирил)-компактина или компактина. Путем применения этих способов из указанного нового штамма был получен мутантный штамм, способный превратить компактин в правастин в более значительной степени, чем это делает родительский штамм.
В ходе экспериментов по оптимизации мы определили состав наиболее полезного инокулята, а также среду, наиболее предпочтительную для биоконверсии компактина его гидроксилированием, и оптимальный способ повторной подачи компактина в более высокой концентрации.
Следовательно, настоящее изобретение основано на том, что штаммы выделенной плесени Mortierella maculata обозначенные как Е-97 и Е-97/15/13 и депонированные под инвентарными номерами NCAIM(P)F 001266 и NCAIM(P)F 001267 соответственно Национальной коллекцией сельскохозяйственных и промышленных микроорганизмов Венгрии (National Collection of Agricultural and Industrial Microorganisms; Department of Microbiology and Biotechnology. University of Horticulture and the Food Industry Budapest), при соответствующих условиях ферментации способны продуцировать правастатин в значительной степени, в то время как нежелательные родственные соединения, такие как кислотные формы 6α -гидроксикомпактин, 2α -гидроксикомпактин, 8-де-(2-метилбутирил)-компактин, 3α ,5β -дигидрокси-5,6-дигидроизокомпактин, 8α ,β -гидроксикомпактин и гидроксилированные производные в положениях 2 и 3 в 2-метилбутирильной боковой цепи компактина получают только в небольших количествах или как примеси в процессе биоконверсии. Таким образом, эти штаммы особенно пригодны для продуцирования правастатина в промышленных масштабах.
Принимая во внимание, что экономное производство активного ингредиента в промышленном масштабе является функцией концентрации субстрата компактина, важно иметь такой штамм, который был бы способен выдерживать высокие концентрации компактина и правастатина. Следовательно, другой важной частью настоящего изобретения является признание того, что гидроксилирующую способность исходного изолята плесени можно повысить применением мутации и селекции, а также способом введения фермента; а, кроме того, разработка подходящего способа подачи субстрата делает возможным осуществить в едином процессе гидроксилирование компактина в правастатин в больших количествах. В итоге новый мутантный штамм, обозначенный как Mortierella maculata n. sp. E-97/15/13, особенно пригоден для получения правастатина.
Ниже суммированы таксономические характеристики выделенного нового вида плесени, сравниваемые с наиболее важными диагностическими атрибутами известных видов Mortierella species.
Таксономическое описание голотипа штамма Mortierella maculata nov.spec. E-97
Указанный воздушный мицелий на среде казеин-солодовый экстракт-агар рос хорошо (толщина покровного слоя над субстратом мицелия составила более 10 мкм). Вначале он появляется в виде плотно сплетенной белой сетки гифов, в которых позднее негусто возникают желтоватые спорулирующие пятна, имеющие диаметр в несколько миллиметров (новое название "maculatus" относится к упомянутому выше: пятнистый). Такое желтоватое окрашивание иногда может занимать более значительные непрерывные площади указанного воздушного мицелия. Окраска субстрата этого мицелия на средах: Czapek-, кровяная-Czapek-, тирозин-, крахмал-казеин-, солодовый экстракт-агар и т.д. чаще всего бесцветная или слегка желтоватая. Окраска сетки субстрата мицелия на среде дрожжевой экстракт-глюкоза-пептон является слегка красноватой. Продуцирование на упомянутых выше средах диффундируемых и растворимых пигментов не наблюдалось, на этих средах иногда возникало незначительное желтоватое окрашивание. За счет своего продуцирования эфирного масла и благодаря схожести с многими другими видами Mortierella (за исключением вида группы Isabellina) колонии штамма Е-97 могут выделять очень характерный сильный запах.
Спорангиеносцы, обозначенные на чертеже цифрами 1-7, часто локально растут на воздушных гифах (но реже на его субстрате) в больших количествах при расстояниях, очень различных друг от друга. Они не сильно разветвленные, а в большинстве своем прямые или изогнутые. Их длина обычно составляет 60-80 мкм. Исходной точкой в подавляющем большинстве случаев является более или менее короткий, но сильно разбухший гифальный участок воздушной сетки, от которой они отделены стенками. Сами по себе спорангиеносцы тоже могут быть разбухшими (иногда сильно), как это показано под номером 6, однако в направлении спорангия они постепенно сужаются от 5,0-9,0 мкм до 1,0-2,0 мкм. Важной таксономической характеристикой является то, что ниже указанных спорангиеносцев они никогда не расширяются (см. ссылку 8).
Спорангии имеют сферическую форму, в некоторых случаях они представляют собой слегка уплощенные сферы. Их диаметр составляет приблизительно 6,0-17,0 мкм, и эта величина сравнительно небольшая по сравнению с размерами спорангия других видов Mortierella. Спорангия может содержать много спор, но несущий спорангий содержит только одну спору. Споры 9 цилиндрические или менее овальные. Их размер составляет 3,0-5,0× 1,5-2,0 мкм. Внутри отдельных спор могут находиться одна или две небольшие темные сферические масляные капли 10; за счет очень легкого разрушения стенок указанной спорангии во влажной среде споры должны быстро рассеиваться. После такого разрушения спорангия иногда на конце спорангиоспор может наблюдаться мелкая, похожая на вилы ″ шейка″ и очень короткая рудиментарная (и нетипичная) колумелла. На различных диагностических средах могут появляться почки 15-28 сферической или цилиндрической формы. Обычно их размер составляет 10-25 мкм. В цепочках культур сферической почки 13, привитых клеток, вставной почки 15-23, гифальных объединениях определенного одного гифа со спиральным ростом вокруг других 11 и т.п., также могут быть найдены структуры, подобные анастомотическим, и гигантоциты. В указанном воздушном мицелии также можно увидеть большие (диаметром 50-250 pm) очень плотные гифальные сетки 14 без присутствия выявляемых зигот.
Культуры штамма Е-97 способны восстанавливать нитраты до нитритов, они не гидролизуют крахмал, эскулин, аргинин или желатин, но гидролизуют полисорбаты твина и не разлагают парафиновые углеводороды. Указанные культуры штамма Е-97 обладают уреазной активностью, имеют хороший рост при рН 7,0-9,0; выдерживают максимум 2% NaCl. Воздействие ксантина, гипоксантина, лецитина, тирозина и аденина негативно. Значительное продуцирование кислоты этими культурами было обнаружено из глюкозы, фруктозы, глицерина и галактозы, но очень слабое продуцирование (или отсутствие такого продуцирования) из ксилозы, арабинозы, раффинозы, сорбита, инозита, инулина и т.п. Слабый рост был выявлен на пирувате и ацетате, но никакого роста не наблюдалось в случае бензоата, салицилата, цитрата, лактата, сукцината, тартрата и малоната. Хороший рост наблюдался с глюкозой и фруктозой как единственными источниками углерода в среде. Было доказано, что проба на применение ксилозы, арабинозы, рамнозы, сахарозы, раффинозы, маннита и инозита была негативна. Указанные культуры не разлагают целлюлозу.
Систематизированное положение: штамм Е-97 принадлежит к семейству Mortierellaceae и является типичным представителем вида Monierella: спорангия обычно содержит много спор, колумелла значительно сокращена, часто присутствуют почки, присутствие зигот не было выявлено, а колонии имеют очень характерный сильный запах. Внутри вида Monierell штамм Е-97 является типичным представителем группы Альпина ("Section Alpina"). Последнюю можно охарактеризовать очень короткими неразветвленными спорангиеносцами (максимальная длина до 200 мкм), а также мельчайшей спорангией (Zycha, H. und Siepmann, R, Mucorales. Erne Beschreibung aller Gattungen und Arten dieser Pilzgmppe. D-3301 Lehre, Verl. von J. Cramer. 1969). Среди членов указанной группы Альпина штамм Е-97 больше всего сходен с видами М. thaxteri Bjorling 1936 и М. renispora Dixon-Stewart 1932. Однако данные из Таблицы 1 ясно показывают разницу между диагностическими свойствами штамма Е-97 и этих двух видов. Следовательно, в качестве нового вида мы представляем штамм под названием Mortierella maculata nov.spec. Е-97.

В процессе получения правастатина по настоящему изобретению предпочтительно использовать культуру плесневого штамма, обозначенного Mortierella maculata n.sp. E-97 или его мутанта, обозначенного E-97/15/13. Выбранный штамм препочтителен благодаря своему быстрому росту. В качестве источника углерода легко использовать глюкозу, глицерин, фруктозу или галактозу. В качестве источника азота можно использовать экстракт дрожжей, пептон, казеин, экстракт мяса, соевую муку, жидкость для замачивания кукурузы, нитрат натрия или сульфат аммония.
В культуральной среде, используемой для продуцирования правастатина, помимо указанных выше источников углерода и азота, могут присутствовать минеральные соли, например вторичный кислый фосфат калия, хлорид магния, сульфат магния, примесные элементы (железо, соли марганца), аминокислоты и антипенные добавки.
Согласно предпочтительному воплощению настоящего изобретения суспензию спор, приготовленную на скошенном агаре из культуры штамма Mortierella maculata n.sp. E-97 или его мутанта [NCAIM(P)F 001267], обозначенного Е-97/15/13, засевают в инокулярную среду; затем 10% этой инокулярной культуры, которую культивировали в течение 3 суток при температуре, составляющей приблизительно 25-30° С (предпочтительно приблизительно 24-28° С, а более предпочтительно приблизительно 28° С), переносят в биоконверсионную среду. Затем ее инкубируют в течение 4 суток при температуре, составляющей приблизительно 25-28° С (предпочтительно приблизительно 28° С), после чего в эту культуру вводят глюкозу и натриевую соль компактина. В зависимости от концентрации подаваемого субстрата компактина культивирование продолжают еще 2-12 суток в аэробных условиях; при этом рН поддерживают в пределах 5,5-7,5, предпочтительно на уровне значения 7,0; биоконверсию проводят при перемешивании и аэрировании при скорости подачи воздуха 0,2 vvm и скорости вращения мешалки 400/мин.
В ходе ферментации после биоконверсии субстрата компактина проводили жидкостную хроматографию высокого давления (ВЭЖХ). Согласно этому способу образец бульона разбавили вдвое метанолом и провели центрифугирование, а супернатант использовали для анализа методом ВЭЖХ. Параметры процесса следующие: оборудование аналитической ВЭЖХ; колонка: Nucleosil C1810 мкм; выявляемая длина волны: 238 нм; впрыскиваемый объем: 20 мкл, скорость подачи: 1 мл/мин; использовалось градиентное элюирование, элюенты: А=0,05% водный раствор фосфорной кислоты, В=ацетонитрил.
Градиентное элюирование:
Приблизительное время удержания: правастатин 8,6-9,0 мин; кислота компактина 11,6-12,0 мин; лактон правастатина 15,0-15,5 мин; компактин 16,5-17,0 мин.
В целях продуцирования правастатина водный раствор натриевой соли компактина добавляют на 96 часу культивирования. Для этой процедуры субстрат приготавливают в твердой форме следующим образом. Лактон компактина гидролизуют в 0,2 М растворе гидроксида натрия в течение 2 ч при 40° С, затем с помощью соляной кислоты рН реакционной смеси доводят до значения 7,5 и полученный нейтрализованный раствор помещают в поглотительную колонку Diaion HP-20. Хлорид натрия, образовавшийся в процессе нейтрализации, удаляют, осуществляя промывку указанной колонки водой, а затем натриевую соль компактина вымывают из колонки 50% водным ацетоном. После этого элюат перегоняют в вакууме, а полученный водный остаток лиофилизируют. После нейтрализации полученный водный раствор щелочного гидролизата компактина можно также непосредственно использовать в качестве субстрата. В этом случае содержание кислой натриевой соли компактина в указанном гидролизате определяют методом ВЭЖХ, а получившийся раствор сохраняют при 20° С вплоть до использования.
Чем более высокого значения достигает величина рН на 4 день ферментации, тем это предпочтительнее для гидроксилирования субстрата компактина. Подачу субстрата компактина допускается начать, когда рН указанного бульона превышает значение 6,3. На 4 день ферментации добавляют такое количество стерильного отфильтрованного водного раствора кислой натриевой соли компактина, которое необходимо для получения концентрации 500 мкг/мл. В культуру подают также глюкозу из ее 50% раствора, простерилизованного при 121° С в течение 25 мин по следующей схеме: если рН указанного бульона выше 6,7, то добавляют 1% глюкозу в количестве, соответствующем объему этого бульона, а если рН составляет 6,3-6,7, то количество подаваемой глюкозы составляет 0,5%. Кислую натриевую соль компактина используют из указанного бульона спустя 24 ч, ее превращение анализируют методом ВЭЖХ. В этом случае на каждый мл указанного бульона добавляют еще 500 мкг компактина. Помимо субстрата компактина, подают также и глюкозу так, как это было описано выше. Морфология полученной 120 ч культуры характеризуется ростом небольших шариков (с диаметром 0,5-3,0 мм). Спустя 24 ч используют также и вторую дозу субстрата из этого бульона, таким образом, параллельно с подачей глюкозы, зависимой от рН бульона в целом, добавляется еще одна порция, продуцирующая кислую натриевую соль компактина в концентрации 500 мкг/мл бульона. Начиная с 4 дня ферментации, повторяют подачу указанного субстрата и глюкозы, проводя ее ежедневно так, как это описано выше, вплоть до 17-18 дня ферментации.
Для извлечения продукта из бульона предпочтительно принимать во внимание тот факт, что в процессе биоконверсии правастатин образуется в своей кислой форме, поэтому его можно выделить из фильтрата бульона путем абсорбции на колонке с анионообменной смолой. Для выделения указанного продукта предпочтительно использовать сильно основную анионообменную смолу, которая представляет собой полистиролдивинилбензольный полимер с активными группами четвертичного аммония. Этот продукт можно непосредственно абсорбировать из фильтрата бульона путем примешивания к нему анионообменной смолы, находящейся в гидроксильной форме. Продукт, абсорбированный на указанной анионообменной смоле, можно элюировать из колонки, используя уксусную кислоту или водно-ацетоновую смесь, содержащую хлорид натрия (предпочтительно 1% хлорид натрия) в водно-ацетоновой (1:1) смеси. Фракции, содержащие правастатин, объединяют, а ацетон, присутствующий в элюате, отгоняют в вакууме. С помощью 15% серной кислоты рН концентрата доводят до значения 3,5-4,0, а водный раствор экстрагируют этилацетатом. Полученный этилацетатный экстракт промывают водой и высушивают безводным сульфатом натрия. Потом из правастатина получают его лактон. Замыкание лактонового цикла осуществляют в высушенном растворе этилацетата при комнатной температуре и постоянном перемешивании, процесс образования лактона проводят с помощью трифторуксусной кислоты, присутствующей в каталитических количествах. Происходящее превращение проверяют методом тонкослойной хроматографии. После того как процесс образования лактона завершен, раствор этилацетата промывают сначала 5% водным раствором кислого карбоната, а затем водой, после чего высушивают с помощью безводного сульфата натрия и упаривают в вакууме. Упаренный остаток в ацетоновом растворе обрабатывают древесным углем, а потом упаривают снова и перекристаллизовывают из алифатического спирта, содержащего 1-4 атома углерода, предпочтительно из этанола. Упаренный после перекристаллизации остаток маточника очищают на колонке с силикагелем, применяя в качестве элюента смесь этилацетат-н-гексан, при постепенном увеличении содержания этилацетата.
Из лактона правастатина, полученного после перекристаллизации и хроматографической очистки, правастатин получают гидролизом эквивалентным количеством гидроксида натрия при комнатной температуре в ацетоне. Когда образование натриевой соли правастатина завершено, реакционную смесь разбавляют водой и нейтрализуют, а ацетонную составляющую перегоняют в вакууме. Правастатин адсорбируют из полученного водного остатка на колонке Diaion HP-20, содержащей смолу, промывают деионизированной водой и элюируют из этой колонки смесью ацетон-деионизированная вода. Потом фракции, содержащие правастатин, объединяют, ацетоновую составляющую отгоняют и после лиофилизации водного остатка можно получить высокой степени чистоты правастатин, который можно перекристаллизовать из смеси этилацетат-этанол.
В ходе этой процедуры может быть адсорбировано все количество правастатина. При замыкании лактонового цикла правастатина также могут образовываться 3β -гидроксиизокомпактин и другие побочные продукты. Хотя последние реакции снижают выход, но указанные соединения можно отделить описанными выше методами очистки и, следовательно, этим способом можно получить правастатин, качество которого приемлемо с фармацевтической точки зрения.
После завершения биоконверсии правастатин можно экстрагировать или из ферментационного бульона, или из фильтрата, полученного после отделения клеток нитевидной плесени. Нитевидные клетки плесени можно удалить или фильтрованием, или центрифугированием; однако предпочтительно, особенно в промышленных масштабах, проводить экстракцию всего бульона целиком. Перед экстракцией рН или ферментационного бульона, или фильтрата, полученного из бульона, доводят до значения 3,5-3,7 при использовании минеральной кислоты (предпочтительно - разбавленной серной кислоты). Экстракцию проводят эфиром уксусной кислоты и алифатического спирта, содержащим 24 атома углерода, предпочтительно этилацетатом или изобутилацетатом. Стадии экстрагирования следует проводить быстро, чтобы предотвратить образование при кислом значении рН лактонового производного правастатина.
Из экстракта органического растворителя правастатин в кислой форме в виде соли натрия можно перенести в водную фазу. Например, из экстракта этилацетата правастатин можно экстрагировать при использовании 1/10 и 1/20 объемного соотношения 5% кислого карбоната натрия или слабо щелочной водой (рН 7,5-8,0). Было установлено, что в чистой форме правастатин можно извлечь из полученного выше щелочного водного экстракта колоночной хроматографией с применением неионогенных смол. Предпочтительный способ заключается в том, что вначале перегонкой в вакууме надо удалить растворитель, растворенный в водной фазе, а затем полученный водный экстракт загружают в колонку Diaion HP-20.
Натриевую соль правастатина, адсорбированную на колонке, очищают путем промывания, постепенно увеличивая содержание ацетона в растворе, потом фракции, содержащие правастатин, объединяют и сгущают в вакууме. Полученный водный концентрат очищают далее хроматографией на другой колонке Diaion HP-20, получая при этом элюат, содержащий чистый правастатин, из которого после осветления древесным углем и лиофилизации можно получить правастатин, качество которого приемлемо с фармацевтической точки зрения.
Эта методика выделения включает меньшее количество стадий, чем предыдущая, поскольку в нее не входят образование лактона правастатина и его гидролиз. При изоляции правастатин подвергают воздействию кислых условий только на ограниченное время, во время которого он менее стабилен, чем в нейтральных или щелочных растворах, следовательно, при такой методике выделения практически не образуется артефактов.
Кроме того, было найдено, что для очистки правастатина предпочтительно использовать хроматографию на геле Sephadex LH-20 Dextran (гидрокиспропилированное производное). Путем применения такого способа можно получить правастатин сверхвысокой чистоты, составляющей 99,5% (по измерениям ВЭЖХ).
В ходе нашего эксперимента было установлено, что правастатин может быть осажден в виде кристаллической соли вместе со вторичными аминами из экстракта органического растворителя (предпочтительно из этилацетатного или изобутилацетатного экстракта) указанного бульона или из нитрата бульона нитевидной плесени или штаммов нитевидных бактерий, среди них штамм Mortierella maculata n. sp., способный к 6β -гидроксилированию соединения общей формулы (II). Далее было обнаружено, что для указанного солеобразования пригодны некоторые вторичные амины, содержащие алкильные, циклоалкильные, аралкильные или арильные заместители. Соответственно были выбраны нетоксичные вторичные амины и среди них диоктиламин, дициклогексиламин, дибензиламин. Выделение указанных промежуточных солей органических вторичных аминов, а именно соли дибензиламина, проводили путем добавления дибензиламина в количестве 1,5 эквивалента относительно содержания правастатина в указанном экстракте. После этого экстракт сгущают перегонкой в вакууме до 5% его исходного объема, затем к этому концентрату добавляют еще дибензиламин при соотношении в 0,2 эквивалента. Кристаллическую соль дибензиламина осаждают из этого концентрата. Неочищенный кристаллический продукт отфильтровывают и высушивают в вакууме. Затем его осветляют древесным углем и перекристаллизовывают в ацетоне.
В упомянутой выше методике, которая включает экстракцию органического растворителя и повторную экстракцию при щелочном значении рН, в целях замены очистки колоночной хроматографией можно также использовать способ выделения, основанный на образовании соли вторичного амина. В этом случае предпочтительно осаждать дибензиламиновую соль правастатина из изобутилацетатного экстракта, полученного после подкисления водного щелочного экстракта.
Органические соли вторичного амина правастатина можно превратить в правастатин с помощью гидроксида натрия или алкоксида натрия (предпочтительно этоксида натрия).
Это превращение детализировано для случая дибензиламиновой соли правастатина. Перекристаллизованную дибензиламиновую соль правастатина суспендируют в смеси изобутилацетат-вода, затем к полученной суспензии добавляют эквивалентное количество гидроксида натрия в водном растворе, поддерживая при перемешивании значение рН в пределах 8,0-8,5. После исчезновения суспензии полученные фазы разделяют и содержащий правастатин водный раствор дважды промывают изобутилацетатом. Полученный водный раствор осветляют активированным углем и лиофилизируют, получив при этом правастатин фармацевтически приемлемого качества.
Одним предпочтительным способом превращения дибензиламиновой соли правастатина в правастатин является суспендирование перекристаллизованной дибензиламиновой соли правастатина в этаноле с последующим добавлением к этой суспензии при перемешивании эквивалентного количества этоксида натрия (или его небольшой избыток), сгущение полученной реакционной смеси в вакууме и осаждение в кислой форме правастатина из этого концентрата путем дополнительного введения ацетона.
Другой предпочтительный способ превращения дибензиламиновой соли правастатина в правастатин состоит в растворении в смеси этилацетат-вода перекристаллизованной дибензиламиновой соли правастатина и в осаждении правастатина путем добавления к полученному раствору эквивалентного количества (или небольшого избытка) гидроксида натрия в этаноле.
Выделение правастатина через промежуточную соль вторичного амина представляет собой более простую методику, чем любая из известных методик выделения. При ее осуществлении не происходит образования никаких артефактов и отделение правастатина от побочных продуктов биоконверсии и от различных метаболитов, синтезированных гидроксилирующими микроорганизмами, можно решить без использования какой-либо из хроматографических методик.
Структуры правастатина, лактона правастатина и изолированной соли вторичного амина правастатина были подтверждены методами УФ-анализа, инфракрасного анализа, 1H-ЯМР, 13С-ЯМР и масс-спектрометрии.
ПРИМЕРЫ
Настоящее изобретение будет более полно описано и будет лучше понятно со ссылкой на следующие далее примеры, которые приведены для иллюстрации, но не предназначены, чтобы каким-либо образом ограничить настоящее изобретение.
Пример 1
Суспензию спор получили с помощью 5 мл 0,9% раствора хлорида натрия из 7-10-дневной культуры штамма Mortierella maculata nov.spec. E-97 [NCAIM(P)F 001266] на скошенном агаре экстракта солода-дрожжевого экстракта, способного к 6β -гидрокислированию компактина. Полученную суспензию использовали для инокулирования 100 мл посевной среды PI, простерилизованной в 500 мл колбе Эрленмейера.
Состав среды PI:
глюкоза 50 г;
соевая мука 20 г
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН этой среды довели до значения 7,0, после чего среду стерилизовали при 121° С в течение 25 мин. Культуру встряхивали в ротационном шейкере (250 об/мин, амплитуда 2,5 см) в течение 3 дней при 28° С. Затем 10 мл полученной культуры перенесли в 100-100 мл среды для биоконверсии MUM, которую стерилизовали в 500 мл колбе Эрленмейера в течение 25 мин при 121° С.
Состав среды MUM:
глюкоза 40 г
соевая мука 20 г
казеин-пептон 1 г
аспарагин 2 г
вторичный кислый фосфат
калия 0,5 г
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН этой среды довели до значения 7,0, после чего среду стерилизовали при 121° С в течение 25 мин.
Колбы встряхивали в ротационном шейкере (250 об/мин, амплитуда 2,5 см) в течение 4 дней при 25° С, затем в них из этих культур в стерильной и отфильтрованной форме ввели 50-50 мг субстрата компактина (натриевая соль кислоты компактина) и продолжили культивирование. Аналогично на 5 день в культуру плесени добавили еще 50-50 мг субстрата компактина и продолжали ферментацию еще 24 ч. Содержание правастатина в бульоне определяли методом ВЭЖХ. Ферментацию продолжали 168 ч. В конце биоконверсии средняя концентрация правастатина в ферментационном бульоне составила 620 мкг/мл.
Пример 2
В лабораторном ферментере с рабочим объемом в 5 л была приготовлена биоконверсионная культуральная среда MU/S. Компоненты этой среды добавляли в количествах, соответствующих 5 л объему, но произвели загрузку только до 4,5 л. Потом среду стерилизовали в течение 45 мин при 121° С и посев 500 мл инокулята проводили в соответствии с примером 1.
Состав среды MU/S:
глюкоза 20 г
глицерин 20 г
соевая мука 20 г
пептон 5 г
вторичный кислый фосфат калия 0,5 г
полипропиленгликоль 2000 1 г
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН этой среды довели до значения 7,0. Ферментацию проводили в течение 4 дней при 28° С и скорости перемешивания 400 об/мин и при скорости аэрирования в направлении от дна, составляющей 60 л/ч. На второй день после переноса началось сильное пенообразование культуры, которое можно снизить добавлением полипропиленгликоля 2000. В начале ферментации (16-20 ч) рН от своего первоначального значения в 6,5 снизился до величины 5,0-5,5; затем с 3 дня его значение стало увеличиваться и на 4 день достигло величины 6,3-7,5. Подача субстрата компактина допускается, если рН бульона выше 6,3. На 4 день ферментации добавляют 2,5 г субстрата компактина в стерильном отфильтрованном растворе. В полученную культуру параллельно с подачей субстрата добавили вычисленное для этого объема бульона количество 0,5-1,0% глюкозы в зависимости от рН в виде 50% раствора, простерилизованного при 121° С в течение 25 мин. Спустя 24 ч используют субстрат компактина из этой культуры, его выявляют методом ВЭЖХ в образцах, отобранных из ферментера. В этом случае добавили еще 2,5 г субстрата компактина и глюкозу так, как это было описано выше, и биоконверсию продолжали еще 24 ч, в течение которых указанный субстрат был превращен в правастатин.
По окончании ферментации 5,1 л бульона, содержащего 630 мкг/мл правастатина, отфильтровали через фильтровальную ткань. К отделенному мицелию добавили 2 л воды, потом суспензию перемешивали 1 ч и отфильтровали. Оба эти фильтрата объединили и со скоростью 500 мл/ч пропустили через колонку, содержащую 138 г (250 мл) смолы Dowex A1 400 (ОН) (диаметр указанной колонки 3,4 см, высота слоя смолы 28 см), и затем слой смолы промыли 300 мл деионизированной воды. Затем проводили элюирование этой смолы с помощью 1 л смеси ацетон-вода (1:1), содержащей 10 г хлорида натрия. Объем каждой фракции составил 100 мл. Полученный элюат анализировали тонкослойной хроматографией по следующей схеме: адсорбент: Kieselgel 60 F 254 DC (Merck) алюминиевая фольга: проявитель: смесь ацетон-бензол-уксусная кислота (50:50:3); выявление: фосфомолибденовым кислым реагентом. Величина Rf для правастатина составила 0,5. Фракции, содержащие продукт, объединили и ацетон выпарили в вакууме. Величину рН полученного 400 мл концентрата довели до значения 3,5-4,0 с помощью 15% серной кислоты, потом его трижды экстрагировали 150 мл этилацетата. Этилацетатные экстракты объединили и высушили с помощью безводного сульфата натрия. После этого из кислоты правастатина получают лактон правастатина путем добавления при комнатной температуре с непрерывным перемешиванием каталитических количеств трифторуксусной кислоты. Образование лактона правастатина контролируют методом тонкослойной хроматографии (величина Rf лактона правастатина для этой системы тонкослойной хроматографии составила 0,7). После завершения образования лактона этилацетат промыли 2× 50 мл 5% водного раствора кислого карбоната натрия, затем промыли 50 мл воды, высушили с помощью безводного сульфата натрия и упаривали в вакууме. Остаток, полученный после упаривания, в количестве 3 г растворили в 100 мл ацетона и провели осветление 0,3 г древесного угля. Потом древесный уголь отфильтровали, а ацетон выпарили в вакууме. Полученный сырой продукт кристаллизовали из 20 мл этанола. Осажденный кристаллический лактон правастатина отфильтровали, промыли на фильтре 30 мл н-гексана и высушили при комнатной температуре в вакууме. Таким способом было получено 1,5 г хроматографически чистого лактона правастатина. Температура плавления 140-142° С, [α ]D=+194° (с=0,5, метанол). Маточный раствор от кристаллизации упарили в вакууме и получили 1,2 г остатка после упаривания, который подвергли хроматографии на 24 г колонке Kieseigel 60, содержащей адсорбент (диаметр колонки: 1,6 см; высота слоя: 20 см). Сырой продукт, растворенный в 5 мл бензола, поместили в эту колонку. Для элюирования использовали смеси этилацетат-н-гексан, в которых содержание этилацетата постепенно увеличивалось. Лактон правастатина можно элюировать из колонки смесью 60% этилацетат-40% н-гексан. Фракции контролировали методом тонкослойной хроматографии при использовании в качестве проявителя смеси этилацетат-н-гексан (9:1). Фракции, содержащие лактон правастатина, объединили и упарили в вакууме. Согласно этому способу было получено 0,3 г чистого продукта, качество которого идентично качеству лактона правастатина, полученного кристаллизацией.
Эти две партии лактона правастатина объединили и натриевую соль приготовили согласно следующему способу: 1,8 г лактона правастатина растворили в 20 мл ацетона и при перемешивании добавили 4,5 мл 1 М водного гидроксида натрия, потом полученный раствор перемешивали еще полчаса при комнатной температуре. Когда образование соли завершилось, в полученную смесь добавили 20 мл воды и раствор нейтрализовали, а ацетон выпарили в вакууме. Водный концентрат подвергли хроматографии на колонке, заполненной 150 мл смолы Diaion HP 20 (диаметр колонки: 2,6 см; высота слоя: 30 см). В качестве элюирующего средства использовали смесь ацетон-деионизированная вода, где концентрация ацетона увеличивалась ступенчато на 5%. Правастатин можно элюировать из этой колонки с помощью смеси ацетон-деионизированная вода, содержащей 15% ацетона. Фракции анализировали методом тонкослойной хроматографии. Фракции, содержащие искомый продукт, объединили, а ацетон выпарили в вакууме. Путем лиофилизации водного остатка было получено 1,3 г правастатина. Хроматографически чистый продукт перекристаллизовывали из смеси этанола и этилацетата.
Температура плавления: 170-173° С (с разложением)
[α ]D 20=+156° , (с=0,5 в воде). УФ-спектр поглощения (20 мкг/мл в метаноле): λ mах=231,237,245 нм
(log ε -4.263; 4.311; 4.136)
Инфракрасный спектр поглощения (KBr): υ ОН 3415, υ CH 2965, υ C=O 1730, υ COO 1575 см-1.
1H-ЯМР спектр (D2O, δ , ppm): 0.86, d, 3Н (2-СН3); 5.92, dd, J=10,0 and 5,4 Hz, 1 H (3-H); 5.99, d, J=10,0 Hz, 1 H (4-H); 5.52, br 1 H (5-H); 4.24, m 1 H (6-H); 5.34, br, 1 H (8-H); 4.06, m, 1 H (β -H), 3.65, m, 1 H (δ -H); 1.05, d, 3 Н (2’-СН3); 0.82, t, 3Н (4’-Н3).
13С-ЯМР спектр (D2O, δ , ppm): 15.3, q (2-СН3); 139:5, d (C-3); 129.5, d, (C-4); 138.1, s (C-4a), 127.7, d (C-5); 66.6, d (C-6); 70.1, d (C-8); 182.6 s (COO-); 72.6. d (C-β ); 73.0, d (C-δ ); 182.0, s (С-1’) 18.8; g(2’-СН3); 13.7, g (С-4’).
Положительный масс-спектр FAB (характеристические ионы):
[M+Na]- 469; [М+Н]+ 447.
Отрицательный масс-спектр FAB (характеристические ионы):
[М-Н]- 445, [M-Na]- 423, m/z 101 [2-метилмасляная кислота-Н]-.
Пример 3
В лабораторном ферментере рабочим объемом на 5 л приготовили биоконверсионную культуральную среду MU/4 так, как это описано в примере 1, хотя его загрузили на 4,5 л, но состав культуральной среды подсчитывали для объема в 5 л. Среду стерилизовали 45 мин при 121° С и инокулировали 500 мл посевной культуры, приготовленной согласно примеру 1. Ферментацию осуществляли в течение 4 дней при 25° С при использовании скорости перемешивания 300 об/мин и при скорости аэрации 50 л/ч. После этого в указанную культуру осуществляли подачу 5 г субстрата компактина, а биоконверсию проводили согласно примеру 2.
После окончании биоконверсии 4,9 л бульона, содержащего 660 мкг/мл правастатина, отфильтровали и отделенный мицелий промыли суспензией в 2× 1 л деионизированной воды. рН объединенных 5,6 л фильтрата бульона с помощью 20% серной кислоты довели до значения 3,5-3,7; а потом подкисленный фильтрат перемешивали вместе с 2750 мл этилацетата в течение 30 мин. После этого образовавшиеся фазы разделили, водную фазу экстрагировали еще раз 2× 1375 мл этилацетата. К объединенному этилацетатному экстракту в 4740 мл добавили 470 мл деионизированной воды, потом рН водной этилацетатной смеси с помощью 1М гидроксида натрия довели до значения 7,5-8,0. После 20 мин перемешивания фазы разделили и затем этилацетатный экстракт экстрагировали 2× 235 мл деионизированной воды, как это было описано выше. Потом объединенный слабощелочной водный раствор объемом 1080 мл сгустили в вакууме до объема в 280 мл. Указанный концентрированный водный раствор поместили в хроматографическую колонку (соотношение высота: диаметр=6,5), заполненную 280 мл неионогенной смолы Diaion HP-20 (Mitsubishi Co., Japan). Поглощение в колонке осуществлялось при скорости потока 250-300 мл/ч, потом эту колонку промыли 840 мл деионизированной воды. После этого колонку элюировали водой, содержащей ацетон, в таком порядке: 800 мл 5% воды, 1000 мл 10% воды, 500 мл 15% воды и 500 мл 20% воды. При элюировании были собраны 50 мл фракции, которые проанализировали методом тонкослойной хроматографии, описанным в примере 2. Фракции, которые в качестве основного компонента содержали правастатин, объединили, а полученный раствор сгустили в вакууме до объема в 260 мл. Этот водный раствор подвергли хроматографии на колонке, содержащей смолу Diaion HP-20 в объеме 260 мл. После адсорбции правастатина колонку промыли 790 мл деионизированной воды, а затем провели элюирование порциями 260-260 мл водно-ацетоновых растворов, в которых содержание ацетона постепенно увеличивалось следующим образом: 2,5; 5,0; 7,5; 10,0; 12,5; 15,0 и 20,0%. В ходе колоночной хроматографии были собраны 25 мл фракции, а содержание правастатина в этих фракциях определяли так, как это было описано выше. Фракции, содержащие правастатин в качестве единственного компонента, объединили при тонкослойной хроматографии и упарили в вакууме. После этого к сконцентрированному водному раствору (приблизительно 30 мл) добавили 0,3 г древесного угля и при комнатной температуре правастатин осветляли в течение 30 мин. Затем древесный уголь отделили от раствора фильтрованием, а фильтрат подвергли лиофилизации. Таким способом было получено 1,62 г правастатина в лиофилизированной форме.
Пример 4
Из культуры штамма Mortierella maculata nov.spec. E-97 [NCAIM(P)F 001266] на скошенном агаре, культивируемой 10-12 дней, приготовили суспензию спор, используя 5 мл стерильного раствора 0,9% хлорида натрия. Эту суспензию применяли для инокулирования 500 мл среды VHIG, простерилизованной в 3000 мл колбе Эрленмейера.
Состав среды VHIG:
глюкоза 30 г
мясной экстракт 8 г
дрожжевой экстракт 1 г
Твин-80 (полиэтиленгликоль (20)
сорбитанмоноолеат) 0,5 г
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН среды довели до значения 7,0; а саму стерилизацию проводили в течение 25 мин при 121° С. Культуру культивировали 3 дня на ротационном шейкере (250 об/мин, амплитуда 2,5 см), а затем полученную среду использовали для посева в лабораторный ферментер, который имел 5 л рабочий объем и содержал биоконверсионную культуральную среду РК.
Состав среды РК:
глюкоза 40 г
пептон 5 г
соевая мука 20 г
К2НРO4 2 г
КН2РO4 1 г
NaNO3 2 г
КСl 0,5 г
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН указанной среды довели до значения 7,0. После инокуляции и культивирования подачу субстрата и биоконверсию проводили в соответствии с примером 2. После этого из полученного бульона выделили правастатин, концентрация которого к концу ферментации составляла 650 мкг/мл.
По окончании ферментации рН 4,9 л бульона, содержащего 650 мкг/мл правастатина, при непрерывном перемешивании с 2 М гидроксидом натрия довели до значения 9,5-10,0; а потом после 1 ч перемешивания значение рН довели до величины 3,5-3,7 используя для этого 20% серную кислоту. После этого полученный кислый раствор экстрагировали 2,45 л этилацетата. Образовавшиеся фазы разделили, а из эмульгированной органической фазы при центрифугировании получили прозрачный экстракт. Бульон еще раз экстрагировали 2× 1,22 л этилацетата согласно изложенному выше способу. Этилацетатные экстракты объединили и добавили 0,4 л деионизированной воды, а потом рН полученной смеси с помощью 1 М гидроксида натрия довели до значения 8,0-8,5. Фазы разделили и этилацетатную фазу экстрагировали 2× 0,2 л деионизированной воды с рН 8,0-8,5 так, как это было описано выше. рН объединенного правастатина, содержащего слабощелочной водный раствор, при перемешивании довели до значения 3,5-3,7, используя для этого 20% серную кислоту. Полученный кислый раствор экстрагировали 4× 0,2 л этилацетата. Объединенные этилацетатные экстракты промыли 2× 0,2 л деионизированной воды; потом в полученный этилацетатный раствор добавили 150 мол.% дибензамина (количество, вычисленное для содержания правастатина согласно методу ВЭЖХ). Полученный этилацетатный раствор сгустили в вакууме до объема в 0,2 л. К полученному концентрату после этого добавили 20 мол.% дибензамина и осажденный раствор выдерживали в течение ночи при температуре 0-5° С. Осажденную дибензаминовую соль правастатина отфильтровали, потом осадок промыли на фильтре сначала холодным этилацетатом, а потом - дважды н-гексаном; в завершении соль высушивали в вакууме при 40-50° С. Полученный сырой продукт (3,9 г) растворили в 100 мл метанола при комнатной температуре, а раствор осветляли с помощью 0,45 г древесного угля. После этого метанольный фильтрат сгустили в вакууме. Упаренный остаток растворили в 120 мл ацетона при внешней температуре в 62-66° С, затем полученный раствор охладили до комнатной температуры. После этого перекристаллизацию продолжали в течение ночи при 0-5° С. Осажденные кристаллы отфильтровали, их промывали на фильтре дважды холодным ацетоном и дважды н-гексаном. Перекристаллизованную дибензиламиновую соль правастатина суспендировали в смеси 160 мл изобутилацетата и 80 мл деионизированной воды. Затем к суспензии при перемешивании добавили эквивалентное количество гидроксида натрия. После исчезновения суспензии фазы разделили, водный раствор, содержащий правастатин, промыли 2× 30 мл изобутилацетата. Полученный водный раствор осветляли с помощью древесного угля. Затем водный фильтрат сгустили до объема, составляющего приблизительно 20 мл. Полученный водный раствор загрузили в хроматографическую колонку (высота: диаметр=22), заполненную 0,4 л геля Sephadex LH-20 (поставщик: Pharmacia, Sweden). В ходе проведения хроматографии в качестве элюента использовали деионизированную воду и были собраны 20 мл фракции. Последние анализировали методом тонкослойной хроматографии, а фазы, содержащие правастатин, анализировали также и методом ВЭЖХ, используя при этом описанный выше способ. Фракции, содержащие чистый правастатин, объединили и лиофилизировали. Таким образом было получено 1,75 г правастатина, чистота которого (по данным ВЭЖХ) была выше 99,5%.
Пример 5
Суспензию спор получили из культуры на скошенном агаре штамма Mortierella maculata n.spec. E-97 [NCAIM(P)F 001266], которую культивировали в течение 10-12 дней вместе с 5 мл стерильного 0,9% раствора хлорида натрия, а затем инокулировали с 500 мл среды так, как это описано в примере 4. В лабораторном ферментере с рабочим объемом в 5 л биоконверсионную культуральную среду РС/4 стерилизовали в течение 45 мин при 121° С, а затем инокулировали культурой для посева.
Состав среды РС/4:
солодовый экстракт 5,0%
соевая мука 1,0%
пептон 1,0%
жидкость для замачивания кукурузы 1,0%
MgSO4·7H2O 0,1%
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН указанной среды довели до значения 7,0. После инокуляции и культивирования подачу субстрата и биоконверсию проводили в соответствии с примером 2. После этого было получено 5,1 л бульона с концентрацией правастатина, составляющей 650 мкг/мл.
Из этого бульона способом, описанным в примере 4, было в качестве неочищенного продукта получено 3,7 г дибензиламиновой соли правастатина. Из последней путем ее перекристаллизации было получено 2,9 г дибензиламиновой соли правастатина. Перекристаллизованную дибензиламиновую соль правастатина суспендировали в 45 мл этанола, после этого при подаче 1 М этанольного раствора гидроксида натрия было добавлено 110 мол.% гидроксида натрия. Перемешивание раствора продолжали еще 1/2 ч, потом в него добавили 0,3 г древесного угля и проводили перемешивание еще 1/2 ч. Полученный раствор отфильтровали, а фильтрат сгустили до объема 15 мл. Потом к полученному концентрату при 56-60° С добавили 60 мл ацетона. Полученный раствор охладили до комнатной температуры и выдерживали в течение ночи при +5° С. После этого осадок отфильтровали, затем промыли 2× 20 мл ацетона, 2× 20 мл этилацетата и 2× 20 мл н-гексана и высушили в вакууме. Полученные в результате 1,7 г неочищенного правастатина растворили в этаноле, осветлили с помощью древесного угля и перекристаллизовывали из смеси этанол-этилацетат. Таким способом было получено 1,54 г правастатина, идентичного продукту, полученному в примере 2.
Пример 6
Как описано в примере 4, из культуры штамма Mortierella maculata n.spec. E-97 [NCAIM(P)F 001266] на скошенном агаре, культивированной в течение 7-10 дней, в 500 мл посевной среды Ml, простерилизованной в 3000 мл колбе Эрленмейера, инокулировали и инкубировали в течение 3 дней при 28° С на ротационном шейкере.
Состав среды MI:
глюкоза 40 г
казеин 5 г
КСl 0,5 г
NaNO3 3 г
КН2РO4 2 г
MgSO4·7Н2О 0,5 г
FeSO4·7H2О 0,01 г
в 1000 мл водопроводной воды.
Перед проведением стерилизации рН среды довели до значения 6,0, а саму стерилизацию проводили в течение 25 мин при 121° С. Полученную культуру для посева инокулировали в 5 л ферментер, который содержал биоконверсионную культуральную среду Р12.
Состав среды Р12:
глюкоза 10 г
солодовый экстракт 50 г
дрожжевой экстракт 5 г
жидкость для замачивания кукурузы 5 г
MgSO4·7Н2О 1 г
твин-80 0,5 г
в 1000 мл водопроводной воды
Перед проведением стерилизации рН указанной среды довели до значения 7,0; а саму стерилизацию проводили в течение 45 мин при 121° С. После ферментации подачу субстрата и биоконверсию проводили в соответствии с примером 2. После завершения биоконверсии был выделен правастатин с концентрацией, составляющей 650 мкг/мл, по следующей схеме:
рН бульона объемом 5,15 л, содержащего 620 мкг/мл правастатина, довели до значения 9,5 при помощи 2 М гидроксида натрия, а потом перемешивали 1 ч при комнатной температуре. Бульон отфильтровали, а мицелий вначале промыли суспензией воды 1× 2 л, а затем 1× 0,5 л. Фильтраты объединили, рН полученного водного раствора довели до значения 3,7 с помощью 20% серной кислоты и потом экстрагировали вначале 2,5 л этилацетата, а потом 1,5 л этилацетата. Этилацетатные экстракты объединили, промыли 2× 0,5 л воды и добавили 1,95 г дициклогексиламина. Этилацетатный экстракт сгустили при 40° С до объема в 200 мл под сниженным давлением и к полученному концентрату снова добавили 1,95 г дициклогексиламина. После этого продолжали перемешивание еще 6 ч при 15° С. Осажденное кристаллическое вещество отфильтровали, промыли сначала 20 мл этилацетата, а затем 15 мл этилацетата и высушивали при 40° С. Таким способом было получено 3,51 г неочищенного продукта. После его перекристаллизации из смеси ацетон-этанол было получено 3,05 г дициклогексиламиновой соли правастатина (температура плавления: 162-168° С), которую превратили в правастатин согласно примеру 5.
Пример 7
Ферментацию, подачу субстрата и биоконверсию штамма Mortierella maculata n. spec. E-97 [NCAIM(P)F 001266] проводили так, как это описано в примере 2. Правастатин, полученный в результате конверсии, выделяли из бульона следующим образом:
5 л культурального бульона, содержащего правастатин в концентрации 650 мкг/мл, отфильтровали через фильтровальную ткань. Мицелий плесени перемешивали в 2 л 0,1 М раствора гидроксида натрия в течение 1 ч, а потом отфильтровали. Полученные в результате два фильтрата объединили и рН довели до значения 3,5-4,0 с помощью 15% серной кислоты. После этого полученный раствор экстрагировали 2× 1,8 л этилацетата. Объединенные этилацетатные фазы промыли 800 мл воды. После этого добавили 400 мл деионизированной воды и значение рН полученной смеси довели до величины 8,0-8,5 с помощью 1 М гидроксида натрия. Смесь перемешивали 15 мин, после чего образовавшиеся фазы разделили. К этилацетатной фазе добавили 300 мл воды и значение рН довели до 8,0-8,5. После перемешивания в течение 15 мин фазы разделили. Снова к этилацетатной фазе добавили 300 мл воды и значение рН довели до 8,0-9,5. Затем смесь перемешивали в течение 15 мин. Образовавшиеся две фазы вновь разделили. Все водные фазы объединили и значение рН с помощью 15% серной кислоты довели до 3,5-4,0. После этого провели экстракцию 3× 300 мл этилацетата. Объединенные этилацетатные экстракты промыли 150 мл воды, высушили безводным сульфатом натрия и отфильтровали. Затем к полученному этилацетатному экстракту добавили 150 мол.% диоктиламина (вычисленного по содержанию правастатина). Этилацетат упарили приблизительно до 1/10 объема и добавляли ацетон вплоть до осаждения. Полученную смесь выдерживали в течение ночи при +5° С. Осадок отфильтровали на фильтре G-4, промыли сначала 20 мл ацетона, а затем 20 мл н-гексана и высушивали в вакууме при комнатной температуре. Полученные 3,3 г диоктиламиновой соли правастатина перекристаллизовали из 20 мл ацетона, получив при этом 2,7 г чистой диоктиламиновой соли правастатина. Температура плавления: 143-146° С. Указанную диоктиламиновую соль правастатина превратили в правастатин с помощью способа, описанного в примере 5.
Пример 8
При разработке гидроксилирующей способности выделенного из естественной среды обитания штамма Mortierella maculata n.spec. E-97, способного к 6β -гидроксилированию компактина в детально обсуждавшихся выше экспериментах по мутации, селекции и введению фермента, был продуцирован мутантный штамм Mortierella maculata n.sp. E-97/15/13 [NCAIM(P)F001267].
Выделенный нами штамм Mortierella maculata n.sp. E-97 [NCAIM(P)F 001266] культивировали на среде MS скошенного агара при 28° С в течение 7 дней.
Состав среды агара MS:
глюкоза 4 г
солодовый экстракт 10 г
дрожжевой экстракт 4 г
агар 20 г
в 1000 мл дистиллированной воды.
Споры отмыли от культуры скошенного агара, используя при этом 5 мл 0,9% раствора хлорида натрия. После перенесения суспензии спор в стерильную чашку Петри ее облучали в течение 1 мин ультрафиолетовым излучением. После этого к указанной суспензии спор добавили N метил N’ нитро-N-нитрозогуанидин в конечной концентрации, составляющей 2000 мкг/мл. Полученную суспензию перенесли в 100 мл колбу Эрленмейера и встряхивали при 28° С со скоростью 150 об/мин в течение 20 мин. Затем споры осадили при центрифугировании со скоростью 4000 об/мин в течение 10 мин, после чего провели суспендирование и стерильном 0,9% растворе хлорида натрия. Полученную суспензию нанесли на пластинки агара MU-VB, содержащие 10 мкг/мл беномила и 1% дефибринированной крови.
Состав среды агара MU-VB:
Глюкоза 40 г
Аспарагин 2 г
Пептон 2,5 г
Вторичный кислый фосфат калия 0,5 г
Агар 20 г
в 990 мл дистиллированной воды; после стерилизации в указанную среду ввели дополнительно 10 мл бычьей крови и 10 мг беномила.
Агаровые пластины инкубировали при 28° С в течение 7 дней, а потом выросшие колонии методом случайного отбора перенесли в лабораторные пробирки, содержащие агаровую среду PS.
Состав среды агара PS:
глюкоза 40 г
микологический пептон 10 г
агар 15 г
в 1000 мл дистиллированной воды.
Перед проведением стерилизации рН указанной среды доведи до величины 5,6-5,7. Стерилизацию осуществляли при 121° С в течение 20 мин.
Культуры скошенного агара инкубировали при 28° С в течение 12 дней, а их продуктивность в отношении правастатина оценивали экспериментально во встряхиваемой колбе так, как это описано в примере 1. Этим способом был выбран мутантный штамм Mortierella maculata n.sp. E-97/15/13, который дает выход правастатина, превышающий 60% норму конверсии из используемого субстрата натриевой соли компактина, имеющего концентрацию 1000 мкг/мл.
Гидроксилазу штамма Mortierella maculata n.sp. E-97/15/13 вводили при культивировании на среде агара MU-VB, содержащей 8-де-(2-метилбутирил)компактин и/или компактин в количестве 100 мкг/мл. После случайного выбора растущих колоний их перенесли в индуктор, содержащий скошенный агар MU-VB. Продуктивность в отношении правастатина выросших культур скошенного агара определяли способом, описанным в примере 1, с той разницей, что подачу субстрата компактина в количестве 500 мкг/мл проводили, начиная с 4 дня ферментации, на протяжении последующих 11 дней, а также субстрат натриевого компактина добавляли постепенно на протяжении 12 дней и он был полностью превращен в правастатин. По окончании биоконверсии, проводимой в 50 культурах во встряхиваемой колбе, образование 18,5 г правастатина из 30 г натриевого субстрата компактина было измерено с помощью метода ВЭЖХ. Извлечение правастатина из объединенных ферментационных бульонов проводили таким способом.
После окончания ферментации рН 5,5 л бульона при концентрации правастатина, составляющей 3360 мкг/мл, довели с помощью раствора 20% серной кислоты до значения 3,5-3,7. Потом полученный кислый раствор экстрагировали 2,75 л этилацетата. Фазы разделили и из эмульгированной органической фазы получили четкий экстракт. Бульон экстрагировали еще дважды 1,37 л этилацетата так, как это было описано выше. Объединенные этилацетатные экстракты промыли 2× 1,15 л деионизированной воды, потом к этому этилацетатному раствору добавили 150 мол.% дибензиламина (вычисленного для количества правастатина, измеренного методом ВЭЖХ). Указанный этилацетатный раствор сгустили в вакууме приблизительно до объема в 0,23 л. После этого к полученному концентрату добавили 20 мол.% дибензиламина и раствор, из которого выпадал осадок, выдерживали в течение ночи при температуре 0-5° С. Осажденную кислую дибензиламиновую соль правастатина отфильтровали, затем осадок промыли путем его суспендирования в охлажденном этилацетате и дважды - в н-гексане. В завершении провели высушивание в вакууме при 40-50° С. Полученный неочищенный продукт (22,4 г) растворили в 0,67 л ацетона при 62-66° С, а образовавшийся раствор осветляли с помощью 2,2 г древесного угля. После осветления ацетоновый фильтрат сгустили в вакууме до объема в 0,56 л. Кристаллы, осажденные из этого концентрата, снова растворили при указанной выше температуре, после чего раствор охладили до комнатной температуры. Затем перекристаллизацию продолжали в течение ночи при температуре 0-5° С. Осажденные кристаллы отфильтровали и промыли в суспензии дважды в охлажденном ацетоне и дважды в н-гексане. Перекристаллизованную кислую дибензиламиновую соль правастатина высушили в вакууме при температуре 40-50° С. Полученную кислую дибензиламиновую соль правастатина (14,8 г) растворили при 40-44° С в 740 мл смеси этилацетат-этанол (9:1), затем к этому раствору добавили 110 мол.% гидроксида натрия в форме 1 М этанольного раствора. Перемешивание полученного раствора продолжали в течение 1/2 ч при комнатной температуре, полное осаждение было достигнуто в результате применения охлаждения на льду в течение 1-1,5 ч. После этого осадок отфильтровали и промыли 2× 150 мл охлажденным этилацетатом и 2× 150 мл н-гексаном, в заключении высушили в вакууме при 40-50° С. Полученный правастатин растворили в этаноле, осветлили с помощью 1,0 г древесного угля, а потом кристаллизовали из смеси этанол-этилацетат. Таким способом было получено 9,4 г правастатина, физические константы которого соответствую данным, приведенным в примере 2.
Несмотря на то, что здесь описаны определенные предпочтительные воплощения настоящего изобретения, люди, квалифицированные в той области, к которой имеет отношение настоящее изобретение, не отступая от сущности и границ настоящего изобретения, могут сделать изменения и модификации. Следовательно, это означает, что настоящее изобретение ограничено только до той степени, которая требуется приведенными пунктами формулы изобретения и применимыми правилами законодательства.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИДРОКСИЛИРОВАНИЕ КОМПАКТИНА ДО ПРАВАСТАТИНА С ПОМОЩЬЮ MICROMONOSPORA | 2000 |
|
RU2235780C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ОЧИСТКИ ПРАВАСТАТИНА | 2012 |
|
RU2522806C1 |
| Способ получения комплексного антибиотика циклоспорина и/или его компонентов и штамм грибка ТоLYросLаDIUм VаRIUм | 1989 |
|
SU1836425A3 |
| ФЕРМЕНТАЦИОННАЯ СРЕДА ДЛЯ ПОЛУЧЕНИЯ ПРАВАСТАТИНА И СПОСОБ ПОЛУЧЕНИЯ ПРАВАСТАТИНА | 2009 |
|
RU2595380C2 |
| ПРОМЫШЛЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ КОМПАКТИНА | 2013 |
|
RU2585233C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЛИДНОГО СОЕДИНЕНИЯ И ШТАММЫ STREPTOMYCES SP., MORTIERELLA SP. И MICROMONOSPORACEAE | 2003 |
|
RU2330069C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПАКТИНА | 2013 |
|
RU2585234C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВЫХ СОЛЕЙ СТАТИНОВ | 2000 |
|
RU2246481C2 |
| ФЕРМЕНТАЦИОННАЯ СРЕДА И СПОСОБ ПОЛУЧЕНИЯ РЕКОМБИНАНТНЫХ БЕЛКОВ | 2009 |
|
RU2556120C2 |
| СПОСОБ БИОТРАНСФОРМАЦИИ КОЛХИКОНОВОГО СОЕДИНЕНИЯ В СООТВЕТСТВУЮЩЕЕ 3-0-ГЛИКОЗИЛЬНОЕ ПРОИЗВОДНОЕ | 1998 |
|
RU2218409C2 |
Изобретение относится к биотехнологии, в частности получению антибиотика правастатина из компактина с использованием метода микробиологической трансформации. Микробиологическое превращение компактина в правастатин осуществляют культивированием штамма Mortierella maculata n. sp. E-97, депонированного под номером (NCAIM(P)F 001266 или 001267, способного к 6β-гидроксилированию компактина, в погруженных аэробных условиях. Питательная среда для культивирования штамма содержит усвояемые источники углерода и азота, минеральные соли. Правастатин выделяют из культурального бульона и очищают. Способ позволяет получать Правастатин в промышленном масштабе. В формуле изобретения приведены структурные формулы компактина (соединение общей формулы (I) и правастатина (формула (II). 3 н. и 53 з.п. ф-лы, 1 ил., 1 табл.
из субстрата соединения формулы (II),
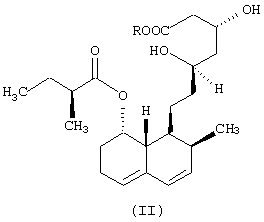
в которой R означает ион щелочного металла или аммония; включающего стадии
а) культивирования штамма нитевидных плесеней вида Mortierella maculata, способных к 6β-гидроксилированию соединения формулы (II) на или в питательной среде, содержащей усвояемые источники углерода и азота, а также минеральные соли;
(б) подачи в указанный культуральный бульон со стадии а) соединения формулы (II) в качестве субстрата для биоконверсии с помощью культуры Mortierella maculata;
(в) ферментации указанного субстрата вплоть до окончания биоконверсии;
(г) отделения соединения формулы (I) от культурального бульона, а также
(д) выделения указанного соединения формулы (I).
(б) контактирование водного раствора гидроксида натрия с указанной суспензией;
(в) промывку указанного водного раствора изобутилацетатом;
(г) осветление указанного водного раствора активированным углем;
(д) лиофилизацию указанного раствора в целях получения соединения формулы (I).
(а) суспендирование осажденной соли вторичного амина правастатина в спирте, содержащем 1 -4 атома углерода;
(б) добавление к суспензии достаточного количества этанольного раствора гидроксида натрия в целях растворения соли вторичного амина правастатина;
(в) добавление ацетона к полученному раствору в целях осаждения соединения формулы (I), а также
(г) выделение соединения формулы (I).
(в) выделение соединения формулы (I).
(в) очистку соединения формулы (I) посредством хроматографии на неионогенной адсорбирующей смоле.
(а) фильтрование культурального бульона;
(б) загрузку отфильтрованного культурального бульона на анионообменную смолу;
(в) элюирование свободной кислоты правастатина от указанной смолы;
(г) лактонизацию свободной кислоты правастатина;
(д) выделение лактона правастатина;
(е) гидролиз лактона гидроксидом натрия в целях получения соединения формулы (I),
(ж) очистку соединения формулы (I) хроматографией на неионогенной адсорбирующей смоле.
| WO 9736996 А2, 09.10.1997 | |||
| WO 9845410 А1, 15.10.1998. |

