Данное изобретение относится к новому микробному способу получения правастатина.
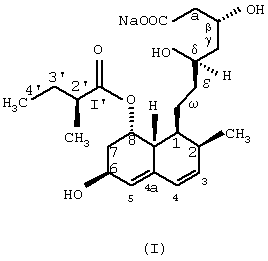
Точнее, данное изобретение относится к микробному способу получения правастатина формулы (I)
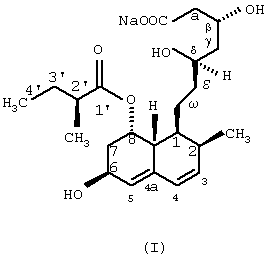
из соединения формулы (II)
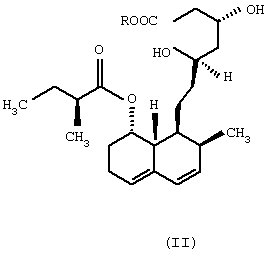
в которой R означает щелочной металл или ион аммония, посредством микроорганизма, где названный микроорганизм представляет собой прокариот из рода Micromonospora, который способен гидроксилировать соединение общей формулы (II) по 6β -положению.
Гиперхолестеринемию рассматривают как основной фактор риска в отношении атеросклеротического заболевания, особенно ишемического заболевания сердца. В течение последних двух десятилетий интенсивно изучали 3-гидрокси-3-метилглутарил-коэнзим А-редуктазу (HMG-CoA-редуктаза КФ 1.1.1.34), главный фермент, лимитирующий скорость в биосинтезе холестерина. Как было обнаружено, мевинолин и родственные соединения, биологически синтезированные определенными штаммами различных грибковых видов, являются конкурентными ингибиторами упомянутого фермента [Endo A. et al., J. Antibiotics 29, 1346-1348 (1976); Endo A. et al., FEBS Lett. 72, 323-326 (1976); Kuo, C.H. et al., Org. Chem. 48, 1991-1998 (1983)].
Правастатин также является представителем семейства ингибиторов HMG-CoA-редуктазы. Сначала, в ходе исследований метаболизма компактина [Arai M. et al., Sankyo Kenkyusho Nempo, 40 1-38 (1988)], правастатин был обнаружен как минорный мочевой метаболит компактина у собаки [Tanaka M. et al., неопубликовано].
Главной характерной особенностью правастатина как гидроксилированного продукта компактина является его тканевая избирательность. Названное лекарственное средство сильно ингибирует синтез стеролов в печени и кишечнике, но слабо - в других органах. Преимущество заключается в том, что правастатин проявляет более низкую токсичность, чем другие ингибиторы HMG-CoA-редуктазы.
Описано, что микробное гидроксилирование компактина может осуществляться в разной степени некоторыми штаммами видов, принадлежащих к различным родам грибов и штаммами видов актиномицет, принадлежащих к родам Nocardia, Actinomadura и Streptomyces, среди прочих Streptomyces roseochromogenes и Streptomyces carbophilus (патент США №5179013, патент США №4448979, патент США 4346227, патент США №4537859, патент Японии №58010572).
Проблема использования грибов для получения правастатина из компактина заключается в том, что упомянутые микроорганизмы в основном не допускают более высоких концентраций компактина в жидкой культуральной среде, по-видимому, вследствие их противомикробной активности [Serizawa N. et al., J. Antibiotics 36, 887-891(1983)]. Показано, что у Streptomyces carbophilus система цитохрома Р450 является необходимой для гидроксилирования компактина до правастатина [Matsuoka, Т. Et al., Eur. J. Biochem. 184, 707-713 (1989)]. Трудность генетического улучшения способности гидроксилирования с использованием такого фермента заключается в том, что он представляет собой комплекс белков, а не один белок.
Исследования заявителей были сфокусированы на обнаружении штамма актиномицет, который бы продуцировал правастатин из солей кислой формы компактина с более высоким выходом и в результате применения более высоких концентраций субстрата при биоконверсии, чем концентрации, известные из предшествующих патентных заявок.
Во время скрининга, охватывающего приблизительно 6000 актиномицет, большей частью собственные штаммы, а также аутентичные штаммы из международной коллекции штаммов, пять Streptomyces и пять Micromonospora были выбраны для дальнейших исследований, так как они оказались способными гидроксилировать натриевую соль кислой формы компактина в правастатин. Выбранные десять штаммов актиномицет, из которых восемь оказались таксокомически идентичными на видовом уровне, представляют собой следующие:
Streptomyces violaceus (согласно Kampfer et al., 1991), штамм №1/43.
Streptomyces rochei (Berger et al., 1949; Waksman and Lechevalier, 1953), штамм №1/41.
Streptomyces resistomyificus (Lindenbein, 1952), штамм №1/44.
Streptomyces sp., штамм №1/28.
Streptomyces lanatus, (Frommer, 1959), штамм №1/16.
Micromonospora sp., штамм №IDR-Р3.
Micromonospora purpurea (Luedemann and Brodsky, 1964), штамм №IDR-P4.
Micromonospora echinospora (Luedemann and Brodsky/ 1964), штамм №IDR-P5.
Micromonospora megalomicea (Weinstein et al., 1969), штамм №IDR-P6.
Micromonospora rosaria (Horan and Brodsky, 1986), штамм №IDR-P7.
Так как вплоть до настоящего времени в литературе нет данных относительно способности Micromonospora превращать соли кислой формы компактина в правастатин, основательно изучали не только эту специфическую энзиматическую способность, а также таксономическое положение вышеперечисленных штаммов Micromonospora.
Таксономическое положение штаммов IDR-P3, -Р4, -P5, -Р6 и –P7 на родовом уровне
Все названные штаммы продуцировали хорошо развитый мицелий, составленный из разветвленных гиф приблизительно 0,4-0,7 мкм в диаметре. Воздушный мицелий отсутствует или имеется только в небольших количествах. Неподвижные споры располагаются на спорофорах поодиночке. Гифы субстратного мицелия являются грамположительными и неустойчивыми в кислой среде. Штаммы №№IDR Р3-Р7 являются аэробными, хемоорганотрофическими и чувствительными к рН ниже 6,0. Стенки содержат мезодиамино-пимелиновую кислоту. Перечисленные выше отличительные свойства - как ключевые признаки - четко демонстрируют, что названные штаммы моноспоровых актиномицет являются типичными представителями рода Micromonospora.
Таксономическое описание Micromonospora sp., штамм №IDR-Р3
Микроморфологические свойства: субстратный мицелий составлен из хорошо развитых, скорее извитых, чем прямых, моноподиально разветвленных филаментов. Споры на спорофорах оказываются одиночными, сферическими, приблизительно 1,8 мкм в диаметре, и более или менее равномерно диспергированными на гифовых филаментах. Споры или прикреплены, или находятся на концах коротких спорофор. В бульонных культурах споры не были обнаружены на гифах вероятно потому, что высвобождение зрелых спор происходит очень быстро.
Культуральные макроморфологические свойства:
агар Чапека с сахарозой: рост в среде, колонии имели красноватый цвет, покрыты подобными точкам, черными спекулирующими областями.
Глюкозный агар с аспарагином: рост регистрировали как подобные точкам и приподнятые, красновато-коричневые или черные колонии. Красноватые диффузные пигменты.
Питательный агар: прекрасный рост, приподнятые, красновато-коричневые или черные колонии. Красновато-коричневый экзопигмент в среде.
Агар с дрожжевым экстрактом и экстрактом солода (ISP Мед.2): хорошо растущие, приподнятые и морщинистые, коричневые колонии, частично покрытые черными спорулирующими областями или “псевдо-воздушным мицелием” (это проявляется как ограниченный белесоватый или сероватый налет). Коричневатый или коричневато-красный растворимый пигмент.
Крахмальный агар с неорганическими солями (ISP Мед. 4): рост в среде красновато-коричневых, приподнятых и морщинистых колоний. Ярко-красноватый растворимый пигмент.
Агар с глицерином и аспарагином (ISP Мед. 5): рост только в небольших количествах не совсем белых или ярко-оранжевых, плоских колоний, ярко-розовый растворимый пигмент.
Утилизация источников углерода: наблюдали хороший рост и позитивную утилизацию L-арабинозы, D-галактозы, D-фруктозы, D-глюкозы, D-ксилозы, лактозы, мелибиозы, сахарозы, D-маннита, дульцита, глицерина и инозита. Рост в присутствии L-рамнозы, D-рафинозы и инулина был несколько лучше, чем на негативной контрольной среде. Утилизация источников азота: хороший рост отмечен в присутствии экстракта дрожжей и NZ-амина, наблюдали слабую утилизацию NаNО3 или отсутствие утилизации NаNО3.
Другие физиологические и биохимические характеристики:
целлюлоза и крахмал гидролизовались, молоко эффективно усваивалось. Тест нитратного восстановления был отрицательным. Не наблюдали никакого роста на срезах картофеля в отсутствие карбоната кальция (рН 5,8-6,0). Не было никакой продукции меланоидного пигмента.
Описанный штамм №IDR-Р3 Micromonospora sp. был выделен из образца ила из озера Балатон (Венгрия).
Положение в систематике: дальнейшие систематические исследования оказались необходимыми для выяснения точного таксономического положения описанного штамма среди видов из рода Micromonospora. На основании определенных свойств можно, по-видимому, предположить, что штамм IDR-Р3 представляет собой новый вид в роду Micromonospora.
Дифференциально-диагностическое описание и идентификация штаммов Micromonospora IDR-P4, -P5, -Р6 и -P7
Штамм IDR-P4
Обычно на вышеперечисленных диагностических средах наблюдали хороший рост окрашенных в оранжевый до оранжево-красного, красный, иногда желтоватый или розовый цвет колоний. Растворимые пигменты и воздушный мицелий не продуцируются. Количество одиночных спор является относительно низким. Споры встречаются на спорофорах ограниченно. Субстратный мицелий состоит из хорошо разветвенных гиф. Воздушный мицелий отсутствует. Нет никакого роста на D-мелибиозе, рафинозе, манните, глицерине, лактозе, L-рамнозе, но имеет место хороший рост на D-арабинозе, глюкозе, D-ксилозе, и слабый рост - на D-галактозе и D-фруктозе. На основании перечисленных диагностических свойств идентифицировали описанный штамм как представителя вида (Micromonospora purpurea (Luedemann and Brodsky, 1964).
Штамм IDR-P5
Данный штамм продуцирует большей частью одиночные спорофоры и сферические темно-коричневые до черных споры (0,8-1,5 мкм в диаметре), которые остаются прочно прикрепленными к спорофорам до созревания. Согласно электромикроскопическим исследованиям, на поверхности названных спор можно наблюдать бородавчатые структуры или выросты ("тупые шипы" согласно Vol. 4 Bergey's Manual of Syst. Bact. 1989, pages 2448), которые являются очень характерными спорами Micromonospora echinosora. Иначе говоря, культурально-морфологические и физиологические диагностические свойства штамма также очень похожи на свойства М. echinospora. Цвет хорошо развитых колоний на стандартной диагностической среде является оранжево-коричневым или темно-пурпурным. Спорулирующий слой оказывается черным или багрово-черным, восковым. Воздушный мицелий отсутствует. Меланиновый пигмент не продуцируется. Молоко усваивается. Хороший рост отмечен на D-ксилозе, D-арабинозе, D-глюкозе и сахарозе, но нет роста на L-рамнозе, рафинозе, D-галактозе, D-фруктозе, D-мелибиозе и глицерине. Данный штамм считают типичным представителем Micromonospora echinospora.
Штамм IDR-Р6
На большинстве из диагностических сред отмечен умеренный до слабого рост. Оранжевые или оранжево-красные колонии состоят из длинных разветвленных филаментов (приблизительно 0,6 мкм в диаметре) и ограниченного количества одиночных сферических, темного цвета спор (0,6-1,0 мкм в диаметре). Не продуцируется воздушный мицелий. В определенных средах образуются слабоокрашенные, красноватые или розовые, растворимые пигменты. На агаре с тирозином не продуцируются меланоидные пигменты. На основной среде описанный штамм утилизировал следующие источники углерода: D-ксилозу и D-фруктозу; только слабо: D-мелибиозу, маннит и галактозу, но никакого роста спор не наблюдали в присутствии глицерина, L-рамнозы, лактозы и рафинозы (см. также Kawamoto I. et al.: Agric. Biol. Chem., 47, 203-215, 1983). Штамм IDR-Р6 продемонстрировал значительное сходство с видом Micromonospora megalomicea (Weinstein, 1972) и данный штамм считают представителем упомянутого вида.
Штамм IDR-P7
Хороший рост на агаре Беннетта (Bennett), агаре Чапека с сахарозой, глюкозном агаре с аспарагином, питательном агаре, агаре из овсяной муки, картофельном агаре с декстрозой и так далее. Окраска растительных пигментов колеблется от красновато-коричневого до пурпурно-коричневого. На определенных средах образуются темно-красные диффузные пигменты. На поверхности колоний часто продуцируются черные споры. Растительные гифы (средний диаметр 0,5 мкм) интенсивно разветвляются. Споры (1,4-1,7 мкм в диаметре), прикрепленные или на коротких спорофорах, располагаются поодиночке, и встречаются по длине гиф. Рост и споруляция происходят по типу открытого сплетения по Luedemann. Данный штамм утилизировал следующие соединения только как источник углерода в среде: D-глюкозу, лактозу, D-маннит, L-рамнозу, сахарозу и D-ксилозу. Дульцит, глицерин, D-мелибиоза и D-рафиноза не утилизировались. Штамм №IDR-P7 идентифицирован как типичный представитель Micromonospora rosaria (Horan and Brodsky, 1986).
Представленные выше штаммы Micromonospora были депонированы в Национальной коллекции сельскохозяйственных и промышленных микроорганизмов (National Collection of Agricultural and Industrial Microorganisms; NCAIM), Будапешт, Венгрия, под нижеприведенным номерами - указателями:
Micromonospora sp. IDR-Р3 NCAIM (P) В 001268
Micromonospora purpurea IDR-P4 NCAIM (P) В 001271
Micromonospora echinospora
ssp. echinospora IDR-P5 NCAIM (P) В 001272
Micromonospora megalomicea
ssp. nigra IDR-P6 NCAIM (P) В 001273
Micromonospora rosaria IDR-P7 NCAIM (P) В 001274
На основании вышеизложенного изобретение относится к новому микробному способу получения правастатина формулы (I)
из соединения общей формулы (II)
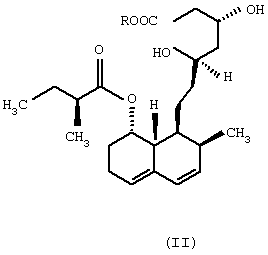
в которой R означает щелочной металл или ион аммония,
в результате культивирования штамма под слоем жидкости, который способен 6β -гидроксилировать соединение формулы (II) при аэробной ферментации и посредством выделения и очистки соединения формулы (I), образованного в ходе биоконверсии, состоящей из стадий
a) культивирования штамма вида, относящегося к роду Micromonospora, который способен 6β -гидроксилировать соединение формулы (II), в которой R является таким, как определено выше, на питательной среде, содержащей ассимилированные источники углерода и азота и минеральные соли, при 25-32° С, после этого
b) подачи субстрата, который трансформируется в растущей культуре, затем
c) гидроксилирования субстрата до окончания биоконверсии, потом
d) выделения соединения формулы (I) из бульона культуры и, если необходимо, очистки соединения.
Сфера действия изобретения распространяется на дикие штаммы и любые мутанты вида, относящегося к роду Micromonospora, которые способны гидроксилировать натриевые соли кислой формы компактина до правастатина.
В соответствии с предпочтительным аспектом данного изобретения, правастатин продуцируется штаммом Micromonospora, выбранным из группы, состоящей из Micromonospora sp. IDR-Р3 [NCAIM (Р) В 001268], Micromonospora purpurea IDR-P4 [NCAIM (P) В 001271], Micromonospora echinospora IDR-P5 [NCAIM (Р) В 001272], Micromonospora megalomicea IDR-P6 [NCAIM (Р) В 001273] и Micromonospora rosaria IDR-P7 [NCAIM (Р) В 001274].
Согласно наиболее предпочтительному аспекту изобретения правастатин продуцируется штаммом IDR-Р3 Micromonospora sp. [NCAIM (P) В 001268].
Данное изобретение может быть выполнено in situ с помощью способа ферментации, то есть когда гидроксилирование достигается при участии активно растущей культуры Micromonospora.
Гидроксилирование можно проводить, применяя перемешивание культуры в качалочной колбе или аэрирования и перемешивания в ферментерах, после того как соединение формулы (I) добавляли в растущие культуры. В таких случаях можно использовать пеногаситель. Адекватная плотность культуры названного штамма может быть достигнута в результате применения соответствующей среды, содержащей подходящие источники углерода и азота, неорганических солей, а также микроэлементов.
Оказывается, что, например, глюкоза, глицерин, декстрин, крахмал, рамноза, ксилоза, сахароза и растворимый крахмал являются ассимилируемыми источниками углерода, в то время как соевая мука, кукурузный экстракт, пептон, экстракт дрожжей, мясной экстракт, цитрат аммония и сульфат аммония являются хорошими источниками азота. Такие неорганические соли как карбонат кальция, фосфаты натрия, фосфаты калия и так далее, могут быть добавлены в культуральную среду. Предпочтительными средами для роста данного выбранного штамма являются среды, описанные в примерах.
Биоконверсию компактина до правастатина можно осуществить с помощью различных способов ферментации, например, в периодической культуре или подпитываемой культуре. Предпочтительно использовали перемешиваемую погруженную культуру. Предпочтительная температура составляет приблизительно 25° С - 37° С, наиболее предпочтительно - приблизительно от 25° С до 32° С.
Предпочтительный рН соответствует приблизительно 6,0-9,0, наиболее предпочтительно - приблизительно 7,0-8,5. Предпочтительные условия перемешивания соответствуют приблизительно 200 оборотам в минуту до 400 оборотов в минуту, наиболее предпочтительно приблизительно 250 оборотов в минуту.
Изобретение предусматривает способ превращения натриевой соли кислой формы компактина до правастатина. Натриевую соль кислой формы компактина можно использовать в данном изобретении при любой концентрации, которая приведет к продукции правастатина. Предпочтительно концентрация компактина находится между 0,1 и 10 г/литр, более предпочтительно составляет приблизительно между 0,3 и 3,0 г/литр.
В изобретении предусматривается рассмотрение любого процента превращения компактина до правастатина посредством штаммов Micromonospora spp., по меньшей мере, 30% и наиболее предпочтительно, по меньшей мере, приблизительно 90%.
Во время ферментации состав культуральной среды контролировали с помощью способа высокоэффективной жидкостной хроматографии (HPLC; ВЭЖХ). В соответствии со способом ВЭЖХ, образец среды разбавляли вдвое метанолом, центрифугировали, и супернатант использовали для анализа. Параметры системы ВЭЖХ, использованные для анализа, составляли: аналитическое оборудование ВЭЖХ Waters; колоночная упаковка: Waters Novapack C18 5 мкм; измерение при 237 нм; введение объема 10 мкл; скорость течения 0,6-0,9 мл/мин, линейный градиент; использовали градиентную элюцию: растворитель А = ацетонитрил - 0,1 М NаН2РO4 в воде (25:75), растворитель В + ацетонитрил - вода (рН 2 посредством Н3РO4) (70:30).
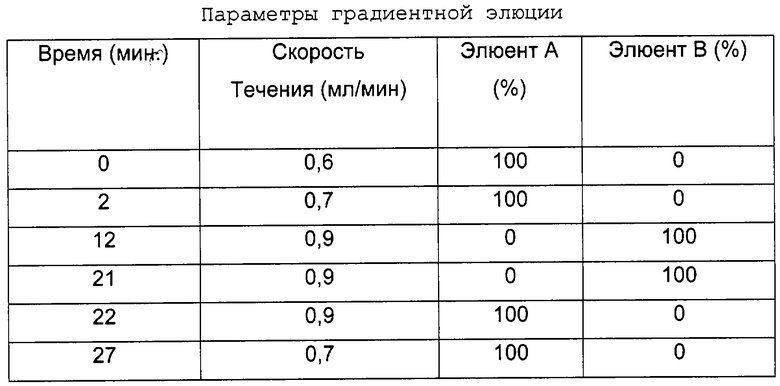
Время удерживания: правастатина (соль Na) 10,6 мин; компактина (Na-соль кислоты) 19,5 мин; правастатина (лактонная форма) 17,3 мин, компактина (лактонная форма) 23,5 мин.
Для выделения правастатина может быть использован любой известный способ выделения, например, экстракция - реэкстракция, анионообменная хроматография, осаждение.
Для извлечения продукта из бульона полезно принимать во внимание тот факт, что во время биоконверсии правастатин образуется в его кислой форме, таким образом, он может быть выделен из фильтрата среды в результате его абсорбции на колонке с анионообменной смолой. Для выделения продукта полезно применение сильной основной анионообменной смолы, которая представляет собой полимер полистилол-дивинилбензола, несущий активные группы четвертичного аммония, например, смолы Dowex А1 400 (ОН-), Dowex 1× 2 (ОН-), Dowex 2× 4 (ОН-), амберлит IRA 900 (ОН-). Продукт, абсорбированный на ионообменной смоле, можно элюировать с колонки водной уксусной кислотой или хлоридом натрия, содержащим смесь ацетон-вода, предпочтительно 1% хлоридом натрия, содержащим смесь ацетон-вода (1:1). Фракции, содержащие правастатин, объединяли, а ацетон, присутствующий в элюате, отгоняли под вакуумом. рН концентрата устанавливали с помощью 15% серной кислоты в области 3,5-4,0, и подкисленный водный раствор экстрагировали этилацетатом. Из этилацетатного экстракта правастатин можно экстрагировать 5% бикарбонатом натрия или слабо щелочной водой при объемном отношении 1/10 и 1/20 (рН 7,5-8,0). Известно, что правастатин можно получить в чистом виде из пролученного выше щелочного водного экстракта с помощью колоночной хроматографии на неионной адсорбционной смоле. Благоприятный способ заключается в том, что прежде всего растворенный в водной фазе этилацетат удаляют из щелочного водного экстракта с помощью вакуумной перегонки, а затем водный экстракт наносят на колонку с Diaion HP-20. Адсорбированный на колонке правастатин очищали путем элюции водным ацетоном, в котором содержание ацетона постепенно увеличивалось, затем хроматографические фракции, содержащие правастатин в виде одного компонента, объединяли и концентрировали в вакууме. Концентрат осветляли углем и лиофилизировали, потом кристаллизовали из смеси этанол - этилацетат, получая правастатин, по качеству приемлемый для фармацевтического применения.
После завершения биоконверсии правастатин можно экстрагировать из ферментационного бульона или из фильтрата, полученного после выделения из массы мицелия. Последний может быть удален или фильтрацией, или центрифугированием, однако - особенно в промышленном масштабе - целесообразно производить экстракцию цельного бульона. До экстракции рН или ферментационного бульона или фильтрата бульона приводили к 3,5-3,7 минеральной кислотой, предпочтительно разбавленной серной кислотой. Экстракцию производили сложным эфиром уксусной кислоты, содержащим алифатический спирт из 2-4 атомов углерода, предпочтительно этилацетат или изобутилацетат. Этилацетатный экстракт промывали водой и высушивали безводным сульфатом натрия. Затем из правастатина получали лактонное производное. Замыкание лактонного кольца проводили в растворе высушенного этилацетата при комнатной температуре при непрерывном перемешивании, индуцируя образование лактона каталитическим количеством трифторуксусной кислоты. Замыкание лактонного кольца проверяли с помощью анализа методом тонкослойной хроматографии (TLC; ТСХ). После завершения образования лактона раствор этилацетата промывали сначала 5% водным раствором бикарбоната натрия, а затем водой, и раствор лактона высушивали безводным сульфатом натрия и упаривали в вакууме. Остаток очищали способом колоночной хроматографии на силикагеле, используя в качестве элюирующих смесей этилацетат - н-гексан при постепенном увеличении содержания этилацетата. Правастатин получали из лактона правастатина в результате гидролиза при комнатной температуре в ацетоне с эквивалентным количеством гидроокиси натрия. Когда образование натриевой соли правастатина было закончено, правастатин осаждали ацетоном. Затем осадок отфильтровывали и промывали ацетоном и н-гексаном и высушивали в вакууме, потом кристаллизовали из смеси этанол - этилацетат.
Установлено, что хроматография через гель сефадекса LH-20 благоприятно подходит для очистки правастатина. В результате применения упомянутого способа может быть получен правастатин, превышающий чистоту 99,5% (измерена с помощью ВЭЖХ).
В ходе экспериментов сделано следующее открытие: из экстракта органического растворителя, предпочтительно из этилацетатного или изобутилацетатного экстракта бульона или бульонного фильтрата штамма IDR-Р3 Microiaonospora, который способен 6β -гидроксилировать соединение общей формулы (II), правастатин можно осаждать в виде кристаллической соли вторичными аминами. Кроме того, было обнаружено, что для образования соли подходят некоторые вторичные амины, содержащие алкил-, циклоалкил-, аралкил- или арил-заместители. Соответственно, среди них были выбраны нетоксичные вторичные амины, например диоктиламин, дициклогексиламин, дибензиламин. Выделение органических интермедиатов солей вторичных аминов, например соли дибензиламина, осуществляли путем добавления дибензиламина в количестве, эквивалентном 1,5 относительно содержания правастатина в экстракте, затем экстракт концентрировали с помощью вакуумной перегонки до 5% исходного объема, потом в концентрат добавляли другое количество дибензиламина в эквивалентном отношении 0,2. Кристаллическую соль дибензиламина осаждали из концентрата. Кристаллический неочищенный продукт отфильтровывали и высушивали в вакууме, а продукт осветляли углем в метаноле или растворе ацетона. Затем в результате перекристаллизации осветленного продукта из ацетона получали хроматографически чистый интермедиат соли правастатин-дибензиламина.
Правастатиновые соли органических вторичных аминов можно трансформировать в правастатин гидроокисью натрия или алкоксидом натрия, предпочтительно этоксидом натрия.
Выделение правастатина через интермедиат в виде соли вторичного амина является более простым способом, чем любой из когда-либо известных способов выделения. В процессе выделения не возникали артефакты, и таким образом отделение правастатина от побочных продуктов биоконверсии и от различных метаболических продуктов, биосинтезированных гидроксилирующим микроорганизмом, может быть благоприятно решено.
Способ согласно изобретению представлен следующими примерами.
Пример 1
Споры получали с поверхности 7-10 дневной культуры штамма Micromonospora sp. IDR-Р3 [NCAIM (P) В 001268, выращенной на скошенном растворимом крахмальном агаре (SM), и суспендировали в 5 мл стерильной дистиллированной воды. Затем полученную суспензию использовали для засевания 100 мл стерильной посевной среды TI в 500-мл колбе Эрленмейера (Erlenmeyer).
Состав среды SM
Растворимый крахмал 10,0 г
Na2HPO4 1,15 г
KH2PO4 0,25 г
КСl 0,2 г
MgSO4×7Н2O 0,2 г
Агар 15,0 г
в 1000 мл дистиллированной воды
рН среды доводили до 7,0 до стерилизации и смесь стерилизовали при 121° С в течение 25 минут.
Состав среды TI
Растворимый крахмал 20,0 г
Экстракт дрожжей 10,0 г
в 1000 мл водопроводной воды
рН доводили до 7,0 перед стерилизацией и производили термообработку при 121° С в течение 25 минут.
Растущую культуру встряхивали на роторном шейкере (250 оборотов в мин и амплитуда 2,5 см) в течение 3 дней при 32° С, затем 5 мл из культуры использовали для засевания 10 колб Эрленмейера объемом 500 мл каждая, содержащих 100 мл среды ТТ, стерилизованной при 121° С в течение 25 минут.
Состав среды ТТ
Картофельный крахмал 30,0 г
Соевая мука 30,0 г
СаСО3 5,0 г
CoCl2 x 6H2O 2,0 мг
Пальмовое масло 2,0 г
в 1000 мл водопроводной воды
рН доводили до 7,0 перед термостерилизацией.
Инкубацию проводили при 32° С в течение 72 часов, затем 50 мг натриевой соли кислой формы компактина в дистиллированной воде добавляли в каждую колбу, и культивирование проводили в течение 96 часов. Степень превращения натриевой соли кислой формы компактина в правастатин, определяемая с помощью ВЭЖХ, составила 82%.
После окончания ферментации культуры объединяли, а из полученного совокупного ферментационного бульона, который содержал 410 мг правастатина, выделение последнего проводили следующим образом: ферментационный бульон центрифугировали при 2500 оборотах в мин в течение 20 мин. Супернатант бульона и массу мицелия разделяли, затем последний повторно суспендировали в 250 мл воды и полученную суспензию перемешивали в течение одного часа и фильтровали. рН объединенного отцентрифугированного бульона и фильтрата приводили к 4,0 15% серной кислотой, потом кислый фильтрат экстрагировали этилацетатом 3× 300 мл. Объединенные этилацетатные экстракты промывали 300 мл воды, высушивали безводным сульфатом натрия и концентрировали в вакууме до объема 100 мл. Лактон правастатина получали из правастатина добавлением трифторуксусной кислоты в каталитическом количестве при комнатной температуре при непрерывном перемешивании. Образование лактона правастатина контролировали способом ТСХ: абсорбент: алюминиевая фольга Kieselgel 60 F254 DC (Merck); проявляющий растворитель: смесь ацетон-бензол-уксусная кислота (50:50:1,5); выявление: с помощью реагента фосфомолибденовой кислоты. Величина Rf (фактор удерживания) лактона правастатина составляла 0,7. После завершения образования лактона этилацетат промывали 5% водным бикарбонатом натрия 2× 20 мл, затем промывали 20 мл воды, высушивали безводным сульфатом натрия и упаривали в вакууме. После упаривания получали 0,5 г остатка, который хроматографировали на колонке, содержащей 10 г адсорбента Kieselgel 60 (диаметр колонки: 1,2 см, высота слоя адсорбента: 17 см). Для элюции использовали смеси этилацетат - н-гексан, в которых содержание этилацетата постепенно увеличивали. Лактон правастатина элюировали с колонки смесью 60% этилацетат - 40% н-гексан. Фракции, содержащие лактон правастатина, объединяли и выпаривали в вакууме. Полученный остаток, который содержал 230 мг лактона правастатина, растворяли в 5 мл ацетона, а потом при перемешивании добавляли 110 моль/% гидроокиси натрия в 1 М растворе этанола. Перемешивание раствора продолжали в течение получаса при комнатной температуре. Впоследствии, раствор концентрировали до объема 2 мл и к концентрату добавляли 4 мл ацетона. Смесь сохраняли при +5° С в течение ночи.
Осадок фильтровали, промывали 2 мл ацетона, а затем 2 мл н-гексана и высушивали в вакууме при комнатной температуре. Полученный неочищенный правастатин растворяли в этаноле, осветляли углем, затем кристаллизовали из смеси этанол-этилацетат. Таким образом получено 170 мг правастатина.
Точка плавления 170-173° С (разл.)
[α ]
Спектр поглощения в ультрафиолете (20 мкг/мл, в метаноле): λ макс.=231, 237, 245 нм (logε =4,263; 4,311; 4,136).
Спектр поглощения в инфракрасной области (КВr):ν ОН 3415, ν CH 2965, ν C=O 1730, ν COO- 1575 см-1.
1H-ЯМР спектр (D2O, δ , м.д.): 0,86, д, 3Н (2-СН3); 5,92 дд, J=10,0 и 5,4 Гц), 1Н (3-Н); 5/99, д, J=10,0 Гц, 1Н (4-Н); 5,52, шир., 1Н (5-Н); 4,24, м, 1Н (6-Н); 5,34, шир., 1Н (8-Н); 4,06, м, 1Н (β -Н), 3,65, м, 1Н (δ -Н); 1,05, д, 3Н (2'-СН3); 0, 82, т, 3Н (4'-Н3).
13С-ЯМР спектр (D2O, δ , м.д.): 15,3, кв. (2-СН3); 139,5, д (С-3); 129,5, д (С-4); 138,1, с (С-4а); 127,7, д (С-5); 66,6, д (С-6); 70,1, д (С-8); 182,6, с (СOO-); 72,6, д, (С-β ), 73,0, д, (C-δ ); 182,0, с (С-1'); 18,8 кв. (2'-СН3); 13,7, кв. (С-4').
Положительный масс-спектр FAB (характерные ионы):
[M+Na]+469; [M+H]+447.
Отрицательный масс-спектр FAB (характерные ионы): [М-Н]-445, [M-Na]-423, m/z 101 {2-метилмасляная кислота-Н]-.
Пример 2
10 колб Эрленмейера объемом 500 мл, каждая из которых содержала 100 мл биоконверсионной среды МТ1, засевали культурой, полученной, как описано в примере 1, затем инкубировали при 28° С в течение 96 часов, и в каждую колбу добавляли 50 мг натриевой соли кислой формы компактина в дистиллированной воде, потом проводили гидроксилирование в течение 72 часов, затем в культуры добавляли еще 50-50 мг субстрата в дистиллированной воде, и ферментацию продолжали еще в течение 72 часов. Состав биоконверсионной среды MT1
Картофельный крахмал 10,0 г
Декстроза 20,0 г
Соевая мука 10,0 г
Экстракт дрожжей 10,0 г
СаСО3 5,0 г
Подсолнечное масло 2,0 г
в 1000 мл водопроводной воды
рН биоконверсионной среды доводили до 7,0 перед стерилизацией. Смесь стерилизовали при 121° С в течение 25 минут.
После окончания периода биоконверсии культуры объединяли, и правастатин выделяли из совокупного бульона в соответствии со следующим способом:
объединенный бульон, который содержал 750 мг правастатина согласно анализу ВЭЖХ, центрифугировали при 2500 оборотах в мин в течение 20 мин. Выделенную массу мицелия перемешивали с 250 мл воды в течение часа, затем фильтровали. Отцентрифугированный бульон и фильтрат объединяли, и рН полученного раствора приводили к 3,5-4,0 15% серной кислотой, потом раствор экстрагировали этилацетатом 3× 300 мл. Затем к этилацетатному экстракту добавляли 150 моль % дибензиламина, рассчитанного относительно содержания правастатина. Этилацетатный экстракт упаривали до объема приблизительно 30 мл, и суспензию выдерживали в течение ночи при 0-5° С. Осажденную соль дибензиламина кислой формы правастатина фильтровали и промывали на фильтре охлажденными этилацетатом и н-гексаном, в конце концов высушивали в вакууме. 1,1 г неочищенной соли дибензиламина кислой формы правастатина растворяли в 33 мл ацетона при температуре 62-66° С, и раствор осветляли 0,1 г угля в течение получаса. Затем уголь удаляли из раствора путем фильтрации. Кристаллы, выпавшие в осадок из осветленного раствора, снова растворяли при указанной выше температуре, затем раствор сохраняли при 5° С в течение ночи. Осадок отфильтровывали, промывали охлажденными ацетоном и н-гексаном и высушивали в вакууме. Полученную соль дибензиламина кислой формы правастатина (0,7 г) суспендировали в 10 мл этанола, потом к раствору добавляли 110 моль % гидроокиси натрия посредством подачи 1М водного раствора. Перемешивание щелочного раствора продолжали в течение получаса при комнатной температуре. После завершения образования натриевой соли добавляли 30 мл воды, и рН раствора доводили до нейтральных значений, потом отгоняли этанол в вакууме. Водный концентрат хроматографировали на колонке, заполненной 50 мл смолы Diaion HP 20 (диаметр колонки 1,5 см, высота слоя смолы 28 см). Колонку элюировали смесями ацетон-деионизированная вода, в которых концентрацию ацетона увеличивали ступенчато по 5%. Правастатин можно элюировать с колонки 15% ацетоном, заключающим в себе смесь ацетон-деионизированная вода. Фракции анализировали посредством ТСХ, представленной в примере 1. Величина Rf правастатина составляла 0,5. Фракции, содержащие правастатин, объединяли, и ацетон выпаривали в вакууме. В результате лиофилизации водного остатка получено 390 мг хроматографически чистого правастатина.
Пример 3
В лабораторном ферментере 4,5 литра среды ТТ/2 стерилизовали при 121° С в течение 45 минут и засевали 500 мл культурой, выращенной при встряхивании, как описано в примере 1, затем инкубировали при 32° С, аэрировали 250 л стерильного воздуха/час и перемешивали с помощью мешалки с плоскими лопастями при 300 оборотах в мин. Инкубацию продолжали в течение 72 часов, и в культуру добавляли 2,5 г натриевой соли кислой формы компактина. После 48-часового периода биоконверсии компактин-субстрат был полностью израсходован из ферментационного бульона, затем снова в культуру добавляли дополнительные 2,5 г натриевой соли кислой формы компактина. Вторая доза компактина была израсходована в течение 24 часов. Степень конверсии натриевой соли кислой формы компактина в правастатин составляла приблизительно 90% в способе биоконверсии.
Состав биоконверсионной среды ТТ/2
Глюкоза 75,0 г
Растворимый крахмал 50,0 г
Соевая мука 50,0 г
Экстракт дрожжей 50,0 г
Пептон 5,0 г
NаNО3 20,0 г
СаСО3 25,0 г
в 4500 мл водопроводной воды
Пример 4
В лабораторном ферментере стерилизовали 4,5 литра ферментационной среды ТТ/1 при 121° С в течение 45 минут и засевали 500 мл посевной культурой, выращенной при встряхивании, полученной как описано в примере 1, затем инкубировали при 28° С, аэрировали 200 л стерильного воздуха/час и перемешивали с помощью мешалки с плоскими лопастями при 400 оборотах в мин.
Состав биоконверсионной среды ТТ/1
Глюкоза 125,0 г
Картофельный крахмал 25,0 г
Соевая мука 50,0 г
Экстракт дрожжей(GISТЕХ) 50,0 г
Пептон 50,0 г
CoCl2×6Н2О 10,0 мг
Подсолнечное масло 10,0 г
в 4500 мл водопроводной воды
рН биоконверсионной среды приводили к 7,0 перед стерилизацией.
Культивирование продолжали при 28° С в течение 96 часов. В это время в культуру добавляли 2,5 г натриевой соли кислой формы компактина в стерильном профильтрованном водном растворе. На 5-ый день ферментации натриевая соль кислой формы компактина была полностью израсходована из ферментационного бульона. Затем подачу субстрата повторяли ежедневно в течение дальнейших 3 дней порциями по 2,5 г/день. Субстрат, натриевую соль кислой формы компактина, постепенно расходовали в течение четырех дней и полностью превратили в правастатин. В соответствии с результатами определений ВЭЖХ в конце периода ферментации из 10 г компактина-субстрата получено 9 г правастатина.
После окончания биоконверсии правастатин, полученный в концентрации 1800 мкг/мл, выделяли следующим образом:
5 литров культурального бульона центрифугировали при 2500 оборотах в мин в течение 20 мин. Затем к отделенной массе мицелия добавляли 2 литра воды, и суспензию перемешивали в течение одного часа и фильтровали. Полученные два фильтрата объединяли и пропускали со скоростью 500 мл/час через колонку, заполненную 300 г (540 мл) смолы Dowex AI 400 (ОН-) (диаметр колонки 4 см, высота слоя смолы 43 см), затем слой смолы промывали 1 литром деионизированной воды. После этого колонку элюировали 1 литром смеси ацетон-вода (1:1), содержащей 10 г хлорида натрия, собирая фракции по 50 мл. Фракции анализировали с помощью ТСХ, как описано в примере 1. Фракции, содержащие продукт, объединяли, и ацетон отгоняли в вакууме. pН концентрата приводили к значению 3,5-4,0 15% серной кислотой, затем концентрат экстрагировали этилацетатом 3× 250 мл. К объединенному этилацетатному экстракту добавляли 40 мл деионизованной воды, потом рН корректировали 1 М гидроокисью натрия до значения 7,5-8,0. После 15 мин перемешивания разделяли водную и этилацетатную фазы, затем раствор этилацетата экстрагировали деионизованной водой 2× 40 мл, как описано раньше. Затем объединенный щелочной водный раствор концентрировали до объема 50 мл и хроматографировали на колонке, заполненной 600 мл неионной адсорбирующей смолы Diaion HP 20 (Mitsubishi Co., Japan) (диаметр колонки 3,8 см, высота слоя смолы 53 см). Колонку промывали 600 мл деионизированной воды, потом элюировали смесями ацетон-деионизированная вода, в которых концентрация ацетона увеличивалась ступенчато по 5%, собирая фракции по 50 мл. Элюат анализировали с помощью ТСХ, описанной в примере 1. Правастатин элюировали с колонки смесью ацетон-деионизированная вода, содержащей 15% ацетона. Фракции, содержащие правастатин в виде единственного компонента, объединяли, а раствор концентрировали в вакууме до объема 150 мл. Впоследствии к концентрированному водному раствору добавляли 0,6 г угля, и правастатин осветляли при комнатной температуре в течение 1 часа. Затем уголь отфильтровывали и фильтрат лиофилизировали. Полученные 6,5 г лиофилизированного правастатина дважды кристаллизовали из смеси этанола и этилацетата. Осадок отфильтровывали и промывали 20 мл этилацетата и 20 мл н-гексана и высушивали в вакууме при комнатной температуре. Таким образом получено 4,6 г хроматографически чистого правастатина.
Пример 5
Суспензию спор получали с использованием 5 мл стерильной дистиллированной воды с поверхности 10-дневной культуры, выращенной на растворимом крахмальном скошенном агаре, как описано в примере 1, штамма Micromonospora echinospora ssp. echinospora IDR-P5 [NCAIM (P) 001272], способного 6(β -гидроксилировать натриевую соль кислой формы компактина, и полученную суспензию спор использовали для пассирования 100 мл посевной культуральной среды TI, стерилизованной в 500-мл колбах Эрленмейера. Состав среды TI также описан в примере 1. Культуральную среду встряхивали на роторном шейкере (250 оборотов в мин, амплитуда 2/5 см) в течение 3 дней при 28° С, затем аликвоты по 5 мл растущей культуры переносили в 100-100 мл биоконверсионной среды ТТ/1, стерилизовали в 500-мл колбах Эрленмейера в течение 25 мин при 121° С. Состав среды ТТ/1 описан в примере 4. Колбы встряхивали на роторном шейкере (250 оборотов в мин, 2,5 см амплитуда) в течение 3 дней при 25° С, затем в культуры добавляли 10-10 мг компактина-субстрата (натриевая соль кислой формы компактина) в стерильном отфильтрованном водном растворе, потом ферментацию продолжали в течение 168 часов.
В конце биоконверсии содержание правастатина ферментационного бульона определяли с помощью способа ВЭЖХ. В это время средняя концентрация правастатина составляла 40 мкг/мл.
Пример 6
Ферментацию, подачу субстрата и биоконверсию проводили со штаммом IDR-P6, [NCAIM (Р) В 001273] Micromonospora megalomicea ssp. nigra, как описано в примере 5. Содержание правастатина ферментационного бульона определяли с помощью способа ВЭЖХ. В конце биоконверсии содержание правастатина бульона составляло 50 мкг/мл.
Пример 7
5 мл посевной культуры штамма IDR-P4 [NCAIM (Р) В 001271] Micromonospora purpurea, полученного, как описано в примере 1, использовали для засевания 100-100 мл среды ТТ/14, распределенной по 500-мл колбам Эрленмейера, и стерилизовали в течение 25 мин при 121° С.
Состав среды ТТ/14
Картофельный крахмал 5,0 г
Глюкоза 25,0 г
Экстракт дрожжей (GISTEX) 15,0 г
Пептон 15,0 г
СаСО3 1,0 г
в 1000 мл водопроводной воды
рН биоконверсионной среды доводили до 7,0 перед стерилизацией.
Колбы встряхивали на роторном шейкере (250 оборотов в мин, амплитуда 2,5 см) в течение 3 дней. Подачу субстрата, биоконверсию и определение содержания правастатина проводили, как описано в примере 5. В конце биоконверсии содержание правастатина в ферментационном бульоне составляло 40 мкг/мл.
Пример 8
Ферментацию, подачу субстрата и биоконверсию проводили со штаммом IDR-P7, [NCAIM (Р) В 001274] Micromonospora rosaria, как описано в примере 1. В конце биоконверсии было определено 350 мкг/мл правастатина в ферментационном бульоне с помощью способа ВЭЖХ.
| название | год | авторы | номер документа |
|---|---|---|---|
| МИКРОБНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРАВАСТАТИНА | 2000 |
|
RU2252258C2 |
| ПРОИЗВОДНЫЕ БОРРЕЛИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2247723C2 |
| ПЕПТИДИЛАРГИНАЛИ И СПОСОБЫ ЛЕЧЕНИЯ СИНДРОМА ДИССЕМИНИРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ | 2002 |
|
RU2312856C2 |
| ПРОМЫШЛЕННЫЙ СПОСОБ ПОЛУЧЕНИЯ КОМПАКТИНА | 2013 |
|
RU2585233C2 |
| ПОЛИСУЛЬФАТИРОВАННЫЕ ГЛИКОЗИДЫ И ИХ СОЛИ | 2005 |
|
RU2413734C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПАКТИНА | 2013 |
|
RU2585234C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВЫХ СОЛЕЙ СТАТИНОВ | 2000 |
|
RU2246481C2 |
| Способ получения комплексного антибиотика циклоспорина и/или его компонентов и штамм грибка ТоLYросLаDIUм VаRIUм | 1989 |
|
SU1836425A3 |
| СОЕДИНЕНИЕ WK-5344А И СОЕДИНЕНИЕ WK-5344B, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОДУЦИРУЮЩИЙ ИХ ШТАММ STREPTOMYCES SP. WK-5344 FERM BP-6668 | 1999 |
|
RU2201964C2 |
| Способ получения метаболита "а 27 106 | 1974 |
|
SU539538A3 |
Изобретение относится к области биотехнологии и касается нового микробного способа получения соединения формулы (I) из соединения общей формулы (II), (формулы I и II приведены в формуле изобретения), в которой R означает щелочной металл или ион аммония, с помощью погруженной культуры штамма, который способен 6β-гидроксилировать соединение формулы (II) при аэробной ферментации и в результате выделения и очистки продукта формулы (I), образованного во время биоконверсии. Биоконверсия включает в себя культивирование штамма Micromonospora, который способен 6β-гидроксилировать соединение общей формулы (II), в которой R является таким как описано выше, при 25-32°С на питательной среде, содержащей подходящие источники углерода и азота и минеральные соли, затем снабжение субстратом для трансформации в растущей культуре, потом гидроксилирование субстрата до окончания биоконверсии, затем выделение соединения формулы (I) из культуральной среды и, если необходимо, очистку соединения. Использование способа позволяет повысить выход правастатина при его биоконверсии из солей кислой формы компактина. 8 з.п. ф-лы.
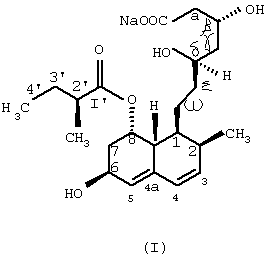
из соединения общей формулы (II)
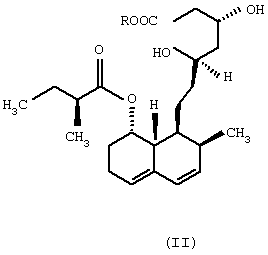
в которой R означает щелочной металл или ион аммония,
включающий в себя стадии а) культивирования штамма Micromonospora, который способен 6β-гидроксилировать соединение формулы (II), в которой R является таким, как определено выше, при 25-32°С на питательной среде, содержащей подходящие источники углерода и азота и минеральные соли, затем b) подачи субстрата для трансформации в растущей культуре, затем с) гидроксилирования субстрата вплоть до окончания биоконверсии, а затем d) выделения соединения формулы (I) из культурального бульона и, если необходимо, очистки этого соединения.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| ЭФИРНЫЕ ПРОИЗВОДНЫЕ ГЕКСАГИДРОНАФТАЛИНА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2114101C1 |
| US 4537859 A, 27.08.1985 | |||
| US 4346227 A, 24.08.1982 | |||
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |

