Область техники
Настоящее изобретение направлено в основном на аминоциклогексилово-эфирные соединения, фармацевтические композиции и наборы, содержащие аминоциклогексилово-эфирные соединения, и их применения в терапии.
Предпосылки изобретения
Аритмия является колебанием нормального ритма сердцебиения и обычно представляет результат аномалий в структуре, количестве или функции ионных каналов. Известны как атриальная аритмия, так и вентрикулярная аритмия. Главной причиной летальных исходов, обусловленных сердечными аритмиями, является разновидность вентрикулярной аритмии (аритмии желудочков), известная как вентрикулярная фибрилляция (фибрилляция желудочков) (ФЖ). По скромным подсчетам, в одних только США каждый год у более чем одного миллиона американцев имеет место новый или повторный коронарный приступ (определяемый как инфаркт миокарда или ишемическая болезнь сердца с летальным исходом). Около 650000 из них являются первыми сердечными приступами и 450000 - повторными приступами. Около одной трети людей, испытавших эти приступы, умирают. По меньшей мере, 250000 человек в год умирают от ишемической болезни сердца в течение 1 часа после появления симптомов и, не успев попасть в больницу. Причиной этих скоропостижных смертельных исходов является остановка сердца, возникающая, как правило, в результате фибрилляции желудочков.
Фибрилляция предсердий или атриальная фибрилляция (ФП) является более частой формой аритмии, наблюдаемой в клинической практике и являющейся причиной болезненности многих людей (Pritchett E.L., N. Engi. J. Med. 327(14):1031 Oct. 1, 1992, обсуждение 1031-2; Kannel and Wolf, Am. Heart J. 123(1):264-7 Jan. 1992). Весьма вероятно, что ее распространенность увеличивается с увеличением возраста населения, и у 3-5% пациентов в возрасте более 60 лет имеет место ФП (Kannel W.B., Abot R.D., Savage D.D., McNamara P.M., N. Engi. J. Med. 306(17):1018-22, 1982; Wolf P.A., Abbot R.D., Kannel W.B. Stroke. 22(8):983-8, 1991). Хотя ФП редко приводит к смертельному исходу, она может ухудшать деятельность сердца и является главной причиной инсульта (Hinton R.C., Kistler J.P., Fallon J.T., Friedlich A.L., Fisher C.M., American Journal of Cardiology 40 (4): 509-13, 1977; Wolf P.A., Abbot R.D., Kannel W.B., Archives of Internal Medicine 147(9):1561-4, 1987; Wolf P.A., Abbot R.D., Kannel W.B. Stroke. 22 (8):983-8, 1991; Cabin H.S., Clubb K.S., Hall C., Perlmutter R.A., Feinstein A.R., American Journal of Cardiology 65(16):1112-6, 1990).
Разработаны средства против аритмии для того, чтобы предотвратить или облегчить состояние при сердечной аритмии. Например, антиаритмические соединения класса I используются для лечения суправентрикулярных аритмий и вентрикулярных или желудочковых аритмий. Очень важно лечение желудочковой аритмии, так как такая аритмия может быть летальной. Имеющие тяжелые последствия желудочковые аритмии (желудочковая тахикардия и фибрилляция желудочков) наиболее часто происходят при наличии ишемии миокарда и/или инфаркта. Фибрилляция желудочков часто происходит на фоне острой ишемии миокарда прежде, чем развивается полностью инфаркт. В настоящее время не существует удовлетворительной фармакотерапии для лечения и/или предотвращения фибрилляции желудочков во время острой ишемии. В действительности, многие антиаритмические соединения класса I могут фактически повышать смертность пациентов с инфарктом миокарда.
Антиаритмические лекарственные препараты класса Iа, Iс и III используются для перевода острого приступа ФП в синусовый ритм и предотвращения повторения аритмии (Fuch and Podrid, 1992; Nattel S., Hadjis T., Talajic M., Drugs 48(3):345-71, 1994). Однако, лекарственная терапия часто ограничена побочными действиями, включающими возможность повышенной смертности, и неадекватной эффективностью (Feld G.K., Circulation. 83 (6):2248-50, 1990; Copien S.E., Antman E.M., Berlin J.A., Hewitt P., Chalmers T.C., Circulation 1991; 83(2):714 и Circulation 82 (4):1106-16, 1990; Flaker G.C., Blackshear J.L., McBride R., Kronmal R.A., Halperin J.L., Hart R.G., Journal of the American College of Cardiology 20 (3):527-32, 1992; CAST, N. Engl. J. Med. 321:406, 1989; Nattel S., Cardiovascular Research. 37 (3):567-77, 1998). Показатели перевода для антиаритмических средств класса I колеблются между 50-90% (Nattel S., Hadjis Т., Talajic M., Drugs 48 (3):345-71, 1994; Steinbeck G., Remp Т., Hoffmann E., Journal of Cardiovascular Electrophysiology. 9(8 Suppi):S104-8, 1998). Антиаритмические средства класса III, по-видимому, более эффективны для прекращения трепетания предсердий, чем для ФП, и обычно считаются менее эффективными, чем лекарственные препараты класса I для прекращения ФП (Nattel S., Hadjis Т., Talajic M., Drugs 48 (3):345-71, 1994; Capucci A., Aschieri D., Villani G.Q., Drugs and Aging, 13 (1):51-70,1988). Примеры таких лекарственных средств включают ибутилид, дофетилид и соталол. Показатели перевода этими лекарственными средствами составляют между 30-50% для острого приступа ФП (Capucci A., Aschieri D., Villani G.Q., Drugs and Aging, 13 (1):51-70,1988), и их применение также связано с риском индукции желудочковой тахиаритмии Торсадеса де Поинтеса (Torsades de Pointes). В случае ибутилида риск проаритмии желудочков оценивается в ~4,4%, при этом ~1,7% пациентам необходима электроимпульсная терапия рефракторной желудочковой аритмии (Kowey Р.R., VanderLugt J.T., Luderer J.R., American Journal of Cardiology 78(8A):46-52,1996). Такие события особенно трагичны в случаях ФП, так как данная аритмия сама по себе редко приводит к летальному исходу.
Таким образом, в данной области существует потребность в определении новых антиаритмических курсов лечения как для аритмий желудочков, так и для аритмий предсердий. Данное изобретение удовлетворяет данную потребность и, кроме того, предоставляет другие связанные с этим преимущества.
Краткое описание изобретения
В одном из вариантов данного изобретения предоставляются аминоциклогексилово-эфирные соединения, имеющие формулу (I), или их сольваты, или фармацевтически приемлемые соли:
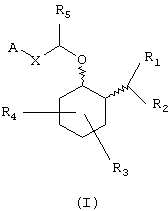
в которой независимо в каждом случае
Х выбран из прямой связи, -С (R6, R14) -Y-и -С (R13)=CH-;
Y выбран из прямой связи, О, S и С1-С4алкилена;
R13 выбран из водорода, C1-С6алкила, С3-С8циклоалкила, арила и бензила;
R1 и R2 независимо выбраны из водорода, C1-С8алкила, С3-С8алкоксиалкила, C1-С8гидроксиалкила и С7-С12аралкила; или
R1 и R2, взятые вместе с атомом азота, с которым они непосредственно связаны в формуле (I), образуют кольцо, обозначаемое формулой (II):
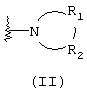
причем, кольцо формулы (II) образовано азотом, как показано, а также тремя-девятью дополнительными кольцевыми атомами, независимо выбранными из углерода, азота, кислорода и серы; любые два смежных атома кольца могут быть связаны вместе одинарной или двойной связями, и любой один или более дополнительных кольцевых атомов углерода могут быть замещены одним или двумя заместителями, выбранными из водорода, гидрокси, C1-С3гидроксиалкила, оксо-, С2-С4ацила, C1-С3алкила, C2-С4алкилкарбокси, C1-С3алкокси, С1-С20алканоилокси, или могут быть замещены с образованием пяти- или шестичленного спирогетероциклического кольца, содержащего один или два гетероатома, выбранных из кислорода и серы; и любые два смежных дополнительных кольцевых атомов углерода могут быть сконденсированы с С3-С8карбоциклическим кольцом, и любой один или более дополнительных кольцевых атомов азота могут быть замещены заместителями, выбранными из водорода, C1-С6алкила, С2-С4ацила, С2-С4гидроксиалкила и С3-С8алкоксиалкила; или
R1 и R2, взятые вместе с атомом азота, с которым они непосредственно связаны в формуле (I), могут образовывать бициклическую кольцевую систему, выбранную из 3-азабицикло-[3.2-2]нонан-3-ила, 2-азабицикло[2.2.2]октан-2-ила, 3-азаби-цикло[3.1.0]-гексан-3-ила и 3-азабицикло[3.2.0]гептан-3-ила;
R3 и R4 независимо связаны с кольцом циклогексана, показанным в формуле (I), в положениях 3-, 4-, 5- или 6- и независимо выбраны из водорода, гидрокси, C1-С6алкила и C1-C6aлкокси, и когда R3 и R4 связаны с одним и тем же атомом циклогексанового кольца, они могут образовывать пяти- или шестичленное спирогетероциклическое кольцо, содержащее один или два гетероатома, выбранных из кислорода и серы;
R5, R6 и R14 независимо выбраны из водорода, C1-С6алкила, арила и бензила, или R6 и R14, взятые вместе с углеродом, с которым они связаны, могут образовывать спироС3-С5циклоалкил;
А выбран из С5-С12алкила, С3-С13карбоциклического кольца, и циклических систем, выбранных из соединений формул (III), (IV), (V), (VI), (VII) и (VIII):

где R7, R8, и R9 независимо выбраны из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-C7алкоксикарбонила, C1-С6тиоалкила, арила и N(R15, R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила;

где R10 и R11 независимо выбран из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-С6тиоалкила и N(R15, R16), где R15 и R16 независимо выбран из водорода, ацетила, метансульфонила и C1-С6алкила;
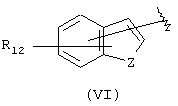
где R12 выбран из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-С6тиоалкила, и N(R15, R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила; и Z выбран из СН, СН2, О, N и S, где Z может быть непосредственно связан с "X", как показано в формуле (I), когда Z представляет СН или N, или Z может быть непосредственно связан с R17, когда Z представляет N, и R17 выбран из водорода, C1-С6алкила, С3-С8циклоалкила, арила и бензила;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.
В другом варианте данное изобретение предоставляет композицию или лекарственное средство, которое включает соединение, соответствующее формуле (I), в сочетании с фармацевтически приемлемым носителем, разбавителем или наполнителем (эксципиентом), и далее предоставляет способ производства композиции или лекарственного средства, которое содержит соединение, соответствующее формуле (I).
В других вариантах данное изобретение предоставляет фармацевтические композиции, которые содержат, по меньшей мере, одно соединение формулы (1) в количестве, эффективном для лечения заболевания или состояния теплокровного животного, подверженного или являющегося носителем заболевания или состояния, и/или для профилактики заболевания или состояния теплокровного животного, которое может иным способом наступить, и, кроме того, содержат, по меньшей мере, один фармацевтически приемлемый носитель, разбавитель или наполнитель. Кроме того, изобретение предоставляет способы лечения заболевания или состояния теплокровного животного, подверженного, или являющегося носителем заболевания или состояния, и/или профилактики возникновения заболевания или состояния у теплокровного животного, при которых терапевтически эффективное количество соединения формулы (I) или композиции, содержащей соединение формулы (I), вводится теплокровному животному, нуждающемуся в нем. Заболеваниями и состояниями, к которым применимы соединения, композиции или способы данного изобретения, являются следующие: аритмия, заболевания центральной нервной системы, судороги, эпилептические спазмы, депрессия, страх, шизофрения, болезнь Паркинсона, респираторные нарушения, муковисцидоз, астма, кашель, воспаление, артрит, аллергии, желудочно-кишечные расстройства, недержание мочи, слизистый колит, сердечно-сосудистые заболевания, церебральная ишемия или ишемия миокарда, гипертензия, синдром удлиненного интервала QT, инсульт, мигрень, глазные болезни, сахарный диабет, миопатии, миотония Беккера, миастения беременных, врожденная парамиотония, злокачественная гипертермия, гиперкалиемический периодический паралич, миотония Томсена, аутоиммунные заболевания, отторжение трансплантата при трансплантации органа или при трансплантации костного мозга, сердечная недостаточность, гипотензия, болезнь Альцгеймера, или другие психические расстройства, и алопеция.
В другом варианте данное изобретение предоставляет фармацевтическую композицию, содержащую количество соединения формулы (I), эффективное для осуществления локальной анальгезии или анестезии у теплокровного животного, которое в этом нуждается, и фармацевтически приемлемый носитель, разбавитель или наполнитель. Кроме того, изобретение предоставляет способ осуществления локальной анальгезии или анестезии у теплокровного животного, который включает введение теплокровному животному, нуждающемуся в этом, эффективного количества соединения формулы (1) или фармацевтической композиции, содержащей соединение формулы (I). Эти композиции и способы могут использоваться для ослабления или предотвращения болевого ощущения у теплокровных животных.
В другом варианте данное изобретение предоставляет фармацевтическую композицию, содержащую количество соединения формулы (I), эффективное для усиления либидо у теплокровного животного, которое в этом нуждается, и фармацевтически приемлемый носитель, разбавитель или наполнитель. Кроме того, изобретение предоставляет способ усиления либидо у теплокровного животного, который включает введение теплокровному животному, которое в этом нуждается, эффективного количества соединения формулы (I) или фармацевтической композиции, содержащей соединение формулы (I). Эти композиции и способы могут использоваться, например, для лечения половой дисфункции, напр. импотенции у самцов, и/или усиления полового влечения у пациентов с половой дисфункцией. В качестве еще одного примера, терапевтически эффективное количество может вводиться быку (или другому разводимому домашнему животному), чтобы содействовать повышенной эякуляции спермы, причем эякулированная сперма собирается и хранится для использования по мере необходимости для оплодотворения коров для развития животноводческой программы.
В другом варианте данного изобретения предоставляется соединение формулы (1) или композиция, содержащая соединение формулы (I), для применения в способах либо модулирования активности ионных каналов у теплокровного животного, либо модулирования активности ионных каналов in vitro.
Эти и другие варианты данного изобретения будут очевидными при обращении к следующим чертежам и подробному описанию.
Краткое описание чертежей
Фиг.1 иллюстрирует последовательность реакций получения соединения аминоциклогексилового эфира данного изобретения, описанных далее в примере 1.
Фиг.2 иллюстрирует процедуру, с помощью которой могут быть получены либо цис-, либо транс- аминоциклогексиловые эфиры данного изобретения.
Фиг.3 иллюстрирует методику синтеза, которая может применяться для получения либо цис-, либо транс- стереоизомеров соединений данного изобретения.
Фиг.4А и 4В иллюстрируют методику синтеза, описанную в примере 15.
Подробное описание изобретения
Как отмечалось выше, настоящее изобретение направлено на соединения аминоциклогексиловых эфиров, фармацевтические композиции, содержащие соединения аминоциклогексиловых эфиров, и различные виды применения соединения и композиций. Такие применения включают блокирование ионных каналов in vitro или in vivo, лечение аритмий, проведение анестезии и другие описанные здесь области применения. Пониманию данного изобретения может способствовать обращение к следующим определениям и пояснениям используемых здесь условных обозначений.
Определения и пояснения
Аминоциклогексилово-эфирные соединения изобретения имеют атом кислорода простого эфира в положении 1 циклогексанового кольца и аминовый атом азота в положении 2 циклогексанового кольца с нумерацией других положений в следующем порядке, показанном ниже в структуре (А):
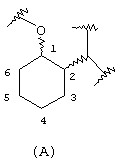
В указанной выше формуле связи от циклогексанового кольца к атомам 1-кислорода и 2-азота могут быть расположены относительно друг друга или в цис- или транс- конфигурации. В предпочтительном варианте данного изобретения стереохимия аминовых и простых эфирных заместителей циклогексанового кольца представляет либо (R,R)-транс, либо (S,S)-транс. В другом предпочтительном варианте стереохимия представляет либо (R,S)-цис, либо (S,R)-цис.
В изображенных здесь формулах связь с заместителем и/или связь, которая соединяет фрагмент молекулы с остальной частью соединения, может быть указана в виде пересекающих одной или более связей в структуре кольца. Это указывает на то, что связь может быть образована с любым одним из атомов, составляющих кольцевую структуру, а в ином случае у этого атома мог бы присутствовать атом водорода. Когда в конкретном положении в структуре не указано никаких конкретных заместителей, тогда в данном положении (положениях) присутствует водород.
Например, подразумевается, что соединения изобретения, содержащие А-Х-СН (R5)-группу, в которой А соответствует формуле (III)
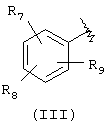
охватывают соединения, имеющие группу (В)
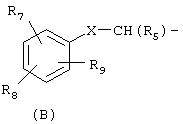
в которой имеется в виду, что группа (В) охватывает группы, в которых любой атом кольца, который мог бы в ином случае иметь в качестве заместителя водород, может быть вместо этого замещен группой R7, R8 или R9 при условии, что каждый из R7, R8 и R9 представлен в кольце один раз и только один. Атомы кольца, которые не замещены каким-либо из R7, R8 или R9, замещены водородом. В тех случаях, когда в изобретении указывается, что неароматическое кольцо замещено более, чем одной R группой, и показано, что эти R группы связаны с неароматическим кольцом связями, которые делят пополам кольцевые связи, тогда R группы могут присутствовать у различных атомов кольца, или у одного и того же атома кольца, поскольку в ином случае этот атом мог бы быть замещен атомом водорода.
Аналогичным образом, когда в изобретении даются соединения, содержащие А-Х-СН(R5)-группу, в которой А соответствует арильной группе (VI)
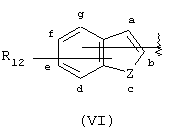
подразумевается, что изобретение охватывает соединения, в которых группа -X-CH(R5)-связана через Х с арильной группой (VI) по любому атому, который образует арильную группу (VI), поскольку в ином случае этот атом группы (VI) мог бы быть замещен атомом водорода. Таким образом, имеется семь положений (обозначенных буквами от "а" до "g") в структуре (VI), в которых может быть присоединена группа -X-CH(R5)-, и она присоединятся в одном из этих семи положений. Группа R12 будет занимать одно и только одно из оставшихся шести положений, а атомы водорода будут присутствовать в каждом из пяти оставшихся положений. Следует понимать, что когда Z представляет двухвалентный атом, например кислород или серу, тогда Z не может быть непосредственно связан с -X-CH(R5)-.
Когда изобретение указывает положение асимметричного двухвалентного радикала, тогда этот двухвалентный радикал может быть расположен любым возможным образом, который обеспечивает стабильную химическую структуру. Например, для соединений, содержащих А-Х-СН (R5)-группу, в которой Х представляет С(R14,R6)-Y-, изобретение предоставляет соединения, имеющие как A-C(R14,R6)-Y-CH(R5)-группу, так и A-Y-C(R14,R6)-CH(R5) группу.
Волнистая связь от заместителя к центральному циклогексановому кольцу указывает на то, что группа может быть расположена с любой стороны плоскости центрального кольца.
Соединения данного изобретения содержат, по меньшей мере, два асимметричных атома углерода и поэтому существуют в виде энантиомеров и диастереомеров. Если не оговорено особо, данное изобретение включает все энантиомерные и диастереомерные формы аминоциклогексилово-эфирных соединений изобретения. В данное изобретение включены чистые стереоизомеры, смеси энантиомеров и/или диастереомеров, и смеси различных соединений изобретения. Таким образом, соединения данного изобретения могут быть в виде рацематов, рацемических смесей и в виде индивидуальных диастереомеров или энантиомеров во всех изомерных формах, включенных в данное изобретение. Рацемат или рацемическая смесь не означает, что это смесь стереоизомеров 50:50.
Фраза “независимо в каждом случае” означает ситуацию (i), когда в соединении изобретения встречается какая-либо переменная более одного раза, определение этой переменной независимо от ее определения в любом другом случае; и (ii) идентичность какой-либо одной из двух различных переменных (например, R1 в комплекте R1 и R2) выбрана безотносительно идентичности другого члена комплекта. Однако комбинации заместителей и/или переменных допустимы, только если такие комбинации дают в результате стабильные соединения.
Следующие термины, в соответствии с данным изобретением используемые здесь, определяются следующим образом.
Термин “соли присоединения кислот” или кислотно-аддитивные соли относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не являются биологически или иным образом нежелательными, образуемым с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и аналогичные, или с органическими кислотами, такими как уксусная, пропионовая, гликолевая, пировиноградная, щавелевая, малеиновая, малоновая, янтарная, фумаровая, винная, лимонная, бензойная, коричная, миндальная кислоты, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфокислота, салициловая кислота и аналогичные.
"Ацил" относится к разветвленным или неразветвленным углеводородным фрагментам, заканчивающимся карбонильной -(С=O)-группой, содержащий указанное число атомов углерода. Примеры включают ацетил [СН3С=O-, С2-ацил] и пропионил [СН3СН2С=О-, С3ацил].
"Алканоилокси" относится к сложноэфирному заместителю, в котором кислород эфира является точкой соединения с молекулой. Примеры включают пропаноилокси [(СН3СН2С=O-O-, С3алканоилокси] и этаноилокси [СН3С=O-O-, С2алканоилокси].
"Алкокси” относится к O-атому, замещенному алкильной группой, например метокси [-ОСН3, С1алкокси].
"Алкоксиалкил" относится к группе алкилена, замещенной алкоксигруппой. Например, метоксиэтил [СН3ОСH2СН2-] и этоксиметил [СН3СН2ОСН2-] являются С3алкоксигруппами.
“Алкоксикарбонил” относится к сложноэфирному заместителю, в котором углерод карбонила является точкой присоединения к молекуле. Примеры включают этоксикарбонил [СН3СН2OC=O-, С3алкоксикарбонил] и метоксикарбонил [СН3ОС=O-, С2алкоксикарбонил].
“Алкил” относится к разветвленному или неразветвленному углеводородному фрагменту, содержащему указанное число атомов углерода и имеющему одну точку присоединения. Примеры включают n-пропил (С3алкил), изопропил (также С3алкил) и трет-бутил (С4алкил).
“Алкилен” относится к двухвалентному радикалу, который представляет разветвленный или неразветвленный углеводородный фрагмент, содержащий указанное число атомов углерода и имеющий две точки присоединения. Примером является пропилен [-CH2CH2CH2-, С3алкилен].
“Алкилкарбокси” относится к разветвленному или неразветвленному углеводородному фрагменту, оканчивающемуся карбоново-кислотной группой [СООН]. Примеры включают карбоксиметил [HOOC-CH2-, С2алкилкарбокси] и карбоксиэтил [НООС-СН2СН2-, С3алкилкарбокси].
“Арил” относится к ароматическим группам, которые имеют, по меньшей мере, одно кольцо, имеющее сопряженную пиэлектронную систему, и включает карбоциклические арильные, гетероциклические арильные (называемые также гетероарильными группами) и биарильные группы.
Обычно, предпочтительными группами в соединениях данного изобретения являются карбоциклические арильные группы, и предпочтительными карбоциклическими арильными группами являются фенил и нафтил.
“Аралкил” относится к группе алкилена, в которой одна из точек присоединения идет к арильной группе. Примером аралкильной группы является бензильная группа [С6Н5СН2-, С7аралкильная группа].
“Циклоалкил” относится к кольцу, которое может быть насыщенным или ненасыщенным и моноциклическим, бициклическим или трициклическим, целиком образованным атомами углеродами. Примером циклоалкильной группы является циклопентильная группа (C5H7-), которая представляет пятиуглеродную (C5) ненасыщенную циклоалкильную группу.
“Карбоциклический” относится к кольцу, которое может быть или арильным, или циклоалкильным кольцом, оба из которых определены выше.
“Карбоциклический арил” относится к ароматическим группам, в которых атомами, образующими ароматическое кольцо, являются атомы углерода. К карбоциклическим арильным группам относятся моноциклические карбоциклические арильные группы, такие как фенил, и бициклические карбоциклические арильные группы, такие как нафтил.
“Гетероатом” относится к неуглеродному атому, причем предпочтительными гетероатомами являются бор, азот, кислород, сеpa и фосфор, и особенно предпочтительными гетероатомами в соединениях данного изобретения являются азот, кислород и сера.
“Гетероарил” относится к арильным группам, имеющим от 1 до 9 атомов углерода, а остальные атомы представляют гетероатомы и включают гетероциклические системы, описанные в "Handbook of Chemistry and Physics, 49th edition, 1968, R.C.Weast, editor; The Chemical Rubber Co., Cleveland, ОН. Смотри, в частности, Section С, Rules for Naming Organic Compounds, B. Fundamental Heterocyclic Systems. К подходящим гетероарильным группам относятся фуранил, тиенил, пиридил, пирролил, пиримидил, пиразинил, имидазолил и аналогичные.
“Гидроксиалкил” относится к разветвленному или неразветвленному углеводородному фрагменту, несущему гидрокси (-ОН) группу. Примеры включают гидроксиметил (-СН2ОН, С1гидроксиалкил) и 1-гидроксиэтил (-СНОНСН3, С2гидроксиалкил).
“Тиоалкил” относится к атому серы, замещенному алкильной группой, например тиометил (CH3S-, С1тиоалкил).
Термин “модулирование или модулирующая” в связи с активностью ионного канала означает, что активность ионного канала может либо увеличиваться, либо уменьшаться в ответ на введение соединения или композиции, или в результате применения способа данного изобретения. Таким образом, ионный канал может быть активирован так, чтобы транспортировать больше ионов, или может быть блокирован так, что меньше ионов транспортируется или вообще не транспортируется каналом.
“Фармацевтически приемлемые носители” для терапевтического применения хорошо известны в области фармацевтики и описаны, например, в Remingtons Pharmaceutical Science, Mack Publishing Co. (A.R.Gennaro edit. 1985). Например, могут использоваться стерильный солевой физиологический раствор и забуференный фосфатом солевой раствор при физиологических значениях рН. В фармацевтической композиции могут быть предусмотрены консерванты, стабилизаторы, красители и даже ароматизирующие или вкусовые агенты. Например, в качестве консервантов могут добавляться бензоат натрия, сорбиновая кислота и сложные эфиры параоксибензойной кислоты. Там же на 1449. Кроме того, могут использоваться антиоксиданты и суспендирующие агенты. Там же.
“Фармацевтически приемлемая соль” относится к солям соединений данного изобретения, получаемым в результате сочетания таких соединений и органических или неорганических кислот (соли присоединения кислот) или органических или неорганических оснований (соли присоединения оснований). Соединения данного изобретения могут применяться либо в форме свободного основания, либо в форме соли, причем обе формы охватываются объемом настоящего изобретения.
“Терапевтически эффективное количество” соединения изобретения зависит от способа введения, типа теплокровного животного, подлежащего лечению, и физических особенностей рассматриваемого конкретного теплокровного животного. Эти факторы и их взаимосвязь для определения этого количества соединения хорошо известны специалистам практикам в области медицины.
Данное количество и способ введения могут быть подобраны с целью достижения оптимальной эффективности и зависят от таких факторов, как вес, режим питания, сопутствующая лекарственная терапия и другие факторы, которые известны специалистам в области медицины.
Композиции, описанные здесь как “содержащие соединение, имеющее формулу (I)”, охватывают композиции, которые содержат более, чем одно соединение, имеющее формулу (I).
Соединения данного изобретения
Соединениями данного изобретения являются амины, которые могут быть представлены формулой (I):
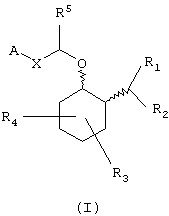
Соединения формулы (I) являются аминоциклогексиловыми эфирами. Более конкретно, эти аминоциклогексиловые эфиры замещены в положении 2 циклогексильного кольца аминогруппой -NR1R2-. Циклогексильное кольцо может быть также замещено дополнительными заместителями (обозначенными R3 и R4), описанными более детально ниже. Примеры конкретных соединений, представленных формулой (I), описаны ниже.
В зависимости от выбора заместителей R1 и R2 соединения формулы (I) могут быть третичными аминами (ни один из R1 и R2 не являются водородом). Когда амин является третичным, он может быть циклическим амином. Заместители амина R1 и R2 могут быть независимо выбраны из заместителей, которые включают алкоксильные группы, содержащие от трех до восьми атомов углерода (т.е. С3-С8алкоксиалкил), алкильные группы, содержащие от одного до восьми атомов углерода, в которых один из атомов углерода замещен гидроксильной группой (т.е. C1-С8гидроксиалкил), и аралкильные группы, содержащие от семи до двенадцати атомов углерода (т.е. С7-С12аралкил).
Альтернативно R1 и R2 вместе с атомом азота, с которым они непосредственно связаны в формуле (I), могут образовывать кольцо, обозначенное формулой (II):
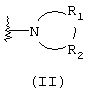
причем кольцо формулы (II) образовано из азота, как показано, а также трех-девяти дополнительных кольцевых атомов, независимо выбранных из углерода, азота, кислорода и серы, где любые два смежных атома кольца могут быть связаны одинарной или двойной связями и где один или более дополнительных атомов углерода в кольце могут быть замещены одним или двумя заместителями, выбранными из водорода, гидрокси, C1-С3гидроксиалкила, оксо, С2-С4ацила, C1-С3алкила, С2-С4алкилкарбокси, C1-С3алкокси, С1-С20алканоилокси, или могут быть замещены с образованием пяти- или шестичленного спирогетероциклического кольца, содержащего один или два гетероатома, выбранных из кислорода и серы (например, ацетальная, тиоацетальная, кетальная или тиокетальная группа); и любые два смежных дополнительных атомов углерода кольца могут быть сконденсированы с С3-С8карбоциклическим кольцом, и один или более дополнительных атома азота в кольце могут быть замещены заместителями, выбранными из водорода, C1-С6алкила, С2-С4ацила, С2-С4гидроксиалкила и С3-С8алкоксиалкила. Примерами заместителей, содержащих сконденсированную кольцевую систему, являются пергидроиндолил и 1,2,3,4-тетрагидроизохинолинил.
Что касается кольца формулы (II), любые два смежных атома кольца могут быть связаны вместе одинарной или двойной связями. Таким образом, кольцо формулы (II) может быть насыщенным или ненасыщенным, и ненасыщенное кольцо может содержать один или более одного участков ненасыщенности. Другими словами кольцо формулы (II) может содержать одну или более двойных связей, однако имеется в виду, что ненасыщенное кольцо формулы (II) является химически стабильным.
Альтернативно R1 и R2 вместе с 2-азотом амина в формуле (I) могут завершать бициклическое кольцо. Бициклические кольца включают, например, 3-азабицикло[3.2.2]нонан, 2-азабицикло[2.2.2]октан, 3-азабицикло[3.1.0]гексан и 3-азабицикло [3.2.0]гептан. В этих производных 2-заместителями циклогексиловых эфиров формулы (I) являются следующие группы: 3-азабицикло[3.2.2.]нонан-3-ил, 2-азабицикло[2.2.2]октан-2-ил, 3-азабицикло[3.1.0]гексан-3-ил и 3-азабицикло[3.2.0]-гептан-3-ил.
Предпочтительно R1 и R2, взятые вместе, содержат только один гетероатом. К предпочтительным гетероатомам относятся азот, кислород и сера. Примером кольца, в котором R1 и R2 вместе включают гетероатом кислорода, является морфолинил. Примером кольца, в котором R1 и R2 вместе включают еще один гетероатом азота, является пиперазинил.
Заместители циклогексана R3 и R4 могут быть независимо связаны с кольцом в положениях 3, 4, 5 или 6 (т.е. и R3, и R4 могут быть присоединены в одном и том же положении кольца, или каждый из них связан с кольцом в различных положениях). R3 и R4 независимо выбраны из водорода, гидрокси, C1-С6алкила и C1-С6алкокси, и когда и R3, и R4 связаны с одним и тем же атомом циклогексанового кольца, они могут вместе образовать пяти- или шестичленное спирогетероциклическое кольцо, содержащее один или два гетероатома, выбранных из кислорода и серы. Предпочтительные гетероциклические заместители содержат либо единственный кольцевой атом кислорода, либо единственный кольцевой атом серы.
В зависимости от идентичности X эфирная боковая цепь -CH(R5)-X-A в формуле (I) может принимать несколько форм. Например, соединение формулы (I) может иметь Х в виде группы -С(R6,R14)-Y-, в которой Y может быть любым из прямой связи, атома кислорода (О), атома серы (S) или С1-С4алкиленовой группы. R6 и R14 независимо выбраны из водорода, C1-С6алкила, арила и бензила, или R6 и R14, взятые вместе с углеродом, с которым они связаны, могут образовывать спиро С3-С5циклоалкил. Таким образом, соединения изобретения включают соединения формулы (I), в которой R6 и R14 представляют водород и Y представляет прямую связь, так что Х может представлять CH2.
Альтернативно Х может быть алкениленовым фрагментом, например цис- или транс-алкениленовым фрагментом, C(R13)=CH, в котором R13 может быть любым из водорода, C1-С6алкила, С3-С8циклоалкила, арила или бензила. Для соединений формулы (I), в которой Х является алкениленовым фрагментом, Х предпочтительно является транс-алкениленовым фрагментом.
Альтернативно Х может быть прямой связью. Независимо от выбора А, Х и других переменных R5 выбран из водорода, C1-С6алкила, арила и бензила.
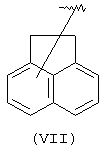
Компонент А эфирной боковой цепи обычно является гидрофобным фрагментом. В типичном случае гидрофобный фрагмент составлен из неполярных химических групп, таких как углеводороды или углеводороды, замещенные галогенами, или простые эфиры, или гетероциклические группы, содержащие кольцевые атомы азота, кислорода или серы. Подходящими углеводородами являются С5-С12алкил и С3-С13карбоциклические кольца. Особенно предпочтительные циклические углеводороды включают выбранные ароматические группы, такие как фенил, 1-нафтил, 2-нафтил, инденил, аценафтил и флуоренил и представлены соответственно формулами (III), (IV), (V), (VI), (VII) или (VIII).
Подходящей "А" группой в соединениях данного изобретения является фенильное кольцо, представленное формулой (III):
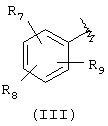
в которой R7, R8 и R9 независимо выбраны из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-C6-тиоалкила, арила и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила.
Для соединений формулы (I), в которой Х является прямой связью или CH2, по меньшей мере, один из R7, R8 и R9 предпочтительно выбран из следующих: амин (-NR15R16, где R15 и R16 независимо являются водородом, ацетилом, метансульфонилом и C1-С6алкилом), бром, хлор, фтор, карбокси, водород, гидрокси, гидроксиметил, нитро, трифторметил, С2-С7алканоилокси, C1-C6aлкил, C1-С6алкокси, С2-С7алкилкарбонил, C1-С6тиоалкил или арил. Для соединений формулы (I), в которой Х является СН=СН, и R3 и R4 являются водородом, по меньшей мере, один из R7, R8 и R9 предпочтительно является заместителем, отличным от водорода.
Другими подходящими "А" группами в соединениях данного изобретения являются группы 1-нафтила, представленные формулой (IV):
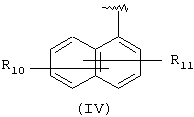
в которой R10 и R11 независимо выбраны из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-C6-тиоалкила, и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила.
Другой подходящей группой "А" в соединениях данного изобретения является группа 2-нафтила, представленная формулой (V):
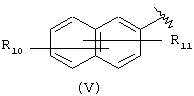
в которой R10 и R11 независимо выбраны из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-С6тиоалкила, и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила, определенных выше.
Другими подходящими группами "А" в соединениях данного изобретения являются ароматические группы, представленные формулой (VI):
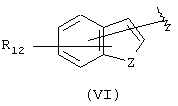
в которой R12 выбран из брома, хлора, фтора, карбокси, водорода, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-С6тиоалкила, и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила; и Z выбран из СН, СН2, О, N и S, где 2 может быть непосредственно связан с "X", как показано в формуле (I), когда Z является СН или N, или Z может быть непосредственно связан с R17, когда Z представляет N, и R17 выбран из водорода, C1-С6алкила, С3-С8циклоалкила, арила и бензила.
Арильные группы формулы (VI) являются производными индена, индола, бензофурана и тианафтена, когда Z является метиленом, азотом, кислородом и серой соответственно. Предпочтительные гетероциклические группы формулы (VI) включают индол, в котором Z является NH, бензофуран, в котором Z является О, и тианафтен, в котором Z является S. Как описано ниже, в предпочтительном варианте Z является О, S или N-R17 и в особенно предпочтительном варианте Z является О или S.
Другими подходящими "А" группами в соединениях данного изобретения являются аценафтильные группы, представленные формулой (VII):
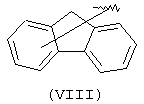
Еще одной подходящей "А" группой в соединениях данного изобретения является группа флуоренила, представленная формулой (VIII):
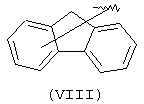
Предпочтительно компонент А эфирной боковой цепи является аценафтильной или флуоренильной группой, только когда Х представляет прямую связь или СН2. В дополнительных предпочтительных вариантах аценафтильная группа представляет 1-аценафтильную группу, а флуоренильная группа представляет 9-флуоренильную группу.
Как упомянуто выше, настоящее изобретение предоставляет аминоциклогексиловые эфиры, представленные формулой (I). В предпочтительном варианте Х является (CH2)-Y. Для этих вариантов Y предпочтительно представляет прямую связь, атом кислорода или атом серы. В особенно предпочтительном варианте Y представляет прямую связь или атом кислорода. В другом предпочтительном варианте Y представляет прямую связь и Х является C(R6,R14), где R6 и R14 имеют значения, указанные выше. В еще одном предпочтительном варианте, в котором Х является C(R13)=CH, R13 представляет атом водорода. В этих вариантах R3 и R4 предпочтительно независимо связаны с циклогексановым кольцом в положениях 4 или 5.
В предпочтительном варианте изобретение предоставляет соединения формулы (IX), или их сольваты, или фармацевтически приемлемые соли:
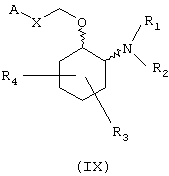
в которой независимо в каждом случае
Х выбран из прямой связи -СН=СН- и -С(R6,R14)-Y-;
Y выбран из прямой связи О и S; и
R1, R2, R3, R4, R6, R7, R8, R9, R10, R11, R12, R14, А и Z имеют значения, определенные выше для соединения формулы (I).
В другом предпочтительном варианте изобретение предоставляет соединение, имеющее формулу (X), или его сольват, или фармацевтически приемлемую соль:

где независимо в каждом случае
Х выбран из прямой связи -СН=СН- и -С(R6,R14)-Y-;
Y выбран из прямой связи О и S; и
R1, R2, R6 и R14 имеют значения, определенные выше для соединений формулы (I);
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и C1-С6алкокси; и
А выбран из С5-С12алкила, С3-С8циклоалкила и любой из формул (III), (IV), (V) и (VI), определенных выше для соединений формулы (I), в которых Z, R7, R8, R9, R10, R11 и R12 имеют те же значения, которые указаны выше для соединений формулы (I).
В другом предпочтительном варианте изобретение предоставляет соединения, имеющие формулу (XI), или их сольваты, или фармацевтически приемлемые соли:
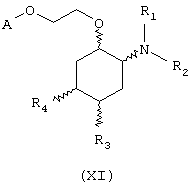
где независимо в каждом случае
R1 и R2 имеют значения, указанные выше для соединений формулы (I);
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и метоксигруппы; и
А выбран из С5-С12алкила, С3-С8циклоалкила и любой из формул (III), (IV), (V) и (VI), определенных выше для соединений формулы (I), в которых Z, R7, R8, R9, R10, R11 и R12 имеют те же значения, что указаны выше для соединений формулы (I).
В другом предпочтительном варианте изобретение предоставляет соединения формулы (XII), или их сольваты, или фармацевтически приемлемые соли:
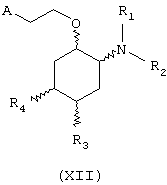
где независимо в каждом случае
R1 и R2 имеют значения, указанные выше для соединений формулы (I);
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и метокси; и
А выбран из С5-С12алкила, С3-С8циклоалкила и любой из формул (III), (IV), (V) и (VI), определенных выше для соединений формулы (I), в которых Z, R7, R8, R9, R10, R11 и R12 имеют те же значения, что указаны выше для соединений формулы (I).
В другом предпочтительном варианте изобретение предоставляет соединения формулы (XIII), или их сольваты, или фармацевтически приемлемые соли:
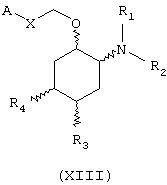
где независимо в каждом случае
Х выбран из прямой связи и -СН=СН-;
R1 и R2 имеют значения, определенные выше для соединений формулы (I);
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и метокси; и
А выбран из С3-С8циклоалкила и любой из формул (III), (IV), (V), (VI), (VII) и (VIII), которые определены выше для соединений формулы (I); где R8 и R9 имеют значения, которые указаны выше для соединений формулы (I); R7, R10, R11 и R12 являются водородом, и Z выбран из О, S и N-R17, где R17 выбран из водорода и метила; при условии, что А может быть выбран из формул (VII) и (VIII), только когда Х представляет прямую связь.
В другом предпочтительном варианте изобретения предоставляются соединения, имеющие формулу (XIV), или их сольваты, или фармацевтически приемлемые соли:
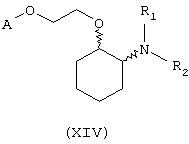
где независимо в каждом случае
R1 и R2 имеют значения, указанные выше для соединений формулы (I);
А выбран из любой из формул (III), (IV), (V) и (VI), которые определены выше для соединений формулы (I), где R7, R10, R11 и R12 представляют водород, R8 и R9 независимо выбраны из водорода, гидрокси, фтора, хлора, брома, метансульфонамидо, метаноилокси, метоксикарбонила, нитро, сульфамила, тиометила, трифторметила, метила, этила, метокси, этокси и NH2 при условии, что, по меньшей мере, один из R8 и R9 не является водородом; и Z выбран из О и S.
В другом предпочтительном варианте изобретения предоставляются соединения, имеющие формулу (XV), или их сольваты, или фармацевтически приемлемые соли:

где независимо в каждом случае
R1 и R2 имеют значения, указанные выше для соединений формулы (I);
А выбран из любой из формул (III), (IV), (V) и (VI), определенных выше для соединений формулы (I), где R7, R10, R11 и R12 представляют водород, R8 и R9 независимо выбраны из водорода, гидрокси, фтора, хлора, брома, метансульфонамидо, метаноилокси, метоксикарбонила, нитро, сульфамила, тиометила, трифторметила, метила, этила, метокси, этокси и NН2 при условии, что, по меньшей мере, один из R8 и R9 не является водородом; и Z выбран из О и S.
В другом предпочтительном варианте изобретения предоставляются соединения, имеющие формулу (XVI), или их сольваты, или фармацевтически приемлемые соли:
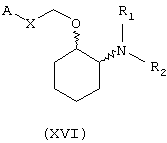
где независимо в каждом случае
Х выбран из прямой связи, транс-СН=СН-, -СН2-и -СН2-О-;
R1 и R2 оба являются метоксиэтилом или вместе с атомом азота, с которым они связаны, завершают кольцо, выбранное из пирролидинила, кетопирролидинила, ацетоксипирролидинила, гидроксипирролидинила, тиазолидинила, пиперидинила, кетопиперидинила, ацетилпиперазинила, 1,4-диокса-7-азаспиро[4.4]нон-7-ила, гексагидроазепинила, морфолинила, N-метилпиперазинила и 3-азабицикло[3.2.2]нонанила; и
А выбран из циклогексила, монохлорфенила, 2,6-дихлорфенила, 3,4-дихлорфенила, 2-бромфенила, 2,4-дибромфенила, 3-бромфенила, 4-бромфенила, 1-нафтила, 2-нафтила, 3-бензо(b)-тиофенила, 4-бензо(b)тиофенила, (2-трифторметил)фенила, 2,4-ди(трифторметил)фенила и (4-трифторметил)фенила.
Следующие соединения являются наиболее предпочтительными соединениями данного изобретения:
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(2-нафтенэтокси)]циклогексан
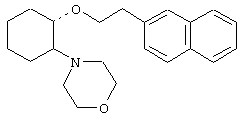
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(1-нафтенэтокси)]циклогексан
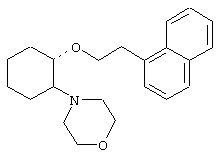
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(4-бромфенэтокси)]циклогексан
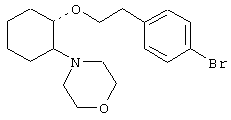
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-[2-(2-нафтокси)этокси]]циклогексан
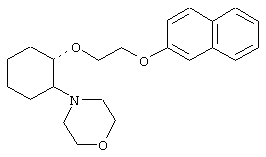
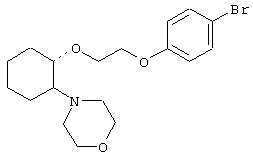
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-[2-(4-бромфенокси)этокси]]циклогексан
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(3,4-диметоксифенэтокси)]циклогексан
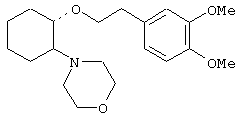
(1R,2R)/(1S,2S)-[2-(1-пирролидинил)-1-(1-нафтенэтокси)]циклогексан
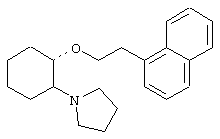
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(2-(бензо[b]тиофен-3-ил)]циклогексан
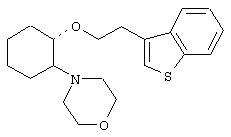
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(2-(бензо[b]тиофен-4-ил)]циклогексан
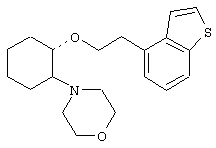
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(3-бромфенэтокси)]циклогексан
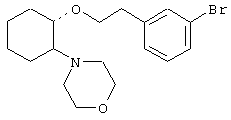
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(2-бромфенэтокси)]циклогексан
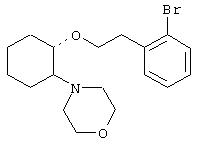
(1R,2R)/(1S,2S)-[2-(4-морфолинил)-1-(3-(3,4-диметоксифенил)пропокси)]циклогексан
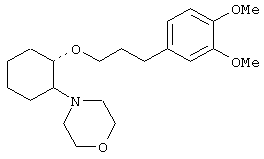
(1R,2R)/(1S,2S)-[2-[бис(2-метоксиэтил)аминил]-1-(2-нафтенэтокси)]циклогексан
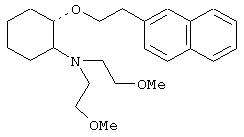
(1R,2R)/(1S,2S)-2-(4-морфолинил)-1-(3,4-дихлорфенэтокси)циклогексан
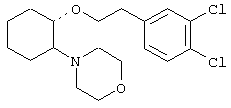
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексан
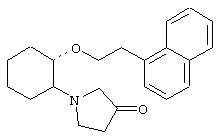
(1R,2R)/(1S,2S)-2-(1-ацетилпиперазинил)-1-(2-нафтенэтокси)циклогексан
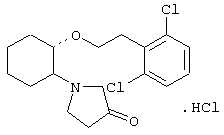
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,6-дихлорфенэтокси)циклогексан
(1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(1-нафтенэтокси)циклогексан
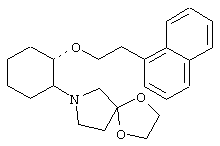
моногидрохлорид (1R,2S)/(1S,2R)-2-(4-морфолинил)-1-[(2-трифторметил)фенэтокси]циклогексана
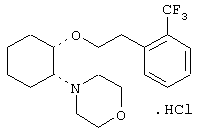
моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[3-(циклогексил)пропокси]циклогексана
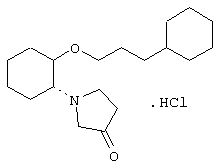
моногидрохлорид (1R,2R)/(1S,2S)-2-(3-ацетоксипирролидинил)-1-(1-нафтенэтокси)циклогексана
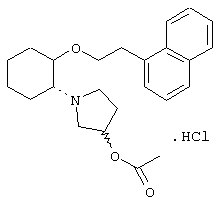
моногидрохлорид (1R,2R)/(1S,2S)-2-(4-морфолинил)-1-[(2,6-дихлорфенил)метокси]циклогексана
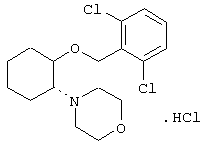
моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[2,6-дихлорфенил)метокси]циклогексана
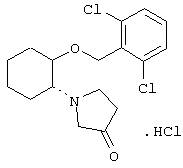
моногидрохлорид (1R,2R)/(1S,2S)-2-(3-гидроксипирролидинил)-1-(2,6-дихлорфенэтокси)циклогексана
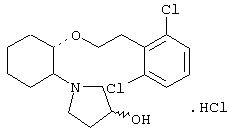
моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,2-дифенилэтокси)циклогексана
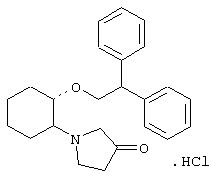
моногидрохлорид (1R,2R)/(1S,2S)-2-(3-тиазолидинил)-1-(2,6-дихлорфенэтокси)циклогексана
моногидрохлорид (1R,2S)/(1S,2R)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексана
Описание способа получения соединений изобретения
Аминоциклогексилово-эфирные соединения данного изобретения содержат амино- и простые эфирные боковые цепи, расположенные в положениях 1,2 циклогексанового кольца. Соответственно амино- и эфирные боковые цепи относительно друг друга и относительно плоскости циклогексанового кольца могут быть либо в цис, либо в трансвзаимосвязи. Настоящее изобретение предоставляет методологию синтеза, посредством которой могут быть получены цис и транссоединения.
Транссоединения данного изобретения могут быть получены по аналогии с известными методиками синтеза (смотри, например, Shanklin, Jr. et al., U.S. Patent, 5130309). На фигуре 1 в общих чертах показано получение транссоединения изобретения, причем данное получение более полно описано в примере 1. Как отмечено на фигуре 1, получение транссоединения изобретения может быть достигнуто с помощью следующей четырехстадийной процедуры.
На первой стадии (обозначенной "i)" на фигуре 1) эпоксид циклогексена подвергается реакции раскрытия кольца с амином. Смотри, например, Szmuszkovicz, U.S. Patent 4145435. Хотя реакция может происходить при комнатной температуре, обычно предпочтительной является повышенная температура, чтобы довести реакцию до завершения в течение желательного в промышленных масштабах промежутка времени. Обычно реакция проводится в растворителе, таком как вода, и температура дефлегмации растворителя обеспечивает подходящую температуру. Удовлетворительные результаты обычно обеспечиваются при использовании равных молярных количеств амина и эпоксида циклогексена. В любом случае, азот амина реагирует с эпоксидной группой с образованием 1-гидрокси 2-аминоциклогексана, причем обычным является взаимное трансрасположение гидрокси- и аминогрупп.
В этой общей реакции может использоваться широкое многообразие аминовых соединений и замещенных оксидов циклогексена, а фигура 1 иллюстрирует данную реакцию для случая, когда амин представляет морфолин и оксид циклогексена не замещен. Для других аминов или замещенных эпоксидов циклогексена, которые могут содержать другие реакционноспособные функциональные группы, перед осуществлением стадии i) вводятся подходящие защитные группы. Подходящие защитные группы описаны, например, в Green, Protective Groups in Organic Chemistry, John Wiley and Sons, New York NY (1991).
На второй стадии (обозначенной "ii)" на фигуре 1) гидроксигруппа, которая получена из эпоксида, превращается в активированную форму. Термин “активированная форма”, используемый здесь, означает, что гидроксигруппа превращается в легко отщепляемую группу. Отщепляемая группа, показанная на фигуре 1, представляет группу мезилата, и она является предпочтительной отщепляемой группой. Однако гидроксигруппа может превращаться в другие отщепляемые группы в соответствии с методами, хорошо известными в данной области. В типичной реакции соединение аминоциклогексанола обрабатывается метансульфонилхлоридом в присутствии основания, такого как триэтиламин, как показано на фигуре 1. Реакция удовлетворительно проводится при около 0° С. Обычно предпочтителен избыток метансульфонилхлорида относительно аминоциклогексанола, чтобы максимально превратить более ценный аминоциклогексанол в активированную форму.
Для некоторых других соединений аминоциклогексанола может быть необходимым введение подходящих защитных групп перед выполнением стадии ii). Подходящие защитные группы описаны, например, в Green, Protective Groups in Organic Chemistry, John Wiley and Sons, New York NY (1991).
На третьей стадии (обозначенной "iii)" на фигуре 1) спирт подвергается реакции с сильным основанием для получения соли алкоксида. Превращение спирта в алкоксид (известный также как алкоголят) с использованием сильного основания является обычной реакцией, и она будет протекать с широким многообразием гидроксисодержащих соединений. В некоторых случаях соединение спирта может иметь другие реакционно-способные функциональные группы, которые желательно защитить перед контактом спирта с сильным основанием. Подходящие защитные группы описаны, например, в Green, Protective Groups in Organic Chemistry, John Wiley and Sons, New York NY (1991). Такие спирты являются или промышленно доступными, либо могут быть получены с помощью процедур, описанных в этой области, или адаптированных для данного случая, причем подходящие процедуры могут быть определены с помощью Chemical Abstracts и индексов (Indicies), разработанных и опубликованных Американским химическим обществом.
На четвертой стадии (обозначенной "iv)" на фигуре 1) алкоголят стадии "iii)" подвергается реакции с активированным аминоциклогексанолом стадии "ii)". Таким образом, в общем говоря, соединения данного изобретения могут быть получены по реакции активированной формы соответствующего 1,2-аминоциклогексанола (1 моль) с алкоголятом (1,25 моль), полученным с помощью обработки выбранного спирта (1,25 моль), например, гидридом натрия (1,3 моль). 1,2-Аминоциклогексанол (1 моль) может быть активирован с помощью образования соответствующего мезилата в присутствии метансульфонилхлорида (1,25 моль) и триэтиламина (1,5 моль). Мезилат быстро добавляется к алкоголяту в подходящем растворителе, таком как диметилформамид. За температурой реакции тщательно следят для того, чтобы избежать нежелательных побочных реакций, таких как β -элиминирование. Вообще для образования соединений изобретения обычно подходящей является температура реакции 80-90° С в течение 2 часов. Когда реакция проходит по существу до завершения, желаемый продукт выделяется из реакционной смеси с помощью традиционных приемов органической химии и очищается в основном с помощью хроматографии на колонках с последующей перекристаллизацией. Защитные группы могут удаляться на соответствующей стадии последовательности реакций. Подходящие способы описаны, например, в Green, "Protective Groups in Organic Chemistry", John Wiley and Sons, New York NY (1991).
Последовательность реакций, описанных выше (и показанных на фигуре 1), дает аминоциклогексиловый эфир в виде свободного основания. С помощью препаративной хиральной ВЭЖХ могут быть получены чистые энантиомерные формы. Свободное основание при желании может с помощью известных способов превращаться в моногидрохлоридную соль и впоследствии при желании в другие соли присоединения кислоты с помощью реакций с неорганическими или органическими солями. Соли присоединения кислоты могут быть также получены метатетически по реакции одной соли присоединения кислоты с кислотой, которая сильнее, чем кислота аниона исходной соли.
Цис- и транс-соединения изобретения могут быть получены в соответствии с химическими процессами, показанными на фигуре 2. Как показано на фигуре 2, 1,2-аминоциклогексаноны могут быть получены путем окисления по Сверну (Swern) соответствующих соединений транс-1,2-аминоциклогексанола (которые могут быть получены, как описано выше) с использованием оксалилхлорида/диметилсульфоксида (смотри, например, Synthesis 1980, 165). Последующее восстановление аминоциклогексанона алюмогидридом лития или борогидридом натрия дает смесь цис- и транс-аминоциклогексанолов. Смесь аминоспиртов может этерифицироваться соответствующей карбоновой кислотой с помощью азеотропной перегонки в толуоле в присутствии каталитического количества пара-толуолсульфокислоты, давая диастереомерную смесь цис- и транс-сложных эфиров. Смесь диастереомерных сложных эфиров может разделяться с помощью препаративной хроматографии с использованием приемов, известных специалистам в данной области. Затем полученные рацемические цис- или транссложные эфиры могут восстанавливаться боргидридом натрия в присутствии кислоты Льюиса в соответствующие рацемические цис- или транс-простые эфиры (смотри, например, J. Org. Сhem. 25, 875, 1960 и Tetrahedron 18, 953, 1962). Рацемический простой цис-эфир можно разделить с помощью препаративной хиральной ВЭЖХ, как обсуждалось выше для транс-соединения.
Альтернативно цис- и транс-соединения изобретения могут быть получены согласно химическим реакциям, указанным на фигуре 3. Как показано на фигуре 3, оксид циклогексена может реагировать со спиртом (ROH) в присутствии Мg(ClO4)2 (смотри, например, М. Chini et al., Synlett, 673-676, 1992), давая 1,2-гидроксициклогексиловые эфиры. Окисление дихроматом пиридиния (смотри, например, R. Oshima et al., J.Org. Chem., 50, 2613-2621, 1985) дает соответствующий 1,2-алкоксициклогексанон. Последующее восстановительное аминирование (R.F.Borch et al., J.Am.Chem.Soc., 93(12), 2897-2904, 1971) дает смесь цис- и транс-аминоциклогексиловых эфиров. Смесь диастереомерных эфиров может разделяться с помощью хроматографии специалистами в данной области. Полученные таким образом рацемические цис- или транс-эфиры могут затем разделяться с помощью хорошо известных в данной области классических способов перекристаллизации или с помощью препаративной хиральной ВЭЖХ, давая индивидуальные энантиомеры: транс-(1R,2R), транс-(1S,2S), цис-(1R,2S) или цис-(1S,2R)аминоэфиры.
Описанные здесь процедуры синтеза, особенно с учетом общих знаний в данной области, предоставляют достаточное руководство специалистам в данной области для проведения синтеза, выделения и очистки соединений данного изобретения.
Композиции и способы введения
В еще одном варианте данное изобретение предоставляет композиции, которые включают описанное выше соединение циклогексиламина в смеси или ином сочетании с одним или более инертными носителями, эксципиентами и разбавителями, а также при желании с необязательными дополнительными ингредиентами. Эти композиции полезны, например, в качестве стандартов для анализа, удобных средств доставки в виде бестарных партий или фармацевтических композиций. Анализируемое количество соединения изобретения представляет количество, которое может легко измеряться с помощью процедур анализа и приемов, хорошо известных специалистам в данной области. Определяемые количества соединения изобретения, как правило, варьируют примерно от 0,001% по массе до 75% по массе относительно общей массы композиции. Инертные носители включают любой материал, который не вызывает деградации или не взаимодействует ковалентно иным способом с соединением изобретения. Примерами подходящих инертных носителей являются вода; водные буферные системы, такие как обычно используются в анализах высокоэффективной жидкостной хроматографии (ВЭЖХ); органические растворители, такие как ацетонитрил, этилацетат, гексан и аналогичные (которые являются подходящими для диагностики или анализов in vitro, но обычно не подходят для введения теплокровному животному); и фармацевтически приемлемые носители, такие как физиологический раствор соли.
Таким образом, настоящее изобретение предоставляет фармацевтическую или ветеринарную композицию (в дальнейшем называемую просто фармацевтической композицией), содержащую соединение циклогексиламина, описанное выше, в смеси с фармацевтически приемлемым носителем, наполнителем или разбавителем. Изобретение, кроме того, предоставляет фармацевтическую композицию, содержащую эффективное количество соединения циклогексиламина, описанного выше, в сочетании с фармацевтически приемлемым носителем.
Фармацевтические композиции данного изобретения могут быть в любой форме, которая позволяет вводить композицию пациенту. Например, композиция может быть в форме твердого вещества, жидкости или газа (аэрозоль). Типичные способы введения включают без ограничения пероральный, местный, парентеральный, подъязычный, ректальный, вагинальный и интраназальный. Термин “парентеральный”, используемый здесь, включает подкожные инъекции, внутривенные, внутримышечные, эпидуральные, интрастернальные (внутригрудинные) инъекции или инфузии. Фармацевтическая композиция изобретения изготавливается по такой рецептуре, которая позволяет содержащимся в ней активным ингредиентам быть биодоступными при введении композиции пациенту. Композиции, которые будут вводиться пациенту, имеют форму одной или более дозированных единиц или единичных доз, в которых, например, таблетка, капсула или крахмальная облатка может быть разовой дозой, а контейнер с соединением циклогексиламина в аэрозольной форме может вмещать множество единичных доз.
Материалы, применяемые для получения фармацевтических композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах. Композиции изобретения могут включать одно или более соединений (активных ингредиентов), известных для конкретного желательного воздействия. Например, эпинефрин может комбинироваться с соединением аминоциклогексилового эфира изобретения, давая композицию, полезную для индуцирования локальной анестезии. Специалистам в данной области очевидно, что оптимальная дозировка активного ингредиента(тов) в фармацевтической композиции будет зависеть от множества факторов. К существенным факторам без ограничения относятся тип субъекта (например, человек), конкретная форма активного ингредиента, способ введения и применяемая композиция.
В общем, фармацевтическая композиция включает соединение циклогексиламина, описанное здесь, в смеси с одним или более носителями. Носитель(ли) может быть в виде частиц, так что композиция имеет форму, например, таблеток или порошка. Носитель(ли) может быть жидким, а композиция представляет собой, например, сироп для перорального введения или инъецируемую жидкость. Кроме того, носитель(ли) может быть газообразным, так чтобы давать аэрозольную композицию, полезную, например, для введения путем ингаляции.
Когда композиция предназначена для перорального введения, она предпочтительно представлена либо в твердой, либо в жидкой форме, при этом полутвердая, полужидкая, суспензионная и гелевая формы включены в категорию форм, которые рассматриваются здесь как твердые или жидкие.
В виде твердой композиции для перорального введения композиция может быть приготовлена в форме порошка, гранул, прессованных таблеток, пилюль, капсул, крахмальных капсул, жевательных резинок, облаток, лепешек или подобной им форме. Такие твердые композиции обычно содержат один или более инертных разбавителей или съедобных носителей. Кроме того, может присутствовать один или более следующих адъювантов: связующие вещества, такие как сиропы, камедь акации, сорбит, поливинилпирролидон, карбоксиметилцеллюлоза, этилцеллюлоза, микрокристаллическая целлюлоза, камедь трагаканта или желатин и их смеси; наполнители (эксципиенты), такие как крахмал, лактоза или декстрины, дезинтегрирующие средства, такие как альгиновая кислота, альгинат натрия, Primogel, кукурузный крахмал и аналогичные; смазывающие вещества, такие как магния стеарат или Sterotex; наполнители, такие как лактоза, манниты, крахмал, фосфат кальция, сорбит, метилцеллюлоза и их смеси; смазывающие вещества, такие как магния стеарат, полимеры с высокой молекулярной массой, такие как полиэтиленгликоль, жирные кислоты с высокой молекулярной массой, такие как стеариновая кислота, кремнезем, увлажняющие средства, такие как лаурилсульфат натрия, вещества, способствующие скольжению (глотанию), такие как коллоидный диоксид кремния; вещества, придающие сладкий вкус, такие как сахароза или сахарин, корригенты запаха и вкуса, такие как мятное масло, метилсалицилат или апельсиновый корригент, и красящие агенты.
Когда композиция имеет форму капсулы, например желатиновой капсулы, она может содержать кроме материалов указанного выше типа, жидкий носитель, такой как полиэтиленгликоль или жирное масло.
Композиция может быть в жидкой форме, например в виде эликсира, сиропа, раствора, водной или масляной эмульсии или суспензии, или даже в виде сухих порошков, которые могут реконституироваться с помощью воды и/или другой жидкой среды перед применением. В качестве двух примеров можно привести пероральное введение жидкости или доставку жидкости путем инъекции. Когда композиция предназначена для перорального введения, предпочтительная композиция содержит в дополнение к данным соединениям один или более подсластителей, загустителей, консервантов (например, алкил пара-гидроксибензоат), красителей/подкрашивающих веществ и усилителей вкуса или запаха (корригентов). В композицию, предназначенную для введения путем инъекции, могут быть включены один или более поверхностно-активных веществ, консервантов (например, алкил пара-гидроксибензоат), увлажняющих средств, диспергирующих средств, суспендирующих агентов (например, сорбит, глюкоза или другие сахарные сиропы), буферных веществ, стабилизаторов и изотонических агентов. Эмульгирующий агент может выбираться из лецитина или моноолеата сорбита.
Жидкие фармацевтические композиции изобретения, являются ли они растворами, суспензиями или другой подобной формой, могут включать один или более следующих адъювантов: стерильные разбавители, такие как вода для инъекций, солевой раствор, предпочтительно физиологический раствор Рингеpa, изотонический хлорид натрия, жирные масла, такие как синтетические моно или диглицериды, которые могут служить растворяющей или суспендирующей средой, полиэтиленгликоли, глицерин, пропиленгликоль или другие растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферные вещества, такие как ацетаты, цитраты или фосфаты, и агенты для доведения осмотического давления, такие как хлорид натрия или декстроза. Парентеральные препараты могут быть заключены в ампулы, одноразовые шприцы или флаконы с многократными дозами из стекла или пластика. Предпочтительным адъювантом является физиологический солевой раствор. Инъецируемые фармацевтические композиции предпочтительно являются стерильными.
Жидкие композиции, предназначенные либо для парентерального, либо для перорального введения, должны содержать такое количество соединения изобретения, чтобы получалась подходящая дозировка. Обычно это количество в композиции составляет, по меньшей мере, 0,01% соединения изобретения. Когда предназначено для перорального введения, данное количество может варьировать между 0,1 и примерно 70% массы композиции. Предпочтительные композиции для перорального введения содержат примерно от 4% до 50% активного соединения циклогексиламина. В соответствии с данным изобретением предпочтительные композиции и препараты готовятся таким образом, чтобы парентеральная дозированная единица содержала между 0,01 и 10% по массе активного соединения.
Фармацевтическая композиция может быть предназначена для местного или топического введения, в таком случае приемлемой композицией является такая, в которой носитель может включать раствор, эмульсию, мазь, крем или гелевую основу. Например, основа может включать одно или более следующих веществ: вазелин, ланолин, полиэтиленгликоли, пчелиный воск, минеральное масло, разбавители, такие как вода и спирт, и эмульгаторы и стабилизаторы. В фармацевтической композиции для местного введения могут присутствовать загустители. Если композиция предназначена для трансдермального введения, она может включать трансдермальный пластырь или прибор для лекарственного электрофореза. Топические готовые формы могут содержать соединение изобретения в концентрации примерно от 0,1 до 25% вес./об. (вес на единицу объема).
Композиция может быть предназначена для ректального введения, например, в форме суппозитория, который будет расплавляться в прямой кишке и высвобождать лекарственное средство. Композиция для ректального введения может содержать в качестве подходящего не вызывающего раздражения наполнителя маслянистую основу. К таким основам без ограничения относятся ланолин, масло какао и полиэтиленгликоль. Для приготовления суппозитория предпочтительны низкоплавкие воски, при этом подходящими восками являются смеси глицеридов жирных кислот и/или масло какао. Воски могут расплавляться, и в них гомогенно диспергируется соединение циклогексиламина путем перемешивания. Расплавленную гомогенную смесь затем разливают в формы подходящего размера, дают ей остыть и тем самым затвердеть.
Композиция может включать различные материалы, которые видоизменяют физическую форму твердой или жидкой единичной дозы. Например, композиция может включать материалы, которые образуют покрывающую оболочку вокруг активных ингредиентов. Материалы, которые образуют покрывающую оболочку, обычно инертны и могут быть выбраны, например, из сахара, шеллака и других энтерических покрывающих агентов. Альтернативно активные ингредиенты могут быть заключены в желатиновую капсулу или крахмальную облатку.
Композиция в твердой или жидкой форме может включать агент, который связывает соединение циклогексиламина и таким образом способствует доставке активных компонентов. Подходящие агенты, которые могут обладать такой способностью, включают моноклональные и поликлональные антитела, белки или липосомы.
Фармацевтическая композиция данного изобретения может состоять из газообразных дозированных единиц, например она может быть в форме аэрозоля. Термин аэрозоль используется для обозначения множества систем в диапазоне от систем коллоидной природы до систем, состоящих из упаковок под давлением. Может осуществляться доставка сжиженного или сжатого газа или доставка с помощью подходящей насосной системы, которая распределяет активные ингредиенты. Аэрозоли соединений изобретения могут поставляться в однофазных, двухфазных или трехфазных системах для доставки активного ингредиента(тов). Система доставки аэрозоля включает необходимый контейнер, активаторы, клапаны, субконтейнеры и т.п., которые вместе могут составлять набор. Предпочтительные аэрозоли могут быть определены специалистами в данной области без чрезмерного экспериментирования.
Будь то твердая, жидкая или газообразная форма, фармацевтическая композиция данного изобретения может содержать одно или более известных фармакологических агентов, используемых либо в способах модулирования активности ионных каналов у теплокровного животного, либо в способах модулирования активности ионных каналов in vitro, или применяемых для лечения аритмии, заболеваний центральной нервной системы, судорожного состояния, эпилептических спазмов, депрессии, страха, шизофрении, болезни Паркинсона, респираторных нарушений, муковисцидоза, астмы, кашля, воспаления, артрита, аллергических состояний, желудочно-кишечных расстройств, недержания мочи, слизистого колита, сердечно-сосудистых заболеваний, церебральной ишемии или ишемии миокарда, гипертензии, синдрома удлиненного QT, инсульта, мигрени, глазных болезней, сахарного диабета, миопатий, миотонии Беккера, миастении беременных, врожденной парамиотонии, злокачественной гипертермии, гиперкалиемического периодического паралича, миотонии Томсена, аутоиммунных заболеваний, отторжения трансплантата при трансплантации органа или при трансплантации костного мозга, сердечной недостаточности, гипотензии, болезни Альцгеймера и других психических расстройств, и алопеции. С соединениями данного изобретения могут комбинироваться другие средства, известные как средства, вызывающие усиление либидо, средства локальной анальгезии или анестезии.
Фармацевтические композиции могут получаться по методике, хорошо известной в фармацевтической области. Аминоциклогексильные соединения изобретения могут быть в форме сольватов в фармацевтически приемлемом растворителе, таком как вода или физиологический раствор соли. Альтернативно соединения могут быть в форме свободного основания или в форме фармацевтически приемлемой соли, такой как гидрохлорид, сульфат, фосфат, цитрат, фумарат, метансульфонат, ацетат, тартрат, малеат, лактат, манделат, салицилат, сукцинат и другие соли, известные в этой области. Соответствующая соль выбирается для усиления биодоступности или стабильности соединения при соответствующем способе применения (например, пероральном или парентеральном пути введения).
Композиции, предназначенные для введения инъекцией, могут получаться с помощью комбинирования соединения циклогексиламина с водой и предпочтительно с буферными агентами, так чтобы получить раствор. Вода предпочтительно представляет стерильную свободную от пирогенов воду. Могут добавляться поверхностно-активные вещества для облегчения образования гомогенного раствора или суспензии. Поверхностно-активные вещества представляют собой соединения, которые нековалентно взаимодействуют с соединением циклогексиламина, тем самым облегчая растворение или гомогенное суспендирование соединения циклогексиламина в водной системе доставки. Желательно, чтобы поверхностно-активные вещества присутствовали в водных композициях изобретения, поскольку соединения циклогексиламина данного изобретения обычно являются гидрофобными. Другие носители для инъекции включают без ограничения стерильный свободный от перекиси этилолеат, дегидратированные спирты, пропиленгликоль, а также их смеси.
Подходящие фармацевтические адъюванты для инъекционных растворов включают стабилизирующие агенты, солюбилизирующие агенты, буферные вещества и регуляторы вязкости. Примеры таких адъювантов включают этанол, этилендиаминтетрауксусную кислоту (EDTA), тартратные буферы, цитратные буферы и высокомолекулярные полиоксиэтиленовые регуляторы вязкости. Эти фармацевтические композиции могут инъецироваться внутримышечно, эпидурально, внутрибрюшинно или внутривенно.
Фармакологическое тестирование
Как отмечалось выше, данное изобретение предоставляет соединения, описанные выше, для применения в in vitro и in vivo способах. В одном варианте блокируются in vitro и in vivo ионные каналы, такие как натриевые каналы сердца.
Ионные каналы представляют собой вездесущие мембранные белки в клетках теплокровных животных, таких как млекопитающие. Их существенная физиологическая роль заключается в контроле трансмембранного электрического потенциала, опосредовании ионного и жидкостного баланса, облегчении нервно-мышечной и нейронной передачи, быстрой трансмембранной сигнальной трансдукции и регуляции секреции и сократительной способности.
Следовательно, соединения, которые способны модулировать активность или функционирование соответствующих ионных каналов, будут полезны для лечения или профилактики многих заболеваний или нарушений, вызываемых дефектным или неадекватным функционированием ионных каналов. Обнаружено, что соединения изобретения обладают существенной активностью в модулировании активности ионных каналов как in vivo, так и in vitro.
Таким образом, данное изобретение предоставляет способы лечения заболеваний или состояний теплокровных животных, страдающих или являющихся носителем этого заболевания или состояния, и/или предотвращения возникновения заболевания или состояния у теплокровного животного, при которых терапевтически эффективное количество соединения формулы (I), или композиции, содержащей соединение формулы (I), вводится теплокровному животному, которое в этом нуждается. Соединения, композиции и способы данного изобретения могут использоваться при следующих заболеваниях и состояниях: аритмия, заболевания центральной нервной системы, судороги, эпилептические спазмы, депрессия, страх, шизофрения, болезнь Паркинсона, респираторные нарушения, муковисцидоз, астма, кашель, воспаление, артрит, аллергии, желудочно-кишечные расстройства, недержание мочи, слизистый колит, сердечно-сосудистые заболевания, церебральная ишемия или ишемия миокарда, гипертензия, синдром удлиненного QT, инсульт, мигрень, глазные болезни, сахарный диабет, миопатии, миотония Беккера, миастения беременных, врожденная парамиотония, злокачественная гипертермия, гиперкалиемический периодический паралич, миотония Томсена, аутоиммунные заболевания, отторжение трансплантата при трансплантации органа или при трансплантации костного мозга, сердечная недостаточность, гипотензия, болезнь Альцгеймера, или другие психические расстройства, и алопеция.
Кроме того, данное изобретение предоставляет способ осуществления локальной анальгезии или анестезии у теплокровного животного, который включает введение теплокровному животному, которое в этом нуждается, эффективного количества соединения формулы (1) или фармацевтической композиции, содержащей соединение формулы (I). Эти способы могут применяться с целью облегчения или предотвращения болевого ощущения у теплокровного животного.
Кроме того, данное изобретение предоставляет способ, при котором обеспечивается контакт препарата, содержащего ионные каналы, с эффективным количеством соединения аминоциклогексилового эфира изобретения, или эффективное количество этого соединения вводится теплокровному животному (например, млекопитающему, такому как человек). Подходящие препараты, содержащие сердечные натриевые каналы, включают клетки, выделенные из ткани сердца, а также культивируемые клеточные линии. Стадия, на которой осуществляется контакт, включает, например, инкубирование ионных каналов с соединением при условиях и в течение периода времени, достаточных для того, чтобы обеспечить модулирование активности каналов этим соединением.
В другом варианте, описанные выше соединения предоставляются для лечения аритмии. Используемый здесь термин “лечение аритмии” относится как к терапии аритмии, так и к профилактике аритмий, возникающих в случае предрасположенности сердца к аритмии. Для лечения аритмии у теплокровного животного, такого как человек, применяется эффективное количество композиции данного изобретения. Способы введения эффективных количеств антиаритмических средств хорошо известны в данной области и включают введение пероральных или парентеральных дозированных форм. Такие дозированные формы включают формы для парентерального введения, но не ограничиваются ими. Такие формы включают, но не ограничиваются ими, растворы для парентерального введения, таблетки, капсулы, имплантаты замедленного высвобождения и системы трансдермальной доставки. В общем, предпочтительным является пероральное и внутривенное введение. Дозируемое количество и частота введения выбраны так, чтобы создать эффективный уровень средства без вредного воздействия. В общем, при пероральном или внутривенном введении с целью антиаритмического воздействия доза будет составлять в пределах примерно от 0,1 до 100 мг/кг/сутки и обычно примерно от 0,1 до 10 мг/кг.
Введение композиций данного изобретения может сочетаться с введением других средств. Например, желательным может быть введение антагониста опиоида, такого как налоксон, если соединение проявляет опиоидную активность там, где такая активность может быть нежелательной. Налоксон может противодействовать опиоидной активности введенного соединения, не мешая его антиаритмическому действию. В качестве другого примера соединение аминоциклогексилового эфира изобретения может вводиться совместно с эпинефрином для того, чтобы вызвать локальную анестезию.
Для того чтобы оценить, обладает ли соединение желательной для данного изобретения фармакологической активностью, его подвергают серии тестов. Определение теста, который следует выполнить, зависит от интересующей физиологической ответной реакции. В опубликованной литературе имеются многочисленные протоколы тестирования эффективности потенциальных терапевтических средств, и эти протоколы могут быть применимы к данным соединениям и композициям.
Например, в связи с лечением или предотвращением аритмии может проводится серия из четырех тестов. В первом из этих тестов соединение данного изобретения каждые 8 минут дается в виде возрастающих (удваиваемых с каждой дозой) внутривенных болюсов анестезированным пентобарбиталом крысам. Через 30 секунд, 1, 2, 4 и 8 минут после каждой дозы измеряют влияние соединения на кровяное давление, частоту сердечных сокращений и ЭКГ. Увеличивающиеся дозы дают до тех пор, пока животные не погибнут. Идентифицируются смертельные случаи либо респираторного, либо сердечного происхождения. Этот тест показывает, модулирует ли соединение активность натриевых каналов и/или калиевых каналов, и, кроме того, дает информацию об острой токсичности. Показателями блокирования натриевых каналов являются увеличенный интервал P-R и уширение комплекса QRS на ЭКГ. Результатом блокирования калиевых каналов является удлинение интервала Q-T на ЭКГ.
Второй тест включает введение соединения в виде инфузии анестезированным пентобарбиталом крысам, левый желудочек которых подвергается электростимуляции прямоугольными импульсами, выполняемой в соответствии с заранее установленным протоколом, описанным ниже более подробно. Этот протокол включает определение порогов индукции экстрасистол и фибрилляции желудочков. Кроме того, с помощью методики введения одиночных экстрасистол оценивается влияние на электрическую рефрактерность. Кроме того, регистрируется влияние на кровяное давление, частоту сердечных сокращений и ЭКГ. В этом тесте блокаторы натриевых каналов дают изменения ЭКГ, ожидаемые на основании первого теста. Кроме того, блокаторы натриевых каналов также повышают пороги индукции экстрасистол и фибрилляции желудочков. Блокирование калиевых каналов выявляется по увеличению рефрактерности и уширению интервалов Q-T на ЭКГ.
Третий тест включает подвержение изолированных сердец крыс воздействию соединения в возрастающих концентрациях. В изолированном сердце в присутствии варьируемых концентраций соединения регистрируются внутрижелудочковое давление, частота сердечных сокращений, скорость проводимости и ЭКГ. Тест предоставляет свидетельство прямого токсического влияния на миокард. Кроме того, в условиях стимулирования ишемии может быть установлена селективность, активность и эффективность действия соединения. Предполагается, что обнаруженные эффективные в данном тесте концентрации будут эффективными в электрофизиологических исследованиях.
Четвертый тест представляет собой оценку антиаритмической активности соединения, направленной против аритмий, индуцированных путем окклюзии коронарной артерии у анестезированных крыс. Ожидается, что хорошее антиаритмическое соединение при нормальных условиях будет обладать антиаритмической активностью в случае использования доз, которые оказывают минимальное влияние либо на ЭКГ, кровяное давление, либо на частоту сердечных сокращений.
Все вышеупомянутые тесты выполняются с использованием ткани крыс. Чтобы удостовериться, что соединение обладает не только специфичным для ткани крыс действием, дальнейшие эксперименты выполняются на собаках и приматах. Чтобы оценить возможное блокирующее действие на натриевые каналы и калиевые каналы in vivo у собак, соединение тестируют, определяя его влияние на ЭКГ, скорость проводимости эпикарда желудочка и ответы на электростимуляцию. Анестезированной собаке вскрывают грудную клетку, чтобы обнажить эпикард левого желудочка. После удаления перикарда сердца в эпикардиальный слой левого желудочка вшивают регистрирующий/стимулирующий электрод. Используя такой порядок и подходящие протоколы стимулирования, можно оценить скорость проводимости эпикарда, а также чувствительность к электростимуляции. Эта информация в сочетании с измерениями ЭКГ позволяет определить, происходит ли блокирование натриевых и/или калиевых каналов. Как и в первом тесте на крысах, соединение дается в виде серии возрастающих болюсных доз. Вместе с тем определяется возможное токсичное действие соединения на сердечно-сосудистую систему собак.
Влияние соединения на ЭКГ и ответы на электростимуляцию оценивали также на интактных анестезированных галотаном бабуинах (Papio anubis). В ходе подготовки в этом случае канюлю для определения кровяного давления и ЭКГ-электроды соответствующим образом вводили анестезированному бабуину. Кроме того, в правый желудочек помещали стимулирующий электрод вместе с электродом для регистрации монофазного потенциала действия. Также как в тесте, описанном выше, реакция на соединение, проявляемая на ЭКГ и при электростимуляции, свидетельствует о возможном блокировании натриевых и/или калиевых каналов. Монофазный потенциал действия также показывает, приводит ли соединение к расширению потенциала действия, эффект, который ожидается от агента, блокирующего калиевые каналы.
В качестве другого примера в связи с ослаблением или предотвращением чувства боли может быть проведено следующее тестирование. Чтобы определить влияние соединения данного изобретения на реакцию животных на острое болевое ощущение оценивали действие слабого укола шприцем на 7,5 г, оснащенным иглой 23G, который наносился на бритое место на спине морской свинки (Cavia porcellus) после подкожного введения солевого раствора (50 мкл, 10 мг/мл), достаточного для вздутия видимого волдыря на коже. Каждый тест проводили в центральной области волдыря, а также на его периферии, чтобы проконтролировать диффузию тестируемого раствора из точки введения. Если животное вздрагивало в ответ на стимул, это свидетельствовало об отсутствии блокирования болевого ощущения. Тестирование проводили с интервалами времени до 4 часов после введения. Участки образования пузырей проверяли через 24 часа и не наблюдали аномалий кожи в результате локального введения тестируемых веществ или солевого раствора, носителя, используемого для приготовления тестируемых растворов.
Другие композиции
Данное изобретение также предоставляет наборы, содержащие фармацевтическую композицию, которая включает одно или более соединений указанной выше формулы. Набор включает также инструкции по применению фармацевтической композиции для модулирования активности ионных каналов, для лечения аритмии или осуществления локальной анальгезии и/или анестезии, и по другим применениям, которые здесь заявлены. Предпочтительно коммерческая упаковка содержит одну или более единичных доз фармацевтической композиции. Например, такая единичная доза может быть количеством, достаточным для внутривенной инъекции. Специалистам в данной области очевидно ясно, что соединения, которые чувствительны к свету и/или воздуху, требуют специальной упаковки и/или технологии приготовления. Например, может использоваться упаковка, не пропускающая свет, и/или герметичная, исключающая контакт с окружающим воздухом, и/или в рецептуре приготовления применяются приемлемые покрытия или наполнители.
Следующие примеры предлагаются с целью иллюстрации, но не с целью ограничения. В примерах, если не оговорено особо, исходные материалы закупались у хорошо известных коммерческих фирм поставщиков, например Aldrich Chemical Company (Milwaukee, WI), и были стандартного качества и чистоты. Термины “эфир” и “этиловый эфир” относятся к диэтиловому эфиру; “час” относится к часам; “мин” относится к минутам; “ГХ” относится к газовой хроматографии; “об./об.” относится к объему на объем; и соотношения являются соотношениями по массе, если не оговорено особо.
Примеры
Пример 1
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(2-нафтенэтокси)]циклогексана
(Соединение №1)
(i) Морфолин (5 мл, 57 ммоль), оксид циклогексена (5,8 мл, 57 ммоль) и воду (3 мл) нагревали с обратным холодильником в течение 1,5 час. ГХ анализ показал, что реакция была завершена. Охлажденную смесь распределяли между насыщенным раствором NaOH (50 мл) и эфиром (75 мл). Водный слой повторно промывали эфиром (30 мл) и объединенные эфирные слои сушили над сульфатом натрия. Эфир удаляли в вакууме, оставляя желтое масло (9,83 г). Неочищенный продукт (± )-транс-[2-(4-морфолинил)]циклогексанол очищали с помощью вакуумной перегонки (т.кип. 75-80° С в условиях полного вакуума), получая прозрачную жидкость (8,7 г). Выход 82,5%.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (6,0 г, 32,4 ммоль) и триэтиламина (6,8 мл, 48 ммоль) в дихлорметане (100 мл) через канюлю добавляли раствор метансульфонилхлорида (3,10 мл, 40 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь перемешивали в течение часа при 0° С и затем при комнатной температуре в течение 4 часов. Дихлорметановую смесь промывали водой (2× 50 мл) и объединенные водные промывки экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 8,5 г (100% выход) неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной дисперсии, предварительно промытой гексанами (3× 20 мл), (1,24 г, 51,6 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 2-нафтенэтанола (6,8 г, 40 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, и по мере того как реакционную смесь перемешивали при комнатной температуре, она превращалась в гель. Мезилат, полученный выше в пункте (ii), растворяли в диметилформамиде (50 мл) и полученный раствор быстро добавляли через канюлю к суспензии алкоголята. Реакционную смесь нагревали до 80° С и затем температуру понижали до 40° С. Полученный в результате желтый раствор выливали в ледяную воду (1500 мл) и экстрагировали этилацетатом (3× 300 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (500 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 13,4 г янтарного масла, которое растворяли в воде (150 мл) и рН раствора доводили до рН 2 водным 1М НСl. Кислый водный раствор экстрагировали этиловым эфиром (2× 100 мл) и затем подщелачивали до рН 10 50% водным раствором гидроокиси натрия. Основный водный раствор экстрагировали этиловым эфиром (2× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 7,16 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - хлороформа (1:1 об./об.), и получали 4,37 г очищенного свободного основания. Продукт растворяли в этиловом эфире (80 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (80 мл). Из раствора выступало масло, растворитель выпаривали в вакууме и остаток растворяли в минимальном количестве теплого этилового спирта, при добавлении большого количества этилового эфира начиналась кристаллизация. Кристаллы собирали, что давало 3,83 г (31% выход) указанного в заголовке соединения, т.пл. 158-160° С, элементный состав которого указан в таблице 1.
Пример 2
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(1-нафтенэтокси)]циклогексана
(Соединение №2)
(i) Исходный транс-аминоциклогексанол получают согласно примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (6,0 г, 32 ммоль) и триэтиламина (6,8 мл, 48 ммоль) в дихлорметане (100 мл) через канюлю добавляли раствор метансульфонилхлорида (3,10 мл, 40 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 4 часов. Дихлорметановую смесь промывали водой (2× 50 мл) и объединенные водные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 9,0 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 20 мл), (1,30 г, 51,6 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 1-нафтенэтанола (6,8 г, 40 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Мезилат, полученный выше в пункте (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к суспензии алкоголята. Реакционную смесь нагревали до 80° С в течение 3 часов, затем температуру понижали до 35° С в течение ночи при перемешивании. Реакционную смесь выливали в ледяную воду (1500 мл) и экстрагировали этилацетатом (3× 300 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (500 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 12,0 г масла, которое растворяли в эфире (80 мл) и обрабатывали насыщенным раствором НСl в эфире. Из раствора выступало масло, растворитель выпаривали в вакууме и полученную в результате неочищенную гидрохлоридную соль растворяли в воде (200 мл). Кислый водный раствор экстрагировали этиловым эфиром (2× 100 мл) и затем подщелачивали до рН 10 50% водным раствором гидроокиси натрия. Основный водный раствор экстрагировали этиловым эфиром (2× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 7,20 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - дихлорметана (1:1 об./об.), и получали очищенное свободное основание. Продукт растворяли в этиловом эфире (80 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (80 мл). Осаждали белый продукт и твердый осадок собирали и растворяли в минимальном количестве теплого этилового спирта, при добавлении большого количества этилового эфира начиналась кристаллизация. Кристаллы собирали, что давало 2,30 г указанного в заголовке соединения, т.пл. 198-200° С, имеющего элементный состав, указанный в таблице 1.
Пример 3
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(4-бромфенэтокси)]циклогексана
(Соединение №3)
(i) Исходный транс-аминоциклогексанол получают согласно примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (25 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (25 мл). Добавление завершали за 5 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли дихлорметаном (50 мл) и промывали водой (2× 50 мл), объединенные водные промывки экстрагировали дихлорметаном (25 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 4,7 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,62 г, 25,8 ммоль) в сухом диметилформамиде (25 мл) через канюлю добавляли раствор 4-бромфенэтилового спирта (4,0 г, 20 ммоль) в диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к суспензии алкоголята. Реакционную смесь нагревали до 80° С в течение 2 часов, затем температуру понижали до 35° С и реакционную смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (150 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 7,4 г масла, которое растворяли в эфире (80 мл) и обрабатывали насыщенным раствором НСl в эфире. Масло выступало из раствора, растворитель выпаривали в вакууме и остаток растворяли в воде (100 мл). Кислый водный раствор экстрагировали этиловым эфиром (2× 50 мл) и затем подщелачивали до рН 10 50% водным раствором гидроокиси натрия. Основный водный раствор экстрагировали этиловым эфиром (2× 50 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 3,67 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - дихлорметана (1:1 об./об.), и получали очищенное свободное основание. Продукт растворяли в этиловом эфире (30 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (30 мл). Растворитель выпаривали, и остаток растворяли в минимальном количестве этилового спирта, при добавлении большого количества этилового эфира начиналась кристаллизация. Кристаллы собирали, что давало 1,31 г указанного в заголовке соединения, т.пл. 148-151° С, имеющего элементный состав, который указан в таблице 1.
Пример 4
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-[2-(2-нафтокси)этокси)]циклогексана
(Соединение №4)
(i) Исходный транс-аминоциклогексанол получают согласно примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 4 часов. Дихлорметановую смесь промывали водой (2× 50 мл) и объединенные водные промывки экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 4,3 г (100% выход) неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,7 г, 29 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 2-(2-нафтокси)этанола (3,76 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к реакционной смеси. Полученную в результате реакционную смесь нагревали до 90° С в течение ночи и затем охлаждали при комнатной температуре. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 7,8 г желтого масла, которое растворяли в эфире (100 мл) и обрабатывали насыщенным раствором НСl в эфире (100 мл). Полученный в результате осадок собирали, частично растворяли в воде (200 мл) и гетерогенный водный раствор экстрагировали эфиром (2× 100 мл). Оставшийся нерастворимый материал собирали и перекристаллизовывали в кипящем этаноле (75 мл), получая первую партию нужного продукта. Кислый водный раствор подщелачивали до рН 10 50% водным раствором NaOH и экстрагировали эфиром (2× 50 мл). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме, получая 1,6 г неочищенного свободного аминоэфира. Продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - дихлорметана, и получали 0,73 г бледно-желтого масла. Затем очищенное свободное основание растворяли в эфире (50 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в эфире (50 мл).
Белый осадок собирали и перекристаллизовывали в кипящем этаноле (40 мл), получая вторую партию. В результате объединения двух партий получали 1,03 г указанного в заголовке соединения, т.пл. 235-237° С, имеющего элементный состав, который указан в таблице 1.
Пример 5
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-[2-(4-бромфенокси)этокси)]]циклогексана
(Соединение №5)
(i) Исходный транс-аминоциклогексанол получают согласно примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 4 часов. Дихлорметановую смесь промывали водой (2× 50 мл) и объединенные водные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 3,95 г (92% выход) неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,63 г, 26 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 2-(4-бромфенокси)этанола (4,34 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 90° С в течение 90 мин и затем температуру понижали до 40° С, и реакционную смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 8,35 г желтого масла, которое растворяли в эфире (100 мл) и обрабатывали насыщенным раствором НСl в эфире (100 мл). Полученное в результате белое твердое вещество собирали и перекристаллизовывали в кипящем этаноле (150 мл), получая 3,7 г (54% выход) чистого указанного в заголовке соединения, т.пл. 228-230° С, имеющего элементный состав, указанный в таблице 1.
Пример 6
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(3,4-диметоксифенэтокси)]циклогексана
(Соединение №6)
(i) Исходный транс-аминоциклогексанол получают согласно Примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 4 часов. Дихлорметановую смесь промывали водой (2× 50 мл) и объединенные водные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 4,18 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,64 г, 27 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 3,4-диметоксифенэтилового спирта (3,64 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 80° С в течение 90 мин и затем температуру понижали до 40° С и перемешивание продолжали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 7,18 г неочищенного продукта, который растворяли в эфире (100 мл) и обрабатывали насыщенным раствором НСl в эфире (100 мл). Растворитель выпаривали в вакууме, и остаток масла смывали водой (100 мл) и экстрагировали эфиром (2× 50 мл). Водный слой подщелачивали до рН 10 50% водным раствором NaOH и экстрагировали эфиром (2× 50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата и дихлорметана (1:1 об./об.), и получали 2,8 г бледно-желтого масла. Свободное основание растворяли в эфире (80 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в эфире (80 мл). Клейкий осадок собирали, растворяли в минимальном количестве этанола и добавляли большой избыток эфира, инициируя кристаллизацию 2,24 г (36% выход) указанного в заголовке соединения, т.пл. 148-150° С, имеющего элементный состав, указанный в таблице 1.
Пример 7
Моногидрохлорид (± )-транс-[2-(1-пирролидинил)-1-(1-нафтенэтокси)]циклогексана
(Соединение №7)
(i) Пирролидин (25 мл, 300 ммоль), оксид циклогексена (30 мл, 297 ммоль) и воду (10 мл) нагревали с обратным холодильником в течение 3 час. ГХ анализ показал, что реакция была завершена. Охлажденную смесь распределяли между насыщенным раствором NaOH (10 мл) и эфиром (150 мл). Водный слой промывали эфиром (2× 100 мл) и объединенные эфирные слои сушили над сульфатом натрия. Эфир удаляли в вакууме, получая в остатке желтое масло. Сырой продукт очищали с помощью вакуумной перегонки (т.кип. 66-69° С в условиях полного вакуума), получая прозрачную жидкость (43,9 г). Выход 87%.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(пирролидинил)]циклогексанола (2,74 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь промывали водой (2× 50 мл) и объединенные водные промывки экстрагировали повторно дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 3,24 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,64 г, 27 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 1-нафтенэтанола (3,64 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 80° С в течение 90 мин и затем температуру понижали до 40° С и смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 9,00 г неочищенного продукта, который растворяли в эфире (50 мл) и обрабатывали насыщенным раствором НСl в эфире (50 мл). Растворитель выпаривали в вакууме, и остаток масла смывали водой (100 мл) и экстрагировали эфиром (2× 50 мл). Водный слой подщелачивали до рН 10 50% водным раствором NaOH и экстрагировали эфиром (2× 50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилметанола и хлороформа (2:8 об./об.). Свободный аминоэфир частично растворяли в эфире (80 мл), нерастворимые материалы отфильтровывали, и затем к фильтрату добавляли насыщенный раствор НСl в эфире (80 мл). Растворитель выпаривали в вакууме, остаток растворяли в ацетоне, и добавление аликвот эфира индуцировало медленную кристаллизацию. Собирали 2 партии указанного в заголовке соединения (0,88 г), т.пл. 103-105° С, имеющего элементный состав, указанный в таблице 1.
Пример 8
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(2-(бензо-[b]тиофен-3-ил)этокси)]циклогексана
(Соединение №8)
(i) Исходный транс-аминоциклогексанол получают согласно Примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 5 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 3 часов. Реакционную смесь промывали водой (3× 30 мл) и объединенные водные промывки экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 5,25 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,60 г, 25 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 2-(бензо[b]тиофен-3-ил)этанола (3,56 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (2 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 75° С в течение 2 часов, затем температуру понижали до 65° С и перемешивание продолжали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 7,7 г масла, которое растворяли в эфире (100 мл) и обрабатывали насыщенным раствором НСl в эфире (100 мл). Масло осаждали из раствора, растворитель выпаривали в вакууме, и полученную в результате неочищенную гидрохлоридную соль растворяли в воде (200 мл). Кислый водный раствор экстрагировали этиловым эфиром (2× 100 мл) и затем подщелачивали до рН 10 50% водным раствором гидроокиси натрия. Основный водный раствор экстрагировали этиловым эфиром (3× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 3,30 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата и дихлорметана (1:1 об./об.), чтобы получить свободное основание. Продукт растворяли в этиловом эфире (100 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (100 мл). Растворитель выпаривали в вакууме и остаток растворяли в минимальном количестве кипящего метанола, получая при охлаждении первую партию (0,7 г) кристаллического продукта. При добавлении диэтилового эфира к метанольному фильтрату получали вторую партию (0,55 г). Обе партии объединяли, получая при этом 1,25 г указанного в заголовке соединения, т.пл. 158-160° С, имеющего элементный состав, указанный в таблице 1.
Пример 9
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(2-(бензо-[b]тиофен-4-ил)этокси)]циклогексана
(Соединение №9)
(i) Исходный транс-аминоциклогексанол получают согласно Примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 5 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 3 часов. Реакционную смесь промывали водой (2× 30 мл) и объединенные водные промывки экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 4,24 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,60 г, 25 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 2-(бензо[b]тиофен-4-ил)этанола (3,56 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (2 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 85° С в течение 2 часов, затем температуру понижали до 40° С и реакционную смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 8,2 г масла, которое растворяли в эфире (100 мл) и обрабатывали насыщенным раствором НСl в эфире (100 мл). Масло осаждали, и растворитель выпаривали в вакууме, и полученную в результате неочищенную гидрохлоридную соль растворяли в воде (200 мл). Кислый водный раствор экстрагировали этиловым эфиром (2× 100 мл) и затем подщелачивали до рН 10 водным раствором гидроокиси натрия (50% вес/объем). Основный водный раствор экстрагировали этиловым эфиром (3× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 3,0 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - дихлорметана (1:1 об./об.), и получали чистое свободное основание. Продукт растворяли в этиловом эфире (50 мл) и переводили в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (50 мл). Растворитель выпаривали в вакууме, и остаток растворяли в минимальном количестве холодного этанола, и добавлением эфира инициировали образование кристаллов (1,17 г), т.пл. 178-180° С, имеющих элементный состав, указанный в таблице 1.
Пример 10
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(3-бромфенэтокси)]циклогексана
(Соединение №10)
(i) Исходный транс-аминоциклогексанол получают согласно Примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 5 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 3 часов. Реакционную смесь промывали водой (2× 30 мл) и объединенные водные промывки экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 5,4 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,60 г, 25 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 3-бромфенэтилового спирта (4,0 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (2 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 85° С в течение 2 часов, затем температуру понижали до 45° С, и реакционную смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 8,0 г масла, которое растворяли в эфире (100 мл) и обрабатывали насыщенным раствором НСl в эфире (100 мл). Масло осаждали, растворитель выпаривали в вакууме, и полученную в результате неочищенную гидрохлоридную соль растворяли в воде (200 мл). Кислый водный раствор экстрагировали этиловым эфиром (2× 100 мл) и затем подщелачивали до рН 10 водным раствором гидроокиси натрия (50% вес/объем). Основный водный раствор экстрагировали этиловым эфиром (3× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 2,9 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - дихлорметана (1:1 об./об.), и получали чистое свободное основание. Продукт растворяли в этиловом эфире (50 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (50 мл). Растворитель выпаривали в вакууме, и остаток растворяли в минимальном количестве холодного этанола, и добавлением эфира инициировали образование кристаллов (0,53 г), т.пл. 145-148° С, имеющих элементный состав, указанный в таблице 1.
Пример 11
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(2-бромфенэтокси)циклогексана
(Соединение №11)
(i) Исходный транс-аминоциклогексанол получают согласно Примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 5 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 3 часов. Реакционную смесь промывали водой (2× 30 мл) и объединенные водные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 5,9 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,60 г, 25 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 2-бромфенэтилового спирта (4,0 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (2 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 85° С в течение 2 часов, затем температуру понижали до 45° С, и реакционную смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 8,4 г масла, которое растворяли в 1,0 М водном растворе НСl (50 мл), объем доводили до 200 мл водой и рН доводили до рН 2 1,0 М водным раствором НСl. Кислый водный раствор экстрагировали этиловым эфиром (3× 100 мл) и затем подщелачивали до рН 10 50% водным раствором гидроокиси натрия. Основный водный раствор экстрагировали этиловым эфиром (3× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 2,8 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата - дихлорметана (1:1 об./об.), и получали чистое свободное основание. Продукт растворяли в этиловом эфире (50 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в этиловом эфире (50 мл). Растворитель выпаривали в вакууме, и остаток растворяли в минимальном количестве холодного этанола, и добавлением эфира инициировали образование кристаллов, которые собирали двумя порциями (0,74 г), т.пл. 140-142° С, имеющих элементный состав, указанный в таблице 1.
Пример 12
Моногидрохлорид (± )-транс-[2-(4-морфолинил)-1-(3-(3,4-диметоксифенил)-1-пропокси)]циклогексана
(Соединение №12)
(i) Исходный транс-аминоциклогексанол получают согласно Примеру 1.
(ii) К охлажденному (0° С) раствору (± )-транс-[2-(4-морфолинил)]циклогексанола (3,0 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Добавление завершали за 10 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 4 часов. Дихлорметановую смесь промывали водой (2× 50 мл) и объединенные водные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая неочищенный мезилат.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,6 г, 27 ммоль) в сухом диметилформамиде (50 мл) через канюлю добавляли раствор 3-(3,4-диметоксифенил)-1-пропанола (3,93 г, 20,0 ммоль) в сухом диметилформамиде (50 мл). Добавление сопровождалось выделением газа, реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 90° С в течение 90 мин и затем температуру понижали до 45° С и перемешивание продолжали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 8,5 г неочищенного продукта, который растворяли в 15% водном растворе НСl (200 мл) и экстрагировали эфиром (2× 100 мл). Водный слой подщелачивали до рН 10 50% водным раствором NaOH и экстрагировали эфиром (2× 100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле 60 (70-230 меш), используя в качестве элюента смесь этилацетата и дихлорметана (1:1 об./об.), и получали свободное основание, которое растворяли в эфире (80 мл) и превращали в моногидрохлоридную соль добавлением насыщенного раствора НСl в эфире (80 мл). Клейкий осадок собирали, растворяли в минимальном количестве теплого этанола и добавляли большой избыток эфира для инициирования кристаллизации указанного в заголовке соединения, т.пл. 175-177° С, имеющего элементный состав, указанный в таблице 1.
Пример 13
Моногидрохлорид (± )-транс-[2-[бис(2-метоксиэтил)амино]-1-(2-нафтенэтокси)]циклогексана
(Соединение №13)
(i) Бис-(2-метоксиэтил) амин (25 мл, 169 ммоль) и оксид циклогексена (17,2 мл, 170 ммоль) смешивали в воде (5 мл) и полученную смесь нагревали с обратным холодильником в течение 30 часов. Охлажденную реакционную смесь распределяли между 10% водным раствором NaOH (200 мл) и диэтиловым эфиром (200 мл). Водный слой дважды экстрагировали дополнительным количеством диэтилового эфира (2× 100 мл), объединенные органические слои промывали водой (8 мл) и сушили над сульфатом натрия. Растворитель выпаривали в вакууме, получая в остатке неочищенный продукт, который подвергали вакуумной перегонке, получая 26,4 г очищенного бесцветного масла.
(ii) К охлажденному (0° С) раствору (± )-транс-2-[бис(2-метоксиэтил)амино] циклогексанола (4,63 г, 20,00 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) через канюлю добавляли раствор метансульфонилхлорида (1,55 мл, 20,00 ммоль) в дихлорметане (50 мл). Добавление завершали за 5 мин, реакционную смесь перемешивали в течение следующего часа при 0° С и затем при комнатной температуре в течение 4 часов. Реакционную смесь промывали водой (2× 30 мл) и объединенные водные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая 4,87 г неочищенного мезилата.
(iii) К гидриду натрия, 80% масляной суспензии, предварительно промытой гексанами (3× 10 мл), (0,6 г, 25,00 ммоль) в безводном диметилформамиде (50 мл) через канюлю добавляли раствор 2-нафтенэтанола (3,4 г, 20,00 ммоль) в безводном диметилформамиде (50 мл). Добавление сопровождалось выделением пузырьков водорода, реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Мезилат, полученный выше на стадии (ii), растворяли в сухом диметилформамиде (50 мл) и полученный в результате раствор быстро добавляли (3 мин) через канюлю к реакционной смеси. Реакционную смесь нагревали до 90° С в течение 2 часов, затем температуру понижали до 40° С и реакционную смесь перемешивали в течение ночи. Реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлорида натрия (300 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 8,1 г масла, которое растворяли в 1 М водном растворе НСl (50 мл) и объем доводили до 200 мл водой. Кислый водный раствор экстрагировали диэтиловым эфиром (2× 100 мл) и затем подщелачивали до рН 10 50% водным раствором гидроокиси натрия. Основный водный раствор экстрагировали этиловым эфиром (2× 100 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 3,58 г неочищенного свободного аминоэфира. Сырой продукт очищали с помощью хроматографической колонки на силикагеле 60, 70-230 меш, полученном от BDH Inc., используя в качестве элюента смесь метанола и дихлорметана (2:8 об./об.), и получали чистое свободное основание. Продукт растворяли в диэтиловом эфире (50 мл) и превращали в моногидрохлоридную соль добавлением раствора НСl в эфире (50 мл). Растворитель выпаривали в вакууме, и получали 0,75 г указанного в заголовке соединения (не перекристаллизованного).
Пример 14
Моногидрохлорид (1R,2R)/(1S,2S)-2-(4-морфолинил)-1-(3,4-дихлорфенэтокси)циклогексана
(Соединение №14)
В целом общий подход, применяемый для синтеза этого соединения, аналогичен показанному на фигуре 1.
(i) (1R,2R)/(1S,2S)-2-(4-морфолинил)циклогексанол: смесь оксида циклогексена (206,5 мл, 2 моль, 98%) и морфолина (175 мл, 2 моль) в воде (60 мл) нагревали с обратным холодильником в течение 3,5 час. К реакционной смеси добавляли морфолин (5,3 мл), затем смесь нагревали с обратным холодильником еще 1,5 час для завершения реакции. Затем охлажденную реакционную смесь распределяли между 40% водным раствором NaOH (100 мл) и диэтиловым эфиром (200 мл). Водный слой отделяли от органического слоя и дважды экстрагировали дополнительным количеством диэтилового эфира (2× 100 мл). Объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме. Вакуумная перегонка давала 342,3 г (92,4%) указанного в заголовке соединения.
(ii) К охлажденному (0° С) раствору (1R,2R)/(1S,2S)-2-(4-морфолинил)циклогексанола (40,76 г, 0,22 моль) и триэтиламина (36,60 мл, 0,26 моль) в дихлорметане (400 мл) по каплям добавляли раствор метансульфонилхлорида (20,53 мл, 0,26 моль) в дихлорметане (50 мл). Реакционную смесь перемешивали при 0° С в течение 45 мин и затем при комнатной температуре в течение 3 часов. Затем реакционную смесь промывали водой (2× 100 мл); объединенные промывки повторно экстрагировали дихлорметаном (100 мл). Объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая неочищенный мезилат, подходящий для следующей стадии без какой-либо дополнительной очистки.
(iii) 3,4-Дихлорфенэтиловый спирт: К раствору алюмогидрида лития (7,79 г, 195 ммоль) в безводном диэтиловом эфире (435 мл) медленно через капельную воронку для твердых веществ добавляли в виде порошка 3,4-дихлорфенилуксусную кислоту (27,20 г, 130 ммоль). После завершения добавления реакционную смесь нагревали с обратным холодильником в течение 12 часов. Реакцию гасили осторожным добавлением насыщенного водного раствора сульфата натрия (20 мл), затем полученный в результате нерастворимый материал отфильтровывали и фильтрат концентрировали в вакууме, получая 25,09 г нужного спирта.
(iv) К NaH (6,00 г, 0,2 моль, 80% дисперсия в масле) в безводном этиленгликоля диметиловом эфире (200 мл) добавляли раствор 3,4-дихлорфенэтилового спирта (38,87 г, 0,2 моль) в безводном этиленгликоля диметиловом эфире (100 мл). Полученную в результате смесь перемешивали в течение 3 часов при температуре окружающей среды в атмосфере аргона.
(v) Мезилат (ii) в безводном этиленгликоля диметиловом эфире (100 мл) быстро добавляли к алкоксиду (iv) и полученную в результате реакционную смесь сразу же нагревали с обратным холодильником в течение 16 часов. К охлажденной реакционной смеси добавляли воду (200 мл) и органический растворитель выпаривали в вакууме. Полученный в результате водный раствор затем разбавляли водой (200 мл) и рН доводили до рН 1,5 10% водным раствором НСl. Кислый водный слой экстрагировали диэтиловым эфиром (500 мл) для удаления непрореагировавшего 3,4-дихлорфенэтилового спирта. Дальнейшее подщелачивание водного слоя 5 М водным раствором NaOH до рН 5,7 с последующей экстракцией диэтиловым эфиром давало неочищенное указанное в заголовке соединение, загрязненное некоторым количеством оставшегося мезилата (ii). Растворитель из органического экстракта с рН 5,7 выпаривали в вакууме, затем остаток нагревали с обратным холодильником в смеси этанол - вода (1:1 об./об., 200 мл) в присутствии гидрида натрия (4,12 г, 0,1 моль) в течение 2 часов для гидролиза оставшегося мезилата. Охлажденную реакционную смесь разбавляли водой (300 мл) и органический растворитель выпаривали в вакууме. рН оставшегося водного раствора доводили до рН 5,7 6 М водным раствором НСl с последующей экстракцией диэтиловым эфиром (700 мл). Органический экстракт концентрировали в вакууме, получая чистый аминоэфир. Затем оставшийся продукт распределяли между 1 М водным раствором НСl (300 мл) и дихлорметаном (300 мл). Кислый водный раствор дважды экстрагировали дополнительным количеством дихлорметана (2× 300 мл). Объединенные органические слои сушили над сульфатом натрия, растворитель выпаривали в вакууме и остаток перекристаллизовывали из смеси этанол - гексаны (3:7 об./об., 700 мл), получая 49,3 г указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 15
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексана
(Соединение №15)
При синтезе соединения №15 руководствуются последовательностью реакций, показанных на фигуре 4А и фигуре 4В, синтез детально описан ниже.
(i) N-Безилоксикарбонил-3-пирролидинол: К охлажденному (-60° С) раствору (R)-(+)-3-пирролидинола (20,0 г, 98%, 224,9 ммоль) и триэтиламина (79,2 мл, 99%, 562 ммоль) в дихлорметане (200 мл) по каплям добавляли раствор бензилхлорформиата (33,8 мл, 95%, 224,9 ммоль) в дихлорметане (80 мл). После завершения добавления, продолжавшегося в течение 45 мин, реакционной смеси (желтая суспензия) давали возможность подогреться до комнатной температуры и перемешивали в атмосфере аргона при комнатной температуре в течение ночи. Затем реакционную смесь гасили 1М водным раствором НСl (350 мл) и собирали органический слой. Кислый водный слой экстрагировали дихлорметаном (2× 150 мл) и объединенные органические слои сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало 59,62 г бледно-желтого масла, которое затем выдерживали в условиях высокого вакуума в течение 15 мин, получая 58,23 г (17% превышение теоретически рассчитанного выхода) неочищенного указанного в заголовке соединения, подходящего для следующей стадии без какой-либо дополнительной очистки.
(ii) N-Безилоксикарбонил-3-пирролидинон: К охлажденному (-60° С) раствору оксалилхлорида (23 мл, 98%, 258,6 ммоль) в дихлорметане (400 мл) по каплям добавляли раствор безводного диметилсульфоксида (36,7 мл, 517,3 ммоль) в дихлорметане (20 мл) с такой скоростью, чтобы сохранять температуру ниже -40° С. Затем реакционную смесь перемешивали при -60° С в течение 15 мин. Затем добавляли по каплям раствор N-бензилоксикарбонил-3-пирролидинола (58,22 г, стадия (i), не больше 224,9 ммоль) в дихлорметане (80 мл), сохраняя температуру реакционной смеси ниже -50° С. Затем реакционную смесь перемешивали при -60° С в течение 30 мин перед добавлением триэтиламина (158,3 мл, 99%, 1,125 моль). Полученной в результате смеси давали возможность подогреться до комнатной температуры и затем промывали водой (600 мл), 1 М водным раствором НСl (580 мл) и водой (400 мл). Органический слой сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 54,5 г янтарного масла, которое затем выдерживали в условиях высокого вакуума при перемешивании при комнатной температуре в течение 25 мин, и получали 52,08 г (5,6% превышение теоретически рассчитанного выхода) неочищенного указанного в заголовке соединения, подходящего для следующей стадии без какой-либо дополнительной очистки.
(iii) 7-Бензилоксикарбонил-1,4-диокса-7-азаспиро[4.4]нонан: Смесь N-бензилоксикарбонил-3-пирролидинона (51,98 г, стадия ii, не более 224,9 ммоль) и этиленгликоля (18,8 мл, 99+%, 337,4 ммоль) в толуоле (180 мл) в присутствии каталитического количества моногидрата пара-толуолсульфокислоты (1,04 г, 5,4 ммоль) нагревали с обратным холодильником в аппарате Дина и Старка в течение 16 часов. Затем реакционную смесь разбавляли дополнительным количеством толуола (250 мл) и промывали насыщенным водным раствором бикарбоната натрия (150 мл) и насыщенным водным раствором хлорида натрия (2× 150 мл). Объединенные водные слои повторно экстрагировали толуолом (100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме, получая в остатке 79,6 г темного масла. Неочищенный продукт растворяли в этаноле (500 мл) и пропускали через слой активированного угля (80 г), обесцвечивая раствор. Уголь промывали дополнительно этанолом (1000 мл) и толуолом (500 мл). Фильтрат концентрировали в вакууме и далее выдерживали в условиях высокого вакуума в течение 1 часа, получая 63,25 г (6,8% превышение теоретически рассчитанного выхода) неочищенного указанного в заголовке соединения, подходящего для следующей стадии без какой-либо дополнительной очистки.
(iv) 1,4-диокса-7-азаспиро[4.4]нонан: Смесь 7-бензилоксикарбонил-1,4-диокса-7-азаспиро[4.4]нонана (34,79 г, стадия iii, не более 123,7 ммоль) и 10% Pd-C (13,9 г) в этаноле (90 мл) подвергали гидрогенолизу (60 фунтов на кв. дюйм, 0,41 МПа) во встряхивающем аппарате Парра при комнатной температуре в течение 1,5 часов. Катализатор отфильтровывали, растворитель выпаривали в вакууме, и остаток выдерживали при откачивании, при высоком вакууме в течение 20 мин, получая 15,86 г указанного в заголовке соединения (выход 99,3%).
(v) (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)циклогексанол: смесь 1,4-диокса-7-азаспиро[4.4]нонана (23,54 г, стадия iv, не более 182 ммоль), оксида циклогексена (22,6 мл, 98%, 219 ммоль) и воды (7,8 мл) нагревали при 80° С в течение 2 часов. Затем реакционную смесь распределяли между 40% водным раствором гидроокиси натрия (60 мл) и диэтиловым эфиром (120 мл). Основный водный слой дважды экстрагировали дополнительным количеством диэтилового эфира (2× 120 мл). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. Затем остаток выдерживали при откачивании в условиях высокого вакуума при 50° С в течение 1 часа при перемешивании (для удаления избытка оксида циклогексена) и получали 32,79 г неочищенного указанного в заголовке соединения (выход 79,3%).
(vi) К охлажденному (0° С) раствору (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)циклогексанола (27,47 г, 120 ммоль, стадия v) и триэтиламина (15,86 г, 156 ммоль) в дихлорметане (240 мл) по каплям добавляли метансульфонилхлорид (18,23 г, 156 ммоль). Реакционную смесь перемешивали при 0° С в течение 45 мин и затем при комнатной температуре в течение 3 часов. Затем реакционную смесь промывали смесью воды - насыщенного водного раствора бикарбоната натрия (1:1 об./об., 120 мл). Промывочные слои собирали и повторно экстрагировали дихлорметаном (120 мл). Объединенные органические экстракты сушили над сульфатом натрия, растворитель выпаривали в вакууме, и остаток выдерживали при откачивании в условиях высокого вакуума в течение 4 часов, на выходе получали неочищенный мезилат, пригодный для следующей стадии без какой-либо дополнительной очистки.
(vii) К гидриду натрия (4,32 г, 144 ммоль), суспендированному в безводном этиленгликоль диметиловом эфире (80 мл), добавляли раствор 1-нафтенэтанола (25,31 г, 144 ммоль) в безводном этиленгликоль диметиловом эфире (80 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 4 часов.
(viii) (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(1-нафтенэтокси)циклогексан: Раствор мезилата (vi) в безводном этиленгликоль диметиловом эфире (80 мл) быстро добавляли к алкоксиду (vii) и полученную в результате реакционную смесь сразу же нагревали до кипячения с обратным холодильником в атмосфере аргона в течение 66 часов. Охлажденную реакционную смесь гасили водой (200 мл) и органический растворитель выпаривали в вакууме. Оставшийся водный раствор разбавляли водой (500 мл) и подкисляли 10% водным раствором НСl до рН 0,5. Кислый водный слой экстрагировали диэтиловым эфиром (2× 500 мл), чтобы экстрагировать непрореагировавший 1-нафтенэтанол. рН водного раствора доводили до рН 4,8 5 М водным раствором NaOH и затем экстрагировали диэтиловым эфиром (600 мл). Затем водный раствор подщелачивали до рН 5,7 и экстрагировали диэтиловым эфиром (600 мл). Эту же процедуру повторяли при рН 6,5 и 12,1. Анализ различных эфирных экстрактов с помощью газовой хроматографии показал, что органические экстракты при рН 4,8; 5,7 и 6,5 содержат указанное в заголовке соединение, тогда как эфирный экстракт при рН 12,1 содержит только неидентифицированные включения. Органические экстракты, полученные при рН 4,8; 5,7 и 6,5, объединяли и сушили над сульфатом натрия. Растворитель выпаривали в вакууме, и остаток выдерживали при откачивании в условиях высокого вакуума в течение 3,5 часов, и получали 35,82 г (75% выход) указанного в заголовке соединения, подходящего для следующей стадии без какой-либо дополнительной очистки.
(ix) моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексана: Раствор (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(1-нафтенэтокси)циклогексана (13,73 г, 36,0 ммоль, стадия vi) с 6 М водным раствором НСl (50 мл) в 2-бутаноне (200 мл) перегоняли с обратным холодильником в течение 12 часов. Бутанон выпаривали в вакууме и оставшийся водный раствор доводили до 250 мл водой. Водный раствор экстрагировали диэтиловым эфиром (2× 200 мл) и затем дихлорметаном (2× 200 мл). Объединенные дихлорметановые экстракты сушили над сульфатом натрия, и растворитель выпаривали в вакууме. Оставшееся масло подвергали азеотропной сушке с толуолом. Полученный клейкий продукт энергично перемешивался в течение ночи в диэтиловом эфире (500 мл) с потиранием время от времени для инициирования кристаллизации продукта реакции. Полученный в результате твердый материал собирали и растворяли в небольшом количестве дихлорметана (~10 мл), добавление большого количества диэтилового эфира (~400 мл) индуцировало перекристаллизацию. Твердый материал собирали, сушили в вакууме в течение 3 часов и получали 9,3 г (76% выход) указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 16
Моногидрохлорид (1R,2R)/(1S,2S)-2-(1-ацетилпиперазинил)-1-(2-нафтенэтокси)циклогексана
(Соединение №16)
Соединение №16 получали согласно процедуре, сходной с процедурой, указанной на фигуре 1, и более детально описанной в Примере 14.
(i) (1R,2R)/(1S,2S)-2-(1-ацетилпиперазинил)-1-циклогексанол: смесь 1-ацетилпиперазина (5 г, 39 ммоль) и оксида циклогексена (3,95 мл, 39 ммоль) в воде (1,2 мл) нагревали с обратным холодильником в течение 16 часов. Охлажденную реакционную смесь распределяли между 40% водным раствором NaOH (20 мл) и диэтиловым эфиром (2× 20 мл). Объединенные органические слои сушили над сульфатом натрия, и растворитель выпаривали в вакууме, получая 7,63 г указанного в заголовке соединения в виде белых кристаллов (выход 87%).
(ii) К охлажденному (0° С) раствору (1R,2R)/(1S,2S)-2-(1-ацетилпиперазинил)-1-циклогексанола (3,65 г, 16,2 ммоль) и триэтиламина (3,4 мл, 24 ммоль) в дихлорметане (50 мл) по каплям добавляли раствор метансульфонилхлорида (1,55 мл, 20 ммоль) в дихлорметане (50 мл). Реакционную смесь перемешивали при 0° С в течение одного часа и затем позволяли ей подогреться до температуры окружающей среды. Затем реакционную смесь промывали водой (2× 50 мл) и объединенные промывки повторно экстрагировали дихлорметаном (50 мл). Объединенные органические слои сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая неочищенный мезилат, пригодный для следующей стадии без какой-либо дополнительной очистки.
(iii) К суспензии гидрида натрия (0,8 г, 24 ммоль), предварительно промытого гекасанами (2× 15 мл) в безводном диметилформамиде (50 мл), добавляли раствор 2-нафтенэтанола в безводном диметилформамиде (50 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 30 мин.
(iv) Моногидрохлорид (1R,2R)/(1S,2S)-2-(1-ацетилпиперазинил)-1-(2-нафтенэтокси)циклогексана: раствор мезилата (ii) в безводном диметилформамиде (50 мл) быстро добавляли к алкоксидной смеси (iii) и полученную в результате смесь нагревали до 80° С в течение 16 часов. Охлажденную реакционную смесь выливали в ледяную воду (800 мл) и экстрагировали этилацетатом (3× 200 мл). Объединенные органические экстракты повторно промывали солевым раствором (200 мл) и растворитель выпаривали в вакууме. В оставшееся масло вливали воду (80 мл) и полученный в результате водный раствор подкисляли до рН 2 с помощью 6 М водного раствора НСl. Кислый водный раствор экстрагировали диэтиловым эфиром (3× 40 мл), чтобы экстрагировать непрореагировавший 2-нафтенэтанол. рН водного слоя доводили до рН 10 50% водным раствором NaOH и экстрагировали диэтиловым эфиром (3× 40 мл). Объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая неочищенный свободный аминоэфир. Очистка с помощью хроматографии на колонке с силикагелем при использовании в качестве элюента смеси этилацетата - дихлорметана (1:1 об./об.) давала чистое свободное основание. Превращение в гидрохлоридную соль проводили с помощью раствора НСl в эфире с последующей перекристаллизацией в смеси этанола - диэтилового эфира, при этом получали указанное в заголовке соединение, имеющее элементный состав, указанный в таблице 1.
Пример 17
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,6-дихлорфенэтокси)циклогексана
(Соединение №17)
Соединение №17 получали в 10 стадий в соответствии с процедурой, описанной в Примере 15. Стадии (i)-(v) идентичны стадиям в Примере 16.
(vi) К охлажденному (0° С) раствору (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)циклогексанола (27,77 г, 120 ммоль) и триэтиламина (22 мл, 156 ммоль) в дихлорметане (240 мл) добавляли метансульфонилхлорид (12,32 мл, 156 ммоль). Реакционную смесь перемешивали при 0° С в течение 45 мин и затем при комнатной температуре в течение 3 часов. Реакционную смесь промывали водой (2× 100 мл) и объединенные промывки повторно экстрагировали дихлорметаном (120 мл). Объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая неочищенный мезилат, который затем выдерживали при откачивании в условиях высокого вакуума в течение 4 часов перед использованием на стадии ix.
(vii) 2,6-Дихлорфенэтиловый спирт: К суспензии алюмогидрида лития (13,75 г, 365,75 ммоль) в безводном диэтиловом эфире (500 мл) через воронку для добавления сыпучих веществ добавляли 2,6-дихлорфенилуксусную кислоту (50 г, 243,75 ммоль). Полученную в результате реакционную смесь нагревали с обратным холодильником в течение 16 часов и затем гасили медленным добавлением насыщенного водного раствора сульфата натрия (25 мл). Полученную в результате суспензию перемешивали в течение 3 часов и затем фильтровали, нерастворимый материал осторожно промывали диэтиловым эфиром (2× 100 мл). Объединенные эфирные фильтраты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая на выходе 38,6 г (выход 85%) указанного в заголовке соединения.
(viii) К гидриду натрия (144 ммоль, 4,32 г, 80% масляная дисперсия) в безводном этиленгликоль диметиловом эфире (80 мл) добавляли раствор 2,6-дихлорфенэтилового спирта (27,65 г, 144 ммоль) в безводном этиленгликоль диметиловом эфире (80 мл). Полученную в результате смесь перемешивали при комнатной температуре в атмосфере аргона в течение 4 часов.
(ix) (1R,2R)/(1S,23)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(2,6-дихлорфенэтокси)циклогексан: мезилат (vi) в безводном этиленгликоль диметиловом эфире (80 мл) быстро добавляли к алкоксидной смеси (viii) и полученную в результате смесь сразу же нагревали с обратным холодильником в течение 66 часов. Охлажденную реакционную смесь выливали в воду (200 мл) и органический растворитель выпаривали в вакууме. Оставшийся водный раствор разбавляли дополнительным количеством воды до объема 700 мл, подкисляли до рН 0,5 с помощью 6 М водного раствора НСl и экстрагировали диэтиловым эфиром (2× 600 мл). рН водного слоя доводили до рН 5,9 и затем водный раствор экстрагировали диэтиловым эфиром (700 мл). Органический экстракт сушили над сульфатом натрия, и растворитель выпаривали в вакууме, получая 34,0 г указанного в заголовке соединения (выход 70%).
(х) Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,6-дихлорфенэтокси)циклогексана: Смесь (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(2,6-дихлорфенэтокси)циклогексана (15,85 г, 38,9 ммоль, стадия ix) и 6 М водного раствора НСl (100 мл) в 2-бутаноне (400 мл) нагревали с обратным холодильником в течение 16 часов. Охлажденную реакционную смесь разбавляли водой (100 мл) и органический растворитель выпаривали в вакууме. Органический слой дополнительно разбавляли водой (400 мл), экстрагировали диэтиловым эфиром (500 мл) и дихлорметаном (2× 600 мл). Объединенные дихлорметановые экстракты сушили над сульфатом натрия, и растворитель выпаривали в вакууме. В результате азеотропной перегонки с толуолом получали указанное в заголовке соединение, которое затем сушили в условиях высокого вакуума в течение 15 мин. Гидрохлоридную соль кристаллизовали путем растирания в диэтиловом эфире, кристаллы собирали и подвергали перекристаллизации из смеси этанола - диэтилового эфира, получая 11,85 г чистого продукта (выход 77%), имеющего элементный состав, указанный в таблице 1.
Пример 18
Моногидрохлорид (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро-[4.4]нон-7-ил]-1-(1-нафтенэтокси)циклогексана (Соединение №18)
(1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(1-нафтенэтокси)циклогексан (1,2 г, 3,14 ммоль, из Примера 15, стадия (viii)) в диэтиловом эфире (80 мл) обрабатывали раствором НСl в эфире. Растворитель выпаривали в вакууме и в остаток вливали диэтиловый эфир, в результате растирания получали твердой вещество, которое собирали и осаждали из смеси дихлорметана - диэтилового эфира, получая 0,85 г указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 19
Моногидрохлорид (1R,2S)/(1S,2R)-2-(4-морфолинил)-1-[(2-трифторметил)фенэтокси]циклогексана
(Соединение №19)
(i) 2-(4-Морфолинил)циклогексанон: К охлажденному (-70° С) раствору оксалилхлорида (20 мл, 0,23 моль) в дихлорметане (500 мл) по каплям добавляли раствор безводного диметилсульфоксида (34 мл, 0,48 моль) в дихлорметане (50 мл) и полученную в результате смесь перемешивали в течение 5 мин при температуре ниже -60° С. Затем добавляли по каплям раствор (1R,2R)/(1S,2S)-2-(4-морфолинил)циклогексанола (37,05 г, 0,2 моль) в дихлорметане (50 мл) для поддержания температуры реакции ниже -60° С и реакционную смесь перемешивали в течение 15 мин. Триэтиламин (140 мл) по каплям добавляли к реакционной смеси, сохраняя температуру реакции ниже - 50° С, и затем реакционной смеси давали возможность подогреть до комнатной температуры. Реакционную смесь выливали в воду (600 мл) и водный слой отделяли и экстрагировали дихлорметаном (2× 500 мл). Объединенные органические слои сушили над сульфатом натрия и растворитель удаляли в вакууме. Вакуумная перегонка давала 35,1 г (выход 96%) указанного в заголовке соединения.
(ii) 2-(4-морфолинил)циклогексанол: К охлажденной (0° С) суспензии борогидрида натрия (2,14 г, 56 ммоль) в изопропаноле (120 мл) добавляли раствор 2-(4-морфолинил)циклогексанола (24,7 г, 135 ммоль, стадия i) в изопропаноле (80 мл). Полученную в результате реакционную смесь перемешивали при 0° С в течение 10 мин и затем в течение 30 мин при температуре окружающей среды. К реакционной смеси добавляли воду (200 мл) и органический растворитель выпаривали в вакууме. Затем оставшийся водный раствор экстрагировали этилацетатом (4× 50 мл), объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая 22,48 г указанного в заголовке соединения, подходящего для следующей стадии без какой-либо дополнительной очистки.
(iii) (1S,2R)/(1R,2S)-2-(4-морфолинил)циклогексил-2-(трифторметил)фенилацетат: Смесь 2-(4-морфолинил)циклогексанола (7,41 г, 40 ммоль, стадия ii), 2-(трифторметил)фенилуксусной кислоты (10,21 г, 49 ммоль) и моногидрата пара-толуолсульфокислоты (40 мг) в толуоле (60 мл) нагревали с обратным холодильником в аппарате Дина и Старка в течение 48 часов. К охлажденной реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия (40 мл), водный слой отделяли и экстрагировали этилацетатом (3× 50 мл). Объединенные органические слои сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая смесь (1S,2R)/(1R,2S)-2-(4-морфолинил)циклогексил 2-(трифторметил)фенилацетата и (1R,2R)/(1S,2S)-2-(4-морфолинил)циклогексил 2-(трифторметил)фенилацетата. Хроматография цис/транс смеси на “сухой” колонке с использованием в качестве элюента смеси этилацетата - гексанов (+0,5% изопропиламина, об./об.) давала 3,19 г неочищенного указанного в заголовке соединения с примесью исходного материала 2-(4-морфолинил)циклогексанола. Неочищенный продукт распределяли между дихлорметаном (30 мл) и 0,5 М водным раствором НСl (7 мл). Водный слой отделяли и дополнительно экстрагировали дихлорметаном (2× 18 мл). Объединенные органические слои сушили над сульфатом натрия и растворитель выпаривали в вакууме. В результате перекристаллизации из смеси этанола - гексанов получали 2,78 г указанного в заголовке соединения.
(iv) Моногидрохлорид (1S,2R)/(1R,2S)-2-(4-морфолинил)-1-[(2-трифторметил)фенэтокси]циклогексана: К смеси (1S,2R)/(1R, 2S)-2-(4-морфолинил)циклогексил 2-(трифторметил) фенилацетата (1,64 г, 4,28 ммоль, стадия iii) и борогидрида натрия (332 мг, 8,70 ммоль) в безводном тетрогидрофуране (35 мл) при нагревании с обратным холодильником добавляли раствор диэфирата трехфтористого бора (8,2 мл, 65 ммоль) в течение 1,5 часов. Реакционную смесь гасили добавлением воды (~70 мл), органический растворитель выпаривали в вакууме и рН оставшегося водного раствора доводили до рН 9,6. Водный слой экстрагировали диэтиловым эфиром (2× 70 мл), объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме. Затем остаток распределяли между 0,5 М водным раствором НСl (50 мл) и диэтиловым эфиром (2× 50 мл). Водный раствор подщелачивали до рН 5,9 и экстрагировали диэтиловым эфиром (50 мл). Органический слой собирали, сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая неочищенный свободный аминоэфир. Свободное основание переводили в моногидрохлоридную соль путем распределения между 0,5 М водным раствором НСl (10 мл) и дихлорметаном (10 мл). Кислый водный раствор экстрагировали еще раз дихлорметаном (10 мл), объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме. Перекристаллизация из смеси этанола - гексанов давала 636 мг (выход 38%) указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 20
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[3-(циклогексил)пропокси]циклогексана
(Соединение №20)
(i) 3-Циклогексил-1-пропил бромид: К охлажденному (0° С) 3-циклогексил-1-пропанолу (5 г, 35,15 ммоль) медленно добавляли раствор трехбромистого фосфора (1,1 мл, 17,6 ммоль) в дихлорметане (2 мл). После завершения добавления реакционной смеси давали подогреться до комнатной температуры и перемешивали в течение 4 часов. Реакцию гасили добавлением насыщенного водного раствора бикарбоната натрия (5 мл) и 10% NaOH (10 мл). Полученную в результате смесь экстрагировали диэтиловым эфиром (3× 50 мл), объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая масло. Вакуумная перегонка давала 3,4 г (выход 47%) указанного в заголовке соединения.
(ii) (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-[3-(циклогексил)пропокси]циклогексан: К суспензии гидрида натрия (200 мг, 8,33 ммоль) в безводном диметилформамиде (20 мл) добавляли раствор (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил]циклогексанола (1,5 г, 6,6 ммоль) в безводном диметилформамиде (10 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 30 мин и затем быстро добавляли раствор 3-(циклогексил)пропил бромида (1,67 г, 8,15 ммоль) в безводном диметилформамиде. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь выливали в воду (200 мл) и затем экстрагировали этилацетатом (3× 50 мл). Объединенные органические экстракты повторно промывали солевым раствором (50 мл), и растворитель выпаривали в вакууме. Остаток смывали водой (50 мл) и рН доводили до рН 1,0 с помощью 6 М водного раствора НСl. Кислый водный раствор экстрагировали диэтиловым эфиром (2× 50 мл), затем подщелачивали до рН 5,0-5,5 с помощью 5 М водного раствора NaOH и экстрагировали диэтиловым эфиром (3× 50 мл). Объединенные органические экстракты при рН 5,0-5,5 концентрировали в вакууме, получая неочищенное указанное в заголовке соединение, подходящее для следующей стадии без какой-либо дополнительной очистки.
(iii) Моногидрохлорид (1R,2S)/(1S,2R)-2-(3-кетопирролидинил)-1-[3-(циклогексил)пропокси]циклогексана: (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-[3-(циклогексил)пропокси]циклогексан (ii) в смеси 6 М водного раствора НСl - бутанона (1:4 об./об., 100 мл) нагревали с обратным холодильником в течение 16 часов. Охлажденную реакционную смесь концентрировали в вакууме и оставшийся водный раствор разбавляли водой (~50 мл). Кислый водный раствор экстрагировали этиловым эфиром (50 мл) и затем дихлорметаном (3× 50 мл). Дихлорметановые экстракты сушили над сульфатом натрия, и растворитель выпаривали в вакууме, получая неочищенное указанное в заголовке соединение. Гидрохлоридную соль кристаллизовали путем растирания в смеси диэтилового эфира - гексанов (1:1 об./об., ~200 мл) и затем осаждали из смеси дихлорметана - диэтилового эфира - гексанов, получая 0,8 г указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 21
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-ацетоксипирролидинил)-1-(1-нафтенэтокси)циклогексана
(Соединение №21)
(i) Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-гидроксипирролидинил)-1-(1-нафтенэтокси)циклогексана: К охлажденному (-0° С) раствору борогидрида натрия в изопропаноле (20 мл) добавляли раствор моногидрохлорида (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексана (1,4 г, 3,75 ммоль) в изопропаноле (30 мл). Полученную в результате смесь перемешивали при 0° С в течение 15 мин и затем в течение 30 мин при комнатной температуре. Реакцию гасили добавлением воды, реакционную смесь выпаривали досуха и остаток промывали дихлорметаном (2× 20 мл). Дихлорметановые промывки сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая указанное в заголовке соединение.
(ii) Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-ацетоксипирролидинил)-1-(1-нафтенэтокси)циклогексана: затем промежуточный продукт спирт (i) нагревали с обратным холодильником в уксусном ангидриде (15 мл) в течение 2 часов. Избыток уксусного ангидрида удаляли в вакууме; остаток брали в воду (100 мл) и экстрагировали диэтиловым эфиром (2× 30 мл). Водный раствор подщелачивали до рН 8,0 и экстрагировали диэтиловым эфиром (3× 50 мл). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. Оставшееся масло растворяли в небольшом количестве дихлорметана и добавляли большой объем диэтилового эфира для инициирования кристаллизации 1,0 г (выход 65%) указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 22
Моногидрохлорид (1R,2R)/(1S,2S)-2-(4-морфолинил)-1-[(2,6-дихлорфенил)метокси]циклогексана
(Соединение №22)
Соединение №22 получали согласно процедуре синтеза эфиров Вильямсона. К суспензии гидрида натрия, 80% масляной дисперсии (337 мг, 11 ммоль) в этиленгликоль диметиловом эфире (20 мл) добавляли раствор (1R,2R)/(1S,2S)-2-(4-морфолинил)-1-циклогексанола (2,0 г, 10,8 ммоль) в этиленгликоль диметиловом эфире (10 мл). Полученную в результате реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 3 часов, затем добавляли раствор 2,6-дихлорбензилбромида в этиленгликоль диметиловом эфире (10 мл), и реакционную смесь нагревали с обратным холодильником в течение 16 часов. Охлажденную реакционную смесь выливали в воду (40 мл) и органический растворитель выпаривали в вакууме. Оставшийся водный раствор разбавляли дополнительным количеством воды (60 мл) и подкисляли до рН 0,5 с помощью 6 М водного раствора НСl. Кислый водный раствор экстрагировали диэтиловым эфиром (2× 40 мл), и затем рН доводили до рН 5,5. Экстракция диэтиловым эфиром (3× 50 мл) с последующей сушкой над сульфатом натрия и концентрированием в вакууме давала чистый аминоэфир. Гидрохлоридную соль осаждали с помощью обработки свободного основания раствором НСl в эфире. Перекристаллизация из смеси ацетона - метанола - диэтилового эфира давала 2,6 г (выход 68%) указанного в названии соединения, имеющего элементный состав, указанный в таблице 1.
Пример 23
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[(2,6-дихлорфенил)метокси]циклогексана
(Соединение №23)
Соединение №23 получали в 7 стадий согласно процедуре, детально описанной в Примере 15. Стадии (i)-(v) были идентичны стадиям, описанным в Примере 15. Синтез эфира (стадия vi) выполняли согласно процедуре синтеза эфиров Вильямсона, как в Примере №22.
(vi) (1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-[(2,6-дихлорфенил)метокси]циклогексан: К суспензии гидрида натрия, 80% масляной дисперсии (222 мг, 7,25 ммоль) в этиленгликоль диметиловом эфире (20 мл) добавляли раствор (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)циклогексанола (1,5 г, 6,60 ммоль, стадия (v) Примера 15) в этиленгликоль диметиловом эфире (10 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов и затем добавляли раствор 2,6-дихлорбензил бромида (1,9 г, 7,9 ммоль) в этиленгликоль диметиловом эфире (10 мл). Реакционную смесь нагревали с обратным холодильником в течение 16 часов в атмосфере аргона, растворитель выпаривали в вакууме и остаток брали в воду (70 мл). Водный раствор подкисляли до рН 0,5 с помощью 6 М водного раствора НСl и затем экстрагировали диэтиловым эфиром (2× 40 мл). Подщелачивание водного раствора до рН 4,5-5,5, последующая экстракция диэтиловым эфиром (4× 40 мл), сушка объединенных органических экстрактов над сульфатом натрия и выпаривание растворителя в вакууме давали указанное в заголовке промежуточное соединение.
(vii) Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[(2,6-дихлорфенил)метокси]циклогексана: промежуточный продукт кеталь (стадия vi) в смеси 6 М HCl - бутанона (1:4 об./об., 100 мл) нагревали с обратным холодильником в течение 16 часов. Бутанон выпаривали в вакууме и оставшийся водный слой разбавляли дополнительным количеством воды (100 мл). Кислый водный слой экстрагировали диэтиловым эфиром (2× 40 мл) и затем дихлорметаном (3× 40 мл). Объединенные дихлорметановые экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая указанное в заголовке неочищенное соединение. Продукт кристаллизовали путем растирания в диэтиловом эфире и повторно осаждали из смеси дихлорметана - диэтилового эфира, получая 1,8 г (выход 72%) указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 24
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-гидроксипирролидинил)-1-(2,6-дихлорфенэтокси)циклогексана
(Соединение №24)
К раствору соединения №17 (5,0 г, 12,7 ммоль) в изопропаноле (120 мл) добавляли порошок боргидрида натрия (2,0 г, 52,8 ммоль) и полученную в результате смесь перемешивали при комнатной температуре до завершения реакции. Реакцию гасили водой (40 мл) и затем концентрировали досуха. Остаток промывали дихлорметаном (50 мл); фильтрат сушили над сульфатом натрия, концентрировали в вакууме, получая указанное в заголовке соединение, которое кристаллизовалось через 3 часа в условиях высокого вакуума. Результаты элементного анализа продукта показаны в таблице 1.
Пример 25
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,2-дифенилэтокси)циклогексана
(Соединение №25)
Соединение №25 получали в 10 стадий в соответствии с процедурой, идентичной процедуре, описанной в Примерах 15 и 17. Стадии (i)-(v) идентичны стадиям в Примере 15.
(vi) К охлажденному (0° С) раствору (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)циклогексанола (2,0 г, 8,8 ммоль) и триэтиламина (2,1 мл, 15 ммоль) в дихлорметане (30 мл) добавляли метансульфонилхлорид (0,9 мл, 11,44 ммоль). Реакционную смесь перемешивали при 0° С в течение 45 мин и затем при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли дихлорметаном (25 мл), промывали водой (2× 25 мл) и объединенные промывки повторно экстрагировали дихлорметаном (25 мл). Объединенные органические экстракты сушили над сульфатом натрия и растворитель выпаривали в вакууме, получая неочищенный мезилат, который затем выдерживали при откачивании в условиях высокого вакуума в течение 30 мин перед использованием на стадии ix.
(vii) (2,2-Дифенил)этиловый спирт: К алюмогидриду лития (2,85 г, 23,56 ммоль) в безводном диэтиловом эфире (150 мл) добавляли в виде порошка дифенилуксусную кислоту (5,0 г, 56 ммоль). Полученную в результате реакционную смесь осторожно нагревали с обратным холодильником в течение одного часа. Реакцию гасили насыщенным водным раствором сульфата натрия и полученный в результате осадок отфильтровывали. Фильтрат концентрировали в вакууме, получая 4,0 г (выход 86%) указанного в заголовке соединения.
(viii) К гидриду натрия, предварительно промытому гексанами (253 мг, 10,56 ммоль) в виде суспензии в этиленгликоль диметиловом эфире (15 мл) добавляли раствор 2,2-дифенилэтилового спирта (2,09 г, 10,56 ммоль, стадия vii) в этиленгликоль диметиловом эфире (15 мл). Полученную в результате смесь перемешивали при комнатной температуре в атмосфере аргона в течение 30 мин.
(ix) (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(2,2-дифенилэтокси)циклогексан: мезилат (vi) в этиленгликоль диметиловом эфире (20 мл) быстро добавляли к алкоксиду (viii) и реакционную смесь нагревали с обратным холодильником в течение 5 дней. Охлажденную реакционную смесь концентрировали в вакууме, остаток брали в воду (50 мл) и рН доводили до рН 1,0 с помощью 6 М водного раствора НСl. Кислый водный раствор экстрагировали диэтиловым эфиром (2× 50 мл), водный слой собирали и подщелачивали до рН 6,0. Экстракция диэтиловым эфиром (2× 50 мл) с последующей сушкой над сульфатом натрия и выпариванием растворителя в вакууме давали 1,55 г (выход 43%) указанного в заголовке соединения.
(х) Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,2-дифенилэтокси)циклогексана: Смесь (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(2,2-дифенилэтокси)циклогексана (1,55 г, 3,8 ммоль) в 6 М HCl-бутаноне (1:4 об./об., 50 мл) нагревали с обратным холодильником в течение 2 часов. Бутанон выпаривали в вакууме, и остаток брали в воду (50 мл). Водный раствор экстрагировали диэтиловым эфиром (2× 50 мл); водный слой собирали и экстрагировали дихлорметаном (2× 50 мл). Объединенные дихлорметановые экстракты сушили над сульфатом натрия и концентрировали в вакууме, получая указанное в заголовке неочищенное соединение. Продукт кристаллизовали путем растирания в диэтиловом эфире и повторным осаждением из смеси дихлорметана - диэтилового эфира, получая 1,21 г (выход 80%) указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 26
Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-тиазолидинил)-1-(2,6-дихлорфенэтокси)циклогексана
(Соединение №26)
(i) (1R,2R)/(1S,2S)-2-(3-тиазолидинил)циклогексанол: К безводному перхлорату магния (12,93 г, 53,3 ммоль) добавляли раствор оксида циклогексена (6,1 мл, 58,6 ммоль) в безводном ацетонитриле (25 мл), и полученную в результате смесь перемешивали при комнатной температуре в течение 20 мин. Затем добавляли раствор тиазолидина (5,16 г, 55,0 ммоль) в безводном ацетонитриле, и реакционную смесь нагревали при 35° С в течение 16 часов. Реакционную смесь концентрировали в вакууме, и остаток распределяли между водой (350 мл) и диэтиловым эфиром (350 мл). Водный слой отделяли и экстрагировали еще раз диэтиловым эфиром (350 мл). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме, получая неочищенный продукт. Неочищенный аминоспирт очищали с помощью хроматографии на “сухой” колонке, используя в качестве элюента смесь этилацетата - гексанов (1:1 об./об.), и получали 4,83 г (выход 47%) указанного в заголовке соединения.
(ii) К охлажденному (0° С) раствору (1R,2R)/(1S,2S)-2-(3-тиазолидинил)циклогексанола (3,17 г, 16,9 ммоль) и триэтиламина (3,08 мл, 22 ммоль) в дихлорметане (30 мл) по каплям добавляли метансульфонилхлорид (1,74 мл, 22 ммоль). Реакционную смесь перемешивали при 0° С в течение одного часа и затем при температуре окружающей среды в течение 3 часов. Реакционную смесь разбавляли дихлорметаном (20 мл) и промывали водой (2× 30 мл). Объединенные промывки повторно экстрагировали дихлорметаном (25 мл) и объединенные органические экстракты сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало мезилат, пригодный для следующей стадии без какой-либо дополнительной очистки.
(iii) К гидриду натрия, 80% дисперсии в масле (608 мг, 20,28 ммоль) в этиленгликоль диметиловом эфире (30 мл) добавляли раствор 2,6-дихлорфенэтилового спирта (3,87 г, 20,28 ммоль, Пример 4, стадия vii) в этиленгликоль диметиловом эфире (15 мл). Полученную в результате смесь перемешивали при комнатной температуре в атмосфере аргона в течение 2 часов.
(iv) Моногидрохлорид (1R,2R)/(1S,2S)-2-(3-тиазолидинил)-1-(2,6-дихлорфенэтокси)циклогексана: Мезилат (ii) в этиленгликоль диметиловом эфире (15 мл) быстро добавляли к алкоксиду (iii) и реакционную смесь нагревали с обратным холодильником в течение 40 часов. Охлажденную реакционную смесь выливали в воду (100 мл), и органический растворитель выпаривали в вакууме. Оставшийся водный раствор разбавляли дополнительным количеством воды (100 мл) и рН доводили до рН 1,5. Кислый водный слой экстрагировали диэтиловым эфиром (3× 100 мл), объединенные органические экстракты сушили над сульфатом натрия и растворитель удаляли в вакууме, получая неочищенное свободное основание. Продукт очищали с помощью хроматографии на “сухой” колонке, используя в качестве элюента смесь этилацетата - гексанов (1:10 об./об.), получая 2,4 г чистого свободного аминоэфира. Чистый продукт (1,0 г) переводили в гидрохлоридную соль обработкой раствором НСl в эфире, и полученную в результате соль перекристаллизовывали из смеси ацетона - диэтилового эфира, при этом получили 0,69 г указанного в заголовке соединения, имеющего элементный состав, указанный в таблице 1.
Пример 27
Моногидрохлорид (1R,2S)/(1S,2R)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексана
(Соединение №27)
Соединение №27 получали в 8 стадий в соответствии со схемой синтеза, указанной на фигуре 3. Стадии с (i) no (iv) были идентичны стадиям, описанным в Примере 15.
(v) (1R,2R)/(1S,2S)-1-(1-нафтенэтокси)-2-циклогексанол: К безводному перхлорату магния (270 мг, 1,2 ммоль) в безводном ацетонитриле (1,7 мл) добавляли оксид циклогексена (0,12 г, 1,2 ммоль). Полученную в результате смесь перемешивали в течение 15 мин при комнатной температуре и затем добавляли 1-нафтенэтанол (2,7 г, 10,15 ммоль). Реакционную смесь нагревали с обратным холодильником и к нагреваемой реакционной смеси добавляли дополнительно оксид циклогексена (2,0 мл, 2,0 г, 20 ммоль) со скоростью 0,4 мл/час. Нагревание прекращали спустя 16 часов и охлажденную реакционную смесь распределяли между диэтиловым эфиром (50 мл) и насыщенным водным раствором бикарбоната натрия (30 мл). Водный слой отделяли и дважды экстрагировали дополнительным количеством диэтилового эфира (2× 40 мл). Объединенные органические экстракты повторно промывали водой (15 мл), солевым раствором (15 мл) и сушили над сульфатом натрия. Выпаривание растворителя в вакууме давало указанное в заголовке неочищенное соединение, пригодное для следующей стадии без какой-либо дополнительной очистки.
(vi) 1-(1-Нафтенэтокси)-2-циклогексанон: К раствору (1R,2R)/(1S,2S)-2-(1-нафтенэтокси)-1-циклогексанола (1,0 г, стадия v) в диметилформамиде (20 мл) небольшими порциями добавляли дихромат пиридиния (5,0 г, 13,2 ммоль) и полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь выливали в воду (100 мл) и полученную в результате суспензию экстрагировали диэтиловым эфиром (3× 50 мл). Объединенные органические экстракты повторно промывали 1 М водным раствором NaOH (30 мл), солевым раствором (30 мл) и сушили над сульфатом натрия. Выпаривание растворителя давало 1,0 г неочищенного указанного в заголовке соединения, подходящего для следующей стадии реакции.
(vii) (1R,2S)/(1S,2R)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(1-нафтенэтокси)циклогексан: К раствору (1,4-диокса-7-азаспиро[4.4]нонана (5,17 г, 40 ммоль) и 1-(1-нафтенэтокси)-2-циклогексанона (1,79 г, 6,58 ммоль, стадия vi, 77% чистоты) в безводном метаноле (10 мл) добавляли 5 N раствор НСl в метаноле (2,7 мл) и затем цианоборогидрид натрия (397 мг, 6 ммоль). Затем реакционную смесь разбавляли безводным метанолом (7 мл) и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили добавлением 6 М водного раствора НСl (40 мл), органический растворитель выпаривали в вакууме, оставшийся водный раствор разбавляли до 100 мл водой и рН доводили до рН 0,5 с помощью 6 М водного раствора НСl. Кислый водный слой экстрагировали диэтиловым эфиром (100 мл); водный слой отделяли и подщелачивали до рН 6,7 с помощью 5 М водного раствора NaOH. Экстракция диэтиловым эфиром (100 мл) с последующей сушкой над сульфатом натрия и выпариванием растворителя в вакууме, после очистки с помощью хроматографии на “сухой” колонке с использованием в качестве элюента смесей этилацетата - гексанов (от 1:9 до 1:6 об./об., + 0,5% об./об. изопропиламина) давала 1,28 г неочищенных (1R,2S)/(1S,2R)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(1-нафтенэтокси)циклогексана и (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(1-нафтенэтокси)циклогексана. Отделение (1R,2S)/(1S, 2R)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(1-нафтенэтокси)циклогексана от (1R,2R)/(1S,2S)-2-(1,4-диокса-7-азаспиро-[4.4]нон-7-ил)-1-(1-нафтенэтокси)циклогексана выполняли с помощью препаративной ВЭЖХ (Waters Delta Prep 4000, PrePak картридж 40× 100 мм, изопропанолгексаны (2:98 об./об., +0,05% об./об. диэтиламина), получая 590 мг указанного в заголовке соединения.
(viii) Моногидрохлорид (1R,2S)/(1S,2R)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексана: Смесь (1R,2S)/(1S,2R)-2-(1,4-диокса-7-азаспиро[4.4]нон-7-ил)-1-(1-нафтенэтокси)циклогексана (480 мг, 1,23 ммоль, стадия vii) в 6 М водном растворе НСl - бутаноне (1:4 об./об., 40 мл) нагревали с обратным холодильником в течение 2 часов. Органический растворитель выпаривали в вакууме, остаточный водный раствор разбавляли до 50 мл водой и дважды экстрагировали диэтиловым эфиром (2× 50 мл) и затем трижды дихлорметаном (3× 50 мл). Объединенные дихлорметановые экстракты сушили над сульфатом натрия, и растворитель выпаривали в вакууме, оставшееся масло далее сушили с помощью азеотропной отгонкой толуола. Указанное в заголовке соединение кристаллизовали растиранием в гексанах (430 мг, выход 93%), соединение имело элементный состав, указанный в таблице 1.
Пример 27-А
Моногидрохлорид (±)-транс-[2-морфолино-1-93-(2,4-диметокси)циннамил)]циклогексана
i) Исходный транс-аминоциклогексанол получали в соответствии с примером 1 заявки РСТ/СА99/00280.
ii) К охлажденному до 0° С раствору (±)-транс-(2-морфолино)циклогексанола (3,0 г, 16,20 ммоль) и триэтиламина (3,4 мл, 24,00 ммоль) в дихлорметане (50 мл) через канюлю прибавляли раствор метансульфонилхлорида (1,55 мл, 20,00 ммоль) в дихлорметане (50 мл). Прибавление завершали в течение 5 минут, после чего реакционную смесь дополнительно перемешивали в течение 45 минут при 0° С, а затем при комнатной температуре еще 1 час. Реакционную смесь промывали водой (2× 30 мл) и объединенные водные промывные фракции подвергали обратной экстракции дихлорметаном (30 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с образованием 4,71 г неочищенного мезилата.
iii) К перемешиваемому раствору этил транс-2,4-диметоксициннамата (7,5 г, 34 ммоль) в толуоле (100 мл) при комнатной температуре прибавляли раствор DIBAL (100 мл, 1 М в гекснах). Прибавление продолжалось в течение приблизительно 2 часов, затем реакционную смесь оставляли на ночь при перемешивании при комнатной температуре. Реакционную смесь обрабатывали, медленно при 0° С добавляя воду (10 мл), затем раствор перемешивали еще 2 часа и фильтровали. Фильтрат сушили над сульфатом натрия и растворитель удаляли в вакууме.
iv) Гидрид натрия, 80% дисперсию, предварительно промытую гексанами (3× 10 мл) (0,60 г, 25,00 ммоль) в безводном диметилформамиде (50 мл) через канюлю прибавляли к раствору 2,4-диметоксикоричного спирта (4,2 г, 21,62 ммоль) в безводном диметилформамиде (50 мл). После прибавления барботировали водород, реакционную смесь перемешивали при комнатной температуре в течение 1 часа.
v) Через канюлю быстро (3 минуты) прибавляли раствор мезилата в диметилформамиде (50 мл). Реакционную смесь нагревали до 80° С в течение 0,5 часа, затем температуру снижали до 50° С и реакционную смесь оставляли при перемешивании на ночь. Реакционную смесь выливали в смесь льда и воды (800 мл) и экстрагировали этилацетатом (3× 200 мл) и сушили над сульфатом натрия. После выпаривания расторителя в вакууме получали темное красное масло. Неочищенный продукт очищали на хроматографической колонке с силикагелем 60, 70-230 меш производства BDH Inc., используя в качестве элюента смесь этилацетат-дихлорметан (1:1 об./об.) с получением чистого свободного основания (1,4 г). Продукт растворяли в дихлорметане (50 мл), фильтровали и смешивали с уксусной кислотой (3 мл). Растворитель выпаривали в вакууме с получением 1,4 г указанного в заголовке соединения.
Элементный анализ. Вычислено для C21H31NO4Cl (М.м. 481,59): С 62,35%, Н 8,16%, N 2,91%. Найдено: С 62,87%, Н 8,35%, N 2,92%.
Биологические показатели моногидрохлорида (±)-транс-[2-морфолино-1-93-(2,4-диметокси)циннамил)]циклогексана были получены с использованием методики, раскрытой в примере 28 заявки РСТ/СА99/00280. Определяли значение объема вливания соединения (микромоль/кг мин), необходимого для снижения на 50% показателя аритмии у животных, подвергшихся действию лекарства (ED50AA) по сравнению с контрольными животными, получившими чистый носитель. Значение ED50AA для данного соединения равно 0,82, и оно находится в пределах, установленных для типичных соединений изобретения в Таблице 3. Параметры ЭКГ при использовании данного соединения были определены, как описано в примере 29 заявки РСТ/СА99/00280. Интервалы P-R, QRS и Q-T имели следующие значения: P-R=18, QRS=16 и Q-Т=3,5. Каждое из значений находится в пределах, установленных для типичных соединений изобретения в Таблице 4.
Также были определены модулирующие свойства в отношении ионных каналов для соединения, оцениваемые величинами ED25 для трех показателей (It, ERP и VFT) блокирования сердечных натриевого и калийного каналов. Величина ED25 соответствует объему вливания (микромоль/кг мин), необходимого для достижения 25% увеличения измеряемого параметра по сравнению с контрольными животными. Подробности опыта изложены в примерах 29 и 30 РСТ/СА99/00280. Для данного соединения были определены следующие значения: It=4, VFT=3 и ERP=3, каждое из которых находится в пределах, установленных для типичных соединений изобретения в Таблице 5 описания данного изобретения.
Пример 28
Оценка антиаритмической эффективности
Антиаритмическую эффективность оценивали путем исследования влияния соединения на частоту возникновения аритмии сердца у находящихся в сознании крыс, подверженных окклюзии коронарной артерии. Крыс весом 200-300 г подвергали подготовительной хирургической операции и распределяли на группы случайным образом. В каждом случае во время подготовки к операции животных анестезировали галотаном. В левую бедренную артерию вводили канюлю для измерения среднего артериального кровяного давления и отбора образцов крови. В левую бедренную вену также вводили канюлю для инъекции лекарственных препаратов. Открывали полость грудной клетки, и полиэтиленовый обтуратор свободно размещали вокруг левой передней нисходящей коронарной артерии. Затем полость грудной клетки закрывали. ЭКГ записывали путем введения электродов, размещенных вдоль анатомической оси сердца. Все подводки к канюлям и электродам выводили наружу в межлопаточной области. Случайным и двойным слепым способом примерно через 0,5-2 часа после операции инфузировали наполнитель или тестируемое соединение. Через 15 минут после начала инфузии обтуратор затягивали таким образом, чтобы получить окклюзию коронарной артерии. В течение 30 минут после окклюзии следили за ЭКГ, аритмией, кровяным давлением, частотой сердечных сокращений и смертностью. Аритмию регистрировали в виде желудочковой пароксизмальной тахикардии (ЖТ) и фибрилляции желудочков (ФЖ) и оценивали в соответствии с Curtis, M.J. and Walker, M.J.A., Cardiovasc. Res. 22:656(1988) (смотри таблицу 2).
ЖТ = желудочковая тахикардия
ФЖ = фибрилляция желудочков
Крыс исключали из исследования, если концентрация калия в их сыворотке перед окклюзией не была в пределах 2,9-3,9 мМ. Окклюзии соответствует увеличение высоты зубца R и поднятия сегмента "S-T"; и зона окклюзии (измеренная после смерти с помощью перфузии красителя кардиогрин) в пределах 25%-50% общего веса левого желудочка.
В таблице 3 описаны результаты тестирования соединений, описанных здесь, в виде значений скорости инфузии в микромоль/кг/мин. (ЭД50АА), которая будет снижать оценку аритмии у обработанных животных до 50%, от оценки аритмии у животных, обработанных только наполнителем, в котором растворяют тестируемое лекарственное средство.
Пример 29
Измерение параметров ЭКГ
В этом примере использовали крыс весом 200-250 г. Животных анестезировали введением 60 мг/кг пентобарбитона в/б. В сонную артерию и яремную вену вводили канюли для измерения кровяного давления и инъекции лекарственного средства соответственно. ЭКГ записывали с помощью электродов, размещенных вдоль анатомической оси сердца. Все соединения давали в виде болюсных инъекций.
Измеряли различные параметры ЭКГ. В таблице 4 описаны результаты тестов в виде ЭД25 (микромоль/кг), которые представляют собой дозы, необходимые для получения увеличения измеряемого параметра на 25% (НД = нет данных). Увеличение интервала P-R и увеличение интервала QRS свидетельствует о блокировании натриевых каналов сердца, в то время как увеличение интервала Q-T свидетельствует о дополнительном блокировании калиевых каналов сердца, что характерно для антиаритмического средства типа 1а.
Пример 30
Оценка блокирования натриевых каналов
Крыс подготавливали согласно предыдущей процедуре. Два серебряных стимулирующих электрода вставляли через грудную клетку и имплантировали в левый желудочек. Чтобы определить пороговый ток для захвата, пороговый ток желудочковой фибрилляции и эффективный рефракторный период, использовали стимуляцию прямоугольными импульсами (Howard, P.G. and Walker, M.J.A., West. Pharmacol. Soc. 33:123-127 (1990)). Таблица 5 содержит значения ЭД25 для этих показателей блокирования натриевых каналов сердца, где ЭД25 представляет собой скорость инфузии соединения в микромоль/кг/минуту, необходимого для того, чтобы вызвать 25% увеличение по сравнению с контролем. Случаи увеличения рефрактерности указывают на дополнительное блокирование калиевых каналов. Пороговый ток для захвата представлен значениями "It". Пороговый ток фибрилляции представлен значениями “ФЖП”. Эффективный рефракторный период представлен значениями “ЭРП”.
Пример 31
Модель ФП, вызванной стимуляцией вагуса у собак
Общая методика
Беспородных собак любого пола весом 15-49 кг анестезировали морфином (2 мг/кг вначале с последующим в/в введением 0,5 мг/кг каждые 2 час) и α -хлоралозом (120 мг/кг в/в с последующей инфузией 29,25 мг/кг/час; St.-Georges et al., 1997). Вентиляцию легких собакам проводили механически комнатным воздухом с добавлением кислорода через эндотрахеальную трубку при скорости от 20 до 25 вдохов/минуту с объемом дыхания, полученным на основании номограммы. Измеряли количество газов в артериальной крови и поддерживали в физиологических пределах (SАО2>90%, рН 7,30-7,45). В бедренную артерию вводили катетер для регистрации кровяного давления и измерения содержания газов в крови, в обе бедренные вены вводили катетеры для введения лекарственного средства и отбора проб венозной крови. Просвет катетеров поддерживали открытым с помощью гепаринизированного 0,9% солевого раствора. Температуру тела поддерживали на уровне 37-40° С с помощью согревающего одеяла.
Сердце открывали с помощью срединной торакотомии и создавали перикардиальную подвеску. Для регистрации и стимуляции в правое предсердие вводили три биполярных электрода из нержавеющей стали, покрытые тефлоном (Teflon™ ), и один электрод для регистрации вводили в ушко левого предсердия. Программируемый стимулятор (Digital Cardiovascular Instruments, Berkeley, CA) использовали для стимуляции правого предсердия 2 мс импульсами, в два раза превышающими диастолический порог. Два электрода из нержавеющей стали, покрытые тефлоном (Teflon™ ), вводили в левый желудочек, один для регистрации и другой для стимуляции. Желудочковый пейсмекер, включаемый по требованию (GBM 5880, Medtronics, Minneapolis, MN), использовали для стимуляции желудочков в режиме 90 ударов/минуту в том случае, когда (особенно во время ФП, вызванной стимуляцией вагуса) скорость сокращения желудочков становилась крайне низкой. Для регистрации отведений II и III ЭКГ, электрограмм предсердий и желудочков, кровяного давления и сигналов, искусственно вызванных в результате стимуляции, использовали датчик Р23 ID, электрофизиологический усилитель (Bloom Associates, Flying Hills, PA) и бумажный самописец (Astromed МТ-95000, Toronto, ON, Kanada). Блуждающие нервы на шее отделялись, дважды лигировались и рассекались, и электроды вводились в каждый нерв (смотри ниже). Для блокирования изменений β -адренэргических воздействий на сердце вводили надолол с начальной дозой 0,5 мг/кг в/в с последующими 0,25 мг/кг в/в каждые два часа.
Модель фибрилляции предсердий
Оценивали действие лекарственных средств на прекращение длительной ФП, поддерживаемой в ходе непрерывной стимуляции блуждающего нерва. Монополярные электроды в виде крючка (нержавеющая сталь, изолированная тефлоном Teflon™ , покрытые кроме дистального участка длиной 1-2 см) вводили посредством иглы 21 калибра в каждый нерв и параллельно стволу каждого нерва. В большинстве экспериментов применяли монополярные стимулы, создаваемые с помощью стимулятора (модель DS-9F, Grass Instruments, Quincy, MA.), настроенного так, чтобы подавать 0,1 мс прямоугольные импульсы с частотой 10 Гц и при напряжении, составляющем 60% от того, которое необходимо для получения асистолии. В некоторых экспериментах использовалась биполярная стимуляция. Напряжение, необходимое для получения асистолии, находится в пределах между 3-20 вольтами. При контрольных условиях подавали короткий импульс навязывания быстрого ритма предсердию (10 Гц, в четыре раза выше диастолического порога) для индуцирования ФП, которая обычно продолжалась более чем 20 минут. Напряжение для стимуляции вагуса устанавливали в контрольных условиях и затем регулировали при каждой обработке, чтобы поддерживать один и тот же брадикардический эффект. ФП определялась как быстрая (>500 в минуту при контрольных условиях) при нерегулярном ритме сокращений предсердий с изменяющейся структурой электрограммы.
Измерение электрофизиологических параметров и ответа на стимуляцию вагуса
Диастолический пороговый ток определяли при продолжительности основного цикла 300 мс путем увеличения тока добавлением по 0,1 мА вплоть до получения стабильного захвата. Для последующих протоколов ток устанавливали на уровне, превышающем диастолический порог в два раза. ЭРП предсердий и желудочков измеряли с помощью метода экстрастимуляции в пределах S1S2 интервалов при продолжительности основного цикла 300 мс. Ранний экстрастимул S2 вводили через каждые 15 основных стимулов. Интервал S1S2 увеличивали, добавляя по 5 мс. вплоть до наступления захвата, самый продолжительный интервал S1S2 последовательно уменьшали, чтобы получить распространяющийся сигнал, определяющий ЭРП. Диастолический порог и ЭРП определяли повторно и усредняли для получения единого значения. Как правило, эти значения были в пределах 5 мс. Интервал между искусственным стимулом и пиком локальной электрограммы измеряли в качестве показателя скорости проводимости. Продолжительность цикла ФП (ПЦФП) измеряли во время вызванной стимуляцией вагуса ФП путем подсчета количества циклов (количество систол-1) в 2-секундном интервале в каждом участке предсердий, где проводилась регистрация. Три измерения ПЦФП усредняли для получения итогового значения ПЦФП для каждого экспериментального состояния.
В большинстве части экспериментов определяли взаимосвязь между напряжением стимула и частотой сердечных сокращений при стимуляции блуждающего нерва в контрольных условиях. Блуждающие нервы стимулировали, как описано выше, при различных значениях напряжения для определения напряжения, которое вызывало асистолию (определяемую как синусовая пауза продолжительностью более 3 секунд). При каждом экспериментальном условии убеждались в ответе на стимуляцию блуждающего нерва, и напряжение регулировали для поддержания скорости сердечных сокращений в ответ на стимуляцию блуждающего нерва постоянной. В тех случаях, когда было невозможно получить асистолию, стимуляцию блуждающего нерва доводили до напряжения, которое позволяло поддерживать два 20-минутных эпизода ФП, вызванной стимуляцией вагуса в контрольных условиях (смотри ниже).
Протоколы экспериментов
Изучаемые экспериментальные группы суммированы в таблице 6. Каждая собака получала только одно лекарственное средство в дозах, указанных в таблице 6. В первой серии экспериментов исследовался интервал доз с последующим слепым испытанием, в котором давали 1-3 дозы. Все лекарственные средства вводили в/в с помощью инфузионного насоса с растворами лекарственных средств, свежеприготовленными в пластиковых контейнерах в день эксперимента. Параметры стимуляции вагуса определяли в контрольных условиях, как описано выше, и проверяли поддержание ФП в течение 20 минут при стимуляции блуждающего нерва в контрольных условиях. После окончания ФП определяли диастолический порог и ЭРП предсердия и желудочка. Впоследствии эти параметры повторно оценивались в предсердии при стимуляции блуждающего нерва. Электрофизиологическое тестирование обычно занимало 15-20 минут. Убеждались в ответе на стимуляцию блуждающего нерва, выраженном в частоте сердечных сокращений, и протокол вызванной стимуляцией вагуса ФП/электрофизиологического тестирования повторяли. Получали образец крови перед введением препарата и повторно начинали вызванную стимуляцией вагуса ФП. Спустя пять минут, одно из средств обработки вводили в дозах, показанных в таблице 6. Всю дозу вливали за 5 минут и сразу после этого отбирали образец крови. Поддерживающей инфузии не проводили. Если ФП прекращалась в пределах 15 минутного периода времени, повторяли электрофизиологические измерения в контрольных условиях и отбирали образец крови. Если ФП не прекращалась при введении первой дозы (в течение 15 минут), отбирали образец крови и прекращали стимуляцию вагуса, чтобы дать возможность вернуться к синусовому ритму. Повторяли электрофизиологические измерения и отбирали третий и последний образец крови для этой дозы. ФП инициировали повторно и повторяли протокол вызванной стимуляцией вагуса ФП/инфузии препарата/электрофизиологического тестирования до тех пор, пока ФП не прекращалась при действии лекарственного средства.
Статистический анализ
Данные, полученные для группы, выражали в виде среднего ± стандартное отклонение. Статистический анализ проводили для эффективных доз для ПЦФП и ЭРП с помощью t-теста с поправкой Бонферроини для множественных сравнений. Действие лекарственного препарата на кровяное давление, частоту сердечных сокращений, диастолический порог и интервалы ЭКГ оценивали при срединной дозе, необходимой для прекращения ФП. Использовали два последних теста и брали значение р<0,05 для указания статистической значимости.
Экспериментальные группы и дозы лекарственных средств
1-10
Отдельное лекарственное средство вводили каждой собаке в пределах заданных доз вплоть до прекращения ФП. Показано количество собак, у которых ФП прекращалась при каждой дозе (количество собак - доза, в мкмоль/кг). Показаны средняя ± стандартное отклонение, а также срединная доза, необходимая для прекращения ФП. Каждая собака получала только одно лекарственное средство.
Данным способом оценивался ряд соединений изобретения. Результаты показывают, что все тестируемые соединения являются эффективными в качестве средств прекращения ФП в модели, вызванной стимуляцией вагуса ФП у собак. Степени перевода ритма сходны с показателями, о которых сообщалось для множества других лекарственных средств класса I и класса III в этой модели. Эффективность флекаинида, взятого в качестве контроля в данном исследовании, была сравнима с эффективностью, сообщавшейся ранее. Все лекарственные средства удлиняли ПЦФП перед прекращением ФП; действия, которые, в общем, согласуются с длиной волны в модели повторного входа импульса (ри-энтри) для прекращения ФП. Тестируемые соединения данного изобретения не снижали кровяное давление или частоту сердечных сокращений при срединной дозе, необходимой для прекращения вызванной стимуляцией вагуса ФП. Частота сердечных сокращений в ответ на стимуляцию блуждающего нерва была сходной во всех группах и не изменялась при действии каких-либо тестируемых соединений. Стимуляция блуждающего нерва при напряжении, составляющем 60% от напряжения, необходимого для получения асистолии (10±1 вольт), вызывала паузу продолжительностью 1,3±0,1 секунды.
Пример 32
Модель стерильного перикардита собак
Данная модель использовалась для характеристики механизмов ФП и трепетания предсердий (ТП). Waldo с коллегами обнаружили, что ФП зависит от циркуляции возбуждения и что местом окончания ФП обычно является область замедленной проводимости. Данная модель собак подготавливалась путем опыления открытого предсердия тальком с последующим “импульсным” навязыванием ритма предсердию в течение нескольких дней после восстановления. ФП способна индуцироваться через два дня после хирургической операции, но не позже четвертого дня после хирургической подготовки; преимущественным индуцируемым ритмом является выдерживаемое трепетание предсердия. Индуцируемость ФП на второй день несколько варьирует, так что только 50% собак может выдерживать ФП (обычно <60 минут) в течение требуемых 30 минут. Однако выдерживаемое трепетание предсердий, которое проявляется к четвертому дню, способно индуцироваться в большинстве случаев. Трепетание предсердий легче “картируется” с целью определения механизмов действия лекарственных средств. Индуцируемость ФП понижается после четвертого дня после хирургической операции аналогично ФП, которая часто развивается после хирургической операции на сердце, которую имитирует модель стерильного перикардита. В этиологию постхирургической ФП может быть вовлечен компонент воспаления, который будет давать степень селективности по отношению к ишемии или к селективному к кислоте лекарственному средству. Аналогично хотя операция артериально-коронарного шунтирования (CABG) проводится для облегчения состояния ишемии желудочков, у таких пациентов может также существовать риск возникновения легкой формы ишемии предсердия вследствие артериально-коронарной болезни (CAD). Хотя инфаркты предсердий являются редкими, существует взаимосвязь между стенозом артерии атриовентрикулярного узла и риском ФП после CABG хирургической операции. Нарушение автономной иннервации предсердия в результате хирургического вмешательства может также играть некоторую роль в ФП после создания CABG.
Методы
Исследования на модели стерильного перикардита собак проводились с целью определения потенциальной возможности и эффективности соединения 1 по прекращению фибрилляции/трепетания предсердия. Трепетание предсердий или фибрилляцию индуцировали через 2-4 дня после создания стерильного перикардита у взрослых беспородных собак весом от 19 кг до 25 кг. Во всех случаях фибрилляция или трепетание предсердий длились дольше 10 минут. Все исследования выполняли в соответствии с руководствами, установленными нашим Институциональным комитетом по защите и использованию животных (Institutional Animal Care and Use Committee), политикой Американской ассоциации кардиологов по использованию животных в научных исследованиях (the American Heart Association Policy on Research Animal Use) и политикой Службы здравоохранения по применению лабораторных животных (the Public Health Service Policy on Use of Laboratory Animals).
Создание модели фибрилляции/трепетания предсердий при стерильном перикардите
Модель стерильного перикардита собак создавалась, как описано ранее. Во время хирургической операции в ушко правого предсердия, пучок Бахмана и задненижнюю часть левого предсердия вблизи проксимальной части венечной пазухи вшивались пары проволочных электродов из нержавеющей стали, покрытых за исключением кончика полимером FEP (эластомерный сополимер тетрафторэтилена и гексафторпропилена) (О Flexon, Davis and Geсk). Расстояние между каждым электродом в каждой паре составляло примерно 5 мм. Эти проволочные электроды выводились через грудную стенку наружу сзади в межлопаточной области для последующего применения. По окончании хирургической операции собакам давали антибиотики и анальгетики и затем давали возможность восстановиться. Послеоперационный уход включал введение антибиотиков и анальгетиков.
У всех собак, начиная со 2 дня после операции, предпринимались попытки индукции стабильной фибрилляции/трепетания предсердий в состоянии сознания без применения седативных средств, чтобы убедиться в индуцируемости и стабильности фибрилляции/трепетания предсердий, для тестирования эффективности лекарственных средств. Стимуляцию предсердий выполняли с помощью электродов, вшитых во время начальной хирургической операции. На 4 день после операции, когда индуцировалось стабильное трепетание предсердия, выполняли исследование при открытой грудной клетке.
Для исследования на открытой грудной клетке каждую собаку анестезировали пентобарбиталом (30 мг/кг в/в) и проводили механическую вентиляцию легких 100% кислородом с помощью наркозного аппарата Бойля модели 50 (Harris-Lake, Inc.). Температуру тела каждой собаки в течение исследования поддерживали в нормальных физиологических пределах с помощью согревающего одеяла. У анестезированных собак перед тем, как вскрывалась грудная клетка, проводили высокочастотное отсечение пучка Гиса для создания полного атриовентрикулярного (АВ) блока с помощью стандартного электродно-катетерного способа. Это делалось для минимизации наложения комплексов предсердия и желудочка во время последующей регистрации монополярных электрограмм предсердия после индукции трепетания предсердий. После того как был создан полный АВ-блок, эффективная частота сокращения желудочков поддерживалась стимуляцией желудочков со скоростью от 60 до 80 ударов в минуту с помощью генератора импульсов Medtronic 5375 Pulse Generator (Medtronic Inc.), подающего стимулы через электроды, введенные в правый желудочек во время начальной хирургической операции.
Определение порогов стимулов и рефракторных периодов во время электростимуляции
Для индукции ФП/ТП применяли один из двух ранее описанных способов: (1) введение одной или двух ранних экстрасистол предсердий после ряда 8 стимулированных систол предсердия при длине цикла 400 мс, 300 мс, 200 мс или 150 мс или (2) быстрая электростимуляция предсердия в течение периодов от 1 до 10 сек, при наращивании частоты, чтобы она превышала спонтанный синусовый ритм на 10-50 ударов в минуту до тех пор, пока не индуцировалось трепетание предсердия или не наблюдалась потеря 1:1 распространения ретроградного импульса на предсердия. Стимуляцию предсердий выполняли либо с помощью электродов в ушке правого предсердия, либо электродов в задненижней части левого предсердия. Все процедуры стимулирования проводили, используя стимулы, в два раза превышающие порог для каждого основного навязываемого ритма, с помощью модифицированного программируемого стимулятора Medtronic 5325, питающегося от батареек, используя импульс шириной 1,8 мс.
После индукции стабильной фибрилляции/трепетания предсердий (продолжительностью более 10 минут) измеряли продолжительность цикла фибрилляции/трепетания предсердий и проводили начальное картирование и анализ для определения локализации циркуляции повторного возбуждения при фибрилляции/трепетании предсердия. Трепетание предсердия определяли как быстрый ритм предсердий (частота >240 ударов в минуту), который характеризовался постоянной продолжительностью цикла между ударами, полярностью, морфологией и амплитудой регистрируемых биполярных электрограмм.
Протокол тестирования эффективности лекарственных средств
1. Эффективные рефракторные периоды (ЭРП) измеряли в трех местах: ушко правого предсердия (УПП), задняя часть левого предсердия (ЗЛП), пучок Бахмана (ПБ), при двух продолжительностях основного цикла 200 и 400 мс.
2. Индукция ФП или ТП стимуляцией. Эти попытки проводили в течение одного часа. Если не происходило никакой индукции аритмии, дальнейшие исследования в этот день не проводили.
3. Если индукция удавалась, ФП должна была продолжаться в течение 10 минут. Затем предоставляли период ожидания для спонтанного прекращения или 20 минут, даже если прекращение наступало раньше.
4. Затем ФП индуцировали повторно и предоставляли 5 минут перед началом инфузии лекарственного средства.
5. Затем проводили инфузию лекарственного средства в виде болюсов в течение 5 минут.
6. Если ФП прекращалась с введением первой дозы, то отбирали образец крови и повторяли измерения ЭРП.
7. Пять минут предоставляли для прекращения ФП веществом. Если не происходило прекращения, то в течение 5 минут давали вторую дозу.
8. После прекращения и измерения ЭРП предпринимали повторную попытку индуцировать ФП в течение десятиминутного периода.
9. Если повторная индукция удавалась и продолжалась в течение 10 минут, отбирали образец крови и исследование повторяли, начиная с пункта №3, указанного выше.
10. Если повторная индукция не удавалась, то исследование заканчивали.
Пример 33
Определение блокирования болевых ощущений
Морских свинок брили (только спину) и 6 аликвот (50 мкл) раствора соединения (10 мг/мл) инъецировали непосредственно под кожу, чтобы образовались 6 волдырей, которые очерчивали стойким маркером. Болевые ответы оценивали, как указано выше, в каждом волдыре в регулярные промежутки времени вплоть до 4 часов после инъекции и регистрировали продолжительность блокирования болевых ощущений у трех животных для каждого тестируемого раствора.
Этим способом оценивали ряд соединений данного изобретения. Результаты показали, что все тестируемые соединения эффективны в эпизодах прекращения фибрилляции/трепетания предсердий на этой модели. Во время обработки лекарственным средством не наблюдалось проаритмии или побочных сердечно-сосудистых нарушений.
Все публикации и патентные заявки, упомянутые в данном описании, включены в него для сведения, как если бы каждая отдельная публикация или заявка на патент была конкретно и отдельно включена в описание.
Из вышесказанного должно быть ясно, что хотя здесь в целях иллюстрации описаны конкретные варианты изобретения, могут быть осуществлены различные модификации без отступления от сущности и, не выходя за рамки изобретения. Соответственно изобретение не ограничивается ничем, кроме прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ПРОСТОГО АМИНОЦИКЛОГЕКСИЛОВОГО ЭФИРА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2008 |
|
RU2478617C2 |
| СОЕДИНЕНИЯ ПРОСТОГО АМИНОЦИКЛОГЕКСИЛОВОГО ЭФИРА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2330017C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТАНА | 2010 |
|
RU2572555C2 |
| 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1996 |
|
RU2178409C2 |
| ЦИКЛИЧЕСКОЕ УГЛЕВОДОРОДНОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2676328C2 |
| ГИДРОКСИЭТИЛАМИНОСУЛЬФОНАМИДЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ РЕТРОВИРУСНЫХ ПРОТЕАЗ, СПОСОБ ЛЕЧЕНИЯ РЕТРОВИРУСНЫХ ИНФЕКЦИЙ, СПОСОБ ЛЕЧЕНИЯ СПИДА | 1993 |
|
RU2173680C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| 2,3-ТРАНСДИЗАМЕЩЕННОЕ ТРОПАНОВОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167876C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
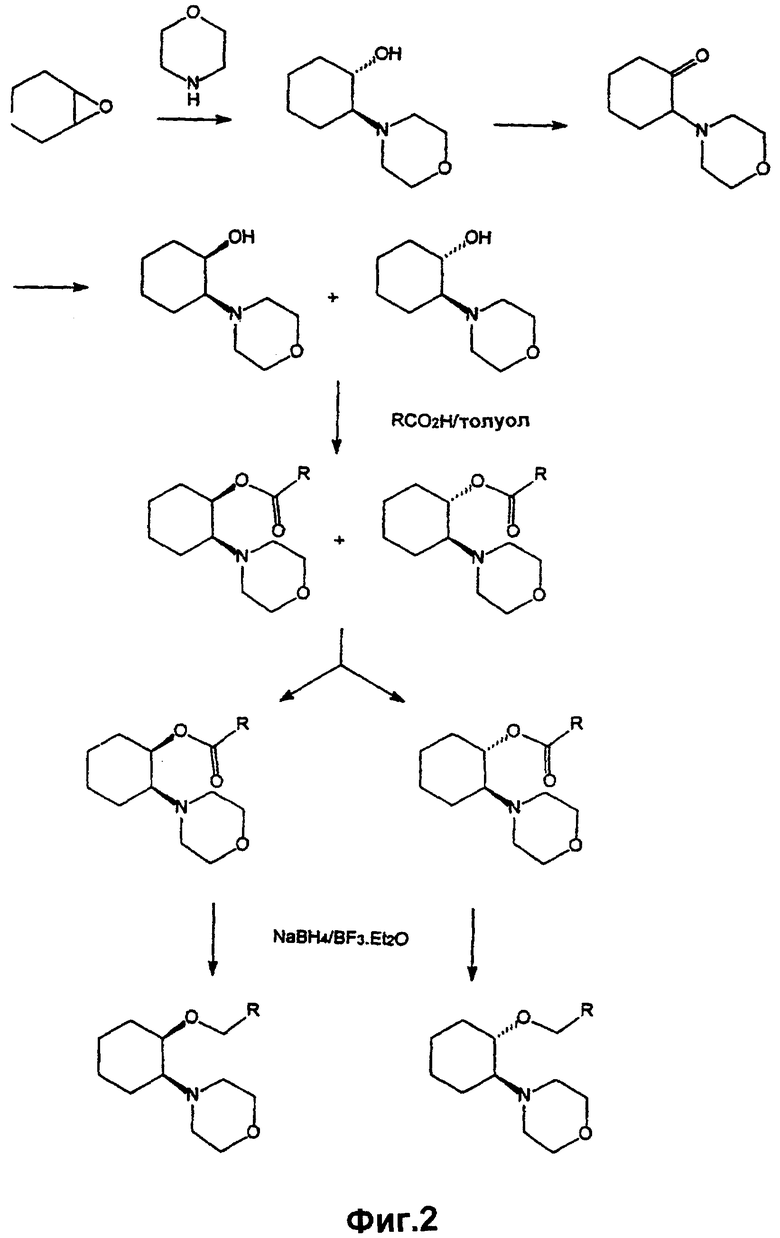
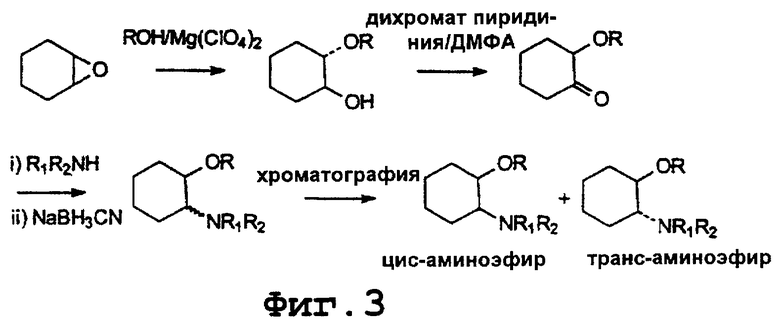
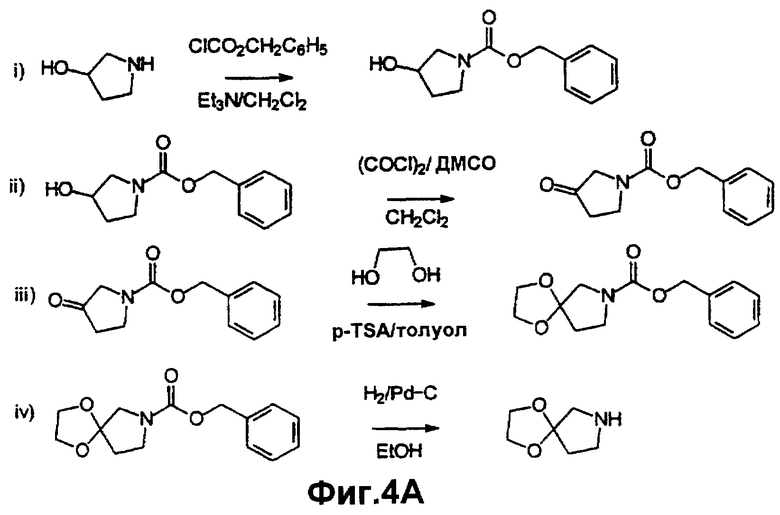
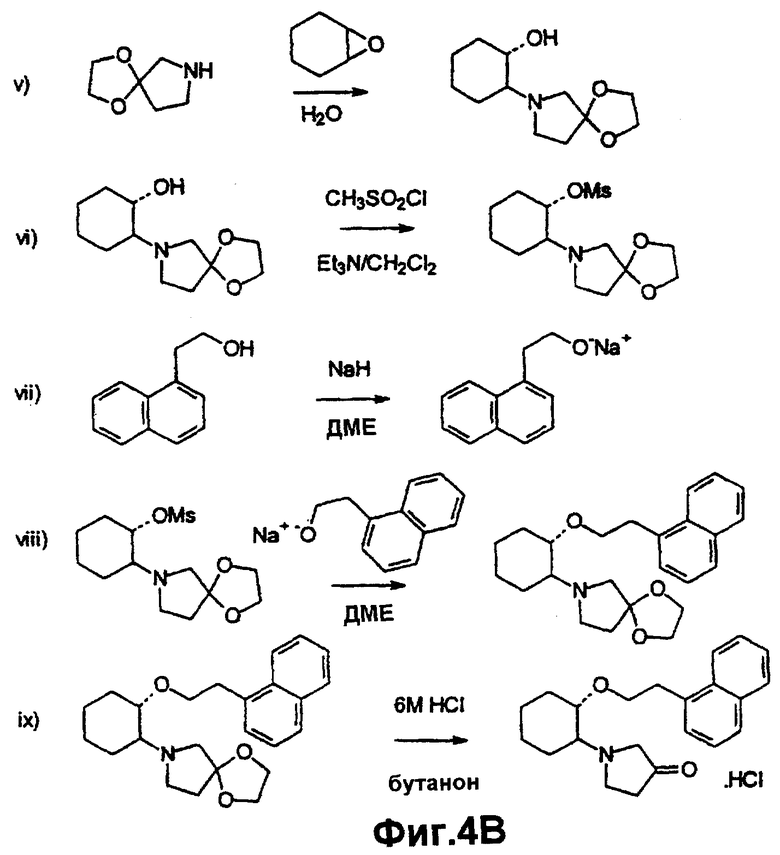
Изобретение относится к новым соединениям формулы (I), или их сольватам и фармацевтически приемлемым солям, которые обладают антиаритмической активностью, включающей аритмию предсердий, желудочковую аритмию, фибрилляцию предсердий и фибрилляцию желудочков, а также фармацевтическим композициям на их основе. Соединения могут быть использованы для лечения или предотвращения аритмии, при модулировании активности ионных каналов и при лечении или предотвращении заболеваний, которые отвечают на модулирование активности ионных каналов, для проведения местной или локальной анестезии, для усиления либидо. В общей формуле (I)
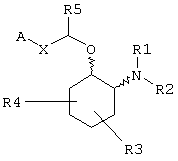
Х независимо в каждом случае выбран из прямой связи, -С(R6,R14)-Y- и -C(R13)=CH-; Y выбран из прямой связи, О, S и С1-С4алкилена; R13 выбран из водорода, C1-С6алкила, С3-С8циклоалкила, незамещенного арила и бензила; или R1 и R2 независимо выбраны из С3-С8 алкоксиалкила, С1-С8гидроксиалкила и С7-С12аралкила; или R1 и R2, взятые вместе с атомом азота, с которым они непосредственно связаны в формуле (I), образуют кольцо формулы (II):
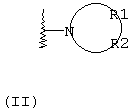
где кольцо формулы (II) образовано азотом, как показано, а также дополнительными тремя-девятью кольцевыми атомами, независимо выбранными из углерода, азота, кислорода и серы; и др.; R3 и R4 независимо связаны с кольцом циклогексана, показанным в формуле (I) в положениях 3-, 4-, 5- или 6- и независимо выбраны из водорода, гидрокси, C1-С6алкила и C1-С6алкокси, и когда и R3, и R4 связаны с одним и тем же атомом циклогексанового кольца, они вместе могут образовывать пяти– или шестичленное спирогетероциклическое кольцо, содержащее один или два гетероатома, выбранных из кислорода и серы; А выбран из С5-С12алкила, С3-С13карбоциклического кольца, и кольцевых систем формул, указанных в формуле изобретения. 13 н. и 17 з.п. ф-лы, 5 ил, 7 табл.

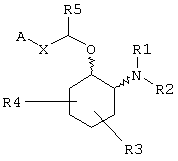
где независимо в каждом случае Х выбран из прямой связи, -С(R6, R14)-Y- и -C(R13)=CH-;
Y выбран из прямой связи, О, S и С1-С4алкилена;
R13 выбран из водорода, C1-С6алкила,С3-С8циклоалкила, незамещенного арила и бензила;
R1 и R2 независимо выбраны из С3-С8 алкоксиалкила, С1-С8гидроксиалкила и С7-С12аралкила; или
R1 и R2, взятые вместе с атомом азота, с которым они непосредственно связаны в формуле (I), образуют кольцо, обозначаемое формулой (II):
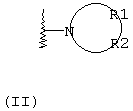
где кольцо формулы (II) образовано азотом, как показано, а также дополнительными тремя-девятью кольцевыми атомами, независимо выбранными из углерода, азота, кислорода и серы; где любые два смежных атома кольца могут быть связаны одинарной или двойной связями, и любой один или более дополнительных атомов углерода в кольце могут быть замещены одним или двумя заместителями, выбранными из водорода, гидрокси, C1-С3 гидроксиалкила, оксо, C2-С4ацила, C1-С3 алкила, C2-С4алкилкарбокси, C1-С3алкокси, C1-С20алканоилокси, или могут быть замещены с образованием пяти- или шестичленного спирогетероциклического кольца, содержащего один или два гетероатома, выбранных из кислорода и серы, и любые два смежных дополнительных атома углерода кольца могут быть сконденсированы с C3-С8карбоциклическим кольцом, любой один или более дополнительных атомов азота кольца могут быть замещены заместителями, выбранными из водорода, C1-С6алкила, C2-С4ацила, C2-С4гидроксиалкила и C3-С8алкоксиалкила; или
R1 и R2, взятые вместе с атомом азота, с которым они непосредственно связаны в формуле (I), могут образовывать бициклическую кольцевую систему, выбранную из следующих: 3-азабицикло[3.2.2]нонан-3-ил, 2-азабицикло[2.2.2]октан-2-ил, 3-азабицикло[3.1.0]гексан-3-ил и 3-азабицикло[3.2.0]гептан-3-ил;
R3 и R4 независимо связаны с кольцом циклогексана, показанным в формуле (I) в положениях 3-, 4-, 5- или 6-, и независимо выбраны из водорода, гидрокси, C1-С6алкила и C1-С6алкокси и, когда R3 и R4 связаны с одним и тем же атомом циклогексанового кольца, они вместе могут образовывать пяти- или шестичленное спирогетероциклическое кольцо, содержащее один или два гетероатома, выбранных из кислорода и серы;
R5, R6 и R14 независимо выбраны из водорода, C1-С6алкила, арила и бензила, или R6 и R14, взятые вместе с углеродом, с которым они связаны, могут образовывать спироC3-С5циклоалкил;
А выбран из С5-С12алкила, С3-С13карбоциклического кольца и кольцевых систем, выбранных из формул (III), (IV), (V), (VI), (VII) и (VIII):
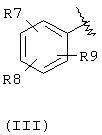
где R7, R8 и R9 независимо выбраны из водорода, брома, хлора, фтора, карбокси, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-С6 тиоалкила, арила и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6 алкила;

где R10 и R11 независимо выбраны из водорода, брома, хлора, фтора, карбокси, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, C1-С6тиоалкила и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и C1-С6алкила;
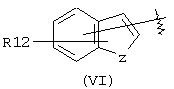
где R12 выбран из водорода, брома, хлора, фтора, карбокси, гидрокси, гидроксиметила, метансульфонамидо, нитро, сульфамила, трифторметила, С2-С7алканоилокси, C1-С6алкила, C1-С6алкокси, С2-С7алкоксикарбонила, С1-С6тиоалкила и N(R15,R16), где R15 и R16 независимо выбраны из водорода, ацетила, метансульфонила и С1-С6алкила; Z выбран из СН, СН2, О, N и S, где Z может быть непосредственно связан с Х, как показано в формуле (I), когда Z является СН или N, или Z может быть непосредственно связан с R17, когда Z является N, и R17 выбран из водорода, С1-С6алкила, С3-С8 циклоалкила, незамещенного арила и бензила;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры и их смеси.
где независимо в каждом случае
Х выбран из прямой связи, -СН=СН- и -С(R6,R14)-Y-;
Y выбран из прямой связи, О и S;
R1, R2, R3, R4, R6, R7, R8, R9, R10, R11, R12, R14, А и Z имеют значения, определенные в п.1;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры и их смеси.
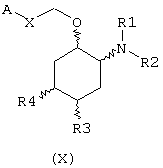
где независимо в каждом случае
Х выбран из прямой связи, -СН=СН- и -С(R6,R14)-Y-;
Y выбран из прямой связи, О и S;
R1, R2, R6 и R14 имеют значения, как в п.1;
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и C1-C6алкокси;
А выбран из С5-С12алкила, С3-С8циклоалкила и любой из формул (III),(IV),(V) и (VI), определенных в п.1, где Z, R7, R8, R9, R10, R11 и R12 имеют значения, определенные в п.1;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры и их смеси,
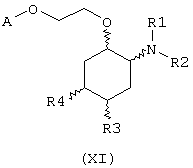
где независимо в каждом случае
R1 и R2 имеют значения, определенные в п.1;
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и метокси;
А выбран из С5-С12алкила, С3-С8циклоалкила и любой из формул (III), (IV), (V) и (VI), определенных в п.1, где Z, R7, R8, R9, R10, R11 и R12 имеют значения, определенные в п.1; включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.
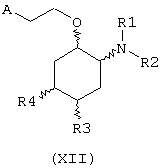
где независимо в каждом случае
R1 и R2 имеют значения, определенные в п.1;
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и метокси;
А выбран из С5-С12алкила, С3-С8циклоалкила и любой из формул (III), (IV), (V) и (VI), охарактеризованных в п.1, где Z, R7, R8, R9, R10, R11 и R12 имеют значения, определенные в п.1;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.
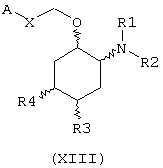
где независимо в каждом случае
Х выбран из прямой связи и -СН=СН-;
R1 и R2 имеют значения, определенные в п.1;
R3 и R4 независимо связаны с циклогексановым кольцом в положениях 4 или 5 и независимо выбраны из водорода и метокси;
А выбран из С3-С8циклоалкила и любой из формул (III), (IV), (V), (VI), (VII) и (VIII), охарактеризованных в п.1, где R8 и R9 имеют значения, определенные в п.1, R7, R10, R11 и R12 являются водородом, Z выбран из O,S и N-R17, где R17 выбран из водорода и метила; при условии, что А может быть выбран из формул (VII) и (VIII) только, когда Х представляет прямую связь;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.

где независимо в каждом случае R1 и R2 имеют значения, определенные в п.1; А выбран из любой из формул (III), (IV), (V) и (VI), охарактеризованных в п.1, где R7, R10, R11 и R12 представляют водород, R8 и R9 независимо выбраны из водорода, гидрокси, фтора, хлора, брома, метансульфонамидо, метаноилокси, метоксикарбонила, нитро, сульфамила, тиометила, трифторметила, метила, этила, метокси, этокси и NH2, при условии, что по меньшей мере один из R8 и R9 не является водородом; Z выбран из О и S;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.
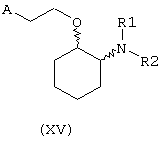
где независимо в каждом случае
R1 и R2 имеют значения, определенные в п.1;
А выбран из любой из формул (III), (IV), (V) и (VI), охарактеризованных в п.1, где R7, R10, R11 и R12 представляют водород, R8 и R9 независимо выбраны из водорода, гидрокси, фтора, хлора, брома, метансульфонамидо, метаноилокси, метоксикарбонила, нитро, сульфамила, тиометила, трифторметила, метила, этила, метокси, этокси и NH2, при условии, что по меньшей мере один из радикалов R8 и R9 не является водородом; Z выбран из О и S;
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.
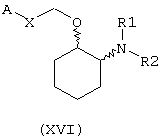
где независимо в каждом случае Х выбран из прямой связи, транс-СН=СН-, -СН2- и -СН2-О-;
R1 и R2 оба являются метоксиэтилом или, взятые вместе с атомом азота, с которым они связаны, образуют кольцо, выбранное из пирролидинила, кетопирролидинила, ацетоксипирролидинила, гидроксипирролидинила, тиазолидинила, пиперидинила, кетопиперидинила, ацетилпиперазинила, 1,4-диокса-7-азаспиро[4.4]нон-7-ила, гексагидроазепинила, морфолинила, N-метилпиперазинила и 3-азабицикло[3.2.2]нонанила; А выбран из циклогексила, монохлорфенила, 2,6-дихлорфенила, 3,4-дихлорфенила, 2-бромфенила, 2,4-дибромфенила, 3-бромфенила, 4-бромфенила, 1-нафтила, 2-нафтила, 3-бензо-(b)-тиофенила, 4-бензо(b)тиофенила,(2-трифторметил)фенила, 2,4-ди(трифторметил)фенила и (4-трифторметил)фенила,
включая их отдельные энантиомерные, диастереомерные и геометрические изомеры, и их смеси.
(+)-транс-[2-(4-морфолинил)-1-(2-нафтенэтокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(2-нафтенэтокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(1-нафтенэтокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(1-нафтенэтокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(4-бромфенэтокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(4-бромфенэтокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-[2-(2-нафтокси)этокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-[2-(2-нафтокси)этокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-[2-(4-бромфенокси)этокси]]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-[2-(4-бромфенокси)этокси]]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(3,4-диметоксифенэтокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(3,4-диметоксифенэтокси)]циклогексан;
(+)-транс-[2-(1-пирролидинил)-1-(1-нафтенэтокси)]циклогексан;
(-)-транс-[2-(1-пирролидинил)-1-(1-нафтенэтокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(2-(бензо[b]тиофен-3-ил)-этокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(2-(бензо[b]тиофен-3-ил)-этокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(2-(бензо[b]тиофен-4-ил)-этокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(2-(бензо[b]тиофен-4-ил)этокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(3-бромфенэтокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(3-бромфенэтокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(2-бромфенэтокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(2-бромфенэтокси)]циклогексан;
(+)-транс-[2-(4-морфолинил)-1-(3-(3,4-диметоксифенил)-1-пропокси)]циклогексан;
(-)-транс-[2-(4-морфолинил)-1-(3-(3,4-диметоксифенил)-1-пропокси)]циклогексан;
(+)-транс-[2-[бис(2-метоксиэтил)аминил]-1-(2-нафтенэтокси)]-циклогексан;
(-)-транс-[2-[бис(2-метоксиэтил)аминил]-1-(2-нафтенэтокси)]-циклогексан;
(1R,2R)/(1S,2S)-2-(4-морфолинил)-1-(3,4-дихлорфенэтокси)циклогексан;
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексан;
(1R,2R)/(1S,2S)-2-(1-ацетилпиперазинил)-1-(2-нафтенэтокси)-циклогексан;
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,6-дихлорфенэтокси)циклогексан;
(1R,2R)/(1S,2S)-2-[1,4-диокса-7-азаспиро[4.4]нон-7-ил]-1-(1-нафтенэтокси)циклогексан;
(1R,2S)/(1S,2R)-2-(4-морфолинил)-1-[(2-трифторметил)фенэтокси]циклогексан;
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[3-(циклогексил)пропокси]циклогексан;
(1R,2R)/(1S,2S)-2-(3-ацетоксипирролидинил)-1-(1-нафтенэтокси)циклогексан;
(1R,2R)/(1S,2S)-2-(4-морфолинил)-1-[(2,6-дихлорфенил)метокси]циклогексан;
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-[(2,6-дихлорфенил)-метокси]циклогексан;
(1R,2R)/(1S,2S)-2-(3-гидроксипирролидинил)-1-(2,6-дихлорфенэтокси)циклогексан;
(1R,2R)/(1S,2S)-2-(3-кетопирролидинил)-1-(2,2-дифенилэтокси)-циклогексан;
(1R,2R)/(1S,2S)-2-(3-тиазолидинил)-1-(2,6-дихлорфенэтокси)циклогексан;
(1R,2S)/(1S,2R)-2-(3-кетопирролидинил)-1-(1-нафтенэтокси)циклогексан;
и их фармацевтически приемлемые соли.
Приоритеты:
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА ИЛИ ТЕТРАГИДРОПИРАНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ГИДРАТЫ, ИЛИ СОЛЬВАТЫ ЭТИХ СОЕДИНЕНИЙ, ИЛИ ИХ СОЛЕЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2084439C1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

