Изобретение относится к фармацевтической промышленности и касается способа получения антибиотика карминомицина или его гидрохлорида.
Карминомицин (формула 1, R=R1=R2=Н) относится к группе антрациклиновых антибиотиков, зарекомендовавших себя как высокоэффективные противоопухолевые и противолейкозные препараты, широко используемые в клиниках РФ и за рубежом. Антибиотик карминомицин является оригинальным отечественным антибиотиком, который был впервые получен путем биосинтеза в Институте по изысканию антибиотиков АМН СССР. Карминомицин успешно прошел клинические испытания, и был разрешен к применению в СССР и РФ. Карминомицин рекомендован при следующих заболеваниях: саркома мягких тканей (лейкомиосаркома, рабдомиосаркома, липосаркома и др.); ретикулосаркома и лимфосаркома; рак молочной железы и хорионэпителиома матки; острые миелобластные и лимфобластные лейкозы. Антибиотик имеет широкие показания при лечении злокачественных новообразований у взрослых и детей и применяется как индивидуальный препарат, так и в комбинациях с другими лекарственными средствами.
Известен способ биосинтетического получения карминомицина [а.с. "Способ получения карминомицина" №508076 от 11.11.1975 г, а.с. "Способ получения карминомицина" №543257 от 21.09.1976 г.]. Недостатком этого метода является присутствие сопутствующей примеси - 13-дигидрокарминомицина, низкая продуктивность штамма-продуцента Actinomadura carminata по сравнению с продуктивностью штаммов противоопухолевых антибиотиков антрациклиновой группы, сложность процесса выделения и очистки.
Известен способ получения карминомицина химическим синтезом [Cassinelli G., Grein A., Masi P., Suarato A., Bemardi L., Arcamone F.Н. Preparation and biological evaluation of 4-0-demethyldaunorubicin (carminomycin I) and its 13-dihydro derivative -Journal of Antibiotics, 31, 178-184, 1978]. Гидролизом гликозидной связи даунорубицина получают агликон дауномицинон (формула 2, R=СН3) и моносахарид - даунозамин (формула 3, R1=R2=R3=H). Дауномицинон подвергают деметилированию реакцией с AlClз в кипящем хлористом метилене. Полученный с выходом 62% агликон карминомицинон (формула 2, R=H) гликозилируют 2,3,6-тридезокси-3-трифторацетамидо-4-O-трифторацетил-α-L-лмксо-гексапиранозил хлоридом в присутствии трифторметансульфоната серебра в смеси хлористый метилен-диметилформамид. Защищенный гликозид деблокируют 0.13н NaOH, получая карминомицин гидрохлорид с выходом 10% в расчете на дауномицинон. Недостатком описанного химического синтеза является низкий выход - 10% в расчете на дауномицинон, необходимость получения чистого аномера 2,3,6-тридезокси-3-трифторацетамидо-4-O-трифторацетил-α-L-ликсо-гексапиранозил хлорида и его нестабильность.
Техническая задача изобретения - увеличение выхода карминомицина при получении его путем химического синтеза.
Техническая задача решается тем, что предложен способ получения карминомицина или его гидрохлорида, включающий гликозилирование карминомицинона гликозил-донором 1,4-ди-O-замещенным-3-N-ацилдаунозамином в присутствии катализатора в смеси безводных апротонных органических растворителей, удаление блокирующих групп и выделение карминомицина в виде основания или гидрохлорида, при этом в качестве катализатора используется триметилсилилтрифторметансульфонат, в качестве гликозилдонора используется 1,4-ди-O-замещенный-3-N-ацилдаунозамин (2,3,6-тридезокси-3-амино-L-ликсо-гексапираноза),

где R1=трифторацетил или N-(9-Н-флуорен-2-илметокси)карбонил или аллилоксикарбонил, преимущественно N-(9-Н-флуорен-2-илметокси)карбонил или трифторацетил, R2=ацетил или трифторацетил или 4-нитробензоил или аллилоксикарбонил, преимущественно ацетил, R3=трихлорацетамидил или триалкилсилил или ацетил или трифторацетил или 4-нитробензоил или аллилоксикарбонил, преимущественно ацетил, в качестве безводных апротонных органических растворителей используют диоксан-метиленхлорид или диоксан-хлороформ или диоксан-метиленхлорид-диэтиловый эфир или диоксан-ацетон или диоксан-хлороформ-диэтиловый эфир или диоксан-тетрагидрофуран-метиленхлорид, преимущественно диоксан-метиленхлорид, при этом реакция проводится в присутствии молекулярных сит (преимущественно 4Å), удаление блокирующих групп проводят щелочным агентом.
В качестве гликозил-донора используют смесь α и β изомеров или индивидуальные α или β изомеры. Реакцию проводят при пониженной температуре, преимущественно не выше 10°С. Реакцию проводят в атмосфере осушенного воздуха или осушенного инертного газа. Удаление защитных групп проводят в присутствии гидроксидов щелочных металлов в водно-органической среде: вода - метанол или вода - диоксан или вода - тетрагидрофуран или вода - ацетон.
Для защиты 3-амино и 4-O-гидроксильной групп даунозамина используют сочетание блокирующих групп, устойчивых в слабокислых средах. Введение защитных и активирующих групп проводили по стандартным методикам, используемых в химии углеводов [H.M.I.OSBORN, Best Synthetic methods, Carbohydrates, Academic press, 2003; Handbook of Reagents for Organic Synthesis "Activating Agents and Protective Groups", edited by A.J.Pearson, W.J. Roush, JOHN WILEY & SONS, 1999]. Частный случай в виде использования 1,4-ди-O-замещенного-N-ацилдаунозамина, содержащего в положениях 1-O- и 4-O- одну и ту же группу ацильного типа (формула 3, где R2=R3=ацильная группа) преимущественно ацетильную, позволяет упростить синтез гликозил-донора. Используя эти соединения (формула 3, где R2=R3=ацильная группа), также возможна замена активирующей ацильной группы R3 на другую активирующую группу путем ее селективного гидролиза до гидроксильной группы (с сохранением защитных групп R1, R2), с последующим введением другой активирующей группы (R3) [H.M.I.OSBORN, Best Synthetic methods. Carbohydrates, Academic press, 2003, p.164].
Предлагаемый способ позволяет увеличить выход карминомицина в 3 раза по сравнению с прототипом.
Вспомогательные средства:
Триметилсилилтрифторметансульфонат, диэтиламин, молекулярные сита 4Å, триэтиламин, 4-(диметиламино)пиридин были коммерческим продуктами фирмы Aldrich (США). N-(9Н-Флуорен-2-илметоксикарбонил)сукцинимид был коммерческим продуктом фирмы Merck (Германия).
Тонкослойную хроматографию осуществляли на пластинках с силикагелем G60 (Merck) в смесях растворителей: петролейный эфир-этилацетат, 1:1 (А); хлороформ-метанол-муравьиная кислота, 20:1:0.1 (Б); хлороформ-метанол-25%-ный водный аммиак, 7:3:0.01 (В); хлороформ-метанол-муравьиная кислота, 7:1:0.1 (Г). Для препаративной очистки использовали колоночную хроматографию на силикагеле Merck G60 с размером частиц 0.040-0.063 mm.
ВЭЖХ проводили на приборе Shimadzu HPLC LC 10 на колонке Diaspher С 18 №1914, элюент: 0.01н Н3PO4 - MeCN, pH 2.6, в градиенте ацетонитрила от 10% до 90%, скорость потока 1.1 мл/мин. Регистрация велась на длине волны 254 нм, при температуре 20°С.
Оптическое вращение [α]D изучали на поляриметре "Perkin - Elmer-241".
Пример 1.
Карминомицин гидрохлорид.
Карминомицинон (0.500 г, 1.30 ммоль) растворяли в смеси сухой хлористый метилен - сухой диоксан (4:1, 375 мл), добавляли 1,4-ди-O-ацетил-3-N-(9-Н-флуорен-2-илметокси)карбонилдаунозамин (α:β=4:1) (0.706 г, 1.56 ммоль) и молекулярные сита (10 г). Раствор помешивали в токе аргона, охлаждали до -10°С и прибавляли по каплям раствор триметилсилилтрифторметансульфоната (0.35 мл, 1.95 ммоль) в сухом хлористом метилене (10 мл). Реакционную смесь перемешивали при температуре не выше 10°С в течение 40 мин, затем выливали в насыщенный раствор NaHCO3 (150 мл). Органический слой отделяли, промывали водой до рН6, высушивали и упаривали. Остаток растворяли в диоксане (20 мл), приливали 0.2 н NaOH (20 мл). Реакционную смесь перемешивали в течение 3 часов при 4°С в атмосфере аргона, затем подкисляли до рН 5 0.5н HCl. Реакционную смесь разбавляли Н2O и экстрагировали хлороформом агликон (5×70 мл), затем значение рН водной фракции доводили до 7.5 5%-ным раствором К2СО3 и экстрагировали хлороформом основание карминомицина (4×50 мл). Органические фракции упаривали до минимального объема. Раствор подкисляли до рН 3.5 0.5н раствором HCl в сухом МеОН и добавляли эфир (30 мл). Выпавший осадок отфильтровывали и высушивали.
Выход 250 мг (35% в расчете на карминомицинон), Rf(Г) 0.22, Rt 16.20.
Тпл 185-187°С (с разложением)
1Н-ЯМР (Py-d5): 7.835 (d, 1Н, 7.33 Hz, H-1); 7.62 (t, 1H, 7.81 Hz, H-2); 7.325 (d, 1H, 8.30 Hz, H-3); 5.82 (s, 1H, W˜7 Hz, Н-1'); 5.35 (m, 1H, H-7); 4.70 (q, 1H, 6.65 Hz, H-5'); 4..50 (s, 1H, W˜7 Hz, H-4'); 4.39 (m, 1H, H-3'); 3.60d, 3.42d (2H, 17 Hz, H-10), 2.75 (m, 1H, H-2'); 2.75 (m, 2H, 2H-8); 2.57 (s, 3Н, 3Н-14); 2.80m, 2.45dd, (2H, Jgem 14 Hz, 4.3Hz, 2H-H-2'); 1.43(d, 3H, 6.65 Hz, 3H-6')
λmax 236, 256, 293, 464, 478, 492, 512, 525 нм (в этаноле)
[α]D+289° (с 0.15 МеОН)
Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [α]D) и спектральным характеристикам (УФ-, ЯМР-спектры) не отличается от стандартного образца, полученного путем биосинтеза на опытной установке ГУ НИИНА им. Г.Ф.Гаузе РАМН.
Пример 2.
Карминомицин гидрохлорид.
Карминомицинон (0.520 г, 1.35 ммоль) растворяли в смеси сухой хлористый метилен - сухой диоксан (4:1, 600 мл), добавляли 1,4-ди-O-ацетил-3-N-трифторацетилдаунозамин (α:β=1:2) (0.531 г, 1.62 ммоль) и молекулярные сита (12 г). Раствор смесь перемешивали в токе аргона, охлаждали до -10°С и прибавляли по каплям раствор триметилсилилтрифторметансульфоната (0.24 мл, 1.35 ммоль) в сухом хлористом метилене (10 мл). Реакционную смесь перемешивали при температуре не выше 10°С в течение 40 мин, затем выливали в насыщенный раствор NaHCO3 (150 мл). Органический слой отделяли, промывали водой до рН 6, высушивали и упаривали. Остаток растворяли в смеси диоксан - МеОН (30 мл, 2:1), охлаждали до 4°С и приливали 0.4 н NaOH (10 мл). Реакционную смесь перемешивали в течение 30 часов при 4°С атмосфере аргона, затем подкисляли до рН 5 0.5н HCl. Реакционную смесь разбавляли H2O и экстрагировали хлороформом (2×40 мл). Значение рН водной фракции доводили до 7.5 5%-ным раствором К2СО3 и экстрагировали хлороформом (4×30 мл). Органические фракции объединяли, высушивали и упаривали до минимального объема. Раствор подкисляли до рН 3.5 0.5н раствором HCl в сухом МеОН и добавляли эфир (30 мл). Выпавший осадок отфильтровывали и высушивали.
Выход 230 мг (31% в расчете на карминомицинон), Rf(Г) 0.22, Rt 16.201.
Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [α]D) и спектральным характеристикам (УФ-, ЯМР-спектры) не отличается от стандартного образца, полученного путем биосинтеза на опытной установке ГУ НИИНА им. Г.Ф.Гаузе РАМН.
Пример 3.
Карминомицин гидрохлорид.
Синтез ведется аналогично предыдущему примеру, исходя из 100 мг (0.26 ммоль) карминомицинона, 102 мг (0.31 ммоль, 1.2 экв) 1,4-ди-O-ацетил-N-трифторацетилдаунозамина (чистый α-аномер по данным 'Н-ЯМР), 56.5 μl триметилсилилтрифторметансульфоната. Выход гидрохлорида карминомицина - 38 мг (28% в расчете на карминомицинон).
Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [α]D) и спектральным характеристикам (УФ-, ЯМР-спектры) не отличается от стандартного образца, полученного путем биосинтеза на опытной установке ГУ НИИНА им. Г.Ф.Гаузе РАМН.
Пример 4.
1,4-Ди-O-ацетил-3-N-(9-Н-флуорен-2-илметокси)карбонилдаунозамин (α:β=4:1)
Даунозамин гидрохлорид (1.00 г, 5.45 ммоль) растворяли в смеси диоксан - вода (14 мл, 1:1), добавляли Et3N до рН 8, порциями добавляли сухой Н-(9Н-флуорен-2-илметоксикарбонил)сукцинимид (2.20 г, 6.54 ммоль), поддерживая рН 8 добавлением Et3N. Реакционную смесь перемешивали 20 часов, добавляли 30 мл воды, частично упаривали диоксан. Выпавший осадок отфильтровывали и высушивали. Осадок растворяли в этилацетате и добавляли петролейный эфир. Выпавший осадок отфильтровывали, упаривали с пиридином и использовали в следующей стадии без дополнительной очистки. 3-N-(9-Н-Флуорен-2-илметокси)карбонилдаунозамин [˜2 г, Rf=0.75 (В)] растворяли в сухом пиридине (20 мл) и добавляли уксусный ангидрид (1.03 мл, 10.9 ммоль) и каталитическое количество 4-(диметиламино)пиридина (30 мг). Реакционную смесь перемешивали в течение 20 часов, добавляли 1 мл метанола, перемешивали в течении 1 часа и упаривали досуха. Остаток растворяли в этилацетате и добавляли петролейный эфир. Выпавший осадок отфильтровывали, высушивали. Выход 1.97 г (80% в расчете на даунозамин). Соотношение аномеров α:β=4:1 определены методом 1Н-ЯМР - спектроскопии. Rf α ≈Rf β=0.68 (Б).
Пример 5.
1,4-Ди-O-ацетил-3-N-трифторацетилдаунозамин (α:β=1:2), и индивидуальные α- и β-изомеры.
Даунозамин гидрохлорид (4) (1.26 г, 6.87 ммоль) растворяли в сухом МеОН (20 мл), добавляли Et3N (2.87 мл, 2.6 ммоль) и этиловый эфир трифторуксусной кислоты (1.63 мл, 13.73 ммоль).
Реакционную смесь перемешивали в течение 3 ч, затем упаривали. Остаток растворяли в горячем ацетоне (40 мл), выпавший осадок отфильтровывали. Раствор упаривали, остаток повторно упаривали из сухого пиридина и использовали без дополнительной очистки в следующей стадии. 3-N-Трифторацетилдаунозамин (1.42 г, Rf 0.70, В) растворяли в абсолютном пиридине (30 мл), добавляли Ас2О (1.8 мл) и каталитическое количество 4-(диметиламино)пиридина (20 мг). Реакционную смесь перемешивали в течение 48 ч, после чего охлаждали до 0°С и добавляли МеОН (1 мл). Реакционную смесь перемешивали в течение 1 ч, затем упаривали. Остаток делили флэш-хроматографией на силикагеле, петролейный эфир - этилацетат, используя градиент этилацетата от 20% до 50%. Выделяли три фракции: α-аномер, смесь аномеров (α+β) и β-аномер. Для каждой фракции удаляли растворитель, к остатку приливали эфир (40 мл), выпавший осадок отфильтровывали и высушивали. Выход: α-аномер 164 мг Rf(A) 0.62, [α]D 20-92.8°; смесь аномеров (α+β) 1.23 г; β-аномер 430 мг, Rf (А) 0.53, [α]D 20-50.4°. Суммарный выход (α+β) 1.82 г (81% в расчете на даунозамин).
Пример 6.
Карминомицинон.
К раствору дауномицинона (2.50 г, 6.28 ммоль) в сухом дихлорэтане (300 мл) добавляли AlCl3 (2.51 г, 18.84 ммоль). Реакционную смесь кипятили в течение 1.5 ч, затем добавляли свежую порцию AlCl3 (2.51 г, 18.84 ммоль) и кипятили в течение 1.5 ч, процедуру повторяли еще раз (всего добавляли 8 г AlCl3), после чего выливали реакционную смесь в воду (400 мл), подкисляли водную фракцию 4н HCl до рН 3. Отделяли водный слой и нагревали его до кипения. Выпавший осадок отфильтровывали, промывали водой, высушивали. Органическую фракцию высушивали, частично упаривали и оставляли кристаллизоваться сначала при комнатной температуре в течение 4 часов, а затем при 4°С в течение 20 часов. Выпавший осадок отфильтровывали, объединяли с осадком из водной фракции и высушивали до постоянного веса. Выход 2.05 г (85%), Rf(A) 0.37, Rf(B) 0.47.
Пример 7.
Карминомицин гидрохлорид.
Карминомицинон (2.60 г, 6.77 ммоль) растворяли в смеси сухой хлористый метилен - сухой диоксан (4:1, 1.8 л), добавляли 1,4-ди-O-ацетил-3-N-трифторацетилдаунозамин (α:β=1:2) (2.655 г, 8.12 ммоль) и молекулярные сита (50 г). Раствор смесь перемешивали в токе аргона, охлаждали до -10°С и прибавляли по каплям раствор триметилсилилтрифторметансульфоната (1.20 мл, 6.75 ммоль) в сухом хлористом метилене (20 мл). Реакционную смесь перемешивали при температуре не выше 10°С в течение 40 мин, затем выливали в насыщенный раствор NaHCO3 (700 мл). Органический слой отделяли, промывали водой до рН 6, высушивали и упаривали. Остаток растворяли в смеси диоксан - МеОН (150 мл, 2:1), охлаждали до 4°С и приливали 0.4 н NaOH (50 мл). Реакционную смесь перемешивали в течение 30 часов при 4°С атмосфере аргона, затем подкисляли до рН 5 0.5н HCl. Реакционную смесь разбавляли H3O и экстрагировали хлороформом (3×100 мл). Значение рН водной фракции доводили до 7.5 5%-ным раствором K2CO3 и экстрагировали хлороформом (4×100 мл). Органические фракции объединяли, высушивали и упаривали до минимального объема. Раствор подкисляли до рН 3.5 0.5н раствором HCl в сухом МеОН и добавляли эфир (100 мл). Выпавший осадок отфильтровывали и высушивали.
Выход 1.23 г (33% в расчете на карминомицинон), Rf(Г) 0.22, Rt 16.203.
Образец карминомицина гидрохлорида по своим физико-химическим (Т.пл., Rf, Rt и [α]D) и спектральным характеристикам (УФ-, ЯМР-спектры) не отличается от стандартного образца, полученного путем биосинтеза на опытной установке ГУ НИИНА им. Г.Ф.Гаузе РАМН.
Способ получения антибиотика карминомицина или его гидрохлорида
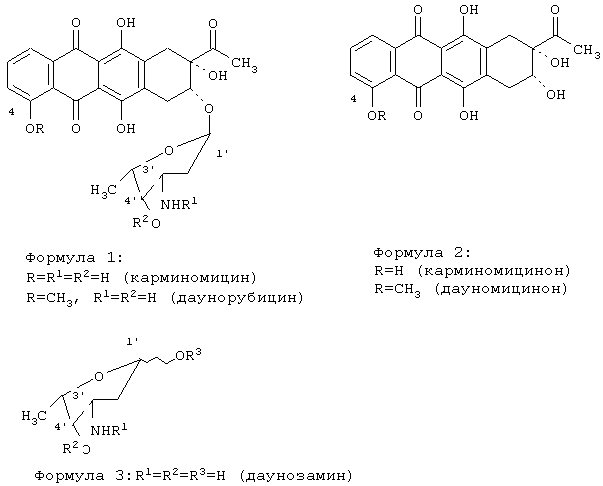
R1=трифторацетил, N-(9-Н-флуорен-2-илметокси)карбонил, аллилоксикарбонил; R2=ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил; R3=трихлорацетамидил, триалкилсилил, ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил.
Изобретение относится к способу получения антибиотика карминомицина или его гидрохлорида. Способ включает гликозилирование карминомицинона 1,4-ди-O-замещенным-N-ацилдаунозамином (2,3,6-тридезокси-3-амино-L-ликсо-гексапиранозой) формулы 3
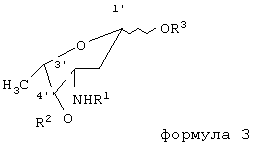
где R1=трифторацетил, N-(9-Н-флуорен-2-илметокси)карбонил, аллилоксикарбонил, R2=ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил, R3=трихлорацетамидил, триалкилсилил, ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил, в присутствии триметилсилилтрифторметансульфоната в смеси безводных апротонных органических растворителей, удаление блокирующих групп и выделение карминомицина в виде основания или гидрохлорида. Гликозил-донор используют в виде смеси α и β изомеров или в виде индивидуальных α или β изомеров. В качестве безводных апротонных органических растворителей используют смеси диоксана с хлороформом, метиленхлоридом, диэтиловым эфиром, ацетоном и тетрагидрофураном. Реакцию проводят в присутствии молекулярных сит, при температуре не выше 10°С, в атмосфере осушенного воздуха или осушенного инертного газа. Удаление защитных групп проводят в присутствии гидроксидов щелочных металлов в водно-органической среде: вода - метанол, вода - диоксан, вода - тетрагидрофуран, вода - ацетон. Способ позволяет увеличить выход карминомицина в 3 раза по сравнению с прототипом. 4 з.п. ф-лы.
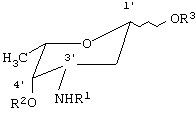
где R1 - трифторацетил, N-(9-Н-флуорен-2-илметокси)карбонил, аллилоксикарбонил,
R2 - ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил,
R3 - трихлорацетамидил, триалкилсилил, ацетил, трифторацетил, 4-нитробензоил, аллилоксикарбонил,
в качестве безводных апротонных органических растворителей используют диоксан-метиленхлорид, диоксан-хлороформ, диоксан-метиленхлорид-диэтиловый эфир, диоксан-ацетон, диоксан-хлороформ-диэтиловый эфир, диоксан-тетрагидрофуран-метиленхлорид, при этом реакцию проводят в присутствии молекулярных сит, удаление блокирующих групп проводят щелочным агентом.
| BAER H.H | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Can.J.Chem | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| KIMURA, Y | |||
| et al | |||
| Trimethylsilyl trifluoromethanesulfonate (trimethylsilyl triflate) as an excellent glycosidation reagent for anthracycline synthesis | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

