Данное изобретение относится к новым соединениям, которые являются лигандами рецептора тироидных гормонов и предпочтительно селективны относительно β-рецептор а тироидного гормона, и к способам применения таких соединений, например, для регуляции метаболизма.
Хотя исключительная роль тироидных гормонов для регуляции процесса обмена веществ у человека хорошо известна, создание и совершенствование новых специфических лекарственных препаратов для более успешного лечения гипертиреоза и гипотиреоза осуществляется очень медленно. Это также ограничивает создание тироидных агонистов и антагонистов для лечения других важных клинических показаний, например гиперхолистеринемии, ожирения и сердечной аритмии.
Тироидные гормоны влияют на метаболизм практически каждой клетки тела. При нормальных уровнях эти гормоны поддерживают вес тела, скорость обмена веществ, температуру тела и настроение и влияют на уровни липопротеида низкой плотности (ЛПНП, LDL) в сыворотке. Так, при гипотиреозе увеличивается вес тела, характерны высокие уровни LDL холестерина и депрессия. При избытке в случае гипертиреоза эти гормоны вызывают потерю веса, гиперметаболизм, понижение уровней сывороточного LDL, аритмию, сердечную недостаточность, мышечную слабость, остеопороз у женщин в постклимактерический период и чувство тревоги.
В настоящее время тироидные гормоны применяются, прежде всего, в качестве заместительной терапии у больных с гипотиреозом. Терапия с помощью L-тироксина возвращает нормальные метаболические функции и может легко.контролироваться с помощью обычных измерений уровней тиреотропина (ТТГ, TSH), тироксина (3,5,3',5'-тетраиод-L-тиронин, или Т4) и трииодтиронина(3,5,3'-трииод-L-тиронин, или Т3). Однако заместительная терапия, особенно у пожилых людей, ограничена из-за вредного действия тироидных гормонов.
Кроме того, некоторые эффекты тироидных гормонов могут быть терапевтически полезными при нетироидных нарушениях, если вредные побочные явления свести к минимуму или устранить. Эти потенциально применимые эффекты включают уменьшение веса, снижение уровней сывороточного LDL, ослабление депрессии и стимуляция формирования костей. Предшествующие попытки фармакологического применения тироидных гормонов для лечения этих нарушений были ограничены проявлениями гипертиреоза и в особенности сердечно-сосудистой токсичностью.
Разработка специфичных и селективных агонистов рецепторов тироидных гормонов могла бы привести к специфическим средствам для лечения этих обычных расстройств и при этом избегнуть сердечно-сосудистой и других видов токсичности природных тироидных гормонов. Тканеселективные агонисты тироидных гормонов можно получать селективным поглощением или вытеснением ткани, местной или локальной доставкой, нацеливанием на клетки с помощью других лигандов, присоединенных к агонисту, и нацеливанием рецепторных подтипов. Агонисты рецепторов тироидных гормонов, которые селективно взаимодействуют с β-формой рецептора тироидного гормона, «предлагают» особенно привлекательный способ, как избежать кардиотоксичности.
Рецепторы тироидных гормонов (TRs) представляют собой подобно другим ядерным рецепторам одиночные полипептидные цепи. Различные рецепторные формы, по-видимому, являются «продуктами» двух различных генов α и β. Дополнительные различия изоформ вызваны тем, что дифференциальный процессинг РНК приводит, по меньшей мере, к двум изоформам каждого гена. Изоформы TRα1, TRβ1 и TRβ2 связывают тироидный гормон и ведут себя как лигандрегулируемые факторы транскрипции. У взрослого человека изоформа TRβ1- наиболее широко распространенная форма в большинстве тканей, особенно в печени и в мышцах. TRα- форма преобладает в гипофизе и других областях центральной нервной системы, не связывает тироидные гормоны и во многих ситуациях ведет себя как ингибитор транскрипции. TRα1 -изоформа также широко распространена, хотя ее уровни значительно ниже, чем уровни TRβ1-изоформы.Эта изоформа может быть особенно важна для совершенствования. Хотя в гене TRβ обнаружены многие мутации и они вызывают синдром общей резистентности к тироидному гормону, мутации, приводящие к ослабленной TRα-функции, не найдены. Многочисленные данные говорят о том, что многие или большая часть эффектов тироидных гормонов, действующих на сердце и, в частности на частоту и ритмичность сердечных сокращений, опосредуется через α-форму изоформы TRα1, тогда как влияние гормона, например, на печень, мышцы и другие ткани опосредуется в основном β-формой рецептора. Следовательно, TRβ-селективный агонист, по-видимому, не выявляет влияние гормонов на частоту и ритмичность сердечных сокращений, но выявляет многие другие действия гормонов. Полагают, что α-формарецептора является основной формой, влияющей на частоту сердечных сокращений, по следующим причинам:
тахикардия является обычным явлением при синдроме общей устойчивости (резистентности) к тироидному гормону, при котором дефектны TR β-формы и высоки расчетные уровни Т4 и Т3;
описан только один случай тахикардии у больного с двойной делецией в TRβ-гене (Takeda et al., J. Clin. Endocrinol. And Metab. 1992, vol.74, p.49);
TRα-ген(но не β-ген) с двойным нокаутированном у мышей имеет замедленный пульс по сравнению с контрольными мышами;
вестерн-блоттинг TRs миокарда показывает присутствие белков TR α1, TRα2 и TRβ2, но не TRβ1.
Если эти признаки корректны, тогда TR β-селективный агонист можно использовать для подражания ряду действий тироидного гормона, хотя он оказывает более слабое действие на сердце. Такое соединение можно использовать при (1) заместительной терапии у пожилых людей с гипотиреозом с повышенным риском сердечно-сосудистых осложнений; (2) заместительной терапии у пожилых людей с бессимптомным гипотиреозом с повышенным риском сердечно-сосудистых осложнений; (3) ожирении; (4) холистеринемии, вызванной повышением уровней LDL в плазме; (5) депрессии; (6) остеопорозе в комбинации с ингибитором резорбции костей.
В соответствии с данным изобретением созданы соединения, представляющие собой лиганды тироидных рецепторов и имеющие общую формулу

где
X обозначает кислород (-O-);
Y обозначает -(СН2)n-, где n обозначает целое число от 1 до 3, или -С=С-, который представляет собой цис- или транс-изомер;
R1 алкил, содержащий 3-6 атомов углерода;
R2 и R3 одинаковы или различны и обозначают водород, галоген, алкил, содержащий 1-4 атома углерода, при этом, по меньшей мере, один из R2 и R3 не обозначает водород;
R4 обозначает водород или низший алкил;
R5 обозначает водород;
R6 обозначает карбоновую кислоту или ее эфир;
R7 обозначает водород;
включая все его стереоизомеры или его фармацевтически приемлемую соль.
Кроме того, объектом данного изобретения является способ предупреждения, ингибирования или лечения заболевания, обусловленного дисфункцией обмена веществ или зависящей от экспрессии Т3-регулируемого гена, при котором соединение формулы I вводится в терапевтически эффективном количестве. Соединение формулы I предпочтительно является агонистом, предпочтительно селективным в отношении бета-рецептора тироидного гормона. Примеры таких заболеваний, обусловленных дисфункцией обмена веществ или зависящих от экспрессии Т3-регулируемого гена, представлены далее и включают ожирение, гиперхолистеринемию, атеросклероз, сердечную аритмию, депрессию, остеопороз, гипотиреоз, зоб, рак щитовидной железы, а также глаукому и застойную сердечную недостаточность.
Соединения формулы I могут быть в виде солей, в частности в виде фармацевтически приемлемых солей. Если в соединении формулы I имеется, например, по меньшей мере, один основной центр, они могут образовывать соли присоединения кислот. Эти соли образуются, например, с сильньми неорганическими кислотами, такими как минеральные кислоты, например серная кислота, фосфорная кислота или галоидоводородная кислота, с сильными карбоновыми кислотами, такими как алканкарбоновые кислоты с 1-4 атомами углерода, незамещенные или замещенные, например, на галоген, например уксусная кислота, такими как насыщенные или ненасыщенные дикарбоновые кислоты, например щавелевая, малоновая, янтарная, малеиновая фумаровая, фталевая или терефталевая кислота, такими как гидроксикарбоновые кислоты, например аскорбиновая, гликолевая, молочная, яблочная, винная или лимонная кислота, такими как аминокислоты (например, аспарагиновая кислота, или лизин, или аргинин), или бензойная кислота, или с органическими сульфоновыми кислотами, такими как (С1-С4)-алкил- или арилсульфоновые кислоты, незамещенными или замещенными, например, на галоген, например метансулифоновая кислота или п-толуолсульфоновая кислота. Также, если требуется, можно получать соответствующие соли присоединения кислот, имеющие дополнительный центр основности. Соединения формулы I, содержащие, по меньшей мере, одну кислотную группу (например, СООН), могут также образовывать соли с основаниями. Соответствующие соли с основаниями представляют собой, например соли металлов, такие как соли щелочных металлов или соли щелочноземельных металлов, например, соли натрия, калия или магния, или соли аммония или соли с органическими аминами, такими как морфолин, тиоморфолин, пиперидин, пирролидин, моно-, ди- и три-низший алкиламин, например этил-, трет-бутил-, диэтил-, диизопропил-, триэтил-, трибутил- или диметилпропиламин, ди- или тригидроксинизший алкиламин, например моно-, ди-, или триэтаноламин. Кроме того, могут образовываться соответствующие внутренние соли.
Предпочтительные соли соединений формулы I, которые содержат основную группу, включают моногидрохлорид, гидросульфат, метансульфонат, фосфат или нитрат.
Предпочтительные соли соединений формулы I, которые содержат кислотную группу, включают соли натрия, калия и магния и соли фармацевтически приемлемых органических аминов.
Предпочтительными являются соединения формулы I, где
Х обозначает О;
Y обозначает цис- или транс-этилен;
R1 обозначает изопропил;
R2 и R3 независимо обозначает независимо бром, хлор или метил;
R4 обозначает водород или метил;
R5 обозначает водород;
R6 обозначает карбоксил;
R7 обозначает водород.
Другими предпочтительными соединениями являются соединения формулы I, в которых Х обозначает О;
Y обозначает -(CH2)n-, где n обозначает 1 или 2;
R1 обозначает алкил, содержащий 1-3 атомов углерода;
R2 и R3 обозначают независимо бром, хлор или метил;
R4 обозначает водород или метил;
R5 обозначает водород;
R6 карбоксил;
R7 обозначает водород.
Наиболее предпочтительными являются соединения формулы I по изобретению, в которых Х обозначает О;
Y обозначает -(СН2)n-, где n обозначает 1;
R1 обозначает алкил, содержащий 1-3 атомов углерода;
R2 и R3 независимо обозначают бром или хлор;
R4 обозначает водород или метил;
R5 обозначает водород;
R6 обозначает карбоксил;
R7 обозначает водород.
Следовательно, предпочтительные соединения по изобретению будут иметь строение
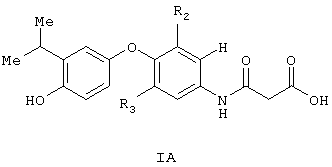
или
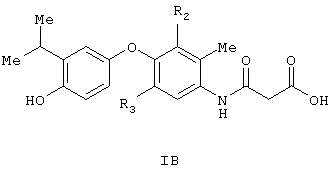
Предпочтительные соединения имеют следующую структуру:


или

Наиболее предпочтительные соединения по изобретению имеют строение

или
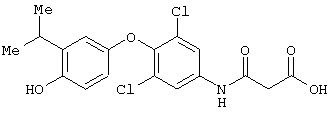
Соединения формулы I можно получать методами, примеры которых приведены на представленных ниже реакционных схемах, а также опубликованными в литературе релевантными методами, которые используются специалистами в данной области техники. Примеры реагентов и методов проведения этих реакций представлены ниже и в разделе Примеры. Защиту и снятие защиты в приведенных ниже схемах можно осуществлять методами, хорошо известньми из уровня техники (см., например, T.W.Green and P.G.M.Wuts, «Protecting Groups in Organic Synthesis», 3rd Edition, Wiley, 1999).
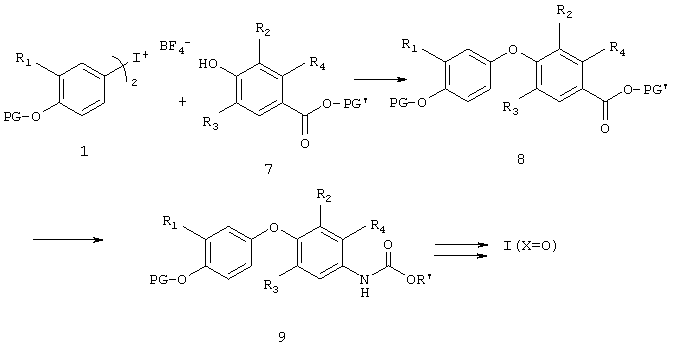
На схеме 1 показан общий синтетический подход к соединениям формулы I, где Х=O, который использует присоединение соответствующей замещенной иодониевой соли 1 к соответствующему фенолу 2 с образованием интермедиата 3. В формуле 1 и во всех прилагаемых формулах, содержащихся в описываемых ниже схемах, PG (ЗГ) относится к защитной группе для этой функциональной группы (в данном примере для фенольного кислорода). Специфические защитные группы для каждого конкретного интермедиата хорошо известны специалистам в данной области техники (см. также приведенную выше ссылку «Protecting Groups in Organic Synthesis»). Соответствующая манипуляция с защитной и с функциональной группой дает заданное соединение формулы I. Например, интермедиат 2 может представлять собой нитрофенол (R' и R'' обозначают кислород) и образующийся продукт присоединения является соответствующим нитропроизводным диарилового эфира 3, где R'=R''=O. Это промежуточное нитросоединение можно легко восстановить до соответствующего ариламина (см. обсуждение ниже). Образующийся ариламин можно затем легко ацилировать и получить нужные соединения формулы I (Х=O). Интермедиат 2 может также представлять собой соединение с защищенной аминогруппой, например R'=R5 и R''=PG. Защитная группа (PG, ЗГ) может быть карбаматной группой, такой как трет-бутилоксикарбонильная (ВОС) или бензилоксикарбонильная (CBZ), которую позже можно снять ацидолизом и/или гидрогенолизом в стандартных условиях. Ацилированием образующегося ариламина также хорошо известными специалистам в данной области техники способами получают искомые соединения формулы I. Кроме того, ариламин (интермедиат 3, где R'=R"=H), образующийся при восстановлении продукта присоединения - аналога нитробензола, может реагировать с альдегидом по реакции восстановительного аминирования, таким образом из альдегидного фрагмента вводится группа R5. Методы восстановительного аминирования, например, с помощью цианоборгидрида натрия или триацетоксиборгидрида натрия хорошо известны специалистам в данной области техники. Образующийся продукт можно затем ацилировать обычными методами, получая соединения формулы I.
Схема 1
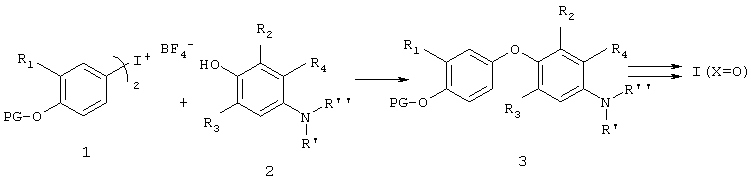
Метод, использующий иодониевую соль, иллюстрированный Схемой 1, подробно описан в литературе по синтезу аналогов тироидных гормонов («Novel Thyroid Receptor Ligands and Methods», Y.-L.Li, Y.Liu, A.Hedfors, J.Malm, C. Mellin, M. Zhang, Международная заявка WO 9900353 Fl 990107; D.M.B.Hickey et al., J. Chem. Soc. Perkin Trans. I, 3103-3111, 1988; N. Yokoyama et al., J. Med. Chem., 38, 695-707, 1995) и по диариловым эфирам в целом (E.A.Couladouros, V.I. Moutsos, Tetrahedron Lett., 40, 7023-7026, 1999).
На Схеме 2 показан другой синтетический подход к соединениям формулы I, где Х=O, при котором соответствующий замещенный нитробензольный интермедиат 5 алкилируют соответствующим замещенным фенолом 4, получая нитроинтермедиат 6. Нитрогруппу в интермедиате 6 можно восстановить до аминогруппы хорошо известными из уровня техники методами, такими как каталитическое гидрирование в присутствии, например, никеля Ренея или палладия на угле в полярном растворителе, таком как ледяная уксусная кислота или этанол. Или же восстановление можно проводить с применением порошка железа в разбавленной уксусной кислоте при комнатной температуре. Соответствующие манипуляции с защитной и функциональной группами дают нужные соединения формулы I.
Схема 2
 Этот способ, иллюстрированный на Схеме 2 как общий метод синтеза, хорошо описан в литературе (P.D.Leeson, J.C.Emmet, J.Chem. Perkm Trans. 1,3085-3096,1988; N.Yokoyama et al., J. Med., 38, 695-707, 1995).
Этот способ, иллюстрированный на Схеме 2 как общий метод синтеза, хорошо описан в литературе (P.D.Leeson, J.C.Emmet, J.Chem. Perkm Trans. 1,3085-3096,1988; N.Yokoyama et al., J. Med., 38, 695-707, 1995).
Другие способы синтеза соединений формулы I, где Х=O, NH, S, СО или СН2 в целом описаны в литературе (Х=O: D.M.B. Hickey et al., J. Chem. Soc. Perkin Trans. I, 3097-3102, 1988; Z.-W. Guo et al., J. Org. Chem., J. Org. Chem., 62, 6700-6701, 1997; D.M.T. Chan et al., Tetrahedron Lett., 39, 2933-2936, 1998; D.A.Evans et al., Tetrahedron Lett., 39,2937-2940, 1998; G.M.Salamonczyk et al., Tetrahedron Lett., 38, 6965-6968, 1997; J.F.Marcoux, J. Am. Chem. Soc., 119, 10539-10540; A.V.Kalinin et al., J. Org. Chem., 64, 2986-2987, 1999; X=N: D.M.T. Chan et al., Tetrahedron Lett., 39, 2933-2936, 1998; J.P.Wolfe et al., J. Am. Chem. Soc., 118, 7215, 1996; M.S.Driver, J.F.Hartwig, J. Am. Chem. Soc., 118, 7217, 1996; см. ссылки в обзоре C.G. Frost, P. Mendonca, J. Chem. Soc. Perkin I, 2615-2623, 1998; для X=S: C.R. Harrington, Biochem. J., 434-437, 1948; A. Dibbo et al., J. Chem. Soc., 2890-2902, 1961; N. Yokoyama et al., Патент США 54017726, 1995; Х=СО или СН: L. Homer, H.H.G. Medem, Chem. Ber., 85, 520-530, 1952; G. Chiellini et al., Chemistry and Biology, 5, 299-306, 1988).
Способы синтеза соединений формулы I, в которых Х=O, a R2 и R3 независимо варьируются, принимая значения: водород, галоген и алкил,- описаны в « Novel Thyroid Receptor Ligands and Methods», Y.-L. Li, Y. Liu, A. Hedfors, J. Malm, C. Mellin, M. Zhang, Международная заявка WO 9900353 A1 990107.
Другой общий метод синтеза соединений формулы I, в которых Х=O, показан на Схеме 3. По этому методу соответствующая замещенная иодониевая соль 1 присоединяется к соответствующему интермедиату 7 - замещенной 4-гидроксибензойной кислоте. Затем удаляют защитную карбоксильную группу (PG') в полученном продукте присоединения 8. Образующаяся промежуточная свободная карбоновая кислота затем претерпевает перегруппировку Курциуса в присутствии известных реагентов, таких как дифенилфосфорилазид (DPPA). Промежуточные соединения, образующиеся при перегруппировке Курциуса, можно улавливать либо трет-бутанолом, либо бензиловым спиртом, получая продукт 9, анилин с защитной, трет-бутоксикарбонильной (ВОС) или бензилоксикарбонильной (CBZ) группой соответственно. Эти защитные группы можно удалять хорошо известными в технике методами, получая соответствующую свободную аминогруппу. Затем амин можно ацилировать с образованием соединений формулы I с Х=O любым из хорошо известных методов, например ацилированием свободной карбоновой кислотой в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид (DCC) или (-1-[3-(диметиламино)пропил]-3-этилкарбодиимид (EDCI).
Или же свободный амин можно ацилировать производным хлорангидрида карбоновой кислоты в присутствии эквивалентного количества третичного амина, такого как триэтиламин или N-метилморфолин.
Схема 3
Рассматриваются все стереоизомеры соединений по данному изобретению либо в виде смеси, либо в чистом, либо в практически чистом виде. Соединения по данному изобретению могут иметь асимметрические центры при любом атоме углерода, включая любой из атомов заместителей R. Следовательно, соединения формулы I могут существовать в виде энантиомеров, или стереомеров, или в виде их смесей. В процессе получения в качестве исходных веществ можно использовать рацематы, энантиомеры или стереомеры. Когда получают диастереомерные или энантиомерные продукты, их можно разделять соответствующими способами, например хроматографией или фракционной кристаллизацией.
Соединения по изобретению являются агонистами, которые предпочтительно селективны в отношении бета-рецептора тироидного гормона и в качестве таковых пригодны для лечения ожирения, гиперхолистеринемии и атеросклероза за счет понижения уровней LDL в сыворотке, одни или в комбинации с липидмодулирующим лекарственным веществом, таким как ингибитор HMG-CoA- редуктазы, фибрат (fibrate), ингибитор МТР, ингибитор сквален-синтетазы и/или другой гиполипидемический агент, и/или при необходимости в комбинации с антидиабетическим агентом; пригодны для уменьшения депрессии, одни или в комбинации с антидепрессантом, таким как флуоксетин и дезипрамин; и применимы для стимуляции формирования костей при лечении остеопороза, одни или в комбинации с любым известным ингибитором резорбции костей, таким как натрия алендронат. Кроме того, соединения по изобретению могут применяться в качестве заместительной терапии у пожилых пациентов с гипотиреозом или бессимптомным гипотиреозом с повышенным риском сердечно-сосудистых осложнений, при терапии престарелых с целью придать им ощущение здоровья и при лечении нетоксического зоба, при лечении палиллярного или фолликулярного рака щитовидной железы (одни или с Т4), при лечении поражений кожи, таких как псориаз, глаукомы, сердечно-сосудистые заболевания, например для предупреждения или лечения атеросклероза и застойной сердечной недостаточности.
Соединения по изобретению можно применять одни или в сочетании с препаратами для подавления аппетита, такими как сибутрамин, и/или в сочетании с препаратами против ожирения, и/или в сочетании с β3-агонистом для лечения ожирения.
Соединения по изобретению можно также применять для лечения поражений кожи или заболеваний, сопровождающихся дермальной атрофией, включая излечение дермальной атрофии, вызываемой местными глюкортикоидами, включая «восстановление» дермальной атрофии, вызванной местными глюкокортикоидами, предупреждение дермальной атрофии, местными глюкокортикоидами (например, при одновременном лечении местным глюкокортикоидом или фармакологическим продуктом, содержащим глюкокортикоид и соединение по изобретению), восстановление/предупреждение дермальной атрофии, вызванной системным лечением глюкокортикоидами, восстановление/предупреждение атрофии дыхательной системы, вызванной местным применением глюкокортикоидов, УФ- индуцированной дермальной атрофии или дермальной атрофии, связанной со старением (морщины и т.д.), заживление ран, келоида, атрофированных полосок кожи, целлюлита, бугристой кожи, фотохимического повреждения кожи, плоского лишая, ихтиоза, угрей, лечение псориаза, болезни Дернье (Dernier), экземы, диффузного нейродермита, хлоракне, питириаза и кожных рубцов.
При лечении кожных поражений и заболеваний, описанных выше, соединения по изобретению можно применять одни или в комбинации с ретиноидами, таким как третиноин или аналог витамина Д в количествах, предписываемых PDR (настольным врачебным справочником).
Гиполипидемический агент, который при необходимости можно применять в комбинации с соединениями формулы I по изобретению, может включать тиазолидиндионы, ингибиторы МТР, ингибиторы HMG СоА редуктазы, ингибиторы сквален-синтетазы, производные фибриновой кислоты, ингибиторы АСАТ, ингибиторы всасывания холестерина, ингибиторы котранспортера Na+/желчной кислоты в подвздошной кишке, вещества, усиливающие экскрецию желчных кислот, и/или никотиновая кислота и ее производные.
Ингибиторы МТР, применяемые в данном описании, включают ингибиторы, раскрываемые в патентах США 5595872, 5739135, 5712279, 5760246, 5827875, 5885983 и 5962440. Предпочтительными являются каждый из предпочтительных ингибиторов, описываемых в каждом из вышеуказанных патентов.
Наиболее предпочтительные ингибиторы МТР, применяемые по данному изобретению, включают предпочтительные ингибиторы МТР, указанные в патентах США 5739135 и 5712279 и 5760246.
Наиболее предпочтительным ингибитором является
9-[4-[4-[[2-(2,2,2-Трифторэтокси)бензоил]амино]-1-пиперидинил]бугил]-N-(2,2,2-трифторэтил)-9Н-флуорен-9-карбоксамид

Гиполипидемический агент может представлять собой ингибитор HMG СоА -редуктазы, который включает без ограничения мевастатин и родственные соединения, описанные в патенте США 3983140, ловастатин (мевинолин) и родственные соединения, описанные в патенте США 4231938, правастатин и родственные соединения, описанные, например, в патенте США 4346227, симвастатин и родственные соединения, описанные в патентах США 4448784 и 4450171. Другие ингибиторы HMG СоА-редуктазы, которые можно применять по данному изобретению, включают, но без ограничения флувастатин, описанный в патенте США 5354772, церивастатин, раскрытый в патентах США 5006530 и 5177080, аторвастатин, раскрытый в патентах США 4681893, 5273995, 5385929 и 5686104, пиразольные аналоги производных мевалонолактона, раскрытые в патенте США 4613610, инденовые аналоги производных мевалонолактона, раскрытые в международной заявке WO 86/03488, 6-[2-(замещенные-пиррол-1-ил)-алкил)пиран-2-оны и их производные, раскрытые в патенте США 4647576, дихлорацетат соединения SC-45355 фирмы Searle (производное 3-замещенной пентандиовой кислоты), имидазольные аналоги мевалонолактона, раскрытые в международной заявке WO 86/07054, производные 3-карбокси-2-гидроксипропанфосфоновой кислоты, раскрытые во французском патенте 2596393, производные 2,3-дизамещенного пиррола, фурана и тиофена, раскрытые в Европейской патентной заявке 0221025, нафтильные аналоги мевалонолактона, раскрытые в патенте США 4686237, октагидронафталины, такие, которые раскрыты в патенте США 4499289, кетоаналоги мевинолина (ловастатина), раскрытые в Европейской патентной заявке 0142146 А2, а также другие известные ингибиторы HMG СоА -редуктазы.
Кроме того, фосфиновые кислоты, применимые для ингибирования HMG СоА-редуктазы, пригодные для применения по данному изобретению, описаны в английском патенте GB 2205837.
Ингибиторы сквален-синтетазы, пригодные для применения по данному изобретению, включают без ограничения α-фосфонсульфонаты, раскрытые в патенте США 5712396, фосфонсульфонаты, описанные Biller et al., J. Med. Chem., 1988, vol., 31, № 10, pp.1869-1871, включая изопреноидные (фосфинилметил)фосфонаты, а также другие ингибиторы сквален-синтетазы, раскрытые в патентах США 4871 и 4924024 и в Biller, S.A., Neuenschwander, К., Ponpipom, M.M., and Poulter, C.D., Current Pharmaceutical Design, 2, 1-40 (1996).
Кроме того, другие ингибиторы сквален-синтетазы, пригодные для применения по данному изобретению, включают терпеноидные пирофосфаты, раскрытые Р. Ortiz de Montellano et. al., J. Med. Chem., 1977, 20, 243-249, аналог А фарнезилдифосфата и аналоги прескваленпирофосфата (PSQ-PP), раскрытые Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293, фосфинилфосфонаты, о которых сообщает McClard, R.W. et al., J. А. С. S., 1987, 109, 5544, и циклопропаны, описываемые Capson, T.L., Ph. D dissertation, June, 1987, Dept. Med. Chem. U of Uta, Abstract, Table of Contents, pp. 16, 17, 40-43, 48-51, Summary.
Другие гиполипидемические агенты, пригодные для применения по данному изобретению, включают, но без ограничения производные фибриновой кислоты, такие как фенофибрат, гемфиброзил, клофибрат, безафибрат, ципрофибрат, клинофибрат и т.п., пробукол и родственные соединения, описываемые в Патенте США 3674836, причем пробукол и гемфиброзил являются предпочтительными, вещества, усиливающие экскрецию желчных кислот, такие как холестирамин, колестипол и DEAE-Sephadex (Secholex®, Policexide®), атакже липостабил (Rhone-Poulenc), Eisai e-5050 (производное замещенного этаноламина), иманиксил (НОЕ- 402), тетрагидролипстатин (THL), истигмастанилфосфорилхолин (SPC, Roche), аминоциклодекстрин (Tanabe Seiyoku), Ajinomoto AJ-814 (производное азулена), мелинамид (Sumitomo), Sandoz 58-035, CL-277082 и CL-283546 фирмы American Cyanamid (производные дизамещенной мочевины), никотиновая кислота, аципимокс, ацифран, неомицин, п-аминосалициловая кислота, аспирин, производные поли(диаллилметиламина), такие как раскрытые в патенте США 4759923, соль четвертичного аммониевого основания поли(диаллилдиметиламмонийхлорид) и иононы, такие как раскрытые в патенте США 4027009, и другие известные вещества, снижающие сывороточный холестерин.
Другой гиполипидемический агент может быть ингибитором АСАТ, таким как описанный в Drugs in Future 24, 9-15 (1999), (Avasimbe); « The ACAT inhibitor. Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters», Nicolosi et al., Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; «The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hipolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB100-containing lipoprotein», Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; «RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor». Smith, C., et al., Bioorg. Med. Chem. Lett. (1996), 6 (1), 47-50; «ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals», Krause et al., Editor(s)" Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation: Mediators P(1995), 173-98, Publisher: CRC, Boca Raton, Fla.; «ACAT inhibitors: potential anti-atherosclerotic agents», Slivcovic et al., Curr. Med. Chem. (1994), 1(3), 204-25; «Inhibitors of acyl-CoA: cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA: cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(1-phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity», Stout et al., Chemtracts; Org. Chem. (1995), 8(6), 359-62.
Гиполипидемический агент может представлять собой ингибитор абсорбции (всасывания), предпочтительно SCH48461 фирмы Schering-Plough, а также ингибитор адсорбции, раскрытый в Atherosclerosis 115, 45-63 (1995) и J. Med. Chem. 41, 973 (1998).
Гиполипидемический агент может быть ингибитором котранспортера Na+/желчных кислот в подвздошной кишке, таким как описанный в Drugs of the Future, 24, 425-430 (1999).
Предпочтительными гаполипидемическими агентами являются правастатин, ловастатин, симвастатин, аторвастатин, флувастатин и церивастатин.
Соединения формулы I по изобретению применяются с гиполипидемическим агентом, антидепрессантом и/или ингибитором резорбции костей, и/или веществом, подавляющим аппетит (при его наличии), в весовом соотношении примерно 500:1 - 0,005:1, предпочтительно примерно 300:1-0,01:1.
Антидиабетический агент, который, при необходимости, применяется в комбинации с соединениями формулы I по изобретению, может включать бигуаниды, сульфонилмочевины, ингибиторы глюкозидазы, тиазолидиндионы и/или ингибиторы аР2, и/или агонисты PPARα, агонисты PPARγ или двойные α/γ агонисты, и/или ингибиторы SGLT или меглитинид.
Антидиабетический агент может быть пероральным антигипергликемическим агентом, предпочтительно бигуанидом, таким как метформин или фенформин или их соли.
Если антидиабетический агент представляет собой бигуанид, соединения формулы I применяются в весовом отношении к бигуаниду примерно 0,01:1-100:1, предпочтительно около 0,05:1-2:1.
Антидиабетический агент предпочтительно может также представлять собой сульфонилмочевину, такую как глибурид (также известную как глибенкламид), глиперимид (описанный в патенте США 4379785), глипизид, гликлазид или хлорпропамид, другие известные сульфонилмочевины или другие антигипергликемические агенты, которые влияют на АТФ-зависимый канал β-клеток, при этом предпочтительными являются глибурид и глипизид.
Соединения формулы I применяются в весовом соотношении с мочевиной примерно 0,01:1-100:1, предпочтительно около 0,2:1-10:1.
Соединения формулы I применяются с сульфонилмочевиной в весовом соотношении примерно 0,2:1-10:1.
Пероральный антидиабетический агент может также быть ингибитором глюкозидазы, таким как акарбоза (описанная в патенте США 4639436), которую можно принимать в виде отдельной лекарственной формы.
Соединения формулы I можно применять с ингибитором глюкозидазы в весовом соотношении около 0,01:1-100:1, предпочтительно около 0,5:1-50:1.
Соединения формулы I можно применять в комбинации с тиазолидиндионовым пероральным антидиабетическим агентом или другими сенсибилизаторами инсулина (которые влияют на чувствительность к инсулину у больных NIDDM), такими как троглитазон (Rezulin® фирмы Warner-Lambert, описанный в патенте США 4572912), розиглитазон (SKB), проглитазон (Takeda), MCC-555 фирмы Mitsubishi (описанный в патенте США 5594016), GI-262570 фирмы Glaxo-Welcome, эглитазон (СР-68722, Pfizer) или дарглитазон (СР-86325, Pfizer).
Соединения формулы I можно применять с тиазолидиндионом в весовом соотношении около 0,01:1-100:1, предпочтительно около 0,5:1-5:1.
Сульфонилмочевина и тиазолидиндион в количествах менее примерно 150 мг перорального антидиабетического агента могут применяться в виде единой таблетки с соединениями формулы I.
Соединения формулы I можно также применять в комбинации с непероральным антигипергликемическим агентом, таким как инсулин, или с глюкагоноподобным пептидом - 1 (GLP-1), таким как GLP-1 (1-36) амид, GLP-1 (7-36) амид, GLP-1 (7-37) (описаны в патенте США 5614492, который можно вводить в виде инъекции интраназально или с помощью трансдермальных или трансбуккальных устройств.
Метформин, сульфонилмочевины, такие как глибурид, глимепирид, глипирид, глипизид, хлорпропамид и гликлазид и ингибиторы глюкозидазы акарбоза, или маглитол, или инсулин, если они присутствуют, могут применяться в препаратах, как описано выше, и в количествах и дозах, указанных во врачебных справочниках.
Метформин или его соль, если они присутствуют, могут применяться в количествах около 500-2000 мг в день, и их можно вводить в виде разовой дозы или раздельных доз один-четыре раза в день.
Тиазолидиндионовый антидиабетический агент, если он присутствует, может применяться в количествах около 0,01-2000 мг/день, и его можно вводить в виде разовой или раздельных доз один-четыре раза в день.
Если присутствует инсулин, то он применяется в препаратах, количествах и дозах, указанных во врачебных справочниках.
При их наличии пептиды GLP-1 можно вводить виде пероральных трансбуккальных препаратов назально или парентерально, как описано в патентах США 5346701,5614492 и 5631224.
Антидиабетический агент может также представлять собой α/γ двойной агонист PPAR, такой как описанный Murakami et al., «A Novel Insulin Sensitizer Acts as a Coligand for Peroxisome Proliferation - Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid Metabolism in Liver of Zucker Fatty Rats», Diabets 476 1841-1847.
Антидиабетический агент может быть ингибитором аР2, таким как описанный в заявке на патент США 09/391053, поданной 7 сентября 1999 года, и в предварительной заявке на патент США 60/127745, поданной 5 апреля 1999 года, и применяется в указанных в данном описании дозах.
Антидиабетический агент может быть ингибитором SGTL2, таким как описанный в предварительной заявке на патент США 60/158773, поданной 12 октября 1999 года.
Соединения формулы I применяются в весовом соотношении с PPAR α-агонистом, PPAR γ-агонистом, PPAR α/γ двойными агонистами, ингибитором SGTL2 и/или ингибитором аР2 примерно 0,01:1-100:1, предпочтительно около 0,5:1-5:1.
Назначаемые дозы следует точно корректировать в соответствии с возрастом, весом тела и состоянием пациента, а также в соответствии со способом введения, лекарственной формой и схемой и требуемым результатом.
Дозы и рецептуры гиполипидемического агента и антидиабетического агента описаны во многих патентах и патентных заявках, обсуждающихся выше, и в PDR (НВС).
Применяемые дозировки и рецептуры другого гиполипидемического агента, антидепрессанта, ингибитора резорбции костей, вещества, подавляющего аппетит, и агента против ожирения, где они требуются, берутся из врачебных справочников.
В случае перорального применения удовлетворительные результаты можно получить, применяя ингибитор МТР в количестве около 0,01-100 мг/кг и предпочтительно около 0,1-75 мг/кг один-четыре раза в день.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор МТР в количестве около 1-500 мг, предпочтительно около 2-400 мг и более предпочтительно около 5-250 мг один-четыре раза в день.
Для парентерального введения ингибитор МТР можно применять в количестве примерно 0,005-10 мг/кг и предпочтительно около 0,005-8 мг/кг один-четыре раза в день.
Для перорального применения удовлетворительный результат можно получать, применяя ингибитор HMG СоА редуктазы, например правастатин, ловастатин, симвастатин, аторвастатин, флувастатин или церивастатин, например, в количестве около 1-2000 мг и предпочтительно около 4-200 мг.
Ингибитор сквален-синтетазы можно применять в дозировках примерно 10-2000 мг и предпочтительно около 25-200 мг.
Предпочтительная пероральная лекарственная форма, например таблетки или капсулы, содержит ингибитор HMG СоА редуктазы в количестве примерно 0,1-100 мг, предпочтительно около 5-80 мг и более предпочтительно около 10-40 мг.
Предпочтительная пероральная лекарственная форма, такая как таблетки или капсулы, содержит ингибитор сквален-синтетазы в количестве около 10-500 мг, предпочтительно около 25-200 мг.
Соединения формулы I и гиполипидемический агент, антидепрессант или ингибитор резорбции костей можно применять совместно в одной и той же пероральной лекарственной форме или в раздельных пероральных лекарственных формах, принимаемых одновременно.
Вышеописанные композиции можно назначать в виде лекарственных форм, как указано выше, в виде однократной дозы или раздельных доз один-четыре раза в день. Можно посоветовать пациенту начинать с комбинации низких доз и постепенно переходить к комбинации высоких доз.
Предпочтительным гиполипидемическим агентом является правастатин, симвастатин, ловастатин, аторвастатин, флувастатин или церивастатин.
Соединения формулы I по изобретению можно вводить перорально или парентерально, например подкожно или внутривенно, а также назально, ректально или подъязычно различным видам млекопитающих, о которых известно, что они подвержены таким заболеваниям, например человеку, кошкам, собакам и т.п., в количестве около 0,1-100 мг/кг, предпочтительно около 0,2-50 мг/кг и более предпочтительно около 0,5-25 мг/кг (или около 1-2500 мг, предпочтительно около 5-2000 мг) по схеме в виде однократной дозы или 2-4 раздельных суточных доз.
Активное вещество можно применять в композициях, таких как таблетка, капсула, мазь, гидрофильная мазь, крем, лосьон, раствор или суспензия или в носителях другого рода, таких как трансдермальные устройства, приборы для электрофореза, ректальные суппозитории, приборы для ингаляции и т.п. Композиция или носитель может содержать около 5-500 мг соединения формулы I на единицу дозы. Они могут смешиваться соответствующим образом с физиологически приемлемым наполнителем или носителем, эксципиентом, связующим, консервантом, стабилизирующим веществом и вкусовой добавкой в соответствии с принятой фармацевтической практикой.
Следующие рабочие примеры представляют предпочтительные варианты данного изобретения.
Пример 1
3-[[3,5-дибром-[4-гидрокси-3-(1-метилэтил)фенокси]-фенил]амино]-3-оксопропановая кислота

Соединение 1а
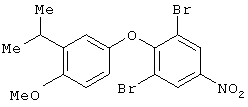
Бис (3-изопропил-4-метоксифенил)иодонийтетрафторборат (32,8 г, 64 ммол), 2,6-дибром-4-нитрофенол (12,6 г, 42 ммол) и порошок Cu [Lancaster, 300 меш (6,8 г, 108 ммол)] суспендируют в 400 мл СН2Cl2 в колбе, покрытой алюминиевой фольгой. При кипячении добавляют триэтиламин и реакционную смесь перемешивают под аргоном в темноте в течение 4 дней. Сырую реакционную смесь упаривают примерно до 70 мл и затем хроматографируют двумя порциями, на 1,8 литрах каждая, силикагеля Merk 3-5%-ным раствором этилацетата в смеси гексанов. Общий выход соединения составляет 15,4 г (81,9%).
Соединение 1b

Соединение 1a (15,2 г, 34,15 ммол) растворяют в 129 мл ледяной уксусной кислоты и 13 мл воды. Добавляют железный порошок (Aldrich <10 микрон, 12 г, 215 ммол) и реакционную смесь перемешивают под аргоном в течение ночи. Реакционную смесь фильтруют через слой целита и промывают целит 50 мл уксусной кислоты. Фильтрат упаривают примерно до 60 мл и выливают в 400 г Na2 СО3. Добавляют воду (400 мл) и продукт экстрагируют этилацетатом (3×500 мл каждая порция). Этилацетат упаривают в вакууме и остаток (13,2 г хроматографируют на 1,8 литрах силикагеля Merk смесью этилацетат: гексан 8:2. Соединение 1b (8,75 г) получают с выходом 61,7% в виде твердого вещества. Протонный и углеродный ЯМР-спектры соответствуют заданной структуре.
Соединение 1c

Соединение 1b (8,1 г, 19,7 ммол) растворяют в 20 мл хлористого метилена и этот раствор по каплям добавляют к предварительно охлажденному (около -60°С) раствору BBr3 (18 мл, около 10 эквивалентов) хлористого метилена под аргоном. При этой низкой температуре выпадает твердое вещество. Смесь медленно доходит до 0° и затем перемешивают один час при этой температуре. Реакционную смесь разбавляют 200 мл CH2Cl2 и «гасят», выливая в холодный, энергично перемешиваемый насыщенный водный раствор Na2СО3 и CH2Cl2 (300 мл). Органический слой отделяют, разбавляют 100 мл МеОН и упаривают в вакууме, растворяют в МеОН (100 мл) и снова упаривают трижды. Остаток растворяют в 400 мл EtOAc, промывают 2Х насыщенным раствором NaHCO3, рассолом, сушат (Na2SO4), фильтруют и упаривают в вакууме, получая 1с в виде твердого вещества (7,2 г, выход 91% ). Протонный и углеродный ЯМР-спектры соответствуют заданной структуре.
Соединение 1d

Соединение 1с (6,23 г, 15,5 ммол), моноэтилмалоновый эфир (2,9 г, 22 ммол), N-метилморфолин (1,75 мл, 15,8 ммол) и гпдрокси-7-азабензотриазол (3,1 г, 23 ммол) частично растворяют в 200 мл СН2Cl2. Эту реакционную смесь охлаждают до 0° и добавляют гидрохлорид (1-[3-(диметиламино)пропил]-3-этилкарбодиимида (4,4 г, 23 ммол) и реакционную смесь перемешивают 2 часа. Реакционную смесь разбавляют 200 мл СН2Cl2 и промывают водой, насыщенным водньм раствором NaHCO3, рассолом, сушат Na2SO4, фильтруют и упаривают Сырой продукт хроматографируют двумя партиями на 300 гсиликагеля Merk каждую 30%-ным этилацетатом в смеси гексанов. Промежуточные фракции объединяют и упаривают, получая 5,3 г (66,7%) чистого продукта. Первые и последние фракции объединяют, из них получают 0,96 г продукта Id, содержащего исходный малонат и неизвестную примесь.
Пример 1
Малоновый эфир 1d (5,180 г, 9,91 ммол) растворяют в 29,5 мл метанола и охлаждают до 0°. Затем, через 5 минут, к реакционной смеси добавляют 1N гидроксид натрия (29,73 ммол, 3 эквив.) и реакционную смесь доводят до комнатной температуры. Через 15 минут метанол удаляют под вакуумом. Затем оставшийся щелочной раствор разбавляют 29,7 мл воды и охлаждают в бане со льдом. К основному раствору по каплям добавляют 1N соляную кислоту до получения рН 1. Образующееся белое «полутвердое» вещество собирают в большой воронке со стеклянным фильтром. Твердое вещество 5 раз промывают холодной водой, затем сушат 3 дня гидроксидом калия в вакууме. Конечный вес заглавного соединения составляет 5,01 г (выход 99%). Масс-спектр соответствует заданной структуре.
1ЯМР (500 мГц, ацетон - D6, δ):8,07 (с, 2Н), 6,75 (м, 2Н ), 6,37 (дд, 1Н, J=8,8,3,3 Гц), 3,51 (с, 2Н), 3,28 (кв, 1Н, J=6,5 Гц), 1,17 (д, 6Н, J=7,1 гГц).
13С ЯМР (500 мГц, метанол - D3, δ): 171,06; 167,36; 151,54; 150,59; 147,15; 138,30; 137,47; 125,04; 119,54; 116,34; 114,14; 113,14; 41,98; 28,15; 22,80.
Элементный анализ соответствует формуле C18H17Br2NO5. 1,75 Н2О: С, 41,68%; Н, 3,89%; N, 2,63%; Br, 30,70%.
Пример 2
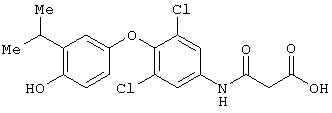
3-[[3,5-дихлор-[4-гидрокси-3-(1-метилэтил)фенокси]-фенилаллиламино]-3-оксопропановая кислота.
Соединение 2а
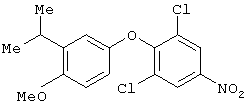
Бис-(3-изопропил-4-метоксифенил)иодонийтетрафторборат (15,0 г, 29,4 ммол), 2,6-дихлор-4-нитрофенол (4,16 г, 20 ммол) и Cu - порошок [Lancaster 300 меш (3,2 г, 50 ммол) суспендируют в 200 мл СН2Cl2 в колбе, покрытой алюминиевой фольгой. При перемешивании добавляют триэтиламин (8,4 мл, 100 ммол) и реакционную смесь перемешивают под аргоном в темноте в течение 5 дней. Сырую реакционную смесь упаривают примерно до 50 мл, а затем хроматографируют через слой силикагеля Merk, 2 л, 3% этилацетатом в смеси гексанов. Общий выход соединения 2а составляет 4,9 г (68,8%). Спектр протонного ЯМР соответствует структуре соединения.
Соединение 2b

Соединение 2а (4,9 г, 13,8 ммол) растворяют в 80 мл ледяной уксусной кислоты и 8 мл воды. Добавляют порошок железа (Aldrich <10 микрон, 4,6 г, 81,6 мл) и реакционную смесь перемешивают под аргоном в течение ночи. Реакционную смесь фильтруют через целит и слой целита промывают 50 мл метанола. Фильтрат упаривают в вакууме. Добавляют насыщенный раствор Na2СО3 (400 мл) и продукт экстрагируют этилацетатом (3Х 500 мл каждая). Этилацетатные вытяжки упаривают в вакууме и остаток хроматографируют через 1,8 л силикагеля Merk смесью этилацетат: смесь гексанов (8:2). Соединение 2b (2,2 г) получают с выходом 49,9% в виде твердого вещества. Протонный и углеродный ЯМР-спектры подтверждают заданную структуру.
Соединение 2c
Соединение 2b (1,8 г, 5,65 ммол) растворяют в 15 мл хлористого метилена и этот раствор под аргоном добавляют по каплям к предварительно охлажденному (около - 60°) раствору BBr3 (5,3 мл, 56,5 ммол) в 40 мл хлористого метилена. При этой низкой температуре выпадает твердый осадок. Реакционную смесь медленно нагревают до 0° и перемешивают при этой температуре один час. Реакционную смесь разбавляют 200 мл CH2Cl2 и гасят, выливая в холодную энергично перемешиваемую смесь насыщенного водного раствора Na2СО3 (200 мл) и СН2Cl2 (200 мл). Органический слой отделяют, разбавляют 100 мл МеОН и упаривают в вакууме и 3Х из МеОН (50 мл каждый). Остаток растворяют в 300 мл EtOAc, промывают 2Х насыщенным NaHCO3, рассолом, сушат (Na2SO4), фильтруют и упаривают в вакууме, получая соединение 2с в виде твердого вещества (1,77 г, выход 99%). Протонный и углеродный ЯМР-спектры подтверждают заданную структуру.
Соединение 2d

К смеси соединения 2с (700 мг, 2,24 ммол), моноэтилмалоната (440 мг, 3,32 ммол), 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида (632 мг, 3,33 ммол), 1-гидроксибензотриазола (450 мг, 3,40 ммол) в CH2Cl2 (24 мл), охлаждаемой в бане со льдом, добавляют N-метилморфолин (41 мкл, 4,46 ммол). Доводят до комнатной температуры и перемешивают под аргоном в течение ночи (примерно 18 час). Смесь разбавляют 30 мл СН2Cl2 и затем последовательно промывают Н2О (3×100 мл), 1N HCl (3×150 мл), насыщенным NaHCO3 (3×120 мл) и рассолом (1×150 мл). СН2Cl2 слой сушат (Na2SO4) и упаривают в вакууме, получая 632 мг белого пенистого вещества. Сырой продукт очищают хроматографией (75 г силикагеля, 20% EtOAc в гексане), получая 500 мг (52%) очищенного соединения 2d в виде белого вещества. Протонный и углеродный ЯМР-спектры и LC/MS подтверждают строение продукта.
Пример 2
К раствору соединения 2d (330 мг, 0,78 ммол) в метиловом спирте (3,9 мл) добавляют 1N водный раствор гидроксида натрия (2,3 мл, 2,3 ммол). Через 20 минут смесь упаривают в вакууме до водного раствора, который разбавляют 3,2 мл дистиллированной воды. Раствор охлаждают до 0° и подкисляют, прибавляя по каплям 1N HCl до рН 1. Белый осадок собирают и сушат в вакууме над гидроксидом калия в течение 18 часов, получая 288 мг (74%) заглавного соединения в виде белого твердого вещества. Протонный и углеродный ЯМР-спектры и спектр LC/MS подтверждают строение заданного продукта.
Пример 3
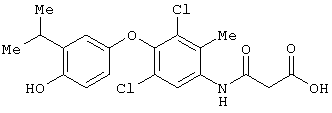
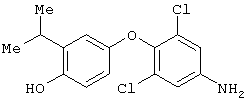
3-[[3,5-дихлор-[4-гидрокси-3-(1-метилэтил)фенокси]-2-метилфенил]амино]-3-оксопропановая кислота.
Соединение 3а

К раствору 4-амино-2,6-дихлор-3-метилфенола (0,70 г, 3,64 ммол) в безводном ТГФ (18 мл), охлаждаемому в бане со льдом, добавляют трифторуксусный ангидрид (0,92 мг, 0,62 мл, 4,39 ммол). Смесь доводят до комнатной температуры. Через час к смеси добавляют EtOAc (50 мл), а затем промывают рассолом (2×25 мл). Экстракт EtOAc сушат над Na2SO4, фильтруют, упаривают и сушат в вакууме, получая 1, 07 г сырого продукта. Сырой продукт очищают хроматографией (50 г силикагеля, 20% EtOAc в гексане), получая 0,93 мг (89%) соединения За в виде твердого вещества светло-оранжевого цвета.
1Н ЯМР (500 мГц, CD3OD, δ) 7,22 (с, 1Н), 2,22 (с, 3Н)
LC - MS ESI- [M-Н]-=286, 288, 290 (100: 64:10)
Соединение 3b

К смеси бис (3-изопропил-4-метоксифенил)иодонийтетрафторбората (3,11 г, 6,07 ммол) и меди (0,31 г, 4,86 ммол) в CH2Cl2 (12 мл) добавляют раствор соединения За (0,70 г, 2,43 ммол) и триэтиламина (0,49 г, 0,68 мл, 4,88 ммол) в CH2Cl2 (12 мл). Смесь перемешивают в темноте при комнатной температуре под азотом в течение 92 часов. Смесь фильтруют через небольшой слой целита и фильтрат упаривают в вакууме. Сырой продукт очищают хроматографией (200 г силикагеля, 10% EtOAc в гексане), получая 0,42 г (40%) соединения 3b в виде светло-оранжевого твердого вещества.
1Н ЯМР (500 мГц, CDCl3, δ): 7,83 (с, 1Н), 7,74 (уш. с., 1Н), 6,86 (д, 1Н, J=2,6 Гц), 6,68 (д, 1Н, J=8,7 Гц), 6,4 (дц, 1Н=8,7, 3 Гц), 3,77 (с, 3Н), 3,27 (М, 1Н), 2,36 (с, 3Н), 1,18 (д, 6Н, 1=7 Гц).
LC- MS ESI- [М-Н]-=434, 436, 438 (100:64:10)
Соединение 3c

К раствору соединения 3b (243 мг, 0,557 ммол) в ледяной уксусной кислоте добавляют водный 48% раствор HBr (5 мл). Смесь нагревают до 120° С и выдерживают при этой температуре 2 часа. Смесь охлаждают до комнатной температуры, а затем упаривают в вакууме. К остатку добавляют EtOAc (75 мл), а затем доводят рН до 7, добавляя водный раствор NaHCO3. Слой EtOAc промывают рассолом (2×25 мл), сушат (MgSO4), фильтруют, упаривают и сушат в вакууме, получая 179 мг твердого сырого продукта фиолетового цвета. Сырой продукт очищают хроматографией (25 г силикагеля, 25% EtOAc в гексане), получая 117,2 мг (64%) соединения 3с в виде белого твердого вещества.
1Н ЯМР (500 мГц, CD3OD, δ) 6,78 (с, 1Н), 6,60 (д, 1н, J=3,3 Гц) 6,58 (д, 1Н, J=8,8 Гц), 6,28 (дд, 1Н, J=8,8, 3,3 Гц), 3,21 (м, 1Н), 1,14 (д, 6Н, J=6,6 Гц).
LC-MS ESI- [М-Н]-=324, 326, 328 (100:64:10)
Соединение 3d

К смеси соединения 3с (78 мг, 0,24 ммол), моноэтилмалоната (47 мг, 0,36 ммол), 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида (69 мг, 0,36 ммол), 1-гидроксибензотриазола (48 мг, 0,36 ммол) в СН2Cl2 (5 мл), охлаждаемой в бане со льдом, добавляют N-метилморфолин (25 мг, 27 мкл, 0,24 ммол). Доводят до комнатной температуры и перемешивают под N2 в течение ночи (около 18 час). Растворяют смесь в EtOAC (50 мл), а затем промывают последовательно Н2О (2×20 мл), 1N HCl (2×20 мл), насыщенным раствором NaHCO3 (2×25 мл) и рассолом (2×25 мл). Слой EtOAc сушат (MgSO4), фильтруют и упаривают в вакууме, получая 136 мг сырого продукта в виде слегка розоватого густого масла. Сырой продукт очищают хроматографией (25 г силикагеля, 30% EtOAc в гексане), получая 82 мг (78%) соединения 3d в виде белого твердого вещества.
1Н ЯМР (500 мГц, CDCl3, δ) 9,56 (с, 1Н), 8,10 (с, 1Н), 6,82 (д, 1Н, J=3,3 Гц), 6,60 (д, 1Н, J=8,3 Гц), 6,34 (дд, 1Н, J=8,8; 2,8 Гц), 4,53 (с, 1Н), 4,28 (кв, 2Н, J=7,1 Гц), 3,53 (с, 2Н), 3,15 (м, 1Н), 2,38 (с, 3Н), 1,34 (т, 3Н, J 7,2 Гц), 1,22 (д, 6Н, J=6,6 Гц)
LC-MS ESI- [М-Н]-=438, 440, 442 (100:64610)
Пример 3
К раствору соединения 3d (70 мг, 0,16 ммол) в ТГФ (1,5 мл) добавляют 1N водный раствор гидроксида лития (0,5 мл, 0,5 ммол). Через час смесь подкисляют 1N HCl, а затем экстрагируют EtOAc (50 мл). Экстракт EtOAc промывают рассолом (2×20 мл), сушат (Na2SO4), фильтруют и упаривают в вакууме, получая 57 мг желтоватого твердого вещества. Сырой продукт содержит следы примеси, поэтому его чистят препаративной обращенно-фазовой хроматографией ПОФХ [градиентная система растворителей, от 50%В: 50%А до 0%А: 100%В (А=90% Н2О/10% МеОН+0,1% ТФК, В=90% МеОН/10% Н2О+0,1% ТФК) за 10 минут, YMC ODS 20×100 мм колонка ], получая 40 мг (61%) заглавного соединения в виде белого твердого вещества.
1Н ЯМР (500 мГц, CD3OD, δ) 7,59 (с, 1Н), 6,69 (д, 1Н, J=2,7 Гц), 6,60 (д, 1Н, J=8,8 Гц), 6,30 (дц, 1Н, J=8,8; 3,3 Гц), 3,50 (с, 2Н), 3,23 (м, 1Н), 2,34 (с, 3Н), 1,16 (д,6H, J=6,6 Гц)
MS ESI-=410, 412, 414 (100:64:10)
Пример 4
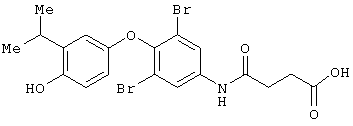
4-[[3,5-дихлор-[4-гидрокси-3-(1-метилэтил)фенокси]-фенил]амино]-4-оксобутановая кислота..
Соединение 4а

К. предварительно охлажденному до -78° раствору соединения 1с (50 мг, 0,125 ммол) в СН2Cl2 (500 мкл) добавляют триэтиламин (26 мкл, 0,18 ммол). Затем по каплям добавляют 3-карбометоксипропионилхлорид (16 мкл, 0,14 ммол). Реакционную смесь перемешивают два часа. Раствор доводят до комнатной температуры и упаривают в вакууме, получая 26 мг (выход 63 %) соединения 4а в виде желтого масла. Протонный ЯМР-спектр и LC/MS соответствуют продукту с примесью диацелированного побочного продукта.
Пример 4
К раствору соединения 4а (23 мг, 0,045 ммол) в метаноле (1,5 мл) добавляют 1N раствор гидроксида натрия (0,08 мл, 0,08 ммол). Через 3 часа смесь упаривают в вакууме. Реакционную смесь охлаждают в бане со льдом и добавляют 1N HCl до достижения значения рН 1. Водный раствор экстрагируют этилацетатом (3×30 мл). Объединенные этилацетатные вытяжки промывают рассолом и сушат Na2SO4. Упаривают в вакууме, получая 15 мг белого полужидкого вещества. Сырой продукт очищают препаративной обращенно-фазовой хроматографией (ПОФХ, HPLC) [градиентная система растворителей, от 50%А: 50%В до 0%а: 100%В (А=90% H2O/10% МеОН+0,1% ТФК, В=90% МеОН/10% Н2О+0,1% ТФК) за 15 минут, колонка YMC ODS 20×100 мм], получая 8,0 мг (36%) заглавного соединения в виде белого твердого вещества. Протонный ЯМР и LC/MS-спектры соответствуют заданному продукту.
Пример 5
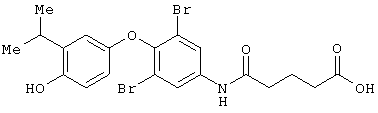
5-[[3,5-дихлор-[4-гидрокси-(1-метилэтил)фенокси]-фенил]амино]-5-оксопентановая кислота
По методике, описанной выше в Примере 4, получают 15 г (выход 36%) заглавного соединения в виде белого твердого вещества. Протонный ЯМР-спектр и спектр LC/MS соответствуют заданной структуре.
Пример 6
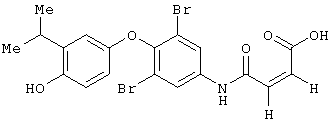
Соединение 6а

К смеси соединения 1с (40 мг, 0,10 ммол), монометилового эфира малеиновой кислоты (36 мкл, 0,29 ммол), гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (72 мг, 0,38 ммол), 1-гидроксибензотриазола (54 мг, 0,40 ммол) в CH2Cl2 (50 мкл), охлаждаемой в бане со льдом, добавляют триэтиламин (46 мкл, 0,028 ммол). Оставляют нагреваться до комнатной температуры и перемешивают под азотом в течение ночи (около 18 часов). Приливают EtOAc (50 мл) и затем последовательно промывают Н2О (2×20 мл), 1N HCl (2×20 мл), насыщенным раствором NaHCO3 и рассолом (2×25 мл). Слой EtOAc сушат (Na2SO4) и упаривают в вакууме, получают 30 мг (58%) сырого продукта в виде светло-розового вязкого масла. Этот продукт пускают на стадию гидролиза. Протонный ЯМР-спектр и спектр LC/MS соответствуют заданному продукту.
Пример 6
К раствору соединения 6а (15 мг, 0,029 ммол ) в метиловом спирте (1,5 мл) добавляют 1N водный раствор гидроксида натрия (0,08 мл, 0,08 ммол). Через 3 часа реакционную смесь упаривают в вакууме для удаления метанола. Образующийся раствор охлаждают в бане со льдом и добавляют 1N HCl до рН 1. Водный раствор экстрагируют этилацетатом (3×30 мл). Объединенные этилацетатные вытяжки промывают рассолом (2×30 мл) и сушат над Na2SO4. Этилацетатные вытяжки упаривают в вакууме, получают 12 мг белого полужидкого продукта. Сырой продукт очищают препаративной обращенно-фазовой хроматографией ПОФХ [градиентная система растворителей, от 50%А: 50%В до 0%А: 100%В (А=Н2О/10% МеОН+0,1% ТФК, В=90% МеОН/10% Н2О+0,1% ТФК) за 15 минут, колонка YMC ODS 20×100 мм], получают 7,9 мг (53%) заглавного соединения в виде белого вещества. Протонный ЯМР-спектр и спектр LC/MS соответствуют заданной структуре.
Пример 7
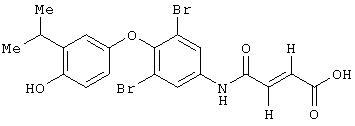
По методике, описанной выше в Примере 6, получают 17,9 мг (46%) заглавного соединения в виде белого твердого вещества. Протонный ЯМР-спектр и спектр LC/MS соответствуют заданной структуре.
Примеры 8-19
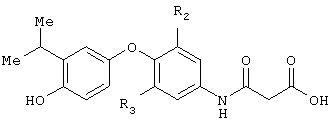
Применяя соответствующие описанные методы наряду с методами, описанными в "Novel Thyroid Receptor Ligands and Methods", Y.-L. Li., Y. Liu, A. Hedfors, J.Malm, C.Mellin, M.Zhang, Международной заявке WO 9900353 A1, 990107, получены соединения в Примерах 8-19, описанные в нижеприведенной таблице.
В приведенной ниже таблице представлены биологические данные для Примеров 1,2 и 3 в патенте.
- in vitro доказательство активности агониста тиреоидного гормона:
- демонстрация прочного рецепторного связывания 150
- демонстрация высокой функциональной активности в клеточном функциональном анализе;
- in vivo демонстрация метаболического действия:
- понижение активности холестерина на крысиной модели показывает возможность лечения дислипидемии, атеросклероза, метаболического синдрома
- увеличение потребления кислорода демонстрирует способность повышать скорость обмена веществ и, следовательно, влиять на уменьшение веса тела при лечении ожирения
2 Связывание с очищенным человеческим рецептором тиреоидного гормона альфа (TR- Е), определяемое замещение меченого радиоактивной меткой L- тиронина (L-T3),
меченного радиоактивной меткой.
3 Функциональная активность, определяемая анализом трансактивации в клетках яичников китайского хомячка, устойчиво трансфецированных с помощью полноразмерного человеческого TR-a рецептора при использовании секретированной
щелочной фосфатазы в качестве контроля. ЕС50 обозначает концентрацию соединения, вызывающую 50% максимальной активности, индуцированной L- тиронином (L-Т3).
4 Функциональная активность, определяемая анализом трансактивации в клетках яичников китайского хомячка, устойчиво трансфецированных с помощью полноразмерного человеческого TR-E рецептора при использовании секретированной щелочной фосфатазы в качестве контроля. ЕС50 обозначает концентрацию соединения, вызывающую 50% максимальной активности, индуцированной L-тиронином (L-T3).
5 Снижение активности холестерина определяют на подкармливаемых
холестерином тощих крысах Sprague-Dawley (самцы). Через 2 недели кормления холестерином животным дают п.о. (перорально) носитель или в возрастающих дозах одно из соединений всего в течение 7 дней, затем (в это время) определяют уровни холестерина в плазме крови. ED50 обозначает концентрацию соединения, вызывающую снижение уровня холестерина на 50% по сравнению с носителем.
6 Увеличение скорости обмена веществ (метаболизма) измеряют, определяя потребление кислорода (мл/кг/час) у подкармливаемых холестерином тощих крыс Sprague- Dawley (самцы). Через 2 недели кормления холестерином животным дают п.о.
(перорально) носитель или в возрастающих дозах одно из соединений всего в течение 7 дней, затем (в это время) определяют потребление кислорода. ED5 обозначает концентрацию соединения, вызывающую снижение скорости обмена веществ на 5% по сравнению с носителем.
| название | год | авторы | номер документа |
|---|---|---|---|
| О-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2269540C2 |
| С-АРИЛГЛЮКОЗИДНЫЕ ИНГИБИТОРЫ SGLT2 | 2000 |
|
RU2262507C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| ПРОИЗВОДНЫЕ БЕНЗАМИДА ИЛИ ФЕНИЛАЦЕТАМИДА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ЛИГАНДОВ ТИРЕОИДНОГО РЕЦЕПТОРА | 2003 |
|
RU2317287C2 |
| ПРОИЗВОДНЫЕ АЛЬФА-(N-СУЛЬФОНАМИДО)АЦЕТАМИДА КАК ИНГИБИТОРЫ БЕТА-АМИЛОИДА | 2002 |
|
RU2300518C2 |
| МОДУЛЯТОРЫ PPAR | 2007 |
|
RU2449999C2 |
| ОКСА- И ТИАЗОЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИДИАБЕТИЧЕСКИХ АГЕНТОВ И АГЕНТОВ ПРОТИВ ОЖИРЕНИЯ | 2000 |
|
RU2279427C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ ИНГИБИТОРА SGLT2 | 2013 |
|
RU2643764C2 |
| С-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2746132C1 |
| С-АРИЛ ГЛЮКОЗИДНЫЕ SGLT2 ИНГИБИТОРЫ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2800510C1 |
Получены новые лиганды тироидных рецепторов общей формулы  где Х обозначает кислород (-O-); Y обозначает -(СН2)n-, n обозначает целое число от 1 до 3, или -С=С-, который представляет собой цис- или транс-изомер; R1 обозначает алкил, содержащий 3-6 атомов углерода; R2 и R3 одинаковы или различны и обозначают водород, галоген, алкил, содержащий 1-4 атома углерода, при этом, по меньшей мере, один из R2 и R3 не обозначает водород; R4 обозначает водород или низший алкил; R5 обозначает водород; R6 обозначает карбоновую кислоту или ее эфир; R7 обозначает водород, включая все его стереоизомеры или его фармацевтически приемлемую соль. Новые соединения могут использоваться для предупреждения, подавления или лечения заболевания, обусловленного дисфункцией обмена веществ или зависящего от экспрессии Т3 регулируемого гена, например, таких заболеваний, как ожирение, гиперхолистеринемия, атеросклероз, сердечная аритмия, депрессия, остеопороз, гипотиреоз, зоб, рак щитовидной железы, а также глаукому, застойную сердечную недостаточность и поражения кожи. 3 н. и 16 з.п. ф-лы, 2 табл.
где Х обозначает кислород (-O-); Y обозначает -(СН2)n-, n обозначает целое число от 1 до 3, или -С=С-, который представляет собой цис- или транс-изомер; R1 обозначает алкил, содержащий 3-6 атомов углерода; R2 и R3 одинаковы или различны и обозначают водород, галоген, алкил, содержащий 1-4 атома углерода, при этом, по меньшей мере, один из R2 и R3 не обозначает водород; R4 обозначает водород или низший алкил; R5 обозначает водород; R6 обозначает карбоновую кислоту или ее эфир; R7 обозначает водород, включая все его стереоизомеры или его фармацевтически приемлемую соль. Новые соединения могут использоваться для предупреждения, подавления или лечения заболевания, обусловленного дисфункцией обмена веществ или зависящего от экспрессии Т3 регулируемого гена, например, таких заболеваний, как ожирение, гиперхолистеринемия, атеросклероз, сердечная аритмия, депрессия, остеопороз, гипотиреоз, зоб, рак щитовидной железы, а также глаукому, застойную сердечную недостаточность и поражения кожи. 3 н. и 16 з.п. ф-лы, 2 табл.

где
X обозначает кислород (-O-);
Y обозначает -(СН2)n-, где n обозначает целое число от 1 до 3, или -С=С-, который представляет собой цис- или транс-изомер;
R1 обозначает алкил, содержащий 3-6 атомов углерода;
R2 и R3 одинаковы или различны и обозначают водород, галоген, алкил, содержащий 1-4 атома углерода, при этом, по меньшей мере, один из R2 и R3 не обозначает водород;
R4 обозначает водород или низший алкил;
R5 обозначает водород;
R6 обозначает карбоновую кислоту или ее эфир;
R7 обозначает водород;
включая все его стереоизомеры или его фармацевтически приемлемую соль.

или

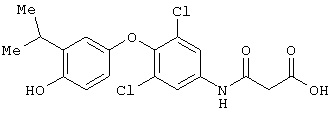
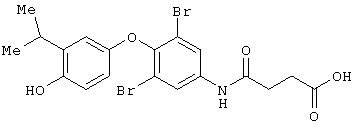 или
или 
или

| RU 95108547 A1, 10.02.1997 | |||
| WO 9900353 A1, 07.01.1999 | |||
| N.YOKIYAMA | |||
| Synthesis and structure activity relationship ofoxamic acid acetic acid derivatives related to L-thyronine | |||
| Journal of medicinal chemistry | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |

