Область изобретения
Настоящее изобретение относится к новым соединениям, имеющим сродство к одному или более чем одному GABAВ (γ-аминомасляная кислота) рецептору, а также их фармацевтически приемлемым солям, сольватам и стереоизомерам. Изобретение также относится к способам их получения, фармацевтическим композициям, содержащие указанные терапевтически активные соединения, и к применению указанных активных соединений в терапии.
Предшествующий уровень техники
Рефлюкс
Гастроэзофагеальная рефлюксная болезнь (GORD) является наиболее распространенным заболеванием верхнего отдела желудочно-кишечного тракта. Современная терапия стремится к уменьшению секреции желудочной кислоты или к уменьшению экспозиции кислоты в пищеводе путем повышения пищеводного клиренса, тонуса нижнего пищеводного сфинктера и опорожнения желудка. Ранее считалось, что главный механизм, лежащий в основе рефлюкса, зависит от гипотонического состояния нижнего пищеводного сфинктера. Однако недавнее исследование (например, Holloway & Dent (1990) GastRoenteRol.Clin. N. Amer. 19, 517-535) показало, что большинство приступов рефлюкса происходит во время преходящих расслаблений нижнего пищеводного сфинктера, ниже упоминаемых как TLOSR, то есть расслаблении, не инициируемых глотаниями. Также было показано, что у пациентов с GORD секреция желудочной кислоты обычно является нормальной.
Следовательно, существует потребность в соединениях, которые снижают число случаев TLOSR и таким образом предотвращают рефлюкс.
Фармацевтические композиции, содержащие местный анестетик, применяемые для ингибирования расслабления нижнего пищеводного сфинктера, раскрыты в WO 87/04077 и в US 5036057. Недавно было показано, что агонисты ОАВАв-рецепторов ингибируют TLOSR, что раскрыто в WO 98/11885.
Агонисты GABAB рецепторов
GABA (4-аминомасляная кислота) является эндогенным нейромедиатором в центральной и периферической нервной системах. Рецепторы к GABA традиционно разделяют на подтипы GABAA и GABAB рецепторы. GABAB рецепторы относятся к надсемейству рецепторов, связанных с G-белками. Агонисты GABAB рецепторов описаны как полезные при лечении нарушений ЦНС (центральной нервной системы), таких как мышечное расслабление при спинальной спастичности, сердечно-сосудистых нарушений, астмы, нарушений кишечной моторики, таких как синдром раздраженной толстой кишки (IBS), и в качестве прокинетических и противокашлевых агентов. Агонисты GABAB рецепторов также были описаны как пригодные для лечения рвоты (WO 96/11680) и недавно, как указано выше, для ингибирования TLOSR (WO 98/11885).
Наиболее изученным агонистом GABAB рецепторов является баклофен (4-амино-3-(хлорфенил)масляная кислота), раскрытый в швейцарском патенте №СН 449046. Баклофен в течение нескольких лет применяли в качестве антиспазматического агента. ЕР 0356128 описывает применение отдельного соединения (3-аминопропил)метилфосфиновой кислоты, в качестве сильного агониста GABAB рецепторов, в терапии. ЕР 0181833 раскрывает замещенные 3-аминопропилфосфиновые кислоты, которые, как обнаружено, имеют очень высокое сродство к местам связывания GABAB рецепторов. По аналогии с баклофеном, эти соединения можно применять в качестве, например, мышечных релаксантов. ЕР 0399949 раскрывает производные (3-аминопропил)метилфосфиновой кислоты, которые описаны как сильные агонисты GABAB рецепторов. Установлено, что эти соединения пригодны в качестве мышечных релаксантов. Обе заявки, ЕР 0463969 и FR 2722192, относятся к производным 4-аминомасляной кислоты, имеющим разные гетероциклические заместители на 3-углероде бутиловой цепи. Взаимосвязи структура-активность некоторых аналогов фосфиновой кислоты по отношению к их сродству к GABAB рецептору, а также их действие на мышечное расслабление, обсуждаются в J. Med. Chem. (1995), 38, 3297-3312. Заключением в данной статье является то, что, по сравнению с баклофеном, значительно более сильного мышечного расслабления можно достичь сприменением (S)-энантиомера 3-амино-2-гидроксипропилметилфосфиновой кислоты, и без нежелательных влияний на ЦНС.
В литературе фосфиновые кислоты, имеющие атом водорода, присоединенный к фосфору, также называются фосфонистые кислоты. Это два названия для одних и тех же соединений, и оба названия можно использовать. Однако для соединений по настоящему изобретению авторы выбрали для использования название фосфиновые кислоты.
Краткое изложение сущности изобретения
В настоящем изобретении предложены новые соединения формулы I
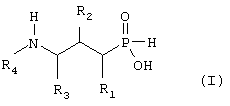
где
R1 представляет собой водород, гидрокси, низший алкил, низший алкокси или галоген;
R2 представляет собой гидрокси, меркапто, галоген или оксогруппу;
R3 представляет собой водород или низший алкил (возможно замещенный гидрокси, меркапто, низшим алкокси, низшим тиоалкокси или арилом);
R4 представляет собой водород, низший алкил (возможно замещенный арилом), или арил;
и их фармацевтически приемлемые соли, сольваты и стереоизомеры, за исключением:
1) рацемата (3-амино-2-гидроксипропил)фосфиновой кислоты, и
2) (2R/S, 3R)-(3-амино-2-гидроксибутил)фосфиновой кислоты.
В предпочтительном воплощении
R1 представляет собой водород, низший алкил или галоген;
R2 представляет собой галоген, гидрокси или оксогруппу;
R3 представляет собой водород; и
R4 представляет собой водород;
за исключением рацемата (3-амино-2-гидроксипропил)фосфиновой кислоты.
Еще более предпочтительными соединениями являются (3-амино-2-фторпропил)фосфиновая кислота, (2R)-(3-амино-2-фторпропил)фосфиновая кислота, (2S)-(3-амино-2-фторпропил)фосфиновая кислота, (3-амино-2-фтор-1-метилпропил)фосфиновая кислота, (3-амино-2-оксопропил)фосфиновая кислота, (2S)-(3-амино-2-гидроксипропил)фосфиновая кислота, (2R)-(3-амино-2-гидроксипропил)фосфиновая кислота и (3-амино-1-фтор-2-гидроксипропил)фосфиновая кислота.
В рамках объема данного изобретения следует понимать, что когда R1 является оксогруппой, связь между R2 и углеродом является двойной связью.
В рамках объема данного изобретения под "низшими" радикалами и соединениями следует понимать, например, соединения, имеющие до и включая 7, особенно до и включая 4 углеродных атома. Также основные выражения имеют следующие значения:
Низший алкил представляет собой, например, С1-С4алкил, такой как метил, этил, н-пропил или н-бутил, а также изопропил, изобутил, вторичный бутил или третичный бутил, но также может быть С5-С7алкильной группой, такой как пентильная, гексильная или гептильная группа.
Низший алкокси представляет собой, например, С1-С4алкокси, такой как метокси, этокси, н-пропокси или н-бутокси, также изопропокси, изобутокси, вторичный бутокси или третичный бутокси, но также может быть С5-С7 алкоксигруппой, такой как пентокси, гексокси или гептоксигруппа.
Низший тиоалкокси представляет собой, например, С1-С4тиоалкокси, такой как тиометокси, тиоэтокси, н-тиопропокси или н-тиобутокси, также тиоизопропокси, тиоизобутокси, вторичный тиобутокси или третичный тиобутокси, но также может быть С5-С7 тиоалкоксигруппой, такой как тиопентокси, тиогексокси или тиогептоксигруппа.
Галоген представляет собой галоген с атомным номером до и включая 35, такой как фтор или хлор, и менее предпочтительно бром.
Соединения формулы I по изобретению имеют амфотерный характер и могут существовать в форме внутренних солей. Они также могут образовывать соли присоединения кислот и соли с основаниями. Такие соли представляют собой в частности фармацевтически приемлемые соли присоединения кислот, а также фармацевтически приемлемые соли, образованные с основаниями. Подходящие кислоты для образования таких солей включают в себя, например, минеральные кислоты, такие как соляная, бромистоводородная, серная или фосфорная кислота, или органические кислоты, такие как сульфоновые кислоты и карбоновые кислоты. Соли с основаниями представляют собой, например, соли щелочных металлов, например соли натрия или калия, или соли щелочноземельных металлов, например соли кальция или магния, а также соли аммония, такие как соли аммония или органических аминов. Эти соли можно получить традиционными способами.
Когда в молекуле имеется один или более чем один стереоцентр, соединения формулы I могут находиться в форме смеси стереоизомеров, то есть смеси диастереомеров и/или рацематов, или в форме отдельных стереоизомеров, то есть отдельного энантиомера и/или диастереомера. Эти соединения также могут находиться в форме сольватов, например гидратов.
Все соединения формулы I можно применять для ингибирования TLOSR, и таким образом для лечения гастроэзофагеальной рефлюксной болезни. Указанное ингибирование TLOSR также предполагает, что указанные соединения формулы I можно применять для лечения срыгивания у младенцев. Эффективная терапия срыгивания у младенцев была бы важным путем лечения, недостаточно успешной из-за чрезмерной потери проглоченого питательного вещества. Кроме того, новые соединения можно применять для лечения связанных с GORD или не связанных с GORD астмы, отрыжки, кашля, боли, привыкания к кокаину, икоты, IBS, диспепсии, рвоты и ноцицепции.
В противоположность тому, что установлено в уровне техники (J. Med. Chem. (1995) 3297-3312 и The GABA Receptors: Second Edition. Edited by S. J. Enna and Norman Bowery, Humana PRess (1997) особенно стр.281-282), соединения по изобретению обладают неожиданно высокой метаболической стабильностью, несмотря на присутствие Р-Н связи. Данные соединения также обладают неожиданно высоким терапевтическим индексом.
Получение
Соединения формулы I по настоящему изобретению можно получать одним из следующих способов.
А) Соединение формулы II,
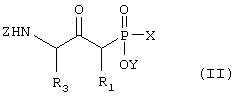
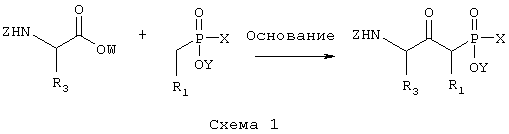
где R1 и R3 такие, как определено выше в формуле I, X представляет собой водород или защитную группу, такую как -ССН3(ОСН2СН3)2, Z представляет собой защитную группу, такую как трет-бутилоксикарбонил, и Y представляет собой водород или защитную группу, такую как низший алкил, соединение формулы II, причем данное соединение возможно синтезировали реакцией конденсации согласно Схеме I с использованием подходящего N-защищенного аминокислотного сложного эфира, в котором R3 такой, как определено выше, W является защитной группой, такой как низший алкил, и Z такой, как определено в формуле II, и подходящего защищенного производного фосфиновой кислоты, в котором R1 такой, как определено выше в формуле I, X и Y такие, как определено в формуле II, и основания, такого как диизопропиламид лития,
а) возможно превращают путем реакции N-алкилирования для введения R4, если желательно, чтобы R4 не представлял собой водород, и затем гидролитической реакции с получением соединения формулы III
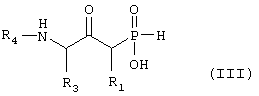
где R1, R3 и R4 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение III в другое химическое соединение формулы III, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы III и/или в другую соль, и/или превращают полученное свободное соединение формулы III в соль в соответствии с вышеуказанным определением, или б) превращают путем восстановительной реакции, возможно реакции N-алкилирования, если желательно, чтобы R4 не представлял собой водород, и, наконец, гидролитической реакции с получением соединения формулы IV
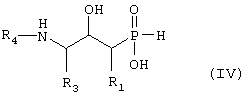
где R1, R3 и R4 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение IV в другое химическое соединение формулы IV, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы IV и/или в другую соль, и/или превращают полученное свободное соединение формулы IV в соль в соответствии с вышеуказанным определением, или
в) превращают путем восстановительной реакции с последующей реакцией дезоксигалогенирования, возможно реакции N-алкилирования для введения R4, если желательно, чтобы R4 не представлял собой водород, и, наконец, гидролитической реакции с получением соединения формулы V
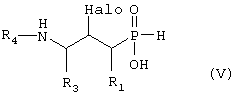
где R1, R3 и R4 такие, как определено выше в формуле I, и Halo представляет собой атом галогена, и возможно превращают полученное выше соединение V в другое химическое соединение формулы V, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы IV и/или в другую соль, и/или превращают полученное свободное соединение формулы V в соль в соответствии с вышеуказанным определением, или
Б) соединение формулы VI
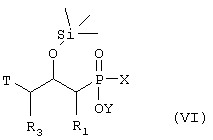
где R1 и R3 такие, как определено выше в формуле I, X представляет собой водород или защитную группу, такую как - ССН3(ОСН-СН3)2, Т представляет собой группу, которую можно превратить в - NH2 группу, и Y представляет собой водород или защитную группу, такую как низший алкил, причем соединение формулы VI возможно синтезировали реакцией конденсации согласно Схеме 2 с использованием 2,3-эпоксипропильного производного, такого как подходящее N-защищенное производное 2,3-эпоксипропиламина или эпихлоригидриновое производное, в котором R1 и R3 такие, как определено выше в формуле I, и подходящего защищенного производного фосфиновой кислоты, активированного O-силилированием, в котором Х и Y такие, как определено в формуле VI, и кислоты Льюиса, такой как безводный ZnCl2,
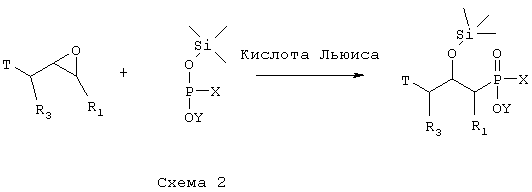
а) превращают путем реакции, при которой триметилсилильную группу замещают атомом водорода, реакции, при которой Т группу, как она определена в формуле VI, превращают в -NHR4, где R4 такой, как определено выше в формуле I, и, наконец, гидролитической реакции с получением соединения формулы IV
где R1, R3 и R4 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение IV в другое химическое соединение формулы IV, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы IV и/или другую соль, и/или превращают полученное свободное соединение формулы IV в соль в соответствии с вышеуказанным определением,
или
б) превращают путем реакции, при которой триметилсилильную группу замещают водородом, окислительной реакции, реакции, при которой Т группу, как она определена в формуле IV, превращают в - NHR4, где R4 такой, как определено выше в формуле I, и, наконец, гидролитической реакции с получением соединения формулы III
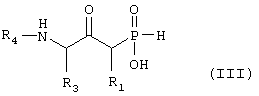
где R1, R3 и R4 такие, как определено выше в формуле I, и, возможно, превращают полученное выше соединение III в другое химическое соединение формулы III, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы III и/или в другую соль, и/или превращают полученное свободное соединение формулы III в соль в соответствии с вышеуказанным определением, или
в) превращают путем реакции, при которой триметилсилильную группу замещают водородом, реакции дезоксигалогенирования, реакции, при которой Т группу, как она определена в формуле VI, превращают в -NHR4, где R4 такой, как определено выше в формуле I, и, наконец, гидролитической реакции с получением формулы V,
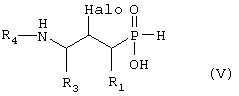
где R1, R3 и R4 такие, как определено выше в формуле I, и Halo представляет собой атом галогена, и возможно превращают полученное выше соединение V в другое химическое соединение формулы V, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы V и/или в другую соль, и/или превращают полученное свободное соединение формулы V в соль в соответствии с вышеуказанным определением, или В) соединение формулы VII,
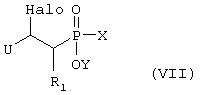
где R1 такой, как определено выше в формуле I, X представляет собой водород или защитную группу, такую как - ССН3(ОСН2СН3)2, U представляет собой электрон-акцепторную группу, такую как, например, -CN или - CO2Et, которую можно превратить в -CH2NH2 группу, и Y представляет собой водород или защитную группу, такую как низший алкил, и Halo представляет собой атом галогена, причем соединение формулы VII возможно синтезировали реакцией присоединения согласно Схеме 3 с использованием ненасыщенного соединения, в котором R1 такой, как определено выше в формуле I, U и Halo такие, как определено в формуле VII, и подходящего защищенного производного фосфиновой кислоты, активированного O-силилированием, где Х и Y такие, как определено в формуле VII,
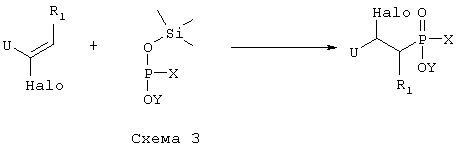
превращают путем реакции, при которой U группу превращают в -NHR4, где R4 такой, как определено выше в формуле I, и гидролитической реакцией с получением соединения формулы VIII,
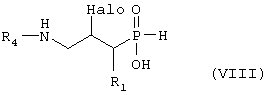
где R1 и R4 такие, как определено выше в формуле I, и Halo представляет собой атом галогена, и возможно превращают полученное выше соединение VIII в другое химическое соединение формулы VIII, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы VIII и/или в другую соль, и/или превращают полученное свободное соединение формулы VIII в соль в соответствии с вышеуказанным определением; или
Г) соединение формулы IX, возможно в виде отдельного стереоизомера,
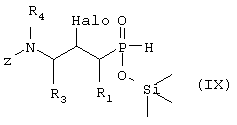
где R1, R3 и R4 такие, как определено выше в формуле I, Z представляет собой защитную группу, такую как трет-бутилоксикарбонил, и Halo представляет собой атом галогена, причем соединение формулы IX возможно синтезировали реакцией замещения согласно Схеме 4 с использованием электрофильного соединения, в котором R1, R3 и R4 такие, как определено выше, L является уходящей группой, такой как йодо, Z и Halo такие, как определено выше, и фосфиновой кислоты, активированной O-силилированием,
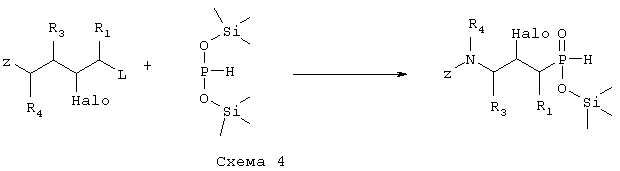
превращают путем гидролитической реакции в соединение формулы V
где R1, R3 и R4 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение V в другое химическое соединение формулы V, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы V и/или другую соль, и/или превращают полученное свободное соединение формулы V в соль в соответствии с вышеуказанным определением; или
Д) соединение формулы XI,
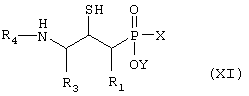
где R1, R3 и R4 такие, как определено выше в формуле I, X представляет собой водород или защитную группу, такую как - ССН3(ОСН2СН3)2, и Y представляет собой водород или защитную группу, такую как низший алкил, причем соединение формулы XI возможно синтезировали при помощи реакции присоединения согласно Схеме 4, обрабатывая ненасыщенное производное фосфиновой кислоты, в котором R1, R3 и R4 такие, как определено выше в формуле I, H2S, меркаптидионом (HS-) или защищенным меркаптосоединением, таким как бензилтиол, в случае чего защитную группу после этого удаляют,
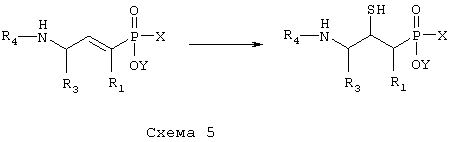
превращают путем гидролитической реакции с получением соединения формулы XII,
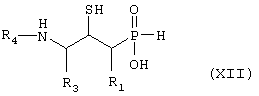
где R1, R3 и R4 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение XII в другое химическое соединение формулы XII, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы XII и/или в другую соль, и/или превращают полученное свободное соединение формулы XII в соль в соответствии с вышеуказанным определением.
Подробное описание изобретения.
Изобретение описано более детально следующими неограничивающими примерами.
Пример 1. (3-Амино-2-Фторпропил)фосфиновая кислота
К охлажденному на ледяной бане раствору этил-(3-амино-2-фтор-3-оксопропил)(диэтоксиметил)фосфината в THF (тетрагидрофуране) добавляли 1 М ВН3-THF в атмосфере аргона. Через 10 минут раствор нагревали с обратным холодильником в течение 2,5 часов. Раствор охлаждали до комнатной температуры, и добавляли 6 н. HCl (200 мл). THF удаляли выпариванием на роторном испарителе, и водный слой кипятили с обратным холодильником в течение 2,5 часов. Раствор охлаждали и упаривали. Остаток очищали ионообменной колоночной хроматографией (DOWEX® 50WX-8-200, Н+ форма, 3,5×4,0 см). Ионообменную смолу предварительно промывали смесью 2:1 метанол/вода (400 мл). Неочищенный продукт, растворенный в смеси 1:1 метанол/вода, наносили на колонку и промывали смесью 1:1 метанол/вода (400 мл). Элюент заменяли на смесь 3:1 метанол/концентрированный гидроксид аммония. Объединяли две фракции (всего 150 мл) и упаривали с получением 645 мг (34%) (3-амино-2-фторпропил)фосфиновой кислоты в виде белого твердого вещества. Данные: точка плавления 203-207°С, Rf=0,35 (60:40:1 метанол, метиленхлорид, концентрированный гидроксид аммония); 1H ЯМР (300 МГц, D2O) δ 7.11 (d, J=528 Гц, 1Н), 5.18 (dm, J=54 Гц, 1Н), 3.28-3.45 (m, 2H). 1.65-2.23 (m, 2H); 13С ЯМР (125 МГц, D2O + Диоксан) δ 87.8 (d, J=170 Гц), 44.3 (dd, J=12.6, 21,6 Гц), 35.6 (dd. J=20,2, 86,5 Гц); APIMS (масс-спектрометрия с ионизацией при атмосферном давлении): m/z=142 (М+Н)+.
Пример 2.
(2S)-(3-Амино-2-гидроксипропил)фосфиновая кислота
Смесь этил-(2S)-(3-амино-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината (1,0 г, 3,5 ммоль) и концентрированной HCl (50 мл) нагревали с обратным холодильником в течение 2 часов. Раствор охлаждали до комнатной температуры и упаривали. Остаток растворяли в метаноле (100 мл) и обрабатывали пропиленоксидом (2 мл) при комнатной температуре. После перемешивания смеси в течение 5 часов, осевшее твердое вещество собирали, декантируя растворитель. Твердое вещество сушили струей аргона с получением 220 мг (45%) (2S)-(3-амино-2-гидроксипропил)фосфиновой кислоты в виде белого твердого вещества. Данные: 1H ЯМР (300 МГц, D2O) δ 7.1 (d, J=540 Гц, 1Н), 4.2 (m, 1H), 2.9-3.2 (m, 2H), 1.7-2.0 (m, 2H); 31P ЯМР (121 МГц, D2O) δ 24.2 (d, J=522 Гц); FABMS (масс-спектрометрия с бомбардировкой быстрами атомами): m/z=140 (М+Н)+;[α]D при 20°С=+8° (0,5% в 0,1 M HCl).
Пример 3.
(2R)-(3-Амино-2-гидроксипропил)фосфиновая кислота
Смесь этил-(2R)-(3-амино-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината (0,9 г, 3,2 ммоль) и концентрированной HCl (50 мл) нагревали с обратным холодильником в течение 2 часов. Раствор охлаждали до комнатной температуры и упаривали. Остаток растворяли в метаноле (50 мл) и обрабатывали пропиленоксидом (3 мл) при комнатной температуре. После перемешивания смеси в течение 5 минут осажденное твердое вещество собирали декантацией растворителя. Твердое вещество сушили потоком аргона с получением 260 мг (59%) (2R)-(3-амино-2-гидроксипропил)фосфиновой кислоты в виде белого твердого вещества. Данные: 1H ЯМР (300 МГц, D2O) δ 7.1 (d. J=540 Гц, 1H). 4.2 (m, 1H), 2.9-3.2 (m, 2H), 1.7-2.0 (m, 2H); 31P ЯМР (121 МГц, D2O) δ 23.9 (d, J=525 Гц); FABMS: m/z=140 (М+Н)+; [α]D при 20°С=-8° (0,5% в 0.1 М HCl).
Пример 4.
(3-Амино-2-оксопропил)фосфиновая кислота
Образец этил-[3-[N-(трет-бутоксикарбонил)амино]-2-оксопропил](1,1-диэтоксиэтил)фосфината (8,11 г, 21,0 ммоль) растворяли в 3 н. HCl (400 мл), которую предварительно обескислороживали барботированием N2 через раствор. Смесь перемешивали в течение 14 часов при комнатной температуре и затем концентрировали. Остаток упаривали совместно с метанолом. Остаток затем растворяли в метаноле (10 мл), и добавляли пропиленоксид (10 мл). Смесь перемешивали в течение 6 часов, и полученный в результате осадок выделяли фильтрованием. Твердое вещество промывали холодным метанолом и сушили под вакуумом при 50°С с получением 2,1 г (73%) (3-амино-2-оксопропил)фосфиновой кислоты в виде желтоватого твердого вещества.
Данные: Точка плавления 126-127°С; Rf=0,64 (85:15 метанол, вода); 1H ЯМР(300 МГц, D2O) δ 7.13 (d, J=551 Гц, 1Н), 4.14 (s, 2H), 3.14 (d, J=18 Гц, 2Н); 13С ЯМР (75 МГц, D2O+Диоксан) (199.5, 49.2, 47.3 (d, J=69 Гц); FABMS: т/г=138 (М+Н)+.
Пример 5.
(2R)-(3-Амино-2-фторпропил)фосфиновая кислота
Гипофосфит аммония (73,8 г, 0,89 моль) добавляли в 3-горлую 2-литровую колбу, снабженную механической мешалкой, термометром, воронкой для добавления и барботером аргона. Колбу помещали в водяную баню при комнатной температуре, и добавляли N,O-бис-(триметилсилил)ацетамид (215 мл, 0,87 мол - BSA) с такой скоростью, чтобы поддерживать температуру внутри ниже 38°С (приблизительно 30 минут), используя охлаждение льдом. По окончании добавления BSA реакционную смесь нагревали до 45-48°С и поддерживали при этой температуре в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, и к реакционной смеси добавляли раствор трет-бутил-(2R)-2-фтор-3-йодпропилкарбамата (27,3 г, 0,09 моль) в метиленхлориде (300 мл). Реакционную смесь затем оставляли перемешиваться при комнатной температуре в течение 18 часов. Реакционную смесь охлаждали до 0°С и осторожно гасили метанолом (275 мл) и затем водой (32 мл). Реакционную смесь перемешивали в течение 30 минут, после чего ее фильтровали, и твердые вещества промывали метанолом. Фильтрат концентрировали, и остаток помещали в глубокий вакуум (0,1 мм рт. ст. (13,33 Па)) на ночь. Неочищенный остаток растирали со смесью метиленхлорида, метанола, концентрированного раствора аммиака (80:20:1) и фильтровали. Фильтрат концентрировали при пониженном давлении, и растирание повторяли. Неочищенный концентрат переносили в 2-литровую колбу, растворяли в метаноле (375 мл) и помещали на водяную баню при комнатной температуре. Добавляли насыщенный раствор газообразного хлористого водорода в этилацетате (500 мл), и смесь перемешивали в течение 3 часов. Реакционную смесь фильтровали, и твердые вещества промывали смесью метанола и этилацетата (90:10). Фильтрат концентрировали при пониженном давлении, и неочищенный продукт пропускали через колонку с Dowex® 50WX8-200 меш в Н+ форме (500 г, 8×15 см), элюируя смесью 1:1 метанол/вода, до тех пор, пока вещество более не обнаруживалось посредством ТСХ (тонкослойная хроматография) анализа. Требуемый неочищенный продукт затем элюировали смесью 1:3 концентрированный раствор гидроксида аммония/метанол. Продукт затем очищали колоночной хроматографией, элюируя смесью хлороформа, метанола, концентрированного раствора гидроксида аммония (6:3:1) с получением (2R)-(3-амино-2-фторпропил)фосфиновой кислоты в виде белого твердого вещества (3,12 г, 24%). 1H ЯМР (300 МГц, D2O) δ 7.90 (s. 0,5 Н), 6.15 (s, 0.5 Н), 5.12-5.29 (m, 0,5 Н). 4.92-5.10 (m, 0,5 Н), 3.12-3.42 (m. 2H), 1.74-2.26 (m. 2Н).
Пример 6.
(2S)-(3-Амино-2-фторпропил)фосфиновая кислота
Гипофосфит аммония (58,1 г, 0,70 моль) добавляли в 3-горлую 2-литровую колбу, снабженную механической мешалкой, термометром, воронкой для добавления и барботером аргона. Добавляли N,O-бис-(триметилсилил)ацетамид (175,9 мл, 0,71 моль - BSA) с такой скоростью, чтобы поддерживать температуру внутри между 35-40°С. По окончании добавления BSA реакционную смесь поддерживали при 35-40°С в течение 45 минут. Добавляли метиленхлорид (150 мл), и смесь перемешивали при 35-40°С в течение еще 45 минут. Реакционную смесь охлаждали до комнатной температуры, и к реакционной смеси добавляли раствор трет-бутил-(2S)-2-фтор-3-йодпропилкарбамата (42,5 г, 0,14 моль) в метиленхлориде (300 мл). Реакционную смесь затем оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь охлаждали до 0°С и осторожно гасили метанолом (150 мл) и затем водой (60 мл). Реакционную смесь концентрировали, и остаток помещали в глубокий вакуум (0,1 мм рт. ст. (13,33 Па)). Остаток доводили до приблизительно рН 8 добавлением концентрированного гидроксида аммония (50 мл), затем добавляли метиленхлорид (400 мл) и метанол (250 мл). Полученные твердые вещества отфильтровывали, и фильтрат концентрировали. Остаток растирали со смесью метиленхлорида, метанола, концентрированного раствора аммиака (80:20:1, 400 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении, и неочищенный концентрат растворяли в метаноле (400 мл). Добавляли насыщенный раствор газообразного хлористого водорода в этилацетате (600 мл), и смесь перемешивали в течение 3 часов. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении.
Неочищенный продукт пропускали через колонку с Dowex® 50WX8-200 меш в Н+ форме (450 г), элюируя смесью 1:1 метанол/вода, до тех пор пока вещество более не обнаруживалось посредством ТСХ анализа. Требуемый неочищенный продукт затем элюировали смесью 1:3 концентрированный раствор гидроксида аммония/метанол. Продукт затем очищали колоночной хроматографией, элюируя смесью метиленхлорида, метанола, концентрированного раствора гидроксида аммония (6:3:1) с получением (2S)-(3-амино-2-фторпропил)фосфиновой кислоты в виде белого твердого вещества (3,46 г, 17%). 1H ЯМР (300 МГц, D2O) δ 7.90 (s. 0,5 Н), 6.15 (s, 0,5 Н), 5.12-5.29 (m. 0,5 Н), 4.92-5.10 (m. 0.5 Н), 3.12-3.42 (m. 2H), 1.74-2.
Пример 7.
(3-Амино-1-фтор-2-гидроксипропил)фосфиновая кислота
Этил-(3-(N-(трет-бутоксикарбонил)амино)-1-фтор-2-гидроксипропил)(1,1-диэтоксиэтил)фосфинат (180 мг, 4,5 ммоль) растворяли в метаноле (2 мл), обрабатывали 3 н. соляной кислотой (20 мл, 60,0 ммоль, пробарботированной аргоном непосредственно перед использованием). Эту смесь перемешивали при комнатной температуре в течение 6 часов в атмосфере аргона. Реакционную смесь концентрировали при пониженном давлении, неочищенный продукт повторно растворяли в метаноле (5 мл);
остаточную воду удаляли совместным выпариванием при пониженном давлении с метанолом. Неочищенный продукт (70 мг) очищали колоночной хроматографией (1×10 см колонка), элюируя смесью метиленхлорида, метанола, концентрированного раствора гидроксида аммония (6:3:1). Фракции, содержащие продукт, концентрировали при пониженном давлении, упаривали совместно с ацетонитрилом (2×10 мл), затем с метанолом (1×10 мл), и сушили в течение ночи в глубоком вакууме (0,1 мм рт. ст. (13,33 Па). В результате этой процедуры получили (3-амино-1-фтор-2-гидроксипропил)фосфиновую кислоту в виде белого твердого вещества (40 мг, 56%). 1H ЯМР (300 МГц, D2O) δ 7.93 (s, 0.5 Н), 6.11 (s, 0.5 Н), 4.60-4.20 (m. 2 Н), 3.42-3.08 (m, 2H).
Пример 8.
(3-Амино-2-фтор-1-метилпропил)фосфиновая кислота
К охлажденному в ледяной бане раствору этил-3-амино-2-фтор-1-метил-3-оксопропил(диэтоксиметил)фосфината (1,6 г, 5,3 ммоль) в THF (15 мл) добавляли 1 М ВН3-THF (12,3 мл, 12,3 ммоль) в атмосфере аргона. Через 10 минут раствор нагревали с обратным холодильником в течение 3 часов. Раствор охлаждали до комнатной температуры и по каплям добавляли 6 н. HCl (100 мл). THF удаляли выпариванием на роторном испарителе и добавляли еще одну порцию 6 н. HCl (100 мл). Смесь кипятили с обратным холодильником в течение 3 часов. Раствор охлаждали, упаривали и выпаривали совместно с водой и затем с этанолом. Остаток очищали ионообменной колоночной хроматографией (DOWEX® 50WX-8-200, H+ форма. 3,5×4,0 см). Ионообменную смолу предварительно промывали смесью 2:1 метанол/вода. Неочищенный продукт, растворенный в смеси 1:1 метанол/вода, наносили на колонку и промывали смесью 1:1 метанол: вода. Элюент заменяли на смесь 3:1 метанол/концентрированный гидроксид аммония. Соответствующие фракции объединяли и упаривали с получением 150 мг (15%) смеси диастереоизомеров (3-амино-2-фтор-1-метилпропил)фосфиновой кислоты в виде масла. Данные: 1H ЯМР (400 МГц, D2O) (6.2-7.8 (m, 1Н), 4.8-5.2 (m, 1H), 3.2-3.5 (m, 2H), 1.8-2.2 (m, 1H); 1.0-1.2 (m, 3 H); m/z=156 (М+Н)+.
Следующие промежуточные соединения использовали при получении соединений по изобретению.
Промежуточные соединения
Пример 11.
Этил-3-[(диэтоксиметил)(этокси)фосфорил]-2-фторпропаноат (промежуточное соединение для соединения по Примеру 1)
Смесь этил-(диэтоксиметил)фосфината (26,0 г, 133 ммоль) и 1,1,1,3,3,3-гексаметилдисилазана (28 мл, 133 ммоль) нагревали с обратным холодильником в течение 2 часов в атмосфере аргона. Смесь охлаждали до комнатной температуры и добавляли фторакрилат (10,5 г, 89,0 ммоль). Реагенты нагревали до 60°С в течение трех суток в атмосфере аргона. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (300 мл), промывали 1 н. HCl (2×150 мл) и насыщенным хлоридом натрия (100 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением 32,0 г желтого масла. Остаток очищали колоночной хроматографией на колонке с мокрой набивкой силикагеля (6×30 см), элюируя смесью 97:3 метиленхлорид/метанол. Соответствующие фракции объединяли и упаривали с получением 16,0 г (57%) этил-3-[(диэтоксиметил)(этокси)фосфорил]-2-фторпропаноата в виде прозрачного масла. Данные: 1H ЯМР (300 МГц, CDCl3) δ 5.32 (dm, 1H), 4.67-4.77 (m. 1H), 4.18-4.32 (m, 2H), 3.58-3.91 (m, 4H), 2.30-2.62 (m. 2Н); 1.20-1.41 (m. 9H).
Пример 12.
Этил-(3-амино-2-фтор-3-оксопропил)(диэтоксиметил)фосфинат (промежуточное соединение для соединения по Примеру 1)
К раствору этил-3-[(диэтоксиметил)(этокси)фосфорил]-2-фторпропаноата (16,0 г, 51,1 ммоль) в этаноле (22 мл) добавляли концентрированный гидроксид аммония (14,8 н, 3,5 мл, 51,1 ммоль). Раствор перемешивали в течение 16 часов и упаривали. Остаток очищали хроматографией на колонке с мокрой набивкой силикагеля (7×37 см), элюируя смесью 96,5:3,5 метиленхлорид/метанол. Соответствующие фракции объединяли и упаривали с получением 3,43 г (27%) этил-(3-амино-2-фтор-3-оксопропил)(диэтоксиметил)фосфината в виде прозрачного масла. Данные: 1H ЯМР (300 МГц, CDCl3) δ 6.43 (s, 1H), 5.70 (s, 1H), 5.21-5.49 (dm. 1H), 4.7 (dd, 1H), 4.18-4.31 (m, 2H), 3.65-3.91 (m, 4H), 2.21-2.81 (m, 2H); 1.30-1.40 (m, 3Н), 1.20-1.28 (m, 6H).
Пример 13.
Этил-(2R)-(3-хлор-2-гидроксипропил)(1,1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 2)
После того как смесь этил-(диэтоксиэтил)фосфината (15,0 г, 71 ммоль) и толуола упарили досуха, остаток и 1,1,1,3,3,3-гексаметилдисилазан (13,2 г, 82 ммоль) нагревали с обратным холодильником в течение 3 часов в атмосфере аргона. Смесь охлаждали до комнатной температуры и упаривали. Добавляли (R)-эпихлоргидрин (6,6 г, 71 ммоль) и безводный хлорид цинка (2,5 г, 18 ммоль), и реагенты нагревали до 60°С в течение ночи в атмосфере аргона. Смесь охлаждали до комнатной температуры, разбавляли метиленхлоридом и водой. Органический слой промывали водой, сушили над MgSO4, фильтровали и упаривали с получением 20,7 г желтого масла. Остаток растворяли в метаноле (150 мл), содержащем 1% уксусной кислоты, и раствор перемешивали в течение ночи. Растворитель удаляли с получением 17,7 г (82%) этил-(2R)-(3-хлор-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината в виде прозрачного масла. Данные: 1H ЯМР (500 МГц, CDCl3) δ 4.3-4.4 (m, 1H), 4.1-4.3 (m, 2H), 3.5-3.8 (m, 4H), 1.9-2.4 (m, 2H); 1.5 (dd, J=2.3, 11,4 Гц, ЗН), 1.32-1.37 (m, 3H), 1.18-1.24(m, 6H).
Пример 14.
Этил-(2S)-(3-амино-2-гидроксипропил)(1,1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 2)
Раствор этил-(2R)-(3-хлор-2-гидроксипропил)(1,1 -диэтоксиэтил)фосфината (5,0 г, 17 ммоль) в этаноле, содержащий 9% аммиака, перемешивали в автоклаве при комнатной температуре в течение 4 суток и при 60°С в течение еще одного дня. Раствор упаривали, и остаток очищали хроматографией на колонке с мокрой набивкой силикагеля, элюируя смесью метиленхлорид/метанол (5-8% МеОН), содержащей 5% триэтиламина. Соответствующие фракции объединяли, упаривали и разбавляли метиленхлоридом и водой. В водном слое рН подводили добавлением нескольких миллилитров 10%-ного водного Na2СО3 и повторно экстрагировали метиленхлоридом. Объединенные органические слои сушили над Na2SO4 и упаривали с получением 1,2 г (26%) этил-(2S)-(3-амино-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината в виде прозрачного масла. Данные: 1H ЯМР (300 МГц, CDCl3) δ 4.40-4.55 (b, 1H), 4.10-4.30 (m, 2Н), 3.55-3.80 (m, 4H), 3.20-3.30 (m, 1H), 3.00-3.10 (m, 1H), 2.00-2.40 (m, 2H); 1.45-1.53 (dd. J=3,4. 11,7 Гц, 3Н). 1.30-1.40 (m, 3H),1.15-1.25(m, 6H).
Пример 15.
Этил-(2S)-(3-хлор-2-гидроксипропил)(1.1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 3)
После того, как смесь этил-(диэтоксиэтил)фосфината (15,0 г, 71 ммоль) и толуола упарили досуха, остаток и 1,1,1,3,3,3-гексаметилдисилазан (13,2 г, 82 ммоль) нагревали с обратным холодильником в течение 3 часов в атмосфере аргона. Смесь охлаждали до комнатной температуры и упаривали. Добавляли (S)-эпихлоргидрин (6,6 г, 71 ммоль) и безводный хлорид цинка (2,5 г, 18 ммоль), и реагенты нагревали до 60°С в течение ночи в атмосфере аргона. Смесь охлаждали до комнатной температуры, разбавляли метиленхлоридом и водой. Органический слой промывали водой, сушили над MgSO4, фильтровали и упаривали с получением 20,7 г желтого масла. Остаток растворяли в метаноле (150 мл), содержащем 1% уксусной кислоты, и раствор перемешивали в течение ночи. Растворитель удаляли с получением 16,8 г (79%) этил-(2S)-(3-хлор-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината в виде прозрачного масла. Данные: 1H ЯМР (500 МГц, CDCl3) δ 4.4 (m, 1H), 4.2-4.3 (m, 2H). 3.6-3.8 (m, 4H). 1.9-2.4 (m, 2H); 1.5 (dd. J=2.3, 11,4 Гц, 3Н), 1.32-1.37 (m, 3H), 1.18-1.24 (m, 6H).
Пример 16.
Этил-(2R)-(3-амино-2-гидроксипропил)(1.1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 3)
Раствор этил-(2S)-(3-хлор-2-гидроксипропил)(1,1 -диэтоксиэтил)фосфината (5,0 г, 17 ммоль) в этаноле, содержащий 9% аммиака, перемешивали в автоклаве при комнатной температуре в течение 6 суток и при 55°С в течение еще одного дня. Раствор упаривали, и остаток очищали хроматографией на колонке с мокрой набивкой силикагеля, элюируя смесью метиленхлорид/метанол (5-8% МеОН), содержащей 5% триэтиламина. Соответствующие фракции объединяли, упаривали и разбавляли метиленхлоридом и водой. В водном слое подводили рН добавлением нескольких миллилитров 10%-ного водного Na2СО3 и повторно экстрагировали метиленхлоридом. Объединенные органические слои сушили над Na2SO4 и упаривали с получением 0,9 г (19%) этил-(2R)-(3-амино-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината в виде прозрачного масла. Данные: 1H ЯМР (300 МГц, CDCl3) δ 4.1-4.3 (m, 2H), 4.05 (b, 1H), 3.60-3.80 (m, 4H). 2.4-2.9 (m, 2H). 1.7-2.1 (m, 2H). 1.4-1.5 (dd, 3Н), 1.3-1.4 (m, 3Н), 1.2 (m, 6H).
Пример 17.
Этил-[3-[N-(трет-бутоксикарбонил)амино]-2-оксопропил](1.1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 4)
К раствору диизопропиламина (3,0 мл, 21 ммоль) в THF (5 мл) при -10°С добавляли по каплям н-BuLi (н-бутилитий) (2,5 М в гексанах, 8,6 мл, 21 ммоль). Через 10 минут реакционную смесь охлаждали до -78°С и по каплям добавляли раствор этил-(1,1-диэтоксиэтил)(метил)фосфината (4,80 г, 21,0 ммоль) в THF (5 мл). После добавления раствор перемешивали при -78°С в течение 1 часа. По каплям добавляли раствор метилового эфира N-Вос-глицина (N-бутоксикарбонилглицина) (810 мг, 4,3 ммоль) в THF (15 мл). После окончания добавления реакционную смесь перемешивали в течение 45 минут. Добавляли уксусную кислоту (1,2 мл, 21 ммоль), и реакционную смесь подогревали до комнатной температуры. Реакционную смесь разделяли между метиленхлоридом и водой, и слои разделяли. Водный слой экстрагировали один раз метиленхлоридом. Объединенные органические экстракты сушили над MgSO4, фильтровали и упаривали с получением 4,89 г масла. Остаток очищали хроматографией на 100 г силикагеля, элюируя этилацетатом. Соответствующие фракции собирали с получением 1,2 г (74%) этил-[3-[N-(трет-бутоксикарбонил)амино]-2-оксопропил)(1,1-диэтоксиэтил)фосфината в виде масла. Данные: 1H ЯМР (300 МГц, CDCl3) δ 5.48 (s. 1H), 4.10-4.30 (m, 2H), 4.17 (d, 2H), 3.60-3.80 (m, 4Н), 3.01-3.30 (m, 2H), 1.52 (d, 3Н), 1.43 (s, 9H), 1.32 (t. 3H), 1.19 (t, 6H).
Пример 18.
(2R)-3-Дибензиламино)-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 5)
Боргидрид лития (5,3 г, 0,24 моль) суспендировали в THF (200 мл) в атмосфере азота и охлаждали до -15°С при перемешивании. Метил-(2R)-3-(дибензиламино)-2-фторпропаноат (56,6 г, 0,19 моль) суспендировали в THF (250 мл) и добавляли по каплям к смеси в течение 1 часа; внутреннюю температуру во время добавления поддерживали ниже -10°С. По окончании добавления реакционную смесь оставляли подогреваться до комнатной температуры и перемешивали при этой температуре в течение 17 часов. Реакционную смесь охлаждали до 0°С и осторожно гасили насыщенным водным раствором хлорида аммония (300 мл). Реакционную смесь экстрагировали этилацетатом (2×200 мл), и органическую фазу концентрировали при пониженном давлении. Неочищенный остаток растворяли в 2 н. соляной кислоте (200 мл, рН 2 приблизительно), и водную фазу промывали эфиром (2×200 мл). Водную фазу подщелачивали (приблизительно рН 10) 80%-ным гидроксидом аммония в рассоле, экстрагировали этилацетатом (3×200 мл), сушили над безводным сульфатом натрия (10 г), фильтровали и концентрировали при пониженном давлении с получением (2R)-3-(дибензиламино)-2-фтор-1-пропанола (48 г, 93%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 7.15-7.38 (m, 10Н), 4.65-4.78 (m, 0.5H), 4.48-4.58 (m, 0.5Н), 3.50-3.82 (m, 6H), 2.70-2.88 (m, 2H).
Пример 19.
(2R)-3-Амино-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 5)
(2R)-3-(Дибензиламино)-2-фтор-1-пропанол (29,2 г, 0,11 моль) растворяли в этаноле (300 мл). Добавляли десятипроцентный по массе гидроксид палладия (II) на угле (5,0 г), и смесь помещали на Parr® шейкер, и встряхивали в атмосфере водорода (55 фунт/кв. дюйм (379,225 кПа)) в течение 6 часов. Когда больше не наблюдалось поглощения водорода, смесь фильтровали через набивку Celite® (20 г). К этанольной смеси добавляли свежую порцию гидроксида палладия (II) (5 г) и снова подвергали режиму гидрирования, описанному выше, в течение 17 часов. Неочищенную реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении с получением (2R)-3-амино-2-фтор-1-пропанола в виде бледно-желтого масла (9,6 г, 96%).
1H ЯМР (300 МГц, CDOD) δ 4.78-5.00 (br s, 3H), 4.49-4.62 (m. 0.5H), 4.32-4.46 (m, 0.5H), 3.54-3.70 (m. 2Н), 2.70-2.96 (m, 2H).
Пример I10.
Трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамат (промежуточное соединение для соединения по Примеру 5)
(2R)-3-амино-2-фтор-1-пропанол (4,6 г, 49 ммоль) растворяли в 25%-ном водном диоксане (160 мл), добавляли карбонат калия (7,1 г, 51 ммоль), и смесь охлаждали до 0°С. Ди-трет-бутилдикарбонат (11,6 г, 53 ммоль) добавляли двумя порциями. Смесь затем оставляли нагреваться до комнатной температуры в течение ночи. Неочищенную реакционную смесь концентрировали досуха, добавляли воду (150 мл), затем насыщенный водный гидросульфат калия (до рН 3 приблизительно). Органическое вещество экстрагировали метиленхлоридом (2 х 150 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамата (9,5 г, 100%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ 4.82-5.04 (br s. 1H). 4.62-4.72 (m, 0.5H), 4.48-4.58 (m. 0.5H), 3.62-3.72 (m, 2H), 3.32-3.62 (m, 2H), 3.20-3.44 (br s. 1H), 1.48 (s, 9H).
Пример I11.
Трет-бутил-(2R)-2-фтор-3-йодпропилкарбамат (промежуточное соединение для соединения по Примеру 5)
Имидазол (26,6 г, 0,39 моль) растворяли в метиленхлориде (400 мл) при комнатной температуре. Добавляли йод (102,5 г, 0,39 моль), и реакционную смесь перемешивали в течение 10 минут при комнатной температуре, и затем охлаждали до 0°С. Трифенилфосфин (102,5 г, 0,39 моль) добавляли частями в течение 10 минут таким образом, чтобы внутренняя температура оставалась ниже 10°С. По каплям добавляли раствор трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамата (60,4 г, 0,31 моль) в метиленхлориде (100 мл). По окончании добавления трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамата, добавляли дополнительно метиленхлорид (200 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжали в течение 17 часов. Реакционную смесь фильтровали через набивку Celite® (50 г) и промывали дополнительно метиленхлоридом. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле, элюируя метиленхлоридом. В результате этой процедуры получали трет-бутил-(2R)-2-фтор-3-йодпропилкарбамат в виде белого твердого вещества (64,7 г, 68%).
1H ЯМР (300 МГц, CDCl3) δ 4.80-5.10 (br s, 1H). 4.58-4.72 (m, 0.5H), 4.42-4.56 (m, 0,5H), 3.48-3.70 (m, 1H), 3.20-3.46 (m, 3Н), 1.48 (s, 9H).
Пример I12.
Метил-(2S)-3-(дибензиламино)-2-фторпропаноат (промежуточное соединение для соединения по Примеру 6)
Метил-(2R)-2-(дибензиламино)-3-гидроксипропаноат (231,7 г, 0,77 моль) растворяли в THF (850 мл) и медленно по каплям добавляли раствор DAST (трифторида (диэтиламино)серы) (196 г, 1,2 моль) в THF (400 мл). Как только добавление было закончено, реакционную смесь перемешивали в течение еще 1,5 часов. ТСХ анализ показывал расход исходного вещества. Реакционную смесь затем охлаждали до 0°С и гасили медленным добавлением воды (1,5 л) с последующей нейтрализацией добавлением твердого бикарбоната натрия. Сразу после нейтрализации добавляли смесь 1:1 концетрированного гидроксида аммония/насыщенного раствора хлорида натрия, и реакционную смесь экстрагировали этилацетатом и концентрировали при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат, гексаны (1:4) с получением желаемого соединения (188,3 г, 62%) в виде масла.
1H ЯМР (300 МГц, CDCl3) δ 7.18-7.38 (m. 10Н), 5.12-5.17 (m, 0,5H), 4.95-5.00 (m, 0.5H), 3.81-3.87 (m. 2H), 3.69 (s, 3Н), 3.49-3.55 (m, 2H), 2.90-3.12 (m. 2H).
Пример I13.
(2S)-3-(Дибензиламино)-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 6)
Боргидрид лития (17,7 г, 0,81 моль) суспендировали в THF (400 мл) в атмосфере азота и охлаждали до -15°С при перемешивании. Метил-(2S)-3-(дибензиламино)-2-фторпропаноат (188,3 г, 0,62 моль) суспендировали в THF (400 мл) и добавляли по каплям к смеси. По окончании добавления реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при этой температуре в течение 3 часов. ТСХ анализ показал полный расход исходного вещества. Реакционную смесь охлаждали до 0°С и осторожно гасили насыщенным водным раствором хлорида аммония (300 мл). Добавляли дополнительно воду (400 мл), затем реакционную смесь экстрагировали этилацетатом, и органическую фазу концентрировали при пониженном давлении. Неочищенный остаток растворяли в 2 н. соляной кислоте, и водную фазу промывали дважды эфиром. Водную фазу подщелачивали (приблизительно рН 10) 80%-ным гидроксидом аммония в рассоле, экстрагировали этилацетатом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (2S)-3-(дибензиламино)-2-фтор-1-пропанола (156,6 г, 92%) в виде желтого масла. 1H ЯМР (300 МГц, CDCl3) δ 7.15-7.38 (m, ЮН), 4.65-4.78 (m, 0.5Н), 4.48-4.58 (m, 0.5H), 3.50-3.82 (m, 6H), 2.70-2.88 (m, 2H).
Пример I14.
(2S)-3-Амино-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 6)
(2S)-3-(дибензиламино)-2-фтор-1-пропанол (39,1 г, 0,14 моль) растворяли в этаноле (300 мл). Добавляли десятипроцентный по массе гидроксид палладия (II) на угле (5,0 г), и смесь помещали на Parr® шейкер и встряхивали в атмосфере водорода (55 фунт/кв.дюйм (379,225 кПа)) в течение ночи. Когда не наблюдалось больше поглощения водорода, смесь фильтровали через набивку Celite®. К этанольной смеси добавляли свежую порцию гидроксида палладия (II) (5 г), и снова подвергали режиму гидрирования, описанному выше, в течение 12 часов. Снова, когда не наблюдалось больше поглощения водорода, смесь фильтровали через набивку Celite®. К этанольной смеси добавляли свежую порцию гидроксида палладия (II) (5 г) и снова подвергали режиму гидрирования, описанному выше, в течение 12 часов. Неочищенную реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении с получением (2S)-3-амино-2-фтор-1-пропанола в виде бледно-желтого масла (13.3 г, 100%). 1H ЯМР (300 МГц, CDCl3) δ 4.78-5.00 (bR s, 3H), 4.49-4.62 (m. 0.5H), 4.32-4.46 (m, 0,5H), 3.54-3.70 (m, 2H), 2.70-2.96 (m, 2H).
Пример I15.
Трет-бутил-(2S)-2-фтор-3-гидроксипропилкарбамат (промежуточное соединение для соединения по Примеру 6)
(2S)-3-Амино-2-фтор-1-пропанол (38,6 г, 0,41 моль) растворяли в 25%-ном водном диоксане (1,4 л), добавляли карбонат калия (60,1 г, 0,43 моль), затем ди-трет-бутилдикарбонат (99,5 г, 0,46 моль). Смесь перемешивали в течение ночи. ТСХ анализ показал полный расход исходного вещества. Неочищенную реакционную смесь концентрировали досуха, добавляли воду (300 мл), затем насыщенный водный гидросульфат калия (до рН 3 приблизительно). Органическое вещество экстрагировали дважды метиленхлоридом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-(2S)-2-фтор-3-гидроксипропилкарбамата (79,5 г, 99%) в виде бледно-желтого масла. 1H ЯМР (300 МГц, CDCl3) δ 4.82-5.04 (bR s, 1H), 4.62-4.72 (m, 0.5H). 4.48-4.58 (m, 0.5Н), 3.62-3.72 (m, 2H), 3.32-3.62 (m, 2H), 3.20-3.44 (br s, 1H), 1.48 (s, 9H).
Пример I16.
Трет-бутил-(2S)-2-фтор-3-йодпропилкарбамат (промежуточное соединение для соединения по Примеру 6)
Имидазол (19,8 г, 0,29 моль) растворяли в метиленхлориде (900 мл) при комнатной температуре. Добавляли йод (73,9 г, 0,29 моль), и реакционную смесь перемешивали в течение 10 минут при комнатной температуре и затем охлаждали до 0°С. Трифенилфосфин (76,3 г, 0,29 моль) добавляли частями в течение 10 минут таким образом, чтобы внутренняя температура оставалась ниже 10°С. По каплям добавляли раствор трет-бутил-(2S)-2-фтор-3-гидроксипропилкарбамата (45,0 г, 0,23 моль) в метиленхлориде (300 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжали в течение 12 часов. Реакционную смесь фильтровали через набивку Celite® и промывали дополнительно метиленхлоридом. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле, элюируя метиленхлоридом. В результате этой процедуры получали трет-бутил-(2S)-2-фтор-3-йодпропилкарбамат в виде бесцветного масла (42,5 г, 62%). 1H ЯМР (300 МГц, CDCl3) δ 4.80-5.10 (br s, 1H), 4.58-4.72 (m, 0.5H), 4.42-4.56 (m, 0.5H), 3.48-3.70 (m, 1H), 3.20-3.46 (m, 3Н), 1.48 (s, 9H).
Пример I17.
Этил-(фторметил)(1.1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 7)
Гидрид натрия (1,4 г, 57,1 ммоль) суспендировали в THF (50 мл) в колбе для реакций под давлением в атмосфере азота и охлаждали до -10°С при перемешивании. Этил-(1,1-диэтоксиэтил)фосфинат (10,0 г, 47,6 ммоль) в THF (20 мл) добавляли по каплям к смеси в течение 10 минут; внутреннюю температуру поддерживали ниже 0°С во время добавления. По окончании добавления реакционную смесь оставляли перемешиваться при этой температуре в течение 90 минут. Колбу охлаждали до -78°С, и газообразный хлорфторметан (9,7 г, 142,8 ммоль) конденсировали в реакционную смесь. Перегородку удаляли, и колбу герметически закрывали пробкой с винтовой нарезкой. Колбу затем оставляли нагреваться до комнатной температуры и затем нагревали при 50°С в течение 24 часов. Реакционную смесь охлаждали до 0°С и осторожно гасили водой (25 мл). К реакционной смеси добавляли метиленхлорид (50 мл), и эмульсию фильтровали через набивку Celite® (20 г). Водную фазу экстрагировали метиленхлоридом (2×100 мл), сушили над безводным сульфатом магния, и органическую фазу концентрировали при пониженном давлении с получением неочищенного продукта в виде бледного желтого масла (6,93 г). Неочищенный продукт очищали хроматографией на колонке с силикагелем (6×25 см колонка), элюируя 20%-ным ацетоном в гексанах. В результате этой процедуры получали этил-(фторметил)(1,1-диэтоксиэтил)фосфинат в виде прозрачного бесцветного масла (4,4 г, 42%). 1H ЯМР (300 МГц, CDCl3) δ 4.94-4.54 (m, 2Н), 4.32-4.20 (m, 2H), 3.82-3.54 (m, 4H), 1.60-1.44 (m, 3Н), 1.40-1.28 (m, 3Н), 1.26-1.08 (m, 6H).
Пример 118.
Этил-(3-(N-(трет-бутоксикарбонил)амино)-1-фтор-2-оксопропил)(1,1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 7)
К раствору диизопропиламина (2,5 мл, 14,5 ммоль, 3,5 эквивалента) в THF (30 мл) при -10°С добавляли по каплям (около 10 минут) н-BuLi (1,4 M в гексанах, 9,0 мл, 14,5 ммоль). Через 10 минут реакционную смесь охлаждали до -78°С, и по каплям в течение 10 минут добавляли раствор этил-(фторметил)(1,1-диэтоксиэтил)фосфината (2,0 г, 8,26 ммоль, 2 эквивалента) в THF (10 мл). После добавления реакционную смесь перемешивали при -78°С в течение 1 часа. По каплям добавляли раствор метилового эфира N-Вос-глицина (0,8 г, 4,1 ммоль) в THF (10 мл) в течение 10 минут таким образом, чтобы поддерживать внутреннюю температуру ниже -70°С. Когда добавление было закончено, реакционную смесь перемешивали при -78°С в течение 1 часа. Реакцию гасили уксусной кислотой (1 мл, 14,5 ммоль) и затем подогревали до комнатной температуры. Насыщенный водный хлорид натрия (75 мл) добавляли к реакционной смеси, и органическую фазу отделяли. Водную фазу затем экстрагировали этилацетатом (2×75 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта в виде бледно-желтого масла (2,69 г). Неочищенный продукт очищали колоночной хроматографией (2×35 см колонка), элюируя 40%-ным этилацетатом в гексане. В результате этого способа получали этил-(3-(N-трет-бутоксикарбонил)амино)-1-фтор-2-оксопропил)(1,1-диэтоксиэтил)фосфинат в виде прозрачного бесцветного масла (0,73 г, 44%). 1H ЯМР (300 МГц, CDCl3) δ 5.78-5.24 (m, 2H), 4.52-4.08 (m, 4Н), 3.94-3.50 (m. 4H), 1.62-1.51 (m, 3H). 1.50-1.32 (m, 3H), 1.42 (s. 9H), 1.30-1.12 (m, 6H).
Пример I19.
Этил-(3-(N-(трет-бутоксикарбонил)амино)-1-фтор-2-гидроксипропил)(1.1-диэтоксиэтил)фосфинат (промежуточное соединение для соединения по Примеру 7)
К раствору этил-(3-(N-(трет-бутоксикарбонил)амино)-1-фтор-2-оксопропил)(1,1-диэтоксиэтил)фосфината (0,7 г, 1,8 ммоль) в метаноле (30 мл) при -5°С в атмосфере азота добавляли боргидрид натрия (76 мг, 2,0 ммоль) одной порцией. Происходило небольшое выделение теплоты; однако внутреннюю температуру поддерживали ниже -2°С. Реакционную смесь перемешивали при 0°С в течение 1 часа. Реакционную смесь гасили насыщенным водным гидрокарбонатом натрия (5 мл). Неочищенную смесь концентрировали при пониженном давлении. Неочищенный остаток экстрагировали этилацетатом (30 мл), промывали насыщенным водным раствором хлорида натрия (5 мл) и сушили над безводным сульфатом магния. После удаления растворителя при пониженном давлении получали неочищенный продукт в виде бледно-желтого масла (580 мг). После очистки колоночной хроматографией получали 2 фракции, которые оказались соответствующими разным диастереомерам этил-(3-(N-трет-бутоксикарбонил)амино)-1-фтор-2-гидроксипропил)(1,1-диэтоксиэтил)фосфината. Оказалось, что менее полярная фракция являлась смесью 1:1 двух диастереомеров, (210 мг, 29%), тогда как более полярная фракция представляла собой преимущественно один диастереомер, как показал 1H ЯМР анализ (190 мг, 26%). 1H ЯМР менее полярного соединения (300 МГц, CDCl3) δ 5.32-5.04 (br s, 1H). 4.88-4.82 (m, 0.5H), 4.72-4.68 (m, 0.5H), 4.40-4.08 (m, 4H), 3.90-3.26 (m. 6H), 1.66-1.52 (m, 3H), 1.50-1.32 (m, 3H), 1.44 (s. 9Н), 1.30-1.12 (m, 6H).
Пример I20.
Этил-3-[(диэтоксиметил)(этокси)фосфорил1-2-фторбутаноат (промежуточное соединение для соединения по Примеру 8)
Смесь этил-(диэтоксиметил)фосфината (21,7,0 г, 110 ммоль) и 1,1,1,3,3,3-гексаметилдисилазана (23,3 мл, 110 ммоль) нагревали с обратным холодильником в течение 2 часов в атмосфере аргона. Смесь охлаждали до комнатной температуры, и добавляли смесь диастереомеров этил-2-фторбут-2-еноата (14,6 г, 110 ммоль). Реагенты нагревали до 80°С в течение одних суток и 120°С в течение 2 часов в атмосфере аргона. Смесь охлаждали до комнатной температуры, и добавляли другую порцию активированного триметилсилилом этил-(диэтоксиметил)фосфината (его получали из этил-(диэтоксиметил)фосфината (21.7,0 г, 110 ммоль) и 1,1,1,3,3,3-гексаметилдисилазана (23,3 мл, 110 ммоль) таким же образом, как выше). Смесь нагревали до 100°С в течение трех суток, и добавляли еще одну порцию активированного триметилсилилом этил-(диэтоксиметил)фосфината. Смесь нагревали до 100°С в течение трех суток в атмосфере аргона, охлаждали до комнатной температуры и затем разбавляли этилацетатом (300 мл). Раствор промывали 1 н. HCl (2×200 мл) и насыщенным хлоридом натрия. Органический слой сушили над MgSO4, фильтровали и упаривали с получением 42,0 г желтого масла. Остаток очищали колоночной хроматографией на колонке с мокрой набивкой силикагеля, элюируя метиленхлоридом и затем смесью 98:2 метиленхлорид/метанол. Соответствующие фракции объединяли и упаривали с получением 3,6 г (10%) этил-3-[(диэтоксиметил)(этокси)фосфорил]-2-фторбутаноата в виде прозрачного масла. 1H ЯМР (300 МГц, CDCl3 ) δ 4.9-5.6 (m, 1H), 4.7-4.8 (m. 1H), 4.2-4.4 (m. 4H). 3.6-4.0 (m. 4H), 2.6-2.9 (m, 1H); 1.2-1.4 (m, 12H).
Пример I21.
Этил-3-амино-2-фтор-1-метил-3-оксопропил(диэтоксиметил)(фосфинат (промежуточное соединение для соединения по Примеру 8)
К раствору этил-3-[(диэтоксиметил)(этокси)фосфорил]-2-фторбутаноата (1,8 г, 5,5 ммоль) в этаноле (3 мл) добавляли концентрированный гидроксид аммония (14,8 M, 0,5 мл, 7,4 ммоль). Раствор перемешивали в течение 24 часов при 40°С и затем выпаривали с получением 1,6 г (97%) смеси диастереомеров этил-3-амино-2-фтор-1 -метил-3-оксопропил(диэтоксиметил)фосфината в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3) δ 5.7-6.7 (m, 2H), 4.9-5.5.6 (m, 1H), 4.7-4.8 (m, 1H), 4.1-4.4 (m, 2H), 3.8-4.0 (m, 4H), 2.8-3.0 (m. 1H); 1.2-1.4 (m, 12H).
Фармацевтические препараты
Соединение формулы I по настоящему изобретению можно использовать в качестве активного ингредиента в фармацевтическом препарате для перорального, ректального, эпидурального, внутривенного, внутримышечного, подкожного, назального введения и введения путем инфузии, или для любого другого подходящего пути введения. Предпочтительный путь введения представляет собой пероральный или путем инъекции/инфузии.
Фармацевтические препараты содержат соединение по настоящему изобретению в комбинации с одним или более чем одним фармацевтически приемлемым ингредиентом. Готовые лекарственные формы изготавливают при помощи известных фармацевтических способов. Обычно количество активных соединений составляет между 0,1-95% по массе от препарата, предпочтительно между 0,2-20% по массе в препаратах для парентерального применения, и предпочтительно между 1-50% по массе в препаратах для перорального введения.
При приготовлении фармацевтических препаратов, содержащих соединение по настоящему изобретению, в форме твердых единиц дозировки для перорального введения, выбранное соединение можно смешивать с твердыми фармацевтически приемлемыми ингредиентами (среди них, например, разрыхляющие агенты и смазывающие агенты). Смесь затем перерабатывают в гранулы, таблетки, капсулы или саше.
Единицы дозировки для ректального введения можно приготовить в форме суппозиториев; в форме желатиновых ректальных капсул; в форме готовых микроклизм; или в форме сухого препарата для микроклизмы для разведения в подходящем растворителе непосредственно перед введением.
Жидкие препараты для перорального введения можно приготовить в форме сиропов или суспензий, или в форме сухой смеси для разведения подходящим растворителем перед применением.
Растворы для парентерального введения можно приготовить в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, и их разливают в ампулы или флаконы. Их можно также приготовить в виде сухого препарата для разведения подходящим растворителем непосредственно перед применением.
Обычная суточная доза активного соединения будет зависеть от различных факторов, таких как, например, индивидуальная потребность каждого пациента, путь введения и заболевание. Обычно дозировки будут в пределах от 1 мкг до 100 мг в сутки и на кг массы тела, предпочтительно от 10 мкг до 20 мг в сутки и на кг массы тела.
Биологические исследования
[3H]GABA радиолигандный анализ связывания
Синаптические мембраны крыс готовили из целого мозга самцов крыс Sprague Dawley. по существу аналогично описанному ранее (Zukin, et al. (1974) PRoc. Natl. Acad. USA 71, 4802-4807). [3H]GABA конкурентный анализ, модифицированный OIpe et al ((1990) Eur. J. Pharmacol. 187,27-38) выполняли в 200 мкл TCI (трис-кальций изогувацинового) буфера (50 мМ Трис (три(гидроксиметил)аминометан), рН 7,4, 2,5 мМ CaCl2 и 40 мкМ изогувацина), содержащем 20 нМ [3H]GABA (специфическая активность: 3 тера-беккерель (ТБк)/ммоль), испытуемое соединение или растворитель, и 80 мкг белка синаптических мембран, используя 96-луночные планшеты. После инкубации в течение 12-20 минут при комнатной температуре, инкубирования заканчивали путем быстрой фильтрации через фильтр из стекловолокна (Printed filtermat В filters, Wallac), который был предварительно обработан 0,3%-ным полиэтиленимином, используя 96-луночный харвестер клеток (Skatron или Tomtec). Фильтры промывали буфером, содержащим 50 мМ Трис (трис(гидроксиметил)аминометан) и 2,5 мМ CaCl2, рН 7,4 при 4°С, и затем сушили при 55°С. MeltiLex B/HS сцинтилляторный лист (Wallac) растапливали на фильтр, и радиоактивность определяли в сцинтилляционном счетчике Microbeta (Wallac).
Результаты и обсуждение
Было обнаружено, что соединения по настоящему изобретению имеют высокое сродство и активности в отношении GABAв рецепторов, как выявлено низкими IC50 (средняя ингибирующая концентрация) и EC50 (средняя эффективная концентрация) в анализах связывания и подвздошной кишки соответственно. Также было обнаружено, что соединения снижают TLOSR при введении внутривенно, а также перорально в моделях на животных. Вопреки тому, что было заявлено в литературе для производных 3-аминопропилфосфиновой кислоты, имеющих Р-Н связь, авторы обнаружили, что соединения по настоящему изобретению имеют высокую метаболическую стабильность в моделях на животных. Кроме того, побочные эффекты на ЦНС (как определено по снижению температуры тела у мышей) не наблюдались или наблюдались лишь при очень высоких дозах. Таким образом, разница между терапевтической дозой (ингибирование TLOSR в модели на собаках) и дозой, вызывающей побочные эффекты (в модели на мышах), была неожиданно высокой.
В качестве показателя метаболической стабильности соединений по изобретению использовали показатель биодоступности соединений, который представляет собой отношение количества соединения, которое достигло мишени, не претерпев при этом трансформации, к количеству введенного соединения.
Ниже в таблице, приведены результаты испытаний на биодоступность, полученные в in vivo модели на крысах, для пяти конкретных представителей соединений по изобретению (в скобках указан номер примера получения).
Результаты, представленные в таблице, демонстрируют неожиданно высокую степень биодоступности (порядка 80-100%) и, соответственно, высокую метаболическую стабильность заявленных соединений.
Для оценки профиля безопасности соединений по изобретению определяли их терапевтическую и токсическую дозу. Терапевтическая доза соответствует количеству лекарственного соединения, которое надо ввести для получения целевого терапевтического эффекта, а именно для ингибирования TLOSR. Терапевтическую дозу соединений по изобретению определяли по ингибированию тестируемым соединением преходящих расслаблении нижнего пищеводного сфинктера (TLOSR), которое оценивали в in vivo модели на собаках.
Токсическая доза соответствует количеству соединения, при котором начинается развитие нежелательных, побочных или токсических эффектов. При этом профиль безопасности соединения выражается его терапевтическим индексом, который представляет собой отношение дозы соединения, вызывающей нежелательное побочное действие, к дозе, обеспечивающей указанный терапевтический эффект. Нежелательные эффекты соединений по изобретению на центральную нервную систему (ЦНС) оценивали традиционным для агонистов ОАВАв-рецепторов способом - по их способности снижать температуру тела экспериментального животного (в in vivo модели на мышах). Известно, что снижение температуры тела взаимосвязано с такими побочными эффектами на ЦНС лекарственного средства как седативное действие и гипотермия.
Баклофен и 3-аминопропил(метил)фосфиновые кислоты, описанные в The GABA Receptors", second edition, Humana press, 1997, page 281 и J. Med. Chem (1995), 3297-3312, оказывали побочные действия на ЦНС, такие как седативный эффект и гипотермия, при дозах близких к терапевтическим, и их терапевтический индекс был не очень высоким по сравнению с соединениями по изобретению.
Например, для ингибирования на 45% TLOSR в in vivo модели на собаках требовалось 3 мкмоль/кг соединения по изобретению - [(R)-3-амино-2-фторпропил]фосфиновой кислоты, и для снижения температуры тела мыши на 2°С требовалось 690 мкмоль/кг этого соединения. В то же время баклофен в количестве 1,4 мкммоль/кг ингибировал на 45% TLOSR в in vivo модели на собаках, при этом количество баклофена 41,5 мкмоль/кг вызывало снижение температуры тела мышей на 2°С.
Таким образом, терапевтический индекс [(R)-3-амино-2-фторпропил]фосфиновой кислоты составлял 690/3=230, а терапевтический индекс баклофена составлял 41,5/1,4=29,6, т.е. терапевтический индекс соединения по изобретению был в 7,77 раз выше, чем у баклофена.
Терапевтический индекс всех остальных соединений по изобретению или был таким же как у [(R)-3-амино-2-фторпропил]фосфиновой кислоты или лучше.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ (АМИНОПРОПИЛ)МЕТИЛФОСФИНОВЫЕ КИСЛОТЫ | 2000 |
|
RU2267495C2 |
| (6S,9AS)-N-БЕНЗИЛ-6-[(4-ГИДРОКСИФЕНИЛ)МЕТИЛ]-4,7-ДИОКСО-8-({ 6-[3-(ПИПЕРАЗИН-1-ИЛ)АЗЕТИДИН-1-ИЛ]ПИРИДИН-2-ИЛ} МЕТИЛ)-2-(ПРОП-2-ЕН-1-ИЛ)-ОКТАГИДРО-1H-ПИРАЗИНО[2,1-C][1,2,4]ТРИАЗИН-1-КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2669805C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| ПРОИЗВОДНЫЕ АДАМАНТИЛА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2626890C2 |
| ИНГИБИТОРЫ VLA-4 | 2001 |
|
RU2290403C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| АГОНИСТЫ TLR7 | 2019 |
|
RU2817014C2 |
| СИНЕРГИЧЕСКОЕ ЛЕЧЕНИЕ ПАРКИНСОНИЗМА | 1995 |
|
RU2176145C2 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ | 2013 |
|
RU2642066C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2298550C2 |
Изобретение относится к новым соединениям формулы (1), имеющим сродство к одному или более чем одному GABAB рецептору, их фармацевтически приемлемые соли, сольваты и стереоизомеры, за исключением рацемата (3-амино-2-гидроксипропил)фосфиновой кислоты. Технический результат: увеличение терапевтического индекса. 3 н. и 11 з.п. ф-лы, 1 табл.
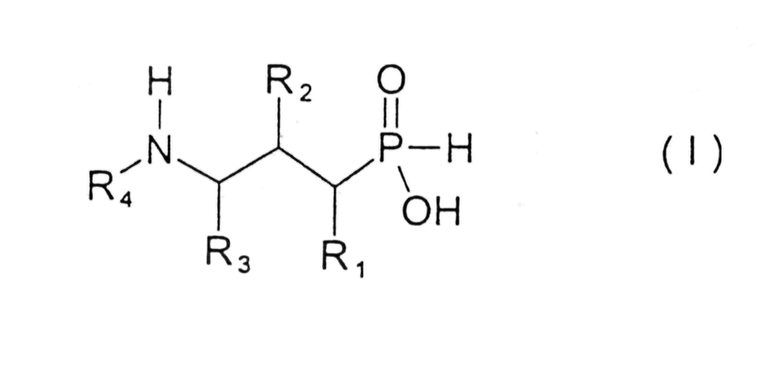
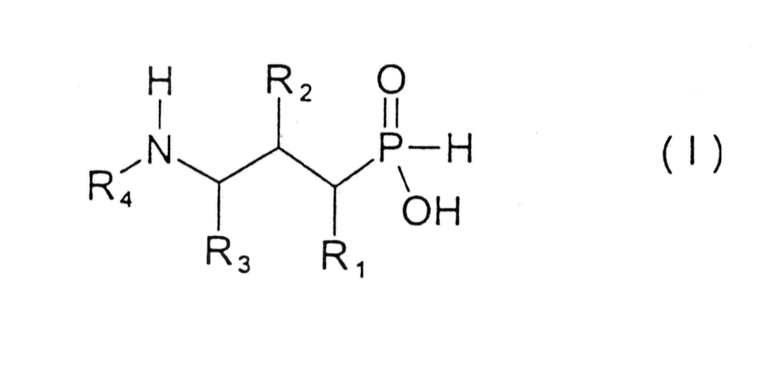
где R1 представляет собой водород,
R2 представляет собой гидрокси, фторо- или оксогруппу;
R3 представляет собой водород;
R4 представляет собой водород;
и его фармацевтически приемлемые соли, сольваты и стереоизомеры,
за исключением рацемата (3-амино-2-гидроксипропил)фосфиновой кислоты.
| WO 9811885 A1, 26.03.1998.EP 0356128 A2, 28.02.1990.SU 643510 A, 25.01.1976.SU 589756 A, 07.10.1982. |

