Область изобретения
Настоящее изобретение относится к новым соединениям, имеющим сродство к одному или более чем одному ОАВАB (γ-аминомасляная кислота) рецептору, а также их фармацевтически приемлемым солям, сольватам и стереоизомерам. Изобретение также относится к способам их получения, фармацевтическим композициям, содержащим указанные терапевтически активные соединения, и к применению указанных активных соединений в терапии.
Предшествующий уровень техники
Рефлюкс
Гастроэзофагеальная рефлюксная болезнь (GORD) является наиболее распространенным заболеванием верхнего отдела желудочно-кишечного тракта. Современная терапия стремится к уменьшению секреции желудочной кислоты или к уменьшению экспозиции кислоты в пищеводе путем повышения пищеводного клиренса, тонуса нижнего пищеводного сфинктера и опорожнения желудка. Ранее считалось, что главный механизм, лежащий в основе рефлюкса, зависит от гипотонического состояния нижнего пищеводного сфинктера. Однако недавнее исследование (например Holloway & Dent (1990) Gastroenterol., Clin. N. Amer. 19, 517-535) показало, что большинство приступов рефлюкса происходит во время преходящих расслаблений нижнего пищеводного сфинктера, ниже упоминаемых как TLOSR, то есть расслаблении, не инициируемых глотаниями. Также было показано, что у пациентов с GORD секреция желудочной кислоты обычно является нормальной.
Следовательно, существует потребность в соединениях, которые снижают число случаев TLOSR и таким образом предотвращают рефлюкс.
Фармацевтические композиции, содержащие местный анестетик, применяемые для ингибирования расслабления нижнего пищеводного сфинктера, раскрыты в WO 87/04077 и в US 5036057. Недавно было показано, что агонисты GABAB-рецепторов ингибируют TLOSR, что раскрыто в WO 98/11885.
Агонисты GABAB рецепторов
GABA (4-аминомасляная кислота) является эндогеным нейромедиатором в центральной и периферической нервной системах. Рецепторы к GABA традиционно разделяют на подтипы GABAA и GABAB рецепторы. GABAB рецепторы относятся к надсемейству рецепторов, связанных с G-белками. Агонисты GABAB рецепторов описаны как полезные при лечении нарушений ЦНС (центральной нервной системы), таких как мышечное расслабление при спинальной спастичности, сердечно-сосудистых нарушений, астмы, нарушений кишечной моторики, таких как синдром раздраженной толстой кишки (IBS), и в качестве прокинетических и противокашлевых агентов. Агонисты GABAB рецепторов также были описаны как пригодные для лечения рвоты (WO 96/11680) и недавно, как указано выше, для ингибировании TLOSR (WO 98/11885).
Наиболее изученным агонистом GABAB рецепторов является баклофен (4-амино-3-(хлорфенил)масляная кислота), раскрытый в швейцарском патенте №СН 449046. Баклофен в течение нескольких лет применяли в качестве антиспазматического агента. ЕР 0356128 описывает применение отдельного соединения (3-аминопропил)метилфосфиновой кислоты, в качестве сильного агониста GABAB рецепторов, в терапии. ЕР 0181833 раскрывает замещенные 3-аминопропилфосфиновые кислоты, которые, как обнаружено, имеют очень высокое сродство к местам связывания GABAB рецепторов. По аналогии с баклофеном эти соединения можно применять в качестве, например, мышечных релаксантов. ЕР 0399949 раскрывает производные (3-аминопропил)метилфосфиновой кислоты, которые описаны как сильные агонисты GABAB рецепторов. Установлено, что эти соединения пригодны в качестве мышечных релаксантов. Обе заявки, ЕР 0463969 и FR 2722192, относятся к производным 4-аминомасляной кислоты, имеющим разные гетероциклические заместители на 3-углероде бутиловой цепи. Взаимосвязи структура-активность некоторых аналогов фосфиновой кислоты по отношению к их сродству к GABAB рецептору, а также их действие на мышечное расслабление, обсуждаются в J. Med. Chem. (1995), 38, 3297-3312. Заключением в данной статье является то, что, по сравнению с баклофеном, значительно более сильного мышечного расслабления можно достичь с применением (S)-энантиомера 3-амино-2-гидроксипропилметилфосфиновой кислоты, и без нежелательных влияний на ЦНС.
Краткое изложение сущности изобретения
В настоящем изобретении предложены новые соединения формулы I
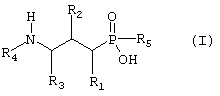
где
R1 представляет собой водород, гидрокси, низший алкил, низший алкокси или галоген;
R2 представляет собой гидрокси, меркапто, галоген или оксогруппу;
R3 представляет собой водород или низший алкил (возможно замещенный гидрокси, меркапто, низшим алкокси, арилом или низшим тиоалкокси);
R4 представляет собой водород, низший алкил (возможно замещенный арилом) или арил;
R5 представляет собой метил, фторметил, дифторметил или трифторметил;
и их фармацевтически приемлемые соли, сольваты и стереоизомеры, за исключением:
1) рацемата (3-амино-2-гидроксипропил)метилфосфиновой кислоты;
2) (S)-(3-амино-2-гидроксипропил)метилфосфиновой кислоты;
3) (R)-(3-амино-2-гидроксипропил)метилфосфиновой кислоты;
4) (3-амино-2-гидроксипропил)дифторметилфосфиновой кислоты;
5) (3-амино-2-оксопропил)метилфосфиновой кислоты.
Предпочтительно соединение представляет собой одно из (3-амино-2-фторпропил)(метил)фосфиновой кислоты, (2R)-(3-амино-2-фторпропил)(метил)фосфиновой кислоты, (2S)-(3-амино-2-фторпропил)(метил)фосфиновой кислоты, (3-амино-2-фтор-1-метилпропил)(метил)фосфиновой кислоты или их фармацевтически приемлемых солей, сольватов или стереоизомеров.
В рамках объема данного изобретения следует понимать, что когда R2 является оксогруппой, связь между R2 и углеродом является двойной связью.
Кроме того, в рамках объема данного изобретения под "низшими" радикалами и соединениями следует понимать, например, соединения, имеющие до и включая 7, особенно до и включая 4 углеродных атома. Также основные выражения имеют следующие значения:
Низший алкил представляет собой, например, С1-С4алкил, такой как метил, этил, н-пропил или н-бутил, а также изопропил, изобутил, вторичный бутил или третичный бутил, но также может быть С5-С7алкильной группой, такой как пентильная, гексильная или гептильная группа.
Низший алкокси представляет собой, например, С1-С4алкокси, такой как метокси, этокси, н-пропокси или н-бутокси, также изопропокси, изобутокси, вторичный бутокси или третичный бутокси, но также может быть С5-С7 алкоксигруппой, такой как пентокси, гексокси или гептоксигруппа.
Низший тиоалкокси представляет собой, например, С1-С4тиоалкокси, такой как тиометокси, тиоэтокси, н-тиопропокси или н-тиобутокси, также тиоизопропокси, тиоизобутокси, вторичный тиобутокси или третичный тиобутокси, но также может быть С5-С7 тиоалкоксигруппой, такой как тиопентокси, тиогексокси или тиогептоксигруппа.
Галоген представляет собой галоген с атомным номером до и включая 35, такой как фтор или хлор, и менее предпочтительно бром.
Соединения формулы I по изобретению имеют амфотерный характер и могут существовать в форме внутренних солей. Они также могут образовывать соли присоединения кислот и соли с основаниями. Такие соли представляют собой в частности фармацевтически приемлемые соли присоединения кислот, а также фармацевтически приемлемые соли, образованные с основаниями. Подходящие кислоты для образования таких солей включают в себя, например, минеральные кислоты, такие как соляная, бромистоводородная, серная или фосфорная кислота или органические кислоты, такие как сульфоновые кислоты и карбоновые кислоты. Соли с основаниями представляют собой, например, соли щелочных металлов, например соли натрия или калия, или соли щелочноземельных металлов, например соли кальция или магния, а также соли аммония, такие как соли аммония или органических аминов. Эти соли можно получить традиционными способами.
Когда в молекуле имеется один или более чем один стереоцентр, соединения формулы I могут находиться в форме смеси стереоизомеров, то есть смеси диастереомеров и/или рацематов, или в форме отдельных стереоизомеров, то есть отдельного энантиомера и/или диастереомера. Эти соединения также могут находиться в форме сольватов, например гидратов.
Все соединения формулы I можно применять для ингибирования TLOSR, и таким образом для лечения гастроэзофагеальной рефлюксной болезни. Указанное ингибирование TLOSR также предполагает, что все указанные соединения формулы I можно применять для лечения срыгивания у младенцев. Эффективная терапия срыгивания у младенцев была бы важным путем лечения, недостаточно успешной из-за чрезмерной потери проглоченого питательного вещества. Кроме того, новые соединения можно применять для лечения связанных с GORD или не связанных с GORD астмы, отрыжки, кашля, боли, привыкания к кокаину, икоты, IBS, диспепсии, рвоты и ноцицепции.
В настоящем изобретении предложены соединения, имеющие неожиданно высокие активности и/или терапевтический индекс.
Получение
Соединения формулы I по настоящему изобретению можно получать одним из следующих способов.
А) Соединение формулы II
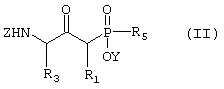
где R1, R3 и R5 такие, как определено выше в формуле I, Z представляет собой защитную группу, такую как трет-бутилоксикарбонил, и Y представляет собой водород или защитную группу, такую как низший алкил, причем соединение формулы II возможно синтезировали реакцией конденсации согласно Схеме I, используя подходящий N-защищенный аминокислотный сложный эфир, в котором R3 такой, как определено выше в формуле I, W представляет собой защитную группу, такую как низший алкил, и Z такой, как определено в формуле II, и подходящее защищенное производное фосфиновой кислоты, в котором R1 и R5 такие, как определено выше в формуле I, Y такой, как определено в формуле II, и основание, такое как диизопропиламид лития,
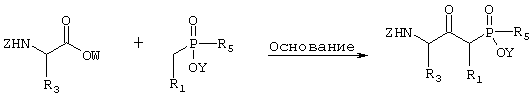
Схема 1
а) возможно превращают путем реакции N-алкилирования для введения R4, если желательно, чтобы R4 не представлял собой водород, и затем гидролитической реакции с получением соединения формулы III,
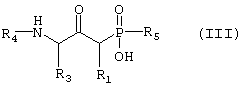
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение III в другое химическое соединение формулы III, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы III и/или другую соль, и/или превращают полученное свободное соединение формулы III в соль в соответствии с вышеуказанным определением, или
б) превращают путем восстановительной реакции, возможно реакции N-алкилирования, если желательно, чтобы R4 не представлял собой водород, и наконец гидролитической реакции с получением соединения формулы IV
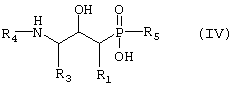
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение IV в другое химическое соединение формулы IV, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы IV и/или в другую соль, и/или превращают полученное свободное соединение формулы IV в соль в соответствии с вышеуказанным определением, или
в) превращают путем восстановительной реакции с последующей реакцией дезоксигалогенирования, возможно реакции N-алкилирования для введения R4, если желательно, чтобы R4 не представлял собой водород, и наконец гидролитической реакции с получением соединения формулы VI,
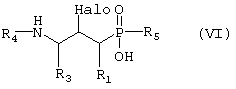
где R1, R3 и R4 такие, как определено выше в формуле I, и Halo представляет собой атом галогена, и возможно превращают полученное выше соединение VI в другое химическое соединение формулы VI, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы VI и/или в другую соль, и/или превращают полученное свободное соединение формулы VI в соль в соответствии с вышеуказанным определением;
или Б) соединение формулы VII
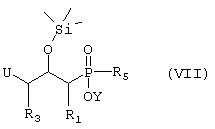
где R1, R3 и R5 такие, как определено выше в формуле I, U представляет собой группу, которую можно превратить в -NH2 группу, и Y представляет собой водород или защитную группу, такую как низший алкил, причем соединение формулы VII возможно синтезировали реакцией конденсации согласно Схеме 2, используя 2,3-эпоксипропильное производное, такое как подходящее N-защищенное производное 2,3-эпоксипропиламина, или эпихлоригидриновое производное, где R1 и R3 такие, как определено выше в формуле I, и подходящее защищенное производное фосфиновой кислоты, активированное O-силилированием, где R5 и Y такие, как определено в формуле VII, и кислоту Льюиса, такую как безводный ZnCl2,

Схема 2
а) превращают путем реакции, при которой триметилсилильную группу замещают атомом водорода, реакции, при которой U группу, как она определена в формуле VII, превращают в -NHR4, где R4 такой, как определено выше в формуле I, и наконец гидролитической реакции с получением соединения формулы IV,
где R1, R3, R4 и R5 такие, как определены выше в формуле I, и возможно превращают полученное выше соединение IV в другое химическое соединение формулы IV, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы IV и/или другую соль, и/или превращают полученное свободное соединение формулы IV в соль в соответствии с вышеуказанным определением, или
б) превращают путем реакции, при которой триметилсилильную группу замещают водородом, окислительной реакции, реакции, при которой U группу, такую как определена в формуле VII, превращают в -NHR4, где R4 такой, как определено выше в формуле I, и наконец гидролитической реакции с получением соединения формулы III
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение III в другое химическое соединение формулы III, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы III и/или в другую соль, и/или превращают полученное свободное соединение формулы III в соль в соответствии с вышеуказанным определением, или
в) превращают путем реакции, при которой триметилсилильную группу замещают водородом, реакции дезоксигалогенирования, реакции, при которой U группу, как она определена в формуле VII, превращают в -NHR4, где R4 такой, как определено выше в формуле I, и наконец гидролитической реакции с получением формулы VI,
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и Halo представляет собой атом галогена, и возможно превращают полученное выше соединение VI в другое химическое соединение формулы VI, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы VI и/или в другую соль, и/или превращают полученное свободное соединение формулы VI в соль в соответствии с вышеуказанным определением;
или В) соединение формулы VIII,
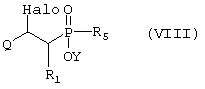
где R1 и R5 такие, как определено выше в формуле I, Q представляет собой электрон-акцепторную группу, такую как, например, -CN или -CO2Et, которую можно превратить в -CH2NH2 группу, и Y представляет собой водород или защитную группу, такую как низший алкил, и Halo представляет собой атом галогена, причем соединение формулы VIII возможно синтезировали реакцией присоединения согласно Схеме 3, используя ненасыщенное соединение, в котором R1 такой, как определено выше в формуле I, Q и Halo такие, как определено в формуле VIII, и подходящее защищенное производное фосфиновой кислоты, активированное O-силилированием, где R5 и Y такие, как определено в формуле VIII,
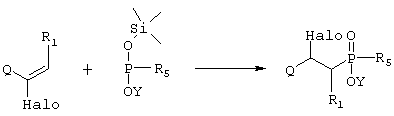
Схема 3
превращают путем реакции, при которой Q группу превращают в -NHR4, где R4 такой, как определено выше в формуле I, и гидролитической реакции с получением соединения формулы IX,
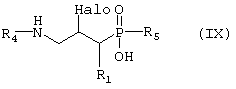
где R1 и R4 такие, как определено выше в формуле I, и Halo представляет собой атом галогена, и возможно превращают полученное выше соединение IX в другое химическое соединение формулы IX, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы IX и/или в другую соль, и/или превращают полученное свободное соединение формулы IX в соль в соответствии с вышеуказанным определением;
или Г) соединение формулы X, возможно в виде отдельного стереоизомера,
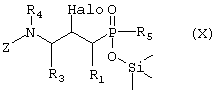
где R1, R3, R4 и R5 такие, как определено в формуле I, Z представляет собой защитную группу, такую как трет-бутилоксикарбонил, и Halo представляет собой атом галогена, причем соединение формулы Х возможно синтезировали реакцией замещения согласно Схеме 4, используя электрофильное соединение, в котором R1, R3 и R4 такие, как определено выше, L представляет собой уходящую группу, такую как йодо, Z и Halo такие, как определено выше, и активированное O-силилированием производное метилфосфиновой кислоты, в котором R5 такой, как определено выше,
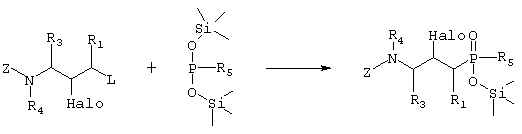
Схема 4
превращают путем гидролитической реакции в соединение формулы VI
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение VI в другое химическое соединение формулы VI, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы VI и/или в другую соль, и/или превращают полученное в результате свободное соединение формулы VI в соль в соответствии с вышеуказанным определением,
или Д) соединение формулы XI, возможно в виде отдельного стереоизомера,
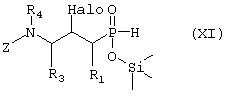
где R1, R3 и R4 такие, как определено в формуле I, Z представляет собой защитную группу, такую как трет-бутилоксикарбонил, и Halo представляет собой атом галогена, причем соединение формулы XI возможно синтезировали реакцией замещения согласно Схеме 5, используя электрофильное соединение, в котором R1, R3 и R4 такие, как определено выше, L представляет собой уходящую группу, такую как йодо, Z и Halo такие, как определено выше, и фосфиновую кислоту, активированную O-силилированием,
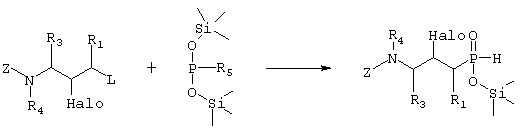
Схема 5
превращают путем гидролитической реакции и затем реакции Р-алкилирования в соединение формулы VI,
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение VI в другое химическое соединение формулы VI, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы VI и/или в другую соль, и/или превращают полученное свободное соединение формулы VI в соль в соответствии с вышеуказанным определением;
или Е) соединение формулы XII,
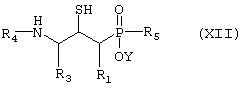
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и Y представляет собой водород или защитную группу, такую как низший алкил, причем соединение формулы XII возможно синтезировали реакциией замещения согласно Схеме 6, обрабатывая ненасыщенное производное фосфиновой кислоты, в котором R1, R3, R4 и R5 такие, как определено выше в формуле I, H2S, меркаптидионом (HS-) или защищенным меркаптосоединением, таким как бензилтиол, в случае чего защитную группу затем удаляют,

Схема 6
превращают путем гидролитической реакции в соединение формулы XIII,
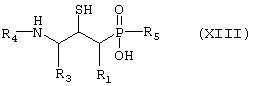
где R1, R3, R4 и R5 такие, как определено выше в формуле I, и возможно превращают полученное выше соединение XIII в другое химическое соединение формулы XIII, и/или разделяют полученную смесь изомеров на отдельные изомеры, и/или превращают полученную соль в свободное соединение формулы XIII и/или в другую соль, и/или превращают полученное свободное соединение формулы XIII в соль в соответствии с вышеуказанным определением.
Данное изобретение описано далее более подробно при помощи следующих примеров, которые не следует рассматриваить как ограничивающие изобретение.
Примеры
Пример 1. (3-Амино-2-фторпропил)(метил)фосфиновая кислота
К раствору ВН3-ТНР(тетрагидрофуран) (1 М, 22,2 мл, 22,2 ммоль) при 0°С добавляли раствор этил-(3-амино-2-фтор-3-оксопропил)(метил)фосфината (2,00 г, 10,1 ммоль) в THF (15 мл). Полученный раствор нагревали с обратным холодильником в течение 2 часов. Раствор охлаждали до комнатной температуры, и медленно добавляли 6 н. HCl (60 мл). THF удаляли выпариванием in vacuo, и добавляли еще одну порцию 6 н. HCl (20 мл). Раствор нагревали с обратным холодильником в течение 2,5 часов, охлаждали до комнатной температуры и упаривали. Остаток очищали ионообменной хроматографией (DOWEX® 50WX 8-200, H+ форма, 3,5×4,0 см). Смолу предварительно промывали смесью 1:1 метанол/вода (200 мл). Неочищенный продукт растворяли в смеси 1:1 метанол/вода и наносили на колонку. Колонку промывали смесью 1:1 метанол/вода (400 мл). Элюент заменяли на смесь 2:1:1 метанол/вода/концентрированный гидроксид аммония. Объединяли две фракции (150 мл) и упаривали с получением 1,28 г вязкого масла. Остаток очищали хроматографией на колонке с мокрой набивкой силикагеля (4×28 см), элюируя смесью 50:50:1 метанол/метиленхлорид/гидроксид аммония. Соответствующие фракции объединяли и упаривали с получением 730 мг (46%) (3-амино-2-фторпропил)(метил)фосфиновой кислоты в виде белого твердого вещества. Данные: точка плавления 78-84°С, Rf=0,26 (60:40:1 метанол, метиленхлорид, концентрированный гидроксид аммония); 1Н ЯМР (300 МГц, D2O) δ 5.01 (dm, J=49,5 Гц, 1Н), 3.09-3.32 (m, 2H), 1.76-2.18 (m, 2H); 1.28 (d, J=14 Гц); 13С ЯМР (75 МГц, D2O+Диоксан) δ 91.3 (d, J=169 Гц), 47.1 (d, J=12 Гц), 37.2 (dd, J=20,9, 89 Гц), 19.8 (d, J=95 Гц); APIMS (масс-спектрометрия с ионизацией при атмосферном давлении): m/z=156 (М+Н)+.
Пример 2. (3-Амино-2-фтор-1-метилпропил)(метил)фосфиновая кислота
К охлажденному на ледяной бане раствору этил-3-амино-2-фтор-1-метил-3-оксопропил(метил)фосфината (2,1 г, 10 ммоль) в THF (25 мл) добавляли 1 М ВН3-THF (23 мл, 23 ммоль) в атмосфере аргона. Через 10 минут раствор нагревали с обратным холодильником в течение 2 часов. Раствор охлаждали до комнатной температуры, и по каплям добавляли 6 н. HCl (30 мл). THF удаляли выпариванием на роторном испарителе, и добавляли еще одну порцию 6 н. HCl (30 мл). Смесь кипятили с обратным холодильником в течение 2 часов и затем перемешивали при комнатной температуре в течение ночи. Раствор охлаждали, упаривали, упаривали совместно с водой и затем с этанолом. Остаток растворяли в метаноле (25 мл), и затем по каплям добавляли пропиленоксид (4 мл). Через 3 часа при комнатной температуре раствор концентрировали и продукт очищали хроматографией на колонке с мокрой набивкой силикагеля, элюируя смесью вода/метанол (2%-4% воды). Соответствующие фракции объединяли и упаривали с получением масла, которое осаждалось при добавлении диэтилового эфира. Продукт отделяли фильтрацией и получали 580 мг (35%) смеси диастереомеров (3-амино-2-фтор-1-метилпропил)(метил)фосфиновой кислоты в виде белого твердого вещества. 1H ЯМР (400 МГц, D2O) δ 4.8-5.2 (m, 1H), 3.2-3.5 (m, 2H), 1.8-2.2 (m, 1H), 1.0-1.3 (m, 6H); MS: m/z=170 (М+Н)+.
Пример 3. (2R)-(3-Амино-2-фторпропил)(метил)фосфиновая кислота
Суспензию соединения (2R)-(3-амино-2-фторпропил)фосфиновой кислоты (1,0 г, 7,1 ммоль) в HMDS (гексаметилдисилазане) (7,47 мл, 35,4 ммоль) нагревали с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, обрабатывали диглимом (8,0 мл) и нагревали с обратным холодильником в течение 6 часов. После того как смесь охладили до комнатной температуры, добавляли основание Хунига (1,23 мл, 7,1 ммоль), потом по каплям добавляли метилиодид (1,32 мл, 21,2 ммоль). Реакционную смесь перемешивали в течение 23,5 часов, затем разбавляли метиленхлоридом и экстрагировали 2 н. раствором HCl. Водный слой промывали метиленхлоридом и диэтиловым эфиром и затем упаривали при пониженном давлении. Неочищенный продукт пропускали через колонку с Dowex 50WX8-200 меш в Н+ форме, элюируя смесью 1:1 метанол/вода до тех пор, пока вещество более не обнаруживали при ТСХ анализе. Продукт затем элюировали смесью 1:3 концентрированный раствор гидроксида аммония/метанол. После дальнейшей очистки колоночной хроматографией на силикагеле, элюируя смесью метиленхлорида, метанола и концентрированного раствора гидроксида аммония (6:3:1), получали (2R)-(3-амино-2-фторпропил)(метил)фосфиновую кислоту в виде белого твердого вещества (720 мг, 65%). 1Н ЯМР (300 МГц, D2O) δ 5.20 (m, 0.5H); 5.03 (m, 0,5H), 3.20-3.42 (m, 2Н), 1.80-2.22 (m, 2H), 1.30 (d, J=14,0 Гц, 3Н).
Пример 4. (2S)-(3-Амино-2-фторпропил)(метил)фосфиновая кислота
Суспензию соединения (2S)-(3-амино-2-фторпропил)фосфиновой кислоты (1,2 г, 8,5 ммоль) в HMDS (8,96 мл, 42,4 ммоль) нагревали с обратным холодильником в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры, обрабатывали диглимом (9,6 мл) и нагревали с обратным холодильником в течение 7 часов. После охлаждения смеси до комнатной температуры добавляли основание Хунига (1,47 мл, 8,4 ммоль), затем по каплям добавляли метилиодид (1,58 мл, 25,4 ммоль). Реакционную смесь перемешивали в течение ночи, затем разбавляли метиленхлоридом и экстрагировали 2 н. раствором HCl. Водный слой промывали метиленхлоридом и диэтиловым эфиром и затем упаривали при пониженном давлении. Неочищенный продукт пропускали через колонку с Dowex 50WX8-200 меш в H+ форме, элюируя смесью 1:1 метанол/вода до тех пор, пока вещество более не обнаруживали при ТСХ анализе. Продукт затем элюировали смесью 1:3 концентрированный раствор гидроксида аммония/метанол. После дальнейшей очистки колоночной хроматографией на силикагеле, элюируя смесью метиленхлорида, метанола и концентрированного раствора гидроксида аммония (6:3:1), получали (2S)-(3-амино-2-фторпропил)(метил)фосфиновую кислоту в виде белого твердого вещества (504 мг, 38%). 1H ЯМР (300 МГц, D2O) δ 5.20 (m, 0.5H); 5.03 (m, 0,5H), 3.20-3.42 (m, 2H), 1.80-2.22 (m, 2H), 1.30 (d, J=14,0 Гц, 3Н).
Промежуточные соединения
Пример I1. Этил-3-[этокси(метил)фосфорил1-2-фторпропаноат (промежуточное соединение для соединения по Примеру 1)
Этил-метилфосфинат (12,8 г, 118 ммоль) нагревали с обратным холодильником с 1,1,1,3,3,3-гексаметилдисилазаном (HMDS) (24,9 мл, 118 ммоль) в течение 2 часов в атмосфере аргона. Раствор охлаждали до комнатной температуры и затем добавляли к раствору фторакрилат (13,3 г, 113 ммоль). После перемешивания при комнатной температуре в течение 60 часов в атмосфере аргона смесь разбавляли метиленхлоридом (200 мл) и промывали 1 н. HCl (200 мл). Водный слой экстрагировали метиленхлоридом (200 мл). Объединенные органические слои промывали рассолом (150 мл), сушили над Na2SO4, фильтровали и упаривали с получением 20,0 г масла. Остаток хроматографировали на колонке с мокрой набивкой силикагеля (7,5×40 см), элюируя смесью 95:5 метиленхлорид/метанол. Соответствующие фракции объединяли и упаривали с получением 12,6 г (50%) этил-3-[этокси(метил)фосфорил]-2-фторпропаноата в виде желтого масла. Данные: 1Н ЯМР (300 МГц, CDCl3) δ 5.11-5.43 (m, 1H), 4.23-4.34 (m, 2H), 4.03-4.19 (m, 2H), 2.26-2.53 (m, 2H), 1.58 (d, J=14 Гц, 3Н); 1.28-1.39 (m, 3Н).
Пример I2. Этил-(3-амино-2-фтор-3-оксопропил)(метил)фосфинат (промежуточное соединение для соединения по Примеру 1)
Раствор этил-3-[этокси(метил)фосфорил]-2-фторпропаноата (12,6 г, 55,6 ммоль), этанола (15 мл) и концентрированного гидроксида аммония (14,8 н., 5,6 мл, 83 ммоль) перемешивали при комнатной температуре в течение 16 часов. Растворитель выпаривали, и остаток хроматографировали на колонке с мокрой набивкой силикагеля (5,5×31 см), элюируя смесью 90:10 метиленхлорид/метанол. Подходящие фракции объединяли и упаривали с получением 9,5 г (86%) этил-(3-амино-2-фтор-3-оксопропил)(метил)фосфината в виде прозрачного масла. Данные: 1Н ЯМР (300 МГц, CDCl3) δ 6.62 (s, 1H), 6.33 (s, 1H), 5.10-5.42 (m, 1H), 4.02-4.18 (m, 2H), 2.19-2.70 (m, 2H), 1.60 (d, J=14 Гц, 3Н), 1.34 (m, 3H).
Пример I3. Этил-3-[этокси(метил)фосфорил1-2-фторбутаноат (промежуточное соединение для соединения по Примеру 2)
Смесь этил-(метил)фосфината (3,2 г, 30 ммоль) и 1,1,1,3,3,3-гексаметилдисилазана (4,8 г, 30 ммоль) нагревали с обратным холодильником в течение 2 часов в атмосфере аргона. Смесь охлаждали до комнатной температуры, и добавляли смесь диастереомеров этил-2-фторбут-2-еноата (4,0 г, 30 ммоль). Реагенты нагревали до 70°С в течение трех суток и затем при комнатной температуре в течение 4 суток в атмосфере аргона. Смесь разбавляли метиленхлоридом (200 мл). Раствор промывали 1 н. HCl (200 мл), и водный слой экстрагировали метиленхлоридом (200 мл). Объединенные органические растворы промывали насыщенным хлоридом натрия, сушили над MgSO4, фильтровали и упаривали. Остаток очищали хроматографией на колонке с мокрой набивкой силикагеля, элюируя смесью метиленхлорид/метанол (99:1-97:3). Соответствующие фракции объединяли и упаривали с получением 2,9 г (40%) этил-3-[этокси(метил)фосфорил]-2-фторбутаноата в виде прозрачного масла. 1Н ЯМР (400 МГц, CDCl3) δ 4.8-5.6 (m, 1Н), 4.2-4.4 (m, 2Н), 4.0-4.2 (m, 2H), 2.4-2.6 (m, 1H); 1.1-1.5 (m, 12H).
Пример I4. Этил-3-амино-2-фтор-1-метил-3-оксопропил(метил)фосфинат (промежуточное соединение для соединения по Примеру 2)
К раствору этил-3-[этокси(метил)фосфорил]-2-фторбутаноата (3,0 г, 12,4 ммоль) в этаноле добавляли концентрированный гидроксид аммония (14,8 M, 1,3 мл, 19 ммоль). Раствор перемешивали в течение 24 часов при комнатной температуре и затем при 50°С в течение 2 часов. Добавляли еще одну порцию концентрированного гидроксида аммония (14,8 M, 0,5 мл, 7 ммоль), и смесь перемешивали при комнатной температуре в течение трех суток, и затем упаривали с получением 2,4 г (91%) смеси диастереомеров этил-3-амино-2-фтор-1-метил-3-оксопропил(метил)фосфината в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3) δ 5.9-6.7 (m, 2H), 4.9-5.6 (m, 1H), 4.0-4.2 (m, 2H), 2.5-2.8 (m, 1H), 1.2-1.6 (m, 9H).
Пример I5. (2R)-(3-Амино-2-фторпропил)фосфиновая кислота (промежуточное соединение для соединения по Примеру 3)
Гипофосфит аммония (73,8 г, 0,89 моль) добавляли в 3-горлую 2-литровую колбу, снабженную механической мешалкой, термометром, воронкой для добавления и барботером аргона. Колбу помещали в водяную баню при комнатной температуре, и добавляли N,O-бис-(триметилсилил)ацетамид (215 мл, 0,87 мол - BSA) с такой скоростью, чтобы поддерживать внутреннюю температуру ниже 38°С (приблизительно 30 минут), используя ледяное охлаждение. По окончании добавления BSA реакционную смесь нагревали до 45-48°С и поддерживали при этой температуре в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, и к реакционной смеси добавляли раствор трет-бутил-(2R)-2-фтор-3-йодпропилкарбамата (27,3 г, 0,09 моль) в метиленхлориде (300 мл). Реакционную смесь затем оставляли перемешиваться при комнатной температуре в течение 18 часов. Реакционную смесь охлаждали до 0°С и осторожно гасили метанолом (275 мл) и затем водой (32 мл). Реакционную смесь перемешивали в течение 30 минут, после чего реакционную смесь фильтровали, и твердые вещества промывали метанолом. Фильтрат концентрировали, и остаток помещали в глубокий вакуум (0,1 мм рт. ст. (13,33 Па)) на ночь. Неочищенный остаток растирали со смесью метиленхлорида, метанола и концентрированного раствора гидроксида аммония (80:20:1) и фильтровали. Фильтрат концентрировали при пониженном давлении, и растирание повторяли. Неочищенный концентрат переносили в 2-литровую колбу, растворяли в метаноле (375 мл) и помещали на водяную баню при комнатной температуре. Добавляли насыщенный раствор газообразного хлористого водорода в этилацетате (500 мл), и смесь перемешивали в течение 3 часов. Реакционную смесь фильтровали, и твердые вещества промывали смесью метанола и этилацетата (90:10). Фильтрат концентрировали при пониженном давлении, и неочищеный продукт пропускали через колонку с Dowex® 50WX8-200 меш в Н+ форме (500 г, 8×15 см), элюируя смесью 1:1 метанол/вода, до тех пор пока вещество более не обнаруживали посредством ТСХ анализа. Нужный неочищенный продукт затем элюировали смесью 1:3 концентрированный раствор гидроксида аммония/метанол. Продукт затем очищали колоночной хроматографией, элюируя смесью хлороформа, метанола и концентрированного раствора гидроксида аммония (6:3:1) с получением (2R)-(3-амино-2-фторпропил)фосфиновой кислоты в виде белого твердого вещества (3,12 г, 24%). 1H ЯМР (300 МГц, D2O) δ 7.90 (s, 0,5 Н), 6.15 (s, 0,5 Н), 5.12-5.29 (m, 0,5 Н), 4.92-5.10 (m, 0,5 Н), 3.12-3.42 (m, 2H), 1.74-2.26 (m, 2H).
Пример I6. (2R)-3-(Дибензиламино)-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 3)
Боргидрид лития (5,3 г, 0,24 моль) суспендировали в THF (200 мл) в атмосфере азота и охлаждали до -15°С при перемешивании. Метил-(2R)-3-(дибензиламино)-2-фторпропаноат (56,6 г, 0,19 моль) суспендировали в THF (250 мл) и добавляли по каплям к смеси за 1 час; внутреннюю температуру поддерживали ниже -10°С во время добавления. По окончании добавления реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при этой температуре в течение 17 часов. Реакционную смесь охлаждали до 0°С и осторожно гасили насыщенным водным раствором хлорида аммония (300 мл). Реакционную смесь экстрагировали этилацетатом (2×200 мл), и органическую фазу концентрировали при пониженном давлении. Неочищенный остаток растворяли в 2 н. соляной кислоте (200 мл, рН 2 приблизительно), и водную фазу промывали эфиром (2×200 мл). Водную фазу подщелачивали (приблизительно рН 10) 80%-ным гидроксидом аммония в соляном растворе, экстрагировали этилацетатом (3×200 мл), сушили над безводным сульфатом натрия (10 г), фильтровали и концентрировали при пониженном давлении с получением (2R)-3-(дибензиламино)-2-фтор-1-пропанола (48 г, 93%) в виде желтого масла. 1H ЯМР (300 МГц, CDCl3) δ 7.15-7.38 (m, 10Н), 4.65-4.78 (m, 0.5H), 4.48-4.58 (m, 0,5Н), 3.50-3.82 (m, 6H), 2.70-2.88 (m, 2H).
Пример I7. (2R)-3-Амино-2-Фтор-1-пропанол (промежуточное соединение для соединения по Примеру 3)
(2R)-3-(Дибензиламино)-2-фтор-1-пропанол (29,2 г, 0,11 моль) растворяли в этаноле (300 мл). Добавляли десятипроцентный по массе гидроксид палладия (II) на угле (5,0 г), и смесь помещали на Parr® шейкер и встряхивали в атмосфере водорода (55 фунт./кв. дюйм (379,225 кПа)) в течение 6 часов. Когда поглощение водорода больше не наблюдалось, смесь фильтровали через набивку Celite® (20 г). К этанольной смеси добавляли свежую порцию гидроксида палладия (II) (5 г) и снова подвергали режиму гидрирования, описанному выше, в течение 17 часов. Неочищенную реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении с получением (2R)-3-амино-2-фтор-1-пропанола в виде бледно-желтого масла (9,6 г, 96%).
1H ЯМР (300 МГц, CD3OD) δ 4.78-5.00 (br s, 3H), 4.49-4.62 (m, 0,5H), 4.32-4.46 (m, 0,5H), 3.54-3.70 (m, 2H), 2.70-2.96 (m, 2H).
Пример I8. Трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамат (промежуточное соединение для соединения по Примеру 3)
(2R)-3-амино-2-фтор-1-пропанол (4,6 г, 49 ммоль) растворяли в 25%-ном водном диоксане (160 мл), добавляли карбонат калия (7,1 г, 51 ммоль), и смесь охлаждали до 0°С. Ди-трет-бутилдикарбонат (11,6 г, 53 ммоль) добавляли двумя порциями. Смесь затем оставляли нагреваться до комнатной температуры в течение ночи. Неочищенную реакционную смесь концентрировали досуха, добавляли воду (150 мл), затем насыщенный водный гидросульфат калия (до рН 3 приблизительно). Органическое вещество экстрагировали метиленхлоридом (2×150 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамата (9,5 г, 100%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ 4.82-5.04 (br s, 1H), 4.62-4.72 (m, 0,5H), 4.48-4.58 (m, 0,5H), 3.62-3.72 (m, 2H), 3.32-3.62 (m, 2H), 3.20-3.44 (br s, 1H), 1.48 (s, 9H).
Пример I9. Трет-бутил-(2R)-2-фтор-3-йодпропилкарбамат (промежуточное соединение для соединения по Примеру 3)
Имидазол (26,6 г, 0,39 моль) растворяли в метиленхлориде (400 мл) при комнатной температуре. Добавляли йод (102,5 г, 0,39 моль), и реакционную смесь перемешивали в течение 10 минут при комнатной температуре и затем охлаждали до 0°С. Трифенилфосфин (102,5 г, 0,39 моль) добавляли частями в течение 10 минут, таким образом, чтобы внутренняя температура оставалась ниже 10°С. По каплям добавляли раствор трет-бутил-(2R)-2-фтор-3-гидроксипропилкарбамата (60,4 г, 0,31 моль) в метиленхлориде (100 мл). По окончании добавления трет-бутил-(2R)-фтор-3-гидроксипропилкарбамата добавляли дополнительно метиленхлорид (200 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжали в течение 17 часов. Реакционную смесь фильтровали через набивку Celite® (50 г) и промывали дополнительно метиленхлоридом. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле, элюируя метиленхлоридом. В результате этой процедуры получали трет-бутил-(2R)-2-фтор-3-йодпропилкарбамат в виде белого твердого вещества (64,7 г, 68%).
1H ЯМР (300 МГц, CDCl3) δ 4.80-5.10 (br s, 1H), 4.58-4.72 (m, 0,5H), 4.42-4.56 (m, 0,5H), 3.48-3.70 (m, 1H), 3.20-3.46 (m, 3H), 1.48 (s, 9H).
Пример I10. (2S)-(3-Амино-2-фторпропил)фосфиновая кислота (промежуточное соединение для соединения по Примеру 4)
Гипофосфит аммония (58,1 г, 0,70 моль) добавляли в 3-горлую 2-литровую колбу, снабженную механической мешалкой, термометром, воронкой для добавления и барботером аргона. N,O-бис-(триметилсилил)ацетамид (175,9 мл, 0,71 моль - BSA) добавляли с такой скоростью, чтобы поддерживать внутреннюю температуру между 35-40°С. По окончании добавления BSA реакционную смесь поддерживали при 35-40°С в течение 45 минут. Добавляли метиленхлорид (150 мл), и смесь перемешивали при 35-40°С в течение дополнительных 45 минут. Реакционную смесь охлаждали до комнатной температуры, и к реакционной смеси добавляли раствор трет-бутил-(2S)-2-фтор-3-йодпропилкарбамата (42,5 г, 0,14 моль) в метиленхлориде (300 мл). Реакционную смесь затем оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь охлаждали до 0°С и осторожно гасили метанолом (150 мл) и затем водой (60 мл). Реакционную смесь концентрировали, и остаток помещали в глубокий вакуум (0,1 мм рт. ст (13,33 Па)). Остаток доводили до рН приблизительно 8 добавлением концентрированного гидроксида аммония (50 мл), затем добавляли метиленхлорид (400 мл) и метанол (250 мл). Полученные в результате твердые вещества отфильтровывали, и фильтрат концентрировали. Остаток растирали со смесью метиленхлорида, метанола и концентрированного раствора гидроксида аммония (80:20:1; 400 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении, и неочищенный концентрат растворяли в метаноле (400 мл). Добавляли насыщенный раствор газообразного хлористого водорода в этилацетате (600 мл), и смесь перемешивали в течение 3 часов. Реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт пропускали через колонку с Dowex® 50WX8-200 меш в H+ форме (450 г), элюируя смесью 1:1 метанол/вода, до тех пор пока вещество более не обнаруживали посредством ТСХ анализа. Нужный неочищенный продукт затем элюировали смесью 1:3 концентрированный раствор гидроксида аммония/метанол. Продукт далее очищали колоночной хроматографией, элюируя смесью метиленхлорида, метанола и концентрированного раствора гидроксида аммония (6:3:1) с получением (2S)-(3-амино-2-фторпропил)фосфиновой кислоты в виде белого твердого вещества (3,46 г, 17%).
1H ЯМР (300 МГц, D2O) δ 7.90 (s, 0,5 Н), 6.15 (s, 0,5 Н), 5.12-5.29 (m, 0,5 Н), 4.92-5.10 (m, 0,5 Н), 3.12-3.42 (m, 2H), 1.74-2.
Пример I11. Метил-(2S)-3-(дибензиламино)-2-фторпропаноат (промежуточное соединение для соединения по Примеру 4)
Метил-(2R)-2-(дибензиламино)-3-гидроксипропаноат (231,7 г, 0,77 моль) растворяли в THF (850 мл), и медленно по каплям добавляли раствор DAST (трифторида (диэтиламино)серы) (196 г, 1,2 моль) в THF (400 мл). Как только добавление было закончено, реакционную смесь перемешивали в течение еще 1,5 часов. ТСХ анализ показывал расход исходного вещества. Реакционную смесь затем охлаждали до 0°С и гасили медленым добавлением воды (1,5 л) с последующей нейтрализацией добавлением твердого бикарбоната натрия. Сразу после нейтрализации добавляли смесь 1:1 концентрированного гидроксида аммония/насыщенного раствора хлорида натрия, и реакционную смесь экстрагировали этилацетатом и концентрировали при пониженном давлении. Неочищенную смесь очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетата и гексанов (1:4) с получением желаемого соединения (188,3 г, 62%) в виде масла.
1H ЯМР (300 МГц, CDCl3) δ 7.18-7.38 (m, 10Н), 5.12-5.17 (m, 0,5H), 4.95-5.00 (m, 0,5H), 3.81-3.87 (m, 2H), 3.69 (s, 3H), 3.49-3.55 (m, 2H), 2.90-3.12 (m, 2H).
Пример I12. (2S)-3-(Дибензиламино)-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 4)
Боргидрид лития (17,7 г, 0,81 моль) суспендировали в THF (400 мл) в атмосфере азота и охлаждали до -15°С при перемешивании. Метил-(2S)-3-(дибензиламино)-2-фторпропаноат (188,3 г, 0,62 моль) суспендировали в THF (400 мл) и добавляли по каплям к смеси. По окончании добавления реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при этой температуре в течение 3 часов. ТСХ анализ показал полный расход исходного вещества. Реакционную смесь охлаждали до 0°С и осторожно гасили насыщенным водным раствором хлорида аммония (300 мл). Добавляли дополнительно воду (400 мл), затем реакционную смесь экстрагировали этилацетатом, и органическую фазу концентрировали при пониженном давлении. Неочищенный остаток растворяли в 2 н. соляной кислоте, и водную фазу промывали дважды эфиром. Водную фазу подщелачивали (рН 10 приблизительно) 80%-ным гидроксидом аммония в рассоле, экстрагировали этилацетатом, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (2S)-3-(дибензиламино)-2-фтор-1-пропанола (156,6 г, 92%) в виде желтого масла. 1Н ЯМР (300 МГц, CDCl3) δ 7.15-7.38 (m, 10Н), 4.65-4.78 (m, 0.5Н), 4.48-4.58 (m, 0,5H), 3.50-3.82 (m, 6H), 2.70-2.88 (m, 2H).
Пример I13. (2S)-3-Амино-2-фтор-1-пропанол (промежуточное соединение для соединения по Примеру 4)
(2S)-3-(Дибензиламино)-2-фтор-1-пропанол (39,1 г, 0,14 моль) растворяли в этаноле (300 мл). Добавляли десятипроцентный по массе гидроксид палладия (II) на угле (5,0 г), и смесь помещали на Parr® шейкер и встряхивали в атмосфере водорода (55 фунт./кв. дюйм (379,225 кПа)) в течение ночи. Когда поглощение водорода больше не наблюдали, смесь фильтровали через набивку Celite® (20 г). К этанольной смеси добавляли свежую порцию гидроксида палладия (II) (5 г), и снова подвергали режиму гидрирования, описанному выше, в течение 12 часов. Снова, когда поглощение водорода больше не наблюдали, смесь фильтровали через набивку Celite®. К этанольной смеси добавляли свежую порцию гидроксида палладия (II) (5 г) и снова подвергали режиму гидрирования, описанному выше, в течение 12 часов. Неочищенную реакционную смесь фильтровали через Celite® и концентрировали при пониженном давлении с получением (2S)-3-амино-2-фтор-1-пропанола в виде бледно-желтого масла (13,3 г, 100%). 1Н ЯМР (300 МГц, CD3OD) δ 4.78-5.00 (br s, 3Н), 4.49-4.62 (m, 0,5H), 4.32-4.46 (m, 0,5H), 3.54-3.70 (m, 2H), 2.70-2.96 (m, 2H).
Пример I14. Трет-бутил-(2S)-2-фтор-3-гидроксипропилкарбамат (промежуточное соединение для соединения по Примеру 4)
(2S)-3-Амино-2-фтор-1-пропанол (38,6 г, 0,41 моль) растворяли в 25%-ном водном диоксане (1,4 л), добавляли карбонат калия (60,1 г, 0,43 моль), затем ди-трет-бутилдикарбонат (99,5 г, 0,46 моль). Смесь перемешивали в течение ночи. ТСХ анализ показал полный расход исходного вещества. Неочищенную реакционную смесь концентрировали досуха, добавляли воду (300 мл), затем насыщенный водный гидросульфат калия (до рН 3 приблизительно). Органическое вещество экстрагировали дважды метиленхлоридом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-(2S)-2-фтор-3-гидроксипропилкарбамата (79,5 г, 99%) в виде бледно-желтого масла. 1H ЯМР (300 МГц, CDCl3) δ 4.82-5.04 (br s, 1H), 4.62-4.72 (m, 0,5H), 4.48-4.58 (m, 0,5H), 3.62-3.72 (m, 2H), 3.32-3.62 (m, 2H), 3.20-3.44 (brs, 1H), 1.48 (s, 9H).
Пример I15. Трет-бутил-(2S)-2-фтор-3-йодпропилкарбамат (промежуточное соединение для соединения по Примеру 4)
Имидазол (19,8 г, 0,29 моль) растворяли в метиленхлориде (900 мл) при комнатной температуре. Добавляли йод (73,9 г, 0,29 моль), и реакционную смесь перемешивали в течение 10 минут при комнатной температуре и затем охлаждали до 0°С. Трифенилфосфин (76,3 г, 0,29 моль) добавляли частями в течение 10 минут таким образом, чтобы внутренняя температура оставалась ниже 10°С. По каплям добавляли раствор трет-бутил-(2S)-2-фтор-3-гидроксипропилкарбамата (45,0 г, 0,23 моль) в метиленхлориде (300 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжали в течение 12 часов. Реакционную смесь фильтровали через набивку Celite® и промывали дополнительно метиленхлоридом. Фильтрат концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле, элюируя метиленхлоридом. В результате этой процедуры получали трет-бутил-(2S)-2-фтор-3-йодпропилкарбамат в виде бесцветного масла (42,5 г, 62%). 1H ЯМР (300 МГц, CDCl3) δ 4.80-5.10 (br s, 1H), 4.58-4.72 (m, 0,5H), 4.42-4.56 (m, 0,5H), 3.48-3.70 (m, 1H), 3.20-3.46 (m, 3H), 1.48 (s, 9H).
Фармацевтические препараты
Соединение формулы (I) по настоящему изобретению можно использовать в качестве активного ингредиента в фармацевтическом препарате для перорального, ректального, эпидурального, внутривенного, внутримышечного, подкожного, назального введения и введения путем инфузии или для любого другого подходящего пути введения. Предпочтительный путь введения представляет собой пероральный или путем инъекции/инфузии.
Фармацевтические препараты содержат соединение по настоящему изобретению в комбинации с одним или более чем одним фармацевтически приемлемым ингредиентом. Готовые лекарственные формы изготавливают при помощи известных фармацевтических способов. Обычно количество активных соединений составляет между 0,1-95% по массе от препарата, предпочтительно между 0,2-20% по массе в препаратах для парентерального применения, и предпочтительно между 1-50% по массе в препаратах для перорального введения.
При приготовлении фармацевтических препаратов, содержащих соединение по настоящему изобретению, в форме твердых единиц дозировки для перорального введения, выбранное соединение можно смешивать с твердыми, порошкообразными фармацевтически приемлемыми ингредиентами (среди них, например, разрыхляющие агенты и смазывающие агенты). Смесь затем перерабатывают в гранулы, таблетки, капсулы или саше.
Единицы дозировки для ректального введения можно приготовить в форме суппозиториев; в форме желатиновых ректальных капсул; в форме готовых микроклизм; или в форме сухого препарата для микроклизмы для разведения в подходящем растворителе непосредственно перед введением.
Жидкие препараты для перорального введения можно приготовить в форме сиропов или суспензий, или в форме сухой смеси для разведения подходящим растворителем перед применением.
Растворы для парентерального введения можно приготовить в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, и их разделяют на стандартные дозы в форме ампул или флаконов. Их также можно приготовить в виде сухого препарата для разведения подходящим растворителем непосредственно перед применением.
Обычная суточная доза активного соединения будет зависеть от различных факторов, таких как, например, индивидуальная потребность каждого пациента, путь введения и заболевание. Обычно дозировки будут в пределах от 1 мкг до 100 мг в сутки и на кг массы тела, предпочтительно от 10 мкг до 20 мг в сутки и на кг массы тела.
Биологические исследования
[3H]GABA радиолигандный анализ связывания
Синаптические мембраны крыс готовили из целого мозга самцов крыс Sprague Dawley no существу аналогично описанному ранее (Zukin, et al. (1974) Proc. Natl. Acad. USA 71, 4802-4807). [3H]GABA конкурентный анализ, модифицированный OIpe et al. ((1990) Eur. J. Pharmacol. 187, 27-38) выполняли в 200 мкл TCI (трис-кальций изогувацинового) буфера (50 мМ Трис (три(гидроксиметил)аминометан), рН 7,4, 2,5 мМ CaCl2 и 40 мкМ изогувацина), содержащем 20 нМ [3H]GABA (специфическая активность: 3 тера-беккерель (ТБк)/ммоль), испытуемое соединение или растворитель и 80 мкг белка синаптических мембран, используя 96-луночные планшеты. После инкубации в течение 12-20 минут при комнатной температуре, инкубирования заканчивали путем быстрой фильтрации через фильтр из стекловолокна (Printed filtermat В filter, Wallac), который был предварительно обработан 0,3%-ным полиэтиленимином, используя 96-луночный харвестер клеток (Skatron или Tomtec). Фильтры промывали буфером, содержащим 50 мМ Трис (трис(гидроксиметил)аминометан) и 2,5 мМ CaCl2, рН 7,4 при 4°С, и затем сушили при 55°С. MeltiLex B/HS сцинтилляторный лист (Wallac) растапливали на фильтр, и радиоактивность определяли в Microbeta сцинтилляционном счетчике (Wallac).
Результаты и обсуждение
Было обнаружено, что соединения по настоящему изобретению имеют неожиданно высокое сродство и активности в отношении GABAB рецепторов, как выявлено низкими IC50 (средняя ингибирующая концентрация) и EC50 (средняя эффективная концентрация) в анализах связывания и подвздошной кишки соответственно. Активности в отношении ингибирования TLOSR были связаны со сродством и активностью по отношению к GABAB рецептору. Побочные эффекты на ЦНС (как определено по снижению температуры тела у мышей) наблюдались лишь при дозах выше, чем терапевтические дозы, ингибирующие TLOSR в модели на собаках. Следовательно, разница между терапевтической дозой (ингибирование TLOSR) и дозой, вызывающей побочные эффекты, была неожиданно высокой.
Результаты [3H]JGABA радиолигандного анализа связывания
Значение IC50 для 3-аминопропилметилфосфиновой кислоты, замещенной фтором по положению 2, в 1900 раз ниже, чем IC50 значение 3-метилфосфиновой кислоты, замещенной по положению 2 наиболее близким к фтору заместителем - хлором. Это свидетельствует о неожиданном и необычайно высоком сродстве к САВАB-рецепторам 3-аминопропилметифосфиновых кислот, замещенных фтором по положению 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АМИНОПРОПИЛФОСФИНОВЫЕ КИСЛОТЫ | 2000 |
|
RU2260595C2 |
| СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ | 2009 |
|
RU2481329C2 |
| ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ | 2003 |
|
RU2333203C2 |
| ЦИКЛОАЛКИЛ-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ИМИДАЗОЛА, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ TAFIa | 2011 |
|
RU2572814C2 |
| ПРОИЗВОДНЫЕ ДИАМИНА | 2002 |
|
RU2314303C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОВ | 2002 |
|
RU2319699C2 |
| АГОНИСТЫ TLR7 | 2019 |
|
RU2817014C2 |
| СИНТЕТИЧЕСКОЕ ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1-(2-ДЕЗОКСИ-2-ФТОР-4-ТИО-β-D-АРАБИНОФУРАНОЗИЛ)ЦИТОЗИНА, СИНТЕТИЧЕСКОЕ ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ТИОНУКЛЕОЗИДА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2633355C2 |
| ПРОИЗВОДНОЕ 4,4-ДИФТОР-1,2,2,4-ТЕТРАГИДРО-5Н-1-БЕНЗАЗЕПИНА ИЛИ ЕГО СОЛЬ | 2004 |
|
RU2326868C2 |
Предложены (аминопропил)метилфосфиновые кислоты общей формулы
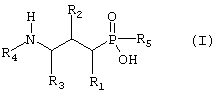
за исключением (1) рацемата (3-амино-2-гидроксипропил)метилфосфиновой кислоты; (2) S-(3-амино-2-гидроксипропил)метилфосфиновой кислоты; (3) R-(3-амино-2-гидроксипропил)метилфосфиновой кислоты; (4) (3-амино-2-гидроксипропил)дифторметилфосфиновой кислоты; и (5) (3-амино-2-оксопропил)метилфосфиновой кислоты, имеющие сродство к одному или более чем одному GABAe (γ-аминомасляная кислота) рецептору, их фармацевтически приемлемые соли, сольваты и стереоизомеры, а также фармацевтические композиции, содержащие указанные терапевтически активные соединения, и применение указанных активных соединений в терапии. 7 н. и 5 з.п. ф-лы.
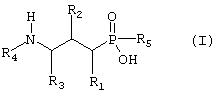
где
R1 представляет собой водород или низший алкил;
R2 представляет собой фторгруппу;
R3 представляет собой водород;
R4 представляет собой водород;
R5 представляет собой метил, фторметил, дифторметил или трифторметил;
и его фармацевтически приемлемые соли, сольваты и стереоизомеры.
| WO 9811885 A1, 26.03.1998 | |||
| J.MED.CHEM., Vol.38, 1995, p.3297-3333 | |||
| Способ получения солей перфторалкилфосфиновых кислот | 1976 |
|
SU589756A1 |
| Способ получения о-аминофениларилфосфиновых кислот | 1975 |
|
SU643510A1 |

