УРОВЕНЬ ТЕХНИКИ
1. Область техники
Изобретение в целом относится к производным биологически активных молекул, которые имеют более высокую биодоступность при пероральном введении и, соответственно, обладают более высокой биологической активностью по сравнению с исходным биологически активным лекарственным средством.
2. Описание уровня техники
Множество эндогенных веществ, природных продуктов и синтетических веществ, обладающих полезными терапевтическими свойствами, при пероральном введении имеют высокий эффект «первого прохождения». Это означает, что указанные молекулы быстро подвергаются метаболизму или выводятся из организма, и, соответственно, для обеспечения целевого биологического эффекта требуется применение относительно высоких доз.
Типовые соединения, обладающие высоким эффектом первого прохождения, представляют собой половые стероиды, такие как эстрогены и андрогены. Например, естественные гормоны эстрадиол и тестостерон имеют низкую биодоступность при пероральном введении, что значительно ограничивает их применение.
Для преодоления указанного недостатка на протяжении многих лет проводятся обширные исследования.
Традиционные сложные эфиры, такие как эстрадиол-17-бензоат, или сложные эфиры жирных кислот, например, те, что присутствуют в тестостерона энантате, характеризуются в некоторой степени улучшенной пероральной биодоступностью.
Было показано, что производные сульфокислоты являются особенно перспективными. В патенте США №5705495 и Европейских патентах ЕР 1273590 и 1284273 были описаны производные сульфокислоты и эстрадиола, обладающие более высокой эстрогенной активностью после перорального введения по сравнению с эстрадиолом.
Для дальнейшей разработки фактически было выбрано одно производное J955 (эстрадиола сульфамат). J995 имел более высокую эстрогенную активность после перорального введения по сравнению с эстрадиолом и этинилэстрадиолом в пробе Аллена-Дойзи у самок крыс после овариэктомии (Walter Elger et al., J. Steroid Biochem. Molec. Biol. Vol 55 395-403, 1995). Разработку J995 были вынуждены прекратить после того, как было определено, что J995 также действовал как ингибитор сульфатазы у женщин.
В патенте США №7507725 и Европейском патенте ЕР 1294402 указанная концепция была расширена и сведена к соединениям, состоящим из трех различных фрагментов: активный ингредиент-(группа-разделитель)-SO2NR1R2. Было выдвинуто предположение о том, что указанные соединения связываются с эритроцитами. Активный ингредиент может представлять собой молекулу стероида, но также и другие лекарственные средства, такие как мочегонные средства, агонисты дофамина и т.д. Описанная группа-разделитель может состоять из углеродной цепи или ароматического кольца или комбинации указанных групп.
Указанная концепция позднее была расширена до определенных классов веществ (патенты США №6841548; 6956031; 6958327; заявка на патент США №2005/2277625; патент США №7534780; публикация международной заявки WO 03/104253).
Несмотря на то, что описанные соединения имели активность, несколько превышающую активность исходных лекарственных средств в определенных биологических исследованиях, природа описанной группы-разделителя и сульфонамидного фрагмента обеспечивает соединения с очень низкой растворимостью в воде, что значительно ограничивает возможность их применения в качестве лекарственных средств.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В первом варианте реализации аминосодержащие производные имеют следующую общую формулу (I):

где n равен 0-4; m равен 0-4; L равен 0-1;
R1 представляет собой Н, алкил, циклоалкил, арил, алкиларил, гетероарил или галоген;
hAr представляет собой арил, гетероарил, алкил, алкиларил или алкилгетероарил;
R2 представляет собой Н, алкил, циклоалкил, арил, гетероарил, галоген, арилсульфонамид, гетероарилсульфонамид, алкиларилсульфонамид или алкилгетероарилсульфонамид;
R1 и R2 могут быть объединены с образованием циклоалкила или 3-7-членного кольца, содержащего до одного гетероатома;
каждый R3 и R4 независимо представляет собой Н, алкил, циклоалкил, арил, гетероарил, ацил, циано, галоген, гидрокси, алкокси;
каждый X и Y независимо представляет собой Н, галоген, циано, гидрокси, алкокси, алкил, арил, гетероарил;
Z представляет собой О или NR1;
O=C-(CH)n-R1 имеет R- или S-конфигурацию.
Во втором варианте реализации кислородсодержащие производные имеют общую формулу (II)

где n равен 0-4; m равен 0-4; L равен 0-1;
R1 представляет собой Н, алкил, циклоалкил, арил, гетероарил или галоген;
hAr представляет собой арил или гетероарил;
каждый R3 и R4 независимо представляет собой Н, алкил, циклоалкил, арил, гетероарил, ацил, циано, галоген, гидрокси, алкокси;
каждый X и Y независимо представляет собой Н, галоген, циано, гидрокси, алкокси, алкил, арил, гетероарил;
Z представляет собой О или NR1;
O=C-(CH)n-R1 имеет R- или S-конфигурацию.
В другом варианте реализации замещенные алкильные производные имеют следующую общую формулу (III):

где n равен 0-1; m равен 1-4;
Z представляет собой О или NR1;
каждый R1 и R2 независимо представляет собой галоген, или R1 и R2 объединены с образованием циклоалкила.
Согласно другому аспекту в некоторых вариантах реализации настоящего изобретения предложена фармацевтическая композиция, содержащая соединения формулы I, II или III в фармацевтически приемлемом носителе.
Согласно другому аспекту в некоторых вариантах реализации настоящего изобретения предложен противозачаточный продукт и продукт для гормонозаместительной терапии, содержащий соединения формул I и II, приведенных далее.


Для лучшего понимания последующего подробного описания в вышеуказанном описании в общих чертах отмечены отличительные признаки настоящего изобретения. Дополнительные отличительные признаки и преимущества, которые описаны далее, составляют предмет, определенный формулой изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ
Следует понимать, что настоящее изобретение не ограничено конкретными устройствами или способами, которые безусловно могут быть различными. Также следует понимать, что терминология, используемая в настоящем описании, предназначена для описания исключительно конкретных вариантов реализации и не является ограничивающей. При использовании в настоящем описании и прилагаемой формуле изобретения формы единственного числа включают ссылки на отдельные объекты и их совокупность, если из контекста явным образом не следует иное. Кроме того, слово «может» используют в настоящем описании в качестве свободного термина (т.е. обладает потенциалом, имеет возможность), но не обязательного термина (т.е. должен). Термин «включает» и их производные обозначают «включает, но не ограничивается ими». Термин «сопряженный» обозначает связанный непосредственно или косвенно.
Если отсутствуют иные определения, то все технические и научные термины, используемые в настоящем описании, имеют значения, общепринятые специалистами в данной области техники.
Термин «алкил», используемый в настоящем описании, в общем случае относится к химическому заместителю, содержащему одновалентную группу CnH2n, где n представляет собой целое число больше нуля. В некоторых вариантах реализации n равен от 1 до 12. Термин «алкил» включает разветвленный или неразветвленный одновалентный углеводородный радикал. Примеры алкильных радикалов включают, но не ограничиваются ими, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, пентил, 3-пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил. Алкильную группу, содержащую 1-6 атомов углерода, называют «низшим алкилом». Подходящие низшие алкильные радикалы включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, 2-пропенил (или аллил), н-бутил, трет-бутил и изобутил (или 2-метилпропил).
Термин «циклоалкил» обозначает группы, содержащие алифатическую цепь, замкнутую с образованием кольцевой структуры. Количество атомов углерода в алифатической цепи может находиться в диапазоне от трех до 12. Типовые циклоалкильные группы включают циклопропил, циклобутил, циклопентил и циклогексил.
Термин «алкокси» в общем случае относится к группе -OR, где R представляет собой низший алкил, замещенный низший алкил, арил, замещенный арил, аралкил или замещенный аралкил. Подходящие алкоксирадикалы включают, но не ограничиваются ими, метокси, этокси, фенокси, трет-бутокси, метоксиэтокси и метоксиметокси.
Термин «галоген» используют в настоящей заявке для описания атомов фтора, брома, хлора и йода.
Термин «гидроксил» используют в настоящей заявке для описания группы -ОН.
Термин «арил» используют для описания ароматического заместителя, который может представлять собой отдельное кольцо или несколько колец, которые являются конденсированными, связанными посредством ковалентной связи или общей группы, такой как этиленовый фрагмент. Ароматическое(-ие) кольцо(-а) включает(-ют), но не ограничивается(-ются) ими, фенил, нафтил, бифенил, дифенилметил и 2,2-дифенил-1-этил. Арильная группа также может быть замещена заместителями, включая, но не ограничиваясь ими, алкильные группы, атомы галогенов, нитрогруппы, карбоксильные группы, алкокси и фенокси, с образованием «замещенной арильной группы». Заместители могут быть присоединены по любому положению арильного радикала, которое в иных случаях занято атомом водорода.
Термин «гетероцикл», используемый в настоящем описании, в общем случае относится к закрытой кольцевой структуре, в которой один или более атомов в кольце представляют собой элементы, отличающиеся от углерода. Гетероцикл может включать ароматические соединения или неароматические соединения. Гетероциклы могут включать кольца, такие как тиофен, пиридин, изоксазол, фталимид, пиразол, индол, фуран или аналоги указанных колец, конденсированные с бензолом. Примеры гетероциклов включают тетрагидрофуран, морфолин, пиперидин, пирролидин и т.д. В некоторых вариантах реализации «гетероцикл» обозначает стабильное 5-7-членное моноциклическое или бициклическое или 7-10-членное бициклическое гетероциклическое кольцо, которое может быть насыщенным или ненасыщенным и состоит из атомов углерода и 1-4 гетероатомов (например, N, О и S), причем гетероатомы азота и серы необязательно могут быть окисленными, а атом азота необязательно может быть четвертичным, и включает любые бициклические группы, в которых какое-либо из определенных выше гетероциклических колец конденсировано с бензольным кольцом. В некоторых вариантах реализации гетероциклы могут включать циклические кольца, содержащие атомы бора. Гетероциклическое кольцо может быть присоединено к соседней группе через любой гетероатом или атом углерода с образованием стабильной структуры. Гетероциклические кольца, описанные в настоящей заявке, могут быть замещены по атому углерода или атому азота, если получаемое соединение является стабильным. Примеры указанных гетероциклов включают, но не ограничиваются ими, 1Н-индазол, 2-пирролидонил, 2Н,6Н-1,5,2-дитиазинил, 2Н-пирролил, 3Н-индолил, 4-пиперидонил, 4аН-карбазол, 4Н-хинолизинил, 6Н-1,2,5-тиадиазинил, акридинил, азоцинил, бензофуранил, бензотиофенил, карбазол, хроманил, хроменил, циннолинил, декагидрохинолинил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, индолинил, индолизинил, индолил, изобензофуранил, изохроманил, изоиндолинил, изоиндолил, изохинолинил (бензимидазолил), изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксазолидинил, оксазолил, фенантридинил, фенантролинил, фенарсазинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, пирролил, хиназолинил, хинолинил, хиноксалинил, хинуклидинил, карболинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, тиантренил, тиазолил, тиенил, тиофенил, триазинил, ксантенил. Также включены конденсированные кольца и спиросоединения, содержащие, например, указанные выше гетероциклы. Термин «гетероарил» имеет значение, эквивалентное гетероциклу, и указанные термины используют взаимозаменяемо.
Термин «фармацевтически приемлемые соли» включает соли, полученные в результате взаимодействия с участием фармацевтически приемлемых нетоксичных оснований или кислот, включая взаимодействие неорганических или органических оснований с неорганическими или органическими кислотами. Фармацевтически приемлемые соли могут включать соли, полученные из неорганических оснований, включая соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца (III), марганца (II), калия, натрия, цинка и т.д. Примеры включают соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-дибензилэтилендиамин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.д.
Список сокращений
AcOH - уксусная кислота
Boc - трет-бутилкарбамат
Cbz - бензилкарбамат
DCC - дициклогексилкарбодиимид
ДХМ - дихлорметан
DIC - диизопропилкарбодиимид
DMAP - N,N-диметил-4-аминопиридин
ДМФ - диметилформамид
ДМСО - диметилсульфоксид
EDCI - гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида
EtOAc - этилацетат
HOBt - гидрат 1-гидроксибензотриазола
ВЭЖХ - высокоэффективная жидкостная хроматография
Основание Хюнига - диизопропилэтиламин
ИКС - инфракрасная спектроскопия
ЖХСД - жидкостная хроматография среднего давления
ЯМР - ядерный магнитный резонанс
пТСК - пара-толуолсульфокислота
PDC - дихромат пиридиния
TBAI - йодид тетрабутиламмония
TBAF - фторид тетрабутиламмония
TBS - трет-бутилдиметилсилил
TBSCl - трет-бутилдиметилсилилхлорид
ТГФ - тетрагидрофуран
ТСХ - тонкослойная хроматография
Варианты реализации, описанные в настоящей заявке, направлены на устранение недостатков соединений, известных из уровня техники, и описывают молекулы пролекарств, обладающих значительно повышенной пероральной активностью по сравнению с исходными молекулами.
Было выдвинуто предположение о том, что введение гетероатомов, таких как азот или кислород, в линкерную группу может увеличивать растворимость и, следовательно, пероральную активность молекул. В указанном контексте особенно эффективными могут быть аминокислоты. Описаны сложные эфиры аминокислот и стероидов. В патентах Китая CN 102127137 и 102079771 раскрыты сложные эфиры аминокислот с эстрогенами, обладающие противоопухолевой активностью и имеющие хорошую растворимость в воде. В патенте Франции FR 2774989 описаны пептиды эстрадиола, обладающие противоопухолевой и цитотоксической активностью. В патенте Германии DE 4029499 описаны дифосфонатные производные эстрадиола для лечения остеопороза. В Европейском патенте ЕР 351561 описаны производные эстратриендиола в качестве неопластических соединений, и в патенте США №4615835 описаны сложные эфиры стероидов и нитрозокарбамоиламинокислот, обладающие высокой противоопухолевой активностью.
В настоящем изобретении заявлены соединения, содержащие следующие фрагменты:
Активный ингредиент - (кислота)-(линкерная группа) - SO2NR1R2.
Активный ингредиент может представлять собой любой активный ингредиент, содержащий по меньшей мере одну гидрокси- или аминогруппу. Активный ингредиент предпочтительно выбран из андрогенов, анаболических агентов, антиандрогенов, эстрогенов, прогестинов или соединений, действующих на ЦНС.
В одном из вариантов реализации активный ингредиент представляет собой андроген, такой как тестостерон, где функциональная группа представляет собой 17-гидроксигруппу. Типовым андрогеном является 7α,11β-диметилэстра-4,9-диен-17-ол, где функциональная группа представляет собой 17-гидроксигруппу.
В другом варианте реализации активный ингредиент представляет собой эстроген. Типовыми эстрогенами являются эстрадиол или эстриол, где функциональная группа представляет собой 16- или 17-гидроксигруппу.
В одном из вариантов реализации активный ингредиент представляет собой прогестин, такой как тримегестон, и функциональная группа представляет собой 21-гидроксигруппу.
Соединения, описанные в настоящей заявке, действуют как пролекарство, которое обеспечивает захват активного агента эритроцитами. Возможность захвата указанных соединений эритроцитами обеспечивается группой формулы SO2NR3NR4; где R3 и R4 независимо друг от друга представляют собой Н, алкил, циклоалкил, арил, гетероарил, ацил, циано, галоген, гидрокси или алкокси. Пролекарства согласно настоящему изобретению обеспечивают возможность эффективного перорального введения или значительного улучшения пероральной активности активных агентов, таких как эндогенные вещества, природные вещества и синтетические вещества, со свойствами, подходящими для терапии, имеющих высокий «эффект первого прохождения».
В первом варианте реализации аминосодержащие производные имеют следующую общую формулу (I):

где n равен 0-4; m равен 0-4; L равен 0-1;
R1 представляет собой Н, алкил, циклоалкил, арил, алкиларил, гетероарил или галоген;
hAr представляет собой арил, гетероарил, алкил, алкиларил или алкилгетероарил;
R2 представляет собой Н, алкил, циклоалкил, арил, гетероарил, галоген; арилсульфонамид, гетероарилсульфонамид, алкиларилсульфонамид или алкилгетероарилсульфонамид;
R1 и R2 могут быть объединены с образованием циклоалкила или 3-7-членного кольца, содержащего до одного гетероатома;
каждый R3 и R4 независимо представляет собой Н, алкил, циклоалкил, арил, гетероарил, ацил, циано, галоген, гидрокси, алкокси;
каждый X и Y независимо представляет собой Н, галоген, циано, гидрокси, алкокси, алкил, арил, гетероарил;
Z представляет собой О или NR1;
O=C-(CH)n-R1 имеет R- или S-конфигурацию.
Во втором варианте реализации кислородсодержащие производные имеют общую формулу (II)

где n равен 0-4; m равен 0-4; L равен 0-1;
R1 представляет собой Н, алкил, циклоалкил, арил, гетероарил или галоген;
hAr представляет собой арил, гетероарил, алкил, алкиларил или алкилгетероарил;
каждый R3 и R4 независимо представляет собой Н, алкил, циклоалкил, арил, гетероарил, ацил, циано, галоген, гидрокси или алкокси;
каждый X и Y независимо представляет собой Н, галоген, циано, гидрокси, алкокси, алкил, арил, гетероарил;
Z представляет собой О или NR1;
O=C-(CH)n-R1 имеет R- или S-конфигурацию.
В другом варианте реализации замещенные алкильные производные имеют следующую общую формулу (III):

где n равен 0-1; m равен 1-4;
Z представляет собой О или NR1;
каждый R1, R2 независимо представляет собой галоген, или R1 и R2 объединены с образованием циклоалкила.
В другом варианте реализации замещенные гетероарильные производные имеют следующую общую формулу (IV):
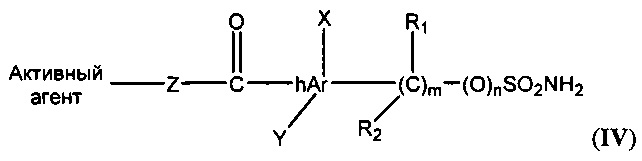
где n равен 0-1; m равен 1-4;
каждый R1 и R2 независимо представляет собой Н, галоген, алкил, алкенил, циклоалкил, алкокси, ацил;
hAr представляет собой арил, гетероарил;
Z представляет собой О или NR1;
каждый X и Y независимо представляет собой Н, галоген, циано, гидроксил, алкокси, алкил, циклоалкил, арил, гетероарил или ОАс.
В другом варианте реализации замещенные гетероарильные производные имеют следующую общую формулу (V):

где n равен 0-2; m равен 0-1; L равен 0-4;
каждый R1, R2 независимо представляет собой Н, галоген, циклоалкил, алкил, арил, гетероарил, ацил, циано, галоген, гидроксил или алкокси;
R1 и R2 могут быть объединены с образованием циклоалкила;
каждый R3 и R4 независимо представляет собой Н, алкил, циклоалкил, арил, гетероарил, ацил, циано, галоген, гидрокси или алкокси;
каждый R5 и R6 независимо представляет собой Н, алкил, арил, галоген, алкокси, циклоалкил;
hAr представляет собой арил, гетероарил;
Z представляет собой О или NR1;
каждый X и Y независимо представляет собой Н, галоген, циано, гидроксил, алкокси, алкил, арил или гетероарил;
Q представляет собой О.
В другом варианте реализации замещенные гетероарильные производные имеют следующую общую формулу (VI):

где n равен 0-1;
каждый R1 и R2 независимо представляет собой Н, галоген, алкил, алкенил, циклоалкил, циано, алкокси или ацил;
hAr представляет собой винил, ацетилен, арил, гетероарил;
hAr' представляет собой арил, гетероарил;
Z представляет собой О или NR1;
каждый X и Y независимо представляет собой Н, галоген, циано, гидроксил, алкокси, алкил, циклоалкил, арил, гетероарил или ацил.
Согласно другому аспекту в некоторых вариантах реализации настоящего изобретения предложена фармацевтическая композиция, содержащая соединения формул I-VI в фармацевтически приемлемом носителе.
В конкретных вариантах реализации настоящего изобретения предложен противозачаточный продукт и продукт для гормонозаместительной терапии, содержащий соединения формул I и II, приведенных ниже.

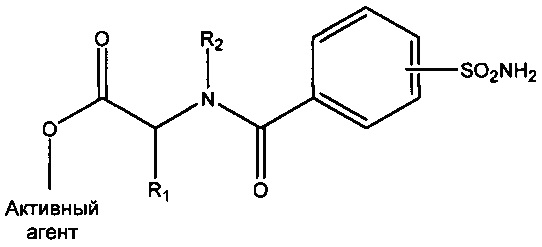
Описанные выше соединения можно вводить в состав фармацевтической композиции, содержащей соединение любой из формул (I)-(VI) и фармацевтически приемлемый носитель. В зависимости от способа введения можно применять дополнительные вспомогательные вещества.
Для обеспечения эффективной дозировки соединений, описанных в настоящей заявке, можно применять любые подходящие способы введения. Например, можно применять пероральный, ректальный, местный, парентеральный, внутриглазной, внутрилегочный, интраназальный способы и т.д. Лекарственные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и т.д. В определенных вариантах реализации пероральное введение композиций, описанных в настоящей заявке, может быть эффективным.
Следующие примеры приведены для демонстрации предпочтительных вариантов реализации изобретения. Специалисты в данной области техники должны понимать, что способы, раскрытые в последующих примерах, представляют собой способы, предложенные авторами настоящего изобретения для эффективной реализации изобретения, и, таким образом, их можно рассматривать как предпочтительные варианты реализации. Тем не менее, специалистам в данной области техники после изучения настоящего изобретения должно быть понятно, что можно проводить множество изменений конкретных раскрытых вариантов реализации и добиваться при этом аналогичных или схожих результатов, не выходя за рамки сущности и объема изобретения.
Экспериментальная часть. Предложенные пролекарства
(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 3-сульфамоилбензойной кислоты 1А синтезировали согласно описанию, приведенному в литературе (патент США №7507725 и Европейский патент ЕР 1294402).
(13S,17S)-17-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-3-иловый эфир сульфаминовой кислоты J995 синтезировали согласно описанию, приведенному в литературе (патент США №5705495 и Европейские патенты ЕР 1273590 и 1284273).
Схема 1

2А можно синтезировать, как показано на схеме 1. Простой эфир эстрадиол-3-TBS 7 обрабатывали бензилоксиацетилхлоридом в присутствии пиридина с получением сложного эфира 2, из которого путем дебензилирования получали соединение 3. Этерификация 3 с использованием 3-хлорсульфонилбензоилхлорида в присутствии пиридина приводила к получению 3-TBS-защищенного Е2, которое подвергали десилилированию с использованием фторида тетрабутиламмония с получением 2А.
3А можно синтезировать, как показано на следующей схеме 2.
Схема 2
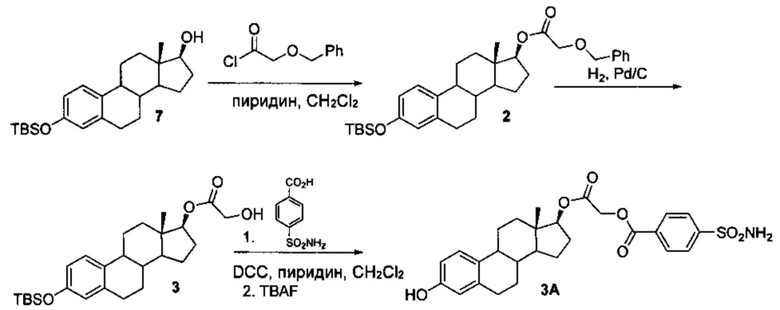
Промежуточные соединения 2 и 3 получали, как показано на схеме 1. Проводили сочетание соединения 3 с 4-сульфамоилбензойной кислотой с использованием DCC и пиридина с получением 3-TBS производного 3А, из которого путем десилилирования с использованием фторида тетрабутиламмония получали 3А.

(13S,17S)-3-(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-иловый эфир 2-(бензилокси)уксусной кислоты (2):
В раствор стероида 7 (0,5 г, 1,29 ммоль) и пиридина (0,13 г, 1,67 ммоль) в безводном дихлорметане (10 мл) при 0°С добавляли бензилоксиацетилхлорид (0,31 г, 1,67 ммоль). После перемешивания указанного раствора в течение 45 минут добавляли воду. Реакционную смесь трижды экстрагировали дихлорметаном. Объединенные органические слои промывали нас. NaHCO3, солевым раствором и сушили над безв. Na2SO4. Удаляли растворитель в вакууме и очищали неочищенный продукт путем колоночной хроматографии (SiO2, гексан-этилацетат) с получением 2 (640 мг, 93%). 1Н ЯМР (δ, CDCl3, 300 МГц): 7,27-7,38 (m, 5Н, ArH), 7,13 (d, J=8,4 Гц, 1Н, ArH), 6,62 (dd, J1=8,4 Гц, J2=2,6 Гц, 1Н, ArH), 6,55 (d, J=2,4 Гц, 1H, ArH), 4,82 (t, J=8,7 Гц, 1Н, -СН), 4,65 (s, 2Н, -ОСН2), 4,11 (s, 2H, -OCH2), 2,81 (m, 2Н, -СН2), 0,97 (s, 9Н, Si-CH3), 0,82 (s, 3Н, -СН3), 0,18 (s, 6Н, Si-СН3).

(13S,17S)-3-(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-иловый эфир 2-гидроксиуксусной кислоты (3):
Раствор соединения 2 (0,64 г, 1,19) в этилацетате перемешивали с 5% палладием на углеродной подложке (0,06 г, 0,6 ммоль) и гидрировали под давлением 20 psi (138 кПа) в течение 1 часа. Фильтровали раствор для удаления катализатора и выпаривали досуха. Полученный неочищенный остаток очищали путем колоночной хроматографии (SiO2, гексан-EtOAc) с получением 3 (0,47 г, 89%). 1Н ЯМР (δ, CDCl3, 300 МГц): 7,21 (d, J=8,4 Гц, 1H, ArH), 6,60 (dd, J1=8,4 Гц, J2=2,6 Гц, 1H, ArH), 6,55 (d, J=2,4 Гц, 1Н, ArH), 4,81 (t, J=8,8 Гц, 1Н, -СН), 4,15 (d, J=2,4 Гц, 2Н, -ОСН2), 2,80 (m, 2Н, -СН2), 0,97 (s, 9Н, Si-СН3), 0,82 (s, 3Н, -СН3), 0,18 (s, 6Н, Si-СН3).

2-((13S,17S)-3-(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 3-сульфамоилбензойной кислоты:
Раствор 3 (0,2 г, 0,45 ммоль) и пиридина (0,05 г, 0,6 ммоль) в дихлорметане (10 мл) при -20°С обрабатывали, добавляя по каплям раствор 3-сульфамоилбензоилхлорида (0,14 г, 0,6 ммоль) в дихлорметане. После перемешивания в течение 30 минут при -20°С реакционную смесь нагревали до 8°С и гасили раствором NH4OH (3 мл) и перемешивали еще 30 минут. Реакционную смесь трижды экстрагировали дихлорметаном. Объединенные органические слои промывали водой, солевым раствором и сушили над безводным Na2SO4. Удаляли растворитель в вакууме и очищали неочищенный остаток путем колоночной хроматографии (SiO2, гексан-EtOAc) с получением 2А, защищенного 3-TBS (0,24 г, 85%). 1Н ЯМР (δ, CDCl3, 300 МГц): 8,64-8,66 (m, 1H, ArH), 8,28-8,31 (m, 1Н, ArH), 8,12-8,16 (m, 1H, ArH), 7,63 (t, J=7,9 Гц, 1H, ArH), 7,09 (d, J=8,4 Гц, 1H, ArH), 6,60 (dd, J1=8,4 Гц, J2=2,6 Гц, 1H, ArH), 6,54 (d, J=2,4 Гц, 1H, ArH), 4,89 (s, 2H, -OCH2), 4,80 (t, J=8,8 Гц, 1H, -CH), 2,80 (m, 2H, -CH2), 0,97 (s, 9H, Si-CH3), 0,79 (s, 3H, -CH3), 0,18 (s, 6H, Si-CH3).
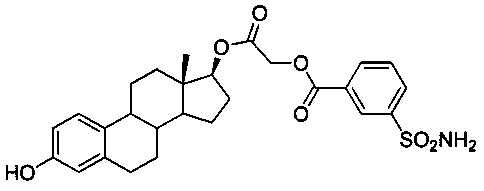
2-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 3-сульфамоилбензойной кислоты (2А)
Раствор простого эфира 3-TBS-2A (0,24 г, 0,38 ммоль) в безводном ТГФ (10 мл) обрабатывали фторидом тетрабутиламмония (0,13 г, 0,42 ммоль). После перемешивания в течение 1 часа при КТ добавляли воду и трижды экстрагировали EtOAc. Объединенные органические слои промывали водой, солевым раствором и сушили над безводным Na2SO4. Удаляли растворитель в вакууме и очищали неочищенное вещество путем колоночной хроматографии (SiO2, гексан-EtOAc) с получением 2А (0,16 г, 82%). ИКС (см-1): 3352, 3257, 2926, 2858, 1727, 1499, 1221, 1159, 1127, 1039, 750. 1Н ЯМР (δ, CDCl3, 300 МГц): 8,65-8,66 (m, 1H, ArH), 8,28-8,31 (m, 1Н, ArH), 8,12-8,16 (m, 1H, ArH), 7,63 (t, J=7,8 Гц, 1H, ArH), 7,12 (d, J=8,4 Гц, 1H, ArH), 6,61 (dd, J1=8,4 Гц, J2=2,6 Гц, 1Н, ArH), 6,54 (d, J=2,4 Гц, 1Н, ArH), 5,08 (s, 2Н, -NH2), 4,95 (s, 1H, -ОН), 4,83 (s, 2Н, -ОСН2), 4,80 (t, J=8,9 Гц, 1Н, -CH), 2,79 (m, 2H, -CH2), 0,73 (s, 3Н, -CH3). 13C ЯМР (δ, CDCl3, 75 МГц): 167,64, 164,50, 153,38, 138,09, 133,79, 132,33, 130,90, 130,44, 129,52, 127,93, 126,48, 115,23, 112,70, 84,23, 61,64, 49,61, 43,66, 43,13, 38,47, 36,81, 29,49, 27,40, 27,09, 26,12, 23,20, 12,03.

2-((13S,17S)-3-(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 4-сульфамоилбензойной кислоты:
В раствор 3 (0,22 г, 0,5 ммоль) и 4-сульфамоилбензойной кислоты (0,24 г, 1,15 ммоль) в пиридине (10 мл) добавляли n-TsOH (0,08 г, 0,5 ммоль), затем 1М раствор DCC в дихлорметане (0,24 г, 1,15 ммоль). После перемешивания при КТ в течение 72 часов добавляли воду и доводили pH реакционной смеси до 7 путем добавления 4н. хлороводородной кислоты. Реакционную смесь трижды экстрагировали этилацетатом и объединенные органические слои промывали водой, солевым раствором и сушили над безводным Na2SO4. Удаляли растворитель в вакууме и очищали неочищенный остаток путем колоночной хроматографии (SiO2, гексан-EtOAc) с получением простого эфира 3-TBS-3A (0,27 г, 86%). 1Н ЯМР (δ, CDCl3, 300 МГц): 8,23 (d, J=8,5 Гц, 2Н, ArH), 8,02 (d, J=8,5 Гц, 2Н, ArH), 7,10 (d, J=8,4 Гц, 1H, ArH), 6,61 (dd, J1=8,4 Гц, J2=2,5 Гц, 1Н, ArH), 6,53 (d, J=2,4 Гц, 1Н, ArH), 4,89 (s, 2Н, -ОСН2), 4,80 (t, J=8,9 Гц, 1H, -СН), 2,80 (m, 2Н, -СН2), 0,97 (s, 9Н, Si-СН3), 0,78 (s, 3Н, -СН3), 0,18 (s, 6Н, Si-СН3), 0,18 (s, 6Н, Si-CH3).

2-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 4-сульфамоилбензойной кислоты (2А):
Согласно описанию способа получения 2A TBS группу удаляли путем обработки простого эфира 3-TBS-3A (0,26 г, 0,4 ммоль) с использованием TBAF⋅3Н2О (0,3 г, 0,45 ммоль) с получением 3А (0,2 г, 95%). ИКС (см-1): 3386, 3265, 2930, 2853, 1731, 1549, 1225, 1167, 1122, 1039, 765. 1Н ЯМР (δ, CDCl3, 300 МГц): 8,15 (d, J=8,5 Гц, 2Н, ArH), 7,95 (d, J=8,5 Гц, 2Н, ArH), 7,05 (d, J=8,4 Гц, 1H, ArH), 6,55 (dd, J1=8,4 Гц, J2=2,5 Гц, 1Н, ArH), 6,49 (d, J=2,4 Гц, 1Н, ArH), 4,91 (s, 2Н, -ОСН2), 4,74 (t, J=8,8 Гц, 1H, -СН), 2,80 (m, 2Н, -СН2), 0,73 (s, 3Н, -СН3). 13С ЯМР (δ, CDCl3, 75 МГц): 167,59, 164,68, 154, 146,91, 137,91, 133,79, 132,40, 131,30, 130,36, 126,23, 115,09, 112,61, 84,17, 61,54, 49,48, 49,38, 49,10, 48,81, 48,68, 43,55, 42,99, 38,42, 36,68, 33,57, 29,39, 27,26, 27,03, 25,99, 25,42, 24,73, 23,06, 11,86.

2-((13S,16R,17R)-16-(трет-бутилдиметилсилилокси)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 3-сульфамоилбензойной кислоты
Раствор 25 (0,7 г, 1,2 ммоль) и пиридина (0,19 г, 2,43 ммоль) в дихлорметане (10 мл) при -20°С обрабатывали, добавляя по каплям раствор 3-сульфамоилбензоилхлорида (0,58 г, 2,43 ммоль) в дихлорметане. После перемешивания в течение 30 минут при -20°С реакционную смесь нагревали до 8°С и гасили раствором NH4OH (3 мл) и перемешивали еще 30 минут. Реакционную смесь трижды экстрагировали дихлорметаном. Объединенные органические слои промывали водой, солевым раствором и сушили над безводным Na2SO4. Удаляли растворитель в вакууме и очищали неочищенный остаток путем колоночной хроматографии (SiO2, гексан-EtOAc) с получением 3- и 16-TBS-защищенного 24А (0,78 г, 84%). Полученное соединение (0,28 г, 0,37 ммоль) обрабатывали TBAF (0,12 г, 0,37 ммоль) с получением простого эфира 16-TBS-24A (0,2 г, 84%). 1Н ЯМР (CDCl3-ДМСО, 300 МГц): 8,66-8,67 (m, 1H, ArH), 8,30-8,33 (m, 1Н, ArH), 8,13-8,15 (m, 1H, ArH), 7,65 (t, J=7,8 Гц, 1Н, ArH), 7,11 (d, J=8,4 Гц, 1H, ArH), 6,59 (dd, J1=8,4 Гц, J2=2,2 Гц, 1Н, ArH), 6,55 (d, J=2,2 Гц, 1Н, ArH), 4,96 (s, 2Н, -ОСН2), 4,52 (d, J=4,7 Гц, 1Н, -СН), 4,06 (m, 1H, -СН), 2,73 (m, 2Н, -СН2), 0,89 (s, 9Н, Si-СН3) 0,74 (s, 3Н, -СН3), 0,05 (s, 6Н, Si-CH3).

2-((13S,16R,17R)-3,16-дигидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 3-сульфамоилбензойной кислоты:
16-TBS-защищенное 24А (0,2 г, 0,3 ммоль) обрабатывали 4н. HCl (0,02 г, 0,5 ммоль) с получением 24А (0,16 г, 97%). ИКС (см-1): 3346, 2933, 2856, 1737, 1728, 1342, 1225, 1168, 1132, 913, 731. 1Н ЯМР (CDCl3-ДМСО, 300 МГц): 8,54-8,56 (m, 1Н, ArH), 8,19-8,22 (m, 1H, ArH), 8,07-8,10 (m, 1Н, ArH), 7,58 (t, J=7,9 Гц, 1H, ArH), 7,03 (d, J=8,6 Гц, 1Н, ArH), 6,56 (dd, J1=8,4 Гц, J2=2,2 Гц, 1H, ArH), 6,49 (d, J=2,2 Гц, 1Н, ArH), 4,90 (s, 2Н, -ОСН2), 4,52 (d, J=4,7 Гц, 1Н, -СН), 4,06 (m, 1Н, -СН), 2,73 (m, 2Н, -СН2), 0,74 (s, 3Н, -СН3).
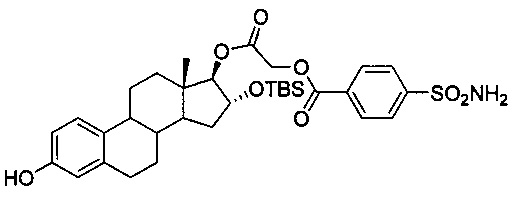
2-((13S,16R,17R)-16-(трет-бутилдиметилсилилокси)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 4-сульфамоилбензойной кислоты:
В раствор 25 (1,1 г, 1,9 ммоль) и 4-сульфамоилбензойной кислоты (0,95 г, 4,4 ммоль) в пиридине (10 мл) добавляли n-TsOH (0,3 г, 1,71 ммоль), затем 1М раствор DCC в дихлорметане (4,4 мл, 1,15 ммоль). После перемешивания при КТ в течение 72 часов добавляли воду и доводили pH реакционной смеси до 7 путем добавления 4н. хлороводородной кислоты. Реакционную смесь трижды экстрагировали этилацетатом и объединенные органические слои промывали водой, солевым раствором и сушили над безводным Na2SO4. Удаляли растворитель в вакууме и очищали неочищенный остаток путем колоночной хроматографии (SiO2, гексан-EtOAc) с получением простого эфира 3- и 16-TBS-25A (0,71 г, 51%). Полученное соединение (0,51 г, 0,67 ммоль) обрабатывали TBAF (0,2 г, 0,67 ммоль) с получением простого эфира 16-TBS-25A (0,4 г, 93%). 1Н ЯМР (CDCl3-ДМСО, 300 МГц): 8,23-8,26 (m, 2Н, ArH), 8,01-8,04 (m, 2Н, ArH), 7,12 (d, J=8,4 Гц, 1Н, ArH), 6,63 (dd, J1=8,1 Гц, J2=2,1 Гц, 1Н, ArH), 6,55 (d, J=2,4 Гц, 1Н, ArH), 4,91-4,93 (m, 5Н, -ОСН2, -ОН, -NH2), 4,68 (s 1Н, -СН), 4,29-,32 (m, 1Н, -СН), 2,81 (m, 2Н, -СН2), 0,89 (s, 9Н, Si-СН3) 0,77 (s, 3Н, -СН3), 0,05 (s, 6Н, Si-СН3).
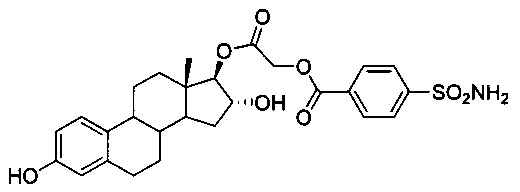
2-((13S,16R,17R)-3,16-дигидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илокси)-2-оксоэтиловый эфир 4-сульфамоилбензойной кислоты:
Согласно описанию способа получения 24А простой эфир 16-TBS-25A (0,45 г, 0,69 ммоль) обрабатывали 4н. HCl (0,07 г, 2,07 ммоль) с получением 25А (0,32 г, 87%). ИКС (см-1): 3485, 3409, 2929, 2859, 1740, 1720, 1424, 1345, 1221, 1178, 1117, 913, 608. 1Н ЯМР (CDCl3-ДМСО, 300 МГц): 8,19 (d, J=8,5 Гц, 2Н, ArH), 7,99 (d, J=8,5 Гц, 2Н, ArH), 7,61 (шир.s, 1Н, ОН), 7,02 (d, J=8,6 Гц, 1H, ArH), 6,51 (dd, J1=8,1 Гц, J2=2,1 Гц, 1H, ArH), 6,42 (d, J=2,4 Гц, 1H, ArH), 5,02 (s, 3H, -OCH2, -OH), 4,72 (d, J=5,5 Гц, 1H, -CH), 4,11 (m, 1H, -CH), 2,68 (m, 2H, -CH2), 0,69 (s, 3H, -CH3).
Схема 5
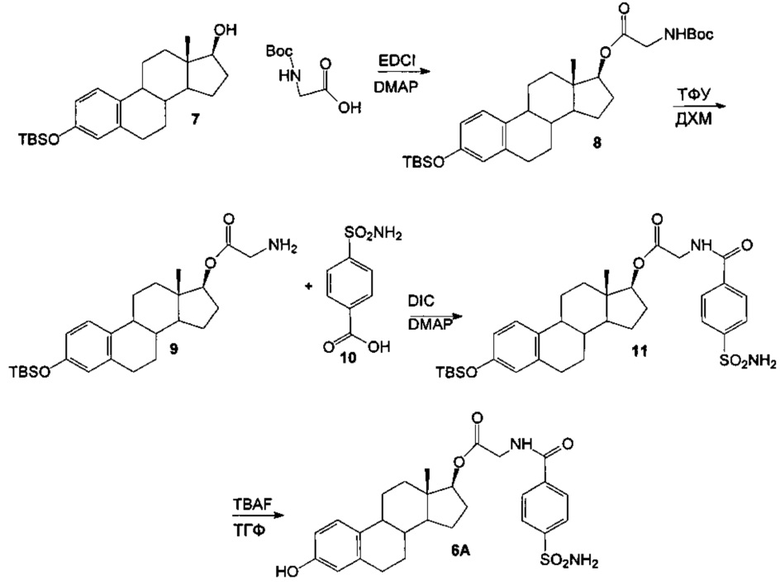
Вос-глицин-ОН (2,0 г, 2 экв.) обрабатывали 1-этил-3-(3-диметиламинопропил)-карбодиимидом (EDCI, 2,2 г, 2 экв.) в ДХМ (30 мл) в течение 1 часа при КТ. Затем добавляли эстрадиол 7 (2,1 г, 1 экв.) и 4-диметиламинопиридин (DMAP, 0,7 г, 1 экв.) и полученную смесь перемешивали при КТ в течение 20 часов. Концентрировали реакционную смесь и очищали остаток путем хроматографии на силикагеле с использованием 60-100% ДХМ в гексанах в качестве элюента с получением сложноэфирного продукта 8 в виде белого твердого вещества (2,1 г, выход 68%).
Сложный эфир 8 (2,0 г) обрабатывали трифторуксусной кислотой (ТФУ, 6 мл) в ДХМ (30 мл) при КТ в течение 24 часов. После завершения взаимодействия реакционную смесь разбавляли толуолом (30 мл) и удаляли трифторуксусную кислоту в безводных условиях с получением аминосодержащего соединения 9 (2,1 г в виде соли ТФУ).
Затем соединение 9 (1,0 г, 1 экв.) обрабатывали п-аминосульфамоилбензойной кислотой 10 (0,54 г, 1,5 экв.) в присутствии DIC (0,42 мл, 1,5 экв.), HOBt (0,41 г, 1,5 экв.) и основания Хюнига (DIEA, 1,3 мл, 4 экв.) в ДХМ (20 мл) при КТ в течение 72 часов. Концентрировали реакционную смесь и очищали остаток путем хроматографии на силикагеле с использованием 15% ацетона в ДХМ в качестве элюента с получением целевого продукта 11 в виде белого твердого вещества (0,4 г, выход 36%).
Проводили взаимодействие соединения 11 (0,4 г, 1 экв.) с тригидратом фторида тетрабутиламмония (TBAF, 0,2 г, 1 экв.) в ТГФ (20 мл) при КТ в течение 60 минут. Реакцию гасили водн. хлоридом аммония, смесь экстрагировали этилацетатом (3×25 мл), объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали путем хроматографии на силикагеле с использованием 5% ацетона в смеси 1:1 гексаны : этилацетат с получением (13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-(4-сульфамоилбензамидо)уксусной кислоты 6А (0,15 г, выход 46%) в виде белого твердого вещества: 1Н ЯМР (δ, ДМСО-d6 300 МГц): 9,15 (t, 1Н, -NH, J=5,82 Гц), 8,01 (d, 2Н, ArH, J=8,56 Гц), 7,91 (d, 2Н, ArH, J=8,54 Гц), 7,50 (шир.s, 1Н, ArOH), 7,02 (d, 1Н, ArH, J=8,46 Гц), 6,49 (dd, 1Н, ArH, J=2,46, 58,37 Гц), 6,42 (d, 1Н, ArH, J=2,42 Гц), 4,67 (t, 1Н, 17-СН, J=7,08 Гц), 4,02 (d, 2Н, J=5,75 Гц), 0,73 (s, 3Н, СН3). ИКС (см-1): 3369, 3278, 2933, 1736, 1656, 1529. [α]D23=+18° (с=0,5, 1,4-диоксан).

(2S)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)-овый эфир 2-(4-сульфамоилбензамидо)пропановой кислоты (12А) получали в виде белого твердого вещества (75 мг) согласно способам, приведенным на схеме 5, с использованием схожего защищенного аланина вместо глицина: 1Н ЯМР (δ, 5:1 CDCl3:ДМСО-d6 300 МГц): 8,54 (s, 1H, ArOH), 8,18 (d, 1Н, NH, J=7,03 Гц), 7,99 (dd, 4Н, ArH, J=2,4, 8,88 Гц), 7,07 (d, 1Н, ArH, J=8,35 Гц), 6,97 (s, 2Н, NH2), 6,62 (dd, 1Н, ArH, J=2,60, 8,39 Гц), 6,54 (d, 1H, ArH, J=2,46 Гц), 4,77 (t, 1H, 17-CH, J=7,99 Гц), 4,71 (t, 1H, 17-CH, J=7,30 Гц), 1,54 (d, 3H, CH3, J=7,29 Гц), 0,75 (s, 3Н, СН3). ИКС (см-1): 3496, 3381, 2930, 22911, 1743, 1706, 1650. [α]D23=+34° (c=0,5, 1,4-диоксан).
Схема 6

Стероид 9 (0,7 г) и диизопропилэтиламин (DIEA, 1 мл) в ТГФ (20 мл) охлаждали до -50°С, затем добавляли м-хлорсульфонилбензоилхлорид 12 (0,9 мл). Полученную желтую смесь медленно нагревали до 0°С в течение 40 минут, а затем добавляли 28% гидроксид аммония (7 мл). Затем полученную смесь нагревали до КТ в течение 30 минут, реакционную смесь разбавляли водн. хлоридом аммония, экстрагировали этилацетатом (3×25 мл), объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали путем хроматографии на силикагеле с использованием 15% ацетона в дихлорметане с получением белого твердого вещества 13 (0,53 г, выход 67%).
Соединение 13 (0,52 г, 1 экв.) обрабатывали тригидратом фторида тетрабутиламмония (TBAF, 0,32 г, 1,2 экв.) в ТГФ (30 мл) при КТ в течение 60 минут. Реакцию гасили водн. хлоридом аммония, экстрагировали этилацетатом (3×25 мл), объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали путем хроматографии на силикагеле с использованием 5% ацетона в смеси 1:1 гексаны : этилацетат с получением (13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-(3-сульфамоилбензамидо)уксусной кислоты 5А (0,31 г, выход 73%) в виде белого твердого вещества: 1Н ЯМР (δ, 5:1 CDCl3:D3COD 300 МГц): 8,25 (s, 1Н, ArH), 7,96 (d, 2Н, ArH, J=7,62 Гц), 7,51 (t, 1Н, ArH, J=7,83 Гц), 7,01 (d, 1Н, ArH, J=8,43 Гц), 6,52 (d, 1Н, ArH, J=8,22 Гц), 6,46 (s, 1Н, ArH), 4,68 (t, 1Н, 17-CH, J=7,74 Гц), 4,11 (s, 2Н, СН2), 0,75 (s, 3Н, СН3). ИКС (см-1): 3416, 3341, 3064, 2934, 2853, 1748, 1642. [α]D23=+14° (с=0,5, 1,4-диоксан).
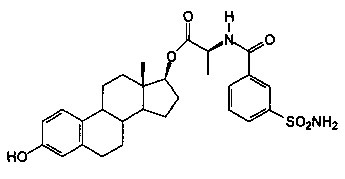
(2S)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)овый эфир 2-(3-сульфамоилбензамидо)пропановой кислоты синтезировали согласно способам, приведенным на схеме 6, получали белое твердое вещество 11A (0,41 г): 1Н ЯМР (δ, CDCl3 300 МГц): 8,33 (s, 1Н, ArH), 7,99 (d, 1H, ArH, J=7,89 Гц), 7,87 (d, 1H, ArH, J=7,89 Гц), 7,64 (d, 1Н, ArH, J=7,14 Гц), 7,49 (t, 1Н, ArH, J=7,8 Гц), 7,13 (d, 1Н, ArH, J=8,16 Гц), 6,63 (m, 1H, ArH), 6,56 (d, 1Н, ArH, J=2,49 Гц), 5,23 (шир.s, 2Н, NH2), 4,91 (m, 2Н), 4,80 (s, 1Н), 2,84 (m, 2Н), 1,58 (d, 3Н, СН3, J=7,14 Гц), 0,87 (s, 3Н, СН3). ИКС (см-1): 3341, 3252, 2923, 2862, 1718, 1646, 1535, 1499. [α]D23=+26° (с=0,5, 1,4-диоксан).

(2S)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)овый эфир 1-(3-сульфамоилбензоил)пирролидин-2-карбоновой кислоты синтезировали согласно способам, приведенным на схеме 6, получали белое твердое вещество 7А (0,91 г): 1H ЯМР ( , CDCl3 300 МГц): 8,12 (s, 1Н, ArH), 7,99 (d, 1Н, ArH, J=7,71 Гц), 7,76 (d, 1Н, ArH, J=7,62 Гц), 7,57 (t, 1Н, ArH, J=7,7 Гц), 7,37 (s, 1H, ArOH), 7,11 (d, 1Н, ArH, J=8,64 Гц), 6,65 (d, 1Н, ArH, J=5,76 Гц), 6,58 (s, 1Н, ArH), 5,85 (шир.s, 2Н, NH2), 4,80 (t, 1Н, 17-CH, J=8,29 Гц), 4,66 (m, 1H), 0,84 (s, 3Н, СН3). ИКС (см-1): 3341, 3249, 2924, 2866, 1727, 1612, 1499. [α]D23=-16° (c=0,5, 1,4-диоксан).
Схема 7
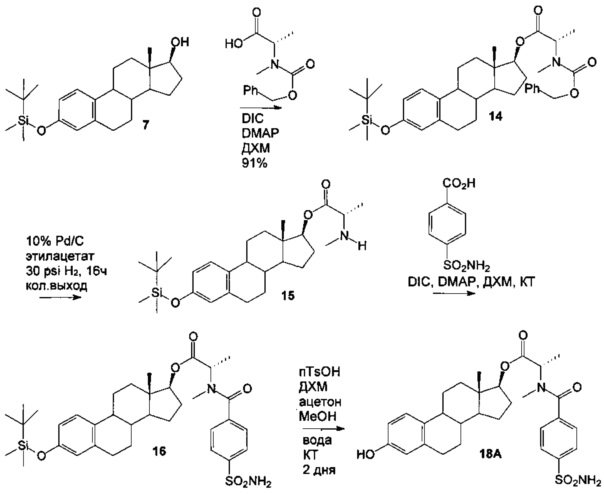
N-Cbz-метилаланин (2,45 г, 2 экв.) обрабатывали DIC (1,6 мл, 2 экв.) в ДХМ (33 мл) в течение 30 минут в атмосфере азота при КТ. Затем добавляли TBS-эстрадиол 7 (2,0 г, 1 экв.) и DMAP (0,063 г, 0,1 экв.) и полученную белую смесь перемешивали в течение 16 часов при КТ. После фильтрования концентрировали фильтрат и очищали остаток путем хроматографии на силикагеле с использованием 5-40% этилацетата в гексанах в качестве элюента с получением белого твердого продукта 14 (2,85 г, выход 91%).
Затем удаляли группу Cbz из сложного эфира 14 (2,85 г) с использованием Pd/C (10% Pd, 0,51 г) и водорода (30 psi (207 кПа)) и этилацетата (35 мл) в качестве растворителя на встряхивателе Парра в течение 16 часов. После фильтрования через целит и концентрирования растворителя получали белый твердый продукт 15 (2,2 г, количественный выход).
Амид получали из амина 15 с использованием п-сульфамоилбензойной кислоты (2,22 г, 2,4 экв.) и DIC (1,7 мл, 2,4 экв.) в ДХМ (75 мл), затем добавляли амин 15 (2,2 г, 1 экв.) и DMAP (56 мг, 0,1 экв.). Реакционная смесь изначально была нерастворимой, поэтому добавляли этилацетат (35 мл) и реакционную смесь перемешивали при КТ в течение 3 дней. Фильтровали реакционную смесь и концентрировали фильтрат. Очищали остаток путем хроматографии на силикагеле с использованием 10% этилацетата в гексанах в качестве элюента с получением белого твердого продукта 16 (1,90 г, выход 63%).
Затем удаляли силильную группу простого эфира 16 (1,90 г) с использованием п-толуолсульфокислоты (1,16 г, 2 экв.) в ДХМ (14 мл), ацетоне (14 мл), метаноле (0,4 мл) и воде (0,3 мл) при КТ в течение 16 часов. Реакцию гасили водн. бикарбонатом натрия и смесь экстрагировали этилацетатом (2×60 мл). Объединенные органические слои сушили сульфатом натрия, фильтровали и концентрировали. Очищали остаток путем хроматографии на силикагеле с использованием 10-30% ацетона в ДХМ в качестве элюента с получением [(13S,17S)-3-гидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]фенантрен-17-ил]ового эфира (2S)-2-[метил-(4-сульфамоилбензоил)амино]-пропановой кислоты 18А в виде белого твердого вещества (1,47 г, выход 94%): 1H ЯМР (δ, CDCl3 300 МГц): 7,96 (d, 2Н, J=8,1 Гц), 7,99 (d, 2Н, J=8,1 Гц), 7,13 (d, 1H, ArH, J=8,1 Гц), 6,62 (dd, 1Н, ArH, J=2,7, 8,4 Гц), 6,54 (d, 1Н, ArH, J=2,7 Гц), 5,30 (s, 1Н), 4,95 (s, 2Н, NH2), 4,80 (m, 1Н), 3,00 (s, 1H, СН3, ротамер), 2,90 (s, 2H, CH3, ротамер), 0,85 (s, 2Н, СН3, ротамер), 0,83 (s, 1H, СН3, ротамер). ИКС (см-1): 3353, 3257, 2924, 2862, 1731, 1617. [α]D24=-10° (с=0,5, 1,4-диоксан).
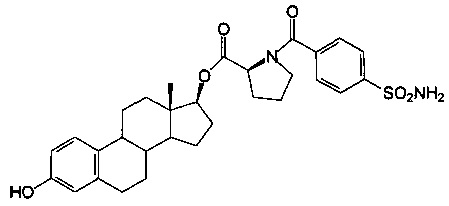
(2S)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а] фенантрен-17-ил)овый эфир 1-(4-сульфамоилбензоил)пирролидин-2-карбоновой кислоты синтезировали согласно способам, приведенным на схеме 7, получали белое твердое вещество 8А (0,51 г): 1Н ЯМР (δ, CDCl3 300 МГц): 7,96 (d, 2Н, ArH, J=7,25 Гц), 7,66 (d, 1,6Н ротамер, ArH, J=7,28 Гц), 7,47 (d, 0,4Н ротамер, ArH, J=7,28 Гц), 7,40 (s, 1H, ArOH), 7,10 (d, 1Н, ArH, J=8,43 Гц), 6,64 (d, 1Н, ArH, J=8,31 Гц), 6,58 (s, 1Н, ArH), 6,04 (шир.s, 2Н, NH2), 4,81 (t, 1Н, 17-CH), 4,67 (m, 1H, CH), 0,84 (s, 3Н, СН3). ИКС (см-1): 3378, 3218, 2925, 1738, 1615, 1498. [α]D23=-12° (c=0,5, 1,4-диоксан).

[(13S,17S)-3-гидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]-фенантрен-17-ил]овый эфир (2S)-3-метил-2-[(4-сульфамоилбензоил)амино]бутановой кислоты синтезировали согласно способам, приведенным на схеме 7, получали белое твердое вещество 19А (1,90 г): 1H ЯМР (δ, 5 CDCl3 300 МГц): 7,97 (d, 2Н, ArH, J=8,7 Гц), 7,91 (d, 2Н, ArH, J=8,4 Гц), 7,13 (d, 1H, ArH, J=8,1 Гц), 6,81 (d, 1Н, J=8,4 Гц), 6,63 (dd, 1H, ArH, J=2,7, 8,4 Гц), 6,56 (d, 1Н, ArH, J=2,1 Гц), 5,01 (s, 2Н, NH2), 4,79 (m, 3Н), 1,04 (dd, 6Н, J=2,7, 6,9 Гц), 0,87 (s, 3Н, СН3). ИКС (см-1): 3338, 3252, 2967, 2924, 1722, 1710, 1657. [α]D24=+40° (с=0,6, 1,4-диоксан).

[(13S,17S)-3-гидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]-фенантрен-17-ил]овый эфир 2-метил-2-[(4-сульфамоилбензоил)амино]пропановой кислоты синтезировали согласно способам, приведенным на схеме 7, получали белое твердое вещество 20А (0,61 г): 1Н ЯМР (δ, ДМСО-d6 300 МГц): 8,98 (s, 1H), 8,78 (s, 1H), 7,97 (d, 2Н, ArH, J=8,4 Гц), 7,89 (d, 2Н, ArH, J=8,1 Гц), 7,48 (s, 2Н, NH2), 7,02 (d, 1Н, ArH, J=8,7 Гц), 6,48 (dd, 1H, ArH, J=2,4, 8,1 Гц), 6,41 (d, 1H, ArH, J=2,1 Гц), 4,55 (t, 1H, J=8,4 Гц), 1,47 (s, 6H, ди-СН3), 0,66 (s, 3Н, СН3). ИКС (см-1): 3504, 3355, 3168, 3067, 2922, 2867, 1727, 1648. [α]D24=+48° (c=0,5, 1,4-диоксан).
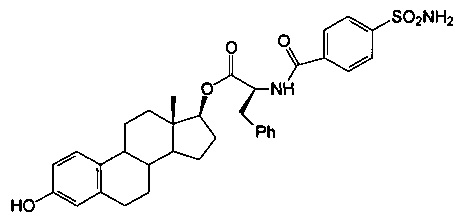
[(13S,17S)-3-гидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]-фенантрен-17-ил]овый эфир (2S)-3-фенил-2-[(4-сульфамоилбензоил)амино]пропановой кислоты синтезировали согласно способам, приведенным на схеме 7, в виде белого твердого вещества 21А (0,44 г): 1Н ЯМР (δ, ДМСО-d6 300 МГц): 9,04 (d, 1H, NH, J=7,5 Гц), 9,00 (s, 1Н, ОН), 7,94 (d, 2Н, ArH, J=8,4 Гц), 7,89 (d, 2Н, ArH, J=8,1 Гц), 7,49 (s, 2Н, NH2), 7,30 (m, 5Н), 7,02 (d, 1Н, ArH, J=8,4 Гц), 6,49 (dd, 1Н, ArH, J=1,8, 8,1 Гц), 6,42 (d, 1Н, ArH, J=1,8 Гц), 4,67 (m, 2Н), 0,68 (s, 3Н, СН3). ИКС (см-1): 3351, 2924, 2868, 1722, 1650. [α]D24=+42° (с=0,5, 1,4-диоксан).

Получение промежуточного соединения 17 - (13S,17S)-3-(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-бромуксусной кислоты: В круглодонную колбу помещали 7 (2,0 г, 5,3 ммоль) и растворяли в 5 мл ДХМ и 2 мл (25 ммоль) пиридина. Смесь охлаждали до -78°С. Затем растворяли бромацетилхлорид (1 мл, 6,3 ммоль) в 10 мл ДХМ и по каплям добавляли в раствор стероида. После завершения добавления смесь удаляли с бани и оставляли нагреваться до комнатной температуры. Через тридцать минут после удаления из ледяной бани анализ ТСХ (1:4 EtOAc : гексаны) указывал на полную конверсию исходного вещества. Смесь обрабатывали ледяной 2М HCl (50 мл), а затем разделяли слои. Органический слой промывали водой (2×50 мл), солевым раствором, а затем сушили над сульфатом натрия. Выделяли продукт путем флэш-хроматографии (5% EtOAc в гексанах) с получением 1,32 г соединения 17 (49%). 1Н ЯМР (δ, CDCl3, 300 МГц): 7,09 (d, 1H, J=4,3 Гц), 6,50 (d, 1Н, J=3,9 Гц), 6,43 (s, 1H), 4,69 (t, J=8,5 Гц, 1H), 4,08 (s, 2Н), 0,98 (s, 9Н), 0,85 (s, 3Н), 0,19 (s, 6Н).

Получение (13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6H-циклопента[а]фенантрен-17-ил)овый эфир 2-(4-сульфамоилбензиламино)уксусной кислоты 26А: 500 мг (2,3 ммоль) гидрохлорида 4-гомосульфаниламида растворяли в 5 мл ДМФ и добавляли 1 эквивалент карбоната калия, а затем 0,5 эквивалента основания Хюнига при комнатной температуре и кристалл йодида третбутиламмония. Через 20 минут в раствор непосредственно добавляли 1 эквивалент бромацетатного сложного эфира эстрадиола 17, а затем реакционную смесь оставляли перемешиваться на 12 часов при комнатной температуре. Затем фильтровали неочищенную смесь и помещали непосредственно в колонку с оксидом кремния и разделяли путем ЖХСД с получением 75 мг целевого промежуточного соединения, которое затем подвергали обработке в описанных условиях десилилирования. 1Н ЯМР (δ, CDCl3, 300 МГц): 7,77-7,74 (d, 2Н, J=8,4 Гц), 7,51-7,48 (d, 2Н, J=8,4 Гц), 7,04-7,02 (d, 1Н), 6,51-6,48 (d, 1H), 6,43-6,42 (s, 1Н, J=2,4 Гц), 5,45 (s, 1Н), 4,67 (t, J=8,9 Гц, 1Н), 4,22-4,06 (m, 2Н), 3,99 (s, 2Н), 3,80 (s, 2Н), 0,73 (s, 3Н). ИКС (см-1): 3419, 3311, 2920, 2832, 1739, 1499, 1221, 1152, 1127, 1039, 750. Оптическое вращение: [α]D20=+42° (С=0,5, МеОН]). Тпл: 201-205°С.
Общий способ удаления TBS-группы - простой силильный эфир растворяли в ТГФ (как правило, поддерживая концентрацию примерно 0,1М) и добавляли один эквивалент TBAF (1,0М в ТГФ). Через один час отмечали полное целевое десилилирование. Затем смесь обрабатывали раствором хлорида аммония и экстрагировали.
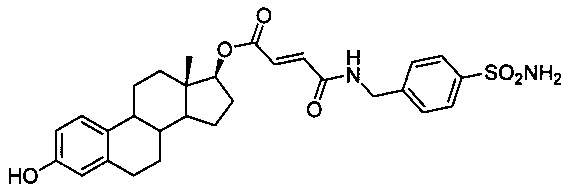
Получение (13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6H-циклопента[а]фенантрен-17-ил)ового эфира 4-оксо-4-(4-сульфамоилбензиламино)бут-2-еновой кислоты 4А: В круглодонную колбу помещали моноэтилфумарат (2,34 г, 16,3 ммоль), который затем растворяли в ДМФ (10 мл). Добавляли HOBt (2,75 г, 17,9 ммоль), а через 5 минут добавляли DCC (4,02 г, 19,5 ммоль). Смесь оставляли перемешиваться на двадцать минут, после чего добавляли 4-гомосульфаниламид-HCl (3,95 г, 17,8 ммоль) и, наконец, основание Хюнига. Через 12 часов взаимодействие считали завершенным согласно анализу ТСХ, затем смесь очищали путем ЖХСД, элюируя с градиентом смесями 10-30% ацетона в ДХМ, с получением 1,2 г целевого промежуточного вещества. Его затем подвергали омылению с использованием 20% раствора NaOH в течение 1 часа при 80°С, затем обрабатывали 50% HCl с получением свободной кислоты в виде белого кристаллического порошка. Свободную кислоту (0,30 г, 1,06 ммоль) растворяли в пиридине (3 мл, 37 ммоль), затем добавляли 3-TBS-эстрадиол (0,30 г, 0,78 ммоль), 0,4 г DCC (1,94 ммоль) и 25 мг (0,13 ммоль) п-ТСК. Смесь оставляли перемешиваться при комнатной температуре на 72 часа, после чего взаимодействие считали завершенным согласно результатам ТСХ. Смесь разбавляли 2 мл воды, 3 мл 10% HCl и 25 мл этилацетата. Фильтровали смесь и отделяли органический слой. Органический раствор промывали насыщенными растворами NaHCO3 и NaCl. Раствор сушили над сульфатом магния, фильтровали и выпаривали растворитель с получением неочищенного продукта 18, который очищали на силикагеле, элюируя с градиентом 5-20% смесями ацетона в ДХМ, с получением целевого промежуточного соединения с выходом 45%. Затем полученное вещество подвергали обработке в общих условиях десилилирования, описанных при получении соединения 26А. 1Н ЯМР (δ, CDCl3, 300 МГц): 7,81-7,78 (d, 2Н, J=8,4 Гц), 7,37-7,35 (d, 2Н, J=8,4 Гц), 7,04-7,02 (d, 1Н), 6,90-6,83 (q, 2Н), 6,52-6,49 (d, 1Н), 6,48 (s, 1Н), 5,92 (s, 1Н), 4,81 (t, J=8,9 Гц, 1H), 4,42 (s, 2Н), 1,98-1,92 (m, 4Н), 0,73 (s, 3Н). ИКС (см-1): 3331, 2920, 2839, 1705, 1658, 1221, 1152, 1127, 1039, 750. Оптическое вращение: [α]D20=+66° (С=0,5, МеОН]). Тпл: 177-180°С.
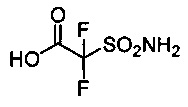
2,2-дифтор-2-сульфамоилуксусную кислоту 19 синтезировали согласно описанию, приведенному в Boyle, et al., Organic Biomolecular Chemistry, 2005, 3, 222-224.
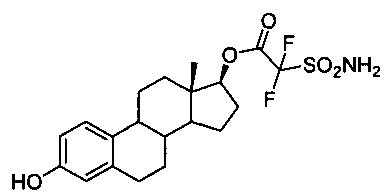
(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 2,2-дифтор-2-сульфамоилуксусной кислоты 9А: Промежуточное соединение 19 (0,35 г, 2,0 ммоль) помещали в высушенную в печи круглодонную колбу и добавляли 3,65 мл POCl3 (40,0 ммоль). Над колбой размещали высушенный в печи холодильник и смесь оставляли кипятиться с обратным холодильником на пять часов, после чего удаляли избыток POCl3 в вакууме (на роторном испарителе в глубоком вакууме в течение 24 часов). Получали кристаллическое твердое вещество (0,376 г, выход 97%). 7 (0,39 г, 1,05 ммоль) растворяли в 2 мл ДХМ и 0,4 мл пиридина (5 мл) и охлаждали до -78°С. Хлорангидрид 19 растворяли в 3 мл ДХМ и добавляли в раствор стероида. Смесь оставляли перемешиваться на десять минут, после чего удаляли из бани и постепенно нагревали до комнатной температуры. После выдерживания в течение одного часа при комнатной температуре взаимодействие считали завершенным и удаляли все летучие вещества в вакууме. Неочищенную смесь очищали путем флэш-хроматографии, элюируя 15% этилацетатом в гексанах, с получением 353 мг целевого промежуточного соединения (выход 65%). Затем из промежуточного соединения удаляли силильную группу в стандартных условиях и очищали путем флэш-хроматографии, элюируя 10% ацетоном в ДХМ, с получением 236 мг аналога 9А (выход 96%): 1Н ЯМР (300 МГц, ДМСО-d6) δ ppm 9,01 (s, 1Н), 8,94 (шир.s, 2Н), 7,04 (d, J=10,6 Гц, 1Н), 6,51 (d, J=8,4 Гц, 1Н), 6,43 (s, 1Н), 4,87 (t, J=7,93 Гц, 1Н), 0,75 (s, 3Н); ИКС (см-1) 3378, 3358, 2921, 2854, 1767, 1494, 1355, 1154; оптическое вращение: [α]D20=+38° (МеОН); Тпл: 209,5-212,0°С.

(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 2-сульфамоилуксусной кислоты 10А: В круглодонную колбу помещали хлорсульфонилацетилхлорид (0,50 г, 2,8 ммоль) и растворяли в 2 мл ДХМ. Смесь охлаждали до 0°С. Затем по каплям добавляли раствор 7 (546 мг, 1,4 ммоль) в 3 мл ДХМ. Анализ ТСХ, который проводили через 1 час при указанной температуре, указывал на полную конверсию исходного вещества, затем смесь оставляли нагреваться до комнатной температуры. Для полного ацилирования 17-спиртовой группы стероида требовалось перемешивание в течение 3,5 часа при комнатной температуре. После этого по каплям добавляли 2 мл 28% гидроксида аммония. Через десять минут профиль ТСХ полностью отличался от аликвоты смеси, которую отбирали непосредственно перед добавлением аммиака. Затем удаляли все летучие вещества в вакууме, получали 1,12 г неочищенного вещества, которое затем очищали путем флэш-хроматографии с использованием 5% ацетона в ДХМ. Получали 0,16 г целевого промежуточного соединения (выход 23%). Затем из вещества удаляли силильную группу в стандартных условиях с получением конечного аналога 10А: 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 9,01 (s, 1Н), 7,18 (s, 2Н), 7,04 (d, J=8,60 Гц, 1H), 6,50 (d, J=7,94 Гц, 1Н), 6,43 (s, 1H), 4,70 (t, J=8,58 Гц, 1H), 4,10 (s, 2Н), 0,801 (s, 3Н). ИКС (см-1): 3429, 3353, 3303, 2925, 2854, 1725, 1607, 1502, 1448, 1347, 1318. Оптическое вращение: [α]D20=+47° (МеОН); Тпл: 218-220°С.
Примечание - при использовании пиридина в качестве протонной губки в указанном превращении (что обычно проводят в случае ацилирования хлорангидридами) региоселективность отсутствовала, и происходила замена 17-спиртовой группы на группу кислоты и сульфонилхлорида. Указанное превращение наблюдали при различных температурах и при использовании различных растворителей.

Синтез 3,3'-азандиилбис(метилен)дибензолсульфонамида 20: 3-цианобензолсульфонамид (1,0 г, 5,49 ммоль) помещали в сосуд для гидрирования совместно с 0,6 г 10% (масс./масс.) палладия на подложке активированного угля. Добавляли 10 мл МеОН и смесь обрабатывали в условиях гидрирования (20 psi (138 кПа)) на встряхивателе Парра. Смесь встряхивали в течение ночи и следующего дня. Анализ ТСХ (30% ацетон в ДХМ) указывал на полную конверсию исходного вещества. Смесь фильтровали через Celite и промывали осадок метанолом с получением белого твердого вещества (0,78 г, 87%). 1Н ЯМР (300 МГц, δ, ДМСО-d6) 7,84 (s, 2Н), 7,70 (d, J=7,43 Гц, 2Н), 7,59-7,49 (m, 4Н), 7,32 (s, 4Н), 3,77 (s, 4Н), 2,90 (шир.s, 1Н).

(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 2-(бис(3-сульфамоилбензил)амино)уксусной кислоты 13А: В круглодонную колбу помещали промежуточное соединение 20 (0,26 г, 0,51 ммоль) и растворяли в ДМФ (5 мл). Добавляли карбонат калия (0,14 г, 1,01 ммоль), затем промежуточное соединение 17 (0,18 г, 0,51 ммоль) и несколько кристаллов TBAI. Смесь оставляли перемешиваться на ночь. На следующий день взаимодействие считали завершенным согласно анализу ТСХ и смесь непосредственно добавляли в колонку с силикагелем и разделяли, элюируя с градиентом от 50 до 70% смесями этилацетата в гексанах. Затем очищенное промежуточное соединение обрабатывали согласно указанному выше протоколу десилилирования с получением аналога 13А; 1Н ЯМР (300 МГц, CDCl3) δ ppm 8,20 (s, 2Н), 7,82-7,79 (m, 2Н), 7,45-7,43 (m, 4Н), 7,14 (d, J=4,34 Гц, 1H), 6,64 (d, J=7,14 Гц, 1Н), 6,57 (s, 1H), 5,49 (s, 4H), 4,78 (t, J=9,00 Гц, 1H), 4,65 (s, 1H), 3,88 (s, 4H), 3,47 (s, 2H), 0,84 (s, 3Н); ИКС (см-1) 3357, 3257, 2925, 1712, 1326, 1150; ИЭР-МС 668,1 (M+H)+; оптическое вращение: [α]D20=+32° (МеОН).
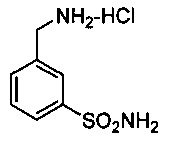
Синтез 3-(аминометил)бензолсульфонамида-HCl 21: В 2-горлую круглодонную колбу помещали 3-цианобензолсульфонамид (1,5 г, 8,23 ммоль) и 0,80 г палладия на подложке активированного угля (10% (масс./масс.)). Добавляли 30 мл МеОН и 5 мл 2М HCl. Одно отверстие закрывали мембраной, а через другое присоединяли регулятор расхода потока с краном. Колбу вакуумировали, затем повторно заполняли азотом. Указанную операцию повторяли дважды и после заключительного вакуумирования через мембрану вводили водород из баллона, колбу заполняли водородом. Удаляли баллон и вакуумировали колбу. Указанную операцию повторяли дважды и после заключительного вакуумирования заменяли баллон. Смесь оставляли перемешиваться до тех пор, пока анализ ТСХ не указывал на полную конверсию исходного вещества (требовалось 4 часа). Затем смесь фильтровали через Celite и удаляли летучие вещества в вакууме с получением 3,2 г кристаллического полутвердого вещества, которое растирали с этилацетатом с получением 1,5 г беловатого твердого вещества 21 (83%); 1H ЯМР (300 МГц, ДМСО-d6) δ ppm 7,96 (s, 1H), 7,84 (d, J=7,07 Гц, 1H), 7,76 (d, J=7,06 Гц, 1Н), 7,63 (d, J=7,73 Гц, 1Н), 7,46 (s, 2Н), 4,09 (s, 2Н); ЖХМС-ИЭР, 170,0 (свободное основание, M-NH2+H)+, 187,1 (свободное основание, М+Н)+.
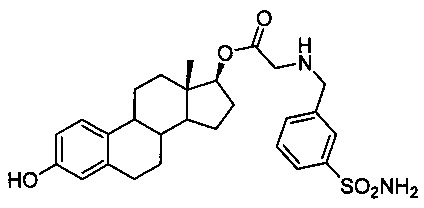
Синтез (13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-(3-сульфамоилбензиламино)уксусной кислоты 14А: В круглодонную колбу помещали 3-TBS-эстрадиол (0,60 г, 1,2 ммоль), промежуточное соединение 21 (0,40 г, 1,8 ммоль), 18-краун-6 (16,0 мг, 0,06 ммоль), TBAI (44,0 мг, 0,12 ммоль), карбонат калия (0,83 г, 6,0 ммоль) и 5 мл ДМФ. Реакционную смесь оставляли перемешиваться на 48 часов, после чего исходное стероидное вещество было полностью израсходовано согласно анализу ТСХ (1:3 EtOAc : гексаны). Смесь разбавляли водой, образовывался осадок, который собирали путем фильтрования в вакууме. Осадок промывали водой (50 мл), а затем сушили в вакууме. Собирали 668 мг неочищенного вещества, которое затем очищали путем флэш-хроматографии с использованием 10% ацетона в ДХМ с получением 217 мг целевого промежуточного соединения (выход 30%). Затем промежуточное соединение обрабатывали согласно указанному выше протоколу десилилирования и после хроматографии с градиентным элюированием 15-50% смесями ацетона в ДХМ собирали 140 мг продукта 14А (выход 96%); 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 9,00 (s, 1H), 7,81 (s, 1Н), 7,71-7,68 (m, 1Н), 7,52-7,50 (m, 2Н), 7,33 (s, 2Н), 7,04 (d, J=9,00 Гц, 1H), 6,502 (d, J=7,80 Гц, 1H), 6,427 (s, 1Н), 4,68 (t, J=8,04 Гц, 1H), 3,803 (s, 2Н), 0,772 (s, 3Н, -СН3); ИКС (см-1): 3412, 3294, 2917, 1721, 1305, 1234, 1146; оптическое вращение: [α]D20=+32° (МеОН); Тпл: 193,0-195,2°С.
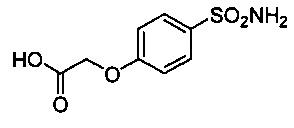
2-(4-сульфамоилфенокси)уксусную кислоту 22 синтезировали согласно способу, опубликованному в Biochemistry, 2009, 4488-4496.

(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 2-(4-сульфамоилфенокси)уксусной кислоты 15А: В круглодонную колбу помещали 22 (0,50 г, 2,16 ммоль) и растворяли в 5 мл ДМФ. Добавляли основание Хюнига (0,5 мл, 2,9 ммоль), а затем DIC (0,7 мл, 4,32 ммоль). Полученную смесь оставляли перемешиваться на тридцать минут, после чего добавляли 3-TBS-эстрадиол 7 (558 мг, 1,44 ммоль), а затем DMAP (88 мг, 0,72 ммоль). Смесь оставляли перемешиваться на 48 часов, после чего разбавляли смесь 2М HCl (50 мл) и экстрагировали диэтиловым эфиром. Затем объединенные органические слои промывали водой, солевым раствором, после чего сушили над сульфатом натрия. Затем выделяли целевое промежуточное соединение путем флэш-хроматографии, элюируя с градиентом 2-10% смесями ацетона в ДХМ, с получением 110 мг (выход 13%) продукта, из которого затем удаляли силильную группу в стандартных условиях с получением 81 мг аналога 15А (выход 93%); 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 9,00 (s, 1Н), 7,81 (s, 1H), 7,71-7,68 (m, 1H), 7,52-7,50 (m, 2H), 7,33 (s, 2H), 7,04 (d, J=9,00 Гц, 1H), 6,50 (d, J=7,80 Гц, 1H), 6,43 (s, 1H), 4,68 (t, J=8,04 Гц, 1H), 3,80 (s, 2H), 0,77 (s, 3Н). ИКС (см-1): 3362, 3257, 2917, 2862, 1733, 1595, 1498, 1209, 1154, 831, 671. Оптическое вращение: [α]D20=+30° (МеОН).

Синтез 1-(N-трет-бутилсульфамоил)циклопропанкарбоновой кислоты 23: N-трет-бутил-1-циклопропилсульфонамид получали согласно описанию, приведенному в Synlett, 2006, 725-728. Указанное исходное вещество (0,78 г, 4,4 ммоль) растворяли в 15 мл безводного ТГФ и охлаждали до -78°С в атмосфере азота. В смесь добавляли 2,5М раствор н-бутиллития в гексанах (3,7 мл, 9,2 ммоль) и оставляли нагреваться до комнатной температуры при перемешивании в течение 2 часов. В фильтровальную колбу помещали сухой лед. Закрывали колбу пробкой, ко входу присоединяли шланг, соединенный с трубкой осушителя, заполненной сульфатом кальция, совместно с холодной ловушкой, погруженной в смесь сухой лед-ацетон, и иглой с большим диаметром. Затем в фильтровальную колбу добавляли воду и заменяли пробку, смесь насыщали диоксидом углерода (в реакционный сосуд через мембрану вводили вентиляционную иглу) в течение 2 часов. Затем добавляли 10 мл 2М HCl и водный слой экстрагировали ДХМ (3×25 мл). Объединенные органические слои промывали солевым раствором и сушили сульфатом натрия. Удаляли растворитель в вакууме с получением 0,81 г (выход 83%) целевого промежуточного соединения 23 в виде бледно-желтого твердого вещества; 1Н ЯМР (300 МГц, CDCl3) δ ppm 5,05 (шир.s, 1Н), 1,92-1,85 (m, 2H), 1,83-1,76 (m, 2H), 1,37 (s, 9H). ИКС (см-1): 3320, 2984, 1691, 1322, 1301, 1131, 1003.

Синтез 1-сульфамоилциклопропанкарбонилхлорида 24: В круглодонную колбу помещали кислоту 23 (0,50 г, 2,3 ммоль) и суспендировали в ДХМ (2 мл) и ТФУ (2 мл, 23 ммоль). Смесь оставляли перемешиваться в атмосфере азота на ночь. На следующий день образовывался осадок, который собирали путем вакуумного фильтрования и промывали ледяной смесью 1:1 ДХМ : гексаны с получением 185 мг чистого сульфонамида с удаленными защитными группами (выход 49%). Удаляли летучие вещества из маточного раствора, получали твердое вещество, которое идентифицировали как сульфонамид с удаленными защитными группами, с чистотой примерно 65% (примесь представляла собой непрореагировавший N-трет-бутил-1-циклопропилсульфонамид, полученный на предыдущей стадии). Затем очищенное вещество помещали в круглодонную колбу, оборудованную вкладышем магнитной мешалки. Добавляли тионилхлорид (2 мл, 27,6 ммоль) и сверху присоединяли холодильник, затем колбу помещали на предварительно нагретую масляную баню. Смесь оставляли кипятиться с обратным холодильником, для полного растворения требовалось примерно двадцать минут. Отбирали аликвоту смеси и удаляли все летучие вещества в вакууме. В анализе ИКС сдвиг карбонила, соответствующего кислоте, полностью исчезал, и появлялся сдвиг нового карбонила. В последующих анализах определяли, что после растворения всего вещества в кипящем тионилхлориде взаимодействие завершалось. Тем не менее, для некоторых порций исходного вещества, если растворение происходило не полностью за тридцать минут, то для завершения конверсии в течение нескольких минут требовалось добавление примерно 5-10 мкл ДМФ. В описанном способе синтеза получали 201 мг желтого кристаллического твердого вещества 24 после удаления тионилхлорида в вакууме: 1Н ЯМР (300 МГц, CDCl3) δ ppm 5,05 (шир.s, 2Н), 2,06 (шир.s, 4Н). ИКС (см-1) 3387, 3265, 1771, 1532, 1330.

(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 1-сульфамоилциклопропанкарбоновой кислоты 16А: В пробирку, закрытую мембраной, помещали 3-TBS-эстрадиол 7 (222 мг, 0,57 ммоль) и DMAP (7,0 мг, 0,057 ммоль) и растворяли в 2 мл ДХМ и 0,23 мл пиридина (2,85 ммоль). Суспендировали промежуточное соединение 24 (315 мг, 1,72 ммоль) в 3 мл ДХМ и добавляли в раствор стероида в виде взвеси. Заменяли крышку, подключали линию подвода азота и смесь оставляли перемешиваться на ночь. На следующий день анализ ВЭЖХ указывал на конверсию примерно 70% исходного вещества. Смесь выпаривали при вращении и наносили на силикагель и проводили флэш-хроматографию с градиентным элюированием 20-50% смесями этилацетата в гексанах с получением 136 мг целевого промежуточного соединения (выход 45%). Из вещества удаляли силильную группу в стандартных условиях с получением 16А; 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 9,00 (s, 1Н), 7,04 (d, J=8,44 Гц, 1H), 6,96 (s, 2Н), 6,50 (dd, J=8,24 Гц, 2,12 Гц, 1Н), 6,43 (d, J=2,42 Гц, 1Н) 4,67 (t, J=8,73 Гц, 1H), 0,789 (s, 3Н). ИКС (см-1): 3408, 3353, 3273, 2934, 1715, 1611, 1498, 1326, 1138. ИЭР-МС: 437,1 (М+H2O)+, 861,3 (2М+Na)+; 418,1 (М-1)-. Оптическое вращение: [α]D20=+42° (МеОН); Тпл: 225-227°С.

Синтез (13S,16R,17R)-3,16-бис(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ола 25: В 2-литровую 3-горлую круглодонную колбу помещали эстриол (20 г, 70 ммоль) и имидазол (20 г, 0,29 моль). Добавляли 0,5 мл безводного ДМФ и в колбу помещали верхнеприводную мешалку. Затем перемешивали смесь и по частям в течение десяти минут добавляли TBSCl (40 г, 0,265 моль). Колбу закрывали и подключали линию подачи азота. Смесь оставалась гомогенной в течение 0,5 часа после завершения добавления силилхлорида, после чего наблюдали образование густой белой смеси. Анализ ТСХ в смеси 1:3 этилацетат : гексаны указывал на полную целевую конверсию исходного вещества. Смесь разбавляли 0,8 л воды и собирали полученный осадок путем вакуумного фильтрования и промывали 2 л воды. Неочищенный остаток очищали путем флэш-хроматографии с использованием одного кг силикагеля, элюируя с градиентом 2-20% смесями этилацетата в гексанах. Получали 17,1 г (выход 49%) целевого промежуточного соединения и 22,5 г простого трис-силильного эфира эстриола (выход 49%). 1H ЯМР (300 МГц, CDCl3) δ ppm 7,12 (d, J=8,4 Гц, 1H), 6,61 (dd, J=8,25, 2,70 Гц), 6,55 (d, J=2,40 Гц), 4,13-4,07 (m, 1H), 3,57 (d, J=5,7 Гц), 0,98 (s, 9H), 0,92 (s, 9H), 0,79 (s, 3H), 0,19 (s, 6H), 0,107-0,094 (m, 6H).

Синтез (13S,16R,17R)-3,17-бис(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-16-ола 26: В круглодонную колбу помещали простой трис-силильный эфир эстриола (31,5 г, 50 ммоль), полученный на предыдущей стадии, и растворяли в 250 мл ДХМ и 250 мл ацетона. Добавляли п-ТСК (9,5 г, 50 ммоль) и смесь оставляли перемешиваться. Через два часа реакцию гасили бикарбонатом натрия (ранее на более мелких порциях было определено, что при увеличенном времени взаимодействия образуются сложные смеси продуктов) и удаляли летучие вещества в вакууме. Концентрированный водный слой экстрагировали ДХМ (3×100 мл). Объединенные органические слои промывали солевым раствором и сушили над сульфатом натрия. Неочищенное вещество очищали путем флэш-хроматографии, элюируя с градиентом 5-10% смесями этилацетата в гексанах, с получением 16,6 г целевого промежуточного соединения 26 (выход 64%). Непрореагировавшее исходное вещество легко выделяли для дальнейшего использования. 1Н ЯМР (300 МГц, CDCl3), δ 7,11 (d, J=8,7 Гц, 1Н), 6,62 (dd, J=8,4 Гц, 2,40 Гц, 1Н), 6,56 (d, J=2,40 Гц, 1Н), 4,18-4,06 (m, 1Н), 3,52 (d, J=5,4 Гц, 1Н), 0,98 (s, 9Н), 0,93 (s, 9Н), 0,77 (s, 3Н), 0,19 (s, 6Н), 0,12-0,098 (m, 6Н).

Синтез (13S,16R,17R)-3,16-бис(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-бромуксусной кислоты 27: Проводили способ согласно описанию способа получения промежуточного соединения 17 со следующими исключениями: хлорангидрид перегоняли перед использованием и добавляли два эквивалента полученного вещества. Вещество выделяли путем флэш-хроматографии с использованием 2% этилацетата в гексанах. Выход составлял 88%: 1Н ЯМР (300 МГц, CDCl3) δ ppm 7,10 (d, 1Н), 6,60 (dd, 1H), 6,52 (d, 1H), 4,91 (d, 1H), 4,36 (t, 1H), 3,87 (d, 2H), 0,97 (s, 9H), 0,88 (s, 9H), 0,83 (s, 3H), 0,18 (s, 6H), 0,036 (d, 6H).

Синтез (13S,16R,17R)-3,17-бис(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-16-илового эфира 2-бромуксусной кислоты 28: Проводили способ согласно описанию способа получения промежуточного соединения 27: 1Н ЯМР (300 МГц, CDCl3) δ ppm 7,10 (d, 1Н), 6,60 (dd, 1H), 6,52 (d, 1H), 5,04-4,92 (m, 1H), 3,83 (s, 2H), 3,79 (d, 1H), 0,97 (s, 9H), 0,88 (s, 9H), 0,81 (s, 3H), 0,18 (s, 6H), 0,045 (d, J=7,18 Гц, 6H).
Синтез (13S,16R,17R)-3,16-дигидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-(4-сульфамоилбензиламино)уксусной кислоты 27А: В круглодонную колбу помещали промежуточный стероид 27 (0,96 г, 1,5 ммоль), 4-гомосульфониламид-HCl (0,67 г, 3,0 ммоль), 16-краун-6 (20 мг, 0,075 ммоль), TBAI (55 мг, 0,15 ммоль), карбонат калия (1,03 г, 7,5 ммоль), молекулярные сита  (128 мг) и, наконец, 7 мл безводного ДМФ. Смесь оставляли перемешиваться в атмосфере азота и периодически проверяли прохождение взаимодействия путем ТСХ (5% EtOAc в гексанах). Для полной конверсии промежуточного стероида требовалось 48 часов. Смесь разбавляли водой (25 мл) и экстрагировали диэтиловым эфиром (50 мл). Органическую фазу промывали водой (2×50 мл), солевым раствором и сушили над сульфатом натрия. Удаляли растворитель в вакууме с получением 0,98 г неочищенного вещества. Целевое промежуточное соединение выделяли путем флэш-хроматографии с использованием 10% ацетона в ДХМ с получением 0,48 г продукта (выход 43%). Затем вещество обрабатывали TBAF согласно приведенному выше описанию, что приводило к удалению 3-TBS-группы с 50% выходом, также получали 133 мг более полярного продукта, который идентифицировали как целевой аналог с удаленной защитой 3-й 16-гидроксильной групп (выход 40%). Промежуточное соединение, содержащее простую эфирную 16-TBS-группу, обрабатывали 2 мл 2М HCl в 10 мл диоксанов; для полного целевого удаления указанной защитной группы согласно анализу ТСХ требовался один час. Смесь разбавляли раствором бикарбоната натрия (25 мл) и экстрагировали водный слой этилацетатом. Объединенные экстракты промывали водой (3×50 мл), солевым раствором и сушили над сульфатом натрия. Выделяли продукт путем флэш-хроматографии с использованием смеси 1:1 ацетон в ДХМ с получением 108 мг целевого аналога 27А (72%); 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 8,99 (s, 1Н), 7,77 (d, ArH J=8,34 Гц, 2Н), 7,51 (d, J=8,32 Гц, 2Н), 7,30 (s, 2Н), 7,02 (d, J=11,22 Гц, 1Н) 6,50 (dd, J=8,24 Гц, 2,12 Гц, 1Н), 6,43 (d, J=2,46 Гц, 1Н), 4,92 (d, J=5,38 Гц, 1H), 4,69 (d, J=5,58 Гц, 1H), 4,112-4,089 (m, 1Н), 3,809 (s, 2Н), 3,413 (s, 2Н) 0,714 (s, 3Н). ИКС (см-1): 3299, 2921, 2858, 1725, 1498, 1448, 1320, 1221, 1158, 818, 676, 587. Оптическое вращение: [α]D20=+10° (МеОН).
(128 мг) и, наконец, 7 мл безводного ДМФ. Смесь оставляли перемешиваться в атмосфере азота и периодически проверяли прохождение взаимодействия путем ТСХ (5% EtOAc в гексанах). Для полной конверсии промежуточного стероида требовалось 48 часов. Смесь разбавляли водой (25 мл) и экстрагировали диэтиловым эфиром (50 мл). Органическую фазу промывали водой (2×50 мл), солевым раствором и сушили над сульфатом натрия. Удаляли растворитель в вакууме с получением 0,98 г неочищенного вещества. Целевое промежуточное соединение выделяли путем флэш-хроматографии с использованием 10% ацетона в ДХМ с получением 0,48 г продукта (выход 43%). Затем вещество обрабатывали TBAF согласно приведенному выше описанию, что приводило к удалению 3-TBS-группы с 50% выходом, также получали 133 мг более полярного продукта, который идентифицировали как целевой аналог с удаленной защитой 3-й 16-гидроксильной групп (выход 40%). Промежуточное соединение, содержащее простую эфирную 16-TBS-группу, обрабатывали 2 мл 2М HCl в 10 мл диоксанов; для полного целевого удаления указанной защитной группы согласно анализу ТСХ требовался один час. Смесь разбавляли раствором бикарбоната натрия (25 мл) и экстрагировали водный слой этилацетатом. Объединенные экстракты промывали водой (3×50 мл), солевым раствором и сушили над сульфатом натрия. Выделяли продукт путем флэш-хроматографии с использованием смеси 1:1 ацетон в ДХМ с получением 108 мг целевого аналога 27А (72%); 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 8,99 (s, 1Н), 7,77 (d, ArH J=8,34 Гц, 2Н), 7,51 (d, J=8,32 Гц, 2Н), 7,30 (s, 2Н), 7,02 (d, J=11,22 Гц, 1Н) 6,50 (dd, J=8,24 Гц, 2,12 Гц, 1Н), 6,43 (d, J=2,46 Гц, 1Н), 4,92 (d, J=5,38 Гц, 1H), 4,69 (d, J=5,58 Гц, 1H), 4,112-4,089 (m, 1Н), 3,809 (s, 2Н), 3,413 (s, 2Н) 0,714 (s, 3Н). ИКС (см-1): 3299, 2921, 2858, 1725, 1498, 1448, 1320, 1221, 1158, 818, 676, 587. Оптическое вращение: [α]D20=+10° (МеОН).

Синтез (13S,16R,17R)-3,16-дигидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-илового эфира 2-(3-сульфамоилбензиламино)уксусной кислоты 28А: Использовали способ, описанный при получении аналога 14А (в настоящем случае использовали 3-сульфамоилбензиламин-HCl). Удаление TBS-групп проводили следующим образом: 386 мг (0,5 ммоль) простого 3,16-бис-силильного эфира растворяли в 10 мл 1,4-диоксана и 5 мл 2М HCl. Полное удаление защитных групп подтверждали при помощи анализа ТСХ в течение одного часа. Реакционную смесь разбавляли раствором бикарбоната натрия и экстрагировали этилацетатом, трижды промывали водой, после чего удаляли растворитель в вакууме. Целевой аналог 28А очищали путем флэш-хроматографии с использованием смеси 1:1 ацетон : ДХМ с получением 150 мг продукта (выход 58%); 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 8,99 (s, 1Н), 7,82 (s, 1Н), 7,71-7,69 (m, 1Н), 7,53-7,50 (m, 2Н), 7,32 (s, 2Н), 7,04 (d, J=11,22 Гц, 1Н) 6,50 (dd, J=8,24 Гц, 2,12 Гц, 1Н), 6,43 (d, J=2,46 Гц, 1H) 4,92 (d, J=5,78 Гц, 1Н), 4,70 (d, J=4,80 Гц, 1Н), 4,117-4,074 (m, 2Н), 3,81 (шир.s, 2Н), 3,41 (s, 2Н), 0,72 (s, 3Н). ИКС (см-1): 3311, 2917, 2854, 1729, 1498, 1330, 1196, 1146. ИЭР-МС: 513,2 (М-1)-, 515,2 (М+1)+. Оптическое вращение: [α]D20=+3° (МеОН); Тпл: 90-92,5°С.

Синтез (13S,16R,17R)-3,17-дигидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-16-илового эфира 2-(4-сульфамоилбензиламино)уксусной кислоты 29А: Использовали способ получения аналога 14А: 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 9,00 (s, 1Н), 7,77 (d, J=8,40 Гц, 2Н), 7,50 (d, J=8,40 Гц, 2Н), 7,30 (s, 2Н), 7,02 (d, J=11,22 Гц, 1H,) 6,50 (dd, J=8,24 Гц, 2,12 Гц, 1Н), 6,43 (d, J=4,92 Гц, 1Н) 5,04 (d, J=4,98 Гц, 1Н), 4,879-4,741 (m, 1Н), 3,79 (шир.s, 2Н), 3,580 (t, J=5,44 Гц, 1Н), 0,719 (s, 3Н). ИКС (см-1): 3282, 2921, 2858, 1721, 1330, 1217, 1154. ИЭР-МС: 514,2 (М-1)-. Оптическое вращение: [α]D20=+17° (MeOH).

Синтез 4-((трет-бутилдиметилсилилокси)метил)бензолсульфонамида 29: 4-(гидроксиметил)бензолсульфонамид (5,5 г, 30 ммоль; получали путем восстановления 4-сульфамоилбензойной кислоты с использованием борана согласно описанию JMC, 4522, 2010) помещали в круглодонную колбу совместно с TBSCl (4,9 г, 9,3 ммоль) и имидазолом (4,1 г, 16,8 ммоль) и растворяли реагенты в 25 мл безводного ДМФ. Смесь оставляли перемешиваться в атмосфере азота, и анализ ТСХ указывал на полную конверсию исходного вещества в течение 2 часов. Затем смесь разбавляли 300 мл этилацетата и промывали водой (5×100 мл), солевым раствором и сушили над сульфатом натрия с получением 10 г неочищенного белого полутвердого вещества. Полученное вещество очищали путем флэш-хроматографии, элюируя с градиентом 20-30% смесями этилацетата в гексанах, с получением продукта 29 в виде белого кристаллического твердого вещества (7,56 г, 84%); 1Н ЯМР (300 МГц, CDCl3) δ ppm 7,90 (d, J=8,44 Гц, 2H), 7,48 (d, J=8,65 Гц, 2H), 4,89-4,73 (m, 4Н), 0,95 (s, 9Н), 0,12 (s, 6Н).

Синтез трет-бутил-4-(гидроксиметил)фенилсульфонилкарбамата 30: В круглодонную колбу помещали промежуточное соединение 29 (7,55 г, 25 ммоль) и DMAP (0,31 г, 2,5 ммоль) и растворяли в 75 мл ДХМ и 4,2 мл ТЭА (30 ммоль) и охлаждали до 0°С. Приготовленный отдельно раствор Вос-ангидрида (6,56 г, 30 ммоль) в 75 мл ДХМ добавляли через капельную воронку в атмосфере азота и оставляли смесь перемешиваться и постепенно нагревали до комнатной температуры в течение ночи. Анализ ТСХ указывал на полную конверсию исходного вещества, и смесь разбавляли 100 мл насыщенного хлорида аммония. Затем водный слой экстрагировали ДХМ и объединенные органические слои промывали солевым раствором и сушили над сульфатом натрия. Затем неочищенное вещество очищали путем флэш-хроматографии с использованием 15% этилацетата в гексанах с получением целевого вещества (8,1 г, выход 80%). Затем удаляли TBS-группу согласно описанию общего способа с получением спирта 30: 1Н ЯМР: (ДМСО-d6) δ ppm 7,82 (d, J=8,35 Гц, 2Н), 7,55 (d, J=8,31 Гц, 2Н), 5,42 (шир.s, 1Н), 4,59 (s, 1H), 1,29 (s, 9H).

Синтез трет-бутил-2-(4-(N-(трет-бутоксикарбонил)сульфамоил)бензилокси)ацетата 31: В сцинтилляционную пробирку, оборудованную резиновой завинчивающейся крышкой с мембраной, помещали гидрид натрия (50 мг, 60% (масс./масс.) в минеральном масле, 1,24 ммоль) и суспендировали в 1 мл безводного ДМФ в атмосфере азота. Пробирку охлаждали до 0°С и по каплям в пробирку добавляли промежуточное соединение 30 (170 мг, 0,59 ммоль) в 1 мл ДМФ. Смесь оставляли перемешиваться на тридцать минут при температуре охлаждения, после чего добавляли чистый трет-бутилбромацетат (0,1 мл, 0,71 ммоль). Через два часа анализ ВЭЖХ указывал на полное израсходование промежуточного соединения 30. Смесь обрабатывали 1 мл 0,2М HCl и экстрагировали этилацетатом. Неочищенное вещество очищали путем флэш-хроматографии с использованием 5% ацетона в ДХМ с получением 110 мг целевого промежуточного соединения 31 (46%): 1Н ЯМР (300 МГц, CDCl3) δ ppm 8,00 (d, J=8,4 Гц, 2H), 7,55 (d, J=8,3 Гц), 4,71 (s, 2H), 4,05 (s, 2H), 1,49 (s, 9H), 1,39 (s, 9H).

Синтез 2-(4-сульфамоилбензилокси)уксусной кислоты 32: В сцинтилляционную пробирку помещали промежуточное соединение 31 (0,15 г, 0,37 ммоль) и растворяли в 2 мл ДХМ. Затем добавляли ТФУ (1 мл, 13 ммоль). Смесь оставляли перемешиваться на 5 часов, после чего удаляли все летучие вещества в вакууме. Неочищенное вещество (в виде светло-коричневой маслянистой жидкости) кристаллизовали из смеси ДХМ : МеОН : диэтиловый эфир : гексаны (1:0,2:5:10) с получением продукта 32 в виде белого кристаллического твердого вещества (65 мг, выход 71%), в качестве единственной примеси получали 25 мг целевого продукта, содержащего непрореагировавшую Boc-группу, присутствовавшего в маточном растворе; продукт: 1Н ЯМР (300 МГц, ДМСО-d6) δ ppm, 7,80 (d, 8,35 Гц, 2Н), 7,52 (d, 8,41 Гц, 2Н), 4,62 (s, 2Н), 4,11 (s, 2Н). ИКС (см-1) 3395, 3265, 1750, 1318, 1146, 1091, 819, 697, 596.

(13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]-фенантрен-17-иловый эфир 2-(4-сульфамоилбензилокси)уксусной кислоты 17А: Промежуточное соединение 32 (240 мг, 1,02 ммоль) суспендировали в 1,56 мл 1М раствора DCC в ДХМ и 10 мл ДХМ и оставляли перемешиваться на тридцать минут, после чего добавляли стероид 7 (0,24 г, 0,63 ммоль) и DMAP (8 мг, 0,0625 ммоль). Смесь оставляли перемешиваться на ночь, а затем разбавляли ДХМ и фильтровали. Удаляли летучие вещества в вакууме и получали 65 мг целевого промежуточного соединения путем ЖХСД, элюируя с градиентом 0-10% смесями ацетона в ДХМ. 3-TBS-группу удаляли путем обработки 2М HCl (2 мл) в 4 мл 1,4-диоксана, отслеживание взаимодействия путем ТСХ показало, что для полного удаления TBS-группы требовалось перемешивание в течение 6 часов. Конечный аналог 17А очищали путем ЖХСД, элюируя с градиентом 0-20% смесями ацетона в ДХМ; 1Н ЯМР (δ, ДМСО-d6, 300 МГц): 9,002 (s, 1Н, ArOH), 7,809 (d, 2Н, ArH, J=4,20 Гц), 7,532 (d, 2Н, ArH, J=4,35 Гц), 7,352 (s, 2Н, -SO2NH2), 7,028 (d, 1Н, ArH, J=4,16 Гц), 6,497 (dd, 1H, ArH, J=4,12 Гц, 1,06 Гц), 6,454 (d, 1H, ArH, J=0,05 Гц), 4,712 (t, J=5,10 Гц, 1H, -CH), 4,640 (s, 2H, -CH2), 4,233 (s, 2H, -CH2), 0,773 (s, 3Н, -CH3). ИКС (см-1): 3362, 3257, 2917, 2862, 1733, 1595, 1498, 1209, 1154, 831, 671. ИЭР-МС: 498,1 (М-1)-, 517,2 (М+H2O)+. Оптическое вращение: [α]D20=+36° (МеОН); Тпл: 167,5-170,3°С.

Синтез (2S)-1-бензил-2-((13S,16R,17R)-3,16-бис(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)овый эфир пирролидин-1,2-дикарбоновой кислоты 33: Использовали способ, описанный на схеме 7 (выход 76%); 1Н ЯМР (300 МГц, CDCl3) δ ppm 7,40-7,28 (m, 5Н), 7,08 (t, J=6,90 Гц, 1H), 6,61 (dd, J=8,10 Гц, 1,80 Гц, 1Н), 6,54 (d, J=1,80 Гц, 1H), 5,09 (dd, J=65,25 Гц, 10,5 Гц, 1H), 5,00 (шир.s, 1H), 4,85 (dd, J=8,7 Гц, 5,4 Гц, 1H), 4,52-4,44 (m, 1H), 4,32-4,24 (m, 1H), 3,65-3,56 (m, 2H), 0,97 (s, 9H), 0,92-0,85 (m, 10H), 0,71 (d, J=2,1 Гц, 3Н), 0,18 (s, 6H), 0,10-0,01 (m, 6H).
Синтез (2S)-((13S,16R,17R)-3,16-бис(трет-бутилдиметилсилилокси)-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)овый эфир пирролидин-2-карбоновой кислоты 34: Использовали способ, описанный на схеме 7 (выход 96%); 1Н ЯМР (300 МГц, CDCl3) δ ppm 7,09 (d, J=8,40 Гц, 1H), 6,60 (dd, 7,65 Гц, 2,4 Гц, 1H), 6,54 (d, J=1,80 Гц), 4,88 (d, J=5,7 Гц, 1H), 4,35-4,28 (m, 1H), 3,84-3,77 (m, 1H), 3,16-3,08 (m, 1H), 2,96-2,88 (m, 1H), 0,97 (s, 9H), 0,87 (s, 9H), 0,79 (s, 3H), 0,18 (s, 6H), 0,033-0,016 (m, 6H).

[(13S,16R,17R)-3,16-дигидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]фенантрен-17-ил]-(2S)-1-(4-сульфамоилбензоил)овый эфир пирролидин-2-карбоновой кислоты 30А: Использовали способ сочетания 4-сульфамоилбензойной кислоты с промежуточным соединением 34, такой как описано на схеме 7 (выход 57%). Бис-TBS аналог 30А (1,8 г, 2,26 ммоль) растворяли в 45 мл 1,4-диоксана и добавляли 22 мл 2М HCl. Анализ ТСХ (1:4 ацетон : ДХМ) указывал на полное удаление защитных групп в течение 2 часов. Смесь переносили в колбу Эрленмейера и обрабатывали, добавляя по частям насыщенный раствор бикарбоната натрия, до достижения нейтрального pH. Водный слой экстрагировали этилацетатом и выделяли конечный аналог 30А путем ЖХСД (от 0 до 50% ацетона в ДХМ), получали 652 мг продукта (выход 51%); 1Н ЯМР (300 МГц, ДМСО-d6) δ 9,02 (s, 1H), 8,02-7,88 (m, 2H), 7,79-7,66 (m, 2H), 7,50 (s, 2H), 7,02 (d, J=8,4 Гц, 1H), 6,50 (d, J=8,5 Гц, 1H), 6,43 (s, 1H), 4,64-4,52 (m, 1H), 4,17 (s, 1H), 3,52 (s, 1H), 0,71 (s, 3Н). ИКС (см-1): 1737, 1611, 1444, 1339, 1158, 710, 613. ИЭР-МС: 567,2 (M-1)-; 569,2 (М+1)+. Оптическое вращение: [α]D20=-30° (МеОН); Тпл: 243,0-245,5°С.

Синтез (S)-1-(4-сульфамоилфенил)пирролидин-2-карбоновой кислоты 35: В круглодонную колбу помещали 4-фторбензолсульфонамид (945 мг, 5,4 ммоль), соль HCl трет-бутилового эфира L-пролина (1,24 г, 6 ммоль) и карбонат цезия (4,2 мг, 12 ммоль) и суспендировали в 10 мл безводного ДМСО. Смесь помещали на предварительно нагретую масляную баню, присоединяли холодильник и грели в атмосфере азота при 168°С в течение ночи. Анализ ВЭЖХ смеси указывал на полную конверсию исходного бензолсульфонамида. Смесь переносили в колбу Эрленмейера, реакционный сосуд промывали 2 мл концентрированной HCl и добавляли к смеси в колбе Эрленмейера и оставляли перемешиваться. Затем водный слой экстрагировали этилацетатом (3×50 мл) и объединенные экстракты промывали водой (3×15 мл). Затем удаляли летучие вещества в вакууме и получали 1,24 г неочищенного остатка. Целевое промежуточное соединение выделяли путем ЖХСД, элюируя с градиентом 0-60% смесями ацетона в ДХМ, получали 35 (0,56 г, 41%); 1Н ЯМР (300 МГц, ДМСО-d6) δ ppm 7,58 (d, J=9,0 Гц, 2Н), 6,98 (s, 2Н), 6,54 (d, J=9,0 Гц, 2Н), 4,28 (d, J=8,4 Гц, 1H), 2,35-2,22 (m, 2H), 2,04-1,96 (m, 2H).

[(13S,17S)-3-гидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]-фенантрен-17-ил]овый эфир 1-(4-сульфамоилфенил)пирролидин-2-карбоновой кислоты 22А: Сочетание стероида 7 с промежуточным соединением 35 проводили согласно описанию, приведенному на схеме 7 (выход 69%). 3-TBS-группу удаляли согласно приведенному выше описанию (62%); 1Н ЯМР (300 МГц, ДМСО-d6) δ 9,02 (s, 1Н), 7,67-7,56 (m, 2Н), 7,14-6,93 (m, 3Н), 6,61-6,39 (шир.s, 4Н), 4,60 (t, J=9,7 Гц, 1H), 4,47-4,38 (m, 1Н), 0,73 (s, 2Н), 0,65 (s, 1Н)- 2 последние сдвига соответствуют 19-метилу ротамеров. ИКС (см-1): 3374, 3252, 1725, 1704, 1591, 1146, 1095, 814, 605. ИЭР-МС: 523,2 (М-1)-. Оптическое вращение: [α]D20=+50° (МеОН).
Синтез 4-формилбензолсульфонамида 36: В круглодонную колбу помещали 4-гидроксиметилбензолсульфонамид (3,75 г, 20 ммоль). Добавляли 40 г активированных молекулярных сит, а затем 150 мл ТГФ. Затем добавляли 37,6 г (0,1 моль) PDC и смесь оставляли перемешиваться на 2 часа, после чего анализ ТСХ в смеси 1:1 ацетон : ДХМ указывал на полную конверсию исходного вещества. Смесь фильтровали через небольшую колонку с оксидом кремния и элюировали ацетоном. Удаляли растворители в вакууме и выделяли целевое вещество путем ЖХСД, элюируя с градиентом 0-60% смесями этилацетата в гексанах, с получением 36 (2,05 г, 55%). 1Н ЯМР (300 МГц, ДМСО-d6) δ ppm 10,09 (s, 1Н), 8,20-7,93 (m, 4H), 7,61 (s, 2H).

[(13S,17S)-3-гидрокси-13-метил-6,7,8,9,11,12,14,15,16,17-декагидроциклопента[а]-фенантрен-17-ил]овый эфир 1-[(4-сульфамоилфенил)метил]пирролидин-2-карбоновой кислоты 23А: В круглодонную колбу помещали альдегид 36 (2,0 г, 10,8 ммоль) и цианоборгидрид натрия (0,45 г, 7,2 ммоль). Добавляли 100 мл ТГФ, затем 0,41 мл АсОН (7,2 ммоль). Добавляли промежуточный пролин-Е2-3-ТВS, полученный согласно описанию, приведенному на схеме 7 (3,49 г, 7,2 ммоль), и 300 мг активированных молекулярных сит. Смесь оставляли перемешиваться в атмосфере азота на ночь. После гашения раствором хлорида аммония и нейтрализации раствором бикарбоната натрия и экстракции этилацетатом начальная попытка выделения целевого промежуточного соединения путем хроматографии была неудачной, так как требовалось удаление избытка альдегида, используемого для взаимодействия. Затем вещество обрабатывали путем суспендирования в 10 мл ТГФ и 1 мл МеОН и охлаждали до 0°С, обрабатывали 50 мг боргидрида натрия. После указанной обработки промежуточное соединение легко выделяли путем ЖХСД (градиент 0-16% ацетона в ДХМ). TBS-группу удаляли в стандартных условиях; 1Н ЯМР (300 МГц, ДМСО-d6) δ 9,02 (s, 1Н), 7,76 (d, J=6,6 Гц, 2Н), 7,49 (d, J=8,6 Гц, 2Н), 7,32 (s, 2Н), 7,03 (d, J=6,4 Гц, 1Н), 6,50 (dd, J=8,0, 2,1 Гц, 1Н), 6,43 (d, J=2,0 Гц, 1Н), 4,73-4,58 (m, 1H), 3,98 (d, J=14,6 Гц, 1Н), 3,59 (d, J=13,8 Гц, 1H), 0,77 (s, 3Н). ИКС (см-1): 3257, 2921, 2850, 1721, 1326, 1158, 684, 575. ИЭР-МС: 537,3 (М-1)-; 539,2 (М+1)+. Оптическое вращение: [α]D20=-2° (МеОН).

Синтез (S)-1-(4-сульфамоилбензоил)пирролидин-2-карбоновой кислоты 37: L-пролин (12,4 г, 0,108 моль) суспендировали в 50 мл метанола и смесь охлаждали на ледяной бане. По каплям в течение двадцати минут добавляли тионилхлорид (8,1 мл, 0,11 моль). После завершения добавления удаляли полученную гомогенную смесь из ледяной бани и оставляли перемешиваться на два часа, после чего удаляли все летучие вещества в вакууме с получением 19,43 г метилового эфира L-пролина, который использовали без какой-либо дополнительной очистки.
Метиловый эфир L-пролина (14,8 г, 86,3 ммоль) помещали в круглодонную колбу совместно с 4-сульфамоилбензойной кислотой (15,6 г, 77,7 ммоль). Добавляли ДМФ (75 мл), затем HOBt (14,3 г, 93 ммоль) и EDCI (20,7 г, 108 ммоль). Смесь оставляли перемешиваться в атмосфере азота на ночь, после чего анализ ТСХ указывал на практически полную конверсию исходного вещества. Затем смесь помещали в 2,5 л воды и охлаждали при 4°С в течение ночи. На следующий день собирали кристаллы и промывали водой (2×100 мл). Собирали кристаллы метилового эфира (23 г) и проводили вторую кристаллизацию из бикарбоната натрия. Собирали кристаллы продукта (11,4 г, выход 47%).
Метиловый эфир (10,3 г, 33 ммоль) подвергали омылению с использованием гидроксида натрия (13,2 г, 0,33 моль) в воде (200 мл) и ТГФ (100 мл). Анализ ТСХ в смеси 1:4 ацетон : ДХМ указывал на полную конверсию исходного вещества (после подкисления и экстракции аликвоты реакционной смеси). pH смеси снижали до 7 с использованием 2М HCl, затем удаляли ТГФ в вакууме. Затем pH снижали до 0 с использованием конц. HCl и экстрагировали этилацетатом. Объединенные экстракты сушили над сульфатом натрия и удаляли летучие вещества с получением кислоты 37 в виде белого твердого вещества (7,82 г, выход 80%); 1Н ЯМР (300 МГц, ДМСО-d6): δ (сложный сигнал ароматики связан с наличием ротамеров) 7,94-7,81 (m, 2Н), 7,70-7,54 (m, 2Н), 7,49 (шир.s, 2Н), 4,47-4,34 (m, 1H), 3,08-2,93 (m, 1H), 2,37 (m, 1H), 1,93-1,86 (m, 2H). ИКС (см-1): 3395, 3290, 1725, 1586, 1560, 1441, 1343, 1167, 835, 705. Оптическое вращение: [α]D20=-80° (МеОН).
Синтез 2-(4-сульфамоилфенил)уксусной кислоты 38: Использовали способ, описанный в международной заявке РСТ №2013029338, поданной 7 марта 2013 года; 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,77 (d, J=7,5 Гц, 2Н), 7,45 (d, J=7,8 Гц, 2Н), 7,34 (шир.s, 2Н), 3,70 (шир.s, 2Н).

(2S)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)ового эфира 1-(2-(4-сульфамоилфенил)ацетил)-пирролидин-2-карбоновой кислоты 31А: В круглодонную колбу помещали пролин-Е2-3-TBS (полученный согласно описанию, приведенному на схеме 7, 1,15 г, 2,38 ммоль), 38 (1,03 г, 4,77 ммоль) и HOBt (730 мг, 4,77 ммоль). Растворяли твердые вещества в ДМФ, после чего добавляли EDCI (1,14 г, 5,95 ммоль) и оставляли перемешиваться в атмосфере азота на 18 часов, после этого анализ ТСХ указывал на полную конверсию исходного вещества. Затем смесь разбавляли водой, что приводило к осаждению целевого продукта. Выделяли 1,51 грамма (выход 93%) целевого промежуточного соединения, 3-TBS-группу удаляли при помощи описанного выше способа; 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,01 (шир.s, 1H), 7,76-7,68 (m, 2Н), 7,50-7,30 (m, 4Н), 7,04 (d, J=8,1 Гц, 1Н), 6,50 (dd, J=8,6,2,4 Гц, 1H), 6,43 (шир.s, 1Н), 4,73-4,58 (m, 1Н), 4,35-4,28 (m, 1Н) 3,78 (шир.s, 2Н), 0,78-0,71 (m, 3Н). ИКС (см-1): 3244, 1725, 1624, 1439, 1339, 1188, 1154, 663. Оптическое вращение: [α]D20=-36° (МеОН).

(2S)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)овый эфир 1-(2-(4-сульфамоилфенил)ацетил)пирролидин-2-карбоновой кислоты 32А: В круглодонную колбу помещали хлорсульфонилацетилхлорид (0,3 мл, 2,8 ммоль) и растворяли в ДХМ (2 мл). Охлаждали раствор до -78°С, а затем по каплям в течение 15 минут добавляли раствор Е2-пролина 1 (1,14 г, 2,36 ммоль) в ДХМ (8 мл). Смесь оставляли перемешиваться на 1,5 часа, после чего добавляли раствор гидроксида аммония (28-30%, 1 мл, 14 ммоль). Смесь оставляли перемешиваться на ночь, постепенно нагревали до комнатной температуры. На следующий день при помощи анализа ТСХ подтверждали полную конверсию исходного вещества и смесь разбавляли 25 мл 2М HCl. Затем водный слой экстрагировали этилацетатом и сушили объединенные органические слои сульфатом натрия. Затем неочищенное вещество очищали путем ЖХСД, элюируя с градиентом 0-36% смесями ацетона в ДХМ, с получением 318 мг целевого промежуточного соединения, из которого затем удаляли защитные группы при помощи стандартного способа с получением целевого продукта 32А: 1Н ЯМР (300 МГц, ДМСО-d6) δ 9,01 (шир.s, 1Н), 7,68 (шир.s, 1H), 7,40 (шир.s, 1Н), 7,03 (d, J=8,4 Гц, 1Н), 6,50 (dd, J=8,4, 1,8 Гц, 1H), 6,43 (шир.s, 1H), 4,67 (t, J=7,5 Гц, 1H), 4,41 (dd, J=8,6, 2,7 Гц, 1H), 4,10 (d, J=13,8 Гц, 1Н), 3,96 (d, J=13,8 Гц, 1Н), 0,79 (s, 3Н). ИКС (см-1): 3437, 3336, 3202, 1729, 1670, 1607, 1334, 1200, 1137. Оптическое вращение: [α]D20=-12° (МеОН).

(2S)-((10R,13S,17S)-10,13-диметил-3-оксо-2,3,6,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1Н-циклопента[а]фенантрен-17-ил)овый эфир 1-(4-сульфамоилбензоил)пирролидин-2-карбоновой кислоты 33А: В круглодонную колбу помещали тестостерон (0,96 г, 3,33 ммоль), кислоту 37 (2,98 г, 10 ммоль) и DMAP (41 мг, 0,33 ммоль). Смесь растворяли в ДМФ (25 мл), а затем добавляли EDCI (1,92 г, 10 ммоль) и смесь оставляли перемешиваться на ночь. Анализ ВЭЖХ указывал на примерно 50% конверсию тестостерона, и затем смесь разбавляли 2,5 л воды, что после фильтрования, промывки водой и сушки в вакууме приводило к получению 1 г твердого вещества. Затем путем ЖХСД, элюируя с градиентом 0-30% смесями ацетона в ДХМ, получали целевой продукт 33А; 1Н ЯМР (300 МГц, ДМСО-d6) δ 7,98-7,81 (m, 2Н), 7,75-7,65 (m, 2Н), 7,49 (шир.s, 1H), 5,63 (шир.s, 1Н), 4,66-4,44 (m, 2Н), 0,86-0,76 (m, 6Н). ИКС (см-1): 3244, 1733, 1613, 1427, 1334, 1161, 608. ИЭР-МС: 569,3 (М+Н)+. Оптическое вращение: [α]D20=+56° (МеОН).
(2R)-((13S,17S)-3-гидрокси-13-метил-7,8,9,11,12,13,14,15,16,17-декагидро-6Н-циклопента[а]фенантрен-17-ил)овый эфир 1-(4-сульфамоилбензоил)пирролидин-2-карбоновой кислоты синтезировали согласно способам, приведенным на схеме 7, с использованием N-Cbz-D-пролина, получали целевой продукт 34А в виде белого твердого вещества (0,45 г); 1Н ЯМР (δ, ацетон-d6 300 МГц): 7,97 (d, 2Н, ArH, J=8,4 Гц), 7,93 (s, 1Н, ArOH), 7,73 (d, 1,6Н ротамер, ArH, J=8,1 Гц), 7,61 (d, 0,4Н ротамер, ArH, J=8,4 Гц), 7,09 (d, 1H, ArH, J=8,1 Гц), 6,70 (шир.s, 2Н, NH2), 6,59 (m, 1Н, ArH), 6,53 (s, 1H, ArH), 4,73 (t, 1H, 17-CH), 4,56 (m, 1H, CH), 0,90 (s, 3Н, СН3). ИКС (см-1): 3248, 2922, 2875, 1737, 1725, 1611. Оптическое вращение: [α]D24=+46,5° (с=0,58, 1,4-диоксан).
Исследовали пероральную биологическую активность (таблица 1); связывание с карбоангидразой II человека (hCA 2) (таблица 2) и ферментативное омыление некоторых типовых соединений (таблица 3).


Е2, EE, J995 (патент США №5705495, Европейские патенты ЕР 1273590 и ЕР 1284273) и 1А (патент США №7507725 и Европейский патент ЕР 1294402) представляют собой стандартные соединения, известные из литературы.
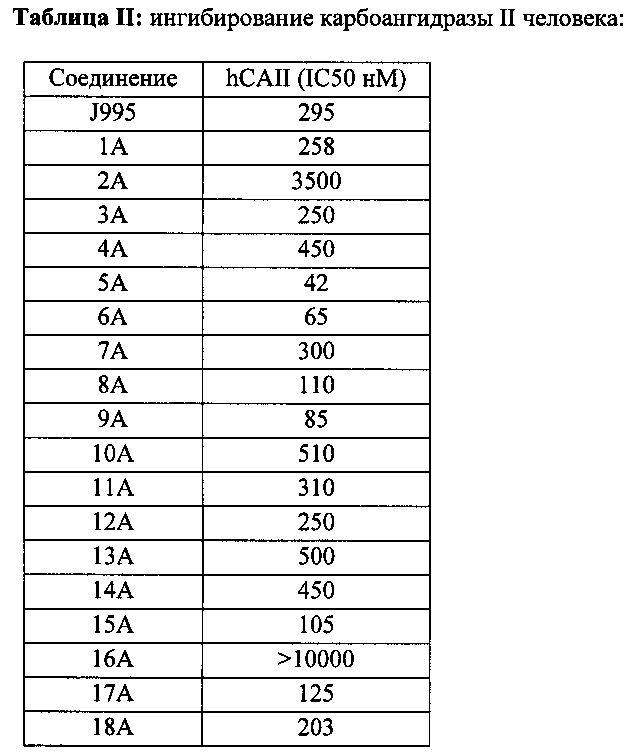

Ссылка: J Biomol Screen, 2006 Oct; 11(7): 782-91.

Ссылки: i) J Endocrinol. 1976 Oct; 71(1):77-85. ii) Clinical guide to laboratory tests, 3e издание, W.B.. Saunders, Co., Philadelphia, 1995: 216-217.
Некоторые из исследуемых соединений имели более высокую пероральную активность по сравнению с исходным лекарственным средством и другими известными возможными пролекарствами, такими как J995 и А1. Также наблюдали, что повышенная биологическая активность при пероральном введении дозы вероятно не зависела от связывания с НСА2.
В то время, как стандартное соединение J995 и другие соединения, такие как 8А, с высокой активностью связывались с hCA2 со значениями IC50, составлявшими 295 и 110 нМ, соответственно, связывание для 16А не наблюдали вообще, и значение IC50 составляло 10000 нМ, но при этом указанное соединение имело высокую пероральную активность, сравнимую с J995 и А8.
Было показано, что соединения формулы (II) согласно настоящему изобретению являются высокоактивными и имеют повышенную биодоступность в пробе Аллена-Дойзи у самок крыс после овариэктомии (Walter Elger et al., J. Steroid Biochem. Molec. Biol. Vol 55 395-403, 1995) и в модели РК у крыс. Хорошие свойства указанных соединений, которые выделяют их среди конкурентов, связаны с растворимостью, превосходным всасыванием, то есть, пролекарства обеспечивают хорошее воздействие при пероральном введении. Преимущество фрагмента (кислота)-(линкер) позволяет свободно регулировать химическую стабильность сложноэфирной связи между активными ингредиентами и фрагментами (кислота)-(линкер) таким образом, чтобы она не была очень слабой или прочной в зависимости от наличия заместителей в альфа-положении кислоты, что обеспечивает неожиданные превосходные терапевтические результаты (8А).
Содержание определенных патентов США, заявок на патент США и других материалов (например, статей) включено в настоящий патент посредством ссылок. Тем не менее, текстовая информация, приведенная в указанных патентах США, заявках на патент США и других материалах, включена посредством ссылки только если отсутствуют противоречия между текстовой информацией и иными утверждениями и чертежами, приведенными в настоящем описании. В случае указанных противоречий какая-либо указанная текстовая информация, содержащаяся в указанных включенных посредством ссылок патентах США, заявках на патент США и других материалах, не включена в указанный патент посредством ссылки.
Дополнительные модификации и альтернативные варианты реализации различных аспектов изобретения будут понятны специалистам в данной области техники после изучения настоящего описания. Соответственно, настоящее описание следует рассматривать исключительно как иллюстративное, и оно предназначено для объяснения специалистам в данной области техники общего способа реализации изобретения. Следует понимать, что формы изобретения, показанные и описанные в настоящей заявке, следует рассматривать как примеры вариантов реализации. Можно заменять элементы и материалы, проиллюстрированные и описанные в настоящей заявке, можно изменять порядок частей и способов, определенные отличительные признаки изобретения можно применять независимо, что должно быть понятно специалистам в данной области техники после изучения преимуществ описания настоящего изобретения. Можно проводить изменения элементов, описанных в настоящей заявке, не выходя за рамки сущности и объема изобретения, описанного в последующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ УЛУЧШЕНИЯ КАЧЕСТВА МЫШЦ | 2015 |
|
RU2724329C2 |
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
| Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | 2017 |
|
RU2731385C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ Е3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2666530C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ E3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2781452C2 |
| Нитроны стероидов для лечения и предотвращения инсульта или ишемии головного мога, болезни Альцгеймера, болезни Паркинсона и бокового амиотрофического склероза | 2014 |
|
RU2668514C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2830163C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
Изобретение описывает соединения-пролекарства, имеющие общую структуру: Активный агент - (кислота)-(линкер) - SO2NR1R2:

где R1 представляет собой Н, С1-С12алкил или С1-С12алкилС6-С10арил; hAr представляет собой С6-С10арил или 5-7-членное моноциклическое гетероциклическое кольцо или 7-10-членное бициклическое гетероциклическое кольцо, содержащее 1-4 гетероатома, выбранных из N, О и S; R2 представляет собой Н или С1-С12алкил; R1 и R2 могут быть объединены с образованием 3-7-членного кольца, содержащего до одного гетероатома; каждый R3 и R4 независимо представляет собой Н или С1-С12алкил; X и Y представляют собой Н; Z представляет собой О; и активный агент представляет собой андроген, эстроген или прогестин. Соединения, имеющие указанную общую структуру (1), имеют более высокую активность при пероральном введении по сравнению с немодифицированной исходной молекулой. 2 н. и 15 з.п. ф-лы, 3 табл.
1. Соединение структурной формулы I:

где R1 представляет собой Н, С1-С12алкил или С1-С12алкилС6-С10арил;
hAr представляет собой С6-С10арил или 5-7-членное моноциклическое гетероциклическое кольцо или 7-10-членное бициклическое гетероциклическое кольцо, содержащее 1-4 гетероатома, выбранных из N, О и S;
R2 представляет собой Н или С1-С12алкил;
R1 и R2 могут быть объединены с образованием 3-7-членного кольца, содержащего до одного гетероатома;
каждый R3 и R4 независимо представляет собой Н или С1-С12алкил;
X и Y представляют собой Н;
Z представляет собой О; и
активный агент представляет собой андроген, эстроген или прогестин.
2. Соединение по п. 1, отличающееся тем, что R1 и R2 объединены с образованием 3-7-членного кольца, содержащего до одного гетероатома.
3. Соединение по п. 1, отличающееся тем, что R1 представляет собой С1-С12алкил.
4. Соединение по п. 1, отличающееся тем, что R1 представляет собой изопропил.
5. Соединение по п. 1, отличающееся тем, что R1 представляет собой метил.
6. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой андроген.
7. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой тестостерон.
8. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой 7α,11β-диметилэстра-4,9-диен-17β-ол.
9. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой эстроген.
10. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой эстрадиол.
11. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой эстриол.
12. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой прогестин.
13. Соединение по любому из пп. 1-5, отличающееся тем, что активный агент представляет собой тримегестон.
14. Соединение по любому из пп. 1-13, отличающееся тем, что hAr представляет собой тиофен, пиридин, изоксазол, фталимид, пиразол, индол или фуран.
15. Соединение по любому из пп. 1-13, отличающееся тем, что hAr представляет собой пиридин или фуран.
16. Соединение по любому из пп. 1-13, отличающееся тем, что hAr представляет собой бифенил.
17. Фармацевтическая композиция, содержащая соединение по любому из пп. 1-16 и один или более фармацевтически приемлемых носителей.
| WO 2008113851 A2 25.09.2008 | |||
| US 20090186869 A1 23.07.2009 | |||
| WO 2005113574 A1 01.12.2005 | |||
| US2005277625, A1, 15.12.2005 | |||
| US2009023896, A1, 22.01.2009. |

