Настоящее изобретение относится к новым лекарственным веществам, предназначенным как для систематического, так и для несистематического применения, а также композициям на их основе для использования при окислительном стрессе и/или эндотелиальных дисфункциях средней интенсивности.
Под окислительным стрессом подразумевается образование свободных радикалов или радикальных соединений, которые вызывают разрушение как клеток, так и окружающей их ткани (Pathophysiology: the biological basis for disease in adults and children, McCance&Huether 1998, pp. 48-54).
Под эндотелиальными дисфункциями подразумевают дисфункции, относящиеся к сосудистому эндотелию. Известно, что повреждение сосудистого эндотелия является одной из важнейших причин, которые могут вызвать серию патологических процессов, затрагивающих различные органы и системы организма, как описано далее (Pathophysiology: the biological basis for disease in adults and children, McCance & Huether 1998, p. 1025).
Как известно, окислительный стресс и/или эндотелиальные дисфункции связаны с различными патологиями, которые описаны далее. Окислительный стресс также может быть обусловлен токсичностью большого числа различных лекарственных веществ, что сильно сказывается на эффективности их действия.
Указанные патологические явления носят хронический характер, подрывающий силы организма, и очень часто являются типичными для людей пожилого возраста. Как уже было сказано, при указанных патологических состояниях эффективность применяемых лекарственных веществ значительно снижается.
Можно привести следующие примеры патологических состояний, обусловленных окислительным стрессом и/или эндотелиальными дисфункциями или характерных для людей пожилого возраста:
- Для сердечно-сосудистой системы: миокардиальная и сосудистая ишемия в целом, гипертония, инсульт, атеросклероз и т.д.
- Для соединительной ткани: ревматоидный артрит и связанные с ним воспалительные заболевания и т.д.
- Для легочной системы: астма и связанные с ней воспалительные заболевания и т.д.
- Для желудочно-кишечной системы: язвенные и неязвенные диспепсии, кишечные воспалительные заболевания и т.д.
- Для центральной нервной системы: болезнь Альцгеймера и т.д.
- Для мочеполовой системы: импотенция, недержание.
- Для кожных покровов: экзема, нейродерматит, угри.
- Инфекционные заболевания в целом (см.: Schwarz, Brady "Oxidative stress during viral infection: a review" Free Radical Biol. Med. 21/5, 641-649, 1996).
Кроме того, сам процесс старения может рассматриваться как действительное патологическое состояние (см. Pathophysiology: the biological basis for disease in adults and children, pp. 71-77).
Известные лекарственные вещества, при введении их пациентам, имеющим патологии, связанные с окислительным стрессом и/или эндотелиальными дисфункциями, проявляют сниженную активность и/или повышенную токсичность.
Это происходит, например, в случае таких лекарственных веществ, как противовоспалительные, сердечно-сосудистые лекарственные вещества, лекарственные вещества для дыхательной системы, лекарственные вещества для центральной нервной системы, лекарственные вещества для костной системы, антибиотики, лекарственные вещества для мочеполовой, эндокринной системы и т.д.
Исследования лекарственных веществ направлены на поиск новых молекул, имеющих улучшенный терапевтический индекс (соотношение эффективность/токсичность), или пониженное соотношение риск/полезное действие, в том числе и для указанных выше патологических состояний, при которых терапевтический индекс значительного числа лекарственных веществ оказывается низким. Фактически при указанных выше состояниях окислительного стресса и/или эндотелиальных дисфункций многие лекарственные вещества проявляют пониженную активность и/или повышенную токсичность.
В частности, противовоспалительные лекарственные вещества, такие как NSAIDs и лекарственные вещества против колик, например 5-аминосалициловая кислота и ее производные, имеют следующие недостатки. NSAIDs проявляют токсические свойства, особенно в тех случаях, когда организм ослаблен или находится под влиянием патологических состояний, связанных с окислительным стрессом. К указанным состояниям можно отнести, например, следующие: преклонный возраст, ранее перенесенная язва, ранее перенесенное желудочное кровотечение, хронические заболевания, подрывающие силы организма, в частности, воздействующие на сердечно-сосудистую систему, почечный аппарат, состояние крови и т.д. ("Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving non-steroidal anti-inflammatory drugs. A randomized, double blind, placebo-controlled trial." F.E. Silverstein et al. Ann. Intern. Med. 123/4, 241-9, 1995; Martindale 31 a ed. 1996, pag. 73, Current Medical Diagnosis and Treatment 1998, pages 431 and 794).
Введение противовоспалительных лекарственных веществ пациентам, находящимся в указанных выше патологических состояниях, можно осуществить, только используя более низкие дозы лекарственного вещества по сравнению с теми, которые обычно используются для терапии, чтобы избежать заметных токсических проявлений. Поэтому противовоспалительная активность проявляется слабо.
Бета-блокаторы, используемые для лечения стенокардии, гипертонии и сердечной аритмии, оказывают побочное воздействие на дыхательную систему (одышка, бронхостеноз), поэтому из-за них могут возникнуть проблемы у пациентов с патологиями указанных органов (астмой, бронхитом). Таким образом бета-блокаторы еще больше ухудшают состояние при таких заболеваниях дыхательной системы, как астма. Поэтому пациентам, страдающим от астмы, следует назначать уменьшенные дозы указанных лекарственных веществ, чтобы не подвергать еще большей опасности их дыхательную систему. В результате эффективность бета-блокаторов значительно снижается.
Антитромботические средства, такие как, например, дипиридамол, аспирин и т.д., используемые для профилактики явлений тромбоза, имеют те же недостатки. У пациентов, имеющих патологии, связанные с окислительным стрессом и/или эндотелиальными дисфункциями, терапевтическое действие или переносимость этих лекарственных средств, в частности аспирина, значительно снижены.
Для лечения астмы и бронхита используют бронходилататоры, например сальбутамол и т. д., а при таких патологиях, как недержание мочи используют лекарственные вещества, действующие на холинэргическую систему. При их введении могут возникнуть аналогичные побочные эффекты, влияющие на сердечно-сосудистую систему и порождающие проблемы у пациентов, страдающих от сердечной недостаточности и гипертонии. Сердечная недостаточность и гипертония являются патологиями, связанными, как упоминалось выше, с окислительным стрессом и/или эндотелиальными дисфункциями. И эти лекарственные вещества тоже имеют такие же недостатки, как и перечисленные ранее.
Отхаркивающие и муколитические лекарственные вещества, которые применяются для лечения воспалительных заболеваний органов дыхания, проявляют те же недостатки у пациентов в описанных выше состояниях. Их введение может вызвать изжогу и раздражение желудка, особенно у людей пожилого возраста.
Ингибиторы костной резорбции, такие как дифосфонаты (например, алендронат и т.д.), являются лекарственными веществами, проявляющими повышенную желудочно-кишечную токсичность. Следовательно, эти лекарственные вещества также могут иметь такие же недостатки, которые описаны выше.
В случае ингибиторов фосфодиэстераз, таких как, например, силденафил, запринаст, используемых для лечения заболеваний сердечно-сосудистой и дыхательной систем, возникают аналогичные проблемы с переносимостью и/или эффективностью при упомянутых выше патологических состояниях окислительного стресса и/или эндотелиальных дисфункций.
Антиаллергические лекарственные вещества, например цетиризин, монтелукаст и т.д., вызывают аналогичные проблемы при упомянутых патологических состояниях, в особенности в отношении эффективности.
Антиангиотензиновые лекарственные вещества, т.е. ингибиторы АСЕ, например эналаприл, каптоприл и т.д., или ингибиторы рецепторов, например лозартан и т.д., используют для лечения сердечно-сосудистых заболеваний. Их недостатком является побочное воздействие на органы дыхания (например, возникновение кашля и т.д.) при указанных выше патологических состояниях.
Противодиабетические лекарственные вещества, как повышающие чувствительность к инсулину, так и снижающие уровень глюкозы, такие как, например, сульфонилмочевины, толбутамид, глипирид, гликлазид, глибурид, никотинамид и т.д., являются неэффективными для профилактики диабетических осложнений. При их введении могут возникать побочные эффекты, например поражение желудка. Эти явления усиливаются при указанных выше патологических состояниях.
В случае антибиотиков, например ампициллина, кларитромицина и т.д., и противовирусных лекарственных веществ, например ацикловира и др., возникают проблемы, связанные с их переносимостью, в частности они вызывают раздражение желудочно-кишечного тракта.
Противоопухолевые лекарственные вещества, например доксорубицин, даунорубицин, цисплатин и др., обладают высокой токсичностью по отношению к различным органам, в том числе к желудку и кишечнику. Это токсическое воздействие еще более усиливается при указанных выше патологических состояниях окислительного стресса и/или эндотелиальных дисфункций.
Лекарственные вещества против слабоумия, например никотин и колиномиметики, характеризуются слабой переносимостью, особенно в случае указанных выше патологий.
Лекарственные вещества стероидной структуры, которые используют для терапии острых (астма и т.д.) или хронических заболеваний (заболеваний кишечника, печени, органов дыхания, заболеваний женской репродуктивной системы, гормональных расстройств, кожных заболеваний, и т.д.), отличаются заметным токсическим воздействием на различные органы, особенно в вышеупомянутом состоянии окислительного стресса.
Класс стероидных лекарственных веществ, в том числе гидрокортизон, кортизон, преднизон, преднизолон, флюдрокортизон, дезоксикортикостерон, метилпреднизолон, триамцинолон, параметазон, бетаметазон, дексаметазон, триамцинолона ацетонид, флуопинолона ацетонид, беклометазон, ацетоксипрегнелон и т.д., оказывает заметное фармакотоксическое влияние на различные органы, и поэтому как их клиническое применение, так и перерывы в приеме приводят к ряду побочных эффектов, некоторые из которых могут быть очень серьезными. См., например, Goodman & Gilman, "The Pharmaceutical Basis of Therapeutics" 9-е изд., стр. 1459-1465, 1996.
Среди указанных токсических эффектов следует отметить воздействие на костную ткань, что приводит к изменению клеточного метаболизма и повышению вероятности возникновения остеопороза; воздействие на сердечно-сосудистую систему, приводящее к гипертонии; воздействие на желудочно-кишечный аппарат, приводящее к желудочным расстройствам. См., например, Martindale "The Extrapharmacopoeia, 30-е изд., стр. 712-723,1993.
К классу стероидных лекарственных веществ также принадлежат желчные кислоты, применяемые в терапии заболеваний печени и при желчных коликах. Урсодезоксихолевая кислота также применяется при некоторых расстройствах функций печени (цирроз печени желчного происхождения и т.д.). Переносимость этих лекарственных веществ значительно ухудшается при наличии желудочно-кишечных осложнений (хронические повреждения печени, язва желудка, кишечные воспаления и т.д.). В случае желчных кислот окислительный стресс также заметно влияет на эффективность действия лекарственного вещества: как эффективность, так и переносимость хенодезоксихолевой и урсодезоксихолевой кислот значительно снижаются. Особенно усиливается нежелательное воздействие на печень. Среди стероидных соединений также можно упомянуть эстрогены, применяемые для лечения дислипидемии, гормональных расстройств, опухолей женских органов. Упомянутые стероиды также оказывают вышеуказанное побочное воздействие, особенно па печень.
Исходя из вышеописанного уровня техники представляется практически невозможным отделить терапевтическую активность от побочных эффектов (см. Goodman et al, как указано выше, стр. 1474).
Таким образом существует необходимость разработки доступных лекарственных веществ, обладающих улучшенным терапевтическим воздействием, т.е. обеспечивающих и пониженную токсичность и/или повышенную эффективность, чтобы их можно было вводить пациентам при патологических состояниях окислительного стресса и/или эндотелиальных дисфункций, без проявления недостатков, характерных для лекарственных веществ, известных из уровня техники.
Неожиданно было обнаружено, что вышеупомянутые проблемы, возникающие при введении лекарственных веществ пациентам, страдающим от окислительного стресса и/или эндотелиальных дисфункций, или пожилым людям в общем случае, можно решить при помощи нового класса лекарственных веществ, описанных далее.
Объектом изобретения являются соединения или их соли, соответствующие следующей общей формуле (I):
A-B-N(O)s (I)
где: s=2
А=R-T1-, где
R представляет собой радикал лекарственного вещества при условии, что лекарственное вещество по формуле R-T1-Z или R-T1-OZ, где T1=(CO) или О, S, NH и Z представляет собой Н или линейный или разветвленный C1-C5 алкил, выбирается из парацетамола, салбутамола, амброксола, алендроновой кислоты, цетиризина, амбрициллина, ацикловира, доксорубицина, симвастатина, дифиллина, такрина, клопидогрела, деметиломепразола, диктофенака, феруловой кислоты, эналаприла, пропранолола, бенфуродил гемисукцината, толрестата или сулиндака;
В=-ТB-Х2-0-, где
ТB=(СО), О или NH;
Х2 представляет собой бивалентный радикал, равный радикалу R1B-X-R2B, где Х представляет собой связь. О, S, NR1C, где R1C представляет собой Н или линейный или разветвленный C1-C5 алкил, R1B и R2B равны или различаются, являются линейными или разветвленными C1-C6 алкиленами, что соответствующий предшественник В не соответствует условиям теста 5 и соответствует условиям теста 4а; причем указанный предшественник имеет формулу -ТB-Х2-ОН, где когда ТB=(СО), а свободная валентность ТB занята
-OZ, где Z как определено выше, когда ТB=0, а свободная валентность ТB занята Н;
лекарственное вещество А=R-T1-, где свободная валентность занята, как определено выше, соответствует условиям по меньшей мере одного из тестов 1-3;
- где тест 1 (NEM) представляет собой тест, который проводят in vivo с четырьмя группами крыс (каждая состоит из 10 крыс), включая контрольные группы (две группы) и опытные группы (две группы), при этом соответственно одной из контрольных и одной из опытных групп подкожно вводят одну дозу 25 мг/кг N-этилмалеимида (NEM), контрольные группы обрабатывают носителем, а опытные группы обрабатывают носителем + лекарственным веществом формулы А=R-T1-, где свободная валентность занята, как указано выше, причем вводимая доза лекарственного вещества эквивалентна дозе, максимально переносимой крысами, которым не вводили NEM, т.е. самой высокой дозе, которую можно ввести животным, чтобы она не вызвала никаких очевидных токсических явлений, т.е. с заметными симптомами; при этом лекарственное вещество соответствует условиям теста 1, т.е. оно может быть использовано для получения соединений общей формулы (I), если у группы крыс, обработанной NEM + носителем + лекарственным веществом, наблюдаются желудочно-кишечные повреждения, или у группы крыс, обработанной NEM + носителем + лекарственным веществом, наблюдаются желудочно-кишечные повреждения в большей степени, чем у группы, обработанной только носителем, или чем у группы, обработанной носителем + лекарственным веществом, или чем у группы, обработанной носителем + NEM;
где тест 2 (CIP) представляет собой in vitro тест, в котором эндотелиальные клетки человека из пупочной вены выращивают в стандартных условиях, затем делят на две группы (каждый опыт воспроизводят пять раз), одну из этих групп обрабатывают смесью лекарственного вещества с концентрацией 10-4 М в культуральной среде, а другую группу обрабатывают носителем; затем к каждой из этих двух групп добавляют гидропероксид кумола (CIP) с концентрацией 5 мМ в культуральной среде; при этом лекарственное вещество соответствует условиям теста 2, т.е. оно может быть использовано для получения соединений общей формулы (I), если не имеет место статистически значимое ингибирование апоптоза (повреждения клеток), вызванного CIP, при р<0,01, по сравнению с группой, обработанной носителем и CIP;
- где тест 3 (L-NAME) представляет собой тест, который проводят in vivo в течение 4 недель с четырьмя группами крыс (каждая состоит из 10 крыс), получающих питьевую воду, включая контрольные группы (две группы) и опытные группы (две группы), при этом соответственно одна из контрольных и одна из опытных групп получает на протяжении этих 4 недель питьевую воду с добавлением метилового эфира N-ω-нитро-L-аргинина (L-NAME) с концентрацией 400 мг/л, контрольным группам в течение четырех недель вводят носитель, а опытным группам в течение четырех недель вводят носитель + лекарственное вещество, причем носитель или носитель + лекарственное вещество вводят один раз в день, а лекарственное вещество вводят в дозе, максимально переносимой группой крыс, которые не были предварительно обработаны L-NAME, т.е. в самой высокой дозе, которую можно ввести животным, чтобы она не вызвала никаких очевидных токсических явлений, т.е. с заметными симптомами; через указанные четыре недели доступ к воде прекращают на 24 часа, а затем животных умерщвляют, измерив кровяное давление за I час до умерщвления, после умерщвления измеряют уровень в плазме глутамат-пируват трансаминазы (ОРТ) и исследуют состояние тканей желудка; лекарственное вещество соответствует условиям теста 3, т.е. оно может быть использовано для получения соединений общей формулы (I), если у группы крыс, обработанных L-NAME + носителем + лекарственным веществом, наблюдаются более сильные повреждения печени (определенные по более высокому уровню содержания GPT), и/или повреждения желудка, и/или сердечно-сосудистые повреждения (определенные по более высоким величинам кровяного давления) по сравнению с группой, обработанной носителем, или с группой, обработанной носителем + лекарственным веществом, или с группой, обработанной носителем + L-NAME, соответственно;
- где тест 4А, условиям которого соответствует предшественник соединения В, представляет собой in vitro тест, при котором часть суспензии эритроцитов, предварительно выдержанной при 4°С в течение 4 дней, причем указанные эритроциты получают стандартным способом от мужских особей крыс Wistar и суспендируют в физиологическом растворе, забуференном при рН 7,4 с помощью фосфатного буфера, центрифугируют при 1000 об./мин в течение 5 минут и 0,1 мл центрифугированных эритроцитов разбавляют натрий-фосфатным буфером с рН 7,4 до 50 мл; из указанной разбавленной суспензии отбирают аликвоты по 3,5 мл (всего 5 образцов) и инкубируют при 37°С в присутствии гидропероксида кумола в концентрации 270 мкМ, и определяют мутность суспензии при 710 нм через каждые 30 минут, чтобы установить, в какой момент времени (Tmax) имеет место максимальная мутность, что соответствует максимальному количеству клеток, подвергшихся лизису под действием гидропероксида кумола (гемолиз принимают равным 100%); затем к 3,5 мл аликвотам разбавленной центрифугированной суспензии эритроцитов добавляют спиртовые растворы предшественников соединений В (тест проводят с 5 образцами для каждого из исследуемых предшественников В) до достижения 2 мМ конечной концентрации предшественника В, а затем полученную суспензию предварительно выдерживают в течение 30 минут, добавляют гидропероксид кумола в количестве, достаточном для достижения такой же конечной концентрации, как указано выше, и при Tmax определяют ингибирование гемолиза в процентах из отношения абсорбции образца, содержащего соответственно эритроциты, предшественник В и гидропероксид кумола, и образца, содержащего эритроциты и гидропероксид кумола, умноженного на 100; предшественники В соответствуют условиям данного теста, если они ингибируют гемолиз, вызванный гидропероксидом кумола, более чем на 15%;
- где тест 5, условиям которого не соответствует предшественник соединения В, представляет собой аналитическое определение, которое выполняют путем добавления аликвот 10-4 М метанольного раствора предшественника В, к раствору, полученному путем смешивания 2 мМ водного раствора дезоксирибозы с 100 мМ фосфатным буфером и 1 мМ раствором соли Fell(NH4)2(SO4)2; после термостатирования этого раствора при 37°С в течение 1 часа добавляют аликвоты 2,8% водного раствора трихлоруксусной кислоты и 0,5 М водного раствора тиобарбитуровой кислоты в указанном порядке, нагревают раствор при 100°С в течение 15 минут и затем измеряют поглощение тестируемых растворов при 532 нм; ингибирование образования радикалов FeII, вызванное предшественниками соединений В, рассчитывают в процентах по следующей формуле:
(1-As/Ac)×100
где As и Ас соответственно представляют собой величины поглощения раствора, содержащего тестируемое соединение и соль железа, и раствора, содержащего только соль железа; соединение соответствует условиям теста 5, если процент ингибирования предшественником В, определенный как указано выше, больше или равен 50%;
при условии, что в формуле (I), когда Х2 в составе В представляет собой линейный или разветвленный C1-C20 алкилен,
лекарственные вещества формулы А=R-T1-, со свободной валентностью, занятой, как указано выше, используемые в соединениях формулы (I), не относятся к следующим веществам: эналаприл (ингибиторы АСЕ) и диклофенак (NSAID).
В формуле -ТB-Х2-О- предшественника соединения В, который соответствует условиям теста 4А и не соответствует условиям теста 5, можно использовать соединения, в которых Х2 представляет собой радикал R1B-X-R2B, где R1B и R2B, одинаковые или разные, представляют собой линейные или разветвленные C1-C6 алкилены, и Х как определено выше.
Другими примерами предшественников соединений В являются: 1,4-бутандиол: ОН-(СН2)4-ОН, 6-гидроксигексановая кислота: ОН-(CH2)5-COOH, 4-гидроксимасляная кислота: ОН-(СН2)3-СООН, N-метилдиэтаноламин: ОН-(CH2)2-N(СН3)-(СН2)2-ОН, диэтиленгликоль: ОН-(СН2)2-O-(CH2)2-ОН, тиодиэтиленгликоль: ОН-(СН2)2-S-(СН3)2-ОН; предпочтительно предшественником В являются N-метилдиэтаноламин, диэтиленгликоль, тиодиэтиленгликоль.
Соединения, являющиеся предшественниками лекарственных веществ и В, получают в соответствии с методами, известными из уровня техники и описанными, например, в "The Merck Index, 12a Ed.", (1996), включенном сюда в качестве ссылки.
Тесты (1-5) более подробно состоят в следующем:
Тест 1 (NEM): оценка желудочно-кишечных повреждений вследствие окислительного стресса, вызванного свободными радикалами, образовавшимися после введения N-этилмалеимида (NEM) (Н. G. Utiey, F. Bernheim, P. Hochstein "Effects of sulphydril reagents on peroxidation in microsomes" Archiv. Biochem. Biophys. 118, 29-32, 1967).
Животных (крыс) распределяют по следующим группам (по 10 животных в группе):
А) Контрольные группы:
группа 1°: обработка: только носитель (водная суспензия 1% мас./об.
карбоксиметилцеллюлозы, доза: 5 мл/кг, если лекарственное вещество вводят через рот, или физиологический раствор, если его вводят парентерально, т.е. подкожно, внутрибрюшинно, внутривенно или внутримышечно),
группа 2°: обработка: носитель, как определено выше, + NEM,
В) Опытные группы, которым вводят лекарственное вещество:
группа I: обработка: носитель + лекарственное вещество,
группа II: обработка: носитель + лекарственное вещество + NEM.
Способы введения являются известными для данного лекарственного вещества; это может быть оральный, подкожный, внутрибрюшинный, внутривенный или внутримышечный способ.
Доза NEM составляет 25 мг/кг в физиологическом растворе (подкожное введение), а лекарственное вещество вводят спустя 1 час в суспензии носителя, в виде единичной дозы, соответствующей максимальной дозе, или наивысшей дозе, переносимой животными из группы крыс, не подвергшихся предварительному воздействию NEM, то есть самой высокой дозе, которую можно вводить указанной группе, чтобы она не вызывала у животных явных токсических явлений, которые можно ясно распознать по заметным симптомам. Животных умерщвляют по истечении 24 часов, после чего приступают к оценке повреждений желудочно-кишечной слизистой оболочки.
Лекарственное вещество соответствует условиям теста 1, то есть может быть использовано для получения соединений общей формулы (I), если у группы крыс, обработанных NEM + носителем -г лекарственным веществом, наблюдаются желудочно-кишечные повреждения, или отмеченные у указанной группы желудочно-кишечные повреждения проявляются в большей степени, чем те, которые наблюдаются у группы, обработанной только носителем, или у группы, обработанной носителем и лекарственным веществом, или у группы, обработанной носителем и NEM, даже если фармакотерапевтическая эффективность лекарственного вещества, исследованная с использованием специальных тестов, существенно не снижается.
Тест 2 (CIP): Показатели защиты эндотелиальных клеток от окислительного стресса, вызванного гидропероксидом кумола (CIP).
Эндотелиальные клетки человека из пупочной вены получают в соответствии со стандартной процедурой. Свежие пупочные вены заполняют 0,1 мас.% раствором коллагеназы и инкубируют при 37°С в течение 5 минут.
После этого вены заливают средой М 199 (GIBCO, Grand Island, NY) с рН 7,4, к которой впоследствии добавляют другие вещества, как описано в примерах. Собирают клетки из перфузата при помощи центрифугирования и выращивают в культуральных колбах Т-75, предварительно обработанных фибронектином человека. Затем выращивают клетки в той же самой среде, к которой дополнительно добавляют 10 нг/мл фактора роста гипоталамуса крупного рогатого скота. Когда клетки первичной клеточной культуры (т.е. полученные напрямую ex-vivo) образуют слитный клеточный монослой (примерно 8000000 клеток на одну колбу), рост культуры останавливают, слои промывают и обрабатывают трипсином. Переносят клеточные суспензии в лунки 24-луночного планшета для клеточных культур, к половине из которых затем добавляют ту же самую культуральную среду, содержащую лекарственное вещество в концентрации 10-4 М, после чего выращивают клетки в термостате при 37°С и при постоянной влажности. Только клетки из указанных первых субкультур используют для экспериментов с гидропероксидом кумола (CIP). Клетки идентифицируют как эндотелиальные клетки путем морфологического анализа, а также по их специфической иммунологической реакции на фактор VIII; указанные культуры не содержат каких-либо загрязнений из миоцитов или фибробластов.
Перед началом теста клеточную культуральную среду удаляют и клеточные слои осторожно промывают физиологическим раствором при температуре 37°С. Лунки культурального планшета затем инкубируют в течение 1 часа с CIP, содержащимся в культуральной среде в концентрации 5 мМ. Оценку клеточных повреждений (апоптоза) проводят путем определения процентного изменения фрагментации ДНК относительно контрольной группы (которую обрабатывали только CIP), оценивая изменение флуоресценции при длинах волн 405-450 нм. Для каждого образца опыт повторяют пять раз.
Лекарственное вещество соответствует условиям теста, то есть может быть использовано для получения соединений общей формулы (I), если статистически значимое ингибирование апоптоза (клеточных повреждений), вызванного CIP по сравнению с группой, которую обрабатывали только CIP, не зарегистрировано при р<0,01.
Тест 3 (L-NAME): оценка эндотелиальных дисфункций, вызванных введением L-NAME (метиловый эфир Nw-нитро-L-аргинина), J. Clin. Investigation 90, 278-281, 1992.
Эндотелиальные дисфункции оценивают путем определения повреждений слизистых оболочек желудочно-кишечного тракта, повреждений печени, а также повышения кровяного давления, вызванных введением L-NAME.
Животных (крыс) делят на группы в соответствии со схемой, изложенной ниже. Группе, получающей L-NAME, вводят в течение 4 недель указанное соединение, растворенное в питьевой воде, в концентрации 400 мг/л. Выделяют следующие группы (по 10 животных в каждой):
A) Контрольные группы:
группа 1°: только носитель (водная суспензия 1 мас.%/об. карбоксиметилцеллюлозы, доза: 5 мл/кг, если лекарственное вещество вводят через рот, физиологический раствор, если его вводят парентерально),
группа 2°: носитель + L-NAME,
B) Опытные группы - которым вводят лекарственное вещество:
группа 3°: носитель + лекарственное вещество,
группа 4°: носитель + лекарственное вещество + L-NAME.
Способы введения являются известными для данного лекарственного вещества, и это может быть оральный или подкожный, внутрибрюшинный, внутривенный или внутримышечный способ. Лекарственное вещество вводят в дозе, соответствующей максимальной дозе, переносимой животными из группы крыс, которым не вводили L-NAME, то есть самой высокой дозе, которую можно вводить, чтобы она не вызывала у животных явных токсических явлений, которые можно распознать по заметным симптомам. Лекарственное вещество вводят один раз в день в течение 4 недель.
К концу четвертой недели обработки прекращают доступ животных к воде, и по истечении 24 часов их умерщвляют.
За один час до умерщвления измеряют кровяное давление, и его повышение принимают в качестве критерия оценки повреждения сосудистого эндотелия. Повреждения слизистой оболочки желудка оценивают так, как проиллюстрировано в тесте 1 (см. Пример F1). Повреждения печени определяют путем оценки содержания глутамат-пируват трансаминазы (увеличение уровня GPT) после умерщвления.
Лекарственное вещество соответствует условиям теста 3, то есть может быть использовано для получения соединений общей формулы (I), если в группах крыс, обработанных L-NAME + лекарственным веществом + носителем, наблюдаются более существенные повреждения печени (GPT), и/или более существенные повреждения желудка, и/или более существенные повреждения сердечно-сосудистой системы (кровяное давление) по сравнению с группой, обработанной только носителем, или группой, обработанной носителем и лекарственным веществом, или группой, обработанной носителем и L-NAME, даже если фармакотерапевтическая эффективность лекарственного вещества, исследованная с использованием специальных тестов, существенно не снижается.
В условиях, указанных для вышеописанных in vivo тестов 1 и 3, терапевтический индекс лекарственного вещества снижается вследствие того, что обычные дозы, при которых это лекарственное вещество может быть эффективным, уже не переносятся.
Тест 4А проводят в соответствии с методом, описанным в R. Maffei Facino, M. Carini G. Aldini, M.T. Calloni, Drugs Exptl. Clin. Res. XXIII (5/8) 157-165, 1997. Тест 4А представляет собой in vitro тест, при котором эритроциты, полученные стандартным способом от мужских особей крыс Wister (Charles River) выдерживают в течение 4 дней при 4°С в суспензии в физиологическом растворе, забуференном при рН 7,4 с помощью фосфатного буфера. По окончании указанного периода отбирают аликвоту суспензии и центрифугируют при 1000 об./мин в течение 5 минут. 0,1 мл центрифугированных эритроцитов разбавляют до 50 мл натрий-фосфатным буфером с рН 7,4, получая 0,2% об. суспензию эритроцитов. К 5 аликвотам по 3,5 мл разбавленной суспензии добавляют 0,1-0,3 мл спиртового раствора гидропероксида кумола до достижения концентрации 270 мкМ и затем инкубируют их при 37°С. Это соединение вызывает лизис клеток, и за счет этого увеличивается мутность суспензии. За лизисом клеток следят при помощи турбидиметрии при 710 нм. Проводя измерение оптической плотности (или пропускания) через каждые 30 минут, определяют момент времени (Тmax), когда мутность суспензии максимальна, что соответствует максимальному количеству клеток в суспензии, подвергшихся лизису. Тmax принимают за время, соответствующее 100% лизиса эритроцитов. Для определения ингибирующего воздействия предшественников В на гемолиз, вызванный гидропероксидом кумола, добавляют по 0,1-0,2 мл раствора каждого из исследуемых соединений-предшественников В в этаноле к 3,5 мл аликвотам центрифугированной суспензии эритроцитов (5 образцов для каждого соединения) до достижения конечной концентрации 2 мМ, и полученную суспензию предварительно выдерживают в течение 30 минут.
Затем добавляют гидропероксид кумола в количестве, достаточном для достижения такой же конечной молярной концентрации, как определено ранее, и ингибирование соединением гемолиза в процентах при Tmax определяют из отношения абсорбции исследуемого образца суспензии, содержащей соответственно эритроциты, предшественник В и гидропероксид кумола, и абсорбции суспензии, содержащей эритроциты и гидропероксид кумола, умноженного на 100; предшественник соединения В соответствуют условиям теста 4А, если он ингибирует гемолиз, вызванный гидропероксидом кумола, более чем на 15%.
Тест 5 представляет собой колориметрический тест, в котором 0,1 мл аликвоты 10-4 М метанольных растворов тестируемых соединений добавляют в пробирки, содержащие раствор, полученный путем смешивания 0,2 мл 2 мМ водного раствора дезоксирибозы, 0,4 мл 100 мМ фосфатного буфера с рН 7,4, и 0,1 мл 1 мМ раствора FeII(NH4)2(SO4)2 в 2 мМ HCl. Затем пробирки выдерживают при 37°С в течение 1 часа. Затем в каждую пробирку добавляют 0,5 мл 2.8% водного раствора трихлоруксусной кислоты и 0,5 мл 0,1 М водного раствора тиобарбитуровой кислоты в указанном порядке. Прозрачный контрольный раствор готовят путем добавления в пробирку, содержащую только указанный выше водный раствор реагентов, 0,1 мл метанола. Пробирки закрывают и нагревают на масляной бане при 100°С в течение 15 минут. Растворы окрашиваются в розовый цвет, причем интенсивность окрашивания пропорциональна количеству дезоксирибозы, подвергшейся радикальному окислительному расщеплению. Растворы охлаждают до комнатной температуры и затем измеряют их поглощение при 532 нм, сравнивая с его прозрачным контрольным раствором. Ингибирование образования радикалов, вызываемое предшественником В относительно образования радикалов солью FeII определяют при помощи следующей формулы:
(1-As/Ac)×100
где As и Ас являются соответственно величинами поглощения раствора, содержащего тестируемое соединение и соль железа, и раствора, содержащего только соль железа; соединение соответствует условиям теста 5 в том случае, если ингибирование предшественником В образования радикалов, выраженное в процентах, как указано выше, больше или равно 50%.
Неожиданно выяснилось, что продукты по изобретению формулы (I) в состоянии окислительного стресса обладают улучшенным терапевтическим индексом по сравнению с лекарственными веществами-предшественниками. Соединения формулы (I) по изобретению, где предшественник соединения В соответствует условиям теста 4А, но не соответствует условиям теста 5, можно использовать, как указано выше, в качестве лекарственных веществ для терапии состояний окислительного стресса средней силы. В этом смысле в соответствии с настоящим изобретением объектом лечения являются состояния окислительного стресса средней силы.
Вышеупомянутые тесты будут проиллюстрированы при помощи следующих соединений (см. Таблицы):
Тест 1: лекарственное вещество-предшественник: индометацин
- Максимально допустимая вводимая доза для крыс: 7,5 мг/кг перорально. При введении более высоких доз проявляется токсичность, характеризующаяся энтеропатией, дрожью, угнетением сознания вплоть до смертельного исхода (в течение 24 часов).
- У группы крыс, обработанных NEM и индометацином в указанной выше дозе, наблюдались желудочно-кишечные повреждения.
Поскольку индометацин вызывает желудочно-кишечные повреждения у крыс, обработанных NEM, он соответствует условиям теста 1.
Тест 2: лекарственные вещества-предшественники: индометацин, парацетамол и мезаламин
Индометацин и парацетамол соответствуют условиям теста 2, поскольку ингибирование клеточных повреждений (апоптоза), вызванных CIP, незначительно отличается от подобного ингибирования в контрольных группах.
Следовательно, вышеупомянутые лекарственные вещества могут быть использованы в качестве лекарственных веществ для получения соединений (I) настоящего изобретения.
В противоположность этому, мезаламин не соответствует условиям теста 2, поскольку ингибирует апоптоз, вызванный CIP. Следовательно, в соответствии с тестом 2 мезаламин не мог быть использован в качестве предшественника для получения соединений (I) настоящего изобретения. Однако было обнаружено, что мезаламин, подвергнутый тесту 1, вызывает желудочно-кишечные повреждения.
Следовательно, мезаламин также может быть использован в качестве предшественника для получения соединений (I) настоящего изобретения.
Тест 3 (L-NAME) лекарственные вещества-предшественники: парацетамол, симвастатин, омепразол
Парацетамол и симвастатин соответствуют условиям теста 3, поскольку их действие наносит повреждения желудку и печени в большей степени, чем действие как системы L-NAME + носитель, так и системы лекарственное вещество + носитель.
Следовательно, они могут быть использованы в качестве предшественников для получения соединений (I) настоящего изобретения.
В противоположность этому было обнаружено, что омепразол не наносит повреждений ни желудку, ни печени, а также не влияет на кровяное давление. В соответствии с тестом 3 омепразол не мог быть использован в качестве предшественника для получения соединений (I) настоящего изобретения.
Тест 4А (тест для предшественника В)
N-метилдиэтаноламин на 54,4% (Таблица V) ингибирует гемолиз, вызванный гидропероксидом кумола. Следовательно, он соответствует условиям теста 4А и может быть использован в качестве предшественника В, если он не соответствует условиям теста 5.
Диэтаноламин не ингибирует гемолиз, вызванный гидропероксидом кумола и не соответствует условиям теста 4А. Следовательно, это соединение не может быть использовано в качестве предшественника В,
Тест 5 (тест для предшественника В)
В Таблице III, относящейся к этому тесту, показано, что N-метилдиэтаноламин не соответствует условиям теста 5, так как он не ингибирует образование радикалов из FeII. Следовательно, это он может быть использован в качестве предшественника В.
Соединения формулы (I) по изобретению можно превратить в соответствующие соли. Например, существует следующий способ образования солей. Если в молекуле соединения формулы (I) присутствует атом азота, достаточно основный для солеобразования, соответствующие соли указанных соединений получают путем их взаимодействия в органическом растворителе, таком как ацетонитрил или тетрагидрофуран, с эквимолярным количеством соответствующей органической или неорганической кислоты.
Примеры органических кислот: щавелевая, винная, малеиновая, янтарная, лимонная кислоты.
Примеры неорганических кислот: азотная, соляная, серная, фосфорная кислоты.
Производные по изобретению могут быть использованы согласно терапевтическим показаниям, при которых применяются лекарственные вещества-предшественники, что позволяет добиться преимуществ, которые приводятся далее в качестве примера для некоторых групп этих соединений:
- Противовоспалительные лекарственные вещества NSAIDs: соединения по изобретению отличаются очень хорошей переносимостью и эффективностью, даже в случаях, когда организм ослаблен и находится в состоянии окислительного стресса. Указанные лекарственные вещества могут быть также использованы при патологиях, при которых воспаление играет важную патогенетическую роль, например, таких как рак, астма, инфаркт миокарда, но не ограничиваясь ими.
- Адренэргические блокаторы α- или β-типа: спектр действия соединений формулы (I) становится более широким, чем у исходных лекарственных веществ: к непосредственному воздействию на гладкую мускулатуру добавляется ингибирование нервных бета-адренэргических сигналов, управляющих сокращением кровеносных сосудов. Уменьшается количество побочных эффектов (затрудненное дыхание, сокращение бронхов), воздействующих на дыхательный аппарат.
- Антитромботические лекарственные вещества: повышается антитромбоцитарная активность, и в случае производных аспирина также улучшается желудочная переносимость.
- Бронходилататоры и лекарственные вещества, действующие на холинэргическую систему: снижаются побочные эффекты, воздействующие на сердечно-сосудистый аппарат (тахикардия, гипертония).
- Отхаркивающие в муколитические лекарственные вещества: улучшается желудочно-кишечная переносимость.
- Дифосфонаты: токсичность по отношению к желудочно-кишечному тракту существенно снижается.
- Ингибиторы фосфодиэстеразы (PDE) (бронходилататоры): улучшается терапевтическая эффективность при тех же дозах; следовательно, становится возможным, используя соединения по изобретению, введение пониженных доз лекарственного вещества и снижение побочных эффектов.
- Антилейкотриеновые лекарственные вещества: улучшенная эффективность.
- АСЕ-ингибиторы: улучшенная терапевтическая эффективность и пониженные побочные эффекты (затрудненное дыхание, кашель), воздействующие на дыхательный аппартат.
- Противодиабетические лекарственные вещества (повышающие чувствительность к инсулину и снижающие уровень глюкозы), антибиотики, противовирусные, противоопухолевые, противоколиковые лекарственные вещества, лекарственные вещества против слабоумия:
улучшенная эффективность и/или переносимость.
Лекарственные вещества, которые могут быть использованы в качестве предшественников соединений по изобретению, - это все те соединения, которые соответствуют условиям по меньшей мере одного их вышеупомянутых тестов 1, 2, 3.
Эффективность соединений настоящего изобретения в качестве лекарственных веществ для использования в состояниях окислительного стресса средней силы также была продемонстрирована в фармакологическом тесте, в котором указанные соединения могли ингибировать цитолитическое воздействие, которое оказывает пероксид водорода на эндотелиальные клетки из пупочной вены человека. Эндотелиальная клетка является одной из первых, которая страдает от патологических процессов (Pathophysiology: the biological basis for disease in adults and children, McCance&Huether, 1998, p. 1025), а пероксид водорода является мягким окислителем и считается основным медиатором при патологиях, связанных с окислительным стрессом (В. Halliwell, J. Gutteridge "Free Radicals in Biology and Medicine", p. 416, 1993). Эффективность нейтрализации собственного цитолитического воздействия считается существенным признаком фармакологической активности соединений для использования в состояниях окислительного стресса (В.Halliwell, J.Gutteridge "Free Radicals in Biology and Medicine", p. 416, 1993).
Соединения формулы (I) получают с помощью реакций, описанных ниже.
В случае, когда реакционноспособная функциональная группа (например, -СООН, -ОН) лекарственного вещества образует ковалентную связь, например сложноэфирную, амидную, простую эфирную, перед получением указанных соединений эта функциональная группа может быть регенерирована при помощи методов, хорошо известных из уровня техники.
Реакции, используемые для получения соединений формулы (I), представляют собой взаимодействия, приводящие к образованию, например, сложноэфирной, амидной или тиоэфирной связи, хорошо известные специалистам в данной области.
Если в двух соединениях, вступающих в реакцию, присутствуют другие функциональные группы СООН и/или НХ, где Х определен выше, они должны быть защищены перед проведением реакции в соответствии с методами, хорошо известными из уровня техники; например, как описано в публикации Th. W. Greene: "Protective groups in organic synthesis", Harward University Press, 1980.
Соединения формулы (I), где s=2, получают, как указано ниже.
IA) - Лекарственное вещество имеет общую формулу R-COOH, а функциональная группа предшественника соединения В, которая связана с карбоксильной функцией лекарственного вещества, имеет формулу XZ, где Х такой, как указано выше, a Z является Н, или функциональной группой ОН, или атомом галогена, одновременно являясь реакционноспособной группой в составе предшественника соединения В для реакции нитрования.
Общая схема синтеза в случае, когда в предшественнике соединения В присутствует ОН-группа, включает в себя первоначальное образование галогенангидрида кислоты R-COHal (Hal=Cl, Br) и его последующее взаимодействие с НХ-группой предшественника соединения В:
RCOOH→RCOHal+Н-Х-Х2-СООН→R-Т1-ТB-X2-СООН (IA.1)
где Х2, T1, ТB такие, как указано выше.
Ацилгалогенид RCOHal получают в соответствии с методами, известными из уровня техники, например при помощи тионил- или оксалилхлорида, РIII или Pv галогенидов, проводя реакцию в инертных растворителях, например, таких как толуол, хлороформ, DMF и т.д. Затем ацилгалогенид вводят во взаимодействие с НХ группой соединения-предшественника В, проводя реакцию в инертных растворителях, например, таких как толуол, тетрагидрофуран, хлороформ и т.д. при температуре в диапазоне от 0°С до 25°С.
В качестве альтернативы предыдущему синтезу лекарственное вещество-предшественник, имеющее формулу R-COOH, может быть обработано агентом, активирующим карбоксильную группу, выбранным из N,N'-карбонилдиимидазола (CDI), N,N'-дициклогексилкарбодиимида, в инертном растворителе, таком, например, как толуол, THF, хлороформ и т. д., при температуре в диапазоне от -5°С до +50°С. Полученное соединение вводят во взаимодействие in situ с предшественником В, после предварительной защиты ОН-функции, например, ацетильной группой, регенерируя исходную функцию на финальной стадии синтеза при помощи методов, хорошо известных из уровня техники. Синтез проводят по следующей схеме:
R-COOH+CDI+НХ-Х2-OG→R-Т1-ТB-X2-OG→R-Т1-ТB-X2-OH (IA.1)
где X2, T1, ТB такие, как указано выше, a G является защитной группой ОН-функции.
Соединение, имеющее формулу (IA.1), затем подвергают реакции галогенирования, например, такими агентами, как PBr3, PCl5, SOCl2, PPh3 и I2 в инертном растворителе, таком как толуол, тетрагидрофуран, хлороформ и т.д., при температуре в диапазоне от -5°С до +50°С. Галогенпроизводное далее взаимодействует с AgNO3 в органическом растворителе, таком как ацетонитрил, тетрагидрофуран, при температуре в диапазоне 25°С-80°С. Синтез проводят по следующей схеме:
R-T1-TB-X2-OH→R-T1-TB-X2-Hal→R-T1-TB-X2-ONO2 (IA.2)
В качестве альтернативы, если Х2 является линейным С4 алкилом, то соответствующая кислота R-COOH взаимодействует с трифенилфосфином в присутствии галогенирующего агента, такого как CBr4 или N-бромсукцинимид в тетрагидрофуране, и полученное соединение (IA.2), где Х2 является бутиленом, нитруют, как указано выше.
С другой стороны, возможно превратить кислоту R-COOH в ее натриевую соль при помощи методов, известных из уровня техники, и затем ввести ее во взаимодействие с галогенпроизводным, имеющим формулу Hal-X2-R3, где R3=ОН, Hal, в инертном растворителе, таком как тетрагидрофуран, хлороформ и т.д., при температуре в диапазоне -5°С - +25°С. Если R3=Hal, то полученное производное нитруют, как указано выше. Реакционная схема такова:
R-COOH→R-COONa+Hal-Х2-R3→R-T1-TB-X2-R3→R-T1-TB-X2-ONO2
IIA) - Лекарственное вещество имеет общую формулу R-XH, а функциональная группа предшественника соединения В, которая связана с НХ-функцией лекарственного вещества, является карбоксильной группой, где Х такой, как указано выше, а функциональная группа ОН или атом галогена одновременно присутствуют в составе предшественника соединения В в качестве реакционноспособных групп для реакции нитрования.
Общая схема синтеза включает реакцию кислоты HOOC-X2-R4, где R4 является Hal или OG, где G - подходящая защитная группа, с активирующим агентом, как указано в IA), с последующим взаимодействием с НХ-группой лекарственного вещества.
HOOC-X2-R4+CDI+HX-R→R-T1-TB-X2-R4 (IIA.1)
где R4, T1, ТB, Х2 такие, как указано выше.
Полученное соединение (IIA.1) далее превращают в соответствующее нитропроизводное, как указано в IA). В случае, если присутствует заместитель OG, то удаляют защитную группу при помощи известных методов.
В качестве альтернативы предыдущему синтезу лекарственное вещество R-OH вводят во взаимодействие с ацилгалогенидом, имеющим формулу Hal-Х2-COHal в условиях реакции, описанных в IA), а полученное галогенпроизводное далее нитруют, как указано выше:
HalOC-Х2-Hal+HX-R→R-T1-TB-X2-Hal→R-T1-TB-X2-ONO2
где Х2, T1, ТB такие, как указано выше.
Соединения формулы I, где s=1, получают при помощи методов, описанных далее.
IB) - Лекарственное вещество имеет общую формулу R-COOH, а функциональная группа предшественника соединения В, которая связана с карбоксильной функцией лекарственного вещества, имеет формулу XZ, где Х такой, как указано выше, а 7 представляет собой Н, и предшественник соединения В содержит функциональную группу ОН, или атом галогена, являющиеся реакционноспособными группами для реакции нитрования.
Соединение, имеющее формулу R-T1-TB-X2-OH (IA.1), полученное, как описано в IA), превращают в нитрозопроизводное путем реакции с нитритом натрия в воде в присутствии соляной кислоты, в соответствии с процедурами, известными из уровня техники.
R-T1-TB-X2-OH+NaNO2→R-T1-TB-X2-ONO
IIB) - Лекарственное вещество имеет общую формулу R-XH, а функциональная группа предшественника соединения В, которая связана с НХ-функцией лекарственного вещества, является карбоксильной группой, где Х такой, как указано выше. Синтетическая схема подобна описанной в IIA).
Соединение, имеющее формулу R-T1-TB-X2-R4 (IIA.1), полученное, как описано в IIA), превращают в нитрозопроизводное методом, описанным в IIA).
Соединения, являющиеся объектом настоящего изобретения, были введены в состав соответствующих фармацевтических композиций для парентерального, перорального и местного применения в соответствии с хорошо известными из уровня техники методами, вместе с обычными эксципиентами; см. например "Remington's Pharmaceutical Sciences 15a Ed."
Молярное количество активного начала в этих композициях является таким же или пониженным по сравнению с количеством используемого соответствующего лекарственного вещества-предшественника.
Дневные вводимые дозы являются такими же или в некоторых случаях пониженными по сравнению с дневными дозами лекарственных веществ-предшественников. Дневные дозы могут быть найдены в соответствующих публикациях на эту тему, например в "Physician's Desk Reference".
Следующие примеры приведены с целью иллюстрации настоящего изобретения и не должны рассматриваться, как ограничивающие его объем.
Пример 1
Получение 4'-ацетиламинофенилового эфира 4-нитроксибутановой кислоты
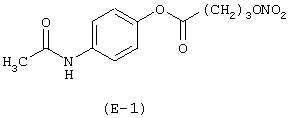
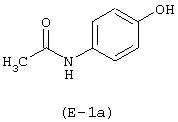
Лекарственным веществом является парацетамол, имеющий формулу:
Предшественником соединения В является 4-гидроксибутановая кислота.
а) Получение 4'-ацетиламинофенилового эфира 4-бромбутановой кислоты
К раствору 4-бромбутановой кислоты (4,6 г, 27,6 ммоль) в хлороформе (45 мл) и N,N'-диметилформамиде (20 мл) добавляют парацетамол (4,17 г, 27,6 ммоль), N,N'-дициклогексилкарбодиимид (8,42 г, 40,8 ммоль) и 4-диметиламинопиридин (0,15 г, 1,25 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 72 часов, фильтруют и упаривают в вакууме. К неочищенному реакционному материалу добавляют этилацетат, после чего промывают насыщенным водным раствором NaCl и затем водой. Органическую фазу высушивают сульфатом натрия и затем упаривают в вакууме. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 4/6 (об./об.). Получают 5,33 г продукта в виде белого твердого вещества. Т. пл. 108-110°С.
б) Получение 4'-ацетиламинофенилового эфира 4-нитроксибутановой кислоты
К раствору 4'-ацетиламинофенилового эфира 4-бромбутановой кислоты (5,33 г, 17,8 ммоль) в ацетонитриле (80 мл) добавляют нитрат серебра (4,56 г, 26,9 ммоль). Реакционную смесь выдерживают без доступа света при температуре 80°С в течение 7 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 4/6. Получают 4,1 г продукта в виде белого твердого вещества. Т. пл. 80-83°С.
Пример 2
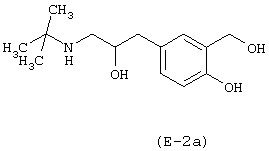
Получение 4-гидрокси-3-(4-нитроксибутаноилоксиметил)-α-[(трет-бутиламино)метил]бензилового спирта

Лекарственным веществом-предшественником является сальбутамол, имеющий формулу:
Предшественником соединения В является 4-гидроксибутановая кислота.
Соединение (Е-2) синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 21%.
Пример 3
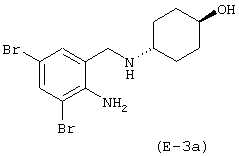
Получение 4-[(2-амино-3,5-дибромфенил)метиламино]-транс-циклогексилового эфира 4-нитроксибутановой кислоты
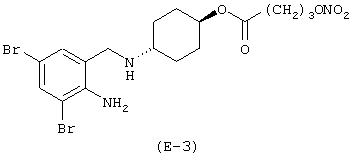
Лекарственным веществом-предшественником является амброксол
Предшественником соединения В является 4-гидроксибутановая кислота.
а) Получение 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил) метиламино]-транс-циклогексанола
К раствору амброксола (5 г, 13,22 моль) в диоксане (35 мл) и воде (50 мл) добавляют триэтиламин (3,31 мл, 23,7 ммоль) и ди-трет-бутилдикарбонат (3,46 г, 15,86 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 24 часов, а затем концентрируют при пониженном давлении. К остатку добавляют порциями 1% раствор HCl до достижения рН 7, затем раствор экстрагируют этилацетатом. Высушивают органическую фазу сульфатом натрия и упаривают в вакууме. Получают 4-[(2-трет-бутилоксикарбониламино-3,5 -дибромфенил)метиламино]-транс-циклогексанол, который используют на следующей стадии без дальнейшей очистки.
б) Получение 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил) метиламино]-транс-циклогексилового эфира 4-нитроксибутановой кислоты
Соединение синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 57%.
с) Получение 4-[(2-амино-3,5-дибромфенил)метиламино]-транс-циклогексилового эфира 4-нитроксибутановой кислоты
К раствору 4-[(2-трет-бутилоксикарбониламино-3,5-дибромфенил) метиламино]-транс-циклогексилового эфира 4-нитроксибутановой кислоты (3,5 г, 5,74 ммоль) в этилацетате (100 мл), охлажденному до 0°С, добавляют 5N раствор HCl в этилацетате (5,95 мл). Перемешивают раствор при 0°С в течение 5 часов, затем фильтруют. Полученное твердое вещество суспендируют в этилацетате и промывают органический слой 5% раствором карбоната натрия. Промывают органическую фазу водой, высушивают сульфатом натрия и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1 (об./об.). Получают 4-[(2-амино-3,5-дибромфенил)метиламино]-транс-циклогексиловый эфир 4-нитроксибутановой кислоты. Выход: 31%.
Пример 4
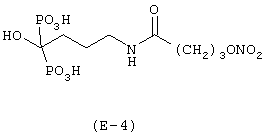
Получение [4-[4-нитроксибутироил]амино-1-гидроксибутилиден]-бис-фосфорной кислоты
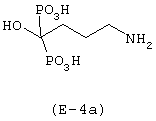
Лекарственным веществом-предшественником является алендроновая кислота, имеющая формулу:
Предшественником соединения В является 4-гидроксибутановая кислота.
Соединение синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 11%
Пример 5
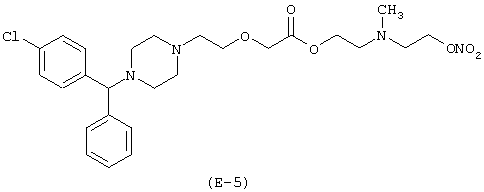
Получение [N-метил-N-(2-нитроксиэтил)]-2-аминоэтилового эфира [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]уксусной кислоты
Лекарственным веществом-предшественником является цетиризин
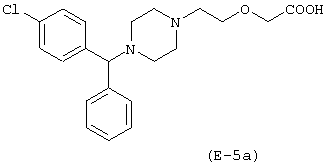
Предшественником соединения В является N-метилдиэтаноламин, имеющий формулу НО-(СН2)2-N(СН3)-(СН2)2-ОН.
а) Получение [N-метил-N-(2-гидроксиэтил)]-2-аминоэтилового эфира[2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси] уксусной кислоты
К раствору цетиризина (5 г, 12,85 ммоль) в N,N-диметилформамиде (5 мл) и толуоле (50 мл), охлажденному до 0°С, медленно добавляют оксалилхлорид (1,1 мл, 25,7 ммоль). Перемешивают смесь при комнатной температуре в течение 12 часов, после чего упаривают в вакууме. К полученному неочищенному продукту, растворенному в тетрагидрофуране (40 мл), добавляют N-метилдиэтаноламин (4,05 г, 38,55 ммоль) и перемешивают смесь при комнатной температуре в течение 6 часов. Упаривают реакционную смесь при пониженном давлении. К остатку приливают этилацетат и промывают раствор водой. Высушивают органическую фазу сульфатом натрия и упаривают. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 3/7 (об./об.). Получают [N-метил-N-(2-гидроксиэтил)]-2-аминоэтиловый эфир [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси] уксусной кислоты.
б) Получение [N-метил-N-(2-хлорэтил)]-2-аминоэтилового эфира [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси] уксусной кислоты
К раствору [N-метил-N-(2-гидроксиэтил)]-2-аминоэтилового эфира [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]уксусной кислоты (3,8 г, 7,75 ммоль) в хлороформе (70 мл), охлажденному до 0°С, добавляют тионилхлорид (0,58 мл, 8,06 ммоль) в хлороформе (30 мл). Перемешивают раствор при 0°С в течение 30 минут, а затем при 40°С в течение 6 часов. После этого промывают реакционную смесь насыщенным водным раствором бикарбоната натрия, а затем водой. Органическую фазу, высушенную сульфатом натрия, упаривают при пониженном давлении. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3 (об./об.). Получают [N-метил-N-(2-хлорэтил)]-2-аминоэтиловый эфир [2-[4-[(4-хлорфенил) фенилметил]-1-пиперазинил]этокси]уксусной кислоты.
c) Получение [N-метил-N-(2-нитроксиэтил)]-2-аминоэтилового эфира [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси] уксусной кислоты
К раствору [N-метил-N-(2-хлорэтил)]-2-аминоэтилового эфира [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси] уксусной кислоты (2,3 г, 4,52 моль) в ацетонитриле (100 мл) добавляют нитрат серебра (1,53 г, 9,04 ммоль). Реакционную смесь выдерживают без доступа света при температуре 80°С в течение 48 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3 (об./об.). Получают [N-метил-N-(2-нитроксиэтил)]-2-аминоэтиловый эфир [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил] этокси]уксусной кислоты. Выход: 23%.
Пример 6
Получение 5-(нитрокси)этилоксиэтилового эфира 6-[D(-)-α-аминофенил-ацетамидо]пенициллановой кислоты
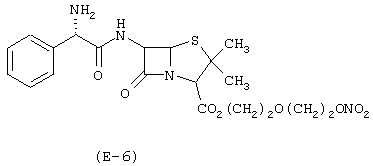
Лекарственным веществом-предшественником является ампициллин, имеющий формулу:
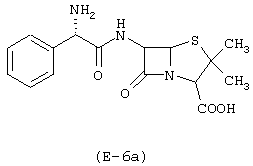
Предшественником соединения В является диэтиленгликоль.
а) Получение 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты
К раствору ампициллина (3 г, 8,58 ммоль) в смеси диоксана (18 мл) и воды (25 мл) добавляют триэтиламин (2,1 мл, 15,3 ммоль) и ди-трет-бутилдикарбонат (2,24 г, 10,29 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 24 часов, затем концентрируют при пониженном давлении. К остатку добавляют порциями 1% раствор HCl до достижения в водной фазе рН 7. Экстрагируют водную фазу этилацетатом. Высушивают органическую фазу сульфатом натрия и затем упаривают в вакууме. Получают 6-[D(-)-α-трет-бутилоксикарбониламино-фенилацетамидо]пенициллановую кислоту, которую используют в последующем синтезе без дополнительной очистки.
б) Получение 5-(гидрокси)этилоксиэтилового эфира 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо]пенициллановой кислоты
К раствору 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты (3,8 г, 8,58 ммоль) в смеси N,N-диметилформамида (5 мл) и толуола (40 мл), охлажденному до 0°С, медленно добавляют оксалилхлорид (0,74 мл, 17,16 ммоль). Перемешивают раствор при комнатной температуре в течение 12 часов, после чего упаривают его в вакууме. Полученный неочищенный продукт растворяют в тетрагидрофуране (70 мл), а затем к раствору добавляют этиленгликоль (2,45 мл, 25,7 ммоль). Полученный раствор перемешивают при комнатной температуре в течение 5 часов, а затем упаривают при пониженном давлении. К остатку приливают этилацетат и промывают органическую фазу водой. Далее органическую фазу высушивают сульфатом натрия и упаривают досуха. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 2/8 (об./об.). Получают 5-(гидрокси)этилоксиэтиловый эфир 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты.
с) Получение 5-(хлор)этилоксиэтилового эфира 6-[D(-}-α-трет-бутилоксикарбониламинофенилацетамидо]пенициллановой кислоты
К раствору 5-(гидрокси)этилоксиэтилового эфира 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты (3 г, 5,58 ммоль) в хлороформе (70 мл), охлажденному до 0°С, добавляют тионилхлорид (0,42 мл, 5,8 ммоль) в хлороформе (30 мл). Перемешивают раствор при 0°С в течение 30 минут, а затем при 40°С в течение 4 часов. После этого смесь промывают насыщенным раствором бикарбоната натрия, а затем водой. Высушивают органическую фазу сульфатом натрия и упаривают при пониженном давлении. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1 (об./об.). Получают 5-(хлор)этилоксиэтиловый эфир 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты.
d) Получение 5-(нитрокси)этилоксиэтилового эфира 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты
К раствору 5-(хлор)этилоксиэтилового эфира 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты (2,1 г, 3,77 ммоль) в ацетонитриле (100 мл) добавляют нитрат серебра (1,28 г, 7,54 ммоль). Реакционную смесь выдерживают без доступа света при температуре 80°С в течение 24 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1 (об./об.). Получают 5-(нитрокси)этилоксиэтиловый эфир 6-[D(-)-α-трет-бутилоксикарбонил-аминофенилацетамидо] пенициллановой кислоты.
е) Получение 5-(нитрокси)этилоксиэтилового эфира 6-[D(-)-α-аминофенилацетамидо] пенициллановой кислоты
К раствору 5-(нитрокси)этилоксиэтилового эфира 6-[D(-)-α-трет-бутилоксикарбониламинофенилацетамидо] пенициллановой кислоты (1,5 г, 2,57 ммоль) в этилацетате (100 мл), охлажденному до 0°С, добавляют 5N раствор HCl в этилацетате (2,67 мл). Перемешивают раствор при 0°С в течение 7 часов, затем фильтруют. Полученное твердое вещество суспендируют в этилацетате и промывают органический слой 5% мас./об. раствором карбоната натрия. Промывают органическую фазу водой, высушивают сульфатом натрия и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1 (об./об.). Получают 5-(нитрокси)этилоксиэтиловый эфир 6-[D(-)-α-аминофенилацетамидо] пенициллановой кислоты. Выход: 13%.
Пример 7
Получение 2-амино-1,9-дигидро-9-[[2-(4-нитроксибутироилокси)этокси]метил]-6Н-пурин-6-она
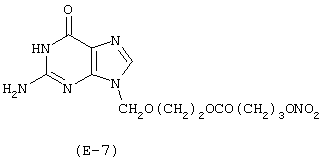
Лекарственным веществом-предшественником является ацикловир, имеющий формулу:
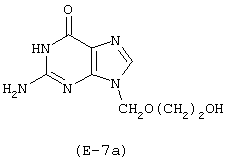
Предшественником соединения А является 4-гидроксибутановая кислота.
Соединение (E-7) синтезируют в соответствии с методикой, описанной в Примере 3. Выход: 14%.
Пример 8
Получение (8S-цис)-10-[(3-амино-2,3,6-тридезокси-α-L-ликсо-гексопиранозил)окси]-7,8,9,10-тетрагидро-6,8,11-тригидрокси-8-[(4-нитрокси-бутироилокси)ацетил]-1-метокси-5,12-нафтацендиона
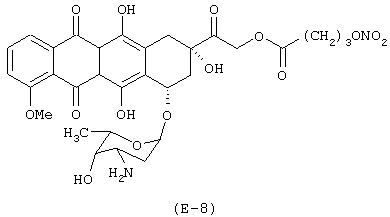
Лекарственным веществом-предшественником является доксорубицин, имеющий формулу (Е-8а)
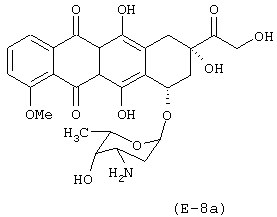
Предшественником соединения В является 4-гидроксибутановая кислота.
Соединение синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 7%.
Пример 9
Получение [1S-[1α,3α,7β,8β(2S*,4S*),8αβ]]-1,2,3,7,8,8α-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-(6-нитроксигексаноилокси)-6-оксо-2Н-пиран-2-ил]этил]-1-нафтилового эфира 2,2-диметилбутановой кислоты
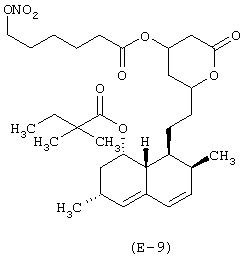
Лекарственным веществом-предшественником является симвастатин, имеющий формулу:
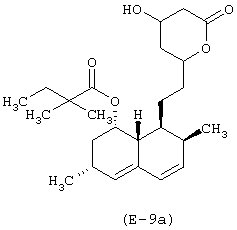
Предшественником мостиковой связи В является 6-гидроксигексановая кислота.
а) Получение [1S-[1α,3α,7β,8β(2S*,4S*),8αβ]]-1,2,3,7,8,8α-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-(6-бромгексаноилокси)-6-оксо-2Н-пиран-2-ил]этил]-1-нафтилового эфира 2,2-диметилбутановой кислоты
К раствору симвастатина (4 г, 9,56 ммоль) в хлороформе (50 мл) и N,N-диметилформамиде (20 мл) добавляют 6-бромгексановую кислоту (1,86 г, 9,56 ммоль), N,N'-дициклогексилкарбодиимид (1,97 г, 9,56 ммоль) и 4-диметиламинопиридин (52 мг, 0,43 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 24 часов, затем разбавляют хлороформом и промывают водой. Органическую фазу высушивают сульфатом натрия и упаривают при пониженном давлении. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1 (об./об.). Получают [1S-[1α,3α,7β,8β(2S*,4S*),8αβ]]-1,2,3,7,8,8α-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-(6-бромгексаноилокси)-6-оксо-2Н-пиран-2-ил]этил]-1-нафтиловый эфир 2,2-диметилбутановой кислоты.
b) Получение [1S-[1α,3α,7β,8β(2S*,4S*),8αβ]]-1,2,3,7,8,8α-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-(6-нитроксигексаноилокси)-6-оксо-2Н-пиран-2-ил]этил]-1-нафтилового эфира 2,2-диметилбутановой кислоты
К раствору [1S-[1α,3α,7β,8β(2S*,4S*),8αβ]]-1,2,3,7,8,8α-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-(6-бромгексаноилокси)-6-оксо-2Н-пиран-2-ил]этил]-1-нафтилового эфира 2,2-диметилбутановой кислоты (1 г, 1,67 ммоль) в ацетонитриле (60 мл) добавляют нитрат серебра (0,57 г, 3,35 ммоль). Реакционную смесь выдерживают без доступа света при температуре 80°С в течение 6 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1 (об./об.). Получают [1S-[1α,3α,7β,8β(2S*,4S*),8αβ]]-1,2,3,7,8,8α-гексагидро-3,7-диметил-8-[2-[тетрагидро-4-(6-нитроксигексаноилокси)-6-оксо-2Н-пиран-2-ил]этил]-1-нафтиловый эфир 2,2-диметилбутановой кислоты. Выход: 13%.
Пример 10
Получение теофиллинового эфира 6-(нитрокси)гексановой кислоты
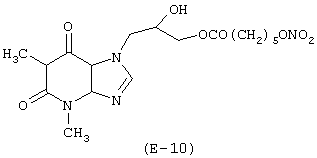
Лекарственным веществом-предшественником является дифиллин, имеющий формулу:
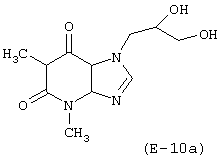
Предшественником соединения В является 6-гидроксигексановая кислота.
Соединение, имеющее формулу (Е-10), синтезируют в соответствии с методикой, описанной в Примере 9. Выход: 23%.
Пример 11
Получение 9-[4-(нитрокси)бутироиламино]-1,2,3,4-тетрагидроакридина
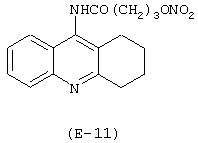
Лекарственным веществом-предшественником является такрин, имеющий формулу:
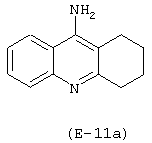
Предшественником соединения В является 4-гидроксибутановая кислота.
a) Получение 9-[4-бромбутироиламино]-1,2,3,4-тетрагидроакридина
К раствору такрина (4 г, 20,17 ммоль) в хлороформе (50 мл) и N,N-диметилформамиде (15 мл) добавляют 4-бромбутироилхлорид (3,5 мл, 30,25 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 6 часов, затем разбавляют хлороформом и промывают водой. Органическую фазу высушивают сульфатом натрия и упаривают при пониженном давлении. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 8/2 (об./об.). Получают 9-[4-бромбутироиламино]-1,2,3,4-тетрагидроакридин.
b) Получение 9-[4-(нитрокси)бутироиламино]-1,2,3,4-тетрагидроакридина
К раствору 9-[4-бромбутироиламино]-1,2,3,4-тетрагидроакридина (3,5 г, 10,56 ммоль) в ацетонитриле (150 мл) добавляют нитрат серебра (2,08 г; 12,68 ммоль). Реакционную смесь выдерживают без доступа света при температуре 80°С в течение 6 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 8/2 (об./об.). Получают 9-[4-(нитрокси)бутироиламино]-1,2,3,4-тетрагидроакридин. Выход: 33%.
Пример 12
Получение 5-(нитрокси)этилтиоэтилового эфира (S)-α-(2-хлорфенил)-6,7-дигидротиено[3,2-с]пиридин-5(4Н) уксусной кислоты
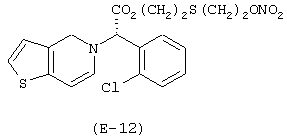
Лекарственным веществом-предшественником является клопидогрель, имеющий формулу:
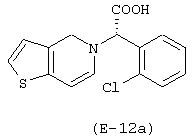
Предшественником соединения В является тиодиэтиленгликоль, имеющий формулу HO-(CH2)2-S-(CH2)2-OH.
Соединение (Е-12) синтезируют в соответствии с методикой, описанной в Примере 5, используя тиодиэтиленгликоль вместо диэтиленгликоля. Выход: 56%.
Пример 13
Получение 5-метокси-2-[[4-(4-нитроксибутироилокси)-3,5-диметил-2-пиридинил)метил]сульфинил]-1Н-бензимидазола
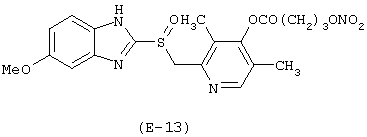
Лекарственным веществом-предшественником является деметиломепразол, имеющий формулу:
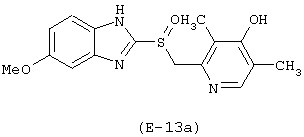
Предшественником соединения В является 4-гидроксибутановая кислота.
Соединение (E-13) синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 22%.
Пример 14
Получение [N-метил-N-(2-гидроксиэтил)1-2-аминоэтилового эфира 2-[(2,6-дихлорфенил)амино]фенилуксусной кислоты (Е-14)
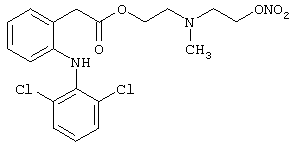
Лекарственным веществом-предшественником является диклофенак, имеющий формулу:
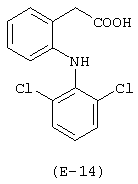
Предшественником соединения В является N-метилдиэтаноламин, имеющий формулу HO-(CH2)2-N(CH3)-(CH2)2-OH.
Соединение синтезируют в соответствии с методикой, описанной в Примере 5. Выход: 52%.
Пример 15
Получение 4-(нитрокси)бутилового эфира 3-(4-гидрокси-3-метоксифенил)-2-пропеновой кислоты
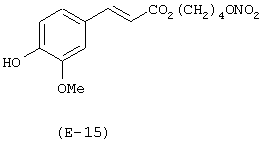
Лекарственным веществом-предшественником является феруловая кислота, имеющая формулу:
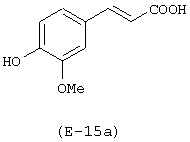
Предшественником соединения В является 1,4-бутандиол.
a) Получение 4-бромбутилового эфира 3-(4-гидрокси-3-метоксифенил)-2-пропеновой кислоты
К раствору феруловой кислоты (10 г, 51,51 ммоль) в тетрагидрофуране (400 мл) добавляют трифенилфосфин (27 г, 103 ммоль) и тетрабромметан (34,1 г, 103 ммоль). Перемешивают реакционную смесь при комнатной температуре в течение 4 часов, затем фильтруют и упаривают при пониженном давлении. Неочищенный продукт очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3 (об./об.). Получают 4-бромбутиловый эфир 3-(4-гидрокси-3-метоксифенил)-2-пропеновой кислоты.
b) Получение 4-(нитрокси)бутилового эфира 3-(4-гидрокси-3-метоксифенил)-2-пропеновой кислоты
К раствору 4-бромбутилового эфира 3-(4-гидрокси-3-метоксифенил)-2-пропеновой кислоты (2,72 г, 6,89 ммоль) в ацетонитриле (25 мл) добавляют нитрат серебра (1,48 г, 8,71 ммоль). Реакционную смесь выдерживают без доступа света при температуре 80°С в течение 7 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 7/3 (об./об.). Получают 4-(нитрокси)бутиловый эфир 3-(4-гидрокси-3-метоксифенил)-2-пропеновой кислоты. Выход: 56%.
ФАРМАКОЛОГИЧЕСКИЕ ТЕСТЫ
ПРИМЕР
Острая токсичность
Острую токсичность оценивали, вводя группе из 10 крыс весом 20 г однократную дозу каждого из тестируемых соединений в ротовое отверстие, через трубку, в составе водной суспензии 2% карбоксиметилцеллюлозы (мас./об.).
За животными вели наблюдение в течение 14 дней. Ни у одного из животных в тестируемой группе не появлялись токсические симптомы, даже после введения 100 мг/кг дозы.
ПРИМЕР F1
Тест 1 - экспериментальная модель in vivo с N-этилмалеимидом (NEM): исследование желудочной переносимости некоторых лекарственных веществ, анализированных в качестве предшественников соединений настоящего изобретения.
Животные (крысы, вес примерно 200 г) были распределены по следующим группам (по 10 животных в каждой группе):
A) Контрольные группы:
1° группа: обработка: только носитель (водная суспензия 1% (мас./об.) карбоксиметилцеллюлозы, доза: 5 мл/кг, если лекарственное вещество вводили через рот, физиологический раствор в случае парентерального способа введения).
2° группа: обработка: носитель + NEM,
B) Опытные группы, которым вводили лекарственное вещество:
группа I: обработка: носитель + лекарственное вещество,
группа II: обработка: носитель + лекарственное вещество + NEM.
Следующие лекарственные вещества были исследованы в этом эксперименте (Таблица I): индометацин, амброксол, мезаламин, алендронат натрия, такрин, омепразол, мизопростол.
Индометацин, амброксол и алендронат вводили через рот, мезаламин вводили в прямую кишку (ректально), а такрин, омепразол и мизопростол - подкожно.
Максимальная переносимая доза, определяемая путем введения каждого вещества указанными способами животным, которым не вводили NEM, указана в Таблице I. При превышении указанных доз у животных появлялись энтеропатия, понос, депрессия, дрожь и заторможенные реакции.
В этой экспериментальной модели животным сначала вводили дозу NEM (25 мг/кг) в физиологическом растворе путем подкожной инъекции. Лекарственное вещество вводили спустя 1 час в суспензии носителя. Спустя 24 часа животных умерщвляли и оценивали ущерб, нанесенный слизистым оболочкам желудочно-кишечного тракта, путем подсчета числа крыс в каждой группе, имеющих видимые повреждения желудка. Суммарное число таких крыс затем делили на суммарное число крыс в группе и умножали на 100. Полученные таким образом процентные величины представлены в Таблице I. Эта таблица показывает, что в группах крыс, которым вводили указанные лекарственные вещества без NEM, не было обнаружено желудочных повреждений.
У всех крыс из группы II (которым вводили NEM) были обнаружены повреждения желудка после введения следующих лекарственных веществ: индометацин, амброксол, мезаламин, алендронат натрия, такрин.
Следовательно, указанные лекарственные вещества могут быть использованы в синтезе продуктов настоящего изобретения.
По результатам теста 1 омепразол и мизопростол, напротив, не могут быть использованы для получения продуктов настоящего изобретения.
ПРИМЕР F2
Тест 2 (in vitro): ингибирование апоптоза (фрагментация ДНК), вызываемого в эндотелиальных клетках соединением CIP, в присутствии некоторых лекарственных веществ, анализированных в качестве предшественников соединений настоящего изобретения.
Были протестированы следующие лекарственные вещества-предшественники (Таблица II): индометацин, парацетамол, клопидогрель, сальбутамол, амброксол, алендронат натрия, дифиллин, цетиризин, эналаприл, никотинамид, ампициллин, ацикловир, мезаламин, такрин, симвастатин, омепразол.
Эндотелиальные клетки человека из пупочной вены получают в соответствии со стандартной процедурой. Свежие пупочные вены заполняют 0,1% (по весу) раствором коллагеназы и инкубируют при 37°С в течение 5 минут.
После этого вены подвергают перфузии в среде М 199 (GIBCO, Grand Island, NY), pH 7,4, содержащей 0,1% (мас./об.) коллагеназы с добавлением 10% фетальной сыворотки крупного рогатого скота (10 мкг/мл), натриевой соли гепарина (50 мкг/мл), тимидина (2,4 мкг/мл), глутамина (230 мкг/мл), пенициллина (100 МЕ/мл), стрептомицина (100 мкг/мл) и стрептомицина В (0,125 мкг/мл). Собирают клетки из перфузата при помощи центрифугирования (800 об/мин) и выращивают в культуральных колбах Т-75, предварительно обработанных фибронектином человека. Затем выращивают клетки в той же самой среде, к которой добавлен фактор роста гипоталамуса крупного рогатого скота (100 нг/мл). Когда клетки первичной клеточной культуры (то есть полученные напрямую ex-vivo из пупочной вены) образуют слитный клеточный монослой (примерно 8000000 клеток на одну колбу), рост культуры останавливают, слои промывают и обрабатывают трипсином. Переносят клеточные суспензии в лунки 24-луночнго планшета для клеточных культур, к половине из которых добавляют ту же самую культуральную среду, содержащую лекарственное вещество в концентрации 10-4 М, после чего выращивают клетки в термостате при 37°С, постоянной влажности (90%) и 5% концентрации СО2. Если лекарственное вещество нерастворимо в культуральной среде, его сначала растворяют в небольшом количестве диметилсульфоксида. Максимальное количество диметилсульфоксида, которое может быть добавлено к культуральной среде, составляет 0,5%. Только клетки, произошедшие из указанных первых субкультур используют для экспериментов с гидропероксидом кумола (CIP). Клетки идентифицируют как эндотелиальные клетки путем морфологического анализа, а также по их специфической иммунологической реакции на фактор VIII; в указанных культурах не обнаруживалось каких-либо загрязнений из миоцитов или фибробластов.
Перед началом теста клеточную культуральную среду удаляют и клеточные слои осторожно промывают стандартным физиологическим раствором, содержащим 0,1М фосфатный буфер (рН 7,0), при температуре 37°С. Содержимое каждой лунки затем инкубируют в течение 1 часа с суспензией CIP в культуральной среде в концентрации 5 мМ. Оценку клеточных повреждений (апоптоз) проводят путем определения процентного изменения фрагментации ДНК в культурах, содержащих лекарственное вещество и CIP, относительно контрольных групп, которые обрабатывали только CIP. Указанное процентное изменение фрагментации ДНК определяют путем оценки изменения флуоресценции при длинах волн 405-450 нм, при помощи микроскопа ВХ60 Olympus (Olympus Co, Рим), сравнивая оптические плотности тестируемых и контрольных образцов. Для каждого образца проводят 5 повторных измерений флуоресценции. Статистическую оценку результатов проводят при помощи t теста Стьюдента (р<0,01).
Результаты, приведенные в Таблице II, показывают, что индометацин, парацетамол, клопидогрель, сальбутамол, алендронат натрия, дифиллин, цетиризин, эналаприл, никотинамид, ампициллин, ацикловир, такрин и омепразол практически не ингибируют апоптоз. Следовательно, эти лекарственные вещества могут быть использованы для получения продуктов настоящего изобретения.
В противоположность этому, амброксол, мезаламин и симвастатин ингибируют апоптоз. Следовательно, по результатам теста 2, эти соединения не могли быть использованы для получения продуктов настоящего изобретения.
ПРИМЕР F3
Тест 3 - экспериментальная in vivo модель, использующая метиловый эфир Nw-нитро-L-аргинина (L-NAME): анализ желудочной переносимости (частоту появления повреждений желудочно-кишечного тракта), переносимости для печени (доза GPT, глутамат-пируват-трансаминазы) и сердечно-сосудистой системы (кровяное давление) некоторых лекарственных веществ, использованных в качестве предшественников соединений настоящего изобретения.
Экспериментальная модель является адаптированной моделью, описанной в J.Clin. Investigation 90, 278-281,1992.
Эндотелиальные дисфункции оценивают путем определения повреждений слизистых оболочек желудочно-кишечного тракта, повреждений печени (увеличение уровня GPT), а также повреждений сосудистого эндотелия и сердечно-сосудистой системы (повышение кровяного давления), вызванных введением L-NAME.
Животных (крысы, средний вес 200 г) делят на группы в соответствии с приведенной ниже схемой. Группе, получающей L-NAME, вводят в течение 4 недель указанное соединение, растворенное в питьевой воде, в концентрации 400 мг/л. Определены следующие группы (по 10 животных в каждой):
A) Контрольные группы:
группа 1°: обработка: только носитель (водная суспензия 1% (мас./об.) карбоксиметилцеллюлозы, доза: 5 мл/кг, когда лекарственное вещество вводится через рот, физиологический раствор, если вводится парентерально),
группа 2°: обработка: носитель + L-NAME,
B) Опытные группы, которым вводили лекарственное вещество:
группа 3°: обработка: носитель + лекарственное вещество,
группа 4°: обработка: носитель + лекарственное вещество + L-NAME.
Были протестированы следующие лекарственные вещества: парацетамол, доксорубицин, симвастатин, омепразол и мизопростол. Каждое из них вводили один раз в день в течение 4 недель.
Максимальную переносимую дозу лекарственного вещества, вводимого животным, определяли в ходе экспериментов с масштабированием отдельной дозы на животных, которым до этого ничего не вводили, путем оценки появления у животных симптомов энтеропатии, поноса, депрессии, дрожи и заторможенной реакции.
В конце четвертой недели животным перестают давать воду и по истечении 24 часов их умерщвляют.
За один час до умерщвления определяют кровяное давление, и его повышение принимают в качестве показателя повреждения сосудистого эндотелия.
Повреждения слизистой оболочки желудка оценивают так, как проиллюстрировано в тесте 1 (см. Пример F1). Повреждения печени определяют путем оценки содержания глутамат-пируват-трансаминазы (увеличение уровня GPT) после умерщвления.
Лекарственное вещество соответствует условиям Теста 3, то есть может быть использовано для получения соединений настоящего изобретения, если в группах крыс, обработанных системой L-NAME + лекарственное вещество + носитель, обнаружены более существенные повреждения печени (более высокие величины GPT) и/или более существенные повреждения желудка и/или сосудов (увеличенное давление крови) по сравнению с группой, обработанной только носителем, или группой, обработанной носителем и лекарственным веществом, или группой, обработанной носителем и L-NAME.
Результаты теста представлены в Таблице IV. Частоту появления (в%) желудочных повреждений определяли так же, как в Тесте 1. Величины (в%) уровня GPT и кровяного давления относятся к соответствующим величинам, найденным у животных 1-й группы из контрольных групп. Среднее значение кровяного давления в этой группе составило 105±8 мм рт. ст.
Полученные результаты свидетельствуют о том, что действие парацетамола, доксорубицина и симвастатина приводит к повреждению печени и гастроэнтеропатии (величины уровня GPT и частоты появления повреждений желудка (в%) превышают аналогичные величины у соответствующих групп, которым вводили лекарственное вещество без L-NAME, и контрольных групп, которым вводили L-NAME).
Следовательно, эти лекарственные вещества могут быть использованы для получения продуктов настоящего изобретения.
По результатам этого теста омепразол и мизопростол, напротив, не могут быть использованы для получения продуктов настоящего изобретения.
ПРИМЕР F4
Тест 4А: активность некоторых веществ, использованных в качестве предшественников В в продуктах настоящего изобретения, в ингибировании гемолиза эритроцитов, вызванного пероксидом кумола.
Тест 4а проводят в соответствии с методом, описанным в публикации R.Maffei Facino, M.Carini G.Aldini, M.Т.Calloni, Drugs Exptl. Clin. Res. XXIII (5/8) 157-165, 1997.
Эритроциты, выделенные при использовании стандартных процедур из мужских особей крыс Wistar (Charles River), суспендируют в физиологическом растворе, содержащем фосфатный буфер с рН 7,4, и уравновешивают при 4°С в течение 4 дней. Затем аликвоту указанной суспензии центрифугируют при 1000 об./мин в течение 5 минут и 0,1 мл центрифугированных эритроцитов разбавляют до 50 мл натрий-фосфатным буфером, имеющим такую же молярную концентрацию. В результате получают суспензию, содержащую 0,2% об. эритроцитов. К порциям указанной разбавленной суспензии по 3,5 мл добавляют по 0,1 мл 9,72 мМ спиртового раствора гидропероксида кумола, который вызывает лизис клеток. Полученную суспензию инкубируют при 37°С. Суспензия становится более мутной. Процесс лизиса клеток наблюдают при помощи турбидиметрии при 710 нм путем определения оптической плотности (или пропускания) через каждые 30 минут. Момент времени, когда максимальное число клеток подверглось лизису, который соответствует максимальной мутности суспензии, принимают за Tmax и считают, что он соответствует 100% лизису клеток. 0,2 мл порции 38 мМ этанольных растворов тестируемых соединений, предполагаемых для использования в качестве предшественников В, добавляют к аликвотам (3,5 мл) разбавленной суспензии эритроцитов, приготовленной, как описано выше, и преинкубируют полученную суспензию в течение 30 минут. Затем к этой суспензии добавляют 0,1 мл 10,26 мМ спиртового раствора гидропероксида кумола и в момент времени Tmax определяют ингибирование гемолиза для данного образца в процентах по отношению между поглощением суспензии образца, содержащего эритроциты, предшественник В и гидропероксид кумола, соответственно, и поглощением суспензии, содержащей эритроциты и гидропероксид кумола, умноженному на 100. Предшественники В соответствуют условиям теста в том случае, если они ингибируют гемолиз, вызываемый гидропероксидом кумола, на 15% и более.
В Таблице V представлены результаты, полученные для следующих веществ: N-метилдиэтаноламин, диэтиленгликоль, тиодиэтиленгликоль, 1,4-бутандиол, бутанол и диэтаноламин.
Из Таблицы V видно, что:
- N-метилдиэтаноламин, диэтиленгликоль, тиодиэтиленгликоль и 1,4-бутандиол соответствуют условиям теста 4, так как они ингибируют гемолиз, вызываемый гидропероксидом кумола, более чем на 15%.
- бутанол и диэтаноламин, напротив, являются неэффективными, поскольку ингибируют гемолиз, вызываемый гидропероксидом кумола, менее чем на 15%, и, следовательно, они не могут быть использованы в качестве предшественников В в синтезе соединений настоящего изобретения.
ПРИМЕР F5
Тест 5: Активность соединений, используемых в качестве предшественников В, в ингибировании образования радикалов из соединений FeII.
0,1 мл аликвоты 10-4 М метанольных растворов соответственно 1,4-бутандиола, N-метилдиэтаноламина, диэтиленгликоля и тиодиэтиленгликоля добавляют в пробирки, содержащие водный раствор, образованный путем смешивания 0,2 мл 2 мМ раствора дезоксирибозы, 0,4 мл 100 мМ фосфатного буфера (рН 7,4) и 0,1 мл 1 мМ раствора FeII (NH4)2(SO4)2 в 2 мМ растворе HCl. Пробирки затем выдерживают при температуре 37°С в течение одного часа. Затем в каждую пробирку добавляют 0,5 мл 2,8% водного раствора трихлоруксусной кислоты и 0,5 мл водного раствора 0,1 М тиобарбитуровой кислоты в указанном порядке. Контрольный прозрачный раствор готовят путем замены указанной выше 0,1 мл аликвоты метанольного раствора тестируемого соединения на 0,1 мл метанола. Пробирки закрывают и нагревают на масляной бане при температуре 100°С в течение 15 минут. Развивается розовое окрашивание, интенсивность которого пропорциональна количеству дезоксирибозы, подвергшейся радикальному окислительному расщеплению. Растворы охлаждают до комнатной температуры и измеряют их оптические плотности при 532 нм относительно прозрачного контрольного раствора.
Ингибирование, вызываемое предшественником В относительно образования радикалов из FeII, определяют в процентах по следующей формуле:
(1-As/Ac)×100
где As и Ас являются, соответственно, величинами поглощений раствора, содержащего тестируемое вещество + соль железа, и раствора, содержащего только соль железа.
Результаты представлены в Таблице III, которая показывает, что протестированные соединения являются неэффективными в ингибировании образования радикалов из ионов железа. Следовательно, эти соединения могут быть использованы в качестве предшественников В для получения соединений настоящего изобретения.
ПРИМЕР F6
Оценивали активность некоторых соединений, являющихся объектом настоящего изобретения, и соответствующих лекарственных веществ-предшественников в ингибировании деградации ДНК (алоптоза) в эндотелиальных клетках, подвергнутых воздействию пероксида водорода.
Пероксид водорода является мягким окислителем и считается важным медиатором в патологиях, связанных с окислительным стрессом (В.Halliwell, J. Gutteridge "Free Radicals in Biology and Medicine", page 416, 1993). Следовательно, фармакологическая активность соединений, предполагаемых для использования в условиях окислительного стресса, оценивается их способностью к нейтрализации повреждающих клетку воздействий пероксида водорода (В.Halliwell, J.Gutteridge "Free Radicals in Biology and Medicine", page 416, 1993).
Данный способ описан Herman et al. (Herman С., Zeiner M.A., Dimeller S, Arterioscler. Thromb. Vase. Biol. 17 (12), 3588-82, 1997).
Эндотелиальные клетки человека из пупочной вены получали в соответствии со стандартной процедурой. Свежие пупочные вены заполняли 0,1% (по весу) раствором коллагеназы и инкубировали при 37°С в течение 5 минут.
После этого вены подвергали перфузии в среде М 199 (GIBCO, Grand Island, NY), pH 7,4, содержащей 20% сыворотки человека. Собирали клетки из перфузата центрифугированием при 800 об./мин и выращивали их в культуральных колбах Т-75, предварительно обработанных фибронектином человека. Затем клетки выращивали в среде с pH 7,4, содержащей 20% сыворотку человека, низкомолекулярный гепаринат натрия (30 мкг/мл), пенициллин (100000 МЕ/мл) и фактор роста гипоталамуса крупного рогатого скота (100 нг/мл). Первичные слитные клеточные монослои (примерно 8,000,000 клеток на одну колбу) промывали и обрабатывали трипсином. Переносили клеточные суспензии в лунки 24-луночного культурального планшета, после чего выращивали клетки в термостате при 37°С, постоянной влажности (90%) и 5% концентрации CO2. Только клетки, произошедшие из указанных первых субкультур, использовали для экспериментов с пероксидом водорода. Клетки идентифицировали как эндотелиальные клетки путем морфологического анализа, а также по их специфической окрашивающей реакции. В указанных культурах не обнаруживалось каких-либо загрязнений из миоцитов или фибробластов.
Для экспериментов с пероксидом водорода клеточную культуральную среду удаляли и клеточные слои осторожно промывали физиологическим раствором, содержащим 0,1 М фосфатный буфер (рН 7,0), при температуре 37°С. Затем клетки инкубировали в течение 18 часов с пероксидом водорода при концентрации последнего 200 мкмоль/л.
Оценку клеточных повреждений (апоптоз) проводили путем определения процентного изменения фрагментации ДНК в образце относительно контрольных групп, которые обрабатывали только пероксидом водорода. Анализируемые продукты тестировали при концентрации 100 мкмоль/л. Если обнаруживалось, что указанные продукты нерастворимы в культуральной среде, их растворяли в небольшом количестве диметилсульфоксида (DMSO), принимая во внимание, что максимальное количество DMSO, которое может быть добавлено к культуральной среде, составляет 0,5% об./об. Для каждого образца было проделано три повторных измерения.
Результаты, представленные в Таблице VI, показывают, что в образцах клеточных культур, обработанных соединениями настоящего изобретения, ингибирование фрагментации ДНК или, в более общем смысле, ингибирование клеточных повреждений по меньшей мере в два раза больше того, что наблюдается в образцах, обработанных соответствующими предшественниками.
ПРИМЕР F7
Желудочные повреждения, вызванные соединениями настоящего изобретения в сравнении с соответствующими лекарственными веществами-предшественниками.
Группам мужских особей крыс Wistar весом 180-200 г (по 10 животных в группе), выдержанных без пищи в течение 17 часов, вводили через трубку в ротовое отверстие 2% водную суспензию карбоксиметилцеллюлозы (носитель), содержащую одно из следующих соединений:
- Диклофенак, доза 20 мг/кг перорально,
- Нитроксиэфирное производное диклофенака (Пр. 14), в той же дозе перорально,
- Амброксол, доза 100 мг/кг перорально,
- Нитроксиэфирное производное амброксола (Пр. 3), в той же дозе перорально,
- Алендронат, доза 100 мг/кг перорально,
- Нитроксиэфирное производное алендроновой кислоты (Пр. 4), в той же дозе перорально.
Такрин и соответствующий нитроксиэфир, полученный в соответствии с Примером 11, вводили крысам подкожно в физиологическом растворе в дозе 10 мг/кг.
Животных умерщвляли через 6 часов после введения веществ. Слизистую оболочку желудочно-кишечного тракта удаляли и исследовали. Степень желудочно-кишечных повреждений оценивали так же, как это описано в эксперименте F1.
Результаты, представленные в Таблице VII, показывают, что соединения настоящего изобретения либо не вызывают желудочных повреждений, либо степень этих повреждений, наблюдавшихся в ряде случаев, оказывалась значительно ниже той, которую вызывают лекарственные вещества-предшественники.
ПРИМЕР 16
Синтез [2-(N-метил-N'-(2-нитрокси)этил)амино]этилового эфира (S)-1-[N-[1-(этоксикарбонил)-3-фенилпропил]-L-аланил]-L-пролина формулы
Предшественником является эналаприл, имеющий формулу:
Предшественником В является N-метил-диэтаноламин:
Соединение (Е-16) синтезируют в соответствии с методикой, описанной в Примере 5. Выход: 19%. Элементный анализ:
ПРИМЕР 17
Синтез 1-[(1-метилэтил)амино]-3-(1-нафтилокси)-2-пропилового эфира (4-нитрокси)бутановой кислоты, имеющего формулу:
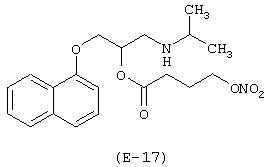
Предшественником является пропранолол, имеющий формулу:
Предшественником В является 4-гидроксибутановая кислота.
Соединение (Е-17) синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 25%. Элементный анализ:
ПРИМЕР 18
Синтез 1-[5-(2,5-дигидро-5-оксо-3-фуранил)-3-метил-2-бензофуранил]этилового [(2-нитрокси)этокси]этилового диэфира бутандиовой кислоты
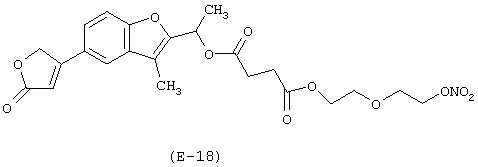
Лекарственным веществом-предшественником является бенфуродил гемисукцинат, имеющий формулу:
Предшественником В является диэтиленгликоль:
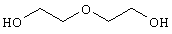
Соединение (Е-18) синтезируют в соответствии с методикой, описанной в Примере 6. Выход: 16%.
Элементный анализ:
ПРИМЕР 19
Синтез [2-(N-метил-N'-(2-нитроксиэтил)амино]этилового эфира N-[[6-метокси-5-(трифторметил)-1-нафтил]тиоксометил]-N-метилглицина, имеющего формулу:
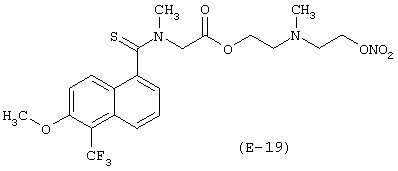
Лекарственным веществом-предшественником является толрестат, имеющий формулу:
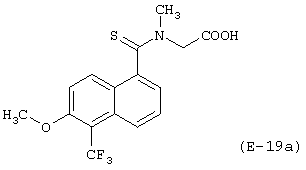
Предшественником В является N-метил-диэтаноламин:
Соединение (Е-19) синтезируют в соответствии с методикой, описанной в Примере 5. Выход: 12%.
Элементный анализ:
ПРИМЕР 20
Синтез (8S-цис)-10-[(3-амино-2,3,6-тридезокси-α-L-ликсо-гексопиранозил)окси]-7,8,9,10-тетрагидро-6,8,11-тригидрокси-8-[[3-метокси-4-(4-нитроксибутаноилокси]метилоксо]-1-метокси-5,12-нафтацендиона формулы
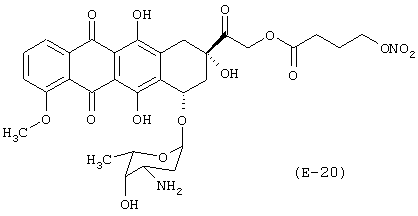
Лекарственным веществом-предшественником является доксорубицин, имеющий формулу:
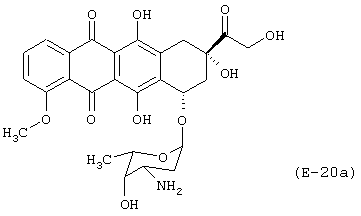
Предшественником В является 4-гидроксибутановая кислота.
Соединение (Е-20) синтезируют в соответствии с методикой, описанной в Примере 1. Выход: 12%.
Элементный анализ:
ПРИМЕР 21
Синтез (4-нитрокси)бутилового эфира (Z)-5-фтор-2-метил-1-[[4-(метилсульфинил)фенил]метилен]-1Н-инден-3-уксусной кислоты, имеющего формулу:
Лекарственным веществом-предшественником является сулиндак, имеющий формулу:
Предшественником В является 1,4-бутандиол.
а) Получение 4-бромбутилового эфира цис-5-фтор-2-метил-1-[n-(метилсульфинил)бензилиден]инден-3-уксусной кислоты
К раствору сулиндака (5,17 г, 14,5 ммоль) в диметилформамиде (50 мл) добавляют EtONa (1,18 г, 16,4 ммоль). Перемешивают реакционную смесь в течение 1 часа, затем добавляют раствор 1,4-дибромбутана в диметилформамиде (20 мл). Перемешивают реакционную смесь при комнатной температуре в течение 8 часов, затем разбавляют диэтиловым эфиром и промывают водой. Органическую фазу высушивают сульфатом натрия и затем упаривают при пониженном давлении. Полученный неочищенный продукт очищают при помощи колоночной хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 3/7 (об./об.). Получают 4-бромбутиловый эфир цис-5-фтор-2-метил-1-(n-(метилсульфинил)бензилиден]инден-3-уксусной кислоты.
b) Получение 4-(нитрокси)бутилового эфира цис-5-фтор-2-метил-1-(n-(метилсульфинил)бензилиден]инден-3-уксусной кислоты
К раствору 4-бромбутилового эфира цис-5-фтор-2-метил-1-(n-(метилсульфинил)бензилиден]инден-3-уксусной кислоты (5,01 г, 10,18 ммоль) в ацетонитриле (60 мл) добавляют нитрат серебра (3,5 г, 20,6 ммоль). Реакционную смесь перемешивают без доступа света при температуре 80°С в течение 48 часов, охлаждают до комнатной температуры, фильтруют для удаления солей серебра и упаривают при пониженном давлении. Остаток очищают при помощи колоночной хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 3/7 (об./об.). После упаривания растворителя получают (4-нитрокси)бутиловый эфир (Z)-5-фтор-2-метил-1-[[4-(метилсульфинил)фенил]метилен]-1Н-инден-3-уксусной кислоты (т. пл. 93-97°С). Выход: 40%.
Элементный анализ:
Рассчитано, %: С 60,87% Н 5,11% F 4,01 N 2,96% S 6,77
Найдено, %: С 60,85% Н 5,13% F 3,93 N 2,94% S 6,75
ПРИМЕР F8
Пример F1 повторили с тремя группами крыс (по 10 животных в каждой группе), которым перорально вводили NEM в соответствии со следующей схемой:
а) контрольная группа: носитель, образованный водной суспензией 1% (мас./об.) карбоксиметилцеллюлозы,
b) одна группа (группа сравнения b), животным из которой в то же самое время вводили 10 мг/кг (0,034 ммоль/кг) диклофенака, а также 4 мг/кг (0,034 ммоль/кг) N-метилдиэтаноламина в вышеупомянутом носителе,
с) одна группа (группа сравнения с), животным из которой вводили 15 мг/кг (0,034 ммоль/кг) эфирного производного диклофенака по изобретению (см. Пр. 14) в вышеупомянутом носителе,
Результаты, представленные в Таблице VIII, показывают, что смесь, вводимая животным группы b (группа сравнения), была гораздо менее эффективна в уменьшении частоты появления повреждений желудка, чем соединение настоящего изобретения, вводимое животным группы с.
ПРИМЕР F9
Противовоспалительная и обезболивающая активность 4-(N-ацетиламино)фенилового эфира 4-(нитрокси)бутановой кислоты (NO-парацетамол) и его предшественника парацетамола.
Введение
Главный терапевтический эффект NSAIDs (нестероидных противовоспалительных лекарственных веществ) связан с их способностью ингибировать производство простагландинов ("Goodman & Gilman's, The Pharmacological Basis of Therapeutics" 9й Ed. 1996, McGraw Hill page 620), и эти агенты классифицируют в соответствии с данным принципом. Сулиндак и парацетамол имеют механизмы действия, отличные от большинства используемых в настоящее время NSAIDs с точки зрения их весьма незначительной способности к ингибированию производства простагландинов. Оба этих вещества взаимодействуют со свободными радикалами кислорода.
Противовоспалительную и обезболивающую активность измеряли в соответствии с методами измерения отека лапок крыс под действием каррагинана и подсчета судорог мышей под действием уксусной кислоты. Были использованы мужские особи крыс (Wistar, 100-150 г) и мышей (LACA, 22-35 г). NO-парацетамол, парацетамол или носитель вводили в составе суспензии карбоксиметилцеллюлозы (0,5% мас./об.) в количестве 1 мг/кг.
Отек лапок под действием каррагинана
Эксперименты проводили так, как это описано в публикации Al-Swayeh et al, Brit. J. Pharmacol. 129, 343-350, 2000. Объем задней лапки определяли посредством плетизмографии до интерплантарной инъекции каррагинана (100 мкл, 2% мас./об.) и спустя 3 часа после нее. Соединения вводили внутрибрюшинно в объеме 15 мл перед инъекцией каррагинана. По окончании эксперимента животных умерщвляли путем смещения шейных позвонков и обескровливания. Результаты, показанные в Таблице IX, выражены в величинах процентного ингибирования отека лапок, т. е. путем вычитания из объема лапок у контрольных животных (носитель) объема лапок у животных, которым вводили вещества, с последующим делением полученной разности на объем лапок у контрольных животных.
Подсчет судорог под действием уксусной кислоты
Эксперименты были проведены в соответствии с тем, как это описано в публикации Moore et al., Brit. J. Pharmacol. 102, 198-202, 1990. Соединения вводили перорально за 15 минут до внутрибрюшинного введения уксусной кислоты (2% мас./об. в солевом растворе с рН 2,7, 10 мл/кг). Сразу после этого мышей переносили в клетки для индивидуального наблюдения и в течение последующих 30 минут подсчитывали число брюшных сокращений. По окончании периода наблюдения животных умерщвляли путем смещения шейных позвонков и обескровливания. Результаты выражены как число брюшных сокращений (судорог) в течение 30-минутного периода наблюдений, в процентном соотношении к числу сокращений в контрольных группах.
Результаты, представленные в Таблице, демонстрируют, что NO-парацетамол является гораздо более активным в обоих тестах, чем парацетамол.
ПРИМЕР F10
Безопасность для печени введения NO-парацетамола и парацетамола
Крысам вводили NO-парацетамол (1,4 г/кг внутрибрюшинно), или парацетамол (1,16 г/кг внутрибрюшинно), или носитель (0,9% мас./об. NaCl, содержащий 20% об./об. твин-20). Через 6 часов животных умерщвляли путем смещения шейных позвонков, собирали кровь из артерии и анализировали плазму на аспартат-аминотрансферазную (AST) и аланин-аминотрансферазную активность, концентрацию в печени глутатиона и билирубина.
Уменьшение содержания глутатиона, вызываемое парацетамолом, считается признаком окислительного стресса (В. Halliwell, J. Gutterbridge "Free radicals in biology and medicine" 1993, Clarendon Press, pages 334-335).
Результаты выражены в виде процентной величины, рассчитанной относительно соответствующих величин в контрольных (носитель) группах (100%).
Результаты демонстрируют, что введение парацетамола вызывает повреждения печени по результатам измерения величин как AST и ALT трансаминаз, так и билирубина относительно соответствующих величин в контрольных группах.
Введение NO-парацетамола вызывает значительно меньшие увеличения AST и ALT, в то время как концентрация билирубина оказывается пониженной по сравнению с контрольной группой.
Таким образом, в отличие от парацетамола NO-парацетамол обладает более щадящим действием на печень, даже в условиях окислительного стресса (т.е. содержание печеночного глутатиона в равной степени снижается под действием парацетамола и NO-парацетамола).
Изобретение относится к новым биологически активным соединениям. Описываются соединения или их соли, имеющие следующую общую формулу (I):
A-B-N(O)s (I)
где: s=2; A=R-T1-, где R представляет собой радикал лекарственного вещества при условии, что лекарственное вещество по формуле R-T1-Z или R-T1-OZ, где Z представляет собой Н или C1-C5 алкил выбирается из парацетамола, салбутамола, амброксола, алендроновой кислоты, цетиризина, ампициллина, ацикловира, доксорубицина, симвастатина, дифиллина, такрина, клопидогрела, диметиломепразола, диклофенака, феруловой кислоты, эналаприла, пропранолола, бенфуродил гемисукцината, толрестата или сулиндака; T1=(СО), О или NH; В=-ТB-Х2-0-, где ТB=(СО) или О; Х2 представляет собой бивалентный радикал, равный R1B-X-R2B, в котором R1B и R2B равны или различаются, являются линейными или разветвленными C1-С6 алкиленами и Х представляет сбой связь, О, S или NR1C, где NR1C представляет собой Н или линейный или разветвленный C1-С6 алкил; соответствующий предшественник В представляется по формуле -ТB-Х2-ОН, где ТB=(СО), а свободная валентность ТB занята -OZ, где Z как определено выше или ТB=O, а свободная валентность ТB занята Н; при условии, что в формуле (I), когда Х2 предшественника В является линейным или разветвленным С2-С20 алкиленом, лекарственное вещество по формуле R-T1-Z или R-T1-OZ, используемое в формуле (I), не принадлежит следующим веществам: эналаприл (ингибиторы АСЕ) и диклофенак (NSAID). Также описываются фармацевтические композиции для использования в случаях окислительного стресса и 4'-ацетиламино фениловый эфир 4-нитроксибутановой кислоты. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 4 н. и 3 з.п. ф-лы, 8 табл.
A-B-N(O)s, (I)
где s=2;
A=R-T1-, где R представляет собой радикал лекарственного вещества при условии, что
лекарственное вещество по формуле R-T1-Z или R-T1-OZ, где Z представляет собой Н или C1-C5 алкил, выбирается из парацетамола, салбутамола, амброксола, алендроновой кислоты, цетиризина, ампициллина, ацикловира, доксорубицина, симвастатина, дифиллина, такрина, клопидогрела, деметиломепразола, диклофенака, феруловой кислоты, эналаприла, пропранолола, бенфуродил гемисукцината, толрестата или сулиндака;
T1=(СО), О или NH;
В=-ТB-X2-O-, где ТB=(СО) или О;
Х2 представляет собой бивалентный радикал, равный R1B-X-R2B, в котором R1B и R2B равны или различаются, являются линейными или разветвленными C1-C6 алкиленами и Х представляет сбой связь, О, S или NR1C, где NR1C представляет собой Н или линейный или разветвленный C1-С6 алкил;
соответствующий предшественник В представляется по формуле -ТB-Х2-ОН, где ТB=(СО), а свободная валентность ТB занята -OZ, где Z как определено выше или ТB=0, а свободная валентность ТB занята Н;
при условии, что в формуле (I), когда Х2 предшественника В является линейным или разветвленньм С2-С20 алкиленом, лекарственное вещество по формуле R-T1-Z или R-T1-OZ, используемое в формуле (I), не принадлежит следующим веществам: эналаприл (ингибиторы АСЕ) и диклофенак (NSAID).
1,4-бутандиол: ОН-(СН2)4-ОН,
6-гидроксигексановую кислоту: ОН-(СН2)5-СООН,
4-гидроксимасляную кислоту: ОН-(СН2)3-СООН,
N-метилдиэтаноламин:ОН-(СН2)2-N(СН3)-(СН2)2-ОН,
диэтиленгликоль: ОН-(СН2)2-O-(СН2)2-ОН,
тиодиэтиленгликоль: OH-(CH2)2-S-(CH2)2-OH.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ПРОИЗВОДНЫЕ 2-(2,6-ДИГАЛОФЕНИЛАМИНО)ФЕНИЛУКСУСНОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109009C1 |

