Изобретение относится к новым продуктам, обладающим противотромбозной активностью.
Подавляющие циклооксигеназу (ЦОК) противовоспалительные продукты известны из предшествующих патентных заявок под авторством заявителя. См., в частности, опубликованные патентные заявки WO 94/04484, WO 94/12463, WO 95/09831, WO 95/30641. Эти патентные заявки относятся к нестероидным противовоспалительным продуктам с некислотным окончанием и к продуктам с кислотным окончанием, упоминаемым как известные продукты.
Указанные продукты проявляют значительно более низкий уровень токсичности по сравнению со ссылочными продуктами, не содержащими группу -ON2.
В WO 95/30641 описаны соединения, обладающие ингибирующей ЦОК активностью и обладающие противотромбозной и антигипертензивной активностью.
ЕР 637583 касается нитроэфиров 1-арилокси-3-алкиламино-2-пропанола общей формулы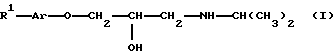
где R1 представляет собой - (CH2)m-Z-R2, m равно 1 или 2, Z представляет собой функциональную группу -О- простого эфира, -CONH амида или -СОО- сложного эфира и R2 представляет собой С2-3 алкил с прямой или разветвленной цепью, имеющий, по меньшей мере, одну нитроксигруппу. Указанные соединения могут использоваться в качестве лекарственных средств в области сердечно-сосудистых заболеваний.
В WO 97/31896 описаны производные формулы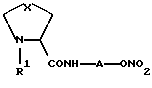
где Х представляет собой -СН2-, -О-, -S-; R1 представляет собой алканоильную группу, имеющую один или несколько заместителей, А представляет собой алкилен или группу -B-D-E- (где В и Е могут быть одинаковыми или разными и каждый представляет собой одинарную связь или алкилен, и D представляет собой циклоалкилен, необязательно замещенный арилом).
Указанные соединения обладают сосудорасширяющим и антиангинальным действием и могут использоваться для профилактики или лечения стенокардии.
Ощущается потребность в доступных продуктах, обладающих противотромбозной активностью в сочетании с низкой токсичностью для продолжительного лечения. В частности, эффективность и безопасность противотромбозных средств тесно связаны, и исследования имеют целью поиск новых соединений с повышенным терапевтическим индексом, т.е. с повышенной эффективностью и пониженной токсичностью (Goodman & Gilman: "The pharmacological basis of therapeutics", Ed. J. Hardman, L. Limbrid, p.1357, 1996).
Неожиданно и к удивлению было обнаружено, что соединения этого изобретения, которые определены ниже, эффективны при ингибировании агрегации тромбоцитов, вызванной различными видами стимулов, в частности коллагеном и тромбином, и в то же время, в основном, проявляют в основном высокую степень безопасности, в частности, высокую безопасность в отношении желудочно-кишечного тракта, не вызывая повреждений слизистой оболочки желудочно-кишечного тракта у получавших лечение животных.
Результаты данного изобретения еще более неожиданны, если принять во внимание то, что новые классы веществ этого изобретения не являются веществами, подавляющими ЦОК, и поэтому никаким образом не могут быть предположены на основе соединений, описанных в литературе данной области, в частности, в вышеуказанных патентах.
Предметом данного изобретения являются соединения, или их композиции, общей формулы:
A-(X-NO2)to
или их соли для применения в качестве лекарственных средств, в частности, в качестве антитромбозных средств, так как они эффективны при ингибировании агрегации тромбоцитов, где:
to является целым числом, равным 1 или 2;
А= RNo, где No=(COXu)t- или COON1, где t является целым числом, равным нулю или 1; и является целым числом, равным 0 или 1;
Х= О, NH, NR1c, где R1c является алкилом с прямой или разветвленной цепью, имеющим от 1 до 10 атомов углерода; N1 является алкилом с прямой или разветвленной цепью, имеющим от 1 до 10 атомов углерода, или водородом;
R выбран из следующих групп;
* Группа А)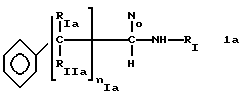
где RIa и RIIa являются одинаковыми или разными и представляют собой Н или алкил с прямой или, где возможно, с разветвленной цепью, имеющий от 1 до 3 атомов углерода, предпочтительно, RIa=RIIa=Н; nIa является целым числом от 1 до 6, предпочтительно, от 2 до 4; RI может быть:


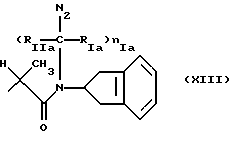
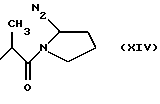
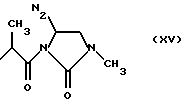
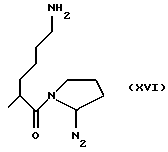
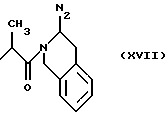
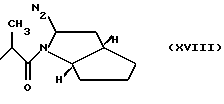



где N2 имеет то же значение, что и No; причем по меньшей мере одна из групп No или N2 имеет одну свободную валентность, способную к связыванию с X1 (то есть, t=1),
где RIa, RIIa, nIa имеют значение, как указано для Ia;
N3 представляет собой Н, (СН3)2СН-СН-ОСОСH2СН3, или свободную валентность, с которой связан X1 (то есть N3 отсутствует);
RIb выбран из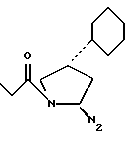
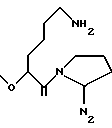
N2 имеет значение, как указано выше, где по меньшей мере одна из групп N3 или N2 имеет свободную валентность, способную к связыванию с X1 (когда он представляет собой N2, t=1);
1с) где t=1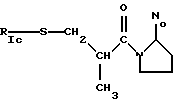
имеют значения, как указано выше, где t=1, т.е. она имеет свободную валентность, способную связываться с XI;
RIc выбран из Н, -СОСН3 или

где N2 имеет значения, как указано выше, и по меньшей мере одна из групп N2 имеет свободную валентность (t=1), способную к связыванию с X1;
когда в группе Iс) RIc представляет собой Н или СОСН3, Х не может быть -NH.
* Группа В
где t=1 u=0;
RIa, RIIa имеют значения, как указано для Iа);
RIIb имеет значение RIa;
RSA выбран из:




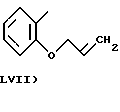
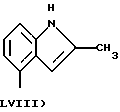

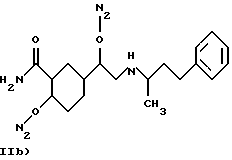
где в группе В) N2 имеет значения, как указано выше, и по меньшей мере одна из N2 групп имеет свободную валентность, способную связываться с X1 (то есть по меньшей мере один заместитель N2 имеет t=1);
X1 представляет собой бивалентный соединяющий мостик, выбранный из следующих:
- YO, где Y представляет собой C1-C20 алкилен с прямой или, где возможно, разветвленной цепью, предпочтительно имеющий от 2 до 5 атомов углерода, или необязательно замещенный циклоалкилен, имеющий от 5 до 7 атомов углерода;
- Y1, выбранный из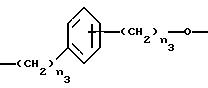
где n3 является целым числом от 0 до 3;

где nf' является целым числом от 1 до 6, предпочтительно от 2 до 4;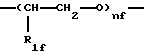
где R1f=Н, СН3, а nf является целым числом от 1 до 6, предпочтительно от 2 до 4.
Соединения, которые могут быть упомянуты и которые являются предпочтительными соединениями, перечислены ниже, где R может быть получен с помощью известных в данной области способов.
Например, соединения и способы, описанные в The Merck Index, Ed. 12, 1996, могут быть указаны в качестве предшественников и близких способов. Предшественники (по номенклатуре Merck), как показано ниже, где различные заместители, указанные в формулах группы А) и группы В), имеют значения, как указано для перечисленных соединений: аласеприл, беназеприл, каптоприл, керонаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фозинаприл, имидаприл, лизиноприл, хинаприл, рамиприл, спираприл, темокаприл, трандолаприл, мовелтилприл, периндоприл, бефунолол, бетаксолол, бупранолол, картеолол, левобунолол, метипранолол, тимолол, окспренолол, мепиндолол, атенолол, лабеталол.
Соединяющие мостики X1, как указано выше, могут быть получены способами, известными специалистам в данной области, или модификацией известных способов путем введения X1 мостиков, которые отличны от соединяющих мостиков, описанных в вышеупомянутых патентах, с помощью известных специалистам способов. Обычно связь между А и X1, как видно, представляет собой сложноэфирную или амидную (NH или NR1c, как указано для X). Для образования этих связей может использоваться любой хорошо известный путь синтеза.
В случае сложных эфиров наиболее прямой путь синтеза включает взаимодействие ацилхлоридов А-СО-Сl или А-(СО-Сl)2 в галогеноспиртах типа HO-Ya-Cl, HO-Ya-Br, HO-Ya-I, где Ya является одинаковым с Y или Y1, как указано выше, без атома кислорода  в экспериментальных условиях, которые являются частью известной методологии.
в экспериментальных условиях, которые являются частью известной методологии.
Реакционные продукты формулы A-CO-O-Ya-Cl(Br, I) могут быть также получены путем взаимодействия натриевых или калиевых солей указанных кислот А-СО-ОН с дигалогенпроизводными общей формулы YaCl2, YaBr или YaI2.
Реакционные продукты преобразуют в конечные продукты путем взаимодействия с AgNO3 в ацетонитриле в соответствии со способами, известными из уровня техники.
Общая схема является следующей:
А-СО-Сl+HO-Ya-Br- -->A-CO-O-Ya-Br+AgNO3- -->A-X1NO2,
где X1=YaO
Общая схема может быть также следующей:
A-CO-ONa+Br2Ya- -->A-CO-O-Ya-Br+AgNO3- -->A-X1NO2,
где X1=YaO.
В случае амидов последовательность синтеза включает взаимодействие тех же ацилхлоридов А-СО-Сl с аминоспиртами общей формулы NH2-Ya-OH или NHR1c-Ya-OH с получением амилов общей формулы;
A-CO-NH-Ya-OH или A-CO-NR1c-Ya-OH
в соответствии с известными способами.
Взаимодействие этих амидов с галогенирующими веществами, такими как, например, PCl5, PBr5, SOCl2 и т.д., дает галогенопроизводные общей формулы:
A-CO-NH-Ya-Br(Cl) и A-CO-NR1c-Ya-Br(Cl).
Путем взаимодействия с AgNO3 в ацетонитриле в соответствии с известными из литературы способами указанные последние продукты дают конечные продукты AX1NO2.
Эта последовательность может быть представлена следующим образом:
где YaO представляет собой X1.
Альтернативный путь образования сложного эфира представляет собой взаимодействие натриевых и калиевых солей кислот с эфирами азотной кислоты и галогеноспиртов общей формулы:
NO2-O-Ya-Cl(Br, I)
с получением непосредственно продуктов по данному изобретению.
Схема этой реакции является следующей:
A-CO-ONa+Br-Ya-ONO2- -->A-CO-O-Ya-ONO2
где YaO представляет собой X1.
Другими синтетическими путями, подобными описанным выше, являются те, при которых дигалогеновое производное Br2Ya взаимодействует с енолятами. Реакционные продукты затем преобразуют путем взаимодействия с AgNO3 в ацетонитриле по вышеуказанной реакции. Общая схема, показанная для -ОН, относящегося к группе А, является следующей: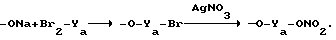
Общий метод для группы -ОН описан в примере 1 только в целях иллюстрации.
Способы получения этих связующих групп X1 описаны в патентной заявке WO 95/30641.
Продукты по данному изобретению, которые описаны выше, являются новыми в качестве лекарственных средств. В частности, они являются новыми в отношении их антитромбозной активности, а также являются новыми в качестве самих соединений.
Дополнительное фармацевтическое применение, которое может быть указано для продуктов по изобретению, представлено, например, их антигипертензивной активностью (например, при артериальной гипертензии, глаукоме) и их кардиопротекторной активностью (например, при стенокардии, сердечной недостаточности, коронарной ишемии).
В отношении антигипертензивной активности следует отметить, что продукты по изобретению продемонстрировали чрезвычайно удовлетворительный фармакотерапевтический профиль с повышенной эффективностью по сравнению с предшественниками, которые не содержат группу -ONO2, и в то же время проявили превосходную безопасность.
Следует также отметить, что продукты по изобретению проявляют антигипертензивную активность в сочетании с антитромбозной активностью. Это дает чрезвычайно ценное преимущество при лечении сердечно-сосудистого заболевания в целом, так как цель любого терапевтического подхода состоит в обеспечении больному, в совокупности, снижения риска сердечно-сосудистого заболевания, такого как инфаркт сердца или мозга, и атеросклероза (Goodman & Gilman "The Pharmacological basis of therapeutics", Ed. J.Hardman, L. Limbrid, pages 747, 1354-7, 1996).
Следующие примеры представлены для объяснения, но не ограничения данного изобретения.
ПРИМЕРЫ
ПРИМЕР 1: Синтез и характеристики NO-тимолола (NO-ТИМ)
Синтез (R)-(4-нитрокси)бутаноата 1-[(1,1-диметил)амино)-3-{ [4-(4-морфолинил)-1,2,5-тиадиазол-3-ил]окси}-2-пропилмалеата
Исходной точкой является тимололмалеат (коммерческий продукт), тимолол, имеющий общую формулу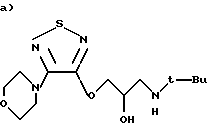
(S)-1-[(1,1-диметилэтил)амино] -3-{ [4-морфолинил)-1,2,5-тиадиазол-3-ил} окси}-2-пропанол.
Тимололмалеат (2,0 г) подвергают взаимодействию с 10% раствором NaOH (30 мл). Добавляют 30 мл СН2Сl2 и затем фазы разделяют. Водную фазу несколько раз экстрагируют CH2Cl2. Объединенные органические фазы сушат (Na2SO4) и растворитель выпаривают при пониженном давлении. Получают 1,4 г чистого продукта (выход 96%).
1Н ЯМР (300 МГц CDCl3): δ 1,05 (9Н, с, 3СН3), 2,7 (2Н, 2дд, CH2-NH), 3,5 (4Н, м, морфолин), 3,8 (4Н, т, морфолин), 3,85 (1H, м, СН), 4,4 (2Н, 2дд, O-СН2).
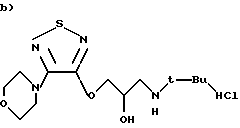
Гидрохлорид (S)-1-[(1,1-диметилэтил)амино]-3-{[4-(4-морфолинил)-1,2,5-тиадиазол-3-ил]окси}-2-пропанола.
0,8 мл 7М раствора НСl в изопропаноле по каплям добавляют к перемешиваемому магнитной мешалкой раствору тимолола (1,4 г) в изопропаноле (30 мл). Раствор перемешивают в течение 30 минут. Реакционную смесь освобождают от растворителя при пониженном давлении. Получают 1,47 г чистого продукта (выход 91%).
1Н ЯМР (300 МГц CDCl3): δ 1,45 (9Н, с, 3СН3), 3,05 (2Н, 2дд, CH2-NH), 3,5 (4Н, т, морфолин), 3,8 (4Н, т, морфолин), 4,5 (2Н, д, O-СН2), 4,55 (1Н, м, СН).

(R)-(4-бром)бутаноат 1-[(1,1-диметилэтил)амино] -3-{[4-(4-морфолинил)-1,2,5-тиадиазол-3-ил)оксил}-2-пропила
4-Бромбутирилхлорид (0,4 мл) по каплям в атмосфере азота добавляют к перемешиваемому магнитной мешалкой раствору гидрохлорида тимолола (0,82 г) в СНСl2, сушат над Р2О5 (20 мл). Перемешивание продолжают в течение 4 дней. Затем реакционную смесь освобождают от растворителя при пониженном давлении. Остаток подвергают хроматографии на силикагеле, используя диэтиловый эфир с 3% Et3N в качестве элюента. Получают 0,830 г чистого продукта из промежуточных фракций (выход 78%).
1H ЯМР (300 МГц CDCl3): δ 1,05 (9Н, с, 3-СН3), 2,05 (2Н, м, COCH2-CH2-CH2-ONO2), 2,5 (2Н, м, COCH2-CH2-CH2-ONO2), 2,8 (2Н, д, СН2-NH),
3,5 (6Н, м, морфолин, СН2-Br), 3,8 (4Н, т, морфолин}, 4,65 (2Н, 2дд, O-СН2), 5,25 (1Н, м, СН).
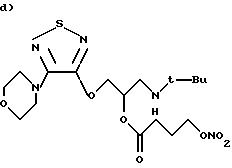
(R)-(нитрокси)бутаноат 1-[(1,1-диметилэтил)амино] -3-{ [4-(4-морфолинил)-1,2,5-тиадиазол-3-ил)окси}-2-пропила.
Раствор AgNO3 (0,450 г) в СН3СН (5 мл) по каплям при комнатной температуре добавляют к перемешиваемому магнитной мешалкой раствору тимолол-(4-бром)бутаноата (0,830 г) в СН3СN (10 мл). Температуру постепенно повышают до 60oС, и реакция продолжается в течение 24 часов. Реакционную смесь освобождают от растворителя при пониженном давлении. Остаток подвергают хроматографии на силикагеле, используя диэтиловый эфир с 3% Et3N в качестве элюента. Получают 0,51 г чистого продукта из первых фракций (выход 64%).
1Н ЯМР (300 МГц СDСl3): δ 1,05 (9Н, с, 3-СН3), 2,05 (2Н, м, COCH2-CH2-CH2-ONO2), 2,5 (2Н, 2т, COCH2-CH2-CH2-ONO2), 2,8 (2Н, д, CH2-NH),
3,5 (4Н, м, морфолин), 3,8 (4Н, т, морфолин), 4,5 (2Н, т, -CH2-ONO2), 4,58 (2Н, 2дд, O-СН2), 5,25 (1Н, м, СН).
МС: М+448
(R)-(нитрокси)бутаноат 1-[(1,1-диметилэтил)амино] -3-{ [4-(4-морфолинил)-1,2,5-тиадиазол-3-ил]окси}-2-пропилмалеата.
Раствор малеиновой кислоты (0,132 г) в ацетоне (5 мл) по каплям добавляют к перемешиваемому магнитной мешалкой раствору тимолол-(4-нитрокси)бутаноата (0,50 г) в ацетоне (10 мл). Перемешивание продолжают в течение 2 часов. Реакционную смесь освобождают от растворителя под пониженным давлением. Неочищенный остаток растирают в диэтиловом эфире с получением 0,5 г белого твердого вещества (т. пл. 133-136oС, выход 70%).
1H ЯМР (300 МГц CDCl3): δ 1,48 (9Н, с, 3-СН3), 2,05 (2Н, м, COCH2-CH2-CH2-ONO2), 2,58 (2Н, 2тд, COCH2-CH2-CH2-ONO2), 3,3 (2Н, 2м, СН2-NH), 3,5 (4Н, м, морфолин), 3,8 (4Н, т, морфолин), 4,5 (2Н, т, -CH2-ONO2), 4,7 (2Н, 2дд, O-СН2), 5,55 (1Н, м, СН), 6,47 (2Н, с, малеиновый).
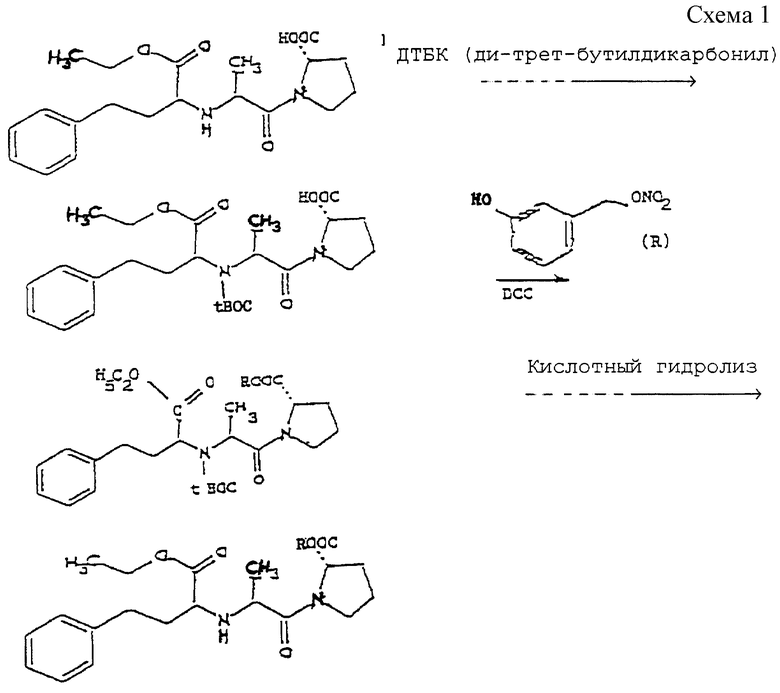
ПРИМЕР 2а: Синтез и характеристики NO-эналаприла (NO-ЭHA)
Схема 1 реакции приведена в конце текста.
Стадия 1
3 г ди-трет-бутилдикарбонила (ДТБК) при комнатной температуре добавляют к раствору 5 г эналаприла в 100 мл диметилформамида (ДМФ) и триэтиламина (ТЭА) (2,76 г). Раствор перемешивают в течение 16 часов. Затем раствор дважды промывают разбавленной HCl и водой, 3 раза экстрагируют 100 мл порциями эфира. Высушенные и выпаренные органические фазы дают 3 г продукта формулы 2) (масло). В формуле 2) tBOC = трет-бутилдикарбонил.
Стадия 2
1,4 г дихлоргексилкарбодиимида (ДЦК) и затем 30 мл раствора 1,1 г нитроксиметилфенола в CH2Cl2 добавляют к 3 г защищенного по N эналаприла (соединения формулы 2), растворенного в 50 мл метилендихлорида. Смесь перемешивают в течение ночи, дициклогексилмочевину отфильтровывают и растворитель выпаривают досуха. Остаток подвергают хроматографии на силикагеле 60 Merck, используя смесь этилацетата/гексана. Собирают фракцию 2 г промежуточного соединения формулы 3), где R представляет собой остаток нитроксиметилфенола без ОН.
Стадия 3
1 г продукта формулы 3) растворяют при 0oС в 4Н растворе 30 мл сухого газообразного HCl в этилацетате (ACO-Et) и перемешивают в течение 10 часов. Полученный остаток фильтруют и сушат при пониженном давлении, получают 0,5 г продукта 4).
ПРИМЕР 2В: Синтез и характеристики NO-эналаприлата (NO-ЭНП)
Схема 2 реакции приведена в конце текста.
Стадия 1:
3 г ди-трет-бутилкарбонила (ДТБК) добавляют при комнатной температуре к раствору 5 г эналаприлата в 100 мл диметилформамида (ДМФ) и триэтиламина (ТЭА) (2,76 г). Раствор перемешивают в течение 16 часов. Затем раствор дважды промывают разбавленной НСl и водой, 3 раза экстрагируют 10 мл порциями эфира. Высушенные и выпаренные органические фазы дают 3 г продукта 2) в виде масла. В формуле 2) tBOC = t-бутилдикарбонил.
Стадия 2
2,75 г дициклогексилкарбодиимида (ДЦК) и затем 30 мл раствора 2,25 г нитроксиметилфенола добавляют к 3 г защищенного по N эналаприлата, растворенного в 50 мл метилендихлорида. Смесь перемешивают в течение ночи, дициклогексилмочевину отфильтровывают и растворитель выпаривают досуха. Остаток подвергают хроматографии на силикагеле 60 Merck, используя смесь этилацетата/гексана. Собирают фракцию из 3 г промежуточного продукта 3). R имеет значение, которому дано определение в примере 2А.
Стадия 3
1 г продукта 3) растворяют при 0oС в 4Н растворе 30 мл сухого газообразного НСl в ACOEt и перемешивают в течение 10 часов. Полученный осадок отфильтровывают и сушат при пониженном давлении. Получают 0,7 г продукта 4).
ПРИМЕР 3: Фармакологические исследования
Продукты по примерам 1 и 2 применяли in vivo всегда в виде 2% по массе суспензии в карбоксиметилцеллюлозе.
Экспериментальные группы составлялись из 6-8 образцов, чтобы сделать возможной соответствующую статистическую оценку, которую проводили, когда было необходимо.
Что касается острой токсичности соединений, которые являются предметом этого изобретения, ее оценивали после однократного введения дозы соединения перорально группам из 10 мышей каждая.
Уровень гибели и наличие токсических симптомов регистрировали в течение периода наблюдения, равного 14 суткам. Даже после введения 50 мг/кг у животных не выявлено никаких видимых признаков явной токсичности.
ПРИМЕР 3А: Изучение антитромбозной активности
Способность МО-ЭНА и NO-ТИМ подавлять агрегацию тромбоцитов оценивали, используя модель in vivo, которая описана Pinon (J.Pharmacol. Methods 12, 79, 1989). В 5 группах крысам-самцам Wistar (200-250 г) вводили перорально суточную дозу по 10 мг/кг соответственно NO-ЭНА, эналаприла, NO-ТИМ, тимолола или носителя в течение 5 суток. В соответствующее время, на четвертый день, отнимали пищу (но не воду). Через 18-20 часов животные получали последнюю дозу. Через один час животным делали анестезию 10% уретаном (1 мг/кг, внутрибрюшинно) и вставляли канюли в левую яремную вену и правую сонную артерию. Затем внутривенно вводили коллаген (тип 6, Sigma) в дозе 2 мг/кг. Через 3 минуты отбирали два образца крови (А и В) из сонной артерии, используя 2,5 мл пластиковые шприцы, следующим образом: образец А, 0,4 мл крови в 1,6 мл ЭДТУ/формалиновый буфер (тетранатриевая соль ЭДТУ 24 мМ, КН2РО4 1,3 мМ, Na2PO4 13,4 мМ), затем образцы переносили в 5-мл полистирольные пробирки для исследования и давали осесть в течение 15 минут при комнатной температуре. После этого срока агрегацию тромбоцитов в образце А фиксировали формалином, тогда как агрегаты из образца В обрабатывали ЭДТУ. Затем производили подсчет тромбоцитов в каждом образце, используя обычный микроскоп. Подсчет для образца В был общим числом тромбоцитов, тогда как подсчет для образца А считали числом только неагрегированных тромбоцитов. Результаты выражали как процент агрегации, вычисленный следующим образом:
{ [1-(число тромбоцитов в образце А)/(число тромбоцитов в образце В))•100} . Результаты выражены как процент подавления от контрольной группы (носитель, растворитель) и показаны в Таблице 1.
ТАБЛИЦА 1
ИЗУЧЕНИЕ ПРОТИВОТРОМБОЦИТНОЙ АКТИВНОСТИ NO-ЭНА ИЛИ NO-ТИМ ПО СРАВНЕНИЮ С ЭНАЛАПРИЛОМ ИЛИ ТИМОЛОЛОМ У КРЫС
СОЕДИНЕНИЕ - АНТИТРОМБОЦИТНАЯ АКТИВНОСТЬ (%)
NO-ЭНА - 65
NO-ТИМ - 58
ЭНАЛАПРИЛ - 15
ТИМОЛОЛ - 2
Как показано в таблице 1, в отличие от сравниваемых продуктов, нитропроизводные по данному изобретению способны подавлять агрегацию, вызванную коллагеном.
ПРИМЕР 3В: Изучение антитромбозной активности
В 5 группах крысам-самцам Charles River швейцарской линии 15-20 г, вводили ежесуточно перорально дозу, равную 10 мг/кг, соответственно NO-ЭНА, эналаприла, NO-ТИМ, тимолола или носителя (растворителя) в течение 5 дней. В соответствующее время, на четвертый день, пищу (но не воду) отнимали. Через 18-20 часов животные получали последнюю дозу. Через один час животным в хвостовую вену делали инъекцию 0,1 мл смеси коллагена (тип 6, Sigma) плюс гидрохлорид адреналина (100 мкМ), растворенных в 0,154 М растворе хлорида натрия. Как объяснено ранее (Cirino G. et al., Thrombosis Research 79, 73, 1995), инъекция этой смеси вызывает смерть в течение 3 минут у 90% контрольных животных.
Результаты выражены как процент подавления по сравнению с контрольной группой и представлены в таблице 2.
ТАБЛИЦА 2
ИЗУЧЕНИЕ АНТИТРОМБОЗНОЙ АКТИВНОСТИ NO-ЭНА И NO-ТИМ В СРАВНЕНИИ С ЭНАЛАПРИЛОМ И ТИМОЛОЛОМ У КРЫС
СОЕДИНЕНИЕ - АНТИТРОМБОЗНАЯ АКТИВНОСТЬ (%)
NO-ЭНА - 53
NO-ТИМ - 44
ЭНАЛАПРИЛ - 11
ТИМОЛОЛ - 6
Как показано в таблице 2, в отличие от сравниваемых продуктов нитропроизводные по данному изобретению способны подавлять тромбоз, вызываемый коллагеном.
ПРИМЕР 3С: ИЗУЧЕНИЕ АНТИГИПЕРТЕНЗИВНОЙ АКТИВНОСТИ
Способность NO-ЭНА подавлять гипертензию оценивали, используя модель in vivo, которая описана Ribeiro et al. (Hypertension 20, 298, 1992). В 5 группах крысы-самцы Wistar (235-284 г) получали суточную дозу по 10 мг/кг внутривенно соответственно NO-ЭНА, эналаприла, NO-ТИМ, тимолола или носителя в течение 5 дней. Артериальную гипертензию вызывали путем введения НWнитро-L-аргининметилового эфира (L-НАМЭ) с питьевой водой в течение 6 недель. L-HAME растворяли в питьевой воде до концентрации 60-70 мг на 100 мл, так чтобы в сутки вводилось количество, равное примерно 60 мг/кг. Через один час после окончания лечения измеряли системное давление крови с помощью метода с использованием манжетки на хвост (tail-cap method) (Zats, Lab.Anim. Sci. 42, 198, 1990).
ТАБЛИЦА 3
ИЗУЧЕНИЕ АНТИГИПЕРТЕНЗИВНОЙ АКТИВНОСТИ NO-ЭНА В СРАВНЕНИИ С ЭНАЛАПРИЛОМ У КРЫС
СОЕДИНЕНИЕ - СРЕДНЕЕ ДАВЛЕНИЕ КРОВИ (мм Нg)
НОСИТЕЛЬ - 170±7
NO-ЭНА - 115±4*
ЭНАЛАПРИЛ - 163±5
*Р<0,05 в сравнении с другими группами
Как показано в таблице 3, в отличие от сравниваемого продукта нитропроизводное по данному изобретению способно снижать давление крови, вызванное тромбозом, индуцированным L-NAME.
ПРИМЕР 3В: ИЗУЧЕНИЕ ГИПОТЕНЗИВНОЙ АКТИВНОСТИ И БЕЗОПАСНОСТИ В ОТНОШЕНИИ ГЛАЗ NO-ЭНА ИЛИ NO-ТИМ В СРАВНЕНИИ С ЭНАЛАПРИЛОМ ИЛИ ТИМОЛОЛОМ У КРОЛИКОВ
У кроликов местное применение 100 мкг NO-ЭНА или NO-ТИМ давало более выраженное и более продолжительное (более 6 часов) снижение внутриглазного давления (6-7 мм Нg, соответственно), чем сравниваемые продукты тимолол и эналаприл. Кроме того, для NO-ТИМ отношение между концентрациями продукта в плазме (Р) и внутриглазной жидкости (АН) в сравнении с тимололом определяли методом ВЭЖХ. Было обнаружено, что отношение Р/АН для NO-ТИМ было в 5,5 раз ниже, чем отношение для тимолола, что наводит на мысль о том, что системное всасывание нитропроизводного (и, следовательно, какое-либо потенциальное побочное действие указанного производного) было заметно снижено по сравнению с эталонным продуктом.
ПРИМЕР 3Е: ИЗУЧЕНИЕ ДЕЙСТВИЯ NO-ЭНА НА ИНДУЦИРОВАННЫЙ БРОНХОСПАЗМ У МОРСКИХ СВИНОК В СРАВНЕНИИ С ЭНАЛАПРИЛОМ
Бронхоспазм, вызванный капсаицином у морских свинок, является моделью на животных, близко напоминающей способность ингибиторов АПФ (ангиотензин превращающего фермента) (АСЕ) вызывать кашель у пациентов (Subissi et al., J. Cardiovasc. Pharmacol. 20/1, 139-146, 1992).
Принятые условия испытания были такими, которые описаны ранее Del Soldato et al. (J. Pharmacological Methods 5, 279, 1981). Самкам морских свинок, весящим 300-400 г, делали анестезию путем внутрибрюшинной инъекции 5,5-диэтилбарбитурата натрия (200 мг/кг) и поддерживали искусственное дыхание при постоянном положительном давлении. В правую яремную вену вставляли канюлю для введения испытуемого соединения. С помощью срединного сечения брюшины выделяли двенадцатиперстную кишку и через небольшое сечение вставляли кончик подходящей полиэтиленовой канюли и фиксировали его. Другой конец канюли соединяли со шприцом для интрадуоденального введения NO-ЭНА (10 мг/кг), эналаприла (10 мг/кг) или носителя. Через 45 минут животным в яремную вену делали инъекцию 0,1 мл капсаицина (1 мг/кг). Перед инъекцией капсаицина и после нее измеряли изменения в промываемой области с помощью модифицированного аппарата Konzett, соединенного с соответствующим полиграфическим умножителем (Hewlett Packard).
Результаты рассчитаны в виде соотношения ответных реакций, полученных до и после введения испытуемого соединения, выраженны как % ответной реакции, полученной с одним носителем, и показаны в таблице 4.
ТАБЛИЦА 4
ИЗУЧЕНИЕ ДЕЙСТВИЯ NO-ЭНА НА БРОНХОСПАЗМ, ВЫЗВАННЫЙ У МОРСКИХ СВИНОК В СРАВНЕНИИ С ЭНАЛАПРИЛОМ
ЛЕЧЕНИЕ - РЕАКЦИЯ БРОНХОСПАЗМА
НОСИТЕЛЬ - 100
NO-ЭНА - 72
ЭНАЛАПРИЛ - 327
Как показано в таблице 4, нитропроизводное по данному изобретению снижало бронхоспазм, вызванный капсаицином, в отличие от сравниваемого продукта, который на самом деле заметно усиливал реакцию бронхоспазма.
Выводы
Как можно видеть из вышеприведенных примеров, нитропроизводные, которые являются предметом данного изобретения, демонстрируют заметное противотромбозное действие и действие на сердечно-сосудистую систему при превосходной безопасности по сравнению со стандартными продуктами.
ПРИЛОЖЕНИЕ:
Синтез малеата 4-[2-[4-(нитрооксиметил)бензоилокси] -3-[(1-метилэтил)амино]пропокси]бензолацетамида (NO-атенолола).
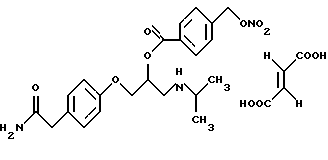
1. Синтез 4-[2-[4-(хлорметил)бензоилокси] -3-((1-метилэтил)амино] пропокси]бензолацетамида
0,8 мл 7М раствора НСl в изопропаноле (5 мл) добавляют по каплям к перемешиваемому магнитной мешалкой раствору атенолола (1,5 г, 5,63 ммоль) в изопропаноле (70 мл). Раствор перемешивают 3 часа, затем удаляют растворитель при пониженном давлении, чтобы получить гидрохлорид 4-[2-гидрокси-3-[(1-метилэтил)амино] пропокси] бензолацетамида (1,75 г) как сырой продукт, который используют в синтезе без дополнительной очистки.
4-(хлорметил)бензоилхлорид (2,2 г, 11,6 ммоль) добавляют к раствору гидрохлорида 4-[2-гидрокси-3-[(1-метилэтил)амино] пропокси] бензолацетамида (5,63 ммоль) в хлороформе (70 мл). Смесь перемешивают при комнатной температуре 6 часов и обрабатывают триэтиламином до достижения рН 7. Органический слой промывают водой (4•50 мл), обезвоживают сульфатом натрия и растворитель удаляют при пониженном давлении. Остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью н-гексан/этилацетат 1/1. 4-[2-[4(хлорметил)бензоилокси] -3-[(1-метилэтил)амино] пропокси] бензолацетамид получают (950 мг) в виде масла. Выход 40%.
2. Синтез 4-[2-[4-(нитрооксиметил)бензоилокси] -3-[(1-метилэтил)амино] пропокси]бензолацетамида
К раствору 4-[2-[4-(хлорметил)бензоилокси] -3-[(1-метилэтил)амино]пропокси] бензолацетамида (900 мг, 2,14 ммоль) в ацетонитриле (30 мл) добавляют нитрат серебра (520 мг, 3,06 ммоль) и смесь нагревают при орошении флегмой в течение 10 часов в темноте. Осадок (хлорид серебра) отфильтровывают, растворитель удаляют при пониженном давлении и остаток очищают путем колоночной хроматографии на силикагеле, используя в качестве элюирующего растворителя этилацетат, чтобы получить 4-[2-[4-(нитрооксиметил)бензоилокси]-3-[(1-метилэтил)амино] пропокси] бензолацетамид (600 мг, 1,3 ммоль) в виде масла. Выход 63%.
3. Синтез малеата 4-[2-[4-(нитрооксиметил)бензоилокси] -3-[(1-метилэтил)амино]пропокси]бензолацетамида (NO-атенолола)
Раствор малеиновой кислоты (180 мг, 1,55 ммоль) в ацетоне (8 мл) добавляют по каплям к раствору 4-[2-[4-(нитрооксиметил)бензоилокси]-3-[(1-метилэтил)амино] пропокси] бензолацетамида (500 мг) в ацетоне (10 мл). Раствор перемешивают при комнатной температуре в течение 1 часа, затем растворитель удаляют при пониженном давлении. Остаток обрабатывают простым диэтиловым эфиром и твердое вещество отфильтровывают. Образующийся в результате малеат 4-[2-[4-(нитрооксиметил)бензоилокси]-3-[(1-метилэтил)амино]пропокси]бензолацетамида получают с выходом 85% в виде аморфного белого вещества.
Анал. рас. для С22Н27N3O7•С4Н4O4:
Рассчитано: %C 55,61; %H 5,56; %N 7,48; %O 31,34.
Обнаружено: С 55,50; Н 5,58; N 7,46; O 31,40.
Фармакологическое исследование активности против тромбоцитов NO атенолола
(пример 3А на странице 23 заявки)
Способность NO атенолола ингибировать агрегацию тромбоцитов оценивают с помощью модели in vivo, как описано Pinon (J.Pharmacol. Methods 12, 79, 1989). 3 группы самцов крыс Wistar (от 200 до 250 г) получают оральную суточную дозу 10 мг/кг NO атенолола, атенолол или носитель в течение 5 дней. На четвертый день в подходящее время им отказывают в пище, в то время как воду дают по потребности, как прежде. Спустя 18-20 часов, животных анестезируют 10% уретаном (1 мг/кг интраперитонеально) в яремную вену и правую сонную артерию канюлируют. Затем внутривенно вводят коллаген (тип 6, Sigma) при дозе 2 мг/кг. Три минуты спустя, две пробы крови, идентифицированные соответственно как проба А и проба В, отбирают из сонной артерии с помощью пластиковых шприцев на 2,5 мл следующим образом: 0,4 мл крови в 1,6 мл буфера EDTA/формалин (тетранатриевые соли EDTA 24 мМ, КН2РO4 1,3 мМ, Na2PO4 13,4 нМ), затем пробу переносят в полистироловые пробирки для тестирования емкостью 5 мл и оставляют для осаждения на 15 минут при окружающей температуре. После этого времени агрегацию тромбоцитов в пробе А фиксируют в формалине, в то время как в пробе В их обрабатывают EDTA. Затем в каждой пробе делают подсчет тромбоцитов, используя обычный микроскоп. Число для пробы В является общим числом тромбоцитов, в то время как в пробе А учитывают только неагрегированные тромбоциты. Результаты представлены как процентная агрегация, рассчитанная следующим образом: {[1-(число тромбоцитов в пробе А)/(число тромбоцитов в пробе В)]•100}. Результаты представлены как процентное ингибирование контрольной группы (носитель). Результаты приведены в таблице 1.1 ниже:
ТАБЛИЦА 1.1
Соединение - Активность против тромбоцитов (%)
NO-атенолол - 40
Атенолол - 2
Таблица иллюстрирует, что активность против тромбоцитов NO-атенолола почти в 20 раз выше, чем активность предшественника.
Фармацевтический препарат в форме глазных капель, содержащий в качестве активного ингредиента соединение изобретения.
Компоненты: - Процент (маc./об.)
NO-тимолол - 0,445
Хлорид бензалкония - 0,010
Хлорид натрия - 0,490
Мононатрийфосфат - 0,460
Динатрийфосфат - 0,880
Динатриевая соль EDTA - 0,025
Вода для инъекций - до 100 мл
ПРИГОТОВЛЕНИЕ
NO-тимолол получают согласно прим. 1, страница 14 заявки. Лекарственную форму готовят в стерильных условиях. Солюбилизируют 4,450 NO-тимолола в 500 мл воды для инъекций (USP) путем перемешивания. К полученному таким образом раствору добавляют в указанном порядке следующие компоненты и тщательно перемешивают до полной солюбилизации, г:
Хлорид бензалкония - 0,100
Хлорид натрия - 4,900
Мононатрийфосат - 4,600
Динатрийфосфат - 8,800
Динатриевая соль EDTA - 0,250
Когда все компоненты солюбилизируются, раствор переносят в мерную колбу на 1 литр и доводят до этого объема путем добавления воды для инъекций.
Раствор фильтруют с использованием стерилизующего фильтра (0,22 мкм PVDF).
Раствором заполняют стерильные сосуды из РЕ емкостью 10 мл.

Изобретение относится к области органической химии. Описываются нитраты общей формулы 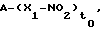 или их соли, где t0 = 1; А=RN0, где N0=(COXu)t или COON1, где t является целым числом, равным 0 или 1; и является целым числом, равным 0 или 1; Х=O, NR1c, где R1c представляет собой алкил с прямой или разветвленной цепью, имеющий от 1 до 10 атомов углерода; N1 представляет собой алкил с прямой или разветвленной цепью, имеющий от 1 до 10 атомов углерода или водород; R выбран из следующих групп:
или их соли, где t0 = 1; А=RN0, где N0=(COXu)t или COON1, где t является целым числом, равным 0 или 1; и является целым числом, равным 0 или 1; Х=O, NR1c, где R1c представляет собой алкил с прямой или разветвленной цепью, имеющий от 1 до 10 атомов углерода; N1 представляет собой алкил с прямой или разветвленной цепью, имеющий от 1 до 10 атомов углерода или водород; R выбран из следующих групп:
* Группа А)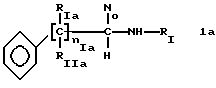
где RIa и RIIa являются одинаковыми или разными и представляют собой Н или линейный или, когда возможно, разветвленный алкил из 1-3 атомов С, предпочтительно RIa= RIIa= Н; nIa является целым числом от 1 до 6, предпочтительно от 2 до 4; RI может быть
где N2 имеет то же значение, что и N0, причем по меньшей мере одна из групп N0 или N2 имеет одну свободную валентность, способную к связыванию с X1 (то есть t=1);
* Группа В), где t=1 и u=0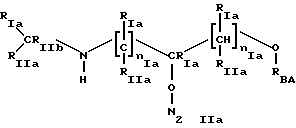
где RIa, RIIa имеют значения, указанные для 1a); RIb имеет значение RIa; nIa имеет значения, указанные для 1а); RBA выбран из: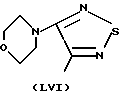

где в группе В) N2 имеет значения, указанные выше, и, по меньшей мере, одна из N2 групп имеет свободную валентность, способную связываться с X1 (то есть, по меньшей мере, один заместитель N2 имеет t=1); X1 представляет собой бивалентный связующий мостик, выбранный из: - Y0, где Y представляет собой C1-C20 алкилен с прямой или, где возможно, разветвленной цепью, предпочтительно имеющий от 2 до 5 атомов углерода; - Y1, выбранного из
где n3 является целым числом от 0 до 3.
Также описывается фармацевтическая композиция, обладающая ингибирующей агрегацию тромбоцитов активностью. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами при превосходной безопасности по сравнению со стандартными продуктами. 2 с. и 7 з.п.ф-лы, 5 табл.
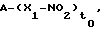
или их соли,
где t0 = 1;
А - RN0, где N0 = (COXu)t или COON1,
где t является целым числом, равным 0 или 1;
u является целым числом, равным 0 или 1;
Х = О, NR1c,
где R1c представляет собой алкил с прямой или разветвленной цепью, имеющий от 1 до 10 атомов углерода;
N1 представляет собой алкил с прямой или разветвленной цепью, имеющий от 1 до 10 атомов углерода или водород;
R выбран из следующих групп:
* Группа А)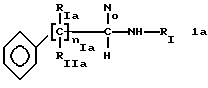
где RIa и RIIa являются одинаковыми или разными и представляют собой Н или линейный или, когда возможно, разветвленный алкил из 1 - 3 атомов С, предпочтительно RIa = RIIa - Н;
nIa является целым числом от 1 до 6, предпочтительно от 2 до 4;
RI может быть
где N2 имеет то же значение, что и N0, причем по меньшей мере одна из групп N0 или N2 имеет одну свободную валентность, способную к связыванию с X1 (то есть t = 1);
* Группа В),
где t = 1 и u = 0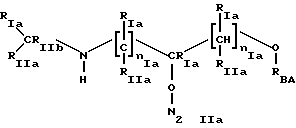
где RIa, RIIa имеют значения, указанные для 1a);
RIb имеет значение RIa;
nIa имеет значения, указанные для 1а);
RBA выбран из:
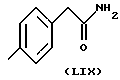
где в группе В) N2 имеет значения, указанные выше, и, по меньшей мере, одна из N2 групп имеет свободную валентность, способную связываться с X1 (то есть, по меньшей мере, один заместитель N2 имеет t = 1);
X1 представляет собой бивалентный связующий мостик, выбранный из: - Y0, где Y представляет собой C1-C20 алкилен с прямой или, где возможно, разветвленной цепью, предпочтительно имеющий от 2 до 5 атомов углерода; - Y1, выбранного из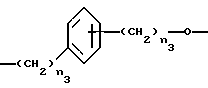
где n3 является целым числом от 0 до 3.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Уплотнение вращающегося вала | 1977 |
|
SU637583A1 |
| Способ получения производных 5-ароил1,2-дигидро-3н-пиррол(1,2-а)-пиррол-1карбоновой кислоты или их солей | 1977 |
|
SU695558A3 |

