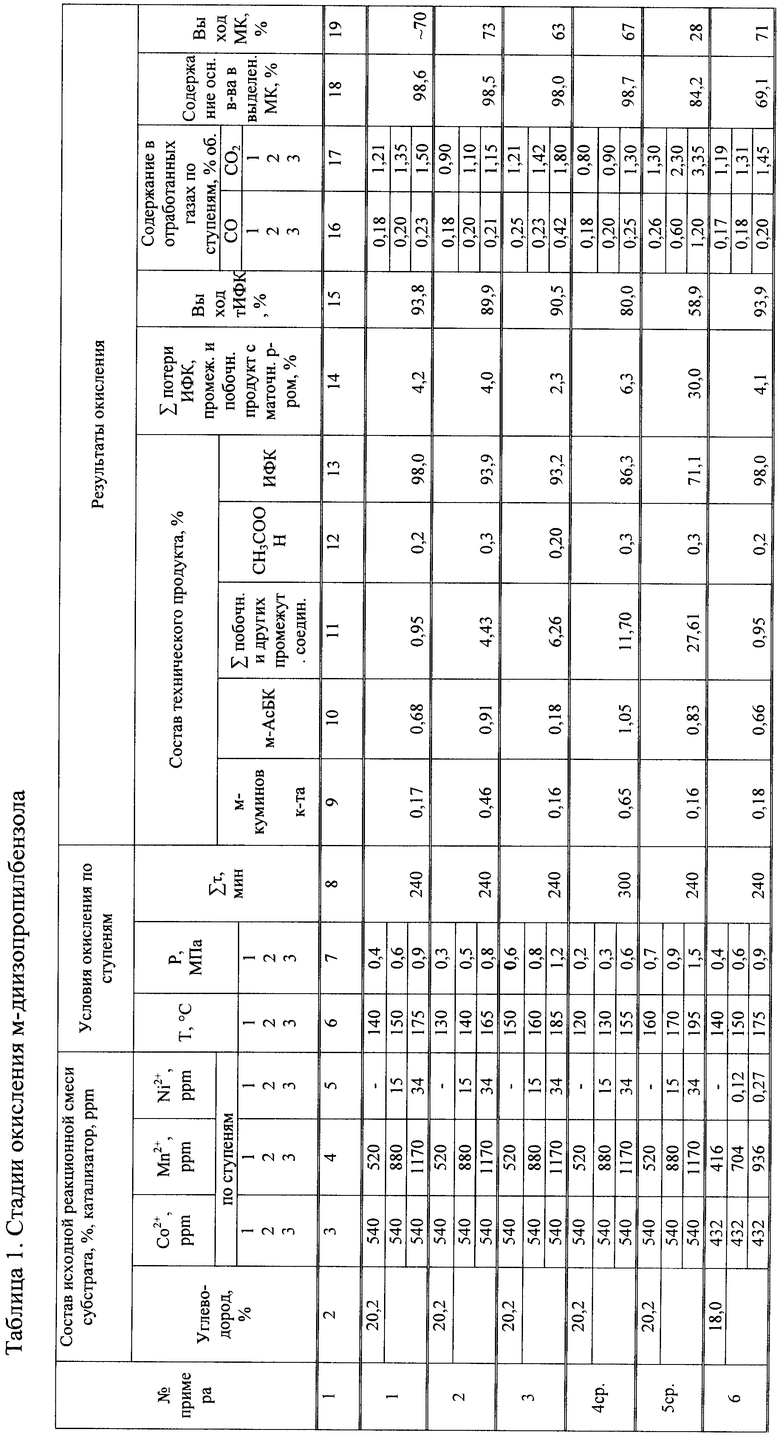
Изобретение относится к технологии органического и нефтехимического синтеза. Конкретно оно касается технологии получения изофталевой кислоты (ИФК) и сопутствующего продукта - муравьиной кислоты (МК) жидкофазным окислением О2-газом м-диизопропилбензола и м-этил-, изопропилбензола. ИФК является широко используемым мономером и полупродуктом в полимерной химии для получения химических волокон, полиэфирных пленок, красителей, пластмасс. МК является сильной карбоновой кислотой, и ее используют при отделке текстиля, бумаги, кожи; для получения пестицидов, лекарственных средств, консервантов зеленых кормов, фруктовых соков; применяется как дезинфицирующее средство, как растворитель. В промышленной практике ее получают разложением формиатов действием H2SO4 или HNO3, а также кислотным гидролизом или аммонолизом метилформиата. В связи с тем, что в промышленных способах получения ИФК используют чистый м-ксилол, выделение которого из ксилольных фракций сопряжено с значительными затратами и техническими сложностями, предложены способы получения ИФК из других альтернативных источников нефтехимического сырья - изомеров диизопропилбензола или этил-, изопропилбензола.
Один из первых известных способов получения фталевых кислот из изомеров диизопропилбензола предложен в Голландском патенте №108519 (1953). Согласно этому способу диизопропилбензол (на примере п-диизопропилбензола) окисляют кислородсодержащим газом в жидкой фазе жирной алифатической кислоты (СН3СООН) в присутствии растворимых в реакционной среде солей кобальта и марганца при температуре 120-300°С, предпочтительно 80-180°С, и повышенном давлении 10-45 кг/см2, предпочтительно при 20-30 кг/см2. Весовую концентрацию катализатора ацетатов Со и Mn по отношению к исходному диизопропилбензолу рекомендуют поддерживать в пределе 0,6-1,0% при соотношении Со:Mn=1:0,1÷1. При продолжительности реакции окисления в интервале 6-12 часов образуется терефталевая кислота (ТФК) с общим выходом 31,2-41,3% на прореагировавший п-диизопропилбензол.
К основным недостаткам предложенного способа следует отнести пониженный выход целевого продукта, не превышающий 41,3%, и значительную продолжительность реакции окисления 10-12 часов.
Кроме того, проведение процесса при повышенном давлении 20-30 кг/см2, значительно превышающем упругость паров растворителя (СН3СООН) и образующихся легкокипящих продуктов реакции - реакционной воды, формальдегида, муравьиной кислоты, а также побочных эфиро-производных соединений в виде метилацетата, приводит в рекомендуемом температурном режиме 80-180°С к их накоплению в жидкой фазе реакционной смеси (в оксидате) и к замедлению реакции. Известно, что завышенное содержание Н2О и муравьиной кислоты снижает активность катализатора вплоть до полной его дезактивации на завершающей стадии реакции, приводит к прекращению процесса окисления значительно раньше, чем это требуется для полного превращения диалкилбензола в бензолдикарбоновую кислоту.
Известен способ получения смеси терефталевой и изофталевой кислот путем жидкофазного окисления пара- и мета-изомеров диизопропилбензола кислородсодержащим газом в среде уксусной кислоты в присутствии ацетатов Со и Mn (патент GB №786930, 1957). Отличительной особенностью этого способа является то, что реакцию окисления осуществляют при более низкой температуре 100-140°С и давлении близком к упругости паров растворителя (СН3СООН) и легкокипящих продуктов реакции (Н2О, формальдегида, муравьиной кислоты и метилацетата) в интервале 2-10 кг/см2. Тепло реакции отводится в основном за счет испарения растворителя и легкокипящих продуктов окисления. В качестве кислородсодержащего газа может быть использован воздух, газовая смесь, обогащенная кислородом в большей степени, чем это имеет место в воздухе, или же чистый кислород. Выход целевого продукта (ТФК-ИФК) при окислении смеси п-, м-диизопропилбензола по предложенному способу достигает 82%.
Вышеизложенный способ получения смеси ИФК и ТФК из смеси м- и п-диизопропилбензолов имеет важное преимущество перед ксилолами. Это связано с тем, что при подборе соответствующих условий реакции алкилирования бензола или кумола происходит образование практически чистых м- и п-изомеров диизопропилбензола, тогда как образование о-изомера может быть подавлено. В дальнейшем м- и п-диизопропилбензолы можно легко выделить ректификацией и подвергнуть их окислению до смеси ИФК и ТФК по предложенному способу.
В то же время процесс получения ИФК и ТФК из м- и п-диизопропилбензола согласно патенту №108519 обладает рядом существенных недостатков, а именно:
1) В процессе окисления в присутствии Со-Mn-катализатора при относительно низкой температуре <150°С образуется смесь сырых ИФК-ТФК с большим содержанием примесей. Образовавшуюся бинарную смесь сырых ИФК-ТФК необходимо в дальнейшем разделить на индивидуальные мономеры и провести их очистку до требуемого качества. Это сильно усложняет схему процесса, так как достигнуть четкого разделения смеси ИФК-ТФК методом кристаллизации является проблематичным из-за повышенной их способности к сокристаллизации, а разделение методом ректификации практически невозможно из-за высоких температур кипения (до 280°С), приводящих к возгонке, температурной деструкции и осмолению разделяемых продуктов.
2) Процесс окисления осуществляют при относительно низкой температуре, что приводит к понижению скорости реакции и как следствие к увеличению продолжительности процесса т.е. к снижению производительности реакционного узла. Повышение качества технической смеси ИФК-ТФК по содержанию вредных примесей - куминовых кислот, ароматических кетокислот и альдегидокислот - путем повышения температуры до 180-200°С (этот температурный интервал обычно используется в известных процессах получения ИФК и ТФК из индивидуальных м- и п-ксилола) невозможен, так как при температуре t≥160°С происходит термическое разложение муравьиной кислоты до СО, СО2 и Н2О, что резко снижает экономическую эффективность способа.
3) Использование других физико-химических методов разделения и очистки смеси ИФК-ТФК, например, путем этерификации их метанолом, последующим разделением смеси диметилизофталата (ДМИ)-диметилтерефталата (ДМТ) и гидролизом последних до ИФК и ТФК в принципе возможен, однако в этом случае значительно усложняется схема процесса, увеличиваются материальные и энергетические затраты на производство, снижается конкурентоспособность предложенного способа по отношению к известным.
Наиболее близким по технической сущности и достигаемым результатам является способ, согласно которому изофталевую и терефталевую кислоту получают окислением индивидуальных м-диизопропилбензола или п-диизопропилбензола кислородсодержащим газом в жидкой фазе соответствующей алифатической карбоновой кислоты при температуре 100-140°С и давлении 2-10 кг/см2 в присутствии солей кобальта и марганца (патент DE №1008279, май 1957 г.). Отличительным признаком этого способа является то, что окисление диизопропилбензола, конкретно м-диизопропилбензола, осуществляют в диапазоне абсолютных давлений от 2 до 10 атм О2-газом с избытком кислорода по отношению к стехиометрии в температурном интервале 100-140°С таким образом, что тепло реакции из реакционной зоны отводят преимущественно испарением уксусной кислоты и легколетучих продуктов, образованных в процессе окисления. Процесс получения ИФК из м-диизопропилбензола согласно этому способу может осуществляться как в аппаратах с мешалкой, так и в реакторах барботажного или газлифтного типа (т.е. без мешалок), в которых перемешивание достигается за счет энергии всплывания газовых (парогазовых) пузырьков. Ниже приводится один из типичных примеров получения ИФК из м-диизопропилбензола в реакторе барботажного типа.
Стеклянный цилиндрический реактор (диаметр 6,5 см, высота 250 см) заполнили предварительно реакционной смесью, содержащую:
250 г м-диизопропилбензола;
460 г м-изопропилбензойной кислоты;
1,8 г ацетата кобальта;
0,6 г ацетата марганца;
2300 г 89%-ной уксусной кислоты
и при температуре 120°С, давлении 6,5 атм. абсолюта, в нижнюю часть реактора вводили 1100 л/час воздуха.
Во время процесса окисления, который длился 10 часов, в реактор непрерывно закачивали 8823 г СН3СООН, а также каждый час подавали по 65 мл (около 55 г) м-диизопропилбензола. Практически одновременно из реакционного устройства отводилось 45 г изофталевой кислоты. Вместе с отработанным воздухом из реактора выводились пары растворителя (СН3СООН) и легкокипящих продуктов реакции.
Путем фильтрации охлажденного, полученного в течение 10 часов оксидата, выделено 1021 г осадка сырой ИФК и 2396 г фильтрата. Осадок промыли ацетоном, а затем высушили до постоянного веса при температуре 140°С и получили 692 г сухой изофталевой кислоты (ИФК). Выход составил ≈80% в пересчете на м-диизопропилбензол.
Указанный способ позволил существенно повысить эффективность процесса получения фталевых кислот из диизопропилбензолов и, в частности, синтез ИФК из м-диизопропилбензола. Прежде всего, повышен выход технического продукта (ИФК) до 82%, значительно уменьшено в зоне окисления содержание продуктов реакции, ведущих к снижению активности (ингибированию) катализатора (реакционной воды до 1,04%, муравьиной кислоты до 0,77%) путем вывода их из реакционной зоны в виде паров с отработанным газом.
Предложена схема эффективного отвода тепла реакции за счет испарения растворителя (СН3СООН) и легкокипящих продуктов окисления м-диизопропилбензола в найденных пределах технологических параметров: по температуре (100-140°С), по давлению (4-8 кг/см2), по избытку кислорода ≥100 мол.%.
К недостаткам предложенного способа следует отнести относительно низкий выход технической ИФК (не более 82%), большая продолжительность реакции окисления (≥7 часов). Это указывает на пониженную удельную производительность реактора и процесса в целом
Кроме того, после 10 часов окисления в жидкой части оксидата после выделения твердых (кристаллических) продуктов содержится более 20% промежуточных (недоокисленных) соединений в виде м-изопропил-бензойной кислоты и более 2% исходного м-диизопропилбензола. Это подтверждает относительно низкую степень конверсии м-диизопропилбензола в целевой продукт (50-60%).
Целью настоящего изобретения является получение изофталевой кислоты (ИФК) и муравьиной кислоты (МК) жидкофазным окислением О2-газом в среде уксусной кислоты в присутствии катализатора солей Со и Mn при повышенной температуре и давлении с последующим выделением ИФК и ее очисткой перекристаллизацией в водно-уксусном растворителе, выделением МК методом дистилляции из обводненного уксуснокислого конденсата, образованного при охлаждении парогазовой смеси (ПГС), выводимой из зоны реакции с отработанным воздухом, где осуществляют окисление м-диизопропилбензола или м-этил-, изопропилбензола осуществляют в три ступени с возрастанием по ступеням температуры в пределах (°С) 130-150; 140-160; 165-185°С, давления (МПа) 0,3-0,6; 0,6-0,8; 0,9-1,2, суммарной концентрации Co-Mn-Ni катализатора (ppm) 800÷1060, 1000÷1435, 1250÷1744 и при протоке воздуха через зоны окисления поддерживают концентрацию СО/СО2 в отработанном газе, после каждой ступени в пределах (об.%) 0,18-0,25/0,9-1,12; 0,2-0,3/1,1-1,42; 0,23-0,42/1,2-1,6 достигают содержания ИФК в продуктах окисления, выделенных из охлажденного оксидата 3-й ступени не ниже 94%, после чего техническую ИФК (тИФК) подвергают очистке методом последовательной перекристаллизации в СН3СООН при температуре нагрева суспензии до 180-200°С, затем в H2O при температуре нагрева суспензии 200-230°С с получением высокочистой ИФК, а параллельно образующуюся в процессе окисления муравьиную кислоту выделяют из парогазовой смеси, выходящей из 1-й и 2-й ступеней окисления, путем охлаждения ПГС до 30-40°С, выделения образовавшегося конденсата (К) и его обработки н-бутилацетатом (Н-БАс) в соотношении К:Н-Бас=1:0,17, после чего методом ректификации из полученной смеси последовательно выделяют реакционную воду в виде ее азеотропной смеси с Н-БАс, затем муравьиную и уксусную кислоты.
Соотношение Со:Mn:Ni в составе катализатора на 1-й, 2-й и 3-й ступенях окисления снижают в пределах: 1:1:0, 1:1,2-1,5:0,01, 1:1,8-2:0,02 соответственно.
Дискретное увеличение суммарной концентрации Co-Mn-Ni на 2-й и 3-й ступенях осуществляют путем индивидуальных вводов уксуснокислых растворов тетрагидратов ацетатов Со, Mn и Ni = до 1:1,5:0,01 и 1:2:0,02 соответственно.
Содержание ИФК в продуктах окисления, выделенных из оксидата 3-й ступени, достигает 93,2-98,8%.
Такие новые приемы как разделение процесса окисления на три ступени с изменением условий их проведения по температуре, концентрации и соотношению компонентов Со-Mn катализатора, модифицированного добавкой Ni, создание условий по температуре и давлению для непрерывного отвода из зон реакции образующейся муравьиной кислоты и последующего ее выделения, как сопутствующего продукта, при получении изофталевой кислоты из мета-изомеров диизопропилбензола или м-этил-, изопропилбензола, привело к неожиданным результатам, обеспечивающим повышение выхода целевого продукта (ИФК) и сопутствующего продукта (МК).
Ниже приводится описание пилотной установки, а также примеры, раскрывающие, но не ограничивающие, сущность предложенного изобретения.
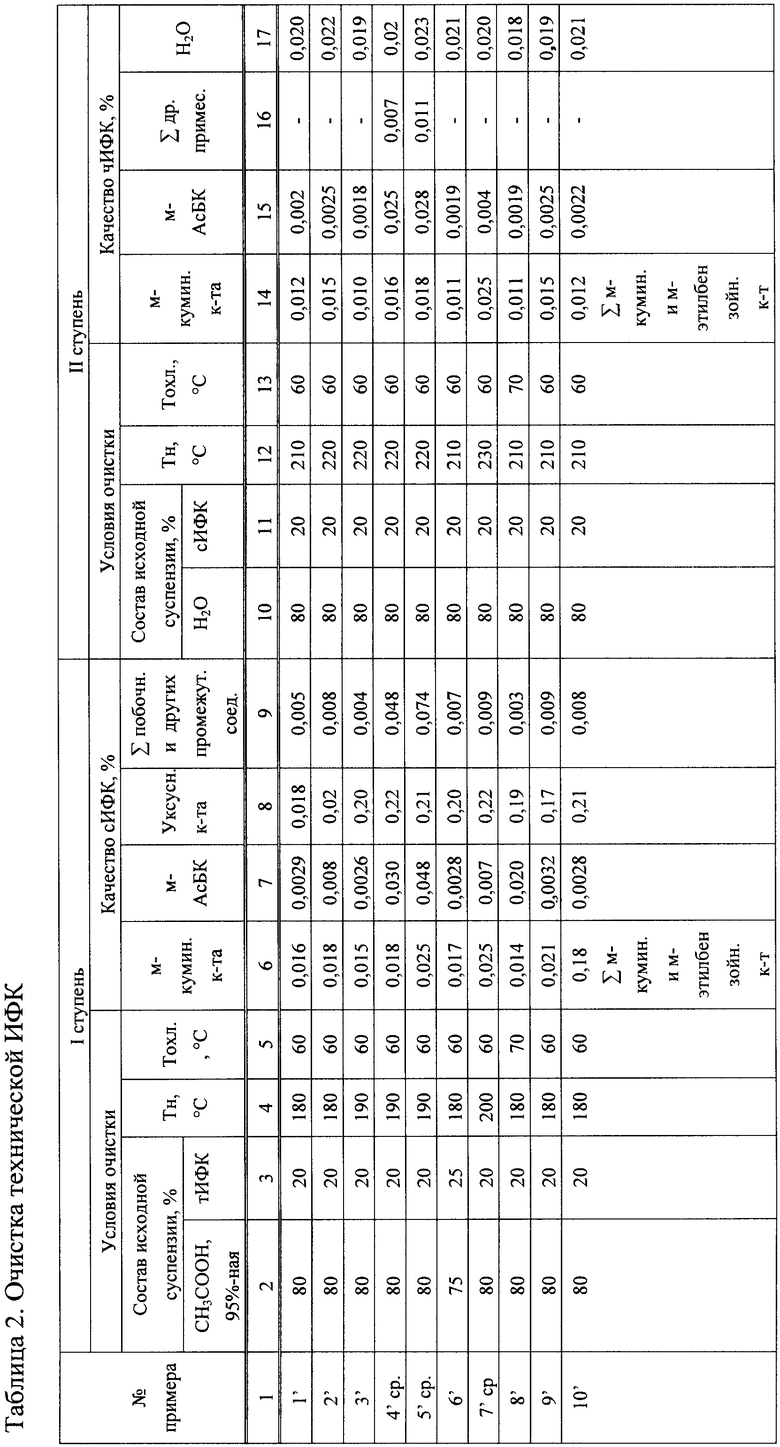
На фигуре 1 представлена технологическая схема пилотной установки окисления концентрата м-диизопропилбензола и м-этил-изопропилбензола до изофталевой кислоты в реакторах с турбинной мешалкой.
В сборник исходной реакционной смеси 1 загружают реагенты для окисления в периодическом или непрерывном режиме. При окислении в периодическом режиме исходными реагентами окисления являются м-диизопропилбензол или м-этил-, изопропилбензол, компоненты катализатора (тетрагидраты ацетатов Со, Mn) и растворитель - СН3СООН, которые дозирующим насосом 2 подаются в реактор окисления 3 по линии исходной реакционной смеси I для последующего окисления воздухом в дискретном 3-ступенчатом по технологическим параметрам режиме. Подача воздуха в реактор окисления 3 осуществляется от компрессора сжатого воздуха 4 через фильтр-поглотитель влаги 4a по линии II. Реактор окисления 3 изготовлен из титана BTI-0 (V=1,2 л), снабжен турбинной мешалкой с переменным числом оборотов, конденсатором-холодильником 5 для охлаждения парогазовой смеси, выходящей из реактора, электронагревателем 14, регулятором поверхности теплопередачи 15, пробоотборником 16, расходомером газа 17, угольным фильтром очистки газа 18, а также газоанализаторами непрерывного измерения концентраций O2, СО2, СО, СН4 в отработанных газах 19 по линии VI. Необходимые изменения концентрации катализатора и соотношения его компонентов на 2-й и 3-й ступенях окисления осуществляли вводом растворов тетрагидратов ацетатов Mn и/или бинарной смеси Mn-Ni через капсулы 13, 13a по линиям VIII. После достижения заданной степени превращения субстрата в режиме 1-й ступени дальнейшее окисление проводили в этом же реакторе, в режимах 2-й и 3-й ступеней путем изменения температуры, суммарной концентрации катализатора и соотношения его компонентов. После завершения процесса оксидат, полученный в условиях 3-ступенчатого окисления, перепускают по переточной линии с механизмом удаления забивок 7 по линии VII в реактор-кристаллизатор 8, где он охлаждается путем снижения давления и прохождением образовавшихся паров через холодильник-конденсатор 9 и возврата охлажденного конденсата в реактор-кристаллизатор 8 по линии V, через сборник флегмы 6a. Охлажденный до 60-80°С оксидат, в виде образовавшейся суспензии сливается в сборник оксидата 10, из которого под давлением направляется на разделение в дру-фильтр 11 по линии III. Полученную при фильтрации пасту технической изофталевой кислоты (далее тИФК) перемещают в сушилку 12 и после сушки до постоянного веса подвергают анализу и выгрузке по линии X. Образовавшийся в нижней части друк-фильтра 11 маточный раствор, содержащий уксусную кислоту, растворенный в ней катализатор и примеси, выводят из процесса для регенерации СН3СООН и катализатора известными приемам по линии IX. Парогазовая смесь (далее ПГС), выходящая из реактора при его работе в периодическом 3-ступенчатом режиме, по линии IV поступает в холодильник-конденсатор 5, где охлаждается до 40°С. Образовавшийся конденсат, состоящий из уксусной и муравьиной кислот, реакционной воды и других легкокипящих продуктов, из конденсатора-холодильника 5 сливается в промежуточный сборник флегмы 6, откуда основное его количество (до 80%) под давлением поступает в сборник конденсата 20 по линии V, а незначительная часть в виде флегмы возвращается в реактор 3.
Для разделения тройной смеси уксусная кислота-муравьиная кислота-реакционная вода используется 2-х колонная схема с применением азеотро-пообразующего реагента - н-бутилацетата (далее Н-БАс). Процесс разделения осуществляют следующим образом.
В сборник конденсата 20 вводят Н-БАс по линии XI в необходимом соотношении с конденсатом, после чего насосом 21 подают в колонну азеотропной ректификации 22 по линии XIII для отделения реакционной воды. Подвод тепла осуществляют электронагревательным устройством 14, расположенным в нижней части колонны. Выходящие пары из верхней части колонны после их конденсации в дефлегматоре 23 перетекают в флорентийский сосуд 24, в котором происходит разделение конденсата на водный и органический слои. Водный слой выводится из процесса по линии XIV, органический слой возвращается в процесс в виде орошения колонны сверху, а также в виде рециклового компонента питающей смеси. Образовавшаяся в нижней части колонны бинарная смесь СН3СООН-НСООН выводится из куба и насосом 25 подается в колонну 26 для ее разделения по линии XV. С верхней части колонны после конденсации паров в дефлегматоре 27 выводится муравьиная кислота по линии XVI, из куба колонны по линии XVII - уксусная кислота. Муравьиную кислоту используют как товарный продукт, уксусную кислоту - как рецикловый растворитель в процессе окисления, а именно для приготовления исходной реакционной смеси, а также для приготовления уксуснокислого раствора тетрагидрата ацетата Mn и/или бинарной смеси Mn-Ni, подаваемой на 2-ю и 3-ю ступени окисления.
Очистка технической ИФК проводится в две ступени методом рекристаллизации в СН3СООН и перекристаллизацией в H2O в реакторах окисления 3 и 8 (фиг.1) при нагреве 20%-ной суспензии в СН3СООН до 180-190°С, в Н2О - до 210-220°С. Схемы процессов фильтрации суспензий в уксуснокислой среде и в воде, а также сушки сИФК и чИФК аналогичны выше описанным при выделении и сушке технической ИФК.
Процесс окисления м-диизопропилбензола или м-этил-, изопропилбензола в непрерывном 3-ступенчатом режиме по варианту схемы, представленной на фиг.1, осуществляют в два приема. Сначала в реакторе окисления 3 в условиях первой ступени в непрерывном режиме нарабатывают партию оксидата-1 в количестве 5-6 литров, который затем загружают в освобожденный сборник исходной реакционной смеси 1 и насосом 2 непрерывно подают в реактор окисления 3, в котором технологические параметры соответствуют режиму 2-й ступени окисления с образованием оксидата-2. Оксидат-2, непрерывно перетекая в реактор окисления 8, подвергается доокислению в условиях технологических параметров, соответствующих режиму 3-й ступени с образованием оксидата-3.
Ниже приводятся примеры, раскрывающие сущность предложенного способа.
Пример 1. Приготавливают исходную реакционную смесь (ИРС) в сборнике 1 (фиг.1), куда загружают:
Содержимое сборника нагревают до 80°С и в условиях перемешивания достигают полного растворения компонентов катализатора. После анализа определен следующий состав исходной реакционной смеси, %:
Приготовленную ИРС указанного состава насосом 2 закачивают в реактор 3, предварительно продутый инертным газом (N2).
В условиях перемешивания реакционную массу нагревают до 130°С при общем давлении 0,3 МПа.
Практически одновременно с этим прекращают подачу азота и вместо него в реактор подают воздух в количестве, обеспечивающем поддержание избытка кислорода в отходящих газах не более 5% объемных.
Процесс окисления проводят в периодическом режиме в три ступени с повышением температуры (°С) 140, 150, 175, давления (МПа) 0,4, 0,6, 0,9 концентрации компонентов катализатора (ppm):
Время реакции на каждой ступени определялось по снижению поглощения кислорода и содержанию СО и СО2 в отработанном газе. Тепло реакции снималось за счет испарения СН3СООН и легкокипящих продуктов реакции. Заданное увеличение концентрации катализатора в режимах 2-й и 3-й ступеней достигалось вводом их в реактор 3 через капсулу 13 в виде уксуснокислых растворов.
Парогазовая смесь, выходящая из реактора, охлаждалась в конденсаторе 5 до 30-40ºС. В результате охлаждения образовавшийся конденсат частично (20%) возвращали в реактор через сборник 6, остальное количество (80%) выводилось из процесса для регенерации СН3СООН и выделения образовавшейся в результате реакции муравьиной кислоты.
После завершения процесса окисления в 3-ступенчатом режиме, оксидат охлаждали до 60°С и проводили его обработку и последующий анализ. Охлажденную суспензию оксидата подвергали разделению методом фильтрации. Пасту выделенного влажного продукта технической ИФК (далее тИФК), высушили до постоянного веса при температуре ≤125ºС. По результатам анализа тИФК имела состав, %:
Выход технической ИФК на стадии окисления составил 93.8%.
Условия и результаты примера 1 и последующих примеров приведены в таблице 1.
Влажную пасту технической ИФК подвергли обработке СН3СООН, сущность которой заключается в следующем. Приготовили 20%-ную суспензию тИФК в 95%-ной СН3СООН. В условиях перемешивания суспензию нагрели до 180°С с выдержкой 5 минут, после чего охладили до 60°С со скоростью понижения температуры 3°С/мин. Охлажденную суспензию ИФК в СН3СООН отфильтровали. Кристаллический осадок ИФК высушили до постоянного веса и подвергли анализу. Получен продукт следующего качества, %:
Из полученных результатов следует, что при обработке технической ИФК уксусной кислотой в вышеприведенных условиях ее качественные показатели значительно улучшились. Данный продукт по существующим нормативам относится к продукту средней степени чистоты (сИФК).
Для достижения требуемых по техническим условиям качественных показателей конечного продукта проведена перекристаллизация сИФК в воде при температуре нагрева 20%-ной суспензии 210°С. Раствор ИФК охлаждали до 60°С со скоростью 3°С/мин.
В результате перекристаллизации получена очищенная ИФК (далее чИФК) с показателями качества, %:
Муравьиную кислоту, образовавшуюся: при окислении изопропильных групп м-диизопропилбензола, выделили из конденсата парогазовой смеси, накопленной в сборнике 6 в количестве 540 г (505 мл) (фиг.1). Конденсат имел следующий состав, %:
Пробу конденсата указанного состава в количестве 100 мл (107,0 г) смешали с н-бутилацетатом (Н-БАс) в соотношении конденсат: Н-БАс=1:0,17, после чего отогнали азеотропную смесь (Н2О-Н-БАс). Из кубовой части, состоящей из бинарной смеси СН3СООН-НСООН методом ректификации выделили муравьиную кислоту в количестве 9,8 мл (12 г), содержащую 98,6% основного вещества, что соответствует ее выходу (от теории) ~70%.
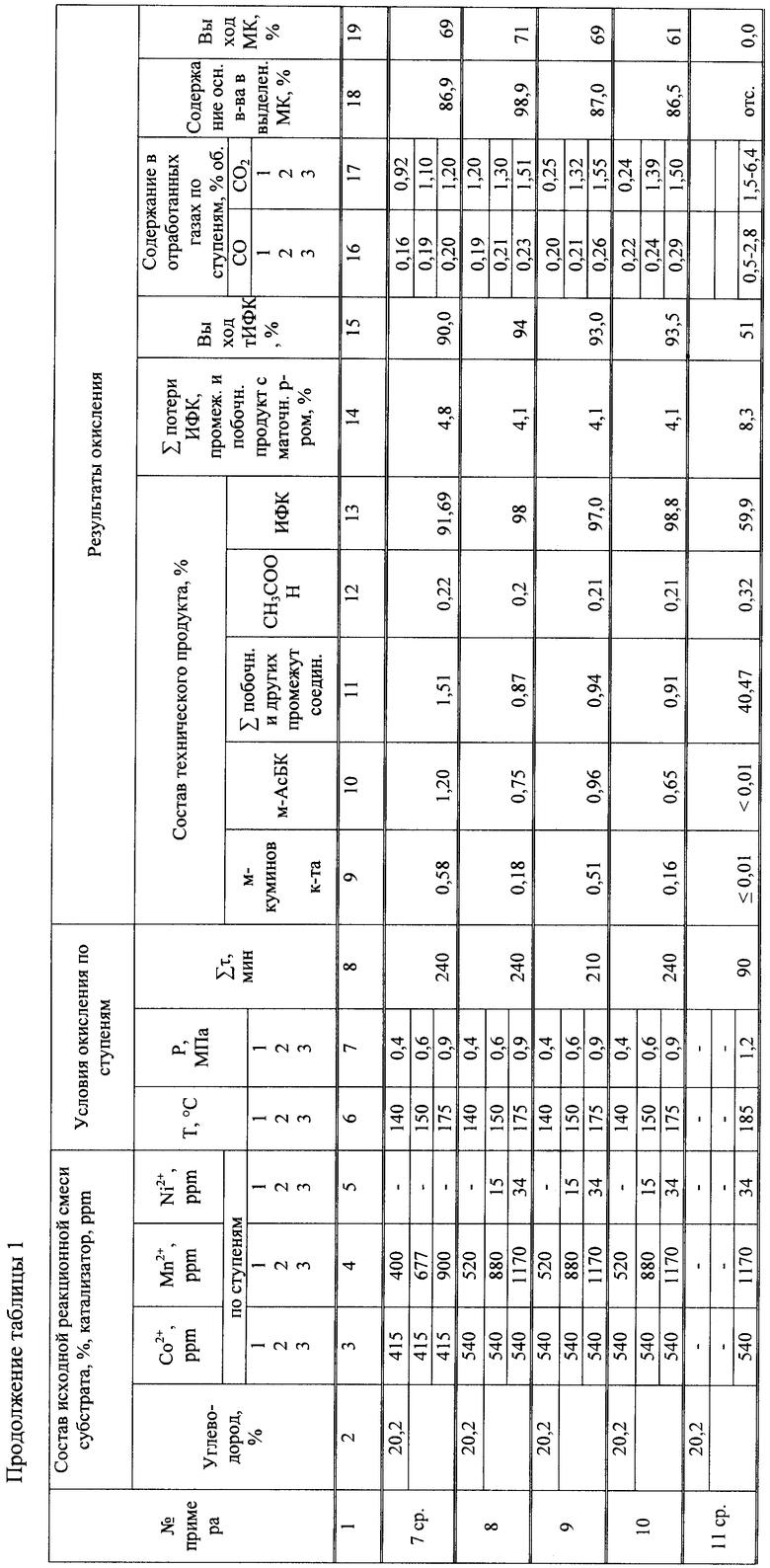
Условия и результаты первого и последующих опытов по очистке технической ИФК в уксусной кислоте и воде приведены в таблице 2.
Пример 2. Опыт проведен в условиях примера 1, с той лишь разницей, что температуру на каждой ступени понизили на 10°С, давление на 0,1МПа, а температуру нагрева 20%-ной суспензии на стадии перекристаллизации в Н2О повысили на 10°С.
Достигнут результат по качеству технической ИФК, %:
Выход технической ИФК составил - 89,9%, муравьиной кислоты ≈73%. Качество чИФК, полученной после очистки в условиях примера 1' (таблица 2) достигло показателей, %:
соответствующих техническим требованиям.
Из приведенных данных следует, что снижение температуры на 3-х ступенях окисления на 10°С приводит к снижению выхода ИФК на 3,9%, что указывает на min допустимый температурный режим окисления на 3-х ступенях 130°С, 140°С, 165°С соответственно. Требуемое качество чИФК достигнуто при повышенной температуре нагрева суспензии 220°С в процессе перекристаллизации в Н2О.
Пример 3. Опыт проведен в условиях примера 1, с той лишь разницей, что температуру на каждой ступени повысили на 10°С.
Достигнут следующий результат по качеству технической ИФК, %:
Выход ИФК составил - 90,5%, муравьиной кислоты ≈63,0%.
Пониженный выход ИФК и МК в сравнении с примером 1 обусловлен повышенным температурным режимом окисления на 10°С, что привело к возрастанию скоростей побочных реакций окислительного декарбонилирования и декарбоксилирования: [СО] возросла с 0,23% до 0,42 об.%, [СО2] с 1,5% до 1,8 об.%;
Качество очищенной ИФК достигло показателей, %:
удовлетворяющих техническим требованиям.
Пример 4 (сравнительный). Опыт проведен в условиях примера 1, с той лишь разницей, что температура по ступеням понижалась на 20°С.
Результат: продолжительность реакции до момента прекращения поглощения кислорода возросла с 240 мин до 300 мин, что указывает на снижение производительности процесса.
Качество технической ИФК значительно ухудшилось и достигло значений, %:
Потери ИФК, промежуточных и побочных продуктов с маточным раствором - 6,3%.
Понизились также выходы технической ИФК до 80%, муравьиной кислоты до 67%. Качество очищенной ИФК не достигло требуемых значений: содержание м-куминовой кислоты и м-АсБК превышало допустимые нормы на порядок.
Пример 5 (сравнительный). Опыт проведен в условиях примера 1, с той лишь разницей, что температура по ступеням повышалась на 20ºС.
В результате значительного повышения температуры возросло содержание побочных продуктов декарбоксилирования ([CO2] повысилось с 1,5 об.% до 3,35 об.%), произошло осмоление оксидата, качество технической ИФК резко ухудшилось и по суммарному содержанию побочных соединений.
Состав технической ИФК достиг следующих значений, %:
Потери ИФК, промежуточных и побочных продуктов с маточным раствором - 30%. Выход ИФК на стадии окисления снизился с 93,8% до 58,9% (в 1,5 раза), а выход муравьиной кислоты понизился с 70% до 28% (более, чем в 2 раза).
Содержание примесей в очищенной ИФК превысило более чем на порядок от допустимых значений.
Пример 6. Опыт проведен в условиях примера 1, с той лишь разницей, что концентрацию углеводорода в исходной реакционной смеси уменьшили до 18%, а суммарные концентрации Co-Mn-Ni катализатора на 3-х ступенях окисления понизили на 25% до (ppm): 800, 1000 и 1250 соответственно.
Получены результаты.
Качество технической ИФК на стадии окисления достигло значений, близких к опыту 1, %:
На стадии очистки достигнуто качество очищенной ИФК, соответствующей техническим требованиям.
Содержание, %:
Содержание основного вещества в муравьиной кислоте 99,1%, выход - 71%.
Пример 7 (сравнительный). Опыт проведен в условиях примера 1, с той лишь разницей, что суммарную концентрацию Со-Mn катализатора снижают в 1,3 раза и исключают Ni, а температуру нагрева суспензий ИФК при последовательной перекристаллизации в уксусной кислоте и в воде повышают на 20°С до 200°С и 230°С соответственно.
Получены результаты.
Качество технической ИФК на стадии окисления значительно понизилось и достигло значений, %:
В процессе очистки полученной технической ИФК последовательной перекристаллизацией в уксусной кислоте и воде требуемое качество чИФК не достигнуто даже в условиях повышенной температуры нагрева суспензий в уксусной кислоте 200°С, в воде 230°С. Заметно понизилось качество муравьиной кислоты (содержание основного вещества 86,9%) и ее выход (69%).
Пример 8. Опыт проведен в условиях примера 1, с той лишь разницей, что окисление м-диизопропилбензола ведут в 3 ступени в непрерывном режиме с суммарным временем пребывания 240 минут, а температуры охлаждения суспензий ИФК в СН3СООН и в Н2О на стадии перекристаллизации повысили на 10°С.
Получен следующий результат.
Качество технического продукта, %:
приемлемое для дальнейшей очистки до требуемого качества.
Выход технической ИФК достиг - 94,0%, муравьиной кислоты - 71%. Полученные результаты указывают на то, что проведение процесса окисления в непрерывном режиме при прочих равных условиях обеспечивают наиболее высокие результаты по качеству и выходу как целевого продукта - ИФК, так и выходу сопутствующего продукта - муравьиной кислоты. Это объясняется фактором установления стационарности концентраций компонентов реакционных смесей на каждой ступени вместо текущих концентраций (от min к max) в периодическом процессе.
Пример 9. Опыт проведен в условиях примера 8, с той лишь разницей, что суммарное время пребывания на трех ступенях сократили на 30 минут с 240 до 210 мин.
Получен следующий результат.
Качество технического продукта, %:
Выход технической ИФК достиг 93,0%, выход муравьиной кислоты -69%.
Качество очищенной ИФК на стадиях перекристаллизации в СН3СООН и Н2О достигло показателей, %:
удовлетворяющее техническим требованиям.
Из приведенных данных следует, что суммарное время окисления (время пребывания) 210 мин является минимально допустимым для достижения требуемого качества чИФК и приемлемого выхода ИФК и МК.
Пример 10. Опыт проведен в условиях примера 1, с той лишь разницей, что вместо м-диизопропилбензола используют м-этил-, изопропилбензол.
Получены следующие результаты.
На стадии окисления качество технической ИФК достигло следующих значений, %:
 - 0,16;
- 0,16;
Потери ИФК, промежуточных и побочных продуктов с маточным раствором 4,1%. Выход технической ИФК 93,5%, муравьиной кислоты - 61%.
Приведенные результаты показывают, что при прочих равных условиях окисление другого по природе алкилбензола - м-этил-, изопропилбензола приводит к сопоставимым результатам по качеству и выходу ИФК, однако выход муравьиной кислоты понижается более чем на 10%. Это может быть объяснено различием продуктов окисления этильной и изопропильной групп в этой молекуле.
Пример 11 (сравнительный). Опыт проведен в условиях примера 3, с той лишь разницей, что реакцию окисления м-диизопропилбензола проводят в одну стадию в режиме 3-й ступени.
Результаты. Поглощение О2 прекратилось через 90 минут. При проведении процесса в одну стадию в режиме 3-й ступени окисления резко возросли скорости как основной, так и побочных реакций окислительной деструкции декарбоксилирования и декарбонилирования. Содержание СО2 в отработанных газах возросло более чем в 3 раза (от 1,81 до 6,4%), СО - более чем в 6 раз (от 0,42 до 2,8%).
Оксидат представлял собой черную смолообразную суспензию. Выделенная паста кристаллического продукта имела темно-коричневый цвет. Она содержала ≈59% ИФК, остальное ≈40% побочные продукты окислительной конденсации, которые разделить и очистить не представлялось возможным. В маточном растворе муравьиной кислоты не обнаружено.
Таким образом, предложенное техническое решение позволяет решить поставленные задачи по расширению сырьевой базы, получению двух товарных продуктов и повышению выхода целевых продуктов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ И СОПУТСТВУЮЩИХ ПРОДУКТОВ ИЗ ИЗОМЕРОВ ЦИМОЛА И ДИИЗОПРОПИЛБЕНЗОЛА | 2009 |
|
RU2415836C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ И СОПУТСТВУЮЩИХ ПРОДУКТОВ ИЗ КСИЛОЛЬНЫХ ФРАКЦИЙ | 2009 |
|
RU2430911C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОФТАЛЕВОЙ КИСЛОТЫ С ВЫСОКОЙ СТЕПЕНЬЮ ЧИСТОТЫ | 2004 |
|
RU2266277C2 |
| СПОСОБ СЕЛЕКТИВНОГО ПОЛУЧЕНИЯ МЕТА-ДИАЛКИЛБЕНЗОЛОВ | 2011 |
|
RU2459796C1 |
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ | 2003 |
|
RU2254324C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 1997 |
|
RU2137753C1 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 2010 |
|
RU2458042C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 1998 |
|
RU2163592C2 |
| Способ получения тере- или изофталевой кислоты | 1983 |
|
SU1171452A1 |
| СПОСОБ ПОЛУЧЕНИЯ ВНУТРИМОЛЕКУЛЯРНЫХ АНГИДРИДОВ БЕНЗОЛПОЛИКАРБОНОВЫХ КИСЛОТ | 2009 |
|
RU2412178C1 |
Изобретение относится к технологии органического и нефтехимического синтеза, конкретно - к технологии получения изофталевой кислоты (ИФК) и сопутствующего продукта - муравьиной кислоты (МК) жидкофазным окислением О2-газом в среде уксусной кислоты в присутствии катализатора солей Со и Mn при повышенной температуре и давлении с последующим выделением ИФК и ее очисткой перекристаллизацией в водно-уксусном растворителе, выделением МК методом дистилляции из обводненного уксуснокислою конденсата. образованного при охлаждении парогазовой смеси (ПГС). выводимой из зоны реакции с отработанным воздухом, где осуществляют окисление м-диизоиропилбензола или м-этил-, изоиропилбензола в три ступени с возрастанием по ступеням температуры в пределах (°С) 130-150; 140-160; 165-185°С, давления (МПа) 0,3-0,6: 0,6-0,8; 0,9-1,2. суммарной концентрации Со-Mn-Ni катализатора (ppm) 800÷1060, 1000÷1435, 1250÷1744 и при протоке воздуха через зоны окисления поддерживают концентрацию СО/СО2 в отработанном газе, после каждой ступени в пределах (об.%) 0,16, 0,17, 0,18-0,25, 0,26/0,24. 0.25. 0,9-1,12, 1,19, 1,20, 1,21; 0,18, 0,2-0,3/0,9, 1,1-1,42; 0,2, 0,23-0,42/1,15. 1,2-1,6. 1,8 достигают содержания ИФК в продуктах окисления, выделенных из охлажденного оксидата 3-й ступени 93,2-98,8%, после чего техническую ИФК (тИФК) подвергают очистке методом последовательной перекристаллизации в СН3СООН при температуре нагрева суспензии до 180-200°С, затем в Н2О при температуре нагрева суспензии 200-230°С с получением высокочистой ИФК, а параллельно образующуюся в процессе окисления муравьиную кислоту выделяют из парогазовой смеси, выходящей из реактора окисления, путем охлаждения ПГС до 30-40°С, выделения образовавшегося конденсата (К) и его обработки н-бутилацетатом (Н-БАс) в соотношении К:Н-Бас=1:0,17, после чего методом ректификации из полученной смеси последовательно выделяют реакционную воду в виде ее азеотропной смеси с Н-Бас, затем муравьиную и уксусную кислоты. Способ позволяет получить два товарных продукта - ароматическую ИФК и алифатическую МК и повысить выход целевых продуктов. 2 з.п. ф-лы, 1 ил., 2 табл., 11 пр.
1. Способ получения изофталевой кислоты (ИФК) и муравьиной кислоты (МК) жидкофазным окислением О2-газом в среде уксусной кислоты в присутствии катализатора солей Со и Mn при повышенной температуре и давлении с последующим выделением ИФК и ее очисткой перекристаллизацией в водноуксусном растворителе, выделением МК методом дистилляции из обводненного уксуснокислого конденсата, образованного при охлаждении парогазовой смеси (ПГС), выводимой из зоны реакции с отработанным воздухом, где осуществляют окисление м-диизопропилбензола или м-этил-изопропилбензола в три ступени с возрастанием по ступеням температуры в пределах (°С) 130-150; 140-160; 165-185°С, давления (МПа) 0,3-0,6; 0,6-0,8; 0,9-1,2, суммарной концентрации Co-Mn-Ni катализатора (ppm) 800÷1060, 1000÷1435, 1250÷1744 и при протоке воздуха через зоны окисления поддерживают концентрацию СО/СО2 в отработанном газе, после каждой ступени в пределах (об.%) 0,16, 0,17, 0,18-0,25, 0,26/0,24, 0,25, 0,9-1,12, 1,19, 1,20, 1,21; 0,18, 0,2-0,3/0,9, 1,1-1,42; 0,2, 0,23-0,42/1,15, 1,2-1,6, 1,8 достигают содержания ИФК в продуктах окисления, выделенных из охлажденною оксидата 3-й ступени 93,2-98,8%, после чего техническую ИФК (тИФК) подвергают очистке методом последовательной перекристаллизации в СН3СООН при температуре нагрева суспензии до 180-200°С, затем в H2O при температуре нагрева суспензии 200-230°С с получением высокочистой ИФК, а параллельно образующуюся в процессе окисления муравьиную кислоту выделяют из парогазовой смеси, выходящей из реактора окисления, путем охлаждения ПГС до 30-40°С, выделения образовавшегося конденсата (К) и его обработки н-бутилацетатом (Н-БАс) в соотношении К:Н-БАс=1:0,17, после чего методом ректификации из полученной смеси последовательно выделяют реакционную воду в виде ее азеотропной смеси с Н-БАс, затем муравьиную и уксусную кислоты.
2. Способ по п.1, отличающийся тем, что соотношение Со:Mn:Ni в составе катализатора на 1-й, 2-й и 3-й ступенях окисления снижают в пределах: 1:1:0/1:1,2÷1,5:0,01/1:1,8÷2:0,02 соответственно.
3. Способ по п.1 или 2, отличающийся тем, что дискретное увеличение суммарной концентрации Co-Mn-Ni на 2-й и 3-й ступенях осуществляют путем индивидуальных вводов уксуснокислых растворов тетрагидратов ацетатов Со, Mn и Ni до соотношения Со:Mn:Ni в составе катализатора 1:1,5:0,01 и 1:2:0,02 соответственно.
| RU 2009109869 А, 27.09.2010 | |||
| Универсальный анализатор для магнитной сепарации минералов в полевых условиях | 1956 |
|
SU108519A1 |
| US 5110984 А, 05.05.1992 | |||
| RU 95117934 А, 20.09.1997 | |||
| Легирующее покрытие для литейныхфОРМ и СТЕРжНЕй | 1979 |
|
SU833357A1 |
| Устройство для герметизации узлов скважинного прибора | 1986 |
|
SU1408058A1 |
| GB 1336726 А, 07.11,1973 | |||
| Овчинников В.И., Александров В.Н., Гитис С.С | |||
| «Химия и технология мономеров» | |||
| Труды ВНИПИМ | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 3082250 А, 19.03.1963. | |||

