Данное изобретение относится к амидам R-2-аминоарилпропионовых кислот и фармацевтическим композициям, их содержащим, которые могут использоваться для предотвращения и ингибирования рекрутинга и активации лейкоцитов и для лечения патологий, непосредственно зависимых от указанной активации.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Для осуществления многих физиологических процессов требуется, чтобы клетки находились в тесном контакте с популяциями других клеток и/или экстраклеточными матрицами. Эти процессы адгезии, однако, необходимы для активации, миграции, пролиферации и дифференциации клеток. Некоторые семейства молекул клеточной адгезии (МКА), которые играют существенную роль в нормальных и патофизиологических процессах, опосредуют клеточно-клеточно-матриксные взаимодействия.
Процесс клеточной адгезии, существенный для активации нейтрофилов, сопровождается выделением цитокинов, среди которых есть IL-8 и МСР-1, которые дают возможность амплификации воспалительного процесса (см.: Huang C.D. et al., Chang Keng J. Hsueh, 22, 392, 1999; B.Walzog et al., FASEB J., 13, 1855, 1999).
Хемокины, в свою очередь, отличаются по своему действию от других цитокинов клеточной избирательностью их действия: каждый из них регулирует миграцию и функцию единственного вида клеток специфическим образом. Таким образом, тогда как МСР-1 влияет и направляет движение моноцитов, IL-8 выполняет исключительную роль нейтрофильного хемопривлекающего фактора.
Это подтверждается наличием высоких концентраций IL-8 в местах воспаления и в окружающей жидкости, количественно определенных в ходе многих острых патологий, опосредованных нейтрофилами, предотвращением серьезного поражения ткани и сниженной инфильтрацией нейтрофилов, наблюдаемых после введения антител к IL-8 в ходе экспериментов на моделях на животных, на которых демонстрируются зависимые от нейтрофилов патологии (см.: Yang X.D., et al, J. Leukoc. Biol. 66, 401, 1999). Типичными клиническими ситуациями, где активация нейтрофилов играет превалирующую патологическую роль, являются поражения, появляющиеся в результате восстановления церебрального кровотока (Stanimirovic D. and Satoh K., Brain Pathol., 10, 113, 2000) и в результате ишемии и восстановления кровотока в миокарде.
Эти наблюдения подтвердили гипотезу о том, что IL-8 составляет главный медиатор повреждения ткани, вызываемого нейтрофилами, до такой степени, что таким образом можно предложить интерлейкин-8 в качестве оптимальной мишени для терапевтического вмешательства при зависимых от нейтрофилов патологиях (N. Mukaida et al., Inflammation Res. 47 (Suppl. 3) S151, 1998). По этой причине в качестве альтернативы использования анти-IL-8 антител вещества с низкой молекулярной массой могли бы представлять большой интерес для клиники и быть чрезвычайно полезными, причем указанные вещества способны встраиваться в меж- и внутриклеточные системы передачи сигнала и подавлять миграцию человеческих нейтрофилов, стимулированную IL-8 и подобными веществами (GRO-α,β,γ; ENA-78, NAP-2, GCP-2).
В настоящее время все более признается, что становится более ясной определяющая роль того, что активация некоторых киназ и тирозинкиназ играет в динамике явления зависимого от IL-8 хемотаксиса.
В течение длительного времени предполагалось, что активация некоторых тирозинкиназ является запускающим событием феномена хемотаксиса после того, что Yasui et al. (J. Immunol. 152, 5922, 1994) продемонстрировали, что полное подавление хемотаксиса нейтрофилов (PMN) следовало за подавлением активности этих ферментов.
Совсем недавно обнаружено, что резкое снижение хемотаксиса нейтрофилов, вызванное рядом хемокинетических факторов, наблюдаемое у генетически модифицированных крыс, дефектных по ферменту фосфоинозитид-3-киназе (IР3К, Hirsch et al., Science, 287, 1049, 2000; Sasaki et al., Science, 287, 1040, 2000), позволило охарактеризовать этот фермент как ведущую киназу - primus movens последовательного каскада событий - и утвердиться в предположении об избирательной и определяющей роли, выполняемой ферментативными процессами фосфорилирования.
При феномене хемотаксиса, вызванного интерлейкином-8 у нейтрофилов, кроме фосфоинозитид-3-киназы, одинаково важную роль, по-видимому, играет Руk2 (богатая пролином тирозинкиназа), активация которой существенна для развития сопутствующих процессов клеточной адгезии. В свою очередь процесс активации Руk2, вызванный IL-8 в нейтрофилах, является зависимым от IР3К процессом, который, по-видимому, проходит и локализуется в областях клеточной мембраны, предназначенных для фокальной адгезии (Clark et al., 268, 233, 1995; Avraham et al., Blood, 88, 417, 1996) при прямом контакте с цитоструктурными протеинами, участвующими в феномене адгезии (например, винкулин, α-актин и т.д.).
Также в свете недавних открытий считают, что терапевтический потенциал новых молекул с низкой молекулярной массой, предназначенных для того, чтобы препятствовать феномену зависимого от IL-8 хемотаксиса, повышается посредством некоторой подавляющей активности в отношении факторов, которые вызывают адгезию, и/или антагонистической активности в отношении некоторых интегринов, таких как очень поздний антиген 4 (VLA-4), LPAM-1, чтобы блокировать их связывание со своими лигандами для того, чтобы практически предотвратить ab initio начало этого каскада событий, которые, начинаясь с внутриклеточных процессов адгезии, превращаются в активацию нейтрофилов.
Недавно были описаны N-ацилсульфонамиды и амиды R-2-арилпропионовой кислоты (WO 00/24710 и РСТ/ЕР01/01285 соответственно), которые характеризуются избирательностью их подавляющей активности стимулированного IL-8 хемотаксиса человеческих нейтрофилов. В ходе исследований, направленных на выяснение молекулярных механизмов действия, наблюдалось, что они проявляют очень значительную (70-80% подавления), зависимую от дозы подавляющую активность в отношении активности Руk2-тирозинкиназы для концентраций, находящихся в интервале от 10-7 до 10-8 М, отличных от концентраций, которые проявляют эффективное подавление зависимого от IL-8 хемотаксиса.
Кроме того, активность указанных амидов и N-ацилсульфонамидов R-2-арилпропионовых кислот, по-видимому, является полностью независимой от их участия в подавлении (СОХ-1 и/или СОХ-2) зависимых от циклооксигеназы воспалительных процессов.
Накоплены также данные о том, что ингибирование синтеза простагландинов (ПГ) (S)-энантиомерами 2-арилпропионовых кислот и некоторыми 2-арилуксусными кислотами может в течение длительного периода давать отрицательную рефлекторную реакцию на динамику зависимого от нейтрофилов воспалительного процесса, где ингибирование синтеза ПГ и, следовательно, PGE2 удаляет фактор регуляции эндогенного синтеза ФНО-α. Соответственно, при сравнении с тем же IL-8 ФНО-α может способствовать, вместе с цитокинами IL-6 и IL-1 и с молекулами адгезии (Е-селектин, ICAM-1 и С-реактивный белок) ухудшению сущности и тяжести поражения ткани в ходе острого инфаркта миокарда (R.Pudil et al., Clin. Chim. Acta, 280, 127, 1999).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Было обнаружено, что амиды (R)-2-фенилпропионовых кислот, несущие содержащие азот заместители на фенильной группе, обладают неожиданными свойствами подавления вызванного IL-8 хемотаксиса. Примерами таких заместителей являются алкиламино или диалкиламиногруппы и содержащие азот гетероциклы. Соединения по данному изобретению проявляют повышенную растворимость в воде и оптимизированные фармакокинетические свойства по сравнению с другими амидами, такими как описанные в РСТ/ЕР01/01285.
Стереохимия, электронные, поляризационные и пространственные эффекты заместителей у атома азота способствуют модуляции свойств подавления вызванного IL-8 хемотаксиса.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В следующих абзацах представлены определения важнейших химических групп, которые имеются в структурах соединений по данному изобретению и предназначены для употребления по всему описанию и в формуле изобретения, если не оговорено для конкретного значения более широкое определение.
«С1-С3-Алкил» или «С1-С5-алкил» относится к моновалентным алкильным группам, имеющим от 1 до 3 или от 1 до 5 атомов углерода. Примерами для этих терминов служат такие группы, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и тому подобное.
«С3-С7-Циклоалкил» относится к циклоалкильным группам, имеющим от 3 до 7 атомов углерода. Примерами для этих терминов служат такие группы, как циклопропил, циклобутил, циклопентил, циклогексил и тому подобное.
Примерами С3-С7-циклоалкил-С1-С3-алкильных групп являются циклопропилметил или циклопентилметил.
«Арил» относится к ненасыщенной ароматической карбоциклической группе, состоящей из от 6 до 14 атомов углерода, имеющей единственное кольцо (например, фенил) или поликонденсированные кольца (например, нафтил). Предпочтительные арилы включают фенил, бифенил, нафтил, фенантренил и тому подобное.
«Замещенный или незамещенный»: если не оговорено иного для конкретного определения конкретного заместителя, представленные выше группы, такие как «алкильная», «циклоалкильная», «арильная» группы и т.д., могут быть необязательно замещены от 1 до 5 заместителями, выбранными из группы, состоящей из «С1-С3-алкила», «С1-С3-алкиларила», «С1-С3-алкилгетероарила», первичных, вторичных или третичных аминогрупп или групп четвертичного аммония, «ацила», «ацилокси», «ациламино», «аминокарбонила», «алкоксикарбонила», «арила», «гетероарила», карбоксила, циано, галогена, гидрокси, меркапто, нитро, сульфокси, сульфонила, алкокси, тиоалкокси, тригалометила и тому подобного. В рамках данного изобретения указанное «замещение» означает также, что включены ситуации, когда соседние заместители подвергаются закрытию кольца, в частности, когда в этом участвуют соседние функциональные заместители, образуя таким образом, например, лактамы, лактоны, циклические ангидриды или циклоалканы, а также ацетали, тиоацетали, аминали, образуемые закрытием кольца, например, при попытке получить защитную группу.
«Фармацевтически приемлемые соли» относятся к солям или комплексам получивших определение ниже соединений формулы I, которые сохраняют желаемую биологическую активность. Примеры таких солей включают, но не ограничиваются этим, соли присоединения кислот, образованные с неорганическими кислотами (например, хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, азотной кислотой и т.п.), и соли, образованные с органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, фумаровая кислота, малеиновая кислота, аскорбиновая кислота, бензойная кислота, дигалловая кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота и полигалактуроновая кислота. Указанные соединения можно также вводить в виде фармацевтически приемлемых четвертичных солей, известных специалистам. Примеры солей включают также органические основания, такие как трометамин, L-лизин, L-аргинин и тому подобное.
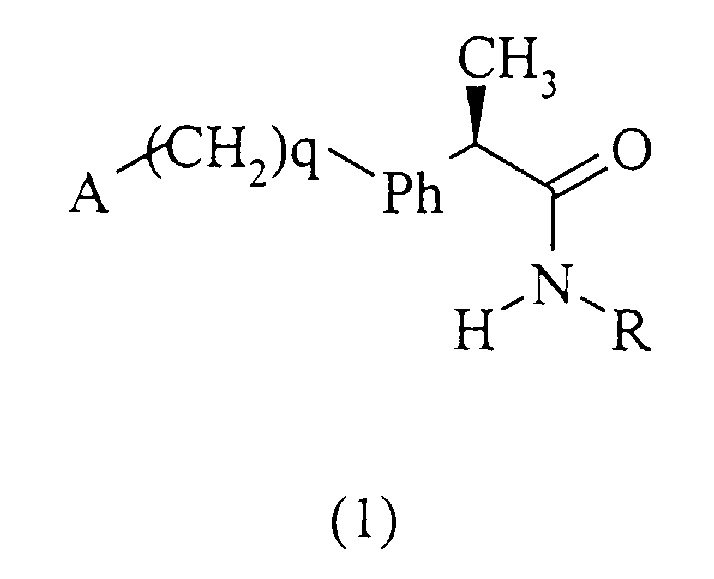
Данное изобретение относится также к амидам (R)-энантиомеров 2-арилпропионовых кислот формулы (1):

и их фармацевтически приемлемым солям, где
q равно нулю или целому числу, равному 1;
Ph представляет собой фениленовую группу, связанную с группой -(СН2)q-А в положении 2, 3 или 4 и необязательно замещенную в остальных положениях одним или несколькими заместителем, одинаковыми или различными, выбранными из С1-С3-алкила, галогенов, С1-С3-алкокси, гидрокси, SH, С1-С3-алкилтио, нитро, галогеналкила.
А представляет собой:
- N-С1-С5-алкиламиногруппу, N,N-С1-С5-диалкиламиногруппу, N-С1-С8-алканоил(циклоалканоил, арилалканоил)-N-С1-С5-алкиламиногруппу;
- насыщенное или ненасыщенное азотосодержащее 5-7-членное гетероциклическое кольцо;
- остаток формулы (2а):
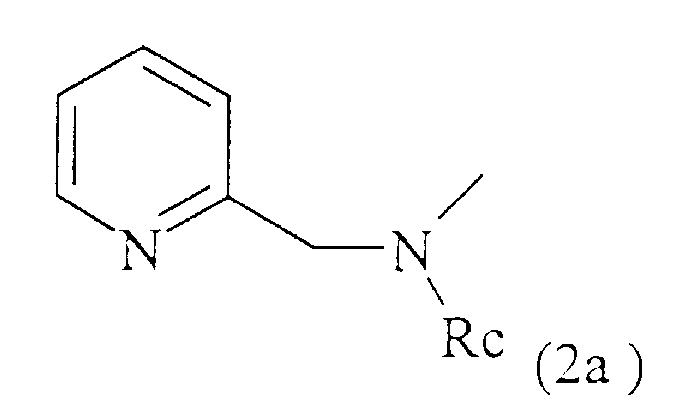
где Rс представляет собой водород, С1-С3-алкил или остаток С1-С3-алкановой кислоты;
R представляет собой:
- Н; -SO2-СН3; С1-С3-алкил, группу формулы -СН2-СН2-Х-(СН2-СН2О)n-Р, где Р представляет собой Н, метил, этил, изопропил;
-СН2СО2R1, где R1 представляет собой Н или С1-С3; n равно целому числу от 0 до 5,
Х представляет собой О или S;
- группа формулы (3)
- где, когда m равно целому числу от 2 до 3, Ф представляет собой незамещенный или замещенный фенилен, как указано выше, 2-(1-метилпирролидил), 2-пиридил, 4-пиридил, 1-имидазолил, 4-имидазолил, 1-метил-4-имидазолил, 1-метил-5-имидазолил или группу -NRaRb, где каждый из Ra и Rb, одинаковые или различные, представляют собой С1-С5-алкил или гидроксиалкил-(СН2)mi-ОН, где mi равно целому числу от 2 до 3, или Ra и Rb вместе с атомом N, с которым они связаны, образуют гетероциклическое кольцо, состоящее из 3-7 членов; когда m равно нулю, Ф выбран из группы, включающей 2- или 4-пиридил, 2- или 4-пиримидинил, 2-пиразинил, 5-метил-2-пиразинил, 1-1,2,4-тиадиазолил, 3-1,2,4-триазолил, 3-1-бензил-1,2,4-триазолил, 2-1,3-тиазолидинил, 2-1,3-тиазолил, 3-изоксазолил, дигидроизоксазол-4-ил, 5-метилизоксазол-4-ил, 2-имидазолил, имидазол-4-ил-5-карбоксиамид, имидазол-2-ил-4,5-дикарбонитрил, 5-инданил, 5-индазолил, 7-азаиндол-3-ил, индол-3-ил, 2- или 3- или 4-хинолил;
- остаток α-аминокислоты, выбранной из группы, включающей аланин, валин, лейцин, изолейцин, нор-лейцин, фенилаланин, п-фторфенилаланин, тирозин, бифенилаланин, 2'-метоксибифенил-аланин, триптофан, 7-азатриптофан, гистидин, S-метилцистеин, карбоксиметилцистеин, метионин, О-метилсерин, О-этилсерин, глицин, фенил- или п-фторфенилглицин;
- остаток кислоты, выбранный из группы, включающей β-аланин, γ-аминомасляную, δ-аминовалериановую, цис-4-аминоциклогексан-карбоновую, транс-4-аминометилциклогексанкарбоновую и 3-амино-1,5-пентадиовую кислоту;
- остаток формулы (3а)
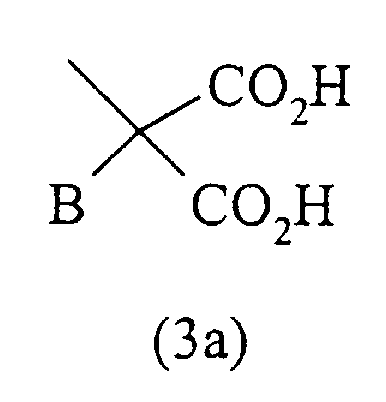
где В представляет собой Н; линейный или разветвленный С1-С5-алкил; (СН2)ni-NH2; -(СН2)ni-NH-трет-бутоксикарбонил; -(СН2)ni-NH-бензилоксикарбонил; (СН2)ni-СО2Н, где ni равно целому числу между 1 и 3; бензил; п-фторбензил; п-фенилбензил; п-(2-метоксифенил)бензил; -СН2О-С2Н5; -СН2-S-СН3; -СН2-S-СН2-СО2Н; индолил-3-метил; 7-азаиндолил-3-метил;
при условии, что, когда q равно нулю, R представляет SO2СН3 и Ph представляет собой 3-хлор-1,4-фенилен, А является группой, отличной от 1-2,5-дигидропирролидино.
Последнее соединение, где А представляет собой 1-2,5-дигидропирролидино, описано в WO 00/24710.
Соли соединений формулы (1) с фармацевтически приемлемыми основаниями и кислотами являются следующим объектом данного изобретения.
Ph предпочтительно связан с группой (-СН2)q-А по положению 3 или более предпочтительно по положению 4. Ph более предпочтительно является 1,4-фениленом, незамещенным или замещенным по положению 3, например хлором.
А предпочтительно является N,N-диметиламино, N,N-диэтиламино, N-метил-N-этиламино, N-ацетил-N-метиламино, N-пивалоил-N-этиламино или азотосодержащее кольцо, выбранное из 1-пирролидина, 1-2,5-дигидропирролидина (или 1-3-пирролина), 1-пиррола, 1-пиперидино, 1-пиперазино-4-незамещенного или 4-замещенного (метил, этил, 2-гидроксиэтил, бензил, бензгидрил или фенил)-4-морфолина, 4-3,5-диметилморфолина, 4-тиоморфолина.
Примеры 3-7-членных гетероциклических колец, образованных -NRaRb группой, включают 4-морфолино, 1-пиперидино, 1-пиперазино и 4-замещенного-1-пиперазина (4-метил, 4-бензил, 4-2-фенилэтил).
Примерами соединений данного изобретения являются:
R-N-2-[4-(пирролидин-1'-ил)метилфенил]пропионилметансульфонамид;
R-N-2-[4-(4'-бензилпиперазин-1'-иламинометил)фенил]пропионилметансульфонамид;
(R,S')-2-[3'-хлор-4-(тиоморфолин-4-ил)фенил]-N-(2-карбоксиэтил)пропионамид;
(R)-2-[3-хлор-4-(тиоморфолин-4-ил)фенил]-N-(2-метоксикарбоксиметил)пропионамид;
(R,S')-2-[(3-хлор-4-(тиоморфолин-4-ил)фенил]-N-[1-(3,5-диметилбензилоксикарбонил)этил-2-(3-индолил)]пропионамид;
(R,S')-2-[(3-хлор-4-(тиоморфолин-4-ил)фенил]-N-[1-(бензилоксикарбонил)этил-2-(4'-фторфенил)]пропионамид;
R-2-[3-хлор-4-(3-пирролидин-1-ил)фенил]пропионамид;
(R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)фенил]-N-(N',N'-диметиламинопропил)пропионамид;
(R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)фенил]-N-(бискарбоксиметил)пропионамид;
R(-)-2-[3-хлор-4-(пиперидин-1-ил)фенил]-N-(2''-гидроксиэтоксиэтил)пропионамид;
(R)-2-(4-морфолинофенил)-N-(4'-пиримидинил)пропионамид;
(R)-2-[4-((N-этил-N-хинол-2'-илметиламино)метил)фенил]-N-(2'-аллилоксикарбонилметил)пропионамид;
(R,R')-2-[3-хлор-4-(пиперидин-1-ил)]-N-[1'-метил-2'-(2''-гидроксиэтокси)этил]пропионамид;
(R)-2-(4-морфолинофенил)-N-(4'-пиримидинил)пропионамид;
R-2-[4-((N-этил-N-хинол-2'-илметиламино)метил)фенил)]-N-(4'-пиридил)пропионамид;
R-2-[3-хлор-4-(пирролидин-1-ил)-фенил)]-N-(2'-пиридил)-пропионамид;
R-2-[3-хлор-4-(3-пирролин-1-ил)-фенил]-N-(4'-пиридил)-пропионамид;
R-2-[3-хлор-4-(1Н-пиррол-1-ил)-фенил]-N-(4'-пиридил)-пропионамид;
R-2-[3-хлор-4-(морфолин-4-ил)-фенил]-N-(1,3-тиазол-2-ил)-пропионамид;
R-2-[4-(морфолин-4-иламинометил)-фенил]-N-(пиразин-2-ил)-пропионамид;
R-2-[3-хлор-4-(3'-пирролин-1-ил)-фенил]-N-(4'-пиримидинил)-пропионамид;
R-2-[4-(пирролидин-1-илметил)-фенил]-N-(4'-пиридил)пропионамид;
R-2-[4-(3-пирролин-1-ил)-фенил]-N-(4'-пиридилэтил)пропионамид;
R-2-(4-пиперидин-1-илфенил]-N-(1-имидазолэтил)-пропионамид;
R-2-[3-хлор-4-(пирролидин-1-ил)-фенил]-N-(2'-пиридил)-пропионамид;
R-2-[3-хлор-4-(3-пирролин-1-ил)-фенил]-N-(4'-пиридил)-пропионамид;
R-2-[3-хлор-4-(1Н-пиррол-1-ил)-фенил]-N-(4'-пиридил)-пропионамид;
R-2-[3-хлор-4-(морфолин-4-ил)-фенил]-N-(1,3-тиазол-2-ил)-пропионамид;
R-2-[4-(морфолин-4-иламинометил)-фенил]-N-(пиразин-2-ил)-пропионамид;
R-2-[3-хлор-4-(3'-пирролин-1-ил)-фенил]-N-(4'-пиримидинил)пропионамид;
R-2-[4-(пирролидин-1-илметил)-фенил]-N-(4'-пиридил)-пропионамид;
R-2-[4-(3'-пирролин-1-ил)-фенил]-N-(4'-пиридилэтил)-пропионамид;
R-2-[4-пиперидин-1-илфенил]-N-(1-имидазолэтил)-пропионамид;
(R)-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)-фенил]-пропионилметансульфонамид;
(R)-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)-фенил]-N-(2-карбоксиэтил)пропионамид;
(R)-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)-фенил]-пропионамид.
Особенно предпочтительными являются соединения формулы (1), где фениленовая группа Ph является незамещенным 1-4 фениленом или С-3 монозамещенным или С-3,С-5 дизамещенным фениленом.
Предпочтительными являются также соединения формулы (1), где R является аминокислотным остатком, таким как глициновый, аминомалоновый, бензил- и п-фторбензиламиномалоновый, или остатком одной из монокарбоновых или дикарбоновых аминокислот.
Особенно предпочтительными являются соединения формулы (1), где R является остатком L-аланином, L-фенилаланином, L-п-фторфенилаланином, L-метионином или L-п-(2'-метоксифенил)-фенилаланином.
Предпочтительными амидами формулы (1) являются те, у которых R представляет Н, группу -SO2-СН3, полиоксиэтиленовый остаток формулы -(СН2-СН2-О)2-Р, где Р представляет Н, метил, этил, изопропил или СН2-СО2R1, где R1 является Н или С1-С3-алкилом.
Предпочтительными амидами формулы (1) являются также те, у которых R является заместителем формулы (3)
где, когда m равно целому числу от 2 до 3, Ф является основным остатком, выбранным из групп: N,N-диэтиламино, 4-морфолил, 1-пиперидил, 1-(4-бензил)пиперазинил, 1-(4-дифенилметил)пиперазинил, 1-(4-(4',4''-дифтордифенил)метил)пиперазинил, где m равно нулю, Ф предпочтительно представляет гетероарильный остаток, такой как 2- или 4-пиридил, 2- или 4-пиримидинил, 2-пиразинил, 2-1,3-тиазолил, 2-1,3-тиазолидинил, 2-имидазолил, более предпочтительно 4-пиридил.
Соединения данного изобретения, которым дано определение в формуле (1), получены с использованием хорошо известных методов, включающих взаимодействие активированной формы кислоты формулы (4):
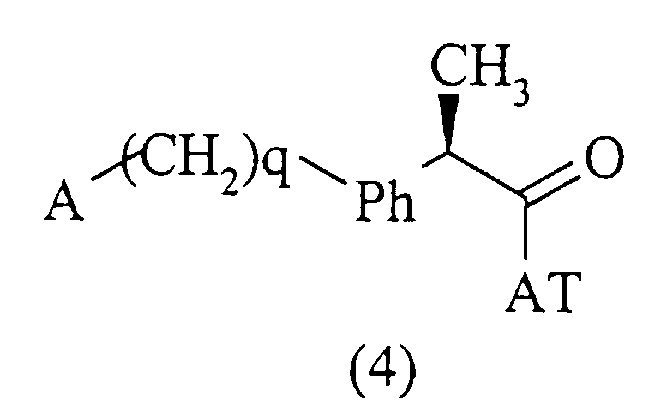
где А и (СН2)q указаны выше, и АТ является остатком, активирующим карбоксильную группу; с амином формулы (5):
в нерацемизирующих условиях в присутствии, если желательно, молярного избытка основания. Примерами активированных форм кислот формулы (4, АТ=Н) являются соответствующие хлориды (АТ = Cl), имидазолиды (АТ = 1-имидазол), сложные эфиры с фенолами, такие как п-нитрофенол (АТ=pNO2-С6Н4О-) или активированные формы, полученные реакцией в присутствии 1-гидроксибензотриазола (ГОБТ) или карбодиимида (такого как циклогексилкарбодиимид).
В первичных аминах формулы (5) R имеет значение, которое указано выше.
Реакции получения амидов формулы (1) обычно осуществляются при комнатной температуре при использовании общепринятых протонных и апротонных растворителей, предпочтительно обезвоженных на молекулярных ситах, или их смесей.
Указанные растворители включают сложные эфиры, такие как этилацетат, метилацетат, этилформиат, нитрилы, такие как ацетонитрил, циклические простые эфиры, такие как диоксан, тетрагидрофуран, этиловый эфир, сульфолан, амиды, такие как диметилформамид, формамид; галогенированные растворители, такие как дихлорметан, ароматические углеводороды, такие как толуол, хлорбензол и гетероароматические углеводороды, такие как пиридин и пиколин.
Данные реакции могут осуществляться в присутствии основания. Предпочтительные неорганические основания являются карбонатами и бикарбонатами щелочных и щелочноземельных металлов, такими как тонкоизмельченные карбонат калия, бикарбонат калия и карбонаты Mg и Са.
Если желательно, соединения формулы (1) могут быть преобразованы в другие соединения формулы (1) путем отщепления защищающих групп и/или путем селективного гидролиза групп сложных эфиров. Особенно предпочтительной сложноэфирной группой является аллил, отщепляемой в высоко селективных условиях, например, на основе переноса аллильной группы на молекулу морфолина, которая в присутствии Pd(0) в качестве катализатора действует как средство переноса Н и как нуклеофильный акцептор в соответствии с процессом, описанным в J. Org. Chem., 54, 751, 1989.
И, наконец, если желательно, соединение формулы (1) может быть превращено в соль с использованием фармацевтически приемлемых кислот или оснований.
Примеры фармацевтически приемлемых кислот включают хлористоводородную, серную, азотную, фосфорную кислоту или моно- или многоосновные органические кислоты, такие как уксусная, бензойная, винная, лимонная, фумаровая, малеиновая, яблочная, миндальная, щавелевая и малоновая кислота.
Примерами фармацевтически приемлемых солей являются соли с катионами щелочных и щелочноземельных металлов, предпочтительно натрия и магния, и соли с органическими основаниями, такими как трометамин, D-глюкозамин, лизин, аргинин, тетраэтиламмоний.
R-энантиомеры 2-(аминоарил)пропионовых кислот формулы (4а)
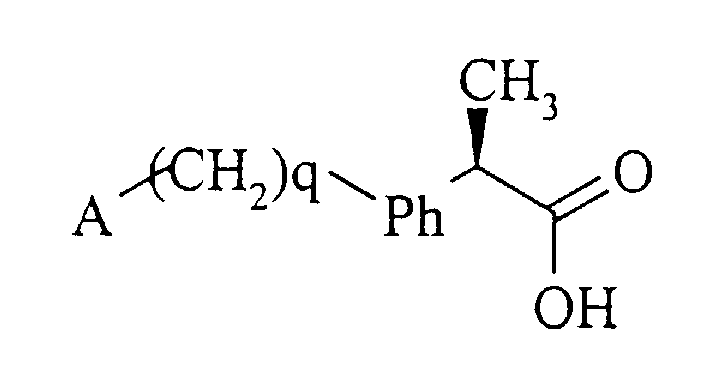
(4а)
являются хорошо известными соединениями, которые от соответствующих S-энантиомеров отличаются тем, что не являются эффективными ингибиторами циклооксигеназных ферментов.
Из R-2-арилпропионовых кислот формулы (4а), где q равно нулю, особенно предпочтительными являются следующие:
R-2-[4-(2,5-дигидро-1Н-пиррол-1)-фенил]пропионовая, R-2-[4-(1Н-пиррол-1)-фенил]пропионовая, R-2-[4-(1Н-пирролидин-1-ил)-фенил]пропионовая, R-2-(4-пиперидинофенил]пропионовая, R-2-(4-морфолинофенил]пропионовая, R-2-(4-тиаморфолинофенил]пропионовая, R-2-[4-(4'-бензилпиперазин-1'-ил)-фенил]пропионовая и ее 3-хлор-производные, R-2-[4-(4'-бензгидрилпиперазин-1'-ил)-фенил]пропионовая и ее 3-хлорпроизводное. Все эти кислоты являются хорошо известными соединениями и получены с использованием хорошо известных методов. Основные указания в отношении синтеза и оптическое разрешение указанных кислот формулы (4а; q=0) найдены в патентах США US 3641040; US 3993763; US 3997669 и US 4337264. Более конкретно описаны в этих патентах синтезы производных метиловых и этиловых эфиров 2-(4-аминофенил)пропионовой кислоты и 2-(3-хлор-4-аминофенил)пропионовой кислоты, которые путем реакции с подходящим α,ω-алканом или алкеном, различно замещенными, преобразовывают в желаемый 1-азациклоалкан или циклоалкен.
Например, в следующей схеме реакции представлен способ получения, использованный в синтезе сложных эфиров 2-(4-пиперазин-1-илфенил)пропионовых кислот формулы (4b):
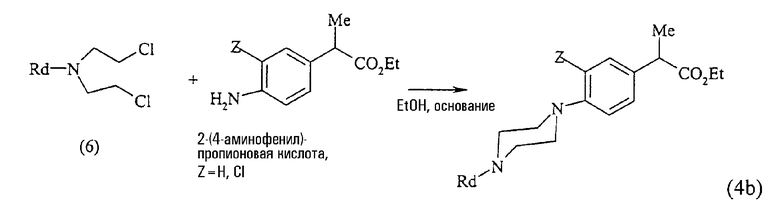
исходя из сложного эфира 2-(4-аминофенил)пропионовой кислоты путем реакции с бис-(2-хлорэтил)амином формулы (6), где R4 представляет Вос, С1-С3-алкил, фенил, бензил, бензгидрил, 4,4'-дифторбензгидрил.
Из R-2-арилпропионовых кислот формулы (4), где q равно целому числу, равному 1, особенно предпочтительными являются следующие:
R-2-[4-(2,5-дигидро-1Н-пиррол-1-метил)фенил]пропионовая, R-2-[4-(1Н-пиррол-1-метил)фенил]пропионовая, R-2-[4-(1Н-пирролидин-1-метил)фенил]пропионовая, R-2-[4-(1Н-пирролидин-3-он-1-илметил)фенил]пропионовая, R-2-[4-(пиперидин-1-илметил)фенил]пропионовая, R-2-(4-пиперидин-4-он-1-илметилфенил)пропионовая, R-2-(4-морфолин-4-илметилфенил)пропионовая, 4-(тиоморфолин-4-илметил)фенилпропионовая, R-2-[4-(4'-бензилпиперазин-1'-илметил)фенил)пропионовая, R-2-[4-(4'-бензгидрилпиперазин-1'-илметил)фенил)пропионовая кислоты.
Их получение основано на превращении трет-бутилового эфира 2-толилпропионовой кислоты в соответствующее Br-метилпроизводное, которое затем трансформируют в трет-бутиловый эфир кислоты формулы (4а, q=1) путем реакции с желаемым амином формулы А-Н (где А имеет значение, которое уже указано выше).
Аллиловый и бензиловый эфиры α- или ω-аминокислот являются хорошо известными доступными для приобретения продуктами, или они могут быть получены с использованием хорошо известных методов; в отношении получения аллиловых сложных эфиров см., например, H. Waldmann and H. Kunz, Liebigs Ann. Chem., 1712, 1983 и J. Org. Chem. 1989, уже цитированные; в отношении получения бензиловых сложных эфиров см., например, Mac Leod A.M. et al., J. Med. Chem., 37, 1269, 1994.
Для оценки фармакологической активности некоторых соединений данного изобретения формулы (1) использовали эксперименты in vitro на человеческих нейтрофилах (здесь ниже называемых как нейтрофилы, выделенные из гепаринизированной человеческой крови, взятой у согласившихся здоровых взрослых субъектов, путем осаждения на декстран по методике, описанной W.J. Ming et al., J. Immunol., 138, 1469, 1987. Каждое из соединений, находящихся на исследовании, предварительно инкубируют в течение 10 минут при температуре 37°С. В экспериментах по хемотаксису, в экспериментах, направленных на количественное определение активности тирозинкиназы, и в направленных на количественное определение уровней цитозольного Са2+ использовали человеческий рекомбинантный интерлейкин-8 (rhIL-8, Pepro Tech): лиофилизированный белок растворяли в (HBSS) ССРХ (сбалансированный солевой раствор Хенкса) в концентрации 100 мкг/мл и затем разбавляли до концентраций 10 нг/мл в экспериментах по хемотаксису и до 25-50 нг/мл при оценке внутриклеточных изменений Са2+ (например, [Са2+]i) и до 400 нг/мл при оценке активности тирозинкиназы.
В процессе изучения хемотаксиса (по W.Falket et al., J. Immunol. Methods, 33, 239, 1980) использовали фильтры без PVP с порами 5 мкм и микрокамеры, пригодные для осуществления репликаций. Соединения оценивали в интервале концентраций от 10-6 до 10-10 М, используя R(-)-2-(4-изобутилфенил)пропионил-метансульфонамид, ЕD50 которого равна 10-9 М, в качестве стандарта для сравнения.
Кроме того, соединения данного изобретения были способны подавлять вызванную IL-8 тирозинкиназную активность, как известно, определяющей для хемотаксиса нейтрофилов (Yasui et al., 1994, уже цитированная). Указанную оценку выполняли, используя метод, описанный L'Heureux et al., Blood, 85, 522, 1995. И, наконец, некоторые из соединений данного изобретения были способны подавлять повышение концентрации ионов внутриклеточного Са2+, вызванного IL-8, причем указанная оценка выполняется на экспериментальной модели, описанной Bizzarri et al, Blood, 86, 2388, 1995.
Как установлено ранее, соединения по данному изобретению не проявили способности подавлять СО ферменты, когда оценивались ex vivo в крови in toto по методике, описанной Patrignani et al, in J. Pharmacol. Exper. Ther., 271, 1705, 1994. Кроме того, почти во всех случаях соединения данного изобретения формулы (I) не влияют на продукцию PGE2, вызванную в мышиных макрофагах стимуляцией липополисахаридами (ЛПС, 1 мкг/мл) при концентрациях в интервале от 10-5 до 10-7 М. Любое подавление продукции PGE2, которое может быть зарегистрировано, находится большей частью на пределе статистического значения и в большинстве случаев менее 15-20% основного значения.
Это незначительное подавление синтеза PGE2 позволяет четко дифференцировать соединения данного изобретения формулы (1) от (S)-энантиомеров 2-арилпропионовых кислот и от их амидов, которые, наоборот, из-за их выраженного подавления синтеза PGE2 создают стимул для некоторых мышиных макрофагов усиливать синтез ФНО-α.
Хорошо известно, что усиление синтеза ФНО-α дает возможность активации нейтрофилов и способствует их хемотаксису, являясь, кроме того, стимулом синтеза IL-8. Соединения данного изобретения формулы (I) не проявляют какого-либо воздействия на ПГ синтез, тогда как регистрируется подавляющее действие для некоторых из них в отношении синтеза ФНО-α, который обычно стимулируется в макрофагах ЛПС, подавляющее действие, которое также регистрируется в отношении синтеза того же цитокина после стимуляции Н2О2.
Принимая во внимание экспериментальные данные, обсужденные выше, и участие IL-8 и его производных как наиболее важных медиаторов и промоторов инфильтрации нейтрофилов при таких патологиях, как псориаз (R.J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991), ревматоидный артрит (M. Selz et al., J. Clin. Invest., 87, 463, 1981), язвенный колит (Y. R. Mahla et al., Clin. Sci., 82, 273, 1992), острая респираторная недостаточность и идиопатический фиброз (E.J. Miller, уже цитированная, и P.C. Carré et al., J. Clin. Invest., 88, 1882, 1991), гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994) и при предотвращении повреждения от ишемии и возобновления кровотока, соединения данного изобретения, как полагают, особенно пригодны для достижения терапевтических целей в этих случаях.
Для этой цели с соединениями данного изобретения соответственно изготавливают фармацевтические композиции, используя общепринятые методы и вспомогательные вещества, такие как описанные в «Remington's Pharmaceutical Sciences Handbook», Mack Publishing, New York, 18° Ed., 1990.
Композиции по данному изобретению могут вводиться внутривенно в виде болюсной инъекции или перорально в виде капсул, таблеток, сиропа и лекарственных форм с регулируемым высвобождением, а также в виде препаратов для дерматологического использования (кремов, лосьонов, распыляемых препаратов и мазей). Средняя суточная дозировка будет зависеть от нескольких факторов, таких как тяжесть заболевания и от данных пациента (возраст, пол и вес). Доза будет меняться в основном от 1 или нескольких мг до 1500 мг соединений формулы (1) в сутки, по желанию разделенных на несколько введений. Также благодаря низкой токсичности соединений данного изобретения можно вводить более высокие дозы в течение длительных периодов времени.
Следующие примеры иллюстрируют данное изобретение.
В описании абсолютной конфигурации одиночных хиральных заместителей, необязательно присутствующих у соединений данного изобретения, апексы (например, R', S', S'' и т.д.) будут использоваться, как правило, для указания абсолютных конфигураций, присутствующих в заместителе R, связанном с атомом азота указанных соединений.
Примерами аббревиатур являются: ТГФ для тетрагидрофурана, ДМФ для диметилформамида, AcOEt для этилацетата, ГОБТ для 1-гидроксибензотриазола, ДЦК для дициклогексилкарбодиимида.
Пример 1
R-N-2-[4-(пирролидин-1'-ил)метилфенил]-пропионилметансульфонамид
N,N-диметиламинопиридин (2,4 г, ˜ 0,02 моль), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (3,73 г, ˜ 0,02 моль) и метансульфонамид (1,85 г, 0,02 моль) добавляют в этом порядке к раствору 2-[(4-(пирролидин-1-ил)метилфенил]пропионовой кислоты (4 г, примерно 0,019 моль) в безводном СН2Cl2 (30 мл). Смесь оставляют перемешиваться в течение ночи. Растворитель выпаривают в вакууме, и остаток обрабатывают этилацетатом (3 х 15 мл). Органические фазы объединяют, промывают водой до нейтральности, сушат сульфатом натрия и выпаривают досуха. Остаток очищают на колонке силикагеля с получением 3,15 г R-(N)-2-[4-(пирролидин-1'-ил)метилфенил]пропионилметансульфонамида.
Пример 2
R-N-2-[4-(4'-бензилпиперазин-1'-иламинометил)фенил]-пропионилметансульфонамид
Метансульфонамид (2,3 г, 0,0243 моль) добавляют к суспензии t-BuOK (2,73 г, 0,0244 моль) в безводном ТГФ (30 мл). Смесь перемешивают в течение 30 мин при комнатной температуре, затем охлаждают при -25°С, затем добавляют предварительно охлажденный раствор имидазола R-2-[4-(4'-бензилпиперазин-1'-ил)аминометилфенил)]пропионовой кислоты (5,45 г, ˜ 0,019 моль) в ТГФ (10 мл). Перемешивание продолжают при этой температуре в течение 2 часов, затем в течение 1 часа при 0°С. Реакционную смесь нейтрализуют добавлением 0,04 молярных эквивалента раствора ледяной АсОН в ТГФ. После отделения фильтрованием неорганических солей, которые выделились, растворитель выпаривают в вакууме и масляный остаток распределяют между дихлорметаном (30 мл) и раствором 2,5% NaHCO3. Органические экстракты объединяют, промывают водой, сушат сульфатом натрия. После выпаривания растворителя и очистки на колонке силикагеля получают 4,05 г R-N-2-[4-(4'-бензилпиперазин-1'-иламинометил)фенил]пропионилметансульфонамида.
Пример 3
(R,S)-2-[3'-хлор-4-(тиоморфолин-4-ил)фенил]-N-(2-карбоксиэтил)пропионамид
К раствору R-2-[3-хлор-4-(тиоморфолин-4-ил)фенил]пропионовой кислоты (6,4 г; ˜ 22,1 нмоль) в ДМФ (20 мл), охлажденному до примерно Т=0°С, добавляют при перемешивании 3 г ГОБТ (22,2 ммоль). Через 15 мин добавляют смесь гидрохлорида метилового эфира L-аланина (3,2 г; ˜ 22,2 ммоль) и триэтиламина (3 мл) в ДМФ (5 мл); и, наконец, добавляют ДЦК последовательными порциями всего 5 г (24,24 ммоль). Смесь выдерживают при перемешивании в течение двух часов при Т=0°С и затем при комнатной температуре в течение ночи. После удаления фильтрованием осадка дициклогексилмочевины фильтрат выпаривают до небольшого объема и распределяют между этилацетатом и насыщенным раствором NaHCO3. Органические фазы объединяют и повторно экстрагируют 2 н. Н2SO4, кислотные экстракты объединяют, добавляют 2 н. NaOH до нейтральной реакции, затем снова экстрагируют AcOEt (50 мл). Органическую фазу промывают 10% раствором сульфата натрия (20 мл). После высушивания Na2SO4 и выпаривания растворителя при пониженном давлении получают остаток, который суспендируют в гексане (60 мл) и выдерживают при перемешивании в течение ночи, что приводит к выделению белого кристаллического осадка, состоящего из (R,S')-2-[3-хлор-4-(тиоморфолин-4-ил)фенил]-N-(2-метоксикарбонилэтил)пропионамида (4,9 г, ˜16,84 ммоль). Добавляют раствор 2 г (6,87 ммоль) данного соединения в диоксане (9 мл) с равным объемом 1 н. NaOH (9 мл) и выдерживают при перемешивании при комнатной температуре в течение ночи. После разбавления водой и льдом (130 мл) эту смесь подкисляют 2 н. Н2SO4 до рН 6-6,5. Водную фазу экстрагируют СН2Cl2 (4 х 20 мл); органические экстракты объединяют, промывают насыщенным раствором NaCl (20 мл) и сушат Na2SO4. После выпаривания растворителя при пониженном давлении остаток, кристаллизованный из этилового эфира (30 мл), дает (R,S')-2-[3'-хлор-4-(тиоморфолин-4-ил)фенил]-N-(2-карбоксиэтил)пропионамид (1,6 г, 6,52 ммоль).
Путем замены метилового эфира L-аланина метиловым эфиром глицина и 3,5-диметилбензилового эфира L-триптофана бензиловым эфиром L-4-фторфенилаланина в процедуре, описанной выше, получают следующие соединения:
(R)-2-[3-хлор-4-(тиоморфолин-4-ил)фенил]-N-(2-метоксикарбонилметил)пропионамид;
(R,S')-2-[3-хлор-4-(тиоморфолин-4-ил)фенил]-N-[1-(3,5-диметилбензилоксикарбонил)-этил-2-(3-индолил)]пропионамид;
(R,S')-2-[3-хлор-4-(тиоморфолин-4-ил)фенил]-N-[1-(бензилоксикарбонил)этил-2-(4'-фторфенил)]пропионамид.
Пример 4
R-2-[3-хлор-4-(3-пирролин-1-ил)фенил]пропионамид
Раствор R(-)-пирпрофена (2 г, 9,69 ммоль) в тионилхлориде (4 мл) нагревают в течение 3 часов при температуре дефлегмации; после охлаждения до комнатной температуры растворитель выпаривают при пониженном давлении, остаток обрабатывают диоксаном последовательно два раза и растворители выпаривают в высоком вакууме для удаления следовых остатков тионилхлорида. Раствор желтого маслянистого остатка R(-)-пирпрофеноилхлорида гидрохлорида (2,16 г; 9,6 ммоль) в безводном ацетонитриле (4 мл) по каплям добавляют к раствору 28% NH4ОН (10 мл), охлажденного до 0-5°С, с такой скоростью, чтобы температура смеси не превышала +5°С. Смесь перемешивают в течение 1 ч при комнатной температуре с получением после выпаривания растворителей остатка, который растворяют в AcOEt (6 мл). После охлаждения выделяется осадок R-2-[3-хлор-4-(3-пирролин-1-ил)фенил]пропионамида.
Пример 5
(R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)фенил]-N-(N',N'-диметиламинопропил)пропионамид
К раствору (R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)]фенилпропионовой кислоты (5 г, 20 ммоль) в безводном CHCl3 (20 мл), охлажденном до примерно Т=0°С при перемешивании добавляют ГОБТ (2,7 г; 20 ммоль). Через 15 мин по каплям добавляют раствор 3-(диметиламино)пропиламина (2,03 г; 20 ммоль) в безводном CHCl3 (5 мл); в конце последовательными порциями добавляют ДЦК (4,13 г, 20 ммоль). Когда добавление заканчивают смесь оставляют при перемешивании на два часа при Т=0°С и затем при комнатной температуре в течение ночи. После отфильтровывания выпавшей в осадок дициклогексилмочевины фильтрат разбавляют дихлорметаном (50 мл). Органическую фазу промывают водой (3 х 15 мл) и насыщенным раствором NaCl (2 х 20 мл). После высушивания над Na2SO4 выпаривание растворителей дает остаток, который растворяют безводным ацетоном (5 мл). Газообразный HCl пропускают через раствор, желаемый продукт осаждается в виде гидрохлоридной соли в форме белого твердого вещества, которое выделяют фильтрованием и сушат в вакууме при Т=40°С с получением 5,37 г (16 ммоль) гидрохлорида (R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)фенил]-N-(N',N'-диметиламинопропил)пропионамида.
Пример 6
(R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)фенил]-N-(бис-карбоксиметил)пропионамид
К раствору (R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)]фенилпропионовой кислоты (5 г, 20 ммоль) в безводном CHCl3 (20 мл), охлажденном до примерно Т=0°С, добавляют при перемешивании раствор ГОБТ (2,7 г; 20 ммоль). Через 15 мин по каплям добавляют раствор гидрохлорида диэтиламиномалоната (4,23 г; 20 ммоль) и триэтиламина (2,78 мл, 20 ммоль) в безводном CHCl3 (5 мл); в конце последовательными порциями добавляют ДЦК (4,13 г; 20 ммоль). Когда добавление заканчивают, смесь оставляют при перемешивании на два часа при Т=0°С, а затем при комнатной температуре на ночь. После отфильтровывания выпавшей в осадок дициклогексилмочевины фильтрат разбавляют дихлорметаном (50 мл), органическую фазу промывают водой (3 х 15 мл) и насыщенным раствором NaCl (2 х 20 мл). После высушивания Na2SO4 и выпаривания растворителей получают остаток, который очищают флэш-хроматографией (элюент СН2Cl2/СН3ОН 95:5) с получением промежуточного соединения диэтилового эфира (4,9 г; 12 ммоль).
К раствору диэтилового эфира (4,5 г; 11 ммоль) в безводном СН2Cl2 (30 мл), охлажденном до Т=-10°С, добавляют раствор 1М BBr3 в СН2Cl2 (12,36 мл) при перемешивании и с помощью шприца. Смесь оставляют при Т=-10°С на 1 час и затем при комнатной температуре на 6 часов до завершения реакции. Затем смесь разбавляют водой (30 мл), две фазы встряхивают и разделяют; водную фазу экстрагируют (2 х 15 мл) СН2Cl2. Органические экстракты объединяют и экстрагируют насыщенным раствором NaHCO3 (3 х 15 мл); основную водную фазу затем подкисляют до рН 6-6,5 насыщенным раствором монозамещенного фосфата натрия и противоэкстрагировали СН2Cl2 (3 х 15 мл). Объединенные органические экстракты промывают насыщенным раствором NaCl, сушат Na2SO4 и выпаривают с получением (R)-2-[3-хлор-4-(2,5-дигидро-1Н-пиррол-1)фенил]-N-(бис-карбоксиметил)пропионамида в виде стекловидного твердого вещества (1,75 г; 4,95 ммоль).
Пример 7
R(-)-2-[3-хлор-4-(пиперидин-1-ил)фенил]-N-(2''-гидроксиэтоксиэтил)пропионамид
Раствор R-2-[3-хлор-4-(пиперидин-1-ил)фенил]пропионил-хлорида (9,5 ммоль) в безводном ацетонитриле (10 мл), полученный по методике примера 4, по каплям добавляют при комнатной температуре к раствору 2-(2-аминоэтокси)этанола (0,97 мл; 9,7 ммоль) и триэтиламина (3 мл) в безводном СН2Cl2 (15 мл). Реакционную смесь перемешивают в течение ночи при комнатной температуре, затем выпаривают досуха. Остаток собирают 5% раствором этилацетата и 5% раствором бикарбоната калия, промывают водой и органическую фазу экстрагируют 2 н. серной кислотой. Кислотные водные фазы объединяют, добавляют лед и водную фазу подщелачивают до рН 8 и повторно снова экстрагируют AcOEt. Эти экстракты объединяют и промывают 10% раствором сульфата натрия. После высушивания Na2SO4 и выпаривания растворителя при пониженном давлении получают остаток, который очищают флэш-хроматографией (элюент СН2Cl2/СН3ОН 98:2) с получением в виде прозрачного масла, 1,87 г R(-)-2-[3-хлор-4-(пиперидин-1-ил)фенил]-N-(2''-гидроксиэтоксиэтил)пропионамида.
Пример 8
Путем использования амина, выбранного из группы п-метоксианилина, (R)-1-метил-2-(2'-гидроксиэтокси)этиламина, метилового эфира L-метионина, метилового эфира L-2'-метоксибифенилаланина и этил-3-окса-5-аминопентаноата в процедуре предшествующего примера, получали
R-2-[3-хлор-4-(пиперидин-1-ил)]-N-(4-метоксифенил)пропионамид;
(R,R')-2-[3-хлор-4-(пиперидин-1-ил)]-N-[1'-метил-2'-(2''-гидроксиэтокси)этил]пропионамид;
(R,S')-2-[3-хлор-4-(пиперидин-1-ил)]-N-[1-метоксикарбонил-3-метилтиопропил]пропионамид;
(R,S')-2-[3-хлор-4-(пиперидин-1-ил)]-N-[1-метоксикарбонил-2-(2'-метоксибифенил)этил)]пропионамид;
R-2-[3-хлор-4-(пиперидин-1-ил)]-N-[2-(этоксикарбонилметокси)этил)]пропионамид;
Пример 9
(R)-2-(4-морфолинофенил)-N-(4'-пиримидинил)пропионамид
К раствору (R)-2-(4-морфолинофенил)пропионовой кислоты (5 г, 21,3 ммоль) в безводном этилацетате (20 мл) добавляют N,N'-карбонилдиимидазол (3,8 г; 23,43 ммоль) при перемешивании при комнатной температуре. Смесь перемешивают в течение трех часов при комнатной температуре и без выделения промежуточного соединения - имидазолида - добавляют раствор 4-аминопиримидина (2,23 г; 23,43 ммоль) в безводном этилацетате (10 мл). Перемешивание при комнатной температуре продолжают в течение 8 часов, затем органическую фазу промывают 5% раствором NaHCO3 (3 х 25 мл). Органическую фазу экстрагируют 2 н. Н2SO4 (5 х 10 мл); кислую водную фазу затем подщелачивают до рН 8,5-9 NaOH и экстрагируют этилацетатом (3 х 15 мл). Экстракты объединяют, промывают насыщенным раствором NaCl (2 х 25 мл) и сушат Na2SO4. После выпаривания растворителя при пониженном давлении получают неочищенный остаток, который очищают флэш-хроматографией с получением (R)-2-(4-морфолинофенил)-N-(4'-пиримидинил)пропионамида в виде чистого белого твердого вещества (5,3 г; 17 ммоль).
Пример 10
R-2-[4-((N-этил-N-хинолин-2'-илметиламино)метил)фенил]-N-(2'-аллилоксикарбонилметил)пропионамид
К раствору R-2-[4-((N-этил-N-хинолин-2-илметиламино)-метил)фенил]пропионовой кислоты (0,47 г, примерно 1,2 ммоль) в безводном этилацетате добавляют 1,1 молярного эквивалента N,N'-карбонилдиимидазола при комнатной температуре и при перемешивании. После 3 ч при комнатной температуре без выделения промежуточного соединения, пропионилимидазолида, добавляют раствор 1,1 молярного эквивалента аллилового эфира глицина в безводном AcOEt. Перемешивание продолжают в течение 6 ч при комнатной температуре, затем реакционную смесь повторно промывают 5% раствором бикарбоната натрия и воды до нейтральности. Органическую фазу повторно экстрагируют 2N водным раствором Н2SO4 (5 х 8 мл) и водой. Кислые водные экстракты объединяют, подщелачивают и снова экстрагируют этилацетатом. Органические фазы объединяют и промывают до нейтральной реакции 10% раствором сульфата натрия и сушат безводным Na2SO4. После выпаривания растворителя получают R-2-[4-((N-этил-N-хинолин-2'-илметиламино)метил)фенил]-N-(2'-аллилоксикарбонилметил)пропионамид.
Пример 11
R-2-[4-((N-этил-N-хинолин-2-илметиламино)метил)фенил]-N-(4'-пиридил)пропионамид
Раствор (R)-2-[4-((N-этил-N-хинолин-2-илметиламино)-метил)фенил]пропионовой кислоты (4 г, примерно 12 ммоль) в тионилхлориде (6,2 мл) нагревают в течение 3 часов при температуре дефлегмации. После охлаждения до комнатной температуры растворитель выпаривают при пониженном давлении, остаток обрабатывают последовательно два раза диоксаном и растворители выпаривают в высоком вакууме, чтобы удалить следовые остатки тионилхлорида. Маслянистый остаток, полученный таким образом, растворяют в безводном СН2Cl2 (10 мл).
Раствор по каплям при комнатной температуре добавляют к раствору 4-аминопиридина (2,25 г; 24 ммоль) в дихлорметане в присутствии избытка триэтиламина. Смесь перемешивают в течение ночи при комнатной температуре, затем разбавляют СН2Cl2 (20 мл). Органическую фазу промывают водой (2 х 15 мл) и затем насыщенным раствором NaCl (15 мл). После высушивания на Na2SO4 и выпаривания растворителей получают продукт в виде белого твердого вещества (3,07 г; 7,8 ммоль).
Пример 12
Используя методы, описанные в примерах 10 и 11, R-2-(аминофенил)пропионовую кислоту формулы (4а) подвергают взаимодействию с гетероциклическим амином, выбранным из группы, включающей: 2-аминопиридин, 4-аминопиридин, 4-аминопиримидин, 2-аминопиразин, 2-амино-1,3-тиазол, 2-(пирид-4-ил)этиламин, 2-(имидазол-1-ил)этиламин, и получают следующие амиды:
R-2-[3-хлор-4-(пирролидин-1-ил)фенил)]-N-(2'-пиридил)пропионамид;
R-2-[3-хлор-4-(3-пирролин-1-ил)фенил)]-N-(4'-пиридил)пропионамид;
R-2-[3-хлор-4-(1Н-пиррол-1-ил)фенил)]-N-(4'-пиридил)пропионамид;
R-2-[3-хлор-4-(морфолин-4-ил)фенил)]-N-(1,3-тиазол-2-ил)-пропионамид;
R-2-[4-(морфолин-4-иламинометил)фенил]-N-(пиразин-2-ил)-пропионамид;
R-2-[3-хлор-4-(3'-пирролин-1-ил)фенил]-N-(4'-пиримидинил)пропионамид;
R-2-[4-(пирролидин-1-илметил)фенил)]-N-(4'-пиридил)пропионамид;
R-2-[4-(3-пирролин-1-ил)фенил]-N-(4'-пиридилэтил)пропионамид;
R-2-(4-пиперидин-1-илфенил]-N-(1-имидазолэтил)пропионамид.
Пример 13
Путем взаимодействия раствора R-2-[3-хлор-4-(4'-бензилпиперазин-1-ил)фенил]пропионилхлорида в диоксане с водным раствором N-метиламина в условиях Шоттена-Баумана получают R-2-[3-хлор-4-(4'-бензилпиперазин-1-ил)фенил]-N-метилпропионамид.
Получение:
А) Смесь 1,5 г этил-2-(4-аминофенил)пропионата, 2,3 г бис(2-хлорэтил)бензиламина и N-этилдиизопропиламина (5 мл) в 25 мл абсолютного этанола нагревают с обратным холодильником в течение 24 часов. Смесь концентрируют в вакууме досуха, продукт-сырец, полученный таким образом, очищают на колонке с силикагелем, используя СН2Cl2/МеОН 98:2 в качестве элюента, с получением 2,21 г этил-2-[4-(4-бензилпиперазин-1-ил)фенил]пропионата.
В) 22 г трет-бутил-2-п-толилпропионат растворяют в 200 мл CCl4 и раствор доводят до температуры кипения. После добавления 0,85 г 1,1'-азо(бисциклогексаннитрил)а добавляют порциями 18,7 г N-бромсукцинимида и температуру дефлегмации поддерживают в течение 1 часа. Смесь охлаждают до 0°С и отфильтровывают сукцинимид. При концентрировании раствора выпадает в осадок 19,8 г трет-бутил-2-(4-бромметилфенил)пропионата.
По описанной методике и исходя из соответствующего трет-бутил-2-о-толилпропионата и трет-бутил-2-м-толилпропионата получают трет-бутил-2-(2-бромметилфенил)пропионата и трет-бутил-2-(3-бромметилфенил)пропионата.
С) К раствору 3-пирролина (2,56 мл; 33,4 ммоль) в безводном СН2Cl2 (20 мл), выдерживаемом при комнатной температуре, по каплям добавляют раствор трет-бутил-2-(4-бромметилфенил)пропионата (5 г; 16,7 ммоль) в безводном СН2Cl2 (10 мл). Когда добавление заканчивают, раствор доводят до температуры дефлегмации и выдерживают при указанной температуре в течение 12 часов до завершения реакции. После охлаждения до комнатной температуры смесь разбавляют равными объемами 5%-ного NaHCO3 и СН2Cl2 (25 мл), встряхивают и две фазы разделяют; органическую фазу дополнительно промывают водой (2 х 20 мл) и насыщенным раствором NaCl (2 х 20 мл). После высушивания Na2SO4 выпаривание растворителя дает трет-бутил-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)фенил]пропионат в виде бледно-желтого масла (4,25 г; 15,03 ммоль).
Сложный эфир (4 г; 14,1 ммоль) растворяют в безводном СН2Cl2 (10 мл). Раствор охлаждают до Т=0°С и медленно, по каплям добавляют трифторуксусную кислоту. Когда прибавление по каплям заканчивают, смесь оставляют при перемешивании на 6 часов при комнатной температуре. Реакцию прекращают выпариванием растворителей досуха и удаляя все присутствующие растворители выпариванием с толуолом (2 х 15 мл). Оставшееся масло выдерживают при перемешивании в гексане-этиловом эфире (1:1, 50 мл) в течение ночи. Образовавшийся осадок отфильтровывают с получением 2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)фенил]пропионовой кислоты в виде трифторацетата, белого твердого вещества (1,95 г; 8,6 ммоль). Оптическое разделение кислоты, полученной таким образом, производят по методу, описанному в Arzneim. Forsch. Vol.46(II), 9, 891-894, 1996 для получения R-пирпрофена.
Пример 14
(R)-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)фенил]пропионилметансульфонамид
Метансульфонамид получают, исходя из оптически активной кислоты (R), соответствующей примеру 2. Окончательная очистка продукта достигается флэш-хроматографией (элюент СН2Cl2/СН3ОН 98:2). Чистый продукт получают в виде белого твердого вещества.
Пример 15
(R)-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)фенил]-N-(2-карбоксиэтил)пропионамид
Данный амид (R) оптически активной кислоты с L-аланином получают в соответствии с примером 3.
Окончательную очистку продукта осуществляют кристаллизацией из этилового эфира.
Пример 16
(R)-2-[4-(2,5-дигидро-1Н-пиррол-1-илметил)фенил]пропионамид
Первичный амид (R) оптически активной кислоты получают в соответствии с примером 4.
Окончательную очистку продукта осуществляют кристаллизацией из этилацетата.
| название | год | авторы | номер документа |
|---|---|---|---|
| (R)-2-АРИЛПРОПИОНАМИДЫ, ПОЛЕЗНЫЕ ПРИ ИНГИБИРОВАНИИ ИЛ-8-ИНДУЦИРОВАННОГО ХЕМОТАКСИСА НЕЙТРОФИЛОВ, СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ХЕМОТАКСИС НЕЙТРОФИЛОВ, ИНДУЦИРОВАННЫЙ ИНТЕРЛЕЙКИНОМ-8 | 2001 |
|
RU2273630C2 |
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| ПРОИЗВОДНЫЕ 2-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2005 |
|
RU2375347C2 |
| 2-АРИЛУКСУСНЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2004 |
|
RU2356887C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ TAFIa | 2005 |
|
RU2375356C2 |
| ПРИМЕНЕНИЕ ХИРАЛЬНЫХ АРИЛКЕТОНОВ В ЛЕЧЕНИИ НЕЙТРОФИЛ-ЗАВИСИМЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2345759C2 |
| 3-ОКСО-3,9-ДИГИДРО-1Н-ХРОМЕНО[2,3-c]ПИРРОЛЫ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ | 2011 |
|
RU2603191C2 |
| ПРОИЗВОДНЫЕ 2-АРИЛПРОПИОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ ВКЛЮЧАЮЩИЕ | 2005 |
|
RU2410372C2 |
| ЗАМЕЩЕННЫЕ ФЕНИЛМОЧЕВИНЫ И ФЕНИЛАМИДЫ В КАЧЕСТВЕ ЛИГАНДОВ ВАНИЛЛОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2553392C2 |
| АЗАИНДОЛИЛПИРИДОНЫ И ДИАЗАИНДОЛИЛПИРИДОНЫ | 2018 |
|
RU2788659C2 |
Изобретение относится к амидам R-2-аминоарилпропионовых кислот и фармацевтическим композициям, их содержащим, которые могут использоваться для предотвращения и ингибирования рекрутинга и активации лейкоцитов и для лечения патологий, непосредственно зависимых от указанной активации. Предложено соединение общей формулы (1).
где А, q, Ph и R имеют соответствующие значения, или его фармацевтически приемлемая соль. Также описаны способ получения амида формулы (I) и фармацевтическая композиция для предотвращения активации лейкоцитов. Достигнутый технический результат заключается в создании фармацевтической композиции, которая может использоваться для профилактики и лечения повреждения тканей из-за усиления активации нейтрофильных гранулоцитов (полиморфноядерных лейкоцитов (ПМЯ)) в местах воспаления. В частности, данное изобретение относится к R энантиомерам 2-(аминоарил)пропиониламидам формулы (1) для использования для подавления хемотаксиса нейтрофилов, вызванного IL-8. Также соединения данного изобретения могут использоваться для лечения псориаза, язвенного колита, гломерулонефрита, острой респираторной недостаточности, идиопатического фиброза и ревматоидного артрита. 3 н. и 5 з.п. ф-лы.
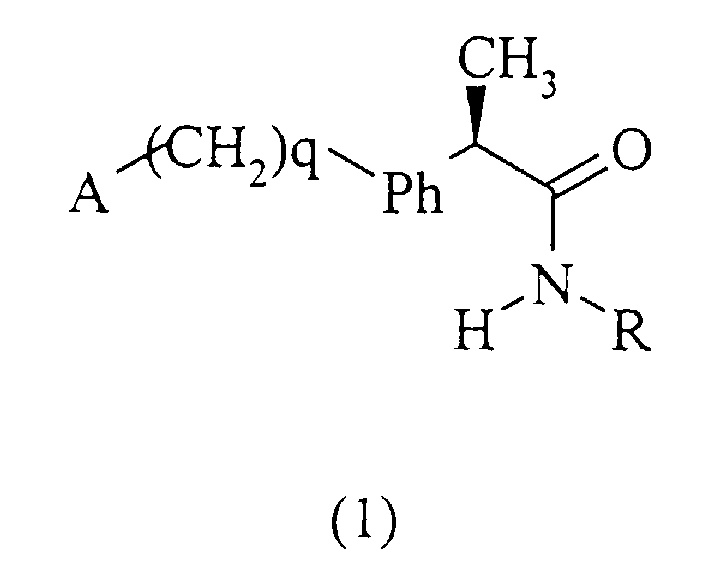
и их фармацевтически приемлемые соли,
где q равно нулю или целому числу, равному 1;
Ph представляет собой фениленовую группу, связанную с группой -(СН2)q-А в положении 2, 3 или 4, и необязательно замещенную в остальных положениях одним или несколькими заместителем, одинаковыми или различными, выбранными из С1-С3-алкила, галогенов;
А представляет собой
- N-С1-С8- арилалканоил-N-С1-С5-алкиламино-группу;
- насыщенное или ненасыщенное азотосодержащее 5-7-членное гетероциклическое кольцо;
R представляет собой
- Н; -SO2-СН3; С1-С3-алкил, группу формулы -СН2-СН2-Х-(СН2-СН2О)n-Р, где Р является Н, метилом, этилом, изопропилом;
-СН2СО2R1, где R1 представляет Н или С1-С3; n равно целому числу от 0 до 5, Х представляет собой О;
- остаток формулы (3)
-(СН2)m-Ф (3)
где, когда m равно целому числу от 2 до 3, Ф представляет собой незамещенный или замещенный фенилен, 2-(1-метилпирролидил), 2-пиридил, 4-пиридил, 1-имидазолил, 4-имидазолил, 1-метил-4-имидазолил, 1-метил-5-имидазолил или группу -NRaRb, где каждый из Ra и Rb, одинаковые или различные, представляют собой С1-С5-алкил или гидроксиалкил-(СН2)mi-ОН, где mi равно целому числу от 2 до 3, или Ra и Rb вместе с атомом N, с которым они связаны, образуют гетероциклическое кольцо, содержащее от 3 до 7 членов; когда m равно нулю, Ф выбран из группы, включающей 2- или 4-пиридил, 2- или 4-пиримидинил, 2-пиразинил, 2-1,3-тиазолил, индол-3-ил;
- остаток формулы (3а)
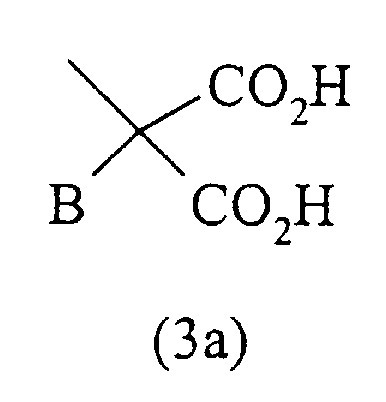
где В представляет собой Н;
при условии, что, когда q равно нулю, R представляет собой -SO2СН3 и Ph представляет собой 3-хлор-1,4-фенилен, А является группой, отличной от 1-2,5-дигидропирролидино.
-(СН2)m-Ф (3)
где, когда m равно целому числу 2 или 3, Ф является остатком основания, выбранным из группы, включающей N,N-диэтиламин, 4-морфолил, 1-пиперидил, 1-(4-бензил)пиперазинил, 1-(4-дифенилметил)пиперазинил, 1-(4-(4',4''-дифтордифенил)- метилпиперазинил; принимая во внимание, что когда m равно 0, Ф предпочтительно представляет собой гетероарильный остаток, такой как 2 или 4-пиридил, 2- или 4-пиримидинил, 2-пиразинил, 2-1,3-тиазолил, 2-1,3-тиазолидинил, 2-имидазолил и, более предпочтительно, 4-пиридил.
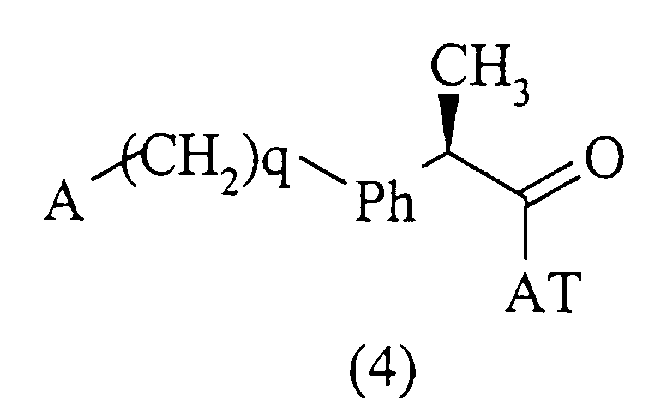
где А и q, как определено в п.1, и АТ является остатком, активирующим карбоксильную группу, с амином R-NH2, где R представляет группу, которая определена в п.1.
| 1971 |
|
SU436049A1 |

