Настоящее изобретение относится, в частности, к применению производных цистеина для получения лекарственного средства, предназначенного для лечения патологий, возникающих вследствие образования гетеротримерного протеина G. Такими заболеваниями являются, в частности, заболевания, связанные со следующими биологическими функциями или нарушениями: обоняние, вкусовые ощущения, световое восприятие, нейротрансмиссия, нейродегенерация, функционирование эндокринных и экзокринных желез, аутокринная и паракринная регуляция, артериальное кровяное давление, эмбриогенез, доброкачественная клеточная пролиферация, онкогенез, вирусная инфекция, иммунологические функции, диабет, ожирение, и доброкачественные и злокачественные пролиферативные заболевания.
Протеины G фактически представляют собой структурную ассоциацию трех различных субъединиц, называемых α, β и γ, однако функционируют как диссоциирующие частицы, образованные, с одной стороны, субъединицами α и, с другой стороны, димерами β/γ.
Протеины G участвуют в передаче сигналов с поверхности клетки за счет своего взаимодействия с рецепторами семи трансмембранных доменов к внутренней части через посредство различных эффекторов, включающих аденилатциклазу, фосфолипазу С или ионные каналы. Фермент аденилатциклаза генерирует циклический аденозинмонофосфат (цАМФ) (см. Gilman A.G., Biosci. Rep., 15, 65-97 (1995)). Также известно, что для активации аденилатциклазы необходимо, чтобы протеины G временно находились в гетеротримерной форме, т.е. форме, в которой мономер, образованный субъединицей α, ассоциирован с димером, образованным субъединицами β и γ. Только в этом состоянии сигнал с поверхности клетки может активировать субъединицу а протеина G, которая после диссоциации может модулировать аденилатциклазу и модулировать продукцию цАТФ.
Также известно, что димеры β/γ могут активировать непосредственно эффекторы, приводящие к активации киназ, регулируемых внеклеточными сигналами (ERKs), или МАР-киназ. Было показано наличие прямой связи между субъединицами β/γ и киназами src или src like (см. Gut kind J.S., J.Biol. Chem., 273, 1839-1842 (1998)).
Кроме того, показано, что бактериальные токсины, такие как Vibrio cholera и Bortella pertussis, пептиды, такие как мастопаран и сурамин, непосредственно модулируют активность протеинов G (см., Freissmuth M., Boehm S., Beindl W. и др., Mol. Pharmacol., 49, 602-611 (1996); Boehm S., Huck S., Motejlek А., и др., Journal of Neurochemistry, 66, 1019-1026 (1966); Cachero T.G., Rigual R., Rocher А. и Gonzaiez С., Eur. J. Neurosci., 8, 2320-2327 (1996); Danilenko M., Worland P., Carlson В., Sausviile E.A. и Sharoni Y., Biochem. Biophys. Res. Commun., 196, 1296-1302 (1993); Beindl W., Mitterauer Т., Hohenegger M., Ijzerman A. P., Nanoff С. и Freissmuth M., Mol. Pharmacol., 50, 415-423 (1996)).
Холерный токсин, например, модифицирует субъединицу αs протеина G за счет присоединения аденозиндифосфатрибозы (АДФ-рибозы), происходящей от никотинамид-аденин-динуклеотида (НАД), к специфичному аргининовому акцепторному сайту. Это полностью блокирует активность гуанозинтрифосфатазы (GTPase), что провоцирует продолжительную стимуляцию ее следующего эффектора, аденилатциклазы, и приводит к сверхпродукции цАМФ.
Негативный эффект повышенного количества цАМФ также известен и, в частности, он проявляется на уровне следующих биологический функций и нарушений: обоняние, вкусовые ощущения, световое восприятие, нейротрансмиссия, нейродегенерация, функционирование эндокринных и экзокринных желез, аутокринная и паракринная регуляция, артериальное кровяное давление, эмбриогенез, доброкачественная клеточная пролиферация, онкогенез, вирусная инфекция и иммунологические функции, диабет и ожирение.
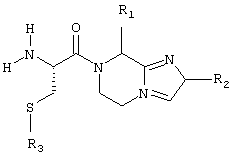
Заявитель в настоящее время нашел, что некоторые производные цистеина, а именно соединения общей формулы (А):
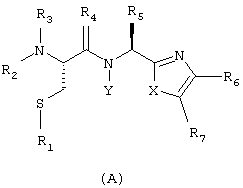
соответствующей частичным формулам (А1) и (А2)
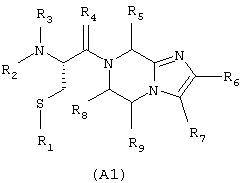
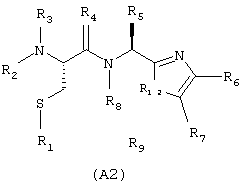
в которых:
Х означает R12 и Y означает R8, или Х и Y входят в состав шестичленного цикла, причем совокупность X-Y означает радикал -СН(R8)-CH(R9)-;
R1 означает Н, низший алкил или низшую алкилтиогруппу;
R2 и R3 независимо означают Н или низший алкил;
R4 означает Н2 или О;
R5 означает Н или один из следующих радикалов: низший алкил, низший алкенил, низший алкинил, арил, низший арилалкил, гетероциклический или низший алкилгетероциклический радикал, причем эти радикалы в случае необходимости могут быть замещены радикалами, выбираемыми из группы, состоящей из низшего алкила, -О-R10, -S(O)mR10 (где m означает 0, 1 или 2), -N(R10)(R11), -N-C(O)-R10, -NH-(SO2)-R10, -CO2-R10, C(O)-N(R10)(R11) и -(SO2)-N(R10)(R11);
R6 и R7 независимо означают Н, радикал -С(О)-NH-CHR13-CO2R14 или один из следующих радикалов: низший алкил, арил, низший арилалкил, гетероциклический или низший алкилгетероциклический радикал, причем эти радикалы в случае необходимости могут быть замещены радикалами, выбираемыми из группы, состоящей из ОН, низшего алкила или алкоксила, N(R10)(R11), COOH, CON (R10)(R11) и галогена;
или R6 и R7 вместе образуют арил или гетероциклический радикал;
R8 и R9 независимо означают Н или один из следующих радикалов: низший алкил, арил, низший арилалкил, гетероциклический или низший алкилгетероциклический радикал, причем эти радикалы в случае необходимости могут быть замещены радикалами, выбираемыми из группы, состоящей из ОН, низшего алкила или алкоксила, N(R10) (R11), COOH, CON(R10)(R11) и галогена;
или R8 и R9 вместе образуют арил или гетероциклический радикал;
R10 и R11 независимо означают Н, арил или гетероциклический радикал, или низший алкил, низший арилалкил или низший алкилгетероциклический радикал;
R12 означает NR9, S или О;
R13 означает низший алкил, возможно замещенный радикалом, выбираемым среди следующих радикалов: низший алкил, -OR10, -S(O)mR10 (где m означает 0, 1 или 2) и -N(R10)(R11);
R14 означает Н или низший алкил;
или соединения общей формулы (В):
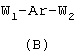
в которой:
W1 означает остаток, происходящий от цистеина в восстановленной или невосстановленной форме;
Ar означает радикал, происходящий от аминобензойной кислоты, ароматическое ядро которой может быть замещено;
W2 означает остаток аминокислоты, предпочтительно алифатической аминокислоты;
или соединения общей формулы (С):
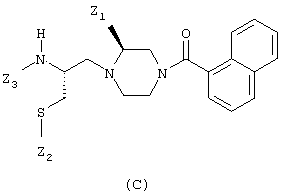
в которой:
Z1 означает низший алкил;
Z2 и Z3 оба означают Н или же Z2 и Z3 вместе образуют цепь из 2-4 звеньев, выбираемых среди радикалов -С(О)-, -CH2-, -CH(NH2)-и -S-, при условии, что два последовательных звена не могут одновременно являться -С(O)-;
при условии, что соединения общей формулы (С) также могут находиться в форме димеров, когда радикал Z2 означает атом водорода, который может быть удален путем окисления;
или фармацевтически приемлемая соль соединения общей формулы (А), (В) или (С);
могут быть использованы для получения лекарственных средств, предназначенных для лечения патологий, возникающих вследствие образования гетеротримерного протеина G.
Под низшим алкилом понимают линейный или разветвленный алкильный радикал с 1-6 атомами углерода и, в частности, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил, пентил, неопентил, изопентил, гексил, изогексил. Под гетероциклическим радикалом понимают радикал, образованный одним или несколькими циклами и включающий по крайней мере один гетероатом. Под низшим арилалкилом, алкилгетероциклическим радикалом, алкилтиогруппой или алкоксилом понимают радикалы, алкильная часть которых имеет вышеуказанное значение.
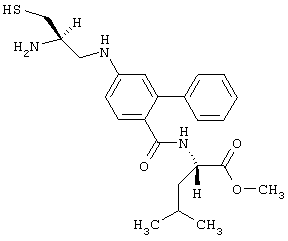
Радикал Ar, входящий в формулу (В), предпочтительно может быть замещен алкилом с 1-6 атомами углерода или арилом, причем эти алкильный и арильный радикалы сами по себе также могут быть предпочтительно замещены алкоксилом с 1-4 атомами углерода, фтором, хлором, бромом. Арил, предпочтительно фенил, сам также может быть замещен алкилом.
Соединения общей формулы (В) предпочтительно еще являются такими, в которых Ar означает радикал, происходящий от аминобензойной кислоты, ароматическое ядро которой замещено фенилом, и W2 означает остаток алифатической аминокислоты.
Для получения лекарственных средств, предназначенных для лечения патологий, возникающих вследствие образования гетеротримерного протеина G, в особенности могут быть использованы следующие соединения:
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-8-(1-метилпропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
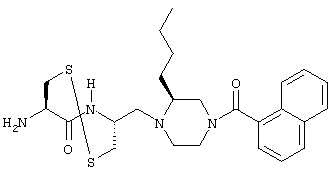
- 1-[2(R)-амино-3-меркаптопропил]-2(S)-н-бутил-4-(1-нафтоил)пиперазин;
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфид;
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфид;
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид;
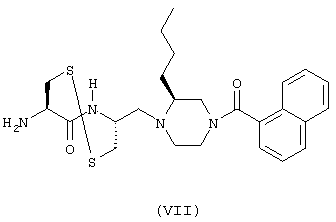
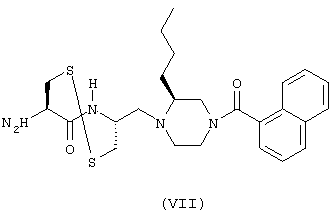
- соединение формулы (VII):
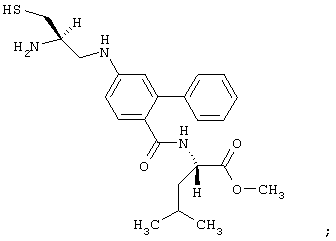
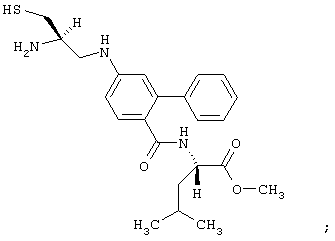
- соединение формулы:
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилметокси)метил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(1-фенилметокси)этил-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин в димерной форме;
- и 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилсульфонилэтил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин;
или фармацевтически приемлемая соль одного из этих соединений.
Предпочтительно используют одно из следующих соединений согласно изобретению:
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфид (I);
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид (II);
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин (III);
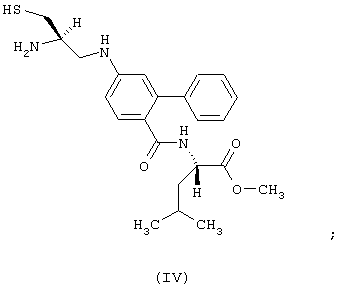
- соединение формулы (IV):
- 7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин (V);
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин]дисульфид (VI);
- соединение формулы (VII):
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-8-(1-метилпропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 1-[2(R)-амино-3-меркаптопропил]-2(S)-н-бутил-4-(1-нафтоил)пиперазин;
- или фармацевтически приемлемую соль одного из этих соединений.
Более предпочтительно используют одно из следующих соединений согласно изобретению:
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимида-зо[1,2а]пиразин]дисульфид (I);
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид (II);
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин (III);
- 7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин (V);
- соединение формулы (VII):
- или фармацевтически приемлемую соль одного из этих соединений.
Наконец, в высшей степени предпочтительными являются следующие соединения:
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфид (I);
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид (II);
- или фармацевтически приемлемая соль одного из этих соединений.
Изобретение, следовательно, относится прежде всего к применению соединений общей формулы (А), (В) или (С), таких, как указанные выше, для получения лекарственного средства, предназначенного для лечения патологий, возникающих вследствие образования гетеротримерного протеина G. В частности, изобретение относится к применению вышеуказанных ингибиторов для получения лекарственных средств в целях лечения заболеваний, связанных со следующими биологическими функциями или нарушениями: обоняние, вкусовые ощущения, световое восприятие, нейротрансмиссия, нейродегенерация, функционирование эндокринных и экзокринных желез, аутокринная и паракринная регуляция, артериальное кровяное давление, эмбриогенез, вирусная инфекция, иммунологические функции, диабет и ожирение.
Более предпочтительно, изобретение относится к применению соединений общей формулы (А), (В) или (С) для получения лекарственного средства, предназначенного для лечения холеры, синдрома приобретенного иммунодефицита (СПИД), диареи путешественника и наследственного мужского преждевременного полового созревания.
Предметом изобретения также являются новые продукты общей формулы (А) под номерами 1-7 и описанные ниже в примерах, а именно:
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилметокси)метил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(1-фенилметокси)этил-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин в димерной форме;
- и 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилсульфонилэтил)-5,6,7,8-тетрагидроимидазо [1,2а]пиразин.
Предметом настоящего изобретения являются также вышеуказанные новые продукты или их фармацевтически приемлемые соли в качестве лекарственных средств, а также их применение для получения лекарственного средства, предназначенного для лечения патологий, возникающих вследствие образования гетеротримерного протеина G. В особенности, изобретение относится к применению вышеуказанных продуктов для получения лекарственных средств, предназначенных для лечения заболеваний, связанных со следующими биологическими функциями или нарушениями: обоняние, вкусовые ощущения, световое восприятие, нейротрансмиссия, нейродегенерация, функционирование эндокринных и экзокринных желез, аутокринная и паракринная регуляция, артериальное кровяное давление, эмбриогенез, доброкачественная клеточная пролиферация, онкогенез, вирусная инфекция, иммунологические функции, диабет, ожирение и доброкачественные и злокачественные пролиферативные заболевания.
Для применения согласно изобретению особенно предпочтительными продуктами являются, таким образом, следующие:
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфид;
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид;
- соединение формулы:
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,1,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилметокси)метил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(1-фенилметокси)этил-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин в димерной форме;
- и 7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил) -8-(фенилсульфонилэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
- или фармацевтически приемлемая соль одного из этих соединений.
Точно также изобретение относится в особенности к применению вышеуказанных соединений для получения лекарственного средства, предназначенного для лечения холеры, синдрома приобретенного иммунодефицита (СПИД), диареи путешественника и наследственного мужского преждевременного полового созревания.
Соединения общей формулы (А) и их получение описываются в Международной заявке на патент 97/30053 или в нижеприводимых примерах. Соединения общей формулы (В) и их получение описываются в Международной заявке на патент 96/21456. Наконец, получение соединений общей формулы (С) описывается в Международной заявке на патент РСТ 95/00497 за исключением соединения формулы (VII), синтез которого описывается в экспериментальной части настоящей заявки.
Фармацевтические композиции, включающие соединение согласно изобретению, могут быть в форме твердых веществ, например, порошков, гранул, таблеток, желатиновых капсул, липосом или суппозиториев. Соответствующими твердыми носителями могут быть, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, поливинилпирролидин и воск.
Фармацевтические композиции, включающие соединение согласно изобретению, также могут находиться в жидкой форме, например, в виде растворов, эмульсий, суспензий или сиропов. Соответствующими жидкими носителями могут быть, например, вода, органические растворители, такие как глицерин или гликоли, а также их смеси, в различных соотношениях, в воде.
Лекарственное средство согласно изобретению можно вводить топическим путем, перорально, парентерально, путем инъекции (внутримышечной, подкожной, внутривенной и т.д.), и т.д. Путь введения зависит, разумеется, от типа заболевания.
Предусматриваемая для лекарственного средства согласно изобретению вводимая доза составляет от 0,1 мг до 10 г в зависимости от типа патологии.
Если не указано иное, все технические и научные термины, используемые в настоящем описании, имеют значения, обычно понимаемые специалистом в области, к которой относится настоящее изобретение. Точно также все публикации, заявки на патенты, все патенты и все другие указанные здесь ссылки включены в настоящее описание в виде ссылки.
ПРИМЕРЫ
Пример 1
7-(2-Амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин: 1
Соединение 1 получают согласно нижеприводимой схеме синтеза:
Схема 1
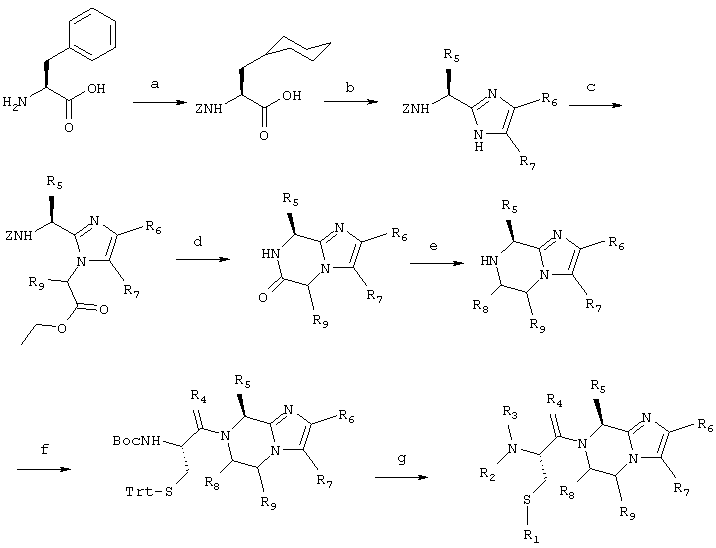
1.а) Карбобензилокси-L-циклогексилаланин
10,0 г (60,6 ммоль) L-Фенилаланина смешивают с 430 мг PtO2 в 60 мл уксусной кислоты и смесь гидрируют при давлении водорода 1,38-3,45 бар в течение ночи. К смеси добавляют водный 5%-ный раствор HCl для получения прозрачного раствора и гидрирование продолжают вплоть до прекращения потребления водорода. Катализатор отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток обрабатывают метанолом и водой и значение рН устанавливают равным 4,4 путем добавления 10%-ного раствора NaOH. Полученный продукт извлекают путем отфильтровывания и используют без дополнительной очистки.
60,6 ммоль L-Циклогексилаланина суспендируют в 100 мл воды, добавляют 8,36 г (60,6 ммоль) К2СО3, затем раствор 15,1 г (60,6 ммоль) N-(бензилоксикарбонилокси)сукцинимида в 150 мл CH3CN и полученную смесь интенсивно перемешивают в течение 45 минут. Смесь концентрируют до получения объема около 100 мл и промывают с помощью 100 мл диэтилового эфира, затем подкисляют с помощью концентрированной соляной кислоты и экстрагируют 2 раза по 50 мл этилацетатом. Объединенные этилацетатные фазы сушат над сульфатом натрия, отфильтровывают и концентрируют, получая 17,27 г (93%) прозрачного масла.
1H-ЯМР (ДМСО-d6 [гексадейтеродиметилсульфоксид]), δ (м.д.): 7,5-7,6 (д, 1Н); 7,2-7,5 (м, 5Н); 5,0-5,1 (с, 2Н); 3,9-4,1 (м, 1Н); 0,7-1,8 (м, 13Н).
1.b) 2-(1-(S)-((Фенилметокси)карбонил)амино-2-(циклогексил)метил)-4-(2-метилфенил)имидазол
4,58 г (15,0 ммоль) Cbz-(L)-Циклогексилаланина и 2,44 г (7,50 ммоль) Cs2СО3 вносят в 75 мл смеси диметилформамида с водой в соотношении 2:1. Полученную смесь перемешивают до тех пор, пока она не станет гомогенной. Растворители удаляют при пониженном давлении, остаток растворяют в 60 мл диметилформамида и добавляют 3,20 г (15,0 ммоль) 2-бром-2'-метилацетофенона в 30 мл диметилформамида. Смесь перемешивают в течение ночи при комнатной температуре, затем отфильтровывают и концентрируют при пониженном давлении. Полученный сложный кетоэфир растворяют в 100 мл смеси ксилолов и добавляют 19,5 г (0,25 моль) ацетата аммония.
Смесь кипятят с обратным холодильником в течение примерно трех часов при удалении избыточного ацетата аммония и высвободившейся воды при использовании ловушки Дина-Старка. Реакционную смесь концентрируют при пониженном давлении, обрабатывают этилацетатом, полученный раствор промывают с помощью 100 мл насыщенного раствора NaHCO3 и 100 мл насыщенного раствора NaCl. Этилацетатную фазу сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Полученный сырой продукт очищают путем флэш-хроматографии на силикагеле при использовании смеси хлороформа с метанолом в соотношении 98:2 в качестве элюента. Содержащие чистый продукт фракции объединяют и концентрируют, получая 2,52 г (40%) продукта в виде слегка коричневого цвета пены, который используют без дополнительной очистки в следующей стадии.
1.с) 2-(1-(S)-((Фенилметокси)карбонил)амино-2-(циклогексил)метил)-1-((2-этокси-2-оксо)этил)-4-(2-метилфенил)имидазол
2,52 г (6,0 ммоль) Промежуточного соединения 1.b) растворяют в 20 мл диметилформамида, обрабатывают с помощью 1,67 г (12,1 ммоль) К2СО3 и добавляют 1,34 мл (12,5 ммоль) этилбромацетата. Полученную смесь нагревают при температуре 45°С в течение полутора часов. Смесь разбавляют с помощью 50 мл диэтилового эфира и раствор промывают с помощью 50 мл насыщенного раствора NaHCO3, затем с помощью 50 мл насыщенного раствора NaCl. Эфирный слой сушат над сульфатом натрия, отфильтровывают и концентрируют, получая масло, которое используют без дополнительной очистки в следующей стадии.
Масс-спектрометрия: 504,3 МН+.
1.d) 8-(Циклогексилметил)-6-оксо-2-(2-метилфенил)имидазо[1,2а]пиразин
Сырое промежуточное соединение, полученное в стадии 1.с, растворяют в 50 мл уксусной кислоты, содержащих 152 мг 10%-ного палладия-на-угле в качестве катализатора, затем гидрируют при давлении водорода 3,45 бар в течение 18 часов при комнатной температуре. Катализатор отфильтровывают и фильтрат нагревают при температуре 70°С в течение двух часов. Полученную смесь концентрируют при пониженном давлении, растворяют в 100 мл дихлорметана и промывают с помощью 100 мл насыщенного раствора NaHCO3. Дихлорметановый слой сушат над сульфатом натрия, отфильтровывают и концентрируют, получая вязкое масло, которое используют без дополнительной очистки в следующей стадии.
Масс-спектрометрия: 324,3 МН+.
1.е) 8-(Циклогексилметил)-2-(2-метилфенил)-4,5,6,7-тетрагидроимидазо[1,2а]пиразин
Сырое промежуточное соединение, полученное в стадии 1.d, растворяют в 25 мл тетрагидрофурана и при комнатной температуре, обрабатывают с помощью 25 мл 1М раствора ВН3 в тетрагидрофуране в течение получаса, затем кипятят с обратным холодильником в течение 1 часа. Смесь охлаждают на бане со льдом и при температуре 0°С прикапывают 40 мл 4 н. соляной кислоты. Смесь доводят до комнатной температуры, затем кипятят с обратным холодильником в течение 1 часа. После этого реакционную смесь охлаждают и концентрируют при пониженном давлении. Остаток обрабатывают 50 мл насыщенного раствора NaHCO3 и экстрагируют три раза по 50 мл дихлорметаном. Дихлорметановые фазы сушат над сульфатом натрия, отфильтровывают и концентрируют, получая 1,63 г (выход составляет 87% по отношению к стадиям 1.c, 1.d и 1.e) масла слегка коричневого цвета.
Масс-спектрометрия: 310,3 МН+.
1.f) 8-(Циклогексилметил)-7-[2-(((1,1-диметилэтокси)карбонил)амино)-1-оксо-3-((трифенилметил)тио)пропил]-2-(2-метилфенил)-4,5,6,7-тетрагидроимидазо[1,2-а]-пиперазин
908 мкл (5,80 ммоль) Диизопропилкарбодиимида и 5,37 г (11,6 ммоль) BocCys(trt)-ОН растворяют в 25 мл дихлорметана, полученную смесь перемешивают в течение 45 минут. Затем добавляют 1,63 г (5,27 ммоль) 8-(циклогексилметил)-2-(2-метилфенил)-4,5,6,7-тетрагидроимидазо[1,2а]пиразина. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Растворитель удаляют при пониженном давлении и полученный продукт очищают путем флэш-хроматографии на силикагеле при использовании смеси дихлорметана с метанолом в соотношении 98:2 в качестве элюента. Чистые фракции концентрируют, получая вязкое масло, которое используют без дополнительной очистки в следующей стадии.
Масс-спектрометрия: 755,6 МН+.
1.g) 7-(2-Амино-1-оксо-3-(меркаптопропил))-8-(циклогексилметил)-2-(2-метилфенил)-4,5,6,7-тетрагидроимидазо[1,2а]пиперазиы: 1:
3,54 г (4,69 ммоль) Полученного в стадии 1.f промежуточного соединения растворяют в 80 мл трифторуксусной кислоты (ТФУК), содержащих 1,92 мл (9,38 ммоль) триизопропил-силана, и реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение часа. Реакционную смесь отфильтровывают и фильтрат концентрируют при пониженном давлении. Остаток экстрагируют путем порошкования с водным 0,1%-ным раствором трифторуксусной кислоты (6 раз по 65 мл) и отфильтровывают. Сырой продукт очищают путем препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) на колонке с C18, используя градиент 0-20% СН3CN в 0,1%-ном водном растворе трифторуксусной кислоты, в течение 30 минут. Чистые фракции продукта объединяют и лиофилизируют. Первоначальный продукт лиофилизируют два раза из разбавленного раствора HCl, получая продукт в виде его гидрохлорида (740 мг; 32%).
Масс-спектрометрия: 413,2 МН+.
1Н-ЯМР (ДМСО-d6), δ (м.д.): 8,5-9,0 (д уш., 3Н); 7,8-8,0 (с, 1Н); 7,5-7,7 (д, 1Н); 7,2-7,5 (м, 3Н); 5,8-6,1 (м, 1Н); 4,65-4,8 (с, 1Н); 4,5-4,7 (д, 1Н); 4,1-4,4 (м, 2Н); 3,8-4,0 (м, 1H); 3,2-3,7 (Н2O); 2,8-3,1 (м, 2Н); 2,35-2,5 (с, 3Н); 2,0-2,2 (м, 1H); 1,8-2,05 (м, 2Н); 1,25-1,4 (с уш., 4Н); 1,3-0,9 (м, 6Н).
Пример 2
7-(2-Амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин: 2:
Соединение 2 получают согласно схеме 1, стадии b-g, по методике, аналогичной таковой примера 1, заменяя в стадии b) 2-бром-2'-метилацетофенон на 2-бромацетофенон.
Масс-спектрометрия: 399,2 МН+.
1H-ЯМР (ДМСО-d6), δ (м.д.): 8,5-8,9 (д уш., 3Н); 8,0-8,2 (с, 1H); 7,8-8,0 (д, 2Н); 7,45-7,56 (т, 2Н); 7,35-7,5 (т, 1H); 5,9-6,05 (с уш., 1H); 4,65-4,8 (с, 1H); 4,5-4,65 (д, 1H); 4,1-4,35 (м, 2Н); 3,8-4,0 (м, 1H); 3,2-3,8 (H2O); 3,25-3,4 (т, 1H); 2,8-3,05 (м, 2Н); 2,05-2,2 (д, 1H); 1,85-2,05 (т, 2Н); 1,55-1,75 (с уш., 4Н); 1,15-1,3 (с уш., 1H); 1,2-0,9 (м, 5Н).
Пример 3
7-(2-Амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилметокси)метил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин: 3:
Соединение 3 получают согласно схеме 1, стадии b-g, по методике, аналогичной таковой примера 1, заменяя в стадии b) Cbz-(L)-циклогексилаланин на Boc-(L)-Ser(Bzl)-ОН и заменяя стадию d) стадией удаления защитной группы при использовании трифторуксусной кислоты и триизопропилсилана, согласно методике, аналогичной таковой стадии 1.g. Продукт получают в виде пары диастерероизомеров в соотношении 2:3.
Масс-спектрометрия: 453,2 МН+.
Времена удерживания для диастереоизомеров составляют соответственно 6,58 и 7,07 минут в следующей системе ВЭЖХ:
Пример 4
7-(2-Амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(1-фенилметокси)этил-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин: 4:
Соединение 4 получают согласно схеме 1, стадии b-g, по методике, аналогичной таковой примера 3, заменяя в стадии b) Boc-(L)-Ser(Bzl)-OH на Boc-(L)-Thr(Bzl)-ОН. Масс-спектрометрия: 467,3 МН+.
1H-ЯМР (ДМСО-d6), δ (м.д.): 8,5-8,9 (д уш., 3Н); 8,0-8,1 (с, 1Н); 7,95-8,1 (д, 2Н); 7,4-7,5 (т, 1Н); 7,15-7,3 (д, 1Н); 7,0-7,2 (м, 3Н); 6,9-7,05 (м, 2Н); 5,85-5,95 (д, 1Н); 4,75-4,85 (с уш., 1Н); 4,65-4,8 (с уш., 1Н); 4,35-4,65 (м, 3Н); 4,1-4,25 (к, 2Н); 3,9-4,0 (с, 3Н); 2,8-3,1 (м, 2Н); 1,2-1,4 (д, 3Н).
Пример 5
7-(2-Амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин: 5:
Соединение 5 получают согласно нижеприводимой схеме синтеза:
Схема 2
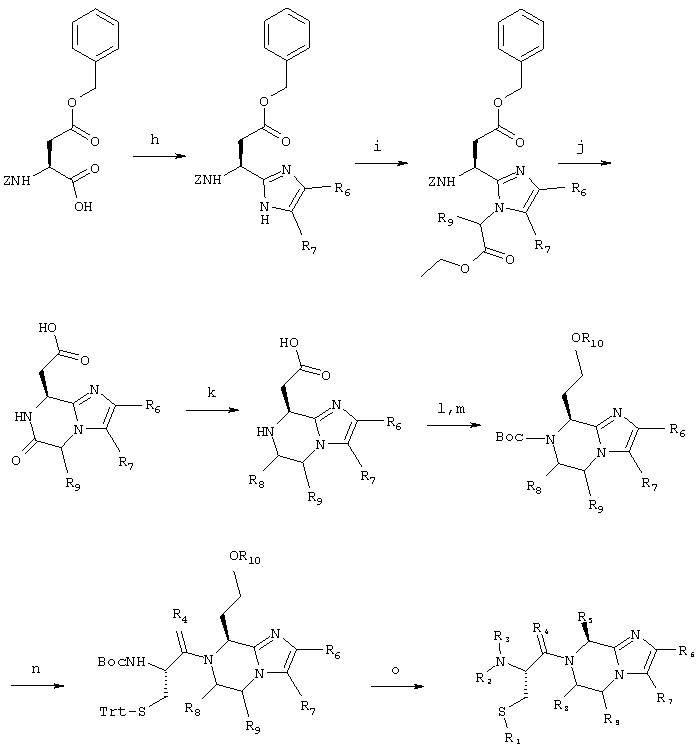
5.h) 2-(1-(S)-((Фенилметокси)карбонил)амино-2-(2-оксо-2-(фенилметокси)этил)-4-(2-метоксифенил)имидазол
5,00 г (14,0 ммоль) Cbz-(L)-Asp(Obzl)-ОН и 2,28 г (7,00 ммоль) Cs2СО3 смешивают с 75 мл смеси диметилформамида с водой в соотношении 1:1. Полученную смесь перемешивают до тех пор, пока она не станет гомогенной. Растворители удаляют при пониженном давлении, остаток растворяют в 60 мл диметилформамида и добавляют 3,21 г (14,0 ммоль) 2-бром-2'-метоксиацетофенона в 30 мл диметилформамида. Полученную смесь перемешивают в течение получаса при комнатной температуре, затем отфильтровывают и концентрируют при пониженном давлении. Полученный сложный кетоэфир порошкуют со смесью диэтилового эфира с гексаном (2 раза по 40 мл), затем суспендируют в 100 мл смеси ксилолов. Добавляют 17,5 г (0,23 моль) ацетата аммония и смесь кипятят с обратным холодильником в течение примерно полутора часов при удалении избыточного количества ацетата аммония и высвободившейся воды, используя ловушку Дина-Старка. Реакционную смесь промывают с помощью 50 мл насыщенного раствора NaHCO3, сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме, получая 6,66 г (98%) целевого продукта.
Масс-спектрометрия: 486,3 МН+.
5.i) 2-(1-(S)-((Фенилметокси)карбонил)амино-2-((2-фенилметокси-2-оксо)этил)-1-((2-этокси-2-оксо)этил)-4-(2-метоксифенил)имидазол
Промежуточное соединение 5.i получают согласно методике, аналогичной таковой стадии 1.c.
Масс-спектрометрия: 572,3 МН+.
5.j) (6-Оксо-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-8-ил)-уксусная кислота
Промежуточное соединение 5.j получают согласно методике, аналогичной таковой стадии 1.d.
Масс-спектрометрия: 302,2 МН+.
1H-ЯМР (ДМСО-d6), δ (м.д.): 8,35-8,5 (д уш., 1Н); 8,0-8,1 (дд, 1Н); 7,45-7,55 (с, 1Н); 7,15-7,25 (м, 1Н); 7,0-7,1 (м, 1Н); 6,9-7,0 (м, 1Н); 4,85-5,0 (с уш., 1Н); 4,55-4,75 (к, 2Н); 3,85-3,95 (с, ЗН); 2,8-2,95 (д, 2Н).
5.k) 8-Гидроксиэтил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин
Промежуточное соединение 5.k получают согласно методике, аналогичной таковой стадии 1.е за исключением того, что используют молярное соотношение ВН3 по отношению к субстрату, составляющее 6:9.
Масс-спектрометрия: 274,3 МН+.
5.1) 7-((1,1-Диметилэтокси)карбонил)-8-гидроксиэтил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
1,36 г (5,0 ммоль) Промежуточного соединения 5.k суспендируют в 5 мл воды и добавляют смесь 1,20 г (5,5 ммоль) ди-трет-бутилдикарбоната в 10 мл п-диоксана. Реакционную смесь интенсивно перемешивают и поддерживают при значении рН, составляющем 8,0-8,4, путем добавления по каплям 2,5 н. раствора NaOH вплоть до окончания реакции (за протеканием реакции следят с помощью тонкослойной хроматографии на силикагеле, элюируя смесью этилацетата с гексанами в соотношении 3:2). Сырой продукт очищают путем флэш-хроматографии на силикагеле с помощью смеси этилацетата с гексанами в соотношении 3:2 в качестве элюента (система Biotage, предварительно заполненные колонки размером 4×15 см). Содержащие продукт фракции объединяют и концентрируют в вакууме, получая 1,60 г (86%) белой пены.
Масс-спектрометрия: 374,3 МН+.
1H-ЯМР (ДМСО-d6), δ (м.д.): 7,95-8,05 (дд, 1Н); 7,45-7,55 (с, 1Н); 7,10-7,25 (м, 1Н); 7,0-7,1 (м, 1Н); 6,9-7,05 (м, 1Н); 5,05-5,15 (т, 1Н); 4,25-4,35 (т, 1Н); 4,05-4,2 (с уш., 1Н); 4,0-4,1 (м, 1Н); 3,9-4,0 (с, 3Н); 3,9-4,0 (м, 1Н); 3,25-3,35 (м, 2Н); 1,9-2,1 (м, 2Н); 1,15-1,25 (с, 9Н).
5.m) 7-((1,1-Диметилэтокси)карбонил)-8-феноксиэтил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
746 мг (2,00 ммоль) Промежуточного соединения 5.1 растворяют в 10 мл тетрагидрофурана, содержащих 550 мг (2,1 ммоль) трифенилфосфина и 198 мг (2,1 ммоль) фенола. Смесь охлаждают до температуры 0°С в атмосфере азота и добавляют по каплям в течение 10 минут 330 мкл (2,1 ммоль) диэтилазодикарбоксилата. Реакционную смесь затем перемешивают в течение двух часов при комнатной температуре. После этого реакционную смесь снова охлаждают до температуры 0°С и добавляют 275 мг (1,05 ммоль) трифенилфосфина и 99 мг (1,05 ммоль) фенола. Затем в течение 10 минут добавляют по каплям 166 мкл (1,05 ммоль) диэтилазодикарбоксилата, после чего смесь перемешивают еще в течение 1 часа при комнатной температуре. Растворители удаляют при пониженном давлении и сырой продукт очищают путем флэш-хроматографии на силикагеле при использовании смеси этилацетата с гексанами в соотношении 3:2 в качестве элюента. Содержащие продукт фракции объединяют и концентрируют в вакууме. После перекристаллизации из этилацетата и гексанов получают 863 г (96%) целевого продукта в виде твердого вещества белого цвета.
Масс-спектрометрия: 450,4 МН+.
5.n) 7-(2-(((1,1-Диметилэтокси)карбонил)амино)-1-оксо-3-((трифенилметил)тио)-пропил)-2-(2-метоксифенил)-8-(феноксиэтил)-4,5,6,7-тетрагидроимидазо[1,2а]пиперазин
850 мг (1,89 ммоль) Промежуточного соединения 5.т обрабатывают с помощью смеси 10 мл трифторуксусной кислоты, содержащей 387 мкл (1,89 ммоль) триизопропилсилана, при комнатной температуре в течение 20 минут. Растворители удаляют при пониженном давлении и сырой продукт распределяют между 15 мл этилацетата и 15 мл насыщенного раствора NaHCO3. Этилацетатную фазу сушат над сульфатом натрия, отфильтровывают и концентрируют при пониженном давлении. Продукт, из которого удалена защитная группа, связывают с Boc-(L)-Cys(Trt)-ОН согласно методике, аналогичной таковой стадии 1.f (1,26 г; 84%).
5.о) 7-(2-Амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-4,5,6,7-тетрагидроимидазо[1,2-а]-пиперазин
Продукт 5 получают из промежуточного соединения 5.n согласно методике, аналогичной таковой стадии 1.g.
Масс-спектрометрия: 274/3 МН+.
1H-ЯМР (ДМСО-d6 при температуре 90°С), δ (м.д.): 8,5-9,2 (с уш., 3Н); 7,95-8,1 (д, 1Н); 7,85-8,0 (с, 1Н); 7,35-7,5 (м, 1Н); 7,15-7,35 (м, 3Н); 7,0-7,15 (т, 1Н); 6,85-7,0 (м, 3Н); 5,9-6,1 (с уш., 1Н); 4,5-4,8 (м уш., 2Н); 4,15-4,45 (м уш., 3Н); 3,9-4,0 (с, 3Н); 3,75-4,0 (муш., 1Н); 2,8-3,05 (м уш., 2Н); 2,55-2,75 (муш., 2Н).
Пример 6
7-(2-Амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(феноксиэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин в виде димера: 6:
467 мг (0,687 ммоль) Соединения 5.0 растворяют в 25 мл воды и значение рН раствора устанавливают равным 7,2 путем добавления водного разбавленного раствора NH4OH. Добавляют ацетонитрил, получая прозрачный раствор, и смесь перемешивают при комнатной температуре в течение ночи. Сырой продукт очищают путем препаративной ВЭЖХ на колонке с C18, используя градиент 15-40% ацетонитрила в 0,1%-ной трифторук-сусной кислоте, в течение 50 минут. Чистые фракции продукта объединяют и лиофилизируют. Первоначальный продукт лиофилизируют два раза из разбавленного раствора HCl, получая продукт в виде гидрохлорида (161 мг; 45%).
Масс-спектрометрия: 903,5 МН+.
1H-ЯМР (ДМСО-d6 при температуре 90°С), δ (м.д.): 8,7-9,3 (с уш., 3Н); 7,95-8,1 (д, 1Н); 7,85-8,0 (с, 1Н); 7,35-7,5 (т, 1Н); 7,1-7,3 (м, 3Н); 7,0-7,15 (т, 1Н); 6,8-7,0 (м, 3Н); 5,85-6,1 (с уш., 1Н); 4,7-4,9 (с уш., 1Н); 4,45-4,7 (м уш., 1Н); 4,1-4,5 (м уш., 4Н); 3,85-4,0 (с, 4Н); 3,3-3,5 (м уш., 2Н); 2,5-2,8 (м уш., 2Н).
Пример 7
7-(2-Амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилсульфонилэтил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин: 7:
Соединение 7 получают согласно нижеприводимой схеме синтеза:
Схема 3
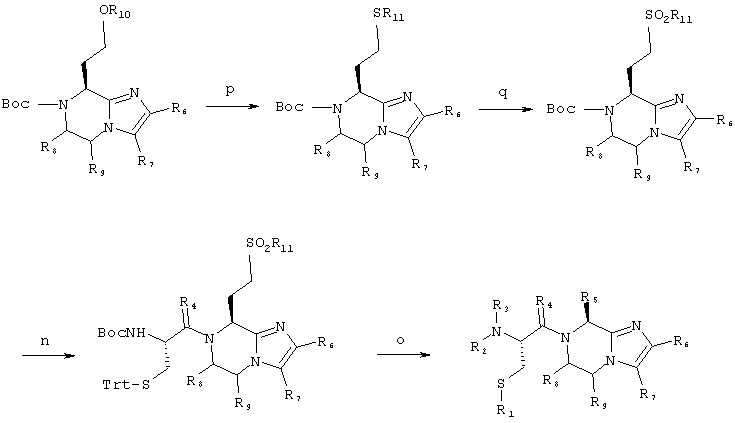
7.p) 7-((1,1-Диметилэтокси)карбонил)-2-(2-метоксифенил)-8-(фенилтиоэтил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
1,23 г (3,30 ммоль) Промежуточного соединения 5.1, 1,64 мл (6,60 ммоль) три-н-бутилфосфина и 1,44 г (6,60 ммоль) фенилдисульфида смешивают в 10 мл тетрагидро-фурана. Смесь перемешивают при комнатной температуре в атмосфере аргона в течение 4-х часов. Растворители удаляют при пониженном давлении и сырой продукт очищают путем флэш-хроматографии на силикагеле при использовании смеси этилацетата с гексанами в соотношении 1:1 в качестве элюента, Содержащие продукт фракции объединяют и концентрируют в вакууме, получая 1,43 г (93%) продукта в виде белой пены.
Масс-спектрометрия: 466/3 МН+.
7.q) 7-((1,1-Диметилэтокси)карбонил)-2-(2-метоксифенил)-8-(фенилсульфонилэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
650 мг (1,40 ммоль) Промежуточного продукта 7.р растворяют в 10 мл дихлорметана и в виде нескольких порций в течение периода 10 минут добавляют 483 мг (2,80 ммоль) 3-хлорпероксибензойной кислоты. Смесь вносят в колонку с диоксидом кремния и элюируют смесью гексанов с этилацетатом в соотношении 7:3, затем смесью гексанов с этилацетатом в соотношении 1:1, получая 220 мг (32%) чистого продукта.
Масс-спектрометрия: 498,3 МН+.
1H-ЯМР (ДМСО-d6 при температуре 30°С), δ (м.д.): 7,9-8,0 (м, 3Н); 7,7-7,85 (м, 1Н); 7,6-7,75 (м, 2Н); 7,45-7,55 (с, 1Н); 7,15-7,25 (м, 1Н); 6,9-7,1 (м, 2Н); 5,1-5,25 (т, 1H); 4,1-4,3 (д уш., 1Н); 4,0-4,15 (м, 1Н); 3,8-4,0 (м, 1H); 3,85-3,95 (с, 3Н); 3,6-3,8 (м, 1H); 3,4-3,6 (м, 1H); 3,2-3,4 (H2O плюс размытый сигнал); 1,9-2,3 (м, 2Н); 1,3-1,5 (с, 9Н).
Стадия 7.n)
Стадию 7.n осуществляют согласно методике, аналогичной стадии 5.n. Сырой продукт без дополнительной очистки используют в следующей стадии.
Стадия 7.о)
Стадию 7.о осуществляют согласно методике, аналогичной стадии 5.o.
Масс-спектрометрия: 501,3 МН+.
Получение соединения формулы (VII):
Это соединение, близкое к описанным в Международной заявке на патент РСТ 97/30053 соединениям, может быть получено согласно нижеприводимой схеме синтеза:
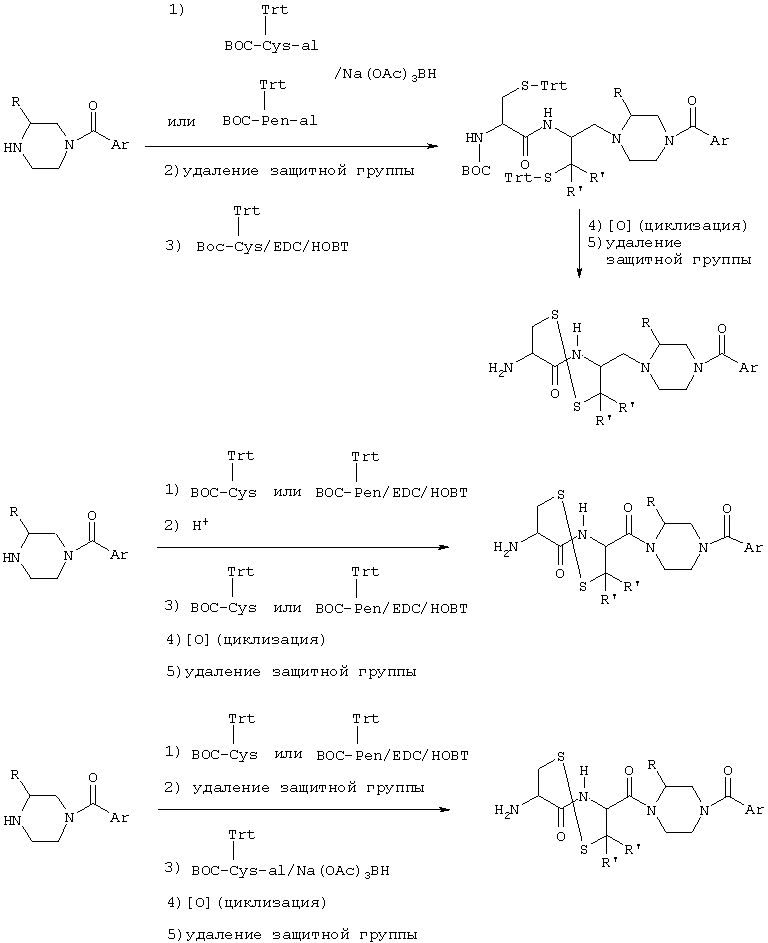
где
R'=H или СН3;
Cys=цистеин; Cys-al=цистеиналь;
Pen=пеницилламин; Pen-al=пеницилламинилаль;
Boc=трет-бутоксикарбонил;
EDC=N-(3-диметиламинопропил)-N-этилкарбодиимид;
HOBT=гидроксибензотриазол;
Trt=трифенилметил.
ФАРМАКОЛОГИЧЕСКАЯ ЧАСТЬ
С целью пояснения полезности изобретения изучают эффект обработки линии человеческих клеток MCF-7 с помощью следующих соединений:
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин]дисульфид, обозначаемый в этой части как соединение (I);
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(3-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид, обозначаемый в этой части как соединение (II);
- 7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин, обозначаемый в этой части как соединение (III);
- соединение формулы:
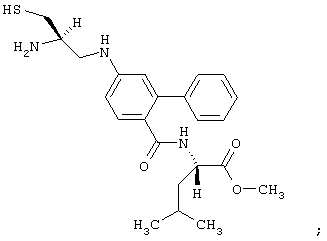
обозначаемое в этой части как соединение (IV);
- 7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидро-имидазо[1,2а]пиразин, обозначаемый в этой части как соединение (V);
- бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфид, обозначаемый в этой части как соединение (VI);
- соединение формулы:
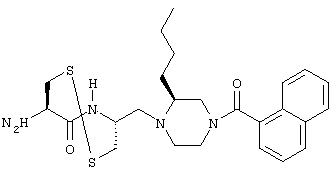
обозначаемое в этой части как соединение (VII).
Методики
Клеточная линия
Клеточные линии MCF-7 (плевральные человеческие клетки; рак молочной железы) были приобретены из Американской коллекции тканевых культур (Роквилл, Мериленд, США).
Определение внутриклеточного количества цАМФ в случае клеток MCF-7
Клетки MCF-7 (2·104 клеток на лунку), посев которых производили в 24-луночные планшеты, культивировали в течение 5 дней в модифицированной по способу Дульбекко среде Игла (Gibco-Brl, Cergy-Pontoise, Франция), дополненной с помощью 10% инактивированной путем нагревания фетальной телячьей сыворотки (Gibco-Brl, Cergy-Pontoise, Франция), 50000 единиц/л пенициллина и 50 мг/л стрептомицина (Gibco-Brl, Cergy-Pontoise, Франция), и 2 ммоль глутамина (Gibco-Brl, Cergy-Pontoise, Франция). Культуральную среду заменяли после двух промывок средой без сыворотки, дополненной или нет определенными агентами во время, указанное на различных фигурах. Затем при температуре 37°С добавляли агенты, активирующие продуцирование цАМФ. Реакцию прекращали по истечении 30 минут путем удаления среды и быстрого добавления 100 мкл 0,1н. раствора HCl. Эти экстракты замораживали при температуре -80°С до их использования. Концентрацию цДМФ определяли при использовании продажного набора для измерения (номер NEK033 NEN, Les Ulis, Франция), следуя инструкции изготовителя. Радиоактивность определяли с помощью гамма-счетчика (Gamma Master-1277, LKB, Turku, Финляндия).
Определение клеточной пролиферации in vitro
Клетки MCF-7 (3000 клеток на лунку) культивировали в 96-луночных планшетах в 80 мкл модифицированной по способу Дульбекко среды Игла (Gibco-Brl, Cergy-Pontoise, Франция), дополненной с помощью 10% инактивированной путем нагревания фетальной телячьей сыворотки (Gibco-Brl, Cergy-Pontoise, Франция), 50000 единиц/л пенициллина и 50 мг/л стрептомицина (Gibco-Brl, Cergy-Pontoise, Франция), и 2 ммоль глутамина (Gibco-Brl, Cergy-Pontoise, Франция), производили посев в 96-луночный планшет в день 0. Клетки обрабатывали в день 1 в течение 96 часов возрастающими концентрациями вплоть до 50 мкмоль каждого из тестируемых соединений. По окончании этого периода количественную оценку клеточной пролиферации осуществляли путем колориметрического теста, базируясь на расщеплении соли тетразолия WST1 митохондриальными дегидрогеназами в жизнеспособных клетках, приводящем к образованию формазана (Boehringer Mannheim, Милан, Франция). Эти тесты осуществляли при двукратном повторении с 8 определениями на тестируемую концентрацию. Для каждого тестируемого соединения учитывали включенные в линейную часть сигмоида значения для анализа путем линейной регрессии и использовали для определения ингибирующей концентрации (ИК50).
Определение активности МАР-киназы
Клетки MCF-7 (5105 клеток на лунку) культивировали в шестилуночных планшетах в модифицированной по способу Дульбекко среде Игла (Gibco-Brl, Cergy-Pontoise, Франция), дополненной с помощью 10% инактивированной путем нагревания фетальной телячьей сыворотки (Gibco-Brl, Cergy-Pontoise, Франция), смесью антибиотиков из 50000 единиц/л пенициллина и 50 мг/л стрептомицина (Gibco-Brl, Cergy-Pontoise, Франция) и 2 ммоль глутамина (Gibco-Brl, Cergy-Pontoise, Франция). После культивирования в течение 24 часов клетки инкубировали в течение 48 часов в среде, не содержащей сыворотки, для приведения клеток в состояние покоя. Клетки тогда обрабатывали в течение 1 часа либо соединением (I), либо PD98059 (Calbiochem, France Biochem, Meudon, Франция), специфическим ингибитором активации МАР-киназы. Клетки затем стимулировали (или нет) в течение 5 минут с помощью 12,5 нг/мл эпидермального фактора роста (EGF). Реакцию прекращали путем двух промывок с помощью забуференного фосфатом физиологического раствора (Gibco-Brl, Cergy-Pontoise, Франция), не содержащим ни кальция, ни магния, при температуре 4°С и путем добавления 150 мкл буфера для лизиса при температуре 4°С, состав которого следующий: 10 ммоль Трис, 150 ммоль NaCl, 2 ммоль этиленбис(оксиэтиленнитрило)тетрауксусной кислоты, 2 ммоль дитиотреитола, 1 ммоль фенил-метилсульфонилфторида, 2 ммоль ортованадата, 10 мкг/мл лейпептина и 10 мкг/мл апротинина. Количество протеинов, содержащихся в экстрактах, определяли методом Бредфорда (реактивы Biorad, Ivry-Sur-Seine, Франция). Эти экстракты замораживали при температуре -80°С до их использования. Активность МАР-киназы определяли с помощью продажного набора для измерения (номер RPN 84, Amersham, Life Science, Les Ulis, Франция), следуя инструкциям изготовителя. Радиоактивность определяли с помощью сцинтилляционного счетчика фирмы Паккард (Tricarb 5000 СА).
Материал
Вазоинтестинальный пептид (VIP) был приобретен у фирмы Bachem (Voisins le Bretonneux, Франция). Холерный токсин, форсколин, изопротеренол, простагландин E2 и PD 98059 были приобретены у фирмы Calbiochem (France Biochem, Meodon, Франция). Соединения формул (I), (II), (III), (IV), (V), (VI) и (VII) были предоставлены фирмой Biomeasure Inc. (Milford, МА, США). Все эти соединения были использованы в соответствии с рекомендациями их изготовителей.
Результаты
На фигуре 1 показано, что активация аденилатциклазы холерным токсином (200 нг/мл) или форсколином (10 мкмоль) приводит к очень значительному повышению количества цАМФ. Предварительная обработка клеток в течение 30 минут с помощью 30 мкмоль соединения (I) не изменяет продукцию цАМФ, индуцированную прямым активатором аденилатциклазы, форсколином. В то же время продукция цДМФ, стимулированная прямым активатором субъединицы, холерным токсином, сильно ингибируется соединением (I). Это показывает, что аденилатциклаза сама по себе не модифицирована соединением (I) и что оно препятствует образованию гетеротримерного комплекса.
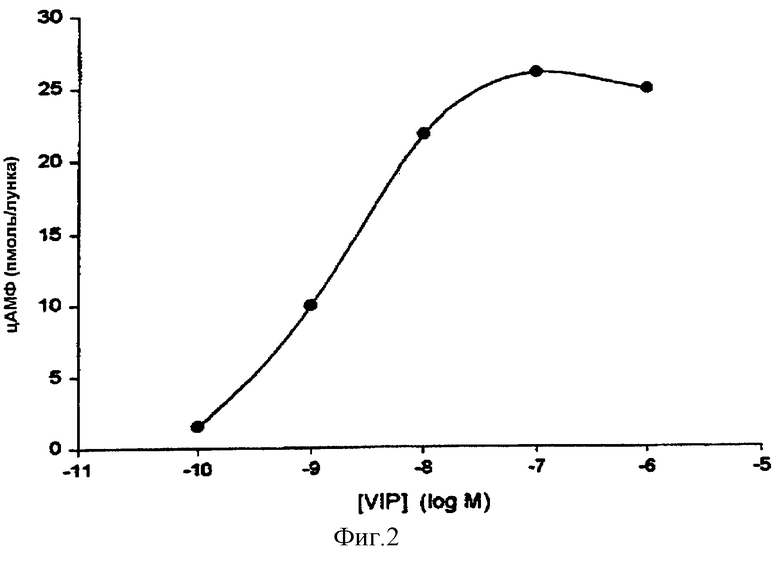
VIP представлен как внеклеточный лиганд рецептора, связанного с протеином G, который стимулирует синтез цАМФ в человеческих клетках рака молочной железы. На фигуре 2 показано, что обработка с помощью VIP человеческих клеток рака молочной железы MCF-7 повышает внутриклеточное количество цАМФ зависящим от концентрации образом. Концентрацию VIP, составляющую 10 нмоль, которая вызывает почти оптимальную продукцию цДМФ, используют для следующих тестов. Эта концентрация согласуется с уже опубликованными данными, касающимися линии человеческих клеток рака молочной железы T47D.
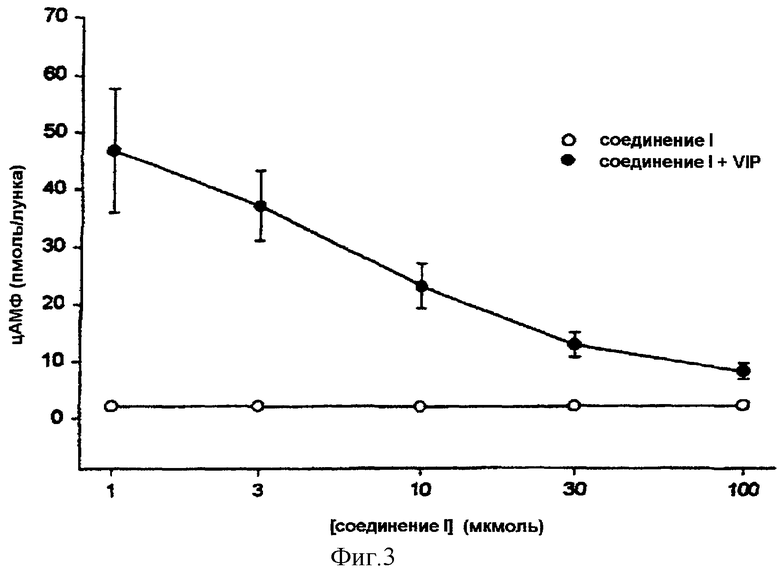
На фигуре 3 показано, что предварительная обработка в течение 30 минут клеток MCF-7, происходящих от культур in vitro, соединением формулы (I) является достаточной для ингибирования зависящим от концентрации образом аккумуляции цАМФ, стимулированной с помощью VIP. Почти полное ингибирование было достигнуто при концентрции 100 мкмоль соединения формулы (I). Эти результаты показывают, что обработка соединением (I) является достаточной для блокирования трансдукции сигнала, на пути прохождения которого используются гетеротримерные протеины G в качестве медиаторов.
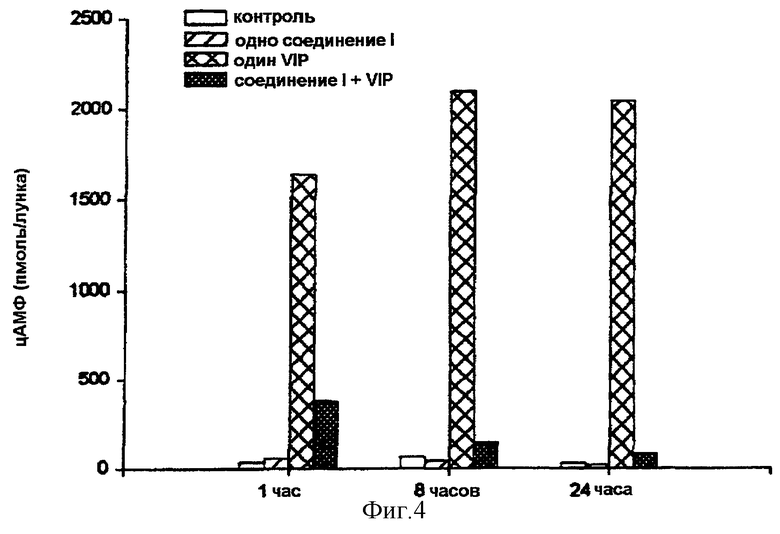
На фигуре 4 показано, что обработка в течение часа соединением формулы (I) является достаточной для модификации ответа на VIP. Обработки в течение более длительного времени (8 часов и 24 часа) продолжают ингибировать цАМФ, но основной эффект достигается очень быстро.
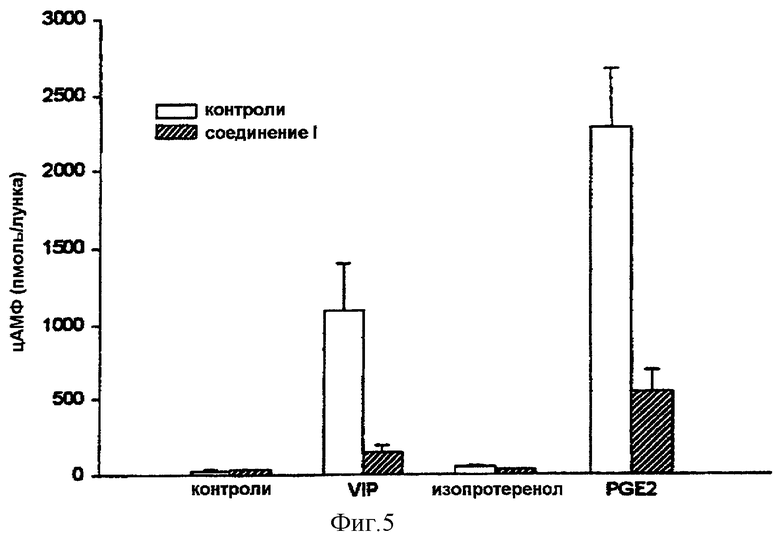
Соединение (I) также способно ингибировать образование цАМФ, индуцированное другими агентами, которые стимулируют рецепторы семи трансмембранных доменов. В клетках MCF-7, например, активность аденилатциклазы, очень сильно повышаемая простагландином Е2, ингибируется путем обработки в течение 30 минут соединением (I). Это позволяет предположить, что обработка клеток соединением (I) модифицирует гетеротримерную форму протеинов G за счет диссоциации субъединицы димера β/γ.
Ингибирование стимуляции с помощью VIP не было уменьшено соединениями структуры, аналогичной таковой соединения формулы (I). Как это показано в таблице I, соединения (II), (III), (IV), (V), (VI) и (VII), тестируемые при использовании одной и той же модели, также способны снижать индуцированное с помощью VIP количество цАМФ.
Совокупность этих результатов позволяет предположить, что тестируемые соединения модулируют активность аденилатциклазы за счет модификации гетеротримерной формы протеинов G. Однако известно, что димеры β/γ могут непосредственно активировать эффекторы, приводящие к активации киназ, регулируемых внеклеточными сигналами (ERKs), или МАР-киназ.
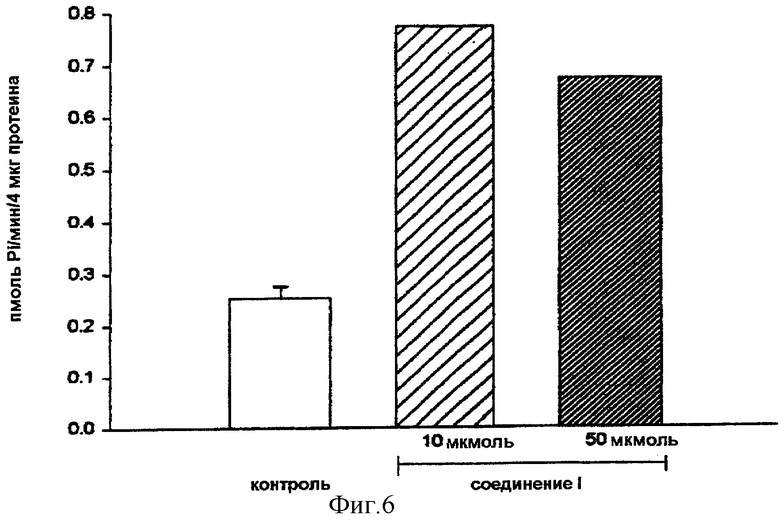
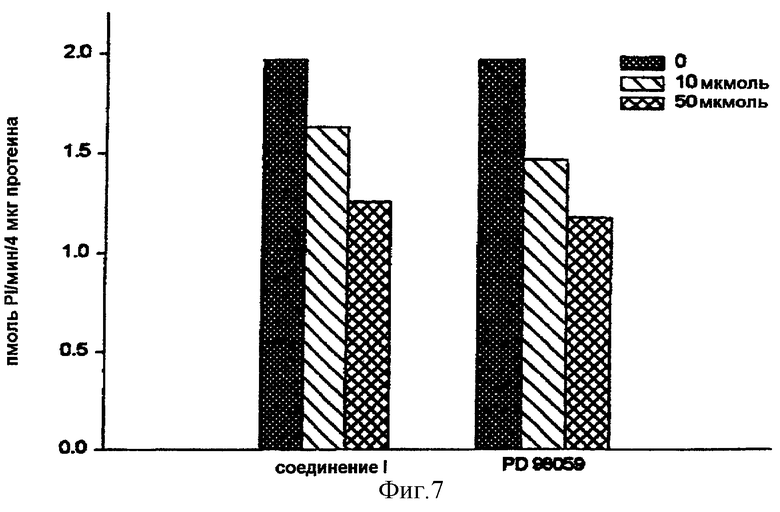
На фигуре 6 показано, что обработка клеток в течение 1 часа соединением (I) повышает вдвое базальную активность МАР-киназы. Это позволяет предположить, что путем препятствования образованию гетеротримерного комплекса соединение (I) высвобождает гетеродимер, который сам остается связанным с мембраной и активирует путь ras. Однако на фигуре 7 показано, что после стимуляции в течение 5 минут МАР-киназы эпителиальным фактором роста (EGF) активность фермента увеличивается примерно в 7 раз. Предварительная обработка в течение 1 часа клеток либо соединением (I), либо с помощью PD98059, специфического ингибитора активации МАР-киназы, уменьшает в 2 раза активность МАР-киназы. Эти результаты позволяют предположить, что соединение (I) стимулирует в базальном состоянии путь ras и ингибирует этот же самый путь, если он стимулирован, проявляя таким образом свое антипролиферативное действие.
Действительно, в таблице II показано, что соединения (I), (II), (III) и (IV) способны ингибировать пролиферацию in vitro человеческих опухолевых клеток MCF-7.
Воздействия соединений (I), (II), (III) и (IV), инкубируемых в течение 30 минут, на стимулированную с помощью VIP продукцию цДМФ в клетках MCF-7
Клетки инкубировали в течение 30 минут в присутствии или нет соединений (I), (II), (III) и (IV) в концентрации 30 мкмоль, которые затем стимулировали с помощью 10-8 моль VIP. Количество цАМФ определяли путем радиоиммунологического анализа. Данные представляют собой среднее значение ± стандартное отклонение (n=5 для контроля и n=1 для различных соединений).
Ингибирование роста in vitro клеток MCF-7 соединениями (I), (II), (III) и (IV)
Результаты ИК50 выражены в мкмоль и представляют собой среднее значение из двух экспериментов.
ФИГУРЫ
Фигура 1: Действие соединения (I) на стимулированную холерным токсином или форсколином продукцию цАМФ в клетках MCF-7.
Клетки инкубировали в течение 30 минут в присутствии или нет соединения (I) в концентрации 30 мкмоль и затем стимулировали либо холерным токсином (200 нг/мл) в течение 90 минут, либо форсколином (10-5 моль) в течение 30 минут. Количество цАМФ определяли путем радиоиммунологического анализа. Данные представляют собой среднее значение ± стандартное отклонение (n=3).
Фигура 2: Действие возрастающих концентраций VIP на продукцию цДМФ в клетках MCF-7.
Клетки стимулировали в течение 30 минут с помощью VIP в указанных концентрациях. Количество цАМФ определяли путем радиоиммунологического анализа.
Фигура 3: Действие различных концентраций соединения (I) на стимулированную с помощью VIP продукцию цАМФ в клетках MCF-7.
Клетки инкубировали в течение 24 часов в присутствии возрастающих концентраций соединения (I) и стимулировали затем в течение 30 минут с помощью 10-8 моль VIP. Количество цАМФ определяли путем радиоиммунологического анализа. Данные представляют собой среднее значение ± стандартное отклонение (n=3).
Фигура 4: Влияние времени инкубации клеток MCF-7 в присутствии соединения (I) на стимулированную с помощью VIP продукцию цАМФ.
Клетки инкубировали в течение 1, 8 или 24 часов в присутствии или нет соединения (I) в концентрации 30 мкмоль и стимулировали затем с помощью 10-8 моль VIP. Количество цАМФ определяли путем радиоиммунологического анализа. Данные представляют собой среднее значение ± стандартное отклонение (n=1).
Фигура 5: Действие соединения (I) на стимулированную с помощью VIP, изопротеренола или простагландина E2 продукцию цАМФ в клетках MCF-7.
Клетки инкубировали в течение 30 минут в присутствии или нет соединения (I) в концентрации 30 мкмоль и стимулировали затем в течение 30 минут либо с помощью 10-8 моль VIP, либо с помощью 10-6 моль изопротеренола, либо с помощью 10-6 моль простагландина Е2. Количество цАМФ определяли путем радиоиммунологического анализа. Данные представляют собой среднее значение ± стандартное отклонение (n=3).
Фигура 6: Действие соединения (I) на базальную активность МАР-киназы в клетках MCF-7.
Клетки, лишенные сыворотки в течение 48 часов, обрабатывали в течение 1 часа соединением (I) (10 и 50 мкмоль). Активность МАР-киназы определяли в клеточных лизатах и выражали в пмоль включенного Pi в минуту и на 4 мкг протеина (n=3 для контрольных образцов и n=2 для обработанных соединением (I) клеток).
Фигура 7: Действие соединения (I), сравниваемого с PD98059, на активность МАР-киназы в клетках MCF-7.
Клетки, лишенные сыворотки в течение 48 часов, обрабатывали в течение 1 часа либо соединением (I) (10 и 50 мкмоль), либо с помощью PD98059 (10 и 50 мкмоль). Клетки затем стимулировали путем добавления 12,5 нг/мл эпидермального фактора роста в течение 5 минут. Активность МАР-киназы определяли в клеточных лизатах и выражали в пмоль включенного Pi в минуту и на 4 мкг протеина (n=4).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОДУКТ, ВКЛЮЧАЮЩИЙ ИНГИБИТОР ТРАНСДУКЦИИ СИГНАЛОВ ГЕТЕРОТРИМЕРНЫХ ПРОТЕИНОВ G В КОМБИНАЦИИ С ДРУГИМ ЦИТОСТАТИЧЕСКИМ СРЕДСТВОМ, ДЛЯ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2000 |
|
RU2298417C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА КАК ИНГИБИТОРЫ ПРЕНИЛТРАНСФЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2241712C9 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРАЗИНА ИЛИ ИМИДАЗОДИАЗЕПИНА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ2 | 2008 |
|
RU2540074C2 |
| СОЛИ ПРОИЗВОДНЫХ ТЕТЕРАГИДРО-ИМИДАЗО[1,5-A]ПИРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2010 |
|
RU2523543C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДОВ ИМИДАЗО(4,5-B)ХИНОЛИНИЛОКСИАЛКАНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИМЕНЯЕМЫХ СОЛЕЙ | 1990 |
|
RU2041210C1 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693629C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
Изобретение относится к области медицины и касается применения производных цистеина для получения лекарственного средства, предназначенного для лечения заболеваний, возникающих вследствие образования гетеротримерного протеина G, к новым производным цистеина и фармкомпозиции на их основе. Производные цистеина, в частности, включают: бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]-дисульфид, бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил]дисульфид. Изобретение обеспечивает высокую эффективность лечения. 3 н. и 3 з.п. ф-лы, 7 ил., 2 табл.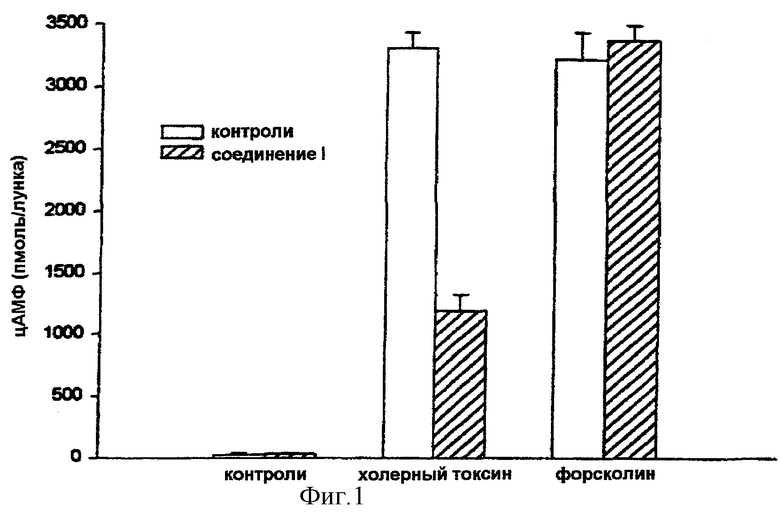
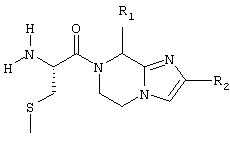
или
в которых R1 означает циклогексилметил, 2-метилпропил, бутил, 1-метилпропил;
R2 означает 2-метоксифенил, 1-нафтил, 2-метилфенил;
R3 означает атом водорода или группу
или их фармацевтически приемлемых солей для получения лекарственного средства, предназначенного для лечения патологий, возникающих вследствие образования гетеротримерного протеина, выбранных среди патологий, связанных со следующими биологическими функциями или нарушениями: обоняние, вкусовые ощущения, световое восприятие, нейротрансмиссия, нейродегенерация, функционирование эндокринных и экзокринных желез, аутокринная и паракринная регуляция, артериальное кровяное давление, эмбриогенез, вирусная инфекция, иммунологические функции, диабет и ожирение.
7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин]дисульфид;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин]дисульфид;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(1-нафтил)-8-(2-метилпропил)-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин-7-ил]дисульфид.
7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин;
7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилметокси)метил-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин;
7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(1-фенилметокси)этил-5,6,7,8-тетрагидроимидазо[1, 2а]-пиразин;
7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(фенилсульфонилэтил)-5,6,7,8-тетрагидроимидазо[1, 2а]пиразин,
или фармацевтически приемлемую соль одного из этих соединений.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Устройство для получения арболита | 2020 |
|
RU2736638C1 |
| US 5705686 А, 06.01.1998 | |||
| ПРОИЗВОДНЫЕ N-АЦИЛ- α -АМИНОКИСЛОТЫ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОСТЫЕ ИЛИ СЛОЖНЫЕ ЭФИРЫ, АМИДЫ ИЛИ ГИДРАТЫ И КОМПОЗИЦИЯ ИНГИБИРУЮЩАЯ СВЯЗЫВАНИЕ АДГЕЗИВНЫХ ПРОТЕИНОВ С ТРОМБОЦИТАМИ И АГРЕГАЦИЮ ТРОМБОЦИТОВ | 1992 |
|
RU2097378C1 |

