Уровень техники
Семейство белков Ras играет важную роль в пути передачи сигнала, модулирующего рост клеток. Белок продуцируется в рибосоме, выделяется в цитозоль и пост-трансляционно модифицируется. Первая стадия в ряду пост-трансляционных модификаций представляет собой алкилирование Cys168 фарнезил или геранил-геранил пирофосфатом в реакции, катализируемой ферментами пренилтрансферазы, такими как фарнезилтрансфераза и геранил-геранилтрансфераза (Hancock, JF, et al., Cell 57:1167-1177 (1989)). Далее отщепляются три С-концевые аминокислоты (Gu-tierrez, L., et al., EMBO J. 8:1093-1098 (1989)), и концевая Cys превращается в сложный метиловый эфир (Clark, S., et al., Proc. Nat'l Acad. Sci. (USA) 85:4643-4647 (1988)). Некоторые формы Ras также обратимо пальмитоилированы на цистеиновых остатках непосредственно от N-концевых до Cys168 (Buss, JE, et al., Mol, Cell Biol. 6:116-122 (1986)). Полагают, что такие модификации повышают гидрофобность С-концевые части Ras, что является причиной их локализации на поверхности мембраны клетки. Локализация Ras на мембране клетки необходима для передачи сигнала (Willumsen, ВМ, et al., Science 310:583-586 (1984).
Онкогенные формы Ras обнаружены в относительно большом количестве случаев рака, включая свыше 50% случае рака прямой кишки и свыше 90% случаев рака поджелудочной железы (Bos, JL, Cancer Research 49:4682-4689 (1989)). Подобные наблюдения позволяют предположить, что вмешательство в функцию Ras медиированной передачи сигнала может быть полезным при лечении рака.
Ранее было показано, что С-концевые тетрапептиды Ras имеют мотив "СААХ" (где С является цистеином, А является алифатической аминокислотой и Х является любой аминокислотой). Было показано, что тетрапептиды, имеющие подобную структуру, являются ингибиторами пренилтрансфераз (Reiss, et al., Cell 62:81-88 (1990)). Слабое действие этих ранее известных ингибиторов фарнезилтрансферазы явилось причиной поиска новых ингибиторов с более благоприятным фармакокинетическим поведением (James, GL, et al., Science 260:1937-1942 (1993); Kohl, NE, et al., Nat'l Acad. Sci. USA 91:9141-9145 (1994), de Solms, SJ, et al. Proc. J. Med. Chem. 38:3967-3971 (1995); Nagasu, Т, et al., Cancer Research 55:5310-5314 (1995); Lerner, EC, et al., J. Biol. Chem. 270:26802-26806 (1995); Lerner, EC, et al., J. Biol. Chem. 270:26770 (1995); и James, et al., Proc. Natl. Acad. Sci. USA 93:4454 (1996)).
Недавно было обнаружено, что ингибитор пренилтрансферазы может блокировать рост Ras-зависимых опухолей у "голых" мышей (Kohl, NE, et al., Proc. Nat'l Acad. Sci. USA 91:9141-9145 (1994)). Кроме того, было показано, что свыше 70% больших образцов линий клеток опухоли ингибируются ингибиторами пренилтрансферазы с селективностью в отношении не трансформированных эпителиальных клеток (Sepp-Lorenzino, I, et al., Cancer Research, 55:5302-5309 (1995)). Ингибирование фарнезилирования описано в качестве способа лечения дельта вирусной инфекции гепатита (Casey, P, et al., WO 97/31641).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
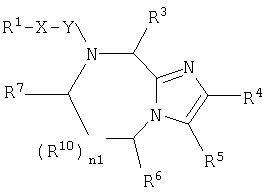
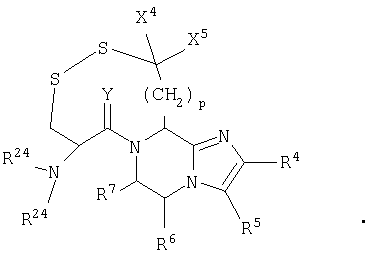
В одном аспекте данное изобретение относится к соединению формулы (I):
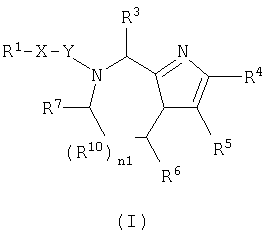
где n1 равно 0 или 1;
Х независимо является (CHR11)n3(CH2)n4Z (CH2)n5;
Z является О, N(R12), S или связью;
n3 независимо равно 0 или 1;
n4 и n5 каждый независимо равен 0, 1, 2 или 3;
Y независимо является СО, CH2, CS или связью;
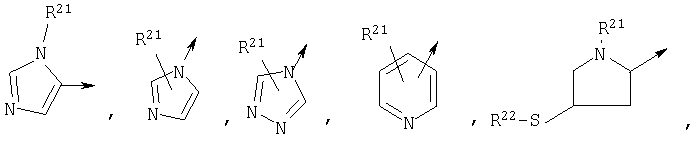
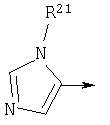
R1 является
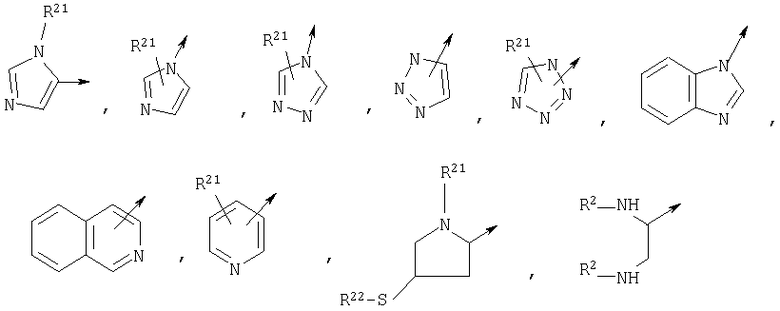
или N(R24R25);
R2, R11 и R12 каждый независимо является Н или необязательно замещенной алкильной группой, выбранной из группы, включающей (С1-6)алкил или арил, где указанная необязательно замещенная группа необязательно замещена одним или более R3 или R30;
R3 независима является Н или необязательно замещенной группой, выбранной из группы, включающей (C1-6)алкил, (С2-6)алкенил, (С2-6)-алкинил, (С3-6)циклоалкил, (С3-6)циклоалкил(C1-6)алкил, (С5-7)циалоалкенил, (C5-7)циклоалкенил(С1-6)алкил, арил, арил(C1-6)алкил, гетероциклил и гетероциклил(С1-6) алкил, где указанная необязательно замещенная группа необязательно замещена одним или более R30;
R4 и R5 каждый независимо является Н или необязательно замещенной группой, выбранной из группы, включающей (C1-6)-алкил, (С3-6)циклоалкил, арил и гетероциклил, где указанная необязательно замещенная группа необязательно замещена одним или более R30, где каждый указанный заместитель выбран независимо, или R4 и R5, взятые вместе с атомом углерода, к которому они присоединены, образуют арил;
R6 независимо является Н или необязательно замещенной группой, выбранной из группы, включающей (C1-6)алкил, (С2-6)-алкенил, (С3-6)циклоалкил, (С3-6)циклоалкил (C1-6)алкил, (C5-7)-циклоалкенил, (C5-7) циклоалкенил(C1-6)алкил, арил, арил(С1-6)-алкил, гетероциклил и гетероциклил(C1-6)алкил, где указанная необязательно замещенная группа необязательно замещена одним или более заместителями, каждый из которых независимо выбран из группы, включающей ОН, (С1-6)алкил, (C1-6)алкокси, N(R8R9), -СООН, CON(R8R9) и галоген, где R8 и R9 каждый независимо является Н, (C1-6)алкилом, (С2-6)алкенилом, (С2-6)алкинилом, арилом или арил (C1-6) алкилом;
R7 независимо является Н, =O, =S или необязательно замещенной группой, выбранной из группы, включающей (С1-6)алкил, (С2-6)алкенил, (С3-6)циклоалкил, (С3-6)циклоалкил(С1-6)алкил, (С5-7) циклоалкенил, (C5-7)циклоалкенил(C1-6)алкил, арил, арил (C1-6)алкил, гетероциклил и гетероциклил (C1-6) алкил, где указанная необязательно замещенная группа необязательно замещена одним или более заместителей, каждый из которых независимо выбран из группы, включающей ОН, (C1-6)алкил, (C1-6)алкокси, -N(R8R9), -СООН, -CON(R8R9) и галоген;
R10 является С;
или если n1=0, R6 и R7, взятые вместе с атомами углерода, к которым они присоединены, могут образовывать арил или циклогексил;
R21 независимо является Н или необязательно замещенной группой, выбранной из группы, включающей (C1-6)алкил и арил (C1-6)алкил, где указанная необязательно замещенная группа необязательно замещена одним или более заместителями, независимо выбранными из группы, включающей R8 и R30;
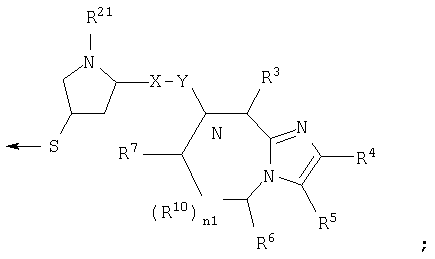
R22 является Н, (C1-6) алкилтио, (С3-6) циклоалкилтио, R8-CO- или заместителем формулы:
R24 и R25 каждый независимо является Н, (C1-6)алкилом или арил (C1-6)алкилом;
R30 независимо является (C1-6)алкилом, -O-R8, -S(O)n6R8, -S(O)n7N(R8R9), -N(R8R9), -CN, -NO2, -CO2R8, -CON(R8R9), -NCO-R8 или галогеном;
n6 и n7 каждый независимо равен 0, 1 или 2;
где указанным гетероциклилом является азепинил, бензимидазолил, бензизоксазолил, бензофуразанил, бензопиранил, бензотиопиранил, бензофурил, бензотиазолил, бензотиенил, бензоксазолил, хроманил, циннолинил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, дигидробензотиопиранилсульфон, фурил, имидазолидинил, имидазолинил, имидазолил, индолинил, индолил, изохроманил, изоиндолинил, изохинолинил, изотиазолидинил, изотиазолил, изотиазолидинил, морфолинил, нафтиридинил, оксадиазолил, 2-оксоазепинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, пиперидил, пиперазинил, пиридил, пиридил N-оксид, хиноксалинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тиаморфолинил, тиаморфолинил сульфоксид, тиазолил, тиазолинил, тиенофурил, тиенотиенил или тиенил; и
где указанным арилом является фенил или нафтил;
при условии, что;
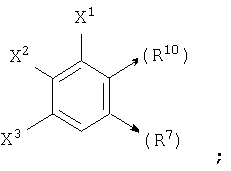
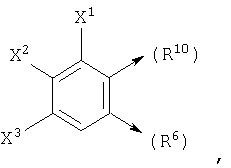
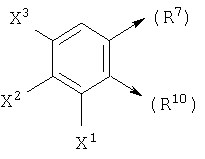
если n1=1, R10 является С и R6 является Н, то R10 и R7, взятые вместе, могут образовывать
или если n1=1, R10 является С и R7 является =O, -Н или =S, то R10 и R6, взятые вместе, могут образовывать
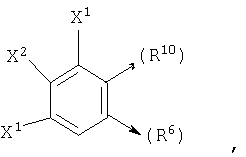
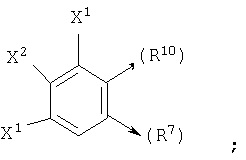
где X1, X2 и X3 каждый независимо является Н, галогеном, -NO2, -NCO-R8, -CO2R8, -CN или -CON(R8R9); и
если R1 является N(R24R25), то n3 равен 1, n4 и n5 каждый равен О, Z является связью и R3 и R11, взятые вместе, образуют
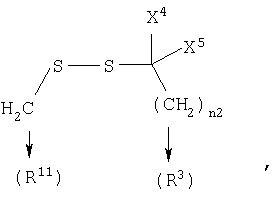
где n2 равен 1-6, и X4 и X5 каждый независимо является Н, (C1-6)алкилом или арилом, или X4 и X5, взятые вместе, могут образовывать (С3-6)циклоалкил;
или его фармацевтически приемлемую соль.
Предпочтительной группой соединений формулы (I), обозначенной как группа А, является группа, в которой
R1 является
или N(R24R25); и
X является CH(R11) n3(СН2) n4 или Z, где Z является O, S или N(R12);
или их фармацевтически приемлемая соль.
Другой предпочтительной группой соединений формулы (I), обозначенной как группа В, является группа, в которой
R1 является
Х является СН (R11)n3(СН2)n4; и
n1 равен 0;
или их фармацевтически приемлемая соль.
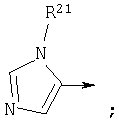
Другой предпочтительной группой соединений формулы обозначенной как группа С, является группа, в которой
R1 является
n3, n4 и n5 каждый равен 0;
Z является связью;
Y является, независимо для каждого случая, СО или CS; и
n1 равен 0;
или их фармацевтически приемлемая соль.
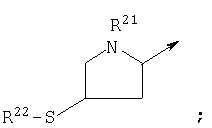
Другой предпочтительной группой соединений формулы (I), обозначенной как группа D, является группа, в которой R1 является
R6 является Н;
n1 равен 1;
R7 и R10, взятые вместе, образуют
n3 равен 1 и R11 является Н;
Z является О или связью;
n5 равен 0; и
Y является СО, СН2 или связью;
или их фармацевтически приемлемая соль.
Другой предпочтительной группой соединений формулы (I), обозначенной как группа Е, является группа, в которой
R1 является N(R24R25);
n1 равен 0;
n3 равен 1;
n4 равен 0;
n5 равен 0;
Y является СО или CS;
z является связью; и
R3 и R11, взятые вместе, образуют
или их фармацевтически приемлемая соль.
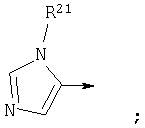
Другой предпочтительной группой соединений формулы (I), обозначенной как группа F, является группа, в которой
R1 является
R7 является Н или=0;
n1 равен 1;
R6 и R10, взятые вместе, образуют
n3 равен 1 и R11 является Н;
n5 равен 0;
Y является СО или СН2; и
Z является О или связью;
или их фармацевтически приемлемая соль.
В другом аспекте, данное изобретение относится к фармацевтической композиции, содержащей одно или более соединений формулы (I), таких, как определены выше, или их фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
В еще одном аспекте, данное изобретение относится к способу ингибирования пренилтрансфераз (например, фарнезилтрансферазы или геранилгеранилтрансферазы) у пациента, например млекопитающего, такого как человек, который включает введение пациенту терапевтически эффективного количества соединения формулы (I), такого как определено выше, или его фармацевтически приемлемой соли. В еще одном аспекте, данное изобретение относится к способу лечения рестеноза или пролиферативных заболеваний тканей (например, опухоли) у пациента введением пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Примеры пролиферативных заболеваний тканей включают заболевания, связанные как с доброкачественной (т.е. не злокачественной) пролиферацией клеток, такие как фиброз, доброкачественная гиперплазия простаты, атеросклероз и рестеноз, так и со злокачественной пролиферацией клеток, такие как рак (например, ras-мутированные опухоли). Примеры лечимых опухолей включают, но не ограничиваются ими, рак груди, толстой кишки, поджелудочной железы, простаты, легких, яичников, эпидермиса и органов кроветворения (Sepp-Lorenzino, I, et al., Cancer research 55:5302 (1995)).
В еще одном аспекте данное изобретение относится к использованию одного или более соединений формулы (I), таких как описано выше, или их фармацевтически приемлемой соли, для связывания с пренилтрансферазой, как представлено в in vitro и in vivo тестах.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В общем, соединения формулы (I) могут быть получены способами, известными в области химии для получения соединений. Определенные способы получения соединений формулы (I) представлены как аспекты данного изобретения и иллюстрированы схемами реакций и примерами, представленными в данном описании.
В представленных выше структурных формулах и в контексте описания данной заявки используемые термины имеют следующие значения, если не указано иначе.
Термин "алкил" включает алкильные группы, имеющие указанное количество атомов углерода, в прямой или разветвленной конфигурации. Примеры таких алкильных групп включают метил, этил, пропил, изопропил, бутил, вторбутил, третичный бутиил, пентил, изопентил, гексил, изогексил и подобные. Если в определение включен термин «С0-алкил», он обозначает простую ковалентную связь.
Термин "циклоалкил" включает моноциклоалкильную группу или бициклоалкильную группу, имеющую указанное количество атомов углерода. Примеры таких циклоалкильных групп включают циклопропил, циклобутил, циклогексил и подобные.
Термин "алкенил" включает углеводородные группы, имеющие одну или более двойных связей и указанное количество атомов углерода, в прямой или разветвленной конфигурации. Примеры таких алкенильных групп включают этенил, пропенил, изопропенил, бутенил, вторбутенил, третичный бутенил, пентенил, изопентенил, гексенил, изогексенил и подобные.
Термин "циклоалкенил" включает моноциклоалкенильную группу или бициклоалкенильную группу с указанным количеством атомов углерода, имеющую одну или более двойную связь, но недостаточно двойных связей для того, чтобы группа стала ароматической. Примеры таких циклоалкенильных групп включают циклопентенил, циклогексенил, циклогептенил и подобные.
Термин "алкинил" включает алкинильные группы, т.е. углеводородные группы, имеющие одну или более тройную связь, имеющие указанное количество атомов углерода, в прямой или разветвленной конфигурации. Примеры таких алкинильных групп включают этинил, пропинил, бутинил, пентинил, изопентинил, гексинил, изогекинил и подобные.
Термин "алкилтио" включает алкилтиогруппы, т.е. углеводородные группы, которые присоединены к молекуле через атом серы, имеющие указанное количество атомов углерода, в прямой или разветвленной конфигурации. Примеры таких алкилтиогрупп включают метилтио, этилтио, пропилтио, изопропилтио, бутилтио, вторбутилтио, третичный бутилтио, пентилтио, изопентилтио, гексилтио, изогексилтио и подобные.
Термин "циклоалкилтио" включает моноциклоалкилтиогруппу и бициклоалкилтиогруппу с указанным количеством атомов углерода. Примеры таких циклоалкилтиогрупп включают циклопентилтио, циклогексилтио и подобные.
Термин "алкокси" включает алкоксигруппы, имеющие указанное количество атомов углерода, в прямой или разветвленной конфигурации. Примеры таких алкоксигрупп включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, третичный бутокси, пентокси, изопентокси, гексокси, изогексокси и подобные.
Термин "арил" включает ароматические кольца, известные в данной области техники, который могут быть моноциклическими или бициклическими, такие как фенил и нафтил.
Термин "гетероциклил" в контексте данного изобретения представляет 5-7-членное моноциклическое, или 8-11-членное бициклическое, или 11-15-членное трициклическое кольцо, которое либо насыщено, либо не насыщено, и которое состоит из атомов углерода и включает от одного до четырех гетероатомов, независимо выбранных из группы, включающей N, O и S. Также термин охватывает любые бициклические группы, в которых определенные выше гетероциклические кольца сконденсированы с бензольным кольцом, где гетероциклическое кольцо может быть присоединено к любому гетероатому или атому углерода. Примеры таких гетероциклических групп включают, но не ограничены ими, азепинил, бензимидазолил, бензизоксазолил, бензофуразанил, бензопиранил, бензотиопиранил, бензофурил, бензотиазолил, бензотиенил, бензоксазолил, хроманил, циннолинил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, дигидробензотиопиранилсульфон, фурил, имидазолидинил, имидазолинил, имидазолил, индолинил, индолил, изохроманил, изоиндолинил, изохинолинил, изотиазолидинил, изотиазолил, изотиазолидинил, морфолинил, нафтиридинил, оксадиазолил, 2-оксоазепинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, пиперидил, пиперазинил, пиридил, пиридил N-оксид, хиноксалинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тиаморфолинил, тиаморфолинил сульфоксид, тиазолил, тиазолинил, тиенофурил, тиенотиенил, тиенил и подобные.
Специалисту в области химии понятно, что определенные сочетания заместителей, содержащих гетероатом, перечисленные в данном изобретении, определяют соединения, которые будут менее стабильными в физиологических условиях. Следовательно, такие соединения менее предпочтительны.
Термин "галоген" или "гало" включает атомы галогена фтор, хлор, бром и йод.
Если в представленной химической структуре имеется стрелка, исходящая из нее, стрелка обозначает место присоединения. Например структура

является пентильной группой. Если рядом со стрелкой строит значение в скобках, данное значение указывает, где в соединении находится место присоединения. Например, в общей формуле (I):
такой как определено выше, если R10 и R7, взятые вместе, образуют
получается следующая структура:
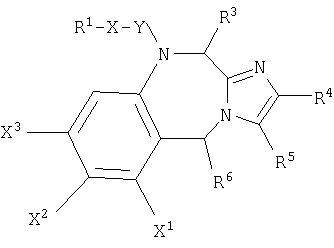
Также, в общей формуле (I), такой как определено выше, если R3 и R11, взятые вместе, образуют
получается следующая структура:
Если линия изображена проходящей через циклическую группу, то данная линия указывает, что заместитель может быть присоединен к циклической группе в любом доступном месте присоединения. Например,
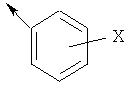
означает, что заместитель «X» может быть присоединен в орто, мета или пара положении. Далее, если линия изображена проходящей через бициклическую или трициклическую группу, такая линия означает, что заместитель может быть присоединен к бициклической или трициклической группе в любом доступном месте присоединения в любом кольце. Также, если стрелка изображена проходящей через циклическую группу, такая стрелка показывает, что место присоединения циклической группы к соединению может быть любым доступным местом присоединения в циклической группе. Далее, если стрелка изображена проходящей через бициклическую или трициклическую группу, такая стрелка показывает, что место присоединения бициклической или трициклической группы к соединению может быть любым доступным местом присоединения колец бициклической или трициклической группы.
Некоторые соединения в соответствии с данным изобретением могут иметь, по крайней мере, один центр асимметрии. Дополнительные центры асимметрии могут присутствовать в молекуле в зависимости от природы различных заместителей в молекуле. Каждый такой центр асимметрии дает два оптических изомера и подразумевается, что все такие оптические изомеры, как разделенные, очищенные или частично очищенные оптические изомеры, их рацемические смеси или диастереомерные смеси, включены в объем данного изобретения.
Соединения в соответствии с данным изобретением обычно могут быть выделены в виде их фармацевтически приемлемых кислотно-аддитивных солей, таких как соли с неорганическими и органическими кислотами. Примеры таких кислот включают соляную, азотную, серную, фосфорную, муравьиную, уксусную, трифторуксусную, пропионовую, малеиновую, янтарную, D-винную, L-винную, малоновую, метансульфоноую и подобные. Кроме того, определенные соединения, содержащие кислотные функциональные группы, такие как карбокси, могут быть выделены в виде их неорганической соли, в которой противоион может быть выбран из натрия, калия, лития, кальция, магния и подобных, а также из органических оснований.
Фармацевтически приемлемые соли могут быть получены взаимодействием приблизительно 1 эквивалента соединения формулы (I) и приблизительно 1 эквивалентом или более подходящей соответствующей кислоты, соль которой желательна. Методы обработки и выделение полученной соли хорошо известны специалисту в данной области.
Соединения в соответствии с данным изобретением могут вводиться перорально, парентерально (например, внутримышечными, внутрибрюшинными, внутривенными или подкожными инъекциями или имплантантом), интраназально, вагинально, ректально, подъязычно или местно и могут быть составлены в препаративную форму с фармацевтически приемлемыми носителями для получения дозированных форм, подходящих для каждого способа введения. Следовательно, в объем данного изобретения включены фармацевтические композиции, содержащие, в качестве активного ингредиента, по крайней мере, одно соединение формулы (I) в сочетании с фармацевтически приемлемым носителем.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В подобных твердых дозированных формах активное соединение смешивают с, по крайней мере, одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы также могут содержать, что является обычной практикой, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие агенты, такие как стеарат магния. Для капсул, таблеток и пилюль, дозированные формы также могут содержать буферные агенты. Таблетки и пилюли могут дополнительно быть приготовлены с энтеросолюбильными оболочками.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры, содержащие инертные разбавители, обычно используемые в данной области техники, такие как вода. Кроме указанных инертных разбавителей, композиции также могут содержать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, и подсластители, вкусовые добавки и ароматизаторы.
Препаративные формы для парентерального введения в соответствии с данным изобретением включают стерильные водные и неводные растворы, суспензии или эмульсии. Примеры неводных растворителей или носителей включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин и органические сложные эфиры, подходящие для инъекций, такие как этилолеат. Указанные дозированные формы также могут содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгаторы и диспергирующие агенты. Они могут быть стерилизованы, например, фильтрацией через улавливающий бактерии фильтр, добавлением стерилизующих агентов в композицию, облучением композиции или нагреванием композиции. Они также могут быть получены в виде стерильных твердых композиций, которые могут быть растворены в стерильной воде или некоторых других стерильных средах для инъекций непосредственно перед использованием.
Композиции для ректального или вагинального введения предпочтительно являются суппозиториями, которые могут содержать, в дополнение к активному ингредиенту, наполнители, такие как кокосовое масло или парафин для суппозиториев.
Композиции для интраназального или подъязычного введения также получают со стандартными наполнителями, которые хорошо известны в данной области техники.
В общем, эффективная доза активного ингредиента в композициях в соответствии с данным изобретением может варьироваться; однако необходимо, чтобы количество активного ингредиента было таковым, чтобы получить подходящую дозированную форму. Выбранная доза зависит от желаемого терапевтического действия, способа введения и продолжительности лечения, что входит в сферу знаний специалиста в данной области. В общем доза составляет от 0,0001 до 100 мг/кг веса тела ежедневно для введения человеку и другим животным, например млекопитающим.
Предпочтительная доза составляет от 0,01 до 10,0 мг/кг веса тела ежедневно и может вводиться единичной дозой или может быть разделена на множество доз.
Соединения в соответствии с данным изобретением могут быть и были оценены в отношении действия на зависимый от якорной подложки рост линий клеток опухоли у человека согласно следующему тесту.
Клетки высевают в 96-ячеечный планшет на 0 день и обрабатывают на 1 день в течение 96 часов следующими концентрациями соединений в соответствии с данным изобретением; 50,25, 12,5, 6,25, 3,12, 1,56, 0,78, 0,39 и 0,00 мкМ. В конце данного периода количественный анализ пролиферации клеток проводят с помощью колориметрического теста, основанного на отщеплении тетразолиевой соли WST1 митохондриальными дегидрогеназами в жизнеспособных клетках, приводящем к образованию формазана. Данные эксперименты повторяют дважды в октупликатах, таким образом, определяя интервал концентраций соединения, включающий значение IC50.
По описанной выше методике проводят анализ со следующими линиями клеток опухоли человека: простата: DU145 аденокарцинома с WT ras, резистентными к L744-832; прямая кишка: НТ29, аденокарцинома с немутантным типом ras (WT), чувствительным к L744-832; поджелудочная железа: MIA РаСа-2, карцинома с Ki-ras мутацией; легкие: А427, карцинома с Ki-ras мутацией. (см. табл.1)
Должно быть понятно, что хотя данное изобретение описано совместно с подробным описанием, представленное ниже описание предназначено для иллюстрации и не ограничивает объем данного изобретения, который определен формулой изобретения.
Примеры
Соединение 1: 8-бутил-7-(3-(имидазол-5-ил)-1-оксопропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Соединение 2: 8-бутил-2-(2-гидроксифенил)-7-(имидазол-4-ил-пропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Соединение 3: 8-бутил-7-(4-имидазолилпропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
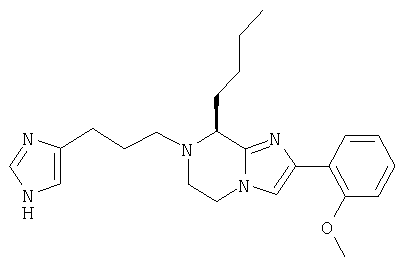
Соединение 4: 7-(2-(имидазол-4-ил)-1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
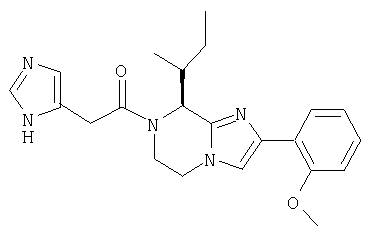
Соединение 5: 2-(2-метоксифенил)-8-(1-метилпропил)-7-(1-оксо-2-(1-фенилметил)имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо [1,2а]пиразин
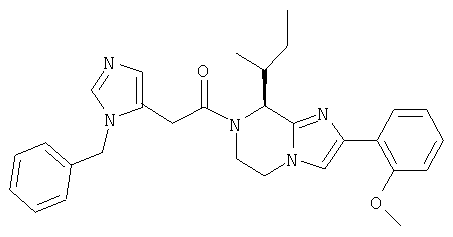
Соединение 6: 2-(2-метоксифенил)-8-(1-метилпропил)-7-(2-(1-фенилметил)имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
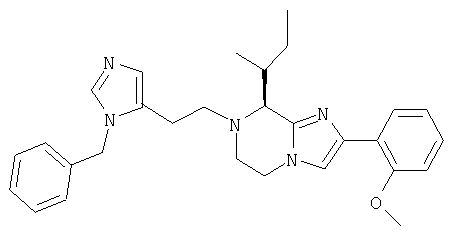
Соединение 7: 7-(2-(1-(4-цианофенилметил)имидазол-5-ил)1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
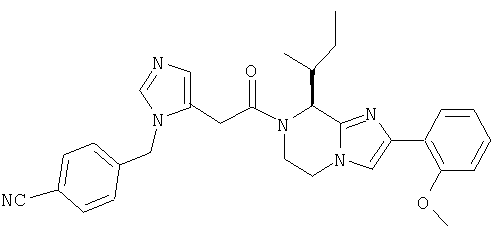
Соединение 8: 7-((1Н-имидазол-4-ил)метил)-2-(2-метоксифенил) -8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
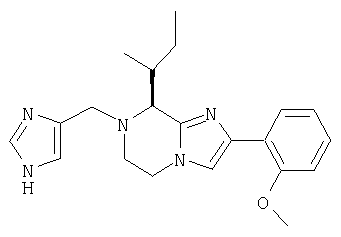
Соединение 9: 7-((4-имидазолил)карбонил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
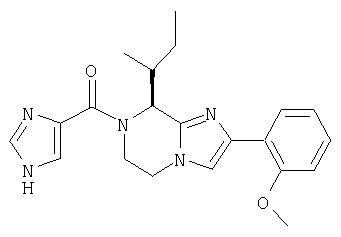
Соединение 10: 7-(1-(4-цианофенилметил)имидазол-5-ил)метил-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
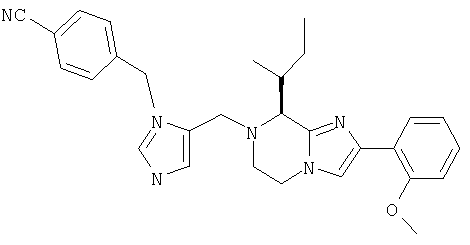
Соединение 11: 5-(2-(1-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-5,6-дигидро-2-фенил-1Н-имидазо[1,2а][1,4]бензодиазепин
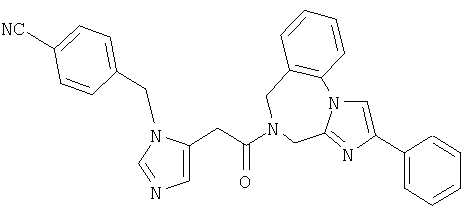
Соединение 12: 7-(2-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,1,8-тетрагидроимидазо[1,2а]-пиразин
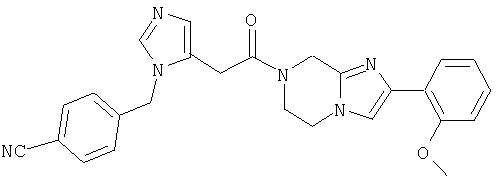
Соединение 13: 7-(2-амино-1-оксо-3-тиопропил)-8-(меркаптоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]-пиразин дисульфид

Соединение 14: 5-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
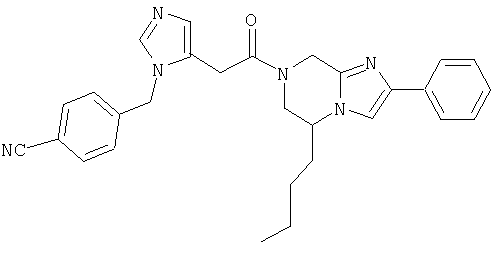
Соединение 15: 6-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
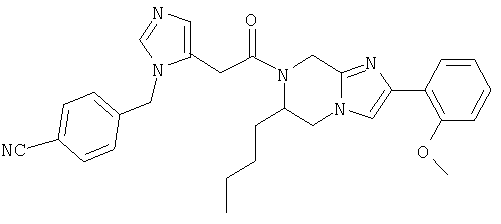
Соединение 16: 6-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
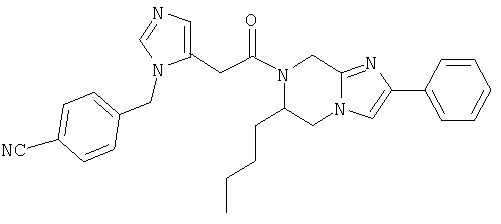
Соединение 17: 5-бутил-7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин

Соединение 18; 7-(2-(1-(4-цианофенилметил)имидазол-5-ил)1-оксоэтил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
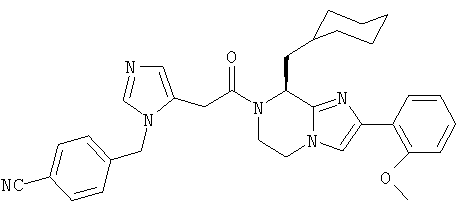
Соединение 19: 5-бутил-7-(2-(1Н-имидазол-5-ил)-1-оксоэтил)2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
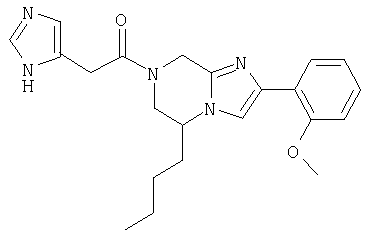
Соединение 20: 7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-фенилметокси)фенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
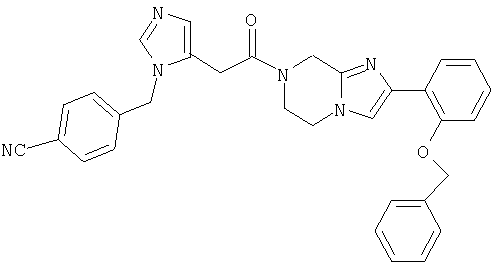
Соединение 21: 2-(2-бутоксифенил)-7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
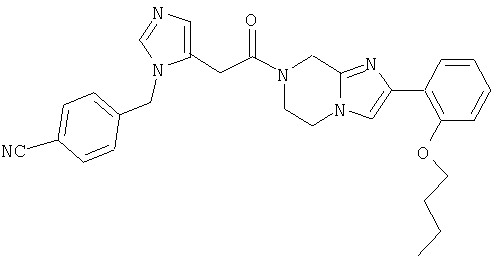
Соединение 22: 1,2-дигидро-1-((1Н-имидазол-4-ил)метил)4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин
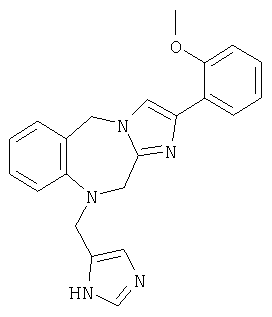
Соединение 23: 1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин
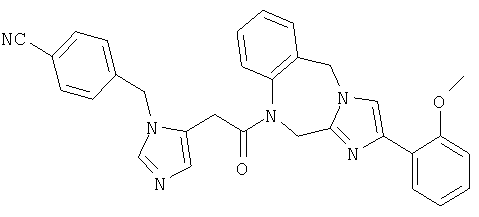
Соединение 24: 9-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с]-[1,4]-бензодиазепин
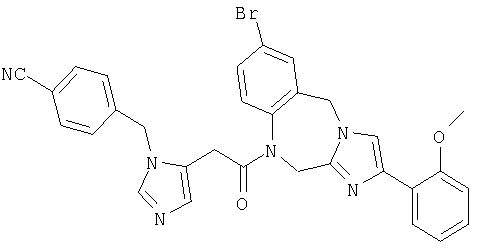
Соединение 25: 9-хлор-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с]-[1,4]-бензодиазепин
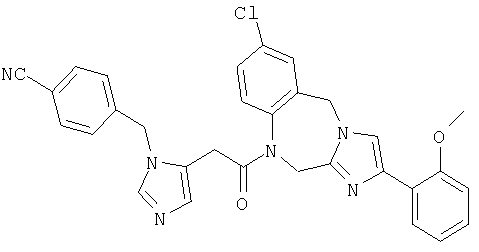
Соединение 26: 10-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с]-[1,4]-бензодиазепин
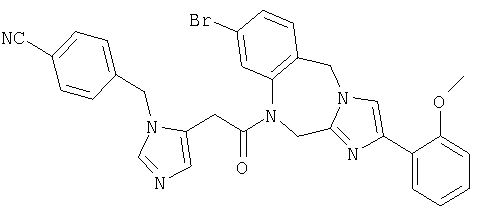
Соединение 27: 1-(2-(1-(4-цианофенилметил)имидазол-4-ил)1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо-[1,2с]-[1,4]бензодиазепин
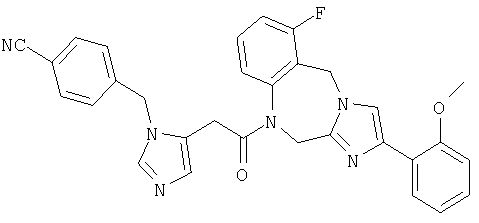
Соединение 28: 1,2-дигидро-1-(2-(имидазол-1-ил)-1-оксоэтил)-4-(2-метоксифенил)имидазо[1,2а][1,4]бензодиазепин
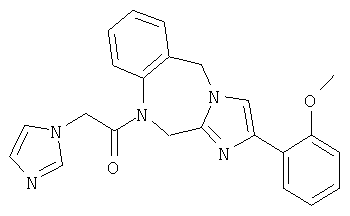
Соединение 29: 1,2-дигидро-4-(2-метоксифенил)-1-(2-(пиридин-3-ил)-1-оксоэтил)имидазо[1,2а][1,4]бензодиазепин
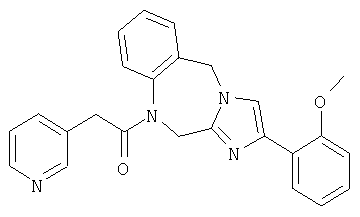
Соединение 30: 1,2-дигидро-4-(2-метоксифенил)-1-(2-(пиридин-4-ил)-1-оксоэтил)имидазо[1,2 а][1,4]бензодиазепин
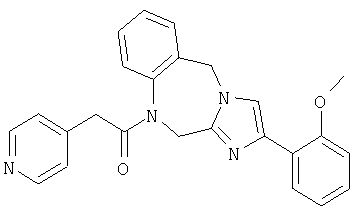
Соединение 31: 1-(2-(1-бензилимидазол-5-ил)-1-оксоэтил)1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2a][1,4]-бензодиазепин
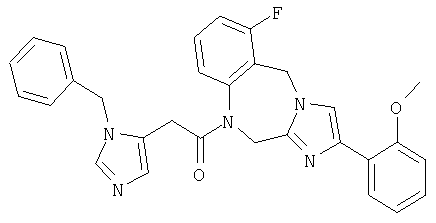
Соединение 32: 1-(2-(1-((4-циано)фенилметил)имидазол-5-ил)-1-оксоэтил)-9,10-дифтор-1,2-дигидро-4-(2-метоксифенил)-имидазо[1,2с][1,4]бензодиазепин
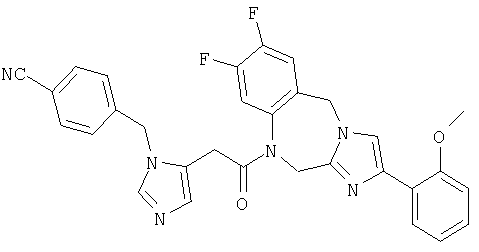
Соединение 33: 4-(2-бромфенил)-1-(2-(1-[(4-циано)фенил-метил]имидазол-5-ил) -1-оксоэтил)-1,2-дигидро-8-фторимидазо-[1,2a][1,4]бензодиазепин
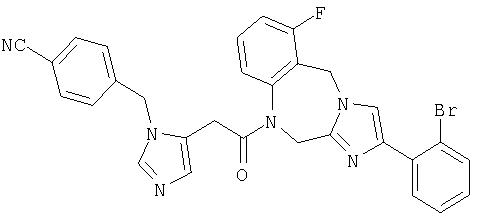
Соединение 34: 1-(2-(1-((4-циано)фенилметил)имидазо-5-ил)1-оксоэтил)-1,2-дигидро-10-фтор-4-(2-метоксифенил)имидазол-[1,2a][1,4]бензодиазепин
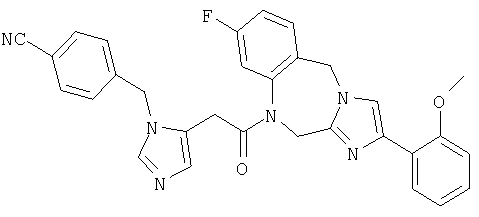
Соединение 35: 1-(2-(1-((4-циано-3-метокси)фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)-имидазол[1,2a][1,4]бензодиазепин
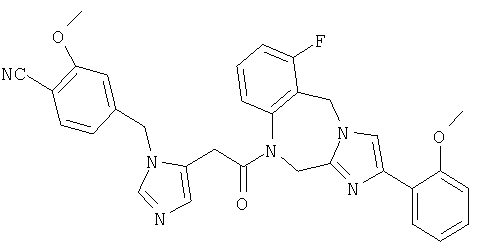
Соединение 36: 10-бром-1-(2-(1-((4-циано-3-метокси)фенилметил) имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)-имидазол[1,2a][1,4]бензодиазепин
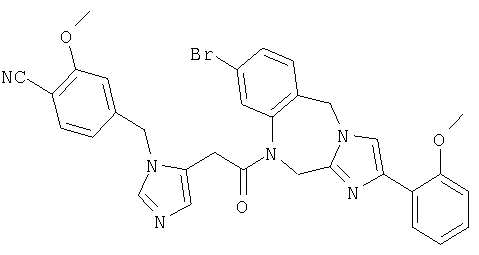
Соединение 37: 1-(2-(1-((4-циано-3-метокси)фенилметил)-имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-фенилимидазол-[1,2a][1,4]бензодиазепин
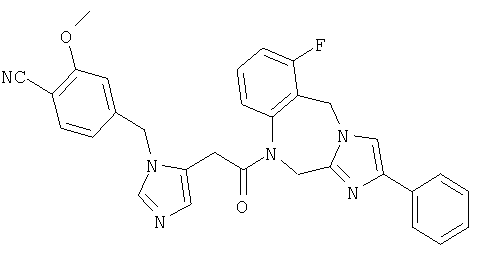
Соединение 38: 4-(2-бромфенил)-1-(2-(1-((4-циано-3-метокси)-фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фторимидазол-[1,2a][1,4]бензодиазепин
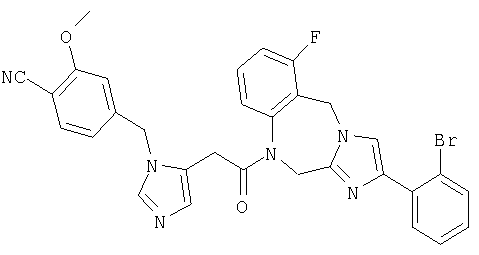
Соединение 39: 1-(2-(1-((3-метокси)фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазол-[1,2a][1,4]бензодиазепин
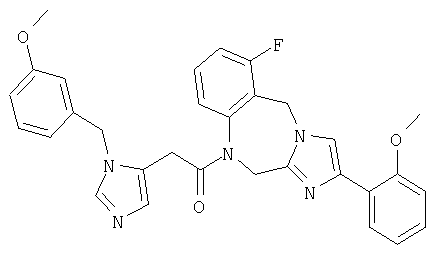
Соединение 40: 1-(2-(5-((4-циано)фенилметил)имидазол-1-ил)-1-оксоэтил)-2,5-дигидро-8-фтор-4-(2-метоксифенил)имидазо-[1,2с][1,4]бензодиазепин
ЭКСПЕРИМЕНТЫ
В следующих примерах и схемах использованы различные заместители, определенные выше. Варианты заместителей в схемах и примерах не обязательно совпадают с определенными в формуле изобретения.
Пример 1. 8-бутил-7-(3-(имидазол-5-ил)-1-оксопропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Синтез в примере 1 проводят по схеме 1 как показано ниже
Схема 1
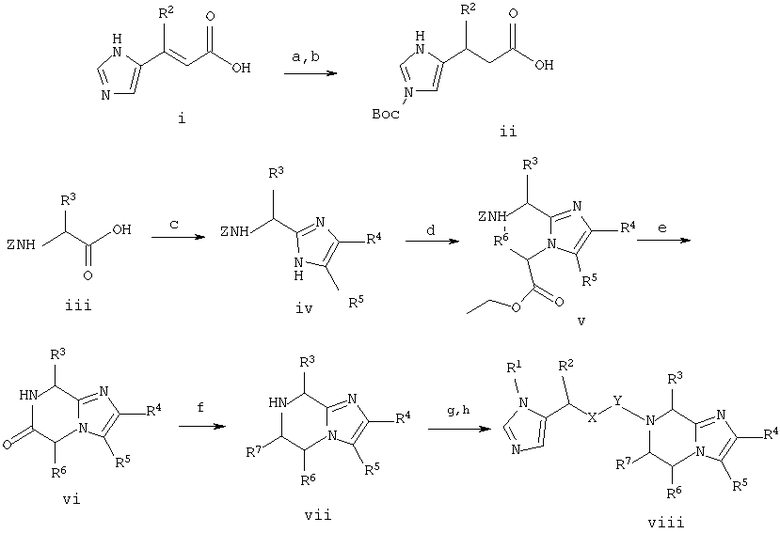
1.a. H2/Pd на угле/НОАс,
1.b. (ВОС)2О/K2СО3/5% водн. HCl/МеОН
1.с. Cs2СО3, Br-CHR5CO R4, затем NH4ОАс/ксилол
1.d. Br-CHR6CO2Et/K2СО3/ДМФ
1.e. H2/Pd на угле/НОАс
1.f. ВН3/ТГФ
1.g. Соединение ii/DCC/HOAt/ДМФ
1.h. ТФА/iPr3SiH
Стадия 1.a. 3-(1Н-имидазол-5-ил)пропионовая кислота
Урокановую кислоту (соединение i, в котором R2 является Н) (1,38 г, 10,0 ммоль) растворяют в 5%-ной водной HCl (20 мл) и МеОН, содержащем 10% Pd на угле (100 мг) и смесь встряхивают в течение ночи при давлении H2 2,109 кг/см2 (30 фунт/кв.дюйм). Катализатор удаляют фильтрацией через 3 см слой диатомовой земли и фильтрат концентрируют до твердого вещества и сушат в течение ночи в вакууме. Неочищенный материал используют без дальнейшей очистки. Масс-спектр 141,4 МН+
Стадия 1.b. 3-(1-((1,1-диметилэтокси)карбонил)имидазол-5-ил)пропионовая кислота
Неочищенный продукт со стадии 1.а. (10,0 ммоль) растворяют в Н2О (10 мл), содержащей K2СО3 (2,76 г, 20 ммоль) и к полученной смеси добавляют ди-трет-бутил дикарбонат (2,18 г, 10,0 ммоль) в ацетонитриле (20 мл). Реакционную смесь энергично перемешивают в течение около 3 часов, затем добавляют Н2О (10 мл) и смесь концентрируют до около 1/2 объема. Смесь подкисляют лимонной кислотой и экстрагируют EtOAc (2×25 мл). Водные экстракты сушат над Na2SO4, фильтруют и концентрируют до твердого вещества, которое сушат при пониженном давлении с получением 1,89 г (79%). ЯМР (300 МГц, ДМСО-d6,в, 30°С) 12, 0-12,2 (1Н, с), 8,0-8,2(1Н, с), 7,2-7,3(1Н, с), 2,65-2/8(2Н, т), 2,45-2, 65(2Н, т), 1,5-1,7(9Н, с).
Стадия 1.c. 2-(1-(S)-(((фенилметокси)карбонил)амино)-пентил)-4-(2-метоксифенил)имидазол
Cbz-(L)-Norleucine (соединение iii, в котором R3 является н-бутилом) (10,6 г, 40,0 ммоль) и Cs2СО3 (6,25 г, 20,0 ммоль) объединяют в 2:1/ДМФ:Н2O (65 мл) и смесь перемешивают до тех пор, пока, она не станет гомогенной. Растворители удаляют при пониженном давлении, остаток растворяют в ДМФ (75 мл) и добавляют 2-бром-2'-метоксиацетофенон (9,16 г, 40,0 ммоль) в ДМФ (50 мл). Смесь перемешивают в течение 15 минут при комнатной температуре, затем концентрируют при пониженном давлении. Полученный кетоэфир растворяют в ксилоле (250 мл), фильтруют, добавляют NH4OAc (50,0 г, 0,36 моль) и смесь кипятят с обратным холодильником в течение около 3 часов с удалением избытка NH4OAc и улавливанием воды с использованием ловушки Дина-Старка. Реакционную смесь концентрируют при пониженном давлении. Добавляют насыщенный раствор NaHCO3 (100 мл) и продукт экстрагируют CH2Cl2 (3×50 мл). Объединенные слои СН2Cl2 сушат над Na2SO2, фильтруют и концентрируют в вакууме с получением первой партии 6.23 г (т.пл.=118=121°С) указанного в заголовке соединения. Маточный раствор очищают флэш-хроматографией на силикагеле с использованием 2:3/EtOAc:гексана в качестве элюента. Очищенные фракции продукта объединяют и концентрируют с получением второй партии 3,26 г (т.пл.=119-122°С), общий выход составляет 9,49 г (60%) указанного в заголовке соединения. ЯМР (300 МГц, ДМСО-d6, 30°С) 11,7-11, 9 (1Н, с), 8,0-8,15 (1Н, д), 7,5-7,7 (1Н, м), 7,4-7,5 (1Н, с), 7,1-7,4 (6Н, м), 6,9-7,1 (2Н, м), 4,95-5,2 (2Н, кв), 4,6-4,8 (1Н, кв), 3,8-4,0 (3Н, с), 1,6-2,0 (2Н, м), 1,1-1,4 (4Н, м), 0,8-1,0 (3Н, т).
Стадия 1.d. 1-(2-этокси-2-оксоэтил)-2-(1-(S)-(((фенилметокси)карбонил)амино)пентил)-4-(2-метоксифенил)имидазол
Продукт со стадии 1.c. (соединение iv, в котором R3 является н-бутилом, R4 является 2-метоксифенилом и R5 является Н) (9,40 г, 23,9 ммоль) растворяют в ДМФ (50 мл) и обрабатывают K2СО3 (6,90 г, 50,0 ммоль) и этилбромацетатом (4,17 мл, 75,0 ммоль), и смесь нагревают до температуры 55°С в течение около 2 часов. Смесь концентрируют и затем растворяют в простом эфире (100 мл) и промывают один раз насыщенным раствором NaHCO3 (50 мл) и один раз насыщенным раствором NaCl (50 мл). Эфирный слой сушат над Na2SO4, фильтруют и концентрируют до масла (11,5 г, 100%), которое используют без дальнейшей очистки. Масс-спектр 480,3 МН+, ЯМР (300 МГц, ДМСО-d6, 30°С) 8,04-8,12 (1H, дд), 7,65-7,85 (1Н, т широкий), 7,5-7,6 (1Н, с), 7,25-7,4(5Н, м), 7,1-7,25(1Н, м), 7,0-7,1(1Н, д), 6,9-7,05 (1Н, т), 4,9-5,15 (2Н, кв), 5,0-5,2 (2Н, с), 4,5-4,7 (1Н, кв), 4,05-4,2 (2Н, кв), 3,8-4,0 (3Н, с), 1,8-2,0 (2Н, м), 1,2-1,4 (4Н, м), 1,15-1,25 (3Н, т), 0,75-0,95 (3Н, т).
Стадия 1.e. 8-бутил-6-оксо-2-(2-метоксифенил)имидазо-[1,2 а]пиразин
Продукт со стадии 1.d. (соединение v в котором R3 является н-бутилом, R4 является 2-метоксифенилом и R5 и R6 являются Н) (11,5 г, 23,9 ммоль) растворяют в НОАс (100 мл), содержащем катализатор 10% Pd на угле (500 мг) и гидрируют при давлении H2 3,515 кг/см2 (50 фунт/кв.дюйм) в течение около 3 часов при комнатной температуре. Катализатор удаляют фильтрацией через 3 см слой диатомовой земли и фильтрат нагревают при температуре около 70°С в течение около 2 часов. Смесь концентрируют до твердого вещества при пониженном давлении, растворяют в CH2Cl2 (100 мл) и промывают насыщенным раствором NaHCO3 (125 мл). Слой СН2Cl2 сушат над Na2SO4, фильтруют и добавляют 275 мл гексана. Продукт фильтруют и сушат до постоянного веса с получением 6,21 г (87%) продукта, Т.пл.=200-202°С, ЯМР (300 МГц, ДМСО-d6, 30°С) 8,5-8,6 (1Н, с), 8,0-8,1 (1Н, д), 7,4-7,6 (1Н, с), 7,1-7,3 (1Н, т), 7,0-7,1 (1Н, д), 6,9-7,1 (1Н, т), 4,55-4,8 (2Н, кв), 4,55-4,7 (1Н, т), 3,8-4,0 (3Н, с), 1,75-1,95 (2Н, м), 1,1-1,5 (4Н, м), 0,8-0,9 (3Н, т).
Стадия 1.f. 8-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидро-имидазо[1,2а]пиразин
Продукт со стадии 1.e. (6,13 г, 20,5 ммоль (соединение vi, где R3 является н-бутилом, R4 является 2-метоксифенилом и R5 и R6 являются Н) растворяют в ТГФ (100 мл) и обрабатывают 1М ВН3/ТГФ (82,0 мл, 82,0 ммоль) при комнатной температуре в течение около 2 часов, и затем кипятят с обратным холодильником в течение около 3 часов. Смесь охлаждают до комнатной температуры и по каплям добавляют 4N HCl (50 мл). Смесь перемешивают при комнатной температуре в течение около 2 часов, затем подщелачивают аккуратным порционным добавлением твердого K2СО3. Продукт экстрагируют EtOAc (3×50 мл). Слои EtOAc объединяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток растворяют в МеОН (около 50 мл), добавляют концентрированную HCl (1,75 мл) и раствор снова концентрируют при пониженном давлении. Продукт кристаллизуют из MeOH/Et2O с получением 6,76 г (92%) желаемого продукта. Масс-спектр 286,3 МН+, ЯМР (300 МГц, ДМСО-d6, 30°С) 10,0-11,5 (2Н, с широкий), 8,05-8,15 (1H, дд), 8,0-8,1 (1Н, с), 7,35-7,5 (1Н, т), 7,15-7,3 (1Н, д), 7,0-7,15 (1Н, т), 4,8-4,95 (1Н, м), 4,4-4,65 (2Н, м), 3,9-4,0 (3Н, с), 3,65-3,8 (1Н, м), 3,5-3,65 (1Н, м), 2,45-2,65 (1Н, м), 2,1-2,35 (1Н, м), 1,5-1,7 (2Н, м), 1,25-1,5 (2Н, м), 0,85-1,0 (3Н, т).
Стадия 1.g. 8-бутил-7-(3-(((1,1-диметилэтокси)карбонил)-имидазол-5-ил)-1-оксопропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
DCC (309 мг, 1,50 ммоль) и продукт со стадии 1.b. (соединение ii, в котором R2 является Н) (720 мг, 3,00 ммоль) растворяют в ТГФ (10 мл) и перемешивают в течение около 30 минут при комнатной температуре. Твердые вещества отфильтровывают, добавляют продукт со стадии 1.f. (соединение vii, в котором R3 является н-бутилом, R4 является 2-метоксифенилом и R5, R6 и R7 являются Н) (358 мг, 1,0 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Смесь концентрируют до смолы и очищают флэш-хроматографией на силикагеле, используя EtOAc в качестве элюента. Фракции продукта объединяют, концентрируют до получения стеклообразной массы и сушат до постоянного веса. Выход = 500 мг, (99%). Масс-спектр 508,2 (МН+).
Стадия 1.h. 8-бутил-7-(3-(имидазол-5-ил)-1-оксопропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 1.g. (450 мг, 0,89 ммоль) растворяют в МеОН (5 мл), добавляют 4N HCl и реакционную смесь перемешивают при комнатной температуре в течение около 1/2 часа. Растворители удаляют при пониженном давлении с получением 420 мг неочищенного продукта. Часть неочищенного продукта (100 мг) очищают препаративной ВЭЖХ на колонке RAININ™C18 (Varian Analytical, Walnut Creek, CA), используя градиент 10-30% СН3CN/водн. HCl (pH 2,0) на протяжении 45 минут с УФ определением при 254 нм. Фракции продукта концентрируют до около 1/2 объема и лиофилизуют с получением чистого продукта (47 мг, 47%) в виде дигидрохлорида. Масс-спектр 508,2 МН.
Пример 2: 8-бутил-2-(2-гидроксифенил)-7-(имидазол-4-ил-пропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 1.h. (соединение viii, где R1, R2, R5, R6 и R7 являются Н, R3 является н-бутилом, R4 является 2-метоксифенилом, Х является -СН2- и Y является -СО-) (320 мг, 0,667 ммоль) растворяют в минимальном количестве ТГФ и по каплям добавляют 1М раствор ВН3 в ТГФ (10,0 мл, 10,0 ммоль) при перемешивании. Смесь кипятят с обратным холодильником в течение 2 часов, затем охлаждают. По каплям добавляют 4N раствор HCl и смесь короткое время кипятят с обратным холодильником и затем подщелачивают осторожным порционным добавлением твердого K2СО3. Продукт экстрагируют EtOAc (3×10 мл) и затем экстрагируют снова 0,5%-ным водным ТФА (4×10 мл). Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 0-25% СН3CN/0,1% водн. ТФА на протяжении 45 минут с УФ определением при 254 нм. Фракции продукта концентрируют до около 1/2 объема и лиофилизуют, затем повторно лиофилизуют дважды из разбавленной HCl с получением чистого продукта (41 мг, 13%) в виде дигидрохлорида. Масс-спектр 380,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,0-9,1 (1Н, с), 7,9-8,05 (2Н, м), 7,45-7,55 (1Н, с), 7,2-7,3 (1Н, т), 7,05-7,15 (1H, д), 6,9-7,0 (1Н, т), 4,5-6,0 (3-4Н, с широкий), 4,0-4,4 (), 1,5-1,7 (2Н, м), 1,25-1,5 (2Н, м), 0,8-1,0 (3Н, т).
Пример 3: 8-бутил-7-(4-имидазолилпропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 1.h. (475 мг, 0,973 ммоль) растворяют в МеОН (10 мл) и добавляют раствор NaOH (80 мг, 2,0 ммоль) в Н2О (1 мл) при комнатной температуре и смесь перемешивают в течение около 1/2 часа и затем концентрируют при пониженном давлении. Остаток растворяют в ТГФ (10 мл), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении до около 2 мл и добавляют 1М ВН3/ТГФ (8,0 мл). Смесь кипятят с обратным холодильником в течение около 3 часов и гасят добавлением 5%-ной водной HCl с кратким нагреванием с обратным холодильником. Реакционную смесь охлаждают до комнатной температуры, подщелачивают осторожным добавлением твердого NaHCO3 и экстрагируют СН2Cl2 (2×10 мл). Слои CH2Cl2 сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 0-25% СН3CN/водн. HCl (рН 2,0) на протяжении 45 минут с УФ определением при 270 нм. Фракции продукта концентрируют до около 1/2 объема и лиофилизуют с получением чистого продукта (64 мг, 15%) в виде дигидрохлорида. Масс-спектр 394,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,0-9,1 (1Н, с), 8,0-8,2 (2Н, д), 7,9-8,1 (1Н, с), 7,4-7,5 (1Н, с), 7,35-7,5 (1Н, т), 7,1-7,3 (1Н, д), 7,0-7,2 (1Н, т), 5,0-7,0 (3Н, с широкий), 4,1-4,5 (3Н, м), 3,9-4,0 (3Н, с), 3,2-3,7 (2Н, д широкий), 2,7-3,0 (2Н, с широкий), 2,6-2,9 (2Н, т), 1,9-2,2 (4Н, м), 1,0-1,7 (4Н, м), 0,8-1,0 (3Н, т).
Пример 4: 7-(2-(имидазол-4-ил)-1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
Синтез в примере 4 проводят по схемам 1 и 4, заменяя стадии 1.а. и 1.b. стадиями 4.а.-4.с.
Схема 4
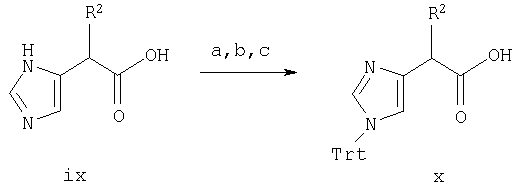
4.a. HCl/MeOH;
4.b. Хлортрифенилметан/Et3N/ДМФ;
4.с. 2,5N NaOH/MeOH
Стадия 4.а. Метил 4-имидазолацетат
Раствор гидрата натриевой соли 4-имидазолуксусной кислоты (соединение ix, в котором R2 является Н) (3,0 г, 18,1 ммоль) в МеОН (50 мл) охлаждают до температуры около 0°С и безводный HCl газ барботируют в смесь в течение около 15 минут, сохраняя температуру реакции ниже 5°С. Реакционную смесь перемешивают в течение около 1/2 часа при комнатной температуре и затем растворители удаляют при пониженном давлении с получением масла, которое затвердевает при выстаивании. Масс-спектр 141,0 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,0-9,2 (1Н, с), 7,4-7,6 (1Н, с), 3,85-3,95 (2Н, 3), 3,6-3,7 (3Н, с).
Стадия 4.b. Метил 1-трифенилметил-4-имидазолацетат
Раствор продукта со стадии 4. а. (3.2 г, 17,5 ммоль) в ДМФ (50 мл) обрабатывают хлортрифенилметаном (4,88 г, 17,5 ммоль) и Et3N (5,4 мл, 38,5 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение около 6 часов. ДМФ выпаривают при пониженном давлении и остаток распределяют между EtOAc и насыщенным раствором NaCl. Слой EtOAc сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением продукта в виде вязкого масла (6,96 г, 104%), которое кристаллизуется при выстаивании. Масс-спектр 383,3 MH+.
Стадия 4.с. 1-трифенилметил-4-имидазолуксусная кислота, натриевая соль
Продукт со стадии 4.b. (2,0 г, 5,24 ммоль) растворяют в МеОН (20 мл) и добавляют 2.5N NaOH (2.1 мл) при комнатной температуре и смесь перемешивают в течение ночи. Растворители удаляют при пониженном давлении с получением неочищенного продукта (2.08 г, 102%), который используют на последующих стадиях без дальнейшей очистки.
Стадии 4.d.-4.i. проводят по методике стадий 1.c.-1.h. примера 1, используя в качестве исходного соединения Cbz-(L)-Ile-OH вместо Cbz-(L)-Nle-ОН и получая (2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидро-(7-(2-(1-трифенилметилимидазол-4-ил)-1-оксоэтил)имидазо[1,2а]пиразин)(соединение viii, в котором R1, R2, R5, R6 и R7 являются Н, R3 является изобутилом и R4 является 2-метоксифенилом).
Стадия 4.j. 7-(2-(имидазол-4-ил)-1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 4.i. (150 мг, 0,24 ммоль) обрабатывают ТФА (1,0 мл), содержащим iPr3SiH (51 мкл, 0,25 ммоль) при комнатной температуре в течение около 2 часов. Добавляют простой эфир (10 мл) и продукт экстрагируют 3×10 мл 0,1%-ным водным раствором ТФА. Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 20% до 70% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта объединяют, концентрируют и повторно лиофилизуют из разбавленной HCl с получением продукта (53 мг, 57%) в виде дигидрохлорида. Масс-спектр 394,3 MH+.
Пример 5: 2-(2-метоксифенил)-8-(1-метилпропил)-7-(1-оксо-2-(1-фенилметил)-имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Синтез в примере 5 проводят по схемам 1 и 5, заменяя стадии 1.а. и 1.b. на стадии 5.а.-5.с.
Схема 5
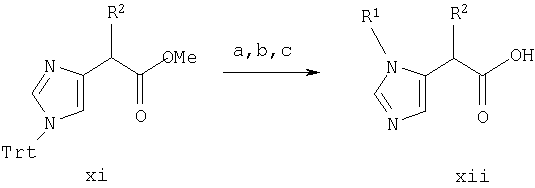
5. a. R1-Br/CH3CN;
5.b. МеОН, кипячение с обратным холодильником;
5.с. 5%-ной водной HCl, кипячение с обратным холодильником
Стадия 5. а. Метил 1-фенилметил-3-трифенилметил-5-имидазол-ацетат
Метил 1-трифенилметил-4-имидазолацетат со стадии 4.b. (1,12 г, 2,93 ммоль) растворяют в СН3CN (15 мл) и добавляют бензилбромид (349 мкл, 2,93 ммоль) при комнатной температуре. Смесь кипятят с обратным холодильником в течение около 3 часов и выстаивают при комнатной температуре в течение ночи. Растворители удаляют при пониженном давлении и остаток используют без дальнейшей очистки на стадии 5.b.
Стадия 5.b. метил 1-фенилметил-5-имидазолацетат
Продукт со стадии 5.а. растворяют в МеОН (20 мл) и смесь кипятят с обратным холодильником в течение 1 часа. Растворители удаляют при пониженном давлении. Остаток растирают с гексаном (2×20 мл) и с EtOAc (2×20 мл). Остаток используют без дальнейшей очистки на стадии 5.с.
Стадия 5.с. 1-фенилметил-5-имидазолуксусная кислота
Продукт со стадии 5.b. растворяют в 5%-ной водной HCl и кипятят с обратным холодильником в течение около 3 часов, затем концентрируют при пониженном давлении. Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 5 до 35% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта объединяют, концентрируют и повторно лиофилизуют из разбавленной HCl с получением продукта (360 мг, 49%) в виде гидрохлорида. Масс-спектр 217,1 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 8,4-8,6 (1Н, с), 7,3-7,5 (3Н, м), 7,1-7,3 (3Н, м), 5,2-5,4 (2Н, с), 3,5-3,7 (2Н, с).
Стадии 5.d.-5.g. проводят по методике стадий 1.c.-1.f. примера 1, используя в качестве исходного соединения Cbz-(L)-Ile-OH вместо Cbz-(L)-Nle-ОН и получая (2-(2-метоксифенил)-8-(1-метилэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин) (соединение vii, в котором R3 является изобутилом, R4 является 2-метоксифенилом и R5, R6 и R7 являются Н).
Стадия 5.h. 2-(2-метоксифенил)-8-(1-метилпропил)-7-(1-оксо-2-(1-фенилметил)имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
DCC (2-6 мг, 1,00 ммоль), HOSu (115 мг, 1,00 ммоль), NMM (220 мкл, 2,0 ммоль), продукт со стадии 5.g. (179 мг, 0,50 ммоль) и продукт со стадии 5.с. (330 мг, 1,00 ммоль) растворяют в ДМФ (10 мл) и перемешивают при комнатной температуре в течение ночи и затем нагревают до температуры около 50°С в течение около 8 часов. Смесь концентрируют до смолы и очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 25 до 40% СН3CN/0,1% водного ТФА на протяжении 30 минут. Фракции продукта объединяют, концентрируют и повторно лиофилизуют из разбавленной HCl с получением продукта (96 мг, 40%) в виде гидрохлорида. Масс-спектр 484,3 МН+.
Пример 6: 2-(2-метоксифенил)-8-(1-метилпропил)-7-(2-(1-фенилметил)имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
Продукт примера 6 получают из продукта со стадии 5.h. по методике примера 3. Масс-спектр 470,4 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,15-9,4 (1Н, с), 8,05-8,2 (2Н, дд), 7,9-8,1 (1Н, д), 7,5-7,6 (1Н, с), 7,25-7,5 (6Н, м), 7,15-7,25 (1H, дд), 7,05-7,15 (1Н, м), 5,5-5,6 (2Н, с), 4,6-5,4 (3Н, с широкий), 4,0-4,3 (2Н, м), 3,8-4,0 (4Н, м), 3,1-3,5 (2Н, м), 2,6-9,95 (4Н, м), 1,95-2,15 (1Н, м), 1,3-1,5 (1Н, м), 1,1-1,3 (1Н, м), 0,85-1,0 (3Н, д), 0,7-0,85 (3Н, т).
Пример 7: 7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,1,8-тетрагидроимидазо[1,2а]пиразин
(2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидро-(7-(2-(1-трифенилметилимидазол-4-ил)-1-оксоэтил)имидазо[1,2а]-пиразин) (соединение viii, в котором R1, R2, R5, R6 и R7 являются Н, R3 является изобутилом и R4 является 2-метоксифенилом) из примера 4, стадия 4.i. (135 мг, 0,21 ммоль) растворяют в СН3CN (3,0 мл) и добавляют α-бром-п-толунитрил (42 мг, 0,21 ммоль) и смесь кипятят с обратным холодильником в течение около 3 часов. Растворители удаляют при пониженном давлении и добавляют МеОН (3,0 мл). Смесь кипятят с обратным холодильником в течение около 1 часа и растворители удаляют при пониженном давлении. Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 5% до 35% СН3CN/0,1% водного ТФА на протяжении 45 минут. Фракции продукта объединяют, концентрируют и повторно лиофилизуют из разбавленной HCl с получением продукта (9,8 мг, 8%) в виде гидрохлорида. Масс-спектр 509,3 МН+.
Пример 8: 7-((1Н-имидазол-4-ил)метил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2 а]пиразин
Синтез в примере 8 проводят по схемам 1 и 8, заменяя стадии 1.а. и 1.b. стадиями 8.а.-8.b. и стадии 1.д. и 1.h. стадиями 8.с. и 8.d.
Схема 8
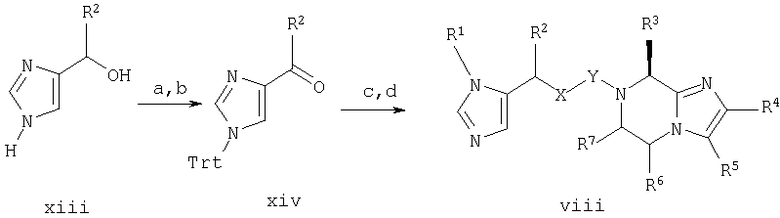
8. а. (С6Н5)3CCl/Et3N/ДМФ;
8.b. SO3-пиридин комплекс/Et3N/ДМСО;
8. с. Соединение vii/NaBH (OAc)3/СН3Cl2;
8.d. ТФА/iPr3SiH
Стадия 8.а. 4-гидроксиметил-1-трифенилметилимидазол
4-Гидроксиметилимидазола гидрохлорид (соединение xiii, в котором R2 является Н) (2,50 г, 18,6 ммоль) и Et3N (2,59 г, 18,6 ммоль) объединяют в ДМФ (30 мл) и перемешивают при комнатной температуре. По каплям добавляют раствор хлортрифенилметана (5.19 г, 18,6 ммоль) в ДМФ (25 мл) при комнатной температуре и полученную смесь перемешивают при комнатной температуре в течение около 23 часов и затем выливают в ледяную воду (300 мл). Продукт отфильтровывают, промывают холодной водой (75 мл) и растирают с п-диоксаном (30 мл). Продукт отфильтровывают и сушат при пониженном давлении с получением продукта (4,96 г, 78%). ЯМР (300 МГц, ДМСО-d6, 30°С) 7,3-7,5 (9Н, м), 7,25-7,35 (1Н, д), 7,0-7,2 (6Н, м), 6,7-6,75 (1Н, с), 4,15-4,2 (2Н, м).
Стадия 8.b. 1-трифенилметилимидазол-4-карбоксальдегид
Продукт со стадии 8.а. (2,04 г, 6,00 ммоль) суспендируют в ДМСО (10,0 мл) и добавляют Et3N (3.34 мл, 24,0 ммоль) и SO3-пиридин комплекс (2,39 г, 15,0 ммоль) при комнатной температуре. Смесь нагревают до температуры около 110°С в течение около 1 часа и затем охлаждают. Смесь выливают в 150 мл H2O и продукт отфильтровывают. Остаток обрабатывают насыщенным раствором NaHCO3 (50 мл) и экстрагируют 2×100 мл CH2Cl3. Объединенные слои CH2Cl2 промывают 5%-ным раствором лимонной кислоты (100 мл), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток кристаллизуют из МеОН и Н2О. Выход = 1,08 г (53%).
Стадия 8.с. 7-((1-трифенилметилимидазол-4-ил)метил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
8-(1-метилпропил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразина дигидрохлорид (соединение vi, в котором R3 является 1-метилпропилом, R4 является 2-метоксифенилом и R5-R7 являются Н) (со стадии 4.g.) (179 мг, 0.50 ммоль) и продукт со стадии 8.b. (соединение xiv, в котором R2 является Н) (338 мг, 1,00 ммоль) объединяют в 1,2-дихлорэтане (2,0 мл). Добавляют NaBH(ОАс)3 (212 мг, 1,00 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение около 1 часа. Реакционную смесь выливают в колонку с силикагелем и продукт элюируют EtOAc элюентом. Фракции продукта объединяют и концентрируют с получением продукта в виде белой пены (150 мг, 49%). Масс-спектр 608,2 МН+.
Стадия 8.d. 7-((1Н-имидазол-4-ил)метил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 8.с. (соединение viii, в котором R1 является трифенилметилом, R3 является изобутилом, R4 является 2-метоксифенилом и R2, R5-R7 являются Н, и Х и Y образуют связь) (160 мг, 0,26 ммоль) обрабатывают ТФА (10 мл), содержащим iPr3SiH (0,20 мл, 1,0 ммоль) в течение около 45 минут при комнатной температуре в атмосфере N2. Растворители удаляют при пониженном давлении и продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 20-40% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта концентрируют до около 1/2 объема и лиофилизуют. Продукт повторно лиофилизуют из разбавленной HCl с получением чистого продукта (77 мг, 66%). Масс-спектр 366,2 MH+.
Пример 9: 7-((4-имидазолил)карбонил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Соединение примера 9 получают по схемам 1 и 4 по методике примера 4, используя в качестве исходного соединения имидазол-4-карбоновую кислоту вместо имидазол-4-уксусной кислоты на стадии 4.а. Масс-спектр 380,3 МН+.
Пример 10: 7-(1-(4-цианофенилметил)имидазол-5-ил)метил-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
Продукт со стадии 8.с. (соединение viii, в котором R1 является трифенилметилом, R3 является изобутилом, R4 является 2-метоксифенилом и R2, R5, R6 и R7 являются Н, и Х и Y образуют связь) (150 мг, 0,25 ммоль) растворяют в CH2Cl2 (2,0 мл). Добавляют α-бром-п-толунитрил (49 мг, 0,25 ммоль) и смесь кипятят с обратным холодильником в течение около 1 часа. Добавляют МеОН (3,0 мл) и смесь снова кипятят с обратным холодильником в течение 1 часа. Растворители удаляют при пониженном давлении, добавляют Et2O (10 мл) и продукт экстрагируют 1,0% ТФА (2×15 мл). Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 20-50% СН3CN/0,1% ТФА на протяжении 40 минут. Фракции продукта концентрируют до около 1/2 объема и лиофилизуют. Продукт повторно лиофилизуют из разбавленной HCl с получением чистого продукта (52 мг, 35%). Масс-спектр 481,4 МН+.
Пример 11: 5-(2-(1-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-5,6-дигидро-2-фенил-1Н-имидазо[1,2а][1,4]бензодиазепин
Синтез в примере 11 поводят по схемам 5 и 11.
Схема 11
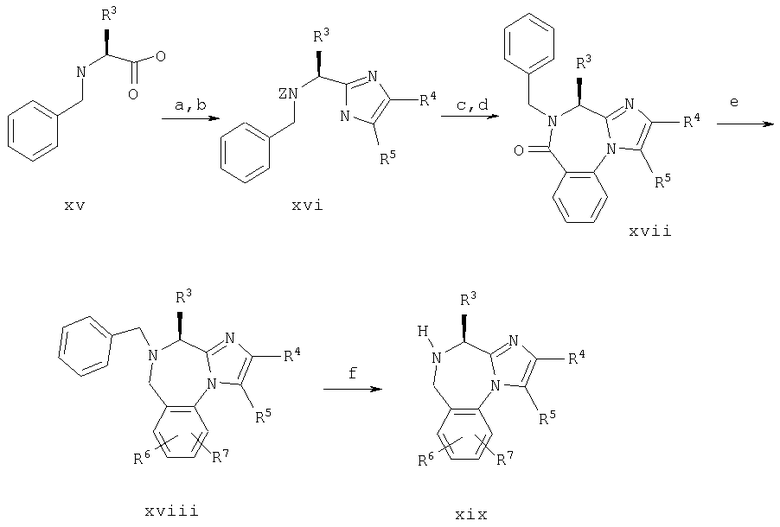
11. а. Cbz-Osu/K2СО3/СН3CN/Н2O;
11.b. Cs2СО3/R4COCHBrR5, затем NH4ОАс/ксилол;
11.с. HBr/НОАс;
11. d. 2-фторбензоилхлорид/Et3N/CH2Cl2/, затем кипячение с обратным холодильником в ДМФ;
11. е. ВН3/ТГФ;
11. f. 1-хлорэтилхлорформиат/СН2Cl2
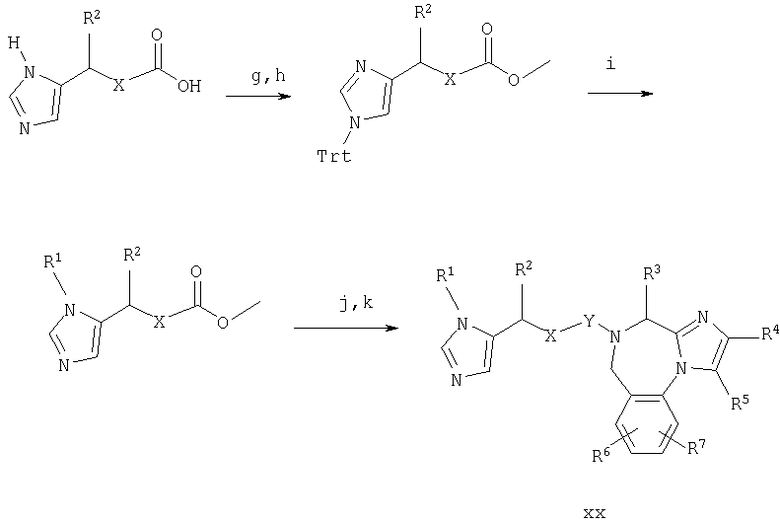
11.g. MeOH/HCl;
11.h. Trt-Cl/Et3N/ДМФ;
11.i. R1-Br/EtOAc, затем МеОН;
11.j. MeOH/H2O/NaOH;
11.k. DCC/HOAt/ДМФ/соединение xix/ Et3N
Стадия 11.a. (N-(фенилметокси)карбонил)-N-(фенилметил)-глицин
Раствор Cbz-Osu (6,18 г, 24/8 ммоль) в CH3CN (55 мл) добавляют к раствору N-бензилглицин гидрохлорида (соединение xv, в котором R3 является Н) (5,00 г, 24,8 ммоль) и K2СО3 (6,84 г, 49,6 ммоль) в H2O (35 мл) и смесь энергично перемешивают в течение около 2 часов. Смесь концентрируют до около 35 мл и промывают Et2O (2×25 мл). Водный слой подкисляют до около pH 1 осторожным добавлением концентрированной HCl и продукт экстрагируют EtOAc (2×50 мл). Слои EtOAc объединяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении до чистого бесцветного масла (7,40 г, 99,7%). ЯМР (300 МГц, ДМСО-d6, 30°С) 12,5-12, 8 (1H, с широкий), 7,1-7,5 (10Н, м), 5,0-5,2 (2Н, с), 4,4-4,6 (2Н, д), 3,8-4,0 (2Н, с).
Стадия 11.b. 2-((N-(фенилметокси)карбонил)-N-(фенилметил)-амино)метил)-4-фенилимидазол
Продукт со стадии 11.а. (7,18 г, 24,0 ммоль) растворяют в ДМФ (50 мл) и добавляют Cs2СО3 (3,91 г, 12,0 ммоль) в Н2О (20 мл) и смесь перемешивают до гомогенности. Растворители удаляют при пониженном давлении, остаток растворяют в ДМФ (25 мл) и добавляют 2-бром-2'-метоксиацетофенон (4,78 г, 24,0 ммоль) в ДМФ (25 мл). Смесь перемешивают в течение около 30 мнут при комнатной температуре, затем концентрируют при пониженном давлении. Полученный кетоэфир растворяют в ксилоле (125 мл) и фильтруют. Добавляют NH4OAc (28,0 г, 0,36 моль) и смесь кипятят с обратным холодильником в течение около 2 часов с удалением избытка NH4OAC и высвобождением Н2О, используя ловушку Дина-Старка. Реакционную смесь охлаждают и промывают насыщенным раствором NaHCO3 (100 мл), сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением 9,70 г (102%) соединения, которое используют без дальнейшей очистки. Масс-спектр 398,2 MH+.
Стадия 11.с. 2-(N-(фенилметил)амино)метил)-4-фенилимидазол
Продукт со стадии 11b. (соединение xvi, в котором R3 и R5 являются Н и R4 является 2-метоксифенилом) (9,7 г, 24,0 ммоль) обрабатывают 30% HBr/НОАс (85 мл)при комнатной температуре в течение 2 часов. К полученной суспензии добавляют Et2O (100 мл) и продукт отфильтровывают, промывают Et2O и сушат при пониженном давлении с получением продукта (7.70 г, 75%) в виде беловато-сероватого твердого вещества. Масс-спектр 264,3 МН+, ЯМР (300 МГц, ДМСО-d6, 30°С) 8,5-11,0 (3Н, с широкий), 8,1-8,2 (1Н, с), 7,8-7,9 (2Н, м), 7,55-7,65 (2Н, м), 7,45-7,55 (2Н, м), 7,35-7,5 (4Н, м), 4,5-4,7 (2Н, с), 4,3-4,5 (2Н, с).
Стадия 11.d. 6-оксо-2-фенил-5-(фенилметил)-1H-имидазо-[1,2а][1,4]бензодиазепин
Продукт со стадии 11.с. (4,25 г, 10,0 ммоль) суспендируют в ТГФ (35 мл) и добавляют Et3N (4,9 мл, 35,0 ммоль) при комнатной температуре. Добавляют 2-фторбензоилхлорид (1,19 мл, 10,0 ммоль) и смесь перемешивают в течение около 1 часа при комнатной температуре. Растворители удаляют при пониженном давлении и остаток помещают в CH2Cl2 (50 мл) и промывают насыщенным раствором NaCl (2×25 мл). Слой СН2Cl2 сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток растворяют в ДМФ (35 мл), добавляют K2СО3 (1,38 г, 10,0 ммоль) и смесь кипятят с обратным холодильником в течение около 1 часа. Растворители удаляют при пониженном давлении, промежуточное соединение растворяют в EtOAc (50 мл) и промывают 2 раза насыщенным NaHCO3 и 1 раз насыщенным раствором NaCl. Слой EtOAc сушат над Na2SO4, фильтруют и концентрируют до около 25 мл. Добавляют Et2O (около 25 мл) и продукт отфильтровывают (1,46 г). Маточные растворы очищают флэш-хроматографией на силикагеле, используя 1:1/гексан:EtOAc в качестве элюента с получением второй партии бледно-оранжевой пены (0,63 г), которую используют на следующих стадиях. Общий выход=57%. Масс-спектр 366,2 MH+, ЯМР (300 МГц, ДМСО-d6, 30°С) 8,25-8,35 (1Н, с), 7, 95-8,05 (1H, дд), 7,6-7,9 (4Н, м), 7,5-7,7 (1Н, т), 7,3-7,5 (2Н, т), 7,2-7,4 (6Н, м), 4,6-5,0 (2Н, с широкий), 4,4-4,6 (2Н, с).
Стадия 11.е. 2-фенил-5-(фенилметил)-1H-имидазо[1,2а]-[1,4]бензодиазепин
Продукт со стадии 11.а. (соединение xvii, в котором R3 и R5 являются Н и R4 является 2-метоксифенилом) (0,63 г, 1,73 ммоль) растворяют в ТГФ и добавляют 1М боран/ТГФ комплекс (16,0 мл, 16,0 ммоль) при комнатной температуре. Смесь кипятят с обратным холодильником в течение около 1 часа и затем охлаждают. Добавляют 4N HCl (12 мл) и смесь кипятят с обратным холодильником в течение 1/2 часа. Смесь охлаждают до комнатной температуры, концентрируют до около 12 мл при пониженном давлении и затем нейтрализуют осторожным добавлением твердого NaHCO3. Продукт экстрагируют 2 раза EtOAc, сушат над Na2SO4, фильтруют, затем концентрируют при пониженном давлении. Остаток растворяют в метаноле (10 мл) и обрабатывают концентрированной HCl (0,5 мл) для превращения продукта в гидрохлорид. Раствор концентрируют и продукт получают кристаллизацией из MeOH/Et2O. Выход=444 мг (60%). Масс-спектр 352,2 МН+, ЯМР (300 МГц, ДМСО-d6, 30°С) 8,5-8,6 (1Н, с), 7,9-8,0 (2Н, д), 7,65-7,9 (5Н, м), 7,4-7,6 (6Н, м), 7,3-7,4 (1Н, т), 4,5-6,5 (Н2O), 4,4-4,6(2Н, с), 4,2-4,3 (2Н, с), 4,1-4,25 (2Н, с).
Стадия 11.f. 2-фенил-1Н-имидазо[1,2а][1,4]бензодиазепин
Продукт со стадии 11.е. (соединение xviii, в котором R3, R5, R6 и R7 являются Н и R4 является 2-метоксифенилом) (382 мг, 0,90 ммоль) распределяют между насыщенным раствором NaHCO3 и CH2Cl2. Слой CH2Cl2 сушат над Na2SO4, фильтруют и добавляют 1-хлорэтилхлорформиат (108 мкл, 1,00 моль) при комнатной температуре. Смесь перемешивают в течение ночи при комнатной температуре. Слой СН2Cl2 промывают насыщенным раствором NaHCO3, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток растворяют в МеОН (5,0 мл) и перемешивают при комнатной температуре в течение около 1 часа. Метанольный раствор обрабатывают концентрированной HCl (0,5 мл) для превращения продукта в гидрохлорид. Раствор концентрируют и продукт получают кристаллизацией из MeOH/Et2O. Выход = 186 мг (62%). Масс-спектр 262,2 МН+, ЯМР (300 МГц, ДМСО-d6, 30°С) 10,7-11,1 (1-2Н, с широкий), 8,5-8,6 (1Н, с), 8,1-8,6 (1-2Н, с широкий), 7,9-8,05 (2Н, дд), 7,7-7,85 (3Н, м), 7,55-7,65 (1Н, м), 7,45-7,55 (2Н, т), 7,3-7,45 (1Н, м), 4,2-4,4 (2Н, с), 4,1-4,3 (2Н, с).
Стадия 11.g. Метил 4-имидазолацетат
Дигидрат натриевой соли 4-имидазолуксусной кислоты (15,3 г, 83,1 ммоль) суспендируют в толуоле (100 мл) и концентрируют для удаления воды, оставшейся от гидрирования. Остаток растворяют в МеОН (235 мл) и раствор охлаждают на бане лед/вода в атмосфере N2. Добавляют газообразный HCl в течение 20 минут, и полученный раствор перемешивают в течение 2 часов при комнатной температуре. Смесь концентрируют досуха. Остаток повторно растворяют в МеОН (235 мл), фильтруют и раствор охлаждают на бане лед/вода в атмосфере N2. Добавляют газообразный HCl в течение 20 минут и полученный раствор перемешивают в течение 2 часов при комнатной температуре. Добавляют толуол (150 мл) и смесь концентрируют досуха и сушат с получением продукта (16,6 г, 113%), который используют на следующей стадии без дальнейшей очистки. Масс-спектр 141,2 MH+, ЯМР (300 МГц, ДМСО-d6, 30°С) 8,8-8,9 (1Н, с), 7,5-7,7 (1Н, с), 3,7-3,9 (2Н, с), 3,6-3,7 (3Н, с).
Стадия 11.h. Метил 1-трифенилметил-4-имидазолацетат
Продукт со стадии 11.g. (неочищенный, 83,1 ммоль) растворяют в ДМФ (70 мл) в атмосфере N2 и в экзотермическую реакционную смесь добавляют Et2N (35,6 мл, 241 ммоль). После охлаждения до комнатной температуры добавляют хлортрифенилметан (23,2 г, 83,1 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Смесь выливают в Н2O (300 мл) и экстрагируют один раз 300 мл EtOAc и один раз 150 мл EtOAc. Объединенные EtOAc слои промывают насыщенным раствором NaHCO3 (300 мл), сушат над Na2SO4, фильтруют и концентрируют до масла, которое кристаллизуется с получением рыжевато-коричневого твердого вещества (30,9 г, 97%). Масс-спектр 382,9 МН+.
Стадия 11.i. метил 1-(цианофенилметил)-5-имидазолацетат
Продукт со стадии 11.h. (15,0 г, 39,2 ммоль) растворяют в EtOAc (80 мл) при нагревании, добавляют α-бром-п-толуонитрил (7,69 г, 39,2 ммоль) и смесь нагревают при температуре 65-70°С в течение 2,5 часов. Первую партию отфильтровывают, промывают EtOAc и сушат до 9,27 г. Фильтрат концентрируют до около 80 мл и нагревают при температуре 65-70°С в течение еще 14 часов. Вторую партию отфильтровывают, промывают EtOAc и сушат до 9,40 г. Фильтрат концентрируют до около 30 мл и нагревают при температуре 65-70°С в течение еще 48 часов. Третью партию отфильтровывают, промывают EtOAc и сушат до 0,87 г. Объединенные промежуточные соединения суспендируют в МеОН (350 мл) и кипятят с обратным холодильником в течение 1/2 часа. Растворитель дистиллируют при пониженном давлении и полученное твердое вещество растирают с EtOAc (250 мл). Полученное твердое вещество суспендируют в CH2Cl2 (500 мл), добавляют насыщенный раствор NaHCO3 (500 мл) и смесь перемешивают в течение 3 часов. Водный слой удаляют и слой СН2Cl2 сушат над Na2SO2, фильтруют и концентрируют при пониженном давлении с получением масла, которое кристаллизуется при выстаивании (8,08 г, 81%) Масс-спектр 256,2 МН+.
Стадия 11.j. 1-(цианофенилметил)-5-имидазола гидрохлорид
Продукт со стадии 11.i. (8,0 г, 31,3 ммоль) растворяют в ТГФ (200 мл) и добавляют 2N NaOH (16,7 мл, 33,4 ммоль). Смесь перемешивают в течение 48 часов при комнатной температуре. Раствор нейтрализуют до pH 2,0 добавлением 2N HCl и раствор концентрируют при пониженном давлении до рыжевато-коричневого твердого вещества. Остаток перемешивают с МеОН (200 мл), твердые вещества удаляют фильтрацией и фильтрат концентрируют при пониженном давлении и сушат с получением продукта (5,85 г, 67%). Масс-спектр 242,1 МН+, ЯМР (300 МГц, ДМСО-d6, 30°C) 9,3-9,4 (1Н, с), 7,8-7,95 (2Н, д), 7,55-7,7 (1Н, с), 7,4-7,6 (2Н, д), 5,5-5,7 (2Н, с), 3,8-4,0 (2Н, с).
Стадия 11.k. 5-(2-(1-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-5,6-дигидро-2-фенил-1Н-имидазо[1,2а][1,4]бензодиазепин
Продукт со стадии 11.j. (77 мг, 0,24 ммоль) объединяют с DCC (49 мг, 0,24 ммоль), HOAt (33 мг, 0,24 ммоль), NMM (110 мкл, 1,0 ммоль) и продуктом со стадии 11.f. (75,0 мг, 0,225 ммоль) в ДМФ (3,0 мл) и реакционную смесь перемешивают в течение 3 дней при комнатной температуре. Растворители удаляют при пониженном давлении и неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 15-40% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта концентрируют до около 1/2 объема и лиофилизуют. Продукт повторно лиофилизуют дважды из разбавленной HCl с получением чистого продукта (71 мг, 61%). Масс-спектр 485,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,3-9,4 (1Н, д), 8,6-8,8 (1Н, д), 8,0-8,15 (2Н, д), 7,3-8,0 (12Н, м), 5,5-5,7 (2Н, с), 4,9-5,1 (1Н, с), 4,75-4,9 (1Н, с), 4,65-4,8 (1Н, с), 4,45-4,6 (1Н, с), 4,25-4,4 (1Н, с), 4,15-4,3 (1Н, с).
Пример 12: 7-(2-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
Стадии 12.а.-12.d. проводят по схеме 1, стадии 1.c.-1.f., используя в качестве исходного соединения Cbz-Gly-OH вместо Cbz-(L)-Ile-OH на стадии 12.а. Стадию 12.f. проводят по схеме 1, стадия 1.д., заменяя продуктом со стадии 11.j. продукт со стадии 1.b. Масс-спектр 453,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,1-9,2 (1Н, д), 7, 95-8,05 (1H, дд), 7,9-8,0 (1Н, с), 7,8-7,9 (2Н, д), 7,55-7,6 (1Н, с), 7,5-7,6 (2Н, д), 7,35-7,5 (1Н, м), 7,15-7,25 (1H, д), 7,0-7,15 (1Н, т), 5,4-5,7 (2Н, с), 4,9-5,1 (2Н, с широкий), 4,15-4,4 (2Н, с широкий), 4,05-4,2 (2Н, с), 3,9-4,1 (2Н, с широкий), 3,9-4,0 (3Н, с).
Пример 13: 7-(2-амино-1-оксо-3-тиопропил)-8-(меркаптоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин дисульфид
Стадии 13.а. - 13.d. проводят согласно схеме 1, стадии 1.c.-1.f., используя Cbz-(L)-Asp(Obz)-ОН вместо Cbz-(L)-Nle-ОН на стадии 13.а. и на стадии 13.d. используют 6,0 ммоль ВН3 на 1,0 ммоль соединения vi (в котором R3 является -СН2CO2Н, R4 является 2-метоксифенилом и R5 и R6 являются Н). Также, высушенный EtOAc слой на стадии 13.d. концентрируют до твердого вещества и используют без превращения в гидрохлорид.
Стадии 13.е.-13.j. проводят по схеме 13:
Схема 13
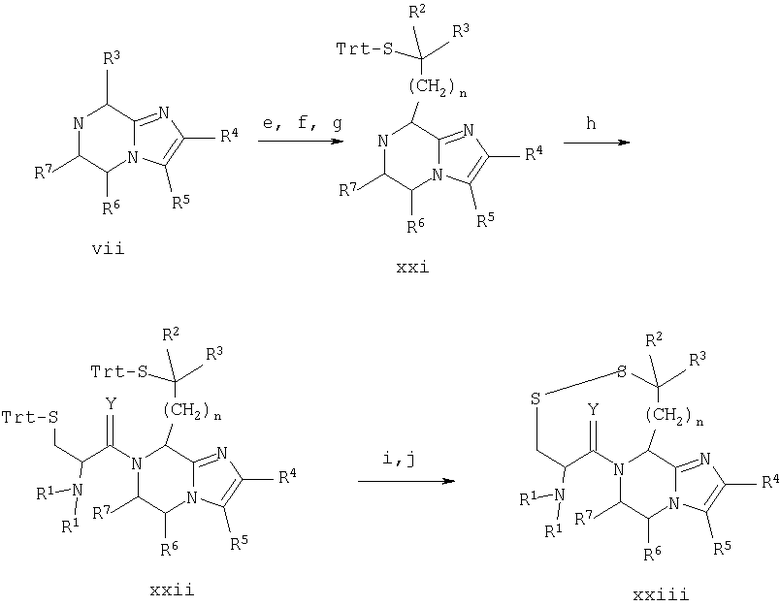
13.e. (Вос)2O/NaOH/ТГФ/Н2O
13.f. (С6Н5)3Р/DEAD/Trt-SH/ТГФ
13.g. 20% ТФА/СН2Cl2
13.h. Boc-(L)-Cys(Trt)-OH/EDC/HOAt/NMM/ТГФ
13.i. ТФА/iPr3SiH/СН2Cl2
13.j. воздух при рН 7,2-7,5
Стадия 13.e. 7-((1,1-диметилэтокси)карбонил)-8-(2-гидрокси-этил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]-пиразин
8-(2-гидроксиэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин (соединение vii, в котором R3 является 2-гидроксиэтилом, R4 является 2-метоксифенилом и R5, R6 и R7 являются H) со стадии 13.d. (3,70 г, 13,6 ммоль) растворяют в ТГФ (50 мл) и добавляют Н2O (10 мл). Добавляют ди-трет-бутилдикарбонат (3.25 г, 14,9 ммоль) и реакционную смесь энергично перемешивают в течение около 3 часов при сохранении рН реакционной смеси около 8,5 добавлением 2,5N раствора NaOH. Растворители удаляют при пониженном давлении, и остаток распределяют между EtOAc (25 мл) и Н2О (25 мл). Продукт экстрагируют 2×25 мл EtOAc и объединенные экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают флэш-хроматографией на силикагеле, используя 3:2/EtOAc:гексан в качестве элюента. Объединенные фракции продукта концентрируют до белой пены и сушат с получением 2,95 г (58%) желаемого продукта. Масс-спектр 374,3 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 7,95-8,05 (1Н, дд), 1,45-7,55 (1H, с), 7,1-7,25 (1Н, м), 7,0-7,1 (1Н, м), 6,9-7,0 (1Н, м), 5,1-5,3 (1Н, м), 4,1-4,4 (1Н, д широкий), 4,0-4,15 (1Н, м), 3,8-4,0 (1Н, м), 3,85-3,9 (3Н, с), 3,5-3,7 (2Н, т), 3,2-3,5 (1Н, т широкий), 3,25-3,25 (1H, с), 1,8-2,2 (2Н, м), 1,35-1,5 (9Н, с).
Стадия 13.f. 7-((1,1-диметилэтокси)карбонил)-2-(2-метоксифенил)-5,6,7,8-тетрагидро-8-(2-((трифенилметил)тио)этил)-имидазо[1,2а]пиразин
Трифенилфосфин (4,08 г, 15.5 ммоль) растворяют в ТГФ (25 мл), охлаждают до около 0°С в атмосфере N2 и по каплям добавляют диэтилазодикарбоксилат (2,45 мл, 15,54 ммоль) таким образом, чтобы температура реакционной смеси сохранялась на уровне <3°С. Перемешивание продолжают в течение 1/2 часа при температуре 0°С. По каплям добавляют смесь трифенилметантиола (4.30 г, 15,54 ммоль) и продукта со стадии 13.е. (2,90 г, 7,77 ммоль) в ТГФ (25 мл). Полученную смесь перемешивают в течение около 1 часа при температуре около 0°С и затем нагревают до комнатной температуры. Смесь концентрируют при пониженном давлении, и остаток растворяют в простом эфире (40 мл) и выстаивают в течение ночи. Твердое вещество отфильтровывают и фильтрат концентрируют, затем очищают флэш-хроматографией на силикагеле, используя 4:1/гексан:EtOAc и затем 4:1/гексан:EtOAc в качестве элюентов. Фракции продукта объединяют и концентрируют при пониженном давлении с получением бледно-желтой пены (5,49 г, 111%), которую используют без дальнейшей очистки. Масс-спектр 632,4 МН+.
Стадия 13.g. 2-(2-метоксифенил)-5,6,7,8-тетрагидро-8-(2-((трифенилметил)тио)этил)имидазо-[1,2 а]пиразин
Продукт со стадии 13.f. (2,50 г, 3,96 ммоль) растворяют в CH2Cl2 (16,0 мл) и обрабатывают ТФА (4,0 мл) при комнатной температуре в атмосфере N2 в течение около 3,5 часов. Реакционную смесь затем осторожно выливают в насыщенный раствор Na-НСО3 (150 мл) и продукт экстрагируют CH2Cl2 (2×50 мл), сушат над Na2SO2, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле, используя 9:1/гексан:EtOAc и затем 100% EtOAc в качестве элюентов. Фракции продукта объединяют и концентрируют при пониженном давлении с получением белой пены (1,35 г, 64%), которую используют без дальнейшей очистки. Масс-спектр 532,4 MH+.
Стадия 13.h. 7-(2-(((1,1-диметилэтокси)карбонил)амино)-1-оксо-3-((трифенилметил)тио)пропил)-2-(2-метоксифенил)-5,6,1,8-тетрагидро-8-(2-((трифенилметил)тио)этил)имидазо-[1,2а]пиразин
Смесь продукта со стадии 13.g. (соединение xxi, в котором R2, R3, R5, R6 и R7 являются Н, R4 является 2-метоксифенилом и n равно 1) (1,30 г, 2,45 ммоль), Boc-(L)-Cys(Trt)-OH (1,14 г, 2,45 ммоль), NMM (270 мкл, 2,45 ммоль) и HOAt (333 мг, 2,45 ммоль) в ТГФ (20 мл) обрабатывают EDC (470 мг, 2,45 ммоль) при комнатной температуре в атмосфере N2. Реакционную смесь перемешивают в течение ночи и затем концентрируют при пониженном давлении. Добавляют насыщенный раствор NaHCO3 (25 мл) и продукт экстрагируют 3:2 смесью гексан:EtOAc. Экстракты вводят в колонку с силикагелем и продукт элюируют 3:2/гексаном:EtOAc. Фракции продукта объединяют и концентрируют при пониженном давлении с получением белой пены (2,31 г, 97%), которую используют без дальнейшей очистки. Масс-спектр 977,6 МН+.
Стадия 13.i. 7-(2-амино-1-оксо-3-тиопропил)-8-(меркапто-этил)-2-(2-метоксифенил)-5,6,1, 8-тетрагидроимидазо-[1,2а]пиразин
Раствор продукта со стадии 13.h. (2,25 г, 2.31 ммоль) (соединение xxii, в котором каждый из R1, R2, R3, R5, R6 и R7 является Н, R4 является 2-метоксифенилом, Y является =O и n равно 1) в CH2Cl2 (16,0 мл) обрабатывают ТФА (4,0 мл) в течение около 1/2 часа. Добавляют (iPr)3SiH (1,42 мл, 6,93 ммоль) и реакционную смесь перемешивают в течение еще 1 часа. Смесь концентрируют при пониженном давлении и затем продукт экстрагируют растиранием с 0,1%-ным раствором ТФА (3×20 мл). Экстракты фильтруют и лиофилизуют с получением 1,40 г (98%) белого твердого вещества с чистотой (ВЭЖХ) около 92%. Неочищенный продукт затем очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 10-30% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта объединяют, концентрируют при пониженном давлении до около 1/2 объема и используют на следующей стадии без дальнейшей очистки. Масс-спектр 393,2 МН+.
Стадия 13.j. 7-(2-амино-1-оксо-3-тиопропил)-8-(меркаптоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин дисульфид
Водный раствор продукта со стадии 13.i. (неочищенный раствор, 0,45 ммоль) разбавляют до 100 мл 0,1% ТФА и нейтрализуют 5%-ным раствором NH4OH. Добавляют метанол (100 мл) для получения гомогенного раствора. Смесь перемешивают в течение ночи, сохраняя рН около 7,2-7,5. Раствор концентрируют до около 25 мл, подщелачивают добавлением чистого NaHCO3 и продукт экстрагируют CH2Cl2 (3×25 мл). Экстракты выливают в колонку с силикагелем и продукт элюируют 90:9:1/СН2Cl2:МеОН:НОАс. Фракции продукта объединяют и концентрируют при пониженном давлении. Остаток растворяют в 0,5%-ном растворе HCl (10 мл) и лиофилизуют и затем повторно лиофилизуют из H2O (10 мл) с получением 43 мг (9%) чистого продукта. Масс-спектр 391,2 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 8,8-9,2(2Н, с широкий), 7,9-8,1 (2Н, м), 7,3-7,5 (1Н, т), 7,15-7,3 (1Н, д), 7,0-7,2 (1Н, т), 6,05-6,25 (1H, м), 4,85-5,0 (1Н, с широкий), 4,7-4,9 (1Н, д), 4,2-4,35 (1Н, д), 4,0-4,2 (1Н, м), 3,9-4,0 (3Н, с), 3,55-3,7 (1Н, д), 3,3-3,55 (1Н, частично перекрытый Н2O пиком), 2,9-3,1 (1Н, т), 2,4-2,8 (4Н, м).
Пример 14: 5-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Стадии 14.а.-14.d. проводят согласно схеме 1, стадии 1.e.-1.f. Используют Cbz-(Gly)-ОН вместо Cbz-(L)-Nle-ОН на стадии 14.а. и этил 2-бромгексаноат вместо этилбромацетата на стадии 14.b. Стадию 14.е. проводят согласно схеме 1, стадия 1.g., заменяя продуктом со стадии 11.j. продукт со стадии 1.b. Масс-спектр 479,3 MH+.
Пример 15: 6-бутил-7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2 а]пиразин
Соединение из примера 15 получают аналогично примеру 14 за исключением того, что стадии 1.d., 1.e. и 1.f. заменяют на стадии 15.а. и 15.b.
Схема 15
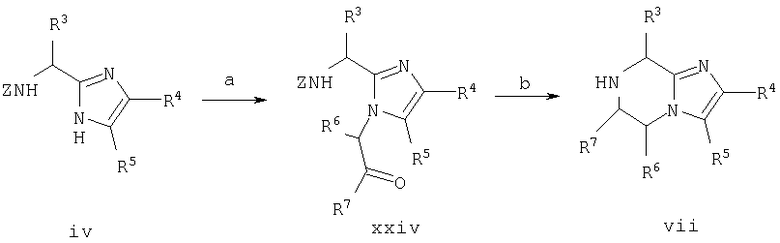
15. а. R7COCHBr6/K2СО3/ДМФ
15.b. H2/Pd на угле/НОАс
Стадия 15.а. 1-(2-оксогексил)-2-(1-(((фенилметокси) карбонил)амино)метил)-4-(2-метоксифенил)имидазол
1-H-2-(1-(((фенилметокси)карбонил)амино)метил)-4-(2-метоксифенил)имидазол (соединение iv, в котором R3 и R5 являются Н и R4 является 2-метоксифенилом) (790 мг, 2.34 ммоль), 1-хлор-2-гексанон (473 мг, 3,51 ммоль) и K2СО3 (469 мг, 4,68 ммоль) объединяют в ДМФ (4 мл) и перемешивают при комнатной температуре в течение около 42 часов. Реакционную смесь разбавляют насыщенным NaHCO3 (25 мл) и экстрагируют Et2O (2×50 мл). Объединенные экстракты Et2O сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают флэш-хроматографией на силикагеле, используя 3% MeOH/CH2Cl2 в качестве элюента. Фракции чистого продукта объединяют и концентрируют при пониженном давлении с получением бледно-желтого масла (650 мг, 64%), которое затвердевает при выстаивании. Масс-спектр 463,3 МН+.
Стадия 15.b. 6-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 15.а. (соединение xxiv, в котором R3, R5 и R6 являются Н, R5 является 2-метоксифенилом и R7 является н-бутилом) (650 мг, 1,49 ммоль) растворяют в НОАс (25 мл), содержащем 10% Pd на угле (65 мг) и смесь гидрируют под давлением Hz 2,109 кг/см2 (30 фунт/кв.дюйм) в течение около 6 часов. Катализатор удаляют фильтрацией через диатомовую землю и фильтрат концентрируют при пониженном давлении с получением бледно-желтого масла, которое кристаллизуется при выстаивании (430 мг, 101%).
Стадия 15.с. 6-бутил-7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
Соединение примера 15 получают на стадии 15.с. по методике стадии 1.д., заменяя продуктом со стадии 15.b. продукт со стадии 1.f. Масс-спектр 509,3 MH+.
Пример 16: 6-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-фенил-5,6,1,8-тетрагидроимидазо[1,2а]пиразин
Соединение примера 16 получают по методике примера 15, используя в качестве исходного соединения 2-бромацетофенон вместо 2-бром-2'-метоксиацетофенона на стадии 1.c. Масс-спектр 509,4 MH+.
Пример 17: 5-бутил-7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин
Соединение примера 17 получают по методике примера 14, используя в качестве исходного соединения 2-бромацетофенон вместо 2-бром-2'-метоксиацетофенона на стадии 1.e. Масс-спектр 509, 3 MH+.
Пример 18: 7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Стадии 18.а.-18.d. проводят аналогично схеме 1, стадии 1.c.-1.f. Используют Cbz-(L)-циклогексилаланин вместо Cbz-(L)-Nle-OH на стадии 18.а. Стадию 18.е. проводят согласно схеме 1, стадия 1.g., заменяя продуктом со стадии 11.j. продукт со стадии 1.b. Масс-спектр 549,4 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,15-9,25 (1Н, с), 8,05-8,15 (1H, д), 8,0-8,1 (1Н, с), 7,8-7,9 (2Н, д), 7,6-7,7 (1Н, с), 7,5-7,6 (2Н, д), 7,4-7,5 (1Н, т), 7,15-7,3 (1Н, д), 7,05-7,15 (1H, т), 5,85-6,05 (1H, дд), 5,5-5,6 (2Н, с), 4,1-4,5 (5Н, м), 3,9-4,1 (3Н, с), 3,75-3,9 (1Н, м), 1,85-2,1 (3Н, м), 1,4-1,8 (41-1, м), 0,8-1,4 (7Н, м).
Соединение 19: 5-бутил-7-(2-(1Н-имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Соединение примера 19 получают по методике примера 4, используя 5-бутил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин (соединение vii, в котором R3, R5 и R7 являются Н, R4 является 2-метоксифенилом и R6 является н-бутилом), как описано в примере 14, вместо 2-(2-метоксифенил)-8-(1-метил-пропил)-5,6,7,8-тетрагидроимидазо[1,2 а]пиразина на стадии 19.е. Масс-спектр 394,3 MH+.
Пример 20: 7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил) -2-(2-фенилметокси)фенил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин
Исходный материал для стадии 20.а. получают аналогично схеме 1, стадии 1.с.-1.f., используя Cbz-(Gly)-OH вместо Cbz-(L)-Nle-ОН. Стадию 20.в. проводят согласно схеме 1, стадия 1.g., заменяя продуктом со стадии 11.j. продукт со стадии 1.b.
Схема 20
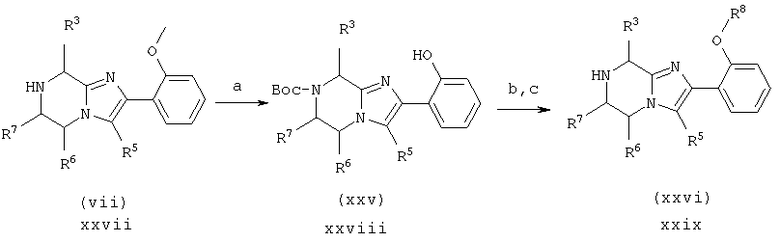
20.а. BBr3/СН2Cl2/гексан, затем (Вос)2O/NaOH/ТГФ/H2О
20.b. NaH/R8-Br/ДМФ
20.с. ТФА/iPr3SiH
Стадия 20.а. 7-((1,1-диметилэтокси)карбонил)-2-(2-гидрокси-фенил)-5,6,1,8-тетрагидроимидазо[1,2а]-пиразин
2-(2-метоксифенил)-5,6,1,8-тетрагидроимидазо[1,2а]пиразин (соединение vii, в котором R3, R5, R6 и R7 являются Н и R4 является 2-метоксифенилом) (5,30 г, 23,1 ммоль) растворяют в СН2Cl2 (100 мл) и по каплям добавляют на протяжении около 15 минут к смеси 1М BBr3/гексана (80,3 мл, 80,3 ммоль) в CH2Cl2 (500 мл) при температуре около 0°С. Смесь нагревают до комнатной температуры и перемешивают в течение ночи. Деметилированное промежуточное соединение экстрагируют Н2О (3×240 мл) и объединенные водные слои промывают Et2O (100 мл). Раствор доводят до рН около 8 добавлением 2,5N NaOH и добавляют ди-трет-бутилдикарбонат (5,55 г, 25,4 ммоль) в ТГФ (200 мл). Раствор энергично перемешивают в течение около 2 часов, сохраняя рН раствора 8,0-8,5. Смесь концентрируют до около 700 мл и экстрагируют СН2Cl2 (2×100 мл). Объединенные слои СН2Cl2 сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением продукта (7,10 г, 97%) в виде рыжевато-коричневого твердого вещества. Масс-спектр 316,2 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 7,65-7,7 (1H, с), 7,6-7,7 (1Н, дд), 7,0-7,15 (1Н, м), 6,75-6,9 (2Н, м), 4,6-4,7 (2Н, с), 4,0-4,2 (2Н, т), 3,75-3,9 (2Н, т), 3,3-3,4 (H2O), 1,4-1,55 (9Н, с).
Стадия 20.b. 7-((1,1-диметилэтокси)карбонил)-2-(фенилметокси)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
60%-ную дисперсию NaH в минеральном масле (76 мг, 1,9 ммоль) промывают гексаном (2×5,0 мл) и добавляют раствор 2-(2-гидроксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразина со стадии 9.а. (500 мг, 1,58 ммоль) в ДМФ (10 мл) при комнатной температуре. Реакционную смесь перемешивают в течение около 10 минут и затем добавляют бензилбромид (188 мкл, 1,58 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение около 2 часов. Смесь выливают в насыщенный раствор NaCl (40 мл) и экстрагируют Et2O (2×50 мл). Слои Et2O объединяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением продукта (430 мг, 67%) в виде вязкого желтого масла, которое кристаллизуется при выстаивании. Масс-спектр 406,3 МН+.
Стадия 20.с. 2-(2-(фенилметокси)фенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин
Продукт со стадии 20.b. (250 мг, 0.62 ммоль) обрабатывают ТФА (15,0 мл), содержащим iPr3SiH (505 мкл, 2,47 ммоль) при комнатной температуре в течение около 1 часа в атмосфере N2. Растворители удаляют при пониженном давлении и остаток растирают с Et2O (2×20 мл), фильтруют и сушат. Остаток используют на следующей стадии без дальнейшей очистки.
Стадия 20.d. 7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксо-этил)-2-(2-фенилметокси)фенил)-5,6,7,8-тетрагидроимидазо[1,2a]-пиразин
Соединение примера 20 получают из продукта со стадии 20.d. и продукта со стадии 11.j. по методике стадии 1.д. Масс-спектр 529,3 МН+.
Пример 21: 2-(2-бутоксифенил)-7-(2-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-5,6,1, 8-тетрагидроимидазо[1,2а]-пиразин
Соединение примера 21 получают по методике примера 20, используя н-бутилиодид вместо бензилбромида на стадии 20.b. Масс-спектр 495,4 MH+.
Пример 22: 1,2-дигидро-1-((1Н-имидазол-4-ил)метил)-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин
Исходный материал для стадии 22.а. получают аналогично схеме 1, стадия 1.c., используя Cbz-(Gly)-ОН вместо Cbz-(L)-Nle-ОН. Стадии 22.d. и 22.с. проводят по методике стадий 8.с. и 8.d..
Схема 22
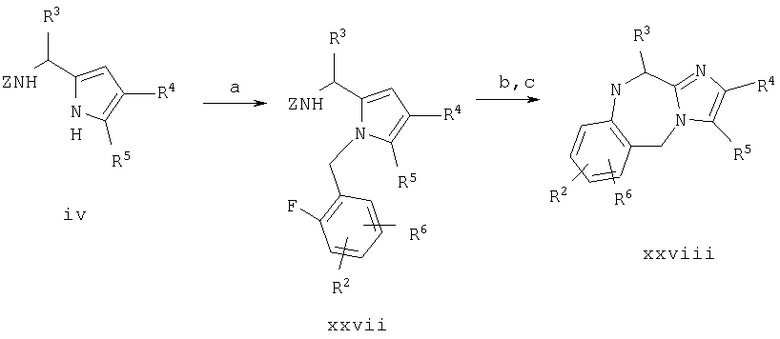
22.а. 2-фторбензилбромид/K2СО3/ДМФ
22.b. HBr/HOAc
22.с. NMP (кипячение с обратным холодильником)
Стадия 22.а. 1-((2-фторфенил)метил)-2-(1-(S)-(((фенилметокси)карбонил)амино)метил)-4-(2-метоксифенил)имидазол
1Н-2-(1-(S)-(((фенилметокси)карбонил)амино)метил)-4-(2-метоксифенил)имидазол (1,69 г, 5,00 ммоль) растворяют в ДМФ (20 мл) и обрабатывают K2СО3 (1,38 г, 10,0 ммоль) и 2-фторбензил-бромидом (1,21 мл, 10,0 ммоль) и смесь нагревают до температуры около 50°С в течение около 2 часов. Смесь концентрируют при пониженном давлении. Остаток помещают в EtOAc (50 мл), промывают один раз насыщенным раствором NaHCO3 (25 мл) и один раз насыщенным раствором NaCl (25 мл). Слой EtOAc сушат над Na2SO4, фильтруют и концентрируют до масла (2,22 г, 99,6%), которое кристаллизуется при выстаивании. Масс-спектр 446,2 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 8,05-8,15 (1H, д), 8,75-8,9 (1Н, т), 7,5-7,6 (1Н, с), 7,1-7,5 (9Н, м), 6,9-7,1 (3Н, м), 5,3-5,45 (2Н, с), 4,9-5,1 (2Н, с), 4,3-4,45 (2Н, с), 3,8-3,9 (3Н, с).
Стадия 22.b. 2-(аминометил)-1-((2-фторфенил)метил)-4-(2-метоксифенил)имидазол
1-((2-фторфенил)метил)-2-(1-(S)-((фенилметокси)карбонил)-амино)метил)-4-(2-метоксифенил)имизадол (соединение xxvi, в котором R2, R3, R5 и R6 являются Н и R4 является 2-метоксифенилом) (2,22 г, 4,99 ммоль) растворяют в 30% HBr/НОАс (25 мл) и реакционную смесь перемешивают в течение около 1 часа при комнатной температуре. Добавляют Et2O (100 мл) и полученную суспензию перемешивают в течение около 15 минут и продукт отфильтровывают и промывают Et2O (100 мл). Продукт нейтрализуют добавлением насыщенного раствора NaHCO3 (50 мл) и продукт экстрагируют CH2Cl2 (2×25 мл). Объединенные слои СН2Cl2 объединяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении до вязкого масла (1,26 г, 81%), которое затвердевает при выстаивании. Масс-спектр 312,2 МН.
Стадия 22.с. 1,2-дигидро-4-(2-метоксифенил)имидазо[1,2-с][1,4]бензодиазепин
2-(аминометил)-1-((2-фторфенил)метил)-4-(2-метоксифенил)-имидазол со стадии 22.b. (1,26 г, 4,05 ммоль) добавляют к NMP (20 мл), содержащему K2СО3 (1,12 г, 8,10 ммоль) и реакционную смесь кипятят с обратным холодильником в течение около 11 часов. Растворители отгоняют при пониженном давлении и добавляют Н2O (50 мл). Неочищенный продукт отфильтровывают и очищают флэш-хроматографией на силикагеле, используя CH2Cl2 и затем 19:1/МеОН:СН2Cl2 в качестве элюентов. Фракции продукта объединяют и концентрируют при пониженном давлении и затем растирают с EtOAc с получением продукта в виде рыжевато-коричневого твердого вещества (329 мг, 28%). Масс-спектр 292,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 7,95-8,1 (1Н, дд), 7,6-7,65 (1H, с), 7,1-7,2 (1Н, м), 6,9-7,1 (4Н, м), 6,5-6,65 (2Н, м), 5,2-5,4 (2Н, с), 4,4-4,5 (2Н, с), 3,8-4,0 (3Н, с).
Стадия 22.d. 1,2-дигидро-4-(2-метоксифенил)-1-((1-(трифенил-метил)имидазол-4-ил)метил)имидазо[1,2-с][1,4]бензодиазепин
По методике стадии 8.с. смесь продукта со стадии 22.с. (соединение xxvii, в котором R2, R3, R5 и R6 являются Н и R4 является 2-метоксифенилом) (146 мг, 0.50 ммоль) и 1-трифенилметилимидазол-4-карбоксальдегида (со стадии 8.b.) (338 мг, 1,00 ммоль) в CH2Cl2 (5,0 мл) обрабатывают уксусной кислотой (1,0 мл) и NaBH(OAc)2 (212 мг, 1,00 ммоль) в течение около 1 часа. Добавляют еще 1-трифенилметилимидазол-4-карбоксальдегида (338 мг, 1,00 ммоль), уксусной кислоты (1,0 мл) и NaBH(OAc)2 (212 мг, 1,00 ммоль) и смесь перемешивают в течение около 1 часа. Добавляют EtOAc (25 мл) и концентрированный NH4OAc (3,0 мл) и смесь перемешивают в течение около 1,5 часов. Добавляют насыщенный раствор NaHCO3 и смесь экстрагируют EtOAc (3×25 мл). Объединенные слои EtOAc сушат, фильтруют и концентрируют при пониженном давлении. Продукт очищают флэш-хроматографией на силикагеле, используя 1:1:1/СН2Cl2:EtOAc:гексан, и затем EtOAc в качестве элюента. Объединенные фракции продукта концентрируют и сушат до 210 мг (68%) при пониженном давлении. Масс-спектр 614,3 MH+.
Стадия 22.е. 1,2-дигидро-4-(2-метоксифенил)-1-((1-(трифенилметил)имидазол-4-ил)метил)имидазо[1,2-с][1,4]бензодиазепин
У продукта со стадии 22.d. снимают защиту по методике стадии 1.h. с получением 104 мг (70%) продукта. Масс-спектр 372,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,0-9,2 (1Н, с), 8,05-8,15 (1Н, с), 7,9-8,0 (1Н, д), 7,7-7,8 (1Н, с), 7,3-7,55 (4Н, м), 7,1-7,3 (2Н, м), 7,0-7,1 (1Н, т), 5,6-5,8 (2Н, с), 4, 55-4,75 (4Н, с), 3,9-4,1 (3Н, с), 2,7-4,0 (Н2O, широкий).
Пример 23: 1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин
Соединение примера 23 получают согласно схеме 22 и схеме 11.
Стадия 23.а. 1-(2-((4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин
1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин (соединение xxvii, в котором R2, R3, R5 и R6 являются Н и R4 является 2-метоксифенилом) (со стадии 22.с.) объединяют с DCC (103 мг, 0,50 ммоль), HOAt (68 мг, 0,50 ммоль), NMM (55 мкл, 0,50 ммоль) и 1-(4-цианофенилметил)-5-имидазолуксусной кислотой (продукт со стадии 11.j.) (160 мг, 0,50 ммоль) в ДМФ (3,0 мл) и реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь нагревают до температуры около 70°С и затем концентрируют при пониженном давлении. Неочищенный материал очищают препаративной ВЭЖХ на колонке RAININ™C18, осуществляя первое пропускание с использованием градиента 10-40% СН3CN/0,1% ТФА на протяжении 45 минут, далее, второй проход с использованием градиента 20-35% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта объединяют и концентрируют досуха. Продукт превращают в дигидрохлорид пропусканием 9:1/Н2O:МеОН раствора через ионообменную колонку (AG 1 Х2, 200-400 меш, 100 мл слой, 0,6 мэкв/мл, хлорид, Biorad, Inc., Hercules, CA). Фракции продукта опять концентрируют при пониженном давлении и лиофилизуют из H2O (10 мл) с получением 21 мг (10%) желаемого продукта. Масс-спектр 515,3 MH+.
Пример 24: 9-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с]-[1,4]-бензодиазепин
Соединение примера 24 получают согласно схеме 22, схеме 24 и схеме 1, стадия 1.g.
Схема 24
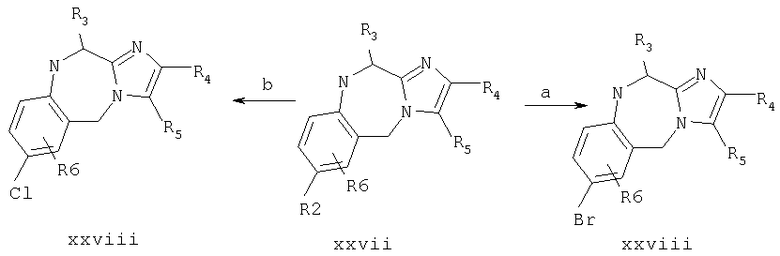
Стадия 24.а. 9-бром-1,2-дигидро-4-(2-метоксифенил)-1Н-имидазо[1,2с][1,4]-бензодиазепин
Продукт со стадии 22.с. (291 мг, 1,00 ммоль) растворяют в уксусной кислоте (10 мл) и по каплям добавляют Br2 (52 мкл) при перемешивании. Добавляют метанол (10 мл) для сохранения раствора гомогенным. По завершении добавления растворители выпаривают при пониженном давлении и остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 20-50% СН3CN/0,1% ТФА на протяжении 45 минут с УФ определением при 254 нм. Фракции продукта концентрируют до 1/2 объема и лиофилизуют. Лиофилизованный продукт превращают в гидрохлорид пропусканием 20% метанольного раствора через ионообменную колонку (BioRad AG 1 Х2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют и лиофилизуют из Н2O с получением чистого продукта (110 мг, 24%) в виде дигидрохлорида. Масс-спектр 370,0 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 8,05-8,15 (1H, с), 7,9-8,0 (1Н, д), 7,4-7,5 (1Н, т), 7,3-7,4 (1Н, с), 7,2-7,3 (1Н, дд), 7,15-7,25 (1H, д), 7,05-7,15 (1H, т), 6,65-6,75 (1Н, д), 5,55-5,7 (2Н, с), 4,8-4,95 (2Н, с), 3,9-4,0 (3Н, с).
Стадия 24.b. 9-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо-[1,2с][1,4]-бензодиазепин
Продукт со стадии 24.а. (72 мг, 0,177 ммоль) растворяют в ДМФ (3 мл). К полученному раствору добавляют NMM (98 мкл, 0,89 ммоль), 1-(4-цианофенилметил)-5-имидазолуксусную кислоту (продукт со стадии 11.j.) (128 мг, 0,36 ммоль), HOAt (49 мг, 0,36 ммоль) и DCC (74 мг, 0,36 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре и затем нагревают до температуры 70°С в течение одного часа. Реакционную смесь нагревают при температуре 70°С в течение 1 часа и концентрируют при пониженном давлении. Остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 25-60% СН3CN/0,1% ТФА на протяжении 45 минут с УФ определением при 254 нм. Требуется вторая очистка с использованием градиента 44-80% СН3CN/0,2% NH4OAc на протяжении 45 минут. Фракции продукта концентрируют и лиофилизуют с получением чистого продукта (11 мг, 9%) в виде ацетата. Масс-спектр 593,2, 595,3 МН+.
Пример 25: 9-хлор-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин
Соединение примера 25 получают согласно схеме 22, схеме 24 и схеме 1, стадия 1.g.
Стадия 25.а. 9-хлор-1,2-дигидро-4-(2-метоксифенил)-1Н-имидазо[1,2с][1,4]-бензодиазепин
Продукт со стадии 22.с. (291 мг, 1,00 ммоль) превращают в дигидрохлорид растворением в метаноле (10 мл), добавляя 5% насыщенную HCl (3,0 мл) и концентрируя до получения твердого вещества. Остаток растворяют в метаноле (5 мл) и добавляют НОАс (1,0 мкл) и N-хлорсукцинимид (133 мг, 1,0 ммоль). Смесь перемешивают при комнатной температуре в течение ночи и концентрируют при пониженном давлении. Остаток распределяют между насыщенным NaHCO3 и CHCl3 и слой CHCl3 вводят в колонку с силикагелем. Колонку элюируют сначала 3:1/CHCl3:EtOAc, затем 1:1/CHCl3:EtOAc с получением неочищенного продукта. Неочищенный материал затем очищают препаративной ВЭЖХ на колонке RAININ™C18 используя градиент 25-60% СН3CN/0,1% ТФА на протяжении 45 минут с УФ определением при 254 нм. Фракции чистого продукта объединяют и лиофилизуют с получением 94 мг (17%) продукта в виде ТФА-соли. Масс-спектр 326,2 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 8,0-8,1 (1Н, с), 7,7-7,8 (1Н, дд), 7,4-7,5 (1Н, т), 7,2-7,3 (1Н, с), 7,15-7,3 (1Н, д), 7,1-7,2 (1Н, дд), 7,05-7,15 (1H, т), 6,7-6,8 (1Н, д), 5,55-5,65 (2Н, с), 4,7-4,8 (2Н, с), 3,9-4,0 (3Н, с).
Стадия 25.b. 9-хлор-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо-[1,2с][1,4]-бензодиазепин
Продукт со стадии 25.а. (72 мг, 0.13 ммоль) растворяют в ДМФ (1,5 мл). К этому раствору добавляют NMM (114 мкл, 1,04 ммоль), 1-(4-цианофенилметил)-5-имидазолуксуную кислоту (соединение xii, в котором R1 является (4-цианофенилметилом) и R2 является Н) (92 мг, 0,26 ммоль) и TFFH (69 мг, 0,26 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре и затем нагревают до температуры 70°С в течение одного часа. Раствор охлаждают до комнатной температуры и добавляют 1-(4-цианофенилметил)-5-имидазолуксусную кислоту (продукт со стадии 11.j.) (92 мг, 0,26 ммоль) и TFFH (60 мг, 0,26 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре, затем концентрируют при пониженном давлении. Остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 25-60% СН3CN/0,1% ТФА на протяжении 45 минут с УФ определением при 254 нм. Фракции продукта концентрируют и лиофилизуют. Лиофилизованный продукт превращают в гидрохлорид пропусканием 30% метанольного раствора через ионообменную колонку (BioRad AG 1-X2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют и лиофилизуют из H2O с получением чистого продукта (59 мг, 73%) в виде дигидрохлорида. Масс-спектр 594,3 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,2-9,3 (1Н, с), 8,0-8,1 (1Н, с), 7,9-8,1 (1Н, д), 7,8-7,9 (2Н, д), 7,7-7,85 (2Н, м), 7,6-7,75 (1Н, дд), 7,5-7,7 (1Н, с), 7,4-7,55 (2Н, д), 7,35-7,5 (1Н, т), 7,15-7,3 (1Н, д), 7,0-7,15 (1Н, т), 4,5-6,3 (2Н, дд), 5,4-5,8 (4Н, м), 3,9-4,05 (3Н, с), 3,5-3,9 (2Н, дд).
Пример 26: 10-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с]-[1,4]бензодиазепин
Стадии 26.а.-26.с. проводят по методике стадий 22.а.-22.с., используя 4-бром-2-фторбензилбромид вместо 2-фторбензил бромида на стадии 26.а. Продукт со стадии 26.с. выделяют в виде дигидрохлорида перемешиванием метанольной суспензии с 20% избытком концентрированной соляной кислоты и фильтрованием полученного твердого вещества. Масс-спектр 370,1, 372,1 МН+.
Стадия 26.d. 10-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин
Продукт со стадии 26.с. (100 мг, 0.23 ммоль) суспендируют в ДМФ (1.5 мл) и добавляют продукт со стадии 11.j. (125 мг, 0,45 ммоль), TFFH (119 мг, 0,45 ммоль) и NMM (110 мкл, 1,0 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение около 45 минут и выстаивают при комнатной температуре в течение ночи. Неочищенную смесь концентрируют при пониженном давлении и остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 20-50% СН3CN/0,1% ТФА на протяжении 45 минут с УФ определением при 254 нм. Проводят вторую очистку, используя изократную систему, содержащую 44% СН3CN/0,2% водный NH4OAc. Фракции продукта концентрируют и лиофилизуют. Лиофилизованный продукт повторно лиофилизуют из 20% СН3CN/Н2O, затем превращают в ТФА соль лиофилизацией из 20% СН3CN/1% ТФА и затем превращают в гидрохлорид пропусканием 30% метанольного раствора через ионообменную колонку (BioRad AG 1-X2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют и лиофилизуют из 20% CH2CN/H2O с получением чистого продукта (45 мг, 30%) в виде дигидрохлорида. Масс-спектр 593,2, 595,2 MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,1-9,3 (1Н, с), 7,9-8,1 (3Н, м), 7,8-7,9 (2Н, д), 7,7-7,8 (1Н, дд), 7,6-7,7 (1Н, д), 7,5-7,6 (1Н, с), 7,4-7,5 (2Н, д), 7,3-7,45 (1Н, т), 7,1-7,25 (1Н, с), 7,0-7,1 (1Н, т), 4,4-6,2 (2Н, дд), 5,4-5,7 (4Н, м), 3,9-4,0 (3Н, с), 3,5-3,9 (2Н, дд).
Пример 27: 1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин
Стадии 27.а.-27.с. проводят по методике стадий 22.а.-22.с., используя 2,6-дифторбензилбромид вместо 2-фторбензилбромида на стадии 27.а.
Стадия 27.d. 1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо-[1,2с][1,4]бензодиазепин
Продукт со стадии 21.с. (124 мг, 0.40 ммоль) суспендируют в ДМФ (2 мл) и добавляют продукт со стадии 11j. (111 мг, 0,40 ммоль), TFFH (106 мг, 0,40 ммоль) и NMM (88 мкл, 0,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение около 1,5 часов. Добавляют вторую порцию продукта со стадии 11.j. (111 мг, 0,40 ммоль), TFFH (106 мг, 0,40 ммоль) и NMM (88 мкл, 0,8 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Неочищенную смесь концентрируют при пониженном давлении и остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 20-50% СН3CN/0,1% ТФА на протяжении 45 минут с УФ определением при 254 нм. Фракции продукта объединяют и лиофилизуют. ТФА соль превращают в гидрохлорид пропусканием 40% метанольного раствора через ионообменную колонку (BioRad AG 1-Х2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют и лиофилизуют из Н2O с получением чистого продукта (126 мг, 59%) в виде дигидрохлорида. Масс-спектр 533,3, МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,2-9,3 (1Н, с), 8,2-8,4 (1Н, с), 7,9-8,1 (1Н, д), 7,8-7,9 (2Н, д), 7,55-7,7 (3Н, м), 7,35-7,55 (4Н, м), 7,15-7,25 (1H, д), 7,0-7,15 (1Н, т), 4,5-6,3 (2Н, дд), 5,5-5,7 (2Н, дд), 3,9-4,1 (3Н, с), 3,6-4,0 (2Н, дд).
Пример 28: 1,2-дигидро-1-(2-(имидазол-1-ил)-1-оксоэтил)-4-(2-метоксифенил)имидазо[1,2a][1,4]бензодиазепин
Раствор продукта со стадии 22.с. (146 мг, 0,50 ммоль) в ДМФ (2 мл) обрабатывают хлорацетилхлоридом (44 мкл, 0,55 ммоль) в ДМФ (0,5 мл) и смесь перемешивают при комнатной температуре в течение 1/2 часа. Добавляют имидазол (204 мг, 3,00 ммоль) и смесь нагревают до температуры 50°С в течение 3 часов и затем выстаивают при комнатной температуре в течение ночи. Неочищенную смесь концентрируют при пониженном давлении и остаток очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 10-50% СН3CN/0,1% ТФА на протяжении 25 минут с УФ определением при 254 нм. Фракции продукта объединяют и концентрируют при пониженном давлении. ТФА соль превращают в гидрохлорид пропусканием 30% метанольного раствора через ионообменную колонку (BioRad AG 1-Х2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют до объема около 10 мл и лиофилизуют из H2O с получением чистого продукта (127 мг, 54%) в виде дигидрохлорида. Масс-спектр 400,2, МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,1-9,2 (1Н, с), 8,1-8,2 (1Н, с), 7,9-8,05 (1Н, д), 7,8-7,9 (1Н, д), 7,5-7,8 (5Н, м), 7,35-7,5 (1Н, м), 7,15-7,25 (1Н, д), 7,0-7,15 (1Н, т), 6,2-6,35 (1Н, д), 6,0-6,15 (1Н, д), 5,5-5,7 (1Н, д), 5,35-5,5 (1Н, д), 4,95-5,15 (1Н, д), 4,6-4,8 (1Н, д), 3,9-4,0 (3Н, с).
Пример 29: 1,2-дигидро-4-(2-метоксифенил)-1-(2-(пиридин-3-ил)-1-оксоэтил)имидазо[1,2a][1,4]бензодиазепин
Раствор продукта со стадии 22.с. (102 мг, 0,35 ммоль) в ДМФ (1,5 мл) обрабатывают 3-пиридинуксусной кислоты гидрохлоридом (69,4 мг, 0,40 ммоль), NMM (110 мкл, 1,00 ммоль) и TFFH (106 мг, 0,40 ммоль) и смесь перемешивают при комнатной температуре в течение 14 часов. К реакционной смеси добавляют дополнительное количество 3-пиридинуксусной кислоты гидрохлорида (26,0 мг, 0,15 ммоль), NMM(33 мкл, 0,30 ммоль) и TFFH (40 мг, 0,15 ммоль) и перемешивание продолжают при комнатной температуре в течение 1,5 часов. Неочищенную смесь концентрируют при пониженном давлении. Добавляют насыщенный раствор NaHCO3 (5 мл) и продукт экстрагируют CH2Cl2 (2×5 мл). CH2Cl2 отгоняют при пониженном давлении и остаток растворяют в 10%-ном водном СН3CN рН которого доведен до 2 добавлением ТФА. Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент 10-50% СН3CN/0,1% ТФА на протяжении 25 минут с УФ определением при 254 нм. Фракции продукта объединяют и концентрируют при пониженном давлении. ТФА соль превращают в гидрохлорид пропусканием 30% метанольного раствора через ионообменную колонку (BioRad AG 1-X2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют до объема около 10 мл и лиофилизуют из Н2О с получением чистого продукта (123 мг, 73%) в виде дигидрохлорида. Масс-спектр 411,1, MH+.
Пример 30: 1,2-дигидро-4-(2-метоксифенил)-1-(2-(пиридин-4-ил)-1-оксоэтил)имидазо[1,2a][1,4]бензодиазепин
Соединение примера 30 получают по методике примера 29 за исключением использования 4-пиридинуксусной кислоты гидрохлорида вместо 3-пиридинуксусной кислоты гидрохлорида на стадии 30.а. Масс-спектр 411,1, МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 8,8-8,9 (2Н, д), 8,1-8,2 (1Н, с), 7,9-8,0 (3Н, м), 7,75-7,9 (1Н, д), 7,65-7,75 (1Н, д), 7,6-7,7 (1Н, т), 7,5-7,65 (1Н, т), 7,35-7,5 (1Н, м), 7,15-7,25 (1Н, д), 7,0-7,15 (1Н, т), 6,2-6,4 (1Н, д), 5,7-5,85 (1Н, д), 5,5-5,65 (1Н, д), 4,5-4,7 (1Н, д), 3,7-4,2 (2Н, м), 3,9-4,0 (3Н, с).
Пример 31: 1-(2-(1-бензилимидазол-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2a][1,4]бензодиазепин
Стадии 31.а.-31.с. проводят по методике стадий 22.а.-22.с., используя 2,6-дифторбензилбромид вместо 2-фторбензилбромида на стадии 31.а.
Стадия 31.d. 1-(2-(1-бензилимидазол-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2a][1,4]-бензодиазепин
Продукт со стадии 31.с. сочетают с продуктом со стадии 5.с. (1-фенилметил-5-имидазолуксусная кислота) и очищают по методике стадии 25. b. Масс-спектр 508,5, MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,1-9,3 (1Н, с), 8,2-8,3 (1Н, с), 7,9-8,0 (1Н, дд), 7,2-7,7 (10Н, м), 7,15-7,25 (1Н, д), 7,0-7,15 (1Н, т), 6,1-6,3 (1Н, д), 5,65-5,8 (1Н, д), 5,3-5,6 (3Н, м), 4,45-4,6 (1Н, д), 3,9-4,0 (3Н, с), 3,6-3,9 (2Н, кв).
Пример 32: 1-(2-(1-((4-циано)фенилметил)имидазол-5-ил)-1-оксоэтил)-9,10-дифтор-1,2-дигидро-4-(2-метоксифенил)имидазо-[1,2с][1,4]бензодиазепин
Стадии 32.а.-32.с. проводят по методике стадий 22.а.-22.с., используя 2,4,5-трифторбензилбромид вместо 2-фторбензилбромида на стадии 32.а.
Стадия 32.d. 1-(2-(1-((4-циано)фенилметил)имидазол-5-ил)-1-оксоэтил)-9,10-дифтор-1,2-дигидро-4-(2-метоксифенил)имидазо-[1,2с] [1,4]бензодиазепин
Продукт со стадии 32.с. сочетают с продуктом со стадии 11.j. (1-(цианофенилметил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 551,4, MH+. ЯМР (300 МГц, ДМСО-d6, 30°С) 9,1-9,3 (1Н, с), 7,9-8,1 (3Н, м), 7,75-7,9 (3Н, м), 7,55-7,65 (1H, с), 7,45-7,55 (2Н, д), 7,35-7,45 (1Н, м), 7,15-7,25 (1H, д), 7,0-7,15 (1Н, т).
Пример 33: 4-(2-бромфенил)-1-(2-(1-[(4-циано)фенилметил]-имидазол-5-ил)-1-оксоэтил)-1,2-дигидро-8-фторимидазо-[1,2a][1,4]бензодиазепин
Стадия 33.а. 2,2'-дибромацетофенон
Бром (40,3 г, 0.25 моль) по каплям добавляют к раствору 2'-бромацетофенона (50,0 г, 0.25 моль) в уксусной кислоте (500 мл) на протяжении 1,5 часов при температуре 15-20°С. Затем раствор нагревают до комнатной температуры и концентрируют при пониженном давлении с получением неочищенного продукта, который используют без дальнейшей очистки.
Стадию 33.b. проводят аналогично стадии 1.c., используя продукт со стадии 33.а. вместо 2-бром-2'-метоксиацетофенона и Cbz-Глицин вместо Cbz-(L)-Норлейцина.
Стадии 33.с.-33.е. проводят по методике стадий 22.а.-22.с., используя 2,6-дифторбензилбромид вместо 2-фторбензил-бромида на стадии 33.с. и 1,8-диазабицикло[5.4.0]ундек-7-ен вместо K2СО3 на стадии 33.е.
Стадия 33.f. 4-(2-бромфенил)-1-(2-(1-[(4-циано)фенилметил]-имидазол-5-ил)-1-оксоэтил)-1,2-дигидро-8-фторимидазо-[1,2a][1,4]бензодиазепин
Продукт со стадии 33.е. сочетают с продуктом со стадии 11.j. (1-(цианофенилметил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 581,1, 583,1 MH+.
Пример 34: 1-(2-(1-((4-циано)фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-10-фтор-4-(2-метоксифенил)имидазол-[1,2a][1,4]бензодиазепин
Стадии 34.а.-34.с. проводят по методике стадий 22.а.-22.с., используя 2.4-дифторбензилбромид вместо 2-фтобензилбромида на стадии 31.а.
Стадия 34.d. 1-(2-(1-((4-циано)фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-10-фтор-4-(2-метоксифенил)имидазол-[1,2a][1,4]бензодиазепин
Продукт со стадии 34.с. сочетают с продуктом со стадии 11.j. (1-(цианофенилметил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 533,2, МН+.
Пример 35: 1-(2-(1-((4-циано-3-метокси)фенилметил)-имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)-имидазол[1,2a][1,4]бензодиазепин
Стадии 35.а.-35.f. проводят согласно схеме 35.
Схема 35
а. МеОН/HCl
b. NaNO2/водн. HCl/KI
с. Zn(CN)2/(Ph3Р)4Pd/ДМФ
d. MeI/NaH/ДМФ
е. LiBH4/ТГФ
f. SOBr2/CH2Cl2
Стадия 35.а. Метил 4-амино-3-гидроксибензоат
Раствор 4-амино-3-гидроксибензойной кислоты (24,5 г, 159 ммоль) в метаноле (650 мл) охлаждают на ледяной бане и обрабатывают газообразным HCl в течение около 20 минут (около 60 г). Перемешивание продолжают в течение ночи и затем раствор концентрируют при пониженном давлении. Остаток растирают с EtOAc (200 мл) и сушат с получением коричневого твердого вещества (24,5 г, 92%), которое используют без дальнейшей очистки. Масс-спектр 168,2, ЯМР (300 МГц, ДМСО-d6, 30°С) 9,3-9,4 (1Н, с), 7,2-7,3 (2Н, м), 6,55-6,65 (1H, д), 5,3-5,4 (2Н, с (широкий)), 3,6-3,8 (3Н, с).
Стадия 35.b. метил 3-гидрокси-4-йодбензоат
Раствор продукта со стадии 35.а. (24,5 г, 146 ммоль) в ТГФ (77 мл) разбавляют 3N HCl (232 мл) и охлаждают на ледяной бане до температуры 8°С с образованием осадка. Добавляют NaNO2 (11,1 г, 161 ммоль) в H2O (75 мл) на протяжении 6 минут при температуре ледяной бани. Перемешивание продолжают в течение 25 минут и затем добавляют раствор KI (97,1 г, 585 ммоль) в H2O (75 мл) одной порцией и перемешивают в течение 15 минут. Добавляют EtOAc (550 мл) и слои разделяют. Слой EtOAc промывают Н2О (500 мл) и насыщенным раствором соли (550 мл), сушат над Na2SO2, фильтруют и концентрируют с получением черного твердого вещества. Неочищенный продукт очищают хроматографией на силикагеле с использованием CH2Cl2 в качестве элюента с получением 19,7 г (48%) беловатого твердого вещества. ЯМР (300 МГц, ДМСО-d6, 30°С) 10, 6-10,8 (1H, с), 7,8-7,9 (1Н, д), 7,4-7,5 (1Н, м), 7,1-7,2 (1Н, м), 3,75-3,85 (3Н, с).
Стадия 35.с. метил 4-циано-3-гидроксибензоат
Раствор продукта со стадии 35.b. (25,3 г, 91,1 ммоль) и ZnCN2 (7,48 г, 63,7 ммоль) в ДМФ (100 мл) обрабатывают, в атмосфере N2, (Ph3P)4Pd (2,0 г, 1,82 ммоль) и нагревают при температуре около 80°С в течение 4 часов. Раствор затем охлаждают до комнатной температуры и распределяют между EtOAc (400 мл) и Н2О (400 мл). Слой EtOAc промывают насыщенным раствором соли (4×200 мл), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают хроматографией на силикагеле, используя СН2Cl2 и затем 2,5% MeOH/CH2Cl2 в качестве элюентов. Фракции концентрируют с получением светло-оранжевого твердого вещества и сушат до постоянного веса в вакууме с получением продукта (13.4 г, 83%), который используют на стадии 35.d. без дальнейшей очистки. ЯМР (300 МГц, ДМСО-d6, 30°С) 7,7-7,8 (1Н, м), 7,5-7,6 (1Н, д), 7,4-7,5 (1Н, м), 3,8-3,9 (3Н, с).
Стадия 35.d. метил 4-циано-3-метоксибензоат
60% NaH в минеральном масле (6,02 г, 151 ммоль) промывают 3 порциями гексана (20 мл) и суспендируют в ДМФ (100 мл) при комнатной температуре. Добавляют продукт со стадии 35.с. (13,3 г, 75,3 ммоль) в ДМФ (100 мл) и полученную смесь обрабатывают йодметаном (9,38 мл, 151 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют EtOAc (400 мл) и промывают 5%-ной лимонной кислотой (2×150 мл) и насыщенным раствором соли (150 мл). Слой EtOAc сушат над Na2SO4, фильтруют и концентрируют с получением твердого продукта (13,4 г, 93%), который используют на стадии 35. е. без дальнейшей очистки. ЯМР (300 МГц, ДМСО-d6, 30°С) 7, 62-7,72 (3Н, м), 4,0-4,1(3Н, с), 3,9-4,0 (3Н, с).
Стадия 35.е. 4-гидроксиметил-2-метоксибензонитрил
Продукт со стадии 35.d. (13,3 г, 70,0 ммоль) в ТГФ (200 мл) обрабатывают 2М LiBH4 (75 мл, 150 ммоль) в атмосфере N2 и полученный раствор кипятят с обратным холодильником в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и обрабатывают осторожно избытком 4N HCl для гашения избыточного реагента. Добавляют Н2О (50 мл) и EtOAc (100 мл) и слои разделяют. Водные слои повторно экстрагируют EtOAc (2×50 мл) и слои EtOAc объединяют и промывают насыщенным раствором соли (3×100 мл), сушат (Na2SO4), фильтруют и концентрируют при пониженном давлении с получением белого твердого вещества (10,7 г, 94%). ЯМР (300 МГц, ДМСО-d6, 30°С) 7,5-7,8 (1Н, д), 7,0-7,1 (1Н, с), 6,94-7,0 (1Н, м), 4,75-4,8 (2Н, с), 3,9-4,0 (3Н, с), 1,9-2.1 (1Н, с (широкий)).
Стадия 35.f. 4-бромметил-2-метоксибензонитрил
Продукт со стадии 35.е. 91,0 г, 6,13 ммоль) суспендируют в CH2Cl2 (3,0 мл), обрабатывают SOBr2 (480 мкл, 6.13 ммоль), перемешивают в течение около 1/2 часа и затем концентрируют. Остаток повторно растворяют в CH2Cl2 (100 мл), промывают насыщенным раствором NaHCO3 (50 мл), сушат (Na2SO4), фильтруют и концентрируют с получением светло-желтого твердого вещества (1,22 г, 88%). ЯМР (300 МГц, ДМСО-d6, 30°С) 7,5-7,6 (1Н, д (j=8 Гц)), 7,02-7,08 (1Н, дд, (j=8 Гц, j=1 Гц)), 7,0-7,02 (1Н, д (j=1 Гц)), 4,45-4,5 (2Н, с), 3,95-4,0 (3Н, с).
Стадии 35.g.-35.i. проводят по методике стадий 5.а.-5.с., используя продукт со стадии 35.f. вместо бензилбромида на стадии 5.а.
Стадия 35.j. 1-(2-(1-((4-циано-3-метокси)фенилметил)-имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)-имидазол[1,2a][1,4]бензодиазепин
Продукт со стадии 31.с. сочетают с продуктом со стадии 35.i. (1-((4-циано-3-метоксифенил)метил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 563,2 MH+.
Пример 36: 10-бром-1-(2-(1-((4-циано-3-метокси)фенилметил)-имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)-имидазол[1,2a][1,4]бензодиазепин
Стадии 36.а.-36.с. проводят по методике стадий 22.а.-22.с., используя 2,4-дибромбензилбромид вместо 2-фторбензилбромида на стадии 36.а.
Стадия 36.d. 10-бром-1-(2-(1-((4-циано-3-метокси) фенилметил) имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)-имидазол[1,2a][1,4]бензодиазепин
Продукт со стадии 36.с. (10-бром-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2-с][1,4]бензодиазепин) сочетают с продуктом со стадии 35.i. (1-((4-циано-3-метоксифенил)метил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 623,1, 625,1 MH+.
Пример 37: 1-(2-(1-((4-циано-3-метокси) фенилметил)-имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-фенилимидазол-[1,2a][1,4]бензодиазепин
Продукт со стадии 31.с. (1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2-с][1,4]бензодиазепин) сочетают с продуктом со стадии 35.i. (1-((4-циано-3-метоксифенил)метил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 533,3 МН+.
Пример 38: 4-(2-бромфенил)-1-(2-(1-((4-циано-3-метокси)-фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фторимидазол-[1,2a][1,4]бензодиазепин
Продукт со стадии 33.е. (4-(2-бромфенил)-1,2-дигидро-8-фтор-имидазо[1,2-с][1,4]бензодиазепин) сочетают с продуктом со стадии 35.i. (1-((4-циано-3-метоксифенил)метил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 611,1, 613,1 MH+.
Пример 39: 1-(2-(1-((3-метокси)фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазол-[1,2a][1,4]бензодиазепин
Стадии 39.а.-39.с. проводят по методике стадий 5.а.-5.с., используя 3-метоксибензилбромид вместо бензилбромида на стадии 5.а.
Стадия 39.d. 1-(2-(1-((3-метокси)фенилметил)имидазо-5-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазол-[1,2a][1,4]бензодиазепин
Продукт со стадии 31.с. (1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2-с][1,4]бензодиазепин) сочетают с продуктом со стадии 39.с. (1-((3-метоксифенил)метил)-5-имидазолуксусной кислоты гидрохлорид) и очищают по методике стадии 25.b. Масс-спектр 538,4 МН+.
Пример 40: 1-(2-(5-((4-циано)фенилметил)имидазол-1-ил)-1-оксоэтил)-2,5-дигидро-8-фтор-4-(2-метоксифенил)имидазо-[1,2с][1,4]бензодиазепин
Стадии 40.а.-40.d. проводят согласно схеме 40 следующим образом:
Схема 40
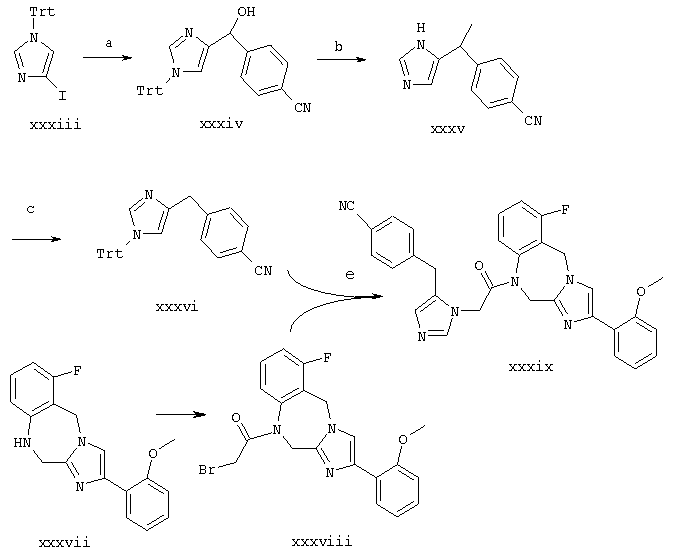
а. EtMgBr/4-цианобензальдегид/Et2O/СН2Cl2
b. ТФА/Et3SiH/(кипячение с обратным холодильником)
с. Trt-Cl/Et3N/ТГФ
d. BrCH2COBr/ДМФ
е. промежуточное соединение xxxvi, затем МеОН
Стадия 40.a. (R,S)-4-(гидрокси-(1-тритил-1Н-имидазол-4-ил)-метил)бензонитрил
Раствор 1-тритил-4-йодимидазола (3,53 г, 8,10 ммоль) в СН2Cl2 (35 мл) охлаждают до температуры около -3°С в атмосфере N2 и по каплям добавляют 3М EtMgBr в Et2O, сохраняя температуру реакционной смеси ниже около 0°С. Раствор перемешивают в течение около 1 часа при температуре около 0°С и затем одной порцией добавляют 4-цианобензальдегид (1,18 г, 9,00 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение около 1 часа. Реакционную смесь затем снова охлаждают до температуры около 0°С и добавляют 5% HCl (30 мл). Реакционную смесь перемешивают в течение около 15 минут и затем экстрагируют CH2Cl2 (2×25 мл). Объединенные слои CH2Cl2 промывают насыщенным NaHCO3, сушат над Na2SO4, фильтруют и затем концентрируют при пониженном давлении. Остаток растирают с EtOAc (25 мл) и продукт отфильтровывают (2,92 г, 82%). Масс-спектр 442,3 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 7,7-7,8 (2Н, д), 7,5-7,6 (2Н, д), 7,3-7,5 (9Н, м), 7,25-7,3 (1Н, с), 7,0-7,15 (6Н, м), 6,75-6,8 (1Н, с). 5,9-6,90 (1Н, м). 5,6-5,7 (1Н, м).
Стадия 40.b. 4-((1Н-имидазол-4-ил)метил)бензонитрил
Раствор продукта со стадии 40.а. (1,10 г, 2,49 ммоль) обрабатывают ТФА (15 мл) и Et3SiH (3,0 мл, 18,8 ммоль). Смесь кипятят с обратным холодильником в течение около 2 часов. Реакционную смесь затем концентрируют для удаления побочных продуктов. Добавляют ТФА (15 мл) и Et3SiH (3,0 мл, 18,8 ммоль) и смесь кипятят с обратным холодильником в течение еще 2 часов и затем концентрируют. Добавляют свежие реагенты и побочные продукты удаляют выпариванием при пониженном давлении сколько потребуется до тех пор, пока не будет израсходован весь исходный материал. Расход исходного материала контролируют с помощью аналитической ВЭЖХ, используя колонку VYDAC C18 (The Nest Group, Southborough, MA) и градиент от 0 до 70% СН3CN/0,1% ТФА на протяжении 25 минут. Неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 0 до 50% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта объединяют, концентрируют до около 1/2 объема и лиофилизуют с получением чистого продукта. Соль продукта подщелачивают добавлением насыщенного раствора NaHCO3, экстрагируют СН2Cl2 (3×25 мл), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Масс-спектр 183,9 МН+. ЯМР (300 МГц, ДМСО-d6, 30°С) 7,55-7,58 (1Н, д (j=1 Гц)), 7,3-7,4 (2Н, д (j=8 Гц)), 6,75-6,8 (1H, д (j=1 Гц)), 3,9-4,1 (2H, с).
Стадия 40.с. 4-((10тритил-1Н-имидазол-4-ил)метил)бензонитрил
Продукт со стадии 40.b. (152 мг, 0,83 ммоль), хлортрифенилметан (231 мг, 0,83 ммоль) и Et3N (139 мкл, 1,0 ммоль) растворяют в ТГФ (4 мл) и перемешивают в атмосфере N2 при комнатной температуре в течение около 2 часов. Добавляют насыщенный раствор NaHCO3 (5 мл) и продукт экстрагируют EtOAc (2×20 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают кристаллизацией из EtOAc и гексана с получением чистого продукта (285 мг, 81%). Масс-спектр 426,4 МН+ (незначительная линия), ЯМР (300 МГц, ДМСО-d6, 30°С) 7,5-7,6 (2Н, д (j=8 Гц)), 7,4-7,45 (1Н, д (j=1 Гц)), 7,3-7,4 (11Н, м), 7,1-7,2 (6Н, м), 6,58-6,62 (1Н, д (j=1 Гц)), 3,9-4,0 (2H, с).
Стадия 40.d. 1-бромацетил-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2а][1,4]бензодиазепин
Продукт со стадии 40.с. (108 мг, 0,35 ммоль) растворяют в ДМФ (1,5 мл) и добавляют бромацетилбромид (65 мкл, 0,75 ммоль) при комнатной температуре при перемешивании. Смесь выстаивают в течение ночи и затем концентрируют при пониженном давлении с получением неочищенного продукта, который используют на стадии 40.е. без дальнейшей очистки. Масс-спектр 430,2, 432,2 МН+.
Стадия 40.e. (2-(5-((4-циано)фенилметил)имидазол-1-ил)-1-оксоэтил)-2,5-дигидро-8-фтор-4-(2-метоксифенил)имидазо-[1,2с][1,4]бензодиазепин
Продукт со стадии 40.d. (0,32 ммоль) распределяют между насыщенным NaHCO3 (2 мл) и EtOAc (5 мл). Водный слой экстрагируют снова EtOAc (5 мл) и слои EtOAc объединяют, сушат (Na2SO4), фильтруют и концентрируют до около 2 мл. Добавляют продукт со стадии 40.с. (134 мг, 0,32 ммоль) и смесь перемешивают при комнатной температуре в течение около 2 дней и затем концентрируют при пониженном давлении. Остаток помещают в метанол (4 мл) и кипятят с обратным холодильником в течение около 1 часа. Реакционную смесь охлаждают и концентрируют при пониженном давлении и неочищенный продукт очищают препаративной ВЭЖХ на колонке RAININ™C18, используя градиент от 0 до 50% СН3CN/0,1% ТФА на протяжении 45 минут. Фракции продукта объединяют и концентрируют при пониженном давлении. ТФА соль превращают в гидрохлорид пропусканием 30% метанольного раствора через ионообменную колонку (BioRad AG 1 Х2, 200-400 меш, 0,6 мэкв/мл, 100 мл, хлорид). Фракции продукта объединяют, концентрируют до объема около 10 мл и лиофилизуют из Н2О с получением чистого продукта (35 мг, 18%) в виде дигидрохлорида. Масс-спектр 533,2 MH+.
Должно быть понятно, что хотя данное изобретение описано с помощью детального описания, такое описание предназначено для иллюстрирования и не ограничивает объем данного изобретения, определенный формулой изобретения. Другие аспекты, преимущества и модификации представлены в формуле изобретения.
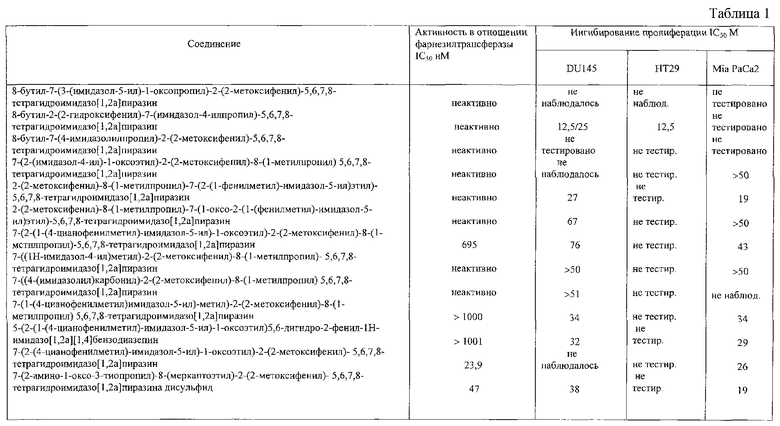
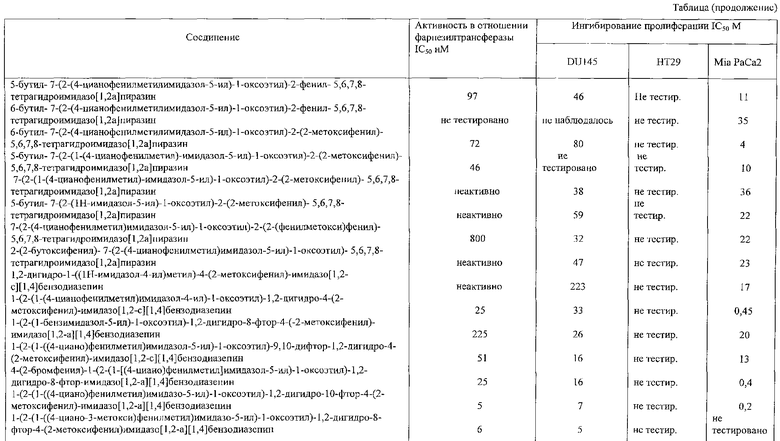

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОДУКТ, ВКЛЮЧАЮЩИЙ ИНГИБИТОР ТРАНСДУКЦИИ СИГНАЛОВ ГЕТЕРОТРИМЕРНЫХ ПРОТЕИНОВ G В КОМБИНАЦИИ С ДРУГИМ ЦИТОСТАТИЧЕСКИМ СРЕДСТВОМ, ДЛЯ ТЕРАПЕВТИЧЕСКОГО ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2000 |
|
RU2298417C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ЦИСТЕИНА ДЛЯ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА, ПРЕДНАЗНАЧЕННОГО ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЙ, ВОЗНИКАЮЩИХ ВСЛЕДСТВИЕ ОБРАЗОВАНИЯ ГЕТЕРОТРИМЕРНОГО ПРОТЕИНА G | 1999 |
|
RU2268889C2 |
| АНАЛОГИ ЦИКЛИЧЕСКОГО СОМАТОСТАТИНА | 1999 |
|
RU2242481C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРАЗИНА ИЛИ ИМИДАЗОДИАЗЕПИНА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ2 | 2008 |
|
RU2540074C2 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2012 |
|
RU2612958C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, ИХ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2007 |
|
RU2474582C2 |
| БИЦИКЛИЧЕСКИЕ 6-АЛКИЛИДЕНПЕНЕМЫ В КАЧЕСТВЕ ИНГИБИТОРОВ β-ЛАКТАМАЗ | 2003 |
|
RU2339640C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 6-АЛКИЛИДЕНПЕНЕМА | 2003 |
|
RU2317297C2 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
Изобретение относится к производным имидазола формулы (I)
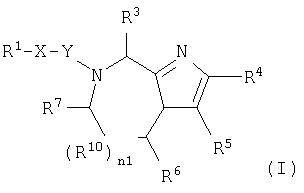
или к его фармацевтически приемлемой соли. Изобретение относится также к фармацевтической композиции, обладающей ингибирующей активностью в отношении пренилтрансферазы, к способам лечения на основе этих соединений. Технический результат - получение новых соединений и фармацевтической композиции на их основе в целях лечения таких заболеваний как фиброз, доброкачественная гиперплазия простаты, атеросклероз, рестеноз, рак груди, рак толстой кишки, рак поджелудочной железы, рак простаты, рак легких, рак яичников, рак эпидермиса и рак органов кроветворения. 5 с. и 13 з.п. ф-лы, 1 табл.
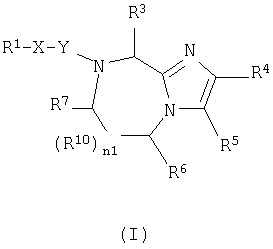
где n1 = 0 или 1;
Х независимо является (CHR11)n3(СН2)n4Z(СН2)n5;
Z является связью;
n3 независимо равно 0 или 1;
n4 и n5 каждый независимо равен 0, 1, 2 или 3;
Y независимо является СО, СН2, CS или связью;
R1 является
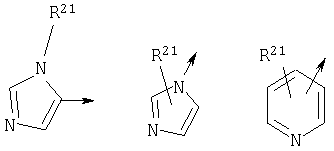
или N(R24R25);
R11 является Н;
R3 независимо является Н или необязательно замещенной группой, выбранной из группы, включающей (C1-C6)алкил, (С3-С6)циклоалкил-(C1-С6)алкил;
R4 и R5 каждый независимо является Н или необязательно замещенным арилом, где указанный необязательно замещенный арил необязательно замещен одним R30;
R6 независимо является Н или необязательно замещенным (C1-C6)алкилом;
где R8 является Н, (C1-С6)алкилом или арил(C1-C6)алкилом;
R7 независимо является для каждого случая Н или необязательно замещенным (C1-С6)алкилом;
R10 является С;
R21 независимо является Н или необязательно замещенным арил(C1-C6)алкилом, где указанная необязательно замещенная группа необязательно замещена одним или более заместителями, независимо выбранными из группы, включающей R8 и R30;
R24 и R25 каждый независимо является Н;
R30 независимо является -O-R8, -CN или галогеном;
где указанным арилом является фенил,
при условии, что если n1=1, R10 является С и R6 является Н, то R10 и R7, взятые вместе, могут образовывать
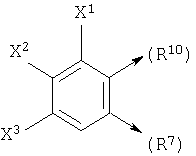
или если n1=1, R10 является С и R7 является -Н, то R10 и R6, взятые вместе, могут образовывать
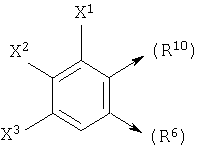
где Х1, X2 и X3 каждый независимо является Н или галогеном,
и если R1 является N(R24R25), то n3 = 1, n4 и n5 каждый равен 0, Z является связью и R3 и R11, взятые вместе, образуют
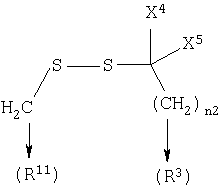
где n2 = 1 и X4 и X5 каждый независимо является Н;
или его фармацевтически приемлемая соль, при условии, что соединение формулы I не является
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]-дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2-(2-метилфенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-2-(2-метоксифенил)-8-(1-метилэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]-дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(1,1-диметилэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин]дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-бутил-2,3-дифенил-5,6,7,8-тетрагидроимидазо[1,2 а]пиразин]дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(1-метилпропил)-2-(2-(фенилметокси)фенил)-5,6,7,8-тетрагидроимидазо[1,2а] пиразин]дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-гексил-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]-дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(циклогексилэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин]дисульфидом;
бис-1,1'-[7-(2-амино-1-оксо-3-тиопропил)-8-(2-(4-метокси-циклогексил)метил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин]дисульфидом;
[S-(2-амино-3-оксо-3-(8-циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин-7-ил)пропил]-S'-циклогексил]дисульфидом;
[S-(2-амино-3-оксо-3-(8-циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил)пропил]-S'-этил]дисульфидом;
S-(диметилэтил)-S'-[2-амино-3-оксо-3-(8-бутил-2-(2-метоксифенил)]-5,6,7,8-тетрагидроимидазо[1,2а]пиразин-7-ил)пропил]дисульфидом.
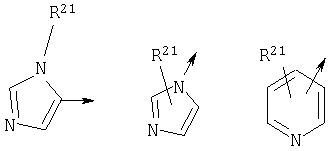
или N(R24R25);
Х является CH(R11)n3(CH2)n4,
или его фармацевтически приемлемая соль.
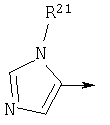
Х является СН(R11)n3(CH2)n4; n1 = 0, или его фармацевтически приемлемая соль.
R6 является Н; n1 = 1; R7 и R10, взятые вместе, образуют
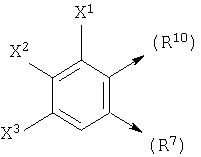
n3 = 1 и R11 является Н; Z является связью; n5 = 0 и Y является СО, СН2 или связью, или его фармацевтически приемлемая соль
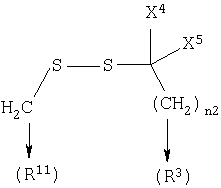
или его фармацевтически приемлемая соль.
R7 является Н; n1 = 1; R6 и R10, взятые вместе, образуют
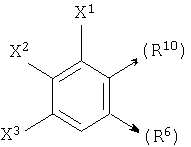
n3 = 1 и R11 является Н; n5 = 0; Y является СО или CH2 и Z является связью, или его фармацевтически приемлемая соль.
8-бутил-7-(3-(имидазол-5-ил)-1-оксопропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
8-бутил-2-(2-гидроксифенил)-7-(имидазол-4-ил-пропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
8-бутил-7-(4-имидазолилпропил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-(2-(имидазол-4-ил)-1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2 а]пиразин;
2-(2-метоксифенил)-8-(1-метилпропил)-7-(1-оксо-2-(1-фенилметил)имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
2-(2-метоксифенил)-8-(1-метилпропил)-7-(2-(1-фенилметил)-имидазол-5-ил)этил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-((1Н-имидазол-4-ил)метил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-((4-имидазолил)карбонил)-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-(1-(4-цианофенилметил)имидазол-5-ил)метил-2-(2-метоксифенил)-8-(1-метилпропил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-(2-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]-пиразин;
5-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
6-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
6-бутил-7-(2-(4-цианофенилметилимидазол-5-ил)-1-оксоэтил)-2-фенил-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
5-бутил-7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин;
7-(2-(1-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-8-(циклогексилметил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо-[1,2а]пиразин;
5-бутил-7-(2-(1Н-имидазол-5-ил)-1-оксоэтил)-2-(2-метоксифенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин;
7-(2-(4-цианофенилметил)имидазол-5-ил)-1-оксоэтил)-2-(2-фенилметокси)фенил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин; или
2-(2-бутоксифенил)-7-(2-(4-цианофенилметил)-имидазол-5-ил)-1-оксоэтил)-5,6,7,8-тетрагидроимидазо[1,2а]пиразин,
или их фармацевтически приемлемые соли.
1,2-дигидро-1-((1Н-имидазол-4-ил)метил)-4-(2-метоксифенил)-имидазо[1,2с][1,4]бензодиазепин;
1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин;
9-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин;
9-хлор-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин;
10-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин;
1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2с][1,4]-бензодиазепин,
или его фармацевтически приемлемая соль.
1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин;
9-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин;
9-хлор-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин;
10-бром-1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-4-(2-метоксифенил)имидазо[1,2с][1,4]бензодиазепин;
1-(2-(1-(4-цианофенилметил)имидазол-4-ил)-1-оксоэтил)-1,2-дигидро-8-фтор-4-(2-метоксифенил)имидазо[1,2с][1,4] бензодиазепин.
1,2-дигидро-1-(2-(имидазол-1-ил)-1-оксоэтил)-4-(2-метоксифенил)имидазо[1,2а][1,4]бензодиазепин;
1,2-дигидро-4-(2-метоксифенил)-1-(2-(пиридин-3-ил)-1-оксоэтил) имидазо[1,2а][1,4]бензодиазепин или
1,2-дигидро-4-(2-метоксифенил)-1-(2-(пиридин-4-ил)-1-оксоэтил) имидазо[1,2а][1,4]бензодиазепин,
или его фармацевтически приемлемая соль.
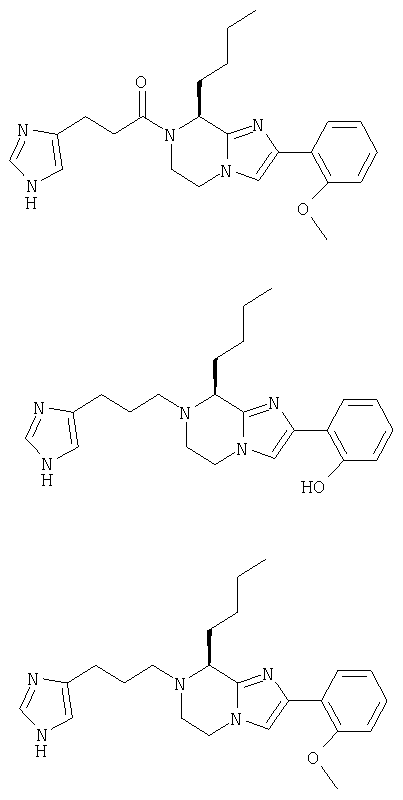
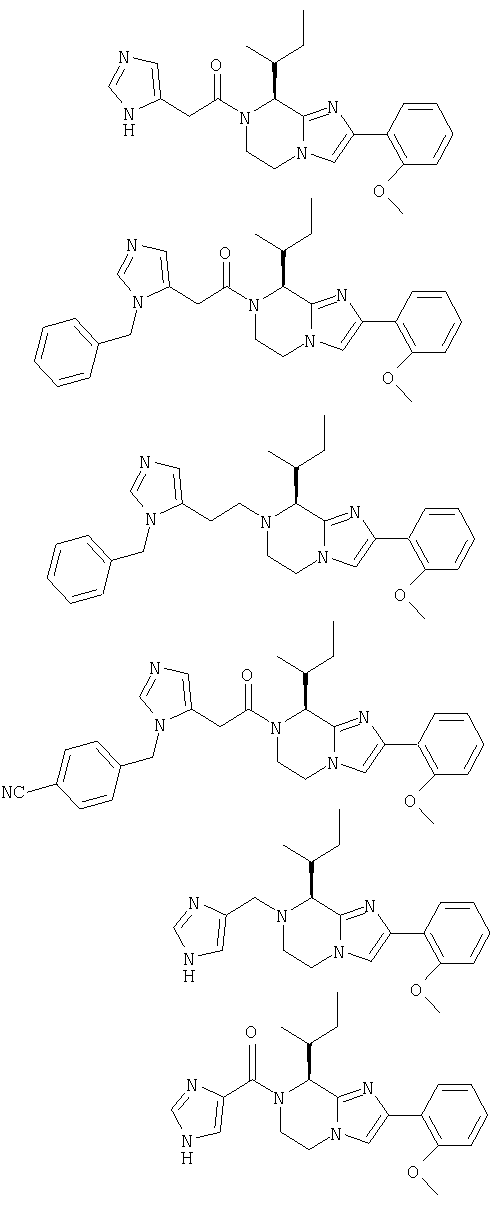
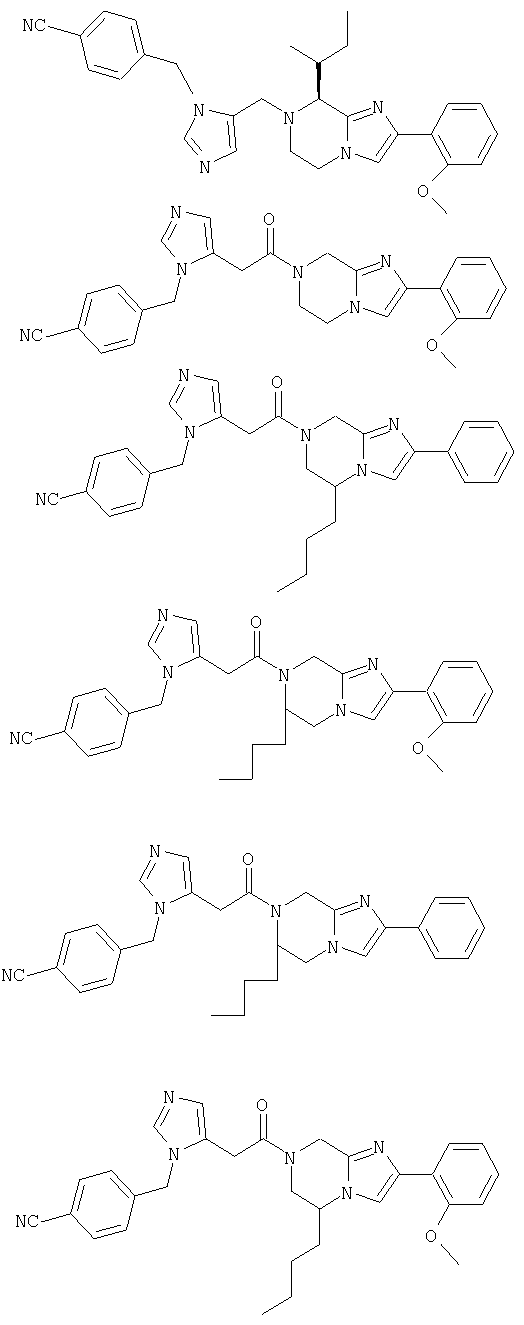
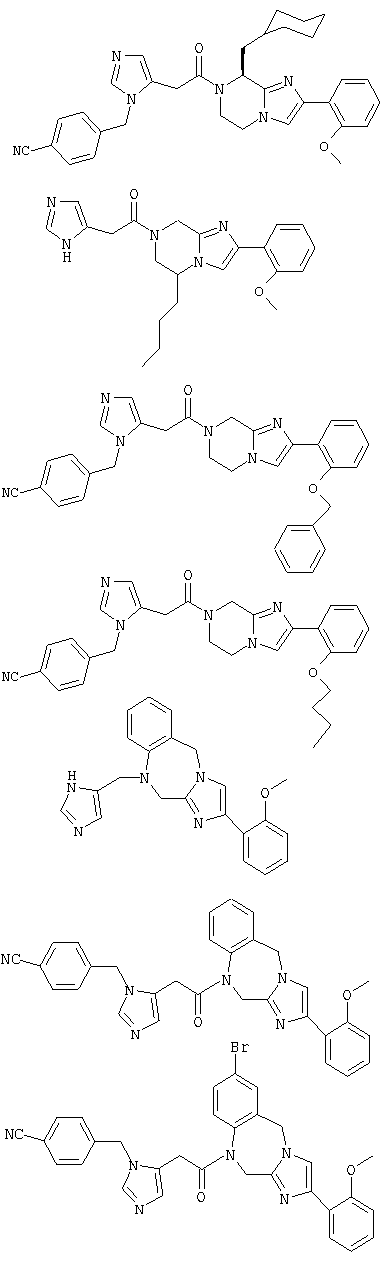
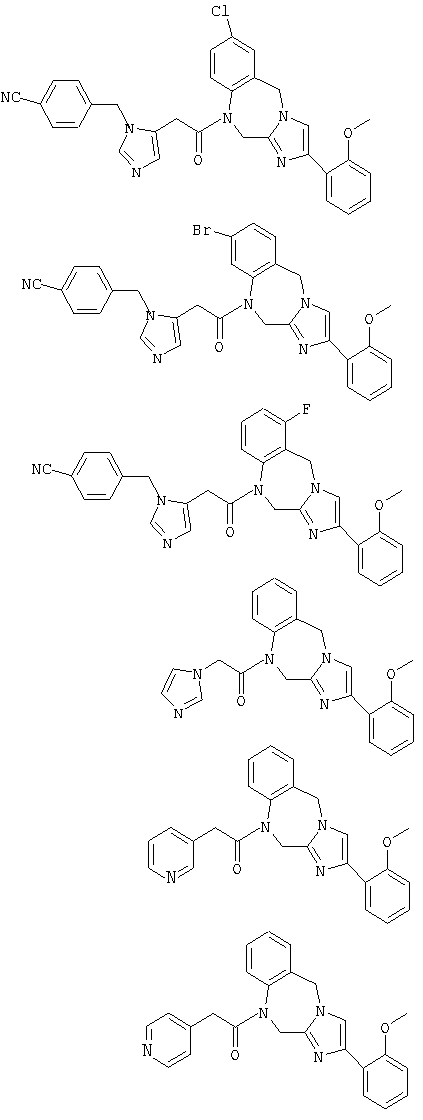
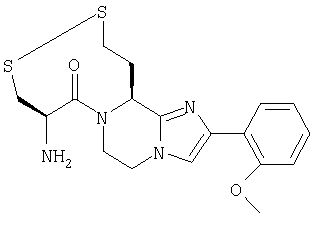
или
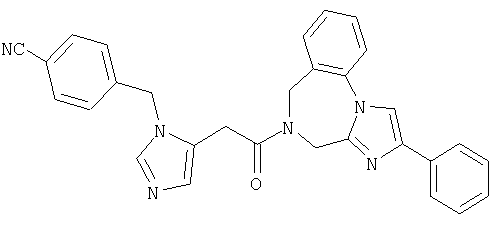
или его фармацевтически приемлемая соль.
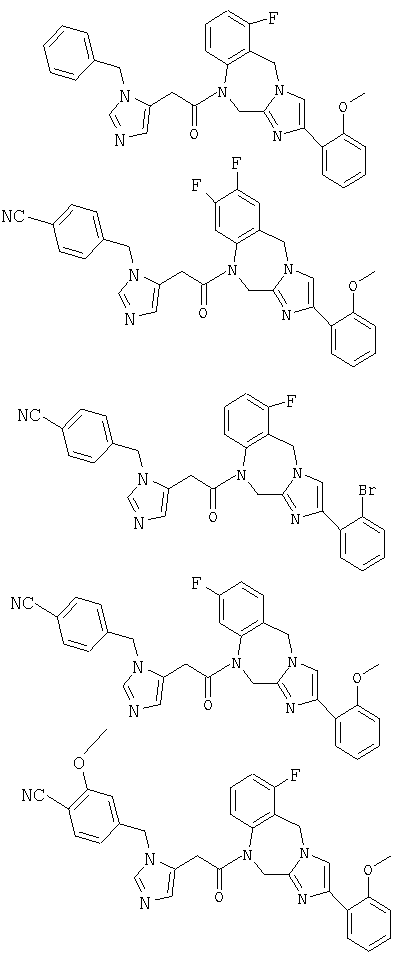
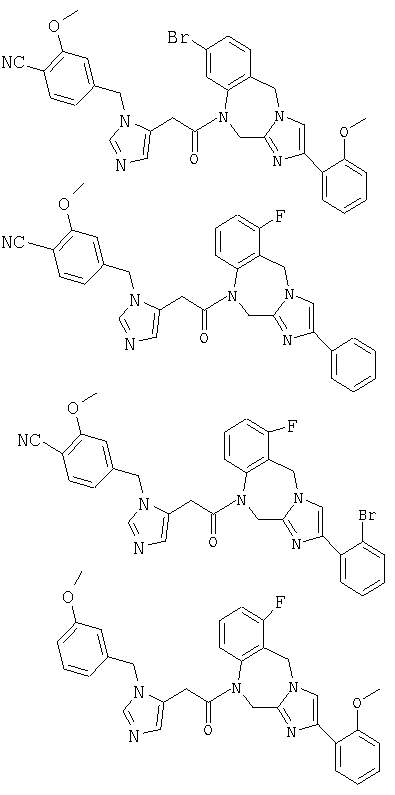
или
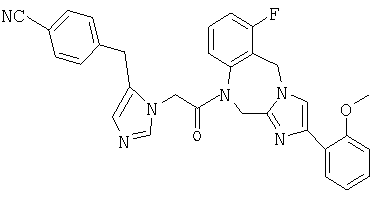
или его фармацевтически приемлемая соль.
| WO 9730053 А1, 21.08.1997 | |||
| Опорное устройство экскаватора | 1972 |
|
SU763537A1 |
| ИМИДАЗОХИНОКСАЛИНЫ | 1992 |
|
RU2092487C1 |
| ПРОИЗВОДНЫЕ 9-ДЕАЗАГУАНИНЫ ИЛИ ИХ ТАУТОМЕРЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ АКТИВНОСТЬ ПУРИННУКЛЕОЗИД ФОСФОРИЛАЗЫ У МЛЕКОПИТАЮЩИХ | 1991 |
|
RU2093513C1 |

