Область техники, к которой относится изобретение
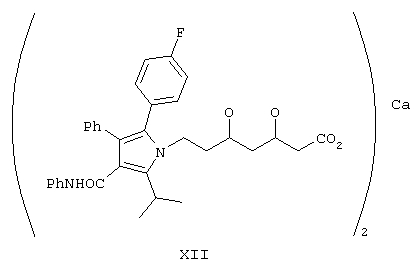
Настоящее изобретение относится к способу получения полукальциевой соли [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты, аторвастатина и новых производных, полученных в процессе синтеза. Указанное соединение используют в качестве ингибитора фермента, ГМГ-СоА-редуктазы (3-гидрокси-3-метилглутарил-СоА) и, следовательно, их можно использовать в качестве гиполипидемических и гипохолестеринемических средств.
Уровень техники
В патенте США No 4681893 описан способ разделения рацемического продукта (рацематов) с использованием R(+)-α-метилбензиламина. В патенте США No 5003080 описан путь синтеза хиральной формы аторвастатина. В патенте описан способ получения лактона или его солей методом конденсации эфира, (4R)-6-(2-аминоэтил)-2,2-диалкил-1,3-диоксан-3-ацетата, и 4-фтор-α-[2-метил-1-оксопропил]-γ-оксо-N-β-дифенилбензолбутанамида, при этом получают продукт после удаления защитных групп и гидролиза. Продукт характеризуется недостатком, так как на одной из стадий синтеза промежуточного аминокеталя требуется использование озонолиза, который является опасным при крупномасштабном производстве. В патенте описана альтернативная методика, согласно которой 4-фтор-α-[2-метил-1-оксопропил]-γ-оксо-N-β-дифенилбензолбутанамид взаимодействует с 3-аминопропинальдегидацеталем, а затем получают аторвастатин по стандартной методике.
В патентах США NoNo 5216174, 5097045, 5103024, 5124482, 5149837, 5155251, 5216174, 5245047, 5273995, 5248793 и 5397792 описаны различные незначительные модификации методики получения кальциевой соли аторвастатина.
Синтез сложных эфиров (4R)-6-(2-аминоэтил)-2,2-диалкил-1,3-диоксан-3-уксусной кислоты является основной стадией получения кальциевой соли аторвастатина. В патенте США No 5155251 описан также путь синтеза эфиров (3R)-4-циано-3-гидроксимасляной кислоты из (S)-3-гидроксибутиролактона, который в свою очередь синтезируют из подходящего углеводородного субстрата.
В других патентах США NoNo 5292939, 5319110 и 5374773 описано получение 3,4-дигидроксимасляной кислоты. Однако не было предпринято попыток выделения такого высокорастворимого в воде соединения или его лактона.
Опубликован другой многостадийный способ с использованием в качестве исходного соединения (S)-яблочной кислоты (J. Оrg. Chem., 1981, т.46, стр.4319). Для синтеза гидроксилактона, включающего восстановление с помощью BMS-NaBH4 с последующей лактонизацией, используют также сложные эфиры (S)-яблочной кислоты (Chem. Lett., 1984, стр.1389). Опубликован также 6-стадийный способ получения D-изоаскорбиновой кислоты (Syn., 1987, стр.570), однако при этом для разделения смеси диастереомеров требуется использование хроматографии на силикагеле.
В патенте США No 5084392 описан метод оптического разделения рацемических гидроксилактонов с использованием липазы, однако, такой метод характеризуется низкой степенью обогащения энантиомера и потерей другого активного изомера.
Таким образом, методы предшествующего уровня техники включают жесткие условия реакции или дорогостоящие исходные материалы, реагенты, которые с трудом поддаются обработке или являются опасными при масштабировании процесса, а также включают многостадийный процесс, что приводит к низкому общему выходу продукта.
Целью настоящего изобретения является разработка недорогостоящего, простого и легко масштабируемого способа синтеза аторвастатина. В находящейся на рассмотрении заявке на выдачу патента РСТ, поданной 28 марта 2000 г. (PCT/IN00/00030), описан способ синтеза кальциевой соли аторвастатина, однако в реакции конденсации используют различные аминокислотные фрагменты.
Сущность изобретения
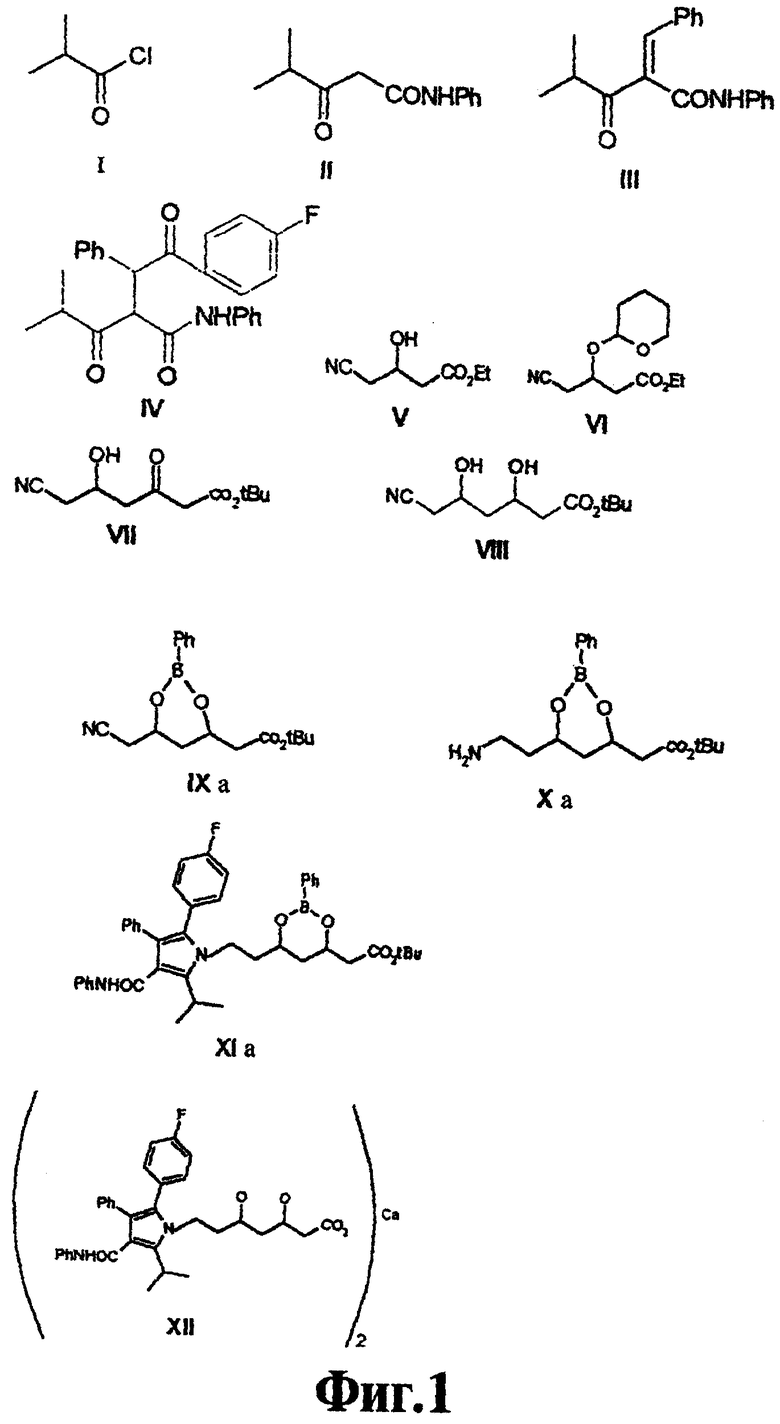
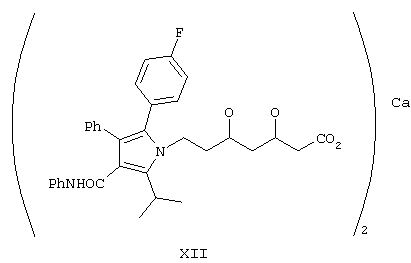
Объектом настоящего изобретения является новый, улучшенный, экономичный и пригодный для промышленного производства способ для получения ингибиторов ГМГ-СоА-редуктазы, в частности аторвастатина формулы XII, которые в связи с этим могут быть использованы в качестве гиполипидемических и гипохолестеринемических средств, причем осуществление способа синтеза показано на схемах Синтез 1-4.
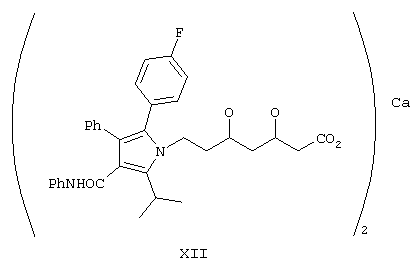
Структура XII
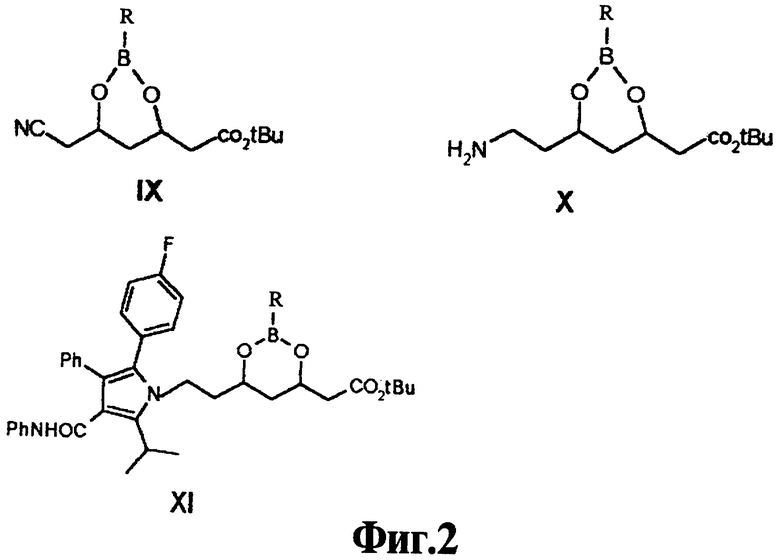
Другим объектом изобретения является фенилборонат (4R)-трет-бутилового эфира 6-циано-3,5-дигидроксигексановой кислоты формулы IX
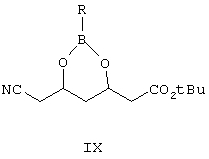
где R выбран из C6H5 или замещенных фенилов,
используемый в качестве промежуточного соединения.
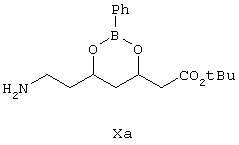
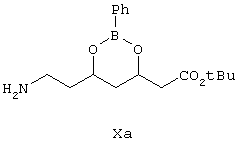
Следующим объектом изобретения является фенилборонат (4R)-трет-бутилового эфира 7-амино-3,5-дигидроксигептановой кислоты формулы Х
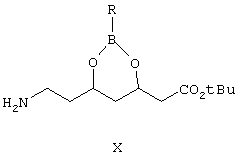
где R выбран из C6H5 или замещенных фенилов,
используемый в качестве промежуточного соединения.
Еще одним объектом изобретения является промежуточное соединение формулы XI
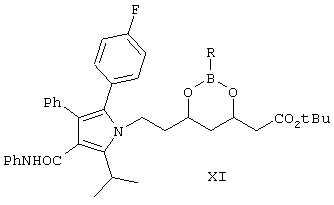
где R выбран из C6H5 или замещенных фенилов,
используемое в качестве промежуточного соединения.
Перечень чертежей и иных материалов
На фиг.1 представлены структурные формулы конкретных соединений I-XIa, используемых в синтезе аторвастатина в соответствии с настоящим изобретением, и структурная формула XII целевого аторвастатина.
На фиг.2 представлены структурные формулы промежуточных соединений IX, Х и XI, предложенных в настоящем изобретении.
Сведения, подтверждающие возможность осуществления изобретения
В настоящем изобретении разработан способ синтеза аторвастатина формулы XII, который включает в себя:
а) взаимодействие соединения формулы Х с соединением формулы IV в смеси растворителей, выбранных из ряда: ксилол, циклогексан, метил-трет-бутиловый эфир, диизопропиловый эфир, ацетонитрил, в присутствии катализатора, выбранного из группы, содержащей пивалиновую кислоту, трифторметилсульфоновую кислоту, метансульфоновую кислоту или п-толуолсульфоновую кислоту, с образованием промежуточного производного формулы XI,
б) гидролиз соединения формулы XI с последующим образованием кальциевой соли.
Соединение формулы X, использованное на стадии (а), где R выбирают из C6H5 или замещенных фенилов, получают следующим образом:
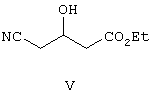
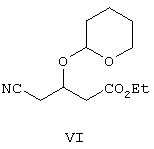
I) осуществляют взаимодействие соединения формулы V с дигидропираном с образованием защищенного простого эфира формулы VI,
II) проводят взаимодействие соединения формулы VI с трет-бутилацетатом и основанием при температуре от -30 до -80°С с образованием соединения формулы VII,
III) восстанавливают соединение формулы VII с образованием соединения формулы VIII,
Iv) превращают соединение формулы VIII в защищенный сложный эфир бороновой кислоты формулы IX,
v) восстанавливают соединение формулы IX с образованием соединения формулы X.
Восстанавливающий агент, использованный на стадии (в), выбирают из группы, включающей в себя борогидрид цинка. Защитную группу, использованную на стадии (г), выбирают из группы, содержащей фенилбороновую кислоту, толилбороновую кислоту или 3-нитробензолбороновую кислоту.
Синтез аминоэфира формулы Ха показан на схеме Синтез 1.
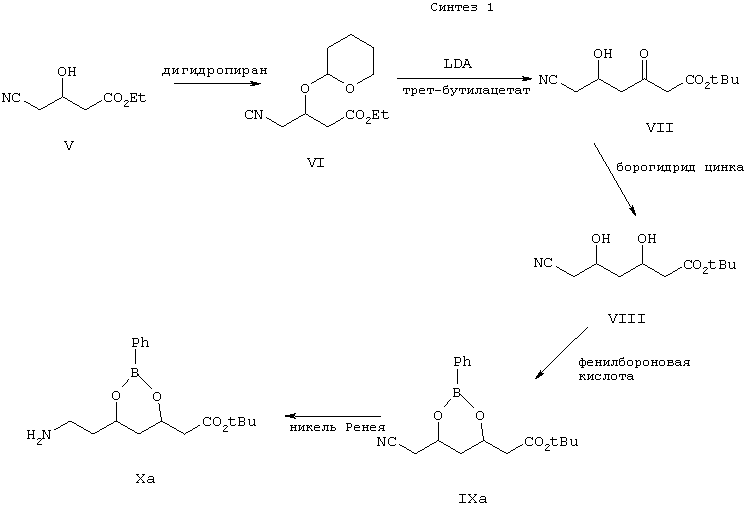
Таким образом, цианогидроксиэфир формулы V обрабатывают дигидропираном в присутствии п-толуолсульфоновой кислоты в растворителе, таком как CH2Cl2, CH3CN, ДМФ и т.п., и получают защищенный простой эфир формулы VI, который затем обрабатывают трет-бутилацетатом в анионной форме, полученным при обработке трет-бутилацетата диизопропиламидом лития в ТГФ, с образованием соединения формулы VII.
Затем β-кетоэфир формулы VII восстанавливают борогидридом цинка в ТГФ и получают дигидроксисоединение формулы VIII.
Затем дигидроксиэфир формулы VIII защищают с использованием бороновой кислоты формулы RB(OH)2, где R выбирают из фенила или замещенного фенила, при этом получают эфир бороновой кислоты формулы IXa. Реакцию предпочтительно проводят в присутствии фенилбороновой кислоты в атмосфере азота.
Затем эфир бороновой кислоты формулы IXa восстанавливают в присутствии никеля Ренея и получают аминоэфир (боронат) формулы Ха.
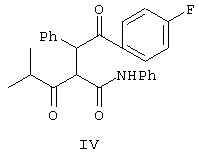
Аминоэфир формулы Ха взаимодействует с дикетоном формулы IV, причем способ получения соединения формулы IV показан на схеме Синтез 2.
Синтез 2
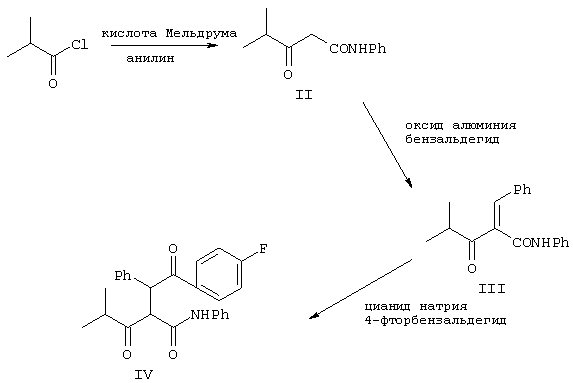
Соединение формулы IV получают, как показано на схеме Синтез 2, причем способ включает взаимодействие изобутирилхлорида и кислоты Мельдрума в присутствии основания, выбранного из ряда: пиридин, триэтиламин, диизопропилэтиламин, диметиланилин и т.п., в СН2Cl2 при 0-5°С в течение 18 ч, при этом получают ацилпроизводное кислоты Мельдрума, которая затем взаимодействует с анилином в растворителе, выбранном из ряда CH2Cl2, ацетонитрил, толуол и т.п., при кипячении с обратным холодильником в течение 12 ч, при этом получают амид формулы II. Реакцию предпочтительно проводят в пиридине и CH2Cl2 при 0°С и в CH2Cl2 при перемешивании при комнатной температуре.
Затем кетоамид формулы II взаимодействует с бензальдегидом в присутствии основания, выбранного из водного NaOH или гидроксида лития и т.п., и с оксидом алюминия в течение 26 ч с образованием метиленфенильного промежуточного производного формулы III.
Соединение формулы III обрабатывают 4-фторбензальдегидом в присутствии катализатора, выбранного из цианида металла, где металл означает Ag, К, Na, Cu, тетраалкиламмоний и т.п., или триметилсилилцианида в полярном растворителе, выбранном из ряда: ДМСО, ДМФ, ацетонитрил и т.п., при кипячении с обратным холодильником, при этом получают соединение формулы IV. Реакцию проводят предпочтительно при взаимодействии 4-фторбенэальдегида и цианида натрия в ДМСО при кипячении с обратным холодильником.
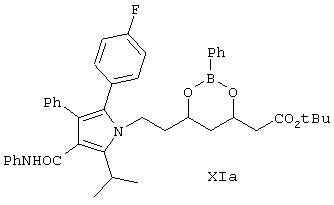
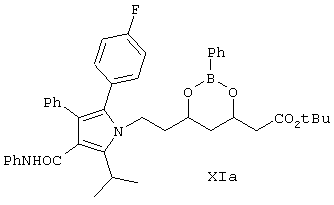
Дикетон формулы IV взаимодействует с аминоэфиром (боронатом) формулы Ха, как показано на схеме Синтез 3, в присутствии катализатора, выбранного из группы, включающей соединение формулы R12SO3H, где R12 выбирают из CF3, СН3, п-СН3С6Н4, уксусную кислоту и триметилуксусную (пивалиновую) кислоту, причем реакцию проводят в растворителе или их смеси, например, в таком как ацетонитрил, ксилол, диизопропиловый эфир, циклогексан, метил-трет-бутиловый эфир и т.п., в течение от 24 до 48 ч при температуре от 5-10°С до приблизительно температуры кипения растворителя (кипячение с обратным холодильником) с удалением воды и образованием соединения формулы XIa. Реакцию проводят предпочтительно в присутствии метансульфоновой кислоты и смеси ксилол/гексан при кипячении с обратным холодильником в течение 48 ч с удалением воды.
Синтез 3
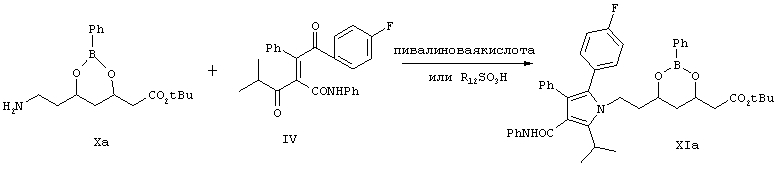
Соединение формулы XIa превращают в кальциевую соль аторвастатина, как показано на схеме Синтез 4.
Синтез 4
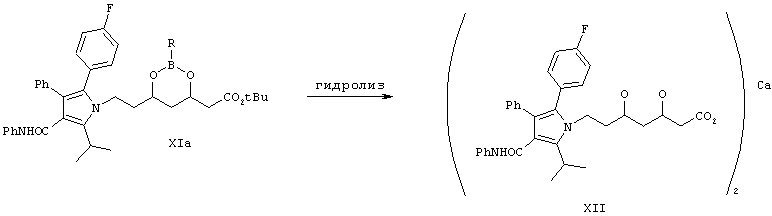
Удаление группы боронатного эфира проводят с помощью гидролиза эфира и получают свободную кислоту, которую превращают в аммониевую соль при взаимодействии с NH4OH, раствором NH3 в метаноле или при пропускании газообразного NH3 через раствор карбоновой кислоты в растворителе, выбранном из смеси EtOAc, гексана, диизопропилового эфира, изопропанола, циклогексана и метанола. В промежуточном соединении формулы XI предпочтительно удаляют защитные группы с использованием водного гидроксида натрия при комнатной температуре в течение 24 ч, затем проводят гидролиз с использованием раствора гидроксида натрия в метаноле и подкисляют с помощью разбавленного раствора HCl, при этом получают свободную кислоту, которую превращают в аммониевую соль при пропускании газообразного NH3 в EtOAc. Затем аммониевую соль обрабатывают ацетатом кальция и получают кальциевую соль аторвастатина.
Настоящее изобретение иллюстрируют следующими примерами.
Пример 1
Получение 4-метил-3-оксо-N-фенилпентанамида (формула II)
К суспензии малоновой кислоты (104 г) в уксусном ангидриде (120 мл) при комнатной температуре добавляют конц. H2SO4 (3 мл). Смесь охлаждают до 20°С и затем по каплям добавляют ацетон (80 мл). Реакционную смесь перемешивают при комнатной температуре (15 мин), выдерживают при 0-5°С в течение ночи и фильтруют. Твердое вещество промывают холодной водой, холодным ацетоном и сушат. Неочищенный материал перекристаллизовывают из смеси ацетон/гексан.
Кислоту Мельдрума (59 г) растворяют в CH2Cl2 (200 мл) и охлаждают при 0°С. Затем по каплям в течение 30 мин добавляют пиридин (73 мл) и смесь перемешивают в течение еще 10 мин. В течение 30 мин по каплям добавляют изобутирилхлорид (44 г) и смесь перемешивают при 0°С в течение 1 ч, затем перемешивают при комнатной температуре в течение ночи. Смесь выливают в 1,5 н. HCl, содержащую измельченный лед. Слои разделяют и водный слой экстрагируют CH2Cl2 (2×100 мл). Объединенные экстракты промывают 1,5 н. HCl (2×100 мл), затем насыщенным раствором NH4Cl (2×100 мл), сушат над Na2SO4 и концентрируют при пониженном давлении, при этом получают неочищенное ацилпроизводное кислоты Мельдрума, которое используют на следующей стадии.
Неочищенное ациллроизводное кислоты Мельдрума (84 г) переносят в бензол (300 мл) и добавляют анилин (111 мл). Смесь кипятят с обратным холодильником в течение 4 ч. Реакционную смесь охлаждают до комнатной температуры, промывают 2 н. HCl (5×100 мл) и бензол удаляют при пониженном давлении, при этом получают соединение формулы II.
Пример 2
Получение 4-метил-3-оксо-N-фенил-2-(фенилметилен)пентанамида (формула III)
Неочищенный амид добавляют к суспензии оксида алюминия, пропитанного гидроксидом лития в тетрагидрофуране. К указанной смеси при комнатной температуре добавляют бензальдегид. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение 2 ч. Смесь фильтруют, тетрагидрофуран удаляют при пониженном давлении и остаток экстрагируют CH2Cl2. Органические экстракты промывают раствором бикарбоната, раствором бисульфита, сушат и концентрируют при пониженном давлении, при этом получают неочищенное соединение формулы III.
Пример 3
Получение 4-фтор-α-[2-метил-1-оксопропил]-γ-оксо-N-β-дифенилбензолбутанамида (формулы IV)
К раствору 4-фторбензальдегида в безводном ДМФ добавляют цианид натрия и смесь кипятят с обратным холодильником в течение 4 ч. К указанной смеси добавляют промежуточное производное, полученное в примере 3, и смесь перемешивают в течение еще 18 ч. После обработки в стандартных условиях получают неочищенное дикетопроизводное формулы IV.
Пример 4
Получение этилового эфира 4-циано-3-(O-тетрагидропиранил)масляной кислоты (формула VI)
Раствор 50 г этилового эфира 4-циано-3-гидроксимасляной кислоты в дихлорметане (1 л) и дигидропирана (53,57 г) в присутствии каталитического количества PPTS (пиридиниевая соль 4-толуолсульфокислоты) (15,9 г) перемешивают при комнатной температуре в течение 24 ч. Затем реакционную смесь промывают бикарбонатом, сушат и растворитель удаляют при пониженном давлении, при этом получают указанное в заголовке соединение.
Пример 5
Получение трет-бутилового эфира 6-циано-5-гидрокси-3-оксогексановой кислоты (формула VII)
К раствору ТГФ (50 мл), диизопропиламина (37,6 мл) при температуре -10°С добавляют трет-бутиллитий (186,5 мл) и смесь выдерживают при -3°С в течение 30 мин. К указанному раствору при температуре от -20 до -25°С добавляют трет-бутилацетат (34,97 мл) в 35 мл ТГФ и температуру поддерживают в течение 1 ч. Затем к смеси при температуре от -20 до -25°С добавляют эфир (14 г), полученный, как описано выше в примере, в 14 мл ТГФ и температуру поддерживают в течение 3 ч. Реакцию останавливают 3 н. HCl при рН 6-7. Органический слой отделяют и водный слой экстрагируют EtOAc. Объединенные органические слои промывают водой, солевым раствором, сушат и концентрируют при пониженном давлении, при этом получают указанное в заголовке соединение формулы VII.
Пример 6
Получение трет-бутилового эфира 6-циано-3,5-дигидроксигексановой кислоты (формулы VIII)
Неочищенный продукт, полученный, как описано выше в примере, переносят в сухой ТГФ и изопропанол в атмосфере азота. Раствор охлаждают до -10°С и добавляют борогидрид цинка. Температуру поддерживают от -10 до -15°С, затем нагревают до комнатной температуры и выдерживают в течение 18 ч. Реакцию останавливают добавлением уксусной кислоты и смесь концентрируют при пониженном давлении, при этом получают масляный остаток.
Пример 7
Получение фенилбороната (4R)-трет-бутилового эфира 6-циано-3,5-дигидроксигексановой кислоты (формулы IXa)
Диол (10 г), полученный, как описано выше в примере, взаимодействует с фенилбороновой кислотой (5,5 г) в толуоле. Реакционную смесь кипятят с обратным холодильником в течение 20 ч и при перегонке азеотропной смеси собирают воду. Толуол удаляют при пониженном давлении и к масляному остатку добавляют петролейный эфир, затем смесь охлаждают до 0°С, при этом боронат выпадает в осадок. Выход продукта данной реакции составляет 10,7 г (78%).
1Н ЯМР (CDCl3): δ 1.508 (s, 9H), 1.712-1.757 (m, 1H), 2.264-2.309 (m, 1H), 2.538-2.754 (m, 4H), 4.593-4.488 (m, 1H), 4.581-4.612 (m, 1H), 7.269-7.796 (m, 5H).
13С ЯМР (CDCl3): δ 25.54, 28.06, 36.85, 43.03, 67.03, 68.25, 81.02, 116.54, 127.54, 131.03, 133.91, 169.63.
Пример 8
Получение фенилбороната (4R)-трет-бутилового эфира 7-амино-3,5-дигидроксигептановой кислоты (формулы Ха)
Боронат эфира (5 г), полученный, как описано выше в примере, добавляют к насыщенному раствору аммиака в метаноле, затем добавляют никель Ренея (5 г). Реакционную смесь гидрируют при давлении (5 кг). Реакционную смесь фильтруют через слой целита, метанол удаляют при пониженном давлении и получают неочищенное, указанное в заголовке соединение формулы Ха. Выход продукта данной реакции составляет 4,8 г (95%).
1Н ЯМР (CDCl3): δ 1.516 (s, 9H), 1.559-1.593 (m, 1H), 1.756-2.109 (m, 5H), 2.456-2.508 (m, 1 H), 2.600-2.637(m, 1 H), 2.979-3.035 (шир., 2Н), 4.282-4.309 (m, 1H), 4.549-4.559 (m, 1H), 7.284-7.782 (m, 5H).
13C ЯМР (CDCI3): δ 28.08, 38.56, 43.56, 46.05, 68.63, 69.99,70.08, 80.77, 27.42, 130.36, 133.65, 170.15.
Пример 9
Получение полукальциевой соли [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-{фениламинокарбонил}-1Н-пиррол-1-гептановой кислоты (формулы XII)
Раствор, содержащий 4,8 г фенилбороната (4R)-трет-бутилового эфира 7-амино-3,5-дигидроксигептановой кислоты (формулы Ха, пример 8) (эквимолярное количество), 4-фтор-α-[2-метил-1-оксопропил]-γ-оксо-N-β-дифенилбензолбутанамид (формулы IV) и пивалиновую кислоту в ксилоле кипятят с обратным холодильником в течение 44 ч. Раствор разбавляют диизопропиловым эфиром и метанолом, промывают разбавленным раствором гидроксида натрия в метаноле, разбавленным раствором HCl и затем растворитель удаляют в вакууме с получением 7,75 г соединения формулы XIa (выход 74%). При использовании других катализаторов, таких как R12SO3H, выход продукта реакции является сопоставимым.
1Н ЯМР (CDCl3): δ 1.494 (s, 9H), 1.577-1.600 (d, 6H), 1.757-1.960 (m, 4H), 2.438-2.458 (m, 1H), 2.561-2.585 (m, 1H), 3.657 (m, 1H), 4.005-4.056 (m, 2H), 4.318-4.490 (m, 2H), 6.884-7.646 (m 19 H).
13С ЯМР (CDCI3): δ 26.17, 28.06,30.80, 37.91, 38.74, 40.98,43.21, 68.58, 69.00, 80.90, 115.31 (141.37, 160.62, 163.91, 164.68, 169.88.
Неочищенное масло перемешивают с влажным диоксидом кремния в CH2Cl2 и перемешивают при комнатной температуре в течение 18 ч. Затем при комнатной температуре добавляют водный раствор NaOH и перемешивают в течение 4 ч. Реакционную смесь разбавляют водой и промывают диизопропиловым эфиром. Водный слой подкисляют HCl и переносят в диизопропиловый эфир. Затем неочищенное промежуточное производное кислоты переносят в EtOAc, через смесь пропускают газообразный NH3. Реакционную смесь перемешивают до полного завершения реакции и после удаления растворителя продукт кристаллизуют. Неочищенную аммониевую соль переносят в смесь диизопропиловый эфир/изопропанол и при комнатной температуре добавляют раствор ацетата кальция с последующим осаждением кальциевой соли из раствора. Продукт фильтруют и сушат в вакууме, при этом получают соединение формулы XII, чистота которого является фармацевтически приемлемой. Выход продукта данной реакции составляет 5,3 г (83%).
Настоящее изобретение описано со ссылкой на определенные варианты воплощения изобретения, которые приведены только для иллюстрации. Для специалистов в данной области техники представляется очевидным, что возможны многочисленные другие варианты воплощения изобретения, которые рассматриваются в объеме следующих пунктов формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТЕНЕЛИГЛИПТИНА | 2013 |
|
RU2654069C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ У МЛЕКОПИТАЮЩИХ | 2004 |
|
RU2344121C2 |
| ЭФИРЫ БОРОНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2325393C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-МЕТОКСИ-10-(1-ГИДРОКСИЭТИЛ)-11-ОКСО-1-АЗАТРИЦИКЛО- (7.2.0.0)-УНДЕКА-2-ЕН КАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ СОЛЕЙ, 4-ТРЕТБУТИЛБЕНЗИЛ- (4S, 8S, 9R, 10S, 12R)-4-МЕТОКСИ-10-(1-ГИДРОКСИЭТИЛ)-11-ОКСО-1-АЗАТРИЦИКЛО- (7.2.0.0)-УНДЕКА-2-ЕН-2-КАРБОКСИЛАТ | 1994 |
|
RU2127735C1 |
| КОНЪЮГАТ ИНГИБИТОРА 3-ГИДРОКСИ-3-МЕТИЛГЛУТАРИЛ-КоА РЕДУКТАЗЫ С ЛИГАНДОМ АСИАЛОГЛИКОПРОТЕИНОВОГО РЕЦЕПТОРА | 2022 |
|
RU2816947C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЛОПРОСТА | 2019 |
|
RU2798239C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2012 |
|
RU2629930C2 |
| ИНГИБИТОРЫ Р38 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2357957C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛИДИН-2-КАРБОКСАМИДЫ | 2011 |
|
RU2564022C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
Изобретение относится к способу синтеза полукальциевой соли [R-(R*,R*)]-2-(4-фторфенил)-β,δ-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановой кислоты, аторвастатина, формулы XII
который заключается во взаимодействии предварительно полученного соединения формулы Ха
с соединением формулы IV 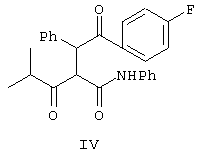
в смеси растворителей, которые выбирают из группы, содержащей ксилол, циклогексан, метил-трет-бутиловый эфир, диизопропиловый эфир, ацетонитрил, в присутствии катализатора, выбранного из группы, содержащей пивалиновую кислоту, трифторметилсульфоновую кислоту, метансульфоновую кислоту или п-толуолсульфоновую кислоту, с образованием промежуточного соединения формулы XIa
с последующим проведением гидролиза соединения формулы XIa, после чего получают кальциевую соль с образованием соединения формулы XII. Полученное соединение может быть использовано в качестве ингибитора фермента, ГМГ-СоА редуктазы, и, следовательно, его можно использовать в качестве гиполипидемических и гипохолестеринемических средств. 4 н. и 3 з.п.ф-лы, 2 ил.
отличающийся тем, что проводят взаимодействие предварительно полученного соединения формулы Ха
с соединением формулы IV
в смеси растворителей, которые выбирают из группы, содержащей ксилол, циклогексан, метилтретбутиловый эфир, диизопропиловый эфир, ацетонитрил, в присутствии катализатора, выбранного из группы, содержащей пивалиновую кислоту, трифторметилсульфоновую кислоту, метансульфоновую кислоту или п-толуолсульфоновую кислоту, с образованием промежуточного соединения формулы XIa
затем осуществляют гидролиз соединения формулы XIa, после чего получают кальциевую соль с образованием соединения формулы XII.
с дигидропираном, с образованием защищенного эфира формулы VI
взаимодействием соединения формулы VI с третбутилацетатом в присутствии основания при температуре от -30 до -80°С с образованием соединения формулы VII
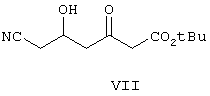
восстановлением соединения формулы VII с образованием соединения формулы VIII
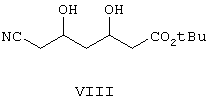
превращением соединения формулы VIII в защищенный боронатный эфир формулы IXa
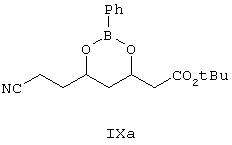
и восстановлением соединения формулы IXa с образованием соединения формулы Ха.
| US 5397792 A, 14.03.1995 | |||
| WO 00/68221 A1, 16.11.2000 | |||
| US 5155251, 13.10.1992 | |||
| RU 96112769 A, 27.02.1998. |

