Объектом настоящего изобретения является способ получения илопроста формулы I посредством новых промежуточных соединений, выделение илопроста в твердой форме, а также получение изомеров - 16(S)-илопроста и 16(R)-илопроста - формул (S)-I и (R)-I и выделение 16(S)-илопроста формулы (S)-I в твердой кристаллической форме.

Предшествующий уровень техники
Илопрост представляет собой производное карбациклина. Скелетом карбациклина является модифицированный простациклин, где атом кислорода кислородсодержащего пятичленного кольца заменен атомом углерода. Карбациклины не содержат очень чувствительной енолэфирной структурной части, поэтому с химической точки зрения они являются более стабильными, чем простациклины. Химические и биохимические свойства карбациклиновой структуры, а также ранее известные способы синтеза подытожены в публикации R. C. Nickolson, M. H. Town, H. Vorbrüggen, Prostacyclin-analogues, Medicinal Research Reviews, VoI. 1985, 5 (1), 1-53.
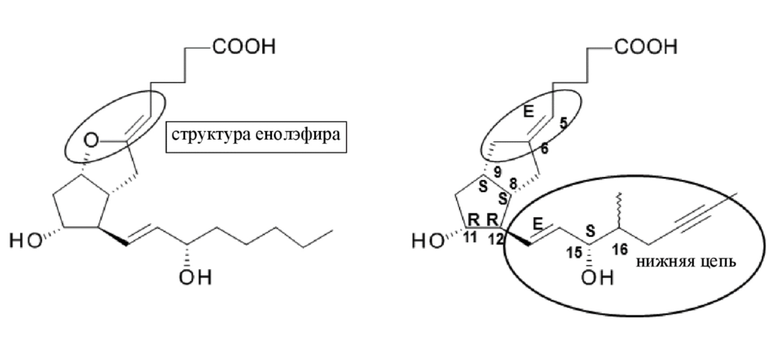
Илопрост содержит 6 асимметричных центров и представляет собой смесь двух диастереоизомеров в соотношении приблизительно 1:1, поскольку конфигурация атома углерода в положении 16 может быть (R) или (S) относительно пространственного положения метильной группы. Хотя в настоящее время в терапии используется смесь 16(R)-илопроста и 16(S)-илопроста в соотношении примерно 1:1, активности двух изомеров различны, причем 16(S)-илопрост является более эффективным (Biochimica et Biophysica Acta, Biomembranes 1988, 942(2), 220-6; Prostaglandins, 1992, 43, 255-261). Контролирующие органы призывают к разработке 16(S)-илопроста как фармацевтического активного ингредиента.
В данном документе указано, что атомы углерода нижней цепи в каждом случае пронумерованы согласно правилам, принятым в химии простагландинов. Эта нумерация в некоторых случаях отличается от названий согласно Химической реферативной службе (CAS) и IUPAC. В примерах для ранее известных соединений использованы названия согласно CAS, тогда как для новых соединений использованы названия согласно IUPAC.

В терапии илопрост применяют для лечения заболеваний периферических артерий (окклюзионного заболевания периферических артерий, PAOD) (1992, Bayer Schering Pharma) и легочной гипертензии (2004, Bayer Schering Pharma).
Ключевой стадией синтеза илопроста является формирование соответствующим образом замещенного бицикла, содержащего два пятичленных кольца.
Ссылки на наиболее часто применяемые способы получения соответствующим образом замещенного бициклического кетона можно найти в самых поздних публикациях, в которых описывается синтез оптически активного 16(S)-илопроста:
S. Chandrasekhar, Ch. Sridhar, P. Srihari, Tetrahedron Asymmetry, 2012, 23, 388-394;
H-J. Gais, G. J. Kramp, D. Wolters, L. R. Reddy, Chem. Eur. J., 2006, 12, 5610-5617;
G. J. Kramp, M. Kim, H-J. Gais, C. Vermeeren, J. Am. Chem. Soc., 2005, 127, 17910-17920.
Бициклический кетон синтезировали путем введения в реакцию глиоксаля с диметил-1,3-ацетодикарбоксилатом
(A. Gawish, U. Weiss, Org. Synth., 1986, 64, 27-38; H. Dahl (Schering AG), DE 3816801, J. A. Caedieux, D. J. Buller, P. D. Wilson, Org. Lett., 2003, 5, 3983-3986). (Схема 1.)
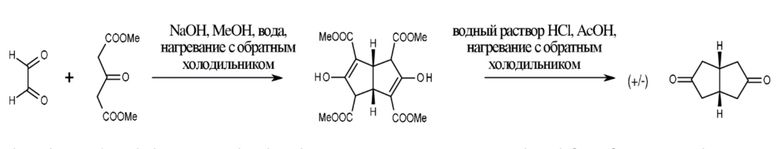
глиоксаль диметил-1,3-ацетодикарбоксилат цис-бицикло[3.3.0]октан-3,7-дион
Схема 1.
Цис-бицикло[3.3.0]октан-3,7-дион подвергали селективному кетализу с помощью неопентилгликоля, монокеталь подвергали реакции с диметилкарбонатом в присутствии гидрида натрия, оксогруппа восстанавливалась, спирт подвергали ацетилированию и ацетат подвергали разложению посредством ферментативного гидролиза (H. Dahl (Schering AG), DE 3816801). (Схема 2.)
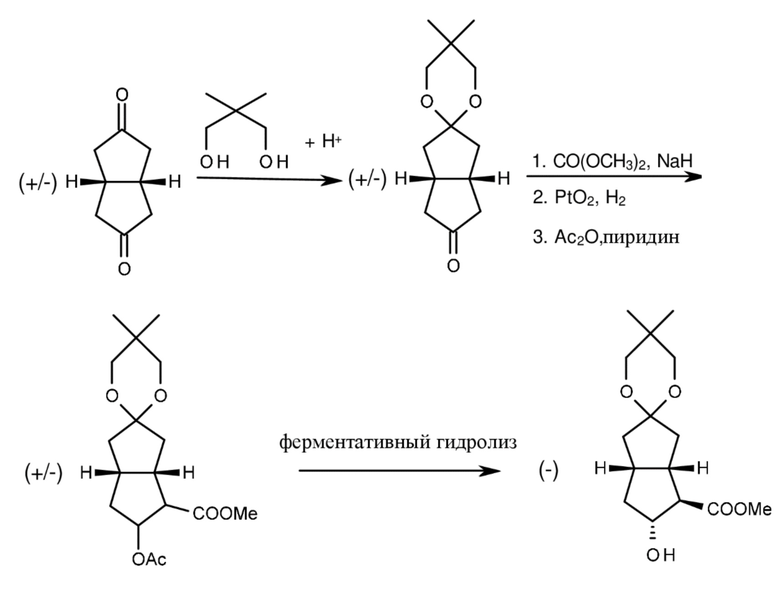 Схема 2.
Схема 2.
Исследователи Schering также работали над ферментативным, селективным монометоксикарбонилированием монокеталя, двузамещенного метоксикарбонильной группой (K. Petzoldt, H. Dahl, W. Skuballa, M. Gottwald, Liebigs. Ann. Chem., 1990, 1087-1091.). (Схема 3.)
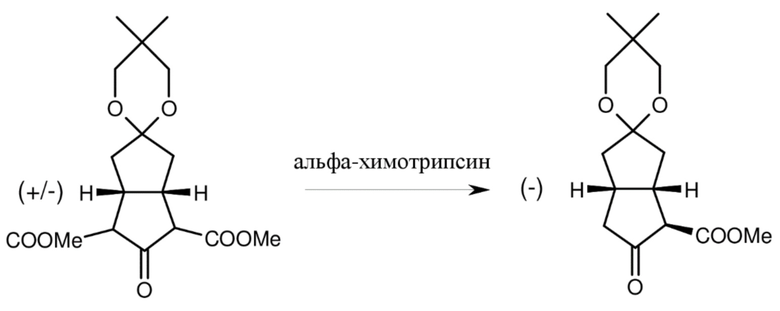
Схема 3.
Соответствующим образом замещенный бицикл также может быть образован из соответствующим образом замещенного хирального лактона Кори, который является наиболее часто используемым исходным материалом для производных простациклина.
В способе Skuballa и Vorbrüggen (Angew. Chem. Int. Ed. Engl., 1981, 20, 1046-1048) первичная гидроксильная группа лактона Кори, содержащего бензоильную защитную группу, была защищена трет-бутилдиметилсилилхлоридом (TBDMSCl).
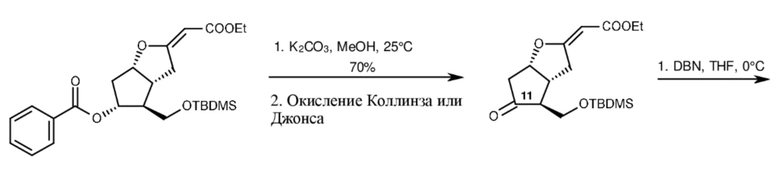
TBDMS-бензоил-лактон Кори подвергали реакции с литированным этилацетатом в THF при (-)70°C, затем из полученного в результате сложного гидроксиэфира удаляли воду. Бензоильную защитную группу TBDMS-бензоил-ненасыщенного сложного эфира расщепляли путем метанолиза в присутствии карбоната калия, вторичную гидроксильную группу превращали в оксогруппу путем окисления с помощью хрома. Реакционноспособное енольноэфирное кольцо TBDMS-ненасыщенного сложного эфира при обработке 1,5-диазабицикло[4.3.0]нон-5-еном (DBN) подвергалось раскрытию кольца и циклизации до кетона. 11-Оксогруппу восстанавливали до группы вторичного спирта при помощи борогидрида натрия в реакции, проводящейся в одном реакционном сосуде. Селективность восстановления обеспечивалась за счет участия соседних групп, но эпимерная чистота не была предоставлена, хотя это имеет ключевое значение для оптической чистоты и для количества оптических изомеров. Суммарный выход касательно раскрытия кольца и восстановления оксогруппы составлял 70%. Этоксикарбонильную группу этоксикарбонильного кетона удаляли путем нагревания с обратным холодильником в водном растворе толуола в присутствии 1,4-диазабицикло[2.2.2]октана (DBO), вторичную гидроксильную группу защищали бензоильной группой, силильную защитную группу первичной гидроксильной группы расщепляли. Суммарный выход данных трех стадий составлял 43%. (Схема 4.)
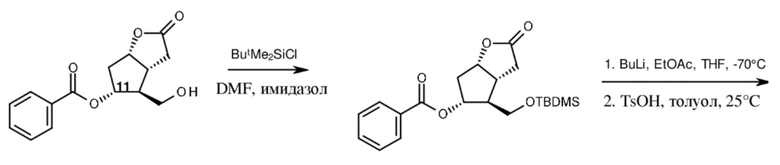
Бензоил-лактон Кори TBDMS-бензоил-лактон Кори
TBDMS-бензоил-ненасыщенный сложный эфир TBDMS-ненасыщенный сложный эфир
 Раскрытие енольного эфира Этоксикарбонилкетон Бензоил-бициклический гидроксикетон
Раскрытие енольного эфира Этоксикарбонилкетон Бензоил-бициклический гидроксикетон
Схема 4.
Из содержащего защитную группу бициклического гидроксикетона могут быть образованы нижняя и верхняя цепи илопроста посредством реакции Виттига или модифицированной реакции Виттига.
Преимущество способа состоит в том, что оптически активное промежуточное соединение, представляющее собой бициклический кетон, необходимое для синтеза карбациклинов, образуется, начиная с оптически активного лактона Кори, который является широко используемым в химии простагландинов. Однако недостатком является то, что в реакции лактона Кори с литиевым соединением этилацетата также образуется множество побочных продуктов, что значительно снижает выход (в упомянутой публикации выход по данной стадии не приведен).
Согласно способу Апджона (P. A. Aristoff, P. D. Johnson, A. W. Harrison, J. Org. Chem., 1981, 46, 1954-1957) содержащий защитную группу лактон Кори, уже содержащий нижнюю цепь, превращается в промежуточное соединение, подходящее для получения карбациклинов.
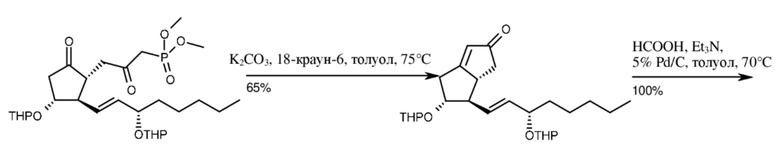
На первой стадии производное бис-THP-лактона Кори (THP=тетрагидропиранил) обрабатывали литиевой солью диметилметилфосфоната (DMMP). Полученный бис-THP-гидроксифосфонат окисляли путем модифицированного окисления Коллинза до бис-THP-кетофосфоната. Посредством внутримолекулярной реакции Хорнера-Уодсворта-Эммонса (HWE) из бис-THP-кетофосфоната получали бис-THP-бициклический енон. Двойную связь енона насыщали путем трансферной гидрогенизации (бис-THP-бициклический кетон), THP-защитные группы затем удаляли путем кислотного гидролиза (бициклический кетон). Верхняя цепь бициклического кетона, уже содержащего нижнюю цепь, образовывалась посредством реакции Виттига. Фосфоран, необходимый для реакции, получали
in situ из 4-карбоксибутилтрифенилфосфония бромида. Благодаря реакции, помимо необходимого продукта с двойной связью E-геометрии (65%), также получали значительное количество Z-изомера (35%) (схема 5).
 производное бис-THP-лактона Кори бис-THP-гидроксифосфонат
производное бис-THP-лактона Кори бис-THP-гидроксифосфонат
бис-THP-кетофосфонат бис-THP-бициклический енон
 бис-THP-бициклический кетон бициклический кетон
бис-THP-бициклический кетон бициклический кетон
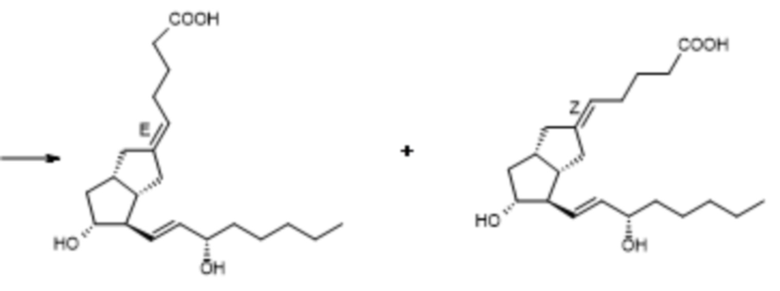
E-карбациклин Z-карбациклин (изомерная примесь)
илопрост примесь Z-изомера
Схема 5.
Процесс включает масштабируемые стадии с хорошими показателями выхода, при этом его недостаток состоит в том, что образование верхней цепи является не селективным, продукт содержит значительное количество загрязняющего вещества в виде Z-изомера. В одной из последних публикаций (P. A. Aristoff, P. D. Johnson, A. W. Harrison, J. Org. Chem., 1983, 48, 5341-5348) авторы описывают вышеуказанные химические стадии с более высокими показателями выхода.
(20% исходного материала)
(20% исходного материала)
(29% исключаемого продукта III)**
(29% исключаемого продукта III)**
(≤ 5% исключаемого продукта)**
(% E-/Z-изомера)
II-VII
*с рециркуляцией (вместе с УФ-изомеризацией)
**структура исключаемого продукта III показана на схеме 9.
Было предпринято множество попыток улучшить селективность реакции Виттига, в ходе которой образуется верхняя цепь.
Исследователи Schering (J. Westermann, M. Harre, K. Nickish, Tetrahedron Letters, 1992, 33, 8055-8056) изучали, как защитные группы бициклического кетона, уже содержащего нижнюю цепь, и условия проведения реакции Виттига влияли на селективность реакции.

бициклический кетон, содержащий нижнюю цепь илопрост примесь в виде Z-изомера
Схема 6.
a. Реакция Виттига; 4-карбоксибутилтрифенилфосфония бромид/трет-бутилат калия
b. Снятие защитных групп, если R1 и/или R2 не являются водородом
На основании экспериментов исследователи заявили, что наихудшее изомерное соотношение (E: Z=60:40) было получено при использовании THP-защитной группы (R1=R2=THP) и смеси растворителей DMSO-THF.
Лучшего изомерного соотношения (E: Z=90:10) добивались тогда, когда бициклический кетон не содержал защитную группу (R1=R2=H), и в качестве растворителя выбирали диметоксиэтан.
H-J. Gais и его коллеги (J. Am. Chem. Soc., 2005, 127, 17910-17920) повторили данную реакцию, начинающуюся с лактона Кори, который содержал хиральную нижнюю цепь илопроста, но они получили гораздо менее удовлетворительное изомерное соотношение (E: Z=62:38).
Изучив литературные данные, H-J. Gais и его коллеги разработали наиболее селективный из ранее известных способ построения верхней цепи (G. J. Kramp, M. Kim, H-J. Gais, C. Vermeeren, J. Am. Chem. Soc., 2005, 127, 17910-17920; H-J. Gais, G. J. Kramp, D. Wolters, L. R. Reddy, Chem. Eur. J., 2006, 12, 5610-5617). Способ был разработан для синтеза илопроста, содержащего хиральную нижнюю цепь.
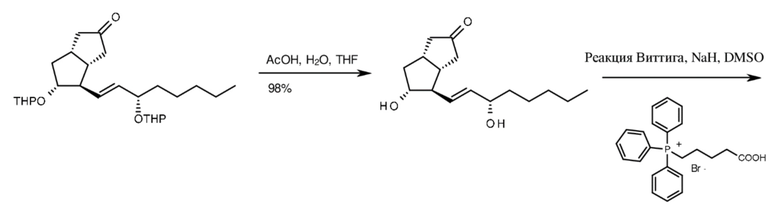
Верхняя цепь формировалась в две стадии. Первую стадию, обеспечивающую селективность, проводили с хиральным фосфонатом. Литиевую соль хирального фосфоната подвергали реакции при
(-)78-(-)62°C с содержащим защитную группу бициклическим кетоном, соответствующим илопросту, но содержащим хиральную нижнюю цепь. Реакцию HWE с хиральным соединением проводили, начиная с 300 мг бициклического кетона. В ходе реакции было образовано только 2% Z-изомера. Сложноэфирную группу восстанавливали до спиртовой с помощью диизобутилалюминия гидрида (DIBAL-H), спиртовую группу защищали ацетильной группой. Верхняя цепь с соответствующим числом атомов углерода формировалась путем обработки ацетильного производного с помощью меднокислого реагента, содержащего защитную трет-бутилдиметилсилильную (TBDMS) группу. Защитную группу первичного спирта удаляли путем протекающего в мягких условиях десилилирования (путем обработки нейтральным оксидом алюминия в гексане), первичный спирт окислялся до альдегида с помощью DMSO-пиридинового реагента. Альдегид окисляли до кислоты с помощью нитрата серебра с получением после удаления защитных групп (16S)-илопроста, который содержит хиральную нижнюю цепь (схема 7).
 R=трет-бутилдиметилсилил
R=трет-бутилдиметилсилил
промежуточное соединение 16(S)-илопроста с хиральной нижней цепью хиральный сложный эфир, содержащий верхнюю цепь

производное 16(S)-ацетила 16(S)-TBDMS-илопрост-спирт
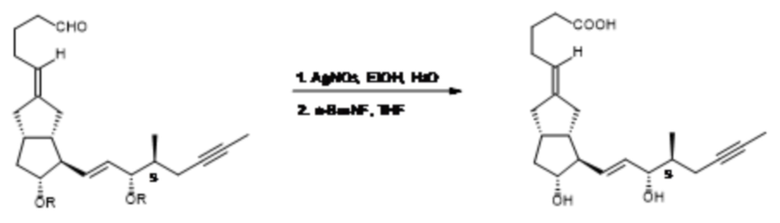
16(S)-илопрост-альдегид 16(S)-илопрост
Схема 7.
Полученное количество 16(S)-илопроста составляло 20 мг, его физическое состояние не было охарактеризовано.
Преимуществом вышеприведенного способа является то, что в ходе формирования верхней цепи образовывалось только 2% примеси в виде Z-изомера. Недостатки: данный способ сложно масштабировать, в нем применяются экстремальные условия реакций и используются дорогостоящие реагенты, полученные в многостадийных синтезах.
В описании патента CN 107324986 описано получение хирального (S)-TBDMS-енона, который получают путем фракционированной кристаллизации рацемического TBDMS-енона (фигура 8).
Из хирального (S)-TBDMS-енона можно получить 16(S)-илопрост.

рацемический TBDMS-енон 16(S)-TBDMS-енон 16(S)-илопрост
TBDMS=трет-бутилдиметилсилильная группа
Схема 8.
Процесс представляет собой формальный синтез 16(S)-илопроста. В патентной заявке предусмотрено только получение хирального енона. Что касается получения и определения физических характеристик 16(S)-илопроста, не приведены ни пример получения, ни физические характеристики.
Цель авторов настоящего изобретения заключалась в разработке альтернативного синтеза илопроста, который обеспечивает лучший выход, чем ранее известные способы.
Описание изобретения
Способ согласно настоящему изобретению, заявленный в данной заявке, представлен на схеме 9.

Схема 9.
В способе согласно настоящему изобретению сначала формируют верхнюю цепь в содержащем защитную группу лактоне Кори формулы II.
В ходе получения соединения III для образования фосфоната требуется сильное основание для депротонирования диметилметилфосфоната. Однако сильное основание, такое как, например, бутиллитий, применяемое в избыточном количестве, будет раскрывать кольцо лактона (скрытый сложный эфир карбоновой кислоты).
Когда применяют сильное основание, например бутиллитий, анион, образованный из раскрытого кольца лактона, не будет алкилирован фосфонатом и после обработки исходного материала будет получен обратно, в зависимости от условий реакции, в количестве вплоть до 20%.
В способе согласно настоящему изобретению применяют диизопропиламид лития или диалкиламиды лития, которые селективно депротонируют метилдиметилфосфонат, кольцо лактона не раскрывается (остается неизменным), поэтому все количество соединения II алкилируется фосфонатом, и образуется сложный эфир фосфоновой кислоты, причем количество исходного материала после обработки остается ниже 2%.
В случае вышеуказанных соединений предпочтительно применяют диизопропиламид лития, поскольку диизопропиламин, высвобожденный из реагента, может быть удален из продукта.
В ходе получения соединения формулы IV
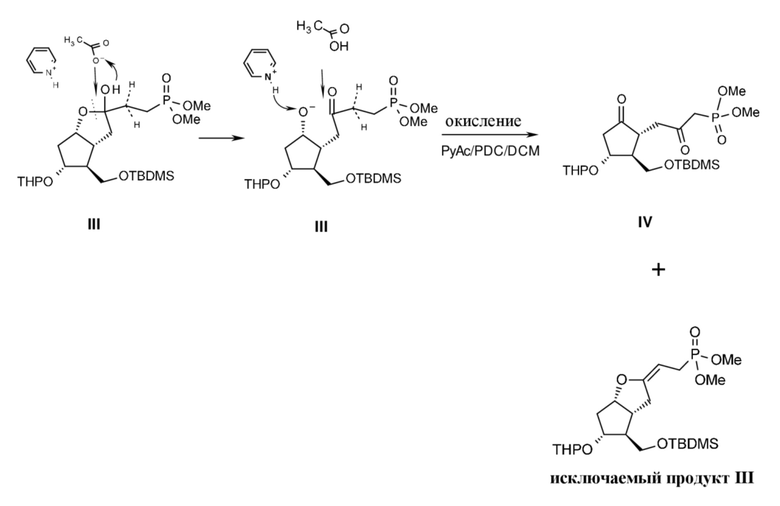
Схема 10.
кольцо лактола должно быть раскрытым (расщепление кеталя), и освобожденная вторичная гидроксильная группа должна быть окислена. Реакция безусловно должна проводиться в кислой среде, поскольку при основных условиях в результате неконтролируемой реакции HWE начнется выработка исключаемого III побочного продукта (схема 10).
В литературе („Synthesis of a carbaprostacyclin intermediate -6β-[(tert-butyldimethylsiloxy)methyl]-7-α-[(tetrahydropyran-2-yl)oxy]bicyclo[3.3.0]octan-3-one", Huaxue Xuebao, 1987, 45(7), 727-9; US4420632 A1, US 9831213, GB 2070596, GB 2070596) для раскрытия кольца применяются просто кислотные условия окислителя Коллинза: CrO3.пиридин, реагент Джонса: CrO3.серная кислота на водной основе, PCC- и PDC-окисления, однако, при сильнокислотных условиях образуется кеталь посредством дегидратации у приблизительно 20-30% так называемого исключаемого соединения III, содержащего двойную связь.
В способе согласно настоящему изобретению кольцо лактола раскрывается при очень мягких кислотных условиях с помощью ацетата пиридиния, и после раскрытия лактола освобожденная гидроксильная группа окисляется при помощи PDC, что происходит при намного более мягких условиях, чем в случае PCC. Окисление происходит медленно, но количество так называемой исключаемой примеси и продуктов разложения намного меньше, менее 5%.
Для получения соединения формулы V аналогично способам, известным из литературы, в способе согласно настоящему изобретению также используют карбонат калия в среде толуола, при этом катализатором внутримолекулярной реакции HWE (Хорнера-Уодсворта-Эммонса) является реагент 18-краун-6. Однако известный способ был доработан авторами настоящего изобретения. На данной стадии реакции происходят одновременно две реакции - внутримолекулярная и межмолекулярная реакции HWE. Подходящие условия, такие как сильное (предпочтительно 30-45-кратное) разбавление, высокая температура (90-110°C) и применяемый способ добавления, будут способствовать внутримолекулярной реакции HWE. В предложенном авторами настоящего изобретения способе добавления к нагревающемуся с обратным холодильником раствору реагентов очень медленно добавляют по каплям раствор IV, таким образом он сразу же реагирует внутримолекулярно. Из-за отсутствия избытка IV является невозможным образование димеров и участие в межмолекулярной реакции Виттига. Результат: соотношение димеров падало от 15% до менее чем 3%. Выход повышался от 35% до 50% (схема 11).

Схема 11.
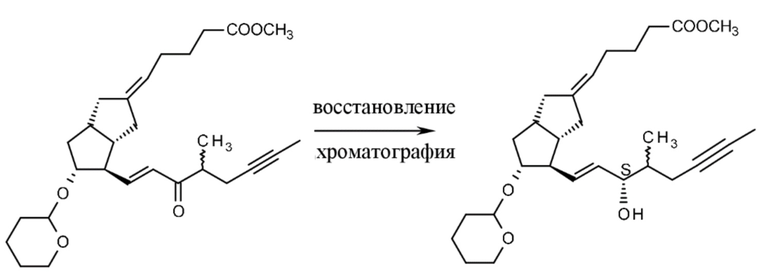
Для получения соединения формулы VI применяют восстановление с помощью палладия на угле, способ, также используемый в литературе, поскольку в качестве растворителя применяют толуол, который содержит триэтиламин для защиты THP-защитных групп. Промежуточное соединение очищают посредством хроматографии. Восстановление можно также проводить с применением никелевого катализатора Ренея.
В способе согласно настоящему изобретению верхняя цепь формируется на стадии, где соединение формулы VII получают путем реакции Виттига. TBDMS-защитную группу необходимого соединения VII и таковую нежелательной изомерной примеси VIIz, полученной как побочный продукт, удаляют при помощи тетрабутиламмония фторида (Bu4NF), полученное соединение VIII (E-изомер) и примесь VIIIz в виде Z-изомера разделяют посредством гравитационной хроматографии. За счет высокоэффективной хроматографической очистки количество примеси в виде Z-изомера может быть снижено до 0,2-0,5% (оптическая чистота, диастереомерный избыток (de)=99,0-99,6%). Для сравнения: чистота, достигаемая в стереоселективной реакции Виттига, способе, описанном в литературе, составляла только de=96%. (Схема 12.)

Схема 12.
В способе согласно настоящему изобретению нежелательный Z-изомер (VIIIz) можно рециркулировать в синтез. В предложенном авторами настоящего изобретения способе рециркуляции нежелательный Z-изомер подвергают изомеризации в УФ-реакторе, в среде толуола в присутствии сенсибилизатора (10 мол. % диметилдисульфида) и из смеси, содержащей 10-30% E-изомера, получают продукт с содержанием E-изомера, составляющим прибл. 55%, который после хроматографической очистки обеспечивает E-изомер (VIII) с выходом 41%, который можно рециркулировать в способ очистки. Выход промежуточного соединения VIII, начиная с промежуточного соединения VI, составляет 65%. Сравнивая техническую возможность и стоимость предложенного авторами настоящего изобретения способа с таковыми способа H-J. Gais и его коллег, где в реакции HWE применяют хиральный фосфонат, можно утверждать, что способ согласно настоящему изобретению является более рентабельным, он не требует дорогостоящего хирального фосфоната, получаемого в ходе большого количества стадий.
Соединение формулы VIII эстерифицируют известным способом в ацетоне с помощью метилйодида в присутствии карбоната калия с получением сложного эфира формулы IX.
Для получения соединения формулы X из соединения IX были разработаны три способа, окисление Пфитцнера-Моффата с помощью DMSO и фосфорной кислоты с содержанием DCC или DIC, а также окисление Анелли (гипохлорит натрия, катализатор TEMPO). Из полученного соединения формулы X формировалась нижняя цепь при помощи способа HWE.
В случае применения окисления Пфитцнера-Моффата предпочтительно в ходе проводящейся в одном реакционном сосуде реакции HWE выделяется фосфонат. После окисления соединение формулы X находится в толуольном растворе, к раствору добавляют небольшое количество THF, затем ILO-фосфонат, а затем твердый KOH. Полезный эффект KOH состоит в том, что он не растворяется в реакционной смеси, таким образом он вступает в реакцию исключительно с ILO-фосфонатом и практически не взаимодействует с альдегидом X, поэтому количество примеси, образованной из альдегида, является очень низким.
Преимущество окисления с помощью катализатора TEMPO по сравнению с вариантами окисления Пфитцнера-Моффата состоит в том, что оно не дает большого количества карбамидного производного, которое сложно удалить.
Соединения формул X и XI являются новыми соединениями.
В способе согласно настоящему изобретению соединения формулы XI восстанавливают.
Были испытаны четыре разных восстанавливающих средства (хлорид церия(III)/NaBH4, катализатор катехолборан-CBS-оксазаборолидин, диизоборнеолоксидиизопропилалюминат, DIBAL-F). Влияния других параметров (например, температуры) также исследовали.
Наиболее подходящим восстанавливающим средством оказался DIBAL-F, причем данный реагент получали путем проведения реакции диизобутилалюминия гидрида с 2,6-ди-трет-бутил-4-метилфенолом.
Основная примесь представляет собой побочный продукт восстановления - 11-THP-15-эпиилопроста метиловый сложный эфир. (Схема 13. )
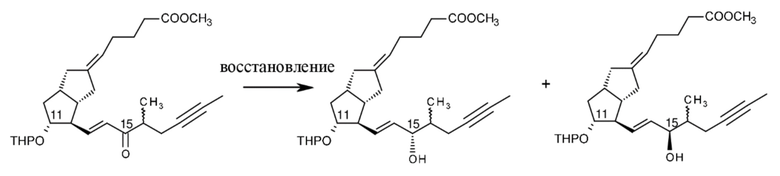
XI XII 15-эпи-XII
11-THP-15-эпиилопроста метиловый сложный эфир
Схема 13.
Соотношение продукта и 15-эпимера при восстановлении составляло 75:25 при использовании восстанавливающего средства DIBAL-F (в первых экспериментах с использованием реагентов хлорид церия/боргидрид натрия данное изомерное соотношение составляло 60:40).
После разложения восстанавливающего средства и удаления водорастворимых побочных продуктов THP-группу расщепляли с помощью метанола и пара-толуолсульфоновой кислоты. В предпочтительном способе толуольный раствор не обрабатывают, но защитную группу удаляют без выделения соединения формулы XII.
Соединение формулы XII является новым.
Соединения формулы XIII можно очищать посредством хроматографии. В ходе нормально-фазовой гравитационной хроматографии (н-гексан:этилацетат) очистка от основного побочного продукта восстановления, эпимерной примеси, происходит на прибл. 95%, кроме того, количество некоторых примесей, которые затрудняют очистку илопроста, также значительно снижается.
Соединение формулы XIII можно также очищать способами обращенно-фазовой и препаративной ВЭЖХ. Как на C18 силикагеле, так и на полистирольной смоле посредством использования элюентов, применимых для обращенной фазы, главным образом элюентов ацетонитрил:вода или метанол:вода, соединение XIII (метиловый сложный эфир илопроста) можно успешно очищать от родственных и других примесей. Для удаления родственных примесей наиболее эффективным способом является очистка илопроста посредством препаративной ВЭЖХ.
В способе согласно настоящему изобретению соединение XIIIb, полученное как побочный продукт, можно рециркулировать в синтез, причем при окислении оно обеспечивает получение соединения XIb, которое после введения защитной группы для 11-OH с помощью THP обеспечивает получение промежуточного соединения формулы XI. (Схема 14.)

XIIIb XIb XI
Схема 14.
Соединение XIIIb растворяют в этилацетате и фильтруют через насадку из активированного диоксида марганца. Фильтрат повторно выливают на насадку и фильтруют. После второй фильтрации продукт XIb вымывают из насадки этилацетатом, насыщенным водой, жидкий фильтрат объединяют с промывными водами и выпаривают. Выпаренный неочищенный продукт сушат от воды путем добавления и отгонки толуола.
Дигидропиран и катализатор, пара-толуолсульфоновую кислоту, добавляют в толуольный раствор для защиты 11-гидроксильной группы с помощью THP-группы. Реакцию останавливают с помощью триэтиламина и реакционную смесь выпаривают. Полученный концентрат рециркулируют на стадию стереоселективного восстановления с получением XII.
Путем рециркуляции промежуточного соединения XI, полученного из побочного продукта XIIIb, выход селективного восстановления повышается с 56% до 63% (XI-XIII).
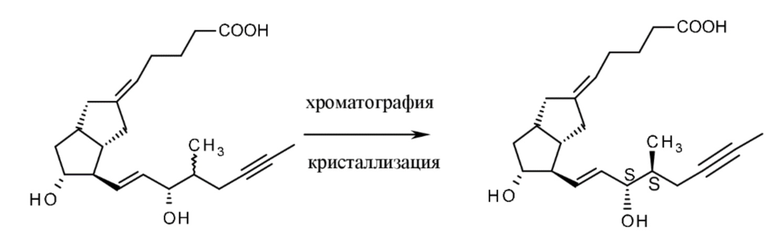
Гидролиз метилового сложного эфира формулы XIII обеспечивает получение илопроста, причем гидролиз проводят в THF с помощью водного раствора NaOH при интенсивном перемешивании.
Илопрост, полученный в конце реакции, все еще содержит на измеримом уровне родственные примеси, 15-эпиилопрост и 15-оксоилопрост, и побочные продукты в результате окисления. Очистку илопроста проводят посредством препаративной ВЭЖХ. Были разработаны два способа: для материала, не загрязненного 15-оксоилопростом, применяли элюент, содержащий ацетонитрил:воду, а для илопроста, в высокой степени загрязненного 15-оксо-илопростом, - элюент, содержащий спирт:воду. В ходе очистки Z-изомер илопроста также удаляли.
Предложенный авторами настоящего изобретения способ в равной степени применим для получения 16(S)-илопроста и 16(R)-илопроста, если вместо рацемического ILO-фосфоната реакцию HWE проводят с применением хирального (S)-ILO-фосфоната или хирального (R)-ILO-фосфоната.
16(S)-Илопрост, полученный в энантиоселективном синтезе с применением хирального (S)-ILO-фосфоната, можно также выделять в кристаллической форме.
Согласно вышесказанному настоящее изобретение относится к способу получения илопроста формулы I,
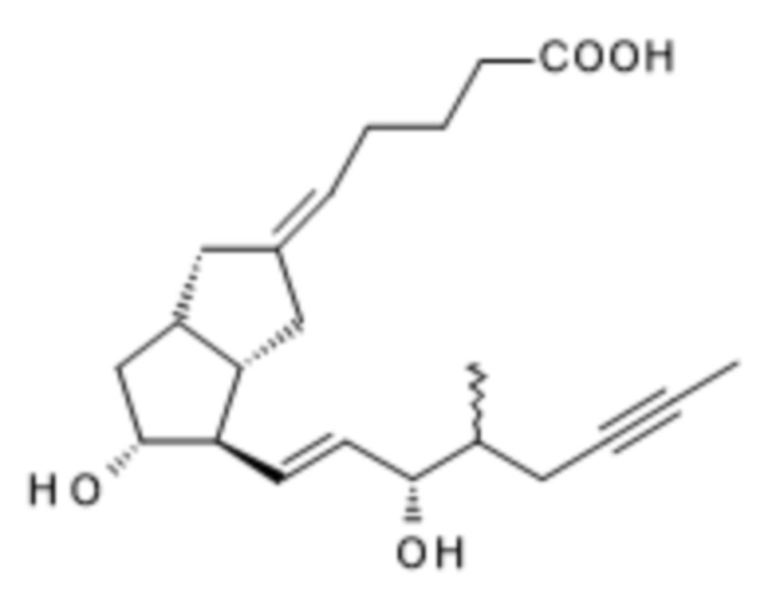 I,
I,
таким образом, что
a.) лактон Кори формулы II,
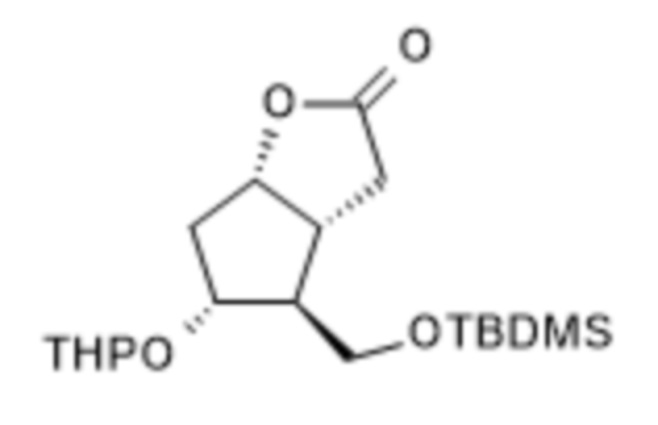 II,
II,
подвергают селективному алкилированию с помощью диметилметилфосфоната в присутствии диалкиламида лития,
b.) кольцо полученного лактола формулы III,

III,
подвергают раскрытию с помощью ацетата пиридиния в слабокислой среде, затем полученную вторичную гидроксильную группу окисляют дихроматом пиридиния,
c.) полученное соединение формулы IV,

IV,
приводят в реакцию с карбонатом калия в присутствии реагента 18-краун-6,
d.) полученное соединение формулы V,

V,
восстанавливают,
e.) полученное соединение формулы VI,

VI,
приводят в реакцию с карбоксибутилтрифенилфосфония бромидом в присутствии трет-бутилата калия,
f.) TBDMS-защитную группу полученных E- и Z-изомеров формулы VII,

VII,
удаляют, изомеры разделяют посредством гравитационной хроматографии, при необходимости Z-изомер (VIIIz) подвергают изомеризации до E-изомера,
g.) полученное соединение формулы VIII,

VIII,
подвергают эстерификации,
h.) полученное соединение формулы IX,

IX,
подвергают окислению,
i.) полученное соединение формулы X,
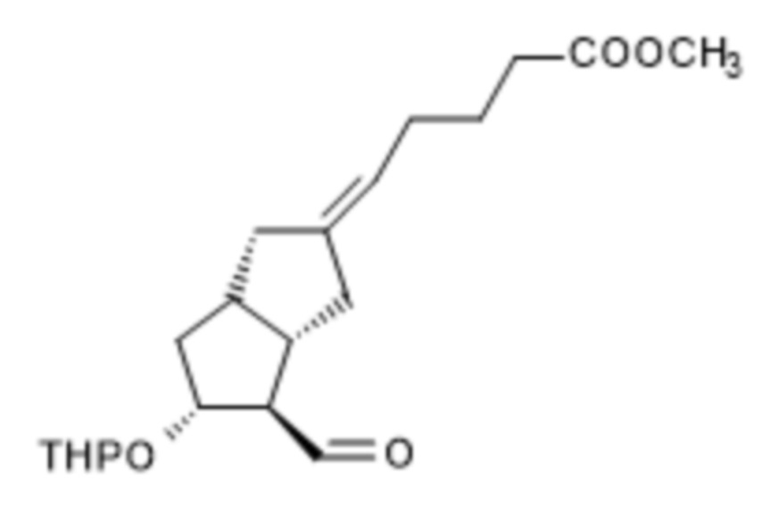
X,
подвергают превращению в реакции HWE в присутствии твердого гидроксида калия в соединение формулы XI,
j.) оксогруппу полученного таким образом соединения формулы XI,

XI,
восстанавливают с помощью DIBAL-F,
k.) тетрагидропиранильную защитную группу соединения формулы XII,

XII,
удаляют и соединение очищают посредством гравитационной колоночной хроматографии, при необходимости дополнительно очищают посредством препаративной ВЭЖХ,
l.) сложноэфирную группу соединения формулы XIII,
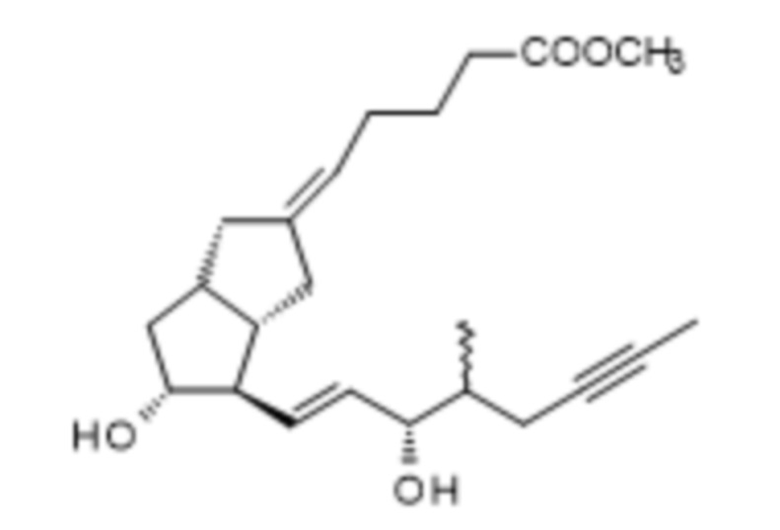
XIII,
удаляют и полученное соединение формулы I очищают.
В предпочтительном варианте осуществления способа согласно настоящему изобретению в качестве диалкиламида лития используют диизопропиламид лития или дициклогексиламид лития. Для получения соединения формулы V реакцию HWE проводят при сильном разбавлении и при высокой температуре, предпочтительно при 90-110°C, причем таким образом, что раствор соединения формулы IV добавляют по каплям в нагреваемый с обратным холодильником раствор реагентов.
Разделение E- и Z-изомеров формулы VIII проводят с применением ступенчато-градиентных смесей
толуол:метил-трет-бутиловый эфир в качестве элюента.
Изомеризацию Z-изомера проводят путем облучения в присутствии сенсибилизатора диметилдисульфида, расщепление силильной защитной группы проводят при помощи тригидрата тетрабутиламмония фторида.
Окисление соединения формулы IX проводят путем окисления Пфитцнера-Моффата с помощью смеси фосфорная кислота-DMSO с содержанием DCC или DIC или путем окисления Анелли (гипохлорит натрия, катализатор TEMPO).
В предпочтительном варианте способа согласно настоящему изобретению соединения формул X и XII не выделяют.
В другом предпочтительном варианте осуществления способа согласно настоящему изобретению 15R-изомер формулы XIIIb выделяют посредством хроматографии, а после окисления затем THP-защиту 11-OH-группы рециркулируют в синтез.
Неочищенный готовый продукт формулы I очищают посредством гравитационной хроматографии и/или препаративной ВЭЖХ.
Соединения формул X, XI и XII являются новыми соединениями.
Для готового продукта - илопроста - чистота является наиболее важным параметром, поскольку продукты различной чистоты ведут себя по-разному. Из илопроста, достигающего определенного уровня чистоты, можно получить твердофазный продукт.
Дополнительным объектом настоящего изобретения является получение твердой формы илопроста путем растворения очищенного маслянистого соединения формулы I в таком же количестве полярного растворителя, что и масса маслянистого продукта, добавления к полученному такого количества алкана, что раствор становится опалесцирующим, затем отверждения раствора до стекловидной массы при температуре от (-)60°C до (-)20°C и удаления растворителя в высоком вакууме.
Предпочтительно илопрост, уже очищенный от его примесей, подвергают специальной обработке, его растворяют в ацетоне, затем добавляют пентан до тех пор, пока раствор не станет опалесцирующим, после этого раствор быстро охлаждают до (-)60°C, где масло полностью осаждается из раствора ацетона-пентана и полученный осадок отверждается, превращается в полупрозрачное, стекловидное, аморфное твердое вещество. После отверждения илопрост выдерживают при (-)20°C; в течение этого периода в среде ацетона-пентана стекловидное твердое вещество превращается в белый, комковатый твердый материал. В ходе отверждения растворитель несколько раз удаляют декантированием и в смесь добавляют н-пентан для снижения содержания ацетона.
После последнего декантирования растворители удаляют из суспензии в высоком вакууме при (-)20°C, при этом продукт превращается в твердый порошок.
A. Неочищенный илопрост (доступная чистота: 93%)
Метиловый сложный эфир илопроста растворяют в THF и подвергают гидролизу при помощи 1 M раствора NaOH до кислоты. После гидролиза илопрост находится в водной щелочной фазе в растворе в виде его натриевой соли. Смесь экстрагируют метил-трет-бутиловым эфиром для снижения количества неполярных примесей, затем илопрост выделяют из его натриевой соли при помощи гидросульфата натрия и экстрагируют из водного раствора метил-трет-бутиловым эфиром. После сушки органической фазы и выпаривания неочищенный илопрост получают в виде масла, которое не может отверждаться, превращаться в его твердую форму, из-за высокого уровня примесей. Чистота полученного неочищенного илопроста: 93% (масло).
Для очистки неочищенного илопроста, полученного в способе согласно настоящему изобретению, авторы разработали несколько способов.
2. Отверждение
2. Фильтрация на силикагеле
3. Отверждение
2. Препаративная хроматография
3. Фильтрация на силикагеле
4. Отверждение
B. Очистка неочищенного илопроста посредством гравитационной хроматографии и отверждение (достигаемая чистота: 95,0%)
Неочищенный илопрост очищают посредством гравитационной хроматографии с применением ступенчато-градиентных смесей-элюентов и силикагеля с частицами неправильной формы размером 0,063-0,2 мм и размером пор 60 ангстрем в качестве насадки. Материал растворяют в ацетоне, затем к нему добавляют такое количество алкана, что он становится опалесцирующим. Раствор выливают в колонку, промывают элюентом, и происходит элюирование. Элюенты представляют собой смеси алкан:ацетон, алкан:метилэтилкетон, алкан:этилацетат или алкан:изопропанол, где алкан представляет собой н-пентан, н-гексан, циклогексан или гептан.
Основной продукт гравитационной хроматографии после выпаривания растворителей имеет чистоту выше 95,0%, данный продукт уже подходит для получения твердого илопроста путем применения методик, описанных ниже.
Масло илопроста, очищенное гравитационной хроматографией, растворяют в таком же количестве ацетона (или этилацетата, или метилэтилкетона, или изопропанола), что и масса масла, в раствор добавляют такое количество нормального пентана (или гексана, или циклогексана, или гептана), что он становится опалесцирующим, после этого раствор хранят при (-)60°C без перемешивания. Через 6 часов раствор отверждается в виде стекловидного продукта. Данный стекловидный, отвержденный материал далее хранят при (-)20°C в течение по меньшей мере 16 часов, при этом он превращается в белую, комковатую твердую форму. Основную часть растворителя удаляют декантированием. В конце отверждения оставшийся растворитель удаляют в высоком вакууме при температуре от (-)10°C до (-)30°C.
Полученный очищенный илопрост представляет собой белый порошок с чистотой 95,0%.
C. Очистка неочищенного илопроста посредством препаративной ВЭЖХ с последующими фильтрацией на силикагеле и отверждением (достигаемая чистота: 98,0%)
Неочищенный илопрост непосредственно очищают методом препаративной ВЭЖХ. Материал растворяют в таком же количестве ацетонитрила, что и его масса, затем к нему добавляют воду. Раствор фильтруют через обращенно-фазовую предколонку, изготовленную из C18 силикагеля с размером частиц 10 микронов и размером пор 120 ангстрем. Очистку профильтрованного исходного раствора проводят посредством препаративной жидкостной хроматографии высокого давления, с обращенной фазой, с применением C8- или C18-насадки или насадки из полистирольной смолы и смесей растворителей вода-органическое вещество в качестве элюентов. Компонент элюента - органический растворитель - представляет собой ацетонитрил, метанол, этанол или изопропанол.
Объединенную основную фракцию после хроматографии концентрируют в вакууме при 40°C, концентрированный раствор экстрагируют метил-трет-бутиловым эфиром, объединенную органическую фазу промывают насыщенным солевым раствором, сушат над сульфатом натрия и концентрируют в вакууме при 30°C.
К концентрированному раствору добавляют сначала ацетон, а затем осторожно, пока раствор не станет опалесцирующим, добавляют н-пентан. Полученный таким образом раствор затем дополнительно очищают посредством нормально-фазовой хроматографии с применением слоя силикагеля с частицами неправильной формы размером 0,063-0,2 мм и размером пор 60 ангстрем и ступенчато-градиентных смесей н-пентан:ацетон в качестве элюента. Основную фракцию выпаривают в высоком вакууме (0,1 мбар) при 30°C. Масло илопроста, профильтрованное через силикагель, превращается в твердый продукт, как описано в способе B.
Илопрост, очищенный указанным выше способом, представляет собой белый порошок с чистотой 98,0%.
D. Очистка неочищенного илопроста посредством гравитационной и препаративной ВЭЖХ с последующими фильтрацией на силикагеле и отверждением
Неочищенный илопрост очищают посредством гравитационной хроматографии, как описано в способе B. Полученное масло илопроста дополнительно очищают с помощью способа препаративной ВЭЖХ, как описано в способе C. К полученному концентрированному раствору добавляют ацетон, а затем осторожно, пока раствор не станет слегка опалесцирующим, добавляют н-пентан. Полученный раствор затем дополнительно очищают посредством нормально-фазовой хроматографии на слое, изготовленном из силикагеля с частицами неправильной формы размером 0,063-0,2 мм и размером пор 60 ангстрем, с применением ступенчато-градиентных смесей н-пентан:ацетон в качестве элюента. Основную фракцию выпаривают в высоком вакууме при 30°C. Полученное масло илопроста, профильтрованное через силикагель, затем превращается в твердый продукт, как описано в способе B.
Илопрост, очищенный указанным выше способом, представляет собой белое кристаллическое вещество с чистотой 98,5%.
Из диастереомеров, разделенных посредством препаративной ВЭЖХ, может быть выкристаллизован 16(S)-илопрост. Предпочтительно растворением в ацетоне и осаждением с помощью пентана его выделяют в кристаллической форме.
Дополнительный объект настоящего изобретения 16(S)-илопрост в кристаллической форме является новым.
В соответствии с вышеизложенным при помощи способа согласно настоящему изобретению можно получать следующее:
изомер 16(S)-илопрост в виде кристаллической фазы.
Илопрост в виде маслянистой или твердой порошкообразной фазы с чистотой по меньшей мере 98,5%, при этом общее количество родственных примесей составляет не более 1,6%, общее количество неидентифицированных примесей составляет не более 0,5%, и количество каждой из неидентифицированных примесей составляет не более 0,1%.
Илопрост в виде маслянистой или твердой порошкообразной фазы с чистотой по меньшей мере 98,5% удовлетворяет следующим требованиям в отношении качества.
Z-изомеры илопроста, общее количество
другие примеси, общее количество
из которых
- 15-эпиилопрост
- 15-оксоилопрост
- метиловый сложный эфир илопроста
- этиловый сложный эфир илопроста
- димер 1 илопроста
- димер 2 илопроста
- неидентифицированные примеси, каждая
≤ 1,0%
≤ 0,10%
≤ 0,20%
≤ 0,20%
≤ 0,05%
≤ 0,10%
≤ 0,10%
≤ 0,10%
Илопрост в маслянистой или твердой порошкообразной фазе с чистотой по меньшей мере 98,0%, при этом общее количество родственных примесей составляет не более 1,6%, общее количество неидентифицированных примесей составляет не более 1,0%, и количество каждой из неидентифицированных примесей составляет не более 0,1%.
Илопрост в маслянистой или твердой порошкообразной фазе с чистотой по меньшей мере 98,0% удовлетворяет следующим требованиям в отношении качества.
Z-изомеры илопроста, общее количество
другие примеси, общее количество
из которых
- 15-эпиилопрост
- 15-оксоилопрост
- метиловый сложный эфир илопроста
- этиловый сложный эфир илопроста
- димер 1 илопроста
- димер 2 илопроста
- неидентифицированные примеси, каждая
≤ 1,0%
≤ 0,20%
≤ 0,20%
≤ 0,10%
≤ 0,10%
≤ 0,20%
≤ 0,20%
≤ 0,10%
Илопрост в маслянистой или твердой порошкообразной фазе с чистотой по меньшей мере 95,0%, при этом общее количество родственных примесей составляет не более 3,5%, и общее количество неидентифицированных примесей составляет не более 2,5%.
16(S)-Илопрост в виде кристаллической фазы можно также получать путем продемонстрированного химического пути
m.), если альдегид формулы X превращается в соединение формулы (S)-XI посредством реакции с (S)-ILO-фосфонатом в присутствии твердого гидроксида калия,
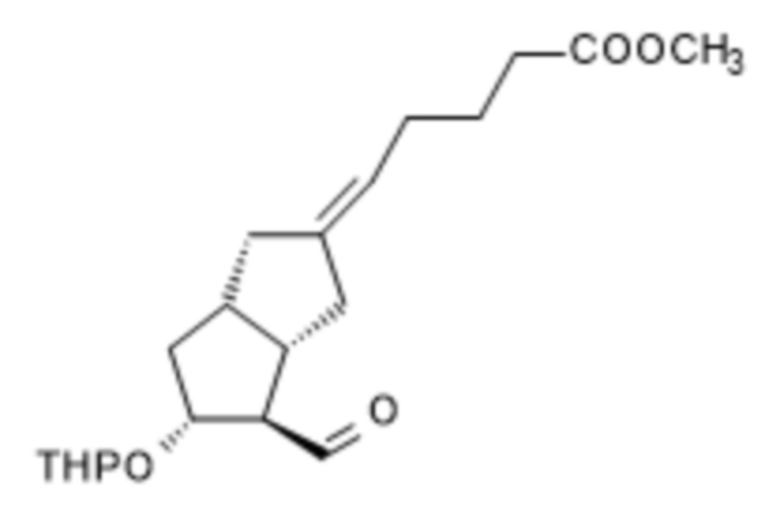

X альдегид (S)-ILO-фосфонат
n.) оксогруппу полученного соединения формулы (S)-XI

(S)-XI
восстанавливают с помощью DIBAL-F,
o.) тетрагидропиранильную защитную группу соединения формулы (S)-XII
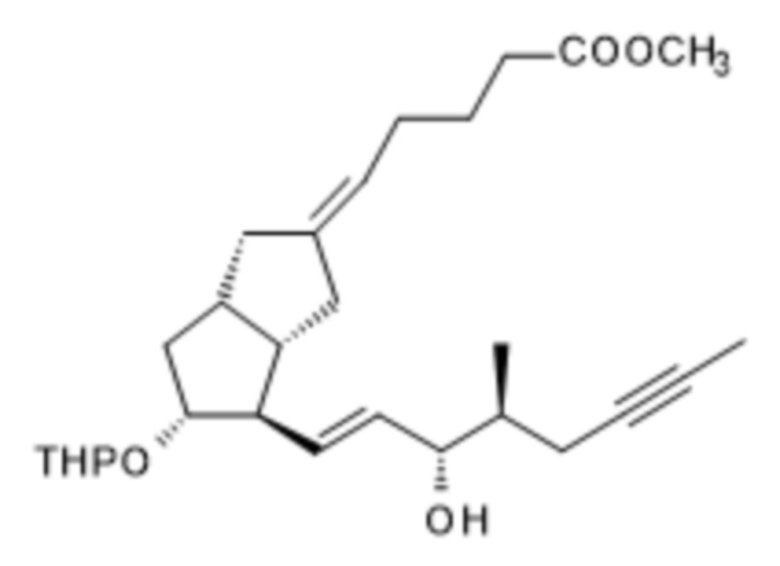
(S)-XII
расщепляют и соединение очищают посредством хроматографии,
p.) сложноэфирную группу полученного соединения формулы (S)-XIII
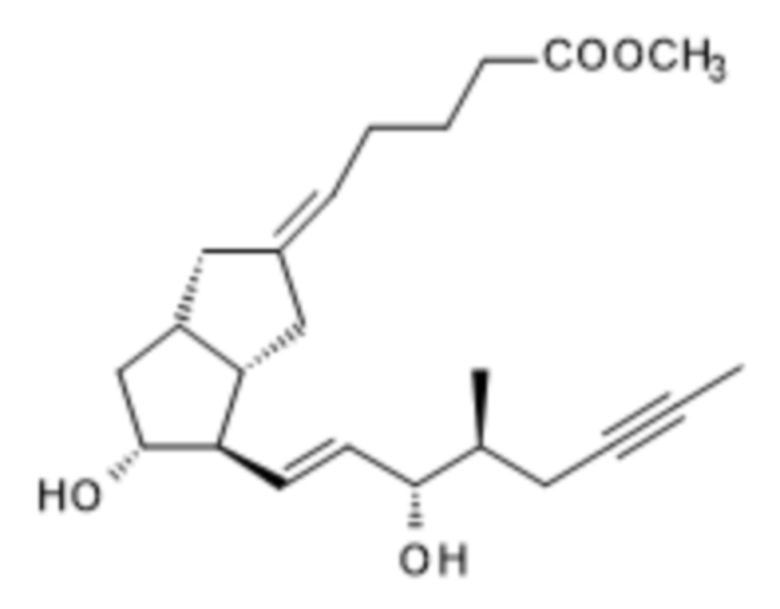
(S)-XIII
удаляют и полученное соединение формулы (S)-I очищают.
q.) Если альдегид формулы X превращают в соединение формулы (R)-XI посредством реакции с (R)-ILO-фосфонатом в присутствии твердого гидроксида калия,
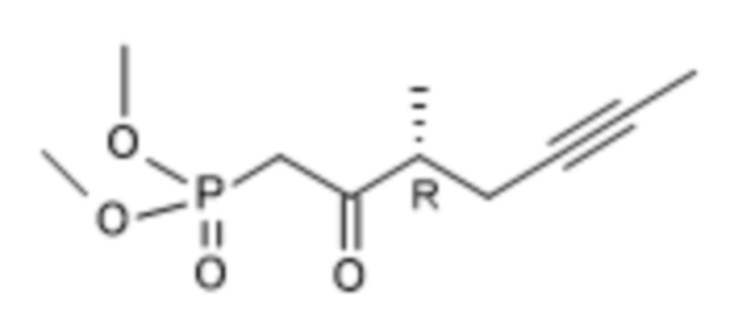
X альдегид (R)-ILO-фосфонат
r.) оксогруппу полученного соединения формулы (R)-XI
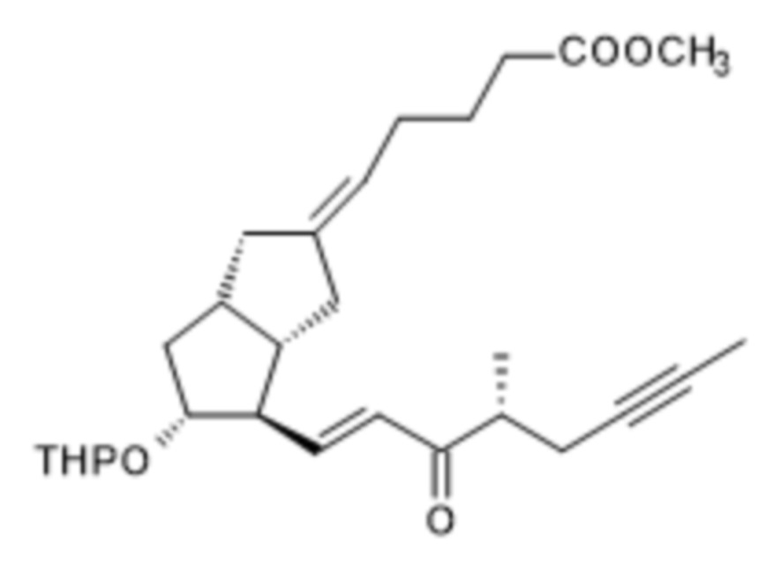
(R)-XI
восстанавливают с помощью DIBAL-F,
s.) тетрагидропиранильную защитную группу соединения формулы (R)-XII

(R)-XII
расщепляют и соединение очищают посредством хроматографии,
t.) сложноэфирную группу полученного соединения формулы (R)-XIII удаляют
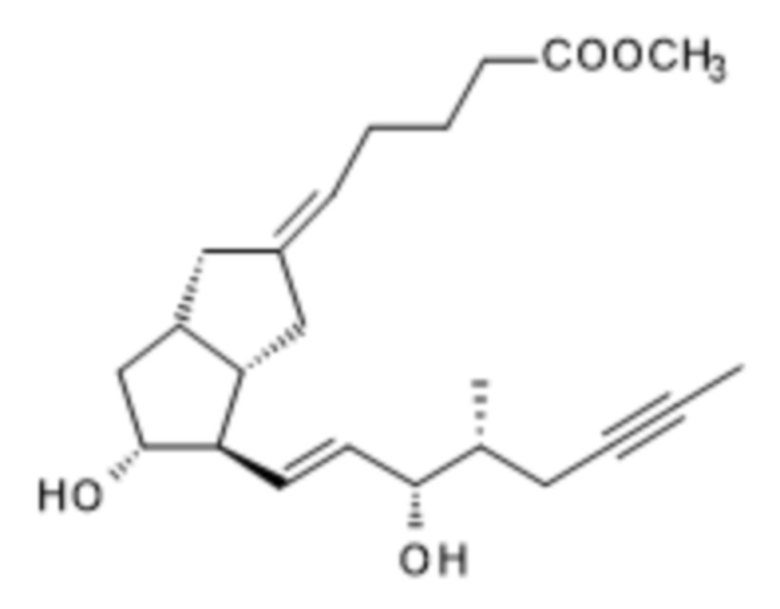
(R)-XIII
и полученное соединение формулы (R)-I очищают, после чего также может быть получен изомер 16-(R)-илопрост.
Краткое описание графических материалов/фигур
Фигура 1: рентгенограмма XRPD илопроста (пример 1m).
Фигура 2: рентгенограмма XRPD 16-(S)-илопроста (пример 1n).
Фигура 3: ДСК-кривая илопроста (пик: 67,58°C, пример 1m).
Фигура 4: ДСК-кривая 16-(S)-илопроста (пик: 104,62°C, пример 1n).
Фигура 5: данные 13C- и 1H-ЯМР илопроста, полученные при 500 МГц в DMSO (пример 1m).
Фигура 6: данные 13C- и 1H-ЯМР соединения формулы XI, полученные при 500 МГц в DMSO (пример 1h).
Фигура 7: данные 13C- и 1H-ЯМР соединения формулы (S)-XI, полученные при 500 МГц в DMSO (пример 1o).
Фигура 8: данные 13C- и 1H-ЯМР соединения формулы (S)-XII, полученные при 500 МГц в DMSO (пример 1 p).
Фигура 9: данные 13C- и 1H-ЯМР 16-(S)-илопроста, полученные при 500 МГц в DMSO (пример 1n).
Примеры
Объект настоящего изобретения подробно описан в примерах без ограничения формулы изобретения вариантами способов, описанных в примерах.
Условия измерений, проводимых в способах согласно настоящему изобретению.
Рентгеновская дифракция:
Исходное положение [°2тета]: 2,0074
Конечное положение [°2тета]: 39,9854
Температура проведения измерения [°C]: 25,00
Материал анода: Cu
K-альфа1 [Ĺ]: 1,54060
K-альфа2 [Ĺ]: 1,54443
ДСК:
Прибор: система METTLER TOLEDO DSC1 STARe, Stare basic V9.30
Методика: Начальная температура: 30°C
Конечная температура: 200°C
Скорость нагрева: 5°C/мин.
Навеска: 5-9 мг, перфорированный алюминиевый тигель (40 мкл)
ЯМР:
Прибор: Bruker Avance III 500 МГц
Растворитель: ДМСО
1a/ Получение [[4-[[[(1,1-диметилэтил)диметилсилил]окси]метил]гексагидро-2-гидрокси-5-[(тетрагидро-2H-пиран-2-ил)окси]-2H-циклопента[b]фуран-2-ил]метил]фосфоновой кислоты диметилового сложного эфира ( III )

C19H34O5Si C22H43O8PSi
Mr: 370,6 Mr: 494,6
II III
В 50 л безводного тетрагидрофурана, в инертной атмосфере, растворяли 4,2 л диметилметилфосфоната и реакционную смесь охлаждали до (-)75°C. При поддержании заданной температуры добавляли 22,5 л н-бутиллития в 1,6 M растворе гексана, а затем 7,5 кг II в 15 л раствора безводного тетрагидрофурана. В конце реакции реакционную смесь гасили 1 M гидросульфатом натрия. Погашенную реакционную смесь экстрагировали толуолом, органическую фазу промывали раствором гидрокарбоната натрия, содержащим хлорид натрия, и толуольный раствор выпаривали в вакууме при 50°C.
Выход: 9,6 кг (96%), масло.
1a/B
Готовили раствор LDA: к 28,7 кг тетрагидрофурана добавляли 5,8 кг диизопропиламина, раствор охлаждали до 0±5°C, затем в течение периода времени, составляющего 1 час, в потоке азота, при непрерывном перемешивании и охлаждении туда добавляли 21 кг 1,6 M раствора бутиллития, поддерживая при этом температуру на уровне 0±5°C. После добавления охлаждение прекращали и смесь перемешивали при 5±10°C в течение 1 часа.
Во втором устройстве в 32,3 кг тетрагидрофурана растворяли 7,5 кг II, к раствору добавляли 4,2 л диметилметилфосфоната, смесь охлаждали до (-)5±5°C в потоке азота и при перемешивании, затем к смеси добавляли ранее полученный раствор LDA, поддерживая при этом температуру на уровне от (-)5°C до (+)5°C.
За реакцией наблюдали с помощью TLC. Ожидаемое время реакции: 60 минут.
В конце реакции смесь переносили посредством отсасывания в 1 M раствор гидросульфата натрия и туда же добавляли толуол. Водную фазу дважды экстрагировали толуолом, объединенную органическую фазу промывали последовательно 15% раствором хлорида натрия и 1 н. раствором гидрокарбоната натрия. Органическую фазу выпаривали в вакууме.
Выход (в пересчете на содержание сухого материала): 9,6 кг (96%). Бесцветное масло.
1b. Получение [1R-(1α,2β,3α)]-[3-[2-[[[(1,1-диметилэтил)диметилсилил]окси]метил]-5-оксо-3-[(тетрагидро-2H-пиран-2-ил)окси]циклопентил]-2-оксопропил]фосфоновой кислоты диметилового сложного эфира ( IV )
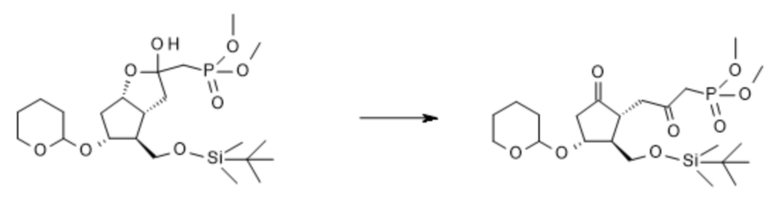
C22H43O8PSi C22H41O8PSi
Mr: 494,6 Mr: 492,6
III IV
Получение реагента ацетата пиридиния.
К 170 кг дистиллированного дихлорметана отвешивали 11,5 кг пиридина и при перемешивании добавляли 6,9 кг уксусной кислоты. Смесь охлаждали при перемешивании до 25±5°C.
Гидролиз и окисление кеталя
К раствору ацетата пиридиния добавляли 9,6 кг III, растворенного в 14 л дихлорметана. Смесь перемешивали в атмосфере азота.
После 30 минут перемешивания добавляли 9,6 кг дихромата пиридиния и смесь перемешивали при 25±5°C до достижения заданной степени превращения. За реакцией наблюдали с помощью TLC. Ожидаемое время реакции: 24-48 часов. При достижении необходимой степени превращения реакционную смесь нагревали до 40±5°C, перемешивали в течение 30 минут, охлаждали снова до 25±5°C, а затем добавляли толуол и перфил. Твердые материалы удаляли центрифугированием. Фильтрат, который содержит продукт, промывали 2 M раствором гидросульфата натрия, фазы разделяли. Водную фазу экстрагировали толуолом, объединенную органическую фазу промывали последовательно раствором гидрокарбоната натрия, содержащим хлорид натрия, и 20% раствором хлорида натрия, сушили над сульфатом натрия и концентрировали в вакууме при 50°C до определенного объема. Концентрированный остаток очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей диизопропиловый эфир:ацетон в качестве элюента.
Выход: 6,0 кг (62,8%), масло.
1c. Получение [5R-(5α,6β,6aα)]-6-[[[(1,1-диметилэтил)диметилсилил]окси]метил]-4,5,6,6a-тетрагидро-5-[(тетрагидро-2H-пиран-2-ил)окси]-2(1H)-пенталенона ( V )
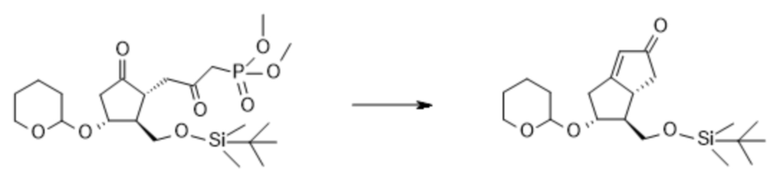
C22H41O8PSi C20H34O4Si
Mr: 492,6 Mr: 366,6
IV V
К 50 л безводного толуола в инертной атмосфере отвешивали 0,34 кг 18-краун-6 и 3,4 кг карбоната калия. Реакционную смесь нагревали до 90°C и добавляли к ней раствор 6 кг IV в безводном толуоле.
Реакционную смесь перемешивали при поддержании указанной температуры. После достижения необходимой степени превращения смесь охлаждали до комнатной температуры, отфильтровывали карбонат калия, фильтрат концентрировали в вакууме при 45°C.
Выход: 4,4 кг (98,5%), масло. Маслянистый продукт можно применять на следующей стадии без очистки.
При необходимости продукт можно очистить посредством хроматографии с применением градиентных смесей гексан:диизопропиловый эфир, выход основной фракции: 2,23 кг масла (50%).
1d. Получение [3aS-(3aα,4α,5β,6aα)]-4-[[[(1,1-диметилэтил)диметилсилил]окси]метил]гексагидро-5-[(тетрагидро-2H-пиран-2-ил)окси]-2(1H)-пенталенона ( VI )

C20H34O4Si C20H36O4Si
Mr: 366,6 Mr: 368,6
V VI
К 6,5 кг V, растворенного в 50 л толуола, добавляли 100 мл триэтиламина и смесь гидрогенизировали при комнатной температуре под давлением 3,5 бара с применением катализатора, представляющего собой палладий на угле и содержащего 10% палладия. В конце реакции катализатор отфильтровывали, промывали толуолом и фильтрат концентрировали в вакууме при 45°C. Концентрированный остаток очищали посредством хроматографии на колонке с силикагелем с применением смесей н-гексан:диизопропиловый эфир и диизопропиловый эфир:ацетон в качестве элюентов. К смеси растворителей н-гексан: диизопропиловый эфир, применяемой для подготовки колонки, добавляли триэтиламин в количестве 0,03% по объему.
Выход: 1,5 кг (23%), масло.
1e. Получение [3aS-(2E,3aα4α,5β,6aα)]-5-[4-[[[(1,1-диметилэтил)диметилсилил]окси]метил]гексагидро-5-[(тетрагидро-2H-пиран-2-ил)окси]-2(1H)-пенталенилиден]пентановой кислоты ( VII )
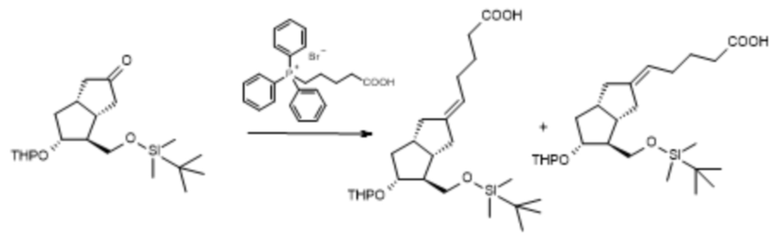
C20H36O4Si C25H44O5Si
Mr: 368,6 Mr: 452,7
VI VII VIIz
7,0 кг карбоксибутилтрифенилфосфония бромида отвешивали в 35 л безводного тетрагидрофурана, реакционную смесь охлаждали до 5°C и добавляли 3,8 кг трет-бутилата калия. Смесь перемешивали при комнатной температуре в течение 15 минут, затем охлаждали до 5°C и добавляли раствор 1,7 кг VI в 8 л тетрагидрофурана. Перемешивание продолжали, поддерживая при этом заданную температуру, до достижения необходимой степени превращения, затем добавляли воду и смесь концентрировали в вакууме при 45°C. Концентрированную реакционную смесь охлаждали до 10°C, осажденный твердый материал отфильтровывали, жидкий фильтрат разбавляли водой, промывали смесью метилэтилкетон:н-гексан. Регулировали pH водной фазы до pH=7-7,5 с помощью 1 н. раствора гидросульфата натрия. Водную фазу экстрагировали диизопропиловым эфиром. Органическую фазу промывали 20% раствором хлорида натрия и после добавления триэтиламина ее выпаривали в вакууме при 45°C.
Выход: 1,71 кг (82%, смесь VII и VIIz), масло.
1f/A . Получение [3aS-(2E,3aα,4α,5β,6aα)]-5-[гексагидро-4-(гидроксиметил)-5-[(тетрагидро-2H-пиран-2-ил)окси]-2(1H)-пенталенилиден]пентановой кислоты ( VIII )
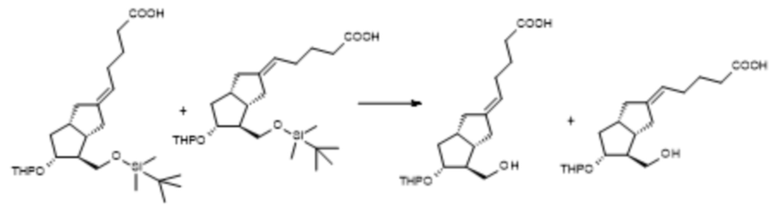
C25H44O5Si C19H30O5
Mr: 452,7 Mr: 338,4
VII VIII VIIIz
3 кг тригидрата тетрабутиламмония фторида суспендировали в 10 л толуола и сушили от воды путем отгонки толуола. Безводную суспензию охлаждали до 20°C и добавляли 2,3 кг VII, растворенной в 24 л тетрагидрофурана. Смесь перемешивали при 60°C. При достижении необходимой степени превращения к реакционной смеси добавляли воду, толуол и триэтиламин. После перемешивания фазы разделяли, водную фазу промывали толуолом, объединенную органическую фазу экстрагировали водой. Регулировали pH объединенной водной фазы до pH=4-6 с помощью 1 М раствора гидросульфата натрия. Подкисленную водную фазу экстрагировали диизопропиловым эфиром. Объединенную органическую фазу промывали 20% раствором хлорида натрия и после добавления пиридина ее выпаривали в вакууме при 45°C. Остаток, полученный в результате выпаривания, сушили от воды путем азеотропной перегонки с толуолом, а затем очищали посредством хроматографии.
В ходе хроматографической очистки нежелательный Z-изомер (VIIIz) также отделяли, его затем подвергали изомеризации по двойной связи в УФ-реакторе.
Элюирование VIII и VIIIz проводили с применением ступенчато-градиентных смесей толуол:метил-трет-бутиловый эфир. Смеси-элюенты содержат пиридин в количестве 0,5% по объему.
В ходе хроматографии нежелательный Z-изомер отмывали ацетоном, содержащим уксусную кислоту в количестве 0,2% по объему. К фракции, которая содержит Z-изомер, добавляли триэтиламин и раствор концентрировали. Концентрат затем разбавляли 10-кратным количеством метил-трет-бутилового эфира и для удаления неорганических солей из продукта его экстрагировали водой и насыщенным солевым раствором. Органическую фазу выпаривали (изомер VIIIz).
Концентрированную основную фракцию соединения VIII, полученную после хроматографической очистки, экстрагировали 1 M раствором карбоната калия, объединенную органическую фазу промывали метил-трет-бутиловым эфиром. Регулировали pH водной фазы до pH=4-6 с помощью 1 М раствора гидросульфата натрия. Подкисленный раствор экстрагировали метил-трет-бутиловым эфиром. Объединенную органическую фазу промывали 20% раствором хлорида натрия и после добавления триэтиламина ее выпаривали в вакууме при 45°C.
Выход: 0,7 кг (41%) VIII, масло. Путем рециркуляции VIIIz можно получить дополнительные 0,41 кг промежуточного соединения VIII, таким образом выход промежуточного соединения VIII составляет 65%.
1f/B. Получение VIII путем рециркуляции VIIIz УФ-облучением

C19H30O5 C19H30O5 C19H30O5
Mr: 338,4 Mr: 338,4 Mr: 338,4
VIIIz VIII VIIIz
Изомер VIIIz подвергали изомеризации посредством УФ-излучения и из него получали VIII (E-изомер) в толуольном растворе в присутствии сенсибилизатора, представляющего собой диметилдисульфид. Реакция протекает до тех пор, пока не будет достигнуто равновесное соотношение, и в результате не будет получена смесь с отношением 1:1. Обработку реакционной смеси и очистку проводили посредством колоночной хроматографии.
Облучение.
Облучение проводили в многогорлой колбе в атмосфере азота при 17-19°C. В колбу отвешивали 0,99 кг VIIIz, затем добавляли 130,7 мл метанола и 19,8 л толуола, а после полного растворения - 99 мл сенсибилизатора, представляющего собой диметилдисульфид. Начинали охлаждение, включали ртутную лампу среднего давления и реакционную смесь подвергали облучению в течение 1,5 часа. За реакцией наблюдали каждые 15 минут при помощи TLC. Когда соотношение изомеров достигало 50: 50%, реакцию останавливали. Раствор выпаривали при температуре макс. 45°C в вакууме при давлении макс. 10 мбар. Концентрированный остаток очищали посредством колоночной хроматографии.
Элюирование проводили с применением ступенчато-градиентных смесей толуол:метил-трет-бутиловый эфир. Смеси-элюенты содержат пиридин в количестве 0,5% по объему.
Выпаренную основную фракцию хроматографической очистки экстрагировали 1 M раствором карбоната калия, объединенную водную фазу дважды промывали метил-трет-бутиловым эфиром. Регулировали pH водной фазы до pH=4-6 с помощью 1 М раствора гидросульфата натрия. Подкисленный раствор экстрагировали метил-трет-бутиловым эфиром. Объединенную органическую фазу промывали 20% раствором хлорида натрия и после добавления триэтиламина ее выпаривали в вакууме при 45°C.
Выход: 0,41 кг (41%), масло.
1g. Получение [3αS-(2E,3α,4,5,6α)]5-[гексагидро-4-(гидроксиметил)-5-[(тетрагидро-2H-пиран-2-ил)окси]-2(1H)-пенталенилиден]пентановой кислоты метилового сложного эфира ( IX )

C19H30O5 C19H32O5
Mr: 338,4 Mr: 352,5
VIII IX
К раствору ацетона добавляли 0,7 кг VIII, 0,75 кг карбоната калия и 1,4 кг метилйодида, смесь нагревали до 45°C и перемешивали при данной температуре. При достижении необходимой степени превращения реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали метил-трет-бутиловым эфиром. Объединенную органическую фазу промывали 20% раствором хлорида натрия и после добавления триэтиламина ее выпаривали в вакууме при 45°C.
Выход: 0,69 кг (95%), масло.
1h. Получение (5E)-5-[(3aS,4R,5R,6aS)-5-[(тетрагидро-2H-пиран-2-ил)окси]-4-[(1E,3S,4RS)-4-метил-3-оксооктен-6-ин-1-ил]гексагидропентален-2(1H)-илиден]пентановой кислоты метилового сложного эфира ( XI )
A. Способ: окисление Пфитцнера-Моффата с последующей реакцией HWE, проводящейся в одном реакционном сосуде.
ILO-фосфонат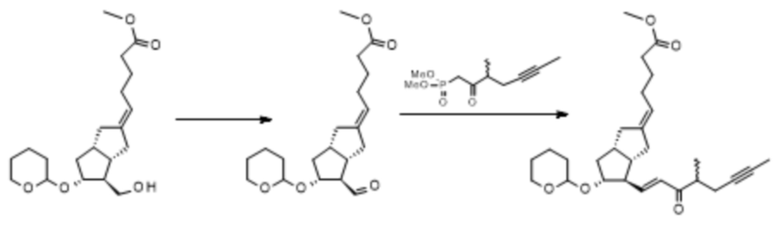
C20H32O5 C20H30O5 C28H40O5
Mr: 352,5 Mr: 350,4 Mr: 456,6
IX X XI
282 г IX растворяли в 1 л дистиллированного толуола в инертной атмосфере. Реакционную смесь охлаждали до 13°C, добавляли 473 г дициклогексилкарбодиимида, растворенного в 1,5 л толуола, а затем 238 мл 1 M фосфорной кислоты в растворе DMSO. Реакционную смесь нагревали до 45°C и перемешивали при данной температуре. После достижения необходимой степени превращения реакционную смесь, которая содержит полученный альдегид* X, охлаждали до комнатной температуры и в инертной атмосфере добавляли 78 г гидроксида калия и 218 г ILO-фосфоната, растворенного в 1 л тетрагидрофурана. Реакционную смесь перемешивали, поддерживая при этом заданную температуру. При достижении необходимой степени превращения к реакционной смеси добавляли перфил, его затем отфильтровывали, твердый фильтрат промывали толуолом, жидкий фильтрат концентрировали в вакууме при 50°C. Концентрированный остаток после добавления н-гексана очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей толуол:диизопропиловый эфир. Выпаренную основную фракцию дополнительно очищали посредством повторной хроматографии.
Выход: 282 г (77%), масло.
*При необходимости альдегид X можно выделять посредством хроматографической очистки.
Данные 13C- и 1H-ЯМР соединения формулы XI показаны на фигуре 6.
B. Способ: окисление DMSO-фосфорной кислотой и DIC с последующей реакцией HWE, проводящейся в одном реакционном сосуде
Растворяли 28 г IX в 100 мл дистиллированного толуола в инертной атмосфере. Реакционную смесь охлаждали до 13°C, добавляли 50 г диизопропилкарбодиимида, растворенного в 150 мл толуола, и 24 мл 1 M фосфорной кислоты в растворе DMSO. После добавления реакционную смесь нагревали до 45°C и перемешивали при данной температуре. После достижения необходимой степени превращения реакционную смесь охлаждали до комнатной температуры, в инертной атмосфере добавляли 8 г гидроксида калия и 22 г ILO-фосфоната, растворенного в 100 мл тетрагидрофурана. Реакционную смесь перемешивали, поддерживая при этом заданную температуру. При достижении необходимой степени превращения к реакционной смеси добавляли перфил, затем отфильтровывали и профильтрованное твердое вещество промывали толуолом. Жидкий фильтрат концентрировали в вакууме при 50°C, остаток, после добавления н-гексана, очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей толуол:диизопропиловый эфир. Выпаренную основную фракцию дополнительно очищали посредством повторной хроматографии.
Выход: 27 г (74%), масло.
C. Способ: окисление Анелли (TEMPO и гипохлорит натрия)
Раствор окислителя получали со 100 мл воды, 100 мл 5% раствора гипохлорита натрия и 36 г бикарбоната натрия. pH раствора составляет 9,4 ± 0,2. Если pH > 9,6, его регулируют с помощью бикарбоната натрия.
Растворяли 6 г IX в 70 мл дихлорметана (DCM), затем добавляли 0,01 г катализатора TEMPO и 0,2 г бромида калия. Смесь охлаждали до 0°C и в нее добавляли раствор окислителя со скоростью, при которой температура оставалась ниже 10°C. Ожидаемое время реакции - 30 минут.
Реакционную смесь затем гасили с помощью 10% раствора тиосульфата натрия, перемешивали при 10-15°C в течение 30 минут. Водную фазу экстрагировали 3 раза с помощью DCM. Органические фазы объединяли и промывали 15% раствором хлорида натрия.
В инертной атмосфере добавляли 20 мл 1 M раствора гидроксида натрия и 4 г ILO-фосфоната, растворенного в 20 мл тетрагидрофурана. Реакционную смесь перемешивали при поддержании указанной температуры. В конце реакции фазы разделяли, органическую фазу промывали последовательно 1 M раствором гидросульфата натрия, 15% раствором хлорида натрия и насыщенным солевым раствором. Органическую фазу концентрировали в вакууме при 45°C. Остаток после добавления н-гексана очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей толуол:диизопропиловый эфир. Выпаренную основную фракцию дополнительно очищали посредством повторной хроматографии.
Выход: 5,75 г (74%), масло.
Получение (5E)-5-[(3aS,4R,5R,6aS)-4-формил-5-тетрагидропиран-2-илокси-3,3a,4,5,6,6a-гексагидро-1H-пентален-2-илиден]пентановой кислоты метилового сложного эфира ( X )
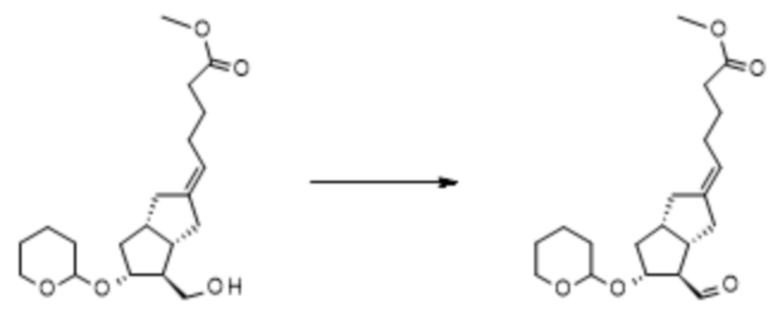
C20H32O5 C20H30O5
Mr: 352,5 Mr: 350,4
IX X
Растворяли 28,2 г IX в 100 мл дистиллированного толуола в инертной атмосфере. Реакционную смесь охлаждали до 13°C, затем добавляли 47,3 г дициклогексилкарбодиимида, растворенного в 150 мл толуола, и 23,8 мл 1 M раствора фосфорной кислоты в DMSO. После добавления реакционную смесь нагревали до 45°C и перемешивали при данной температуре. После достижения необходимой степени превращения реакционную смесь охлаждали до комнатной температуры, промывали водой (2×300 мл), органическую фазу сушили от воды путем концентрирования в вакууме при 50°C до объема, составляющего прибл. 80 мл. Концентрат в толуоле очищали посредством хроматографии с применением колонки с силикагелем и толуола, смесей-элюентов толуол:диизопропиловый эфир=3:1 и 1:1. Фракции, содержащие альдегид X, объединяли и выпаривали.
Выход: 23,82 г (85%).
1i. Получение метил-(5E)-5-[(3aS,4R,5R)-5-гидрокси-4-[(E,3S)-3-гидрокси-4-метил-окт-1-ен-6-инил]-3,3a,4,5,6,6a-гексагидро-1H-пентален-2-илиден]пентаноата ( XIII )

C28H40O5 C28H42O5 C28H42O5 C23H34O4
Mr: 456,6 Mr: 458,6 Mr: 458,6 Mr: 374,5
XI XII XIIb XIII
Для получения реагента DIBAL-F растворяли 350 г ди-трет-бутилметилфенола в 650 мл дистиллированного толуола в инертной атмосфере, при комнатной температуре, и к полученному раствору добавляли толуольный раствор, содержащий 102,8 г диизобутилалюминия гидрида (DIBAL-H). Реагент готовили при 0°C, но в конце добавления реакционную смесь перемешивали в течение 1 часа при комнатной температуре, затем в течение 6 часов при 45°C в инертной атмосфере. Смесь реагентов затем охлаждали до 5°C и в инертной атмосфере добавляли 94 г XI в толуольном растворе. В ходе добавления температура повышалась. Реакционную смесь перемешивали при комнатной температуре до достижения необходимой степени превращения, затем гасили 2 M раствором гидросульфата натрия. Погашенную смесь экстрагировали толуолом с получением содержащих защитные группы енольных изомеров XII* и XIIb, которые без проведения выделения подвергаются дальнейшему взаимодействию. К объединенной органической фазе добавляли метанольный раствор, содержащий 7,05 г п-толуолсульфоновой кислоты. Реакционную смесь перемешивали при комнатной температуре. После достижения необходимой степени превращения регулировали pH реакционной смеси до pH ≥ 7,5 при помощи триэтиламина и концентрировали в вакууме при 45°C. Концентрированный остаток растворяли в н-гексане и очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей н-гексан: этилацетат.
Выход: 43,1 г (56%), масло.
*При необходимости содержащий защитную группу енол XII может быть выделен посредством хроматографической очистки.
Очистка XIII посредством препаративной ВЭЖХ
Растворяли 50 г XIII в 100 мл ацетонитрила, к раствору добавляли по каплям воду до достижения соотношения ацетонитрил:вода=3:1. Исходный раствор фильтровали через предколонку, выполненную из 5 г C18 обратно-фазового силикагеля с размером частиц 10 микрон и размером пор 120 ангстрем. Профильтрованный исходный раствор очищали посредством препаративной жидкостной хроматографии высокого давления с применением 400 г насадки, представляющей собой C18 обратно-фазовый силикагель с размером частиц 10 микрон и размером пор 120 ангстрем, и смесей-элюентов вода:ацетон. Объединенную основную фракцию после хроматографии концентрировали в вакууме при 40°C, концентрированный раствор экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали солевым раствором, сушили над сульфатом натрия и выпаривали в вакууме при 30°C.
Выход согласно препаративной ВЭЖХ: 32 г (64%), масло.
Получение XI из XIIIb
Окисление XIIIb до XIb и введение THP-защитной группы

C23H34O4 C23H32O4 C28H40O5
Mr: 374,5 Mr: 372,5 Mr: 456,6
XIIIb XIb XI
Растворяли 7 г XIIIb в 75 мл этилацетата и фильтровали через фильтрационный слой, выполненный из 110 г активированного MnO2, смачивали этилацетатом. Фильтрат повторно пропускали через фильтрационный слой. Слой MnO2 дважды промывали этилацетатом, предварительно насыщенным водой.
Степень превращения проверяли при помощи TLC: если она была недостаточной, фильтрат повторно фильтровали через свежий фильтрационный слой MnO2. Фильтрат выпаривали, полученный неочищенный продукт сушили от воды путем отгонки толуола. К концентрату добавляли 100 мл толуола, 4 г дигидропирана и 0,01 г пара-толуолсульфоновой кислоты, за введением THP-защитной группы наблюдали при помощи TLC. В конце реакции ее останавливали добавлением триэтиламина, реакционную смесь выливали в воду, органическую фазу дважды экстрагировали водой, затем органическую фазу выпаривали.
Выход: 4,8 г (56,3%), масло. Продукт можно применять на стадии селективного восстановления.
Получение (5E)-5-[(3aS,4R,5R,6aS)-4-[(E)-3S-гидрокси-4-метилокт-1-ен-6-инил]-5-тетрагидропиран-2-илокси-3,3a,4,5,6,6a-гексагидро-1H-пентален-2-илиден]пентановой кислоты метилового сложного эфира ( XII )
C28H40O5 C28H42O5
Mr: 456,6 Mr: 458,6
XI XII
Для получения реагента DIBAL-F растворяли 35 г ди-трет-бутилметилфенола в 65 мл дистиллированного толуола в инертной атмосфере при комнатной температуре. К полученному раствору добавляли толуольный раствор, содержащий 10,3 г диизобутилалюминия гидрида (DIBAL-H). Реагент готовили при 0°C, но в конце добавления реакционную смесь перемешивали в течение 1 часа при комнатной температуре, затем в течение 6 часов при 45°C в инертной атмосфере. Смесь реагентов охлаждали до 5°C и в инертной атмосфере добавляли 9,4 г XI в толуольном растворе. В ходе добавления температура смеси повышалась. Реакционную смесь перемешивали при комнатной температуре до достижения необходимой степени превращения, затем ее гасили 2 M раствором гидросульфата натрия. Погашенную реакционную смесь экстрагировали толуолом. Толуольную фазу концентрировали в вакууме при 50°C до приблизительно 30 мл. Концентрированный толуольный остаток очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей н-гексан:этилацетат. Фракции, содержащие защищенный енол XII, объединяли, объединенную основную фракцию выпаривали.
Выход: 7,22 г (76,5%).
1j. Получение неочищенного илопроста
Растворяли 43,1 г XIII в 22 мл тетрагидрофурана в инертной атмосфере при комнатной температуре, затем к полученному добавляли 520 мл 1 M раствора гидроксида натрия со скоростью, при которой температура реакционной смеси оставалась на уровне 20-30°C. После достижения необходимой степени превращения фазы разделяли, водную фазу дважды экстрагировали метил-трет-бутиловым эфиром, органические фазы объединяли и дважды промывали 1 M раствором гидроксида натрия. Объединенную щелочную фазу разбавляли метил-трет-бутиловым эфиром и при перемешивании регулировали pH до pH ≤ 3 с помощью 2 M раствора гидросульфата натрия. Подкисленную водную фазу экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали 20% раствором хлорида натрия и выпаривали в вакууме при 30°C.
Чистота продукта составляла 93%, общее количество родственных примесей составляло не более 5%, общее количество других, неидентифицированных примесей составляло не более 4%.
Из данного материала невозможно получить твердую форму илопроста из-за уровня чистоты, подробно описанного выше.
1k. Очистка неочищенного илопроста посредством гравитационной хроматографии и отверждения
Очистка неочищенного илопроста, способ B.
Растворяли 64,5 г неочищенного илопроста в 25 мл дист. ацетона, к раствору добавляли прибл. 50 мл н-пентана, пока раствор не становился опалесцирующим, затем его очищали на гравитационной колонке, заполненной нормально-фазовым силикагелем Si60 (размер частиц 0,063-0,2 мм), с применением ступенчато-градиентных смесей н-пентан:ацетон. Фракции объединяли после проверки при помощи TLC. Раствор основной фракции фильтровали через 5-микронную тефлоновую мембрану, а затем выпаривали в вакууме при температуре бани макс. 35°C.
К 50 г подверженного хроматографированию продукта, представляющего собой фазу илопроста, добавляли 200 мл профильтрованного, дист. ацетона. Смесь встряхивали при 20-25°C до завершения растворения, затем при непрерывном встряхивании к раствору добавляли 1080 мл профильтрованного пентана, при этом илопрост полностью отстаивался из смеси в виде масла.
Смесь охлаждали без перемешивания до (-)60°C, через 6 часов обеспечивали ее нагревание до (-)20°C и выдерживали при данной температуре без перемешивания в течение по меньшей мере 16 часов. Растворитель затем удаляли из маслянистой кристаллической массы декантированием, 650 мл профильтрованного пентана выливали на продукт, а кристаллическую массу выдерживали при (-)20°C в течение минимум 2 часов. Растворитель снова удаляли декантированием.
Растворитель отгоняли из продукта в высоком вакууме при (-)30°C, температуру поддерживали на уровне (-)20 - (-)30°C. В ходе удаления растворителя твердый продукт время от времени перемешивали. Удаление растворителя проводили в инертной атмосфере, это занимало прибл. 120 часов.
Чистота продукта составляла 95,0%, общее количество родственных примесей составляло не более 3,5%, общее количество других, неидентифицированных примесей составляло не более 2,5%.
1l. Очистка неочищенного илопроста посредством препаративной ВЭЖХ с последующими фильтрацией через силикагель и отверждением
Очистка неочищенного илопроста, способ C.
Растворяли 64,5 г неочищенного илопроста в 100 мл ацетонитрила, к раствору добавляли по каплям воду до достижения соотношения ацетонитрил:вода=3:1. Исходный раствор фильтровали через предколонку, выполненную из 5 г C18 обратно-фазового силикагеля с размером частиц 10 микрон и размером пор 120 ангстрем. Очистку профильтрованного исходного раствора проводили посредством препаративной жидкостной хроматографии высокого давления с применением 400 г насадки, представляющей собой C18 обратно-фазовый силикагель с размером частиц 10 микрон и размером пор 120 ангстрем, и
смесей-элюентов вода:ацетонитрил. Объединенную основную фракцию хроматографии концентрировали в вакууме при 40°C, концентрированный раствор экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме при 30°C до 100 мл. Концентрированный раствор доводили ацетоном до 150 г, осторожно добавляли н-пентан, пока раствор не станет слегка опалесцирующим. Полученный раствор дополнительно очищали путем фильтрации через силикагель с применением ступенчато-градиентных смесей н-пентан:ацетон. Основную фракцию выпаривали при 30°C в высоком вакууме.
К 50 г продукта, представляющего собой фазу илопроста, очищенного посредством препаративной ВЭЖХ и профильтрованного через силикагель, добавляли 200 мл профильтрованного, дист. ацетона. Смесь встряхивали при комнатной температуре до полного растворения, затем добавляли 1080 мл профильтрованного пентана, при этом илопрост полностью отстаивался из смеси в виде масла.
Смесь охлаждали без перемешивания до (-)60°C, через 6 часов обеспечивали ее нагревание до (-)20°C и выдерживали при данной температуре без перемешивания в течение по меньшей мере 16 часов. Растворитель затем удаляли из маслянистой кристаллической массы декантированием, выливали на нее 650 мл профильтрованного пентана и кристаллическую массу выдерживали при (-)20°C в течение минимум 2 часов. Растворитель снова удаляли декантированием.
Растворитель отгоняли из продукта в высоком вакууме при (-)30°C, температуру поддерживали на уровне (-)20 - (-)30°C. В ходе данного удаления растворителя твердый продукт время от времени перемешивали. Удаление растворителя проводили в инертной атмосфере, это занимало прибл. 120 часов.
Чистота продукта составляла 98,0%, общее количество родственных примесей составляло не более 1,6%, общее количество других, неидентифицированных примесей составляло не более 1,0%.
Z-изомеры илопроста, общее количество
другие примеси, общее количество
из которых
- 15-эпиилопрост
- 15-оксоилопрост
- метиловый сложный эфир илопроста
- этиловый сложный эфир илопроста
- димер 1 илопроста
- димер 2 илопроста
- неидентифицированные примеси, каждая
≤ 1,0%
≤ 0,20%
≤ 0,20%
≤ 0,10%
≤ 0,10%
≤ 0,20%
≤ 0,20%
≤ 0,10%
1m. Очистка неочищенного илопроста посредством гравитационной и препаративной ВЭЖХ, фильтрации через силикагель и отверждения
Очистка неочищенного илопроста, способ D.
Растворяли 80 г неочищенного илопроста в 40 мл дист. ацетона, добавляли прибл. 70 мл н-пентана, пока раствор не становился опалесцирующим, затем его очищали на гравитационной колонке, заполненной нормально-фазовым силикагелем Si60 (размер частиц 0,063-0,2 мм), с применением ступенчато-градиентных смесей н-пентан:ацетон. Фракции объединяли после проверки при помощи TLC. Раствор основной фракции фильтровали через 5-микронную тефлоновую мембрану и выпаривали в вакууме при температуре бани макс. 35°C.
Растворяли 60 г неочищенного продукта, представляющего собой фазу илопроста, очищенного посредством гравитационной хроматографии, в 110 мл ацетонитрила, к раствору добавляли по каплям воду до достижения соотношения ацетонитрил:вода=3:1. Исходный раствор фильтровали через предколонку, выполненную из 5 г C18 обратно-фазового силикагеля с размером частиц 10 микрон и размером пор 120 ангстрем. Очистку профильтрованного исходного раствора проводили посредством препаративной жидкостной хроматографии высокого давления с применением 400 г насадки, представляющей собой C18 обратно-фазовый силикагель с размером частиц 10 микрон, размером пор 120 ангстрем, и смесей вода:ацетонитрил в качестве элюента. Объединенную основную фракцию хроматографии концентрировали в вакууме при 40°C, концентрированный раствор экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали до 100 мл в вакууме при 30°C. Концентрированный раствор доводили ацетоном до 150 г, затем осторожно добавляли н-пентан, пока раствор не становился слегка опалесцирующим, а затем фильтровали его через силикагель. Основную фракцию выпаривали в высоком вакууме при 30°C.
К 50 г продукта, представляющего собой фазу илопроста, очищенного посредством препаративной ВЭЖХ и профильтрованного через силикагель, добавляли 200 мл профильтрованного, дист. ацетона. Смесь встряхивали при комнатной температуре до полного растворения, затем добавляли 1080 мл профильтрованного пентана, при этом илопрост полностью отстаивался из смеси в виде масла.
Смесь охлаждали без перемешивания до (-)60°C, через 6 часов обеспечивали ее нагревание до (-)20°C и выдерживали при данной температуре без перемешивания в течение по меньшей мере 16 часов. Растворитель затем удаляли из маслянистой кристаллической массы декантированием, 650 мл профильтрованного пентана выливали на продукт, а кристаллическую массу выдерживали при (-)20°C в течение минимум 2 часов. Растворитель снова удаляли декантированием.
Растворитель отгоняли из продукта в высоком вакууме при (-)30°C, поддерживая температуру на уровне (-)20 - (-)30°C. В ходе удаления растворителя твердый продукт время от времени перемешивали. Удаление растворителя проводили в инертной атмосфере, это занимало прибл. 120 часов.
Полученный твердый продукт представляет собой порошок, который кристаллизуется при выстаивании.
Чистота полученного продукта составляла 98,5%, общее количество родственных примесей составляло не более 1,6%, общее количество других, неидентифицированных примесей составляло не более 0,5%.
Рентгеновская порошковая дифрактограмма представлена на фигуре 1, ДСК-кривая - на фигуре 3, данные 13C- и 1H-ЯМР - на фигуре 5.
Рентгенограмма XRPD продукта содержит характеристические пики при следующих значениях (градусы ± 0,2° 2-тета): 5,43, 7,51, 7,81, 15,19, 15,57, 15,85, 16,25, 16,84, 17,08, 17,26, 18,14, 18,59, 19,17, 20,32, 20,53, 21,69, 22,12 и 23,28.
Z-изомеры илопроста, общее количество
другие примеси, общее количество
из которых
- 15-эпиилопрост
- 15-оксоилопрост
- метиловый сложный эфир илопроста
- этиловый сложный эфир илопроста
- димер 1 илопроста
- димер 2 илопроста
- неидентифицированные примеси, каждая
≤ 1,0%
≤ 0,10%
≤ 0,20%
≤ 0,20%
≤ 0,05%
≤ 0,10%
≤ 0,10%
≤ 0,10%
1n. Получение 16(S)-илопроста ((S)- I )
A: путем хроматографического разделения и кристаллизации
A1: хроматография с применением смеси-элюента ацетонитрил:вода
C22H32O4 C22H32O4
Mr: 360,49 Mr: 360,49
I (S)-I
Растворяли 50 г неочищенного I (содержание (S)-I: 20 г) в 100 мл ацетонитрила и к раствору добавляли по каплям воду до достижения соотношения ацетонитрил:вода=3:1. Исходный раствор фильтровали через
предколонку, выполненную из 5 г C18 обратно-фазового силикагеля с размером частиц 10 микрон и размером пор 120 ангстрем. Очистку профильтрованного исходного раствора проводили посредством препаративной жидкостной хроматографии высокого давления с применением 400 г насадки, представляющей собой C18 обратно-фазовый силикагель с размером частиц 10 микрон и размером пор 120 ангстрем, и смесей-элюентов вода:ацетонитрил.
Среди диастереомеров илопроста диастереомер 16(S)-илопрост характеризуется более высоким временем удерживания. Посредством препаративной ВЭЖХ его можно отделять с высокой эффективностью от диастереомера 16(R)-илопроста, элюируемого перед ним.
Объединенную основную фракцию 16(S)-илопроста по итогам хроматографии концентрировали в вакууме при 40°C, концентрированный раствор экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и выпаривали в вакууме при 30°C. Остаток растворяли в 32 мл ацетона, затем осторожно добавляли н-пентан, пока раствор не становился слегка опалесцирующим. Раствор, полученный таким образом, дополнительно очищали фильтрацией через силикагель с применением ступенчато-градиентных смесей н-пентан:ацетон. Основную фракцию выпаривали в высоком вакууме при 30°C.
К 15 г продукта, представляющего собой фазу 16(S)-илопроста, очищенного посредством препаративной ВЭЖХ и профильтрованного через силикагель, добавляли 15 мл профильтрованного, дист. ацетона. Смесь встряхивали при комнатной температуре до полного растворения, затем при непрерывном встряхивании добавляли 100 мл профильтрованного пентана, пока раствор не становился опалесцирующим. Смесь охлаждали до (-)40°C и при поддержании данной температуры ее перемешивали в инертной атмосфере в течение 16 часов. Выпавший в осадок кристаллический материал отфильтровывали.
Продукт сушили при температуре от (-)10 до (+)10°C в высоком вакууме в инертной атмосфере. Сушка занимала прибл. 120 часов.
Чистота продукта составляла 98,0%, общее количество родственных примесей составляло не более 1,6%, общее количество других, неидентифицированных примесей составляло не более 1,0%.
Рентгеновская порошковая дифрактограмма представлена на фигуре 2, ДСК-кривая - на фигуре 4, данные 13C- и 1H-ЯМР - на фигуре 9.
Рентгенограмма XRPD продукта содержит характеристические пики при следующих значениях (градусы ± 0,2° 2-тета): 7,46, 7,80, 12,69, 14,91, 15,58, 16,82, 17,27, 20,31, 20,62, 23,30, 28,13, 31,38, 32,05, 34,88 и 38,88.
A2: хроматография с применением смеси-элюента метанол:2-пропанол:вода
Растворяли 64,5 г неочищенного I в 100 мл 2-пропанола, к раствору по каплям добавляли воду до
достижения соотношения 2-пропанол:вода=1:1. Исходный раствор фильтровали через предколонку, выполненную из 5 г C18 обратно-фазового силикагеля с размером частиц 10 микрон и размером пор 120 ангстрем. Очистку профильтрованного исходного раствора проводили посредством препаративной жидкостной хроматографии высокого давления с применением 400 г насадки, представляющей собой C18 обратно-фазовый силикагель с размером частиц 10 микрон и размером пор 120 ангстрем, и
смесей-элюентов вода:метанол: 2-пропанол.
Объединенную основную фракцию по итогам хроматографии концентрировали в вакууме при 40°C, концентрированный раствор экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом натрия и выпаривали в вакууме при 30°C. Остаток растворяли в 32 мл ацетона и осторожно добавляли н-пентан, пока раствор не становился слегка опалесцирующим. Раствор, полученный таким образом, дополнительно очищали фильтрацией через силикагель с применением ступенчато-градиентных смесей н-пентан:ацетон. Основную фракцию выпаривали в высоком вакууме при 30°C.
К 15 г продукта, представляющего собой фазу 16(S)-илопроста, очищенного посредством препаративной ВЭЖХ и профильтрованного через силикагель, добавляли 15 мл профильтрованного, дист. ацетона. Смесь встряхивали при комнатной температуре до полного растворения, затем добавляли 100 мл профильтрованного пентана, пока раствор не становился опалесцирующим. Смесь охлаждали до (-)40°C и при поддержании данной температуры его перемешивали в инертной атмосфере в течение 16 часов. Выпавший в осадок кристаллический материал отфильтровывали.
Продукт сушили при температуре от (-)10 до (+)10°C в высоком вакууме в инертной атмосфере. Сушка занимала прибл. 120 часов.
Чистота продукта составляла 98,0%, общее количество родственных примесей составляло не более 1,6%, общее количество других, неидентифицированных примесей составляло не более 1,0%.
1o. Получение 16(S)-илопроста путем химического синтеза
(5E)-5-[(3aS,4R,5R,6aS)-5-[(тетрагидро-2H-пиран-2-ил)окси]-4-[(1E,4S)-4-метил-3-оксооктен-6-ин-1-ил]гексагидропентален-2(1H)-илиден]пентановой кислоты метилового сложного эфира ((S)- XI )
B: путем химического синтеза
B1: окисление Пфитцнера-Моффата с последующей реакцией HWE, проводящейся в одном реакционном сосуде
(S)-ILO-фосфонат

S-ILO-фосфонат
C20H32O5 C20H30O5 C28H40O5
Mr: 352,5 Mr: 350,4 Mr: 456,6
IX X (S)-XI
Растворяли 140 г IX в 1 л дистиллированного толуола в инертной атмосфере. Реакционную смесь охлаждали до 13°C и добавляли раствор 235 г дициклогексилкарбодиимида в 0,75 л толуола и 118 мл 1 M фосфорной кислоты в растворе DMSO. После добавления реакционную смесь нагревали до 45°C и перемешивали при данной температуре. После достижения необходимой степени превращения реакционную смесь, содержащую полученный альдегид X, охлаждали до комнатной температуры, в инертной атмосфере добавляли 39 г гидроксида калия и 109 г (S)-ILO-фосфоната (оптически активный), растворенного в 0,5 л тетрагидрофурана. Реакционную смесь перемешивали при поддержании указанной температуры. При достижении необходимой степени превращения к реакционной смеси добавляли перфил, его затем отфильтровывали, отфильтрованное твердое вещество промывали толуолом, жидкий фильтрат концентрировали в вакууме при 50°C. Концентрированный остаток после добавления н-гексана очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей толуол:диизопропиловый эфир. Выпаренную основную фракцию дополнительно очищали посредством повторной хроматографии.
Выход: 140 г (77%), масло.
Данные 13C- и 1H-ЯМР соединения формулы (S)-XI представлены на фигуре 7.
B2: окисление Анелли (TEMPO и гипохлорит натрия)
Раствор окислителя получали со 100 мл воды, 100 мл 5%-го гипохлорита натрия и 36 г бикарбоната натрия. pH раствора составляет 9,4 ± 0,2. Если pH > 9,6, его регулируют с помощью бикарбоната натрия.
Растворяли 6 г IX в 70 мл дихлорметана (DCM), затем добавляли 0,01 г катализатора TEMPO и 0,2 г бромида калия. Смесь охлаждали до 0°C и в нее добавляли раствор окислителя со скоростью, при которой температура оставалась ниже 10°C. Ожидаемое время реакции - 30 минут.
Реакционную смесь затем гасили с помощью 10% раствора тиосульфата натрия, перемешивали при 10-15°C в течение 30 минут. Водную фазу экстрагировали 3 раза с помощью DCM. Органические фазы объединяли и промывали 15% раствором хлорида натрия.
В инертной атмосфере добавляли 20 мл 1 M раствора гидроксида натрия и 4 г (S)-ILO-фосфоната, растворенного в 20 мл тетрагидрофурана. Реакционную смесь перемешивали, поддерживая при этом заданную температуру. В конце реакции фазы разделяли, органическую фазу промывали последовательно 1 M раствором гидросульфата натрия, 15% раствором хлорида натрия и насыщенным солевым раствором. Органическую фазу концентрировали в вакууме при 45°C. Остаток после добавления н-гексана очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей
толуол: диизопропиловый эфир. Выпаренную основную фракцию дополнительно очищали посредством повторной хроматографии.
Выход: 5,75 г (74%), масло.
1p. Получение метил-(5E)-5-[(3aS,4R,5R)-5-гидрокси-4-[(E,3S,4S)-3-гидрокси-4-метилокт-1-ен-6-инил]-3,3a,4,5,6,6a-гексагидро-1H-пентален-2-илиден]пентаноата ((S)- XIII )

C28H40O5 C28H42O5 C28H42O5 C23H34O4
Mr: 456,6 Mr: 458,6 Mr: 458,6 Mr: 374,5
(S)-XI (S)-XII 15-эпи-(S)-XII (S)-XIII
Для получения реагента DIBAL-F растворяли 350 г ди-трет-бутилметилфенола в 650 мл дистиллированного толуола при комнатной температуре в инертной атмосфере и к полученному раствору добавляли толуольный раствор, содержащий 102,8 г диизобутилалюминия гидрида (DIBAL-H). Реагент готовили при 0°C, но в конце добавления реакционную смесь перемешивали в течение 1 часа при комнатной температуре, затем в течение 6 часов при 45°C в инертной атмосфере. Смесь реагентов затем охлаждали до 5°C и в инертной атмосфере добавляли 94 г (S)-XI в толуольном растворе. В ходе добавления температура повышалась. Реакционную смесь перемешивали при комнатной температуре до достижения необходимой степени превращения, затем гасили 2 M раствором гидросульфата натрия. Погашенную реакционную смесь экстрагировали толуолом с получением содержащих защитные группы енольных изомеров (S)-XII* и 15-эпи-(S)-XII, которые без проведения выделения подвергаются дальнейшему взаимодействию. К объединенной органической фазе добавляли метанольный раствор, содержащий 7,05 г п-толуолсульфоновой кислоты. Реакционную смесь перемешивали при комнатной температуре. При достижении необходимой степени превращения pH реакционной смеси регулировали до pH ≥ 7,5 с помощью триэтиламина и концентрировали ее в вакууме при 45°C. Остаток растворяли в н-гексане и очищали посредством хроматографии с применением колонки с силикагелем и ступенчато-градиентных смесей н-гексан:этилацетат.
Выход: 43,1 г (56%), масло.
*При необходимости содержащий защитную группу енол (S)-XII может быть выделен посредством хроматографической очистки.
Данные 13C- и 1H-ЯМР соединения формулы (S)-XII представлены на фигуре 8.
Получение неочищенного 16(S)-илопроста

C23H34O4 C22H32O4
Mr: 374,5 Mr: 360,49
(S)-XIII (S)-I
Растворяли 43,1 г (S)-XIII в 50 мл тетрагидрофурана при комнатной температуре в инертной атмосфере. К раствору добавляли 520 мл 1 M раствора гидроксида натрия со скоростью, при которой температура реакционной смеси оставалась на уровне 20-30°C. После достижения необходимой степени превращения фазы разделяли, водную фазу дважды экстрагировали метил-трет-бутиловым эфиром, органические фазы объединяли и дважды промывали 1 M раствором гидроксида натрия. Объединенную щелочную фазу разбавляли метил-трет-бутиловым эфиром и при перемешивании регулировали pH до pH ≤ 3 с помощью 2 M раствора гидросульфата натрия. Подкисленную водную фазу экстрагировали метил-трет-бутиловым эфиром, объединенную органическую фазу промывали 20% раствором хлорида натрия и выпаривали в вакууме при 30°C.
Чистота продукта составляла 93%, общее количество родственных примесей составляло не более 5%, общее количество других, неидентифицированных примесей составляло не более 4%.
Чистота данного материала уже позволяет получать твердый 16(S)-илопрост.
Очистка неочищенного 16(S)-илопроста посредством гравитационной хроматографии и кристаллизации
Растворяли 40,2 г неочищенного 16(S)-илопроста в 60 мл ацетона и осторожно добавляли н-пентан, пока раствор не становился слегка опалесцирующим. Полученный раствор очищали путем фильтрации через силикагель с применением ступенчато-градиентных смесей н-пентан:ацетон. Основную фракцию выпаривали в высоком вакууме при 30°C.
К 37 г продукта, представляющего собой фазу 16(S)-илопроста, профильтрованного через силикагель, добавляли 12 мл профильтрованного дист. ацетона. Смесь встряхивали при комнатной температуре до полного растворения, затем при непрерывном встряхивании добавляли 90 мл профильтрованного пентана, пока раствор не становился опалесцирующим. Смесь охлаждали до (-)40°C и при поддержании данной температуры ее перемешивали в инертной атмосфере в течение 16 часов. Выпавший в осадок кристаллический материал отфильтровывали.
Продукт сушили в высоком вакууме при температуре от (-)10°C до (+)10°C в инертной атмосфере. Сушка занимала прибл. 120 часов.
Чистота продукта составляла 98,0%, общее количество родственных примесей составляло не более 1,6%, общее количество других, неидентифицированных примесей составляло не более 1,0%.
1q. Получение (R)-илопроста ((R)- I ) путем химического синтеза
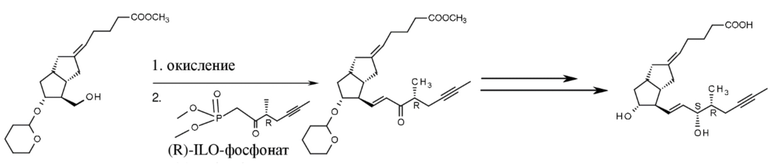
R-ILO-фосфонат
C20H32O5 C28H40O5 C22H32O4
Mr: 352,5 Mr: 456,6 Mr: 360,49
IX (R)-XI (R)-I
Начиная с толуольного раствора, содержащего 140 мг (R)-IX, выполняя описанные выше химические стадии, применяя (R)-ILO-фосфонат, получали 51,1 мг 16(R)-илопроста.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДЕКАГИДРОИЗОХИНОЛИНОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2117661C1 |
| СПОСОБ И СОЕДИНЕНИЕ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНТАГОНИСТА РЕЦЕПТОРА ОРЕКСИНА-2, И ЛЕМБОРЕКСАНТ, СОДЕРЖАЩИЙ НЕБОЛЬШОЕ КОЛИЧЕСТВО ПРИМЕСЕЙ | 2020 |
|
RU2823998C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ МИЗОПРОСТОЛА | 2018 |
|
RU2774634C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАВОПРОСТА | 2012 |
|
RU2631316C2 |
| Способ получения оптически активных соединений ряда простагландинов или их рацематов | 1974 |
|
SU662008A3 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОТИОФЕН-2-ИЛБОРОНАТА | 2018 |
|
RU2790014C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКОГО ИНГИБИТОРА ПРОТЕАЗЫ ВИРУСА ГЕПАТИТА С | 2008 |
|
RU2483067C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-АЦИЛ-4-ФЕНИЛСУЛЬФОНИЛПРОЛИНАМИДА И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2615997C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОПРОСТА И ЕГО ТРОМЕТАМИНОВОЙ СОЛИ | 2016 |
|
RU2729626C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОАЛКАНО-ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2194698C2 |

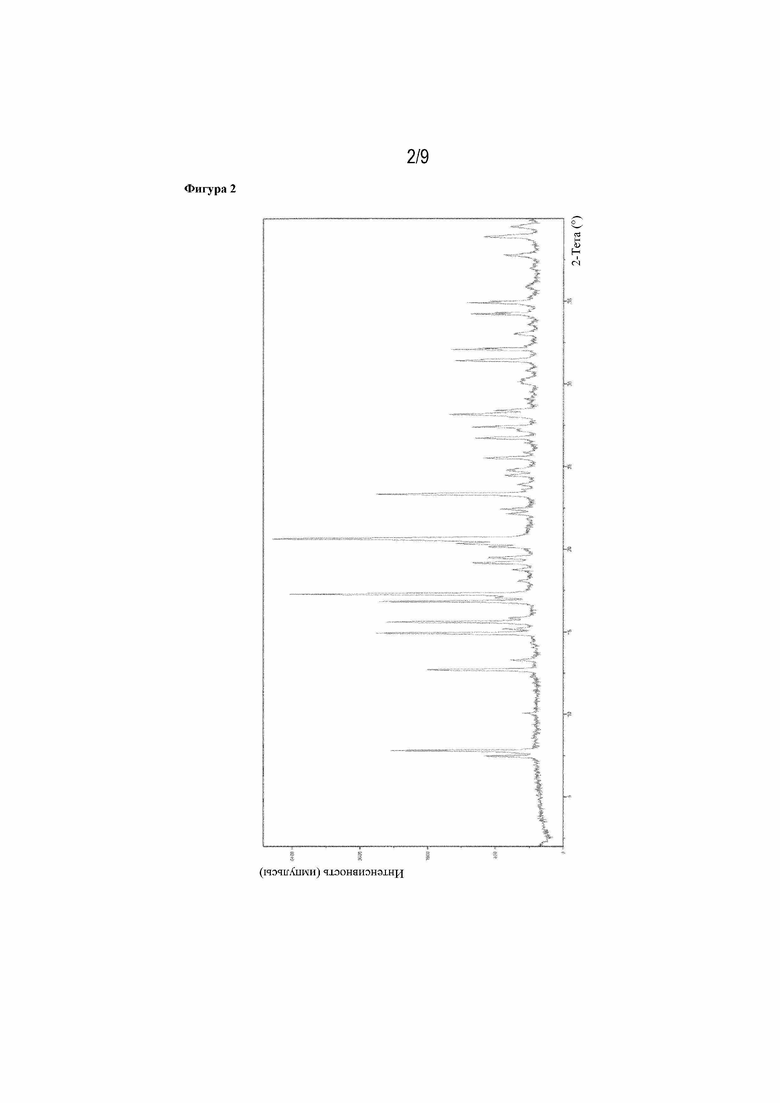
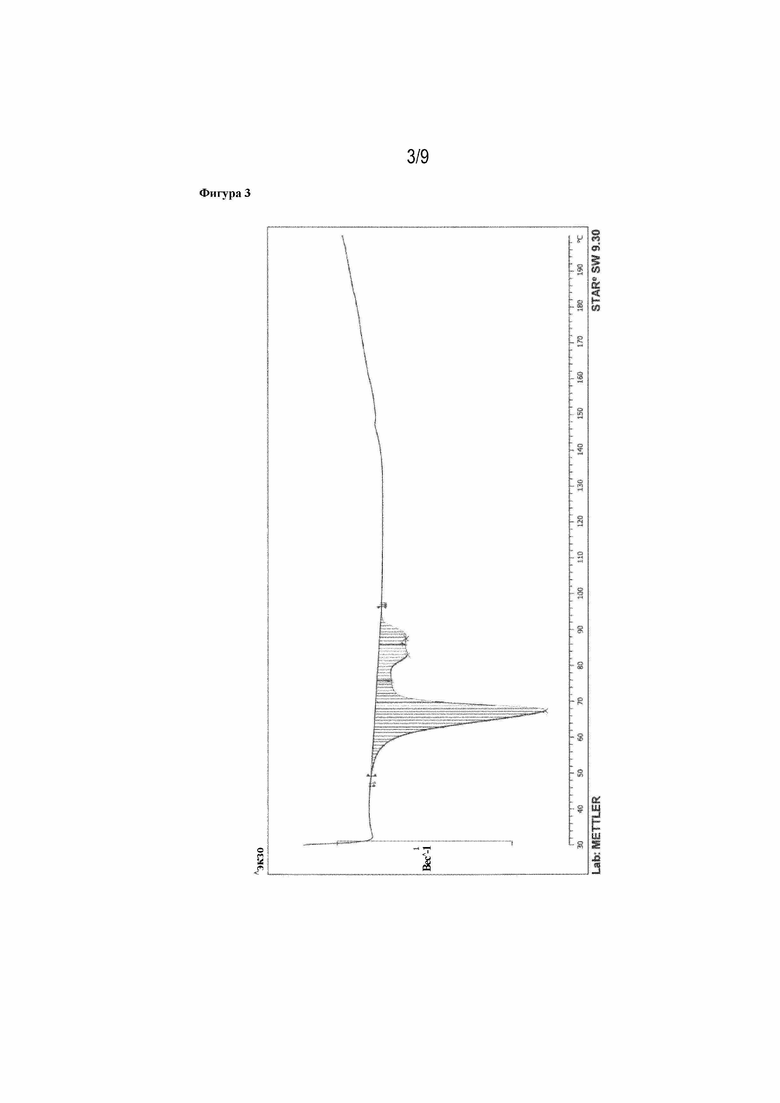
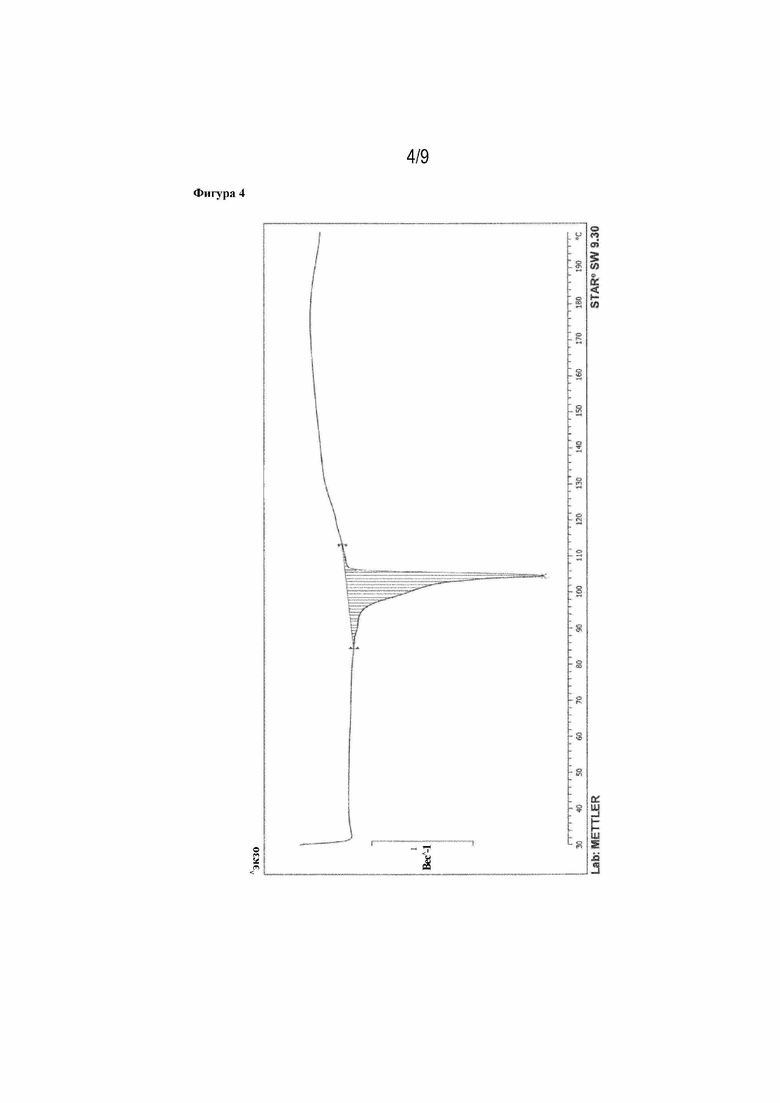

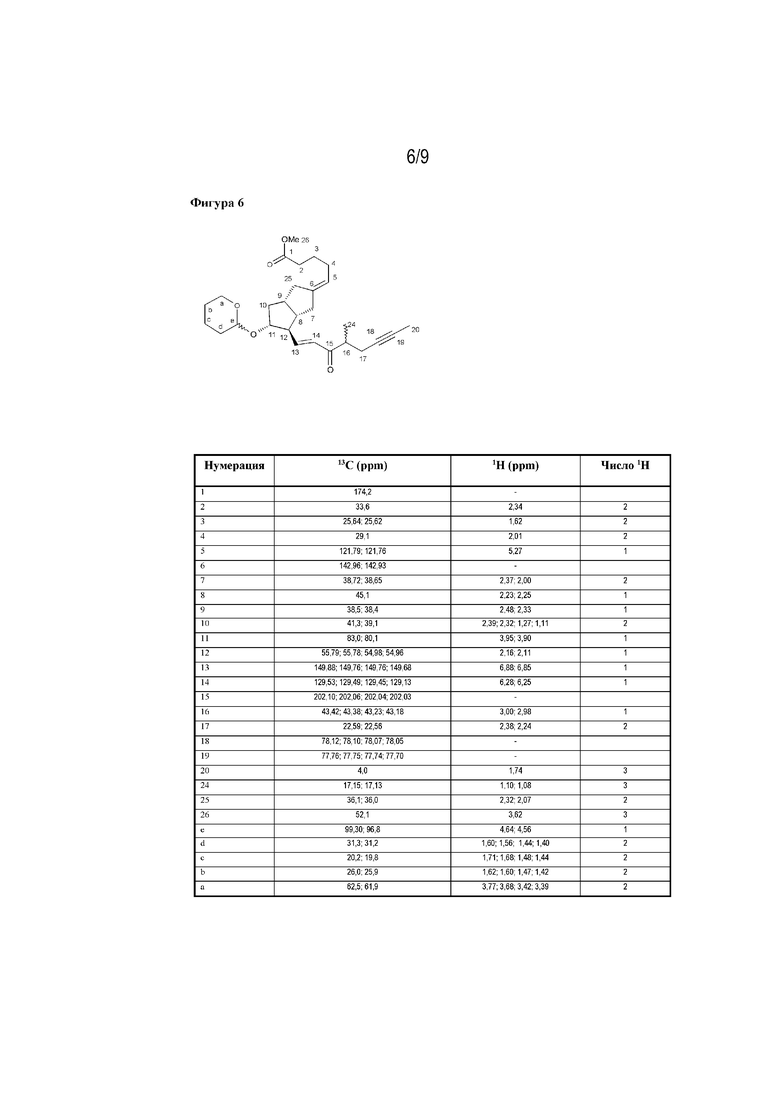



Изобретение относится к новому способу получения илопроста посредством новых промежуточных соединений, а также к соединениям формул XI, XII, (S)-XII и (R)-XII. Технический результат: разработан новый способ получения илопроста, который обеспечивает лучший выход целевого продукта. 5 н. и 13 з.п. ф-лы, 9 ил.



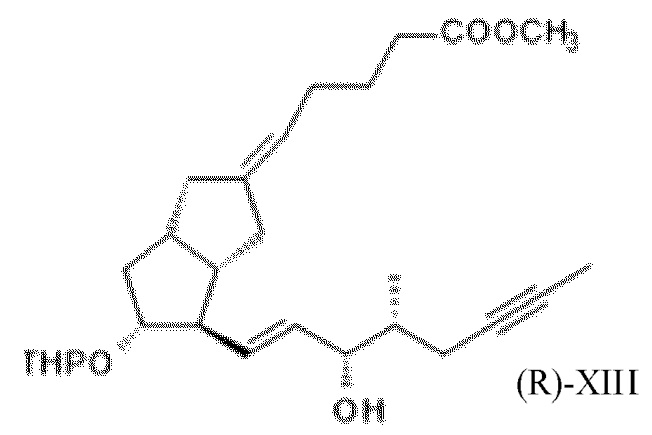
1. Способ получения илопроста формулы I,
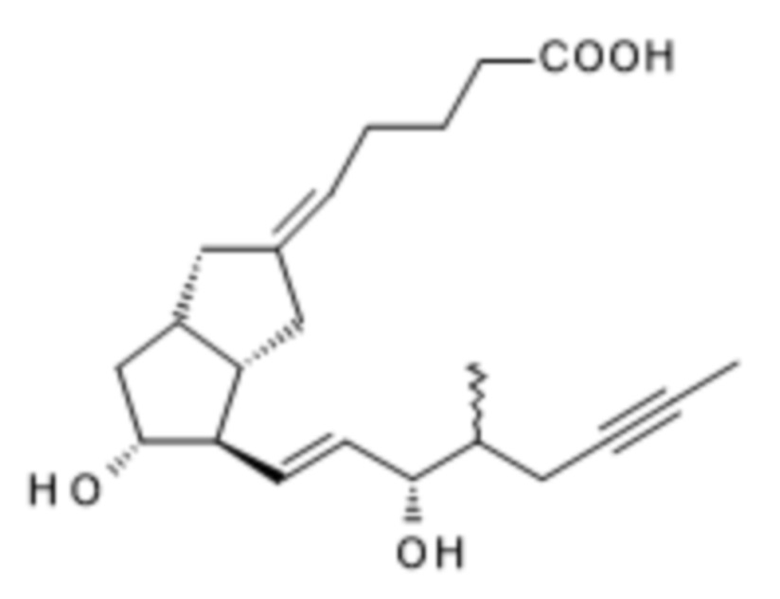 I,
I,
включающий следующее:
a) лактон Кори формулы II
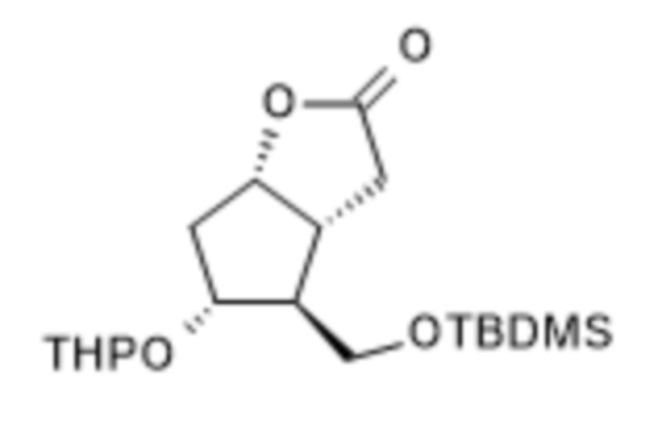 II
II
подвергают селективному алкилированию с помощью диметилметилфосфоната в присутствии диалкиламида лития,
b) кольцо полученного лактола формулы III
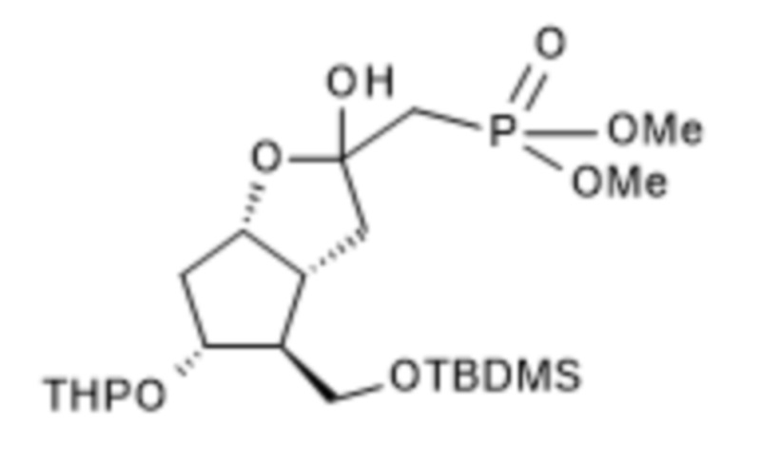 III
III
подвергают раскрытию с помощью ацетата пиридиния в слабокислой среде, затем полученную вторичную гидроксильную группу окисляют дихроматом пиридиния,
c) полученное соединение формулы IV
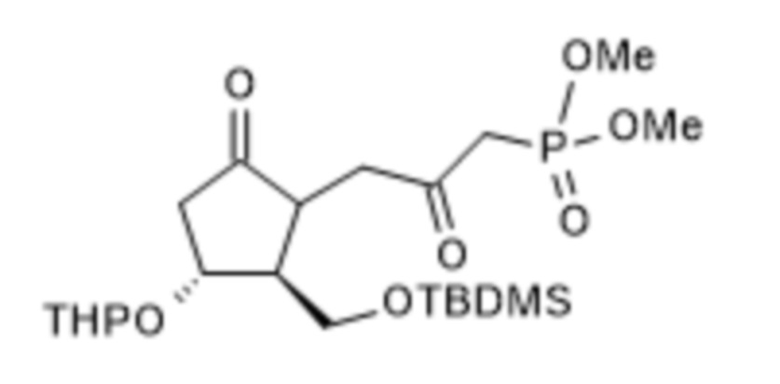
IV
приводят в реакцию с карбонатом калия в присутствии реагента 18-краун-6,
d) полученное таким образом соединение формулы V
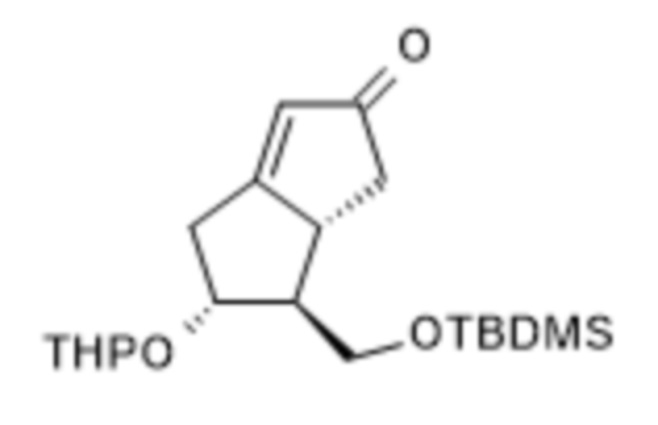
V,
восстанавливают,
e) полученное соединение формулы VI
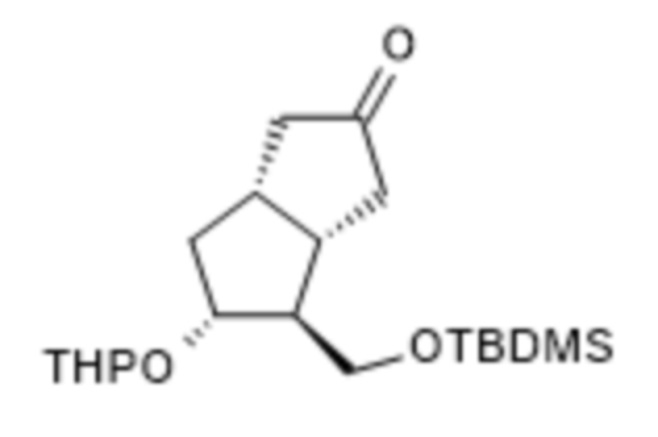
VI
приводят в реакцию с карбоксибутилтрифенилфосфония бромидом в присутствии трет-бутилата калия,
f) TBDMS-защитную группу полученных E- и Z-изомеров формулы VII
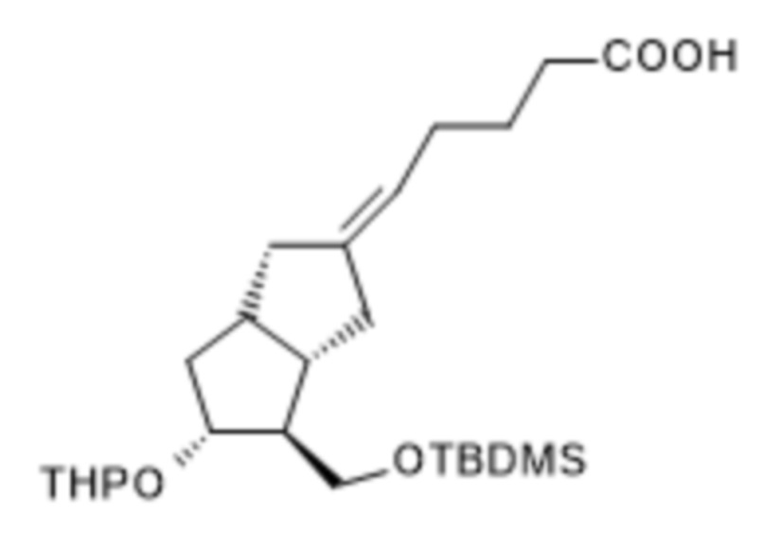
VII
удаляют, изомеры разделяют посредством гравитационной хроматографии, при необходимости Z-изомер (VIIIz) подвергают изомеризации до E-изомера,
g) полученное соединение формулы VIII

VIII
подвергают эстерификации,
h) полученное соединение формулы IX
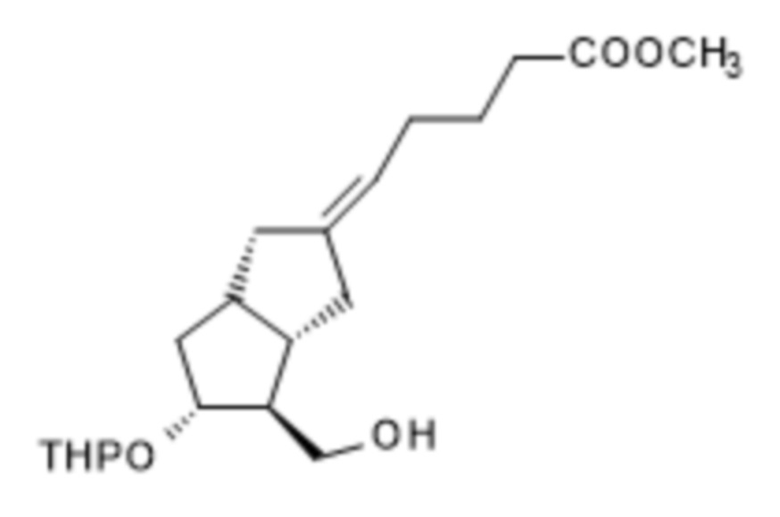
IX
подвергают окислению,
i) полученное соединение формулы X

X
подвергают превращению в реакции HWE в присутствии твердого гидроксида калия в соединение формулы XI,
j) оксогруппу полученного соединения формулы XI

XI
восстанавливают с помощью DIBAL-F,
k) тетрагидропиранильную защитную группу полученного таким образом соединения формулы XII
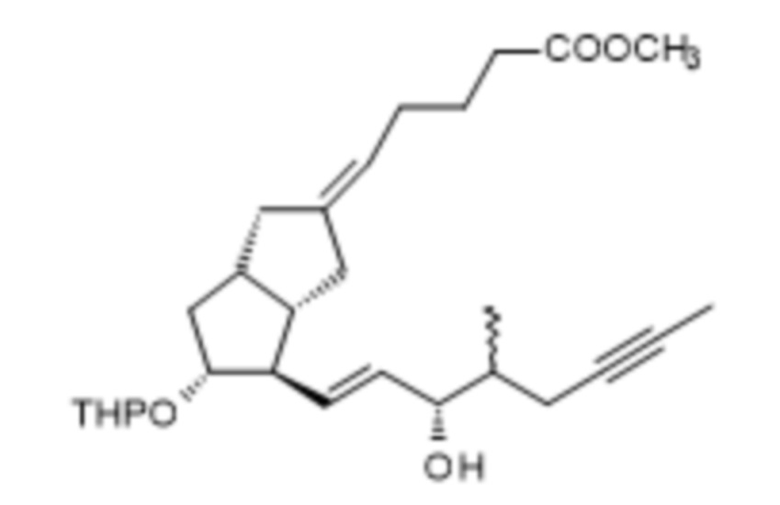
XII
удаляют и соединение очищают посредством гравитационной колоночной хроматографии, при необходимости дополнительно очищают посредством препаративной ВЭЖХ,
l) сложноэфирную группу полученного соединения формулы XIII

XIII
удаляют и полученное соединение формулы I очищают.
2. Способ по п. 1, где диалкиламид лития представляет собой диизопропиламид лития или дициклогексиламид лития на стадии a).
3. Способ по п. 1 или 2, где реакцию на стадии c) проводят при сильном разбавлении, предпочтительно при 30-45-кратном разбавлении, при высокой температуре, предпочтительно при 90-110°C.
4. Способ по любому из пп. 1-3, где реакцию на стадии c) проводят таким образом, что раствор соединения формулы IV добавляют по каплям в нагреваемый с обратным холодильником раствор реагентов.
5. Способ по любому из пп. 1-4, где удаление силильной защитной группы на стадии f) проводят с помощью тригидрата тетрабутиламмония фторида.
6. Способ по любому из пп. 1-5, где разделение E- и Z-изомеров на стадии f) проводят с применением ступенчато-градиентных смесей-элюентов с применением в качестве элюента смеси толуол:метил-трет-бутиловый эфир.
7. Способ по любому из пп. 1-6, где изомеризацию Z-изомера на стадии f) проводят путем облучения в присутствии сенсибилизатора, представляющего собой диметилдисульфид.
8. Способ по любому из пп. 1-7, где окисление на стадии h) проводят путем окисления Пфитцнера-Моффата с помощью смеси фосфорная кислота-DMSO, содержащей DCC или DIC, или путем окисления Анелли (гипохлорит натрия, катализатор TEMPO).
9. Способ по любому из пп. 1-8, где соединение формулы X подвергают превращению на стадии i) в соединение формулы XI без выделения.
10. Способ по любому из пп. 1-9, где на стадии i) 15R-изомер формулы XIIIb, выделенный посредством хроматографии, после окисления и введения THP-защитной группы для 11-OH-группы рециркулируют в синтез.
11. Способ по любому из пп. 1-10, где соединение формулы XII не выделяют на стадии k).
12. Способ по любому из пп. 1-11, где неочищенный готовый продукт формулы I очищают посредством гравитационной хроматографии и/или препаративной ВЭЖХ.
13. Соединение формулы XI, имеющее следующую структуру
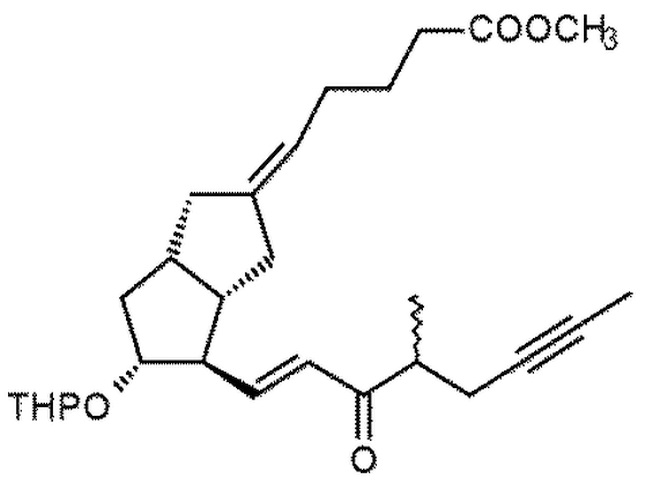 XI.
XI.
14. Соединение формулы XII, имеющее следующую структуру
 XII.
XII.
15. Соединение формулы (S)-XII, имеющее следующую структуру
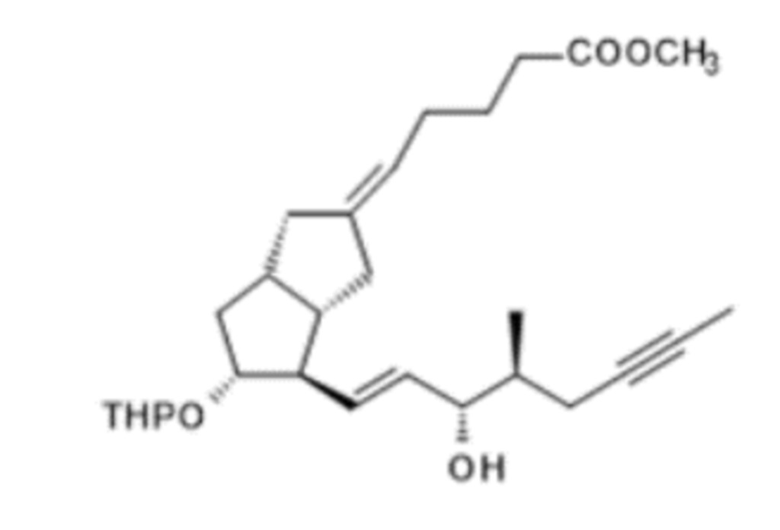 (S)-XII.
(S)-XII.
16. Соединение формулы (R)-XII, имеющее следующую структуру
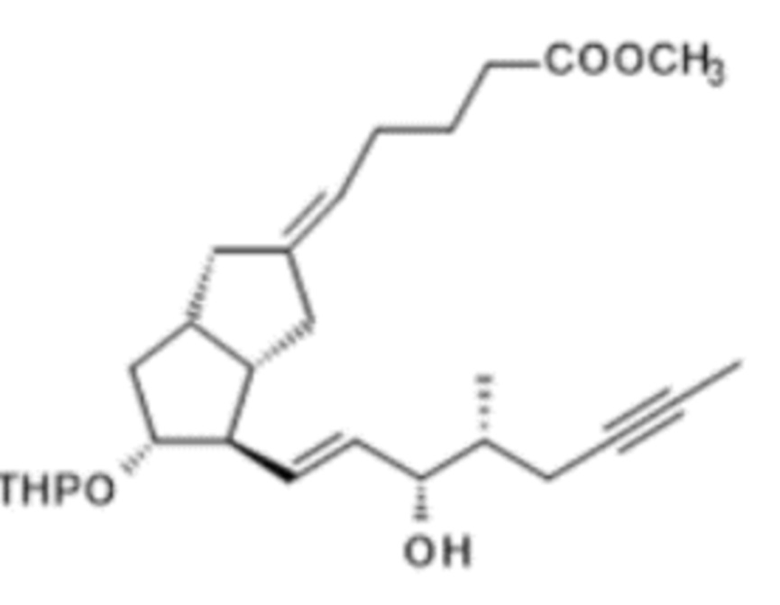 (R)-XII.
(R)-XII.
17. Способ по п. 1 получения 16(S)-илопроста, включающий введение в реакцию соединения формулы X
X
с (S)-ILO-фосфонатом
 (S)-ILO-фосфонат.
(S)-ILO-фосфонат.
18. Способ по п. 1 получения 16(R)-илопроста, включающий введение в реакцию соединения формулы X
X
с (R)-ILO-фосфонатом
 (R)-ILO-фосфонат.
(R)-ILO-фосфонат.
| Устройство для высокочастотного нагрева и сушки неметаллических материалов | 1948 |
|
SU78919A1 |
| R | |||
| C | |||
| NICKOLSONM | |||
| H | |||
| TOWNH | |||
| VORBRIIGGEN: | |||
