Область изобретения
Настоящее изобретение относится к новой фармацевтической композиции в форме эмульсионного предварительного концентрата, к стандартной лекарственной форме, содержащей указанную композицию, к ее применению в терапии, а также к способу ее приготовления.
Предшествующий уровень техники
Нестероидные противовоспалительные лекарства, обычно сокращенно называемые НСПВЛ (NSAID, non-steroidal anti-inflammatory drug), представляют собой хорошо известные лекарства для лечения боли и воспаления. Один из основных недостатков, связанных с НСПВЛ, заключается в том, что они имеют тяжелые желудочно-кишечные побочные действия. Пациенты, подвергающиеся лечению НСПВЛ, такими как напроксен, в течение более длительного периода времени, часто имеют проблемы, связанные с побочными действиями на желудок в желудочно-кишечном тракте.
Недавно обнаружено, что высвобождающие окись азота соединения-НСПВЛ (в дальнейшем: NO-высвобождающие НСПВЛ) имеют улучшенный профиль в плане побочного действия, см., например, WO 94/04484, WO 94/12463, WO 95/09831 и WO 95/30641.
NO-высвобождающие НСПВЛ представляют собой липофильные соединения с плохой растворимостью в воде. Они могут быть классифицированы как класс 2 в соответствии с Биофармацевтической Системой Классификации (Biopharmaceutical Classification System), предложенной Амидоном и др. (Amidon et al., Pharm. Res. 12 (1995) pp.413-420). Лекарства этого класса характеризуются низкой растворимостью в воде, но достаточно хорошей проницаемостью. Биофармацевтическая проблема этих соединений состоит в том, что их абсорбция из желудочно-кишечного тракта (ЖКТ) может ограничиваться скоростью растворения, приводя к плохой биодоступности при пероральном введении.
В WO 95/08983 описана самоэмульгирующаяся композиция для перорального введения, которая образует микроэмульсию in situ при контакте с биологическими жидкостями. Эта композиция может быть охарактеризована как самомикроэмульгирующаяся система доставки лекарств (self-microemulsifying drug delivery system, SMEDDS) и содержит, по меньшей мере:
- активное соединение,
- липофильную фазу, состоящую из смеси глицеридов и сложных эфиров жирных кислот,
- поверхностно-активное вещество,
- вторичное поверхностно-активное вещество и
гидрофильную фазу, которая образуется после приема внутрь под действием физиологической жидкости пищеварительной среды.
Настоящее изобретение отличается по нескольким аспектам от WO 95/08983 и других SMEDDS. В то время как композиции, раскрытые в WO 95/08983, образуют микроэмульсию in situ, композиции по настоящему изобретению образуют эмульсию. SMEDDS из WO 95/08983 требует присутствия липофильной фазы для солюбилизации активного соединения. Такая липофильная солюбилизирующая фаза не требуется для настоящего изобретения, поскольку активное соединение, NO-высвобождающее НСПВЛ, способно самостоятельно составить масляную фазу эмульсии in situ. Композиции WO 95/08983 содержат inter alia вторичное поверхностно-активное вещество в дополнение к поверхностно-активному веществу. Наличие вторичного поверхностно-активного вещества не является необходимым для композиций по настоящему изобретению, что снижает токсикологическую проблему до минимума.
В ЕР 274 870 раскрыта фармацевтическая композиция, содержащая нестероидное противовоспалительное лекарство (НСПВЛ) и поверхностно-активное вещество, которая при пероральном введении образует мицеллы, содержащие НСПВЛ. Обнаружено, что эти мицеллы представляют собой особенно подходящую форму для введения НСПВЛ перорально, смягчая их неблагоприятные действия на желудочно-кишечный тракт. Мицеллы представляют собой агрегаты, в которых молекулы поверхностно-активного вещества, как правило, располагаются в виде сферической структуры с гидрофобной областью внутри, отгороженной в водном растворе от воды покровом из внешних гидрофильных областей. Обычно лекарство солюбилизировано в поверхностно-активном веществе. Мицеллы следует сопоставить в плане их структуры с эмульсиями, которые образуют композиции по настоящему изобретению. В то время как мицеллы представляют собой термодинамически стабильные однофазные системы (согласно правилу фаз Гиббса), в которых агрегаты обычно имеют диаметр, равный приблизительно двум длинам молекулы поверхностно-активного вещества, образующей его, то есть порядка от нескольких десятков до сотен Ангстрем (А), эмульсии представляют собой гораздо более крупные агрегаты, порядка от нанометров до микрометров в диаметре, состоящие из маслянистой внутренней части, которая окружена одним или несколькими слоями поверхностно-активных веществ. Эмульсии представляют собой, как правило, двухфазные системы, и они термодинамически нестабильны (но могут быть кинетически стабильными). Другим существенным различием между композициями из ЕР 274870 и настоящим изобретений является природа активного соединения. В то время как НСПВЛ по своей природе являются кристаллическими порошками, NO-высвобождающие НСПВЛ или смеси NO-высвобождающих НСПВЛ, используемые в настоящем изобретении, находятся в масляной форме или представляют собой терморазмягчающееся полутвердое вещество. Более того, для мицелл обычно требуется гораздо более высокое соотношение лекарство: поверхностно-активное вещество по сравнению с соотношением масло: поверхностно-активное вещество, требующимся для образования эмульсии.
Одной из уникальных особенностей NO-высвобождающих НСПВЛ является то, что многие из этих соединений представляют собой масла или терморазмягчающиеся полутвердые вещества, которые практически нерастворимы в воде. При высокой дозе NO-высвобождающих НСПВЛ, например, когда доза выше чем приблизительно 350 мг, трудно приготовить препарат в виде таблетки приемлемого размера с большим количеством масла или полутвердого вещества. Однако липофильные NO-высвобождающие НСПВЛ могут быть приготовлены в препарате в виде эмульсий типа масло-в-воде, где такое соединение составляет масляную фазу или представляет собой часть масляной фазы, при этом указанная масляная фаза эмульгирована в воде с помощью одного или более чем одного поверхностно-активного вещества.
В фармакокинетических исследованиях на животных неожиданно было обнаружено, что такие эмульсии NO-высвобождающих НСПВЛ типа масло-в-воде показывают гораздо лучшую биодоступность по сравнению с неэмульгированным веществом. Однако существует проблема, связанная с эмульсиями, которая состоит в том, что они являются термодинамически нестабильными и имеют низкую стабильность при длительном хранении, поскольку часто проявляют тенденцию к коалесценции, расслоению/осаждению или разделению фаз. Кроме того, они не являются удобной лекарственной формой для перорального введения, поскольку зачастую для включения одной дозы требуются большие объемы, а также существенную проблему может составлять неприятный горький или мыльный вкус. Наполнить эмульсиями типа масло-в-воде желатиновые капсулы inter alia невозможно, поскольку большое содержание воды в такой эмульсии несовместимо с оболочкой капсулы и могло бы растворить ее.
Сущность изобретения
Проблемы, указанные выше, в настоящее время решены предложением новой самоэмульгирующейся системы доставки лекарств, обычно известной как SEDDS, подходящей для перорального введения. Более конкретно, настоящее изобретение относится к фармацевтической композиции, пригодной для перорального введения, в форме эмульсионного предварительного концентрата, содержащей
(1) одно или более чем одно NO-высвобождающее НСПВЛ;
(2) одно или более чем одно поверхностно-активное вещество;
(3) возможно масло или полутвердый жир;
при этом указанная композиция образует in situ эмульсию типа масло-в-воде при контакте с водными средами, такими как желудочно-кишечные жидкости.
Кроме того, композиция по настоящему изобретению возможно может дополнительно содержать один или более чем один спирт с короткой цепью.
Такая композиция образует in situ эмульсию типа масло-в-воде в виде небольших капель размером от нанометров до микронов при контакте с желудочно-кишечными жидкостями, причем данные капли состоят из одного или более чем одного NO-высвобождающего НСПВЛ, образующего внутреннюю часть капли, которая покрыта одним или несколькими слоями поверхностно-активного вещества. Образованная in situ эмульсия типа масло-в-воде обеспечивает хорошую биодоступность NO-высвобождающих НСПВЛ при пероральном введении. Проблемы стабильности эмульсии при хранении не возникает, поскольку эмульсия не образуется до тех пор, пока предварительный концентрат не принят внутрь пациентом, то есть до момента введения. Возможный неприятный вкус предварительного концентрата не является проблемой, когда он заключен в капсулы.
Фармацевтическая композиция по настоящему изобретению представляет собой эмульсионный предварительный концентрат во время введения пациенту. Эмульсионным предварительным концентратом могут быть наполнены разовые стандартные лекарственные формы, такие как капсулы, ампулы для питья и дозированные подушки (dose cushions), или он альтернативно может быть сформирован в виде других подходящих лекарственных форм, таких как жевательные мягкие пилюли и лепешки с жевательной основой.
При контакте с водными средами, такими как желудочно-кишечные жидкости, эмульсионный предварительный концентрат превращается в эмульсию типа масло-в-воде. Таким образом, данная композиция будет образовывать in situ эмульсию типа масло-в-воде в желудочно-кишечном тракте (ЖК-тракте). Скорость высвобождения лекарства из композиции определяется размером капель эмульсии in situ и полярностью капель эмульсии, причем последнее обуславливается гидрофильно-липофильным балансом (HLB) смеси лекарство/поверхностно-активное вещество и концентрацией поверхностно-активного вещества. Как правило, малый размер капель и высокая полярность дают высокую скорость высвобождения лекарства (N.H. Shah et al., Int. J. Pharm. 106 (1994), pp.15-23).
Обозначение "НСПВЛ" определяется как нестероидное противовоспалительное лекарство, то есть любое лекарство, обладающее противовоспалительным действием, но при этом такое соединение не принадлежит к классу соединений "стероиды". Специалисту в данной области техники понятно, подпадает ли соединение под определение НСПВЛ. Примерами конкретных НСПВЛ являются напроксен, диклофенак, ацеклофенак, индометацин, кеторолак, сулиндак, мелоксикам, пироксикам, теноксикам, ибупрофен, кетопрофен, напроксен, азапропазон, набуметон, карпрофен, тиапрофеновая кислота, супрофен, индопрофен, этодолак, фенопрофен, фенбуфен, флурбипрофен, бермопрофен, пиразолак, залтопрофен, набуметон, бромфенак, ампироксикам и лорноксикам. Однако этот перечень не следует рассматривать как в каком-либо отношении исчерпывающий. Предполагается, что выражение "NO-высвобождающий НСПВЛ" охватывает любое нестероидное противовоспалительное лекарство (НСПВЛ), его соль или энантиомер, которой обладает способностью высвобождать оксид азота.
NO-высвобождающие НСПВЛ представляют собой липофильные соединения с плохой растворимостью в воде. Они могут быть классифицированы как класс 2 в соответствии с Биофармацевтической Системой Классификации (Biopharmaceutical Classification System), предложенной Amidon et al. (Pharm. Res. 12 (1995) 413-420). Лекарства этого класса характеризуются низкой растворимостью в воде, но достаточно хорошей проницаемостью. Биофармацевтическая проблема этих соединений состоит в том, что их абсорбция из желудочно-кишечного тракта (ЖКТ) может быть ограничена скоростью растворения, что приводит к плохой биодоступности при пероральном введении.
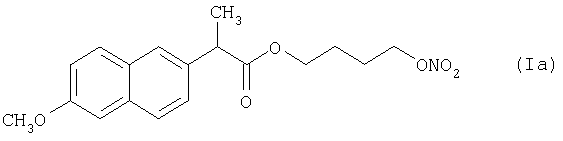
Предпочтительные NO-высвобождающие НСПВЛ в соответствии с настоящим изобретением представляют собой соединения формулы I
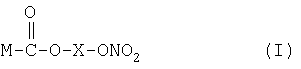
где
X представляет собой спейсер, то есть соединение, образующее мостик между группой, служащей донором оксида азота, и НСПВЛ; и
М выбран как любое из следующих:
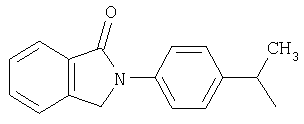
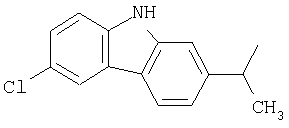
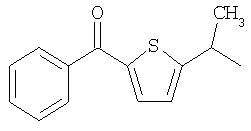
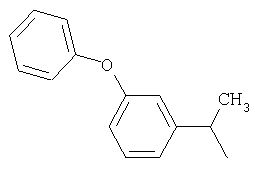
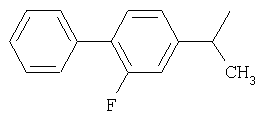
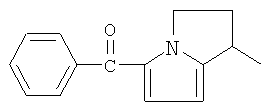
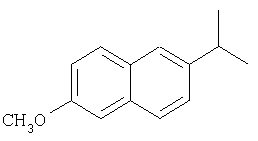
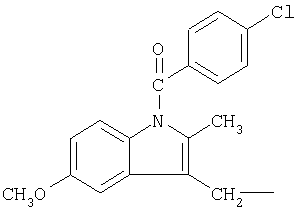
 и
и 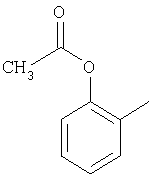 .
.
В предпочтительном воплощении изобретения спейсер Х выбран из линейной, разветвленной или циклической алкиленовой группы -(CH2)-n, где n является целым числом от 2 до 10; -(CH2)m-O-(CH2)p-, где m и р являются целыми числами от 2 до 10; и -CH2-pC6H4-CH2-.
В одном воплощении данного изобретения NO-высвобождающие НСПВЛ, рассматриваемые в качестве активного(ых) соединения(й) в препарате SEDDS по изобретению, представляют собой соединения, раскрытые и заявленные в WO 94/04484, WO 94/12463, WO 95/09831 и WO 95/30641, которые включены в материалы заявки посредством ссылки.
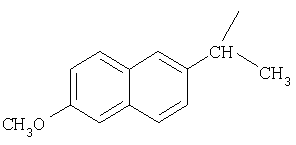
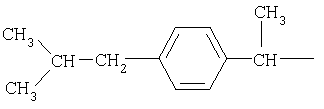
Конкретные NO-высвобождающие вещества, полезные в соответствии с настоящим изобретением, представляют собой:
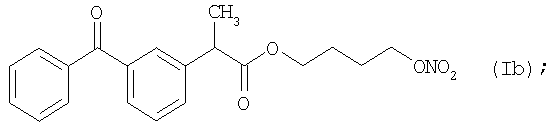
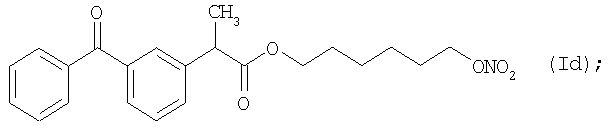
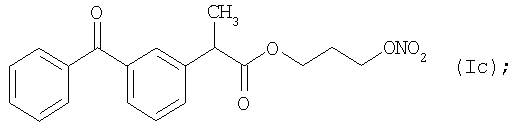
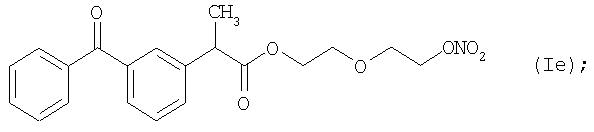
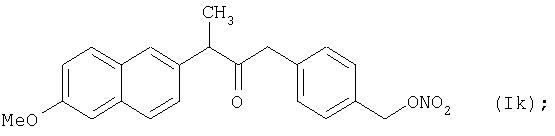
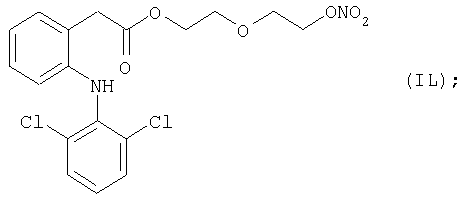
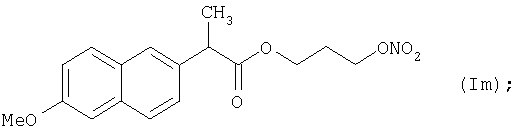
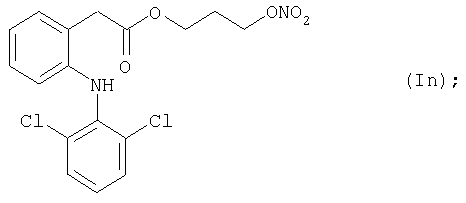
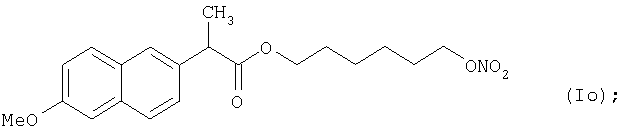
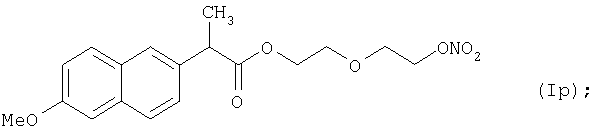 и
и
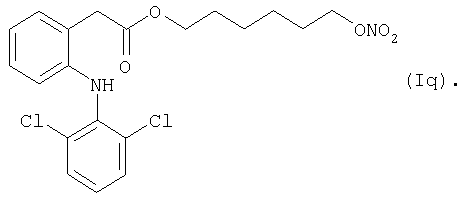
НСПВЛ по природе существуют в форме порошка, в то время как NO-высвобождающие НСПВЛ преимущественно обуславливает, благодаря спейсеру, соединение в полутвердой или масляной форме как таковое. Эта уникальная особенность дает то преимущество, что нет необходимости добавлять к эмульсионному предварительному концентрату липофильную масляную или полутвердую основу извне, поскольку это свойство является неотъемлемой особенностью указанного лекарства. Дополнительно к фармацевтической композиции могут быть добавлены фармакологически инертные масло или полутвердый жир наполнителем или регулятором вязкости. Наполнитель может требоваться, чтобы увеличить точность дозировки для соединений в низкой дозе. Регулятор вязкости может требоваться для того, чтобы отрегулировать вязкость, оптимальную для наполнения композицией, например, капсул. В частности, высокоскоростное наполнение капсул жидкостью требует тщательной регулировки вязкости в пределах интервала, для которого на конце, соответствующем низкой вязкости, предотвращается разбрызгивание, а на конце, соответствующем высокой вязкости, предотвращается нитеобразование. Кроме того, этот интервал вязкости должен быть выбран так, чтобы дать препарат, годный для перекачки насосом. Обычно требуемый для наполнения капсул жидкостью интервал вязкости составляет от 0,1 до 25 Па·с.
Общее количество NO-высвобождающих НСПВЛ, используемое в композиции по данному изобретению, предпочтительно находится в интервале 50-1500 мг на стандартную дозу. Еще в одном предпочтительном воплощении количество NO-высвобождающего(их) НСПВЛ, используемое в данной композиции, составляет 125-500 мг на стандартную дозу.
Термин "стандартная доза" определяется как количество активного соединения, введенное в одной разовой капсуле или растворенное в одном стакане воды.
Выражение "поверхностно-активное вещество" определяется как поверхностно-активные амфифильные соединения, такие как блоксополимеры. Предпочтительными поверхностно-активными веществами в соответствии с настоящим изобретением являются неионогенные поверхностно-активные вещества, например те, что содержат полиэтиленгликолевые (ПЭГ) цепи, в особенности блоксополимеры такие как полоксамеры.
Примерами подходящих полоксамеров являются Полоксамер 407 (Pluronic F127®); Полоксамер 401 (Pluronic L121®); Полоксамер 237 (Pluronic F87®); Полоксамер 338 (Pluronic F138®); Полоксамер 331 (Pluronic L101®); Полоксамер 231 (Pluronic L81®); тетрафункциональный полиоксиэтилен-полиоксипропиленовый блоксополимер этилендиамина, известный как Полоксамин 908 (Tetronic 908®); Полоксамин 1307 (Tetronic 1307®); Полоксамин 1107; полиоксиэтилен-полиоксибутиленовый блоксополимер, известный как Polyglycol BM45®. Этот перечень предназначен только для того, чтобы служить в качестве примера поверхностно-активных веществ, которые могут быть использованы в соответствии с настоящим изобретением, и его не следует никоим образом рассматривать как исчерпывающий или как ограничивающий изобретение.
Все поверхностно-активные вещества, описанные выше, имеются в продаже, например, у BASF, Dow Chemicals и Gattefosse.
Общее количество поверхностно-активных(ого) веществ(а) в соответствии с данным изобретением может находиться в пределах интервала 12,5-6000 мг, предпочтительно 100-500 мг.
Соотношение NO-высвобождающее НСПВЛ: поверхностно-активное вещество может варьировать от 1:0,1 до 1:10, предпочтительно от 1:0,3 до 1:3.
Если к фармацевтической композиции добавлено дополнительное масло, то оно может быть любым маслом, если является инертным и совместимым с материалом капсулы, а также приемлемым для использования в фармацевтических препаратах. Специалисту в данной области техники будет понятно, какое масло выбрать для планируемой цели. Примерами подходящих масел, которые могут быть использованы в соответствии с настоящим изобретением, являются растительные масла, такие как кокосовое масло, кукурузное масло, соевое масло, рапсовое масло, сафлоровое масло и касторовое масло. Кроме этого для целей настоящего изобретения пригодными являются животные масла, такие как рыбий жир и триглицериды.
Если в качестве наполнителя для фармацевтической композиции используется полутвердый жир, он предпочтительно может быть выбран из моно-, ди- и триглицеридов и спирта жирной кислоты, как, например, стеариловый спирт, Gelucires 33/01®, 39/01®, 43/01®, глицерилпальмитостеарат, такой как Precirol АТ05®. Gelucire представляет собой смесь, полученную в результате смешивания моно-, ди- и триэфиров глицерина, моно- и диэфиров ПЭГ или свободного ПЭГ.
В соответствии с одним аспектом изобретения в качестве активного ингредиента используют маслянистое (липофильное) или полутвердое NO-высвобождающее НСПВЛ.
Если в фармацевтической композиции по изобретению использовано дополнительное масло или полутвердый жир, они могут служить в качестве наполнителя или регулятора вязкости.
Выражение "спирты с короткой цепью", используемое в соответствии с настоящим изобретением, определяется здесь как линейные или разветвленные одно-, двух- или трехатомные спирты, имеющие 1-6 углеродных атомов. Примерами таких спиртов с короткой цепью, полезных в соответствии с данным изобретением, являются этанол, пропиленгликоль и глицерин.
Если к фармацевтической композиции по изобретению добавлен спирт с короткой цепью, то это приводит к повышению растворимости и к тому, что требуется меньшее количество поверхностно-активного вещества.
В соответствии с еще одним аспектом настоящего изобретения в качестве активных ингредиентов используются два или более NO-высвобождающих НСПВЛ, причем любое из указанных лекарств может находиться в виде масла или полутвердого вещества, либо по меньшей мере одно из указанных лекарств находиться в виде масла или полутвердого вещества, а другое(ие) может(могут) находиться в виде твердого вещества, которое растворено или суспендировано в указанном маслянистом или полутвердом соединении. Комбинации двух или более NO-высвобождающих НСПВЛ могут быть полезны в том случае, когда желательно, чтобы большая NO-загрузка высокой дозы низкоэффективного NO-высвобождающего НСПВЛ была дополнена низкой дозой высокоэффективного NO-высвобождающего НСПВЛ.
В соответствии с еще одним аспектом данного изобретения предложена комбинация одного или более чем одного NO-высвобождающего НСПВЛ и чувствительного к кислоте соединения - ингибитора протонного насоса (proton pomp inhibitor, PPI). NO-НСПВЛ должны быть приготовлены в виде такого препарата, который эмульгируется в желудке, то есть в виде препарата SEEDS, как он описан выше, в то время как чувствительный к кислоте ингибитор протонного насоса (PPI) должен быть защищен от контакта с кислотным желудочным соком, например, энтеросолюбильным покрытием. PPI с нанесенным на него слоем энтеросолюбильного покрытия остается не нетронутым, пока не достигнет кишечника, где этот PPI высвобождается. Отдельно приготовленные единицы ингибитора протонного насоса (PPI) с нанесенным слоем энтеросолюбильного покрытия могут быть смешаны с расплавом SEDDS. Альтернативно, PPI могут быть загружены в капсулу, наполненную отвердевшим SEDDS, где между SEDDS и приготовленными гранулами PPI может быть необходим слой защитного парафина. В другом альтернативном воплощении приготовленные гранулы PPI могут быть смешаны с жидким препаратом SEDDS.
Таким образом, комбинация может быть либо связанной комбинацией, то есть в виде препарата, где NO-высвобождающее(ие) НСПВЛ и чувствительный к кислоте ингибитор протонного насоса смешаны и после этого загружены в подходящую единицу дозировки. В другом воплощении изобретения чувствительным к кислоте ингибитором протонного насоса может быть наполнена капсула с уже отвердевшим препаратом SEDDS одного или более чем одного NO-высвобождающего НСПВЛ - в этом случае между препаратом SEDDS и чувствительным к кислоте ингибитором протонного насоса может требоваться слой защитного парафина или другого инертного материала. В еще одном воплощении чувствительный к кислоте ингибитор протонного насоса смешан с жидким препаратом SEDDS NO-высвобождающего(их) НСПВЛ.
В другом воплощении изобретения NO-высвобождающее(ие) НСПВЛ и PPI могут быть представлены в форме набора, при этом NO-высвобождающее НСПВЛ и PPI вводят последовательно, то есть один после другого. Порядок введения не является важным, что означает, что любой из NO-высвобождающего НСПВЛ либо PPI может быть введен до другого. Таким образом, одно воплощение данного изобретения представляет собой комбинированное лечение, при котором пациенту, нуждающемуся в лечении, вводят одно или более чем одно NO-высвобождающее НСПВЛ, после чего вводят PPI, или наоборот.
Примерами ингибиторов протонного насоса, подходящих для комбинации с NO-высвобождающим НСПВЛ в соответствии с настоящим изобретением, как раскрыто выше, являются соединение общей формулы I или его фармацевтически приемлемая щелочная соль либо один из его индивидуальных энантиомеров или щелочная соль индивидуального энантиомера:
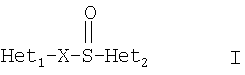
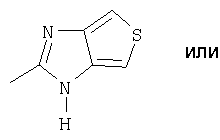
где Het1 представляет собой
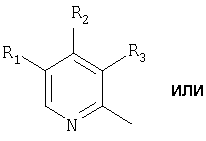
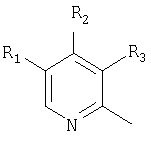 или
или 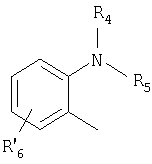
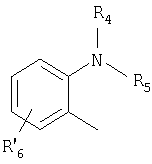
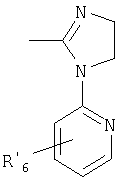
Het2 представляет собой
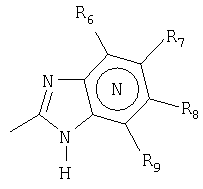 или
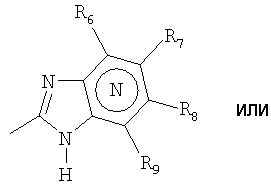
или 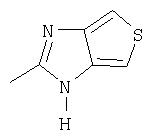 или
или 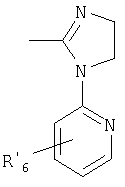
X=
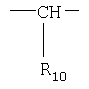 или
или 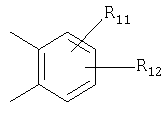
где
N в бензимидазольной группировке означает, что один из углеродных атомов, замещенных R6-R9, возможно может быть заменен на атом азота без каких-либо заместителей;
R1, R2 и R3 являются одинаковыми или разными и выбраны из водорода, алкила, алкокси, возможно замещенного фтором, алкилтио, алкоксиалкокси, диалкиламино, пиперидино, морфолино, галогена, фенила и фенилалкокси;
R4 и R5 являются одинаковыми или разными и выбраны из водорода, алкила и аралкила;
R6 представляет собой водород, галоген, трифторметил, алкил и алкокси;
R6-R9 являются одинаковыми или разными и выбраны из водорода, алкила, алкокси, галогена, галогеноалкокси, алкилкарбонила, алкоксикарбонила, оксазолила, трифторалкила, либо соседние группы R6-R9 образуют кольцевые структуры, которые могут быть дополнительно замещены;
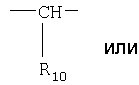
R10 является водородом или образует алкиленовую цепь вместе с R3, и
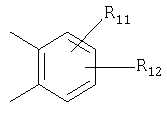
R11 и R12 являются одинаковыми или разными и выбраны из водорода, галогена или алкила;
алкильные группы, алкоксигруппы и их группировки могут быть разветвленными или прямыми С1-С9-цепями либо включают циклические алкильные группы, такие как циклоалкил-алкил.
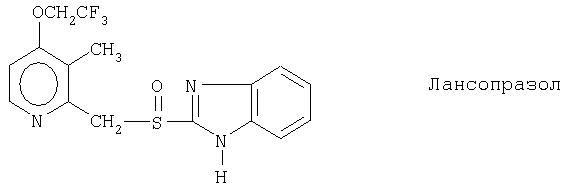
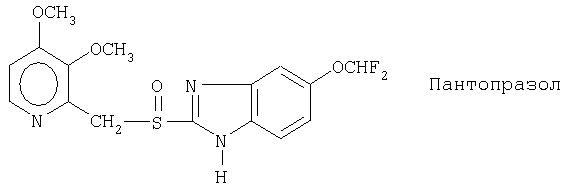
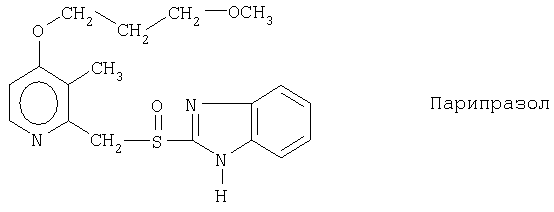
Примерами конкретных ингибиторов протонного насоса, подходящих по изобретению, являются
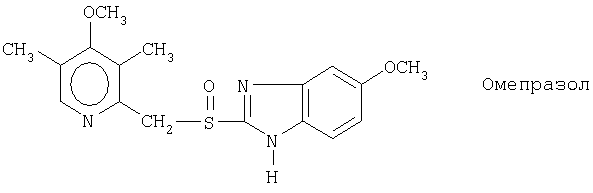
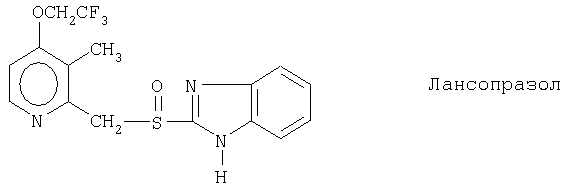
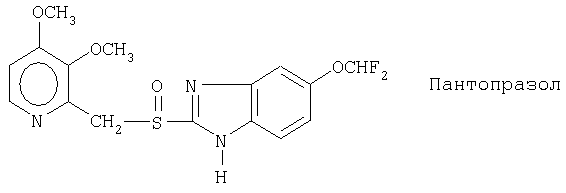
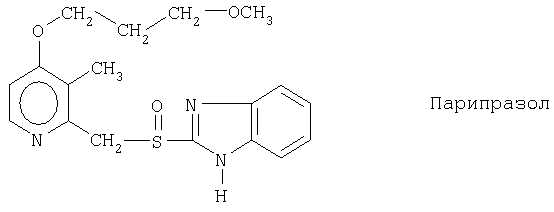
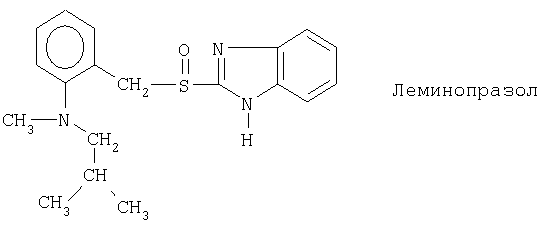
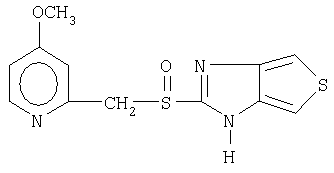
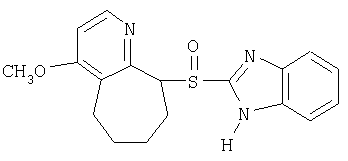
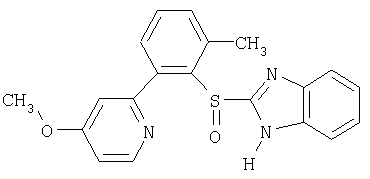
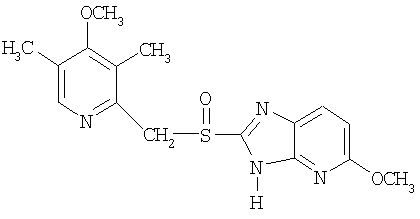
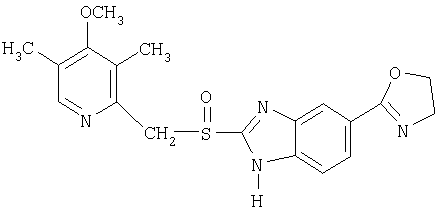
Чувствительные к кислоте ингибиторы протонного насоса, применяемые в лекарственных формах по изобретению, могут быть использованы в своей нейтральной форме или в форме щелочной соли, как, например, Mg2+, Са2+, Na+, К+ или Li+-солей, предпочтительно Mg2+-солей. Кроме того, где применимо, перечисленные выше соединения могут быть использованы в форме рацематов, и в форме их по существу чистого энантиомера, или в форме щелочных солей индивидуальных энантиомеров.
Подходящие ингибиторы протонного насоса описаны, например, в ЕР-А1-0005129, ЕР-А1-174726, ЕР-А1-166287, GB 2163747 и WO 90/06925, и, дополнительно, особенно предпочтительные соединения описаны в WO 95/01977 и WO 94/27988.
Ингибиторы протонного насоса, используемые в комбинации в соответствии с настоящим изобретением, предпочтительно представлены в виде гранул с нанесенным слоем энтеросолюбильного покрытия, содержащих чувствительный к кислоте ингибитор протонного насоса. Для композиции гранул с нанесенным слоем энтеросолюбильного покрытия и их приготовления дается ссылка на WO 96/01623, включенную сюда посредством такой ссылки.
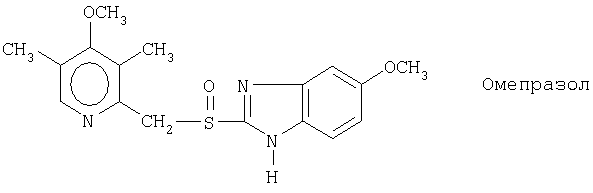
Подходящими комбинациями по настоящему изобретению являются, например, NO-высвобождающее НСПВЛ формулы Ia и омепразол, или щелочная соль омепразола, (S)-омепразол, или щелочная соль (S)-омепразола; либо NO-высвобождающее НСПВЛ формулы Ii и омепразол, или щелочная соль омепразола, (S)-омепразол, или щелочная соль (S)-омепразола.
Фармацевтической композицией по настоящему изобретению наполняют разовые лекарственные формы, пригодные для перорального введения, такие как капсулы, ампулы для питья и дозированные подушки, либо их могут готовить в виде других подходящих пероральных лекарственных форм, таких как жевательные мягкие пилюли и лепешки с жевательной основой.
В предпочтительном воплощении данного изобретения фармацевтической композицией наполняют твердые желатиновые капсулы, но могут быть использованы и капсулы из альтернативных материалов, такие как оболочки на основе метилцеллюлозы и мягкие желатиновые капсулы.
В другом воплощении данного изобретения фармацевтическая композиция может быть растворена, например, в стакане воды, что позволяет предварительному концентрату образовать эмульсию, которая может быть введена как таковая. Композициями, предназначенными для растворения перед введением, могут быть наполнены, например, мягкие желатиновые капсулы, пластиковые или алюминиевые подушки либо пластиковые или стеклянные ампулы. Эта особенность особенно благоприятна для композиций с большой дозой, для которых потребовались бы большие капсулы, для пациентов, испытывающих трудности при глотании капсул, и для детей.
В предпочтительном воплощении фармацевтической композицией по настоящему изобретению наполняют капсулы. Предпочтительными капсулами являются желатиновые капсулы, которые могут быть мягкими или твердыми. Твердые желатиновые капсулы состоят из двух частей, крышечки и тела - одна вставляется внутрь другой. Твердые желатиновые капсулы производят пустыми и наполняют на отдельной операционной стадии. Мягкая желатиновая капсула представляет собой капсулу, которую изготавливают и наполняют на в одну однократную операцию.
Как упомянуто выше, эмульсионный предварительный концентрат при контакте с желудочно-кишечными жидкостями превращается в эмульсию типа масло-в-воде, посредством чего высвобождается активное лекарство.
Таким образом, композиция образует in situ эмульсию типа масло-в-воде в желудочно-кишечном тракте (ЖК-тракте).
Фармацевтическая композиция по изобретению особенно полезна при лечении боли и воспаления. Подразумевается, что термин "боль" включает ноцицептивную и невропатическую боль или их комбинации; острую, прерывистую и хроническую боль; боль раке; мигрень и головные боли схожего происхождения, но не ограничивается этим. Подразумевается, что термин "воспаление" охватывает ревматоидный артрит; остеоартрит и ювенильный артрит, но не ограничивается этим.
Способы приготовления
Фармацевтическая композиция по настоящему изобретению может быть приготовлена главным образом следующими альтернативными способами:
I. Смешивание
а) Маслянистое или полутвердое NO-высвобождающее НСПВЛ помещают в сосуд, добавляют твердое или полутвердое поверхностно-активное вещество и твердый/маслянистый жир (не обязательно). Смесь нагревают до температуры, соответствующей точке плавления эксципиентов, делая препарат текучим, тщательно перемешивают до гомогенности (контролируя визуально) и этим предварительным концентратом наполняют капсулы, пригодные для перорального введения.
б) Альтернативно, маслянистое NO-высвобождающее НСПВЛ помещают в сосуд и добавляют текучее поверхностно-активное вещество. Смесь тщательно перемешивают до гомогенности (контролируя визуально) и предварительным концентратом наполняют капсулы, пригодные для перорального введения.
в) В еще одном альтернативном способе маслянистое NO-высвобождающее НСПВЛ помещают в сосуд, добавляют тонко размолотое (размер частиц <177 мкм) твердое поверхностно-активное вещество. Жидкую смесь тщательно перемешивают до гомогенности (контролируя визуально) и этим предварительным концентратом наполняют капсулы, пригодные для перорального введения.
г) Еще в одном альтернативном способе полутвердое/твердое поверхностно-активное вещество(а) помещают в сосуд и добавляют один или более чем один спирт. Смесь нагревают до температуры, соответствующей точке плавления эксципиентов, делая препарат текучим, тщательно перемешивают до гомогенности (контролируя визуально). Добавляют NO-НСПВЛ и смесь тщательно перемешивают до гомогенности (контролируя визуально). Предварительным концентратом наполняют капсулы, пригодные для перорального введения.
д) Еще в одном альтернативном способе жидкое поверхностно-активное вещество(а) помещают в сосуд и добавляют один или более чем один спирт. Смесь тщательно перемешивают до гомогенности (контролируя визуально). Добавляют NO-НСПВЛ и смесь тщательно перемешивают до гомогенности (контролируя визуально). Предварительным концентратом наполняют капсулы, подходящие для перорального введения.
Для того чтобы наполнить состоящую из двух частей капсулу или мягкую желатиновую капсулу жидкостью, вязкость препарата должна находиться в определенных пределах, как определено производителем, при температуре наполнения, подходящей для процесса. Для состоящей из двух частей капсулы максимальная температура наполнения составляет приблизительно 70°С. Вязкость препарата в норме должна находиться в диапазоне 50-1000 сантипуаз (=0,05-1 Па·с) при температуре, подобранной для процесса наполнения. Для наполнения препаратом мягких желатиновых капсул не допускается, чтобы температура процесса превышала 30-40°С (точная температура зависит от производителя). Препарат должен быть жидким и иметь вязкость, которая делает его годным для перекачки насосом при температуре наполнения. Для того чтобы сделать препарат жидким с приемлемой вязкостью, можно использовать некоторые добавки, например Cremophor EL®.
II. Наполнение
Для процедуры наполнения требуется, чтобы композиция находилась в жидкой форме при температуре наполнения. Наполнение полутвердыми терморазмягчающимися композициями ввиду этого производят при температуре выше температуры разжижения. Мягкие желатиновые капсулы изготавливают и наполняют за одну операцию, и они могут быть наполнены при температурах вплоть до 40°С, тогда как твердые желатиновые капсулы могут быть наполнены при температурах вплоть до 70°С. Твердые желатиновые капсулы, наполненные композициями, которые остаются жидкими при температуре хранения, требуют герметизации, например, полоской желатина, для предотвращения утечки. Процессы наполнения жидкостью твердых желатиновых капсул и требования к продукту описаны, например, в W.J. Bowtle, Pharmaceutical Technology Europe, October 1998; V.M. Young, Pharmaceutical Manufacturing and Packaging Sourcer, March 1999; и E. T.Coole, Pharmaceutical Technology International, September/October 1989. Использование состоящих из двух частей капсул допускает загрузку в одну капсулу более чем одной фазы, что может быть желательно для би- или мультифазного высвобождения лекарства (W.J. Bowtle et al., Int. J. Pharm. 141 (1996), pp.9-16). Несколько фаз отвердевающего материала могут быть загружены на отдельных стадиях. Завершающая фаза может быть жидкой, если требуется. Количество фаз ограничено только размером капсулы и объемом отдельных фаз. Эта частная особенность также может давать возможность регулируемого высвобождения или разделения разных лекарственных субстанций, включенных в состав препарата в одной и той же капсуле. Кроме того, капсулы могут быть дополнительно обработаны, например, энтеросолюбильным покрытием.
III. Комбинация с PPI
Маслянистое или полутвердое NO-высвобождающее НСПВЛ помещают в сосуд, добавляют твердое или полутвердое поверхностно-активное вещество и твердый/маслянистый жир (не обязательно). Смесь нагревают до температуры, соответствующей точке плавления эксципиентов, делая препарат текучим, тщательно перемешивают до гомогенности (контролируя визуально) и добавляют к этой смеси приготовленные гранулы с нанесенным слоем энтеросолюбильного покрытия, содержащие чувствительный к кислоте ингибитор протонного насоса. Этим предварительным концентратом с суспендированными гранулами PPI наполняют капсулы, подходящие для перорального введения, где он отвердевает.
Альтернативно, маслянистое или полутвердое NO-высвобождающее НСПВЛ помещают в сосуд, добавляют твердое поверхностно-активное вещество и твердый/маслянистый жир (не обязательно). Смесь нагревают до температуры, соответствующей точке плавления эксципиентов, делая препарат текучим, тщательно перемешивают до гомогенности (контролируя визуально). Предварительным концентратом наполняют капсулы, пригодные для перорального введения, где он отвердевает. Добавляют защитный слой парафина или любой другой инертной терморазмягчающейся основы, пригодной для перорального введения, и дают ей затвердеть. Поверх парафина добавляют приготовленные гранулы PPI.
В еще одном альтернативном способе маслянистое NO-высвобождающее НСПВЛ помещают в сосуд и добавляют текучее поверхностно-активное вещество. Смесь тщательно перемешивают до гомогенности (контролируя визуально) и к этой смеси добавляют приготовленные гранцлы PPI. Предварительным концентратом с суспендированными гранулами PPI наполняют капсулы, подходящие для перорального введения.
IV. Характеристика препаратов
Для того, чтобы охарактеризовать препараты, определяют время, необходимое для образования препаратом эмульсии типа масло-в-воде при контакте с жидкостью, имитирующей желудочную жидкость, (simulated gastric fluid, SGF) (без ферментов) и образовавшуюся эмульсию характеризуют.SGF содержит 7 миллилитров концентрированной соляной кислоты, 2 грамма хлорида натрия и дистиллированную воду, дающую раствор с общим объемом 1 л. "Тест на образование эмульсии" проводят в пробирках (химическом стакане) с перемешиванием магнитной мешалкой. Пробирку, содержащую маленький магнит, заполняют 12,5 мл SGF без ферментов, что соответствует одной десятой от среднего объема желудочной жидкости у людей, и добавляют препарат, соответствующий одной десятой дозы активного соединения. Если препарат, который нужно охарактеризовать, представляет собой комбинацию с PPI, проверяют, чтобы на гранулы PPI не оказывала воздействие SGF, что делается посредством визуального контроля. Если на энтеросолюбильное покрытие гранул PPI оказывается воздействие, то на PPI может оказываться негативное воздействие при рН 1,2, и это можно наблюдать как заметное изменение окраски.
Время образования эмульсии будет варьировать от 30 секунд до 15 минут в зависимости от состава препарата. Если добавлен один или более чем один спирт с короткой цепью, то время образования эмульсии будет варьировать от 2-3 секунд до 3-4 минут. Кроме этого с использованием лазерной дифракции (Laser Diffraction, LD) или корреляционной спектроскопии фотонов (Photon Correlation Spectroscopy, PCS) изучают средний размер частиц образовавшейся эмульсии. В зависимости от размера частиц может быть использован любой из этих двух способов.
Подробное описание изобретения
Изобретение теперь будет описано более подробно с помощью следующих примеров, которые не должны истолковываться как ограничивающие изобретение.
Готовят следующие эмульсионные предварительные концентраты.
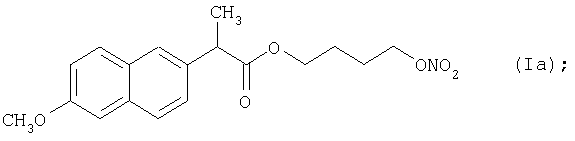
В Примерах 1-7, ниже, используемое в препаратах активное соединение представляет собой соединение формулы (Ia), как оно описано выше.
Пример 1
количество [г]
Полутвердый препарат получают плавлением 1 кг Pluronic F127® (Полоксамер 407) путем нагревания до 62°С. Расплав тщательно перемешивают, обеспечивая отсутствие каких-либо твердых частиц.
К расплавленному Pluronic F127® добавляют 1 кг соединения формулы (Ia) и смеси дают достичь температуры 62°С. Жидкий препарат перемешивают до гомогенности (контролируя визуально). Полученным в результате жидким препаратом затем наполняют твердые желатиновые капсулы. Препарат становится полутвердым при охлаждении (в капсуле).
Характеристика
150 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают магнитной мешалкой. Получают следующие результаты:
Вязкость измеряют с помощью плоскоконического вискозиметра Stress Tech, измерительная система С 40 4 PC, при скорости сдвига 20 с-1. Поток является более или менее ньютоновским.
Пример 2
количество [г]
Жидкий препарат готовят путем смешивания 1 кг жидкого поверхностно-активного вещества Полоксамера 401 с 1 кг соединения формулы (Ia) при комнатной температуре. Жидкий препарат перемешивают до гомогенности (контролируя визуально). Полученным в результате жидким препаратом затем наполняют твердые желатиновые капсулы.
Характеристика
150 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают магнитной мешалкой. Получают следующие результаты:
Пример 3
количество [г]
Препарат готовят смешением 1 кг Polyglycol BM 45® (Полоксамин 1107), 40 граммов натрия додецилсульфата, действующего как вторичное поверхностно-активное вещество, и 1 кг соединения формулы (Ia). Жидкий препарат перемешивают до гомогенности (контролируя визуально). Полученным жидким препаратом затем наполняют твердые желатиновые капсулы.
Характеристика
150 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают магнитной мешалкой. Получают следующие результаты:
Пример 4
количество [г]
Чтобы было возможно наполнить полутвердым препаратом мягкие желатиновые капсулы, температура процесса не должна превышать 30-40°С (конкретная температура зависит от производителя). Это означает, что препарат должен быть текучим и годным для перекачки насосом при температуре ниже 30-40°С. Для получения текучего при этой температуре препарата некоторую часть поверхностно-активного вещества заменяют на Cremophor EL®. Расплав готовят, как описано в Примере 1, за исключением того, что заменяют 0,5 кг поверхностно-активного вещества таким же количеством Cremophor EL®.
Характеристика
150 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают магнитной мешалкой. Получают следующие результаты:
Пример 5
количество [г]
Для того чтобы обеспечить хорошую точность наполнения небольшими дозами препарата и свести к минимуму количество присутствующего воздуха при заполнении капсул определенного объема, к активному соединению может быть добавлена до полного объема аликвота кокосового масла. Полутвердый препарат получают плавлением 1,500 кг Pluronic F127® (Полоксамер 407) путем нагревания до 62°С. Расплав тщательно перемешивают, обеспечивая отсутствие каких-либо твердых частиц. К расплавленному Pluronic F127® добавляют 1,250 кг соединения формулы (Ia) и 1,880 кг фракционированного кокосового масла и смеси дают достичь температуры 62°С. Жидкий препарат перемешивают до гомогенности (контролируя визуально). Полученным жидким препаратом затем наполняют твердые желатиновые капсулы.
Характеристика
Одну десятую часть препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают магнитной мешалкой. Получают следующие результаты:
Пример 6
количество [г]
Препарат готовят, как описано в Примере 5 выше.
Характеристика
Делают характеристику, как указано в Примере 5 выше. Получают следующие результаты:
Пример 7
количество [г]
Препарат готовят, как описано в Примере 5 выше.
Характеристика
Осуществляют характеристику как для Примера 5, выше. Получают следующие результаты:
Пример 8
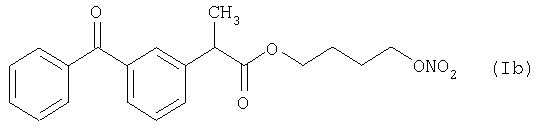
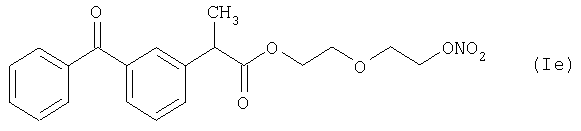
Активное соединение формулы (Ib), описано выше, используют в препарате настоящего Примера 8.
количество [г]
Препарат готовят таким же, как и в предыдущих Примерах.
Характеристика
Пример 9
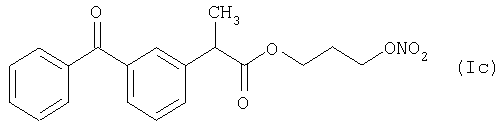
Активное соединение формулы (Ic), как оно описано выше, используют в препарате настоящего Примера 9.
количество [г]
Препарат готовят так же, как и в предыдущих Примерах.
Характеристика
Пример 10
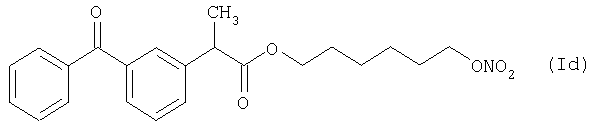
Активное соединение формулы (Id), как оно описано выше, используют в препарате настоящего Примера 10.
количество [г]
Препарат готовят таким же способом, как и в предыдущих Примерах.
Характеристика
Пример 11
Активное соединение формулы (Ie), как оно описано выше, используют в препарате настоящего Примера 11.
количество [г]
Препарат готовят таким же способом, что и в предыдущих Примерах.
Характеристика
Пример 12
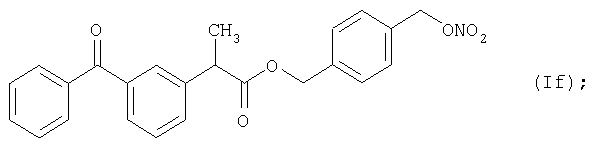
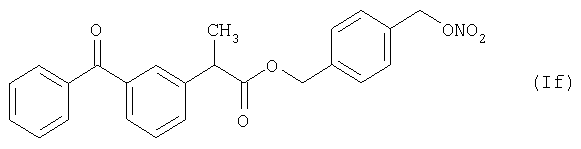
Активное соединение формулы (If), описано выше, используют в препарате настоящего Примера 12.
количество [г]
Препарат готовят таким же способом, что и в предыдущих Примерах.
Характеристика
Пример 13
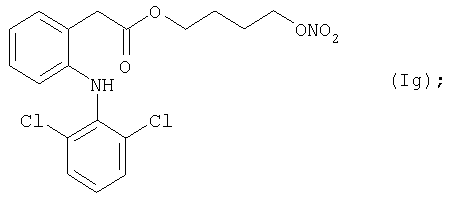
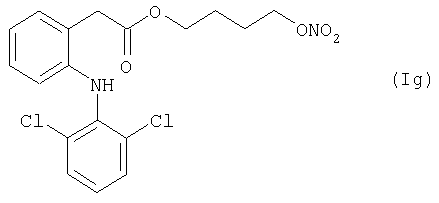
Активное соединение формулы (Ig), описано выше, используют в препарате настоящего Примера 13.
количество [г]
Препарат готовят так же, как в предыдущих Примерах.
Характеристика
Пример 14
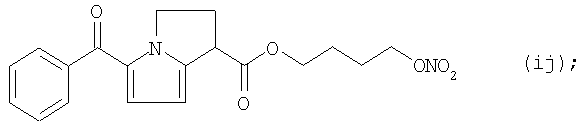
Активные соединения формул (Ia) и (1k), как они описаны выше, используют в препарате настоящего Примера 14.
количество [г]
Препарат готовят путем растворения соединения формулы (Ik) в соединении формулы (Ia), после чего к этой смеси добавляют Pluronic L121® (Полоксамер 401). Жидкий препарат перемешивают до гомогенности (контролируя визуально).
Характеристика
Препарат помещают в 20 мл SGF (без ферментов) с перемешиванием на магнитной мешалке. Определяют время образования эмульсии. Получают следующие результаты:
Пример 15
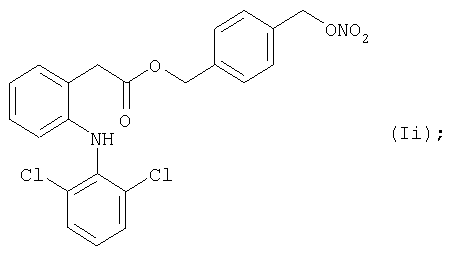
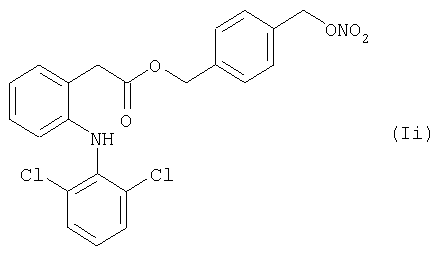
Активные соединения формул (Ia) и (li), выше, используют в препарате настоящего Примера 15.
количество [г]
Препарат готовят, как описано для Примера 14.
Характеристика
Осуществляют, как в предыдущем, вышеописанном Примере 14.
Пример 16
количество [г]
Полутвердый препарат получают в результате плавления 450 г Pluronic F127® (Полоксамер 407) путем нагревания до 62°С. Расплав тщательно перемешивают, обеспечивая отсутствие каких-либо твердых частиц. К расплавленному Pluronic F127® добавляют 750 г соединения формулы (Ia), выше, и смеси дают достичь температуры 62°С. Добавляют 20 г омепразола в форме гранул, содержащих Mg-соль омепразола, с нанесенным слоем энтеросолюбильного покрытия, приготовленных, как описано в WO 96/01623.
Пример 2. Жидкий препарат перемешивают до гомогенности (контролируя визуально) и им наполняют твердые желатиновые капсулы. Препарат становится полутвердым при охлаждении (в капсуле).
Характеристика
120 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) при 37°С и перемешивают магнитной мешалкой. SEDDS образует эмульсию при контакте с SGF, а гранулы PPI остаются не тронутыми SEEDS и при рН 1,2, что видно по отсутствию изменения окраски. Время образования эмульсии составляет 12 минут.
Пример 17
количество [г]
Жидкий препарат готовят путем смешения 450 г жидкого поверхностно-активного вещества Полоксамера 401 с 750 г соединения формулы (Ia), выше, при комнатной температуре. К смеси добавляют 20 г омепразола в форме гранул, содержащих Mg-соль омепразола, с нанесенным слоем энтеросолюбильного покрытия, приготовленных, как описано в WO 96/01623, Пример 2. Жидкий препарат перемешивают до гомогенности (контролируя визуально) и им наполняют твердые желатиновые капсулы.
Характеристика
120 миллиграммов препарата помещают в 12,5 мл SGF (без ферментов) при 37°С и перемешивают магнитной мешалкой. SEDDS образует эмульсию при контакте с SGF, а гранулы PPI остаются не тронутыми SEEDS и при рН 1,2, что видно по отсутствию изменения окраски. Время образования эмульсии составляет 0,5 минуты.
Пример 18
количество [г]
Полутвердый препарат получают путем плавления 0,843 грамма Pluronic F127® (Полоксамер 407), 0,282 грамма сорбитанмонолаурата и 0,375 грамма глицерина нагреванием до 62°С. Расплав тщательно перемешивают, обеспечивая отсутствие каких-либо твердых частиц. К смеси добавляют 3 грамма соединения формулы (Ia). Смеси дают достичь температуры 62°С. Жидкий препарат перемешивают до гомогенности (контролируя визуально). Полученному в результате жидкому препарату дают охладиться до температуры 30°С и затем им наполняют мягкие желатиновые капсулы. Препарат становится полутвердым при охлаждении (в капсуле).
Характеристика
112 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают на магнитной мешалке. Получают следующий результат:
Пример 19
количество [г]
Полутвердый препарат получают путем плавления 0,843 грамма Pluronic F127® (Полоксамер 407), 0,282 грамма сорбитанмонолаурата и 0,375 грамма пропиленгликоля нагреванием до 62°С. Расплав тщательно перемешивают, обеспечивая отсутствие каких-либо твердых частиц. К смеси добавляют 3 грамма соединения формулы (Ia). Смеси дают достичь температуры 62°С. Жидкий препарат перемешивают до гомогенности (контролируя визуально). Полученному жидкому препарату дают охладиться до температуры 30°С и затем им наполняют мягкие желатиновые капсулы. Препарат остается жидким при охлаждении (в капсуле).
Характеристика
112 миллиграммов препарата помещают в 12,5 миллилитрой SGF (без ферментов) и перемешивают на магнитной мешалке. Получают следующий результат:
Пример 20
количество [г]
Готовят жидкий препарат. Раствор 0,506 грамма Pluronic L101® (Полоксамер 331), 0,169 грамма сорбитанмонолаурата и 0,225 грамма этанола перемешивают до гомогенности (контролируя визуально). К смеси добавляют 3 грамма соединения формулы (Ia) при комнатной температуре. Полученным жидким препаратом наполняют мягкие желатиновые капсулы.
Характеристика
97 миллиграммов препарата помещают в 12,5 миллилитров SGF (без ферментов) и перемешивают на магнитной мешалке. Получают следующий результат:
Исследование препаратов in vivo на мини-свинках
Исследование биодоступности препаратов по настоящему изобретению проводят после перорального введения голодным мини-свинкам.
В данном исследовании используют 6 самцов мини-свинок Göttingem SPF. В начале акклиматизационного периода возраст животных составляет 4 месяца, а вес - от 7,7 до 10,1 кг. Животным не дают есть в течение 12 часов перед лечением и до тех пор, пока через 4 часа после лечения не отберут образец крови. Также ежедневно дают в качестве корма автоклавированное сено. Дважды в день животным предлагают питьевую воду бытового качества.
Каждому животному вводят фармацевтическую композицию по изобретению, загруженную в подходящую стандартную лекарственную форму в соответствии с данным изобретением. Уровни доз составляют приблизительно 15 мкмоль/кг массы тела. Для облегчения проглатывания капсулы или соответствующей стандартной дозировки дают 10 мл водопроводной воды.
Ежедневно регистрируют все видимые признаки болезненности или любые изменения поведения. Любые отклонения от нормы регистрируют в плане времени начала, продолжительности и серьезности. В ежедневное обследование здоровья включены наблюдения за консистенцией фекалий. Всех животных взвешивают по прибытии и в первый день каждого лечения.
В пробирки Vacutainer, содержащие гепарин, производят отбор образцов крови (5 мл) из яремной вены. Отбор образцов крови производят перед лечением (0) и в моменты времени 15, 30 и 45 минут, 1, 1,5, 2, 4, 7 и 24 часа после лечения.
Фармакокинетические параметры
В результате описанного выше исследования на мини-свинках для препарата, полученного по методике, описанной в Примере 5, были рассчитаны основные фармакокинетические параметры: Cmax - максимальная концентрация и tmax - время достижения максимальной концентрации, приведенные в таблице ниже.
Из полученных данных видно, что вводимая композиция из Примера 5, содержащая соединение la, обеспечивает быстрое попадание и значительные концентрации лекарственного средства в кровотоке.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ САМОЭМУЛЬГИРУЮЩАЯСЯ СИСТЕМА ДОСТАВКИ ЛЕКАРСТВ | 2001 |
|
RU2275908C2 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НИТРООКСИПРОИЗВОДНЫХ НЕСТЕРОИДНЫХ ПРОТИВОВОСПАЛИТЕЛЬНЫХ СРЕДСТВ | 2006 |
|
RU2406482C2 |
| ТВЕРДАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОИЗВОДНЫЕ БЕНЗОФУРАНА | 1998 |
|
RU2191578C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ N-СУЛЬФОНИЛИНДОЛИНА | 1997 |
|
RU2174394C2 |
| ПЕРОРАЛЬНО ВВОДИМЫЕ ЖИДКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ГВАЙФЕНЕЗИН И БЛОК-СОПОЛИМЕРЫ ПОЛИОКСИАЛКИЛЕНА | 2002 |
|
RU2281087C2 |
| ТВЕРДЫЕ ДОЗИРОВАННЫЕ ФОРМЫ, ВКЛЮЧАЮЩИЕ ФИБРАТ И СТАТИН | 2004 |
|
RU2343905C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ С ИСПОЛЬЗОВАНИЕМ ПРОИЗВОДНЫХ МАЛЫХ ДИКАРБОНОВЫХ КИСЛОТ В СОЧЕТАНИИ С PPI | 2007 |
|
RU2467747C2 |
| ТВЕРДЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ 5-АМИНОЛЕВУЛИНОВУЮ КИСЛОТУ | 2010 |
|
RU2527328C2 |
| ПОЛУТВЕРДЫЕ СИСТЕМЫ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ АЗЕТИДИНА | 2004 |
|
RU2343915C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ОТ HELICOBACTER PYLORI | 2014 |
|
RU2671400C2 |
Изобретение относится к области фармакологии и касается фармацевтической композиции, обладающей противовоспалительным действием, пригодной для перорального введения, в форме эмульсионного предварительного концентрата, содержащей NO-высвобождающее нестероидное противовоспалительное лекарство, поверхностно-активное вещество, масло или полутвердый жир, образующей in 'situ эмульсию типа масло-в-воде при контакте с водными средами, такими как желудочно-кишечные жидкости, лекарственной формы на ее основе, пероральной эмульсии, набора на ее основе и способа лечения воспаления и боли. Композиции обладают улучшенной биодоступностью. 6 н. и 34 з.п. ф-лы, 1 табл.
1) одно или более чем одно NO-высвобождающее нестероидное противовоспалительное лекарство (НСПВЛ), представляющее собой соединение формулы I
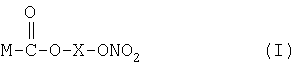
где Х представляет собой спейсер, который выбран из линейной или разветвленной алкиленовой группы -(СН2)-n, где n является целым числом от 2 до 10, или С3-C10циклоалкилена; -(СН2)m-O-(СН2)p-, где m и p являются целыми числами от 2 до 10: и -СН2-рС6Н4-СН2-,
М выбран как любое из следующих
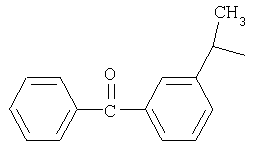
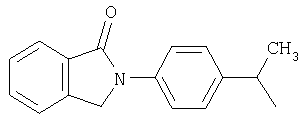
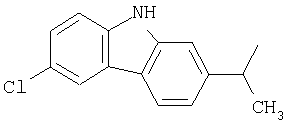
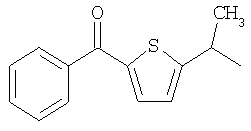
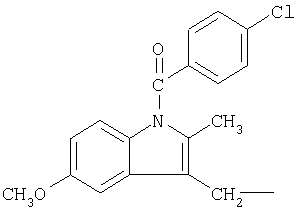
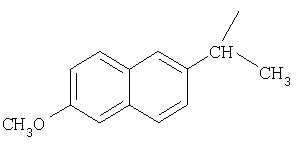
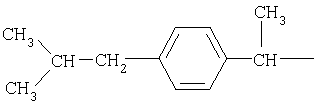 и
и 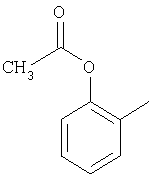
2) одно или более чем одно поверхностно-активное вещество;
3) возможно масло или полутвердый жир;
при этом указанная композиция образует in situ эмульсию типа масло-в-воде при контакте с водными средами, такими, как желудочно-кишечные жидкости.
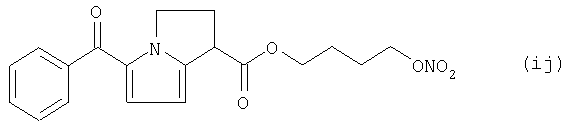
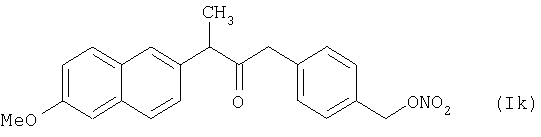
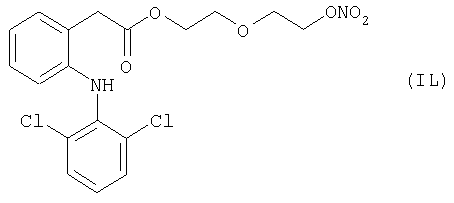
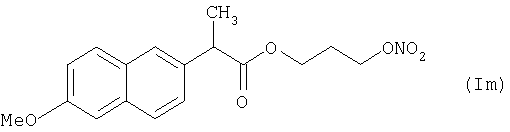
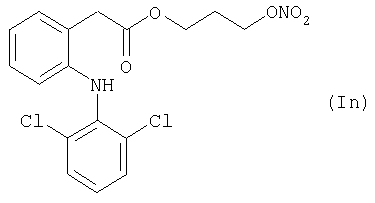
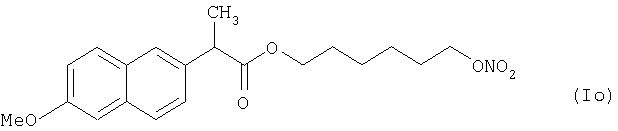
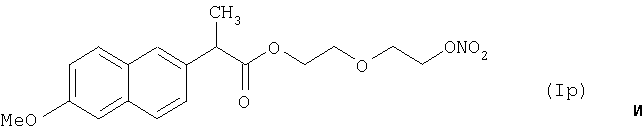
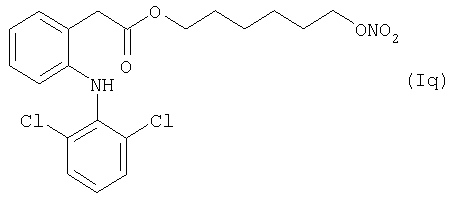
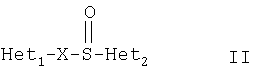
где Het1 представляет собой
Het2 представляет собой
X=
где
N в бензимидазольной группировке означает, что один из углеродных атомов, замещенных R6-R9, возможно может быть заменен на атом азота без каких-либо заместителей;
R1, R2 и R3 являются одинаковыми или разными и выбраны из водорода, алкила, алкокси, возможно замещенного фтором, алкилтио, алкоксиалкокси, диалкиламино, пиперидино, морфолино, галогена, фенила и фенилалкокси;
R4 и R5 являются одинаковыми или разными и выбраны из водорода, алкила и аралкила;
R6 ' представляет собой водород, галоген, трифторметил, алкил и алкокси;
R6-R9 являются одинаковыми или разными и выбраны из водорода, алкила, алкокси, галогена, галогеноалкокси, алкилкарбонила, алкоксикарбонила, оксазолила, трифторалкила, либо соседние группы R6-R9 образуют кольцевые структуры, которые могут быть дополнительно замещены;
R10 является водородом или образует алкиленовую цепь вместе с Р3, и
R11 и R12 являются одинаковыми или разными и выбраны из водорода галогена или алкила;
алкильные группы, алкоксигруппы и их группировки могут быть разветвленными или прямыми C1-C9-цепями либо включают циклические алкильные группы, такие, как циклоалкил-алкил.
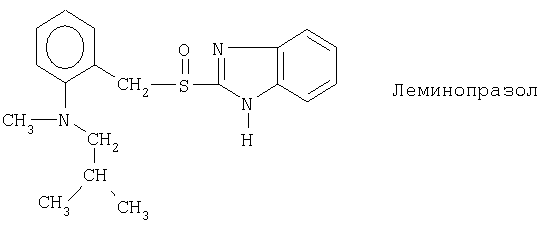
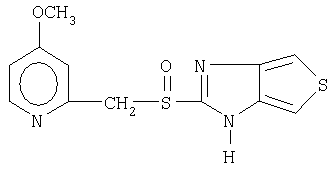
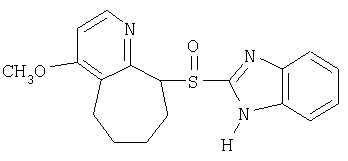
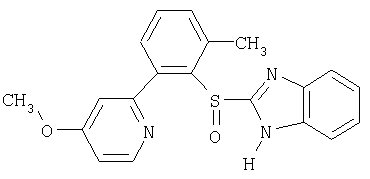
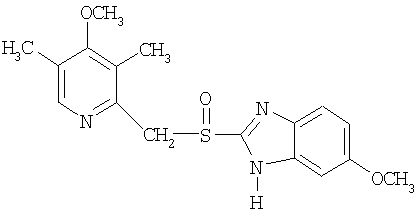
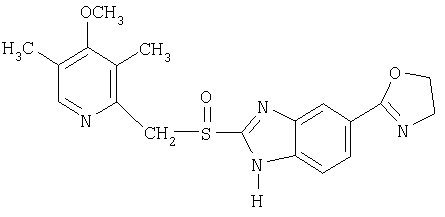
Полоксамер 237; Полоксамер 338; Полоксамер 331; Полоксамер 231; Полоксамин 908; Полоксамин 1307; Полоксамин 1107 и полиоксиэтилен-полиоксибутиленовый блоксополимер.
| WO 9509831 А1, 13.04.1995 | |||
| WO 9508983 А1, 06.04.1995 | |||
| WO 9956727 А2, 11.11.1999 | |||
| ДИФФУЗИОННО-ОСМОТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ СИСТЕМА С РЕГУЛИРУЕМЫМ ВЫДЕЛЕНИЕМ ЛЕКАРСТВА (ВАРИАНТЫ) И СПОСОБ ЕЕ ИЗГОТОВЛЕНИЯ (ВАРИАНТЫ) | 1994 |
|
RU2133605C1 |

