ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому соединению, его фармацевтически приемлемым солям, композициям, содержащим данное соединение, и их применению для лечения медицинских нарушений вследствие дефицита гормона роста.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Гормон роста представляет собой гормон, который стимулирует рост всех способных расти тканей. Кроме того, известно, что гормон роста оказывает ряд воздействий на метаболические процессы, например стимуляцию синтеза белков и мобилизацию свободных жирных кислот, и вызывает включение в обмен энергии метаболизма углеводов до жирных кислот. Дефицит гормона роста может вызвать ряд тяжелых медицинских нарушений, например карликовость.
Гормон роста высвобождается из гипофиза. Его высвобождение находится под жестким контролем ряда гормонов и нейротрансмиттеров, непосредственным или опосредованным. Высвобождение гормона роста можно стимулировать при помощи гормон роста-высвобождающего гормона (GHRH) и ингибировать соматостатином. В обоих случаях гормоны высвобождаются из гипоталамуса, но их действие, в первую очередь, опосредовано специфическими рецепторами, расположенными в гипофизе. Описаны другие соединения, которые стимулируют высвобождение гормона роста из гипофиза. Например, аргинин, L-3,4-дигидроксифенилаланин (L-Dopa), глюкагон, вазопрессин, РАСАР (пептид, активирующий аденилилциклазу гипофиза), агонисты мускариновых рецепторов и синтетический гексапептид, GHRP (гормон роста-высвобождающий пептид) высвобождают эндогенный гормон роста, действуя непосредственно на гипофиз или влияя на высвобождение GHRH и/или соматостатина из гипоталамуса.
При нарушениях или состояниях, когда требуются повышенные уровни гормона роста, белковая природа гормона роста делает нежизнеспособным все введения, кроме парентерального. Кроме того, другие непосредственно действующие стимулирующие секрецию природные средства, например GHRH и РАСАР, являются более длинными полипептидами, поэтому предпочтительно парентеральное введение.
Ранее предлагалось применение некоторых соединений для повышения уровней гормона роста у млекопитающих, например, в ЕР 18072, ЕР 83864, WO 8302272, WO 8907110, WO 8901711, WO 8910933, WO 8809780, WO 9118016, WO 9201711, WO 9304081, WO 9413696, WO 9517423, WO 9514666, WO 9615148, WO 9622997, WO 9635713, WO 9700894, WO 9722620, WO 9723508, WO 9740023 и WO 9810653.
Композиции соединений, высвобождающих гормон роста, важны вследствие их способности высвобождать гормон роста, а также биодоступности. Следовательно, целью настоящего изобретения является обеспечение нового соединения, обладающего свойствами высвобождения гормона роста. Кроме того, целью настоящего изобретения является обеспечение нового соединения, высвобождающего гормон роста (средства, стимулирующего секрецию гормона роста), которое является специфичным и/или селективным и не имеет или по существу не имеет побочных эффектов, таких как, например, высвобождение LH, FSH, TSH, АСТН, вазопрессина, окситоцина, кортизола и/или пролактина. Целью настоящего изобретения является также соединение, которое обладает хорошей биодоступностью при пероральном введении.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому соединению, которое действует непосредственно на клетки гипофиза в обычных экспериментальных условиях in vitro для высвобождения из них гормона роста.
Соединение, высвобождающее гормон роста, можно применять in vitro как уникальный исследовательский инструмент для понимания среди прочего того, как регулируется секреция гормона роста на уровне гипофиза.
Кроме того, соединение, высвобождающее гормон роста, настоящего изобретения можно также вводить in vivo для повышения высвобождения эндогенного гормона роста.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Таким образом, настоящее изобретение относится к соединению, полученному способом, описанным в примере 1, или его фармацевтически приемлемой соли.
Настоящее изобретение относится также к соединению, полученному по методике, описанной в примере 1, которое представляет собой 2-амино-N-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамид

или его фармацевтически приемлемую соль.
Настоящее изобретение относится также к соединению 2-амино-N-[(1R)-2(3R)-3-бензил-3(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамиду
или его фармацевтически приемлемой соли.
Структуру соединения, полученного по методике, описанной в примере 1, можно подтвердить, например, методом анализа дифракции рентгеновских лучей (например, как описано в Remington: The Science and Practice of Pharmacy, 19 th Edition (1995), особенно страницы 160 и 561-562).
В объем настоящего изобретения включена любая возможная комбинация двух или большего количества описанных здесь вариантов изобретения.
Общий способ
Методика, использованная в данном патенте, основана на сочетаниях пептидов, хорошо известных в данной области, и ни коим образом не должна быть интерпретирована как ограничение данного изобретения.
В данной методике перед сочетанием аминокислотных или пептидных остатков подходящую защитную группу, такую как трет-бутилоксикарбонил (Вос), можно удалить хорошо известными специалистам в данной области способами. Можно также избежать использования защитных групп. В подходящие аминокислоты можно ввести защитные группы и удалить защитные группы способами, известными в данной области и описанными, например, в работе Т.W.Green (Protective Groups in Organic Synthesis, 2 Ed., John Wiley and Sons, New York 1991).
В примере 1 подробно описана методика.
При разделении рацемической смеси 1-трет-бутилового эфира 3-бензилпиперидин-1,3-дикарбоновой кислоты с получением одного из энантиомерных соединений полученное по данной методике конечное соединение является диастереомером 2-амино-N-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксо-этил]-2-метилпропионамидом
вместо смеси двух диастереомеров.
Соединение настоящего изобретения демонстрирует улучшенную устойчивость к протеолитическому расщеплению ферментами, так как не является природным, в частности, потому, что природные амидные связи заменены синтетическими миметиками амидных связей. Ожидают, что повышенная устойчивость к протеолитическому расщеплению соединения настоящего изобретения по сравнению с известными гормон-высвобождающими пептидами улучшит его биодоступность по сравнению с биодоступностью пептидов, предложенных в литературе.
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ
Соединение настоящего изобретения необязательно может быть в виде фармацевтически приемлемой соли, такой как фармацевтически приемлемые кислотно-аддитивные соли соединения настоящего изобретения, которые включают соли, полученные при взаимодействии соединения формулы I с неорганической или органической кислотой, такой как соляная, бромистоводородная, серная, уксусная, фосфорная, молочная, малеиновая, миндальная, фталевая, лимонная, глутаровая, глюконовая, метансульфоновая, салициловая, янтарная, винная, толуолсульфоновая, трифторуксусная, сульфаминовая или фумаровая кислота, и/или водой.
Соединение настоящего изобретения можно вводить в виде фармацевтически приемлемой кислотно-аддитивной соли или, если подходит, в виде соли щелочного металла, щелочноземельного металла или (низший алкил)аммониевой соли. Считают, что такие формы солей имеют активность примерно такого же порядка, как формы свободных оснований.
В другом аспекте настоящее изобретение касается фармацевтической композиции, включающей в качестве активного ингредиента соединение настоящего изобретения или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем или разбавителем.
Фармацевтические композиции, содержащие соединение настоящего изобретения, можно получить общеизвестными способами, например, как описано в Remington's Pharmaceutical Sciences, 1985 или Remington: The Science and Practice of Pharmacy, 19th Edition (1995). Композиции могут быть в виде обычных форм, например капсул, таблеток, аэрозолей, растворов, суспензий или форм для местного применения.
Используемый фармацевтический носитель или разбавитель может быть обычным твердым или жидким носителем. Примерами твердых носителей являются лактоза, белая глина, сахароза, циклодекстрин, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния, стеариновая кислота или простые эфиры низших алкилов целлюлозы. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен или вода.
Аналогично, носитель или разбавитель может включать любой известный в данной области материал замедленного высвобождения, такой как глицерилмоностеарат или глицерилдистеарат, сам по себе или в смеси с воском.
Если для перорального введения используют твердый носитель, то препарат может быть получен в виде таблеток, помещен в твердую желатиновую капсулу в виде порошка или гранул или может быть в виде пастилки или лепешки. Количество твердого носителя варьируется в широких пределах, но обычно составляет примерно от 25 мг до 1 г. Если используют жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы или стерильной жидкости для инъекций, такой как водная или неводная жидкая суспензия или раствор.
Типичная таблетка, которую можно получить по известным методикам таблетирования, может содержать, мг:
*Ацилированный моноглицерид, используемый в качестве пластификатора для пленочного покрытия.
Препарат для назального введения может содержать соединение настоящего изобретения, растворенное или суспендированное в жидком носителе, в частности в водном носителе, для аэрозольного применения. Носитель может содержать добавки, такие как агенты, способствующие растворению, например пропиленгликоль, поверхностно-активные вещества, усилители абсорбции, такие как лецитин (фосфатидилхолин) или циклодекстрин, или консерванты, такие как парабены.
Обычно соединения настоящего изобретения готовят в виде стандартной дозированной формы, включающей 50-200 мг активного ингредиента вместе с фармацевтически приемлемым носителем на стандартную дозу.
Подходящей является доза соединений данного изобретения 0,01-500 мг/день, например от 5 до 50 мг, примерно 10 мг на дозу, при введении пациентам, например, человеку, в качестве лекарственного средства.
Еще в одном аспекте настоящее изобретение относится к фармацевтической композиции в виде стандартной дозированной формы, включающей в качестве активного ингредиента примерно от 10 до 200 мг соединения общей формулы I или его фармацевтически приемлемой соли.
Показано, что соединение настоящего изобретения обладает способностью высвобождать эндогенный гормон роста in vivo. Следовательно, данное соединение можно применять при лечении состояний, которые требуют повышенных уровней гормона роста в плазме крови, таких как при дефиците гормона роста у людей или при лечении пожилых пациентов или домашнего скота.
Таким образом, в отдельном аспекте настоящее изобретение относится к фармацевтической композиции для стимулирования высвобождения гормона роста из гипофиза, композиции, включающей в качестве активного ингредиента соединение настоящего изобретения или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым носителем или разбавителем.
Еще в одном аспекте настоящее изобретение относится к способу стимулирования высвобождения гормона роста из гипофиза, способу, включающему введение нуждающемуся пациенту эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли.
Еще в одном аспекте настоящее изобретение относится к применению соединения настоящего изобретения или его фармацевтически приемлемой соли для получения лекарственного средства для стимулирования высвобождения гормона роста из гипофиза.
Специалисту в данной области хорошо известно, что настоящие и возможные применения гормона роста у людей разнообразны и многочисленны. Таким образом, соединение настоящего изобретения можно вводить для стимулирования высвобождения гормона роста из гипофиза, и они будут иметь аналогичные эффекты и применения, как гормон роста сам по себе. Соединения настоящего изобретения полезны для: стимулирования высвобождения гормона роста у пожилых пациентов, предупреждения катаболических побочных эффектов глюкокортикоидов, предупреждения и лечения остеопороза, лечения синдрома хронической усталости (CFS), лечения синдрома острой усталости и потери мышечной массы после избирательной хирургической операции, стимулирования иммунной системы, ускорения заживления ран, ускорения восстановления перелома кости, ускорения заживления при осложненных переломах (например, остеогенез с растяжением), лечения истощения как побочного результата переломов, лечения задержки роста, лечения задержки роста в результате поражения почек или почечной недостаточности, лечения кардиомиопатии, лечения истощения, связанного с хроническим заболеванием печени, лечения тромбоцитопении, лечения задержки роста, связанной с болезнью Крона, лечения синдрома укороченной тонкой кишки, лечения истощения, связанного с хроническим обструктивным заболеванием легких (COPD), лечения осложнений, связанных с трансплантацией, лечения физиологически малого роста, включая детей с дефицитом гормона роста и малого роста вследствие хронического заболевания, лечения ожирения и связанной с ожирением задержки роста, лечения анорексии, лечения задержки роста, связанной с синдромом Прадера-Вилли и синдромом Тернера; повышения скорости роста пациента с синдромом частичной нечувствительности гормона роста, ускорения выздоровления и восстановления госпитализированных пациентов с ожогами; лечения внутриутробной задержки роста, скелетной дисплазии, гиперкортицизма и синдрома Кушинга; индуцирования пульсирующего высвобождения гормона роста; замещения гормона роста у пациентов в состоянии стресса, лечения остеохондродисплазий, синдрома Нунана, шизофрении, депрессий, болезни Альцгеймера, замедленного заживления ран и психосоциальных деприваций, лечения катаболизма, связанного с легочной дисфункцией и дыхательной зависимостью; лечения сердечной недостаточности или связанной с этим дисфункции сосудов, лечения ослабленной сердечной функции, лечения или профилактики инфаркта миокарда, снижения кровяного давления, защиты против дыхательной дисфункции или предупреждения случаев реперфузии; лечения взрослых пациентов при хроническом диализе; ослабления катаболических реакций белков после обширного хирургического вмешательства, снижения кахексии и потери белковой массы вследствие хронического заболевания, такого как рак или СПИД; лечения гиперинсулинемии, включая гиперплазию панкреатических островков, вспомогательного лечения при индуцированной овуляции; стимулирования тимусного развития и предупреждения возрастного ухудшения тимусной функции, лечения пациентов с подавленным иммунитетом; лечения саркопении, лечения истощения вследствие СПИДа; улучшения силы мышц, подвижности, сохранения толщины кожи, метаболического гомеостаза и почечного гомеостаза у болезненных пожилых пациентов, стимулирования остеобластов, ремоделирования костей и роста хрящей; регулирования потребления пищи; стимулирования иммунной системы у домашних животных и лечения возрастных нарушений у домашних животных, содействия росту домашнего скота и стимулирования роста шерсти у овец, повышения производства молока у домашнего скота, лечения метаболического синдрома (синдром X), лечения устойчивости к инсулину, включая NIDDM, у млекопитающих, например человека, лечения инсулинорезистентности в сердце, улучшения качества сна и коррекции относительного гипосоматотропизма старения вследствие сильного увеличения REM-сна и уменьшения REM-латентного состояния, лечения гипотермии, лечения хрупкости, связанной со старением, лечения застойной сердечной недостаточности, лечения переломов бедра, лечения иммунодефицита у пациентов с подавленным соотношением клеток Т4/Т8, лечения атрофии мышц, лечения мышечно-скелетной слабости у пожилых, повышения активности протеинкиназы В (РКВ), улучшения общей легочной функции, лечения нарушений сна, лечения задержки роста, связанной с астмой, лечения задержки роста, связанной с ювенильным ревматоидным артритом, и лечения задержки роста, связанной с кистозным фиброзом.
Доза для указанных выше показаний варьируется в зависимости от способа введения и необходимой терапии. Однако для получения эффективного высвобождения эндогенного гормона роста пациентам и животным обычно вводят дозы в интервале от 0,0001 до 100 мг/кг веса тела в день. Кроме того, соединение настоящего изобретения не имеет или по существу не имеет побочных эффектов при введении указанных выше уровней доз, таких побочных эффектов, как, например, высвобождение LH, FSH, TSH, АСТН, вазопрессина, окситоцина, кортизола и/или пролактина. Обычно дозированные формы, подходящие для перорального, назального, легочного или трансдермального введения, содержат примерно от 0,0001 до 100 мг, предпочтительно примерно от 0,001 до 50 мг соединений настоящего изобретения в смеси с фармацевтически приемлемым носителем или разбавителем.
Необязательно фармацевтическая композиция настоящего изобретения может включать соединение настоящего изобретения вместе с одним или более соединениями, демонстрирующими различные активности, например с антибиотиком или другим фармакологически активным веществом.
Способ введения может быть любым способом, который эффективно транспортирует активное соединение к подходящему или желаемому месту действия, таким как пероральный, назальный, легочный, трансдермальный или парентеральный, предпочтителен пероральный способ.
Кроме фармацевтического применения соединения настоящего изобретения могут быть полезны в качестве in vitro инструментов для исследования регулирования высвобождения гормона роста.
Соединение настоящего изобретения может также быть полезно в качестве in vivo инструмента для оценки способности высвобождения гормона роста гипофизом. Например, можно исследовать на гормон роста образцы сыворотки, взятые до и после введения данного соединения людям. Сравнение гормона роста в каждом образце сыворотки будет непосредственно определять способность гипофиза пациента высвобождать гормон роста.
Соединение настоящего изобретения можно вводить коммерчески важным животным для повышения скорости и степени их роста и увеличения производства молока.
Еще одним применением соединения настоящего изобретения является применение в комбинации с другими средствами, стимулирующими секрецию, такими как GHRP (2 или 6), GHRH и его аналоги, гормон роста и его аналоги или соматомедины, включая IGF-1 и IGF-2.
Фармакологические методы
Соединение настоящего изобретения можно оценить in vitro на его эффективность и способность высвобождать гормон роста в первичных культурах гипофиза крысы, и такую оценку можно выполнить, как описано ниже.
Способ выделения клеток гипофиза крысы представляет модификацию способа О.Sartor et al., Endocrinology, 116, 1985, pp.952-957. Самцов-альбиносов крыс Sprague-Dawley (250±25 г) приобретают у Mollegaard, Lille Skensved, Denmark. Крыс содержат в клетках группами (четыре животных на клетку) и помещают в комнаты с 12-часовым световым циклом. Температуру в комнатах варьируют от 19 до 24°С и влажность от 30 до 60%.
Крыс обезглавливают и извлекают гипофиз. Удаляют нейроинтермедиатные доли и оставшуюся ткань сразу помещают в охлаждаемый льдом буфер для выделения (среда Гея (Gibco 041-04030), дополненная 0,25% D-глюкозой, 2% несущественными аминокислотами (Gibco 043-01140) и 1% бычьим сывороточным альбумином (BSA) (Sigma А-4503)). Ткань разрезают на маленькие кусочки и переносят в буфер для выделения, дополненный 3,8 мг/мл трипсина (Worthington #3707 TRL-3) и 330 мг/мл ДНКазы (Sigma D-4527). Данную смесь инкубируют при 70 об./мин в течение 35 минут при 37°С в атмосфере О2/СО2 95%/5%. Затем ткань промывают три раза в указанном выше буфере. Далее, используя стандартную пастеровскую пипетку, ткань отсасывают в одиночные ячейки. После диспергирования ячейки фильтруют через нейлоновый фильтр (160 мм) для удаления нерасщепленной ткани. Клеточную суспензию промывают 3 раза буфером для выделения, дополненным ингибитором трипсина (0,75 мг/мл, Worthington #2829), и в заключение повторно суспендируют в культуральной среде; DMEM (Gibco 041-01965), дополненной 25 мМ HEPES (Sigma H-3375), 4 мМ глутамином (Gibco 043-05030H), 0,075% бикарбонатом натрия (Sigma S-8875), 0,1% несущественными аминокислотами, 2,5% фетальной коровьей сывороткой (FCS, Gibco 011-06290), 3% сывороткой лошади (Gibco 034-06050), 10% свежей сывороткой крысы, 1 нМ Т3 (Sigma Т-2752) и 40 мг/л дексаметазона (Sigma D-4902), рН 7,3, до плотности 2×105 клеток/мл. Клетки высевают на планшеты для микротитрования (Nunc, Denmark), 200 мл на ячейку, и культивируют в течение 3 дней при 37°С и 8% СО2.
Исследование соединения
После культивирования клетки дважды промывают буфером для стимулирования (сбалансированный солевой раствор Хэнкса (Gibco 041-04020), дополненным 1% BSA (Sigma А-4503), 0,25% D-глюкозой (Sigma G-5250) и 25 мМ HEPES (Sigma H-3375), рН 7,3), и предварительно инкубируют в течение 1 часа при 37°С. Буфер заменяют 90 мл буфера для стимулирования (37°С). Добавляют десять мл раствора исследуемого соединения и инкубируют планшеты в течение 15 минут при 37°С и 5% CO2. Питательную среду декантируют и анализируют на содержание GH в исследуемой системе rGH SPA.
Данное соединение исследуют при дозах в диапазоне от 10 пМ до 100 мМ. Составляют соотношение доза-реакция, уравнение Хилла (Фиг.Р, Biosoft). Эффективность (максимальная величина высвободившегося GH, Emax) выражают в % от Emax GHRP-6. Активность (EC50) определяют как концентрацию, индуцирующую половину максимального стимулирования высвобождения GH.
Данное соединение показало ЕС50=7,5 нМ и Emax=85%
Соединения настоящего изобретения можно оценить на метаболическую стабильность, используя описанную ниже методику.
Соединение растворяют при концентрации 1 мг/мл в воде. Добавляют 25 мл данного раствора к 175 мл соответствующего раствора фермента (получая соотношение фермент:субстрат (мас./мас.) примерно 1:5). Раствор оставляют при 37°С на ночь. Анализируют по 10 мл различных разлагающих растворов относительно соответствующего нулевого образца, используя масс-спектрометрию с электрораспылением вводимого потока (ESMS) при мониторинге молекулярного иона выбранным ионом. Если сигнал снижается более чем на 20% по сравнению с нулевым образцом, остаток раствора анализируют способами ВЭЖХ и масс-спектрометрии, чтобы точно идентифицировать степень и место(места) разложения.
В исследования стабильности включены некоторые стандартные пептиды (АСТН 4-10, ангиотензин 1-14 и глюкагон), чтобы подтвердить способность различных растворов разрушать пептиды.
Стандартные пептиды (ангиотензин 1-14, АСТН 4-10 и глюкагон) получают от Sigma, МО, USA.
Все ферменты (трипсин, химотрипсин, эластаза, аминопептидаза М и карбоксипептидаза Y и В) получают от Boehringer Mannheim GmbH (Mannheim, Germany).
Смесь ферментов поджелудочной железы: трипсин, химотрипсин и эластаза в 100 мМ бикарбонате аммония, рН 8,0, (все концентрации 0,025 мг/мл).
Смесь карбоксипептидаз: карбоксипептидаза Y и В в 50 мМ ацетате аммония, рН 4,5, (все концентрации 0,025 мг/мл).
Раствор аминопептидазы М: аминопептидаза М (0,025 мг/мл) в 100 мМ бикарбонате аммония, рН 8,0.
Масс-спектрометрический анализ проводят, используя два разных масс-спектрометра: Sciex API III тройной квадрупольный прибор ЖХ-МС (Sciex instruments, Thornhill, Ontario), оборудованный источником ионов с электрораспылением, и Bio-Ion 20 времяпролетный плазменный десорбционный прибор (Bio-Ion Nordic АВ, Uppsala, Sweden).
Количественное определение соединения (до и после разложения) проводят на приборе API III, используя мониторинг отдельного иона для рассматриваемого молекулярного иона при впрыскивании потока исследуемого материала. Поток жидкости (МеОН: вода 1:1) 100 мл/мин регулируют при помощи ВЭЖХ-устройства ABI 140В (Perkin-Elmer Applied Biosystems Divisions, Foster City, CA). Параметры приборов устанавливают для стандартных условий работы, и SIM-мониторинг проводят, используя наиболее интенсивный молекулярный ион (в большинстве случаев он соответствует двухзарядному молекулярному иону).
Идентификация продуктов разложения включает также применение плазменной десорбционной масс-спектрометрии (PDMS) с использованием образцов на мишенях, покрытых нитроцеллюлозой, и стандартных инструментальных параметров. Точность определенных таким образом масс обычно выше 0,1%.
Разделение и выделение продуктов разложения осуществляют, используя ВЭЖХ-колонку 4,6×105 мм HY-TACH С-18 с обращенной фазой (Hewlett-Packard Company, Palo Alto, CA) и стандартным градиентом для деления ацетонитрил:TFA. Используемая ВЭЖХ-система представляет собой HP1090М (Hewlett-Packard Company, Palo Alto, CA).
-: Нестабилен (более 20% разложения в SIM-сигнале через 24 часа в разлагающем растворе)
На чертеже представлены результаты эксперимента нового соединения (NNC 26-1291) in vivo на активность по высвобождению гормона роста из гипофиза (на свиньях).
Чертеж. Быстрое высвобождение гормона роста GH у самок свиней после перорального введения NN703, МК-677, NNC-1187 и NNC 26-1291 при разовой дозе ˜3,5 мг/кг веса (5 мкмол/кг веса). Можно видеть четкое различие между разными GH-соединениями, при этом NNC 26-1291 и МК-677 являются наиболее эффективными при данной дозе, затем идут NNC-1187 и NN703.
Фармакокинетические методы
Соединение настоящего изобретения можно оценить на биодоступность при пероральном введении, такую оценку можно выполнить, как описано ниже.
Фармакокинетику соединения можно исследовать на голодных гончих собаках.
Внутривенное и пероральное введение исследуемого соединения в 5% растворе глюкозы разделяют промывкой в течение одной недели.
Отбирают образцы крови непосредственно перед введением лекарства (нулевой момент времени) и затем через 0,08, 0,25, 0,50, 0,75, 1,0, 1,5, 2,0, 3,0, 4,0, 5,0 и 6,0 часов после введения.
Образцы плазмы в ожидании анализа хранят в замороженном виде (<-18°С).
Для количественного определения соединения в плазме используют способ ВЭЖХ с твердофазной экстракцией и УФ-детектированием.
Доступность соединения настоящего изобретения при пероральном введении составляет около 50%.
Фармакокинетические параметры соединений рассчитывают неизолированными методами, используя персональный компьютер с фармакокинетическим программным обеспечением WinNonlin, версия 1.1 (Scientific Consulting Inc., Apex, NC, USA).
Любые описанные здесь новые отличительные признаки или комбинации отличительных признаков считают существенными для данного изобретения.
ПРИМЕРЫ
Способ получения соединения настоящего изобретения и препаратов, содержащих данное соединение, ниже проиллюстрирован в следующих примерах, которые, однако, не следует считать ограничивающими.
Структуру соединения подтверждают либо высокоэффективной жидкостной хроматографией (ВЭЖХ), либо ядерным магнитным резонансом (ЯМР, Bruker 400 МГц), либо жидкостной хроматографией-масс-спектрометрией (ЖХ-МС). ЯМР-сдвиги (δ) даны в миллионных долях (м.д.) и приведены только для выбранных пиков. Температура плавления обозначена т.пл. и приведена в °С. Колоночную хроматографию проводят, используя методику, описанную в работе W.С.Still et al., J. Org. Chem., 1978, 43, 2923-2925, на силикагеле 60 (Merck, Art 9385). Соединения, используемые в качестве исходных веществ, либо являются известными соединениями, либо их легко получить известными способами. Используемый раствор метанол/аммиак представляет собой 10% раствор аммиака в метаноле.
ВЭЖХ-анализ:
Способ A1.
RP-анализ проводит, применяя УФ-детектирование при 214, 254, 276 и 301 нм, на колонке 218ТР54 4, 6 мм × 250 мм 5 м с силикагелем С-18 (The Seperations Group, Hesperia), которую элюируют со скоростью 1 мл/мин при 42°С. Колонку уравновешивают 5% ацетонитрилом в буфере, состоящем из 0,1 М сульфата аммония, рН которого доводят до 2,5 4 М серной кислотой. После введения образец элюируют с градиентом от 5 до 60% ацетонитрила в том же буфере в течение 50 минут.
Способ В1.
RP-анализ проводят, применяя УФ-детектирование при 214, 254, 276 и 301 нм, на колонке 218ТР54 4,6 мм × 250 мм 5 м с силикагелем С-18 (The Seperations Group, Hesperia), которую элюируют со скоростью 1 мл/мин при 42°С. Колонку уравновешивают 5% смесью (ацетонитрил + 0,1% TFA) в водном растворе TFA в воде (0,1%). После введения образец элюируют с градиентом от 5 до 60% (ацетонитрил + 0,1% TFA) в том же водном буфере в течение 50 минут.
Способ h8:
RP-анализ проводят, применяя УФ-детектирование при 214 и 254 нм, на колонке 218ТР54 4,6 мм × 150 мм с силикагелем С-18, которую элюируют со скоростью 1 мл/мин при 42°С. Колонку уравновешивают смесью 5% ацетонитрила, 85% воды и 10% раствора 0,5% трифторуксусной кислоты в воде и элюируют с линейным градиентом от 5% ацетонитрила, 85% воды и 10% раствора 0,5% трифторуксусной кислоты до 90% ацетонитрила и 10% раствора 0,5% трифторуксусной кислоты в течение 15 минут.
Хиральная ВЭЖХ:
Хиральную ВЭЖХ проводят, применяя УФ-детектирование при 225 и 254 нм, на колонке 4,6 мм × 250 мм Chiracel OJ, смонтированной с предварительной колонкой 4,6 мм × 80 мм Chiracel OJ (обе от Daicel Chemical Industries, LTD), которую элюируют со скоростью 0,7 мл/мин при комнатной температуре. Образец элюируют изократическим элюентом гептан(92): изоPrOH(8): TFA(0,1).
ЖХ-МС-анализ:
ЖХ-МС-анализ проводят на установке РЕ Sciex API 100 LC/MS System, применяя колонку Waters® 3 мм × 150 мм 3,5 м С-18 Symmetry и распыление положительных ионов со скоростью потока 20 мл/мин. Колонку элюируют с линейным градиентом 5-90% ацетонитрила, 85-0% воды и 10% раствора 0,1% трифторуксусной кислоты в воде в течение 15 минут при скорости потока 1 мл/мин.
Сокращения:
Связывающиеся блоки:
Используемые в следующих примерах N-метилированные аминокислоты получают, как в Can. J. Chem., 1977, 55, 906.
N',N'-диметилгидразид муравьиной кислоты
Смесь 50 мл метилформиата и 50 мл 1,1-диметилгидразина перемешивают в течение 3 дней при комнатной температуре, концентрируют в вакууме, получая кристаллы, которые перемешивают в смеси EtOH(5):гептан(95), охлаждают в холодильнике в течение ночи и фильтруют: 50,7 г (575 ммоль) (выход 88%).
N,N,N'-триметилгидразин дигидрохлорид
В трехгорлую круглодонную колбу на 2 л, снабженную магнитной мешалкой и капельной воронкой, загружают 20,4 г LiAlH4, воздух откачивают и продувают сильным потоком азота. Затем капельную воронку снабжают барботером для азота и медленно (экзотермически) добавляют 250 мл сухого тетрагидрофурана. Серую суспензию интенсивно перемешивают и добавляют по каплям в течение 1 часа раствор 40,0 г N',N'-диметилгидразида муравьиной кислоты в 250 мл сухого тетрагидрофурана. Смесь перемешивают в течение ночи при комнатной температуре. За реакцией следят методом ТСХ (CH2Cl2(100):МеОН(10):NH3(1)).
В другую трехгорлую круглодонную колбу на 2 л, снабженную холодильником с сухим льдом, загружают 350 мл 4,8 М HCl/СН3ОН и помещают на баню с сухим льдом (˜70°С). Затем ее соединяют с реакционной колбой через колонку Вигре и помещают реакционную колбу на масляную баню. К реакционной смеси осторожно добавляют 200 мл тетрагидрофурана и 200 мл МеОН. Перегонку продукта и растворителя выполняют при медленном нагревании до 130°С, в результате собирают кристаллическую соль дигидрохлорид триметилгидразина (при -70°С). Удаляют баню с сухим льдом и дают температуре подняться до комнатной. Концентрирование в вакууме дает жидкое бесцветное масло, которое сушат в течение ночи, используя вакуумный насос: 45,2 г (309 ммоль) (выход 68%). Очень гигроскопичный продукт хранят под азотом.
Другие исходные вещества можно получить от Aldrich.
Пример 1
Методика получения соединения, которое является 2-амино-N-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамидом
или
2-амино-N-[(1R)-2-[(3S)-3-бензил-3-(N'N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамидом

Стадия а
1-трет-бутиловый эфир 3-этиловый эфир пиперидин-1,3-дикарбоновой кислоты

В одногорлую круглодонную колбу (1 л), снабженную магнитной мешалкой и капельной воронкой, загружают гранулы NaOH (15,6 г), тетрагидрофуран (400 мл) и этилнипекотат (50 мл, 324 ммоль). К перемешиваемой смеси при комнатной температуре добавляют по каплям раствор Boc2О (84,9 г, 389 ммоль) в тетрагидрофуране (150 мл) (1 час, осаждение белого твердого вещества, растворенные гранулы NaOH, экзотермический процесс). Смесь перемешивают в течение ночи при комнатной температуре. Данную смесь добавляют к EtOAc (500 мл) и Н2О (2000 мл)/ водный слой повторно экстрагируют EtOAc (2×500 мл) и объединенные органические слои промывают насыщенным раствором соли (100 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, получая 1-трет-бутиловый эфир 3-этиловый эфир пиперидин-1,3-дикарбоновой кислоты (82,5 г) в виде жидкого желтого масла.
1H ЯМР (300 МГц, CDCl3): δ 1,25 (т, 3Н, СН3); 1,45 (с, 9Н, 3 × СН3); 2,05 (м, 1Н); 2,45 (м, 1Н); 2,85 (м, 1Н); 3,95 (д (широкий), 1Н); 4,15 (кв, 2H, CH2).
Стадия b
1-трет-бутиловый эфир 3-этиловый эфир 3-бензилпиперидин-1,3-дикарбоновой кислоты (рацемическая смесь)
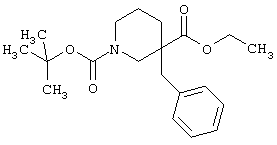
Трехгорлую круглодонную колбу (2 л), снабженную магнитной мешалкой, термометром, барботером для азота и капельной воронкой, откачивают, продувают сильным потоком азота, загружают безводный тетрагидрофуран (500 мл) и охлаждают до -70°С. Затем добавляют литийдиизопропиламин (164 мл 2,0 М раствора в тетрагидрофуране, 327 ммоль). К перемешиваемому раствору при -70°С добавляют по каплям в течение 45 минут раствор 1-трет-бутилового эфира 3-этилового эфира пиперидин-1,3-дикарбоновой кислоты (80 г, 311 ммоль) в безводном тетрагидрофуране (50 мл) (температура от -70°С до -60°С, прозрачный красный раствор). Смесь перемешивают в течение 20 минут и затем добавляют по каплям в течение 40 минут раствор бензилбромида (37 мл, 311 ммоль) в безводном тетрагидрофуране (250 мл) (температура от -70°С до -60°С). Смесь перемешивают в течение 1 часа при -70°С и затем оставляют на ночь при комнатной температуре (светло-оранжевая). Реакционную смесь концентрируют в вакууме примерно до 300 мл, переносят в делительную воронку, разбавляют СН2Cl2 (900 мл) и промывают Н2О (900 мл). Из-за плохого разделения водный слой повторно экстрагируют СН2Cl2 (200 мл), объединенные органические слои промывают водным NaHCO4 (200 мл, 10%), водным NaHCO3 (200 мл, насыщенный), Н2O (200 мл), насыщенным раствором соли (100 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, получая масло, которое растворяют в смеси EtOAc(1):гептан(10) и выдерживают в течение ночи. Образовавшиеся твердые вещества удаляют фильтрованием, промывают гептаном и сушат в вакууме, получая рацемическую смесь 1-трет-бутилового эфира 3-этилового эфира 3-бензилпиперидин-1,3-дикарбоновой кислоты (81,4 г).
Стадия с
1-трет-бутиловый эфир 3-бензилпиперидин-1,3-дикарбоновой кислоты (рацемическая смесь)
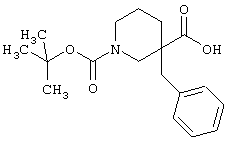
1-трет-бутиловый эфир 3-этиловый эфир 3-бензилпиперидин-1,3-дикарбоновой кислоты (81 г, 233 ммоль) растворяют в EtOH (400 мл) и NaOH (400 мл, 16% водный раствор) в одногорлой круглодонной колбе (1л), снабженной холодильником и магнитной мешалкой. Смесь кипятят с обратным холодильником в течение 10 часов в атмосфере азота, охлаждают до комнатной температуры, концентрируют в вакууме примерно до 600 мл (осаждение твердого вещества), разбавляют H2O (400 мл), охлаждают на бане со льдом и при интенсивном перемешивании подкисляют 4 М H2SO4 до рН 3 (конечная температура 28°С). Смесь экстрагируют EtOAc (2×700 мл) и объединенные органические слои промывают насыщенным раствором соли (200 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, получая масло, которое растворяют в смеси EtOAc(1):гептан(10) и выдерживают в течение ночи. Образовавшиеся кристаллы удаляют фильтрованием, промывают гептаном и сушат в вакууме, получая рацемическую смесь 1-трет-бутилового эфира 3-бензилпиперидин-1,3-дикарбоновой кислоты (66,0 г).
Стадия d
1-трет-бутиловый эфир (3R)-3-бензилпиперидин-1,3-дикарбоновой кислоты или 1-трет-бутиловый эфир (3S)-3-бензилпиперидин-1,3-дикарбоновой кислоты
(разделение 1-трет-бутилового эфира 3-бензилпиперидин-1,3-дикарбоновой кислоты)
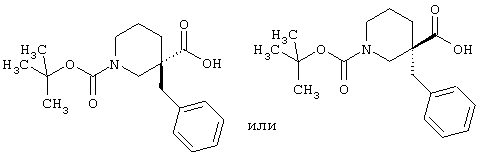
1-трет-бутиловый эфир З-бензилпиперидин-1,3-дикарбоновой кислоты (76 г, 238 ммоль) растворяют в EtOAc (3,0 л) в одногорлой колбе (5 л), снабженной магнитной мешалкой. Затем добавляют Н2О (30 мл), R(+)-1-фенетиламин (18,2 мл, 143 ммоль) и Et3N (13,2 мл, 95 ммоль), и смесь перемешивают в течение ночи при комнатной температуре, получая осаждение белых кристаллов (41,9 г), которые удаляют фильтрованием, промывают EtOAc и сушат в вакууме. Осадок растворяют в смеси водного NaHSO4 (300 мл, 10%) и EtOAc (600 мл), слои разделяют, и водный слой повторно экстрагируют EtOAc (100 мл). Объединенные органические слои промывают насыщенным раствором соли (100 мл), сушат над MgSO4 и фильтруют. Растворитель удаляют в вакууме, получая бесцветное масло, которое растворяют в смеси EtOAc (1):гептан(10) и выдерживают в течение ночи. Образовавшиеся кристаллы удаляют фильтрованием, промывают гептаном и сушат в вакууме, получая одно соединение, которое является либо 1-трет-бутиловым эфиром (3R)-3-бензилпиперидин-1,3-дикарбоновой кислоты, либо 1-трет-бутиловым эфиром (3S)-3-бензилпиперидин-1,3-дикарбоновой кислоты (27,8 г).
Хиральная ВЭЖХ (Chiracel OJ,
гептан(92):изоPrOH(8):TFA(0,1)): Rt = 7,96 мин. 95,8%
Стадия е
Трет-бутиловый эфир (3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-карбоновой кислоты или трет-бутиловый эфир (3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-карбоновой кислоты

Триметилгидразиндигидрохлорид (15,3 г, 104 ммоль) суспендируют в тетрагидрофуране (250 мл) в одногорлой круглодонной колбе (1 л), снабженной большой магнитной мешалкой и капельной воронкой/барботером для азота. Затем колбу помещают на водяную баню (температура 10-20°С), добавляют бром-трис-пирролидинофосфонийгексафторфосфат (40,4 г, 86,7 ммоль) и при интенсивном перемешивании добавляют по каплям диизопропил-этиламин (59 мл, 347 ммоль). Смесь (при обильном образовании осадка) перемешивают 5 минут и медленно в течение 1,5 часов добавляют раствор продукта стадии d, который представляет собой либо 1-трет-бутиловый эфир (3S)-3-бензилпиперидин-1,3-дикарбоновой кислоты, либо 1-трет-бутиловый эфир (3S)-3-бензилпиперидин-1,3-дикарбоновой кислоты (27,7 г, 86,7 ммоль), в тетрагидрофуране (250 мл). Смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют EtOAc (1000 мл), промывают Н2O (500 мл), водным NaHSO4 (200 мл, 10%), водным NaHCO3 (200 мл, насыщенный), насыщенным раствором соли (200 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, получая жидкое оранжевое масло. Смесь растворяют в EtOAc (300 мл), добавляют SiO2 (150 г) и концентрируют в вакууме до сухого порошка, который помещают на фильтр, заполненный SiO2 (150 г), промывают гептаном (1 л) и выделяют желаемое соединение при помощи EtOAc (2,5 л). После концентрирования в вакууме получают продукт, который является либо трет-бутиловым эфиром (3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-карбоновой кислоты, либо трет-бутиловым эфиром (3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-карбоновой кислоты (49 г), в виде оранжевого масла.
ВЭЖХ (h8): Rt = 14,33 мин.
Стадия f
Триметилгидразид (3R)-3-бензилпиперидин-3-карбоновой кислоты или триметилгидразид (3S)-3-бензилпиперидин-3-карбоновой кислоты
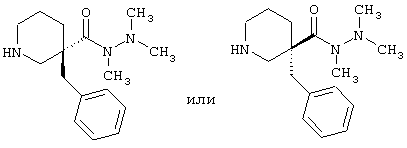
Продукт стадии е, который является либо трет-бутиловым эфиром (3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)-пиперидин-1-карбоновой кислоты, либо трет-бутиловым эфиром (3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-карбоновой кислоты (56,7 г, 100,9 ммоль), растворяют в EtOAc (500 мл) (прозрачный бесцветный раствор) в одногорлой круглодонной колбе (2 л), снабженной магнитной мешалкой. Колбу помещают на водяную баню (температура 10-20°С), пропускают через раствор HCl-газ в течение 5 минут (осаждение в виде пыли). После перемешивания в течение 1 часа (осаждение большого количества белых кристаллов) раствор продувают сильным потоком N2 для удаления избытка HCl. Осадок удаляют осторожным фильтрованием, промывают EtOAc (2×100 мл) и сушат в вакууме при 40°С в течение ночи, получая продукт, который является либо триметилгидразидом (3R)-3-бензил-пиперидин-3-карбоновой кислоты, либо триметилгидразидом (3S)-З-бензилпиперидин-3-карбоновой кислоты (37,0 г).
ВЭЖХ (h8): Rt = 7,84 мин.
Стадия g
Трет-бутиловый эфир [(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-((lH-индол-3-ил)метил)-2-оксоэтил]карбаминовой кислоты или трет-бутиловый эфир [(1R)-2-[(3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)-пиперидин-1-ил]-1-((1H-индол-3-ил)метил)-2-оксоэтил]карбаминовой кислоты

Boc-D-Trp-OH (32,3 г, 106 ммоль) растворяют в диметил-ацетамиде (250 мл) в одногорлой круглодонной колбе (500 мл), снабженной магнитной мешалкой и барботером для азота. Раствор охлаждают до 0-5°С и добавляют 1-гидрокси-7-азабензотриазол (14,4 г, 106 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (20,3 г, 106 ммоль), N-метилморфолин (11,6 мл, 106 ммоль). После перемешивания в течение 20 минут при 0-5°С добавляют продукт стадии f, который является либо триметилгидразидом (3R)-3-бензилпиперидин-3-карбоновой кислоты, либо триметилгидразидом (3S)-3-бензилпиперидин-3-карбоновой кислоты (37,0 г, 106 ммоль), и N-метилморфолин (24,4 мл, 223 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Затем данную смесь добавляют к EtOAc (750 мл) и промывают водным NaHSO4 (300 мл, 10%). Слоям дают разделиться и водный слой повторно экстрагируют EtOAc (500 мл). Объединенные органические слои промывают Н2О (100 мл), водным NaHCO3 (300 мл, насыщенный), Н2О (100 мл), насыщенным раствором соли (300 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, получая продукт, который является либо трет-бутиловым эфиром [(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-((1H-индол-3-ил)метил)-2-оксоэтил]карбаминовой кислоты, либо трет-бутиловым эфиром [(1R)-2-[(3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-((1H-индол-3-ил)-метил)-2-оксоэтил]карбаминовой кислоты (56,7 г), в виде оранжевого масла.
Стадия h
Триметилгидразид 1-[(2R)-2-амино-3-(1H-индол-3-ил)пропионил]-(3R)-3-бензилпиперидин-3-карбоновой кислоты или триметилгидразид 1-[(2R)-2-амино-3-(1H-индол-3-ил)пропионил]-(3S)-3-бензилпиперидин-3-карбоновой кислоты

Продукт стадии g, который является либо трет-бутиловым эфиром [(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-((1H-индол-3-ил)метил)-2-оксоэтил]карбаминовой кислоты, либо трет-бутиловым эфиром [(1R)-2-[(3S)-3-бензил-3-(N,N',N-триметилгидразинокарбонил)-пиперидин-1-ил]-1-((1H-индол-3-ил)метил)-2-оксоэтил]карбаминовой кислоты (56,7 г, 100,9 ммоль) растворяют в EtOAc (500 мл) (прозрачный бесцветный раствор) в одногорлой круглодонной колбе (2л), снабженной магнитной мешалкой. Затем колбу помещают на водяную баню (температура 10-20°С) и пропускают через раствор HCl-газ в течение 10 минут (обильное осаждение масла). Смесь продувают сильным потоком N2 для удаления избытка HCl и затем делят на масло и EtOAc-слой. EtOAc-слой отбрасывают. Масло растворяют в Н2О (500 мл), СН2Cl2 (1000 мл) и добавляют твердый Na2СО3 до рН >7. Слои разделяют и органический слой промывают Н2O (100 мл), насыщенным раствором соли (100 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме, получая продукт, который является либо триметилгидразидом 1-[(2R)-2-амино-3-(1H-индол-3-ил)пропионил]-(3R)-3-бензилпиперидин-3-карбоновой кислоты, либо триметилгидразидом 1[(2R)-2-амино-3(1Н-индол-3-ил)пропионил]-(3S)-3-бензилпиперидин-3-карбоновой кислоты (27 г), в виде оранжевой пены.
ВЭЖХ (h8): Rt = 10,03 мин.
Стадия i
Трет-бутиловый эфир {1-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-три-метилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-ил-метил)-2-оксоэтилкарбамоил]-1-метилэтил}карбаминовой кислоты или трет-бутиловый эфир {1[(1R)-2-[(3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-ил-метил)-2-оксоэтилкарбамоил]-1-метилэтил}карбаминовой кислоты

Boc-Aib-OH (11,9 г, 58,4 ммоль) растворяют в диметил-ацетамиде (125 мл) в одногорлой круглодонной колбе (500 мл), снабженной магнитной мешалкой и барботером для азота. К перемешиваемому раствору при комнатной температуре добавляют 1-гидрокси-7-азабензотриазол (7,95 г, 58,4 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (11,2 г, 58,4 ммоль) и диизопропилэтиламин (13,0 мл, 75,8 ммоль). Через 20 минут (смесь желтого цвета с осадком) добавляют раствор продукта стадии h, который является либо триметилгидразидом 1-[(2R)-2-амино-3-(lH-индол-3-ил)пропионил]-(3R)-3-бензилпиперидин-3-карбоновой кислоты, либо триметилгидразидом 1-[(2R)-2-амино-3-(1H-индол-3-ил)пропио-нил](3S)-3-бензилпиперидин-3-карбоновой кислоты (27,0 г, 58,4 ммоль), в диметилацетамиде (125 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Затем данную смесь добавляют к EtOAc (750 мл) и промывают водным NaHSO4 (300 мл, 10%). Слоям дают разделиться и водный слой повторно экстрагируют EtOAc (500 мл). Объединенные органические слои промывают Н2O (100 мл), водным NaHCO3 (300 мл, насыщенный), Н2О (100 мл), насыщенным раствором соли (300 мл), сушат над MgO4, фильтруют и концентрируют в вакууме примерно до 500 мл. Затем добавляют SiO2 (150 г) и оставшийся EtOAc удаляют в вакууме, получая сухой порошок, который помещают на фильтр, заполненный SiO2 (150 г), промывают гептаном (1 л) и выделяют желаемое соединение при помощи EtOAc (2,5 л). После концентрирования в вакууме получают продукт, который является либо трет-бутиловым эфиром {1-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтилкарбамоил]-1-метилэтил}карбаминовой кислоты, либо трет-бутиловым эфиром {1[(1R)-2-[(3S)-3-бензил-3(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил-карбамоил]-1-метилэтил}карбаминовой кислоты (33,9 г), в виде оранжевой пены.
ВЭЖХ (h8): Rt = 14,05 мин.
Стадия j
Фумарат 2-амино-N-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметил-гидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамида или фумарат 2-амино-N-[(1R)-2-[(3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(lH-индол-3-илметил)-2-оксоэтил]-2-метилпропионамида
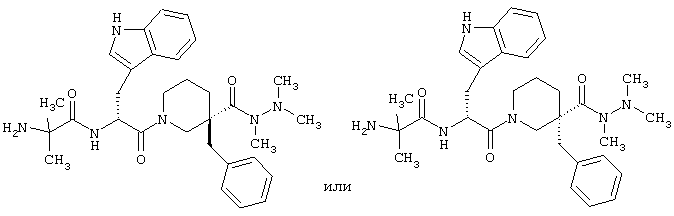
Продукт стадии i, который является либо трет-бутиловым эфиром (1-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтилкарбамоил]-1-метилэтил}карбаминовой кислоты, либо трет-бутиловым эфиром {1-[(1R)-2-[(3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтилкарбамоил]-1-метилэтил} карбаминовой кислоты (23,8 г, 36,8 ммоль), растворяют в EtOAc (800 мл) (прозрачный желтый раствор) в одногорлой круглодонной колбе (1 л), снабженной магнитной мешалкой. Затем колбу помещают на водяную баню (температура 10-20°С) и пропускают через раствор HCl-газ в течение 5 минут (образование осадка, похожего на пыль). После перемешивания в течение 1 часа (осаждение большого количества желтого порошка) раствор продувают сильным потоком N2 для удаления избытка HCl. Осадок удаляют осторожным фильтрованием и сушат в вакууме при 40°С в течение ночи.
Некристаллический осадок растворяют в Н2О (500 мл) и промывают EtOAc (100 мл). Затем добавляют CH2Cl2 (1000 мл) и твердый Na2СО3 до рН >7. 2 слоя разделяют и водный слой повторно экстрагируют CH2Cl2 (200 мл). Объединенные органические слои промывают насыщенным раствором соли (100 мл), сушат над MgSO4 и фильтруют. Растворитель выпаривают при пониженном давлении и смесь снова растворяют в EtOAc (500 мл) в одногорлой круглодонной колбе (1 л), снабженной магнитной мешалкой. Медленно (5 минут) добавляют суспензию фумаровой кислоты (3,67 г) в изопропаноле (20 мл) и EtOAc (50 мл), в результате осаждается белая кристаллическая соль. Через 1 час осадок отделяют фильтрованием и сушат в течение ночи в вакууме при 40°С, получая соль фумарат соединения, которое является либо 2-амино-N-[(1R)-2-[(3R)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамидом, либо 2-амино-N-[(1R)-2-[(3S)-3-бензил-3-(N,N',N'-триметилгидразинокарбонил)пиперидин-1-ил]-1-(1H-индол-3-илметил)-2-оксоэтил]-2-метилпропионамидом (13,9 г), в виде белого порошка.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРИДИНКАРБОНОВЫХ КИСЛОТ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, СПОСОБ СТИМУЛЯЦИИ СЕКРЕЦИИ ГОРМОНА РОСТА | 1999 |
|
RU2243215C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ВЫСВОБОЖДЕНИЯ ГОРМОНА РОСТА, СПОСОБ СТИМУЛЯЦИИ ВЫДЕЛЕНИЯ ГОРМОНА РОСТА ИЗ ГИПОФИЗА МЛЕКОПИТАЮЩЕГО | 1999 |
|
RU2298547C2 |
| ЗАМЕЩЕННЫЕ N-(ИНДОЛ-2-КАРБОНИЛ)-ГЛИЦИНАМИДЫ И ИХ ПРОИЗВОДНЫЕ, СПОСОБЫ ЛЕЧЕНИЯ И ФАРМКОМПОЗИЦИЯ | 1996 |
|
RU2143424C1 |
| ПРОИЗВОДНЫЕ БЕНЗИЛПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2160259C2 |
| СТИМУЛЯТОРЫ СЕКРЕЦИИ ГОРМОНА РОСТА | 1996 |
|
RU2172742C2 |
| 3-БЕНЗИЛПИПЕРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2194047C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| ЗАМЕЩЕННЫЕ N-(ИНДОЛ-2-КАРБОНИЛ)-β-АЛАНИНАМИДЫ И ИХ ПРОИЗВОДНЫЕ, СПОСОБ ЛЕЧЕНИЯ ГЛИКОГЕНФОСФОРИЛАЗОЗАВИСИМЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИШЕМИЧЕСКОГО ПОВРЕЖДЕНИЯ МИОКАРДА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159613C2 |
| ЗАМЕЩЕННЫЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КЕТОГЕКСОКИНАЗЫ | 2016 |
|
RU2696269C1 |
Изобретение относится к новому соединению формулы (I)

или к его фармацевтически приемлемой соли. Указанные соединения стимулируют высвобождение гормона роста из гипофиза и могут найти применение в медицине. 3 н. и 1 з.п. ф-лы, 1 ил., 1 табл.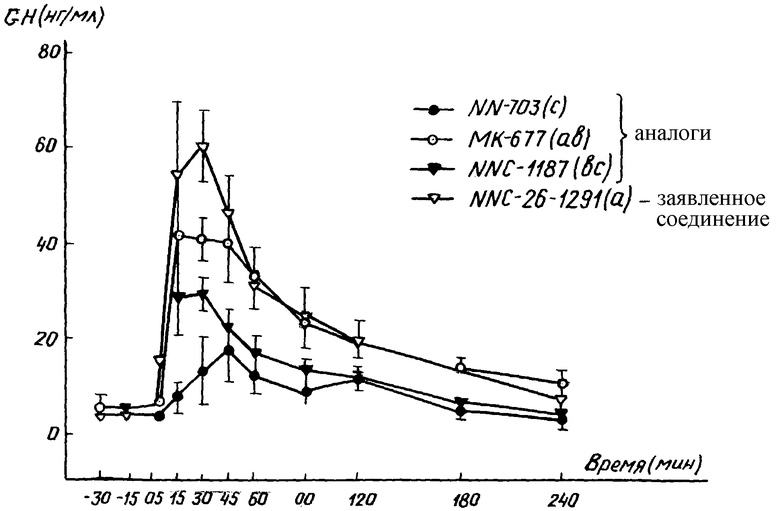

или его фармацевтически приемлемая соль.
| Способ получения производных индола или их солей | 1980 |
|
SU1083910A3 |
| US 5721250 А, 24.02.1998 | |||
| US 5622973 А, 22.04.1997 | |||
| US 5492916 А, 20.02.1996. | |||

