Изобретение относится к области основного органического синтеза, а именно к способу получения метилэтилкетона (МЭК=СН3СОС2Н5) каталитическим окислением н-бутенов кислородом, а также к катализаторам для его осуществления.
Вследствие исключительно высокой растворяющей способности метилэтилкетон (МЭК) находит широкое промышленное применение. Его используют в производстве полиуретановых лаков, служащих для покрытия магнитных лент компьютеров, инструмента, аудио- и видеокассет, является лучшим депарафинизатором смазочных масел, обеспечивая их морозостойкость, является растворителем в производстве пенопластов, различных красок, эпоксидных и глифталевых смол, полихлорвинила, искусственной кожи, служит основой топографических красок и чернил для печатания.
МЭК используют в качестве сырья при получении метилизопропил-кетона, 2,3-бутандиона, оксима метилэтилкетона, предотвращающего образование пленок при хранении масляных красок. Используют для получения этилакриловой и изомерных метилкротоновых кислот, антиоксидантов резин, для пластификации нитроцеллюлозы, используемой в производстве бездымных порохов, находит применение как растворитель для пленочных покрытий таблеток и капсул лекарств, как реагент и экстрагент во многих фармацевтических производствах. Пероксид МЭК`а является инициатором полимеризации ненасыщенных полиэфиров в производстве армированных пластиков.
В промышленном масштабе МЭК получают несколькими способами. Один из них основан на жидкофазном свободнорадикальном окислении н-бутана по схеме (1). В этом способе выход МЭК не превышает 23%, основной же продукт - уксусная кислота. Побочно образуется большое количество отходов, которые сжигаются.

Достоинством этого способа является дешевизна исходного сырья, а недостатком - обилие побочных продуктов, сильно осложняющих и удорожающих процесс выделения МЭК.
Основным промышленным способом синтеза МЭК является трехстадийная переработка бутиленовой фракции, являющейся отходом производства дивинила на заводах синтетического каучука. Способ отличается дешевизной сырья; он включает реакции (2)-(4):
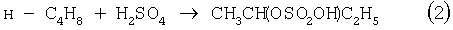
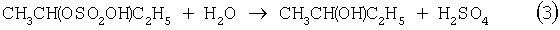
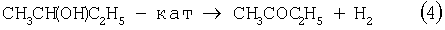
Из них реакции (2) и (3) - жидкофазные, а реакция (4) - гетерогенная каталитическая, а в некоторых вариантах - жидкофазная. Недостатками этого способа являются: обилие вредных необезвреживаемых отходов в виде загрязненной кислыми гудронами серной кислоты; высокая коррозионная активность среды в реакциях (2) и (3); высокая энергоемкость реакции (4); сложность процесса отделения МЭК от многочисленных примесей.
Разновидностью этого способа является процесс, в котором вместо H2SO4 используют уксусную кислоту, а промежуточным продуктом является втор-бутилацетат:
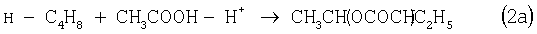
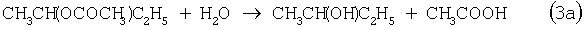
Катализатором стадий (2а) и (3а) служит сульфокатионит, который быстро забивается смолами и потому имеет малый срок службы. Использованный сульфокатионит также является необезвреживаемым отходом производства, серьезно ухудшающим его экологию.
В последнее время были предложены и другие кислые катализаторы этих стадий, например безванадиевые гетерополикислоты [Cai Tianxi, Huang He, Lin Jinlong, He Min, Zhang Suofang, Li Luhui // Shiyou Huagong, 1988, V.17, N.9, P.565-567], однако, они не обеспечивают существенного улучшения способа. Во втор-бутилацетатном способе получения МЭК (2а)+(3а)+(4) отходов стало меньше, чем в сернокислотном. Недостатком его стала неэкономичность, вызванная большими капиталовложениями и текущими затратами на единицу продукции из-за коррозионных свойств водной уксусной кислоты.
В последние годы предпринимались многократные попытки создания новых технологий получения МЭК из н-бутенов. Об этом свидетельствуют многочисленные патенты. Наибольший интерес из них представляют два новых способа: трехстадийный, включающий реакции (5)-(7) с промежуточным получением гидроперекиси втор-бутилбензола [Пат. США 5304684, 1994], и прямое окисление н-бутенов по реакции (8).
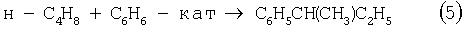
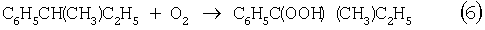
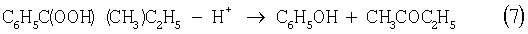
Трехстадийный способ основан на реакциях, подобных используемым в современном промышленном способе получения фенола и ацетона через гидроперекись кумола. Однако получение и разложение гидроперекиси втор-бутилбензола на стадиях (6) и (7) протекает с меньшей избирательностью, чем получение и разложение гидроперекиси кумола. В реакциях (6) и (7), наряду с МЭК и фенолом, образуются карбоновые кислоты, их эфиры, альдегиды, непредельные кетоны, смолы. Побочные продукты удаляют щелочью, что существенно ухудшает экологию производства. Способ (5)-(7) мог стать перспективным для промышленности при условии существенного повышения избирательности на стадиях (6) и (7) получения и разложения гидроперекиси втор-бутилбензола.
В одностадийном способе получения МЭК гомогенная каталитическая реакция (8) аналогична реакции ваккеровского окисления этилена в ацетальдегид [Jira R., Freiesleben W. // in: Organometallic Reactions / Ed.: E.I.Becker, M.Tsutsui. Vol.3 - New York et al.: Wiley Intersci., 1972 - P.1 - 190]:
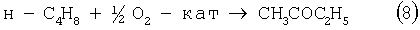
Реакцию (8) можно проводить в одном реакторе, подавая в него стехиометрическую смесь н-C4H8:O2=2:1, которая прежде считалась невзрывоопасной [Справочник химика. 2-е изд. Дополнительный том. - Л.: Химия, 1968 - С.434]. Однако для улучшения селективности и снижения степени риска реакцию следует проводить в две стадии: 1) взаимодействие н-бутилена с раствором промежуточного обратимо действующего окислителя Ох в присутствии соли палладия по уравнению (9); 2) регенерация Ох кислородом по уравнению (10):
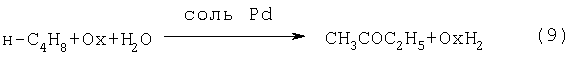
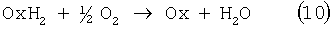
В качестве Ох использовали хлорную медь CuCl2 [Jira R., Freiesleben W., см. выше], восстановленная форма которой - ион CuCl2 - - легко окисляется кислородом. Однако синтез МЭК по реакции (9) в присутствии хлоридной системы (PdCl2+CuCl2) сопровождался образованием большого количества побочных хлорбутанонов (до 25%). В отсутствии Cl-- ионов или при их недостатке ([Cl-]:[Cu2+]<5) медь вообще не способна служить обратимым окислителем. Поэтому для окисления н-бутиленов такая система оказалась совершенно непригодной.
В изобретениях [SU 700973, B 01 J 23/44, 1994; 822417, B 01 J 23/44, 1994; 1669109, B 01 J 23/44, 1994] впервые было предложено использовать в качестве обратимого окислителя (Ох) Mo-V-фосфорные гетерополикислоты структуры Кеггина, имеющие общую формулу H3+nPVnMo12-nO40, обозначенные нами через ГПК1. С их участием бутиленовая реакция описывалась уравнением (9а), а кислородная - уравнением (10а):
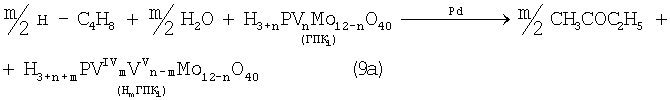
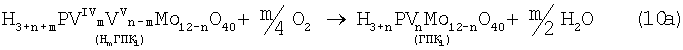
В этом способе (Ох=ГПК1) каталитическая система (PdII+ГПК1) не содержала Cl--ионов и потому обеспечивала полное отсутствие хлорорганических соединений в продуктах реакции. Избирательность системы достигала 95÷98%, а активность в бутиленовой реакции (9а) в 100 раз превышала активность хлоридной системы (PdCl2+CuCl2). Каталитическая система (Pd+ГПК1) могла успешно использоваться как для окисления стехиометрической смеси н-С4Н8:O2=2:1 в МЭК (одностадийный вариант), так и для проведения реакций (9а) и (10а) в разных реакторах (двухстадийный вариант). Последний вариант исключал контакт кислорода или воздуха с бутенами и с МЭК`ом и обеспечивал безопасность производства. Однако в последнем случае происходило глубокое восстановление молекул ГПК1. Это приводило к значительным изменениям химического состава и свойств как ионов Pd2+, так и самих молекул ГПК1. Этот вариант нестационарного катализа оказался связанным со снижением устойчивости каталитической системы (Pd+ГПК1).
Устойчивость катализатора в отношении ионов Pd2+ и молекул ГПК1 по-разному зависела от числа атомов V в молекуле ГПК1 (n) и от отношения m:n,(m - степень восстановления молекулы ГПК1). При малых значениях n(1≤n≤3) и m≈n неустойчивым оказывался палладий, который выделялся на поверхности восстановленного раствора в виде тонких зеркальных пленок, прилипавших к стенкам реактора. При большом числе атомов ванадия (4<n≤6) и m≈n, кроме образования пленок палладия, наблюдалось выпадение из раствора также части ванадия в виде коричневого осадка V3O7·2H2O. После окисления в воздушном реакторе восстановленной формы ГПК1 по реакции (10а) металлический палладий растворялся и возвращался в раствор, но ванадийсодержащий осадок при этом почти не растворялся. Полного же возвращения палладия в раствор в процессе получения МЭК не происходило, так как часть его осаждалась в различных частях пилотной установки. Поэтому катализатор (Pd+ГПК1) нельзя было признать вполне устойчивым ни в отношении палладия, ни в отношении ГПК1 с n≥4. Низкая стабильность такого катализатора оказалась его главным недостатком.
За последние 30 лет было предпринято много попыток стабилизировать палладий в катализаторе (Pd+ГПК1), однако, все они по разным причинам оказались неприемлемыми для технологии. Простейший способ удержать Pd2+ в растворе введением малых концентраций Cl--ионов, соответствующих атомным отношениям Cl-:Pd=5÷50, был предложен в патенте фирмы Catalytica Inc. (США) [Заявка WO 91/13852, 19.09.1991]. Однако даже при таких концентрациях хлор из раствора катализатора быстро переходил в продукты реакции с образованием хлорорганических соединений. Для поддержания стабильности палладия в катализатор приходилось непрерывно добавлять соляную кислоту и одновременно из продуктов реакции удалять и обезвреживать хлорорганические соединения. Поэтому "малохлоридный катализатор" (PdCl2+ ГПК1) также не нашел промышленного применения.
Использование палладия в виде комплексов с пиридинкарбоновыми кислотами (α-пиколиновой или дипиколиновой) повышает его стабильность [SU 1584200, B 01 J 23/44, 1994; 1669109, B 01 J 23/44, 1995]. Однако при этом в 10 и более раз снижается активность катализатора в реакции (9а), и падает его производительность. Это происходит потому, что комплекс Pd с производными пиридина оказывается слишком прочным, и он чрезмерно стабилизирует палладий в высшей степени окисления (Pd2+). Поэтому такой способ оказался нетехнологичным.
В изобретении [Пат. Японии 07-149685, 1995] катализаторы (Pd+ГПК1) использовались в смешанных растворителях, содержащих менее 50% воды. В качестве таковых использовались диоксан, этанол, тетрагидрофуран, γ-бутиролактон или сульфолан. Реакцию (8) проводили одностадийно: растворенный α-бутен при 80°С и давлении O2 9 кг/см2 окислялся в МЭК. Устойчивость палладия обеспечивалась тем, что реакции (9а) и (10а) протекали одновременно (в одном реакторе). Однако конверсия α-бутена в МЭК была невелика: 21% за час в водном диоксане и 14% в водном этаноле. В остальных растворителях она была еще меньше. Этот способ нетехнологичен и небезопасен. Идея использования неводных растворителей для реакции (8) оказалась неперспективной, поскольку эта реакция протекает через стадии (9а) и (10а), в которых вода является сокатализатором реакции (8), и понижение ее содержания резко снижает скорость реакции.
Эта же причина позволяет понять малую активность в реакции (8) гетерогенизированных катализаторов (Pd+ГПК1), нанесенных на твердый носитель [Stobbe-Kreemers A.W., van der Zon M., Makkee M., Scholten J.J.F. // J. Molec. Cat., A: Chem. - 1996 - V.107 (1-3) - P.247-253]. При применении таких катализаторов использовалась не жидкая вода, которая необходима, а водяной пар, который мог давать воду, только конденсируясь в порах носителя. Поэтому стадия (9а), состоящая из этапов (9а`) и (9а''), в гетерогенном варианте реакции (8) протекала очень медленно.
Существенным недостатком известных методов синтеза МЭК на катализаторах (Pd+ГПК) оказался неучет влияния продуктов коррозии аппаратурных материалов на эксплуатационные свойства катализатора. Неучет обусловливался тем, что в прежних разработках в качестве аппаратурного материала намечалось использовать титан, который не корродирует при контакте с катализатором (Pd+ГПК). Однако, исходя из требований технологии, возникла необходимость контактировать катализатор со многими частями арматуры, которые требовалось изготовить из спецсталей.
Наш эксперимент показал, что водные растворы Mo-V-фосфорных гетерополикислот (ГПК), составляющие катализатор (Pd+ГПК), обладают высокой коррозионной активностью в отношении большинства спецсталей. Лишь немногие спецстали оказались достаточно коррозионно-стойкими, пригодными для аппаратуры, контактирующей с катализатором (Pd+ГПК). Однако продукты их коррозии (Me=Fe2+(3+), Cr3+, Ni2+) отрицательно влияли на реакционную способность и устойчивость катализатора.
Главным и наиболее сильно действующим загрязнителем оказались ионы железа Fe3+. В концентрациях [Me]≤0,05 М продукты коррозии снижали реакционную способность и устойчивость молекул ГПК, а также уменьшали устойчивость комплекса Pd0·Pc. Снижение реакционной способности молекул ГПК1 проявлялось в замедлении на 15÷20% скорости кислородной реакции, а уменьшение устойчивости - в выпадении из окисленной формы катализатора (при 190°С в процессе кислородной реакции) ванадийсодержащих осадков с ионами железа. Снижение устойчивости палладия в присутствии Me проявлялось в постепенном выпадении палладиевой черни из восстановленной формы катализатора (при 60÷100°С после бутиленовой реакции).
Ионы железа в концентрациях 0≤[Fe3+]≤0,06 М попадали в катализатор вместе с сырьем (V2O5 х.ч.) еще на стадии его приготовления. Процесс коррозии спецсталей повышал концентрацию Fe3+-ионов и приводил к осадкообразованию. Поэтому предельно допустимое содержание продуктов коррозии Me, при котором катализатор (Pd+ГПК) должен устойчиво работать, удалось ограничить пределами 0≤[Me]≤0,1 М. В этом интервале концентраций Me осадкообразование из окисленной формы катализатора удалось исключить введением в молекулу ГПК небольших добавок ионов натрия и увеличением в ней содержания фосфора. Последняя мера также обеспечивала повышение стабильности восстановленной формы молекулы ГПК. В результате был разработан новый патентоспособный состав (Pd·Рс+ГПК-7Р4), описанный ниже.
Прототипом предлагаемого способа и катализатора получения МЭК является способ [SU 1584200, B 01 J 23/44, 1994], в котором в качестве катализатора используют водный раствора соли палладия, производного пиридина и кислой соли молибдованадофосфорной гетерополикислоты (ГПК1). Реакцию окисления проводят путем взаимодействия катализатора с н-бутиленом при температурах 50-70°С, а после отделения метилэтилкетона окисляют восстановленную форму катализатора кислородом или воздухом при температуре 130-160°С (двухстадийный вариант). В этом способе реакция (8) по-прежнему осуществляется в условиях нестационарного катализа. От глубины и скорости изменений химического состава, окислительных и других физико-химических свойств катализатора зависят его производительность и устойчивость на каждой стадии реакции (8). Оптимизация МЭК-технологии требует, чтобы изменения состава и свойств катализатора при протекании обеих стадий (9а) и (10а) были быстрыми, глубокими и обратимыми. Однако из-за выпадения осадков на стадии (9а) требование обратимости нарушается в отношении как палладия, так и молекул ГПК1 структуры Кеггина. Выпадение палладия в бутиленовом реакторе с неполным возвращением его в раствор в воздушном реакторе (нестабильность палладия) является недостатком каталитической системы (Pd+ ГПК1). Еще более серьезным ее недостатком является нестабильность молекул ГПК1, обусловленная потерей ими части ванадия, выпадавшего в осадок на стадии (9а) и не возвратившегося в раствор на стадии (10а).
Изобретение решает задачу увеличения эффективности процесса за счет повышения стабильности компонентов катализатора (Pd+ГПК), что обеспечит его технологичность, а также позволит значительно повысить производительность катализатора и его активность в обеих реакциях.
Задача решается составом катализатором получения метилэтилкетона окислением н-бутенов кислородом и/или кислородсодержащим газом, который состоит из водного раствора ГПК-7Р4 - молибдованадофосфорной гетерополикислоты или смеси гетерополикислоты и/или ее соли, а также палладия с молярной концентрацией 5 · 10-4 ÷ 1 · 10-2, стабилизированного фталоцианиновым лигандом Рс при мольном отношении [Pd]:[Pc]=0,5÷2. Используют Mo-V-фосфорную ГПК-7Р4 состава H11P4Mo18V7O87 или ее кислую натриевую соль состава Na1,2H9,8P4Mo18V7O87 при концентрации ванадия в водном растворе 0,4-2,2 грамм-атома на литр.
Задача решается также способом получения метилэтилкетона МЭК путем каталитического окисления н-бутенов с использованием катализатора, который состоит из водного раствора ГПК-7Р4-молибдованадофосфорной гетерополикислоты (7 - число атомов ванадия в молекуле ГПК-7, содержащей 4 атома фосфора) или смеси гетерополикислоты и/или ее соли, а также палладия с концентрацией 5·10-4÷1·10-2 М, стабилизированного фталоцианиновым лигандом Рс при мольном отношении [Pd]:[Рс]=0,5÷2. Используют ГПК-7Р4 состава H11P4Mo18V7O87 или ее кислую натриевую соль состава Na1,2H9,8P4Mo18V7O87 при концентрации ванадия в водном растворе 0,4-2,2 грамм-атома на литр.
Способ окисления н-бутенов в МЭК осуществляют непрерывно в двухстадийном режиме, в котором реакцию окисления н-бутенов ведут в интервале температур 15÷90°С, а регенерацию катализатора проводят путем взаимодействия его с кислородом или кислородсодержащим газом в интервале температур 140÷190°С при давлении кислорода 1÷10 ата.
Основным отличием предлагаемого способа синтеза МЭК на катализаторе (Pd+ГПК-7Р4) является то, что были использованы гетерополикислота ГПК-7Р4 нового состава и новые методы стабилизации компонентов катализатора (различные для палладия и ванадия). Новый состав ГПК-7Р4 и новые методы стабилизации были найдены нами в результате анализа причин нестабильности системы (Pd+ГПК1) в присутствии продуктов коррозии аппаратурных материалов, а также на основе исследований кинетики и механизма реакций (9а) и (10а).
Для устранения недостатков известных катализаторов (Pd+ ГПК1) и для создания катализатора с высокой производительностью и устойчивостью ион палладия был стабилизирован комплексообразованием с фталоцианиновым лигандом Рс, а молекулы ГПК1 кеггиновской структуры заменены молекулами ГПК-7Р4 некеггиновского состава. В результате получен новый катализатор (Pd·Pc+ГПК-7P4).
Предлагаемые катализаторы на основе ГПК-7Р4 готовят способом, аналогичным известному [SU 1782934, С 01 В 25/16, 1992].
Такие катализаторы имеют высокую гидролитическую устойчивость и не дают осадков при проведении стадий (9a1), (9a2) и (10а) реакции (8) в условиях промышленного использования катализатора (Pd·Рс+ГПК-7Р4). Активность и производительность катализаторов в широких пределах можно регулировать тремя способами: а) изменениями концентрации ГПК-7Р4 или ее кислых солей; б) вариациями концентраций комплекса Pd·Pc; 3) регулированием температуры реакции (9а).
Для контроля степени восстановления катализатора (или степени его окисленности) впервые использован метод измерения окислительного потенциала (Е) контактного раствора. Найденное значение Е (относительно нормального водородного электрода) сравнивается с полученной заранее кривой зависимости Е от m для катализатора, где m - это степень восстановления молекулы ГПК-7Р4 (m=[V(IV)]/[ГПК-7Р4]). По значению Е находят текущее значение m. В приведенных таблицах примеров имеется отдельная графа, где приведены значения Е окисленных растворов катализаторов. По ним судят о том, насколько хорошо регенерирован катализатор, так как от этого зависит его производительность.
Отличием предлагаемого катализатора является высокая концентрация ванадия (0,4-2,2 грамм-атом на литр), приводящая к повышению производительности катализатора с обеспечением его стабильности.
Следующим отличием способа получения метилэтилкетона является то, что регенерируют катализатор взаимодействием с кислородом или/и кислородсодержащим газом, а повышение производительности способа обеспечивается более высокой температурой регенерации (до 190°С) и парциальным давления кислорода (PO2) до 10 ата.
Другим отличием является то, что бутиленовую реакцию проводят и при более низких, чем в способе-прототипе, температурах (от +15°С), поскольку катализатор (Pd·Pc+ГПК-7P4) обладает значительно большей активностью, чем катализатор способа-прототипа, где палладий стабилизирован дипиколиновой кислотой.
Предлагаемый катализатор (Pd·Рс+ГПК-7Р4) обладает существенно меньшей коррозионной активностью в отношении спецсталей, из которых могут быть изготовлены узлы установки синтеза МЭК, а также значительно меньшей чувствительностью к продуктам коррозии этих сталей (Me=Fe3+, Cr3+, Ni2+), он не снижает своей стабильности в присутствии [Me]≤0,1 М.
Ниже приведен способ синтеза катализатора (Pd·Рс+ГПК-7Р4), а также даны примеры, иллюстрирующие сущность изобретения.
Все катализаторы, описанные в примерах 1-8, готовят известным способом [SU 1782934, С 01 В 25/16, 1992]. Вначале синтезируют раствор ГПК или ее кислой соли заданного состава и заданной концентрации, исходя из стехиометрических количеств компонентов Н3Р04, МоО3 и V2O5. Затем в полученный раствор вводят навески PdCl2 и фталоцианина.
Пример синтеза 250 мл ГПК состава Na1,2H9,8P4Mo18V7087 с концентрацией 0,23 М.
Расчет количеств исходных компонентов ведут по уравнению:
4H3PO4+18MoO3+3,5V2O5+0,6Na2CO3→
→Na1,2H9,8P4Mo18V7O87+0,6CO2+1,1H2O
Используют 149,04 г МоО3 х.ч., 36,63 г V2O5 х.ч., 3,66 г Na2СО3, 32,02 мл 7,183-молярного раствора Н3PO4 и 30%-ную Н2O2 ос.ч. Растворение реагентов проводят в четыре этапа:
1) 149,04 г МоО3+3,66 г Na2CO3 +19,02 мл Н3PO4+1500 мл Н2O+2 мл Н2O2
2) 12,63 г V2O5+700 мл Н2O+50 мл Н2O2+5,0 мл Н3PO4
3) 12,00 г V2O5+700 мл Н2O+50 мл Н2O2+4,0 мл Н3PO4
4) 12,00 г V2O5+700 мл Н2O+50 мл Н2O2+4,0 мл Н3PO4
Для растворения МоО3 в коническую 3-литровую колбу загружают всю навеску МоО3, 1,5 л дистиллированной воды, 3,66 г Na2СО3, 19,02 мл Н3PO4, 2 мл 30%-ной Н2O2 х.ч. Полученную смесь кипятят (она приобретает соломенно-желтую окраску) и постепенно упаривают до ˜0,8 л. В кипящую смесь последовательно вводят растворы, полученные, как описано ниже, растворением V2O5 в воде в присутствии Н2O2 и Н3PO4. Каждую последующую порцию раствора V2O5+Н3PO4 вводят после упаривания смеси в колбе до ˜1 л.
Для операции 2) - растворения V2O5 - в 1-литровый стакан загружают 12,63 г V2O5, 0,7 л дистиллированной воды и 50 мл Н2О2. Полученную смесь перемешивают при +15÷20°С (не выше) в течение 10÷15 мин до получения темно-красно-коричневого раствора. Еще до растворения V2O5 к смеси (при перемешивании) добавляют 5 мл Н3PO4 и ожидают прекращения выделения O2, образующегося за счет разложения перекисных комплексов ванадия. Полученный красно-коричневый (почти черный) прозрачный раствор переливают в колбу с кипящей суспензией МоО3+Н3PO4 и упаривают до ˜1 л.
Аналогично проводят операцию 3) - растворение V2O5 и добавление полученного раствора к суспензии МоО3+V2O5+Н3PO4. Эту смесь кипятят и упаривают до объема ˜1 л, после чего проводят операцию 4), совпадающую с операцией 3). Объединенный раствор упаривают до ˜400 мл. Растворение МоО3 контролируют визуально: после прекращения перемешивания и стояния на дне колбы не должно оставаться белого осадка МоО3.
Полученный раствор ГПК фильтруют через бумажный фильтр с красной полосой и фильтр промывают водой. Если осадок на фильтре превышает 2% от массы исходной V2O5, то его вместе с фильтром обрабатывают в стакане 5÷10%-ной Н2O2 (˜150 мл). Раствор кипятят до полного разложения Н2О2, остатки фильтра отделяют и фильтрат присоединяют к объединенному раствору ГПК с промывными водами. Готовый раствор упаривают до 250 мл.
Раствор H11P4Mo18V7O87 готовят аналогично. При этом синтез ведут без добавления Na2CO3 по уравнению: 4 Н3PO4+18 МоО3+3,5 V2O5→H911P4MO18V7O87+0,5 Н2O.
Катализаторы для опытов, содержащие ГПК, PdCl2 и фталоцианин, обычно готовят в количестве 20 мл. Например, для получения катализатора состава [Na1,2H9,8P4Mo18V7O87]=0,23 М, [Pd2+]=6·10-3 М с мольным соотношением [Pc]:[Pd2+]=1:1 в раствор 0,23 М ГПК вводят навески 0,0212 г PdCl2 и 0,096 г фталоцианина. Раствор нагревают и кипятят 8-10 мин до полного растворения навесок. Затем раствор охлаждают и доводят его объем до 20 мл.
Пример 1. В реактор типа "каталитическая утка" на 120 мл, закрепленный на качалке, вливают 20 мл раствора катализатора состава: [H11P4Mo18V7O87]=0.25 М, [Pd+2]=6·10-3 М мольное отношение [Pc]:[Pd]=1.5. Реактор термостатируют при 60°С, при атмосферном давлении продувают его 10-кратным объемом α-бутилена состава, %: α-С4Н8 - 96,3, сумма цис- и транс-β-C4H8 - 1,4, н-бутан - 2,2, тяжелые примеси - 0,1. Соединяют реактор с бюреткой, заполненной α-бутиленом, и при интенсивном встряхивании реактора проводят окисление C4H8 по реакции (9). За 32 мин раствор катализатора окисляет 325 мл α-бутилена (m=5,24  ) с избирательностью 98,3%. После отпарки МЭК катализатор по реакции (10) окисляют в автоклаве с мешалкой при 190°С и Ро2=4 ата в течение 20 мин. Общее давление в автоклаве 16,4 ати за счет давления водяных паров, составляющих 12,4 ати. С этим катализатором проводят еще 5 циклов, поглощая в каждом из них от 3,4 до 4,0 на моль ГПКх. Избирательность и активность катализатора не меняется. Осадков в растворе нет.
) с избирательностью 98,3%. После отпарки МЭК катализатор по реакции (10) окисляют в автоклаве с мешалкой при 190°С и Ро2=4 ата в течение 20 мин. Общее давление в автоклаве 16,4 ати за счет давления водяных паров, составляющих 12,4 ати. С этим катализатором проводят еще 5 циклов, поглощая в каждом из них от 3,4 до 4,0 на моль ГПКх. Избирательность и активность катализатора не меняется. Осадков в растворе нет.
Примеры 2-3 (по аналогу - RU 2230612).
В первом варианте разработанного нами катализатора (Pd·Pc+ГПК-7) мы остановились на составе ГПК-7=Н10Р3Мо18V7O84. Катализатор обладает хорошими показателями активности в бутиленовой и кислородной реакциях (пример 2, см. табл.1), однако, в процессе исследований коррозионных свойств этого катализатора было установлено, что продукты коррозии спецсталей (Me=Fe3+, Ni2+, Cr3+) существенным образом снижают его стабильность и каталитическую активность. Показано, что наибольшее влияние на катализатор оказывают катионы Fe3+. Это влияние иллюстрирует пример 3 (табл.2).
Пример 2. По примеру 1, отличающемуся тем, что в качестве катализатора используют [Н10Р3Мо18V7O84]=0.25 М, [Pd+2]=6·10-3 М при мольном отношении [Pc]:[Pd]=1.5. Катализатор испытывают в 5 циклах (табл.1). Избирательность его была стабильной 98-99%. Металлический палладий в процессе испытаний не появляется.
Пример 3. По примеру 2, отличающемуся тем, что в качестве катализатора используют [Fe0.2Н9.4Р3Мо18V7O84]=0.25 М. Катализатор также испытан в 5 циклах (табл.2). Избирательность его была 98-98.5%, однако, в процессе испытаний восстановленный раствор выделял заметное количество металлического палладия, а окисленный катализатор постепенно выделяет ванадийсодержащие осадки. Активность катализатора монотонно снижается.
Сравнивая примеры 2 и 3, можно видеть, что введение катионов железа в раствор катализатора (Pd+ГПК-7) ухудшает его активность в отношении бутилена, а также его стабильность как в восстановленном, так и в окисленном состоянии.
Примеры 4-8.
Недостаточную устойчивость восстановленной формы катализатора на основе Н10Р3Мо18V7O84 удалось устранить использованием ГПК-7 нового состава - H11P4Mo18V7O87 (ГПК-7Р4), в молекуле которой содержится четыре атома фосфора. Однако еще более эффективным оказался катализатор на основе кислой натриевой соли этой ГПК (см. пример 4), поскольку дозированное введение катиона Na+ позволяет увеличить стабильность ванадия также и в окисленной форме катализатора.
Катализатор (Pd+ГПК-7Р4) нового состава, где ГПК-7Р4 является кислой солью H11P4Mo18V7O87, отвечающей составу Na1,2H9,8P4Mo18V7O87, полученный дополнительным введением в раствор H10P3Mo18V7O84 фосфорной кислоты и катионов натрия, отличается физико-химическими свойствами от своего предшественника H10P3Mo18V7O84. На чертеже приведены кривые зависимостей окислительного потенциала (Е) и рН этих растворов от степени восстановления ГПК (Зависимости Е (кривые 1-3) и рН (кривые 1`-3`) от m () для 0,25 М растворов катализаторов, содержащих: Н10Р3Мо18V7O84 (1,1`), H11P4Mo18V7O87 (2,2`) и Na1,2H9,8P4Mo18V7O87). Видно, что новый катализатор имеет существенно более высокие значения рН, поэтому он обладает меньшей коррозионной активностью. Несколько меньшие значения Е нового катализатора оказываются, однако, вполне достаточными, чтобы обеспечить требуемую емкость катализатора (3.5-4.0 ) за 1 цикл. Стабильность нового катализатора как в окисленном, так и в восстановленном состоянии оказалась высокой: раствор в процессе исследований сохраняет свою гомогенность на всех стадиях реакции (пример 4, табл.3).
Пример 4. По примеру 2, отличающемуся тем, что в качестве катализатора используют [Na1,2H9,8P4Mo18V7O87]=0.23 М. Катализатор испытан в 12 циклах (табл.3), причем после 10 цикла в раствор вводят катионы Fe3+. Избирательность катализатора остается стабильной 98-98.5%, раствор сохраняет гомогенность. Введение катионов железа в раствор катализатора (Pd+ГПК-7Р4) не ухудшает его активность и стабильность, т.е. он стал значительно менее чувствительным к продуктам коррозии аппаратуры (Me) по сравнению с катализатором на основе H10P3Mo18V7O84. В этом примере температуру регенерации катализатора под давлением кислорода снижают со 190°С до 180°С, несколько увеличивая при этом ее длительность (с 20 мин до 23-25 мин). Снижение температуры регенерации на 10 градусов заметно упрощает решение технологических задач МЭК-процесса.
Пример 5. По примеру 4, отличающемуся тем, что в качестве катализатора используют 0,4 М раствор Na1,2H9,8P4Mo18V7O87 при мольном отношении [Рс]:[Pd]=0,5, за 17 мин окисляют 270 мл α-C4H8 (m=2,81 e) с избирательностью 98,5%. После отпарки МЭК катализатор окисляют в автоклаве при условиях, аналогичных примеру 1. На втором цикле активность и избирательность катализатора не меняется.
Пример 6. По примеру 1, отличающемуся тем, что в качестве катализатора используют [H11P4Mo18V7O87]=0,2 M, [Pd]=5·10-4 при мольном отношении [Pd]:[Рс]=1, за 30 мин окисляют 174 мл α-C4H8 (m=3,06 e) с избирательностью 98,7%. После отпарки МЭК катализатор окисляют в автоклаве при условиях, аналогичных примеру 1. На втором цикле активность и избирательность катализатора не меняется.
Пример 7. По примеру 4, отличающемуся тем, что в качестве катализатора используют 0,3 М раствор Na1,2H9,8P4Mo18V7O87 при мольном отношении [Рс]:[Pd]=2 (или [Pd]:[Pc]=0,5), за 24 мин окисляют 335 мл α-C4H8 (m=4,56 e) с избирательностью 98,2%. После отпарки МЭК катализатор окисляют в автоклаве при условиях, аналогичных примеру 1. На втором цикле его активность и избирательность не меняется.
Пример 8. По примеру 7, отличающемуся тем, что в качестве катализатора используют 0,3 М раствор смеси Na1,2H9,8P4Mo18V7O87 и H11P4Mo18V7O87 в соотношении 1:1, мольное отношение [Рс]:[Pd]=2 (или [Pd]:[Pc]=0,5), за 24 мин окисляют 335 мл α-C4H8 (m=4,56 e) с избирательностью 98,2%. После отпарки МЭК катализатор окисляют в автоклаве при условиях, аналогичных примеру 1. На втором цикле его активность и избирательность не меняется.
Испытания катализатора МЭК-процесса на стабильность в 6 циклах. Состав катализатора: 0,23 М Н10Р3Мо18V7O84, [Pd2+]=6·10-3M, [Рс]=9·10-3М (пример 2). Условия: Vкат=20 мл, tбут=60°С, tрег=190°C, время регенерации (τ)-20 мин (в 5 цикле - 15 мин).)))
(0,22)
(4.70)
(1.50)
(1.50)
(4.30)
(1.15)
(1.15)
(4.32)
(1.28)
(1.28)
(4.85)
(1.26)
(1.26)
(4.86)
(1.86)
*регенерацию вели 15 мин
(1.86)
(5.35)
(1.30)
Испытание катализатора Fe0,2H9,4P3Mo18V7O84, ([Pd]=6·10-3, [Рс]=9·10-3 М) (пример 3) в 5 циклах окисления α-C4H8 в МЭК. Условия: объем катализатора=20 мл; tбут=60°С, tкис=190°С.
(5,23 е-)
(3,48 е-)
(3,86 е-)
(3,39 e-)
(4,48 e-)
Испытания катализатора МЭК-процесса на стабильность в 12 циклах. Состав катализатора: 0,23 М Na1,2H9,8P4Mo18V7O87, [Pd2+]=6·10-3M, [Pc]=9·10-3M (пример 4).
Условия: Vкат=20 мл, tбут=60°C, tрег=180°C. Циклы 11 и 12 проведены с добавкой катиона Fe3+ (0,2 моль на 1 моль Na1,2H9.8P4Mo18V7O87).
Цикла)))
(0,25)
(5.50)
(2.25)
(5.41)
(1.45)
(1.45)
(5.43)
(2.30)
(2.30)
(5.76)
(1.19)
(1.19)
(5.55)
(1.52)
(1.52)
(5.65)
(1.15)
(1.15)
(5.50)
(1.34)
(1.34)
(5.54)
(1.19)
(1.19)
(5.57)
(1.15)
(1.15)
(5.88)
(1.10)
(1.20)
(5,30)
(1.19)
(1.19)
(5.41)
(1.35)
Результаты, полученные в этой серии исследований нового катализатора (Pd+ГПК-7P4) на стабильность, позволяют сделать несколько важных выводов. Главным из них является то, что регенерацию катализатора можно проводить при 180°С, если на ˜15-20% увеличить время кислородной реакции по сравнению с ее временем при 190°С (20 мин). Из таблицы 3 видно, что 20-минутное окисление катализатора при 180°С не обеспечивает требуемых конечных степеней восстановления (mох˜1.6-1.5 ), т.к. окисленный в течение этого времени катализатор имеет mox˜2.25-2.30 . Недоокисление катализатора повлечет за собой снижение его емкости на следующем цикле, поскольку необходимым критерием высокой стабильности катализатора должно быть соблюдение рабочих интервалов значений m, в первую очередь в восстановленном растворе (mвосст≤˜5.6 ). Такое ограничение значений m сверху обеспечит высокую устойчивость палладия в растворе катализатора, а регенерация катализатора при 180°С в течение 25 мин обеспечит его емкость не менее 4-х за проход (см. 5-12 циклы в табл.3).
Полученные результаты показывают также, что введение в раствор катализатора продуктов коррозии Me (катионов Fe2+) практически не изменяет стабильность и активность катализатора (см. 11 и 12 циклы, табл.3). Некоторое снижение окислительного потенциала раствора при добавлении катионов железа (на 11 цикле значение Ео при введении Fe2+ снижается с 1.036 до 1.029 В) практически не меняет рабочий интервал Δm - изменений m при работе катализатора.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ МЕТИЛЭТИЛКЕТОНА | 2003 |
|
RU2243818C1 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ МЕТИЛЭТИЛКЕТОНА | 2003 |
|
RU2230612C1 |
| СПОСОБ ПОЛУЧЕНИЯ КЕТОНОВ И КАТАЛИЗАТОР ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2005 |
|
RU2294322C1 |
| КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ Н-БУТИЛЕНА В МЕТИЛЭТИЛКЕТОН И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 1989 |
|
RU1669109C |
| КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ н-БУТИЛЕНА В МЕТИЛЭТИЛКЕТОН И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 1987 |
|
SU1584200A1 |
| КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ ЭТИЛЕНА В АЦЕТАЛЬДЕГИД ИЛИ БУТИЛЕНА В МЕТИЛЭТИЛКЕТОН | 1979 |
|
SU822417A1 |
| Катализатор и способ получения высших 2-кетонов С5-С10 | 2022 |
|
RU2790246C1 |
| СПОСОБ ОКИСЛИТЕЛЬНОЙ АКТИВАЦИИ БЕСХЛОРИДНОГО КАТАЛИЗАТОРА ДЛЯ ОКИСЛЕНИЯ Н-БУТИЛЕНА | 1987 |
|
SU1578908A1 |
| КАТАЛИЗАТОР ДЛЯ ЖИДКОФАЗНОГО ОКИСЛЕНИЯ ОЛЕФИНОВ В КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ | 1977 |
|
SU700973A1 |
| Способ и установка получения метилэтилкетона | 2022 |
|
RU2796680C1 |
Изобретение относится к области основного органического синтеза, а именно к способу получения метилэтилкетона каталитическим окислением н-бутенов. Описан катализатор получения метилэтилкетона каталитическим окислением н-бутенов кислородом и/или кислородсодержащим газом на основе палладия, стабилизированного комплексообразующим лигандом, и гетерополикислоты и/или ее кислых солей, в качестве гетерополикислоты катализатор содержит ГПК-7P4 - молибдованадофосфорную гетерополикислоту состава H11P4Mo18V7O87 и/или ее кислую соль состава Na1.2H9.8P4Mo18V7O87, а в качестве комплексообразующего лиганда - фталоцианиновый лиганд Рс. Регенерируют катализатор взаимодействием его с кислородом или кислородсодержащим газом при температуре 140÷190°С и давлении кислорода 1÷10 ата. Описан также способ окисления н-бутенов в МЭК, который осуществляют непрерывно в двухстадийном режиме при температуре 15÷90°С в присутствии описанного выше катализатора. Технический результат - увеличение эффективности процесса за счет повышения стабильности катализатора, что позволяет значительно повысить производительность катализатора и его избирательность. 2 н. и 5 з.п. ф-лы, 3 табл., 1 ил.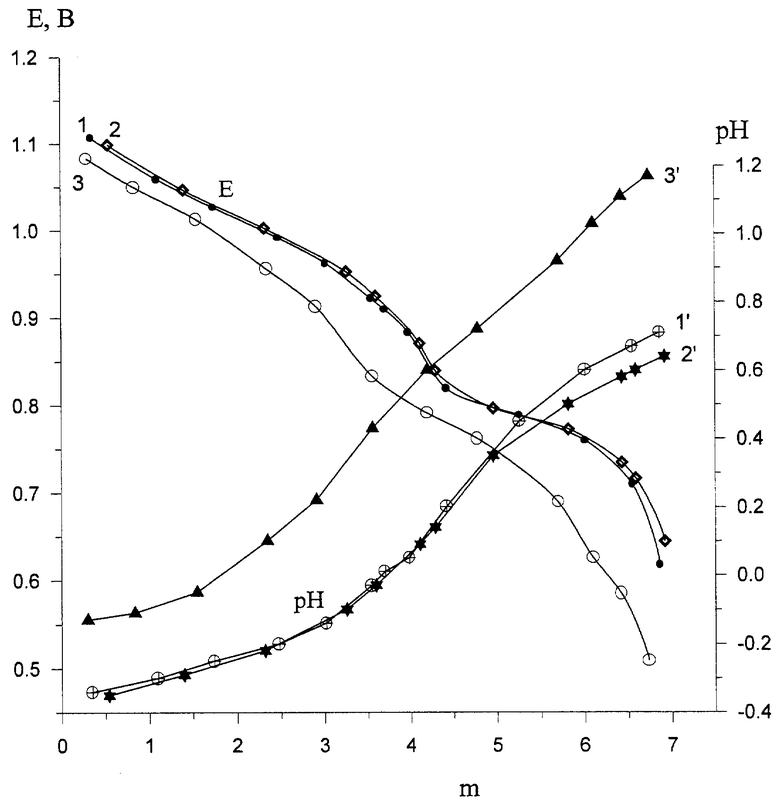
| КАТАЛИЗАТОР ДЛЯ ОКИСЛЕНИЯ н-БУТИЛЕНА В МЕТИЛЭТИЛКЕТОН И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 1987 |
|
SU1584200A1 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ МЕТИЛЭТИЛКЕТОНА | 2003 |
|
RU2230612C1 |
| Твердый сплав | 1973 |
|
SU518977A1 |
| US 5304684 A, 19.04.1994. | |||

