Настоящее изобретение относится к способу получения эфира угольной кислоты из металлоорганического соединения и диоксида углерода. Более конкретно, настоящее изобретение относится к способу получения эфира угольной кислоты, включающему (1) осуществление реакции между металлоорганическим соединением, содержащим металл-кислород-углеродную связь, и диоксидом углерода с получением реакционной смеси, включающей образовавшийся в результате реакции эфир угольной кислоты; (2) выделение эфира угольной кислоты из реакционной смеси с получением остаточной жидкости; и (3) введение во взаимодействие остаточной жидкости со спиртом с получением металлоорганического соединения, содержащего металл-кислород-углеродную связь, и образованием воды и удаление воды из металлоорганического соединения, причем полученное на стадии (3) металлоорганическое соединение рекуперируют для его рециркуляции на стадию (1). Согласно предлагаемому в настоящем изобретении способу, эфир угольной кислоты может быть получен с высоким выходом из металлоорганического соединения, содержащего металл-кислород-углеродную связь, и диоксида углерода. Преимуществом настоящего способа является то, что диоксид углерода не обладает ни токсичностью, ни коррозионной активностью и является недорогостоящим. Далее, способ согласно настоящему изобретению является выгодным не только в том отношении, что металлоорганическое соединение после использования в этом способе может быть рекуперировано и рециркулировано на стадию (1) способа, предотвращая, таким образом, возникновение отходов, получаемых за счет металлоорганического соединения, но и также в том, что в этом способе нет необходимости в использовании большого количества осушителя, избегая, таким образом, отходов, получаемых за счет осушителя. Следовательно, способ согласно настоящему изобретению является пригодным с коммерческой точки зрения и имеет высокую коммерческую ценность.
Уровень техники
Эфир угольной кислоты является полезным соединением. Например, эфир угольной кислоты используют в качестве добавок для различных целей, таких как добавка к бензину для повышения октанового числа бензина и добавка к дизельному топливу для уменьшения количества частиц в выхлопном газе, образующемся при сгорании дизельного топлива. Эфир угольной кислоты также используют в качестве алкилирующего агента, карбонилирующего агента, растворителя и тому подобного в области синтеза органических соединений, таких как поликарбонат, уретан, фармацевтические средства и агрохимикаты. Эфир угольной кислоты также используют в качестве электролита для литиевой батареи, исходного материала для получения смазочного масла и исходного материала для получения восстановителя, который может быть использован для предохранения бойлерных труб от ржавчины.
В качестве обычного способа получения эфира угольной кислоты можно указать способ, согласно которому фосген в качестве источника карбонила вводят во взаимодействие со спиртом, получая таким образом эфир угольной кислоты. Так как используемый в этом способе фосген является чрезвычайно вредным и высококоррозионным продуктом, этот способ является непригодным, поскольку транспортировка и хранение фосгена требуют предельной осторожности и также требуются большие расходы на сохранение производственной аппаратуры и для обеспечения безопасности. Далее, этот способ связан с проблемой, заключающейся в том, что необходимо избавляться от побочно образующейся соляной кислоты в качестве отходов.
В качестве другого обычного способа получения эфира угольной кислоты известен способ окислительного карбонилирования, согласно которому монооксид углерода в качестве источника карбонила вводят во взаимодействие со спиртом и кислородом в присутствии катализатора, такого как хлорид меди, получая таким образом эфир угольной кислоты. Согласно этому способу монооксид углерода (который является чрезвычайно вредным) используют при высоком давлении; следовательно, недостатком этого способа является то, что он требует большие расходы на сохранение производственной аппаратуры и для обеспечения безопасности процесса. Кроме того, недостатком этого способа является то, что протекает побочная реакция - окисление монооксида углерода с образованием диоксида углерода. По этим причинам желательно разработать безопасный способ получения эфира угольной кислоты.
Согласно этим обычным способам, если в качестве исходного материала используют фосген или монооксид углерода, то в самом исходном материале или в используемом катализаторе содержится галоген, такой как хлор. Следовательно, в случае этих способов получаемый эфир угольной кислоты содержит следовое количество галогена, который должен быть полностью удален путем стадии очистки. Когда такой эфир угольной кислоты используют в качестве добавки к бензину, добавки к светлым нефтепродуктам или в качестве материала для производства электронного оборудования, возможно, что содержащийся в эфире угольной кислоты галоген вызывает коррозию оборудования. Для снижения количества галогена в эфире угольной кислоты до крайне ничтожного количества необходимо осуществлять тщательную очистку эфира угольной кислоты. По этой причине желательно разработать способ получения эфира угольной кислоты, в котором нет необходимости использовать галогенсодержащий исходный материал и галогенсодержащий катализатор.
С другой стороны, предложен способ для практического применения, согласно которому диоксид углерода вводят во взаимодействие с этиленоксидом или подобным соединением с получением циклического эфира угольной кислоты и полученный циклический эфир угольной кислоты вводят во взаимодействие с метанолом, получая таким образом диметилкарбонат. Этот способ является выгодным в том отношении, что диоксид углерода в качестве исходного материала является безвредным и по существу в процессе не используется или не выделяется такое коррозионное вещество, как соляная кислота. Однако этот способ связан со следующими проблемами. В данном способе образуется побочный продукт этиленгликоль; следовательно, чтобы снизить издержки, производства необходимо найти пути эффективного использования побочно образующегося этиленгликоля. Далее, затруднительным является осуществление безопасной транспортировки этилена (который представляет собой исходный материал для получения этиленоксида) и этиленоксида. Поэтому, для устранения необходимости транспортировки нужно, чтобы установка для получения эфира угольной кислоты этим способом была сконструирована на участке, который примыкает к установке для получения этилена и этиленоксида.
Также известен способ, в котором диоксид углерода в качестве источника карбонила подвергают равновесной реакции со спиртом в присутствии катализатора, включающего металлоорганическое соединение, содержащее металл-кислород-углеродную связь, получая, таким образом, эфир угольной кислоты и воду. Эта равновесная реакция представлена следующим уравнением (3):
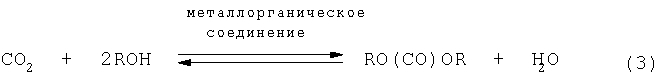
(R означает ненасыщенную или насыщенную углеводородную группу)
Этот способ является выгодным, поскольку диоксид углерода и спирт в качестве исходных материалов являются безвредными. Однако в этом способе используют равновесную реакцию, при которой в качестве продуктов одновременно образуются эфир угольной кислоты и вода. Также в вышеуказанном способе окислительного карбонилирования с использованием монооксида углерода образуется вода. Однако образование воды в равновесной реакции полностью отличается по значительности от образования воды при окислительном карбонилировании, которое не является равновесной реакцией. Равновесие в случае равновесной реакции, при которой используют диоксид углерода в качестве исходного материала, термодинамически смещено в сторону исходной системы. Следовательно, способ с использованием равновесной реакции связан с проблемой, заключающейся в том, что для получения эфира угольной кислоты с высоким выходом необходимо удалять эфир угольной кислоты и воду в качестве продуктов из реакционной системы. Далее, также существует проблема в том, что образующаяся вода разлагает катализатор, так что она не только мешает протеканию реакции, но и также число "оборотов" катализатора (то есть число циклов регенерации и повторного использования) составляет только 2 или 3. Для решения этой проблемы были предложены различные способы удаления воды (которая является продуктом) используя осушитель.
Например, был предложен способ, в котором спирт и диоксид углерода вводят во взаимодействие друг с другом в присутствии алкоголята металла в качестве катализатора, получая таким образом эфир угольной кислоты и воду, причем в качестве осушителя используют большое количество дициклогексилкарбодиимида (DCC) (который является дорогостоящим органическим осушителем) или подобные ему вещества (см. Collect. Czech. Chem. Commun., 60, 687-692 (1995)). Этот способ связан с проблемой, заключающейся в том, что осушитель после использования не может быть регенерирован, поэтому образуется большое количество отходов.
Известен способ получения эфира угольной кислоты, в котором в качестве органического осушителя используют орто-эфир карбоновой кислоты (см. нерассмотренное экспертизой описание изобретения согласно открытой выкладке заявки на патент Японии № Hei 11-35521). (В этом патентном документе написано: "орто-эфир карбоновой кислоты вводят во взаимодействие с диоксидом углерода" и "ацеталь вводят во взаимодействие с диоксидом углерода". Однако в результате последних исследований в уровне техники, в целом, полагают, что действительное направление реакции является следующим: "Спирт и диоксид углерода вводят во взаимодействие друг с другом, получая эфир угольной кислоты и воду. Часть воды реагирует с орто-эфиром карбоновой кислоты. Остаток воды реагирует с ацеталем."). Этот способ связан с проблемой, заключающейся в том, что ортоэфир карбоновой кислоты (который является дорогостоящим соединением) используют в качестве осушителя, и известно, что метилацетат является побочным продуктом (см. "Kagaku Sochi (Chemical Equipment)", 41, №2, 52-54 (1999)). Таким образом, этот способ является также несовершенным, как и вышеуказанный способ.
Далее, предложен способ, в котором в качестве органического осушителя используют большое количество ацеталя (см. патент Германии №4310109). Также известен патентный документ, в котором описывается, что ацеталь и диоксид углерода реагируют друг с другом при использовании в качестве катализатора алкоголята металла или дибутилоловооксида (см. нерассмотренное экспертизой описание изобретения согласно открытой выкладке заявки на патент Японии №2001-31629). (Что касается недавно описанной реакции в результате недавних исследований в уровне техники, в целом, полагают, что действительное направление реакции является следующим: "Спирт и диоксид углерода реагируют друг с другом с образованием эфира угольной кислоты и воды. Часть воды затем реагирует с ацеталем."). Однако в этих патентных документах нет указания или предложения способа получения ацеталя с высоким выходом без образования отходов. Далее, недостатком раскрытых в этих патентных документах способов является то, что при использовании в качестве осушителя ацеталя, образуются большие количества побочных продуктов (отходов), таких как кетон и альдегид.
Таким образом в способах, использующих органический осушитель задачей является повышение числа оборотов катализатора (повторное использование). Однако органический осушитель расходуется в стехиометрическом количестве в соответствии с реакцией образования эфира угольной кислоты (и воды в качестве побочного продукта), поэтому его потребляется большое количество, образуя, таким образом, большое количество продукта "дегенерации" органического осушителя. Следовательно, необходимо осуществлять дополнительную стадию регенерации большого количества подвергшегося "дегенерации" органического осушителя. Далее, несмотря на использование органического осушителя, все еще остается возможность дезактивации катализатора. Причиной является следующее. В традиционном способе получения эфира угольной кислоты, используя равновесную реакцию согласно вышеприведенному уравнению (3), диоксид углерода находится в сверхкритическом состоянии и, следовательно, реакцию осуществляют при использовании диоксида углерода в сверхкритическом состоянии. Обычно, в находящемся в сверхкритическом состоянии диоксиде углерода катализатор проявляет плохую растворимость и частицы катализатора, вероятно, сцепляются друг с другом. Поэтому существует проблема при использовании оловоорганического соединения (которое способно к полимеризации) в качестве катализатора в процессе, в котором диоксид углерода находится в сверхкритическом состоянии, поскольку оловоорганическое соединение в качестве катализатора вероятно дезактивируется вследствие его полимеризации.
Также предложен способ, использующий твердый осушитель (см. Applied Catalysts, 142, L1-L3 (1996)). Однако недостатком этого способа является то, что твердый осушитель не может быть регенерирован, образуя, таким образом, большое количество отходов.
Также известен способ, в котором спирт (метанол) и диоксид углерода вводят во взаимодействие друг с другом в присутствии оксида металла (дибутилоловооксид) с получением реакционной смеси, и полученную реакционную смесь охлаждают и вводят в насадочную колонну, содержащую твердый осушитель, таким образом смещая постепенно равновесие в сторону образования эфира угольной кислоты во время осуществления обезвоживания, получая эфир угольной кислоты (см. нерассмотренное экспертизой описание изобретения согласно открытой выкладке заявки на патент Японии №2001-247519). Этот способ базируется на технологии, в которой обычную методику использования осушителя комбинируют с известным эффектом, что адсорбируемость воды обычным осушителем (таким, как молекулярное сито) зависит от температуры. Осушитель (такой как молекулярное сито) проявляет более низкую адсорбируемость воды при высоких температурах, чем при низких температурах. Следовательно, для удаления следового количества воды (побочный продукт) из реакционной смеси, содержащей большое избыточное количество используемого в качестве растворителя низкомолекулярного спирта, необходимо охлаждать реакционную смесь, в которой достигнуто равновесие в условиях высокой температуры и давления, до введения реакционной смеси в насадочную колонну, содержащую твердый осушитель. Кроме того, для повышения конверсии спирта в качестве исходного материала необходимо, чтобы реакционная смесь, которая была охлаждена и подвергнута осушке в насадочной колонне, была возвращена в условия высокой температуры и давления, требующиеся для реакции. Таким образом, в случае этого способа проблема состоит в том, что необходимо расходовать чрезвычайно большое количество энергии для охлаждения и нагревания реакционной смеси и требуется большое количество твердого осушителя. Этот способ очень широко используют для получения алифатического сложного эфира, имеющего относительно высокую константу равновесия. Однако при получении эфира угольной кислоты из диоксида углерода и спирта, где равновесие реакции в значительной степени смещено в сторону исходной системы, этот способ не достаточно эффективен, поскольку возникает серьезная проблема, заключающаяся в том, что необходимо повторять вышеуказанную операцию, вызывающую необходимость очень большого расхода энергии на охлаждение и нагревание. Далее, для регенерации подвергшегося "дегенерации" осушителя, который адсорбировал воду вплоть до насыщения, обычно его необходимо прокаливать при температуре в несколько сотен градусов Цельсия, что делает, таким образом, этот способ невыгодным в коммерческом отношении. Кроме того, в этом способе удаляют только один (вода) из двух продуктов равновесной реакции и, следовательно, проблема заключается в том, что, когда равновесная реакция протекает в направлении увеличения концентрации эфира угольной кислоты в реакционной системе, реакция становится маловероятной в отношении протекания в какой-либо степени, то есть этот способ прекращается при ограничении (нарушении) равновесной реакции. Кроме того, дибутилоловооксид, который используют в качестве катализатора в этом способе, имеет очень ничтожную растворимость в метаноле и, следовательно, почти весь дибутилоловооксид, используемый в качестве катализатора, остается в твердой форме в реакционной смеси. Следовательно, когда реакционную смесь охлаждают до комнатной температуры на стадии охлаждения, она превращается в суспензию белого цвета, вызывая, таким образом, проблему на последующей стадии обезвоживания, осуществляемой при использовании насадочной колонны, содержащей осушитель, поскольку суспензия вызывает закупорку насадочной колонны.
Вообще, способ обезвоживания, при котором воду удаляют путем отгонки, хорошо известен в области органического синтеза. Однако в области получения эфира угольной кислоты из диоксида углерода и спирта, несмотря на то что в "Study Report of Asahi Glass Association for Promotion of Industrial Technology (Asahi Garasu Kogyogijutsu Shoreikai Kenkyu Hokoku)", 33, 31-45 (1978), утверждают, что "в настоящее время исследовано обезвоживание путем перегонки", отсутствуют сообщения или нечто подобное, в которых констатируют, что способ обезвоживания, использующий перегонку, является совершенным. Причиной, почему обезвоживание путем перегонки не осуществляют согласно уровню техники, является, например, то, что известно, что, когда перегонку проводят при нагревании, протекает обратная реакция, вызывающая потерю эфира угольной кислоты (см. "Journal of the Chemical Society of Japan (Nippon Kagaku Kaishi)", №10, 1789-1794 (1975)). Возможно, что с целью снижения температуры перегонки перегонку осуществляют при пониженном давлении. Однако в области технологии перегонки общеизвестно, что трудно полностью удалить следовое количество воды путем простой отгонки из растворителя, содержащего гидрофильную группу, такого как спирт. Поэтому, в качестве способов обезвоживания неизвестен никакой другой способ обезвоживания, способы обезвоживания, в которых используют большое количество органического осушителя или большое количество твердого осушителя. То есть все известные способы обезвоживания связаны с проблемами, заключающимися в образовании большого количества отходов или неизбежном большом потреблении энергии.
Известно выделение эфира угольной кислоты из реакционной смеси, содержащей алкоголят металла путем отгонки, причем реакционную смесь получают взаимодействием диоксида углерода и спирта друг с другом в присутствии алкоголята металла в качестве катализатора; однако из уровня техники известно, что, когда используют алкоголят металла в качестве катализатора, выделение путем перегонки вызывает обратную реакцию, таким образом, представляется затруднительным извлекать эфир угольной кислоты посредством перегонки (см. "Journal of the Chemical Society of Japan (Nippon Kagaku Kaishi)", №10, 1789-1794 (1975)). По существу, неизвестен способ, с помощью которого эфир угольной кислоты, имеющий высокую температуру кипения, может быть выделен с высоким выходом из содержащей алкоголят металла реакционной смеси.
Алкоголят металла является настолько нестабильным, что он способен дезактивироваться при контакте с влагой воздуха. Следовательно, манипулирование с алкоголятом металла требует осторожности. По этой причине при коммерческом получении эфира угольной кислоты не используют общепринятую технологию, при которой применяют алкоголят металла в качестве катализатора. Алкоголят металла в качестве катализатора является дорогостоящим соединением и неизвестен способ регенерации используемого в качестве катализатора дезактивированного алкоголята металла.
Предложен способ получения эфира угольной кислоты, использующий в качестве катализатора дибутилоловодиалкоголят, причем катализатор образуется в процессе реакции из дибутилоловооксида (который устойчив к влаге), добавляемого в реакционную систему (см. патент Японии №3128576). Недостатком этого способа является то, что, несмотря на то что дибутилоловооксид, загружаемый в реакционную систему, является стабильным, в процессе реакции дибутилоловооксид превращается в нестабильный дибутилоловодиалкоголят. Следовательно, этот способ не позволяет решить вышеуказанную проблему нестабильности алкоголята металла, используемого в качестве катализатора. Далее, для превращения дибутилоловооксида в дибутилоловодиалкоголят в реакционной системе необходимо, чтобы реакционная система находилась в условиях высокой температуры и давления. Причиной этого является следующее. При образовании алкоголята из дибутилоловооксида выделяется вода и воду требуется израсходовать путем гидролиза ацеталя. Однако олово имеет только очень слабую кислотность, так что для катализа вышеуказанного гидролиза ацеталя требуются условия высокой температуры и давления.
Таким образом, в обычных способах получения эфира угольной кислоты путем использования алкоголята металла, диоксида углерода и спирта, алкоголят металла (который является дорогостоящим) теряет свою активность за счет гидролиза или тому подобного, и не существует способа, чтобы легко и эффективно регенерировать и повторно использовать алкоголят металла. Следовательно, обычные способы получения эфира угольной кислоты являются невыгодными в том отношении, что необходимо использование большого количества органического осушителя или твердого осушителя в сочетании с небольшим количеством алкоголята металла.
Как описано выше, способы получения эфира угольной кислоты согласно известному уровню техники связаны с множеством проблем и, следовательно, не могут быть предложены для практического использования.
Краткое изложение сущности изобретения
В этой ситуации, авторы настоящего изобретения провели обширные и интенсивные исследования для решения вышеуказанных проблем известного уровня техники. В результате неожиданно было найдено, что эфир угольной кислоты может быть получен с высоким выходом посредством способа, в котором используют содержащее металл-кислород-углеродную связь металлоорганическое соединение в большом количестве в качестве предшественника эфира угольной кислоты, а не в качестве катализатора, и металлоорганическое соединение подвергают реакции присоединения с диоксидом углерода с получением аддукта, с последующим термическим разложением аддукта, получая таким образом содержащую эфир угольной кислоты реакционную смесь. Далее, авторами настоящего изобретения также обнаружено, что путем осуществления последующей операции, при которой эфир угольной кислоты выделяют из реакционной смеси, получая остаточную жидкость, с последующим проведением реакции остаточной жидкости со спиртом, могут быть получены содержащее металл-кислород-углеродную связь металлоорганическое соединение и вода, причем вода может быть легко отделена от металлоорганического соединения путем перегонки или тому подобным образом. Полученное металлоорганическое соединение может быть рекуперировано и рециркулировано на вышеуказанный путь реакции для получения эфира угольной кислоты. На основании этих полученных данных настоящее изобретение является завершенным.
Таким образом, первым объектом настоящего изобретения является способ коммерческого получения эфира угольной кислоты с высоким выходом, который может быть осуществлен непрерывно и с многократным повторением, без образования отходов, которые образуются при использовании катализатора, и без необходимости использования большого количества осушителя.
Вышеуказанный объект и другие объекты, признаки и преимущества настоящего изобретения следуют из нижеприводимого подробного описания в сочетании с сопровождающими чертежами и прилагаемой формулой изобретения.
Краткое описание чертежей
На чертежах:
фиг.1 представляет собой 119Sn-ЯМР-спектр содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения, используемого на стадии (1) реакции согласно примеру 1;
фиг.2 представляет собой 119Sn-ЯМР-спектр металлоорганического соединения, получаемого сразу после стадии (2) 26-го цикла реакции согласно примеру 2;
фиг.3 представляет собой 119Sn-ЯМР-спектр металлоорганического соединения, получаемого сразу после стадии (3) 26-го цикла реакции согласно примеру 2;
фиг.4 представляет собой 119Sn-ЯМР-спектр содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения, используемого на стадии (1) реакции согласно примеру 3; и
фиг.5 представляет собой 119Sn-ЯМР-спектр металлоорганического соединения, получаемого сразу после стадии (1) реакции согласно примеру 3.
Подробное описание изобретения
Настоящее изобретение относится к способу получения эфира угольной кислоты, включающему:
(1) осуществление реакции между содержащим металл-кислород-углеродную связь металлоорганическим соединением и диоксидом углерода с получением реакционной смеси, содержащей образовавшийся путем реакции эфир угольной кислоты;
(2) выделение эфира угольной кислоты из реакционной смеси с получением остаточной жидкости; и
(3) введение во взаимодействие остаточной жидкости с первым спиртом с образованием по меньшей мере одного, содержащего металл-кислород-углеродную связь металлоорганического соединения и воды и удаление воды из по меньшей мере одного металлоорганического соединения, где по меньшей мере одно металлоорганическое соединение, полученное на стадии (3), рекуперируют для рециркуляции его на стадию (1).
Для удобства понимания настоящего изобретения ниже перечислены существенные признаки и различные предпочтительные варианты осуществления настоящего изобретения.
1. Способ получения эфира угольной кислоты, включающий:
(1) осуществление реакции между содержащим металл-кислород-углеродную связь металлоорганическим соединением и диоксидом углерода с получением реакционной смеси, содержащей образовавшийся путем реакции эфир угольной кислоты;
(2) выделение эфира угольной кислоты из реакционной смеси с получением остаточной жидкости; и
(3) введение во взаимодействие остаточной жидкости с первым спиртом с образованием по меньшей мере одного, содержащего металл-кислород-углеродную связь металлоорганического соединения и воды и удаление воды из по меньшей мере одного металлоорганического соединения, причем по меньшей мере одно металлоорганическое соединение, полученное на стадии (3), рекуперируют для рециркуляции его на стадию (1).
2. Способ согласно вышеуказанному п.1, где, на стадии (1), металлоорганическое соединение используют в количестве, представляющем собой 1/50-1-кратное стехиометрическое количество по отношению к количеству диоксида углерода.
3. Способ согласно вышеуказанному п.2, где реакцию на стадии (1) осуществляют при температуре 20°С или выше.
4. Способ согласно вышеуказанному п.1, где используемое на стадии (1) металлоорганическое соединение включает по меньшей мере одно соединение, выбираемое из группы, состоящей из:
металлоорганического соединения, отвечающего формуле (1):
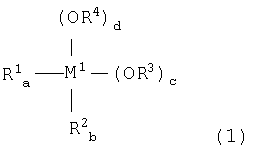
где:
М1 означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния;
каждый из R1 и R2, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью, (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, или незамещенную или замещенную (С6-С20)-арильную группу;
каждый из R3 и R4, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью или (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила; и
каждый из а и b означает целое число от 0 до 2, (а+b) = от 0 до 2; каждый из с и d означает целое число от 0 до 4; и (а+b+c+d)=4; и
металлоорганического соединения, отвечающего формуле (2):
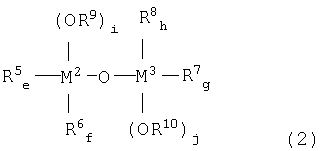
где:
каждый из М2 и М3, независимо, означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния;
каждый из R5, R6, R7 и R8, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью, (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, или незамещенную или замещенную (С6-С20)-арильную группу;
каждый из R9 и R10, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью или (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила; и
(e+f) = от 0 до 2; (g+h) = от 0 до 2; каждый из i и j, независимо, означает целое число от 1 до 3; (е+f+i)=3 и (g+h+j)=3.
5. Способ согласно вышеуказанному п.1, где реакцию на стадии (1) осуществляют в присутствии второго спирта, который является тем же самым или отличным от первого спирта, используемого на стадии (3).
6. Способ согласно вышеуказанному п.1, где выделение эфира угольной кислоты на стадии (2) осуществляют в присутствии третьего спирта, который является тем же самым или отличным от первого спирта, используемого на стадии (3).
7. Способ согласно вышеуказанному п.1, где выделение эфира угольной кислоты на стадии (2) осуществляют с помощью по меньшей мере одного способа выделения, выбираемого из группы, состоящей из перегонки, экстракции и фильтрации.
8. Способ согласно вышеуказанному п.1, где удаление воды на стадии (3) осуществляют путем мембранного отделения.
9. Способ согласно вышеуказанному п.8, где мембранным отделением является испарение через полупроницаемую перегородку.
10. Способ согласно вышеуказанному п.1, где удаление воды на стадии (3) осуществляют путем перегонки.
11. Способ согласно вышеуказанному п.1, где первым спиртом, используемым на стадии (3), является по меньшей мере один спирт, выбираемый из группы, состоящей из алкилового спирта, содержащего (С1-С12)-алкильную группу с линейной или разветвленной цепью; циклоалкилового спирта с (С5-С12)-циклоалкильной группой; алкенилового спирта, содержащего (С2-С12)-алкенильную группу с линейной или разветвленной цепью; и аралкилового спирта с (С7-С20)-аралкильной группой, включающей незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила.
12. Способ согласно вышеуказанному п.11, где каждый из алкилового спирта, циклоалкилового спирта, алкенилового спирта и аралкилового спирта имеет температуру кипения выше, чем температура кипения воды.
13. Способ согласно вышеуказанному п.12, где алкиловый спирт включает по меньшей мере один спирт, выбираемый из группы, состоящей из н-бутилового спирта, изобутилового спирта и алкилового спирта, содержащего (С5-С12)-алкильную группу с линейной или разветвленной цепью, и алкенилового спирта, содержащего (С4-С12)-алкенильную группу с линейной или разветвленной цепью.
14. Способ согласно вышеуказанному п.4, где каждый из R3 и R4 в формуле (1) и R9 и R10 в формуле (2), независимо, означает н-бутильную группу, изобутильную группу, (С5-С12)-алкильную группу с линейной или разветвленной цепью или (С4-С12)-алкенильную группу с линейной или разветвленной цепью.
15. Способ согласно вышеуказанному п.4 или 14, где, на стадии (1), металлоорганическое соединение используют по меньшей мере в одной форме, выбираемой из мономерной формы, олигомерной формы, полимерной формы и ассоциированной формы.
16. Способ согласно вышеуказанному п.4 или 14, где каждый из М1 в формуле (1) и М2 и М3 в формуле (2) означает атом олова.
17. Способ согласно любому из вышеуказанных пп.1-16, который включает, далее, после стадии (3), стадию (4), на которой по меньшей мере одно металлоорганическое соединение, рекуперированное на стадии (3), рециркулируют на стадию (1) с последующим повторением последовательности стадий (1)-(4) один или более раз.
18. Способ согласно вышеуказанному п.17, где металлоорганическое соединение, используемое на стадии (1), получают из органооловооксида и спирта.
Настоящее изобретение описывается подробно ниже.
Как описано выше, в обычных способах получения эфира угольной кислоты используют равновесную реакцию, представляемую следующим уравнением (3):
(R означает ненасыщенную или насыщенную углеводородную группу)
То есть в качестве обычных способов можно указать способ, в котором используют осушитель для реакционной смеси, содержащей равновесную реакционную систему (представленную вышеприведенным уравнением (3)), где равновесная реакционная система содержит смесь продуктов, включающую эфир угольной кислоты и воду; и способ, в котором реакционную смесь, содержащую вышеуказанную равновесную реакционную систему, охлаждают и подвергают осушке, при которой реакционную смесь вводят в насадочную колонну, содержащую твердый осушитель, и подвергают циркуляции через насадочную колонну с тем, чтобы постепенно осушать равновесную реакционную систему, таким образом подавляя реакцию разложения катализатора и накопление образующегося в следовом количестве эфира угольной кислоты.
С другой стороны, техническая концепция способа согласно настоящему изобретению полностью отлична от технической концепции обычных способов.
Способ согласно настоящему изобретению характеризуется тем, что:
в реакции используют в большом количестве содержащее металл-кислород-углеродную связь металлоорганическое соединение в качестве предшественника эфира угольной кислоты, а не в качестве катализатора, причем и металлоорганическое соединение подвергают реакции присоединения с диоксидом углерода с образованием аддукта, с последующим термическим разложением аддукта, получая таким образом содержащую эфир угольной кислоты реакционную смесь (стадия (1));
за стадией (1) следует операция, посредством которой эфир угольной кислоты выделяют из реакционной смеси, получая остаточную жидкость (стадия (2)); и
за стадией (2) следует взаимодействие остаточной жидкости со спиртом, в результате которого получают реакционную смесь, включающую содержащее металл-кислород-углеродную связь металлоорганическое соединение и воду, с последующим удалением воды из реакционной смеси путем перегонки или тому подобным образом, получая таким образом металлоорганическое соединение, с последующей рекуперацией полученного металлоорганического соединения (стадия (3)); и
с последующей рециркуляцией его на стадию (1) для получения эфира угольной кислоты.
Реакция на стадии (1) способа согласно настоящему изобретению представлена в виде нижеприводимого уравнения (4). Реакция на стадии (3) способа согласно настоящему изобретению представлена в виде нижеприводимого уравнения (5).


Таким образом, предлагаемый в настоящем изобретении способ представляет собой способ, согласно которому содержащее металл-кислород-углеродную связь металлоорганическое соединение используют главным образом в качестве предшественника эфира угольной кислоты и металлоорганическое соединение подвергают реакции присоединения с диоксидом углерода с образованием аддукта, после которой следует реакция термического разложения аддукта с получением, таким образом, содержащей эфир угольной кислоты реакционной смеси, после чего эфир угольной кислоты выделяют из реакционной смеси, получая остаточную жидкость, с последующим осуществлением операции, согласно которой остаточную жидкость (содержащую метаморфное металлоорганическое соединение, образовавшееся за счет реакции термического разложения аддукта) вводят во взаимодействие со спиртом для регенерировани, таким образом, содержащего металл-кислород-углеродную связь металлоорганического соединения. Регенерированное металлоорганическое соединение рекуперируют и рециркулируют на стадию получения эфира угольной кислоты, и цикл этих стадий повторяют с тем, чтобы получить эфир угольной кислоты в желательном количестве.
Реакционная смесь, полученная на стадии (1) способа согласно настоящему изобретению, может содержать или нет остаточную часть содержащего металл-кислород-углеродную связь металлоорганического соединения, используемого на стадии (1). Также, остаточная жидкость, полученная на стадии (2) способа согласно настоящему изобретению, может содержать или нет остаточную часть содержащего металл-кислород-углеродную связь металлоорганического соединения, используемого на стадии (1). В любом случае, содержащее металл-кислород-углеродную связь металлоорганическое соединение регенерируют (ресинтезируют) до завершения стадии (3).
Согласно обычным способам, в которых используют равновесную реакцию согласно вышеприведенному уравнению (3), всю реакцию полностью проводят при равновесии. Напротив, согласно способу настоящего изобретения равновесная реакция согласно вышеприведенному уравнению (3) может быть эффективно разделена на последовательные реакции, которые можно легко контролировать, позволяя, таким образом, эффективно получить эфир угольной кислоты при выделении эфира угольной кислоты и воды из реакционной системы. В частности, на стадии (1) способа согласно настоящему изобретению реакция может быть осуществлена в отсутствие воды. На стадии (2) способа согласно настоящему изобретению обратная реакция эфира угольной кислоты и других продуктов термического разложения может быть предотвращена путем отделения эфира угольной кислоты от реакционной смеси. На стадии (3) способа согласно настоящему изобретению после регенерации содержащего металл-кислород-углеродную связь металлоорганического соединения, металлоорганическое соединение может быть рекуперировано путем удаления воды. Далее, на каждой стадии способа согласно настоящему изобретению рабочие условия могут быть легко оптимизированы путем соответствующего использования обычных технологических приемов химического синтеза, таких как охлаждение, нагревание, перемешивание, повышение и снижение давления.
В качестве примера содержащего металл-кислород-углеродную связь металлоорганического соединения, используемого на стадии (1) способа согласно настоящему изобретению, можно указать содержащее алкоксильную группу металлоорганическое соединение. Предпочтительно, что используемое на стадии (1) металлоорганическое соединение включает по меньшей мере одно соединение, выбираемое из группы, состоящей из:
металлоорганического соединения, отвечающего формуле (1):
где:
М1 означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния;
каждый из R1 и R2, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью, (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, или незамещенную или замещенную (С6-С20)-арильную группу;
каждый из R3 и R4, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью или (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, и
каждый из а и b означает целое число от 0 до 2, (а+b) = от 0 до 2; каждый из с и d означает целое число от 0 до 4; и (а+b+c+d)=4; и
металлоорганического соединения, отвечающего формуле (2):
где:
каждый из М2 и М3, независимо, означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния;
каждый из R5, R6, R7 и R8, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью, (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, или незамещенную или замещенную (С6-С20)-арильную группу;
каждый из R9 и R10, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью или (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила; и
(e+f) = от 0 до 2; (g+h) = от 0 до 2; каждый из i и j, независимо, означает целое число от 1 до 3; (е+f+i)=3 и (g+h+j)=3.
Указанная периодическая таблица является такой, как описанная в системе номенклатуры IUPAC (International Union of Pure and Applied Chemistry) (1989).
Вышеуказанное металлоорганическое соединение используют по меньшей мере в одной форме, выбираемой из группы, состоящей из мономерной формы, олигомерной формы, полимерной формы и ассоциированной формы.
Каждый из М1 в формуле (1) и М2 и М3 в формуле (2), независимо, означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния. Предпочтительно, что каждый из М1, М2 и М3 означает атом металла, выбираемый из группы, состоящей из атома титана, атома олова и атома циркония. С точки зрения растворимости в спирте и реакционной способности по отношению к нему, более предпочтительно, что каждый из М1, М2 и М3 означает атом олова.
Примеры R1 и R2 в формуле (1) и R5, R6, R7 и R8 в формуле (2) включают алифатические углеводородные группы с 1-12 атомами углерода и алициклические углеводородные группы с 5-12 атомами углерода, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, 2-бутенил, пентил, гексил, циклопропил, циклобутил, циклопентил, циклопентадиенил, циклогексил и циклогексенил; аралкильные группы с 7-20 атомами углерода, такие как бензил и фенилэтил; и арильные группы с 6-20 атомами углерода, такие как фенил, толил и нафтил. Значения R1, R2, R5, R6, R7 и R8 не ограничиваются этими примерами. Из вышеуказанных групп предпочтительны низшие алкильные группы и более предпочтительно (С1-С4)-алкильные группы с линейной или разветвленной цепью. В качестве R1, R2, R5, R6, R7 и R8 могут быть использованы группы, содержащие большее число атомов углерода, чем указанное выше; однако, когда используют такие группы с гораздо большим числом атомов углерода, иногда может оказаться, что текучесть металлоорганического соединения и/или производительность в отношении эфира угольной кислоты становится низкой. Примеры R3 и R4 в формуле (1) и R9 и R10 в формуле (2) включают алифатические углеводородные группы с 1-12 атомами углерода и алициклические углеводородные группы с 5-12 атомами углерода, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, 2-бутенил, пентил, гексил, циклопропил, циклобутил, циклопентил, циклопентадиенил, циклогексил, циклогексенил, метоксиэтил и этоксиметил; и аралкильные группы с 7-20 атомами углерода, такие как бензил и фенилэтил. Значения R3, R4, R9 и R10 не ограничиваются этими примерами.
Примеры металлоорганических соединений, отвечающих вышеприведенной формуле (1), включают тетраметилат олова, тетраэтилат олова, тетрапропилат олова, тетрабутилат олова, тетрапентилат олова, тетрагексилат олова, тетракис(2-этил-1-гексилокси)-олово, диэтоксидиметоксиолово, тетраметилтитанат, тетраэтилтитанат, тетрапропилтитанат, тетраизопропилтитанат, диметилоловодиметилат, диметилоловодиэтилат,
2-этил-1-гексилоксиметокси-диметилолово, диметилоловодипропилат,
диметилоловодибутилат, диметилоловобис(2-этил-1-бутилат),
диметилоловодипентилат, диметилоловодигексилат,
диметилоловодициклогексилат, диметилоловобис(2-этил-1-гексилат),
диметилоловодипропенилат, диметилоловодибензилат,
метилбутилоловодиметилат, метилбутилоловодиэтилат,
2-этил-1-гексилоксиметоксиметилбутилолово, метилбутилоловодипропилат, метилбутилоловодибутилат,
метилбутилоловобис(2-этил-1-бутилат), метилбутилоловодипентилат,
метилбутилоловодигексилат, метилбутилоловодициклогексилат,
метилбутилоловобис(2-этил-1-гексилат), метилбутилоловодипропенилат, метилбутилоловодибензилат, метил(2-этилгексил)оловодиметилат, метил(2-этил-гексил)оловодиэтилат,
2-этил-1-гексилоксиметоксиметил(2-этил-гексил)олово, метил(2-этилгексил)оловодипропилат, метил(2-этил-гексил)оловодибутилат,
метил(2-этилгексил)оловобис(2-этил-1-бутилат), метил(2-этилгексил)оловодипентилат, метил(2-этил-гексил)оловодигексилат, метил(2-этилгексил)оловодициклогексилат,
метил(2-этилгексил)оловобис(2-этил-1-гексилат),
метил(2-этилгексил)оловодипропенилат,
метил(2-этилгексил)оловодибензилат,
бутил(2-этилгексил)оловодиметилат, бутил(2-этилгексил)-оловодиэтилат, 2-этил-1-гексилоксиметоксибутил(2-этилгексил)-олово, бутил(2-этилгексил)оловодипропилат, бутил(2-этилгексил)-оловодибутилат, бутил(2-этилгексил)оловобис(2-этил-1-бутилат), бутил(2-этилгексил)оловодипентилат,
бутил(2-этилгексил)оловодигексилат,
бутил(2-этилгексил)оловодициклогексилат,
бутил(2-этилгексил)оловобис(2-этил-1-гексилат),
бутил(2-этилгексил)-оловодипропенилат,
бутил(2-этилгексил)оловодибензилат, ди(н-бутил)оловодиметилат, ди(н-бутил)оловодиэтилат, 2-этил-1-гексилоксиметоксиди(н-бутил)олово, ди(н-бутил)оловодипропилат, ди(н-бутил)оловодибутилат, ди(н-бутил)оловобис(2-этил-1-бутилат), ди(н-бутил)оловодипентилат, ди(н-бутил)оловодигексилат, ди(н-бутил)оловодициклогексилат, ди(н-бутил)оловобис(2-этил-1-гексилат), ди(н-бутил)оловодипропенилат, ди(н-бутил)олово-дибензилат, ди(трет-бутил)оловодиметилат, ди(трет-бутил)олово-диэтилат, ди(трет-бутил)оловодипропилат, ди(трет-бутил)олово-дибутилат, ди(трет-бутил)оловодипентилат, ди(трет-бутил)олово-дигексилат, ди(трет-бутил)оловодициклогексилат, ди(трет-бутил)-оловодипропенилат, дифенилоловодиметилат, дифенилоловодиэтилат, дифенилоловодипропилат, дифенилоловодибутилат, дифенилоловобис(2-этил-1-бутилат), дифенилоловодипентилат, дифенилоловодигексилат, дифенилоловобис(2-этил-1-гексилат), дифенилоловодициклогексилат, дифенилоловодипропенилат и дифенилоловодибензилат.
Примеры металлоорганических соединений, отвечающих вышеприведенной формуле (2), включают алкоксидистанноксаны и аралкилоксидистанноксаны, такие как
1,1,3,3-тетрабутил-1,3-диметоксидистанноксан;
1,1,3,3-тетрабутил-1-метокси-3-(2-этил-1-гексилокси)дистанноксан;
1,1,3,3-тетрабутил-1,3-диэтокси-дистанноксан;
1,1,3,3-тетрабутил-1,3-дибутоксидистанноксан;
1,1,3,3-тетрабутил-1,3-бис(2-этил-1-бутокси)дистанноксан;
1,1,3,3-тетрабутил-1,3-дипропоксидистанноксан;
1,1,3,3-тетрабутил-1,3-дипентилоксидистанноксан;
1,1,3,3-тетрабутил-1,3-дигексилоксидистанноксан;
1,1,3,3-тетрабутил-1,3-бис(2-этил-1-гексилокси)дистанноксан;
1,1,3,3-тетрабутил-1,3-дициклогексилоксидистанноксан;
1,1,3,3-тетрабутил-1,3-дибензилоксидистанноксан;
1,1,3,3-тетрафенил-1,3-диметоксидистанноксан;
1,1,3,3-тетрафенил-1,3-диэтоксидистанноксан;
1,1,3,3-тетрафенил-1,3-дибутоксидистанноксан;
1,1,3,3-тетрафенил-1,3-бис(2-этил-1-бутокси)дистанноксан;
1,1,3,3-тетрафенил-1,3-дипропоксидистанноксан;
1,1,3,3-тетрафенил-1,3-дипентилоксидистанноксан;
1,1,3,3-тетрафенил-1,3-дигексилоксидистанноксан;
1,1,3,3-тетрафенил-1,3-бис(2-этил-1-гексилокси)дистанноксан
и 1,1,3,3-тетрафенил-1,3-дициклогексилоксидистанноксан.
Вышеуказанные металлоорганические соединения могут быть использованы индивидуально или в сочетании. Далее, металлоорганические соединения, отличные от указанных выше, могут быть использованы в сочетании с вышеуказанными соединениями. В качестве металлоорганического соединения могут быть использованы коммерчески доступные металлоорганические соединения. Альтернативно, металлоорганические соединения, отвечающие вышеприведенной формуле (1), могут быть получены обычным способом (как, например, способ, описанный в патенте Нидерландов №6612421), согласно которому дибутилоловооксид, спирт с 4 или более атомами углерода и растворитель, проявляющий азеотропию с водой, смешивают вместе для осуществления реакции и продукт реакции подвергают перегонке, получая таким образом фракцию, содержащую металлоорганическое соединение, отвечающее вышеприведенной формуле (1). В вышеуказанном патентном документе (то есть патенте Нидерландов №6612421) описывается, что этот способ не может быть использован для получения металлоорганического соединения, содержащего (С1-С3)-алкоксильную группу, и что металлоорганическое соединение, содержащее (С1-С3)-алкоксильную группу, может быть получено из дибутилоловохлорида и алкоголята натрия. С другой стороны, в результате исследований авторов настоящего изобретения, найдено, что способом, описанным в заявке на патент Японии №2001-396537 и заявке на патент Японии №2001-396545, металлоорганическое соединение формулы (1) или (2), может быть получено из оксида металла и спирта. Этим способом может быть получено металлоорганическое соединение, содержащее (С1-С3)-алкоксильную группу, такую как метоксигруппа. Например, содержащее метоксигруппу металлоорганическое соединение может быть получено из дибутилоловооксида, метанола и гексана. Известно, что метанол и гексан образуют азеотроп с минимальной температурой кипения. Однако способом настоящего изобретения можно осуществлять удаление воды даже несмотря на то, что смесь метанола и гексана имеет температуру кипения ниже температуры кипения воды. Таким образом, авторы настоящего изобретения разработали способ получения металлоорганического соединения из спирта, имеющего температуру кипения ниже температуры воды. Металлоорганическое соединение, получаемое из дибутилоловооксида и спирта с температурой кипения ниже температуры воды, представляет собой металлоорганическое соединение формулы (2). Однако, если желательно получать большое количество металлоорганического соединения формулы (1), вышеуказанное металлоорганическое соединение общей формулы (2) подвергают перегонке, получая, таким образом, фракцию, включающую металлоорганическое соединение формулы (1).
Согласно предлагаемому в настоящем изобретении способу удаление воды на стадии (3) может быть осуществлено любым обычным способом обезвоживания, который обычно используют согласно уровню техники. Удаление воды может быть осуществлено, например, путем использования твердого осушителя (как, например, молекулярные сита), перегонки или мембранного отделения. Однако, если желательно получать большое количество металлоорганического соединения за короткий период времени, предпочтительно, чтобы удаление воды было осуществлено путем перегонки. Перегонку можно осуществлять любым обычным способом перегонки, таким как перегонка при атмосферном давлении, вакуумная перегонка, перегонка при давлении выше атмосферного, перегонка в тонком слое или экстракционная перегонка. Перегонку можно осуществлять при температуре от -20°С до температуры кипения первого спирта, используемого на стадии (3), предпочтительно при температуре от 50°С до температуры кипения первого спирта, используемого на стадии (3). До или во время перегонки к реакционной смеси может быть добавлено любое желательное вещество.
На стадии (1) способа согласно настоящему изобретению необязательно может быть использован второй спирт (что касается предложения в отношении использования второго спирта, объяснение приводится ниже). Когда намереваются использовать второй спирт на стадии (1), можно указать технологическую операцию, согласно которой, до стадии (1), спирт используют для получения металлоорганического соединения формулы (1) и/или металлоорганического соединения формулы (2) и последующую перегонку для удаления воды из полученной в результате реакции смеси осуществляют таким образом, чтобы часть спирта оставалась в остатке от перегонки, содержащем металлоорганическое соединение. Таким путем может быть получено металлоорганическое соединение для использования на стадии (1) в виде его смеси со спиртом, который может быть использован в качестве, по меньшей мере, части второго спирта на стадии (1). В этом случае становится возможным осуществление стадии (1) без дальнейшей добавки спирта в качестве второго спирта.
Согласно предлагаемому в настоящем изобретении способу, первый спирт используют на стадии (3). Кроме того, на стадии (1) необязательно может быть использован второй спирт и на стадии (2) необязательно может быть использован третий спирт. Первый, второй и третий спирты могут быть одними и теми же или разными. Примеры таких спиртов включают алкиловые спирты, содержащие (С1-С12)-алкильную группу с линейной или разветвленной цепью, циклоалкиловые спирты, содержащие (С5-С12)-циклоалкильную группу; алкениловые спирты, содержащие (С2-С12)-алкенильную группу с линейной или разветвленной цепью; и аралкиловые спирты, содержащие (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила. Конкретные примеры этих спиртов включают алифатические спирты с 1-12 атомами углерода, такие как метанол, этанол, пропанол, 2-пропанол, н-бутанол, 2-бутанол, 2-этил-1-бутанол, трет-бутанол, пентанол, гексанол, 2-этил-1-гексанол и гексенол; алициклические спирты с 5-12 атомами углерода, такие как циклопропанол, циклобутанол, циклопентанол, циклогексанол и циклогексенол; и аралкиловые спирты, такие, как бензиловый спирт и фенетиловый спирт. Далее, в качестве первого, второго и третьего спиртов могут быть использованы многоатомные спирты. Примеры многоатомных спиртов включают алифатические многоатомные спирты с 1-12 атомами углерода, такие как этиленгликоль, 1,3-пропандиол и 1,2-пропандиол; алициклические многоатомные спирты с 5-12 атомами углерода, такие как циклогександиол и циклопентандиол; и аралкиловые спирты, такие как бензолдиметанол.
Из вышеуказанных спиртов предпочтительны первичные или вторичные одноатомные спирты с 1-8 атомами углерода, такие как метанол, этанол, пропанол, 2-пропанол, бутанол, 2-бутанол, 2-этил-1-бутанол, пентанол, гексанол, 2-этил-1-гексанол, циклогексанол и гексенол; и первичные или вторичные аралкиловые спирты с 7-8 атомами углерода, такие как бензиловый спирт.
Металлоорганические соединения, отвечающие, соответственно, формуле (1) и формуле (2), можно анализировать с помощью спектроскопии ядерного магнитного резонанса 119Sn-ЯМР (см., например, патент США №5545600). Однако в 119Sn-ЯМР-спектре величина химического сдвига, соответствующая структуре металлоорганического соединения формулы (1), в значительной степени изменяется в зависимости, например, от содержания металлоорганического соединения в образце, который используют для 119Sn-ЯМР-анализа, и от присутствия или отсутствия спирта в образце, который используют для 119Sn-ЯМР-анализа. Следовательно, предпочтительно, чтобы анализ металлоорганического соединения проводили по способу, в случае которого в сочетании с 119Sn-ЯМР-спектроскопией используют спектроскопию протонного ядерного магнитного резонанса (1Н-ЯМР) и спектроскопию ядерного магнитного резонанса углерода-13 (13С-ЯМР). В нижеприводимой таблице 1 представлены данные 119Sn-ЯМР примеров величин химического сдвига, приписываемых структуре металлоорганического соединения, отвечающего формуле (1), которое получено из 2-этил-1-гексанола и дибутилоловооксида.
Содержание металлоорганического соединения, содержащего 2-этил-1-гексилоксигруппу, формулы (1), в растворах образцов и величины химического сдвига, полученные при анализе растворов образцов
Согласно предлагаемому в настоящем изобретении способу используемая на стадии (1) реакционная система может содержать вещества, другие, чем вышеуказанные. Примеры других веществ, которые эффективны при осуществлении стадии (1), включают вещества, которые действуют как осушитель в реакционной системе. Путем использования осушителя на стадии (1) реакционную систему можно поддерживать в неводных условиях. В качестве осушителя может быть использован любой осушитель. Примеры осушителей включают ацетали и сложные орто-эфиры, такие как ортотриметилацетат. Далее, в качестве осушителей могут быть использованы органические осушители, такие как дициклогексилкарбодиимид. Далее, в качестве осушителей могут быть использованы твердые осушители, такие как молекулярные сита. Когда используют твердый осушитель, предпочтительно, чтобы твердый осушитель был удален из реакционной системы до осуществления стадии (3).
На стадии (1) способа согласно настоящему изобретению использование второго спирта является необязательным. Что касается количества второго спирта, используемого на стадии (1), когда в качестве второго спирта используют спирт, содержащий органическую группу, которая является такой же, как алкоксильная или аралкоксильная группа в металлоорганическом соединении, с точки зрения повышения чистоты получаемого эфира угольной кислоты предпочтительно, чтобы второй спирт был использован в количестве, равном 1-100000-кратному стехиометрическому количеству по отношению к количеству металлоорганического соединения. Альтернативно, когда в качестве второго спирта используют спирт, содержащий органическую группу, отличную от алкоксильной или аралкоксильной группы в металлоорганическом соединении, или когда используют одно металлоорганическое соединение формулы (2), количество второго спирта предпочтительно составляет 2-1000-кратное стехиометрическое количество, более предпочтительно 10-1000-кратное стехиомерическое количество, по отношению к количеству металлоорганического соединения. На стадии (1), когда в качестве второго спирта используют спирт, содержащий органическую группу, отличную от органической группы в металлоорганическом соединении, получают асимметричный эфир угольной кислоты. Как указывается ниже, когда на стадии (1) используют второй спирт, в частности в случае, где используют одно металлоорганическое соединение, отвечающее формуле (2), значительно повышается выход эфира угольной кислоты. Таким образом, если используют одно металлоорганическое соединение формулы (2), вышеуказанное предпочтительное количество второго спирта определяют как указано выше.
Если за стадией (3) следуют стадия (4) (для рециркуляции рекуперированного на стадии (3) металлоорганического соединения на стадию (1)) и стадия (1), до ее осуществления, к реакционной системе может быть добавлен второй спирт в таком количестве, чтобы оно находилось в пределах вышеуказанной предпочтительной области. Альтернативно, в том случае, если за стадией (3) следуют стадия (4) и стадия (1), до ее осуществления, спирт может быть удален из реакционной системы.
Что касается каждой стадии способа согласно настоящему изобретению, ниже приводятся подробные объяснения.
Стадия (1) способа согласно настоящему изобретению включает получение аддукта диоксида углерода и металлоорганического соединения, содержащего металл-кислород-углеродную связь, и последующее термическое разложение образовавшегося аддукта с получением эфира угольной кислоты. То есть, на стадии (1), диоксид углерода вступает в реакцию присоединения с металлоорганическим соединением с образованием аддукта, и аддукт подвергают термическому разложению. В отличие от обычных способов стадия (1) способа согласно настоящему изобретению заключается в том, что содержащее металл-кислород-углеродную связь металлоорганическое соединение реагирует с небольшим стехиометрическим количеством диоксида углерода. В традиционных способах диоксид углерода при высоком давлении реагирует со спиртом в присутствии небольшого количества металлического катализатора. В качестве примера такого способа можно указать способ, в котором диоксид углерода реагирует с метанолом в присутствии дибутилоловодиметилата (см. Polyhedron, 19, 573-576 (2000)). Согласно указанному способу диоксид углерода взаимодействует с метанолом при давлении около 30 МПа при температуре 180°С в присутствии нескольких миллимоль дибутилоловодиметилата. В вышеуказанном литературном источнике не указывается точное количество диоксида углерода, используемого в реакции. Однако считают, что даже если вычесть парциальное давление метанола, количество используемого в реакции диоксида углерода должно составлять, по меньшей мере, 100-кратное стехиометрическое количество по отношению к количеству металлоорганического соединения, содержащего металл-кислород-углеродную связь. В вышеуказанных условиях высокого давления равновесие реакции значительно сдвигается в сторону образования эфира угольной кислоты, поэтому эфир угольной кислоты может быть получен с выходом, превышающим ожидаемый в расчете на количество катализатора. Однако в результате реакции диоксида углерода с метанолом также образуется свободная вода, создавая таким образом серьезную проблему за счет гидролиза свободной водой катализатора. Для решения этой проблемы необходимо разработать способ обезвоживания реакционной системы. В вышеуказанном литературном источнике описывается, что в упомянутых реакционных условиях дибутилоловооксид получают в виде продукта гидролиза дибутилоловодиметилата и несмотря на то, что дибутилоловооксид не растворяется в растворителе при комнатной температуре, дибутилоловооксид в вышеуказанных реакционных условиях находится в виде прозрачного раствора. С другой стороны, в способе согласно настоящему изобретению, даже когда реакционную смесь после окончания стадии (1) охлаждают до комнатной температуры, оно обычно остается в виде жидкости. В этом отношении реакция, используемая в способе согласно настоящему изобретению, отличается от реакции, раскрытой в вышеуказанном обычном способе, использующем большое количество диоксида углерода. В традиционном способе, реакционная система содержит высокую концентрацию диоксида углерода и, следовательно, реакцию необходимо осуществлять в условиях высокого давления. Поэтому, когда реакционную смесь, содержащую полученный эфир угольной кислоты, извлекают из реактора, необходимо удалять большое количество диоксида углерода из реактора до извлечения реакционной смеси. Такая необходимость вызывает проблемы не только в том, что бесполезно расходуется большое количество диоксида углерода, но и также в том, что при повторном использовании удаленного диоксида углерода необходимо восстанавливать давление диоксида углерода и, следовательно, расходуется большое количество энергии на восстановление давления диоксида углерода. Далее, согласно обычному (традиционному) способу также вероятна следующая проблема. Известно, что, когда реакционная система содержит высокую концентрацию диоксида углерода, увеличивается плотность слоя газообразного диоксида углерода, таким образом диоксид углерода растворяет не только растворитель и катализатор, но и также полученный эфир угольной кислоты, образуя таким образом реакционную смесь, представляющую собой гомогенную смесь диоксида углерода, растворителя, катализатора и полученного эфира угольной кислоты. Когда реакционную смесь (гомогенная смесь) охлаждают, получая жидкую реакционную смесь, последняя содержит диоксид углерода в виде жидкого диоксида углерода. Таким образом, чрезвычайно трудно выделять полученный эфир угольной кислоты из реакционной смеси.
На стадии (1) способа согласно настоящему изобретению предпочтительно использовать диоксид углерода в количестве, составляющем 1-50-кратное, более предпочтительно 1-20-кратное, стехиометрическое количество по отношению к количеству металлоорганического соединения, содержащего металл-кислород-углеродную связь. При большом количестве диоксида углерода реакция протекает при высоком давлении, поэтому необходимо не только использовать реактор, устойчивый к высокому давлению, но при этом теряется большое количество диоксида углерода во время удаления непрореагировавшего диоксида углерода после окончания стадии (1). Следовательно, все же более предпочтительно использовать диоксид углерода в количестве, составляющем 1-10-кратное стехиометрическое количество по отношению к количеству металлоорганического соединения. Другими словами, на стадии (1) предпочтительно использовать металлоорганическое соединение в количестве, составляющем 1/50-1-кратное, более предпочтительно 1/20-1-кратное, еще более предпочтительно 1/10-1-кратное, стехиометрическое количество по отношению к количеству диоксида углерода. Согласно настоящему изобретению аддукт диоксида углерода и содержащего металл-кислород-углеродную связь металлоорганического соединения может быть легко получен путем введения в контакт металлоорганического соединения с диоксидом углерода. При проведении реакции при комнатной температуре (20°С) аддукт диоксида углерода образуется экзотермически за счет контактирования металлоорганического соединения с потоком диоксида углерода, находящегося при атмосферном давлении. В этом случае аддукт диоксида углерода может быть получен с выходом почти 100%. В соответствии с повышением температуры реакции количество получаемого аддукта диоксида углерода уменьшается; однако, даже когда температура реакции является высокой, уменьшение количества аддукта диоксида углерода можно предотвращать за счет контактирования металлоорганического соединения с диоксидом углерода, находящимся при высоком давлении. На стадии (1), когда металлоорганическое соединение вводят в контакт с диоксидом углерода, находящимся при высоком давлении, трудно определить количество получаемого аддукта диоксида углерода; однако предпочтительным является осуществление реакции металлоорганического соединения с диоксидом углерода при желательном давлении, зависящем от скорости, с которой образуется эфир угольной кислоты, и от количества получаемого эфира угольной кислоты. Давление реакции обычно составляет от атмосферного до 200 МПа. Предпочтительно, чтобы количество получаемого на стадии (1) эфира угольной кислоты составляло 100% или меньше, более предпочтительно 50% или меньше, на основе стехиометрического количества по отношению к количеству металлоорганического соединения, содержащего металл-кислород-углеродную связь. Причиной этого является следующая. Содержащее металл-кислород-углеродную связь металлоорганическое соединение, используемое в способе согласно настоящему изобретению, более способно к гидролизу, чем получаемый эфир угольной кислоты. Следовательно, когда эфир угольной кислоты получают в количестве, составляющем 100% или меньше, предпочтительно 50% или меньше, на основе стехиометрического количества по отношению к количеству металлоорганического соединения воды, которая, вероятно, может гидролизовать получаемый эфир угольной кислоты, преимущественно нет в реакционной смеси. С другой стороны, в случае обычных способов реакцию проводят так, чтобы количество получаемого эфира угольной кислоты было более чем 100%, на основании стехиометрического количества по отношению к количеству металлоорганического соединения. В результате, в случае обычных способов образование свободной воды, которая, вероятно, может гидролизовать получаемый эфир угольной кислоты, вызывает серьезную проблему. Для предохранения получаемого эфира угольной кислоты от гидролиза необходимо добавлять к реакционной системе осушитель или проводить реакцию в присутствии осушителя, где осушитель выбирают из группы, состоящей из осушителя, который более способен гидролизоваться, чем металлоорганическое соединение, и твердого осушителя, обладающего высокой адсорбционной способностью по отношению к воде. Такое использование осушителя невыгодно не только потому, что необходима усложненная стадия, но и также потому, что осушитель является дорогостоящим. Следовательно, обычные способы не могут быть использованы на практике в качестве способа получения эфира угольной кислоты в коммерческом масштабе. Напротив, реакция на стадии (1) способа согласно настоящему изобретению, в основном является реакцией разложения, при которой аддукт диоксида углерода и содержащего металл-кислород-углеродную связь металлоорганического соединения подвергают термическому разложению, получая эфир угольной кислоты. Реакцию термического разложения осуществляют при температуре в интервале от 20 до 300°С. На стадии (1) способа согласно настоящему изобретению реакция обмена спиртовой группы, или реакция переэтерификации, может быть осуществлена вместе с вышеуказанной реакцией разложения. Конкретно, например, когда стадию (1) осуществляют в присутствии второго спирта, реакция обмена спиртовой группы протекает между кислород-углеродной связью второго спирта и кислород-углеродной связью металлоорганического соединения, содержащего металл-кислород-углеродную связь, таким образом может быть получен эфир угольной кислоты соответствующего второго спирта. Альтернативно, после образования эфира угольной кислоты к реакционной системе может быть добавлен второй спирт для осуществления реакции переэтерификации, получая, таким образом, другой эфир угольной кислоты соответствующего второго спирта.
Что касается стадии (1), ниже приводятся более подробные объяснения.
Исследования авторов настоящего изобретения показали, что на стадии (1) эфир угольной кислоты получают путем реакции между металлоорганическим соединением и диоксидом углерода. Следовательно, использование второго спирта на стадии (1) является необязательным. Однако с целью получения эфира угольной кислоты с высоким выходом на стадии (1) предпочтительным является использование второго спирта. Причиной этого является следующее. Реакция термического разложения на стадии (1) является обратимой реакцией. Когда к реакционной системе добавляют второй спирт, возможно, что дополнительно протекает другая равновесная реакция между вторым спиртом и продуктом термического разложения, другим, чем эфир угольной кислоты, повышая таким образом выход эфира угольной кислоты. Добавление второго спирта для повышения выхода эфира угольной кислоты является особенно эффективным, когда металлоорганическое соединение представляет собой главным образом металлоорганическое соединение, отвечающее формуле (2). С другой стороны, когда металлоорганическое соединение представляет собой главным образом металлоорганическое соединение формулы (1), равновесие реакции термического разложения на стадии (1) сдвигается в сторону получаемой системы и, следовательно, выход эфира угольной кислоты значительно повышается, так что, в некоторых случаях, выход эфира угольной кислоты не может быть далее повышен путем добавления второго спирта. Когда второй спирт содержит большое количество воды, выход эфира угольной кислоты снижается. Следовательно, предпочтительно, чтобы количество содержащейся во втором спирте воды составляло не более, чем 0,1-кратное, более предпочтительно не более, чем 0,01-кратное, стехиометрическое количество по отношению к количеству металлоорганического соединения. Когда реакцию на стадии (1) осуществляют при использовании металлоорганического соединения формулы (1), аддукт диоксида углерода и металлоорганического соединения формулы (1) подвергают термическому разложению, получая эфир угольной кислоты. Известно, что эфир угольной кислоты получают из димера металлоорганического соединения формулы (1) (см. ECO INDUSTRY, 6, №6, 11-18 (2001)). Согласно обычному способу, описанному в этом литературном источнике, эфир угольной кислоты, также как дибутилоловооксид, получают из димера металлоорганического соединения формулы (1), где количество получаемого эфира угольной кислоты составляет две молекулы на молекулу димера металлоорганического соединения. Авторами настоящего изобретения проведены обширные и интенсивные исследования в отношении образования эфира угольной кислоты из металлоорганического соединения. В результате неожиданно было найдено, что, когда аддукт диоксида углерода и димера металлоорганического соединения формулы (1) подвергают термическому разложению, быстро удаляют эфир угольной кислоты, причем количество удаляемого эфира угольной кислоты составляет одну молекулу на молекулу аддукта диоксида углерода, так что может быть получено металлоорганическое соединение формулы (2), и/или его аддукт с диоксидом углерода. В этом случае нет необходимости в добавлении спирта. Стадию (2) можно осуществлять тотчас же после получения эфира угольной кислоты и, по меньшей мере, одного соединения, выбираемого из группы, состоящей из металлоорганического соединения формулы (2), и его аддукта с диоксидом углерода. Альтернативно, стадию (2) можно осуществлять после получения далее эфира угольной кислоты из полученного металлоорганического соединения, формулы (2), и/или полученного его аддукта с диоксидом углерода. Как указано выше, предпочтительно, чтобы металлоорганическое соединение, используемое на стадии (1), представляло собой, по меньшей мере, одно соединение, выбираемое из группы, состоящей из металлоорганических соединений соответственно формул (1) и (2). Более предпочтительно, чтобы, по меньшей мере, часть используемого на стадии (1) металлоорганического соединения являлась металлоорганическим соединением формулы (1). Еще более предпочтительно, чтобы используемое на стадии (1) металлоорганическое соединение содержало 5% мол. или более металлоорганического соединения формулы (1), где количество металлоорганического соединения выражено количеством атомов металла, содержащегося в металлоорганическом соединении.
К реакционной системе, используемой на стадии (1), может быть добавлен растворитель для металлоорганического соединения. Используемое согласно настоящему изобретению металлоорганическое соединение обычно находится в виде жидкости. Однако, в некоторых случаях, металлоорганическое соединение находится в виде твердого вещества. Далее, в некоторых случаях металлоорганическое соединение переходит в твердую форму при образовании аддукта металлоорганического соединения с диоксидом углерода на стадии (1), в этом случае в качестве металлоорганического соединения используют дибутилоловодиметилат. Даже если металлоорганическое соединение является твердым веществом, на стадии (1) можно получать эфир угольной кислоты. Однако иногда жидкое состояние металлоорганического соединения является важным, когда осуществляют непрерывное получение эфира угольной кислоты. Далее, для повышения скорости реакции между металлоорганическим соединением и диоксидом углерода иногда предпочтительно, чтобы металлоорганическое соединение находилось в виде жидкости. В таких случаях стадию (1) проводят в растворителе, пригодном для металлоорганического соединения. В качестве растворителя можно использовать спирт с той же самой органической группой, как в получаемом эфире угольной кислоты. Альтернативно, также может быть использован инертный растворитель. Примеры инертных растворителей включают углеводороды и простые эфиры. Конкретные примеры инертных растворителей включают насыщенные углеводороды с 5-20 атомами углерода, такие как пентан, гексан, циклогексан, гептан, октан и декан; ароматические углеводороды с 6-20 атомами углерода (которые могут содержать насыщенную (С1-С14)-алкильную группу и/или (С5-С14)-циклоалкильную группу), такие как бензол, толуол, ксилол и этилбензол; простые насыщенные алкиловые эфиры с 6-20 атомами углерода, такие как дипропиловый эфир, дибутиловый эфир и дигексиловый эфир; простые циклоалкиловые эфиры с 4-20 атомами углерода, такие как тетрагидрофуран и диоксан; и простые фениловые эфиры с 7-28 атомами углерода (включающие фенильную группу, содержащую (С0-С8)-заместитель, и группу, выбираемую из группы, состоящей из (С1-С14)-алкила и (С5-С14)-циклоалкила), такие как анизол, этилфениловый эфир, изопропилфениловый эфир, бензилметиловый эфир и 4-метиланизол.
Температура, используемая для осуществляемой на стадии (1) реакции, обычно находится в пределах от комнатной температуры (20°С) до 300°С. Когда намереваются завершить реакцию за короткий период времени, реакцию предпочтительно проводят при температуре 80-200°С в течение периода времени от 10 минут до 500 часов. Когда металлоорганическое соединение содержит в качестве атома металла атом олова, олово, содержащееся в металлоорганическом соединении, до реакции на стадии (1) значительно различается от атома олова, содержащегося в реакционной смеси после стадии (1). Это можно видеть из сравнения между 119Sn-ЯМР-спектром (см. фиг.4), полученным в случае металлоорганического соединения до реакции на стадии (1), и 119Sn-ЯМР-спектром (см. фиг.5), полученным в случае реакционной смеси после стадии (1), и, следовательно, видно, что металлоорганическое соединение действует как предшественник эфира угольной кислоты. На фиг.5 продемонстрировано, что, когда как металлоорганическое соединение формулы (1), так и металлоорганическое соединение формулы (2) используют на стадии (1), на стадии (1) расходуется металлоорганическое соединение формулы (1). 119Sn-ЯМР-спектр, представленный на фиг.5, имеет пик, приписываемый металлоорганическому соединению формулы (2), и пик, который предположительно приписывают аддукту диоксида углерода и металлоорганического соединения формулы (2).
Когда реакцию на стадии (1) осуществляют при высокой температуре (например, при 200°С или выше), 119Sn-ЯМР-спектр, полученный в случае реакционной смеси после стадии (1), иногда имеет пик, приписываемый некому веществу, около 100 м.д., где тетраметилолово используют в качестве стандарта при 119Sn-ЯМР-анализе. Однако, когда способ согласно настоящему изобретению осуществляют неоднократно, предпочтительным является осуществление реакции на стадии (1) в условиях, где может быть подавлено образование вышеуказанного вещества, дающего пик около 100 м.д., или реакцию на стадии (1) проводят при использовании добавки для подавления образования вышеуказанного вещества, дающего пик около 100 м.д.
Что касается количества диоксида углерода, когда реакцию на стадии (1) осуществляют при комнатной температуре (20°С), достаточно, если его используют в количестве, которое является стехиометрическим по отношению к количеству используемого на стадии (1) металлоорганического соединения. Однако, когда реакцию на стадии (1) проводят при температуре, которая выше, чем комнатная температуре (20°С), в условиях, где количество диоксида углерода является стехиометрическим по отношению к количеству используемого на стадии (1) металлоорганического соединения, скорость связывания диоксида углерода с металлоорганическим соединением иногда становится очень низкой, так что скорость образования эфира угольной кислоты заметно снижается. Используемое для осуществляемой на стадии (1) реакции давление обычно составляет от атмосферного давления до 200 МПа, предпочтительно от атмосферного давления до 100 МПа, где, если желательно, реакция может быть осуществлена при введении дополнительного диоксида углерода в реакционную систему или удалении части диоксида углерода из реакционной системы. Введение дополнительного диоксида углерода в реакционную систему можно осуществлять либо периодически, либо непрерывно.
Когда путем анализа реакционной смеси подтверждается, что желательный эфир угольной кислоты получен, стадию (1) заканчивают. Например, когда эфир угольной кислоты получают в количестве, составляющем 5% или меньше, основываясь на стехиометрическом количестве по отношению к количеству металлоорганического соединения, стадия (1) может быть завершена. Реакционная смесь может быть извлечена из реактора либо после снижения давления в реакторе до атмосферного давления, либо без снижения давления в реакторе. Когда стадию (1), стадию (2) и стадию (3) осуществляют в отдельных реакторах, реакционная смесь может непрерывно циркулировать, например, согласно способу, в случае которого реакционную смесь после стадии (3) подают в реактор стадии (1); реакционную смесь, содержащуюся в реакторе стадии (1), подают в реактор стадии (2); и реакционную смесь, содержащуюся в реакторе стадии (2), подают в реактор стадии (3). Циркуляция реакционной смеси предпочтительна с точки зрения снижения количества диоксида углерода, удаляемого из реактора (для стадии (1)), который в этом случае заполнен диоксидом углерода. Реакционную смесь, получаемую по завершении каждой стадии, можно охлаждать или нагревать. Когда реакционную смесь охлаждают, ее можно принудительно подвергать охлаждению или предоставлять ей самопроизвольно охлаждаться. Как описывается ниже, если желательно, можно одновременно осуществлять стадию (1) синтеза эфира угольной кислоты и стадию (2) выделения синтезированного эфира угольной кислоты.
Стадия (2) способа согласно настоящему изобретению представляет собой стадию, на которой эфир угольной кислоты выделяют из полученной на стадии (1) реакционной смеси. Как описано выше, при получении эфира угольной кислоты из диоксида углерода и спирта обычным способом при использовании реакции по уравнению (3), образуется вода, а также эфир угольной кислоты, и воду вводят в контакт с адсорбентом или осушителем для ее удаления из реакционной системы, сдвигая, таким образом, равновесие реакции в сторону системы образования продукта. Теоретически, количество получаемого эфира угольной кислоты также может быть увеличено за счет непрерывного удаления получаемого эфира угольной кислоты из реакционной системы с тем, чтобы сдвинуть равновесие реакции в сторону образования продукта. Однако в случае обычного способа, когда получаемый эфир угольной кислоты удаляют из реакционной системы, образующаяся за счет реакции вода накапливается в реакционной системе. Как известно из уровне техники, если вода накапливается в реакционной системе, катализатор гидролизуется с помощью воды и теряет свою каталитическую активность. Гидролизованный катализатор обладает ничтожной растворимостью в растворителе и, следовательно, возникает проблема, заключающаяся в том, что на последующей стадии осушки, осуществляемой при использовании абсорбционной колонны, гидролизованный катализатор вызывает закупорку адсорбционной колонны. Другим недостатком является то, что не существует способа регенерации катализатора, который потерял свою каталитическую активность вследствие гидролиза. По этой причине обычными способами невозможно эффективно выделять образовавшийся эфир угольной кислоты из реакционной смеси.
На стадии (2) способа согласно настоящему изобретению может быть использован обычный способ выделения эфира угольной кислоты из реакционной смеси. Например, выделение эфира угольной кислоты из реакционной смеси можно осуществлять путем любой экстракции растворителем, перегонки и мембранного фильтрования, причем каждый из этих способов хорошо известен из уровня техники. В качестве предпочтительного примера экстракционного растворителя можно указать растворитель, нереакционноспособный в отношении эфира угольной кислоты. Примеры таких растворителей включают алифатические и алициклические углеводороды, такие как гексан и циклогексан; галогенированные углеводороды, такие как хлороформ, дихлорметан и трихлорметилен; ароматические углеводороды, такие как бензол, толуол и хлорбензол; и простые эфиры, такие как диэтиловый эфир и анизол.
Когда используют спирт с четырьмя или менее атомами углерода в качестве второго спирта на стадии (1) или когда второй спирт не используют на стадии (1) и металлоорганическое соединение содержит органическую группу с четырьмя или менее атомами углерода, эфир угольной кислоты может быть выделен путем отгонки непосредственно из полученной на стадии (1) реакционной смеси. В этом случае предпочтительно, чтобы получаемым эфиром угольной кислоты являлся эфир угольной кислоты (такой как диметилкарбонат или диэтилкарбонат) с температурой кипения 100°С или ниже. Перегонку можно осуществлять любым обычным способом. Например, перегонка может быть осуществлена путем любого способа перегонки при атмосферном давлении, перегонки при пониженном давлении, перегонки при давлении выше атмосферного и перегонки в тонком слое, каждый из которых хорошо известен в уровне техники. Температура перегонки обычно составляет от -20°С до 200°С, предпочтительно от -20°С до 150°С. Перегонку можно осуществлять либо в присутствии растворителя, либо путем экстрактивной дистилляции.
На стадии (2), если желательно, может быть использован третий спирт. Когда к реакционной системе добавляют третий спирт, между полученным на стадии (1) эфиром угольной кислоты и третьим спиртом протекает реакция переэтерификации, приводя, таким образом, к образованию эфира угольной кислоты, который имеет число атомов углерода, отличное от такового полученного на стадии (1) эфира угольной кислоты, что приводит, таким образом, к легкому выделению эфира угольной кислоты из реакционной смеси. Этот способ с использованием третьего спирта предпочтительно осуществляют тогда, когда получаемый на стадии (1) эфир угольной кислоты содержит девять или более атомов углерода и выделенный на стадии (2) эфир угольной кислоты имеет семь или менее атомов углерода. Этот способ более предпочтительно используют тогда, когда выделенным на стадии (2) эфиром угольной кислоты является диметилкарбонат. Количество третьего спирта, используемого на стадии (2), представляет собой 1-1000-кратное стехиометрическое количество по отношению к количеству металлоорганического соединения, используемого на стадии (1). Температура, используемая в случае реакции переэтерификации, находится предпочтительно в пределах от комнатной температуры (около 20°С) до 200°С. Принимая во внимание желательную скорость реакции переэтерификации и протекание реакции разложения эфира угольной кислоты при высокой температуре, используемая для реакции переэтерификации температура находится более предпочтительно в пределах от 50°С до 150°С. При реакции переэтерификации может быть использован обычный катализатор. Реакцию переэтерификации и выделение эфира угольной кислоты из реакционной смеси можно осуществлять либо периодически, либо одновременно. В качестве способа выделения эфира угольной кислоты из реакционной смеси после реакции переэтерификации может быть использован любой из вышеуказанных способов выделения (такие как экстракция растворителем, перегонка и мембранное фильтрование). Наиболее предпочтительной является реакционноспособная перегонка, при которой одновременно происходит переэтерификация и выделение эфира угольной кислоты из реакционной смеси.
До выделения эфира угольной кислоты из реакционной смеси путем экстракции, перегонки или тому подобным образом оставшееся непрореагировавшим металлоорганическое соединение и продукт термического разложения металлоорганического соединения могут быть удалены из реакционной смеси. Например, эфир угольной кислоты может быть получен следующим способом. Воду или содержащий воду растворитель добавляют к реакционной смеси, получая таким образом суспензию белого цвета. Твердые вещества, находящиеся в суспензии, удаляют путем фильтрации, получая фильтрат. Подвергая таким образом полученный субстрат экстракции, перегонке или тому подобному, эфир угольной кислоты может быть получен с высоким выходом даже тогда, когда эфир угольной кислоты имеет температуру кипения выше чем 100°С. Используемой водой может быть любой тип воды; однако предпочтительным является использование дистиллированной или деионизированной воды.
Количество используемой на стадии (2) воды обычно представляет собой 1-100-кратное стехиометрическое количество по отношению к количеству металлоорганического соединения, используемого на стадии (1). Количество воды, необходимое для отделения непрореагировавшего металлоорганического соединения от реакционной смеси путем разделения фаз, представляет собой самое большее 1-кратное стехиометрическое количество по отношению к количеству металлоорганического соединения, используемого на стадии (1). Однако, так как получаемый на стадии (1) эфир угольной кислоты является гидрофобным, к реакционной смеси предпочтительно добавлять воду в количестве, в несколько раз больше стехиометрического количества по отношению к количеству используемого на стадии (1) металлоорганического соединения, облегчая таким образом выделение не только непрореагировавшего металлоорганического соединения, но и также эфира угольной кислоты из реакционной смеси путем разделения фаз.
Вода, которую используют на стадии (2), не должна затвердевать в реакционной смеси, для чего соответствующим образом подбирают температуру. В частности, температура воды обычно находится в пределах от -20°С до 100°С, предпочтительно от 0°С до 100°С, более предпочтительно от 10°С до 80°С. Для предохранения эфира угольной кислоты от гидролиза температура воды еще более предпочтительно находится в пределах от 10°С до 50°С. В сочетании с водой реакция может протекать в растворителе или в его отсутствие. Когда в сочетании с водой используют растворитель, предпочтительным является использование растворителя, нереакционноспособного в отношении эфира угольной кислоты. Если на стадии (1) используют второй спирт, воду применяют в виде ее раствора в спирте, который является тем же самым, как и используемый на стадии (1) второй спирт, при этом облегчается отделение растворителя от реакционной смеси. Когда на стадии (2) используют третий спирт для осуществления реакции переэтерификации, предпочтительным является, после завершения реакции переэтерификации, добавление воды к реакционной смеси, причем воду используют в виде ее раствора в спирте, который является тем же самым, как и присутствующий в реакционной смеси.
Перегонку реакционной смеси можно осуществлять любым образом путем перегонки при атмосферном давлении, перегонки при пониженном давлении, перегонки при давлении выше атмосферного и дистилляции в тонком слое, причем каждый из этих способов хорошо известен в уровне техники. Перегонку можно осуществлять при температуре от -20°С до температуры кипения эфира угольной кислоты и/или спирта, предпочтительно от 50°С до температуры кипения эфира угольной кислоты и/или спирта. Перегонку можно осуществлять либо в присутствии растворителя, либо путем экстрактивной дистилляции.
Если желательно, можно осуществлять следующую технологическую операцию. К реакционной смеси после стадии (1) добавляют воду и/или экстракционный растворитель с получением смеси, включающей масляную фазу, которая содержит эфир угольной кислоты. Масляную фазу отделяют от смеси с последующей рекуперацией эфира угольной кислоты из масляной фазы.
Согласно способу настоящего изобретения можно получать не только симметричный эфир угольной кислоты, но и также несимметричный эфир угольной кислоты. В случае получения несимметричного эфира угольной кислоты путем использования обычного способа сначала получают симметричный эфир угольной кислоты и затем полученный симметричный эфир угольной кислоты подвергают реакции переэтерификации для получения несимметричного эфира угольной кислоты. С другой стороны, согласно способу настоящего изобретения может быть прямо получен несимметричный эфир угольной кислоты. Следовательно, способ согласно настоящему изобретению является выгодным с точки зрения снижения расхода энергии и уменьшения затрат на оборудование. Согласно способу настоящего изобретения несимметричный эфир угольной кислоты может быть получен следующим образом. Пояснения приводятся ниже, используя в качестве примера случай, где металлоорганическое соединение содержит, по меньшей мере, одну алкоксильную группу. Когда металлоорганическое соединение, используемое на стадии (1), имеет два разных типа алкоксильных групп, несимметричный эфир угольной кислоты может быть получен без использования спиртов (как второй спирт и третий спирт) на стадиях (1) и (2). С другой стороны, когда металлоорганическое соединение, используемое на стадии (1), содержит только один тип алкоксильной группы, несимметричный эфир угольной кислоты может быть получен путем осуществления стадии (1) в присутствии спирта (второй спирт), содержащего органическую группу, которая отлична от алкоксильной группы металлоорганического соединения, или путем осуществления стадии (2) в присутствии спирта (третий спирт), содержащего органическую группу, которая отлична от алкоксильной группы металлоорганического соединения. Далее, в каждом из случаев, где металлоорганическое соединение, используемое на стадии (1), содержит только один тип алкоксильной группы и где металлоорганическое соединение, используемое на стадии (1), содержит два разных типа алкоксильных групп, несимметричный эфир угольной кислоты также может быть получен путем осуществления стадии (1) в присутствии двух разных спиртов (вторых спиртов) или путем осуществления стадии (2) в присутствии двух разных спиртов (третьи спирты). Когда используют два разных спирта, стехиометрическое соотношение двух спиртов изменяется в зависимости от типов обоих спиртов; однако стехиометрическое соотношение обычно находится в пределах от 2:8 до 8:2, где каждое из количеств двух спиртов выражается с точки зрения стехиометрического количества по отношению к количеству металлоорганического соединения. Когда желают получить несимметричный эфир угольной кислоты, предпочтительно, чтобы стехиометрическое соотношение двух спиртов по возможности было близко к 1. В особенности, стехиометрическое соотношение двух спиртов предпочтительно находится в пределах от 3:7 до 7:3, более предпочтительно в пределах от 4:6 до 6:4. Когда несимметричный эфир угольной кислоты получают, используя два разных спирта, которые используют в избыточных количествах (как, например, количества, каждое из которых представляет собой, по меньшей мере, 10-кратное стехиометрическое количество по отношению к количеству металлоорганического соединения), становится возможным получение несимметричного эфира угольной кислоты, содержащего два разных типа алкоксильных групп, соответствующих двум спиртам, независимо от типа алкоксильной группы металлоорганического соединения, используемого на стадии (1). Выделение несимметричного эфира угольной кислоты из реакционной смеси может быть осуществлено любым из способов (таких как экстракция растворителем, перегонка и мембранное фильтрование), описанных выше для стадии (2). Во многих случаях получают не только несимметричный эфир угольной кислоты, но и также симметричный эфир угольной кислоты. В таких случаях можно осуществлять следующую технологическую операцию. Несимметричный и симметричный эфиры угольной кислоты выделяют из реакционной смеси при получении остаточной жидкости. Несимметричный эфир угольной кислоты отделяют от симметричного эфира угольной кислоты. Симметричный эфир угольной кислоты либо смешивают с остаточной жидкостью с последующим осуществлением стадии (3), либо возвращают на стадию (1) или стадию (2).
Стадия (3) представляет собой стадию синтеза (регенерации) металлоорганического соединения с металл-кислород-углеродной связью. Соединение, содержащееся в остаточной жидкости, которую получают после выделения эфира угольной кислоты на стадии (2), обычно находится в виде прозрачной жидкости, но иногда в виде твердого вещества. Независимо от вида соединения оно может быть использовано на стадии (3) для получения металлоорганического соединения. Соединение в остаточной жидкости, получаемой после выделения эфира угольной кислоты на стадии (2), обычно находится в виде жидкости. Например, остаточная жидкость не содержит дибутилоловооксид в виде твердого вещества (следует заметить, что дибутилоловооксид нерастворим едва ли не во всех органических растворителях при комнатной температуре (20°С) и, следовательно, находится в виде твердого вещества в таких условиях). Структура соединения в остаточной жидкости еще не идентифицирована. Однако неожиданно было обнаружено, что путем осуществления реакции на стадии (3) по способу согласно настоящему изобретению можно получать металлоорганическое соединение с металл-кислород-углеродной связью, такое как металлоорганическое соединение формулы (1), и/или металлоорганическое соединение формулы (2).
Стадия (3) включает реакцию полученной на стадии (2) остаточной жидкости с первым спиртом при образовании по меньшей мере одного металлоорганического соединения, содержащего металл-кислород-углеродную связь, и воды и удаление воды из по меньшей мере одного металлоорганического соединения, где по меньшей мере одно металлоорганическое соединение, полученное на стадии (3), рекуперируют для рециркуляции его на стадию (1).
Примеры первых спиртов, используемых на стадии (3), включают таковые, которые приведены в качестве примеров выше. Конкретные примеры первых спиртов включают алифатические спирты с 1-12 атомами углерода и алициклические спирты с 5-12 атомами углерода, такие как метанол, этанол, пропанол, 2-пропанол, н-бутанол, 2-бутанол, 2-этил-1-бутанол, трет-бутанол, пентанол, гексанол, 2-этил-1-гексанол, гексенол, циклопропанол, циклобутанол, циклопентанол, циклогексанол и циклогексенол; и аралкиловые спирты, такие как бензиловый спирт и пентениловый спирт. В качестве первых спиртов также могут быть использованы многоатомные спирты. Примеры многоатомных спиртов включают алифатические многоатомные спирты с 1-12 атомами углерода и алициклические многоатомные спирты с 5-12 атомами углерода, такие как этиленгликоль, 1,3-пропандиол, 1,2-пропандиол, циклогександиол и циклопентандиол; и аралкиловые многоатомные спирты, такие как бензолдиметанол. Если желательно, до использования любого из вышеуказанных спиртов, например, можно осуществлять перегонку спирта для его очистки или установления концентрации спирта. С этой точки зрения, предпочтительным является использование спирта с температурой кипения 300°С или ниже, измеряемой при атмосферном давлении. Для облегчения удаления воды на стадии (3) более предпочтительным является использование, по меньшей мере, одного спирта, выбираемого из группы, состоящей из н-бутанола, изобутанола, алкилового спирта с пятью или более атомами углерода и аралкилового спирта с пятью или более атомами углерода.
Что касается структуры металлоорганического соединения, получаемого путем использования многоатомного спирта в качестве первого спирта на стадии (3), то в этом отношении нет особого ограничения. Например, металлоорганическое соединение может включать по меньшей мере один член, выбираемый из группы, состоящей из сшитого продукта металлоорганического соединения формулы (1) и сшитого продукта металлоорганического соединения формулы (2).
Количество первого спирта, используемого на стадии (3), представляет собой предпочтительно 1-10000-кратное, более предпочтительно 2-100-кратное, стехиометрическое количество по отношению к количеству используемого на стадии (1) металлоорганического соединения. Когда последовательность стадий (1)-(4) повторяют один или более раз, иногда возможно, что спирт находится в остаточной жидкости, получаемой на стадии (2). В таких случаях к остаточной жидкости может быть добавлено соответствующее количество спирта так, чтобы количество спирта в остаточной жидкости находилось в вышеуказанных пределах количества первого спирта. Альтернативно, находящийся в остаточной жидкости спирт может быть удален.
Удаление воды на стадии (3) можно осуществлять любым обычным способом. Например, удаление воды на стадии (3) можно осуществлять любым способом перегонки, способом с использованием осушительной колонны, заполненной твердым осушителем, и способом с использованием мембранного разделения (таким как испарение через полупроницаемую перегородку). Среди этих способов предпочтительны перегонка и способ с использованием мембранного разделения (такой как испарение через полупроницаемую перегородку). Хорошо известно, что испарение через полупроницаемую перегородку может быть использовано для удаления воды, содержащейся в спирте. Согласно настоящему изобретению предпочтительно может быть использовано испарение через полупроницаемую перегородку. В случае спирта с температурой кипения выше, чем таковая воды, удаление воды из спирта также может быть легко осуществлено путем отгонки при нагревании. С другой стороны, в случае спирта с температурой кипения ниже, чем таковая воды, удаление воды из спирта также может быть осуществлено путем способа перегонки, при котором используют растворитель, образующий азеотропную смесь с водой.
Температура для осуществляемой на стадии (3) реакции изменяется в зависимости от типа используемого первого спирта; однако температура обычно составляет от комнатной температуры (20°С) до 300°С. Когда удаление воды на стадии (3) осуществляют путем перегонки, используемая для перегонки температура не очень ограничена до тех пор, пока вода имеет давление пара при этой температуре. Когда намереваются завершить реакцию на стадии (3) за короткий период времени при атмосферном давлении, предпочтительно осуществлять перегонку в условиях, где температура образующегося при перегонке пара является температурой кипения азеотропной смеси воды и первого спирта. Когда вода и первый спирт не образуют азеотропную смесь, предпочтительно осуществлять перегонку при температуре кипения воды. Когда намереваются завершить реакцию на стадии (3) за более короткий период времени, перегонку можно осуществлять при использовании автоклава при температуре выше температуры кипения первого спирта или воды при постепенном переходе воды в паровую фазу. Когда температура, используемая для осуществляемой на стадии (3) реакции, крайне высокая, иногда возможно, что происходит термическое разложение металлоорганического соединения. В таких случаях жидкость, содержащую воду, можно удалять путем перегонки при пониженном давлении или тому подобным образом.
Даже когда первый спирт не образует азеотропную смесь с водой, вода может быть удалена путем азеотропной перегонки, при которой используют растворитель, образующий азеотропную смесь с водой. Этот способ является предпочтительным, так как воду можно удалять при низкой температуре. Примеры растворителей, образующих азеотропную смесь с водой, включают ненасыщенные или насыщенные углеводороды, такие, как гексан, бензол, толуол, ксилол, нафталин; простые эфиры, такие как анизол и 1,4-диоксан; и галогенированные углеводороды, такие как хлороформ.
Для облегчения отделения воды от азеотропной смеси после азеотропной перегонки предпочтительным является использование в качестве растворителя ненасыщенного или насыщенного углеводорода, в котором вода имеет низкую растворимость. Когда используют такой растворитель, его необходимо использовать в количестве, таком, чтобы воду можно было в достаточной степени удалять путем азеотропной перегонки. Для азеотропной перегонки предпочтительным является использование дистилляционной колонны, так как растворитель может быть рециркулирован в реакционную систему после его отделения от азеотропной смеси в дистилляционной колонне и, следовательно, количество растворителя может быть уменьшено до относительно небольшой величины.
Путем осуществления реакции на стадии (3) может быть получено, например, по меньшей мере, одно металлоорганическое соединение, выбираемое из группы, состоящей из металлоорганического соединения, отвечающего формуле (1), и металлоорганического соединения, отвечающего формуле (2).
Когда реакция на стадии (3) достигает фазы, где вода почти не образуется, стадия (3) может быть завершена. Когда последовательность стадий (1)-(4) повторяют один или более раз, количество получаемого на стадии (1) эфира угольной кислоты изменяется в зависимости от количества воды, которую удаляют на стадии (3). Следовательно, предпочтительно, чтобы количество удаляемой на стадии (3) воды было настолько большим, насколько возможно.
Обычно количество воды, удаляемой на стадии (3), составляет 0,01-1-кратное количество, образующееся за счет реакции на стадии (3), где количество образующейся воды теоретически рассчитывают на основании допущения, что путем реакции на стадии (3) получают только металлоорганическое соединение формулы (1). Во многих случаях количество удаляемой на стадии (3) воды меньше, чем однократное вышеуказанному теоретическому количеству воды, образующейся путем реакции на стадии (3). В результате исследований, проведенных авторами настоящего изобретения, обнаружено, что при получении металлоорганического соединения из дибутилоловооксида и спирта и последовательность стадий (1)-4) повторяют один или более раз, количество воды, удаляемой на стадии (3), меньше, чем количество воды, образующейся в процессе реакции, согласно которой получают металлоорганическое соединение из дибутилоловооксида и спирта. Когда на стадии (2) к реакционной системе добавляют воду для отделения эфира угольной кислоты, иногда возможно, что получают содержащее воду твердое вещество белого цвета и количество воды, удаляемой на стадии (3), больше, чем однократное вышеуказанному теоретическому количеству воды, образующейся за счет реакции на стадии (3). Когда последовательность стадий (1)-(4) повторяют один или более раз, трудно рассчитывать теоретическое количество воды, образующейся путем осуществляемой на стадии (3) реакции, так как структура металлоорганического соединения, получаемого после стадии (2), еще не идентифицирована. В этом случае определяют изменение (во времени) количества воды, которую удаляют. Если путем измерения подтверждают, что вода почти более не удаляется, стадия (3) может быть завершена.
После завершения стадии (3), если желательно, может быть удалено избыточное количество спирта. Для повышения чистоты эфира угольной кислоты, получаемого на стадии (1), в случае, где последовательность стадий (1)-(4) повторяют один или более раз, предпочтительным является удаление избыточного количества спирта. Когда на стадии (1) используют такой же спирт, как и применяемый на стадии (3), в случае, где последовательность стадий (1)-(4) повторяют один или более раз, удаление спирта после стадии (3) можно не осуществлять. Далее, к реакционной системе после стадии (3) можно добавлять соответствующее количество спирта.
Удаление избыточного количества спирта может быть осуществлено следующим образом. Когда получаемое на стадии (3) металлоорганическое соединение находится в виде твердого вещества, спирт можно удалять в виде фильтрата, получаемого при фильтрации. С другой стороны, когда получаемое на стадии (3) металлоорганическое соединение находится в виде жидкости, удаление спирта можно осуществлять путем перегонки при пониженном давлении или способом, в случае которого в реактор вводят инертный газ, такой как азот, так что спирт удаляют в количестве, которое соответствует давлению пара спирта. В случае использования инертного газа, когда инертный газ неполностью осушен, вероятно, может происходить ухудшение вследствие того, что металлоорганическое соединение гидролизуется и разлагается на оксид металла и спирт, так что количество эфира угольной кислоты, получаемое путем реакции на стадии (1), в случае, где последовательность стадий (1)-(4) повторяют один или более раз, становится крайней низким. Стадии (1)-(3) можно осуществлять либо непрерывно, либо периодически.
Как описано выше, если желательно, стадии (1) и (2) можно осуществлять одновременно. Также, если желательно, стадии (2) и (3) можно осуществлять одновременно. Далее, стадии (1)-(3) также можно осуществлять одновременно. Что касается такого одновременного осуществления этих стадий, пояснения приводятся ниже.
(Если стадии (1) и (2) осуществляют одновременно)
Что касается реакции, осуществляемой на стадии (1), имеются два случая: одним является случай, где в процессе осуществления реакции на стадии (1) имеются жидкая фаза и паровая фаза, и другим является случай, где диоксид углерода находится в сверхкритическом состоянии в условиях высокой температуры и высокого давления и реакционная смесь образует гомогенную смесь. Стадии (1) и (2) можно осуществлять одновременно в случае, где в процессе осуществления реакции на стадии (1) имеются жидкая фаза и паровая фаза. В этом случае температура реакции и реактивное давление изменяются в зависимости от типа алкоксильной группы, содержащейся в металлоорганическом соединении, и типа спирта, когда его используют. Однако температура реакции обычно составляет 200°С или ниже и реактивное давление составляет 8 МПа или ниже. Эфир угольной кислоты обладает высокой растворимостью в диоксиде углерода, и, следовательно, часть эфира угольной кислоты растворена в паровой фазе. Следовательно, путем осуществления реакции на стадии (1) при удалении части паровой фазы эфир угольной кислоты может быть выделен из реакционной смеси.
(Если стадии (2) и (3) осуществляют одновременно)
Стадии (2) и (3) могут быть осуществлены одновременно, когда металлоорганическое соединение получают из спирта с температурой кипения выше, чем таковая воды, и на стадии (1) или (2) используют алкиловый спирт с 1-3 атомами углерода. Выделение эфира угольной кислоты и воды из реакционной смеси может быть осуществлено способом, в котором получаемая на стадии (1) реакционная смесь находится в токе инертного газа, такого как диоксид углерода, удаляя таким образом эфир угольной кислоты и воду из реакционной смеси за счет увлечения с инертным газом. Выделение эфира угольной кислоты и воды из реакционной смеси также можно осуществлять обычным способом, таким как мембранное разделение. С помощью такого способа эфир угольной кислоты и воду можно непрерывно выделять из реакционной смеси.
(Если стадии (1)-(3) осуществляют одновременно)
Что касается осуществляемой на стадии (1) реакции, имеются два случая: одним является случай, где в процессе осуществления реакции на стадии (1) имеются жидкая фаза и паровая фаза, и другим является случай, где диоксид углерода находится в сверхкритическом состоянии в условиях высокой температуры и высокого давления и реакционная смесь образует гомогенную смесь.
Стадии (1)-(3) можно осуществлять одновременно, если в процессе осуществления реакции на стадии (1) имеются жидкая фаза и паровая фаза, металлоорганическое соединение получают из спирта с температурой кипения выше, чем температура воды, и используют алкиловый спирт с 1-3 атомами углерода (предпочтительно метанол). В этом случае температура реакции и давление, при которой проводят процесс, изменяются в зависимости от типа алкоксильной группы, содержащейся в металлоорганическом соединении, и типа спирта, когда его используют. Однако температура реакции обычно составляет 150°С или ниже и давление процесса составляет обычно 5 МПа или ниже. Вода и эфир угольной кислоты обладают высокой растворимостью в диоксиде углерода и, следовательно, часть эфира угольной кислоты растворена в паровой фазе. Следовательно, путем осуществления реакции на стадии (1) при удалении части паровой фазы эфир угольной кислоты может быть выделен из реакционной смеси при регенерации металлоорганического соединения. Далее, также можно использовать способ, в котором реакцию осуществляют в реакторе с неподвижным слоем, содержащим металлоорганическое соединение, где металлоорганическое соединение нанесено на носитель или находится в твердой форме. Согласно этому способу, диоксид углерода и (С1-С3)-спирт вводят в реактор с неподвижным слоем для осуществления реакции, получая таким образом эфир угольной кислоты и воду в такой форме, как увлекаемая с помощью газообразного диоксида углерода. В качестве носителя для нанесения металлоорганического соединения можно использовать обычный носитель.
Стадия (4) представляет собой стадию, в которой, по меньшей мере, одно металлоорганическое соединение, рекуперированное на стадии (3), рециркулируют на стадию (1). Последовательность стадий (1)-(4) можно повторять один или более раз. До рециркуляции металлоорганического соединения на стадию (1) его можно охлаждать или нагревать. Стадию (4) можно осуществлять либо непрерывно, либо периодически.
Лучший вариант осуществления изобретения
Ниже настоящее изобретение описывается более подробно со ссылкой на следующие примеры и сравнительные примеры, которые не следует истолковывать как ограничивающие объем охраны настоящего изобретения.
В последующих примерах и сравнительных примерах различные измерения и анализы осуществляют следующими методами:
1) Анализ методом ядерного магнитного резонанса (ЯМР) металлоорганического соединения
Аппаратура: система JNM-A-400 FT-NMR (выпускается фирмой JEOL Ltd., Япония) (400 МГц)
(1) Приготовление растворов образцов для 1Н- и 13С-ЯМР-анализов
Взвешивают примерно от 0,1 г до 0,5 г металлоорганического соединения и затем туда добавляют примерно 0,9 г дейтерированного хлороформа, получая таким образом раствор образца для ЯМР-анализа.
(2) Приготовление раствора образца для 119Sn-ЯМР-анализа
Взвешивают примерно от 0,1 г до 1 г жидкости, содержащей металлоорганическое соединение, и затем туда добавляют 0,05 г тетраметилолова и примерно 0,85 г дейтерированного хлороформа, получая таким образом раствор образца для ЯМР-анализа.
2) Газохроматографический (ГХ) анализ эфира угольной кислоты
Аппаратура: система GC-2010 (выпускается фирмой Shimadzu Corporation, Япония).
(1) Приготовление раствора образца
Взвешивают 0,06 г жидкости для определения содержащегося в ней эфира угольной кислоты и затем туда добавляют примерно 2,5 мл обезвоженного диметилформамида или обезвоженного ацетонитрила. Далее, к полученному в результате раствору добавляют примерно 0,06 г дифенилового эфира в качестве внутреннего стандарта.
(2) Условия для ГХ-анализа
колонка: DB-1 (выпускается фирмой J and W Scientific, США);
жидкая фаза: 100% диметилполисилоксан;
длина колонки: 30 м;
диаметр колонки: 0,25 мм;
толщина пленки: 1 мкм;
температура колонки: температуру повышают от 50°С до 300°С со скоростью 10°С/мин;
температура ввода (пробы): 300°С;
температура детектора: 300°С;
детектор: FID (пламенно-ионизационный детектор).
(3) Количественный анализ
Количественный анализ раствора образца осуществляют при использовании калибровочной кривой, полученной для стандартных образцов.
3) Расчет выхода эфира угольной кислоты
Выход эфира угольной кислоты рассчитывают по следующей формуле (6):

Здесь термин "стехиометрическое количество металлоорганического соединения" означает величину, рассчитанную путем деления числа атомов металла металлоорганического соединения на число Авогадро.
Пример 1
Синтез металлоорганического соединения, содержащего 2-этил-1-гексилоксигруппу
В автоклав емкостью 200 мл (выпускается фирмой Toyo Koatsu Co., Ltd., Япония) загружают 29 г (116 ммоль) дибутилоловооксида (выпускается фирмой Aldrich, США) и 75 г (576 ммоль) 2-этил-1-гексанола (выпускается фирмой Aldrich, США). Автоклав продувают газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 192°С. Открывают продувочную линию и перемешивание продолжают в течение 3,5 часов при атмосферном давлении при отгонке воды и 2-этил-1-гексанола. Спустя этот период времени почти никакого дистиллята более не получают. Затем содержимое автоклава охлаждают до температуры около 30°С при продувке автоклава газообразным азотом, и получают реакционную смесь, содержащую металлоорганическое соединение, включающее 2-этил-1-гексилоксигруппу. Количество жидкости (дистиллят), отгоняющееся через продувочную линию в процессе реакции, составляет около 50 г. Количество воды, содержащейся в дистилляте, определяют по методу Карла Фишера. Найдено, что количество воды в дистилляте составляет около 1,7 г. 119Sn-ЯМР-спектр реакционной смеси представлен на фиг.1. Как видно из фиг.1, обнаруживают пик при -45 м.д., приписываемый металлоорганическому соединению формулы (1), и обнаруживают пики, соответственно, при -172 м.д. и -184 м.д., приписываемые металлоорганическому соединению формулы (2).
Стадия (1): Получение диметилкарбоната из содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения, метанола и газообразного диоксида углерода
В вышеуказанный автоклав, содержащий реакционную смесь, загружают 75,5 г (2,4 моль) метанола и все вентили закрывают. Затем из баллона с газообразным диоксидом углерода, газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Жидкий диоксид углерода постепенно вводят по линии для подачи сырья в автоклав с тем, чтобы довести внутреннее давление в автоклаве до 19,6 МПа. После этого осуществляют реакцию в течение 1 часа при поддерживании внутреннего давления в автоклаве 19,6 МПа, получая таким образом реакционную смесь, и содержимое автоклава охлаждают до температуры около 30°С с последующим удалением газообразного диоксида углерода.
Стадия (2): Выделение диметилкарбоната
В вышеуказанном автоклаве осуществляют вакуумную перегонку при температуре 30°С и давлении 13 КПа, выделяя таким образом диметилкарбонат и метанол из реакционной смеси путем отгонки и рекуперируя их по линии для отвода дистиллята из автоклава. Таким образом получают диметилкарбонат с выходом 17%.
Стадия (3): Синтез (регенерация) металлоорганического соединения
75 г (576 ммоль) 2-этил-1-гексанола (выпускается фирмой Aldrich, США) добавляют к остаточной жидкости в автоклаве, получаемой на стадии (2). Автоклав продувают газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 192°С. Открывают продувочную линию и перемешивание продолжают в течение 3,5 часов при атмосферном давлении при отгонке воды и 2-этил-1-гексанола. Спустя этот период времени почти никакого дистиллята более не получают. Затем содержимое автоклава охлаждают до температуры около 30°С при продувке автоклава газообразным азотом и получают реакционную смесь, содержащую металлоорганическое соединение, включающее 2-этил-1-гексилоксигруппу.
Стадия (4): Рециркуляция полученного на стадии (3) металлоорганического соединения на стадию (1)
Потом те же самые операции, как на стадии (1) и стадии (2), осуществляют последовательно следующим образом.
Стадия (1): Получение диметилкарбоната из содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения, метанола и газообразного диоксида углерода
В вышеуказанный автоклав, содержащий реакционную смесь, загружают 75,5 г (2,4 моль) метанола и все вентили закрывают. Затем из баллона с газообразным диоксидом углерода, газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Жидкий диоксид углерода постепенно вводят по линии для подачи сырья в автоклав с тем, чтобы довести внутреннее давление в автоклаве до 19,6 МПа. Затем осуществляют реакцию в течение 1 часа при поддерживании внутреннего давления в автоклаве 19,6 МПа, получая таким образом реакционную смесь, и содержимое автоклава охлаждают до температуры около 30°С с последующим удалением газообразного диоксида углерода.
Стадия (2): Выделение диметилкарбоната
В вышеуказанном автоклаве осуществляют вакуумную перегонку при температуре 30°С и давлении 13 КПа, выделяя таким образом диметилкарбонат и метанол из реакционной смеси путем отгонки и рекуперируя их по линии для отвода дистиллята из автоклава. Таким образом, получают диметилкарбонат с выходом 16%.
Пример 2
Как описывается ниже, диметилкарбонат получают путем осуществления цикла стадий (1)-(4) последовательно 26 раз.
Синтез содержащего гексилоксигруппу металлоорганического соединения
Используют автоклав емкостью 200 мл (выпускается фирмой Toyo Koatsu Co., Ltd., Япония), который, кроме того, соединяют с линией для ввода жидкого диоксида углерода и газообразного диоксида углерода, линией для удаления дистиллята, пробоотборной трубкой и линией для ввода газообразного азота в нижнюю часть автоклава.
В автоклав емкостью 200 мл загружают 15,0 г (60 ммоль) дибутилоловооксида (выпускается фирмой Aldrich, США) и 30,7 г (300 ммоль) гексанола (выпускается фирмой Aldrich, США; безводный). Автоклав герметизируют и все вентили закрывают. Автоклав 3 раза продувают газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Потом перемешивание продолжают в течение 30 минут. После этого открывают вентиль линии для удаления дистиллята и начинают рекуперацию дистиллята и продолжают при введении газообразного азота в нижнюю часть автоклава со скоростью потока 200 мл/мин. Спустя примерно 2 часа после начала рекуперации дистиллята почти никакого дистиллята более не получают. Затем содержимое автоклава охлаждают до температуры около 50°С и получают прозрачную реакционную смесь. Небольшое количество реакционной смеси отбирают и подвергают 119Sn-ЯМР-анализу. 119Sn-ЯМР-анализ подтверждает, что образовались металлоорганическое соединение формулы (1) и металлоорганическое соединение формулы (2). Дистиллят разделяется на два слоя, и количество водного слоя составляет около 0,9 мл.
Затем повторяют цикл вышеописанных стадий (1)-(4).
Стадия (1): Получение диметилкарбоната из содержащего гексилоксигруппу металлоорганического соединения, метанола и газообразного диоксида углерода
В вышеуказанный автоклав, содержащий реакционную смесь, загружают 48,1 г (1,5 моль) метанола и все вентили закрывают. Затем из баллона с газообразным диоксидом углерода, газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Жидкий диоксид углерода постепенно вводят по линии для подачи сырья в автоклав с тем, чтобы довести внутреннее давление в автоклаве до 22 МПа. Затем осуществляют реакцию в течение 16 часов при поддерживании внутреннего давления в автоклаве 22 МПа, получая таким образом реакционную смесь, и содержимое автоклава охлаждают до температуры около 30°С с последующим удалением газообразного диоксида углерода.
Стадия (2): Выделение диметилкарбоната
В вышеуказанном автоклаве осуществляют вакуумную перегонку при температуре 30°С и давлении 13 КПа, выделяя таким образом диметилкарбонат и метанол из реакционной смеси путем отгонки и рекуперируя их по линии для отвода дистиллята из автоклава. Таким образом, получают диметилкарбонат.
Стадия (3): Синтез (регенерация) металлоорганического соединения
В вышеуказанный автоклав загружают около 20 г гексанола (выпускается фирмой Aldrich, США; безводный) и все вентили закрывают. Автоклав продувают 3 раза газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. После этого перемешивание продолжают в течение 30 минут. Затем открывают вентиль линии для удаления дистиллята и начинают рекуперацию дистиллята и продолжают при введении газообразного азота в нижнюю часть автоклава со скоростью потока 200 мл/мин. Спустя 2 часа после начала рекуперации дистиллята содержимое автоклава охлаждают до температуры около 50°С и получают прозрачную реакционную смесь.
Стадия (4): Рециркуляция полученного на стадии (3) металлоорганического соединения на стадию (1)
Выход диметилкарбоната, полученного на стадии (2) каждого цикла, представлен в таблице 2.
После стадии (2) 26-го цикла реакции, полученную на стадии (2) остаточную жидкость подвергают 119Sn-ЯМР-анализу. В 119Sn-ЯМР-спектре остаточной жидкости обнаруживают несколько пиков между -170 м.д. и -500 м.д., в дополнение к пику, приписываемому небольшому количеству металлоорганического соединения формулы (2) (см. фиг.2). После стадии (3) 26-го цикла реакции полученную на стадии (3) реакционную смесь подвергают 119Sn-ЯМР-анализу. В 119Sn-ЯМР-спектре реакционной смеси обнаруживают пик, приписываемый металлоорганическому соединению формулы (2), и больше не обнаруживают вышеуказанных нескольких пиков между -170 м.д. и -500 м.д.(см. фиг.3).
Количества отогнанной на стадии (3) воды с первого по четвертый циклы реакции составляют, соответственно, 0,27 мл, 0,24 мл, 0,22 мл и 0,24 мл.
Пример 3
Синтез содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения
В круглодонную колбу грушевидной формы емкостью 500 мл загружают 105 г (422 ммоль) дибутилоловооксида (выпускается фирмой Aldrich, США) и 277 г (2,1 моль) 2-этил-1-гексанола (выпускается фирмой Aldrich, США). Вышеуказанную колбу соединяют с роторным испарителем (выпускается фирмой EYELA, Япония). Колбу продувают газообразным азотом. Начинают вращение вышеуказанной колбы на масляной бане и масляную баню нагревают до температуры 180°С. Во время нагревания суспензия белого цвета в колбе превращается в прозрачный раствор. Затем вращение колбы продолжают около 30 минут. После этого внутреннее давление в вышеуказанной колбе постепенно снижают от 80,7 КПа до 68,7 КПа в течение 3 часов с помощью вакуумного насоса (выпускается фирмой SATO VAC INC., Япония) и регулятора вакуума (выпускается фирмой OKANO WORKS, LTD., Япония) при отгонке воды и небольшого количества 2-этил-1-гексанола. Спустя этот период времени почти никакого дистиллята более не получают. Затем содержимое колбы охлаждают до температуры около 30°С и внутреннее давление в колбе возвращают к атмосферному с помощью газообразного азота, и получают 310 г реакционной смеси, представляющей собой прозрачный раствор металлоорганического соединения, содержащего 2-этил-1-гексилоксигруппу. 119Sn-ЯМР-спектр полученного прозрачного раствора представлен на фиг.4. Как видно на фиг.4, обнаруживают пик при -14 м.д., приписываемый металлоорганическому соединению формулы (1), и обнаруживают пики, соответственно, при -172 м.д. и -184 м.д., приписываемые металлоорганическому соединению формулы (2).
Стадия (1): Получение диметилкарбоната из содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения, метанола и газообразного диоксида углерода
В автоклав емкостью 500 мл (выпускается фирмой Asahi Shoko Co., Ltd., Япония) загружают 148,8 г (содержащего 202 ммоль атомов Sn) вышеполученного раствора содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения в 2-этил-1-гексаноле и 86,4 г 2-этил-1-гексанола. Автоклав герметизируют и все вентили закрывают. Затем из баллона с газообразным диоксидом углерода газообразный диоксид углерода, давление которого снижают до 3 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 120°С. Внутреннее давление в автоклаве доводят до 3,5 МПа. После этого осуществляют реакцию в течение 4 часов при поддерживании внутреннего давления в автоклаве 3,5 МПа, получая таким образом реакционную смесь, и содержимое автоклава охлаждают до температуры около 30°С с последующим удалением газообразного диоксида углерода. 119Sn-ЯМР-спектр реакционной смеси представлен на фиг.5. Как видно на фиг.5, пик, приписываемый металлоорганическому соединению формулы (1), исчез, и между -170 м.д. и -230 м.д. обнаружено несколько пиков. ГХ-анализ реакционной смеси показывает, что получен ди(2-этилгексил)карбонат с выходом 25%.
Пример 4
Синтез содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения
При использовании автоклава емкостью 200 мл (выпускается фирмой Toyo Koatsu Co., Ltd., Япония) синтезируют металлоорганическое соединение таким же образом, как описывается в примере 1.
Стадия (1): Получение ди(2-этилгексил)карбоната из содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения и газообразного диоксида углерода
Все вентили вышеуказанного автоклава закрывают. Затем из баллона с газообразным диоксидом углерода газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Жидкий диоксид углерода постепенно вводят по линии для подачи сырья в автоклав с тем, чтобы довести внутреннее давление в автоклаве до 19,6 МПа. После этого осуществляют реакцию в течение 2 часов при поддерживании внутреннего давления в автоклаве 19,6 МПа, получая таким образом реакционную смесь, с последующим удалением газообразного диоксида углерода.
Стадия (2): Операция, при которой к реакционной смеси добавляют метанол для осуществления таким образом реакции переэтерификации с тем, чтобы превратить вышеуказанный эфир угольной кислоты в диметилкарбонат, и выделяют образовавшийся диметилкарбонат.
После стадии (1) автоклав продувают газообразным азотом. В автоклав загружают 75,5 г (2,4 моль) метанола (выпускается фирмой Aldrich, США; безводный) и все вентили закрывают. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 120°С. Перемешивание продолжают в течение 2 часов и затем метанол и диметилкарбонат постепенно удаляют путем отгонки из автоклава по продувочной линии. Когда обнаруживают, что более почти не получают никакого дистиллята, содержимое автоклава охлаждают и автоклав продувают газообразным азотом, после чего реакция завершается. В дистилляте диметилкарбонат получают с выходом около 20%.
Стадия (3): Синтез (регенерация) металлоорганического соединения
75 г (576 ммоль) 2-этил-1-гексанола (выпускается фирмой Aldrich, США) добавляют к остаточной жидкости в автоклаве, получаемой на стадии (2). Автоклав продувают газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 192°С. Открывают продувочную линию и перемешивание продолжают в течение 3,5 часов при атмосферном давлении при отгонке воды и 2-этил-1-гексанола. Спустя этот период времени почти никакого дистиллята более не получают. Затем содержимое автоклава охлаждают до температуры около 160°С при продувке автоклава газообразным азотом и получают реакционную смесь, содержащую металлоорганическое соединение, включающее 2-этил-1-гексилоксигруппу.
Стадия (4): Рециркуляция полученного на стадии (3) металлоорганического соединения на стадию (1)
Потом те же самые операции, как на стадии (1) и стадии (2), осуществляют последовательно следующим образом.
Стадия (1): Получение ди(2-этилгексил)карбоната из содержащего 2-этил-1-гексилоксигруппу металлоорганического соединения и газообразного диоксида углерода
Все вентили вышеуказанного автоклава закрывают. Начинают перемешивание содержимого автоклава. Жидкая угольная кислота постепенно вводят по линии для подачи сырья в автоклав с тем, чтобы довести внутреннее давление в автоклаве до 19,6 МПа. После этого осуществляют реакцию в течение 1 часа при поддерживании внутреннего давления в автоклаве 19,6 МПа, получая таким образом реакционную смесь, с последующим удалением газообразного диоксида углерода.
Стадия (2): Операция, при которой к реакционной смеси добавляют метанол для осуществления таким образом реакции переэтерификации с тем, чтобы превратить вышеуказанный эфир угольной кислоты в диметилкарбонат, и выделяют образовавшийся диметилкарбонат.
После стадии (1) автоклав продувают газообразным азотом. В автоклав загружают 75,5 г (2,4 моль) метанола (выпускается фирмой Aldrich, США; безводный) и все вентили закрывают. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 120°С. Перемешивание продолжают в течение 1 часа и затем метанол и диметилкарбонат постепенно удаляют путем отгонки из автоклава по продувочной линии. Когда обнаруживают, что более почти не получают никакого дистиллята, содержимое автоклава охлаждают и автоклав продувают газообразным азотом, после чего реакция завершается. В дистилляте диметилкарбонат получают с выходом около 18%.
Пример 5
Синтез содержащего гексилоксигруппу металлоорганического соединения из дибутилоловооксида и гексанола
В автоклав емкостью 200 мл (выпускается фирмой Toyo Koatsu Co., Ltd., Япония) загружают 24,9 г (100 ммоль) дибутилоловооксида (выпускается фирмой Aldrich, США) и 51,1 г (500 ммоль) гексанола (выпускается фирмой Aldrich, США; безводный). Автоклав продувают газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Перемешивание затем продолжают в течение примерно 30 минут. После этого открывают вентиль продувочной линии автоклава и перемешивание продолжают в течение 2 часов при введении небольшого количества газообразного азота в нижнюю часть автоклава и отгонке воды и гексанола по продувочной линии. Спустя этот период времени почти никакого дистиллята более не получают. Затем содержимое автоклава охлаждают до температуры около 30°С и получают реакционную смесь. Осуществляют 119Sn-ЯМР-анализ реакционной смеси. 119Sn-ЯМР-анализ показывает, что получено около 47 ммоль 1,1,3,3-тетрабутил-1,3-дигексилоксидистанноксана и около 6 ммоль дибутилоловодигексилата.
Стадия (1): Получение дигексилкарбоната из содержащего гексилоксигруппу металлоорганического соединения, гексанола и газообразного диоксида углерода
В вышеуказанный автоклав емкостью 200 мл, содержащий реакционную смесь (включающую содержащее гексилоксигруппу металлоорганическое соединение), загружают 61,5 г (602 ммоль) гексанола (выпускается фирмой Aldrich, США; безводный) и автоклав герметически закрывают. Затем из баллона с газообразным диоксидом углерода газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава. Спустя 10 минут после начала перемешивания закрывают вентиль баллона с газообразным диоксидом углерода. Затем, при перемешивании, температуру внутри автоклава повышают до 180°С. В этот момент внутреннее давление в автоклаве составляет около 7,5 МПа. Затем осуществляют реакцию в течение 6 часов при поддерживании внутреннего давления в автоклаве около 7,5 МПа. После этого содержимое автоклава охлаждают до температуры около 30°С и внутреннее давление в автоклаве возвращают к атмосферному давлению путем осторожного удаления газообразного диоксида углерода, и получают прозрачную реакционную смесь. В реакционной смеси дигексилкарбонат получают с выходом около 14%.
Стадия (2): Операция, при которой содержащий 1% масс. воды гексанол добавляют к полученной на стадии (1) реакционной смеси с образованием таким образом твердых веществ и твердые вещества удаляют путем отфильтровывания, после чего полученный фильтрат подвергают перегонке, так что получают дигексилкарбонат в виде дистиллята.
После стадии (1) к полученной на стадии (1) реакционной смеси осторожно добавляют 10 г гексанола, содержащего 1% масс. воды, и полученную в результате смесь перемешивают в течение около 30 минут. Затем автоклав открывают и обнаруживают, что смесь в автоклаве превратилась в суспензию белого цвета. Суспензию белого цвета подвергают фильтрации при использовании мембранного фильтра (НО20А142С; выпускается фирмой Advantec Toyo Kaisha, Ltd., Япония), получая таким образом твердые вещества белого цвета и фильтрат. Твердые вещества белого цвета промывают 2 раза с помощью 20 мл гексанола. Фильтрат переносят в круглодонную колбу грушевидной формы емкостью 1 л и подвергают перегонке при нагревании на масляной бане при температуре 150°С и давлении 1 КПа. Путем перегонки рекуперируют гексанол и дигексилкарбонат. Выход дигексилкарбоната составляет 13%.
Стадия (3): Синтез (регенерация) металлоорганического соединения
Полученные на стадии (2) твердые вещества белого цвета и остаточную вязкую жидкость, оставшуюся в колбе после осуществляемой на стадии (2) перегонки, загружают в автоклав емкостью 200 мл (выпускаемый фирмой Toyo Koatsu Co., Ltd., Япония). Далее, в автоклав загружают 51,1 г (500 мл) гексанола (выпускаемый фирмой Aldrich, США; безводный) и автоклав герметически закрывают. Автоклав продувают газообразным азотом. Затем начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. Перемешивание затем продолжают в течение около 30 минут. После этого открывают продувочную линию автоклава и перемешивание продолжают в течение 2 часов при введении небольшого количества газообразного азота в нижнюю часть автоклава и отгонке воды и гексанола по продувочной линии. Спустя этот период времени почти никакого дистиллята более не получают. Затем содержимое автоклава охлаждают до температуры около 30°С и получают реакционную смесь. Осуществляют 119Sn-ЯМР-анализ реакционной смеси. 119Sn-ЯМР-анализ показывает, что получено около 47 ммоль 1,1,3,3-тетрабутил-1,3-дигексилоксидистанноксана и около 6 моль дибутилоловодигексилата.
Стадия (4): Рециркуляция полученного на стадии (3) металлоорганического соединения на стадию (1)
Потом те же самые операции, как на стадии (1) и стадии (2), осуществляют последовательно следующим образом.
Стадия (1): Получение дигексилкарбоната из полученного на стадии (3) металлоорганического соединения
В вышеуказанный автоклав, в котором осуществляют стадию (3), загружают 61,5 г (602 ммоль) гексанола (выпускается фирмой Aldrich, США; безводный) и автоклав герметически закрывают. Затем из баллона с газообразным диоксидом углерода газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава. Спустя 10 минут после начала перемешивания закрывают вентиль баллона с газообразным диоксидом углерода. Затем, при перемешивании, температуру внутри автоклава повышают до 180°С. В этот момент внутреннее давление в автоклаве составляет около 7,5 МПа. Затем осуществляют реакцию в течение 6 часов при поддерживании внутреннего давления в автоклаве около 7,5 МПа. После этого содержимое автоклава охлаждают до температуры около 30°С и внутреннее давление в автоклаве возвращают к атмосферному давлению путем осторожного удаления газообразного диоксида углерода по продувочной линии, и получают прозрачную реакционную смесь. В реакционной смеси дигексилкарбонат получают с выходом около 14%.
Стадия (2): Операция, при которой содержащий 1% масс. воды гексанол добавляют к полученной на стадии (1) реакционной смеси с образованием таким образом твердых веществ и твердые вещества удаляют путем отфильтровывания, после чего полученный фильтрат подвергают перегонке, так что получают дигексилкарбонат в виде дистиллята.
После стадии (1) к полученной на стадии (1) реакционной смеси осторожно добавляют 10 г гексанола, содержащего 1% масс. воды, и полученную в результате смесь перемешивают в течение около 30 минут. Затем автоклав открывают и обнаруживают, что смесь в автоклаве превратилась в суспензию белого цвета. Суспензию белого цвета подвергают фильтрации при использовании мембранного фильтра (НО20А142С; выпускается фирмой Advantec Toyo Kaisha, Ltd., Япония), получая таким образом твердые вещества белого цвета и фильтрат. Твердые вещества белого цвета промывают 2 раза с помощью 20 мл гексанола. Фильтрат переносят в круглодонную колбу грушевидной формы емкостью 1 л и подвергают перегонке при нагревании. Путем перегонки рекуперируют гексанол и дигексилкарбонат. Выход дигексилкарбоната составляет 13%.
Пример 6
Получение дигексилкарбоната
(Синтез дибутилоловодигексилата)
В круглодонную колбу грушевидной формы емкостью 200 мл, снабженную холодильником и насадкой Дина-Старка, загружают 12,5 г (50 ммоль) дибутилоловооксида (выпускается фирмой Aldrich, США), 50 мл гексанола (выпускается фирмой Aldrich, США; безводный), 100 мл ксилола и помещают мешалку. Затем вышеуказанную колбу нагревают на масляной бане при перемешивании ее содержимого и температуру колбы повышают до температуры кипения с обратным холодильником ксилола. Кипячение с обратным холодильником ксилола осуществляют в течение около 4 часов при отгонке воды, так что в насадке Дина-Старка собирают около 0,8 мл воды. Насадку Дина-Старка отсоединяют от колбы и ксилол и гексанол удаляют из вышеуказанной колбы путем обычной отгонки. Далее, избыточное количество гексанола из колбы удаляют путем перегонки при пониженном давлении, получая таким образом вязкую прозрачную жидкость. Затем колбу продувают азотом и ее содержимое охлаждают. Полученную в результате реакционную смесь подвергают 119Sn-ЯМР-анализу. На 119Sn-ЯМР-спектре реакционной смеси обнаруживают пик при -134 м.д., приписываемый дибутилоловодигексилату, и обнаруживают пики, соответственно, при -177 м.д. и -187 м.д., приписываемые небольшому количеству 1,1,3,3-тетрабутил-1,3-дигексилоксидистанноксана.
Стадия (1): Получение дигексилкарбоната из дибутилоловодигексилата
В автоклав емкостью 100 мл (выпускается фирмой Toyo Koatsu Co., Ltd., Япония) загружают около 2,2 г металлоорганического соединения, содержащего около 5 ммоль дибутилоловодигексилата (где металлоорганическое соединение содержится в вышеуказанной реакционной смеси), и 25,5 г (250 ммоль) гексанола (выпускается фирмой Aldrich, США; безводный). Автоклав герметически закрывают. Затем из баллона с газообразным диоксидом углерода газообразный диоксид углерода, давление которого снижают до 4 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава. Спустя 10 минут после начала перемешивания закрывают вентиль баллона с газообразным диоксидом углерода. Затем, при перемешивании, температуру внутри автоклава повышают до 120°С. После этого газообразный диоксид углерода постепенно удаляют из автоклава по продувочной линии, с тем, чтобы довести внутреннее давление в автоклаве до 4 МПа. Затем осуществляют реакцию в течение 100 часов при поддерживании внутреннего давления в автоклаве 4 МПа. Спустя этот период времени содержимое автоклава охлаждают до температуры около 30°С и внутреннее давление в автоклаве возвращают к атмосферному давлению путем осторожного удаления газообразного диоксида углерода по продувочной линии, и получают прозрачную реакционную смесь. В реакционной смеси дигексилкарбонат получают с выходом 18%.
Стадия (2): Операция, при которой содержащий 1% масс. воды гексанол добавляют к полученной на стадии (1) реакционной смеси с образованием таким образом твердых веществ и твердые вещества удаляют путем отфильтровывания, после чего полученный фильтрат подвергают перегонке, так что получают дигексилкарбонат в виде дистиллята.
После стадии (1) к полученной на стадии (1) реакционной смеси осторожно добавляют 10 г гексанола, содержащего 1% масс. воды, и полученную в результате смесь перемешивают в течение около 30 минут. Затем автоклав открывают и обнаруживают, что смесь в автоклаве превратилась в суспензию белого цвета. Суспензию белого цвета подвергают фильтрации при использовании мембранного фильтра (НО20А142С; выпускается фирмой Advantec Toyo Kaisha, Ltd., Япония), получая таким образом твердые вещества белого цвета и фильтрат. Твердые вещества белого цвета промывают 2 раза с помощью 20 мл гексанола. Фильтрат переносят в круглодонную колбу грушевидной формы емкостью 1 л и подвергают перегонке при нагревании. Путем перегонки рекуперируют гексанол и дигексилкарбонат. Выход дигексилкарбоната составляет 17%.
Стадия (3): Синтез (регенерация) металлоорганического соединения
Полученные на стадии (2) твердые вещества белого цвета и остаточную вязкую жидкость, оставшуюся в колбе после осуществляемой на стадии (2) перегонки, загружают в круглодонную колбу грушевидной формы емкостью 100 мл, снабженную холодильником и насадкой Дина-Старка. Далее, в эту колбу загружают 20 мл гексанола (выпускается фирмой Aldrich, США; безводный), 30 мл ксилола и помещают мешалку. Затем круглодонную колбу грушевидной формы нагревают на масляной бане при перемешивании содержимого вышеуказанной колбы и температуру колбы повышают до температуры кипения с обратным холодильником ксилола. Кипячение ксилола с обратным холодильником осуществляют в течение около 4 часов при отгонке воды, так что в насадке Дина-Старка собирают около 0,1 мл воды. Насадку Дина-Старка отсоединяют от колбы и ксилол и гексанол удаляют из вышеуказанной колбы путем обычной отгонки. Далее, избыточное количество гексанола из колбы удаляют путем перегонки при пониженном давлении, получая таким образом вязкую прозрачную жидкость. Затем колбу продувают азотом и ее содержимое охлаждают. Полученную в результате реакционную смесь подвергают 119Sn-ЯМР-анализу. На 119Sn-ЯМР-спектре реакционной смеси обнаруживают пик при -134 м.д., приписываемый дибутилоловодигексилату, и обнаруживают пики, соответственно, при -177 м.д. и -187 м.д., приписываемые небольшому количеству 1,1,3,3-тетрабутил-1,3-дигексилоксидистанноксана.
Стадия (4): Рециркуляция полученного на стадии (3) металлоорганического соединения на стадию (1)
Потом те же самые операции, как на стадии (1) и стадии (2), осуществляют последовательно следующим образом.
Стадия (1): Получение дигексилкарбоната из полученного на стадии (3) металлоорганического соединения
В автоклав емкостью 100 мл (выпускается фирмой Toyo Koatsu Co., Ltd., Япония) загружают около 2,2 г металлоорганического соединения, содержащего около 5 ммоль дибутилоловодигексилата (где металлоорганическое соединение содержится в вышеуказанной реакционной смеси) и 25,5 г (250 ммоль) гексанола (выпускается фирмой Aldrich, США; безводный). Автоклав герметически закрывают. Затем из баллона с газообразным диоксидом углерода газообразный диоксид углерода, давление которого снижают до 4 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава. Спустя 10 минут после начала перемешивания закрывают вентиль баллона с газообразным диоксидом углерода. Затем, при перемешивании, температуру внутри автоклава повышают до 120°С. После этого газообразный диоксид углерода постепенно удаляют из автоклава по продувочной линии с тем, чтобы довести внутреннее давление в автоклаве до 4 МПа. Затем осуществляют реакцию в течение 100 часов при поддерживании внутреннего давления в автоклаве 4 МПа. Спустя этот период времени содержимое автоклава охлаждают до температуры около 30°С и внутреннее давление в автоклаве возвращают к атмосферному давлению путем осторожного удаления газообразного диоксида углерода по продувочной линии, и получают прозрачную реакционную смесь. В реакционной смеси дигексилкарбонат получают с выходом 17%.
Стадия (2): Операция, при которой содержащий 1% масс. воды гексанол добавляют к полученной на стадии (1) реакционной смеси с образованием таким образом твердых веществ и твердые вещества удаляют путем отфильтровывания, после чего полученный фильтрат подвергают перегонке, так что получают дигексилкарбонат в виде дистиллята.
После стадии (1) к полученной на стадии (1) реакционной смеси осторожно добавляют 10 г гексанола, содержащего 1% масс. воды, и полученную в результате смесь перемешивают в течение около 30 минут. Затем автоклав открывают и обнаруживают, что смесь в автоклаве превратилась в суспензию белого цвета. Суспензию белого цвета подвергают фильтрации при использовании мембранного фильтра (НО20А142С; выпускается фирмой Advantec Toyo Kaisha, Ltd., Япония), получая таким образом твердые вещества белого цвета и фильтрат. Твердые вещества белого цвета промывают 2 раза с помощью 20 мл гексанола. Фильтрат переносят в круглодонную колбу грушевидной формы емкостью 1 л и подвергают перегонке при нагревании. Путем перегонки рекуперируют гексанол и дигексилкарбонат. Выход дигексилкарбоната составляет 16%.
Пример 7
Синтез диметилкарбоната
Стадия (1): Получение диметилкарбоната из дибутилоловодиметилата и метанола
В работающий под высоким давлением рактор емкостью 10 мл (выпускается фирмой Thar Designs Inc., США), снабженный вентилем, загружают 1,48 г (5 ммоль) дибутилоловодиметилата (выпускается фирмой Aldrich, США), 1,6 г (50 ммоль) метанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; безводный) и шар SUS (который используют для перемешивания содержимого реактора). Содержимое реактора охлаждают до температуры около -68°С с помощью смеси сухого льда и этанола. Затем из баллона с газообразным диоксидом углерода 2,0 г газообразного диоксида углерода высокой чистоты, давление которого снижают до примерно 2 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, осторожно вводят в работающий под высоким давлением реактор. После этого реактор помещают на масляную баню, которую поддерживают при температуре 150°С, и встряхивают в течение 15 часов. Спустя этот период времени содержимое реактора охлаждают до температуры около 20°С и внутреннее давление в реакторе возвращают к атмосферному давлению путем осторожного удаления избыточного количества газообразного диоксида углерода, получая таким образом реакционную смесь. В реакционной смеси диметилкарбонат получают с выходом 30%.
Стадия (2): Операция, при которой к полученной на стадии (1) реакционной смеси добавляют содержащий воду метанол, получая таким образом смесь, содержащую осадившиеся в ней твердые вещества, и содержащую твердые вещества смесь подвергают перегонке.
К реакционной смеси (содержащейся в реакторе), полученной на стадии (1), добавляют 2 мл метанола, содержащего 10% масс. воды, и реактор встряхивают при комнатной температуре (около 20°С) в течение около 5 минут. Затем реактор открывают и обнаруживают, что смесь в реакторе превратилась в суспензию белого цвета. Полученную суспензию белого цвета переносят в круглодонную колбу грушевидной формы емкостью 50 мл и подвергают перегонке при нагревании. Путем перегонки рекуперируют метанол и диметилкарбонат. Выход диметилкарбоната составляет 29%.
Пример 8
Получение метилата металла из дибутилоловооксида и метанола путем азеотропной перегонки воды
2,5 г дибутилоловооксида (выпускается фирмой Aldrich, США), 32,0 г метанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; реактивная чистота) и 100 мл гексана (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; реактивная чистота) загружают в трехгорлую колбу емкостью 200 мл, снабженную транспортирующим жидкость насосом и холодильником Либиха, соединенным с ловушкой. Трехгорлую колбу помещают на масляную баню, которую поддерживают при температуре 80°С, и осуществляют перегонку при нагревании в течение 4 часов при перемешивании содержимого колбы с помощью мешалки. В процессе перегонки определяют количества гексана и метанола, удаляемые из колбы, и в вышеуказанную колбу загружают свежий гексан и метанол с помощью транспортирующего жидкость насоса в количествах, которые являются, соответственно, такими же, как удаляемые из колбы количества гексана и метанола, с тем, чтобы поддерживать постоянными количества гексана и метанола в вышеуказанной колбе. После перегонки содержимое трехгорлой колбы охлаждают до температуры 30°С и гексан и избыточное количество метанола удаляют из колбы путем перегонки при пониженном давлении, получая таким образом вязкую прозрачную жидкость. Полученную жидкость подвергают 119Sn-ЯМР-анализу. В 119Sn-ЯМР-спектре полученной жидкости обнаруживают пики, соответственно, при -174 м.д. и -180 м.д.
Стадия (1): Операция, при которой к вышеполученной жидкости добавляют метанол и реакцию осуществляют в присутствии газообразного диоксида углерода, находящегося под высоким давлением
В работающий под высоким давлением реактор емкостью 10 мл (выпускается фирмой Thar Designs Inc., США), снабженный вентилем, загружают 0,66 г вышеполученной жидкости (содержащей металлоорганическое соединение), 1,6 г метанола и шар SUS316 (который используют для перемешивания содержимого реактора). Содержимое реактора охлаждают до температуры около -68°С с помощью смеси сухого льда и этанола. Затем из баллона с газообразным диоксидом углерода 2,8 г газообразного диоксида углерода высокой чистоты, давление которого снижают до примерно 2 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, осторожно вводят в работающий под высоким давлением реактор. После этого реактор помещают на масляную баню, которую поддерживают при температуре 160°С, и встряхивают в течение 15 часов. Спустя этот период времени содержимое реактора охлаждают до температуры около 20°С и внутреннее давление в реакторе возвращают к атмосферному давлению путем осторожного удаления избыточного количества газообразного диоксида углерода, получая таким образом суспензию белого цвета в качестве реакционной смеси. В реакционной смеси диметилкарбонат получают с выходом 6%.
Стадия (2): Выделение диметилкарбоната
К суспензии белого цвета (содержащейся в работающем под высоким давлением реакторе), полученной на стадии (1), добавляют 10 мл метанола. Затем полученную в работающем под высоким давлением реакторе смесь переносят в круглодонную колбу грушевидной формы емкостью 50 мл. Содержимое вышеуказанной колбы подвергают перегонке при нагревании на масляной бане, которую поддерживают при температуре 90°С. Путем перегонки метанол и диметилкарбонат рекуперируют из остаточной жидкости в колбе. Выход диметилкарбоната составляет 6%.
Стадия (3): Синтез (регенерация) металлоорганического соединения
Остаточную жидкость, которая остается в колбе после рекуперации диметилкарбоната путем осуществляемой на стадии (2) перегонки, переносят в трехгорлую колбу емкостью 100 мл, снабженную транспортирующим жидкость насосом и холодильником Либиха, соединенным с ловушкой. В вышеуказанную колбу помещают мешалку и загружают 30 мл гексана и 30 мл метанола. Трехгорлую колбу помещают на масляную баню, которую поддерживают при температуре 80°С, и осуществляют перегонку при нагревании в течение 4 часов при перемешивании содержимого колбы с помощью мешалки. В процессе перегонки определяют количества гексана и метанола, удаляемые из колбы, и в вышеуказанную колбу загружают свежий гексан и метанол с помощью транспортирующего жидкость насоса в количествах, которые, соответственно, являются такими же, как количества гексана и метанола, удаляемые из колбы, с тем, чтобы поддерживать постоянными количества гексана и метанола в вышеуказанной колбе. После перегонки содержимое колбы охлаждают до температуры 30°С и гексан и избыточное количество метанола удаляют из колбы путем перегонки при пониженном давлении, получая таким образом вязкую прозрачную жидкость. Полученную жидкость подвергают 119Sn-ЯМР-анализу. На 119Sn-ЯМР-спектре полученной жидкости обнаруживают пики, соответственно, при -174 м.д. и -180 м.д.
Пример 9
Стадия (1): Получение эфира угольной кислоты из дибутилоловодибутилата и спирта
В работающий под высоким давлением реактор емкостью 10 мл (выпускается фирмой Thar Designs Inc., США), снабженный вентилем, загружают 1,48 г (4 ммоль) дибутилоловодибутилата (выпускается фирмой Aldrich, США), 2,22 г (30 ммоль) бутанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; безводный), 1,38 г (30 ммоль) этанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; безводный) и шар SUS (который используют для перемешивания содержимого реактора). Содержимое реактора охлаждают до температуры около -68°С с помощью смеси сухого льда и этанола. Затем из баллона с газообразным диоксидом углерода 2,0 г газообразного диоксида углерода высокой чистоты, давление которого снижают до примерно 2 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, осторожно вводят в работающий под высоким давлением реактор. После этого реактор помещают на масляную баню, которую поддерживают при температуре 150°С, и встряхивают в течение 22 часов. Спустя этот период времени содержимое реактора охлаждают до температуры около 20°С и внутреннее давление в реакторе возвращают к атмосферному давлению путем осторожного удаления избыточного количества газообразного диоксида углерода, получая таким образом реакционную смесь. В реакционной смеси этилбутилкарбонат, дибутилкарбонат и диэтилкарбонат получают с выходами соответственно 25%, 10% и 6%.
Стадия (2): Рекуперация эфиров угольной кислоты путем перегонки из полученной на стадии (1) реакционной смеси
Полученную на стадии (1) реакционную смесь переносят в круглодонную колбу грушевидной формы емкостью 50 мл и подвергают перегонке при пониженном давлении. Путем перегонки рекуперируют этанол, бутанол и эфиры угольной кислоты. Что касается выходов рекуперированных эфиров угольной кислоты - этилбутилкарбоната, дибутилкарбоната и диэтилкарбоната, - они составляют соответственно 23%, 8% и 5%.
Пример 10
Стадия (1): Операция, при которой к вязкой прозрачной жидкости (содержащей металлоорганическое соединение), получаемой по существу таким же образом, как в примере 8, добавляют метанол и осуществляют реакцию в присутствии находящегося под высоким давлением газообразного диоксида углерода
В трубчатый реактор SUS316 (объем: 8 мл; наружный диаметр: 12,7 мм; толщина стенки: 2,1 мм), снабженный трубопроводом SUS316 и вентилем, загружают 1,1 г вязкой прозрачной жидкости (содержащей металлоорганическое соединение), которую получают по существу таким же образом, как в примере 8, 2,6 г метанола и шар SUS316 (который используют для перемешивания содержимого реактора). Содержимое реактора охлаждают до температуры около -68°С с помощью смеси сухого льда и этанола. Затем из баллона с газообразным диоксидом углерода 2,8 г газообразного диоксида углерода высокой чистоты, давление которого снижают до примерно 2 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, осторожно вводят в трубчатый реактор. После этого реактор помещают на масляную баню, которую поддерживают при температуре 150°С, и встряхивают в течение 12 часов. Спустя этот период времени содержимое реактора охлаждают до температуры около 20°С и внутреннее давление в реакторе возвращают к атмосферному давлению путем осторожного удаления избыточного количества газообразного диоксида углерода, получая таким образом суспензию белого цвета в качестве реакционной смеси. В реакционной смеси диметилкарбонат получают с выходом около 5%.
Пример 11
Получение содержащего гексилоксигруппу металлоорганического соединения из дибутилоловооксида и н-гексанола
В круглодонную колбу грушевидной формы емкостью 300 мл, снабженную холодильником и насадкой Дина-Старка, загружают 24,9 г дибутилоловооксида (выпускается фирмой Aldrich, США), 40,9 мл н-гексанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; реактивная чистота) и 150 мл толуола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; реактивная чистота). Колбу помещают на масляную баню, которую поддерживают при температуре 120°С, и содержимое колбы подвергают кипячению с обратным холодильником в течение 12 часов при перемешивании. Спустя этот период времени, содержимое колбы охлаждают до температуры 80°С и избыточное количество н-гексанола удаляют из реакционной смеси путем перегонки при пониженном давлении, получая таким образом жидкость, содержащую 75,1 г металлоорганического соединения. Потом полученную жидкость подвергают 119Sn-ЯМР-анализу. На 119Sn-ЯМР-спектре жидкости обнаруживают пик при -130 м.д., приписываемый металлоорганическому соединению формулы (1), и пики, соответственно, при -177 м.д. и -187 м.д., приписываемые металлоорганическому соединению формулы (2).
Стадия (1): Получение дигексилкарбоната из металлоорганического соединения и гексанола при введении газообразного диоксида углерода, находящегося при атмосферном давлении
В двугорлую колбу, снабженную холодильником, вставляют нагнетательную трубку, которая соединена, кроме того, со стеклянным шаровым фильтром (G2) (выпускается фирмой Vidrex Co., Ltd., Япония). Далее, в колбу помещают мешалку. В двугорлую колбу загружают 0,75 г вышеполученной жидкости (содержащей металлоорганическое соединение) и 41 г н-гексанола. Затем начинают введение в колбу газообразного диоксида углерода высокой чистоты через нагнетательную трубку со скоростью потока 100 мл/мин. Колбу нагревают на масляной бане (которую поддерживают при температуре 130°С) при перемешивании содержимого колбы и введении в нее газообразного диоксида углерода высокой чистоты, получая таким образом дигексилкарбонат. Спустя 288 часов после начала нагревания колбы выход дигексилкарбоната составляет 40%.
Пример 12
Получение циклогексилата металла из дибутилоловооксида и циклогексанола путем азеотропной перегонки воды
В круглодонную колбу грушевидной формы емкостью 500 мл, снабженную холодильником и насадкой Дина-Старка, загружают 5,1 г дибутилоловооксида (выпускается фирмой Aldrich, США), 80,1 г циклогексанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; реактивная чистота) и 300 мл толуола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; реактивная чистота). Колбу помещают на масляную баню, которую поддерживают при температуре 130°С, и содержимое колбы подвергают кипячению с обратным холодильником в течение 12 часов при перемешивании. Спустя этот период времени, содержимое колбы охлаждают до температуры 80°С. Избыточное количество циклогексанола удаляют из колбы путем перегонки при пониженном давлении, получая таким образом жидкость, содержащую 15,2 г металлоорганического соединения. Потом полученную жидкость подвергают 119Sn-ЯМР-анализу. На 119Sn-ЯМР-спектре жидкости обнаруживают пики, соответственно, при -176 м.д. и -190 м.д., приписываемые металлоорганическому соединению формулы (2).
Стадия (1): Получение дициклогексилкарбоната, согласно которому циклогексанол добавляют к вышеполученной жидкости и реакцию осуществляют в присутствии газообразного диоксида углерода, находящегося под высоким давлением.
В трубчатый реактор SUS316 (объем: 8 мл; наружный диаметр: 12,7 мм; толщина стенки: 2,1 мм), снабженный трубопроводом SUS316 и вентилем, загружают 0,86 г вышеполученной жидкости, содержащей металлоорганическое соединение, 1,0 г циклогексанола и шар SUS316 (который используют для перемешивания содержимого реактора). Содержимое реактора охлаждают до температуры около -68°С с помощью смеси сухого льда и этанола. Затем из баллона с газообразным диоксидом углерода 2,0 г газообразного диоксида углерода высокой чистоты, давление которого снижают до примерно 2 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, осторожно вводят в реактор. Реактор помещают на масляную баню, которую поддерживают при температуре 130°С, и встряхивают в течение 14 часов. Спустя этот период времени содержимое реактора охлаждают до температуры около 20°С и внутреннее давление в реакторе возвращают к атмосферному давлению путем осторожного удаления избыточного количества газообразного диоксида углерода, получая таким образом прозрачную реакционную смесь. Эту полученную прозрачную реакционную смесь анализируют. В результате найдено, что дициклогексилкарбонат получен с выходом 40%.
Пример 13
Получение метилэтилкарбоната
Стадия (1): Получение метилэтилкарбоната из тетраметилата титана, газообразного диоксида углерода, метанола и этанола
В работающий под высоким давлением реактор емкостью 10 мл (выпускается фирмой Thar Designs Inc., США), снабженный вентилем, загружают 0,9 г (5 ммоль) тетраметилата титана (выпускается фирмой AZmax. co., Япония), около 0,9 г (30 ммоль) метанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; безводный), около 1,4 г (30 ммоль) этанола (выпускается фирмой Wako Pure Chemical Industries Ltd., Япония; безводный) и шар SUS (который используют для перемешивания содержимого реактора).
Содержимое реактора охлаждают до температуры около -68°С с помощью смеси сухого льда и этанола. Затем из баллона с газообразным диоксидом углерода 2,0 г газообразного диоксида углерода высокой чистоты, давление которого снижают до примерно 2 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, осторожно вводят в автоклав. Реактор помещают на масляную баню, которую поддерживают при температуре 150°С, и встряхивают в течение 15 часов. Спустя этот период времени содержимое реактора охлаждают до температуры около 20°С и внутреннее давление в реакторе возвращают к атмосферному давлению путем осторожного удаления избыточного количества газообразного диоксида углерода, получая таким образом суспензию белого цвета в качестве реакционной смеси. В реакционной смеси метилэтилкарбонат, диметилкарбонат и диэтилкарбонат получают с выходами соответственно 25%, 3% и 4%.
Стадия (2): Выделение эфиров угольной кислоты
Вышеуказанную суспензию переносят в круглодонную колбу грушевидной формы емкостью 50 мл и подвергают ее перегонке при пониженном давлении при температуре 30°С. Путем перегонки рекуперируют эфиры угольной кислоты. Что касается выходов рекуперированных эфиров угольной кислоты, то метилэтилкарбонат, диметилкарбонат и диэтилкарбонат получают с выходами соответственно 23%, 2% и 3%.
Сравнительный пример
В этом сравнительном примере получение диметилкарбоната из дибутилоловооксида (который не содержит металл-кислород-углеродной связи), метанола и газообразного диоксида углерода осуществляют как описывается ниже.
Используют автоклав емкостью 200 мл (выпускаемый фирмой Toyo Koatsu Co., Ltd., Япония), который, кроме того, соединен с линией для введения жидкого диоксида углерода и газообразного диоксида углерода, линией удаления дистиллята, пробоотборной трубкой и линией для введения газообразного азота в нижнюю часть автоклава. В автоклав емкостью 200 мл загружают 15,0 г (60 ммоль) дибутилоловооксида (выпускается фирмой Aldrich, США) и 48,1 г (1,5 моль) метанола. Все вентили закрывают. Затем, из баллона с газообразным диоксидом углерода, газообразный диоксид углерода, давление которого снижают до 5 МПа с помощью регулятора давления, связанного с содержащим газообразный диоксид углерода баллоном, вводят в автоклав. Начинают перемешивание содержимого автоклава и температуру внутри автоклава повышают до 160°С. По линии для подачи сырья в автоклав постепенно вводят жидкий диоксид углерода с тем, чтобы довести внутреннее давление в автоклаве до 22 МПа. Затем осуществляют реакцию в течение 16 часов при поддерживании внутреннего давления в автоклаве 22 МПа. Спустя этот период времени, содержимое автоклава охлаждают до температуры около 30°С с последующим удалением газообразного диоксида углерода, получая таким образом суспензию белого цвета в качестве реакционной смеси. Полученную реакционную смесь анализируют путем газовой хроматографии (ГХ). Согласно ГХ-анализу, диметилкарбонат в реакционной смеси не обнаруживают.
Промышленная применимость
По способу согласно настоящему изобретению эфир угольной кислоты может быть получен с высоким выходом из металлоорганического соединения, содержащего металл-кислород-углеродную связь, и диоксида углерода. Преимуществом этого способа является то, что диоксид углерода нетоксичен и не агрессивен и является недорогостоящим. Далее, способ согласно настоящему изобретению выгоден не только в том, что металлоорганическое соединение после использования в этом способе может быть регенерировано и рециркулировано на стадию (1) способа, предотвращая таким образом образование отходов, происходящих за счет металлоорганического соединения, а также в том, что нет необходимости использовать большое количество осушителя, предотвращая таким образом образование отходов, происходящих за счет осушителя. Следовательно, способ согласно настоящему изобретению является коммерчески пригодным и имеет высокую коммерческую ценность.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРА УГОЛЬНОЙ КИСЛОТЫ | 2003 |
|
RU2296117C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКОКСИДОВ АЛКИЛОЛОВА | 2005 |
|
RU2338749C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 2004 |
|
RU2329250C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАЛКОКСИДОВ ДИАЛКИЛОЛОВА | 2007 |
|
RU2414474C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО НИТРИЛЬНОГО СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ КАРБОНАТНОГО СЛОЖНОГО ЭФИРА | 2017 |
|
RU2739444C2 |
| СМЕСЬ ДЛЯ ИЗВЛЕЧЕНИЯ С ЦЕЛЬЮ ИСПОЛЬЗОВАНИЯ ИЛИ ТРАНСПОРТА ДИОКСИДА УГЛЕРОДА | 2007 |
|
RU2396107C2 |
| СПОСОБ ОТДЕЛЕНИЯ И ИЗВЛЕЧЕНИЯ ДИАЛКОКСИДА ДИАЛКИЛОЛОВА | 2007 |
|
RU2401838C2 |
| СПОСОБ АЦИЛИРОВАНИЯ ГЕКСАКИС(ФЕНИЛМЕТИЛ)ГЕКСААЗАИЗОВЮРЦИТАНА | 1998 |
|
RU2182151C2 |
| СПОСОБ ПРЯМОГО АМИНИРОВАНИЯ ВТОРИЧНЫХ СПИРТОВ С ПОМОЩЬЮ АММИАКА ДО ПЕРВИЧНЫХ АМИНОВ | 2011 |
|
RU2593994C2 |
| НОВОЕ АЛИЦИКЛИЧЕСКОЕ ДИОЛЬНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2646220C2 |
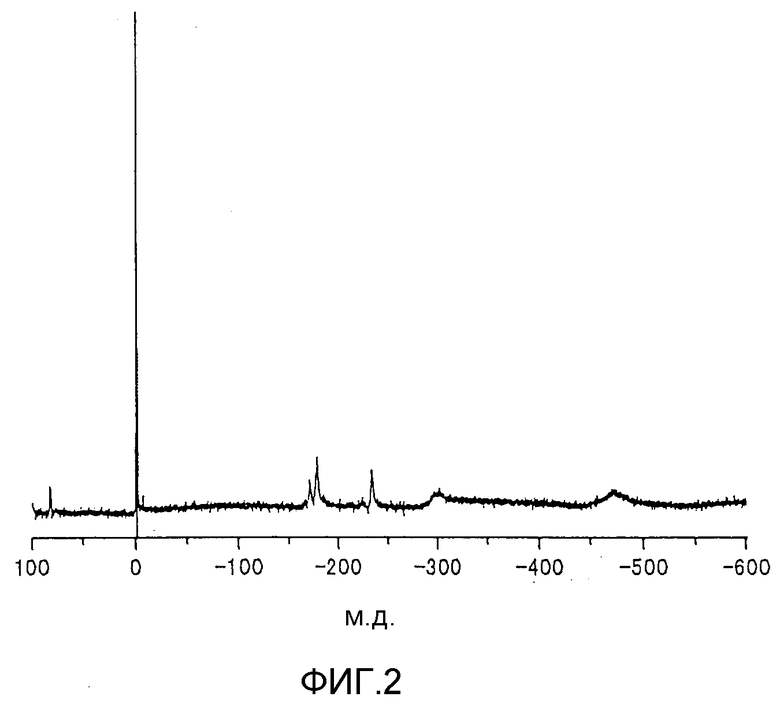
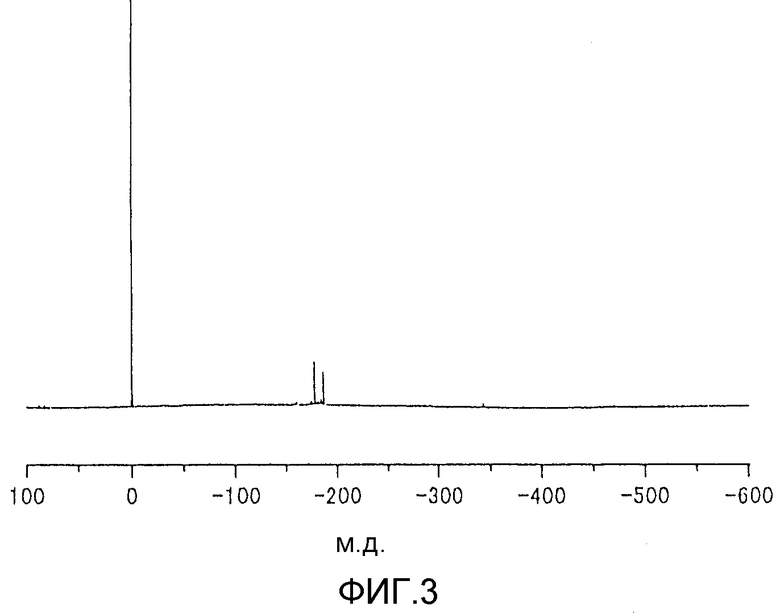
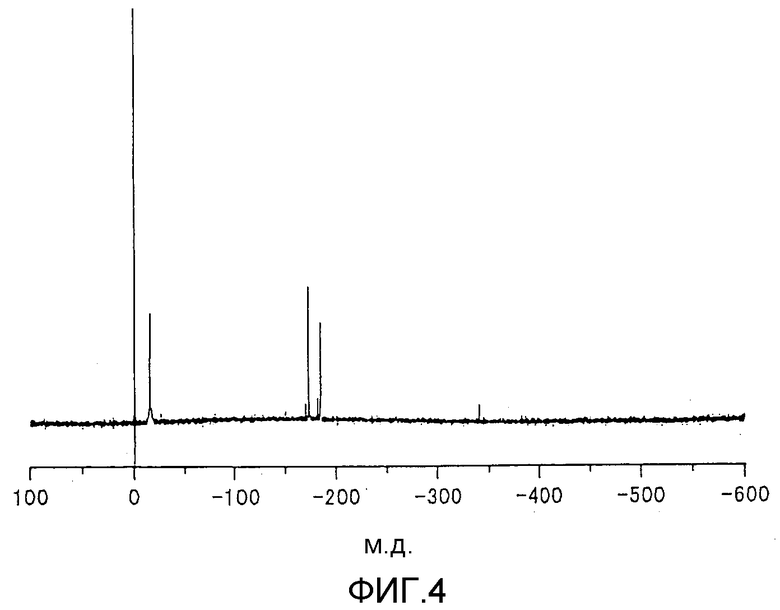
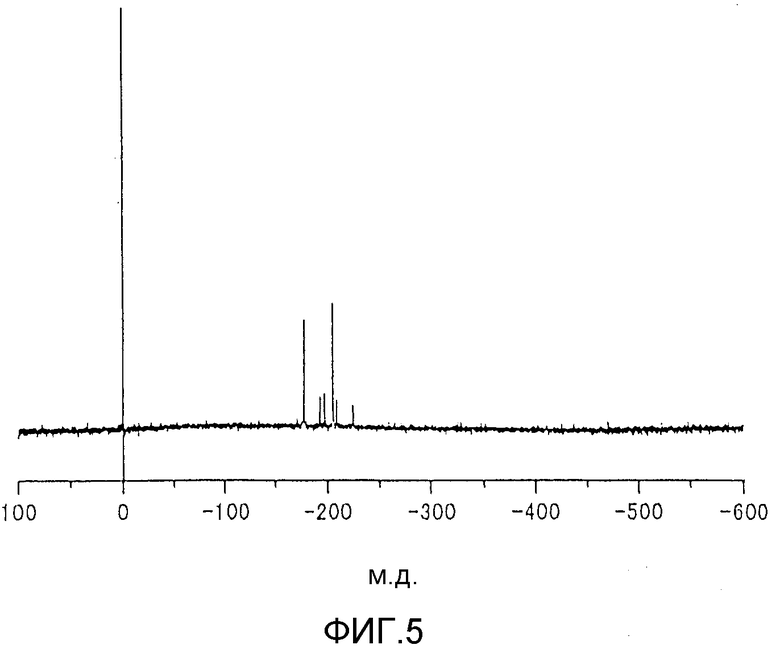
Настоящее изобретение относится к способу получения эфира угольной кислоты из металлоорганического соединения и диоксида углерода. Эфир угольной кислоты используют в качестве добавок для различных целей, в качестве алкилирующего или карбонилирующего агента, растворителя и т.д. в области синтеза органических соединений, в качестве электролита или в качестве исходного материала для получения смазочного масла или получения восстановителя. Предложен способ получения эфира угольной кислоты, включающий: (1) осуществление реакции между содержащим металл-кислород-углеродную связь металлоорганическим соединением и диоксидом углерода с получением реакционной смеси, содержащей образовавшийся путем реакции эфир угольной кислоты; (2) выделение эфира угольной кислоты из вышеуказанной реакционной смеси с получением остаточной жидкости; и (3) введение во взаимодействие вышеуказанной остаточной жидкости с первым спиртом с образованием по меньшей мере одного, содержащего металл-кислород-углеродную связь металлоорганического соединения, где по меньшей мере одно вышеуказанное металлоорганическое соединение, полученное на стадии (3), рекуперируют для рециркуляции его на стадию (1), причем вышеуказанное металлоорганическое соединение, используемое на стадии (1), включает по меньшей мере одно соединение, указанное в материалах заявки. Достигнутый технический результат заключается в получении эфира карбоновой кислоты с высоким выходом и в регенерации металлоорганического соединения. 26 з.п. ф-лы, 5 ил., 2 табл.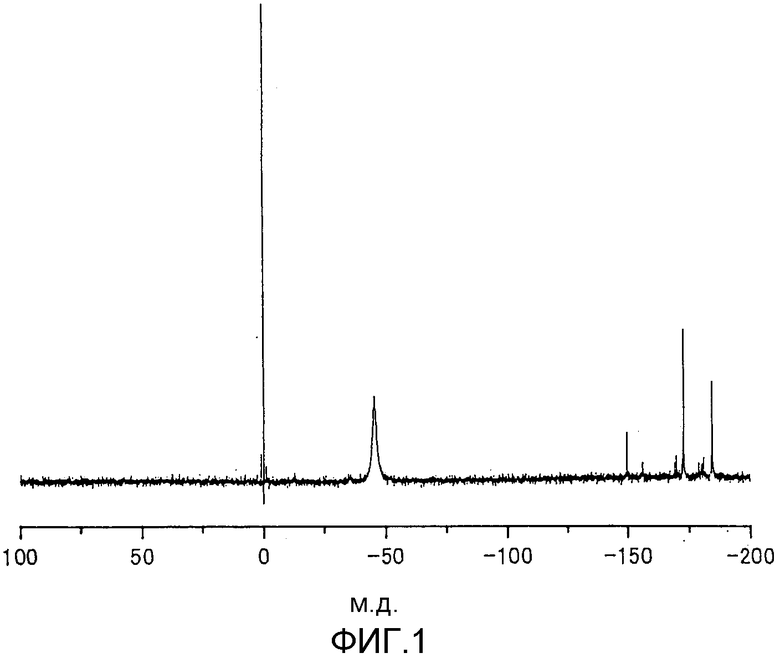
(1) осуществление реакции между содержащим металл-кислород-углеродную связь металлоорганическим соединением и диоксидом углерода с получением реакционной смеси, содержащей образовавшийся путем реакции эфир угольной кислоты;
(2) выделение эфира угольной кислоты из вышеуказанной реакционной смеси с получением остаточной жидкости и
(3) введение во взаимодействие вышеуказанной остаточной жидкости с первым спиртом с образованием по меньшей мере одного, содержащего металл-кислород-углеродную связь металлоорганического соединения и воды и удаление вышеуказанной воды из, по меньшей мере, одного вышеуказанного металлоорганического соединения, где, по меньшей мере, одно вышеуказанное металлоорганическое соединение, полученное на стадии (3), рекуперируют для рециркуляции его на стадию (1), причем вышеуказанное металлоорганическое соединение, используемое на стадии (1), включает по меньшей мере одно соединение, выбираемое из группы, состоящей из
металлоорганического соединения, отвечающего формуле (I)
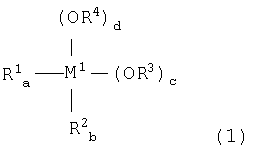
где M1 означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния;
каждый из R1 и R2, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью, (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, или незамещенную или замещенную (С6-С20)-арильную группу;
каждый из R3 и R4, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью или (C7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила; и
каждый из а и b означает целое число от 0 до 2, (a+b) = от 0 до 2; каждый из с и d означает целое число от 0 до 4; и (a+b+c+d)=4, и
металлоорганического соединения, отвечающего формуле (2)
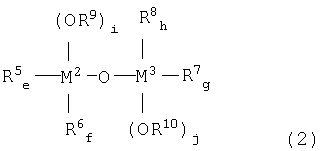
где каждый из М2 и М3, независимо, означает атом металла, выбираемый из группы, состоящей из элементов 4 и 14 групп периодической таблицы, за исключением кремния;
каждый из R5, R6, R7 и R8, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью, (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила, или незамещенную или замещенную (С6-С20)-арильную группу;
каждый из R9 и R10, независимо, означает (С1-С12)-алкильную группу с линейной или разветвленной цепью, (С5-С12)-циклоалкильную группу, (С2-С12)-алкенильную группу с линейной или разветвленной цепью или (С7-С20)-аралкильную группу, включающую незамещенный или замещенный (С6-С19)-арил и алкил, выбираемый из группы, состоящей из (С1-С14)-алкила с линейной или разветвленной цепью и (С5-С14)-циклоалкила и
(e+f) = от 0 до 2; (g+h) = от 0 до 2; каждый из i и j, независимо, означает целое число от 1 до 3; (e+f+i)=3 и (g+h+j)=3.
Приоритет по пунктам и признакам:
| Способ получения эфиров угольной кислоты | 1974 |
|
SU603330A3 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ПОЛИРОВАЛЬНИК К МАШИНЕ ДЛЯ ШЛИФОВАНИЯ И ПОЛИРОВАНИЯ ЛИСТОВОГО СТЕКЛА | 1948 |
|
SU85347A1 |

