Настоящее изобретение касается хемокаталитического жидкофазного способа прямого одностадийного аминирования при необходимости многоатомных и/или содержащих функциональные группы вторичных спиртов до при необходимости имеющих несколько аминогрупп и/или содержащих функциональные группы первичных аминов с помощью аммиака с высоким выходом при помощи гомогенной системы катализаторов.
Превращение кислородсодержащих в азотсодержащие функциональные группы представляет собой незаменимую трансформацию для синтеза большого числа органических соединений. В литературе и технике известен ряд классических методов, чтобы решить указанную задачу.
При этом в основной массе большого числа публикаций первичный или вторичный спирт вводится в реакцию с первичным или вторичным органическим амином. И напротив, взаимодействие первичного или вторичного спирта с аммиаком с получением первичных аминов согласно Схеме 1 было описано только с применением особенных условий проведения процесса, катализаторов и только с некоторыми малочисленными спиртами.

Сложность всех известных способов состоит в достижении высоких селективностей в отношении первичных аминов, поскольку эти амины являются более нуклеофильными, чем аммиак и вследствие этого предпочтительно могут вступать в реакцию с образованием более высокозамещенных аминов. В то время как превращение изолированной гидроксильной функциональной группы в аминную функциональную группу протекает приблизительно термически нейтрально, образование вторичного или третичного амина является экзотермическим примерно из расчета по 30 кДж/моль и, следовательно, также термодинамически предпочтительным по сравнению с образованием первичного амина.
Прямое аминирование в газовой фазе
Одностадийное прямое превращение первичной или вторичной гидроксильной группы с помощью аммиака в первичный амин в случае низших, легко испаряемых спиртов главным образом ограничено газофазными реакциями. При этом соответствующий спирт испаряется и при подходящих условиях (давление, температура, парциальное давление водорода, а при необходимости инертного газа) вводится в реакцию в большинстве случаев на гетерогенном катализаторе. Этот способ проведения процесса описывается, например, в публикациях патентов США US 4314084, US 5530127, US 5932769, французском патенте FR 1347648, патентах США US 3270059, US 4111840, US 4123462, немецком патенте DE 1667193, Fischer с соавт.(J. Catal., 1999, 182, 289-291) или Jenzer с соавт.(Catal. Lett., 1999, 61, 111-114). Недостатком большинства гетерогенно катализируемых газофазных способов является применение высоких температур (вплоть до 400°C) и давлений (до 300 бар), вследствие которых помимо желаемых первичных аминов часто образуются значительные количества более высокозамещенных аминов, аленов и алканов. К тому же в соответствии с характеристичными условиями давления и температуры газофазной реакции с помощью указанного способа с рентабельными выходами в амины могут превращаться исключительно такие вещества, которые могут испаряться и переводиться в другое состояние без потерь, или соответственно при этом могут конденсироваться и ресублимироваться без потерь амины. Поэтому вещества или соответствующие им амины, которые при таких условиях подвергаются разложению, в литературе и технике подвергают взаимодействию в жидкофазном синтезе.
Восстановительное аминирование
Известные специалисту способы получения первичных аминов из спиртов посредством восстановительного аминирования используют многостадийную технологию процесса, которая может сопровождаться изменением состояния окисления атома углерода, имеющего гидроксильную группу. От этого могут разграничиваться способы, которые осуществляются с сохранением степени окисления (прямое аминирование). При изменении степени окисления атома углерода, имеющего гидроксильную группу, (восстановительном аминировании) в классическом способе спирты могут получаться в результате окисления соответствующего карбонильного соединения, последующего образования имина при помощи реакции с аминовым компонентом (первичным, вторичным амином или аммиаком) и последующего гомогенно или соответственно гетерогенно катализируемого восстановления имина водородом. Однако двухстадийный способ проведения процесса с выделением карбонильного соединения является требующим много времени и затрат.
Специальные многостадийные процессы
При сохранении степени окисления атома углерода, имеющего гидроксильную группу, (прямом аминировании) спирты могут превращаться в амины в результате многостадийных реакций замещения. Помимо затрат на выделение промежуточных стадий при соответствующем способе негативно отражается, в частности, работа с часто применяемыми в этой связи взрывоопасными и токсичными азидами.
Исключение из многостадийных способов работы для восстановительного аминирования спиртов с сохранением степени окисления атома углерода, имеющего гидроксильную группу, представляет собой, например, последовательное взаимодействие первичных спиртов с диалкилазодикарбоксилатами, бистретбутилиминодикарбонатом и иммобилизованным трифенилфосфаном, которое согласно Sun с соавт. (Tetrahedron Lett., 2007, 48, 7745-7746) после добавления трифторуксусной кислоты допускает прямой способ доступа к первичным аминам без предварительного выделения промежуточных стадий.
Fabiano с соавт.(Synlett, 1987, 1987, 190-192) для аналогичной цели вместо бистретбутилмонодикарбоната используют ядовитую азотистоводородную кислоту.
Прямое жидкофазное аминирование спиртов
Прямое одностадийное жидкофазное аминирование при необходимости многоатомных первичных спиртов с помощью аммиака уже давно описано в научной и патентной литературе. В некоторых случаях описанные способы не могут однозначно классифицироваться как газо- или жидкофазные способы на основании используемых условий процесса. При температурах около 170°C и давлении 200 бар согласно немецкой заявке на патент DE 19507007 этаноламин может аминироваться на нанесенном на оксид рутениевом катализаторе с получением этилендиамина, причем выходы, которых можно достигнуть, остаются ниже 40%.
Получение первичных моноаминов, при необходимости содержащих функциональные группы, с высокими выходами из соответствующих одноатомных, при необходимости содержащих функциональные группы первичных спиртов описывается в работах Milstein с соавт. (Angew. Chem. Int. Ed., 2008, 47, 8661-8664). Здесь описывается прямое одностадийное аминирование частично замещенного гетероатомами алифатического и бензильного спирта, в результате идущего от 12 до 36 часов взаимодействия с избытком аммиака в растворителе при 7,5 бар и температуре реакции 135-180°C. В качестве катализатора используется стабильный на воздухе пинцерный комплекс на основе акридинила, карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутений(II)], и достигаются выходы между 78 и 96%.
Кроме того, в международной заявке WO 2010018570 описывается использование пинцерных лигандов на хинолинильной основе со сравнимыми выходами.
Недостатком обоих опубликованных способов является то, что с их помощью с получением аминов могут взаимодействовать исключительно первичные спирты; это также соответствует ожиданию, поскольку многократно описано, что катализаторы, подходящие для первичных спиртов, не подходят для вторичных спиртов. Например, Beller, M. с соавт., ChemSusChem, 2009, 2, 551-557 заявляют, что указанный там катализатор селективно превращает реакционноспособные OH-группы диола (первичную OH-группу перед вторичной OH-группой; простую вторичную OH-группу перед стерически затрудненной вторичной OH-группой). Вдобавок Baiker с соавт.(J. Mol. Catal. A: Chem., 1999, 149, 197-204) показывают, что характеристики аминирования первичных диолов ощутимо зависят от расположения заместителей других атомов углерода, находящихся в веществе, что, в свою очередь, наводит на мысль о том, что окружение вторичного спирта, полностью отличающееся от первичного спирта, предусматривает неперспективным использование катализатора, работающего для первичного спирта.
Для имеющих функциональные группы вторичных спиртов в литературе известно снижение селективности образования первичных аминов с увеличивающейся длиной цепи спиртового субстрата. Так, Imm с соавт.(S. Imm, S. Bahn, L. Neubert, H.Neumann, M. Beller, Angew. Chem. 2010, 122(44), 8303-6) описывают значительное снижение селективности для первичного амина с 76 до 58%, когда вместо 3-фенил-2-пропанола в присутствии гомогенного рутениевого катализатора аминируется 4-фенил-2-бутанол. Аналогичным образом, при аминировании алифатических вторичных спиртов для 2-нонанола может наблюдаться заметно более низкий выход амина (51,2%), чем в случае более низкомолекулярного гомолога 2-октанола (67,1%) (D. Pingen, С.Muller, D. Vogt, Angew. Chem. 2010, 122(44), 8307-10). Таким образом, следует исходить из того, что более высокомолекулярные и при необходимости дополнительно имеющие функциональные группы спирты таким способом не могут превращаться с высокими выходами в соответствующие амины.
Прямое одностадийное жидкофазное аминирование имеющих функциональные группы многоатомных спиртов с помощью аммиака было описано исключительно на гетерогенных катализаторах. В немецком патенте DE 3903367 диол простого эфира - диэтиленгликоль аминировали на различных медно-кобальто-никелевых катализаторах (Cu-Co-Ni-катализаторах), нанесенных на диоксид циркония, с помощью жидкого аммиака при 200°C в атмосфере водорода при 30 бар. Однако ни в одном случае в качестве продукта реакции не был выделен диамин простого эфира, а только аминоэтоксиэтанол и морфолин.
С высокими выходами, вплоть до 95,8% согласно немецкому патенту DE 1570542 простые полиэфирдиолы, такие как полипропиленгликоль, могут превращаться прямо в соответствующие диамины, если это взаимодействие осуществляется при 240°C в присутствии никелевых катализаторов Ренея. Однако этот способ проведения процесса также не подходит для неустойчивых к нагреванию веществ, например, являющихся производными углеводов.
Однако согласно патенту США US 4153581 с применением кобальт-медь-цинкового катализатора (Co-Cu-Zn-катализатора) удается получение простых полиэфираминов уже при 140°C, однако это не подходит для вторичных спиртов.
В схожих гетерогенно катализируемых процессах, кроме того, описываются катализаторы на основе кобальта-хрома-марганца (Co-Cr-Mn) в присутствии P2O5 при 140-230°C и давлении водорода 200-300 бар (немецкий патент DE 1543377), на основе Ni/Al2O3 при 200-230°C и давлении водорода 15-20 бар (румынский патент RO 63243) или на основе кальция-алюмосиликатов при 260-300°C и давлении водорода 200 бар (немецкий патент DE 1278432).
При сравнимых условиях аминируют спирты в соответствии со способами, описанными в немецком патенте DE 19859776 (180-230°C на Cu-CuO/TiO2), немецком патенте DE 102006061045 (180-250°C на Ni-Cu/ZrO2), немецком патенте DE 102006061042 (180-220°C на Ni-Cu-Ru/ZrO2), международной заявке WO 2008072428 (180-250°C на Ru/ZrO2) и международной заявке WO 2007077903 (180-250°C на Ru/Al2O3); однако при этом дополнительно необходима атмосфера водорода.
Указанные примеры в качестве образца показывают потребность в способе, чтобы достичь активации спирта также без стехиометрического использования труднодоступных и токсичных вспомогательных веществ. К тому же решающим недостатком всех способов, до сих пор подходящих для прямого жидкофазного аминирования, является то, что для получения и при необходимости требующегося выделения и очистки интермедиатов, возникающих при последовательности синтеза, должны осуществляться дополнительные продолжительные и дорогостоящие технологические операции.
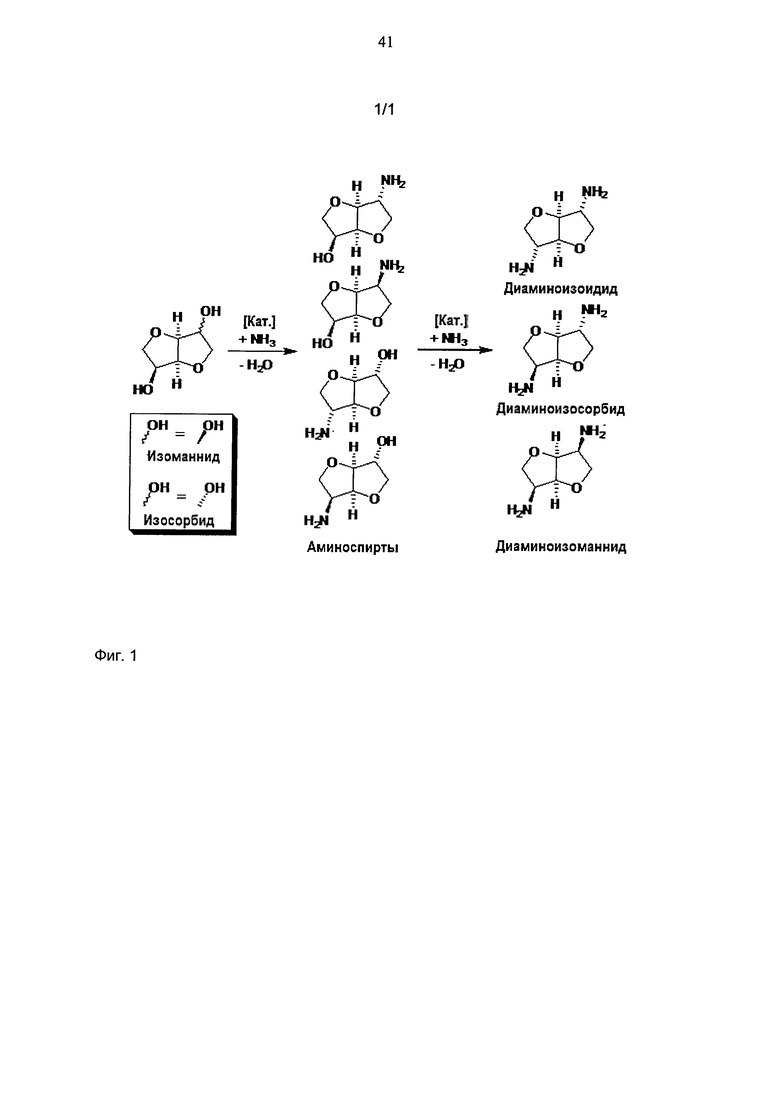
Особенно аминовые производные ангидрогекситолов, такие как, например, изосорбид, изоманнид или изоидид, до сих пор в литературе описаны только как получающиеся при помощи затратного способа.
Так международная заявка WO 2008/145921 описывает получение бисаминоалкильных производных изосорбида, которые получаются из изосорбида в результате присоединения акрилонитрила и последующего гидрирования.
Помимо нередко необходимых при описанных выше способах высоких температур, другой недостаток указанных способов состоит в том, что нужно работать в присутствии высокого парциального давления водорода, чтобы можно было получить целевые продукты с желаемыми выходами.
Согласно описанному уровню техники, неизвестен способ, который описывает прямое, одностадийное, обходящееся без участия водорода жидкофазное аминирование при необходимости многоатомных вторичных и при необходимости содержащих функциональные группы спиртов с помощью аммиака с получением первичных аминов при мягких условиях реакции и с высокими выходами.
Следовательно, задачей настоящего изобретения было предоставить способ получения первичных аминов, исходя из вторичных спиртов, который избегает по меньшей мере одного из указанных недостатков и может проводиться экономически выгодно.
Так, неожиданным образом был разработан способ, который позволяет проводить прямое аминирование вторичных спиртов с помощью аммиака в присутствии катализатора, такого как описан в пункте 1 Формулы изобретения, с высокими выходами, причем аминируется вторичная группа спирта.
Объектом настоящего изобретения, таким образом, является способ, который позволяет осуществлять прямое, гомогенно катализируемое жидкофазное аминирование при необходимости многоатомных и/или содержащих функциональные группы вторичных спиртов с помощью аммиака в количестве, превышающем стехиометрическое, в пересчете на гидроксильные группы, подлежащие аминированию, предпочтительно в отсутствие водорода, причем применяемые условия процесса допускают, в частности, также взаимодействие термически неустойчивых спиртов, например, получаемых из возобновляемого сырья.
Одним преимуществом способа согласно изобретению является то, что при этом взаимодействии предотвращаются необходимые в остальных случаях выделение и очистка на промежуточных стадиях.
Еще одним преимуществом является то, что может предотвращаться использование проблематичных вспомогательных веществ, таких как, например, азиды. Кроме того, еще одним преимуществом является то, что в результате способа согласно изобретению отсутствует образование продуктов сочетания.
Кроме того, предпочтительно то, что спирт вводится в реакцию в растворенном состоянии.
Еще одним преимуществом является то, что аминирование спирта может осуществляться без выделения и/или очистки промежуточных стадий или интермедиатов.
Способ согласно изобретению для получения первичных аминов включает в себя следующие стадии:
A) приготовление раствора вторичного спирта в жидкой, не газообразной фазе,
B) приведение во взаимодействие этой фазы со свободным аммиаком и/или по меньшей мере одним соединением, высвобождающим аммиак, и гомогенным катализатором, и при необходимости
C) выделение первичного амина, образовавшегося на стадии процесса B),
и отличается тем,
что объемное соотношение объема жидкой фазы и объема газовой фазы (Vжид/Vгаз) на стадии процесса B больше или равно 0,25, предпочтительно больше 0,3, особенно больше 2, и/или
что аммиак на стадии процесса B) в пересчете на гидроксильные группы во вторичном спирте используется в мольном соотношении по меньшей мере 5 к 1, предпочтительно 50 к 1, особенно предпочтительно 500 к 1.
Под термином «первичный амин» в связи с настоящим изобретением понимают также его соли, а также смеси этого амина и/или его соли.
Под термином «вторичный спирт» в связи с настоящим изобретением понимают органическое соединение, которое имеет по меньшей мере одну вторичную гидроксильную группу (R-CH(OH)-R', где R и R' не равны H).
Для расчета объемного соотношения в качестве «газовой фазы» понимают внутренний объем аппарата, в котором идет реакция, за вычетом объема жидкой фазы.
В качестве гомогенных катализаторов, которые следует использовать в способе согласно изобретению, рассматривают все гомогенные катализаторы, известные специалисту, которые в состоянии активировать CH-связь атома углерода, содержащего OH-группу, подлежащую аминированию. Примеры таких катализаторов включают алкоксиды щелочных металлов, алюминия и лантанидов, неорганические соединения благородных металлов (например, [RuCl3 * nH2O], IrCl3), моно- или полиметаллические, одно- или многоядерные координационные соединения одного или нескольких благородных металлов, выбираемых среди элементов рутения (например, [RuCl2(PPh3)3], [RuH2(PPh3)4], катализаторы Шво ([(η4-C4Ph4CO)Ru(CO)3]2), [Ru(cod)(cot)], [(PPh3)2Ru(CH3CN)3Cl]BPh4, [Ru(п-цимол)Cl2]2, [Ru(п-цимол)Cl2]2/DPEphos, [Ru(PPh3)3(CO)H2], [Ru3(CO)12], [Ru3(CO)12]/N-фенил-2-(PCl2)пиррол, [RuCl2(ДМСО)4]), родия (например, катализатор Уилкинсона ([RhCl(PPh3)3]), [RhH(PPh3)3]), иридия (например, [IrCl3(ДМСО)3], [Cp*IrCl2]2, [Ir(cod)Cl]2/(dppp)/Cs2CO3, [IrCl2H(cod)]2, активированные KOH фенантролин-иридиевые комплексы) и палладия ([Pd(PPh3)4], [PdCl2(dppe)], [Pd(OAc)2]), а также других платиновых металлов и железа.
В другом предпочтительном варианте исполнения способа согласно изобретению на стадии В) используются катализаторы, которые известны специалисту как катализаторы для гидроформилирования. Для этого могут использоваться карбонильные соединения переходных металлов общей формы HxMyM'y,(CO)zLn, причем может быть n=0 («немодифицированные катализаторы гидроформилирования»), или соответственно n≠0 («модифицированные катализаторы гидроформилирования»), и кроме того, x, y и z принимают целочисленные полные значения, у' может равняться нулю, если используется монометаллический катализатор, или y' может принимать положительное целочисленное значение, если применяется биметаллический катализатор. M и M' могут быть одинаковыми или разными. В качестве переходных металлов M и M' могут использоваться родий, кобальт, иридий, рутений, осмий, платина, палладий, железо, никель, хром, молибден или марганец, предпочтительно применяются родий, кобальт, иридий, рутений, осмий, платина, палладий, железо, никель, хром, молибден или марганец; предпочтительно применяются родий, кобальт, иридий, рутений, осмий или платина. Лиганд L может выбираться из группы фосфанов, фосфаноксидов, фосфитов, аминов, амидов, изонитрилов, арсина или стибина; примерными представителями являются трифенилфосфан, трифенилфосфаноксид, натриевая соль трифенилфосфантрисульфокислоты, трифениламин или трифениларсин. Примерные катализаторы гидроформилирования выбираются из группы, включающей HCo(CO)4, HCo(CO)3PBu3, HRh(CO)(PR3)3, Rh4(CO)12, Rh6(CO)16, Rh2(CO)4Cl2, CoRh(CO)7, Co2Rh2(CO)12, HRh(CO)3.
В этой связи предпочтительным катализатором гидроформилирования является система катализаторов, содержащая по меньшей мере один ксантфосный лиганд общей формулы 1 и соединение переходного металла.
Под термином «ксантфосный лиганд» в контексте настоящего изобретения понимают соединение общей формулы 1,

причем
R1a, R2a, R3a и R4a независимо друг от друга одинаковые или разные выбираются из группы, содержащей, предпочтительно состоящей из фенила, третбутила и изопропила, и
A выбирается из группы, содержащей, предпочтительно состоящей из -C(CH3)2-. -CH2CH2-, -Si(CH3)2-, -S-, -O-, -C(C(CH3)2)-.
Предпочтительно используются ксантфосные лиганды, у которых R1a=R2a=R3a=R4a=фенилу, и A=-C(CH3)2-.
Переходный металл предпочтительно выбирается из группы, содержащей, предпочтительно состоящей из рутения, кобальта, родия, иридия, никеля, палладия и платины, а также других платиновых металлов и железа. Особенно предпочтительно переходный металл выбирается из группы, состоящей из рутения, иридия и палладия; особенно предпочтительно из группы, состоящей из рутения и иридия, особенно рутения.
Следует упомянуть, что в зависимости от выбранной комбинации из описанных образующих катализатор элементов эти катализаторы имеют электрический заряд и могут использоваться в форме соли, образованной с помощью соответствующего противоиона.
В одном особенно предпочтительном варианте катализатор представляет собой координационное соединение на основе ксантена - карбонилхлоргидридо[9,9-диметил-4,5-бис(дифенилфосфино)ксантено]-рутений(II)]:

Карбонилхлоргидридо[9,9-диметил-4,5-бис(дифенилфосфино)ксантено]рутений(II)]
В другом предпочтительном варианте исполнения способа согласно изобретению на стадии B) используются пинцерные катализаторы.
В качестве пинцерных катализаторов, используемых на стадии процесса B), могут применяться координационные соединения переходных металлов общей структуры A).
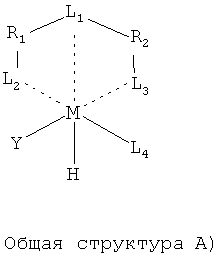
При этом для способа согласно изобретению предпочтительны особенно такие катализаторы, в которых L1 представляет собой служащий в качестве лиганда для центрального атома M, причем M является переходным металлом, атом углерода или гетероатом, предпочтительно азот, к которому через оба двухвалентных органических остатка R1 и R2 ковалентно присоединены другие лиганды L2 и L3.
Центральный металл M предпочтительно выбирается из группы, содержащей рутений, кобальт, родий, иридий, никель, палладий и платину. Особенно предпочтительно центральный металл выбирается из группы, состоящей из рутения, иридия и палладия; особенно предпочтительно из группы, состоящей из рутения и иридия.
Двухвалентные органические остатки R1 и R2 независимо друг от друга при необходимости могут содержать другие замещенные алифатические, эпициклические или ароматические компоненты основы, которые совместно с лигандом L1 при необходимости дают фиксированный по своей конфигурации и конформации молекулярный структурный фрагмент. Предпочтительно при этом лиганд L1 представляет собой часть гетероциклического компонента основы, к которому присоединены остатки R1 и R2. Особенно предпочтительно L1 представляет собой атом азота акридинильного или хинолинильного компонента основы. Этот акридинильный или хинолинильный компонент основы может содержать один, два, три, четыре, пять, шесть или семь заместителей в любом положении, которые вместе с органическими остатками R1 и/или R2 образуют другой ароматический структурный фрагмент, аннелированный с акридинильным или соответственно хинолинильным компонентом основы, и могут выбираться из группы, состоящей из атомов водорода, алкильных, циклоалкильных, арильных, гетероциклильных, гетероарильных, алкилциклоалкильных, алкиларильных, алкилгетероциклильных, алкилгетероарильных, галогеновых, нитро-, сложноэфирных, амидных, циано-, алкоксильных, алкиламиновых или ариламиновых остатков. В одном предпочтительном варианте исполнения R1, R2 и L1 являются компонентами остатка 4,5-диметиленакридина.
Лиганды L2 и L3, ковалентно связанные с описанным структурным фрагментом, образованным из R1, R2 и L1, представляют собой соответственно другие гетероатомы, которые содержатся в молекулярных остатках, которые независимо друг от друга выбираются из группы, включающей фосфин (PRaRb), амин (NRaRb), имин, сульфид (SRa), тиол (SH), сульфоксид (S(=O)Ra), гетероарил, содержащий по меньшей мере один атом, выбираемый среди азота или серы, арсин (AsRaRb), стибин (SbRaRb) и N-гетероциклический карбен, представленный структурами
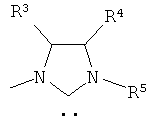 или
или 
Лиганд L4, координированный на описанном центральном металле, представляет собой гетероатом, содержащейся в монодентатном двухэлектронном доноре, выбираемом из группы CO, PRaRbRc, NO+, AsRaRbRc, SbRaRbRc*, SRaRb, нитрила (RaCN), изонитрила (RaNC), N2, PF3, CS, гетероарила (например, тиофена, пиридина), тетрагидротиофена или N-гетероциклического карбена.
Y представляет собой моноанионный лиганд, выбираемый из группы, галогенов, карбоксилата, трифторацетата, сульфоната, трифторметансульфоната, цианида, гидроксида, алкоксида, имида; или нейтральную молекулу сольвата, такую как NH3, NRaRbRc, RaRbNSO2Rc. Предпочтительно Y выбирается из группы галогенида, ацетона, диалкилацетона (например, 2-бутанона), циклического кетона (например, циклогексанона), ТГФ, анизола, ДМСО, ацетонитрила, дихлорметана, толуола, воды, пиридина.
Остатки R3, R4, R5, Ra, Rb и Rc соответственно независимо друг от друга являются одинаковыми или разными, выбираются из группы алкилов, циклоалкилов, арилов, гетероциклилов, гетероарилов, алкилциклоалкилов, аликларилов, алкилгетероциклилов или алкилгетероалкилов. Предпочтительно остатки R3, R4, R5, Ra, Rb и Rc соответственно независимо друг от друга выбираются из группы метила, этила, изопропила, третбутила, циклогексила, циклопентила, фенила и мезитила.
Следует упомянуть, что в зависимости от выбранной комбинации из описанных образующих катализатор элементов эти катализаторы имеют электрический заряд и могут использоваться в форме соли, образованной с помощью соответствующего противоиона.
В одном особенно предпочтительном варианте катализатор представляет собой координационное соединение на основе акридина - карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутений(II)]:
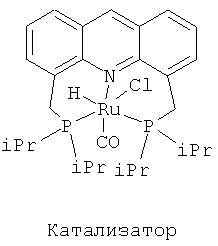
Карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутений(II)]
Способ согласно изобретению может использоваться для прямого аминирования вторичных спиртов с помощью аммиака с получением первичных аминов. Предпочтительно используемые на стадии процесса A) спирты имеют по меньшей мере две вторичные гидроксигруппы. Эти полиолы предпочтительно отличаются тем, что они не могут совсем или не могут в достаточной степени испаряться без разложения и, следовательно, являются неподходящими для газофазного взаимодействия, в частности, эти спирты имеют циклический, предпочтительно полициклический углеродный скелет. Таковыми являются, например, углеводы, сахара, сахарные спирты или соответственно производные, которые могут получаться из них в результате химических превращений (как, например, дегидратации), такие как аминосахара, дезоксисахара, гликали, глицитолы и C- или O-гликозиды.
Особенно предпочтительно спирты, используемые на стадии процесса A), выбираются из группы, состоящей из 2-додеканола, циклододеканола, 4-фенил-2-бутанола, изосорбида, изоманнида, изоидита, полипропиленгликоля, маннитола, сорбитола, галактитола и алкилгликозидов, причем изоманнид, 2-додеканол, циклододеканол и 4-фенил-2-бутанол используются особенно предпочтительно.
Фиг.1 показывает палитру интермедиатов и продуктов, получающихся в результате применения способа согласно изобретению, исходя из трех изомеров 1,4:3,6-диангидрогекситолов, которые представляют собой наиболее предпочтительно используемые в способе согласно изобретению вторичные спирты.
Способ согласно изобретению также предпочтительно следует использовать для вторичных спиртов, которые содержат карбоксильную группу или сложноэфирную группу, особенно карбоксильную группу.
Предпочтительными вторичными спиртами, содержащими карбоксильные группы, являются, в частности, альфа-гидроксикарбоновые кислоты и OH-модифицированные природные жирные кислоты, причем OH-модифицированные природные жирные кислоты, выбираются, в частности, из группы тех, которые являются производными фрагментов кокосового масла, масел из семян и касторового масла.
Примерами таких спиртов, содержащих карбоксильную группу, являются 2-гидроксипропионовая кислота (молочная кислота), 2-гидрокси-3-метил-бутановая кислота, 2-гидрокси-4-метилмеркаптобутановая кислота, 2-гидрокси-4-метилпентановая кислота, 2-гидрокси-3-метилпентановая кислота, 2-гидрокси-3-(3-индолил)пропионовая кислота, 2-гидрокси-3-фенилпропио-новая кислота, 2-гидрокси-6-аминогексановая кислота, 2-гидрокси-5-гуанидинпентановая кислота, 2-гидрокси-3-(1H-имидазол-4-ил)пропановая кислота, 2-гидрокси-3-(4-гидроксифенил)пропановая кислота, 2-гидрокси-4-аминокарбонилбутановая кислота, 2,3-дигидроксибутановая кислота, 2-гидроксипентандикарбоновая кислота, гликолевая кислота, 2,3-дигидроксипропановая кислота, 2-гидрокси-3-меркаптопропановая кислота, 2-гидрокси-3-аминокарбонилпропановая кислота и 2-гидроксиянтарная кислота.
Предпочтительными вторичными спиртами, содержащими сложноэфирные группы, являются, в частности, выбираемые из группы сложных алкиловых эфиров, особенно сложных метиловых эфиров, сложных этиловых эфиров, сложных н-пропиловых эфиров и сложных изопропиловых эфиров гидроксикарбоновых кислот.
Особенно предпочтительными являются спирты, выбираемые из группы сложных эфиров OH-модифицированных природных жирных кислот и сложных эфиров альфа-гидроксикарбоновых кислот. Примерами этих классов соединений являются сложные метиловые эфиры, сложные этиловые эфиры, сложные н-пропиловые эфиры и сложные изопропиловые эфиры 2-гидроксипропионовой кислоты (молочной кислоты), 2-гидрокси-3-метилбутановой кислоты, 2-гидрокси-4-метилмеркаптобутановой кислоты, 2-гидрокси-4-метилпентановой кислоты, 2-гидрокси-3-метилпентановой кислоты, 2-гидрокси-3-(3-индолил)пропионовой кислоты, 2-гидрокси-3-фенилпропионовой кислоты, 2-гидрокси-6-аминогексановой кислоты, 2-гидрокси-5-гуанидинпентановой кислоты, 2-гидрокси-3-(1H-имидазол-4-ил)пропановой кислоты, 2-гидрокси-3-(4-гидроксифенил)пропановой кислоты, 2-гидрокси-4-аминокарбонилбутановой кислоты, 2,3-дигидроксибутановой кислоты, 2-гидроксипентандикарбоновой кислоты, гликолевой кислоты, 2,3-дигидроксипропановой кислоты, 2-гидрокси-3-меркаптопропановой кислоты, 2-гидрокси-3-аминокарбонилпропановой кислоты и 2-гидроксиянтарной кислоты.
Для примера, концентрации спиртов, используемые в способе согласно изобретению, варьируются в интервале между 0,1 и 10000 ммоль/л, предпочтительно между 0,1 и 1000 ммоль/л и особенно предпочтительно между 1 и 100 ммоль/л.
Жидкая фаза, используемая на стадии процесса A), может образовываться растворителем или газом, при условиях процесса, существующих в сжиженной или сверхкритической форме, особенно аммиаком, или смесями из указанных компонентов.
В этом контексте в качестве растворителя могут использоваться вода или органические растворители или смеси этих веществ, эти смеси могут представлять собой гомогенный раствор или также эмульсию. Особенно предпочтительным является использование по меньшей мере одного органического растворителя. Выбор подходящего органического растворителя, который не должен рассматриваться как ограничивающий, включает в себя бензол, толуол, изомеры кислола, мезитилен, диоксан, ТГФ, диметоксиэтан, анизол и циклогексан.
В качестве аммиака или соединения, высвобождающего аммиак, используемого на стадии процесса B), в контексте настоящего изобретения понимают, в частности, также жидкий или сверхкритический аммиак и/или раствор солей аммония в растворителе (как, например, также гидроксид аммония в воде).
Предпочтительно на стадии процесса B) в качестве свободного аммиака используется газообразный или сжиженный аммиак.
В одном предпочтительном варианте исполнения стадия процесса B) проводится при избыточном давлении относительно атмосферного давления. Примерные давления в способе согласно изобретению лежат в области между 1 и 1000 бар, предпочтительно между 5 и 500 бар, особенно предпочтительно между 5 и 100 бар и наиболее предпочтительно между 20 и 50 бар. Давление может создаваться в результате нагнетания аммиака и/или другого газа, в частности, инертного газа, такого как, например, азот или аргон, причем предпочтительным является создание давления при помощи газовых смесей их обоих.
Температуры, описывающие способ согласно изобретению на стадии процесса B), варьируются в таком диапазоне, который до минимума ограничивает реакции разложения вторичного спирта, первичного амина и всех других интермедиатов, возникающих в ходе процесса, приводящие к образованию побочных продуктов в результате термической нагрузки. Например, температуры варьируются в диапазоне между 80 и 220°C, предпочтительно между 100 и 200°C и особенно предпочтительно между 120 и 170°C, при измерении в жидкой фазе.
Согласно изобретению предпочтительно, чтобы способ проводился в отсутствие водорода, причем под отсутствием водорода понимают, что в реакцию дополнительно не подается водород; при необходимости следы водорода, содержащиеся в воздухе, или соответственно водород, образовавшийся из субстрата в условиях реакции, в рамках настоящего изобретения относятся к «отсутствию водорода».
Краткое описание чертежей
Фиг.1: Схема прямого аминирования диангидрогекситолов.
Примеры
Пример 1: Прямое одностадийное аминирование изоманнида аммиаком на гетерогенных катализаторах, пример для сравнения
В реактор высокого давления, снабженный пропеллерной мешалкой и внутренним охлаждающим змеевиком, помещают 1,45 г изоманнида (10 ммоль) и 2,78 г катализатора на основе Ni/Al2O3 и в закрытом реакторе с газовым уплотнением продувают азотом при комнатной температуре. После этого в течение 25 минут добавляют 250 мл жидкого аммиака (10 моль) и реакционную смесь ступенчато сначала нагревают до 150°C (140 бар), потом до 185°C (260 бар). По прошествии 90 минут времени реакции реактор охлаждают, стравливают давление, реакционную смесь извлекают этанолом и фильтруют. С катализатором на основе элементарного никеля нельзя наблюдать никакого превращения изоманнида.
Пример 2: Прямое одностадийное аминирование изоманнида аммиаком на координационных соединениях монодентатных лигандов (Vжид/Vгаз=0,35, пример согласно изобретению)
В атмосфере аргона 1,461 г (10 ммоль) изоманнида, 0,1 ммоль [Ru(п-цимол)Cl2]2/K2CO3 и 25 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона. После этого в автоклав вводят 235,2 ммоль аммиака (в общей сложности имеет место Vжид/Vгаз=0,35). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до 140°C и выдерживают 24 часа при этой температуре. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Было обнаружено образование соответствующего моноаминоспирта.
Пример 3: Прямое одностадийное аминирование 2-додеканола аммиаком на пинцерном комплексе рутения (Vжид/Vгаз=0,3, согласно изобретению)
В атмосфере аргона 1,863 г (10 ммоль) 2-додеканола, 0,030 г (0,05 ммоль) карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутения(II)] в качестве катализатора и 25 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона.
После этого в автоклав вводят 2 г (117,6 ммоль) жидкого аммиака (в общей сложности имеет место Vжид/Vгаз=0,3). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Полученный сырой продукт очищают с помощью перегонки в вакууме в круглодонной колбе. Получают 1,241 г 2-додециламина (выход: 67% от теоретического; интервал кипения 170-180°C температуры воздушной бани при 11 мбар).
Пример 4: Прямое одностадийное аминирование циклододеканола аммиаком на пинцерном комплексе рутения (Vжид/Vгаз=0,3, согласно изобретению)
В атмосфере аргона 1,843 г (10 ммоль) циклододеканола, 0,030 г (0,05 ммоль) карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)-рутения(II)] в качестве катализатора и 25 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона. После этого в автоклав вводят 2 г (117,6 ммоль) жидкого аммиака (в общей сложности имеет место Vжид/Vгаз=0,3). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Полученный сырой продукт очищают с помощью перегонки в вакууме в круглодонной колбе. Получают 1,427 г циклододециламина (выход: 78% от теоретического; интервал кипения 175-180°C температуры воздушной бани при 6 мбар).
Пример 5: Прямое одностадийное аминирование 4-фенил-2-бутанола аммиаком на пинцерном комплексе рутения (Vжид/Vгаз=0,3, согласно изобретению)
В атмосфере аргона 1,502 г (10 ммоль) 4-фенил-2-бутанола, 0,030 г (0,05 ммоль) карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)-рутения(II)] в качестве катализатора и 25 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона. После этого в автоклав вводят 2 г (117,6 ммоль) жидкого аммиака (в общей сложности имеет место Vжид/Vгаз=0,3). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Полученный сырой продукт очищают с помощью перегонки в вакууме в круглодонной колбе. Получают 0,945 г 4-фенил-2-бутиламина (выход: 63% от теоретического; интервал кипения 135-140°C температуры воздушной бани при 8 мбар).
Пример 6: Прямое одностадийное аминирование изоманнида аммиаком на пинцерном комплексе рутения (Vжид/Vгаз=0,35)
В атмосфере аргона 1,461 г (10 ммоль) изоманнида, 0,061 г (0,1 ммоль) карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)-рутения(II)] в качестве катализатора и 25 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона. После этого в автоклав вводят 4 г (235,2 ммоль) жидкого аммиака (в общей сложности имеет место Vжид/Vгаз=0,35). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Полученный сырой продукт очищают с помощью перегонки в вакууме в круглодонной колбе. Получают 1,290 г смеси диаминов: диаминоизоманнида, диаминоизосорбида и диаминоизоидида в соотношении 50:41:9 (выход: 90%от теоретического; интервал кипения 185-190°C температуры воздушной бани при 10 мбар).
Пример 7: Прямое одностадийное аминирование трипропиленгликоля аммиаком на гомогенном рутениевом катализаторе (согласно изобретению; Vжид/Vгаз=0,3)
В атмосфере аргона 0,961 г (5 ммоль) трипропиленгликоля, 0,0305 г (0,05 ммоль) карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино) рутения(II)] в качестве катализатора и 25 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона. После этого в автоклав вводят 2 г (2,95 мл; 117,6 ммоль) жидкого аммиака (в общей сложности имеет место Vжид/Vгаз=0,3). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре, причем давление устанавливается на 45 бар. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Полученный сырой продукт очищают с помощью перегонки в вакууме в круглодонной колбе. Получают диамин трипропиленгликоля с выходом 91% от теоретического (интервал кипения 90-95°C температуры воздушной бани при 10 мбар).
Пример 8: Прямое одностадийное аминирование трипропиленгликоля на гомогенном рутениевом катализаторе (не соответствует изобретению; Vжид/Vгаз=0,17)
В атмосфере аргона 0,4805 г (2,5 ммоль) трипропиленгликоля, 0,01525 г (0,025 ммоль) карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутения(II)] в качестве катализатора и 12,5 мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 100 мл. Автоклав закрывают, трижды каждый раз по 20 бар нагнетают и стравливают аргон и снова нагнетают 15 бар аргона. После этого в автоклав вводят 1 г (1,475 мл; 58,8 ммоль) жидкого аммиака (в общей сложности имеет место Vжид/Vгаз=0,17). Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре, причем давление устанавливается на 45 бар. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси и трехкратного нагнетания 20 бар аргона с последующим стравливанием автоклав открывают, реакционную смесь фильтруют через кизельгур, а фильтрат для удаления растворителя упаривают на роторном испарителе в вакууме. Полученный сырой продукт очищают с помощью перегонки в вакууме в круглодонной колбе. Получают диамин трипропиленгликоля с выходом 90% от теоретического.
Пример 9: Прямое одностадийное аминирование 2-октанола аммиаком на гомогенном рутениевом катализаторе (изменение давления и Vжид/Vгаз)
В атмосфере аргона mO г 2-октанола, mRu г [карбонилхлоргидридо-трис(трифенилфосфан)рутения(II)] в качестве катализатора, mP г ксантфоса и VLM мл 2-метил-2-бутанола в качестве растворителя помещают в стеклянную вставку в автоклаве из сплава Хастеллой объемом 314 мл. Автоклав закрывают, нагнетают 5 бар азота, стравливают давление и охлаждают до -70°C. Затем в автоклаве конденсируют mA г жидкого аммиака, реактор снова нагревают до комнатной температуры и нагнетают p бар азота. Реакционную смесь перемешивают 10 минут при комнатной температуре (600 об/мин), затем при перемешивании нагревают до температуры внутри 170°C и выдерживают 48 часов при этой температуре. После охлаждения до комнатной температуры, осторожного стравливания давления из реакционной смеси, а также нагнетания 5 бар азота с последующим стравливанием давления автоклав открывают и реакционную смесь анализируют с помощью газового хроматографа. Параметры реакции, а также степени превращения и селективности по желаемому первичному амину, 2-октиламину, приведены в Таблице 1. Результаты показывают, что селективность по целевому продукту может повышаться как в результате увеличения соотношения Vжид/Vгаз, так и при помощи повышения давления, а также в результате одновременного повышения обоих параметров.
[г]5
1: масса 2-октанола; 2; масса [карбонилхлоргидридотрис-(трифенилфосфан)рутения(II)]; 3: масса ксантфоса; 4: объем растворителя; 5: масса аммиака; 6: нагнетаемое давление азота перед реакцией; 7: соотношение объема жидкой фазы и объема газовой фазы; 8: степень превращения 2-октанола; 9: селективность по 2-октиламину.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРЕВРАЩЕНИЕ ГЛИКОЛЕВОГО АЛЬДЕГИДА СО СРЕДСТВОМ АМИНИРОВАНИЯ | 2010 |
|
RU2573570C2 |
| СПОСОБ АМИНИРОВАНИЯ | 1998 |
|
RU2215734C2 |
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ ПОСРЕДСТВОМ СОДЕРЖАЩЕГО НИТРОКСИЛЬНЫЙ РАДИКАЛ КАТАЛИЗАТОРА | 2011 |
|
RU2579510C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИКОЛИНОВОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2769693C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОВ ИЗ ГЛИЦЕРИНА | 2008 |
|
RU2480449C2 |
| КАТАЛИЗАТОРЫ И СПОСОБ ГИДРОАМИНИРОВАНИЯ ОЛЕФИНОВ | 2010 |
|
RU2490064C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ АМИНИРОВАНИЯ МОНОЭТАНОЛАМИНА | 1987 |
|
RU2010599C1 |
| СПОСОБ ПОЛУЧЕНИЯ N, N-ЗАМЕЩЕННЫХ 3-АМИНОПРОПАН-1-ОЛОВ | 2009 |
|
RU2522761C2 |
| КАТАЛИЗАТОР ДЛЯ АМИНИРОВАНИЯ И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2836173C1 |
Изобретение относится к усовершенствованному способу получения первичных аминов. Способ включает стадии: A) приготовление раствора вторичного спирта, выбранного из группы, включающей циклические спирты, 2-додеканол, 4-фенил-2-бутанол, полипропиленгликоль, ОН-модифицированные природные жирные кислоты и сложные эфиры ОН-модифицированных природных жирных кислот, во флюидной, не газообразной фазе, B) приведение во взаимодействие этой фазы со свободным аммиаком и гомогенным катализатором. При этом стадию процесса В) проводят при избыточном давлении от 20 до 50 бар и температуре 80-220°С в двухфазной системе, включающей жидкую фазу и газовую фазу, где объемное соотношение объема жидкой фазы и объема газовой фазы на стадии процесса В больше или равно 0,25, и/или аммиак на стадии процесса В) в пересчете на гидроксильные группы во вторичном спирте используют в мольном соотношении по меньшей мере 5 к 1. При необходимости способ включает стадию C) выделение первичного амина, образовавшегося на стадии процесса В). В качестве гомогенного катализатора используют по меньшей мере один катализатор, выбираемый из координационных соединений одного благородного металла, выбираемого среди элементов рутения, иридия, родия, осмия, палладия и платины. Спирт, используемый на стадии процесса А), имеет по меньшей мере две вторичные гидроксигруппы, и выбирается предпочтительно из 2-додеканола, циклододеканола, 4-фенил-2-бутанола, изосорбида, изоманнида, изоидида, полипропиленгликоля, маннитола, сорбитола, галактитола и алкилгликозидов. Предпочтительно на стадии В) используют жидкий аммиак и в качестве гомогенного катализатора, по меньшей мере, один пинцерный катализатор. Предпочтительным гомогенным катализатором является карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутений(II)]. Способ позволяет проводить прямое аминирование вторичных спиртов с получением продуктов с высоким выходом, исключить дополнительные стадии выделения и очистки продуктов, получаемых на промежуточных стадиях, и исключает возможность образования побочных продуктов. 7 з.п. ф-лы, 1 ил., 1 табл., 9 пр.
1. Способ получения первичных аминов, включающий в себя следующие стадии процесса
A) приготовление раствора вторичного спирта, выбранного из группы, включающей циклические спирты, 2-додеканол, 4-фенил-2-бутанол, полипропиленгликоль, ОН-модифицированные природные жирные кислоты и сложные эфиры ОН-модифицированных природных жирных кислот, во флюидной, не газообразной фазе,
B) приведение во взаимодействие этой фазы со свободным аммиаком и гомогенным катализатором, и при необходимости
C) выделение первичного амина, образовавшегося на стадии процесса В),
отличающийся тем, что стадию процесса В) проводят при избыточном давлении от 20 до 50 бар и температуре 80-220°С в двухфазной системе, включающей жидкую фазу и газовую фазу, при этом объемное соотношение объема жидкой фазы и объема газовой фазы на стадии процесса В больше или равно 0,25, и/или что аммиак на стадии процесса В) в пересчете на гидроксильные группы во вторичном спирте используют в мольном соотношении по меньшей мере 5 к 1.
2. Способ по п. 1, отличающийся тем, что в качестве гомогенного катализатора используют по меньшей мере один, выбираемый из координационных соединений одного благородного металла, выбираемого среди элементов рутения, иридия, родия, осмия, палладия и платины.
3. Способ по п. 1, отличающийся тем, что спирт, используемый на стадии процесса А), имеет по меньшей мере две вторичные гидроксигруппы.
4. Способ по п. 1, отличающийся тем, что спирт, используемый на стадии процесса А), имеет циклический или полициклический углеродный скелет.
5. Способ по п. 1, отличающийся тем, что спирт, используемый на стадии процесса А), выбирают из группы, состоящей из 2-додеканола, циклододеканола, 4-фенил-2-бутанола, изосорбида, изоманнида, изоидида, полипропиленгликоля, маннитола, сорбитола, галактитола и алкилгликозидов.
6. Способ по п. 1, отличающийся тем, что на стадии процесса В) используют жидкий аммиак.
7. Способ по одному из пп. 1-6, отличающийся тем, что в качестве гомогенного катализатора используют, по меньшей мере, один пинцерный катализатор.
8. Способ по п. 7, отличающийся тем, что в качестве гомогенного катализатора используют карбонилхлоргидридо[4,5-(диизопропилфосфинометилакридино)рутений(II)].
| WO 2010018570 A1, 18.02.2010 | |||
| GB 2059792 A, 29.04.1981 | |||
| Система электроснабжения с резервированием | 1976 |
|
SU696572A1 |
| US 4855425 A1, 08.08.1989 | |||
| GUANANATHAN CHIDAMBARAM et al | |||
| Selective synthesis of primary amines directly from alcohols and amimonia, ANGEWANDTE CHEMIE | |||
| INTERATIONAL EDDITION, 2008, Bd 47,Nr.45,s.8661-8664 | |||
| S.IMM et al., IMPROVED RUTHENIUM -CATALIZED AMINATION OF | |||

