Область техники, к которой относится изобретение
Настоящее изобретение относится к вновь обнаруженной фазе тригидроксида алюминия. Настоящее изобретение также относится к катализаторам, изготовленным из этой новой фазы тригидроксида алюминия, причем катализаторы могут быть составлены специальным образом, чтобы обеспечить улучшенные эксплуатационные параметры для большого числа процессов переработки углеводородов. Настоящее изобретение также относится к способам получения упомянутой новой фазы тригидроксида алюминия и полученным из нее катализаторам и к способу улучшения активности катализаторов, содержащих носитель диоксид кремния - оксид алюминия.
Известный уровень техники
Область техники, относящаяся к содержащим оксид алюминия носителям, пропитке таких носителей различными каталитически активными металлами, соединениями металлов и/или промотерами и к различным областям использования таких пропитанных носителей в качестве катализаторов, широко и глубоко разработана. В качестве нескольких из множества решений, которые можно привести в качестве примеров, относящихся к указанным областям, можно упомянуть следующие патенты США, которые все включены в настоящее описание в качестве ссылок для всех целей, как они изложены: патенты США №№2838444; 2935463; 2973329; 3032514; 3058907; 3124418; 3152865; 3232887; 3287280; 3297588; 3328122;3493493; 3623837; 3749664; 3778365; 3897365; 3909453; 3983197; 4090874; 4090982; 4154812; 4179408; 4255282; 4328130; 4357263; 4402865; 4444905; 4447556; 4460707; 4503911; 4588706; 4591429; 4595672; 4652545; 4673664; 4677085; 4732886; 4797196; 4861746; 5002919; 5186818; 5232888; 5246569; 5248412 и 6015485.
Хотя известный уровень техники характеризуется постоянной модификацией и усовершенствованием таких катализаторов с улучшением их каталитической активности, и в некоторых случаях действительно достигнут весьма желательный уровень активности, но все же существует растущая потребность в промышленности в еще более высокой активности катализаторов, которая обеспечивается настоящим изобретением.
Основные усилия в разработке катализаторов повышенной активности были направлены на разработку носителей, которые увеличивали бы каталитическую активность осажденных на них металлов. В подавляющем большинстве областей использования выбранным для носителя материалом является оксид алюминия, наиболее часто - γ-оксид алюминия, но в качестве материалов носителей использовали и используют в настоящее время композиты диоксид кремния - оксид алюминия, цеолиты и различные другие неорганические оксиды и их композиты.
В случае оксида алюминия различными исследователями разработаны способы получения носителей, имеющих различные удельные поверхности, объемы пор и распределения пор по размеру, так что при нанесении соответствующих металлов они оказываются особенно подходящими для катализа желательной реакции на конкретном сырье, будь эта реакция, относящаяся к гидродесульфуризации, гидродеметаллированию, гидрокрекингу, реформингу, изомеризации и т.п.
В большинстве случаев носители на основе γ-оксида алюминия получают активированием (обычно прокаливанием) исходного материала на основе псевдобемита (AlOOH). В редких случаях носитель образуется из одного из известных в настоящее время тригидроксидов алюминия (Al(ОН)3), гиббсита, байерита или нордстрендита. Когда в качестве исходного материала используют байерит или нордстрендит, образующийся дегидратированный оксид алюминия имеет структуру, отличную от структуры более типичного γ-оксида алюминия, часто называемую η-оксидом алюминия; для гиббсита продуктом оксида алюминия может быть χ-оксид алюминия. Каждый из этих переходных оксидов алюминия имеет структурные характеристики (пористость и удельную поверхность), отличающиеся от более распространенного γ-оксида алюминия. Однако их типичным недостатком является более низкая теплостойкость, чем у γ-оксида алюминия; для конкретных процессов дегидрирования и прокаливания потеря удельной поверхности для упомянутых оксидов алюминия значительно больше, чем претерпел бы γ-оксид алюминия. Патент США 6015485 показывает путь улучшения структуры катализаторов, отложенных на γ-оксиде алюминия, синтезом in-situ кристаллического оксида алюминия на носителе на основе γ-оксида алюминия. Из этого патента ясно, что образуются катализаторы повышенной активности.
В качестве примера потребности в катализаторах повышенной активности можно упомянуть потребность в катализаторе повышенной активности для первой стадии гидрокрекинга. В типичном процессе гидрокрекинга более высокомолекулярные углеводороды превращаются в более низкомолекулярные фракции в присутствии катализатора гидрокрекинга, которым обычно является пропитанный благородным металлом диоксид кремния - оксид алюминия/цеолит. Известные в настоящее время катализаторы гидрокрекинга обладают очень высокой активностью и способны обеспечивать расщепление большого объема проходящего через установку материала. Однако такие катализаторы очень чувствительны к наличию загрязняющих примесей, таких как сера, металлы и соединения азота, которые, следовательно, должны быть удалены из углеводородного потока перед крекингом. Это осуществляют на первой стадии процессов гидрокрекинга, таких как гидродеазотизация, гидродесульфуризация и гидродеметаллирование. Катализаторы гидрообработки, использованные в этих процессах, обычно представляют пропитанный комбинацией металлов VIB группы и VIII группы субстрат на основе оксида алюминия. Однако существующие катализаторы гидрообработки недостаточно активны, чтобы обеспечить переработку такого же высокого объема проходящего через установку материала, что и перерабатываемый катализаторами гидрокрекинга. Как таковые, первые стадии процесса гидрокрекинга являются узким местом в общем процессе гидрокрекинга, что должно быть компенсировано, например, величиной установки для гидрообработки относительно установки гидрокрекинга.
Существо изобретения
В соответствии с настоящим изобретением один из его аспектов относится к вновь обнаруженной фазе тригидроксида алюминия, которая образуется в результате теплового старения приготовленного и прокаленного носителя диоксид кремния-оксид алюминия, полученного из порошка аморфный оксид алюминия - обогащенный диоксидом кремния оксид алюминия в кислой водной среде. Такую вновь обнаруженную фазу тригидроксида алюминия, далее называемую "каменетсит", можно отличить от трех ранее известных фаз, гиббсита, байерита и нордстрендита, методом рентгеноструктурного анализа. При сушке и прокаливании каменетсит образует материал, который по строению и структуре отличается от других носителей. Катализаторы, полученные из этого материала, проявляют исключительно высокую каталитическую активность во многих реакциях гидрообработки и реакциях, не относящихся к гидрообработке. Действительно, при соответствующем регулировании условий старения, используемых в производстве каменетсита, конечное строение катализатора можно регулировать с учетом конкретных областей применения катализатора. Существует подтверждение того, что катализаторы, содержащие одинаковые активные металлы и те же количества активных металлов, ведут себя по-разному с различным нефтяным сырьем, в зависимости от размера и концентрации частиц кристаллического оксида алюминия, образованных различными содержащими каменетсит предшественниками носителя.
Соответственно изобретение относится к композиции, включающей вышеуказанную новую твердую фазу тригидроксида алюминия, характеризующуюся рентгенограммой, более подробно описанной ниже.
Более конкретно, изобретение относится к композиции, включающей твердую фазу тригидроксида алюминия, причем указанная фаза имеет измеримые полосы рентгенограммы между 2θ=18,15° и 2θ=18,50°, между 2θ=36,1° и 2θ=36,85°, между 2θ=39,45° и 2θ=40,30° и между 2θ=51,48° и 2θ=52,59° и не имеет измеримых полос рентгенограммы между 2θ=20,15° и 2θ=20,65°.
Изобретение относится также к вышеописанной композиции, в которой фаза тригидроксида алюминия дополнительно имеет измеримые полосы рентгенограммы между 2θ=27,35° и 2θ=27,90°, между 2θ=34,75° и 2θ=35,48° и между 2θ=62,40° и 2θ=63,80°.
Данное изобретение относится также к вышеописанной композиции, в которой фаза тригидроксида алюминия дополнительно характеризуется тем, что не имеет измеримых полос на рентгенограмме между 2θ=20,15° и 2θ=20,65° и между 2θ=37,35° и 2θ=37,75°, а также к вышеописанной композиции, в которой фаза тригидроксида алюминия не имеет измеримых дифракционных полос в рентгенограмме между 2θ=18,70° и 2θ=18,90°, между 2θ=20,30° и 2θ=20,50° и между 2θ=40,30° и 2θ=40,70°.
Кроме того, настоящее изобретение относится к предшественникам катализаторов, включающим вышеописанную композицию.
Настоящее изобретение также относится к способу получения упомянутой композиции каменетсита или включающих ее предшественников катализаторов из порошка аморфный оксид алюминия - обогащенный диоксидом кремния оксид алюминия. Этот способ включает технологические стадии, которые аналогичны тем, что указаны в раннем патенте (патент США 6015485). А именно, процесс включает стадии:
(1) смачивание исходного материала, включающего диоксид кремния - оксид алюминия и аморфный оксид алюминия, хелатообразующим агентом и соединением металла в жидком носителе;
(2) старение смоченного таким образом исходного материала, пока он находится в смоченном состоянии;
(3) сушка состаренного таким образом исходного материала при температуре и условиях, способствующих значительному испарению жидкого носителя; и
(4) прокаливание высушенного таким образом материала.
Однако в настоящем изобретении исходный материал отличается от того, который использован в патенте '485, и продукт процесса можно отличить по размеру и концентрации образующихся частиц кристаллического оксида алюминия и по эксплуатационным свойствам катализаторов, полученных из приготовленных носителей.
В другом аспекте настоящее изобретение относится к катализаторам высокой активности, включающим носители на основе каменетсита и пропитанные одним или несколькими соответствующими каталитически активными металлами из VIB группы и VIII группы Периодической таблицы.
Наряду с вышерассмотренным катализатором, настоящее изобретение также относится к способу получения такого катализатора на основе вышеописанного способа получения каменетсита, адаптированного соответствующим образом. Так, исходный материал сначала может быть сформован известными специалистам в данной области методами с получением носителя катализатора желательной определенной формы. Полученный прокаленный носитель затем может быть смочен жидкой системой хелатообразующий агент/носитель перед пропиткой носителя соответствующими каталитически активными металлами и/или одновременно с такой пропиткой, и/или после такой пропитки носителя каталитически активными металлами с последующим осуществлением стадий (2)-(4), как описано выше.
Кроме того, настоящее изобретение также относится к способу повышения активности каталитической композиции, содержащей измельченный пористый носитель, содержащий диоксид кремния - оксид алюминия и аморфный оксид алюминия и пропитанный одним или несколькими каталитически активным металлом, включающему стадии:
(1') смачивание каталитической композиции контактированием с хелатообразующим агентом в жидком носителе;
(2') старение смоченного таким образом субстрата, пока он находится в смоченном состоянии;
(3') сушку состаренного таким образом субстрата при температуре и в условиях, обеспечивающих почти полное испарение жидкого носителя; и
(4') прокаливание высушенного таким образом субстрата.
Указанный способ может быть легко применен к существующим катализаторам, включающим измельченный пористый носитель, содержащий диоксид кремния - оксид алюминия и аморфный оксид алюминия, или может быть использован в процессе получения катализатора одновременно с пропиткой носителя и/или после такой пропитки, содержащего диоксид кремния - оксид алюминия и аморфный оксид алюминия, одним или несколькими каталитически активным металлом и/или его соединениями. Кроме того, способ может быть использован для улучшения активности отработанных катализаторов в процессе регенерации, причем отработанные катализаторы включают измельченный пористый носитель, содержащий диоксид кремния - оксид алюминия и аморфный оксид алюминия, при этом отработанный катализатор смачивают хелатообразующим агентом в жидком носителе, как на вышеописанной стадии (1'), после удаления из него углеродистых отложений с последующими стадиями (2'), (3') и (4').
Настоящее изобретение относится также к способу получения катализатора, специально подготовленного для обработки углеводородного материала, причем данный способ включает: (а) определение концентрации азотсодержащих соединений в углеводородном материале; (b) выбор исходного материала, включающего диоксид кремния - оксид алюминия и аморфный оксид алюминия, содержащего от примерно 4% масс. до примерно 8% масс. диоксида кремния, причем, по меньшей мере, примерно 20% масс. указанного оксида алюминия является аморфным, при этом указанный оксид алюминия имеет подходящую концентрацию диоксида кремния, такую, что, будучи состаренным во влажном состоянии при температуре старения во влажном состоянии, составляющей, по меньшей мере, 20°С, в течение промежутка времени, составляющего, по меньшей мере, 1 день, он образует предшественник катализатора, причем указанный предшественник катализатора включает в достаточной концентрации композицию, содержащую фазу тригидроксида алюминия, имеющую измеримые полосы в рентгенограмме между 2θ=18,15° и 2θ=18,50°, между 2θ=36,1° и 2θ=36,85°, между 2θ=39,45° и 2θ=40,30° и между 2θ=51,48° и 2θ=52,59° и не имеющую измеримых полос рентгенограммы между 2θ=20,15° и 2θ=20,65°, так что катализатор, полученный из указанного предшественника катализатора, будет эффективным для обработки указанного углеводородного материала, причем указанные концентрацию диоксида кремния, температуру старения во влажном состоянии и промежуток времени выбирают так, чтобы они были пропорциональны концентрации указанных азотсодержащих соединений; (с) получение носителя катализатора определенной формы из указанного исходного материала; а затем смачивание полученного носителя катализатора хелатообразующим агентом и соединением металла в жидком носителе, как на вышеописанной стадии (1), с последующими проведением вышеописанных стадий (2), (3) и (4).
Полагают (без намерения быть связанными какой-либо теорией), что при осуществлении упомянутых стадий в указанном порядке происходит взаимодействие между, по меньшей мере, диоксидом кремния - оксидом алюминия, аморфным оксидом алюминия, хелатообразующим агентом и водной кислотой, которое под воздействием температурных и временных условий стадии старения приводит к появлению каменетсита. После высушивания и прокаливания продукта этой реакции образуется кристаллическая фаза оксида алюминия, которую можно отличить от той, что получена в патенте США 6015485 по размеру и концентрации частиц кристаллического оксида алюминия. Размер кристаллитов на поверхности катализатора можно измерить с помощью известных методов, включая трансмиссионную электронную микроскопию.
Одновременно с появлением этой кристаллической фазы также достигается увеличение удельной поверхности катализатора. Кроме того, в предпочтительных вариантах осуществления изобретения образуется структура с пористостью, достигающей пикового значения в первой области размера пор 40Å или меньше, и более предпочтительно - в интервале от 20Å до 40Å, по результатам измерения азотной порометрией с использованием изотермы десорбции.
Образующиеся катализаторы высокой активности находят использование в ряде областей, что детально рассмотрено во многих ранее цитированных ссылках. Особенно предпочтительной областью использования является катализатор первой стадии гидрокрекинга при гидродеазотизации, гидродесульфуризации и гидродеметаллировании.
Соответственно настоящее изобретение относится также к способу обработки углеводородного материала, включающему контактирование указанного углеводородного материала с вышеописанными катализаторами.
Эти и другие отличительные особенности и преимущества настоящего изобретения будут более легко поняты специалистами в данной области при чтении следующего подробного описания.
Краткое описание чертежей
На фиг.1 показаны FTIR спектры тригидроксида алюминия настоящего изобретения, состаренного при 90°С в течение 1 дня и 25 дней, и спектр состаренного в течение 1 дня материала, вычтенный из спектра состаренного в течение 25 дней материала.
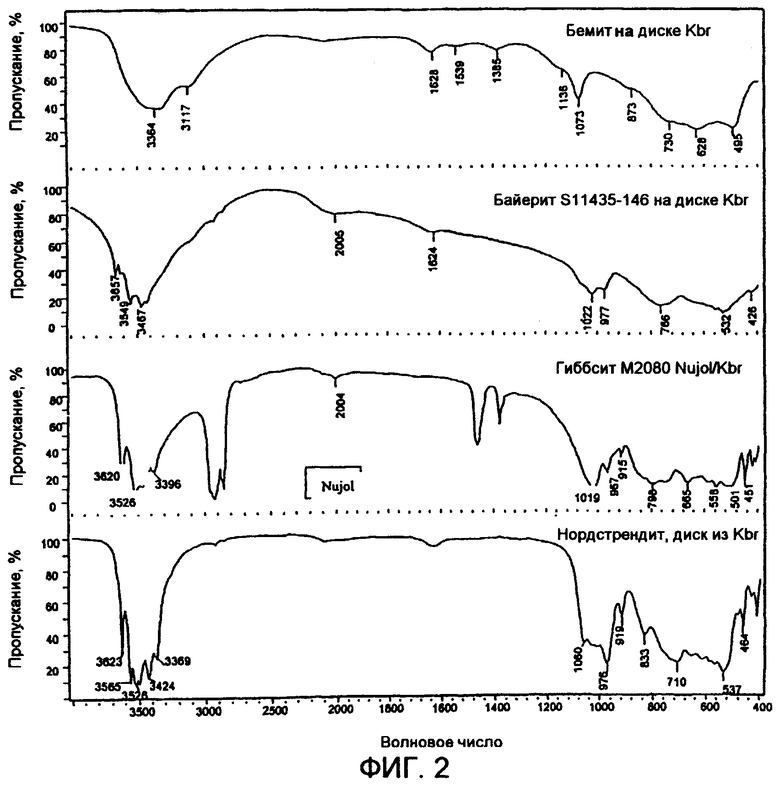
На фиг.2 представлен FTIR спектр бемита, байерита, гиббсита и нордстрендита.
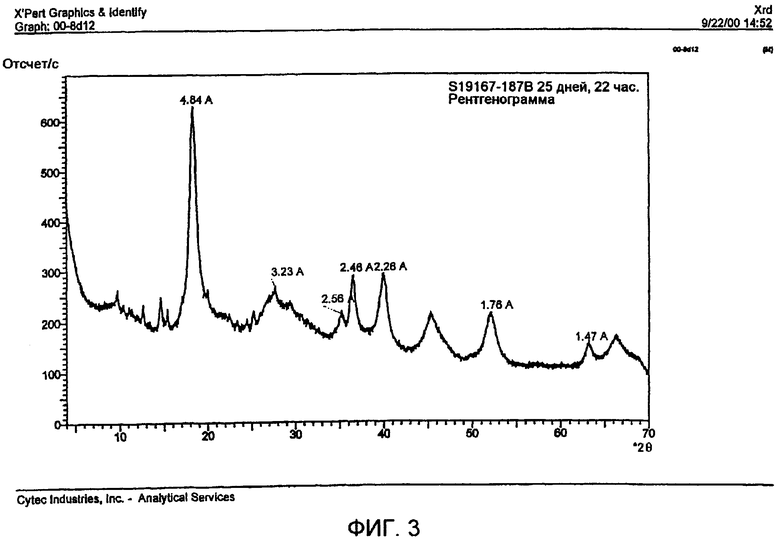
На фиг.3 показана рентгенограмма при 22-х часовом сканировании для образца, состаренного в течение 25 дней при 90°С. Отмеченные полосы относятся к каменетситу. Несколько неотмеченных линий, находящихся ниже d-параметра 5Å, обусловлены органическими соединениями, присутствующими в высушенном в термостате образце. Наблюдаются также широкие дифракционные полосы, приписываемые носителю на основе γ-оксида алюминия и активным оксидам металлов.
Подробное описание изобретения
А. Новая фаза тригидроксида алюминия (каменетсит)
Исходный материал
Предпочтительным исходным материалом для получения каменетсита является порошок диоксид кремния - оксид алюминия, содержащий значительное количество аморфного оксида алюминия. Измеримая концентрация каменетсита может быть получена из порошка, минимально содержащего всего 4% масс. диоксида кремния, а остальное составляет оксид алюминия, по меньшей мере, примерно 20% масс. которого представляет аморфный оксид алюминия, и из порошка, максимально содержащего 8% масс. диоксида кремния, а остальное составляет оксид алюминия, по меньшей мере, примерно 30% масс. которого представляет аморфный оксид алюминия. Предпочтительно исходный материал содержит от примерно 5% масс. до примерно 7% масс. диоксида кремния, а остальное составляет оксид алюминия, причем примерно от 20% масс. до примерно 50% масс. оксида алюминия является аморфными.
Способ получения
Новая фаза гидроксида алюминия настоящего изобретения может быть получена:
(1) смачиванием исходного материала контактированием с хелатообразующим агентом в жидком носителе и кислотном растворе соединения металла;
(2) состариванием смоченного таким образом исходного материала во влажном состоянии в условиях (например, комбинации температуры и продолжительности старения), которые приведут к образованию желательного количества каменетсита, предпочтительно при температурах выше, чем 50°С в течение от 1 до 10 дней;
(3) сушкой состаренного таким образом исходного материала при температуре и условиях, способствующих значительному испарению жидкого носителя; и
(4) прокаливанием высушенного таким образом материала.
Хелатообразующие агенты, подходящие для использования в настоящем способе, включают те, которые, как известно, образуют более стабильные комплексы с переходными металлами и алюминием и, следовательно, обладают высокими константами стабильности по отношению к ним. Особенно предпочтительным для использования в настоящем изобретении является этилендиаминтетрауксусная кислота (ЭDTA) и ее производные, включая, например, N-гидроксиэтилендиаминтетрауксусную кислоту и диаммоний этилендиаминтетрауксусную кислоту. Кроме того, подходящими являются трис(2-аминоэтил)амин и триэтилентетраамин. Другие кандидаты включают диэтилентриаминпентауксусную кислоту, циклогександиаминтетрауксусную кислоту, этиленгликоль-бис-(бета-аминоэтиловый простой эфир)-N,N'-тетрауксусную кислоту, тетраэтиленпентаамин и т.п. Целесообразность применения других хелатообразующих агентов могут легко оценить специалисты в данной области путем обработки образца исходного материала согласно настоящему изобретению, а затем сушки и прокаливания образца, определяя с помощью трансмиссионной электронной микроскопии или рентгеноструктурного анализа, образовался ли каменетсит с соответствующим размером кристаллитов или нет.
Количество используемого хелатообрзующего агента не является определяющим параметром для получения каменетсита, но влияет на количество образующегося продукта. Могут быть использованы количества хелатообразующегося агента, меняющиеся в широком интервале величин, в зависимости от ряда факторов, таких как растворимость в жидком носителе, тип носителя катализатора, и металлы, которыми пропитаны носители или которыми они должны быть пропитаны. Обычно исходный материал должен быть смочен жидким носителем, содержащим хелатообразующий агент в количествах, лежащих в интервале 0,01-1,0 грамм хелатообразующего агента на грамм исходного материала.
Материал может быть смочен любым обычным способом, таким как окунание или распыление. Чтобы обеспечить необходимое проникновение хелатообразующего агента, предпочтительным является окунание с последующим периодом пропитки. Предпочтительным жидким носителем является вода или раствор вода/аммиак.
Продолжительность периода, необходимого для старения смоченного исходного материала, является функцией температуры в процессе старения. При комнатной температуре предпочтительно старить смоченный субстрат в течение, по меньшей мере, 30 дней, более предпочтительно, по меньшей мере, в течение 60 дней. По мере увеличения температуры необходимое время старения снижается. При 80°С предпочтительно старить смоченный материал в течение, по меньшей мере, двух дней, более предпочтительно, по меньшей мере, в течение трех дней. Предпочтительно старение осуществляют при температуре в интервале от 20°С до 90°С.
Далее состаренный материал сушат, чтобы в значительной степени удалить жидкий носитель. Предпочтительно, чтобы сушка происходила сначала медленно, а затем быстро при повышенных температурах в интервале от 100°С до 250°С. Предпочтительно используют термостат с принудительной циркуляцией воздуха, чтобы ускорить сушку до предпочтительной продолжительности менее одного часа.
Высушенный таким образом материал затем прокаливают в условиях, хорошо известных специалистам в данной области. Однако предпочтительно прокаливание происходит в две стадии: первая - более низкотемпературная стадия, на которой температура достаточно высока, чтобы испарились или разложились любые количества оставшегося хелатообразующего агента, но которая недостаточно высока, чтобы хелатообразующие агенты сгорели с образованием углеродистых отложений. Температура этой первой стадии будет меняться в зависимости от конкретного хелатообразующего агента, но обычно достаточно температуры в интервале от 250°С до 350°С. Как только любой оставшийся хелатообразующий агент по существу удален, катализатор может быть затем прокален в условиях нормальной более высокой температуры, обычно используемой для этих целей.
В. Катализаторы
Способ получения содержащих каменетсит катализаторов
Методика получения каменетсита, описанная выше, может быть адаптирована для получения конечного катализатора. Исходный материал сначала может быть сформирован в виде желательного носителя известными специалистам в данной области методами. Полученный прокаленный носитель затем может быть смочен жидкой системой хелатообразующий агент/носитель перед пропиткой носителя соответствующими каталитически активными металлами и/или одновременно с такой пропиткой, и/или после такой пропитки носителя каталитически активными металлами с последующим осуществлением стадий (2)-(4), как описано выше. Важно только обеспечить, чтобы стадия старения протекала в условиях, когда пропитанный носитель смочен жидкостью-носителем для хелатообразующего агента и кислотным раствором металлов для пропитки.
Каталитически активные металлы
Настоящее изобретение применимо к катализаторам, пропитанным одним или несколькими из широкого ряда каталитически активных металлов, хорошо известных специалистам в данной области, что подтверждено примерами, например, многочисленными приведенными ссылками. В контексте настоящего изобретения "каталитически активные металлы" включают как сами металлы, так и соединения металлов. Помимо каталитически активных металлов катализаторы также могут быть пропитаны одним или несколькими хорошо известными промоторами, такими как фосфор, олово, диоксид кремния и титан (включая их соединения).
Обычно каталитически активные металлы представляют переходные металлы, выбранные из группы, включающей металлы группы VIB, группы VIII и их комбинации. Конкретный выбор металла(ов), промотора(ов) и их содержание, безусловно, зависят от желательного конечного использования катализатора, и эти переменные могут легко регулироваться специалистами, исходя из конечного использования. В качестве их конкретных примеров можно упомянуть следующие (% масс. в расчете на общую массу катализатора):
Операции гидрообработки
Операции, не относящиеся к гидрообработке
Такие катализаторы получают пропиткой носителей соответствующими компонентами с последующими стадиями сушки, сульфидирования и/или прокаливания по требованиям соответствующего конечного использования. Такое приготовление катализатора обычно хорошо известно специалистам в данной области, что подтверждается примерами многочисленных, ранее приведенных ссылок, а дополнительные детали могут входить во включенные в данное описание ссылки или многочисленные другие общие ссылочные работы, доступные по данному предмету.
Регенерация катализатора
Как указано выше, способ согласно настоящему изобретению применим не только к предварительно сформированным катализаторам, но также может быть аналогичным образом применен для регенерации катализаторов. В частности, после удаления углеродистого материала из отработанного катализатора хорошо известными методами такие катализаторы обрабатывают затем согласно стадиям с (1) по (4) аналогично тому, как описано выше.
Катализаторы, сформированные с учетом конкретных операций
Тщательным выбором температуры и продолжительности стадии старения можно модифицировать концентрациию и размер кристаллитов каменетсита вместе с его конечной структурой пор.
Тогда модифицированный катализатор по-разному влияет, например, на гидродесульфуризацию каждого из пары газойлей. Одна возможность формирования катализатора настоящего изобретения с определенной целью рассмотрена ниже в примере 9. Пример 9 предназначен пояснить возможности, которые открываются при использовании настоящего изобретения, и не ограничивает его никоим образом в объеме притязаний. Специалисты в данной области могут определить другие такие возможности.
С. Характеристика каменетсита
Рентгеноструктурный анализ вновь обнаруженной фазы тригидроксида алюминия с использованием Cu Кα облучения кристаллов подтверждает, что материал отличается от трех ранее известных фаз тригидроксида алюминия. Как показано ниже в таблице 1, каменетсит дает очень сильный пик при 2θ=18,33°, такой же угол, что и основной пик гиббсита и относительно близко к основным пикам нордстрендита и байерита. Однако в остальной части диаграммы дифракционных полос каменетсит дает существенные пики при углах дифракции, где другие фазы не имеют пиков или не дает пики при углах, где другие фазы их имеют. Положения дифракционных полос каменетсита показаны здесь с относительной точностью 1% (доверительный интервал 95%), и относительные интенсивности - с относительной точностью 10% (доверительный интервал 95%).
(2) Показаны только главные дифракционные полосы для гиббсита, нордстрендита и байерита.
Как показано в таблице 2, размер кристаллитов каменетсита и интегрированная интенсивность рентгеновских дифракционных полос при 2θ=18,33° увеличиваются с увеличением температуры старения и продолжительности старения.
Термогравиметрический анализ (ТГА) и рентгеноструктурный анализ содержащих каменетсит материалов, нагретых до высоких температур, показывают исчезновение основного пика при 2θ=18,33° при температуре примерно 250°С. Поскольку известно, что 250°С представляет собой температуру перехода тригидроксидов алюминия в переходные оксиды алюминия, эти данные подтверждают, что новый материал представляет отдельную новую фазу тригидроксида алюминия.
Кроме того, проведен анализ методом ИК-спектроскопии с трансформацией Фурье (FTIR) при 90°С состаренного в течение 1 дня и состаренного в течение 25 дней продуктов низкотемпературной сушки. Полученные спектры показаны на фиг.1. Ясно видно увеличенное содержание каменетсита в материале 25-дневного старения, когда спектр материала 1-дневного старения вычитают из спектра материала 25-дневного старения, что показано как спетр "разности" в нижней части фиг.1. Полосы поглощения FTIR при волновых числах 3512, 989 и 521 в спектре "разности" подтверждают наличие Al(ОН)3. Для сравнения, на фиг.2 показаны FTIR-спектры бемита, байерита, гиббсита и нордстрендита.
Сравнение с материалами, полученными без диоксида кремния в исходном материале
Появление каменетсита в материале, полученном способом настоящего изобретения, не так очевидно, когда исходный материал содержит менее чем примерно 4% масс. диоксида кремния. Однако установлена корреляция, которая позволяет косвенно определить количество каменетсита, содержащегося в продукте способа настоящего изобретения. Эта корреляция связывает количество каменетсита в продукте с его строением, определяемым через его пористость, измеренную по адсорбции азота. Экстраполируя эту корреляционную зависимость, можно прийти к выводу, что небольшое количество каменетсита вероятно содержится в материале, полученном с использованием в качестве исходного материала не содержащего диоксида кремния оксида алюминия. Данные, показывающие такие экстраполированные значения для каменетсита в материалах, полученных из такого не содержащего диоксида кремния оксида алюминия, представлены в примерах D и Е.
Примеры
Настоящее изобретение, как оно описано выше, далее будет пояснено следующими конкретными примерами, которые представлены только для пояснения и не ограничивают объема притязания изобретения.
Условия испытаний
Условия испытаний примененных для сравнения эксплуатационных свойств катализаторов настоящего изобретения и тех, что описаны в патенте США 6015485, и стандартного катализатора нефтеперерабатывающего производства, следующие:
Испытание типа А
Сырье: газойль прямой перегонки для Североамериканского нефтеперерабатывающего завода
Перегонка, °С:
Условия испытания:
Испытание типа В
Сырье: легкий арабский газойль прямой перегонки для Европейского нефтеперерабатывающего завода
Перегонка, °С:
Условия испытания:
Испытания типов C1, С2, С3
Сырье: смесь газойлей
Условия испытания:
Испытание типа D
сырье: смесь газойлей прямой перегонки/легкого циклического
Условия испытания:
Испытания типов E1, E2
сырье: смесь газойлей прямой перегонки/легкого циклического
Условия испытания:
Испытание типа F
сырье: арабский газойль прямой перегонки
Условия испытаний:
Пример 1
Настоящий пример описывает получение образцов катализаторов настоящего изобретения.
Порошок, включающий частицы оксида алюминия, покрытые 6% масс. диоксида кремния, измельчают на дробильных вальцах, экструдируют в форме скругленного треугольника, сушат и прокаливают обычными средствами. Подробности относительно порошка 6% масс. диоксид кремния - оксид алюминия описаны в открытой литературе (McMillan М., Brinen, J.S., Carruthers, J.D., Haller, G.L., "A 29Si NMR Investigation of the Structure of Amorphous Silica-Alumina Supports", Colloids and Surfaces, 38(1989) 133-148). Использованный в данном случае порошок соответствует критерию стабильности пористости, как описано в цитированной выше публикации.
95,6 г носителя диоксид кремния - оксид алюминия пропитывают до первоначальной влажности 100 мл раствора "А". Раствор, обозначенный в описании как раствор "А", состоит из смеси двух растворов: раствора "С", полученного добавлением 11,3 г раствора гидроксида аммония (28% масс.) к 65,3 г раствора тетрааммонийэтилендиаминтетрауксусной кислоты (38% в расчете на EDTA) от фирмы Dow Versene, и раствора "D". Раствор "D" получают добавлением 4,37 г раствора гидроксида аммония (28% масс.) к 41,0 г раствора "Е". Раствор, обозначенный в данном описании как раствор "Е", получают добавлением 137 г твердого карбоната кобальта к 500 г разбавленного раствора фосфорной кислоты (23,0 г Н3PO4 - 86,0% масс. и 475 г деионизированной воды), нагреванием смеси до 55°С и добавлением затем 300 г Climax МоО3. Затем смесь нагревают до 98°С при перемешивании в течение 1,5 часов, по истечении которых добавляют 100 г раствора азотной кислоты (70% масс.) до полного растворения смеси. Этот раствор, обозначенный в описании как раствор "Е", фосфорной кислоты, содержащий соединения кобальта и молибдена, где массовое отношение Со/Мо составляет 0,258, а рН составляет приблизительно 0,6, охлаждают до комнатной температуры и 41,0 г раствора используют для получения раствора, названного в настоящем описании раствором "D".
Смоченные гранулы оставляют стоять на 2 часа, а затем сушат в термостате тонким слоем при 230°С в течение 1 часа. 122,6 г высушенного продукта окунают затем в емкость с раствором "Е" и обеспечивают циркуляцию 360 г этого раствора, чтобы смочить гранулы. Смоченные гранулы отделяют затем от избытка раствора центрифугированием и помещают в закрытой колбе в термостат при 75°С, и держат там при указанной температуре в течение 3 дней. После этого материал быстро сушат при 230°С в течение 20 минут для испарения жидкого носителя до уровня от начального (LOI) в 30-32% масс. с последующим прокаливанием при 500°С в течение одного часа на воздухе с получением катализатора настоящего изобретения, обозначенного в настоящем описании как катализатор С-2. Катализатор С-2 содержит 5,97% масс. Со, 19,7% масс. Мо и 0,77% масс. Р, где % масс. рассчитан по общей массе катализатора, и имеет удельную поверхность 305 м2/г и установленную интенсивность пиков каменетсита в 3344 отсчета.
Вторую 100-граммовую порцию носителя смачивают до первоначальной влажности раствором, включающим 62,5 г раствора диаммонийэтилендиаминтетрауксусной кислоты (40,0% масс. на EDTA) от фирмы Dow Versene и 77,59 г раствора, обозначенного в настоящем описании как раствор "F". Раствор "F" готовят добавлением 329 г МоО3, 100,0 г Со(ОН)2 и 282,6 г монокристаллогидрата лимонной кислоты к 695 г деионизированной воды и нагреванием от комнатной температуры до 80°С. Затем раствор кипятят приблизительно в течение одного часа до полного растворения всех компонентов, а затем охлаждают до комнатной температуры. Раствор "F" содержит соединения кобальта и молибдена, причем массовое отношение Со/Мо составляет 0,292 при рН приблизительно 0,6. Смоченным гранулам дают пропитаться в течение одного часа, а затем их сушат тонким слоем в сушилке при 230°С в течение одного часа.
После этого высушенные гранулы помещают в 300 г раствора "F" и обеспечивают циркуляцию раствора над гранулами в течение одного часа. Смоченные гранулы отделяют от раствора центрифугированием и помещают в закрытой колбе в термостат, установленный на 75°С, на 3 дня. Затем материал подвергают быстрой сушке при 230°С в течение 1 часа, чтобы выпарить жидкий носитель до LOI 30-32% масс., а затем прокаливают при 500°С в течение 1 часа с получением катализатора настоящего изобретения, обозначенного в настоящем описании как катализатор D-2. Катализатор D-2 содержит 4,11% масс. Со и 16,3% масс. Мо, имеет удельную поверхность 347 м2/г и установленную интенсивность пиков каменетсита в 4320 отсчетов.
Третью 100-граммовую порцию носителя смачивают до первоначальной влажности раствором, включающим 64,7 г раствора диаммонийэтилендиаминтетрауксусной кислоты (40,0% масс. на EDTA) от фирмы Dow Versene с 82,3 г раствора, обозначенного в настоящем описании как раствор "G". Раствор "G" готовят добавлением 300 г МоО3 и 137,5 г СоСО3 к 575 г деионизированной воды с последующим нагреванием до 70-80°С при перемешивании и затем медленным добавлением 225,0 г монокристаллогидрата лимонной кислоты. Затем раствор кипятят до полного растворения всех компонентов в течение 30 минут, а затем оставляют его остывать. Раствор "G", содержащий соединения кобальта и молибдена, в котором массовое отношение Со/Мо составляет 0,321, имеет рН приблизительно 2,0. Смоченные гранулы оставляют стоять в течение 1 часа, а затем их сушат тонким слоем в термостате, установленном на 230°С, в течение одного часа.
После этого высушенные гранулы помещают в 300 г раствора "G" и обеспечивают циркуляцию раствора над гранулами в течение одного часа. Смоченные гранулы отделяют от раствора центрифугированием и помещают в закрытой колбе в термостат, установленный на 75°С, на 3 дня. Затем материал подвергают быстрой сушке при 230°С в течение одного часа, чтобы выпарить жидкий носитель до LOI 30-32% масс., а затем прокаливают при 500°С в течение дополнительного часа с получением катализатора настоящего изобретения, обозначенного в настоящем описании как катализатор Е-2. Катализатор Е-2 содержит 4,53% масс. Со и 14,6% масс. Мо, имеет удельную поверхность 310 м2/г и установленную интенсивность пиков каменетсита в 1082 отсчетов.
Пример 2 (Сравнительный)
Этот пример описывает получение образцов катализаторов патента США 6025485.
Носитель готовят по той же методике, что и в примере 1, за исключением того, что исходный материал не содержит диоксида кремния.
Часть этого носителя обрабатывают таким же образом, как и для катализатора С-2, и получают катализатор С-1. Катализатор С-1 содержит 4,67% масс. Со, 18,1% масс. Мо и 0,61% масс. Р, имеет удельную поверхность 280 м2/г и установленную интенсивность пиков каменетсита в 195 отсчетов.
Вторую порцию этого носителя обрабатывают таким же образом, что и катализатор D-2 с получением катализатора D-1. Катализатор D-1 содержит 4,08% масс. Со и 14,7% масс. Мо, имеет удельную поверхность 230 м2/г и установленную интенсивность пиков каменетсита менее 100 отсчетов.
Пример 3 (Сравнительный)
Настоящий пример описывает получение двух катализаторов, полученных способом настоящего изобретения, но с недостаточным для образования катализатора настоящего изобретения и с близким к минимально достаточному количеством диоксида кремния в исходном материале.
Носитель готовят по той же методике, что и в примере 1, за исключением того, что исходный материал содержит 2% масс. диоксида кремния. Этот носитель обрабатывают тем же методом, что и катализатор Е-2 и получают катализатора Е-1. Катализатор Е-1 содержит 5,91% масс. Со и 19,7% масс. Мо, имеет удельную поверхность 215 м2/г и установленную интенсивность пиков каменетсита в 300 отсчетов.
Второй носитель готовят по той же методике, что и в примере 1, за исключением того, что исходный материал содержит 3,7% масс. диоксида кремния, меньше, чем предпочтительное количество (6% масс.), но выше, чем 2% масс., использованных для катализатора Е-1. Этот носитель обрабатывают таким же образом, что и катализатор D-2 и получают катализатора D-3. Катализатор D-3 содержит 4,08% масс. Со и 15,7% масс. Мо, имеет удельную поверхность 245 м2/г и установленную интенсивность пиков каменетсита в 1880 отсчетов.
Пример 4
В настоящем примере сравнивают эксплуатационные свойства катализатора С-2 в сравнении с катализатором С-1 и стандартного катализатора нефтеперерабатывающего производства ("Стандарт"), полученного традиционными средствами.
Каждый катализатор подвергают испытанию типа А. Полученные результаты представлены в таблице 3.
Это испытание показывает, что катализатор С-2, т.е. катализатор настоящего изобретения, более эффективен при удалении серы, чем любой из двух других катализаторов.
Пример 5
В настоящем примере сравнивают эксплуатационные свойства катализатора D-2, катализатора D-1 и стандартного катализатора нефтеперерабатывающего производства ("Стандарт"), полученного традиционными средствами.
Каждый катализатор подвергали испытанию типа В. Результаты представлены в таблице 4.
Это испытание показывает, что для достижения желательного содержания серы в продукте требуется меньшее количество катализатора D-2 настоящего изобретения, чем любого из двух других катализаторов.
Пример 6
В этом примере сравнивают эксплуатационные свойства катализатора Е-2, катализатора Е-1 и стандартного катализатора нефтеперерабатывающего производства ("Стандарт"), полученного традиционными средствами.
Каждый катализатор подвергали испытанию типа В. Результаты представлены в таблице 5.
Это испытание показывает, что для достижения желательного содержания серы в продукте требуется меньшее количество катализатора Е-2 настоящего изобретения, чем любого из двух других катализаторов. Испытание также показывает, что использование исходного материала, содержащего недостаток диоксида кремния по отношению к методике получения катализатора по настоящему изобретению, приводит к получению катализатора, т.е. катализатора Е-1, который не более эффективен, чем стандартный катализатор нефтеперерабатывающего производства.
Пример 7
Настоящий пример описывает получение образцов катализаторов настоящего изобретения, в которых оба элемента Ni и Со включены в конечный катализатор, и продукты получения подвергают существенно иным условиям старения.
100 г носителя диоксид кремния - оксид алюминия, описанного в примере 1, пропитывают до первоначальной влажности 152,4 г раствора "К". Раствор, обозначенный в настоящем описании как раствор "К", состоит из смеси двух растворов: 68,0 г раствора "L", полученного добавлением 6,66 г твердого ацетата никеля (23,58% масс. на металлический Ni) к 99,54 г раствора диаммонийэтилендиаминтетрауксусной кислоты фирмы Dow Versene (40% масс. на EDTA), и 84,4 г раствора "F", описанного выше в примере 1.
Смоченные гранулы оставляют стоять на 2 часа, как и в предыдущих случаях, а затем сушат в термостате тонким слоем при 230°С в течение 1 часа. 143,8 г высушенного продукта окунают затем в емкость с раствором "F" и обеспечивают циркуляцию 317 г этого раствора для промывки гранул. После этого смоченные гранулы отделяют от избытка раствора центрифугированием и помещют в закрытой колбе в термостат, установленный на 75°С, и держат при указанной температуре в течение 3 дней. Затем материал быстро сушат при 230°С в течение 20 минут, чтобы выпарить жидкий носитель до LOI 30-32% масс., с последующим прокаливанием при 500°С в течение одного часа на воздухе с получением катализатора настоящего изобретения, обозначенного как катализатор А. Катализатор А содержит 4,3% масс. Со, 17,0% масс. Мо и 0,68% масс. Ni, имеет удельную поверхность 347 м2/г и установленную интенсивность пиков каменетсита в 2670 отсчетов.
Второй процесс получения проводят по идентичной для катализатора А схеме, но старение проводят при 90°С в течение 7 дней вместо 3 дней при 75°С. Этот катализатор обозначают в настоящем описании как катализатор В. Катализатор В содержит 4,24% масс. Со, 16,8% масс. Мо и 0,68% масс. Ni, имеет удельную поверхность 340 м2/г и установленную интенсивность пиков каменетсита в 6138 отсчетов.
Пример 8
Настоящий пример показывает, что активность катализатора настоящего изобретения улучшается по сравнению с активностью стандартного катализатора нефтеперерабатывающего производства при интенсификации рабочих условий.
Катализатор А и стандартный катализатор нефтеперерабатывающего производства ("Стандарт"), полученный традиционными средствами, подвергают испытаниям типов C1, C2 и С3, которые идентичны, за исключением того, что рабочая температура повышается от C1 к С3. Результаты испытаний представлены в таблице 6.
Следует обратить внимание на увеличение относительной объемной активности при повышении рабочей температуры от 343°С до 357°С и до 371°С. Представленные данные показывают, что эксплуатационные характеристики катализатора настоящего изобретения в сравнении со стандартным катализатором нефтеперерабатывающего производства повышаются при интенсификации рабочих условий.
Пример 9
Настоящий пример иллюстрирует возможность сформировать катализаторы настоящего изобретения с учетом ожидаемых рабочих условий.
Катализатор А, катализатор В и стандартный катализатор нефтеперерабатывающего производства ("Стандарт"), полученный традиционными средствами, каждый подвергают испытаниям типов D, E1 и E2. Сырье для испытания типа D содержит умеренную концентрацию азота (196 масс. ч/млн), тогда как сырье для испытаний типов E1 и E2 имеет высокое содержание азота (760 масс. ч/млн).
В этом примере эксплуатационные параметры катализатора А согласно изобретению сравнивают с эксплуатационными параметрами катализатора В, полученного со значительно более высокой концентрацией каменетсита в его материале-предшественнике. Это увеличение каменетсита достигается за счет увеличения как температуры, так и продолжительности стадии старения. При таком интенсифицированном старении увеличиваются концентрация и размер кристаллитов каменетсита. Наряду с изменениями в каменетсите претерпевает существенное изменение структура пор конечного катализатора. В результате модифицированный катализатор дает совершенно иной отклик на увеличение температуры в процессе гидродесульфуризации именно одного конкретного газойля. Это можно видеть в следующих результатах испытаний, представленных в таблице 7.
В этой таблице перечислены три катализатора, а именно в минимальном описании стандартный промышленный катализатор сравнения, катализатор А, т.е. катализатор согласно изобретению, полученный так, что он показывает умеренную концентрацию каменетсита в материале предшественнике, и катализатор В, т.е. катализатор, имеющий высокую концентрацию каменетсита в материале-предшественнике. Затем каждый катализатор испытывают наряду с другими при постоянной температуре и давлении с использованием двух смесей газойлей прямой перегонки/легкого циклического, как описано в методах испытаний типов D и Е, G1 и G2.
При испытании типа D оба катализатора настоящего изобретения являются более активными, чем стандартный, причем катализатор с более высокой концентрацией каменетсита несколько лучше другого (130 против 123 RVA). Аналогичные результаты достигаются для испытания типа E1. Однако следует обратить внимание, что когда условия переработки изменяются для трех катализаторов в испытании типа Е2, то вариант с более высоким содержанием каменетсита сохраняет свои эксплуатационные преимущества, а тот, который содержит меньшее количество каменетсита, уступает ему.
Не имея в виду быть связанными какой-либо теорией, авторы полагают, что катализаторы, полученные из материалов с высоким содержанием каменетсита, обладают большим количеством активных центров на единицу объема катализатора, чем традиционно полученные катализаторы. В представленном выше примере два катализатора настоящего изобретения по-разному действуют при увеличении температуры в процессе испытания типа Е2. Сырье для испытания типа Е отличается от газойля для испытания типа D, в первую очередь, концентрацией азотсодержащих молекул.
При низком давлении и в условиях низкой скорости обработки водородом в этих испытаниях удаление азотсодержащих молекул является далеко не полным. Кроме того, неконвертированные азотсодержащие молекулы становятся гидрированными (основными) азотсодержащими молекулами в процессе частичной (неполной) гидродеазотизации газойля. Известно, что такие молекулы снижают активность катализатора десульфуризации за счет адсорбции по его более кислотным центрам. Поэтому можно обоснованно предположить, что катализатор, достигающий более полного удаления азотсодержащих молекул (катализатор В) и обладающий более доступными центрами гидродесульфуризации, будет уменьшать "эффект динамического отравления" остальными азотсодержащими молекулами и поддерживать, таким образом, более высокую активность гидродесульфуризации в катализаторе. Поэтому представленные результаты показывают, что катализаторы настоящего изобретения могут быть сформулированы с учетом оптимальных эксплуатационных свойств, в зависимости от различных концентраций азотсодержащих молекул в сырье.
Пример 10
В настоящем примере сравнивают эксплуатационные характеристики катализатора, полученного с "достаточным" уровнем диоксида кремния в носителе доксид кремния - оксид алюминия, и катализатора, полученного с "близким к минимально достаточному" содержанием диоксида кремния в носителе диоксид кремния - оксид алюминия. Катализатор D-2 сравнивают с катализатором D-3 и стандартным катализатором нефтеперерабатывающего производства ("Стандарт"), полученным традиционными средствами, в стандартном испытании типа F.
Это испытание показывает, что использование исходного материала, содержащего близкое к минимально достаточному количество диоксида кремния в процессе получения катализатора настоящего изобретения, приводит к получению катализатора, т.е. катализатора D-3, который более эффективен, чем стандартный катализатор нефтеперерабатывающего производства, но не так активен, как катализатор с достаточным количеством диоксида кремния в носителе диоксид кремния - оксид алюминия - катализатор D-2.
Изобретение относится к получению новой фазы тригидроксида алюминия и использованию ее при изготовлении катализаторов. Композиция включает твердую фазу тригидроксида алюминия, причем указанная фаза имеет измеримые полосы рентгенограммы между 2θ=18,15° и 2θ=18,50°, между 2θ=36,1° и 2θ=36,85°, между 2θ=39,45° и 2θ=40,30° и между 2θ=51,48° и 2θ=52,59° и не имеет измеримых полос рентгенограммы между 2θ=20,15° и 2θ=20,65°. Способ получения композиции предшественника катализатора включает смачивание исходного материала, содержащего диоксид кремния - оксид алюминия и аморфный оксид алюминия, путем контакта с хелатообразующим агентом в жидком носителе и соединением металла, старение смоченного исходного материала, сушку состаренного исходного материала, прокаливание высушенного материала. Катализатор включает носитель, полученный из композиции или предшественника катализатора, и каталитически активное количество одного или нескольких металлов, соединений металлов или их комбинаций. Способ получения катализатора включает получение носителя катализатора из исходного материала, содержащего диоксид кремния - оксид алюминия и аморфный оксид алюминия, смачивание исходного материала путем контакта с хелатообразующим агентом и каталитически активным количеством одного или нескольких металлов, соединений металлов или их комбинаций в жидком носителе, старение исходного материала, сушку и прокаливание. Способ регенерации использованного материала включает дополнительную стадию удаления материала осевшего на катализаторе в процессе предшествующего использования. Остальные стадии проводят аналогично способу получения катализатора. Способ получения катализатора, специально подготовленного для обработки углеводородного сырья, характеризуется использованием композиции, содержащей фазу тригидроксида алюминия, имеющей измеримые полосы в рентгенограмме между 2θ=18,15° и 2θ=18,50°, между 2θ=36,1° и 2θ=36,85°, между 2θ=39,45° и 2θ=40,30° и между 2θ=51,48° и 2θ=52,59° и не имеющей измеримых полос рентгенограммы между 2θ=20,15° и 2θ=20,65°. Изобретение позволяет улучшить активность и регенерацию катализаторов. 9 н. и 32 з.п. ф-лы, 8 табл., 3 ил.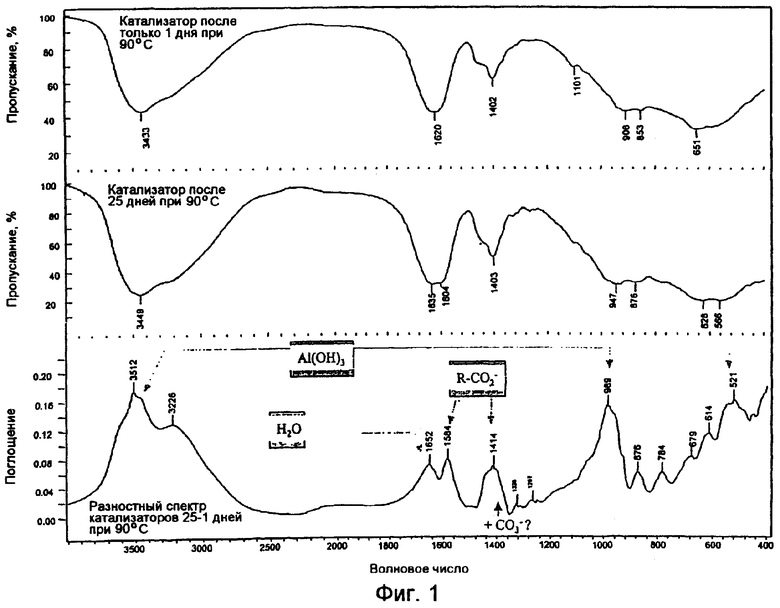
(a) смачивание исходного материала, включающего диоксид кремния-оксид алюминия и аморфный оксид алюминия, содержащий от 4 до 8 мас.% диоксида кремния, причем, по меньшей мере, 20 мас.% указанного оксида алюминия является аморфным, путем контакта с определенным количеством хелатообразующего агента в интервале от 0,1 до 1,0 г на 1 г исходного материала в жидком носителе и соединением металла;
(b) старение смоченного таким образом исходного материала, пока он находится в смоченном состоянии;
(c) сушка состаренного таким образом исходного материала при температуре от 100 до 230°С; и
(d) прокаливание высушенного таким образом материала.
(a) получение носителя катализатора определенной формы из исходного материала, включающего диоксид кремния - оксид алюминия и аморфный оксид алюминия, содержащего от примерно 4 до примерно 8 мас.% диоксида кремния, причем, по меньшей мере, примерно 20 мас.% указанного оксида алюминия является аморфным;
(b) смачивание исходного материала контактированием с хелатообразующим агентом и каталитически активным количеством одного или нескольких металлов, соединений металлов или их комбинаций в жидком носителе;
(c) старение смоченного таким образом исходного материала, пока он находится во влажном состоянии;
(d) сушку состаренного таким образом исходного материала при температуре от 100 до 230°С; и
(e) прокаливание высушенного таким образом материала.
(а) смачивание указанного катализатора контактированием с хелатообразующим агентом в жидком носителе;
(b) старение смоченного таким образом катализатора, пока он находится во влажном состоянии;
(c) сушку состаренного таким образом катализатора при температуре от 100 до 230°С и в условиях, обеспечивающих, по существу, полное улетучивание жидкого носителя; и
(d) прокаливание высушенного таким образом катализатора.
(a) удаление материала, осевшего на указанном катализаторе в процессе предшествующего использования;
(b) смачивание указанного катализатора контактированием с хелатообразующим агентом в жидком носителе;
(c) старение смоченного таким образом катализатора, пока он находится во влажном состоянии;
(d) сушку состаренного таким образом катализатора при температуре от 100 до 230°С и в условиях, обеспечивающих, по существу, полное улетучивание жидкого носителя; и
(e) прокаливание высушенного таким образом катализатора.
(a) определение концентрации азотсодержащих соединений в углеводородном материале;
(b) выбор исходного материала, включающего диоксид кремния-оксид алюминия и аморфный оксид алюминия, содержащего от примерно 4 до примерно 8 мас.% диоксида кремния, причем, по меньшей мере, примерно 20 мас.% указанного оксида алюминия является аморфным, при этом указанный оксид алюминия имеет подходящую концентрацию диоксида кремния, такую, что, будучи состаренным во влажном состоянии при температуре старения во влажном состоянии, составляющей, по меньшей мере, 20°С, в течение промежутка времени, составляющего, по меньшей мере, 1 день, он образует предшественник катализатора, причем указанный предшественник катализатора включает в достаточной концентрации композицию, содержащую фазу тригидроксида алюминия, имеющую измеримые полосы в рентгенограмме между 2θ=18,15° и 2θ=18,50°, между 2θ=36,1° и 2θ=36,85°, между 2θ=39,45° и 2θ=40,30° и между 2θ=51,48° и 2θ=52,59°, и не имеющий измеримых полос рентгенограммы между 2θ=20,15° и 2θ=20,65°, так что катализатор, полученный из указанного предшественника катализатора, будет эффективным для обработки указанного углеводородного материала, где указанные концентрация диоксида кремния, температура старения во влажном состоянии и промежуток времени выбирают так, чтобы они были пропорциональны концентрации указанных азотсодержащих соединений;
(c) получение носителя катализатора определенной формы из указанного исходного материала;
(d) смачивание указанного исходного материала контактированием с хелатообразующим агентом и соединением металла в жидком носителе;
(e) старение смоченного таким образом исходного материала, пока он находится во влажном состоянии, при температуре, выбранной на стадии (b) в течение промежутка времени, выбранного на стадии (b);
(f) сушка состаренного таким образом исходного материала при температуре от 100 до 230°С; и
(g) прокаливание высушенного таким образом материала.
| Journal of Physical Chemestry, 1932, v.36, с.3010-3029 | |||
| WO 9531280 A1, 23.11.1995 | |||
| СПОСОБ ПОЛУЧЕНИЯ СФЕРИЧЕСКИХ ГРАНУЛ АКТИВНОГО ОКСИДА АЛЮМИНИЯ | 1988 |
|
SU1515609A1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ДЛЯ ГИДРООЧИСТКИ НЕФТЯНОГО СЫРЬЯ | 1992 |
|
RU2022644C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ГИДРООЧИСТКИ НЕФТЕПРОДУКТОВ | 1996 |
|
RU2100079C1 |
| RU 94020424 A1, 20.04.1996 | |||
| US 5435986 А, 25.07.1995 | |||
| Устройство для контроля глубины залегания кабеля в грунте | 1970 |
|
SU455307A1 |

