Область техники, к которой относится изобретение
Настоящее изобретение относится к области медицины и биотехнологии и, в частности, касается способов и композиций для лечения и уменьшения интенсивности симптомов заболеваний и других состояний, таких как инфекционные заболевания, аутоиммунные заболевания и аллергические состояния. В способах используются соединения на основе моно- и дисахаридов для селективного стимулирования иммунных реакций у животных и растений.
УРОВЕНЬ ТЕХНИКИ
Врожденная иммунная система координирует воспалительную реакцию на патогены с помощью системы, которая различает «своих» и «чужих» посредством рецепторов, идентифицирующих классы молекул, синтезируемых исключительно микробами. Эти классы иногда называют ассоциированными с патогенами молекулярными структурами, и они включают, например, липополисахарид (ЛПС), пептидогликаны, липотехоевые кислоты и бактериальные липопротеины (БЛП).
Липополисахарид (ЛПС) представляет собой вещество, в большом количестве находящееся на внешней стороне клеточных стенок грамотрицательных бактерий, распознаваемое врожденной иммунной системой. Хотя химическая структура ЛПС известна уже довольно давно, но молекулярная основа распознавания ЛПС белками сыворотки крови и/или клетками только недавно начала проясняться. В ряде последних сообщений мощную врожденную иммунную реакцию на ЛПС и другие микробные компоненты связывают с семейством рецепторов, называемых Толл-подобными рецепторами (TLR). Все члены семейства TLR представляют собой мембранные белки, имеющие один трансмембранный домен. Эти цитоплазматические домены содержат примерно 200 аминокислот и подобны цитоплазматическому домену рецептора IL-1. Внеклеточные домены Толл-семейства белков относительно велики (около 550-980 аминокислот) и могут содержать множественные сайты связывания лигандов.
Значение TLR для иммунного ответа на ЛПС была продемонстрирована по меньшей мере для двух Толл-подобных рецепторов, Тlr2 и Tlr4. Например, в исследованиях по трансфекции на клетках эмбриональных почек было показано, что человеческий Тlr2 отвечает за реактивность на ЛПС (Yang et al., Nature 395:284-288 (1998); Kirschning et al., J. Exp.Med. 11:2091-97 (1998)). Выяснилось, что для сильного ответа на ЛПС требуется наличия как связывающего ЛПС белка (СЛБ), так и CD14, который связывает ЛПС с высокой аффинностью. Прямое связывание ЛПС с Тlr2 наблюдали при сравнительно низкой аффинности, что подтверждает, что вспомогательные белки могут усиливать связывание и/или активацию Тlr2 липополисахаридами (ЛПС) in vivo.
Значение Tlr4 для иммунного ответа на ЛПС (LPS) была продемонстрирована в экспериментах с позиционным клонированием в мутантных по lps линиях мыши. Идентифицировали два мутантных аллеля гена lps мыши: полудоминантный аллель, встречающийся в линии С3Н/HeJ, и рецессивный аллель, присутствующий в линиях C57BL/10SCN и C57BL/10ScCr. Мыши, гомозиготные по мутантным аллелям гена lps, чувствительны к инфицированию грамотрицательными бактериями и устойчивы к индуцированному ЛПС септическому шоку. Локус lps из этих линий был клонирован и оказалось, что в обоих случаях мутации изменили мышиный ген Tlr4 (Portorak et al., Science 282:2085-2088 (1998); Qureshi et al., Exp.Med. 4:615-625 (1999)). Из этих сообщений было сделано заключение, что для ответа на ЛПС требуется Tlr4.
Биологически активная эндотоксическая подструктурная группа липосахарида (ЛПС) представляет собой липид А, фосфорилированный, многократно ацилированный жирными кислотами дисахарид глюкозамин, который служит для заякоривания всей структуры во внешней мембране грамотрицательных бактерий. Авторы настоящего изобретения ранее сообщали, что токсические эффекты липида А можно уменьшить путем селективной химической модификации липида А с получением соединений монофосфорил-липида А (иммуностимулятор MPL®; Corixa Corporation; Seattle, WA). Способы получения и использования иммуностимулятора MPL® и структурно подобных ему соединений в адъювантах для вакцин и для других применений описаны (см., например, патенты США US 4436727, 4877611, 4866034, 4912094 и 4987237; Johnson et al., J. Med. Chem. 42:4640-4649 (1999); Ulrich and Myers, in Vaccine Design: The Subunit and Adjuvant Approach; Powell and Newman, Eds.; Plenum: New York, 495-524, 1995, описания которых включены в настоящую заявку в качестве ссылок во всей их полноте). В частности, в этих и других ссылках показано, что иммуностимулятор MPL® и родственные ему соединения обладают значительной адъювантной активностью при использовании их в составах вакцин с белковыми и углеводными антигенами для усиления гуморального и/или клеточного иммунитета к антигенам.
Кроме того, авторы настоящего изобретения ранее описали класс синтетических моно- и дисахаридных имитаторов монофосфорил-липида А, называемых аминалкилглюкозаминид фосфатами (АГФ), например, в U.S. Ser. №№08/853826, 09/074720, 09/439839 и PCT/US 98/09385, описания которых включены в настоящую заявку в качестве ссылок во всей их полноте. Как и монофосфорил-липид А, эти соединения продемонстрировали сохранение значительных адъювантных свойств при введении их вместе с антигенами в композиции вакцин, и кроме того, они имеют сходные или улучшенные профили токсичности, по сравнению с монофосфорил-липидом А. Значительное преимущество АГФ состоит в том, что их легко получать в коммерческих масштабах с помощью синтетических способов.
Хотя монофосфорил-липид А и соединения АГФ описаны главным образом в связи с их использованием в комбинации с антигенами в составах вакцин, однако об их использовании в качестве монотерапии, в отсутствие антигена, для профилактики и/или лечения болезней и состояний растений и животных, таких как инфекционное заболевание, аутоиммунное заболевание и аллергические состояния, ранее не сообщалось.
В результате возросшего понимания некоторых механизмов активности монофосфорил-липида А и соединений АГФ, настоящее изобретение предлагает описываемые здесь новые терапевтические возможности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
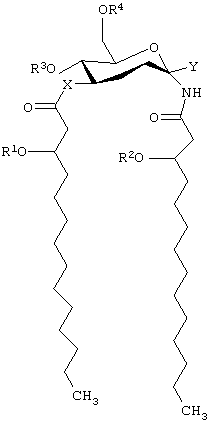
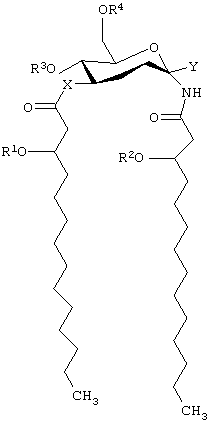
В одном из своих вариантов настоящее изобретение касается способов лечения, облегчения (уменьшения интенсивности симптомов заболевания) или по существу предупреждения болезни или состояния у животного путем введения ему эффективного количества соединения, имеющего формулу:
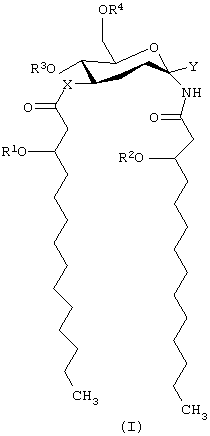
и его фармацевтически приемлемых солей, где Х представляет собой -О- или -NH-; каждый из элементов R1 и R2 независимо представляет собой (C2-C24)ацильную группу, включающую насыщенные, ненасыщенные и разветвленные ацильные группы;
R3 представляет собой -Н или -РО3R11R12, где каждый из элементов R11 и R12 независимо представляет собой -Н или (C1-С4)алкил; R4 представляет собой -Н, -СН3 или -РО3R13R14, причем каждый из элементов R13 и R14 независимо выбирают из -Н и (C1-C4) алкила; и Y представляет собой группу, выбранную из групп формулы:
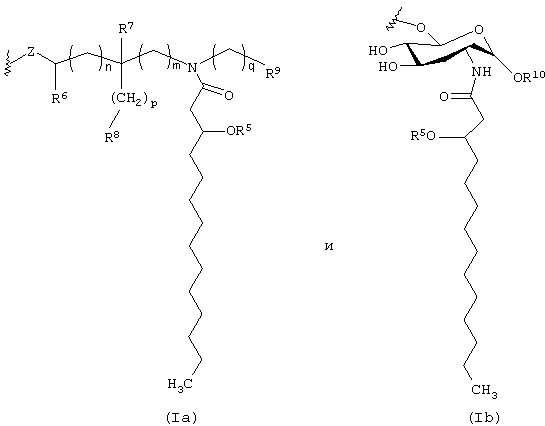
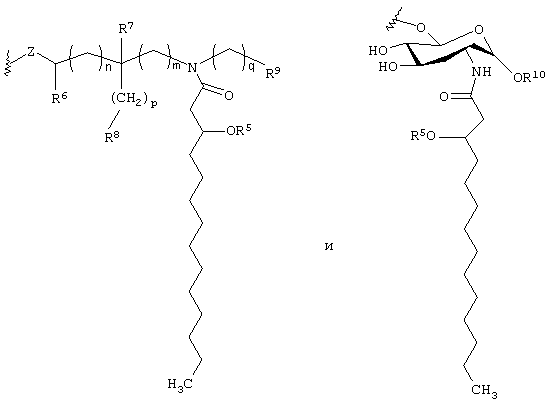
где каждая из букв n, m, p и q независимо означает целое число от 0 до 6; R5 представляет собой (C2-C24) ацильную группу, (включающую, как указано выше, насыщенные, ненасыщенные и разветвленные ацильные группы); R6 и R7 независимо выбирают из Н и СН3; R8 и R9 независимо выбирают из Н, ОН, (C1-C4) алкокси, -РО3Н2, -ОРО3Н2, -SO3Н, -OSO3Н, -NR15R16, SR15, -CN, -NO2, -СНО, -CO2R15, -CONR15R16, -РО3R15R16, -ОРО3R15R16, -SO3R15 и -OSO3R15, где каждый из элементов R15 и R16 независимо выбирают из Н и (C1-C4) алкила;
R10 выбирают из Н, СН3, -РО3Н2, ω-фосфоноокси (С3-С24) алкила и ω-карбокси (С3-С24) алкила; и Z представляет собой -О- или -S-; при условии, что если R3 представляет собой -РО3R11R12, то R4 не является -РО3R13R14.
В некоторых иллюстративных вариантах изобретения вышеупомянутые способы используют для лечения, облегчения или по существу предупреждения инфекционных заболеваний, аутоиммунных заболеваний и аллергических состояний.
В других своих вариантах настоящее изобретение предлагает фармацевтические композиции, включающие одно или более соединений, описанных выше, в подходящем наполнителе, полученных и/или вводимых в отсутствие экзогенного антигена.
ПЕРЕЧЕНЬ ФИГУР
На фигуре 1 приведен график, иллюстрирующий неспецифическую защиту мышей от летального инфицирования гриппом, после или одновременно с введением монофосфорил-липида А (МФЛ).
На фигуре 2 приведена диаграмма, иллюстрирующая клинические симптомы после интраназального введения мышам L-серил-аминоалкилглюкозаминид фосфатов (АГФ).
На фигуре 3 приведена диаграмма, иллюстрирующая клинические симптомы после монотерапии L-серил аминоалкилглюкозаминид фосфатами (АГФ) и заражения гриппом.
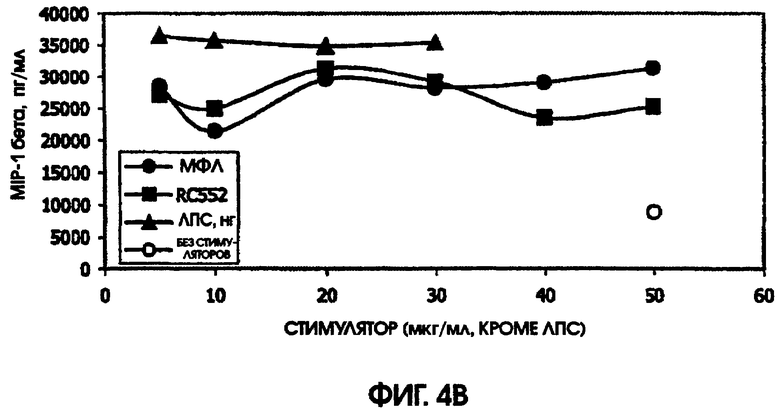
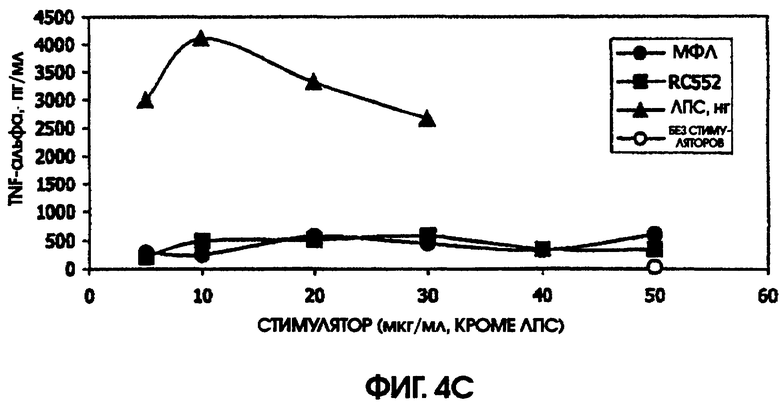
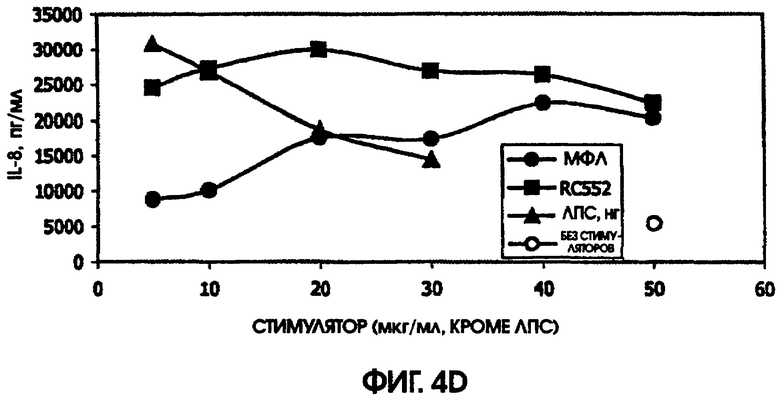
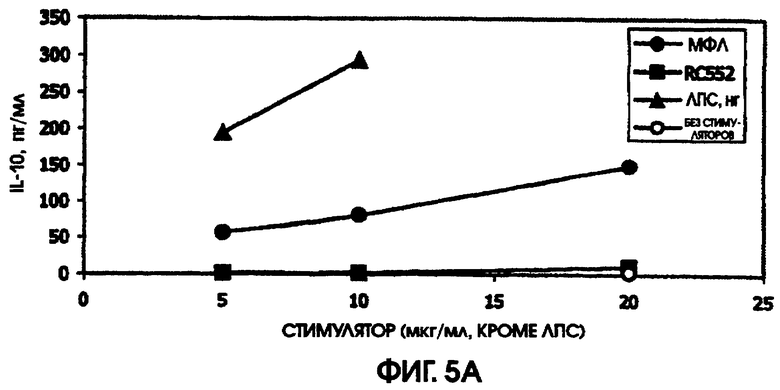
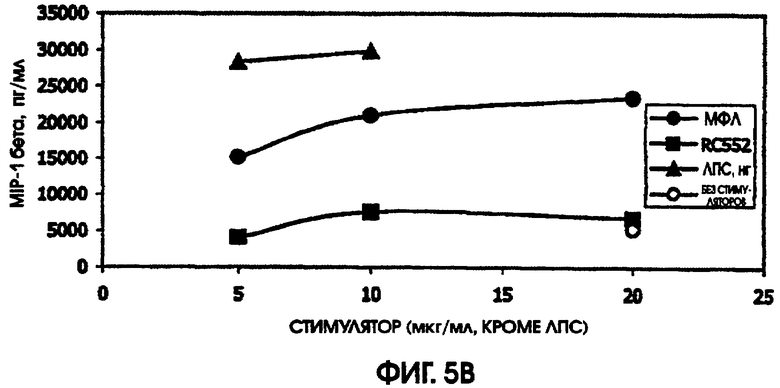
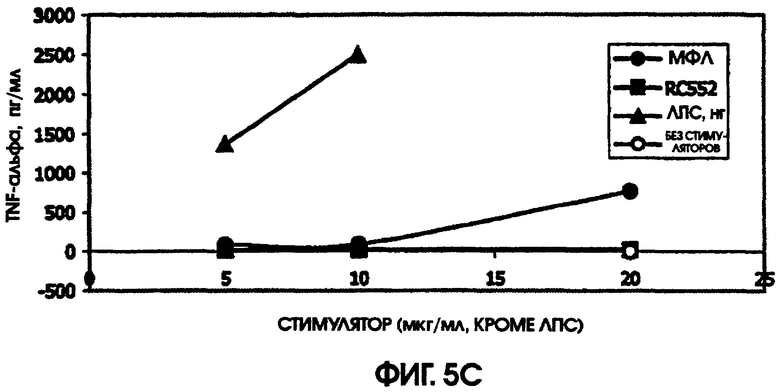
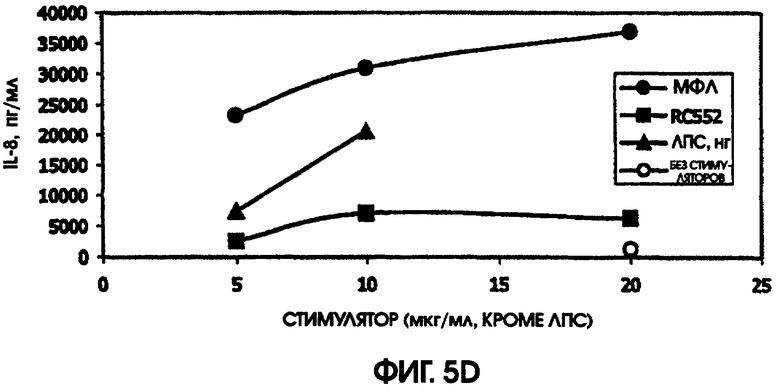
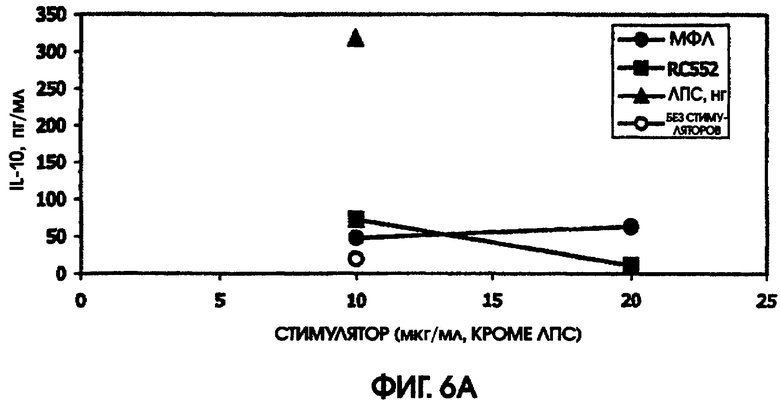
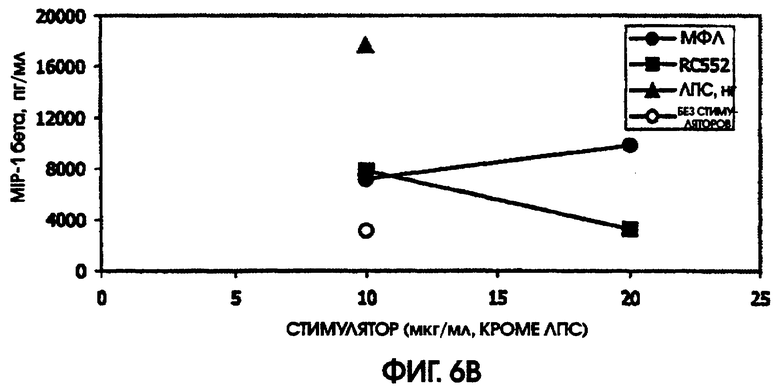
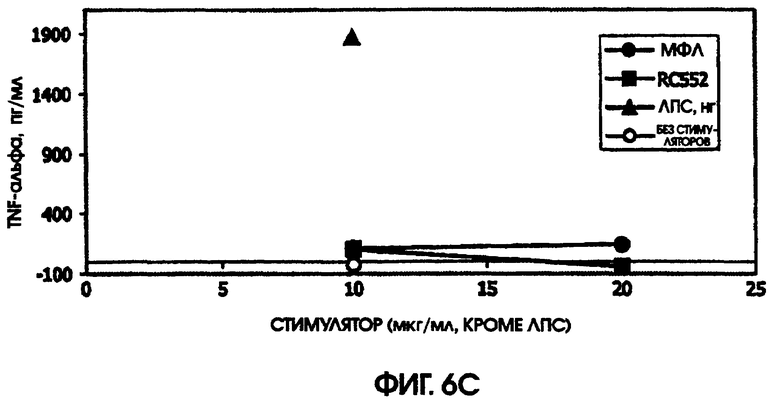
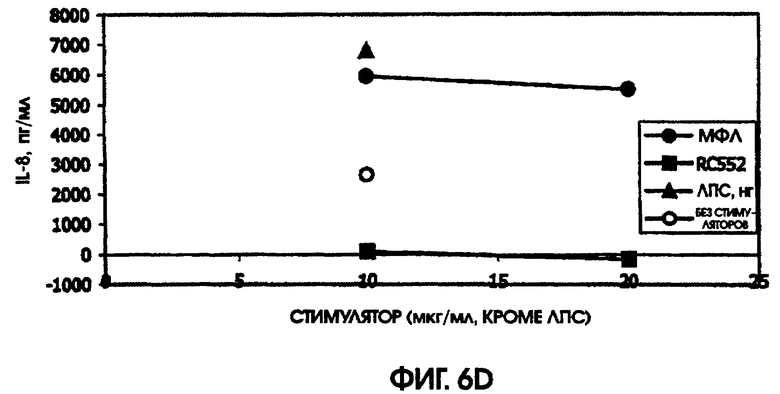
На фигурах 4-6 приведены графики, иллюстрирующие индукцию цитокинов с помощью RC522 по сравнению с МФЛ в суточных культурах цельной крови от трех человеческих доноров (доноры А-С, соответственно).
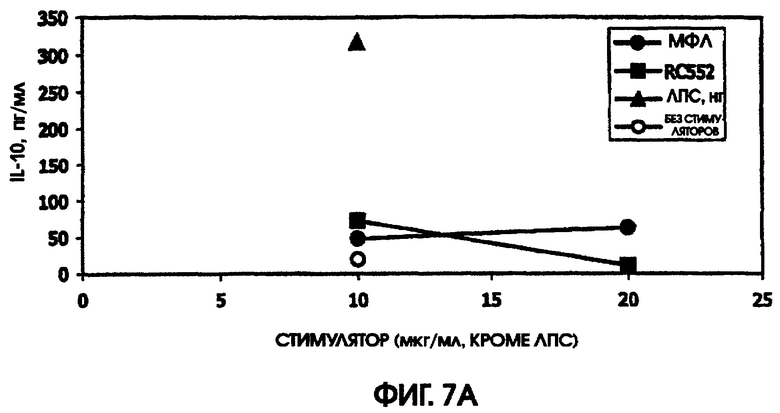
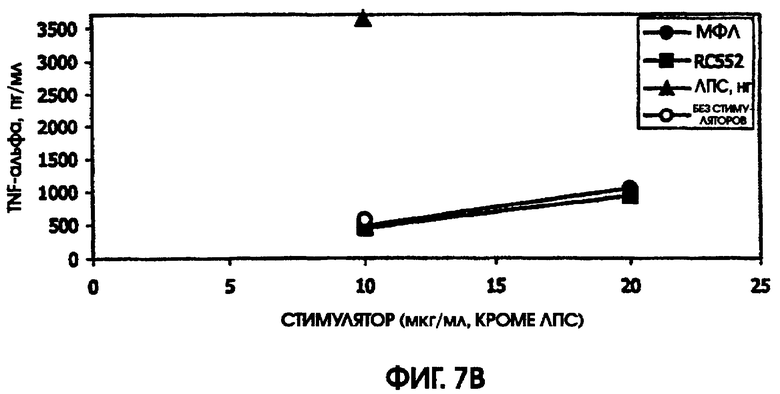
На фигуре 7 приведены графики, иллюстрирующие индукцию цитокинов с помощью RC522 по сравнению с МФЛ в краткосрочных культурах цельной крови от донора А.
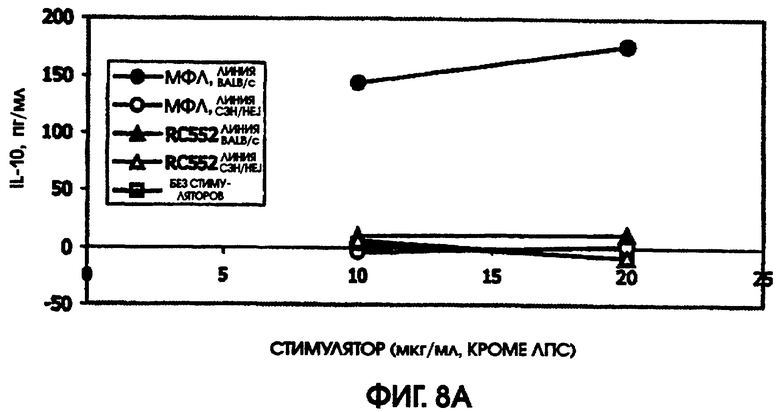
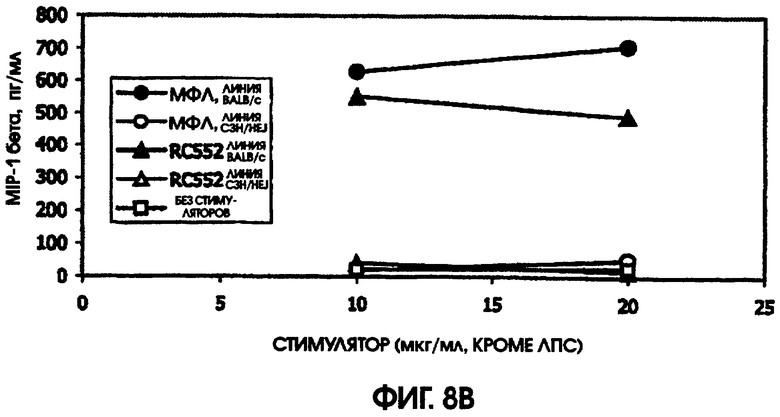
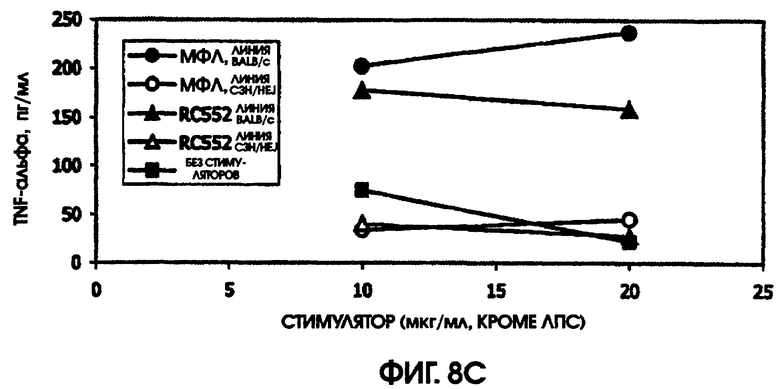
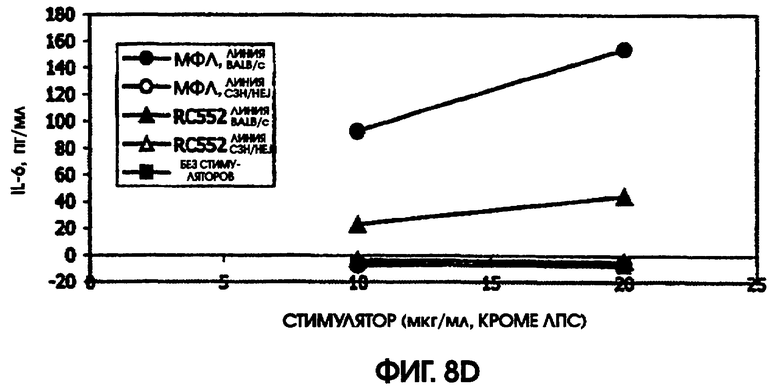
На фигуре 8 приведены графики, иллюстрирующие индукцию цитокинов с помощью RC522 по сравнению с МФЛ в культурах селезенки мышей (линии Ba1b/c и C3H/HEJ).
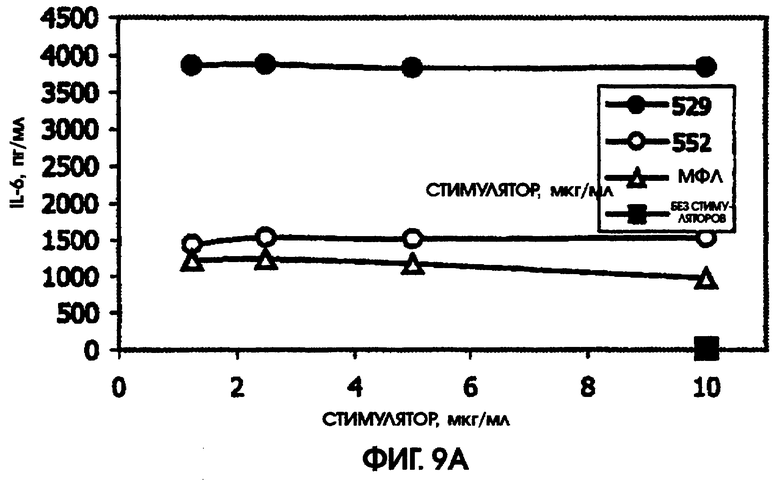
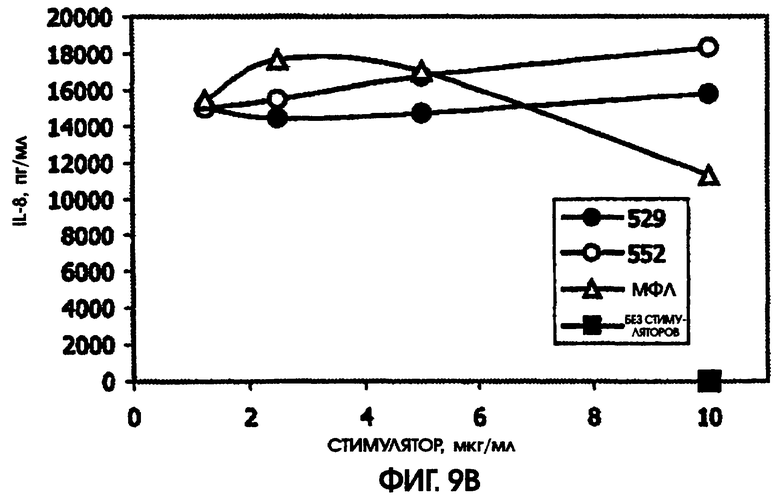
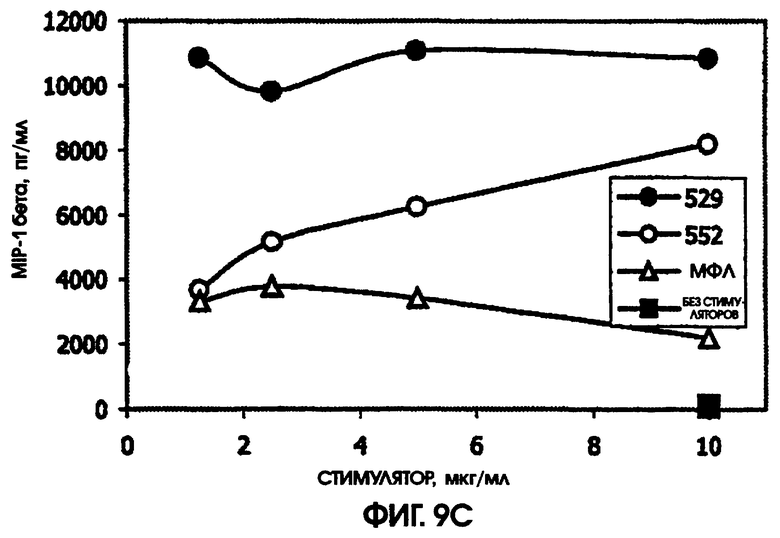
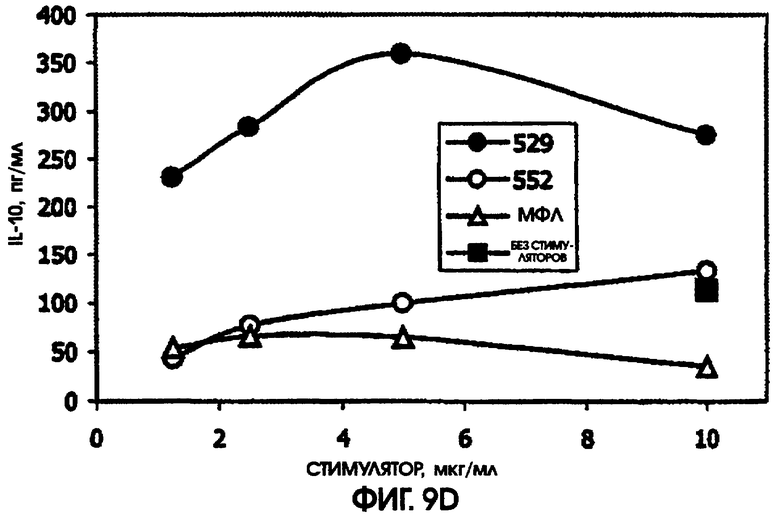
На фигуре 9 приведены графики, иллюстрирующие индукцию цитокинов с помощью RC529 и RC522 по сравнению с МФЛ в периферических одноядерных клетках крови человека (РВМС).
ОПИСАНИЕ ИЛЛЮСТРАТИВНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Иллюстративные варианты профилактики и лечения
Настоящее изобретение в широком смысле касается профилактических и терапевтических способов лечения некоторых заболеваний и других медицинских состояний посредством введения эффективного количества одного или более соединений моно- или дисахаридов или фармацевтической композиции, включающей одно или более таких соединений. В то время как некоторые соединения моно- или дисахаридов ранее описаны для применения в качестве адъювантов в комбинации с экзогенно вводимыми антигенами в составах вакцин, а также для использования в некоторых других применениях, настоящее изобретение предлагает новые терапевтические способы, в которых используются эти соединения, предпочтительно для применения в качестве монотерапии, т.е. в отсутствие экзогенно вводимого антигена.
Таким образом, в одном из своих вариантов настоящее изобретение предлагает способы лечения, уменьшения интенсивности симптомов (облегчения) и/или профилактики инфекционных заболеваний у эукариот, в частности у животных, предпочтительно у человека. Благодаря важности опосредованной TLR передачи сигнала для врожденного иммунного ответа на заражение микробами, возможность стимулирования таких путей селективно и с минимальной токсичностью представляет собой эффективный подход к профилактике и/или лечению, направленным против широкого круга инфицирующих агентов.
Описываемые здесь способы применимы по существу против любого типа инфицирующих агентов, включая бактерии, вирусы, паразиты и грибки. В качестве иллюстрации, изобретение полезно для профилактики и/или лечения бактериальных инфекций, вызываемых видами из Pseudomonas, Escherichia, Klebsiella, Enterobacter, Proteus, Serratia, Candida, Staphylococci, Streptococci, Chlamydia, Mycoplasma и многих других. Примеры вызываемых вирусами состояний, которые можно лечить в соответствии с настоящим изобретением, включают заболевания, вызываемые вирусами гриппа, аденовирусами, вирусами парагриппа, риновирусами, респираторно-синцитиальными вирусами (РСВ), вирусами герпеса, цитомегаловирусами, вирусами гепатита, например вирусами гепатита А и С, и другие. Примеры грибков включают Aspergillis, Candida albicans, Cryptococcus neoformans, Coccidioides immitis и другие.
В одном из примеров своего осуществления настоящее изобретение касается лечения субъектов, в частности субъектов с ослабленным иммунитетом, у которых развились или для которых существует риск развития инфекций, таких как нозокомиальные (внутрибольничные) бактериальные или вирусные инфекции. У около 2 миллионов из 40 миллионов госпитализируемых ежегодно пациентов развиваются нозокомиальные инфекции во время их пребывания в больнице, и у около 1% из них, или у около 400000 пациентов, развивается нозокомиальная пневмония, при этом более 7000 из них умирают. Это делает нозокомиальную пневмонию основной причиной смерти от внутрибольничных инфекций. Таким образом, этот вариант осуществления изобретения отвечает настоятельной потребности в эффективных профилактических подходах к лечению нозокомиальных инфекций.
В связанном с этим варианте своего осуществления настоящее изобретение предлагает способы профилактики для пациентов с ослабленным иммунитетом, таких как ВИЧ-положительные пациенты, у которых развилась или для которых существует риск развития пневмонии, либо в результате воздействия условно-патогенных возбудителей, либо в результате реактивации подавленной или латентной инфекции. В 1992 году сообщалось об около 20000 случаев инфекции, вызванной Pneumocystis carinii, у больных СПИДом пациентов, только в США. Кроме того, у 60-70% от всех больных СПИДом пациентов инфекция, вызванная Р. carinii, развилась в какой-то момент во время их болезни. Таким образом, настоящее изобретение в этом варианте своего осуществления предлагает эффективные способы профилактики для популяции пациентов этой группы риска.
В другом связанном с этим варианте своего осуществления способы по настоящему изобретению используют для лечения другой популяции пациентов, которые могут иметь ослабленный иммунитет и/или для которых существует риск развития инфекционных заболеваний, включая, например, пациентов с муковисцидозом, с хроническим обструктивным легочным заболеванием и других пациентов с ослабленным иммунитетом и/или помещенных в специальные лечебные учреждения пациентов.
В поддержку этих и других вариантов осуществления изобретения авторы продемонстрировали, что введение перед инфицированием приведенного в качестве примера соединения по настоящему изобретению мышам с ослабленным иммунитетам обеспечивает значительную профилактическую защиту против инфекции, вызываемой Pneumocystis carinii (см. пример 1).
В другом варианте данного изобретения описываемые здесь соединения моно- и дисахаридов применяют в способах лечения, облегчения и существенного предупреждения аллергических нарушений и состояний, таких как синусит, хронический риносинусит, астма, атопический дерматит и псориаз. Этот подход основан, по меньшей мере частично, на способности соединений моно- и дисахаридов активировать выработку цитокинов из клеток-мишеней, которые могут конкурировать со стереотипическими, аллергического типа реакциями на цитокины, характеризующимися выработкой IL-4 или гиперчувствительностью к активности IL-4. Введение некоторых описанных здесь соединений моно- и дисахаридов приводит к экспрессии гамма-интерферона и IL-4 осуществляющими процессинг антигена и содержащими антиген клетками, что приводит к регуляции по типу обратной связи цитокинов, связанных с аллергическими реакциями, таких как IL-4, 5, 6, 10 и 13.
Еще в одном варианте данного изобретения описываемые здесь соединения моно- и дисахаридов применяют в способах лечения аутоиммунных болезней и состояний. Соединения моно- и дисахаридов для использования в этом варианте осуществления изобретения, как правило, выбирают из антагонистов, ингибиторов или других негативных модуляторов одного или нескольких Толл-подобных рецепторов, в частности Тlr2 и/или Tlr4, так, что аутоиммунная реакция, связанная с данным состоянием, облегчается или по существу предотвращается. В качестве примеров, способы, предлагаемые этим вариантом осуществления изобретения, можно использовать для лечения таких состояний, как воспалительные заболевания пищеварительного тракта, ревматоидный артрит, хронический артрит, рассеянный склероз и псориаз.
Хотя авторы настоящего изобретения не хотят связывать себя какой-либо теорией, они полагают, что эффективность описанных выше профилактических и терапевтических применений основана, по меньшей мере частично, на том, что соединения моно- и дисахаридов участвуют в модулировании активности Толл-подобных рецепторов. В частности, полагают, что Толл-подобные рецепторы Тlr2, Tlr4 и другие специфически активируются, конкурентно ингибируются или подвергаются иному влиянию нетоксичных производных ЛПС и их имитаторов, описанных здесь. Вследствие этого, способы по настоящему изобретению обеспечивают эффективный и селективный подход к модуляции путей врожденного иммунного ответа у животных, без повышения токсичности, часто связанной с нативными бактериальными компонентами, которые обычно стимулируют эти пути.
ПРИМЕРЫ СОЕДИНЕНИЙ МОНО- И ДИСАХАРИДОВ
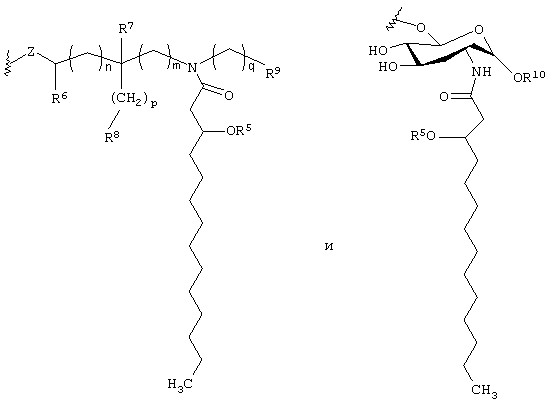
Примеры соединений моно- и дисахаридов, используемых в описанных выше профилактических и терапевтических способах, включают соединения, имеющие формулу:
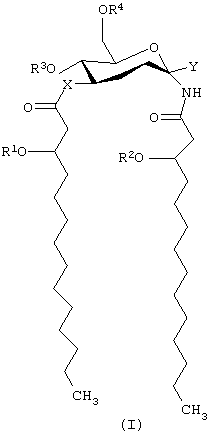
и их фармацевтически приемлемые соли, где Х представляет собой -О- или -NH-; каждый из элементов R1 и R2 независимо представляет собой (С2-С24) ацильную группу, включающую насыщенные, ненасыщенные и разветвленные ацильные группы;
R3 представляет собой -Н или -РО3R11R12, где каждый из элементов R11 и R12 независимо представляет собой -Н или (C1-С4)алкил; R4 представляет собой -Н, -СН3 или -РО3R13R14, причем каждый из элементов R13 и R14 независимо выбирают из -Н и (C1-C4) алкила; и Y представляет собой группу, выбранную из групп формулы:
где каждая из букв n, m, p и q независимо означает целое число от 0 до 6; R5 представляет собой (С2-С24) ацильную группу (включающую, как и выше, насыщенные, ненасыщенные и разветвленные ацильные группы); R6 и R7 независимо выбирают из Н и СН3; R8 и R9 независимо выбирают из Н, ОН, (C1-С4)алкокси, -РО3Н2, -ОРО3Н2, -SO3Н, -OSO3Н, -NR15R16, -SR15, -CN, -NO2, -СНО, -CO2R15, -CONR15R16, -РО3Р15R16, -ОРО3R15R16, -SO3R15 и -OSO3R15, причем каждый из элементов R15 и R16 независимо выбирают из Н и (C1-C4) алкила; R10 выбирают из Н, СН3, -РО3Н2, ω-фосфоноокси (С2-С24) алкила и ω-карбокси (С2-С24) алкила; и Z представляет собой -О- или -S-; при условии, что если R3 представляет собой -РО3R11R12, то R4 не является -РО3R13R14.
Кроме того, если R3 представляет собой -РО3Н2, R4 представляет собой Н, R10 представляет собой Н, R1 представляет собой n-тетрадеканоил, R2 представляет собой n-октадеканоил и R5 представляет собой n-гексадеканоил, то Х не является -О-.
В приведенной выше общей формуле конфигурация 3'-стереогенных центров, к которым присоединены нормальные ацильные остатки жирных кислот, представляет собой R или S, но предпочтительно R. Стереохимия атомов углерода, к которым присоединены R6 и R7, может представлять собой R или S. Все стереоизомеры, энантиомеры, диастереоизомеры и их смеси рассматриваются как входящие в объем настоящего изобретения.
В одной группе предпочтительных вариантов осуществления настоящего изобретения Y имеет формулу:
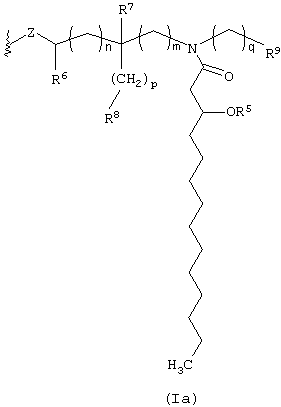
В этой группе вариантов осуществления изобретения ацильные группы R1, R2 и R5 выбирают так, чтобы по меньшей мере две из этих групп представляли собой (С2-С6)ацил. Предпочтительны также такие варианты осуществления изобретения, в которых общее число атомов углерода в R1, R2 и R5 составляет от 6 до 22, более предпочтительно - от 12 до 18. В других предпочтительных вариантах осуществления изобретения Х представляет собой О и Z представляет собой О. Буквы n, m, p и q предпочтительно означают целые числа от 0 до 3, более предпочтительно - от 0 до 2. Из остальных заместителей R6 и R7 предпочтительно представляют собой Н. Настоящее изобретение охватывает также такие варианты его осуществления, в которых предпочтительные заместители объединены в одной молекуле.
В другой группе вариантов осуществления изобретения R1, R2 и R5 выбирают из (C12-C20) ацила, при условии, что общее число атомов углерода в R1, R2 и R5 составляет от 44 до 60. Более предпочтительно, общее число атомов углерода в R1, R2 и R5 составляет от 46 до 52. Еще более предпочтительны те варианты осуществления изобретения, в которых оба элемента Х и Z представляют собой -О-.
В другой группе вариантов осуществления настоящего изобретения Y имеет формулу:
Как и в описанной выше группе вариантов осуществления изобретения, в этой группе ацильные группы R1, R2 и R5 также выбирают так, чтобы по меньшей мере две из этих групп представляли собой (С2-С6)ацил. Предпочтительны также такие варианты осуществления изобретения, в которых общее число атомов углерода в R1, R2 и R5 составляет от 6 до 22, более предпочтительно - от 12 до 18. В других предпочтительных вариантах осуществления изобретения Х представляет собой О. Из остальных заместителей R3 предпочтительно представляет собой фосфоно (-РО3Н2), а R4 предпочтительно представляют собой Н. Настоящее изобретение охватывает также те варианты осуществления, в которых различные комбинации предпочтительных заместителей объединены в одной молекуле.
В другой группе вариантов осуществления изобретения R1, R2 и R5 выбирают из (C12-C24) ацила, при условии, что общее число атомов углерода в R1, R2 и R5 составляет от 44 до 60. Более предпочтительно, общее число атомов углерода в R1, R2 и R5 составляет от 46 до 52. Особо предпочтительно, чтобы жирнокислотные группы для R1, R2 и R5 представляли собой нормальные жирнокислотные группы С14, С16 и С18. Еще более предпочтительны те варианты осуществления изобретения, в которых Х представляет собой -О-. Подобно представленным выше вариантам осуществления изобретения с более короткой ацильной цепью, R3 предпочтительно представляет собой фосфоно (-РО3Н2), а R4 предпочтительно представляет собой Н.
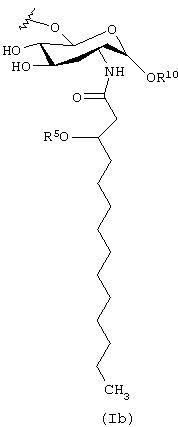
В других предпочтительных вариантах осуществления настоящего изобретения Y представляет собой радикал формулы (Ib), Х представляет собой О, R3 представляет собой фосфоно, R4 представляет собой Н, а R1, R2 и R5 выбирают из (C12-C24) ацила, при условии, что общее число атомов углерода в R1, R2 и R5 составляет от 46 до 52. Еще более предпочтительны такие соединения, в которых R2 представляет собой (C16-C18) ацил.
Термин «алкил» сам по себе или как часть другого заместителя обозначает, кроме особо оговоренных случаев, прямую или разветвленную цепь, или радикал циклического углеводорода, или их комбинацию, которые могут быть полностью насыщенными, моно- или полиненасыщенными и могут включать ди- и мультивалентные радикалы с указанным числом атомов углерода (например, C1-С10 обозначает от одного до десяти атомов углерода). Примеры насыщенных углеводородных радикалов включают такие группы, как метил, этил, n-пропил, изопропил, n-бутил, t-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил, гомологи и изомеры, например, n-пентила, n-гексила, n-гептила, n-октила и т.п. Ненасыщенная алкильная группа представляет собой группу, имеющую одну или более двойных связей или тройных связей. Примеры ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Как правило, алкильная группа имеет от 1 до 24 атомов углерода. «Низший алкил» представляет собой алкильную группу с более короткой цепью, обычно имеющую восемь или менее атомов углерода.
Термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкокси) используются в их обычном смысле, и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно.
Термин «ацил» относится к группе, происходящей из органической кислоты, из которой удалена гидроксильная группа. Примеры ацильных групп включают ацетил, пропионил, додеканоил, тетрадеканоил, изобутирил и т.п. В соответствии с этим подразумевается, что термин «ацил» включает группу, иначе определяемую как -С(O)-алкил.
Подразумевается, что каждый из вышеупомянутых терминов (например, «алкил», «ацил») включает как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикалов приводятся ниже.
Заместителями для алкильных и ацильных радикалов могут быть различные группы, выбранные из OR', =O, =NR', =N-OR', -NR'R'', -SR', - галогена, -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'-C(O)NR''R''', -NR''C(O2)R', -NH-C (NH2)=NH, -NR'C(NH2)=NH, -NHC(NH2)=NR', -S(O)R', -S(O2)R', -S(O)2NR'RR'', -CN и -NO2, в количестве от нуля до (2m'+1), где m' означает общее число атомов углерода в таком радикале. Каждый из элементов R', R'' и R''' независимо относится к водороду и незамещенному (C1-C8)алкилу. Если R' и R'' присоединены к одному и тому же атому азота, они могут быть объединены с атомом азота, образуя 5-, 6- или 7-членное кольцо. Например, подразумевается, что -NR'R'' включает 1-пирролидинил и 4-морфолинил. Из вышеприведенного описания заместителей специалистам в данной области техники понятно, что термин «алкил» включает такие группы, как галоалкил (например, -CF3 и -СН2CF3) и подобные им.
Термин «фармацевтически приемлемые соли» включает соли активных соединений, полученные с относительно нетоксичными кислотами и основаниями, в зависимости от конкретных заместителей, находящихся в описанных здесь соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, то основно-аддитивные соли можно получить путем осуществления контакта нейтральной формы таких соединений с достаточным количеством желаемого основания, либо в чистом виде, либо в подходящем инертном растворителе. Примеры фармацевтически приемлемых основно-аддитивных солей включают соли натрия, калия, кальция, аммония, органические аминосоли, соли магния и подобные им соли. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, то кислотно-аддитивные соли можно получить путем осуществления контакта нейтральной формы таких соединений с достаточным количеством желаемой кислоты, либо в чистом виде, либо в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, азотная, угольная, моногидрокарбоновая, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, гидройодноватая кислота или фосфористые кислоты и тому подобные кислоты, а также соли, полученные из относительно нетоксичных органических кислот, таких как уксусная, пропионовая, изомасляная, малеиновая, малоновая, бензойная, янтарная, субериновая, фумаровая, молочная, миндальная, фталовая, бензолсульфоновая, р-толилсульфоновая, лимонная, винная, метансульфоновая и тому подобные. Примеры включают также соли аминокислот, такие как аргинат и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и тому подобные (см., например, Вегде S.M., et al., "Pharmaceutical salts", Journal of Pharmaceutical Science, 66, 1-19, 1977). Некоторые конкретные соединения по настоящему изобретению содержат как основные, так и кислотные функциональные группы, что позволяет преобразовать эти соединения либо в основно-аддитивные, либо в кислотно-аддитивные соли.
Нейтральные формы этих соединений можно восстановить путем осуществления контакта соли с основанием или кислотой и выделения исходного соединения обычным способом. Исходная форма соединения отличается от различных его солей некоторыми физическими свойствами, такими как растворимость в полярных растворителях, но в остальном соли эквивалентны исходной форме соединения с точки зрения целей настоящего изобретения.
Кроме соединений в форме солей, настоящее изобретение касается также соединений в форме пролекарств. Пролекарства описываемых здесь соединений - это такие соединения, которые легко подвергаются химическим изменениям в физиологических условиях, в результате чего образуются соединения по настоящему изобретению. Кроме того, пролекарства можно преобразовать в соединения по настоящему изобретению с помощью химических или биохимических способов в условиях ех vivo. Например, пролекарства могут медленно превращаться в соединения по настоящему изобретению, будучи помещенными в резервуар трансдермального пластыря вместе с подходящим ферментом или химическим реагентом.
Некоторые соединения по настоящему изобретению могут существовать как в сольватированных, так и в несольватированных формах, включая гидратированные формы. Как правило, сольватированные формы эквивалентны несольватированным формам и охватываются объемом настоящего изобретения. Некоторые соединения по настоящему изобретению могут существовать как в множественно-кристаллической, так и в аморфной форме. Как правило, все физические формы эквивалентны для применений, рассматриваемых в настоящем изобретении, и находятся в пределах объема настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметричные атомы углерода (оптические центры) или двойные связи; рацематы, диастереоизомеры, геометрические изомеры и индивидуальные изомеры все эти соединения находятся в пределах объема настоящего изобретения.
Соединения по настоящему изобретению могут также содержать не являющиеся естественными пропорции атомных изотопов для одного или более атомов, составляющих такие соединения. Например, соединения могут быть мечеными радиоактивными изотопами, например, такими как тритий (3Н), йод-125 (125I) или углерод-14 (14С). Все изотопные вариации соединений по настоящему изобретению, радиоактивные или нерадиоактивные, охватываются объемом настоящего изобретения.
Соединения моно- и дисахаридов можно получить с помощью любых подходящих способов, многие из которых описаны. Например, некоторые соединения, полезные для настоящего изобретения, описаны в одновременно рассматриваемых заявках на изобретение с серийными №№08/853826, 09/439839 (поданных 12 ноября 1999) и PCT/US98/09385, описания которых включены в настоящую заявку в качестве ссылок во всей их полноте. Другие соединения можно получить способом, подобным описанному для RC-552 (L34) в патенте США US 6013640. Другие соединения можно получить с помощью способов, описанных в публикациях Johnson, et al., J. Med. Chem. 42:4640-4649 (1999); Johnson, et al., Biorg. Med. Chem. Lett. 9:2273-2278 (1999) и PCT/US98/50399. Другие соединения также можно получить в соответствии, например, с патентами США US 4436727, US 4877611, US 4866034, US 4912094 и US 4987237. В общем, способы синтеза, описанные в вышеупомянутых ссылках, и другие близкие к ним способы, существующие в данной области техники, широко применимы для получения этих соединений. Например, при получении соединений, имеющих отличающиеся ацильные группы и заместители, специалисту в данной области техники понятно, что описанные здесь сходные способы можно модифицировать с целью использования альтернативных ацилирующих агентов, либо в качестве исходного материала можно использовать коммерчески доступные материалы, имеющие подходящие ацильные группы.
Примеры фармацевтических композиций и их введение
В другом варианте своего осуществления настоящее изобретение касается фармацевтических композиций, включающих одно или более описанных здесь соединений моно- и дисахаридов в фармацевтически приемлемых носителях/наполнителях для введения в клетку, ткань, животное или растение, либо в отдельности, либо в комбинации с одним или более других способов терапии. В предпочтительном варианте осуществления изобретения фармацевтические композиции не включают экзогенного антигена, т.е. используются для монотерапевтических применений. Для многих таких вариантов осуществления изобретения фармацевтические композиции включают одно или более описанных здесь соединений моносахаридов.
Примеры носителей для использования в фармацевтических композициях включают эмульсии масла в воде или воды в масле, водные композиции с включением или без включения органических сорастворителей, пригодных для внутривенного использования, липосомы или содержащие поверхностно-активные вещества носители, микросферы, микрогранулы, микросомы, порошки, таблетки, капсулы, суппозитории, водные суспензии, аэрозоли и другие носители, известные специалистам обычной квалификации в данной области техники.
В некоторых вариантах осуществления изобретения фармацевтические композиции включают один или более буферов (например, нейтральный забуференный физиологический раствор или забуференный фосфатом физиологический раствор); углеводы (например, глюкозу, маннозу, сахарозу или декстраны); маннитол; белки, полипептиды или аминокислоты, такие как глицин; антиоксиданты; бактериостаты; хелатные агенты, такие как ЭДТА или глютатион; адъюванты (например, гидроксид алюминия); растворенные вещества, придающие композициям изотонические, гипотонические или слабо гипертонические свойства в крови реципиента; суспендирующие агенты, загустители и/или консерванты.
Для некоторых применений предпочтительны водные составы, в частности, включающие эффективное количество одного или более поверхностно-активных веществ. Например, композиция может быть в форме мицеллярной дисперсии, включающей по меньшей мере одно подходящее поверхностно-активное вещество, например, фосфолипидное поверхностно-активное вещество. Примеры фосфолипидов включают диацил-фосфатидилглицеролы, такие как димиристоил-фосфатидилглицерол (ДМФГ), дипальмитоил-фосфатидилглицерол (ДПФГ) и дистеароил-фосфатидилглицерол (ДСФГ); диацил-фосфатидилхолины, такие как димиристоил-фосфатидилхолин (ДМФХ), дипальмитоил-фосфатидилхолин (ДПФХ) и дистеароил-фосфатидилхолин (ДСФХ); диацилфосфатидные кислоты, такие как димиристоил-фосфатидная кислота (ДМФК), дипальмитоил-фосфатидная кислота (ДПФК) и дистеароил-фосфатидная кислота (ДСФК); и диацил-фосфатидилэтаноламины, такие как димиристоил-фосфатидилэтаноламин (ДМФЭ), дипальмитоил-фосфатидилэтаноламин (ДПФЭ) и дистеароил-фосфатидилэтаноламин (ДСФЭ). Как правило, молярное соотношение поверхностно-активного вещества и моно/дисахарида в водной композиции составляет примерно от 10:1 до 1:10, более типично примерно от 5:1 до 1:5, однако в водной композиции можно использовать любое эффективное количество поверхностно-активного вещества, наиболее подходящее для конкретной цели применения.
Соединения или фармацевтические композиции по настоящему изобретению можно изготовить таким образом, чтобы они были пригодны по существу для любого пути введения, например, для введения путем инъекции, пероральной или интраназальной ингаляции, для ректальной, вагинальной или внутритрахеальной инстилляции, для приема внутрь, для трансдермального введения или для трансмукозального введения (введения через слизистые оболочки) и т.п. Таким образом, терапевтические воздействия, оказываемые способами и композициями по настоящему изобретению, могут быть, например, системными, местными, ткань-специфичными и др., в зависимости от конкретных потребностей данного применения изобретения.
Приводимые в качестве примеров композиции можно приготовить для парентерального введения, т.е. для внутрибрюшинного, подкожного, внутримышечного или внутривенного введения. Один из примеров носителя для внутривенного использования включает смесь 10% этанола (согласно Фармакопеи США USP), 40% пропиленгликоля или полиэтиленгликоля 600 и воду для инъекций (USP) в количестве, необходимом для доведения до 100%. Другие примеры носителей включают смесь 10% этанола (USP) и воду для инъекций (USP); 0,01-0,1% триэтаноламина в воде для инъекций (USP); или 0,01-0,2% дипальмитоил-дифосфатидилхолина в воде для инъекций, (USP); и 1-10% сквалена или эмульсию растительного масла в воде для парентерального введения. Фармацевтически приемлемые растворители для парентерального введения, как правило, выбирают так, чтобы они обеспечивали получение раствора или дисперсии, которые можно профильтровать через фильтр с размером отверстий в 0,22 микрон, не удалив при этом активный компонент.
Примеры носителей для подкожного или внутримышечного введения включают забуференный фосфатом физиологический раствор (PBS), 5% декстрозу в воде для инъекций и 0,01-0,1% триэтаноламин в 5% декстрозе или 0,9% хлорид натрия в воде для инъекций (USP), или от 1 до 2 либо от 1 до 4% смеси 10% этанола (USP), 40% пропиленгликоля, и в количестве, необходимом для доведения до 100%, приемлемый изотонический раствор, такой как 5% декстроза или 0,9% хлорид натрия; или 0,01-0,2% дипальмитоил-дифосфатидилхолина в воде для инъекций USP и 1-10% сквалена или эмульсии растительного масла в воде для парентерального введения.
Примеры носителей, используемых для введения через слизистые оболочки, зависят от конкретного пути введения, например, парентеральное, сублингвальное, интраназальное введение и др. При пероральном введении примеры носителей включают фармацевтические марки маннитола, крахмала, лактозы, стеарата магния, сахарида натрия, целлюлозы, карбоната магния и т.п., при этом предпочтительным является маннитол. При интраназальном введении примеры носителей включают полиэтиленгликоль, фосфолипиды, гликоли и гликолипиды, сахарозу и/или метилцеллюлозу, суспензии порошков без наполнителей или с наполнителями, такими как лактоза, и консерванты, такие как бензалконийхлорид, ЭДТА. В конкретном примере осуществления изобретения используют фосфолипид 1,2-дипальмитоил-зп-глицеро-3-фосфохолин (ДППХ) в качестве изотонического водного носителя в количестве около 0,01-0,2%, для интраназального введения соединения, являющего предметом настоящего изобретения, в концентрации, составляющей около 0,1-3,0 мг/мл.
Примеры носителей для введения с помощью ингаляции включают полиэтиленгликоль или гликоли, ДПФХ, метилцеллюлозу, порошкообразные диспергирующие агенты и консерванты, при этом предпочтительными являются полиэтиленгликоли и ДПФХ. Во многих случаях предпочтительно, чтобы предназначенные для введения с помощью ингаляции соединения моно- и дисахаридов находились в распыленной форме. В качестве примеров, введение можно осуществлять с помощью одноразового устройства для введения, с помощью аэрозольного распылителя, активируемого дыханием порошкового ингалятора, аэрозольного ингалятора с отмеряемой дозой (MDI), а также с помощью любых других многочисленных аэрозольных ингаляторов, известных в данной области техники. Кроме того, можно также использовать аэрозольные палатки или прямое введение через эндотрахеальные трубки. Внутритрахеальный или назофарингальный пути введения могут быть эффективными для некоторых случаев.
Специалистам в данной области техники понятно, что вышеприведенное описание является иллюстративным, а не исчерпывающим. Действительно, специалистам в данной области техники хорошо известны многие дополнительные способы изготовления композиций, а также другие фармацевтически приемлемые наполнители и растворы носителей, так же, как и разработка подходящих дозировок и режимов лечения для использования описанных здесь конкретных композиций в ряде схем терапии.
Соединения можно оценить с помощью различных методов анализа, чтобы идентифицировать и отобрать те из них, которые обладают наилучшими свойствами, подходящими для использования согласно изобретению. Например, для идентификации и оценки профилей выделения цитокинов в большой круг кровообращения после введения соединений моно- и дисахаридов можно использовать модели на животных. Кроме того, существуют различные in vitro и in vivo модели для изучения изменений в иммунном ответе на различные антигенные компоненты, позволяющие идентифицировать соединения, наиболее подходящие для проявления конкретного представляющего интерес иммунного ответа. Например, соединение можно ввести в контакт с клетками-мишенями, такими как макрофаги, древовидные клетки или клетки Лангерганса in vitro, и измерить вырабатываемые цитокины. Кроме того, для изучения конкретных путей, активируемых или ингибируемых конкретным представляющим интерес моно- или дисахаридом, можно использовать анализ экспрессии генов.
Понятно, что описанные здесь соединения можно по желанию вводить в сочетании с другими лечебными воздействиями, такими как антимикробные, антивирусные и противогрибковые соединения и способы терапии, различные лечебные средства на основе ДНК, лечебные средства на основе РНК, лечебные средства на основе полипептидов и/или в сочетании с другими иммуноэффекторами. Фактически можно также включить по существу любой другой компонент, при условии, что этот дополнительный компонент(ы) не оказывает значительного отрицательного влияния на контакт с клетками-мишенями или тканями хозяина. Таким образом, композиции можно вводить наряду с различными другими агентами, которые требуются или желательны для конкретных вариантов осуществления изобретения.
В качестве примеров, фармацевтические композиции по настоящему изобретению могут включать или использоваться в сочетании с ДНК, кодирующей один или более терапевтических белков, антисмысловыми РНК, рибозимами и тому подобным. ДНК может присутствовать в составе ряда различных систем переноса, известных специалистам обычной квалификации в данной области техники, включая системы экспрессии нуклеиновых кислот, бактериальные и вирусные системы экспрессии. Многочисленные способы переноса генов хорошо известны в данной области техники, такие как описанные в публикации Rolland, Crit. Rev. Therap. Drug Carrier Systems 15:143-198, 1998, и в цитированных в этой публикации ссылках. Подходящие системы экспрессии нуклеиновых кислот содержат последовательности ДНК, необходимые для экспрессии в пациенте (такие как подходящий промотор и сигнал терминации). В предпочтительном варианте осуществления изобретения ДНК можно вводить с помощью вирусной системы экспрессии (например, с помощью вируса коровьей оспы или другого вируса оспы, ретровируса или аденовируса), которая может включать использование непатогенного (дефектного) способного к репликации вируса. Подходящие системы описаны, например, в публикациях Fisher-Hoch et al., Proc. Natl. Acad. Sci. USA 86:317-321, 1989; Flexner et al., Ann. N.Y. Acad. Sci. 569:86-103, 1989; Flexner et al., Vaccine 8:17-21, 1990; патенты США US 4603112, 4769330 и 5017487; WO 89/01973; патент США US 4777127; GB 2200651; ЕР 0345242; WO 91/02805; Berkner, Biotechniques 6:616-627, 1988; Rosenfeld et al., Science 252:431-434, 1991; Kolls et al., Proc. Natl. Acad. Sci. USA 91:215-219, 1994; Kass-Eisler et al., Proc. Natl. Acad. Sci. USA 90:11498-11502, 1993; Guzman etc al., Circulation 88:2838-2848, 1993; и Guzman et al., Cir. Res. 73:1202-1207, 1993. Способы введения ДНК в такие системы экспрессии хорошо известны специалистам обычной квалификации в данной области техники.
ДНК также может быть в чистом виде («naked») (депротеинизированной), как описано, например, в публикации Ulmer et al., Science 259:1745-1749, 1993, и в обзоре Cohen, Science 259:1691-1692, 1993. Введение такой ДНК в клетки можно повысить путем включения ДНК на поверхность биологически расщепляемых гранул, которые эффективно транспортируются в клетки. Ясно, что фармацевтическая композиция по данному изобретению может включать как полинуклеотидный, так и белковый компонент.
В композицию по настоящему изобретению могут быть дополнительно включены любые из существующего разнообразия иммуностимуляторов. Например, цитокины, такие как GM-CSF, интерфероны или интерлейкины могут модулировать желаемый иммунный ответ. Например, в некоторых вариантах осуществления изобретения в композиции могут быть включены дополнительные компоненты, еще более повышающие индукцию высоких уровней цитокинов типа Th1 (например, γ-интерферон, TNF-α, IL-2 и IL-12). В качестве альтернативы, или в дополнение, для некоторых терапевтических применений могут быть желательными высокие уровни цитокинов типа Th2 (например, IL-4, IL-5, IL-6 и IL-10). Уровни содержания этих цитокинов можно легко определить с помощью стандартных анализов. Обзор по семейству цитокинов см. в публикации Mossman and Coffman, Ann. Rev. Immunol. 7:145-173, 1989.
Примеры композиций, предназначенных для использования с целью индукции цитокинов типа Тh1, включают комбинацию CpG-содержащих олигонуклеотидов (в которых динуклеотид CpG является неметилированным), как описано, например, в WO 96/02555, WO 99/33488 и в патентах США US 6008200 и US 5856462. Описаны также иммуностимулирующие последовательности ДНК, например, в публикации Sato et al., Science 273:352, 1996. Другие подходящие иммуностимуляторы включают сапонины, такие как QS21 (Aquila Biopharmaceutical Inc., MA), а также производные сапонина и имитаторы сапонина.
Примеры других иммуностимуляторов, которые можно использовать в сочетании с соединениями по настоящему изобретению, включают Montanide ISA 720 (Seppic, Франция), SAF (Chiron, Калифорния, США), ISCOMS (CSL), MF-59 (Chiron), серию адъювантов SBAS (например, SBAS-2 или SBAS-4, которые можно приобрести в фирме SmithKline Beecham, Rixensart, Бельгия) и иммуностимулятор Enhanzyn™ (Corixa, Hamilton, МТ). Полиоксиэтиленэфирные иммуностимуляторы описаны в WO 99/52549A1.
Основные термины:
Как он используется в настоящем изобретении, термин «эффективное количество» означает количество, которое вызывает реакцию, превышающую реакции, вызываемые носителем или отрицательными контролями. Как описано выше, точная дозировка соединения по настоящему изобретению, которую следует вводить пациенту, зависит от пути его введения, фармацевтической композиции и от состояния пациента.
Фраза «фармацевтически приемлемые» относится к молекулярным частицам и композициям, которые не вызывают аллергической или тому подобной нежелательной реакции при введении их человеку.
Как они используются в настоящем описании, термины «носитель» и «наполнитель» включают любые и все растворители, дисперсионые среды, покрытия, разбавители, антибактериальные и противогрибковые агенты, изотонические и замедляющие поглощение агенты, буферы, растворы носителей, суспензии, коллоиды и тому подобное. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. Изобретение охватывает использование в терапевтических композициях любых обычных сред и агентов, за исключением тех, которые несовместимы с активным компонентом.
Сведения, подтверждающие возможность осуществления изобретения
ПРИМЕР 1
Защита против инфекции, вызываемой Р. carinii, с помощью профилактического введения монофосфорил-липида А
Мышей подвергали предобработке антителом L3T4 против CD4 в течение не менее 2 недель (2 инъекции в неделю, 0,2 мг на инъекцию), или до тех пор, пока количество периферических CD-4 не уменьшилось по меньшей мере примерно на 50%.
Готовили водный состав, содержащий 1 мг/мл 3-0-деацилированного монофосфорил-липида А и 108 мкг/мл поверхностно-активного вещества ДПФХ в воде. Этот состав вводили внутритрахеально через маленькую канюлю через каждые 24 часа, а затем дважды в неделю, в течение остального времени эксперимента. Вводимые концентрации показаны ниже, в таблице 1. В день 0 транстрахеально инокулировали 1 миллион Р. carinii. Введение с периодичностью 2 раза в неделю продолжали в течение 7 недель, затем удаляли легкие и готовили мазки-отпечатки для микроскопического исследования. Препараты окрашивали по Гимзе и серебром и подсчитывали количество Р. carinii следующим образом:
Балл 5 >100/поле зрения микроскопа при увеличении 1000×
4 - 10-100/поле зрения микроскопа
3 - 1-10/поле зрения микроскопа
2 - 1-10/10 полей зрения микроскопа
1 - 1-10/50 полей зрения микроскопа
0 - 0/50 полей зрения микроскопа
Результаты этих экспериментов суммированы в таблице 1:
Исследование повторили и получили следующие результаты (таблица 2):
Эти результаты показывают, что доставка к легким монофосфорил-липида А повышает неспецифическую устойчивость к инфицированию Pneumocystis carinii у мышей с ослабленным иммунитетом. Вдыхание монофосфорил-липида А приводило к активации местных (и периферических) врожденных иммунных реакций, что в свою очередь приводило к повышению неспецифической защиты. Монофосфорил-липид А опосредует эту защиту главным образом через активацию антиген-представляющих клеток, что приводит к повышению активности фагоцитов и к выделению иммуностимулирующих цитокинов. FACS-анализ клеток, смытых из легких, выявил маркеры активированных нейрофилов, но не дал заметных результатов в отношении притока лейкоцитов, характерных для массированной воспалительной реакции (ARDS). Анализ клеток селезенки показал негативную экспрессию CD11b или CD69, что подтверждает, что действие составов с монофосфорил-липидом А и эффекты этого применения не носили системный характер, а были ограничены легкими.
ПРИМЕР 2
Защита против летального заражения гриппом с помощью профилактического введения монофосфорил-липида А
Дозу в 20 мкг монофосфорил-липида А (МФЛ) вводили интраназально (i.n.) группам самок мышей линии BALB, либо за 2 дня до летального инфицирования гриппом, либо в день этого инфицирования. Всех мышей заражали дозой примерно LD50 инфекции гриппа А/НК/68, также вводимой интраназально. Смертность отслеживали в течение 21 суток после заражения гриппом. Результаты этих экспериментов приведены на фигуре 1. Эти данные демонстрируют, что интраназальное введение монофосфорил-липида А усиливает неспецифическую устойчивость к инфицированию летальной дозой гриппом у мышей.
ПРИМЕР 3
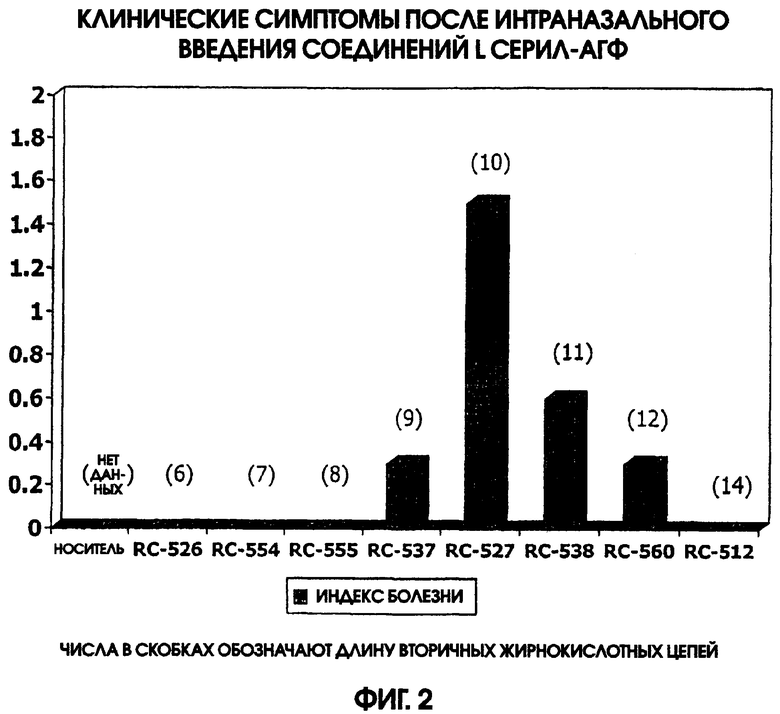
Клинические симптомы после интраназального введения L-серил-аминоалкилглюкозаминид фосфатов (АГФ)
Серию соединений, представляющих собой L-серил-аминоалкил-глюкозаминид фосфаты (АГФ), получили так, как описано в патенте США US 6113918, выданном 5 сентября 2000, и в заявке на патент США US 09/439839, зарегистрированной 12 ноября 1999, каждый из которых включен в настоящее описание в качестве ссылок во всей их полноте.
Дозу в 20 мкг различных соединений L-серил-АГФ (RC-526, RC-554, RC-555, RC-537, RC-527, RC-538, RC-560, RC-512, а также только носитель) вводили интраназально группам самок мышей линии BALB. Во время первых 4 суток после введения АГФ проводили мониторинг мышей, отслеживая три субъективных индикатора болезни (т.е. индекс болезни): взъерошенная шерсть, сгорбленная поза и затрудненное дыхание. Результаты этих экспериментов приведены на фигуре 2. Эти данные показали, что интраназальное введение RC-537, RC-527, RC-538 и RC-560 индуцировало некоторую степень токсичности у мышей в данной дозе 20 мкг.
ПРИМЕР 4
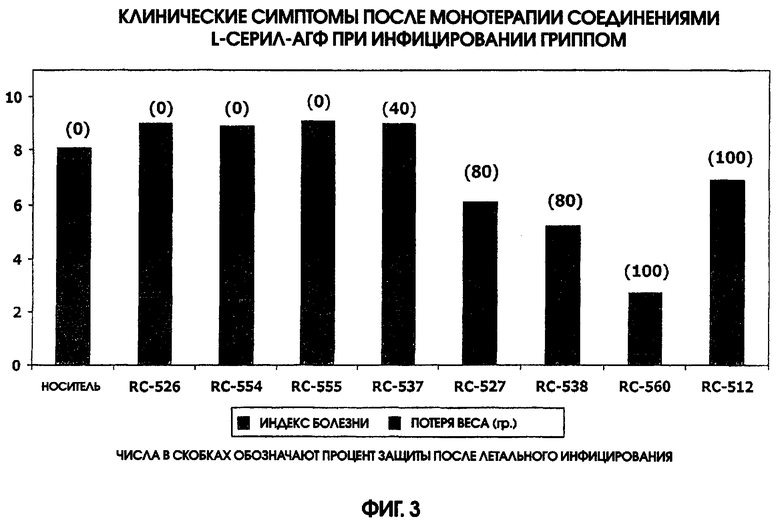
Клинические симптомы после интраназального введения L-серил-аминоалкилглюкозаминид фосфатов (АГФ) и заражения гриппом
Дозу в 20 мкг различных соединений L-серил-АГФ (RC-526, RC-554, RC-555, RC-537, RC-527, RC-538, RC-560, RC-512, а также только носитель) вводили интраназально группам самок мышей линии BALB, либо за 2 дня до летального инфицирования гриппом, либо в день этого инфицирования. Всех мышей заражали дозой примерно LD50 инфекции гриппа А/НК/68, вводимой также интраназально. Показатели индекса болезни (взъерошенная шерсть, сгорбленная поза и затрудненное дыхание) отслеживали в течение дней 4-19 после заражения гриппом. Потерю веса и смертность отслеживали в течение 21 суток после заражения гриппом. Результаты этих экспериментов представлены на фигуре 3.
Эти данные продемонстрировали эффективность соединений RC-538, RC-560 и RC-512 для обеспечения существенной защиты против заражения гриппом.
ПРИМЕР 5
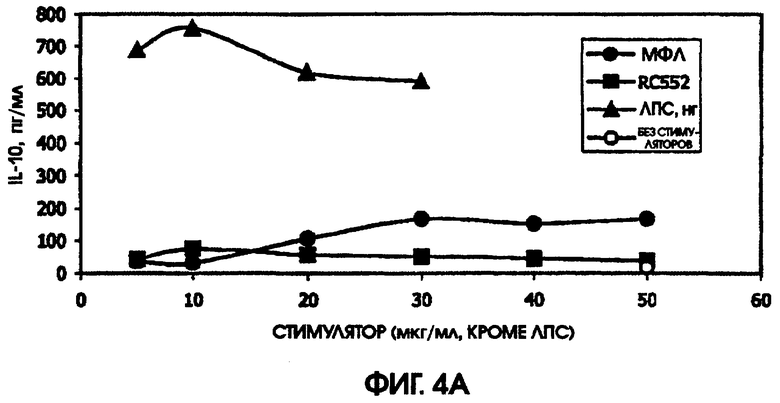
Сравнение RC552 и МФЛ на культурах цельной крови человека и культурах селезенки мышей
Этот пример демонстрирует индукцию цитокинов синтетическим соединением липида A RC552 в сравнении с модифицированным природным монофосфорил-липидом А (МФЛ), на культуре цельной крови человека и культуре селезенки мышей.
Соединения липида А растворяли в 0,2% триэтаноламине в стерильной воде, инкубировали при 56°С и обрабатывали ультразвуком в течение 2×10 минут при 37°С. LPS 055B5 (Sigma-Aldrich; St Louis, МО) растворяли в PBS.
Соединения добавляли к 450 мкл цельной человеческой крови и инкубировали при перемешивании в течение от 5 до 24 часов. Отобрали трех доноров (фигуры 4-6, доноры А-С, соответственно). Надосадочные жидкости собирали путем центрифугирования и разбавляли до 1/2 равным объемом PBS. (Это разбавление не учитывали как фактор разбавления при расчетах цитокинов). Выработку цитокинов измеряли с помощью твердофазного иммуноферментного анализа (ИФА) (R&D Systems; Minneapolis, MN), используя требуемый объем надосадочной жидкости в неразбавленном виде или при разбавлении в десять раз.
Мышей линий BALB/c, DBA/2 и C3H/HEJ получали от Jackson Laboratory (Bar Harbor, ME). Селезенки брали у мышей в период между 2 и 3 часами дня, и для каждой линии мышей получили отдельные одноклеточные суспензии. Эритроциты лизировали раствором трис-аммонийхлорида (Sigma-Aldrich), клетки промывали и подсчитывали, используя исключение с помощью голубого Трипана (Trypan-Blue Sigma-Aldrich). По одному миллиону спленоцитов на лунку культивировали в 1,0 мл культуральной среды. Среда для культуры селезенки (SCM) была предназначена для 5-суточных и более длительных культур спленоцитов мышей и состояла из среды RPMI 1640 (Sigma-Aldrich) с добавкой до 5% фетальной телячьей сыворотки (HyClone; Logan, UT), 100 мкг/мл; 100 нг/мл гентамицина (Sigma-Aldrich), 250 нг/мл амфотерицина (InVitrogen Life Technologies; Carlsbad, California), 1×ITS (бычий инсулин 500 нг/мл, человеческий трансферин (500 нг/мл), селенит натрия 250 нг/мл, (Sigma), бета-меркаптоэтанола 43нМ (Sigma-Aldrich), пуривовой (purivic) кислоты 1 мМ (Sigma-Aldrich), HEPES 10 мМ (Sigma-Aldrich). Данные, полученные в этих экспериментах, приведены на фигуре 8.
В культуре цельной крови человека измерили четыре цитокина: IL-10, MIP-1 бета, TNF альфа и IL-8. Двух доноров тестировали однократно и одного донора тестировали дважды. Стоит отметить, что у донора, которого тестировали дважды, в первом тесте был обнаружен очень высокий фон TNF альфа и очень низкий фон TNF альфа месяц спустя. Показатели в значительной степени зависели от времени культивирования. Высокий фон TNF альфа для 5,5-часовой культуры, а низкий фон TNF альфа обнаружили для культуры, продолжавшейся до следующего дня, около 24 часов. Тем не менее, даже для 5,5-часовой культуры с низким фоном, показатели были выше (около 600 пг/мл), чем полученные в некоторых предыдущих культурах (518,2 пг/мл) в 5-часовом тесте; 417 пг/мл в 4-часовом тесте; ноль пг/мл за 4 часа (линия клеток с низкой иммунологической реактивностью).
RC552 был подобен МФЛ в отношении синтеза TNF альфа в двух из трех 24-часовых культур и в одной 5,5-часовой культуре. Однако индукция IL-8 соединением RC552 отличалась от этого показателя для МФЛ в трех случаях из трех. Индукция IL-8 соединением RC552 была снижена по сравнению с МФЛ для двух 24-часовых культур, но она была больше, чем для МФЛ в одной 24-часовой культуре.
Для 24-часовых культур индукция IL-8 соединением RC552 была меньше, чем для МФЛ. Индукция MIP-1 бета была снижена в одном из трех случаев 24-часовых культур.
Сравнивали ответ на МФЛ и RC552 у линий мышей BALB/c и C3H/HEJ. Мыши линии C3H/HEJ являются генетически гипореактивными по отношению к ЛПС, благодаря мутации в толл-рецепторе 4. В качестве позитивного контроля для культур C3H/HEJ использовали олигонуклеотидный стимулятор. Этот олигонуклеотид индуцировал большие количества IL-6 у мышей линии BALB/c (1000 пг/мл) и линии C3H/HEJ (488,5 пг/мл). Аналогично, в культурах BALB/c и C3H/HEJ наблюдалась индукция MIP-1 (589 пг/мл и 554 пг/мл), так же, как и IL-10 (342 пг/мл и 609 пг/мл) и TNF альфа (204 пг/мл и 30 пг/мл), в ответ на 10 мкг/мл МФЛ или RC552, соответственно.
Ни МФЛ, ни RC552 не индуцировали цитокиновую реакцию при использовании спленоцитов C3H/HEJ. Однако в культурах спленоцитов BALB/c наблюдалась индукция IL-10, MIP-1 бета, TNF альфа и IL-6. RC552 индуцировал меньше MIP-1 бета, TNF альфа и IL-6, чем МФЛ в тех же самых концентрациях. RC552 индуцировал очень мало IL-10 (от 10,4 до 11,6 пг/мл), по сравнению с МФЛ (от 144,1 до 176,6 пг/мл).
При тестировании соединения в 0,2% раствора триэтаноламина RC552 имел сходный, но не идентичный с МФЛ про-воспалительный профиль для индукции TNF альфа в двух из трех 24-часовых культур и в одной краткосрочной культуре цельной крови человека. См. фигуры 4-6 (24-часовые культуры для доноров А-С, соответственно) и фигуру 7 (краткосрочная культура для донора А). Кроме того, индукция MIP-1 бета RC552 была сходной в двух из трех 24-часовых культур. Уменьшенное количество IL-10 наблюдалось при индукции соединением RC552 по сравнению с соединением МФЛ в одной 24-часовой культуре. Индукция IL-8 отличалась от вызываемой МФЛ во всех испытанных случаях.
На модели мышей, дефицитных по рецептору, было обнаружено, что RC552 передает сигналы через толл-подобный рецептор 4. На модели мышей линии BALB/c, реактивных по отношению к липиду А, RC552 индуцировал уменьшенный профиль цитокинов при использованных концентрациях. Интересно, что использованные концентрации находились в диапазоне больших значений конценрации кривой дозозависимого ответа, и RC552 индуцировал слегка увеличенное количество MIP-1 бета и TNF альфа при более низких концентрациях (10 мкг/мл), чем при более высоких концентрациях (20 мкг/мл).
При сравнении профилей цитокина человека и мыши оказалось, что синтетическое соединение липида А RC552 обладало меньшей эффективностью индукции в отношении IL-10 в 2-дневных культурах спленоцитов мышей и в одной из трех 24-часовых культур крови человека при высоких концентрациях стимулятора. Как правило, меньшая индукция TNF альфа в 24-часовых культурах крови человека наблюдалась для RC552 по сравнению с МФЛ. Примерно равная индукция TNF альфа наблюдалась в краткосрочных (5,5 часов) культурах крови человека для RC552 по сравнению с МФЛ. При микроанализе РНК человеческих макрофагов, стимулированных RC552 и МФЛ, было обнаружено наличие ранней (1 час) РНК TNF альфа для обоих соединений и отсутствие поздней РНК TNF альфа для обоих соединений. Однако RC552 индуцировал очень мало 6-часовой TNF альфа, в противоположность МФЛ, для которого было обнаружено измеряемое количество 6-часовой РНК.
ПРИМЕР 6
Стимулирующая способность RC529 по сравнению с МФЛ и RC552
Этот пример демонстрирует, что RC529 обладает более высокой иммуностимулирующей способностью по сравнению с МФЛ, оцениваемой по индукции IL-6, IL-10 и MPI-1 бета в периферических одноядерных клетках крови человека (РВМС). В противоположность этому, синтез IL-8 был аналогичен с этим показателем для МФЛ.
РВМС хранили в замороженном состоянии вплоть до их использования. Донор РВМС имел номер AD112. РВМС при плотности в 6,26×105 высевали в лунки 48-луночного культурального планшета, в 1,0 мл среды. Среда состояла из RPMI-1640, содержащей бикарбонат натрия, 10% фетальной телячьей сыворотки, 4 мМ глютамина, 100 мкг/мл гентамицина и 10 мМ HEPES. РВМС культивировали в течение 22 часов при температуре 37°С в инкубаторе в атмосфере углекислого газа. Надосадочные жидкости собирали и тестировали с помощью ИФА (RScD System) на содержание IL-6, IL-8, IL-10 и MPI-1 бета. Концентрации цитокинов в надосадочных жидкостях сравнивали с концентрациями в надосадочных жидкостях, полученных из культур нестимулированных РМВС, культивированных в идентичных условиях.
При всех использованных дозах RC529 не наблюдалось дозозависимого ответа при самой низкой дозе для IL-6 или IL-8. По сравнению с МФЛ, RC529 индуцировал больше IL-6, IL-10 и MPI-1 бета. Соединение дисахарида, RC552, было, как правило, промежуточным по стимулирующей способности в расчете на массу. См. фигуру 9. Приведенные результаты показывают, что RC529 является сильным индуктором IL-6, IL-10 и MPI-1 бета в замороженных клетках РВМС человека.
Все публикации и заявки на патент, цитированные в данном описании изобретения, включены в него в качестве ссылок так, как если бы каждая отдельная публикация или заявка на патент была конкретно и по отдельности указана как включенная в качестве ссылки. Хотя вышеописанное изобретение было описано с некоторыми подробностями, которые приведены в качестве иллюстрации и в качестве примеров в целях ясности изложения, но для специалистов в данной области техники очевидно, в свете отличительных особенностей настоящего изобретения, что можно произвести в нем некоторые изменения и модификации, не выходя за пределы сущности и объема прилагаемой формулы изобретения.
Настоящее изобретение относится к медицине, в частности к иммунологии, и может быть использовано для лечения и профилактики инфекционных, аутоиммунных или аллергических заболеваний. Для этого вводят в терапевтически эффективных количествах производные моно- и дисахаридов или фармацевтическую композицию на их основе, что обеспечивает эффективную стимуляцию иммунной системы за счет селективного модулирующего воздействия на продукцию цитокинов. 2 н. и 36 з.п. ф-лы, 2 табл., 9 ил.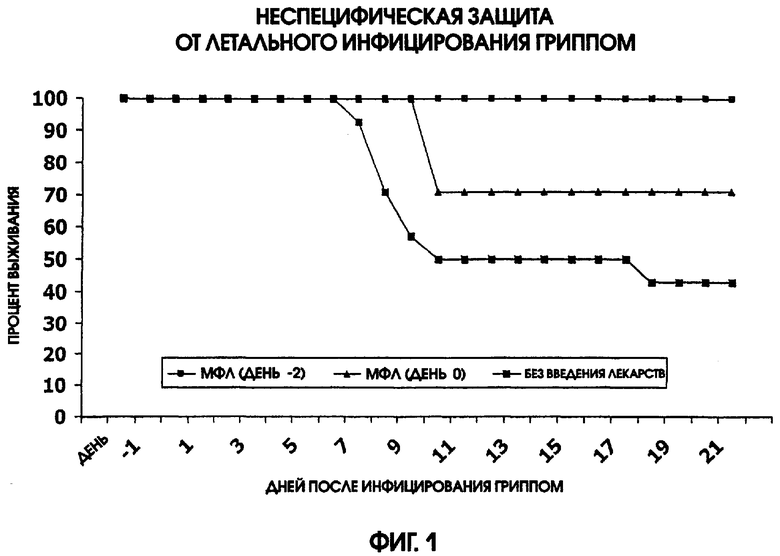
или их фармацевтически приемлемых солей, в которой Х представляет собой фрагмент, выбранный из группы, состоящей из -О- и -NH-;
каждый заместитель R1 и R2 независимо выбирают из группы, содержащей ацил (С2-С24),
R3 представляет собой заместитель, выбранный из группы, состоящей из -Н и- РО3R11R12, где каждый заместитель R11 и R12 независимо выбирают из группы, состоящей из - Н и алкила (С1-С4);
R4 представляет собой заместитель, выбранный из группы, состоящей из - Н, -СН3 и -PO3R13R14 где каждый заместитель R13 и R14 независимо выбирают из группы, состоящей из - Н и алкила (C1-C4); и
Y представляет собой радикал, выбранный из группы, состоящей из
где каждый из подстрочных знаков n, m, p и q независимо означает целое число от 0 до 6;
R5 является ацилом (С2-С24),
R6 и R7 представляют собой заместители, независимо выбранные из группы, состоящей из -Н и -СН3;
R8 и R9 представляют собой заместители, независимо выбранные из группы, состоящей из -Н, -ОН, (С1-С4) алкокси, -РО3Н2, -ОРО3Н2, -SO3H, -OSO3Н, -NR15R16, - SR15, -CN, -NO2, -СНО, -CO2R15, -CONR15R16, -PO3R15R16, -OPO3R15R16, -SO3R15, -и - OSO3R15, где каждый из заместителей R15 и R16 независимо выбирают из группы, состоящей из -Н и алкила (С1-C4);
R10 представляет собой заместитель, выбранный из группы, состоящей из -Н, - СН3, -РО3Н2, ω-фосфонооксиалкила (С2-С24) и ω-карбоксиалкила (C1-C24); и
Z означает -О- или -S-;
при условии, что, когда R3 представляет собой -РО3R11R12, R4 отличается от - РО3R13R14, и при дополнительном условии, что, когда R3 представляет собой -РО3Н2, R4 означает -Н, R10 означает -Н, R1 является н-тетрадеканоилом, R2 является н-октадеканоилом и R5 является н-гексадеканоилом, тогда Х отличается от -O-;
где одно или несколько соединений назначаются в отсутствие экзогенного антигена;
при условии, что когда заболевание является аутоиммунным заболеванием или сепсисом, то соединения монофосфорил липид А и 3-O-деацилированный монофосфорил липид А исключаются.
и их фармацевтически приемлемых солей в качестве единственного (ых) активного(ых) ингредиента(ов), и фармацевтически приемлемый носитель, где
Х представляет собой фрагмент, выбранный из группы, состоящей из -О- и -NH-;
каждый заместитель R1 и R2 независимо выбирают из группы, содержащей ацил (С2-С24);
R3 представляет собой заместитель, выбранный из группы, состоящей из -Н и- РО3R11R12, где каждый заместитель R11 и R12 независимо выбирают из группы, состоящей из - Н и алкила (C1-C4);
R4 представляет собой заместитель, выбранный из группы, состоящей из - Н, -СН3 и -PO3R13R14 где каждый заместитель R13 и R14 независимо выбирают из группы, состоящей из - Н и алкила (C1-C4); и
Y представляет собой радикал, выбранный из группы, состоящей из
где каждый из подстрочных знаков n, m, p и q независимо означает целое число от 0 до 6;
R5 является ацилом (C2-C24);
R6 и R7 представляют собой заместители, независимо выбранные из группы, состоящей из -Н и -СН3;
R8 и R9 представляют собой заместители, независимо выбранные из группы, состоящей из -Н, -ОН, (C1-C4) алкокси, -РО3Н2, -ОРО3Н2, -SO3Н, -OSO3H, -NR15R16, - SR15, -CN, -NO2, -CHO, -CO2R15, -CONR15R16, -РО3R15R16, -ОРО3R15R16, -SO3R15, -и - OSO3R15, где каждый из заместителей R15 и R16 независимо выбирают из группы, состоящей из -Н и алкила (C1-C4);
R10 представляет собой заместитель, выбранный из группы, состоящей из -Н, - СН3, -РО3Н2, ω-фосфонооксиалкила (С2-С24) и ω-карбоксиалкила (C1-C24); и
Z означает -О- или -S-;
при условии, что, когда R3 представляет собой -РО3R11R12, R4 отличается от - РО3R13R14, и при дополнительном условии, что, когда R3 представляет собой -РО3Н2, R4 означает -Н, R10 означает -Н, R1 является н-тетрадеканоилом, R2 является н-октадеканоилом и R5 является н-гексадеканоилом, тогда Х отличается от -O-;
при условии, что когда заболевание является аутоиммунным заболеванием или сепсисом, то соединения монофосфорил липид А и 3-O-деацилированный монофосфорил липид А исключаются.
| ГЛЮКОЗАМИНОВЫЕ ДИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2154068C2 |
| ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ, ОБЛАДАЮЩИЙ ИММУНОМОДЕЛИРУЮЩИМ, ИММУНОКОРРЕГИРУЮЩИМ, ПРОТИВОПАРАЗИТАРНЫМ, ПРОТИВОСКЛЕРОТИЧЕСКИМ, ПРОТИВОВИРУСНЫМ, ПРОТИВОБАКТЕРИАЛЬНЫМ, ПРОТИВОГРИБКОВЫМ, ПРОТИВОВОСПАЛИТЕЛЬНЫМ И ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ, И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2001 |
|
RU2197248C2 |
| WO 9850399, 12 | |||
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| US 4844894, 04.07.1989 | |||
| US 5762943, 09.06.1998 | |||
| CHASE JJ «Effect of monophosphoryl lipid A on host resistance to bacterial infection» | |||
| Infecn Immunol | |||
| Пневматический водоподъемный аппарат-двигатель | 1917 |
|
SU1986A1 |
| JONSON DA et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |

