Это изобретение относится к имидазопиридиновым соединениям, которые имеют замещенную аминогруппу в положении 1, а также к фармацевтическим составам, содержащим такие соединения. Кроме того, это изобретение относится к использованию этих соединений в качестве иммуномодуляторов для стимулирования биосинтеза цитокина в организме животного, а также для лечения различных болезней, включая вирусные и опухолевые заболевания.
Первые надежные данные по 1H-имидазо[4,5-с]хинолиновой циклической системе приведены Бакманом и др., J. Org. Chem., 15, 1278-1284 (1950), которые сообщили о синтезе 1-(6-метокси-8-хинолинил)-2-метил-1Н-имидазо[4,5-с]хинолина для его возможного применения в качестве антималярийного средства. В последующем появились сообщения о синтезе различных замещенных 1H-имидазо[4,5-с]хинолинах. Например, Джайн и др., J. Med. Chem., 11, с.87-92 (1968), синтезировали 1-[2-(4-пиперидил)этил]-1Н-имидазо[4,5-с]хинолин в качестве возможного противосудорожного и сердечно-сосудистого средства. Кроме того, Баранов и др., Chem. Abs., 85, 94362 (1976), сообщили о синтезе нескольких 2-оксоимидазо[4,5-с]хинолинов. Берени и др., J. Heterocvclic Chem., 18, 1537-1540 (1981), также сообщили о синтезе нескольких 2-оксоимидазо[4,5-с]хинолинах.
Позже было установлено, что определенные 1Н-имидазо[4,5-с]хинолин-4-амины и их 1- и 2-замещенные производные могут быть использованы в качестве противовирусных агентов, бронхолитических средств и иммуномодуляторов. Эти соединения описаны среди прочего в патентах США №№4689338, 4698348, 4929624, 5037986, 5268376, 5346905 и 5389640; все эти патенты включены в описание изобретения путем ссылки.
Замещенные соединения 1Н-имидазопиридин-4-амина, являющиеся эффективными модификаторами иммунной реакции, описаны в Патентах США №№5446153; 5494916 и 5644063. Приведенные в этих патентах соединения, однако, не содержат замещенной аминогруппы в положении 1. Определенные 1H-имидазо[4,5-с]хинолин-4-амины, содержащие амидную, сульфонамидную и уретановую группы в положении 1, описаны в РСТ публикациях WO 00/76505, WO 00/76518 и WO 00/76519.
Несмотря на появление в последнее время соединений, являющихся модификаторами иммунной реакции, сохраняется потребность в соединениях, обладающих способностью модулировать иммунную реакцию за счет стимулирования биосинтеза цитокина или с использованием других механизмов.
Краткое изложение сущности изобретения
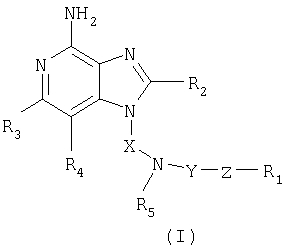
Найден новый класс соединений, способных стимулировать биосинтез цитокина в организме животных. В соответствии с этим данное изобретение предлагает соединения имидазопиридин-4-амина, которые имеют замещенную аминогруппу, содержащую заместитель в 1-положении. Соединения, способные стимулировать биосинтез цитокина, согласно данным ИК-спектроскопии имеют формулу (I), приведенную ниже
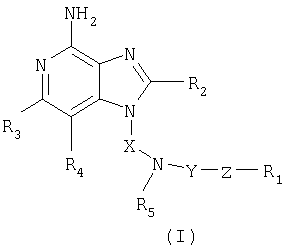
Природа заместителей X, Y, Z, R1, R2, R3, R4 и R5 охарактеризована ниже.
Соединения, имеющие формулу (I), являются полезными модификаторами иммунной реакции благодаря их способности инициировать биосинтез цитокина или иным образом модулировать иммунную реакцию при введении в организм животных. Это делает такие соединения полезными средствами для лечения различных заболеваний, в частности вирусных и опухолевых заболеваний, которые чувствительны к таким изменениям в иммунной реакции.
В изобретении также приводятся фармацевтические составы, содержащие соединения, изменяющие иммунную реакцию, и сообщается о способах стимулирования биосинтеза цитокина в организме животного и лечении вирусной инфекции и/или опухолевого заболевания у животного путем введения в организм животного соединений формулы I.
Кроме того, приводятся способы синтеза соединений, являющихся предметом изобретения, а также промежуточных продуктов, используемых при синтезе этих соединений.
Подробное описание изобретения
Как указывалось выше, мы обнаружили некоторые соединения, которые стимулируют биосинтез цитокина и модифицируют иммунную реакцию в организме животных. Такие соединения имеют формулу (I), показанную ниже
где X - алкиленовая или алкениленовая группы;
Y - -СО-, -CS- или -SO2- группы;
Z представляет собой простую связь или группы -O-, -S- или -NR5-;
R1 представляет собой арильную, гетероарильную, гетероциклильную, C1-20-алкильную или С2-20-алкенильную группы, причем каждая из этих групп может быть незамещенной или содержать один или большее количество заместителей, независимым образом выбранных из группы, включающей
-алкил;
-алкенил;
-арил;
-гетероарил;
-гетероциклил;
-замещенный циклоалкил;
-O-алкил;
-O-(алкил)0-1-арил;
-O-(алкил)0-1-гетероарил;
-O-(алкил)0-1-гетероциклил;
-СООН;
-СО-O-алкил;
-СО-алкил;
-S(O)0-2-алкил;
-S(O)0-2-(алкил)0-1-арил;
-S(O)0-2-(алкил)0-1-гетероарил;
-S(O)0-2-(алкил)0-1-гетероциклил;
-(алкил)0-1-N(R5)2;
-(алкил)0-1-NR5-СО-O-алкил;
-(алкил)0-1-NR5-СО-алкил;
-(алкил)0-1-NR5-СО-арил;
-(алкил)0-1-NR5-СО-гетероарил;
-N3;
-атом галогена;
-галоалкил;
-галоалкоксил;
-СО-галоалкил;
-СО-галоалкоксил;
-NO2;
-CN;
-ОН;
-SH; а в случае алкильной, алкенильной и гетероциклильной групп также оксогруппа;
заместитель R2 выбран из группы, включающей
-атом водорода;
-алкил;
-алкенил;
-алкил-O-алкил;
-алкил-S-алкил;
-алкил-O-арил;
-алкил-S-арил;
-алкил-O-алкенил;
-алкил-S-алкенил, а также
- алкил или алкенил, содержащие один или большее количество
заместителей, выбранных из группы, включающей
-OH;
-галоген
-N(R5)2;
-CO-N(R5)2;
-CS-N(R5)2;
-SO2-N(R5)2;
-NR5-CO-C1-10алкил;
-NR5-CS-C1-10алкил;
-NR5-SO2-C1-10алкил;
-CO-C1-10алкил;
-CO-O-C1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил и
-СО-гетероарил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной, алкенильной, галогеновой, алкоксильной, амино-, алкиламино-, диалкиламино- и алкилтиольной групп, а
каждый заместитель R5 представляет собой независимо атом водорода или C1-10 алкильную группу;
или соль фармацевтического качества на основе этих групп.
Получение соединений
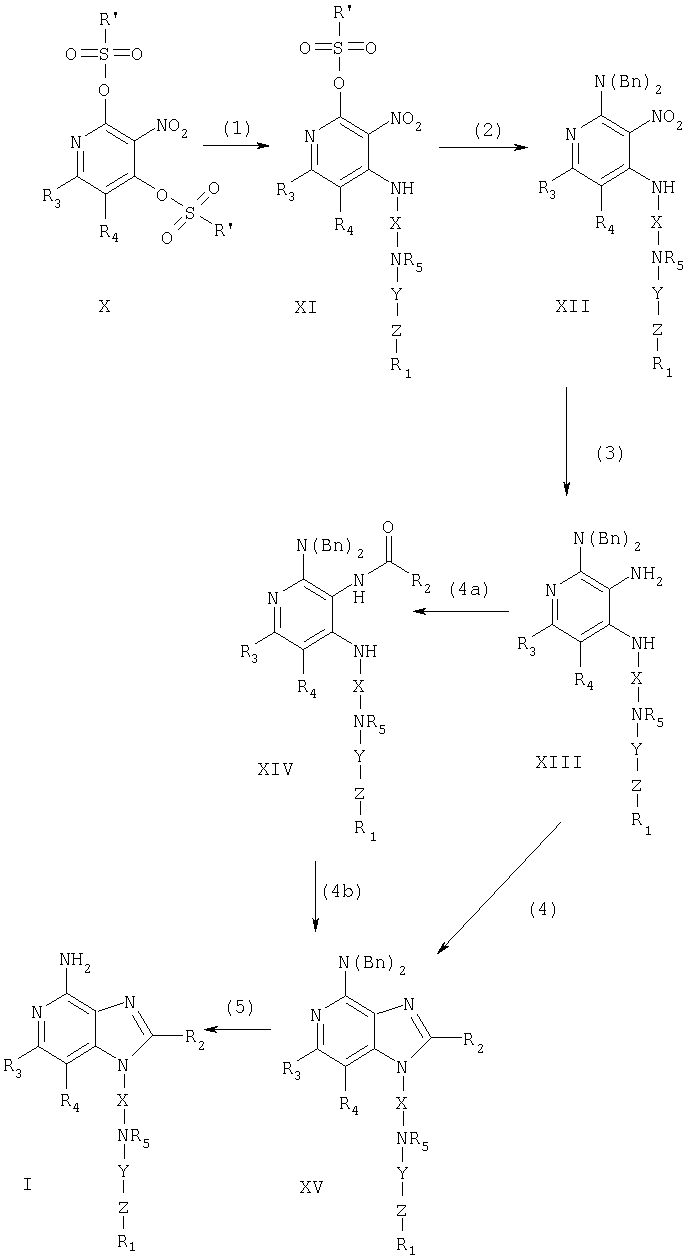
Предлагаемые в изобретении соединения могут быть получены в соответствии со схемой I реакции, в которой значения R1, R2, R3, R4, R5, X, Y и Z определены выше, Bn - бензильная группа, а радикал R' представляет собой алкильную группу, содержащую от одного до четырех атомов углерода, или перфторалкильную группу, содержащую от одного до четырех атомов углерода. В качестве заместителя R' может быть использована также фенильная группа или фенильная группа, содержащая в качестве заместителя атом галогена или алкильную группу, имеющую от 1 до 4 атомов углерода.
На стадии (1) процесса (схема I реакции) проводят реакцию между 3-нитропиридин-2,4-дисульфонатом (формула X) и амином формулы R1-Z-Y-N(R5)-X-NH2, в результате которой образуется 3-нитро-4-аминопиридин-2-сульфонат формулы XI. Наличие двух сульфонатных групп, которые, в принципе, могут быть замещены, обусловливает тот факт, что в результате реакции может образовываться смесь продуктов, которые, однако, можно легко разделить с помощью обычных способов, таких как препаративная хроматография. Реакцию предпочтительно проводят, добавляя амин к раствору соединения формулы Х в подходящем растворителе, таком как дихлорметан, в присутствии третичного амина, например триэтиламина. Поскольку сульфонатная группа является относительно лабильной, реакцию можно проводить при пониженной температуре (0°С). Это способствует уменьшению количества образующихся нежелательных побочных 2-аминированных и 2,4-диаминированных продуктов. 3-Нитропиридин-2,4-дисульфонаты являются хорошо известными продуктами и могут быть легко получены с помощью известных синтетических способов, см., например, Линдстром и др., Патент США №5446153, и приводимых в этом патенте в качестве ссылок.
На стадии (2) процесса (схема I реакции) 1-нитро-4-аминопиридин-2-сульфонат формулы XI обрабатывают дибензиламином, в результате чего получают 2-дибензиламино-3-нитропиридин-4-амин формулы XII. Для проведения реакции соединение формулы XI, дибензиламин и третичный амин, такой как триэтиламин, растворяют в инертном растворителе, таком как бензол, толуол или ксилол, и нагревают полученную смесь.
На стадии (3) процесса (схема 1 реакции) проводят восстановление нитрогруппы 2-дибензиламино-3-нитропиридин-4-амина (соединение формулы XII) до аминогруппы. Восстановление предпочтительно проводят под действием NiB2, который образуется in situ из раствора борогидрида натрия и гидрата хлористого никеля в метаноле. Реакцию предпочтительно проводят при температуре окружающей среды.
На стадии (4) процесса (схема I реакции) 2-дибензиламинопиперидин-3,4-диамин (соединение формулы XIII) обрабатывают карбоновой кислотой или ее эквивалентом для получения 4-дибензиламино-1Н-имидазо[4,5-с]пиридина, имеющего формулу XV. Подходящими эквивалентами карбоновой кислоты являются сложные ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают таким образом, чтобы они обеспечивали введение в соединение формулы XV необходимого заместителя R2. Например, при использовании триэтилортоформиата будет получаться соединение, в котором R2 представляет собой атом водорода, а при использовании триэтилортоацетата R2=СН3. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакцию проводят при достаточном нагревании, чтобы обеспечить удаление любого спирта или воды, образующихся в ходе этой реакции в качестве побочных продуктов. По желанию в реакции можно использовать катализатор, такой как солянокислый пиридин.
Альтернативным образом соединение формулы XV можно получить в две стадии в результате (а) взаимодействия соединения XIII с ацилгалогенидом формулы R2C(O)Cl или R2C(O)Br, при котором образуется соединение формулы XIV, и (b) последующей циклизации полученного продукта. На стадии (4а) ацилгалогенид добавляют к раствору диамина в инертном растворителе, таком как ацетонитрил, пиридин или дихлорметан. Реакцию можно проводить при комнатной температуре. На стадии (4b) продукт, полученный на стадии (4а), нагревают в спиртовом растворителе в присутствии основания. Предпочтительно продукт, полученный на стадии (4а), кипятят в этаноле в присутствии избытка триэтиламина или нагревают в присутствии метанольного раствора аммиака. Другим вариантом проведения стадии (4b) является нагревание продукта, полученного на стадии (4а), в пиридине. Если стадию (4а) проводят в пиридине, стадию (4b) можно проводить путем нагревания реакционной смеси, после того, как согласно данным анализа, стадия (4а) завершится.
На стадии (5) процесса (схема I реакции) осуществляют восстановление 4-дибензиламино-1Н-имидазо[4,5-с]пиридина, имеющего формулу XV, до 4-амино-1H-имидазо[4,5-с]пиридина (соединение формулы I). Предпочтительно соединение формулы XV нагревают в муравьиной кислоте в присутствии гидроксида палладия, нанесенного на активированный уголь. Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
Схема I реакции
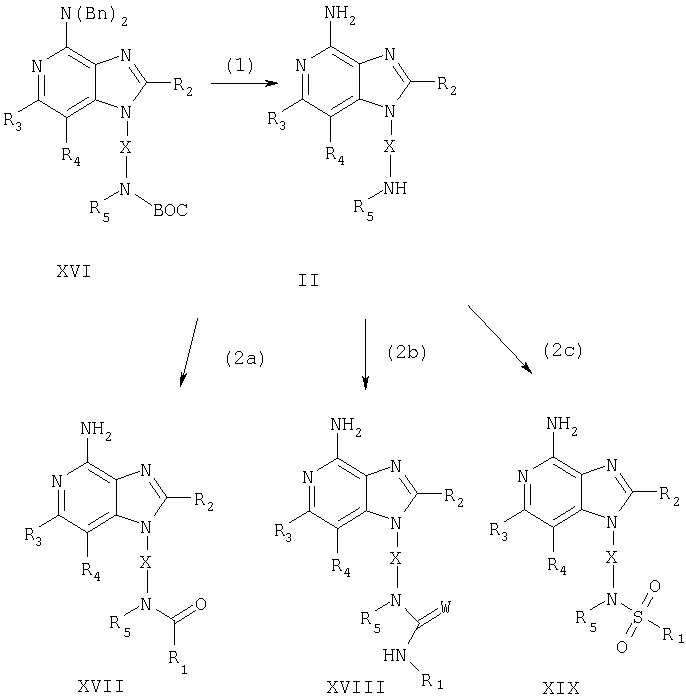
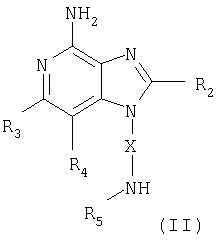
Указанные в изобретении соединения могут быть получены в соответствии со схемой II реакции, в которой R1, R2, R3, R4, R5 и Х определены выше, Bn означает бензильную группу, ВОС является трет-бутоксикарбонильной группой, a W представляет собой атомы О или S.
На стадии (1) процесса (схема II реакции) защитные аминогруппы 1Н-имидазо[4,5-с]пиридина в соединении формулы XVI удаляют и в результате получают 1H-имидазо[4,5-с]пиридин формулы II. Предпочтительно раствор соединения XVI в подходящем растворителе, таком как дихлорметан, обрабатывают при комнатной температуре трифторметануксусной кислотой. Соединения формулы XVI могут быть получены с помощью способа синтеза, используемого для получения продуктов в соответствии со схемой I реакции. На стадии (1) 2,4-дисульфонат формулы Х обрабатывают амином формулы BOC-NR5-X-NH2. Затем проводят стадии (2)-(4), как описано выше, чтобы получить соединение формулы XVI, которое является производным соединения XV.
На стадии (2а) процесса (схема II реакции) проводят реакцию между 1H-имидазо[4,5-с]пиридином формулы II и хлорангидридом кислоты формулы R1-C(O)Cl или ангидридом кислоты формулы R1-C(O)OC(O)-R1 и получают 1H-имидазо[4,5-с]пиридин-1-ил-амид формулы XVII, который является производным соединения I. Реакцию предпочтительно проводят в присутствии основания, такого как триэтиламин, добавляя хлорангидрид кислоты или ангидрид кислоты к раствору соединения формулы II в подходящем растворителе, таком как дихлорметан или ацетонитрил. Реакция может протекать при пониженной температуре (0°С) или при комнатной температуре. Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
На стадии (2b) процесса (схема II реакции) 1Н-имидазо[4,5-с]пиридин формулы II обрабатывают изоцианатом формулы R1-N=C=O или изотиоционатом формулы R1-N=C=S и в результате получают 1H-имидазо[4,5-с]пиридин-1-ил-мочевину или тиомочевину формулы XVIII, которая является производным соединения формулы I. Реакцию предпочтительно проводят при пониженной температуре (0°С), добавляя изоцианат или изотиоцианат к раствору соединения формулы II в подходящем растворителе, таком как дихлорметан. Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
На стадии (2с) процесса (схема II реакции) 1H-имидазо[4,5-с]пиридин формулы II обрабатывают хлорангидридом сульфоновой кислоты формулы R1-S(O)2Cl или ангидридом сульфоновой кислоты R1-S(O)2OS(O)2-R1. В результате получают 1H-имидазо[4,5-с]пиридин-1-ил-сульфонамид формулы XIX, который является производным соединения формулы I. Реакцию предпочтительно проводят, добавляя хлорангидрид сульфоновой кислоты или ангидрид сульфоновой кислоты к раствору соединения формулы II в соответствующем растворителе, таком как дихлорметан, в присутствии основания, например триэтиламина. Реакция может протекать как при пониженной (0°С), так и при комнатной температуре. Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
Схема II реакции
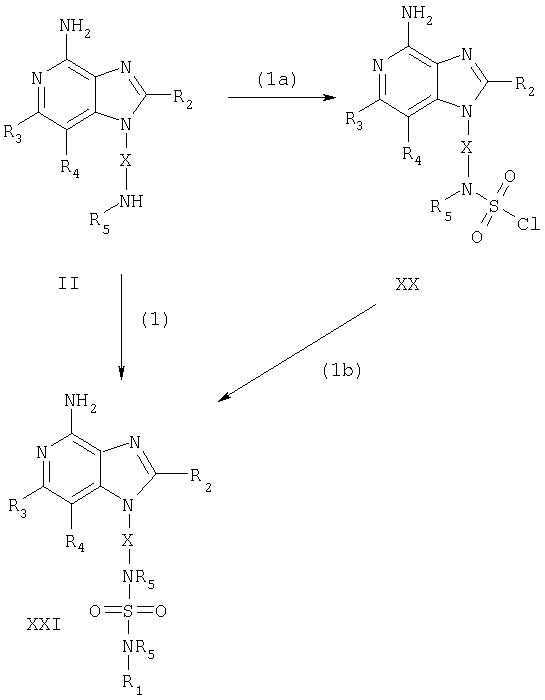
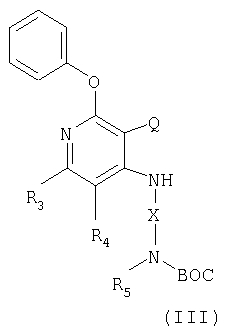
Указанные в изобретении соединения могут быть получены в соответствии со схемой III реакции, в которой R1, R2, R3, R4, R5 и X определены выше.
На стадии (1) процесса (схема III реакции) 1H-имидазо[4,5-с]пиридин формулы II реагирует с сульфамоилхлоридом R1-N(R5)S(O)2Cl с образованием 1Н-имидазо[4,5-с]пиридин-1-ил-сульфамида формулы XXI, который является производным соединения формулы I. Предпочтительно сульфамоилхлорид добавляют к раствору соединения формулы II в подходящем растворителе, таком как 1,2-дихлорэтан, в присутствии основания, например триэтиламина. Реакцию можно проводить при повышенной температуре. Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
В альтернативном варианте соединение формулы XXI можно получить в две стадии в результате (а) взаимодействия 1H-имидазо[4,5-с]пиридина формулы II с хлористым сульфурилом, приводящего к образованию in situ сульфамоилхлорида формулы XX, и (b) последующего взаимодействия сульфамоилхлорида с амином формулы R1-N(R5)H. На стадии (1а) реакцию можно проводить, добавляя раствор хлористого сульфурила в дихлорметане к раствору соединения формулы II в присутствии 1 эквивалента 4-(диметиламино)пиридина. Предпочтительно реакцию проводят при пониженной температуре (-78°С). В некоторых случаях по окончании добавления компонентов реакционную смесь можно нагревать до комнатной температуры. На стадии (1b) раствор, содержащий 2 эквивалента R1-N(R5)H и 2 эквивалента триэтиламина в дихлорметане, добавляют к реакционной смеси, образующейся в результате стадии (1а). Реакцию предпочтительно проводят при пониженной температуре (-78°С). Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
Схема III реакции
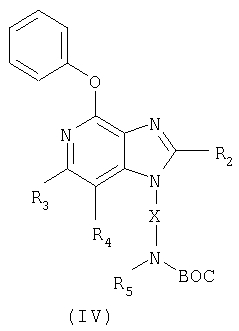
Указанные в изобретении соединения могут быть получены в соответствии со схемой IV реакции, в которой R1, R2, R3, R4, R5 и Х определены выше, а ВОС означает трет-бутоксикарбонильную группу.
На стадии (1) процесса (схема IV реакции) с использованием обычных хлорирующих агентов проводят хлорирование 2,4-дигидрокси-3-нитропиридина формулы XXII, приводящее к получению 2,4-дихлор-3-нитропиридина формулы XXIII. Предпочтительно соединение формулы XXII обрабатывают оксихлоридом фосфора при нагревании. Известны многие 2,4-дигидрокси-3-нитропиридины формулы XXII. Другие подобные соединения можно легко получить с помощью известных синтетических способов, см., например, Линдстом и др., Патент США №5446153, и приводимых в этом патенте в качестве ссылок.
На стадии (2) процесса (схема IV реакции) 2,4-дихлор-3-нитропиридин формулы XXIII обрабатывают амином формулы BOC-NR5-X-NH2, в результате чего получают 2-хлор-3-нитропиридин формулы XXIV. Реакцию предпочтительно проводят, добавляя амин к раствору соединения формулы XXIII в подходящем растворителе, таком как N,N-диметилформамид, в присутствии третичного амина, например триэтиламина.
На стадии (3) процесса (схема IV реакции) проводят реакцию между 2-хлор-3-нитропиридином формулы XXIV и фенолом, приводящую к получению 3-нитро-2-феноксипиридина формулы XXV. Фенол взаимодействует с гидридом натрия в подходящем растворителе, таком как диглим, с образованием феноксида. Феноксид, в свою очередь, реагирует при повышенных температурах с соединением формулы XXIV.
На стадии (4) процесса (схема IV реакции) осуществляют восстановление 3-нитро-2-феноксипиридина формулы XXV до 3-амино-2-феноксипиридина формулы XXVI. Восстановление преимущественно проводят, используя обычные гетерогенные катализаторы гидрирования, такие, например, как нанесенные на активированный уголь платина или палладий. Реакцию предпочтительно проводят в аппарате Парра в подходящем растворителе, таком как изопропиловый спирт или толуол.
На стадии (5) процесса (схема IV реакции) 3-амино-2-феноксипиридин (соединение формулы XXVI) обрабатывают карбоновой кислотой или ее эквивалентом для получения 4-фенокси-1Н-имидазо[4,5-с]хинолина, имеющего формулу IV. Подходящими эквивалентами карбоновой кислоты являются сложные ортоэфиры и 1,1-диалкоксиалкилалканоаты. Карбоновую кислоту или ее эквивалент выбирают таким образом, чтобы они обеспечивали введение в соединение формулы IV необходимого заместителя R2. Например, при использовании триэтилортоформиата будет получаться соединение, в котором R2 представляет собой атом водорода, а при использовании триметилортовалериата R2=C4H9. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакцию проводят при достаточном нагревании, чтобы обеспечить удаление любого спирта или воды, образующихся в ходе этой реакции в качестве побочных продуктов. По желанию в реакции можно использовать катализатор, такой как солянокислый пиридин.
Альтернативным образом стадию (5) можно проводить путем (i) взаимодействия соединения формулы XXVI с ацилгалогенидом формулы R2C(O)Cl или R2C(O)Br и (ii) последующей циклизации полученного продукта. На стадии (i) ацилгалогенид добавляют к раствору соединения формулы XXV в инертном растворителе, таком как ацетонитрил, пиридин или дихлорметан. Реакцию можно проводить при температуре окружающей среды. На стадии (ii) продукт, полученный на стадии (i), нагревают в пиридине.
На стадии (6) процесса (схема IV реакции) проводят отщепление групп ВОС из соединения формулы IV, в результате чего получают 4-фенокси-1H-имидазо[4,5-с]хинолин формулы V. Предпочтительно раствор соединения IV в подходящем растворителе, таком как дихлорметан, обрабатывают при пониженной температуре трифторуксусной кислотой или соляной кислотой.
На стадии (7) процесса (схема IV реакции) 4-фенокси-1H-имидазо[4,5-с]хинолин формулы V превращают в 4-фенокси-1H-имидазо[4,5-с]хинолин-1-ил-сульфонамид формулы VI. Этот процесс проводят таким же образом, как стадию (2с) в схеме реакции II.
На стадии (8) процесса (схема IV реакции) 4-фенокси-1Н-имидазо[4,5-с]хинолин-1-ил-сульфонамид формулы VI подвергают аминированию до получения 4-амино-1H-имидазо[4,5-с]хинолин-1-ил-сульфонамида формулы XIX, который является производным соединения формулы I. Реакцию можно проводить, добавляя к соединению формулы VI ацетат аммония и нагревая полученную смесь в герметичной ампуле при температуре около 150°С. Продукт или соль фармацевтического качества на его основе могут быть выделены с помощью обычных способов.
Схема IV реакции
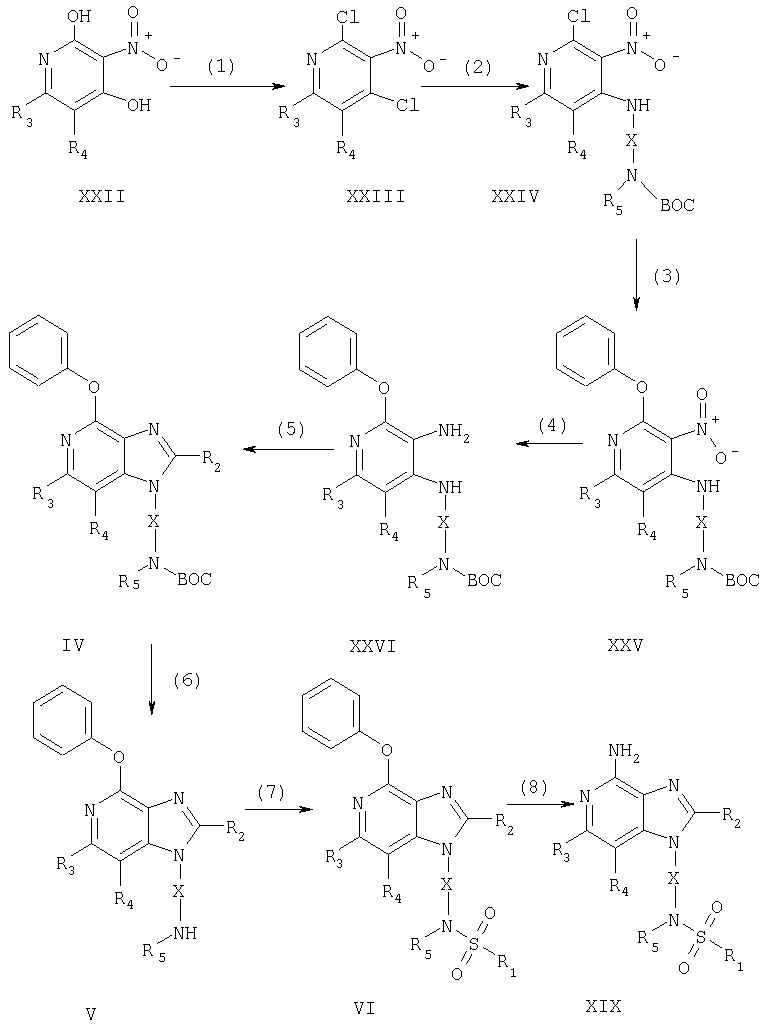
В изобретении также сообщается о синтезе новых соединений, представляющих интерес в качестве промежуточных продуктов, необходимых для получения соединений формулы I. Эти промежуточные соединения имеют структурные формулы II-VI, подробно описанные ниже.
Один класс промежуточных соединений имеет формулу (II)
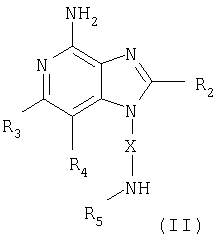
где X - алкиленовая или алкениленовая группа;
заместитель R2 выбран из группы, включающей
-атом водорода;
-алкил;
-алкенил;
-алкил-O-алкил;
-алкил-S-алкил;
-алкил-O-арил;
-алкил-S-арил;
-алкил-O-алкенил;
-алкил-S-алкенил, а также
- алкил или алкенил, содержащие один или большее количество заместителей, выбранных из групп, включающих
-ОН;
-галоген
-N(R5)2;
-CO-N(R5)2;
-CS-N(R5)2;
-SO2-N(R5)2;
-NR5-CO-C1-10алкил;
-NR5-CS-C1-10алкил;
-NR5-SO2-C1-10алкил;
-CO-C1-10алкил;
-CO-O-C1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил и
-СО-гетероарил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной, алкенильной, галогеновой, алкоксильной, амино-, алкиламино-, диалкиламино- и алкилтиольной групп, а
каждый заместитель R5 представляет собой независимо атом водорода или
C1-10 алкильную группу;
или соль фармацевтического качества на основе этих групп.
Другой класс промежуточных соединений имеет формулу III
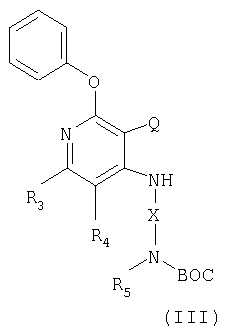
где Q представляет собой NO2 или NH2 группы;
Х - алкиленовая или алкениленовая группа;
заместители R3 и R4 независимым образом выбраны из группы, состоящей из алкильной, алкенильной, галогеновой, алкоксильной, амино-, алкиламино-, диалкиламино- и алкилтиольной группировок, а каждый заместитель R5 представляет собой независимо либо атом водорода или С1-10 алкильную группу;
или соль фармацевтического качества на основе этих групп.
Еще один класс промежуточных соединений имеет формулу (IV)
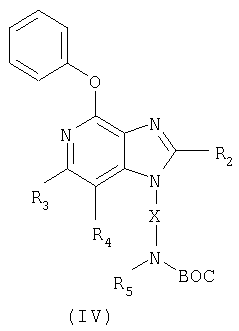
где Х - алкиленовая или алкениленовая группа;
заместитель R2 выбран из группы, включающей
-атом водорода;
-алкил;
-алкенил;
-алкил-O-алкил;
-алкил-S-алкил;
-алкил-O-арил;
-алкил-S-арил;
-алкил-O-алкенил;
-алкил-S-алкенил, а также
- алкил или алкенил, содержащие один или большее количество заместителей, выбранных из группы, включающей
-ОН;
-галоген
-N(R5)2;
-CO-N(R5)2;
-CS-N(R5)2;
-SO2-N(R5)2;
-NR5-CO-C1-10алкил;
-NR5-CS-C1-10алкил;
-NR5-SO2-C1-10алкил;
-CO-C1-10алкил;
-CO-O-C1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил и
-СО-гетероарил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной, алкенильной, галогеновой, алкоксильной, амино-, алкиламино-, диалкиламино- и алкилтиольной групп, а
каждый заместитель R5 представляет собой независимо атом водорода или C1-10 алкильную группу;
или соль фармацевтического качества на основе этих групп.
Другой класс промежуточных соединений имеет формулу (V)
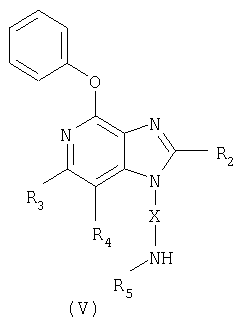
где Х - алкиленовая или алкениленовая группа;
заместитель R2 выбран из группы, включающей
-атом водорода;
-алкил;
-алкенил;
-алкил-O-алкил;
-алкил-S-алкил;
-алкил-O-арил;
-алкил-S-арил;
-алкил-O-алкенил;
-алкил-S-алкенил, а также
- алкил или алкенил, содержащие один или большее количество
заместителей, выбранных из группы, включающей
-ОН;
-галоген;
-N(R5)2;
-CO-N(R5)2;
-CS-N(R5)2;
-SO2-N(R5)2;
-NR5-CO-C1-10алкил;
-NR5-CS-C1-10алкил;
-NR5-SO2-C1-10алкил;
-CO-C1-10алкил;
-СО-О-С1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил и
-СО-гетероарил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной, алкенильной, галогеновой, алкоксильной, амино-, алкиламино-, диалкиламино- и алкилтиольной групп, а
каждый заместитель R5 представляет собой независимо или атом водорода или C1-10 алкильную группу;
или соль фармацевтического качества на основе этих групп.
И, наконец, последний класс промежуточных соединений, представленных в настоящем изобретении, имеет формулу (VI)
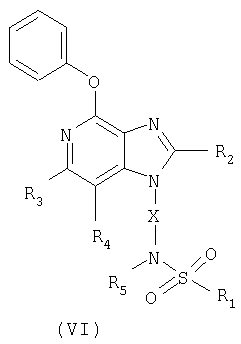
в которой Х - алкиленовая или алкениленовая группа;
R1 представляет собой арильную, гетероарильную, гетероциклильную, C1-20-алкильную или С2-20-алкенильную группы, причем каждая из этих групп может быть незамещенной или содержать один или большее количество заместителей, независимым образом выбранных из группы, включающей
-алкил;
-алкенил;
-арил;
-гетероарил;
-гетероциклил;
- замещенный циклоалкил;
-O-алкил;
-O-(алкил)0-1-арил;
-O-(алкил)0-1-гетероарил;
-O-(алкил)0-1-гетероциклил;
-СООН;
-СО-O-алкил;
-СО-алкил;
-S(O)0-2-алкил;
-S(O)0-2-(алкил)0-1-арил;
-S(O)0-2-(алкил)0-1-гетероарил;
-S(O)0-2-(алкил)0-1-гетероциклил;
-(алкил)0-1-N(R5)2;
-(алкил)0-1-NR5-СО-O-алкил;
-(алкил)0-1-NR5-СО-алкил;
-(алкил)0-1-NR5-СО-арил;
-(алкил)0-1-NR5-СО-гетероарил;
-N3;
-атом галогена;
-галоалкил;
-галоалкоксил;
-СО-галоалкил;
-СО-галоалкоксил;
-NO2;
-CN;
-ОН;
-SH, а в случае алкильной, алкенильной и гетероциклильной групп, также оксогруппа;
заместитель R2 выбран из группы, включающей:-атом водорода;
-алкил;
-алкенил;
-алкил-O-алкил;
-алкил-S-алкил;
-алкил-O-арил;
-алкил-S-арил;
-алкил-O-алкенил;
-алкил-S-алкенил, а также
- алкил или алкенил, содержащие один или большее количество заместителей, выбранных из группы, включающей
-ОН;
-галоген
-N(R5)2;
-CO-N(R5)2;
-CS-N(R5)2;
-SO2-N(R5)2;
-NR5-CO-C1-10алкил;
-NR5-CS-C1-10алкил;
-NR5-SO2-C1-10алкил;
-CO-C1-10алкил;
-CO-O-C1-10алкил;
-N3;
-арил;
-гетероарил;
-гетероциклил;
-СО-арил и
-СО-гетероарил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной, алкенильной, галогеновой, алкоксильной, амино-, алкиламино-, диалкиламино- и алкилтиольной групп, а
каждый заместитель R5 представляет собой атом водорода или C1-10алкильную группу;
или соль фармацевтического качества на основе этих групп.
Используемые здесь термины «алкил», «алкенил» и приставка «алк-» включают как линейные, так и разветвленные цепные группы, а также циклические группы, например циклоалкильные и циклоалкенильные группы. Если не указано иначе, эти группы содержат от 1 до 20 атомов углерода, а алкенильные группы - от 2 до 20 атомов углерода. Предпочтительно эти группы содержат до 10 атомов углерода. Циклические группы могут быть либо моноциклическими, либо полициклическими и преимущественно содержать в цикле от 3 до 10 атомов углерода. Примерами циклических групп являются циклопропильная, циклопропилметильная, циклопентильная, циклогексильная и адамантильная группы.
Термин «галоидоалкил» включает группы, которые в качестве заместителя имеют один или большее количество галоидных атомов, включая перфторированные группы. Такое определение относится также и группам, которые включают приставку «гало». Примерами подходящих галоидоалкильных групп являются хлорметильная, трифторметильная и подобные группы.
Термин «арил», используемый в данном описании, включает в себя карбоциклические ароматические циклы или циклические системы. Примеры арильных групп включают фенильную, нафтильную, бифенильную, флуоренильную и инденильную группы. Термин «гетероарил» относится к ароматическим циклам или циклическим системам, содержащим в кольце, по крайней мере, один гетероатом (например, О, S, N). Подходящие гетероарильные группы включают фурильную, тиенильную, пиридильную, хинолинильную, изохинолильную, индолильную, изоиндолильную, триазольную, пирролильную, тетразолильную, имидазолильную, пиразолильную, оксазолильную, тиазолильную, бензофуранильную, бензотиофенильную, карбазолильную, бензоксазолильную, пиримидинильную, бензимидазольную, хиноксалинильную, бензотиазолильную, нафтиридинильную, изоксазолильную, изотиазолильную, пуринильную, хиназолинильную и подобные группы.
В состав «гетероциклильных» соединений входят неароматические циклы или циклические системы, содержащие в кольце, по крайней мере, один гетероатом (например, О, S, N). К этим соединениям относятся все полностью насыщенные и частично ненасыщенные производные указанных выше гетероарильных групп. Примерами гетероциклических групп являются пирролидинильная, тетрагидрофуранильная, морфолинильная, тиоморфолинильная, пиперидинильная, пиперазинильная, тиазолидинильная и имидазолидинильная группы.
Арильная, гетероарильная и гетероциклильная группы могут быть как незамещенными, так и содержащими один или более заместителей, независимо выбранных из группы, содержащей алкильную, алкоксильную, алкилтионильную, галоидоалкильную, галоидоалкоксильную, галоидоалкилтионильную группы, атом галогена, нитрильную, гидроксильную, меркапто- и цианогруппы, карбоксильную, формильную, арильную, арилоксильную, арилтионильную, арилалкоксильную, арилалкилтионильную, гетероарильную, гетероарилоксильную, гетероарилтионильную, гетероарилалкоксильную, гетероарилалкилтионильную, амино-, алкиламино-, диалкиламино-, гетероциклильную, гетероцикпоалкильную, алкилкарбонильную, алкенилкарбонильную, алкоксикарбонильную, галоалкилкарбонильную, галоалкоксикарбонильную, алкилтиокарбонильную, арилкарбонильную, гетероарилкарбонильную, арилоксикарбонильную, гетероарилоксикарбонильную, арилтиокарбонильную, гетероарилтиокарбонильную, алканоилоксильную, алканоилтионильную, арилкарбонилоксильную, арилкарбонилтионильную, арилкарбониламино-, алкиламиносульфонильную, алкилсульфонильную, арилсульфонильную, гетероарилсульфонильную, арилдиазинильную, алкилсульфониламино-, арилсульфониламино-, арилалкилсульфониламино-, алкилкарбониламино-, алкенилкарбониламино-, арилкарбониламино, арилалкилкарбониламино-, гетероарилкарбониламино-, гетероарилалкилкарбониламино-, алкилсульфониламино-, алкенилсульфониламино-, арилсульфониламино-, арилалкилсульфониламино-, гетероарилсульфониламино-, гетероарилалкилсульфониламино-, алкиламинокарбониламино-, алкениламинокарбониламино-, ариламинокарбониламино-, арилалкиламинокарбониламино-, гетероариламинокарбониламино-, гетероарилалкилкарбониламиногруппы; кроме того, в случае гетероциклильных групп - оксогруппы. В том случае, когда любые другие группы идентифицированы как «замещенные» или «иногда замещенные», эти группы также могут иметь в качестве заместителя одну или большее количество перечисленных выше группировок.
Обычно лишь определенные заместители являются предпочтительными. Например, в качестве предпочтительных групп Y используют -СО- и -SO2- группы; предпочтительными группами Z являются простая связь или группа -NR5-; предпочтительными заместителями R1 являются C1-4 алкильная, арильная или замещенная арильная группа. Предпочтительные R2 группы включают алкильные группы, содержащие от 1 до 4 атомов углерода (т.е., метильная, этильная, пропильная, изопропильная, н-бутильная, втор-бутильная, изобутильная и трет-бутильная группы), метоксиметильную, этоксиметильную и циклопропилметильную группы. Предпочтительным заместителем R3 и R4 является метильная группа. Один или большее количество этих предпочтительных заместителей, если они вообще присутствуют, может содержаться в предлагаемых в изобретении соединениях в любых комбинациях.
Данное изобретение включает описанные выше соединения в любой их фармацевтически доступной форме, включая изомеры, такие как диастереомеры и энантиомеры, соли, сольваты, полиморфные формы и т.п. В частности, если соединение является оптически активным, в изобретении описаны каждый энантиомер этого соединения, а также рацемическая смесь энантиомеров.
Фармацевтические составы и биологическая активность
Предлагаемые в изобретении фармацевтические составы содержат терапевтически эффективные количества описанных выше соединений в сочетании с фармацевтически приемлемым носителем.
Используемый здесь термин «терапевтически эффективное количество» означает количество соединения, достаточное для оказания терапевтического эффекта, такого как стимулирование образования цитокина, противоопухолевое действие и/или противовирусное действие. Хотя точное количество активного вещества, используемого в фармацевтическом составе, предлагаемом в изобретении, широко варьируется в зависимости от различных факторов, таких как физическая и химическая природа вещества, а также природа носителя и предполагаемая доза, полагают, что предлагаемые в изобретении составы будут содержать достаточное количество активного ингредиента, чтобы обеспечить дозу в пределах от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно от приблизительно 10 мкг/кг до приблизительно 5 мг соединения на 1 кг веса пациента. Составы могут использоваться в любых обычных лекарственных формах, таких как таблетки, лепешки, парентеральные составы, сиропы, кремы, мази, аэрозольные составы, различного рода пластыри и т.п.
Предлагаемые в изобретении соединения могут вводиться в организм в виде одного прописанного в рецепте терапевтического агента или в виде комбинации с одним или большим количеством других активных агентов, таких как дополнительные модификаторы иммунной реакции, противовирусные препараты, антибиотики и т.п.
Установлено, что предлагаемые в изобретении соединения стимулируют образование определенных цитокинов в опытах, выполненных в соответствии с приведенными ниже способами испытания. Эти результаты показывают, что предлагаемые соединения представляют собой полезные модификаторы иммунной реакции, т.е. они способны модулировать иммунную реакцию различными способами, тем самым помогая лечению различных заболеваний.
Цитокины, образующиеся при введении предлагаемых в данном изобретении соединений, обычно включают интерферон-α (IFN-α) и/или фактор-α некроза опухоли (TNF-α), а также определенные интерлейкины (IL). В частности, эти соединения вызывают образование IFN-α, TNF-α, IL-1, IL-6, IL-10 и IL-12, а также различных других цитокинов. Помимо других эффектов цитокины замедляют размножение вирусов и рост опухолевых клеток, что делает предлагаемые соединения полезными при лечении опухолей и вирусных заболеваний. Таким образом, настоящее изобретение предоставляет способ стимулирования биосинтеза цитокина при введении в организм животного эффективных количеств соединения или составов, предлагаемых в изобретении.
Установлено, что определенные предлагаемые в изобретении соединения преимущественно стимулируют участие IFN-α в изменении численности кроветворных клеток, таких как РВМС (одноядерные клетки периферийной крови), содержащих pDC2 клетки (предшественников дендритных клеток типа 2), не вызывая значительного воспалительного цитокинеза.
Помимо присущей им способности стимулировать образование цитокинов эти соединения оказывают влияние и на другие аспекты врожденной иммунной реакции. Например, может быть стимулирована естественная активность клетки-убийцы, причем этот эффект может быть обусловлен образованием цитокина. Соединения могут также активировать макрофаги, которые, в свою очередь, стимулируют секрецию оксида азота и образование дополнительных цитокинов. Кроме того, эти соединения могут вызывать разрастание и дифференциацию В-лимфоцитов.
Предлагаемые в изобретении соединения оказывают также влияние на приобретенную иммунную реакцию. Например, хотя, как полагают, соединения не оказывают непосредственного влияния на Т-лимфоциты или на образование Т-лимфоцитных цитокинов, эти соединения оказывают косвенное влияние на образование цитокина IFN-γ из фаг-помощника типа 1 Т-лимфоцита (Th1), а образование Th2 цитокинов IL-4, IL-5 и IL-13 замедляется при введении соединений. Эти результаты показывают, что предлагаемые в данном изобретении соединения оказывают помощь при лечении заболеваний, при которых требуется увеличение количества Th1 и/или уменьшение количества Th2. Учитывая способность соединения замедлять именную реакцию Т-фаг-помощника типа 2, можно ожидать, что эти соединения окажутся полезными при излечении аллергических заболеваний, например атопических дерматитов, астмы, аллергии, аллергических ринитов, системной красной волчанки, а также в качестве вспомогательной вакцины для увеличения иммунитета, а, возможно, и для лечения рецидивов грибковых заболеваний и хламидии.
Модифицирующее воздействие соединений на иммунную реакцию делает их полезными при лечении большого числа различных заболеваний. Благодаря своей способности стимулировать образование цитокинов, таких как IFN-α и/или TNF-α, предлагаемые в данном изобретении соединения особенно полезны при лечении вирусных заболеваний и опухолей. Это иммуномодулирующее действие дает основания полагать, что предлагаемые в изобретении соединения могут быть полезными при лечении таких болезней (помимо прочего), как вирусные заболевания, например остроконечная кондилома, обычные бородавки, подошвенные бородавки, гепатит В, гепатит С, простой герпес типа I и типа II, контагиозный моллюск, оспа, особенно натуральная оспа, ВИЧ, цитомегаловирус, вирус варицелла-зостер, риновирус, аденовирус, коронавирус, грипп и парагрипп, интраэпителиальные неоплазии, такие как цервикальная интраэпителиальная неоплазия, человеческая вирусная папиллома и ассоциированные опухоли; грибковые заболевания, например кандидоз, аспергиллез, криптококковые менингиты; заболевания, связанные с появлением новообразований, например базалиома, лейкемия «волосистых» клеток, рак клеток почечного эпителия, рак клеток простого сквамозного эпителия, лейкемия миелопоэза, саркома Капоши, множественная миелома, меланома, неходжкинская лимфома, кожная лимфома и другие виды онкологических заболеваний; паразитарные заболевания, например пневмоцистоз, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция, лейшманиоз; бактериальные инфекции, например туберкулез и mycobacterium avium. С помощью предлагаемых соединений можно, кроме того, лечить старческий кератоз, экзему, эозинофилию, эссенциальную тромбоцитаэмию, проказу, множественный склероз, синдром Оммена, дискоидную волчанку, болезнь Боуена и боуеноидный папулез, облысение, ингибирование образования келоидных швов после хирургических операций и других типов постхирургических шрамов. Кроме того, эти соединения могут стимулировать заживление ран, включая хронические раны. Они могут быть полезны также для лечения оппортунистических инфекций и опухолей, которые появляются после подавления иммунитета, например, у онкологических и ВИЧ-больных, а также после операций по пересадке органов.
Эффективным количеством введенного соединения, которое предназначено для стимулирования биосинтеза цитокинов, является такое количество, которое оказывается достаточным, чтобы побудить один или большее число типов клеток, таких как моноциты, макрофаги, дендритные клетки и В-клетки, к образованию одного или большего числа цитокинов, таких как, например, IFN-α, TNF-α, IL-1, IL-6, IL-10 и IL-12, в количестве, превышающем фоновый уровень таких цитокинов. Точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения будет находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Изобретение предлагает также способ лечения вирусной инфекции и онкологических заболеваний у животного путем введения в организм животного эффективного количества предлагаемого в изобретении соединения или составов на его основе. Эффективным количеством соединения для лечения или подавления вирусной инфекции является такое его количество, которое будет вызывать уменьшение одного или большего числа симптомов вирусной инфекции, таких как вирусные поражения, вирусная нагрузка, скорость образования вирусов и их смертность по сравнению с ситуацией, наблюдаемой для контрольных животных, не принимающих эти соединения. Точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения должна находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг. Эффективным количеством соединения для лечения онкологического заболевания является такое его количество, которое будет вызывать уменьшение размера опухоли или числа опухолей. И в этом случае точное количество соединения будет изменяться в широких пределах в зависимости от различных факторов, но ожидается, что доза этого соединения должна находиться в интервале от приблизительно 100 нг/кг до приблизительно 50 мг/кг, предпочтительно в интервале между приблизительно 10 мкг/кг до приблизительно 5 мг/кг.
Предлагаемое изобретение иллюстрируется ниже различными примерами. Эти примеры приведены только в качестве иллюстрации и ни в коей мере не ограничивают общие рамки изобретения.
Пример 1
N-[4-(4-Амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]бензамид
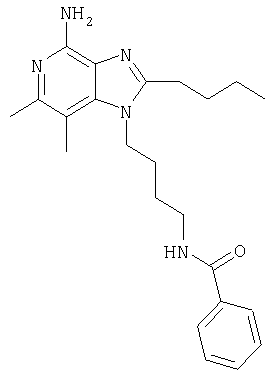
Часть А
К суспензии 4-гидрокси-5,6-диметил-3-нитро-2(1Н)-пиридона (7,6 г, 41,2 ммоля) в 200 мл дихлорметана добавляют 16,8 мл триэтиламина (123,8 ммоля). Полученную смесь охлаждают на ледяной бане, добавляют к ней 13,7 мл (82,5 ммоля) трифторметануксусной кислоты и перемешивают в течение 30 минут. После этого к смеси добавляют одной порцией 7,6 г (41,2 ммоля) моно-трет-бутилкарбонил-1,4-бутиламина и дают ей нагреться до комнатной температуры. Реакцию продолжают в течение 1 часа, а затем смесь дважды промывают 1%-ным водным раствором карбоната натрия (порциями по 100 мл), сушат над сернокислым магнием и концентрируют при пониженном давлении. Полученный неочищенный продукт растворяют в дихлорметане и раствор выливают в колонку, заполненную силикагелем. Силикагель элюируют сначала дихлорметаном для удаления из продукта некоторых примесей, а затем 2-5%-ным раствором этилацетата в дихлорметане для выделения целевого продукта. Фракции, содержащие целевой продукт, собирают вместе и концентрируют при пониженном давлении до получения 12 г 4-({4-[(трет-бутилкарбонил)амино]бутил}амино)-5,6-диметил-3-нитропиридин-2-ил-трифторметансульфоната в виде светло-желтого маслообразного продукта.
Часть В
Продукт, полученный в части А, смешивают с 2,5 г (24,7 ммоля) триэтиламина, 4,8 г (24,7 ммоля) дибензиламина и 150 мл толуола и затем кипятят с обратным холодильником в течение 4 часов. После этого реакционную смесь промывают 1%-ным водным раствором карбоната натрия и концентрируют при пониженном давлении. Полученный неочищенный продукт растворяют в дихлорметане и раствор выливают в хроматографическую колонку, заполненную силикагелем. Силикагель элюируют 2-20%-ным раствором этилацетата в дихлорметане. Фракции, содержащие продукт, собирают вместе и концентрируют при пониженном давлении до получения около 13 г трет-бутил-4-{[2-(дибензиламино)-5,6-диметил-3-нитропиридин-4-ил]амино}бутилкарбамата.
Часть С
К раствору гидрата хлористого никеля (2,9 г, 12,3 ммоля) в метаноле медленно добавляют 1,4 г (36 ммоля) борогидрида натрия и перемешивают полученную смесь в течение 30 минут. После этого к смеси одной порцией добавляют метанольный раствор продукта, полученного в части В. К смеси медленно добавляют борогидрид натрия до тех пор, пока образующаяся пена не становится бесцветной. Смесь фильтруют и фильтрат концентрируют при пониженном давлении. К полученному остатку добавляют дихлорметан и образовавшуюся соль удаляют из раствора фильтрованием. Фильтрат концентрируют при пониженном давлении до получения около 12 г трет-бутил-4-{[3-амино-2-(дибензиламино)-5,6-диметилпиридин-4-ил]амино}бутилкарбамата.
Часть D
К раствору продукта, полученного в части С, в 200 мл ацетонитрила добавляют 3 мл (24,7 ммоля) хлорангидрида валериановой кислоты и реакционную смесь перемешивают при температуре окружающей среды. Через определенное время реакционную смесь концентрируют при пониженном давлении и к остатку добавляют этанол и 5 г (49 ммолей) триэтиламина. Эту реакционную смесь кипятят с обратным холодильником в течение ночи и затем концентрируют при пониженном давлении. Полученный продукт смешивают с дихлорметаном и водой, при этом происходит перераспределение продукта между водной и органической фазами. Слой дихлорметана отделяют и загружают в хроматографическую колонку с силикагелем. Для вымывания целевого продукта из хроматографической колонки в качестве элюента используют смесь 9:90:1 этилацетата : дихлорметана : метанола. Содержащие целевой продукт фракции собирают и концентрируют при пониженном давлении, в результате чего получают 6,5 г маслообразного трет-бутил-4-[2-бутил-4-(дибензиламино)-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил]бутилкарбамата.
Часть Е
Трифторметануксусную кислоту (16 г, 107 ммолей) добавляют к раствору продукта (6,5 г, 11,4 ммоля), полученного в части D, в 250 мл дихлорметана. Смесь перемешивают в течение ночи, после чего добавляют 50 мл гидроксида аммония и 100 мл воды и перемешивание продолжают еще в течение 30 минут. После этого разделяют водный и органический слои и водный слой экстрагируют 100 мл дихлорметана. Органические фракции объединяют, промывают 1%-ным водным раствором карбоната натрия, рассолом и концентрируют при пониженном давлении. К остатку добавляют 30 мл метанола и смесь перемешивают в течение 30 минут, после чего отфильтровывают. Фильтрат концентрируют при пониженном давлении, к остатку добавляют 1%-ный водный раствор карбоната натрия и вновь перемешивают. Смесь экстрагируют гексаном для удаления органических примесей. Водный слой содержал нерастворимое масло, которое экстрагируют с помощью дихлорметана. Органические фракции объединяли, добавляли к ним сульфат магния, перемешивали в течение 5 минут и фильтровали. Фильтрат концентрируют при пониженном давлении до получения твердого продукта, после перекристаллизации которого из толуола получают 1 г 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина.
Часть F
К раствору 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина (150 мг, 0,5 ммоля) в 150 мл дихлорметана добавляют 0,07 мл (0,5 ммоля) триэтиламина. Реакционную смесь охлаждают на ледяной бане и добавляют к ней 0,07 мл (0,5 ммоля) хлористого бензила, после чего удаляют охлаждающую баню. Смесь дважды промывают водой и концентрируют при пониженном давлении. Оставшийся остаток очищают способом флеш-хроматографии, используя 10%-ный раствор метанола в дихлорметане в качестве элюента. В результате получают маслянистый продукт коричневого цвета. Этот продукт растворяют в минимальном количестве изопропанола и затем к раствору при перемешивании добавляют 55 мг (0,5 ммоля) этансульфоновой кислоты. Реакционную смесь перемешивают при температуре окружающей среды приблизительно в течение 1 часа и затем подвергают кратковременному нагреванию на песочной бане до получения гомогенной системы. Полученному раствору позволяют остыть до температуры окружающей среды и затем охлаждают его на ледяной бане. Образовавшийся осадок отфильтровывают и в результате получают 111 мг кристаллического N-[4-(4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-с]пиридин-1-ил)бутил]бензамида. Температура плавления этого продукта составляет 127,8-128,8°С.
Анализ. Рассчитано для С23Н31N5O, %: C 70,20; Н 7,94; N 17,80. Найдено,%: С 69,82; Н 7,70; N 17,68.
Пример 2
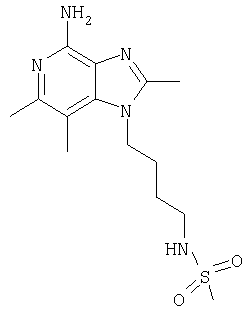
N-[4-(4-Амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]метансульфонамид
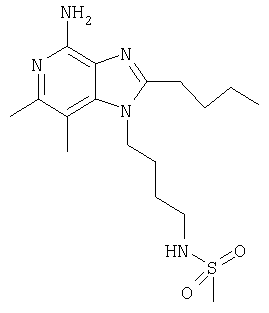
К раствору 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина (150 мг, 0,5 ммоля) в 160 мл дихлорметана добавляют 0,07 мл (0,5 ммоля) триэтиламина и реакционную смесь охлаждают на ледяной бане. К реакционной смеси добавляют 90 мг (0,5 ммоля) ангидрида метансульфоновой кислоты и удаляют смесь из ледяной бани, после чего перемешивают реакционную смесь в течение 35 минут. После этого смесь трижды промывают водой, концентрируют при пониженном давлении и добавляют к остатку минимальное количество метилацетата. Полученный твердый кристаллический осадок отфильтровывают и затем сушат в аппарате Абдерхалдена. В результате получают 94 мг N-[4-(4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-с]пиридин-1-ил)бутил]метан-сульфонамида, температура плавления которого составляет 130-130,5°С.
Анализ. Рассчитано для C17H29N5O2S, %: С 55,56; Н 7,95; N 19,06. Найдено, %: С 55,37; Н 7,89; N 18,03.
Пример 3
Гидрат N-[4-(4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-с]пиридин-1-ил)бутил]-4-фторбензолсульфонамида
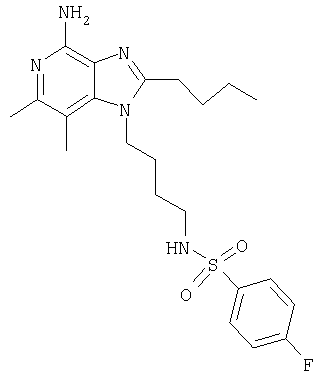
К раствору 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина (150 мг, 0,5 ммоля) в 150 мл дихлорметана добавляют 0,07 мл (0,5 ммоля) триэтиламина и реакционную смесь охлаждают на ледяной бане. К реакционной смеси добавляют 113 мг (0,5 ммоля хлорангидрида фторбензолсульфоновой кислоты и удаляют смесь из ледяной бани. После этого реакционную смесь перемешивают при температуре окружающей среды в течение 48 часов. По окончании перемешивания смесь дважды промывают водой (порциями по 150 мл) и концентрируют при пониженном давлении. Полученный остаток перекристаллизовывают из метилацетата и затем сушат в аппарате Абдерхалдена. В результате получают 50 мг гидрата N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-4-фторбензолсульфонамида в виде белого кристаллического вещества, температура плавления которого составляет 133,1-133,7°С.
Анализ. Рассчитано для C22H30FN5O2S, %: С 56,75; Н 6,93; N 15,04. Найдено, %: С 56,99; Н 6,58; N 15,24.
Пример 4
N-[4-(4-Амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N'-фенилмочевина
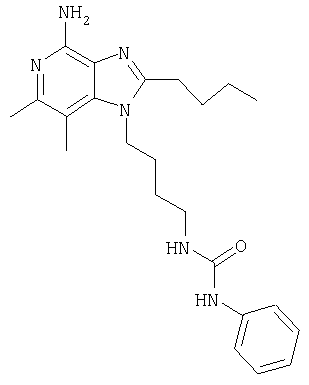
К охлажденному в ледяной бане раствору 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина (150 мг, 0,5 ммоля) в 150 мл дихлорметана добавляют 0,056 мл (0,5 ммоля) фенилизоцианата. Ледяную баню удаляют, через 5 минут после этого образуется белый осадок. Реакционную смесь перемешивают еще в течение 30 минут и затем концентрируют при пониженном давлении до получения беловатого твердого кристаллического вещества. Этот продукт отделяют на фильтре, используя малые количества диэтилового эфира, чтобы перенести продукт на фильтр, и затем сушат в аппарате Абдерхалдена. В результате получают 185 мг N-[4-(4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-с]пиридин-1-ил)бутил]-N'-фенилмочевины, температура плавления которой составляет 195,8-196,8°С.
Анализ. Рассчитано для C23H32N6O, %: С 67,62; Н 7,89; N 20,57. Найдено, %: С 66,84; Н 7,71; N 20,54.
Пример 5
Гидрат N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N'-фенилтиомочевины
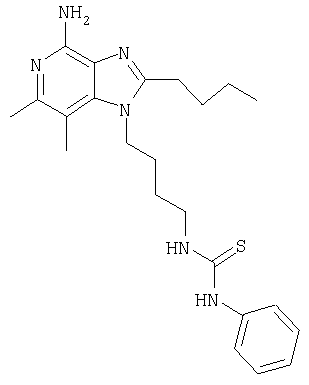
Используя методику, применяемую в примере 4, проводят реакцию 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина (100 мг, 0,35 ммоля) с фенилизотиоцианатом (0,041 мл, 0,35 ммоля), в результате которой получают 97 мг белого твердого кристаллического гидрата N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N'-фенилтиомочевины, температура плавления которого составляет 160,0-160,8°С.
Анализ. Рассчитано для С23Н32N6SH2O, %: С 62,41; Н 7,74; N 18,99. Найдено, %: С 62,39; Н 7,47; N 18,52.
Пример 6
N-[4-(4-Амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N,N-диметилсульфамид
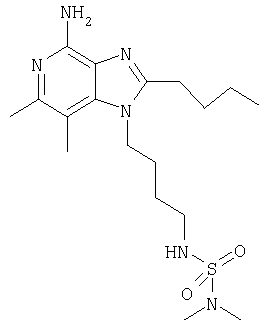
К раствору 1-(4-аминобутил)-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-4-амина (67 мг, 0,23 ммоля) в 45 мл дихлорметана добавляют 0,031 мл (0,23 ммоля) триэтиламина. Реакционную смесь охлаждают на ледяной бане и добавляют к ней 0,025 мл (0,23 ммоля) диметилсульфамоилхлорида. Удаляют ледяную баню и реакционную смесь перемешивают при комнатной температуре в течение приблизительно 113 часов. Согласно данным высокоэффективной жидкостной хроматографии реакция после этого еще не завершена на 100%. Дихлорметан удаляют при пониженном давлении. После этого к остатку добавляют 50 мл 1,2-дихлорэтана и смесь нагревают до 60°С. Через 3 часа добавляют дополнительное количество (2,5 мкл) диметилсульфамоилхлорида и нагревание продолжают. Через 22 часа температуру реакционной смеси доводят до кипения и смесь кипятят с обратным холодильником в течение 100 часов. Смесь дважды экстрагируют водой. Водные фракции соединяют и концентрируют при пониженном давлении. Полученный остаток перекристаллизовывают из метилацетата и получают 10 мг беловатого кристаллического вещества N'-[4-(4-амино-2-бутил-6,7-диметил-1H-имидазо[4,5-с]пиридин-1-ил)бутил]-N,N-диметилсульфамида. Температура плавления этого продукта составляет 129,5-131°С. M/Z=397,1 (М+Н)+.
Пример 7
N-[4-(4-Амино-2,6,7-триметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]метансульфонамид
Часть А
Смесь 5,6-диметил-3-нитропиридин-2,4-диола (60,0 г, 326 ммолей) и 600 мл оксихлорида фосфора кипятят с обратным холодильником в течение 2 часов, после чего концентрируют при пониженном давлении. К остатку добавляют 300 мл этилацетата и смесь отфильтровывают. Фильтрат промывают водным раствором бикарбоната натрия, отделяют водный слой и дважды экстрагируют его этилацетатом. Органические фракции соединяют вместе, сушат над сульфатом магния и концентрируют при пониженном давлении до получения коричневого твердого вещества. Этот материал подвергают хроматографической очистке (силикагель; в качестве элюента - 60/40 этилацетат/гексаны). В результате получают 55 г 2,4-дихлор-5,6-диметил-3-нитропиридина.
Часть В
К смеси 2,4-дихлор-5,6-диметил-3-нитропиридина (50 г, 226 ммолей), безводного N,N-диметилформамида (500 мл) и триэтиламина (50 мл, 339 ммолей) медленно добавляют 60 г (339 ммолей) трет-бутил-4-аминобутилкарбамата. Реакционную смесь перемешивают в течение ночи и затем концентрируют при пониженном давлении до получения маслообразного продукта. Этот продукт растворяют в этилацетате и промывают водой. Органический слой сушат над сульфатом магния и концентрируют при пониженном давлении до получения темного маслянистого продукта. Этот продукт подвергают хроматографической очистке (силикагель; в качестве элюента - 40/60 этилацетат/гексаны). В результате получают 64,5 г трет-бутил-4-(2-хлор-5,6-диметил-3-нитропиридин-4-ил)бутилкарбамата в виде маслообразного продукта ярко-оранжевого цвета. При выдерживании этот продукт затвердевает.
Часть С
Раствор фенола (18,50 г, 196 ммолей) в 50 мл диглима медленно по каплям добавляют к охлажденной до 0°С 60%-ной суспензии в минеральном масле (8,28 г, 207 ммолей) в 50 мл диглима. Через 1 час после окончания добавления реагентов наблюдалось прекращение выделения газа, образующегося в ходе процесса. После этого к реакционной смеси медленно по каплям добавляют раствор трет-бутил-4-(2-хлор-5,6-диметил-3-нитропиридин-4-ил)бутилкарбамата (68,95 г, 185 ммолей) в 200 мл диглима. По завершении добавления реакционную смесь кипятят с обратным холодильником в течение 4 часов, после чего концентрируют при пониженном давлении до получения черного маслообразного продукта. Масло растворяют в этилацетате и затем экстрагируют 1н. раствором гидроксида натрия для удаления избытка фенола. Органический слой сушат над сульфатом магния и концентрируют при пониженном давлении. Полученный остаток очищают на хроматографической колонке (силикагель; в качестве элюента - 30/70 этилацетат/гексаны). В результате получают 40,67 г трет-бутил-4-[(2,3-диметил-5-нитро-6-феноксипиридин-4-ил)]бутилкарбамата в виде маслообразного продукта оранжевого цвета.
Часть D
Смесь трет-бутил-4-[(2,3-диметил-5-нитро-6-феноксипиридин-4-ил)амино]бутилкарбамата (9,17 г, 21,3 ммоля), толуола (50 мл), изопропанола (5 мл) и платины (7,0 г), нанесенной на активированный уголь (содержание платины 5%), выдерживают в течение ночи в аппарате Парра в атмосфере водорода (3,5 кг/см2). После этого катализатор отфильтровывают, а фильтрат упаривают при пониженном давлении. Полученное коричневое масло сушат в вакууме и в результате получают 7,47 г трет-бутил-4-[(3-амино-5,6-диметил-2-феноксипиридин-4-ил)амино]бутилкарбамата.
Часть Е
Смесь продукта, полученного в части D, вместе с триэтилортоацетатом (3,59 мл, 19,58 ммоля), безводным толуолом (75 мл) и солянокислым пиридином (0,75 г) кипятят с обратным холодильником в течение 1 часа и затем концентрируют при пониженном давлении до получения маслообразного продукта коричневого цвета. Масло растворяют в этилацетате и дважды промывают водой, затем рассолом, сушат над сульфатом магния и после этого концентрируют при пониженном давлении. В результате получают 6,74 г трет-бутил-4-[(2,6,7-триметил-4-фенокси-1H-имидазо[4,5-с]пиридин-1-ил)бутилкарбамата, представляющего собой коричневый маслообразный продукт.
Часть F
Раствор трет-бутил-4-[(2,6,7-триметил-4-фенокси-1H-имидазо[4,5-с]пиридин-1-ил)бутилкарбамата (6,70 г, 15,8 ммоля) в 50 мл дихлорметана медленно добавляют к охлажденной до 0°С смеси трифторуксусной кислоты (60 мл) и дихлорметана (100 мл). Реакционную смесь нагревают до температуры окружающей среды и выдерживают при этой температуре в течение ночи. После этого концентрируют полученную смесь при пониженном давлении до коричневого масла. Это масло растворяют в дихлорметане и раствор делают щелочным (доводят рН до 14), добавляя к нему 5%-ный водный раствор гидроксида натрия. Разделяют водный и органический слои и водный слой экстрагируют дихлорметаном. Органические фракции соединяют вместе, сушат над сульфатом магния и затем концентрируют при пониженном давлении. В результате получают 4,50 г 4-(2,6,7-триметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил)бутиламина в виде коричневого масла.
Часть G
Смесь продукта, полученного в части F, вместе с триэтиламином (2,0 мл, 14,6 ммоля) и безводным ацетонитрилом (450 мл) нагревают до тех пор, пока не получают гомогенный раствор. После этого к реакционной смеси медленно добавляют 2,54 г (14,6 ммоля) ангидрида метансульфоновой кислоты. Реакция протекает количественно в течение 10 минут. Реакционную смесь концентрируют при пониженном давлении до получения коричневого маслообразного продукта. Этот продукт растворяют в дихлорметане и промывают 5%-ным водным раствором гидроксида натрия. Водный слой отделяют и экстрагируют дихлорметаном. Органические фракции соединяют вместе, сушат над сульфатом магния и затем концентрируют при пониженном давлении до получения твердого коричневого вещества. После очистки этого вещества хроматографическим способом (силикагель; в качестве элюента используют смесь 95/5 дихлорметан/метанол) получают 4,49 г светло-коричневого твердого вещества, представляющего собой N-[4-(2,6,7-триметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]метансульфонамид.
Часть Н
N-[4-(2,6,7-Триметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]метансульфонамид (4,20 г, 10,4 ммоля) смешивают с ацетатом аммония (42 г) и смесь нагревают в герметичной ампуле при 150°С в течение 36 часов. После этого реакционную смесь охлаждают и растворяют в хлороформе. Полученный раствор экстрагируют 10%-ным водным раствором гидроксида натрия. Водный слой отделяют и несколько раз экстрагируют хлороформом. Органические фракции соединяют вместе, сушат над сульфатом магния и затем концентрируют при пониженном давлении до получения желтого масла. Это масло растворяют в метаноле, соединяют с 1 М раствором хлористого водорода в диэтиловом эфире (10,4 мл). Образующийся на этой стадии реакции осадок отфильтровывают и сушат, после чего растворяют в воде и, добавляя твердый карбонат натрия, доводят рН раствора до 10. Полученный осадок белого цвета отфильтровывают, промывают диэтиловым эфиром и сушат в вакуумной печи при 80°С. В результате получают 2,00 г целевого продукта N-[4-(4-амино-2,6,7-триметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]метансульфонамида, температура плавления которого составляет 228-230°С.
Анализ. Рассчитано для C14H23N5O2S, %: С 51,67; Н 7,12; N 21,52. Найдено, %: С 51,48; Н 6,95; N 21.51.
Пример 8
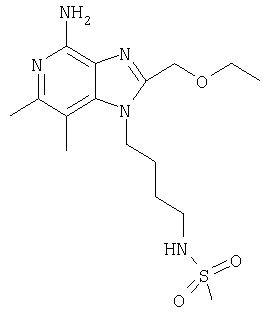
N-{4-[4-Амино-2-(этоксиметил)-6,7-диметил-1H-имидазо[4,5-с]пиридин-1-ил]бутил}метансульфонамид
Часть А
Триэтиламин (3,3 мл, 23,7 ммоля) добавляют к охлажденной до 0°С смеси трет-бутил-4-[(3-амино-5,6-диметил-2-феноксипиридин-4-ил)амино]бутилкарбамата (8,60 г, 21,5 ммоля) и безводного дихлорметана (200 мл). Затем к смеси добавляют 2,76 г (22,5 ммоля) этоксиацетилхлорида, выдерживают смесь в течение 1 часа, нагревают до комнатной температуры и затем перемешивают при этой температуре в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении и получают трет-бутил-4-({3-[(этоксиацетил)амино]-5,6-диметил-2-феноксипиридин-4-ил}амино)бутилкарбамат в виде коричневого маслообразного продукта. К этому маслу добавляют 130 мл пиридина, и смесь кипятят с обратным холодильником в течение ночи, после чего концентрируют до получения коричневого маслообразного продукта. Этот продукт растворяют в дихлорметане и промывают водой. Органический слой сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток растворяют в диэтиловом эфире и вновь концентрируют при пониженном давлении. В результате получают 8,21 г трет-бутил-4-[2-(этоксиметил)-6,7-диметил-4-фенокси-1H-имидазо[4,5-с]пиридин-1-ил]бутилкарбамата.
Часть В
Используя методику, применяемую в части F примера 7, материал, полученный в части А этого примера, подвергают гидролизу, в результате которого получают 5,76 г 4-[2-(этоксиметил)-6,7-диметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил]бутан-1-амина в виде коричневого масла.
Часть С
Используя методику, применяемую в части G примера 7, 4-[2-(этоксиметил)-6,7-диметил-4-фенокси-1H-имидазо[4,5-с]пиридин-1-ил]бутан-1-амин (5,52 г, 15,0 ммолей) обрабатывают ангидридом метансульфоновой кислоты (2,74 г, 15,7 ммоля) и в результате получают 6,26 г N-4-[2-(этоксиметил)-6,7-диметил-4-фенокси-1H-имидазо[4,5-с]пиридин-1-ил]бутил}метансульфонамида в виде твердого вещества коричневого цвета.
Часть D
Используя методику, применяемую в части Н примера 7, проводят аминирование N-{4-[2-(этоксиметил)-6,7-диметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил]бутил}метансульфонамида (5,86 г, 13,1 ммоля), в результате которого получают 1,58 г белого твердого N-{4-(4-амино-2-(этоксиметил)-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил]бутил}метансульфонамида. Температура плавления этого продукта составляет 165-167°С.
Анализ. Рассчитано для C16H27N5O3S, %: С 52,01; Н 7,37; N 18,95. Найдено, %: С 51,83; Н 7,39; N 18,88.
Пример 9
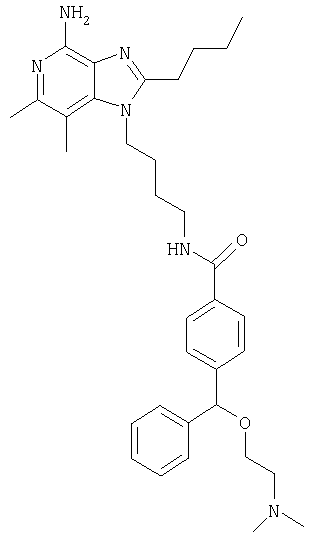
N-[4-(4-Амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-4-[[2-(диметиламино)этокси](фенил)метил]бензамид
Часть А
4-(2-Бутил-6,7-диметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил)бутан-1-амин (122 мг, 0,33 ммоля) в атмосфере азота растворяют в дихлорметане в присутствии триэтиламина (0,093 мл, 0,67 ммоля). Раствор охлаждают на ледяной бане и по каплям добавляют к нему раствор/суспензию 4-[[2-(диметиламино)этокси](фенил)метил]бензоилхлорида (106 мг, 0,33 ммоля) в дихлорметане. Ледяную баню удаляют и реакционную смесь перемешивают в течение 16 часов, после чего выливают ее в 10%-ный водный раствор карбоната натрия. Разделяют водную и органическую фазы и водную фазу экстрагируют дихлорметаном. Органические фракции соединяют вместе, промывают водой и рассолом, сушат над сульфатом натрия, декантируют и упаривают до получения желтого маслообразного продукта. Очистка этого продукта способом флеш-хроматографии (силикагель, градиент элюента от 92:8 дихлорметан/метанол до 95:5 дихлорметан/метанол) дает 101 мг бледно-желтого твердого N-[4-(2-бутил-6,7-диметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-4-[[2-(диметиламино)этокси](фенил)метил]бензамида. Согласно данным высокоэффективной жидкостной хроматографии чистота этого продукта превышает 97%.
MS(Cl): 648 (М+Н).
Часть В
N-[4-(2-Бутил-6,7-диметил-4-фенокси-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-4-[[2-(диметиламино)этокси](фенил)метил]бензамид (101 мг, 0,16 ммоля) и ацетат аммония (1,1 г) помещают в ампулу с мешалкой, предназначенную для работы при высоком давлении. Ампулу герметизируют и в течение 16 часов нагревают при температуре 150°С. Реакционную смесь охлаждают до комнатной температуры и разбавляют водой. Полученную мутную водную смесь подщелачивают 10%-ным водным раствором гидроксида и экстрагируют тремя порциями хлороформа (по 25 мл каждая). Объединенные органические фракции промывают сначала водой и затем рассолом, сушат над сульфатом натрия, декантируют и упаривают до получения желтого маслообразного продукта. Очистка этого продукта способом флеш-хроматографии (силикагель, градиент элюента от 95:5 дихлорметан/метанол до 9:1 дихлорметан/метанол и конечное элюирование смесью 94:5:1 дихлорметан/метанол/триэтиламин)) дает 14 мг желтого маслообразного N-[4-(4-амино-2-бутил-6,7-диметил-4-фенокси-1H-имидазо[4,5-с]пиридин-1-ил)бутил]-4-[[2-(диметиламино)этокси](фенил)метил]бензамида.
1H ЯМР (500 МГц, ДМСО-d6), δ 8,41 (триплет, J=5,5 Гц, 1Н); 7,76 (дублет, J=8,3 Гц, 2Н); 7,43 (дублет, J=8,3 Гц, 2Н); 7,37-7,31 (мультиплет, 4Н); 7,26-7,22 (мультиплет, 1Н); 5,84 (широкий синглет, 2Н); 5,52 (синглет, 1Н); 4,22 (триплет, J=7,7 Гц, 2Н); 3,49 (триплет, J=5,8 Гц, 2Н); 3,29 (двойной дублет, J=6,4, 12,4 Гц, 2Н); 2,76 (триплет, J=7,7 Гц, 2Н); 2,58 (триплет, J=5,7 Гц, 2Н); 2,32 (синглет, 3Н); 2,27 (синглет, 3Н); 2,22 (синглет, 6Н); 1,73-1,65 (мультиплет, 4Н); 1,61-1,55 (мультиплет, 2Н); 1,35 (секстет, J=7,4 Гц, 2Н); 0,86 (триплет, J=7,4 Гц, 3Н).
13С ЯМР (125 МГц, ДМСО-d6): δ 165,9, 153,0, 148,1, 145,4, 142,0, 138,6, 133,5, 128,23, 127,4, 127,3, 127,1, 126,4, 126,1, 124,5, 103,0, 82,0, 66,3, 58,0, 45,2, 43,6, 38,4, 29,3, 28,8, 26,1, 26,0, 21,7, 21,0, 13,6, 12,2.
HRMS(Cl): m/e 571.3763 (M+H), (571.3761 рассчитано для С34H47N6O2, М+Н).
ОБРАЗОВАНИЕ ЦИТОКИНА В КЛЕТКАХ ЧЕЛОВЕКА
Определение активности полученных в изобретении соединений для стимулирования образования цитокина проводили с использованием клеток человека in vitro. Активность оценивали, исходя из количества интерферона (α) и фактора (α) некроза опухоли (IFN и TNF соответственно), определенного по методике, приведенной в статье Тестермана и др. «Индукция кинина с помощью иммуномодуляторов Imiquimod и S-27609», опубликованной в журнале «Journal of Leukocyte Biology», 58, 365-372 (сентябрь, 1995).
Подготовка культуры на основе клеток крови
Цельную кровь собирали венепункцией от здоровых доноров в специальные вакуумные контейнеры, соответствующие требованиям ETDA. Одноядерные клетки периферийной крови (РВМС) отделяли от цельной крови с помощью способа градиентного центрифугирования с использованием аппарата Histopaque®-1077. РВМС дважды промывали раствором сбалансированных солей Ханка (Hank) и затем суспендировали в среде RPMI. Концентрация клеток крови в такой суспензии составляла (3-4)×106 клеток на 1 мл суспензии. Суспензию РВМС помещали в 48 плоскодонных стерильных ячеек (поставляемых компанией Costar, Кэмбридж, шт.Массачусетс, или Becton Dickinson Labware, Линкольн Парк, шт.Нью-Джерси), в которых находились равные объемы среды RPMI, содержащей исследуемое соединение.
Подготовка соединения
Соединения солюбилизировали в диметилсульфоксиде (ДМСО). Конечная концентрация ДМСО в приготовленных образцах, используемых для испытания, не должна превышать 1%. Обычно для испытаний использовали соединения при концентрации в интервале от 0,12 до 30 мкмоль/л.
Инкубация
Сначала в ячейку, в которой находилась среда RPMI, добавляли раствор применяемого для испытания соединения при его концентрации 60 мкмоль/л и затем проводили необходимое троекратное разбавление. После этого добавляли суспензию РВМС в таком количестве, чтобы концентрация исследуемого вещества находилась в заданных пределах (от 0,12 до 30 мкмоль/л). Конечная концентрация суспензии РВМС составляла (1,5-2)×106 клеток/мл. Ячейки закрывали стерильными пластмассовыми крышками, аккуратно перемешивали образцы и выдерживали их в течение 18-24 часов при 37°С в атмосфере, содержащей 5% углекислого газа.
Отделение
После окончания инкубационного периода образцы подвергали центрифугированию в течение 5-10 минут при скорости вращения центрифуги 1000 об/мин (приблизительно 200) и температуре 4°С. Очищенный раствор, не содержащий клеток культуры, удаляли с помощью стерильной полипропиленовой пипетки и переносили в стерильную полипропиленовую ампулу. Образцы до проведения анализа хранили при температуре от -30 до -70°С. Образцы анализировали на интерферон-α и фактор-α некроза опухоли с помощью метода ELISA.
Анализ интерферона (α) и фактора-α некроза опухоли по методу ELISA (иммуноферментный твердофазный анализ)
Концентрацию интерферона (α) определяли по методу ELISA, используя набор человеческих мультичастиц, полученный в PBL биомедицинской лаборатории, Нью-Брунсвик, шт.Нью-Джерси. Этот способ анализа позволяет определить содержание интерферона в единицах пикограмм/мл.
Концентрацию фактора-α некроза опухоли определяли с помощью набора ELISA, поставляемого фирмой Genzyme, Кэмбридж, шт.Массачусетс; RandD Systems, Миннеаполис, шт.Миннесота, или фирмой Pharmingen, Сан-Диего, шт.Калифорния. Этот способ анализа позволяет определить содержание интерферона в единицах пикограмм/мл.
В нижеследующей таблице приведены минимальные концентрации соединений, при которых наблюдается образование интерферона и фактора некроза опухоли. Знак «*» означает, что при любой исследованной концентрации добавленного соединения не наблюдалось образования интерферона и фактора некроза опухоли.
Выше приведено описание лишь нескольких областей применения настоящего изобретения. Подробное описание изобретения и примеры приведены здесь лишь для достижения полной ясности в понимании сути изобретения и областей его применения. Для специалистов в данной области очевиден тот факт, что области применения изобретения могут изменяться с сохранением духа и буквы изобретения. Однако это изобретение не ограничивается только приведенными точными деталями составов и структур, а охватывает все стороны, приведенные в нижеследующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2315049C2 |
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗО-[4,5-с]-ХИНОЛИН-1-ИЛ]-1,1-ДИМЕТИЛЭТИЛ}-МЕТАНСУЛЬФОНАМИД И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2004 |
|
RU2374246C2 |
| ГЕТЕРОЦИКЛИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2351598C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2697090C1 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2304143C2 |
Описывается соединение формулы (I)
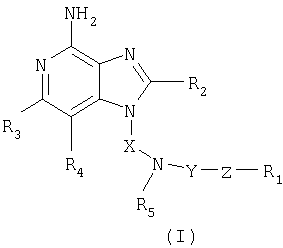
где Х - алкиленовая группа; Y - -СО-, -CS- или -SO2-группа; Z представляет собой простую связь или -NR5-группу; R1 представляет собой фенил незамещенный или замещенный атомом галогена, C1-20-алкильную группу; R2 выбран из алкила, алкил-O-алкила; R3 и R4 представляют собой алкил; R5 представляет собой атом водорода или С1-10алкильную группу. Описываются промежуточные соединения - производные имидазопиридин-4-амина, 2-феноксипиридина, 4-феноксипиридина. Предлагаемые соединения и фармацевтические композиции могут стимулировать биосинтез различных цитокинов и использоваться для лечения вирусных и опухолевых заболеваний. 19 н. и 13 з.п. ф-лы, 1 табл.
где X - алкиленовая группа;
Y - -СО-, -CS- или -SO2- группа;
Z представляет собой простую связь или -NR5-группу;
R1 представляет собой фенил, незамещенный или замещенный атомом галогена, С1-20-алкильную группу,
заместитель R2 выбран из группы, включающей алкил; алкил-O-алкил; заместители R3 и R4 представляют собой алкил; каждый заместитель R5 представляет собой атом водорода или C1-10алкильную группу; или соль фармацевтического качества на основе этих групп.
N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]бензамида;
N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]метансульфонамида;
моногидрата N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-4-фторбензолсульфонамида;
моногидрата N-[-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N'-фенилтиомочевины;
N'-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N,N-диметилсульфамида;
N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-N'-фенилмочевины;
N-[4-(4-амино-2,6,7-триметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-метансульфонамида;
N-[4-(4-амино-2-бутил-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил)бутил]-4-[[2-(диметиламино)этокси](фенил)метил]бензамида и
N-{4-[4-амино-2-(этоксиметил)-6,7-диметил-1Н-имидазо[4,5-с]пиридин-1-ил]бутил}метансульфонамида,
или соли фармацевтического качества на их основе.
где Х - алкиленовая группа;
заместитель R2 выбран из группы, включающей алкил и алкил-O-алкил; заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной группы, и каждый заместитель R5 представляет собой независимо атом водорода или С1-10алкильную группу, или соль фармацевтического качества на основе этих групп.
в которой Q представляет собой NO2 или NH2 группы;
X - алкиленовая группа;
заместители R3 и R4 независимым образом выбраны из алкильной группы;
заместитель R5 представляет собой атом водорода или С1-10алкильную группу;
BOC представляет собой трет-бутоксикарбонильную группу;
или фармацевтически приемлемую соль.
где Х - алкиленовая группа;
заместитель R2 выбран из группы, включающей алкил и алкил-O-алкил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной группы;
каждый заместитель R5 представляет собой атом водорода и C1-10алкильную группу;
ВОС представляет собой третбутоксикарбонильную группу;
или фармацевтически приемлемую соль.
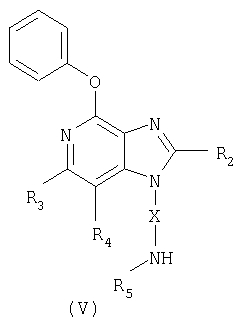
в которой Х - алкиленовая группа;
заместитель R2 выбран из группы, включающей алкил и алкил-O-алкил; заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной группы,
каждый заместитель R5 представляет собой независимо атом водорода или С1-10алкильную группу;
или соль фармацевтического качества на основе этих групп.
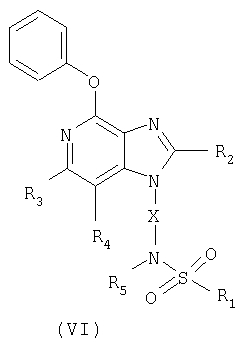
в которой Х - алкиленовая группа;
R1 представляет собой С1-20-алкильную, фенильную группы, или фенил, замещенный атомом галогена;
заместитель R2 выбран из группы, включающей алкил и алкил-O-алкил;
заместители R3 и R4 независимо друг от друга выбраны из группы, состоящей из алкильной группы;
каждый заместитель R5 представляет собой атом водорода или С1-10алкильную группу;
или соль фармацевтического качества на основе этих групп.
| US 5494916 А, 27.02.1996 | |||
| WO 9215582 A1, 17.09.1992 | |||
| US 5389640 A, 14.02.1995 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-ХЛОР-3-НИТРОХИНОЛИНАМИНОВ, СПОСОБ ПОЛУЧЕНИЯ 2-ХЛОР-3,4-ДИАМИНОХИНОЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 4-ХЛОР-1H-ИМИДАЗО (4,5-C)-ХИНОЛИНОВ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 1H-ИМИДАЗО (4,5-C)ХИНОЛИНОВ, 2-ХЛОР-3-НИТРОАМИНОХИНОЛИН, 2-ХЛОР-3,4-ДИАМИНОХИНОЛИН | 1990 |
|
RU2083563C1 |

