Настоящее изобретение относится к соединениям, которые могут использоваться при профилактике и при конструктивном лечении эндотоксинного воздействия, включая сепсис, септицемию, эндотоксикоз и различные формы септического шока.
Предпосылки изобретения
Настоящее изобретение относится к аналогам липида А, которые используются в качестве ингибиторов энодотоксикоза.
Установлено, что частота появления такого заболевания, как бактеремия, в Соединенных Штатах составляет приблизительно 100000-300000 случаев в год, при коэффициенте смертности 30-60%. В качестве первичной химиотерапии против этого заболевания обычно используют антибиотики; однако их бактерицидное действие может привести, в результате, к разрушению бактерий,к сопровождающему этот процесс высвобождению эндотоксина, т. е. липополисахаридного (ЛПС) фрагмента внешней мембраны бактерий. Освобожденный ЛПС вызывает ряд патофизиологических явлений у млекопитающих (которые все относят к грамотрицательному эндотоксикозу или септическому синдрому). Эти явления включают лихорадочное состояние, разлитое воспаление, диссеминированное внутрисосудистое свертывание (ДВС), гипотензию, острую почечную недостаточность, острый респираторный дистресс-синдром (ОРДС), гепатоцеллюлярную деструкцию и сердечную недостаточность.
Хотя эндотоксин и вызывает септический шок, он обладает незначительным или непрямым действием на ткани; на самом деле, он дает начало иммунобиологической реакции, ведущей к каскаду выделения цитокинов, таких как фактор некроза опухоли (ФНО), интерлейкин-1, интерлейкин-6 и интерлейкин-8, и других биологических медиаторов, таких как оксид азота, а также ряда вторичных медиаторов (например, простагландинов, лейкотриенов, интерферонов, фактора активации тромбоцитов, эндорфинов и колониестимулирующих факторов). Наработка патофизиологических концентраций этих цитокинов и медиаторов воспаления влияет на сосудистый тонус, проницаемость капилляров и агрегацию лейкоцитов и тромбоцитов, вызывая синдром, называемый синдромом системной воспалительной реакции (или ССВР), и септический шок.
Молекула бактериального липополисахарида имеет три основные области: длинноцепочечный полисахарид (О-антиген), область ядра и область липида А. Полная молекула липополисахарида, как и некоторые ее отдельные компоненты, обладает описанным выше токсическим действием. Полагают, однако, что большую часть такого токсического действия можно приписать части липида А. Структурно липид А состоит из дифосфорилированного дисахирида, ацилированного длинноцепочечными жирными кислотами.
Лечение в случае связанных с эндотоксинами заболеваний направлено, главным образом, на регулирование воспалительной реакции. Такое лечение включает кортикостероидное лечение, предлагаемое для ослабления опосредованного эндотоксином повреждения клеточной мембраны и для уменьшения продуцирования некоторых биологических медиаторов; введение антител, рассчитанное на нейтрализацию бактериального ЛПС; лечение средствами подавления гипотензии или налоксоном, который, видимо, блокирует гипотензивные эффекты, связанные с септическим синдромом; и лечение нестероидными противовоспалительными лекарственными средствами, имеющее целью блокирование циклооксигеназ и, за счет этого, уменьшение продуцирования некоторых вторичных медиаторов, таких как простагландины и тромбоксан.
Однако ни одна такая терапия на сегодняшний день не приводит к существенному снижению заболеваемости и смертности, являющихся результатом сепсиса и синдрома септического шока. Таким образом, существует давняя потребность в средствах для конструктивного лечения такого нарушения.
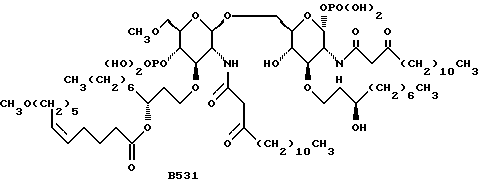
В заявке на патент США, рег. N 07/935050, поданной 25 августа 1992, "Anti-Endotoxin Compounds", Christ, et al., которая включена в настоящее в качестве ссылки, описываются некоторые дисахариды, такие как изображенный ниже В531, используемый при лечении эндотоксикоза.
Некоторые липополисахариды описаны в других литературных источниках, таких как Macher, et al., патент Великобритании 2179945, Meyers, et al., патент Великобритании 2220211, Shiba, et al., европейский патент 172581, Anderson, et al., патент США 4495346, и Shiba, et al., патент США 5066794.
Краткое изложение сущности изобретения
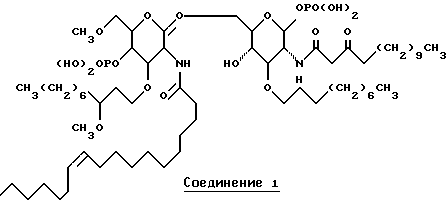
Настоящее изобретение относится к лечению сепсиса, септического шока и родственных нарушений с применением новых липосахаридных аналогов. Соединения по настоящему изобретению обладают такими выгодными для фармацевтического применения свойствами, как повышенная фармакологическая селективность, эффективность и, в особенности, увеличенная устойчивость действия. Представительное соединение по настоящему изобретению - соединение 1 - изображено ниже.
Кроме того, настоящее изобретение относится к профилактике и конструктивному лечению любого опосредованного ЛПС нарушения. Такими нарушениями являются, но не ограничиваются перечисленным, сепсис, септицемия (в том числе, но не только, эндотоксикоз), эндотоксикоз, являющийся результатом грамотрицательной бактериемии (с сопровождающими ее симптомами лихорадки, разлитого воспаления, диссеминированного внутрисосудистого свертывания, гипотензии, острой почечной недостаточности, острого респираторного дистресс-синдрома, респираторного дистресс-синдрома взрослых (ARDS), гепатоцеллюлярной деструкции и/или сердечной недостаточности), и различные формы септического шока (включая, но не ограничиваясь им, эндотоксиновый шок). Соединения по настоящему изобретению также могут быть использованы при профилактике или конструктивном лечении локализованной или системной воспалительной реакции на заражение различными типами микроорганизмов, включая грамотрицательные бактерии, и при заболеваниях, связанных с транслокацией грамотрицательных бактерий или эндотоксина из пищеварительного канала.
Все вместе такие нарушения называются синдромом системной воспалительной реакции или ССВР (обсуждение этих терминов см. в Bone, et al. Chest 1992; 101: 1644-55).
Определения
В соответствии с настоящим изобретением и с использованием при его описании в дальнейшем приводятся значения терминов, если нет других точных указаний.
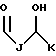
Термин "алкил" относится к алифатическим органическим группам, которые могут быть разветвленными или линейными и которые, необязательно, могут быть замещены одним или несколькими атомами галогена в любом положении алкильной цепи. К алкильным группам относятся как группы, которые имеют одну свободную валентность, например -CH2-CH3, так и алкиленовые группы, которые имеют две свободные валентности, например -CH2-CH2-. Как очевидно для специалистов в этой области техники, одна или две свободные валентности будут использоваться в соответствующем случае для описания соединений, которые химически устойчивы.
Термин "пролекарство", как он используется здесь, относится к любому соединению, которому активность присуща меньше, чем "лекарству", но при введении в биологическую систему оно генерирует "лекарственное" вещество либо в результате спонтанной химической реакции, либо катализированной ферментами или метаболической реакции. Упоминаются различные пролекарства, такие как ацилэфиры, карбонаты, фосфаты и уретаны, включенные сюда в качестве примеров. Иллюстрирующие их группы являются примерами, но не исчерпывающими, и специалист в этой области техники может получить другие известные разновидности пролекарств. Такие пролекарства соединений формулы I входят в объем настоящего изобретения.
Термин "фармацевтически приемлемая соль" относится к солям соединений формулы I, полученным при взаимодействии соединения настоящего изобретения и органической или неорганической кислоты или основания. Соединения формулы I могут использоваться как в неионизированном виде, так и в виде соли. На практике применение соли равнозначно применению в виде основания; оба вида входят в объем настоящего изобретения.
Термин "геометрические изомеры" относится к "транс-" или "цис-" изомерам (или "Е-изомерам", или "Z-изомерам"), как они вообще понимаются специалистами в этой области техники. Все геометрические изомеры входят в объем настоящего изобретения.
Кроме того, соединения по настоящему изобретению могут содержать асимметричные атомы углерода и, следовательно, могут существовать в виде стереоизомеров как энантиомеров, так и диастереомеров. Все стереоизомеры и их смеси рассматриваются как входящие в объем настоящего изобретения. Упомянутые здесь примеры синтеза относятся к наиболее предпочтительному изомеру. Очевидно, что кроме как в сахаристой составляющей, в соединениях формулы I могут присутствовать дополнительные асимметричные атомы углерода, например, в боковых цепях. В таком случае все получающиеся в результате диастереомеры считаются входящими в объем настоящего изобретения.
Краткое описание чертежей
Фиг. 1 иллюстрирует ингибирование выделения α -ФНО соединением 1 с помощью графика ингибирования опосредованной ЛПС индукции фактора нероза опухоли (ФНО) в цельной человеческой крови соединением по настоящему изобретению.
На фиг. 2 приводится общая схема, применяемая при анализе антагонистической эффективности лекарственного средства в цельной крови после инкубации в течение различного времени.
Фиг. 3 отражает зависимость способности испытываемого соединения ингибировать α -ФНО от времени и показывает, что соединение 1 обладает продолжительностью действия в качестве антагониста ЛПС, превосходящей В531. Эти данные являются средними результатами 7 отдельных экспериментов, каждый из которых повторяется трижды.
Подробное описание изобретения
Новые липосахариды
В одном из своих аспектов настоящее изобретение относится к новому применению замещенных липосахаридов, которые включают соединения общей формулы I.
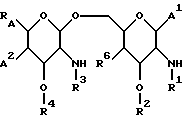
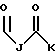
В этой формуле R1 выбран из группы, включающей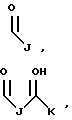
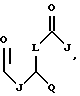
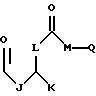
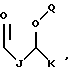
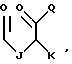
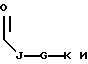
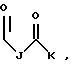
где каждый J, К и Q представляет собой, независимо, линейный или разветвленный C1-C15 алкил; L представляет собой O, NH или CH2; М представляет собой O или NH и G представляет собой NH, О, S, SO или SO2;
R2 является линейным или разветвленным C5 - C15 алкилом;
R3 выбран из группы, включающей линейный или разветвленный C5-C15 алкил,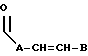
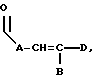
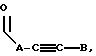
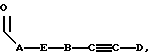
где E представляет собой NH, 0, S, SO или SO2; каждый A, B и D является, независимо, линейным или разветвленным C1-C15 алкилом;
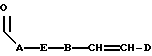
R4 выбран из группы, включающей линейный или разветвленный C4-C20 алкил и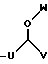
где каждый U и V, независимо, является линейным или разветвленным C2-C15 алкилом, и W представляет собой водород или линейный или разветвленный C1-C5 алкил;
RA представляет собой R5 или R5-O-CH2-, причем R5 выбран из группы, состоящей из водорода, J', -J'-OH, -J'-O-K', -J'-O-K'-OH и -J'-O-PO(OH)2, где каждый J' и К', независимо, представляет собой линейный или разветвленный C1-C5 алкил;
R6 выбран из группы, состоящей из гидрокси, галогена, C1-C5 алкокси и C1-C5 ацилокси;
A1 и A2, независимо, выбраны из группы, включающей
OH,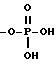
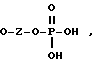
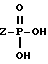
где Z представляет собой линейный или разветвленный C1-C10 алкил;
и их фармацевтически приемлемые соли.
Варианты воплощения приведенной выше формулы включают приведенные ниже значения или их комбинации:
R2 представляет собой линейный или разветвленный C8-C15 алкил;
R2 представляет собой линейный или разветвленный C9-C12 алкил;
R2 представляет собой линейный или разветвленный C10-алкил;
A1 и A2, независимо, являются ОН или -O-РО(ОН)2;
R6 представляет собой гидрокси;
R5 является линейным или разветвленным C1-C5 алкилом;
R1 выбран из группы, включающей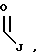
где каждый J, K и Q, независимо, является линейным или разветвленным C1-C15 алкилом;
R3 выбран из группы, включающей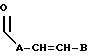
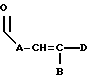
где каждый A, B и D, независимо, является линейным или разветвленным C1-C15 алкилом;
двойные связи R3 являются цис- или Z-;
двойные связи R3 являются транс- или E-;
R4 выбран из группы, включающей линейный или разветвленный C4-C20 алкил и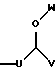
где U является линейным или разветвленным C2-C5 алкилом, V является линейным или разветвленным C5-C12 алкилом и W представляет собой водород или линейный или разветвленный C1-C5 алкил;
RA представляет собой R5; и RA представляет собой R5-O-CH2-.
В других вариантах осуществления изобретения каждый из A1 и A2, независимо, выбраны из группы, состоящей из ОН и -O-PO (ОН)2;
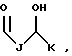
R1 выбран из группы, включающей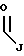
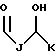
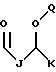
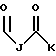
где каждый J, K и Q, независимо, представляет собой линейный или разветвленный C1-C15 алкил;
R2 представляет собой линейный или разветвленный C8-C15 алкил;
R3 выбран из группы, включающей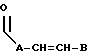
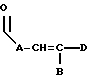
где каждый A, В и D, независимо, является линейным или разветвленным C1-C15 алкилом;
R4 представляет собой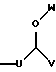
где U является линейным или разветвленным C2-C5 алкилом, V является линейным или разветвленным C5-C12 алкилом и W представляет собой водород или линейный или разветвленный C1-C5 алкил;
и R5 является линейным или разветвленным C1-C5 алкилом; и
R6 представляет собой гидрокси.
В другом варианте осуществления изобретения A1 и A2 представляют собой -O-PO(ОН)2;
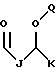
R1 выбран из группы, включающей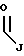
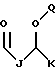
где каждый J и Q, независимо, представляет собой линейный или разветвленный C1-C5 алкил и К представляет собой линейный или разветвленный C8-C15 алкил;
R2 представляет собой линейный или разветвленный C8-C15 алкил;
R3 представляет собой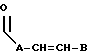
где A является линейным или разветвленным C5-C12 алкилом, а B представляет собой линейный или разветвленный C6-C12 алкил;
R4 представляет собой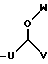
где U является линейным или разветвленным C2-C5 алкилом, V является линейным или разветвленным C5-C12 алкилом и W представляет собой водород или линейный или разветвленный C1-C5 алкил;
R5 является линейным или разветвленным C1-C5 алкилом; и
R6 представляет собой гидрокси.
Еще в одном варианте осуществления изобретения A1 и A2 представляют собой -O-PO(ОН)2;
R4 выбран из группы, включающей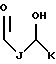
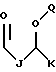
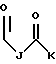
где каждый J и Q, независимо, представляет собой линейный или разветвленный C1-C3 алкил и К представляет собой линейный или разветвленный C10-C12 алкил;
R2 представляет собой линейный или разветвленный C9-C12 алкил;
R3 представляет собой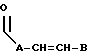
где A является линейным или разветвленным C8-C12 алкилом, а B представляет собой линейный или разветвленный C6-C10 алкил;
R4 представляет собой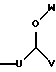
где U является линейным или разветвленным C2-C4 алкилом, V является линейным или разветвленным C5-C10 алкилом и W представляет собой водород или линейный или разветвленный C1-C3 алкил; и
R5 является линейным или разветвленным C1-C3 алкилом; и
R6 представляет собой гидрокси.
Еще в одном варианте осуществления изобретения A1 и A2 представляют собой -O-PO(ОН)2;
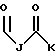
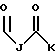
R1 представляет собой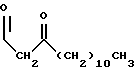
R2 представляет собой (CH2)9CH3;
R3 представляет собой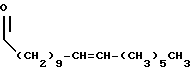
R4 представляет собой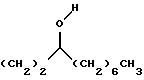
R5 является -CH3; и
R6 представляет собой гидрокси.
Также в объем настоящего изобретения входят соединения, в которых R1 и R3 являются сульфонилами, т.е. соединения, в которых карбонил на этих боковых цепях заменен на SO2. Эти соединения можно получить путем обработки соответствующим образом замещенного спиртового сахара соответствующим алкилсульфонилхлоридом. Таким образом, R1 и R3 также можно выбирать среди следующих групп, в которых A, B, D, E, J, K, L, Q и М имеют установленные выше значения: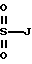
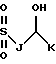
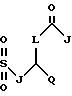
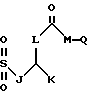
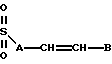
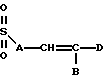
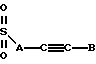
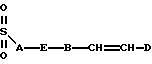
Кроме того, в объем настоящего изобретения входят соединения, в которых ненасыщенной связью в боковой цепи R3 не является двойная или тройная углерод-углеродная связь, а необязательно замещенная ароматическая группа, т. е. соединения, в которых R3 может иметь следующее строение:
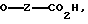
где E представляет собой NH, О, S, SO или SO2; каждый A является линейным или разветвленным C1-C15 алкиленом; D представляет собой линейный или разветвленный C1-C15 алкил; F представляет собой H, -OT, NT1T2, -CO2T или фенил, где каждый из T, T1 и T2 выбраны, независимо, среди водорода или C1-C5 алкила; B представляет собой линейный или разветвленный C1-C15 алкил;
Вообще, предпочтительными являются соединения, в которых R1 выбран из группы, включающей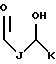
где каждый J, K и Q, независимо, является линейным или разветвленным C1-C15 алкилом;
R2 представляет собой линейный или разветвленный C8-C12 алкил;
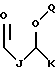
R3 выбран из группы, включающей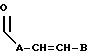
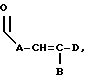
где каждый A, B и D, независимо, является линейным или разветвленным C1-C15 алкилом;
R4 представляет собой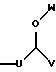
где U является линейным или разветвленным C2-C5 алкилом, V является линейным или разветвленным C4-C10 алкилом, и W представляет собой водород или линейный или разветвленный C1-C5 алкил;
R5 выбран из группы, состоящей из водорода, -J' и -J'-ОН, где J' является линейным или разветвленным C1-C5 алкилом;
R6 выбран из группы, состоящей из гидрокси, галогена и C1-C5 ацилокси;
каждый A1 и A2, независимо, выбраны из группы, включающей OH и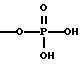
и их фармацевтически приемлемые соли.
Наиболее предпочтительными являются соединения формулы I, в которых
R1 выбран из группы, включающей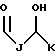
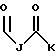
где J представляет собой линейный или разветвленный C1-C5 алкил и K является линейным или разветвленным C9-C14 алкилом;
R2 представляет собой линейный или разветвленный C8-C12 алкил;
R3 представляет собой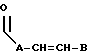
где A представляет собой линейный или разветвленный C6-C12 алкил и B является линейным или разветвленным C4-C8 алкилом;
R4 представляет собой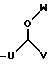
где U является линейным или разветвленным C2-C4 алкилом, V является линейным или разветвленным C5-C9 алкилом и W представляет собой водород или линейный или разветвленный C1-C3 алкил;
R5 является линейным или разветвленным C1-C3 алкилом;
R6 представляет собой гидрокси;
A1 и A2 являются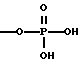
их фармацевтически приемлемые соли.
Общие способы синтеза
Настоящее изобретение также относится к способам получения соединений формулы I. Здесь описываются общие пути синтеза с целью получения различным образом замещенных соединений по настоящему изобретению. Синтез соединения по настоящему изобретению - соединения 1 - описывается ниже.
Большинство реагентов и исходных веществ хорошо известно специалистам в этой области техники. Некоторые реагенты и исходные вещества описаны подробно в заявке на патент США 07/935050, Chist, et al., которая будет опубликована как патент США N 5530113, который включен в настоящее в качестве ссылки.
Один из способов синтеза соединений по настоящему изобретению описан ниже. Хотя в этом примере описывается получение соединения 1, применение других исходных соединений позволит получить другие аналоги настоящего изобретения. Таким образом, этот синтез действительно является общим по характеру.
Например, использование иных алкилирующих агентов на стадии синтеза 22 даст аналоги с заместителями R1, отличающимися по строению.
Характер замещения в R2 регулируется путем применения подходящего алкилирующего агента на стадии 15.
Кроме того, замещение подходящих иных соединений на 25 стадии синтеза даст аналоги, которые отличаются по R3.
Аналоги без окисленной боковой цепи RA можно получить с помощью небольших изменений в схеме синтеза, приведенной ниже, что хорошо известно специалистам в этой области техники. В случае соединения, в котором RA представляет собой метил, например продукт синтеза на стадии 8 - тозилат - может иметь отщепляющуюся группу, замещаемую йодом при реакции Финкельштейна. Иодосодержащие соединения можно дегалогенировать путем обработки металлическим цинком с образованием метильной группы в положении RA.
Характерный синтез боковой цепи RA описан ниже. Получение изменений в этой боковой цепи можно осуществить путем замены исходного вещества на другие подходящие исходные вещества. Например, эту боковую цепь можно получить линейной или разветвленной, исходя из соответствующего исходного вещества. Так, использование различных тозилатов на стадии 6 будет приводить к изменению в R4 (см. схему 1).
Таким образом, коротко описанный ниже синтез обеспечивает универсальный подход к соединениям по настоящему изобретению. (Подробности синтеза см. в приведенных далее экспериментальных примерах, см. схему 2).
Заявители полагают, что показанный выше маршрут - маршрут 1 - является превосходным способом получения соединений по настоящему изобретению благодаря множеству факторов, таких как применение более дешевых исходных веществ, более высокий выход и использование менее токсичных химических агентов, описанный ниже маршрут - маршрут 2 - может быть использован для получения соединений по настоящему изобретению.
Большинство реагентов и исходных веществ хорошо известно специалистам в этой области техники. Некоторые реагенты и исходные вещества для их получения описаны подробно в заявке на патент США 07/935050, Christ, et al., которая включена в настоящее в качестве ссылки. Хотя в этом примере описывается получение соединения 1, использование иных исходных веществ приведет к другим аналогам по настоящему изобретению. Таким образом, указанный синтез действительно является общим.
Например, использование иных алкилирующих агентов при получении промежуточного соединения U даст аналоги с различными по строению заместителями R1.
Картина замещения в R2 регулируется путем использования соответствующего алкилирующего агента при получении промежуточного соединения O.
Кроме того, замещение подходящих иных соединений для промежуточного соединения E при получении промежуточного соединения G будет давать аналоги, которые отличаются в отношении R3.
Пример синтеза боковой цепи R4 описывается ниже. Получение вариаций боковой цепи может достигаться путем замены исходного вещества на другие подходящие исходные вещества. (Подробности синтеза см. в приведенных в схеме 3 экспериментальных примерах).
Характерный пример получения "левой" части описывается в схеме 4.
Характерный пример получения "правой" части соединения 1 описывается в схеме 5.
Две "половинки" молекулы затем соединяют, как описано в схемах 6 и 7, и затем тщательно обрабатывают для получения соединения 1.
Композиции
Аналоги липида A вводят при дозировке, которая обеспечивает соответствующее ингибирование ЛПС-активации клеток-мишеней; как правило, эти дозировки составляют 0,01-50 мг на пациента, предпочтительнее 0,05-25 мг на пациента и наиболее предпочтительно 1-12 мг на пациента. Наиболее предпочтительно вводить дозировки в течение 3 суток в виде непрерывного вливания.
Используемый здесь термин "парентеральный" включает подкожные, внутривенные, внутримышечные и внутриартериальные инъекции с различными методами вливания. Внутриартериальная и внутривенная инъекция здесь включает введение через катетеры. При некоторых показаниях предпочтительными являются способы введения, которые дают возможность быстрого доступа к ткани или органу, которые лечат, например, в случае лечения эндотоксикоза, внутривенные инъекции.
Фармацевтические композиции, содержащие активный ингредиент, могут находиться в любой форме, подходящей для предполагаемого способа введения.
Водные суспензии по настоящему изобретению содержат активные вещества в смеси с добавками, подходящими для изготовления водных суспензий. Такими добавками являются суспендирующий агент, такой как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, и диспергирующие агенты или смачиватели, такие как встречающиеся в природе фосфатиды (например, лецитин), продукты конденсации алкиленоксидов с жирными кислотами (например, полиоксиэтиленстеарат), продукты конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гепта-dea-этиленоксицетанол), продукты конденсации этиленоксида с неполными эфирами, образованными жирными кислотами и гекситолангидридом (например, полиоксиэтиленсорбитанмоноолеат). Водная суспензия также может содержать один или несколько консервантов, таких как этил н-пропил-п-гидроксибензоат.
Фармацевтические композиции по настоящему изобретению находятся, предпочтительно, в форме стерильного, приемлемого для инъекции препарата, такого как стерильная водная или масляная суспензия для инъекций. Такую суспензию можно составить известными в технике методами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые упоминались выше. Стерильный препарат для инъекций может также представлять собой стерильный раствор для инъекций или суспензию в нетоксичном, приемлемом парентерально разбавителе или растворителе, например раствор в 1,3-бутандиоле, или быть полученным в виде лиофилизованного порошка. В число приемлемых носителей и растворителей, которые можно использовать, входят вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, обычно можно использовать в качестве растворителя или суспендирующей среды стерильные нелетучие масла. Для этой цели можно использовать любое мягкое нелетучее масло, в том числе синтетические моно- или диглицериды. Кроме того, при приготовлении композиций для инъекций можно также использовать жирные кислоты, например олеиновую кислоту.
Композиции, пригодные для парентерального введения, включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые придают композиции изотоничность с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут находиться в однодозовых или многодозовых плотно закрытых емкостях, например ампулах и флаконах, и могут храниться в высушенном вымораживанием (лиофилизованном) состоянии, когда требуется только добавить стерильный жидкий носитель, например воду для инъекций, непосредственно перед применением. Приготовленные для немедленного применения растворы и суспензии можно получить из стерильных порошков, описанных ранее.
Следует, однако, иметь в виду, что конкретный уровень дозы для любого конкретного пациента будет зависеть от разных факторов, в том числе от активности конкретно используемого соединения, возраста, массы тела, общего состояния здоровья и пола индивидуума, которого лечат, времени и способа введения, скорости экскреции, других лекарственных средств, которые вводились ранее, и тяжести специфического заболевания, от которого лечат.
Примеры
Далее приводятся примеры применения способа по настоящему изобретению. Соединения по настоящему изобретению и их получение можно представить также с помощью примеров, которые иллюстрируют некоторые процессы, посредством которых эти соединения получают или применяют. Эти примеры не следует истолковывать как определенно ограничивающие изобретение, и вариации изобретения, известные в настоящее время или разработанные впоследствии, считаются входящими в объем настоящего изобретения, определенный далее формулой изобретения.
Соединения по настоящему изобретению обозначаются номерами в соответствии с таблицами 1 и 2.
Химические примеры
Если нет иных указаний, все реакции проводят в инертной атмосфере. При спектральном анализе (например, методом ядерного магнитного резонанса и/или масс-спектроскопии) промежуточные и конечные продукты дают результаты, совместимые с их предполагаемым строением. Протекание реакций контролируют с помощью тонкослойной хроматографии на силикагеле. Препаративную хроматографию, если нет иных указаний, осуществляют на силикагеле.
Получение соединения 1 по пути 1
Все чувствительные к окружающей среде реакции проводят в атмосфере азота и в сухом оборудовании и в качестве осушающего агента используют безводный сульфат натрия, если нет иных указаний. Все продукты дают удовлетворительные спектры ядерного магнитного резонанса.
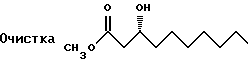
Вещество (5 кг) хроматографируют на силикагеле и элюируют с градиентом гексаном в EtOAc (100-33% гексана). Очищенные фракции объединяют и перегоняют (97-100oC при 0,15 мм рт.ст.). Выход очищенного вещества 4,513 г.
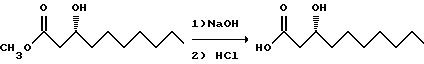
К охлажденному льдом раствору сложного эфира (4500 г, 22,2 моля) в 12,6 л ТГФ добавляют гидроксид натрия (27 молей) в 10,8 л воды. Смесь быстро перемешивают и добавляют 2,5 л конц. соляной кислоты. Слои разделяют и водный слой снова экстрагируют EtOAc. Объединенные органические слои промывают насыщенным солевым раствором, сушат над сульфатом натрия и концентрируют. Продукт постепенно кристаллизуется с образованием 2983 г белого порошка.
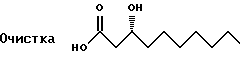
К раствору кислоты (15,8 молей) в 33 л ацетонитрила добавляют дициклогексиламин (16,7 молей). Раствор нагревают до 60oC и оставляют остывать в течение ночи. Кристаллы собирают и промывают дважды растворителем и перекристаллизовывают из ацетонитрила. К суспензии предварительно промытого метанолом амберлита IR-120 Plus (12 кг) в EtOAc (24 л) и воде (24 л) добавляют полученную выше соль. Смесь перемешивают в течение нескольких часов и отделяют органический слой. Водный слой снова экстрагируют EtOAc (12 л) и объединенные органические слои сушат (сульфат натрия) и концентрируют. Получают 2,997 г белого твердого вещества.
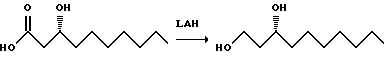
К горячему (~67oC) 1 М раствору алюмогидрида лития (8 л) в ТГФ медленно добавляют раствор кислоты (1 кг) в 4 л ТГФ. Раствор оставляют охлаждаться в течение ночи. Раствор медленно добавляют к 1 М водному раствору HCl (5 л). Смесь экстрагируют толуолом (12 л). Органический слой промывают раствором бикарбоната натрия, сушат (сульфат натрия) и удаляют растворитель в вакууме. Получают сироп, который перегоняют (103oC), и получают 914 г светло-желтого масла.
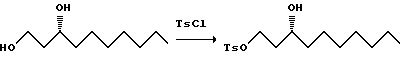
К охлажденному до 0oC раствору диола (913,8 г) в пиридине (3 л) добавляют 3 л триэтиламина, а затем раствор тозилхлорида (1 кг) в пиридине (1,5 л) и триэтиламине (1,5 л). Смесь оставляют нагреваться в течение ночи и выливают в холодный раствор 6 М водного раствора HCl (16 л) и метиленхлорида (8 л). Органический слой отделяют, а водный слой экстрагируют дополнительным количеством метиленхлорида. Объединенные органические слои сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Остаток дважды хроматографируют на силикагеле и элюируют с градиентом гексан: EtOAc (от 9:1 до 1:6), и получают 642 г тозилата.
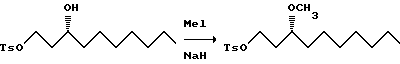
К суспензии 60% дисперсии гидрида натрия в масле (8,68 молей) в 1,15 л ДМФА и 1,1 л ТГФ постепенно добавляют тозилат (1,139 кг) и метилйодид (7,7 кг) в 1,15 л ДМФА и 1,1 л ТГФ. Смесь перемешивают в течение ночи, а затем разбавляют ДМФА (3 л) и постепенно добавляют к насыщенному водному раствору хлорида аммония. Смесь экстрагируют гексаном (8 л), экстракт сушат (сульфат натрия) и удаляют растворитель, и получают оранжевое/коричневое масло. Масло хроматографируют на диоксиде кремния и элюируют с градиентом (гексан: EtOAc, от 100:0 до 6:1), и получают 940 г светло-желтого масла.
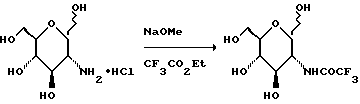
К суспензии аминосахара (1019 г) в 5 л MeOH добавляют 25% раствор NaOMe в MeOH (1080 мл, 5 моль), а затем 610 мл этилтрифторацетата. Смесь перемешивают в течение ночи и удаляют растворитель при пониженном давлении, а остаток обрабатывают изопропанолом. Смесь фильтруют и остаток промывают дополнительным количеством изопропанола, и получают 1369 г продукта.
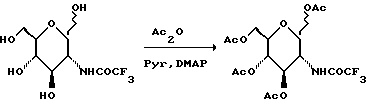
К суспензии оксисахара (1300 г) в пиридине (4 л) добавляют диметиламинопиридин (79 г), а затем уксусный ангидрид (2713 мл). Смесь перемешивают в течение ночи. Растворитель удаляют при пониженном давлении. Добавляют толуол (5х500 мл) и также удаляют при пониженном давлении, получают твердое вещество, которое хроматографируют на диоксиде кремния. Элюирование смесью гексан: EtOAc (1:1) дает 1479 г белого твердого вещества.
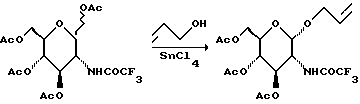
К раствору ацетилированного сахара (1479 г) в 8 л метиленхлорида добавляют аллиловый спирт (764 мл), а затем медленно добавляют тетрахлорид олова (976 мл). Смесь перемешивают в течение ночи и медленно выливают в охлажденную льдом воду (7,5 л). Органический слой отделяют, а водный слой промывают еще раз метиленхлоридом. Объединенные органические слои промывают водным раствором бикарбоната натрия, сушат и концентрируют при пониженном давлении. Остаток хроматографируют на диоксиде кремния (7,5 кг) и элюируют смесью гексан: EtOAc с градиентом (от 4:1 до 1:1), и получают 1327 г бледно-желтого масла.
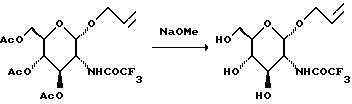
К охлажденному льдом раствору сахара с защитными группами (1322 г) в 8,5 л метанола добавляют в течение одного часа 25% раствор NaOMe в метаноле (437 мл). К этой смеси добавляют 1740 г предварительно промытой смолы амберлит IR-120 Plus. Смесь фильтруют, концентрируют и остаток хроматографируют на диоксиде кремния. Элюирование метанолом дает 907 г продукта.
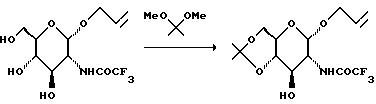
Триол суспендируют в ацетоне (7,5 л) и добавляют камфорсульфоновую кислоту (85 г), а затем медленно добавляют 2,2-диметоксипропан (965 мл). Смесь перемешивают в течение ночи, после чего добавляют триэтиламин (51 мл). Растворитель удаляют при пониженном давлении и получают твердое коричневое вещество, которое хроматографируют на диоксиде кремния. Элюирование смесью гексан: EtOAc с градиентом (от 3:1 до 2:1) дает 842 г не совсем белой смолы.
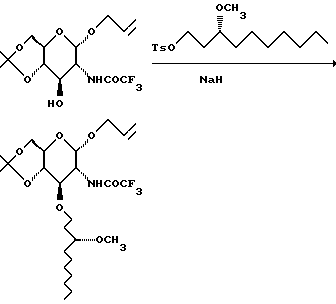
К суспензии 60% дисперсии гидрида натрия в масле (82 г) в 2,2 л ТГФ и 580 мл ДМФА добавляют тозилат (351 г) и раствор свободного спирта (400 г) в смеси 1360 мл ТГФ и 360 мл ДМФА. Смесь перемешивают в течение ночи. Смесь охлаждают на льду и добавляют метанол, а затем воду (2 л). Смесь экстрагируют три раза EtOAc. Объединенные органические слои сушат и концентрируют. Получающуюся в результате смесь хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан: EtOAc (от 19:1 до 1:1) дает 711 г продукта.
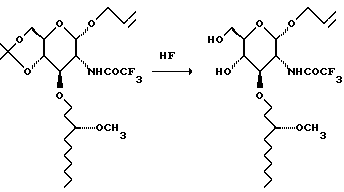
К смеси 48% водной плавиковой кислоты в 1500 мл ацетонитрила в тефлоновой бутыли добавляют раствор исходного вещества (613 г) в 750 мл ацетонитрила и 750 мл метиленхлорида. Смесь перемешивают в течение одного часа и выливают в 8 л воды. Смесь экстрагируют метиленхлоридом (4 х 2 л). Объединенные органические слои промывают водным насыщенным раствором бикарбоната натрия, сушат и концентрируют при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью метиленхлорид: метанол (от 39:1 до 9:1) дает 519 г продукта.
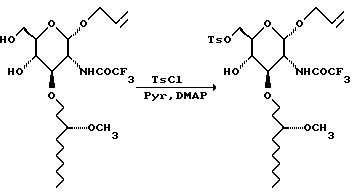
К раствору диола (577 г) в пиридине (5 л) добавляют тозилхлорид (339 г) и N, N-диметиламинопиридин (14,5 г). Смесь перемешивают при комнатной температуре в течение двух суток и затем выливают в 14 л холодной водной 1 М соляной кислоты. Смесь экстрагируют (2х5 л) метиленхлоридом. Объединенные органические слои сушат и концентрируют. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан: EtOAc (от 6:1 до 1:1) дает 632 г желтого сиропа, который при стоянии постепенно кристаллизуется.
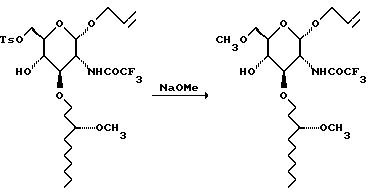
К нагретому до 85oC раствору 25% метилата натрия в метаноле (1825 мл) в ДМФА (1365 мл) в течение 1,25 часа добавляют тозилат (714 г) в ДМФА (1365 мл). Смесь перемешивают 30 минут и охлаждают до 4oC, и выливают ее в охлажденную льдом смесь водной 1 М соляной кислоты и 4,6 кг льда. Смесь перемешивают в течение 30 минут и фильтруют. Фильтрат промывают 2 л воды и объединенные водные слои экстрагируют 2х4 л EtOAc. Объединенные органические слои сушат и концентрируют. Остаток очищают хроматографией на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 3:1 до 1:1) дает 549 г твердого вещества от бледно-желтого до белого цвета.
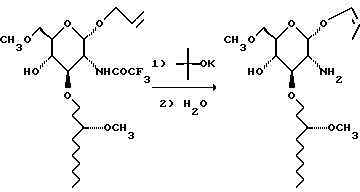
Эту реакцию проводят в атмосфере аргона. К раствору трет-бутилата калия (139 г) в 440 мл ДМСО добавляют раствор сахара (247 г) в 440 мл безводного ДМСО. Смесь нагревают до 85oC в течение 1,5 часов и затем добавляют 250 мл воды. Смесь греют при 85oC в течение ночи и охлаждают на ледяной бане. Смесь выливают в 3,5 л насыщенного солевого раствора и экстрагируют смесь 3х750 мл метиленхлорида. Объединенные органические слои сушат и концентрируют, и получают 560 г коричневого масла.
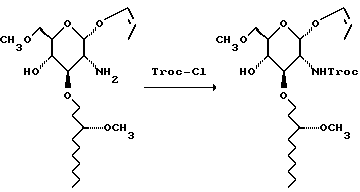
К смеси свободного амина (199 г) в 780 мл ТГФ и 390 мл насыщенного водного раствора бикарбоната натрия добавляют Troc-Cl (157 г). Через 0,5 часа смесь медленно выливают в раствор 500 мл 40% водного метиламина и 3 л воды. Смесь экстрагируют 2 х 1750 мл метиленхлорида. Объединенные органические слои сушат и концентрируют. Остаток хроматографируют на диоксиде кремния. Градиентое элюирование смесью гексан: EtOAc (от 5:1 до 1:1) дает количественный выход - 287 г - твердого вещества от желтого до не совсем белого цвета.
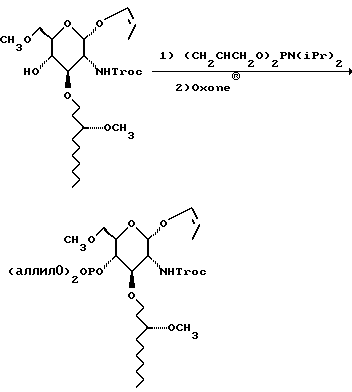
К раствору оксисахара в 2 л метиленхлорида добавляют тетразол (155,6 г), а затем диаллилдиизопропилфосфорамидит (182 мл). Через 0,5 часа смесь выливают в охлажденную льдом смесь Oxone® (пероксимоносульфат калия) (455,6 г), воды (1,25 л) и ТГФ (2,5 л). Через 15 минут смесь выливают в холодный 10% водный раствор тиосульфата натрия. Через 15 минут смесь экстрагируют 2 л метиленхлорида. Органический слой отделяют, водный слой снова экстрагируют метиленхлоридом, объединенные органические слои сушат и удаляют растворитель в вакууме. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 6:1 до 2:1) дает 205,7 г бледно-желтого сиропа.
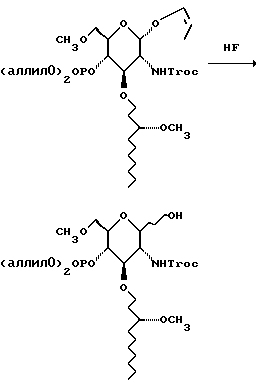
К раствору 400 мл 48% водной плавиковой кислоты в 1,2 л ацетонитрила в тефлоновой емкости добавляют раствор 138,8 г сахара в 500 мл метиленхлорида. Смесь перемешивают в течение ночи, разбавляют 3 л воды и экстрагируют 2,4 л метиленхлорида. Органический слой промывают водным раствором бикарбоната натрия, сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан: этилацетат (от 2: 1 до 1:1) с последующим градиентным элюированием смесью метиленхлорид: метанол (от 19:1 до 9:1) дает 129,2 г воскообразной смолы.
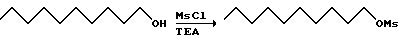
К охлажденному льдом раствору 450 г 1-деканола в 685 мл триэтиламина и 1125 мл метиленхлорида добавляют 330 мл мезилхлорида. Охлаждающую баню удаляют через 1,5 часа и удаляют растворитель при пониженном давлении. К остатку добавляют 2,5 л 1 М соляной кислоты. Эту смесь экстрагируют 3х2 л метиленхлорида. Органические слои объединяют, сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Элюирование смесью гексан:этилацетат (1:1) дает 651 г продукта.
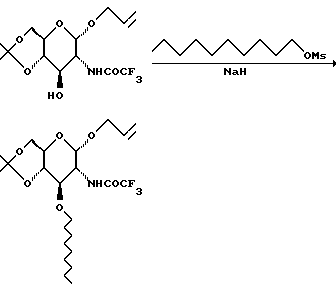
К суспензии 60% дисперсии гидрида натрия в минеральном масле в 1 л ТГФ и 470 мл ДМФД в течение 1 часа добавляют раствор спирта в 280 мл ДМФА и 1 л ТГФ. Затем в течение 15 минут добавляют 470 г мезилата. Через 2 суток добавляют 400 мл метанола, а затем 4 кг льда и 4 л воды. Эту смесь экстрагируют 2х4 л этилацетата. Объединенные слои сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан: EtOAc (от 39:1 до 2:1) дает 618 г продукта.
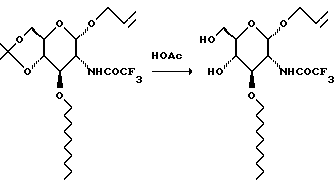
Раствор 520 г сахара в 5,2 л ледяной уксусной кислоты и 1,3 л воды перемешивают в течение ночи. Выливают раствор в 7,5 л воды и фильтруют. Фильтрат сушат азеотропной перегонкой с толуолом (3 х 500 мл) при пониженном давлении и получают 458 г.
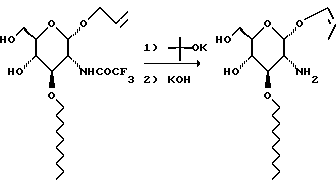
Эту реакцию осуществляют в атмосфере аргона. К суспензии 295 г трет-бутоксилата калия в 1 л ДМСО добавляют раствор 340 г сахара в 1,5 л ДМСО. Смесь нагревают до 85oC в течение 1,25 часа, добавляют 1,4 л 3 М водного раствора гидроксида калия и перемешивают смесь при 85oC в течение ночи. Смесь охлаждают до комнатной температуры и выливают в смесь 3,5 л насыщенного солевого раствора и 3,5 л воды. Смесь экстрагируют три раза метиленхлоридом, сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью метиленхлорид: метанол (от 19:1 до 4:1) дает 740 г продукта.
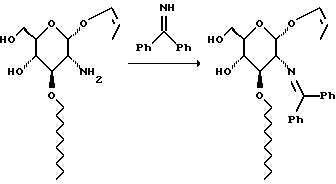
Раствор 740 г аминосахара в 338 г бензофенонимина нагревают при 45oC в течение ночи. Смесь хроматографируют на диоксиде кремния и элюируют смесью гексан: этилацетат с градиентом (от 39:1 до 1:1) и получают 371 г бледно-желтого твердого вещества.
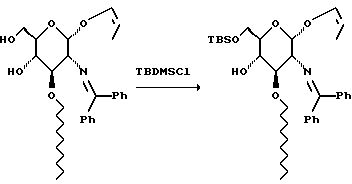
К раствору 366 г диолсахара в 1,3 л ДМФА добавляют 118 г имидазола, а затем 117 г трет-бутилдиметилсилилхлорида. Через 5 минут смесь выливают в 1,4 л водного насыщенного раствора бикарбоната натрия. Смесь экстрагируют три раза этилацетатом. Органические слои объединяют, удаляют растворитель при пониженном давлении и остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 49:1 до 4:1) дает 446 г сиропа.
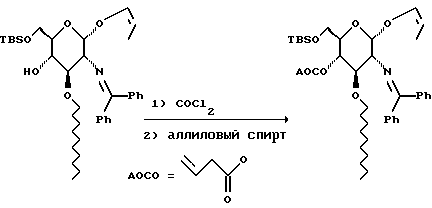
К раствору 437 г спирта в 3 л толуола добавляют 225 мл пиридина и раствор охлаждают на ледяной бане. Добавляют 531 мл 1,9 М раствора фосгена в толуоле и раствор перемешивают в течение 10 минут. Добавляют 469 мл аллилового спирта. Через 40 минут добавляют 2,3 л насыщенного водного раствора бикарбоната натрия и смесь экстрагируют этилацетатом. Органический слой отделяют, сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 49:1 до 4:1) дает 441 г желтого сиропа.
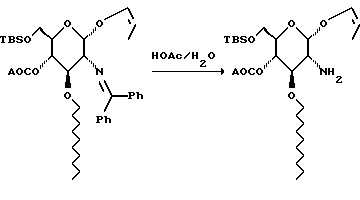
К раствору 431 г сахара в 200 мл ТГФ добавляют 330 мл ледяной уксусной кислоты и 110 мл воды. Смесь перемешивают в течение трех часов, охлаждают на льду и добавляют 6,6 л 1 М водного раствора гидроксида натрия. Смесь экстрагируют метиленхлоридом, 2х2 л. Объединенные органические слои сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью метиленхлорид:метанол (от 19:1 до 4:1) дает 309 г амина в виде сиропа.
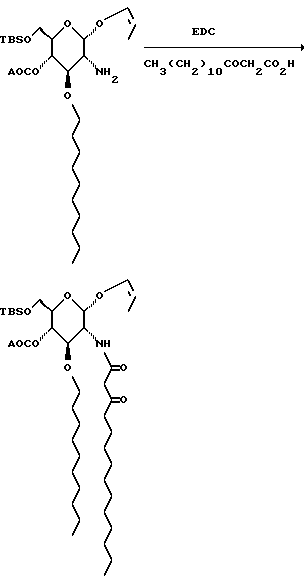
К охлажденному льдом раствору аминосахара, 309 г, в 3 л метиленхлорида добавляют 435 г гидрохлорида 1-(3-диметил- аминопропил)-3-этилкарбодиимида (EDC) и, спустя 10 мин, 275 г
карбоновой кислоты. Через 10 минут смесь экстрагируют насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, водный слой снова экстрагируют метиленхлоридом, объединенные органические слои сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 19:1 до 3:1) дает 338 г бледно-желтого сиропа.
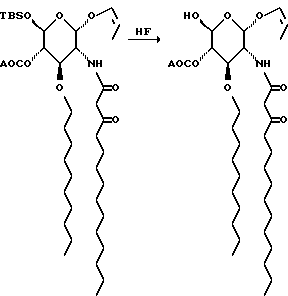
К раствору 11 мл 48% водной плавиковой кислоты в 293 мл ацетонитрила добавляют 4,6 г силикагеля, а затем раствор 146,7 г сахара в 147 мл метиленхлорида. Через полчаса смесь разбавляют 975 мл воды и экстрагируют метиленхлоридом. Органический слой отделяют, а водный слой снова экстрагируют метиленхлоридом. Объединенные органические слои промывают водным раствором бикарбоната натрия, сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан: этилацетат (от 5: 1 до 0:1) дает 110,4 г не совсем белого воскообразного твердого вещества.
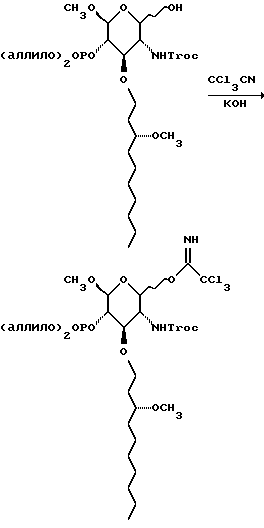
К раствору 129 г сахара в 500 г трихлорацетонитрила добавляют 240 г карбоната калия. Смесь перемешивают в течение получаса и фильтруют через диатомовую землю. Остаток на фильтре промывают метиленхлоридом, фильтраты объединяют и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 1:1 до 0:1) дает 145,7 г желтой смолы.
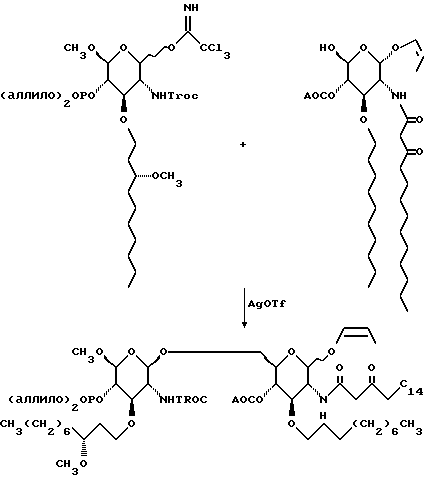
145,7 г сахара, изображенного слева, и 109,2 г сахара, изображенного справа, азеотропно сушат путем испарения толуола (3х200 мл). Раствор двух cахаров в 750 мл метиленхлорида добавляют к охлажденному льдом раствору 62,7 г трифлата серебра в 130 мл метиленхлорида. Смесь нагревают до комнатной температуры и перемешивают в течение ночи. Смесь выливают в смесь насыщенного водного раствора бикарбоната натрия и раствора тиосульфата натрия. Органический слой отделяют, а водный слой промывают метиленхлоридом. Объединенные органические слои сушат и удаляют растворитель при пониженном давлении. Остаток дважды хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан:этилацетат (от 5:1 до 1:1) дает 189,56 г липкой пены.
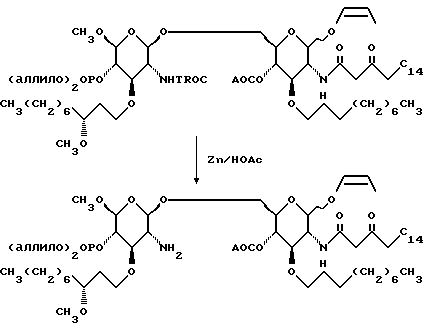
К раствору 188,7 г дисахарида в 590 мл ТГФ добавляют 457,6 г цинковой пыли, а затем 395 мл ледяной уксусной кислоты. Через полчаса смесь фильтруют через диатомовую землю и остаток на фильтре промывают ТГФ. Органические слои объединяют и удаляют растворитель при пониженном давлении. Остаток сушат азеотропно с помощью отгонки растворителя из остатка с бензолом (4х250 мл) и получают 223,1 г розовой смолы.
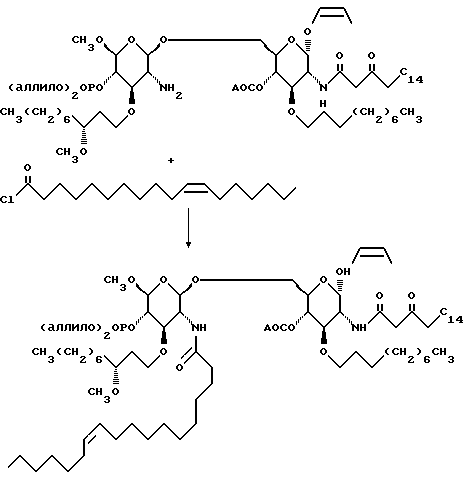
К раствору 223,1 г сахара в 1,3 л ТГФ добавляют раствор 37,5 г бикарбоната натрия в 250 мл воды. Добавляют 67,4 т цис-11- окстадеканоилхлорида. Через 10 минут смесь дважды экстрагируют этилацетатом. Объединенные органические слои сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью гексан: этилацетат (от 2:1 до 0:1) дает 160,2 г бледно-желтого воска.
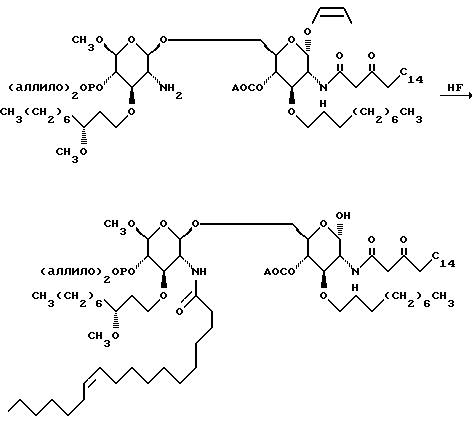
Раствор 161,3 г сахара в 215 мл метиленхлорида добавляют к раствору 150 мл 48% фтористоводородной кислоты в 474 мл ацетонитрила в тефлоновой бутыли. Через четыре часа смесь выливают в 500 мл воды. Смесь дважды экстрагируют метиленхлоридом. Объединенные органические слои промывают насыщенным водным раствором бикарбоната натрия, сушат и удаляют растворитель при пониженном давлении. Остаток хроматографируют на диоксиде кремния. Градиентное элюирование смесью метиленхлорид:этилацетат:метанол (от 500:500:20 до 500:500: 160) дает бледно-желтую воскообразную смолу.
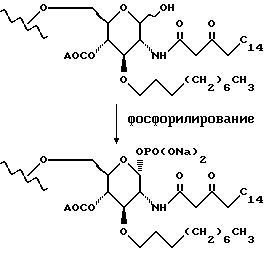
Растворяют 719 мг сахара в метиленхлориде и добавляют сульфат натрия (1,4 г). Добавляют диаллилдиизопропилфосфорамидит (189 мкл) и тетразол (162 мг), смесь перемешивают в течение 10 минут и затем охлаждают до -78oC. Добавляют по каплям раствор м-хлорпероксибензойной кислоты (192 мг) в метиленхлориде (4 мл). Смесь промывают водным раствором тиосульфата натрия и водным раствором бикарбоната натрия, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Остаток хроматографируют и получают 660 мг вещества.
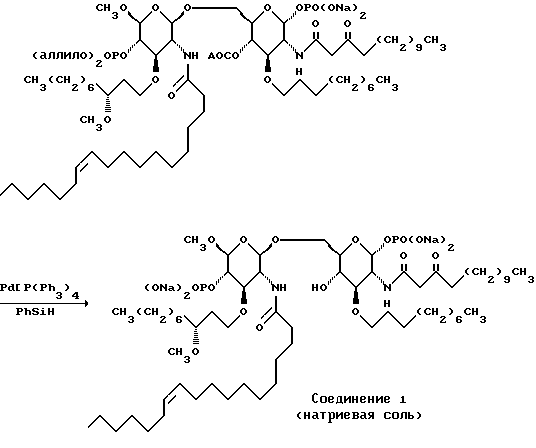
К раствору тетракис(трифенилфосфин)палладия(0) (166 мг) в 2 мл смеси тетрагидрофуран: уксусная кислота (10:1) добавляют раствор промежуточного соединения Z (660 мг) в 3 мл той же смеси растворителей. Через 30 минут добавляют еще тетракис(трифенилфосфин) палладий (0). Еще через 1,5 часа добавляют толуол и удаляют растворитель при пониженном давлении. Смесь очищают с помощью хроматографии на диэтил-аминоэтилцеллюлозе. Очищенную смесь растворяют в 0,1 N водном растворе гидроксида натрия, фильтруют через 0,45-мк стерильный фильтр и очищают с помощью ВЭЖХ на колонке YMC-Pack ODS-AP, и получают 130 мг соединения 1.
Результаты анализа соединения 1, осуществленного способами, описанными выше, приводятся ниже.
Соединение 1: 1H ЯМР (CD3OD) δ : 5,3 (1H, м), 4,6 (1, м), 4,0 (м, м), 3,9 (1H, д), 3,7 (1H, т), 3,6 (1H, т), 3,4 (3H, с), 3,3 (3H, т), 2,6 (2H, т), 2,3 (2H, м), 2,0 (2H, м), 1,7- 1,2 (м, м), 0,9 (6H, т).
31P ЯМР (CD3OD) δ : 4,71, 3,98.
Получение соединения 1 по маршруту 2
Получение соединения 1
Пример 1. Промежуточное соединение В
К суспензии полученного по способу Chirst, et al., заявка на европейский патент 92309057.5, промежуточного соединения А (15 г) в CH2Cl2 (150 мл) и 48% HBF4 (29,2 г), охлажденной на ледяной бане, добавляют TMSCHN2 (165 мл 2М раствора в гексане). Смесь перемешивают до тех пор пока реакция по ТСХ почти завершится, и затем добавляют метанол (20 мл) и вслед за ним уксусную кислоту (10 мл). Добавляют водный раствор бикарбоната натрия и смесь экстрагируют метиленхлоридом. Смесь сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография остатка дает 14,9 г В.
Пример 2. Промежуточное соединение C
К охлажденному (0oC) раствору В (14,9 г) в метиленхлориде (100 мл) медленно добавляют диизобутилалюмогидрид (140 мл 1 М раствора в гексане) до тех пор, пока реакция не завершится, что определяют с помощью ТСХ. Реакционную смесь гасят путем добавления 1 н соляной кислоты (100 мл), а затем конц. соляной кислоты (50 мл). Слоям дают возможность разделиться и водный слой снова экстрагируют CH2Cl2. Объединенные органические слои затем промывают насыщенным солевым раствором, сушат над сульфатом натрия и концентрируют при пониженном давлении. После очистки хроматографией на диоксиде кремния получают 12,06 г промежуточного соединения C.
Пример 3. Промежуточное соединение D
К раствору C (10,64 г) в метиленхлориде (40 мл) добавляют триэтиламин (15,75 мл), п-тоулсульфонилхлорид (11,86 г) и диметиламинопиридин (690 мг). Получающуюся в результате суспензию перемешивают до завершения реакции, что определяют с помощью ТСХ, и затем гасят водой и экстрагируют метиленхлоридом. После очистки хроматографией на диоксиде кремния получают 18,7 г промежуточного соединения D.
Пример 4. Промежуточное соединение E
К раствору D (18,7 г) в 200 мл ацетона добавляют йодид натрия (24,6 г). Смесь кипятят с обратным холодильником в течение 1,5 часов, удаляют растворитель при пониженном давлении, а остаток распределяют между водой и гексаном. Органический слой отделяют, сушат (сульфат натрия) и удаляют растворитель. Хроматография (диоксид кремния) дает 15,4 г E в виде бесцветной жидкости.
Пример 5. Промежуточное соединение F
Это соединение получают с помощью способа, описанного Chirst, et al., заявка на европейский патент 92309057.5.
Пример 6. Промежуточное соединение G
К раствору 18,6 г промежуточного соединения F и 15,4 г промежуточного соединения E в гексане добавляют 23,9 г оксида серебра и смесь кипятят с обратным холодильником в течение ночи. Смесь охлаждают, фильтруют через диатомовую землю, удаляют растворитель и остаток хроматографируют (диоксид кремния). Получают промежуточное соединение G (21 г) в виде бесцветного сиропа.
Пример 7. Промежуточное соединение H
К охлажденному (0oC) раствору промежуточного соединения G (21 г) в метиленхлориде добавляют по каплям 3,5 мл 48% тетрафтороборной кислоты. Через 5 минут смесь промывают водным раствором бикарбоната натрия и насыщенным солевым раствором. Смесь концентрируют при пониженном давлении и хроматографируют (диоксид кремния). Получают 18,7 г промежуточного соединения H в виде бесцветного сиропа.
Пример 8. Промежуточное соединение I
К раствору промежуточного соединения H (17,6 г) в чистом метилйодиде (105 мл) добавляют оксид серебра (83 г). Смесь перемешивают в течение ночи и затем разбавляют гексаном, и фильтруют через диатомовую землю. Смесь концентрируют при пониженном давлении и остаток растворяют в метиленхлориде (40 мл). Смесь охлаждают до 0oC и добавляют к раствору имидазол (2,44 г) и трет-бутилдиметилсилилхлорид (4,7 мл). Все перемешивают в течение ночи и добавляют 150 мл раствора бикарбоната натрия. Органический слой сушат (сульфат натрия) и хроматографируют (диоксид кремния), получают 10,5 г промежуточного соединения I в виде бесцветного сиропа.
Пример 9. Промежуточное соединение J
Промежуточное соединение I растворяют в 100 мл метиленхлорида и к раствору добавляют диаллилдиизопропилфосфорамидит (7,4 мл), а затем тетразол (6,37 г). Смесь охлаждают и перемешивают в течение 20 минут. В течение 15 минут добавляют суспензию метахлорпероксибензойной кислоты (24,2 ммоль) в 50 мл метиленхлорида, в то время как температуру реакции поддерживают ниже -60oC. Добавляют раствор бикарбоната натрия и органический слой отделяют, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография (диоксид кремния) дает 14 г промежуточного соединения J в виде бесцветного сиропа.
Пример 10. Промежуточное соединение К
К суспензии 39,5 г ди(тиофенил) олова (полученного по способу Chirst, et al., заявка на европейский патент 92309057.5) в 235 мл метиленхлорида добавляют тиофенол (12 мл). К этой смеси добавляют по каплям в течение 15 минут триэтиламин. Часть (150 мл) этой смеси "оловосодержащего реагента" добавляют по каплям в течение 15 минут к раствору промежуточного соединения J (12,9 г) в 25 мл метиленхлорида. Оставшуюся часть "оловосодержащего реагента" добавляют в течение 30 минут, чтобы довести реакцию до завершения. Смесь разбавляют этилацетатом и промывают водным I N раствором гидроксида натрия и насыщенным солевым раствором. Органический слой сушат (сульфат натрия), растворитель удаляют и остаток хроматографируют. Получают 11,1 г промежуточного соединения K в виде желтого сиропа.
Пример 11. Промежуточное соединение L
К охлажденному раствору промежуточного соединения К (11,1 г) и пиридина (7,1 мл) в 80 мл метиленхлорида добавляют трихлорэтилхлорформиат (2,9 мл) и смесь перемешивают в течение ночи. Добавляют водный раствор бикарбоната натрия, органический слой отделяют, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография дает 12,96 г промежуточного соединения L в виде светло-желтого твердого вещества.
Пример 12. Промежуточное соединение М
Растворяют 12,96 г промежуточного соединения L в метиленхлориде. К этой смеси добавляют 6 М раствор фтористого водорода в ацетонитриле и перемешивают смесь в течение 4 часов. Добавляют водный раствор бикарбоната натрия, органический слой отделяют, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография дает 10,9 г промежуточного соединения М виде сиропа янтарного цвета.
Пример 13. Промежуточное соединение N
К раствору промежуточного соединения М (9,5 г) в 50 мл трихлорацетонитрила добавляют карбонат калия (15 г) и смесь перемешивают в течение 10 минут. Смесь фильтруют через диатомовую землю и удаляют растворитель при пониженном давлении. Хроматография дает 14,5 г промежуточного соединения N, которое сразу же используют в примере 19.
Пример 14. Промежуточное соединение О
К раствору промежуточного соединения F (160 г) в гексане (475 мл) и йододекане (474 мл) добавляют оксид серебра (723 г). Смесь нагревают при 70oC в темноте в течение 2 часов и фильтруют через диатомовую землю. Раствор концентрируют при пониженном давлении и остаток хроматографируют. Получают 221 г промежуточного соединения O в виде бесцветного масла.
Пример 15. Промежуточное соединение P
К раствору промежуточного соединения О (30 г) в метиленхлориде (90 мл) и ацетонитриле (90 мл) добавляют раствор 48% водной плавиковой кислоты (9 мл) в ацетонитриле (81 мл). Смесь перемешивают в течение 30 минут и добавляют 350 мл водного раствора бикарбоната натрия. Смесь экстрагируют метиленхлоридом. Органический слой сушат (сульфат натрия), растворитель удаляют при пониженном давлении, а остаток хроматографируют и получают 30 г промежуточного соединения P в виде желтого масла.
Пример 16. Промежуточное соединение Q
К охлажденному (0oC) раствору промежуточного соединения P (30 г) и имидазола (10,2 г) в метиленхлориде (500 мл) добавляют трет-бутилдиметилсилилхлорид (10,85 г). Смесь перемешивают в течение 1,5 часов и затем выливают в 400 мл насыщенного водного раствора хлорида аммония. Органический слой отделяют, сушат (сульфат натрия), растворитель удаляют при пониженном давлении, а остаток хроматографируют и получают 34,5 г промежуточного соединения Q в виде бесцветного сиропа.
Пример 17. Промежуточное соединение R
К охлажденному (0oC) раствору промежуточного соединения Q (32,2 г) и пиридина (184 мл) в толуоле (213 мл) добавляют 1,94 М раствор фосгена в толуоле. Через 20 минут добавляют аллиловый спирт (31 мл) и смесь перемешивают в течение 30 минут. Добавляют водный раствор бикарбоната натрия, органический слой отделяют, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография дает 36,9 г промежуточного соединения R в виде бесцветного сиропа.
Пример 18. Промежуточное соединение S
К раствору 2,4 мл 48% водной плавиковой кислоты в 48 мл ацетонитрила добавляют раствор промежуточного соединения R (20 г) в метиленхлориде (24 мл) и смесь перемешивают в течение ночи. Добавляют водный раствор бикарбоната натрия, органический слой отделяют и сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография дает 11 г промежуточного соединения S в виде бесцветного сиропа.
Пример 19. Промежуточное соединение T
Промежуточное соединение S (8,97 г) и промежуточное соединение N (14,5 г) растворяют в толуоле (20 мл) и смесь сушат посредством азеотропного удаления растворителя. Эту процедуру повторяют три раза. Высушенную смесь растворяют в 50 мл метиленхлорида и раствор постепенно добавляют к раствору трифлата серебра (5,8 г) в 50 мл метиленхлорида. Смесь перемешивают в течение 10 минут и добавляют 250 мл водного раствора бикарбоната натрия и 250 мл 10% водного раствора тиосульфата натрия. Органический слой отделяют, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография дает 13 г промежуточного соединения T в виде бледно-желтого сиропа.
Пример 20. Промежуточное соединение U
К раствору промежуточного соединения T в метиленхлориде (10 мл) медленно добавляют комплекс олово (II) трис-бензолтиолата и триэтиламина (12 мл 0,5 М раствора в метиленхлориде). Через 10 минут добавляют еще такое же количество оловосодержащего реагента. Еще через 15 минут добавляют еще такое же количество. Через 15 минут добавляют этилацетат (250 мл) и смесь экстрагируют 1 N водным раствором гидроксида натрия (250 мл). Смесь сушат (сульфат натрия) и концентрируют при пониженном давлении. Добавляют толуол и удаляют растворитель при пониженном давлении. Получают масло, которое используют на следующей стадии трансформации без дополнительной очистки.
Пример 21. Промежуточное соединение V
К охлажденному (0oC) раствору промежуточного соединения U (2 ммоль) в метиленхлориде (5 мл) добавляют 3-кетотетрадекановую кислоту (997 мг), полученную по способу Christ, et al., заявка на европейский патент 92309057.5, а затем гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,5 г) и смесь перемешивают приблизительно 30 минут. Смесь разбавляют метиленхлоридом (150 мл), промывают 1 N водным раствором гидроксида натрия, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Хроматография на диоксиде кремния и последующая хроматография на основном оксиде алюминия дает 1,64 г промежуточного соединения V.
Пример 22. Промежуточное соединение W
Раствор промежуточного соединения V (1,45 г) в ледяной уксусной кислоте (5 мл) добавляют к суспензии тщательно перемешанных цинка и меди (14 г) в уксусной кислоте (10 мл). Смесь перемешивают в течение 15 минут и добавляют еще пару цинк/медь (10 г). Еще через 15 минут смесь фильтруют через диатомовую землю, которую затем промывают этилацетатом. Объединенные промывные жидкости разбавляют толуолом и удаляют растворитель при пониженном давлении. Остаток хроматографируют на двухслойной смеси основного оксида алюминия и диоксида кремния и получают промежуточное соединение W, которое используют без дополнительной очистки.
Пример 23. Промежуточное соединение X
Три раза раствор промежуточного соединения W (1,02 ммоль) и цис-вакценовую кислоту (575 мг) растворяют в толуоле (5 мл) и удаляют растворитель при пониженном давлении. Высушенный остаток растворяют в метиленхлориде (3 мл), добавляют гидрохлорид 1-(3- диметиламинопропил)-3-этилкарбодиимида (780 мг) и перемешивают смесь в течение 3 часов. Смесь разбавляют метиленхлоридом и непосредственно хроматографируют. Получают 734 г промежуточного соединения X. Дополнительная хроматография неочищенных фракций дает еще 58 мг вещества.
Пример 24. Промежуточное соединение Y
К раствору промежуточного соединения X (785 мг) в метиленхлориде (10 мл) добавляют раствор 48% водной плавиковой кислоты в ацетонитриле (15 мл). Смесь перемешивают в течение 90 минут, разбавляют метиленхлоридом (50 мл), промывают водой и водным раствором бикарбоната натрия. Смесь сушат (сульфат натрия) и хроматографируют, получают 719 мг промежуточного соединения Y.
Пример 25. Промежуточное соединение Z
Промежуточное соединение Y (719 мг) растворяют в метиленхлориде и добавляют сульфат натрия (1,4 г). Добавляют диаллилдиизопропилфосфорамидит (189 мкл) и тетразол (162 мг), смесь перемешивают в течение 10 минут и затем охлаждают до -78oC. Добавляют по каплям раствор м-хлоропероксибензойной кислоты (192 мг) в метиленхлориде (4 мл). Смесь промывают водным раствором тиосульфата натрия и водным раствором бикарбоната натрия, сушат (сульфат натрия) и удаляют растворитель при пониженном давлении. Остаток хроматографируют и получают 660 мг промежуточного соединения Z.
Пример 26. Соединение 1
К раствору тетракис(трифенилфосфин) палладия (0) (166 мг) в 2 мл смеси тетрагидрофуран: уксусная кислота (10:1) добавляют раствор промежуточного соединения Z (660 мг) в 3 мл той же смеси растворителей. Через 30 минут добавляют еще тетракис(трифенилфосфин) палладий (0). Еще через 1,5 часа добавляют толуол и удаляют растворитель при пониженном давлении. Смесь очищают с помощью хроматографии на диэтиламиноэтилцеллюлозе. Очищенную смесь растворяют в 0,1 N водном растворе гидроксида натрия, фильтруют через 0,45-мк стерильный фильтр и очищают с помощью ВЭЖХ на колонке YMC-Pack ODS-AP. Получают 130 мг соединения 1.
Результаты анализа некоторых соединений и промежуточных соединений, осуществленного описанными выше методами, приводятся далее.
Соединение 1: 1H ЯМР (CD3OD) δ : 5,3 (1H, м), 4,6 (1, м), 4,0 (м, м), 3,9 (1H, д), 3,7 (1H, т), 3,6 (1H, т), 3,4 (3H, с), 3,3 (3H, т), 2,6 (2H, т), 2,3 (2H, м), 2,0 (2H, м), 1,7- 1,2 (м, м), 0,9 (6H, т).
31P ЯМР (CD3OD) δ : 4,71, 3,98.
Соединение 1: (М + Na)+ = 1333
Соединение 2: (М + 3Na)+ = 1363
Соединение 3: (М + 3Na)+ = 1365
Соединение 5: (М + Na)+ = 1303
Соединение 6: (М + Na)+ = 1359
Соединение 7: (М + Na)+ = 1305
Соединение 8: (М + 3Na)+ = 1393
Соединение 10: (М + Na)+ = 1425
Промежуточное соединение G: 1H ЯМР (CDCl3) δ : д, (1H), 3,9-3,7 (м, множ.), 3,65 (т, 1H), 3,37 (с, 3H), 3,2 (м, 2H), 1,75 (к, 2H), 1,52 (с, 3H), 1,4 (с, 3H), 1,3 (уш.м, множ.), 0,95 (с, 9H), 0,9 (т, 3H) и 0,2 (д, 6H).
Промежуточное соединение H: 1H ЯМР (CDCl3) δ : 4,58 (д, 1H), 4,09 (м, 2H), 3,9 (дд, 1H), 3,75 (дд, 1H), 3,7 (м, 1H), 3,5 (т, 1H), 3,37 (с, 3H), 3,23 (т, 1H), 3,05 (т, 1H), 1,8 (м, 2H), 1,68 (м, 1H), 1,5 (м, 1H), 1,3 (уш. м, множ.), 0,95 (с, 9H), 0,9 (т, 3H), 0,2 (д, 6H).
Промежуточное соединение I: 1H ЯМР (CDCl3) δ : 4,52 (д, 1H), 4,05 (м, 2H), 3,75 (м, 1H), 3,67 (т, 1H), 3,5 (т, 1H), 3,45 (с, 3H), 3,35 (с, 3H), 3,25 (т, 1H), 3,05 (т, 1H), 1,8 (м, 2H), 1,65 (м, 1H), 1,5 (м, 1H), 1,3 (уш. с, м), 0,95 (с, 9H), 0,9 (т, 3H), 0,2 (с, 6H).
Промежуточное соединение J: 1H ЯМР (CDCl3) δ : 5,95 (м, 2H), 5,35 (д, 1H), 5,22 (д, 1H), 4,6 (к, 2H), 4,5 (д, 1H), 4,32 (к, 1H), 3,9-3,75 (м, 3H), 3,7 (дд, 1H), 3,65 (дд, 1H), 3,45 (м, 1H), 3,38 (с, 3H), 3,33 (с, 3H), 3,27 (т, 1H), 3,2 (т, 1H), 1,9-1,75 (м, 3H), 1,5 (м, 1H), 1,3 (уш.м, множ.), 0,95 (с, 9H), 0,9 (т, 3H), 0,2 (с, 6H).
Промежуточное соединение L: 1H ЯМР (CDCl3) δ : 5,95 (д, 1H), 5,4 (д, 2H), 5,25Z (д, 2H), 4,95 (д, 1H), 4,7 (к, 2H), 4,55 (к, 2H), 4,32 (к, 1H), 3,9-3,75 (м, 3H), 3,7 (дд, 1H), 3,65 (дд, 1H), 3,55 (м, 1H), 3,4 (м, 1H), 3,4 (с, 3H), 3,3 (с, 3H), 3,25 (м, 1H), 1,75 (м, множ.), 1,5-1,4 (м, 2H), 1,3 (уш.с, множ.), 0,95-0,9 (уш.с, 12H), 0,2 (д, 6H).
Промежуточное соединение М: 1H ЯМР (CDCl3) δ : 5,95 (м, 2H), 5,75 (д, 1H), 5,4 (д, 1H), 5,25 (д, 2H), 4,75-4,65 (дд, 2H), 4,6 (к, 1H), 4,3 (к, 1H), 4,1 (м, 2H), 3,9 (м, 2H), 3,65 (м, 1H), 3,4 (с, 3H), 3,25 (с, 3H), 1,75 (уш. м, 2H), 1,55-1,4 (м, 2H), 1,3 (уш. с, множ.), 0,9 (т, 3H).
Промежуточное соединение O: 1H ЯМР (CDCl3) δ : 4,5 (д, 1H), 3,8 (дд, 1H), 3,78 (м, 2H), 3,6 (м, множ.), 3,2 (м, 2H), 1,5 (с, 3H), 1,4 (с, 3H), 1,3 (уш. с, множ.), 0,95 (с, 9H), 0,9 (т, 3H), 0,18 (д, 6H).
Промежуточное соединение P: 1H ЯМР (CDCl3) δ : 4,5 (д, 1H), 3,75 (дд, 2H), 3,6 (к, 2H), 3,5 (т, 1H), 3,3 (м, 1H), 3,2 (т, 1H), 3,0 (т, 1H), 1,6 (м, 2H), 1,25 (уш.с, множ.), 0,95 (с, 9H), 0,9 (т, 3H), 0,18 (д, 6H).
Промежуточное соединение Q: 1H ЯМР (CDCl3) δ : 4,5 (д, 1H), 3,82 (т, 2H), 3,7 (м, 2H), 3,6 (т, 1H), 3,3 (м, 1H), 3,2 (т, 1H), 3,05 (к, 2H), 1,6 (м, 2H), 1,3 (уш.с, множ.), 0,95 (с, 9H), 0,88 (с, 9H), 0,85 (т, 3H), 0,2 (д, 6H), 0,1 (д, 6H).
Промежуточное соединение R: 1H ЯМР (CDCl3) δ : 5,9 (м, 1H), 5,4-5,25 (дд, 2H), 4,75 (т, 1H), 4,6 (д, 2H), 4,45 (д, 1H), 3,75 (к, 1H), 3,7 (д, 2H), 3,53 (к, 1H), 3,38 (м, 1H), 3,25 (т, 1H), 3,15 (т,1H), 1,5 (т, 2H), 1,25 (с, множ.), 0,95 (с,9H), 0,85 (м, 12H), 0,2 (с, 6H), 0,07 (с, 6H).
Промежуточное соединение S: 1H ЯМР (CDCl3) δ : 5,9 (м, 1H), 5,4-5,25 (дд, 2H), 4,75 (т, 1H), 4,6 (д, 2H), 4,52 (д, 1H), 3,7 (м, множ.), 3,65-3,6 (дд, 2H), 3,55 (к, 1H), 3,4 (м, 1H), 3,28 (т, 1H), 3,2 (т, 1H), 1,5 (т, 2H), 1,3 (с, множ.), 0,9 (с, 9H), 0,85 (т, 3H), 0,2 (с, 6H).
Промежуточное соединение T: 1H ЯМР (CDCl3) δ : 5,9 (м, 3H), 5,6 (д, 1H), 5,4-5,2 (м, 6H), 4,8 (д, 1H),4,7-4,6 (м, 2H), 4,55 (к, 1H), 4,5 (д, 1H), 4,3 (к, 1H), 3,8-3,7 (м, множ.), 3,6 (дд, 1H), 3,5 (м, множ.), 3,35 (с, 3H), 3,2 (с, 3H), 3,15 (т, 1H), 1,7 (м, 2H), 1,5 (м, 2H), 1,3 (с, множ.), 0,95 (т, 6H), 0,2 (т, 6H).
Промежуточное соединение V: 1H ЯМР (CDCl3) δ : 7,3 (д, 1H), 5,95 (м, 3H), 5,6 (д, 1H), 5,4-5,2 (м, 6H), 4,95 (д, 1H), 4,8 (д, 1H), 4,7-4,5 (м, множ. ), 4,3 (к, 1H), 3,9-3,65 (м, множ.), 3,6 (м, множ.), 3,45 (т, 1H), 3,4 (т, 3H), 3,35 (с, 2H), 3,28 (3H), 2,5 (т, 2H), 1,8 (м, 2H), 1,6 (м, 2H), 1,45 (м, 2H), 1,3 (уш.с, множ.), 0,95-0,8 (м, 18H), 0,15 (д, 6H).
Промежуточное соединение X: 1H ЯМР (CDCl3) δ : 7,3 (д, 1H), 5,95(м, 4H), 5,4-5,2 (м, 8H), 4,95 (д, 1H), 4,8 (д, 1H), 4,7 (т, 1H), 4,6 (д, 1H), 4,55 (к, 1H), 4,3 (к, 1H), 4,1 (т, 1H), 3,9 (к, 1H), 3,8 (т, 1H), 3,75-3,5 (м, множ. ), 3,45 (т, 1H), 3,35 (с,3H), 3,3 (с, 3H), 3,28 (с,3H), 2,5 (т, 2H), 2,2 (т, 1H), 2 (д, 1H), 1,7 (к, 2H), 1,6 (м, 2H), 1,3 (с, множ.), 0,95-0,8 (м, 21), 0,15 (д, 6H).
Промежуточное соединение Y: 1H ЯМР (CDCl3) δ : 6,65 (д, 1H), 6,55 (д, 1H), 5,905 (м, 5Н), 5,7 (м, 1H), 5,4-5,2 (м, 12H), 4,8 (м, 2H), 4,6 (д, 1H), 4,5 (м, 10H), 4,3 (к, 1H), 4,1 (м, 1H), 3,85-3,45 (м, множ.), 3,4 (с, 3H), 3,35 (с, 3H), 3,25 (с, 3H), 3,2 (т, 1H), 2,5 (дд, 2H), 2,2 (т, 2H), 2 (м, множ.), 1,7-1,2 (м, множ.), 0,9 (т, 12H).
Биологические примеры
Как бактериальный ЛПС, так и бактериальный липид A выявляют продуцирование фактора некроза опухоли (ФНО), IL-1β, IL-6 и IL-8, а также других цитокинов и клеточных медиаторов в цельной человеческой крови и в человеческих макрофаговых клеточных линиях. Обнаружено, что генерация патофизиологических количеств таких цитокинов играет существенную роль в инициировании синдрома системной воспалительной реакции и септического шока. Аналоги липосахаридов, описанные здесь, ингибируют такую опосредованную ЛПС и/или липидом A индукцию цитокинов, как показывают приведенные далее эксперименты.
Пример A. Ингибирование in vitro индуцированного ЛПС продуцирования цитокинов
Подготавливают цельную человеческую кровь и испытывают так, как описано в Rose et al. (1995), Infection and Immunity, 63, 833-839. Клетки HL-60 культивируют в среде RPMI с добавлением 10% фетальной телячьей сыворотки и антибиотиков, и индуцируют для дифференцировки в макрофагах путем обработки 0,1 мкМ 1,25- дигидрооксихолекальциферола (витамин D3; Biomol Research Laboratories, Plymouth Meeting, Пенсильвания), и испытывают на опосредованную ЛПС генерацию IL-8. Коротко, бактериальный ЛПС (т.е. от E. coli 0111:B4; Sigma Chemicals, Сент-Луис, Миссури) в количестве 10 нг/мл или липид A (Daiichi Chemicals, Токио, Япония) добавляют в виде 10-кратно сконцентрированных растворов в свободный от Ca++ и Mg++ сбалансированный солевой раствор Хэнкса (CMF-HBSS; Gibco). В экспериментах с участием аналогов настоящего изобретения аналог добавляют непосредственно перед добавлением ЛПС или липида A в CMF-HBSS (например, от 0 до 100 мкМ в виде 10х концентрированной аликвоты). После инкубации в течение трех часов из цельной крови получают плазму или удаляют культуральный супернатант и анализируют на присутствие указанного цитокина с использованием набора для анализа методом ELISA от Genzyme (Кэмбридж, Миннесота), следуя указаниям изготовителя, однако, можно использовать любой стандартный набор для ELISA. Эксперименты осуществляют по меньшей мере дважды при трехкратном повторе.
Аналоги липида A ингибируют индуцируемое ЛПС продуцирование ФНО в цельной крови человека в зависимости от концентрации. Обнаружено, что из испытанных аналогов одним из наиболее эффективных соединений является соединение 1. Результаты этого испытания приводятся на фиг. 1. Соединение 1 ингибирует индуцируемое ЛПС образование ФНО, демонстрируя IC50 приблизительно в 1,5 нМ. Обнаружено, что другими аналогами, которые ингибируют индуцируемое ЛПС образование ФНО, являются соединение 2, соединение 3, соединение 4, соединение 5, соединение 6, соединение 7, соединение 8, соединение 9 и соединение 10. Эти соединения обнаруживают величину IC50 от 1,5 нМ до 159 нМ.
Соединение 1 также ингибирует опосредованную ЛПС индукцию IL-8 в клетках HL-60 (человеческие макрофагоподобные клетки). Ингибирование генерации IL-8 осуществляется при концентрациях соединения 1 в 1 нМ и выше, когда в качестве агониста используют либо ЛПС, либо липид A.
Соединения по настоящему изобретению подобным образом ингибируют индуцируемое ЛПС продуцирование других цитокинов в человеческой цельной крови, хотя некоторые из этих цитокинов генерируются даже через несколько часов после добавления ЛПС. Например, для достижения максимального уровня IL-1β и IL-6 требуется четыре или больше часов, в то время как максимальный уровень IL-8 достигается через десять или больше часов после добавления ЛПС. Используя описанные выше способы, соединения по настоящему изобретению добавляют до концентрации от 0 до 10 мкМ, а ЛПС добавляют в количестве 10 нг/мл. Ингибирование продуцирования ФНО, IL-1β, IL-6 и IL-8 измеряют как функцию времени после добавления ЛПС. Также обнаружено, что такое ингибирование генерации цитокинов зависит от концентрации, но во всех случаях подавление синтеза всех цитокинов составляет > 90% при концентрациях соединения 1 10 нМ и выше в течение 24 часов после добавления ЛПС.
Пример В. Персистенция соединений в человеческой цельной крови
Хотя некоторые из соединений по настоящему изобретению не являются быстро удаляемыми из кровообращения, их активность снижается со временем полужизни 1-3 часа. Чтобы сохранить антагонистическую эффективность при такой быстрой дезактивации, может потребоваться непрерывное введение. Исследование дезактивации привело к разработке анализа для измерения дезактивации лекарственных средств in vitro в цельной человеческой крови. Это осуществляется посредством предварительной инкубации антагонистов липида A с кровью в течение различного времени с последующим добавлением "нагрузки" ЛПС, как описано выше, инкубации в течение трех часов и анализа выделенных цитокинов. Схема такого анализа приводится на фиг. 2.
Используя такой анализ, можно показать, что B531, как описано в Chirst, et al., заявка на патент США 07/935050, "дезактивируется" (активность теряется при возрастании времени предварительной инкубации). Как показано на фиг. 3, соединение 1 также дезактивируется, но его превосходящая активность и пониженная скорость дезактивации делают его таким же активным через 6 часов, каким является B531 непосредственно после его введения. Эти данные являются средними результатами 7 отдельных экспериментов, повторенных трижды.
Пример C. Ингибирование продуцирования ФНО или IL-6 in vitro в системах животных моделей
Индуцированное ЛПС продуцирование ФНО или IL-6 в цельной крови или макрофагах, выделенных из морских свинок, крыс и мышей, ингибируется соединениями по настоящему изобретению. Макрофаги морской свинки Hartley-White (Elm Hill Breeders, Chelmsford, Миннесота) и мыши C57BL/6 (Jackson Labs, Bar Harbor, Мэн) выделяют из брюшного отдела примированных животных. Примирование осуществляют посредством интраперитонеальной инъекции 2 мг бациллы Кальметта-Герэна (БЦЖ; RIBI Immunochemical Research, Inc., Гамильтон, Монтана) при концентрации 10 мг/мл в физиологическом растворе в случае мышей и 2 мг БЦЖ при концентрации 2 мг/7 мл в минеральном масле в случае морских свинок. Через три дня после инъекции из брюшной полости животных стандартными методами выделяют перитонеальные макрофаги. Клеткам дают возможность прилипать к культуральным планшетам в течение двух-трех часов, а затем культивируют со средой RPMI 1640, содержащей 10% фетальной телячьей сыворотки, и добавляют ЛПС (конечная концентрация 10 нг/мл), как описано выше. Для проверки ингибирования соединения по настоящему изобретению (при концентрации от 0 до 100 мкМ) добавляют к культуральной среде непосредственно перед добавлением ЛПС. После трехчасового инкубационного периода анализируют уровни ФНО и/или IL-6 у морской свинки, мыши и крысы методом ELISA или с помощью цитолитического биоанализа, описанного в Lymphokines 2:235, 1981 для ФНО, выделенного из макрофагов морской свинки. В мышиных перитонеальных макрофагах соединение 1 обеспечивает эффективное ингибирование (IC50=16 нМ для IL-6 и 20 нМ для ФНО соответственно); в макрофагах морской свинки IC50 в случае выделения ФНО составляет 0,3 нМ и в перитонеальных крысиных макрофагах IC50 для выделения ФНО составляет 11 нМ.
Пример D. Анализы in vivo
В качестве системы анализа in vivo для контроля за ингибирующим действием аналогов липида A на (1) индуцированное ЛПС продуцирование ФНО и (2) индуцированную ЛПС летальность используют следующим образом БЦЖ-примированных мышей (Vogel, S. et al., J. Immunology, 1980, 124, 2004-2009).
Самцов мышей C57BL/6 (см. выше) в возрасте пяти недель примируют посредством внутривенной инъекции в хвостовую вену 2 мг БЦЖ. Через десять суток после инъекции БЦЖ-примированным мышам вводят внутривенно, через хвостовую вену, ЛПС E. coli (см. выше) в апирогенном 5% растворе глюкозы (Otsuka Pharmaceuticals Inc., Токио, Япония). При исследованиях как продуцирования ФНО, так и смертности вводят 1-3 мкг ЛПС на мышь. Испытываемое соединение вводят в качестве компонента инъецируемого раствора ЛПС при концентрации от 3 до 300 мкг/мышь. Плазму получают через час после инъекции ЛПС, и ФНО анализируют методом ELISA, описанным выше. Смертность в результате септического шока регистрируют в течение 36 часов после инъекции ЛПС. Соединения по настоящему изобретению эффективно подавляют продуцирование ФНО, следующее за введением ЛПС.
Соединение 10 и соединение 1 эффективно ингибируют продуцирование ФНО in vivo у мышей (ED50 = 5 и 10,6 мкг/мышь соответственно). Соединение 2, соединение 3, соединение 4, соединение 5, соединение 6, соединение 7, соединение 8 и соединение 9 также ингибируют продуцирование ФНО с ED50 от 10 до 200 мкг/мышь, причем соединения 5, 6 и 7 дают величину ED50 > 100.
В параллельных экспериментах, осуществленных на морских свинках, эти аналоги также являются эффективными ингибиторами индуцируемого ЛПС образования ФНО in vivo (оптимальные значения ED50 для соединения 1, соединения 7 и соединения 10 составляют от 2,3 до 6,1 мкг на морскую свинку).
Терапия
Описанные здесь аналоги липида A дают полезный терапевтический эффект в случае лечения или предупреждения любого опосредованного ЛПС воспаления или нарушения. К таким нарушениям относятся, но не ограничиваются перечисленным, эндотоксикоз (или синдром сепсиса), вызываемый грамотрицательными бактериями (с сопровождающими его симптомами лихорадки, развитого воспаления, диссеминированной внутрисосудистой коагуляции, гипотензии, острой почечной недостаточности, острого респираторного дистресс-синдрома, гепатоцеллюлярной деструкции и/или сердечной недостаточности); и передаваемое ЛПС обострение латентных или активных вирусных инфекций (например, при инфекции ВИЧ-1 цитомегаловирусами, простым герпесом и вирусом гриппа).
Аналог липида A, как правило, вводят в составе фармацевтически приемлемой композиции, например, растворенным в физиологическом растворе (который может содержать 5% глюкозы). Когда аналог липида A предназначен для лечения вирусной инфекции, его можно вводить в сочетании с соответствующими вироцидными средствами. Аналоги липида A можно хранить в виде высушенной вымораживанием композиции.
Аналоги липида A вводят при дозировке, которая обеспечивает соответствующее ингибирование активации ЛПС клеток-мишеней; обычно дозировки составляют предпочтительно 0,01- 50 мг/пациент/сутки, предпочтительнее 0,05-25 мг/пациент/сутки и наиболее предпочтительно 1-12 мг/пациент/сутки.
Лекарственное средство следует ввести инъекцией или внутривенным вливанием как можно скорее, когда можно диагностировать ССВР с использованием таких прогностических факторов, как показатель APACHE (Knaus, et al., 1991, Chest 100: 1619-36, и Knaus, et al., 1993, JAMA: 1233-41) или других клинических показателей. Кроме того, инъекции или вливание нужно начинать как можно скорее после воздействия эндотоксина или выявления общей грамотрицательной бактериальной инфекции, особенно если становится доступным более быстрый или ранний диагностический определитель системной грамотрицательной инфекции.
Профилактические показания для применения указанных лекарственных средств могут включать их применение, когда можно предвидеть воздействие эндотоксина. Это может иметь место в следующих случаях.
1) Существует возросшая вероятность повышения общего (переносимого кровью) эндотоксина в результате общей или локализованной грамотрицательной бактериальной инфекции (например, во время операции).
2) Существует повышенная вероятность того, что уровень эндотоксина в крови может возрасти. При нормальном физиологическом состоянии эндотоксин только в минимальном количестве транслоцируется через кишечный эндотелий во внутренностное кровообращение. Этот транслоцированный эндотоксин затем выводится обычно через печень (а возможно, через другие клетки и органы). Возрастание уровня эндотоксина в крови может происходить, когда уменьшается скорость клиренса эндотоксина печенью (или другими отделяющими эндотоксин клетками или органами). Нарастание кишечной транслокации может происходить после кишечной ишемии, гипоксии, травмы или другого повреждения целостности кишечной выстелки (или лекарственным средством, или алкогольной токсичностью). Уровень эндотоксина в крови возрастает, когда функция печени ставится под угрозу болезнью (цирроз), повреждением (операция или травма) или временным удалением (как при пересадке печени).
3) Существует острое или хроническое воздействие произведенного снаружи эндотоксина, проявляющееся в воспалительной реакции; это воздействие может быть вызвано ингаляцией или другими способами поглощения эндотоксина. Одним из примеров поглощения индуцирующего SIRS эндотоксина является лихорадка от кукурузной пыли (Schwartz, et al., 1994, Am. J. Physiol., 267: L609-17), которая поражает рабочих зерновой отрасли, например, на американском Среднем Западе. Этих рабочих можно лечить превентивно, например ежедневно, путем ингаляции композиции лекарственного средства в виде аэрозоля перед началом работы, например, в поле или на зерновых элеваторах.
В большинстве случаев другого профилактического или терапевтического применения будет использоваться внутривенное вливание IV или инъекция ударной дозы вещества. Наиболее предпочтительна инъекция, но в некоторых случаях фармакокинетическими требованиями может быть оправдано внутривенное вливание.
Лечение должно начинаться как можно скорее после выявления CCBP и должно продолжаться по меньшей мере в течение трех суток или до тех пор, когда оценка угрозы для жизни снизится до приемлемого уровня.
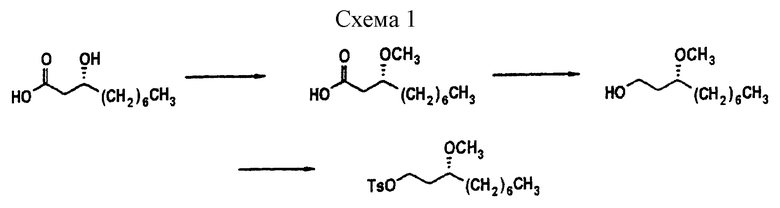
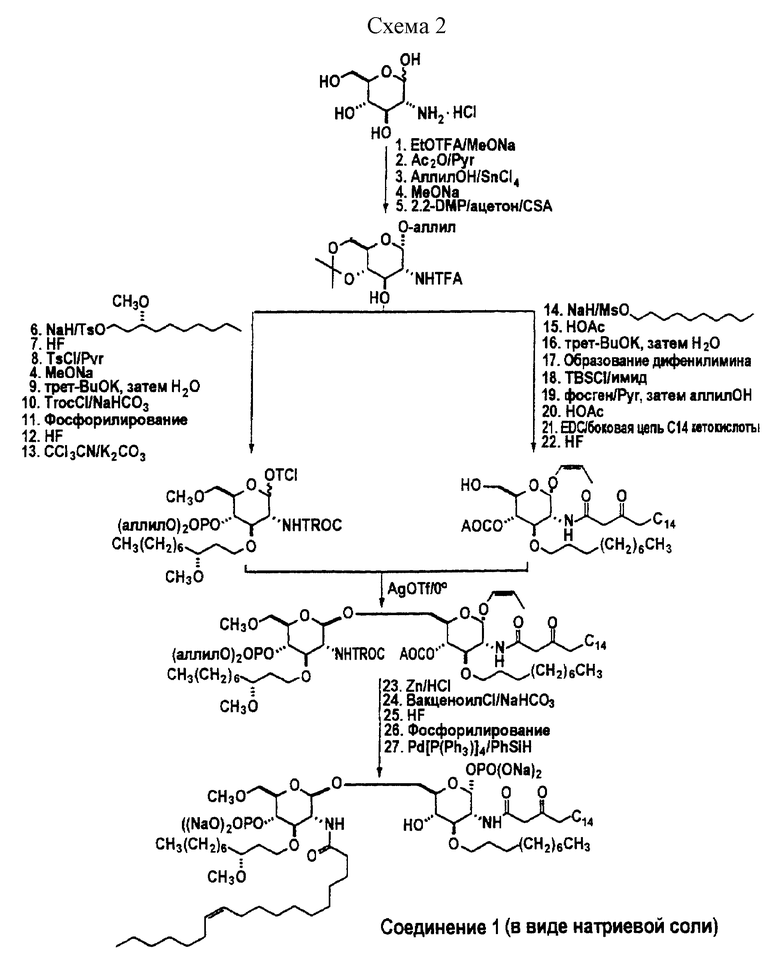
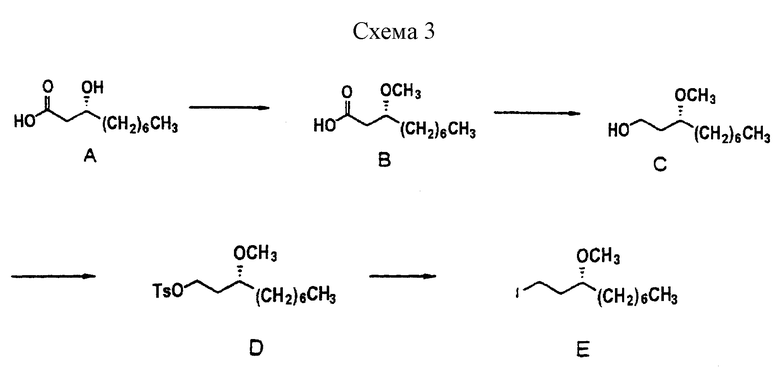
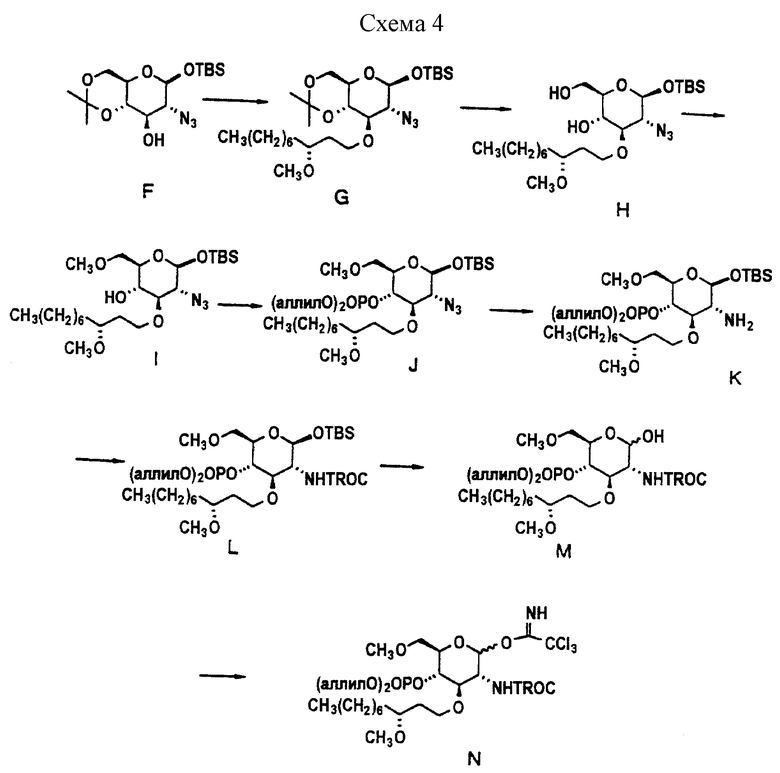
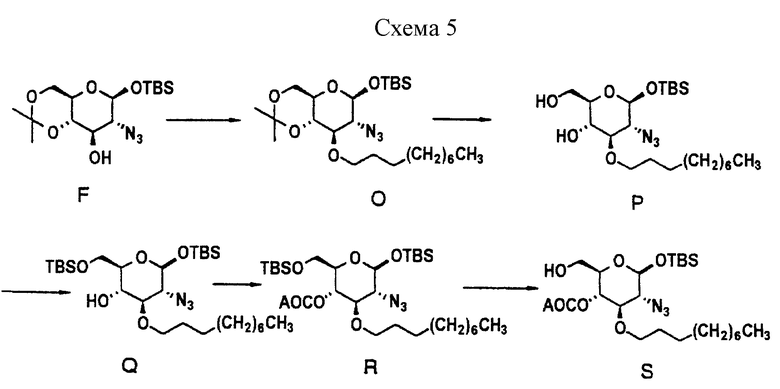
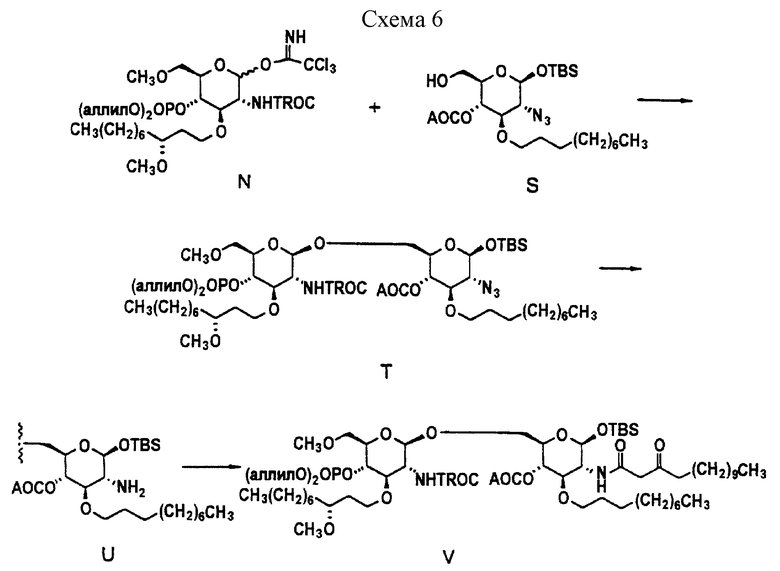
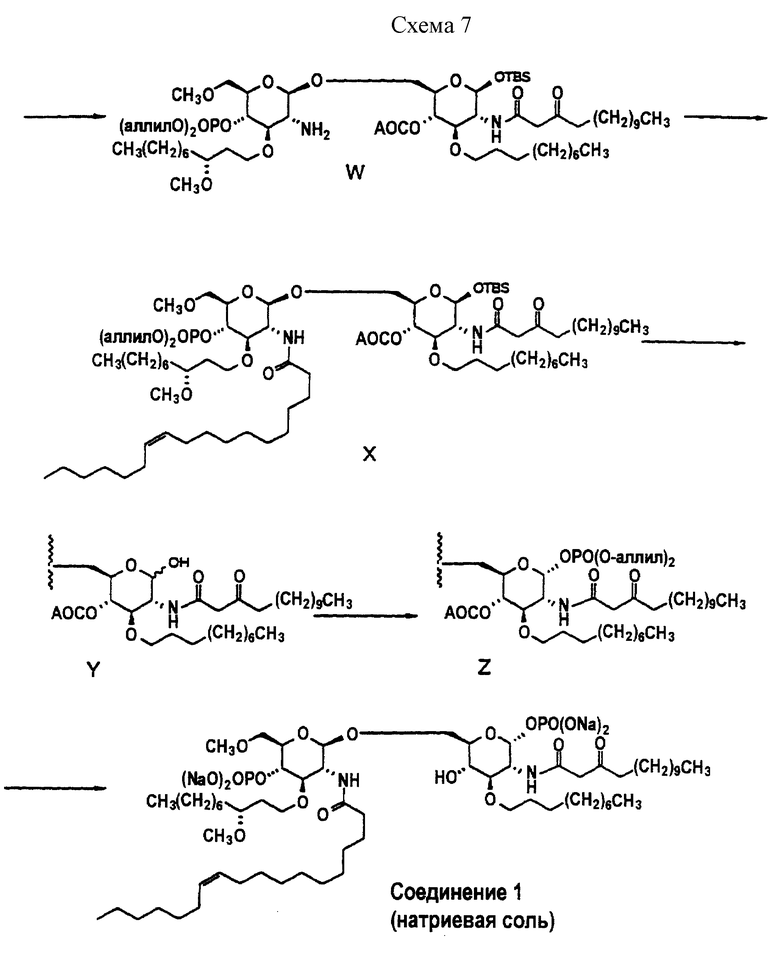
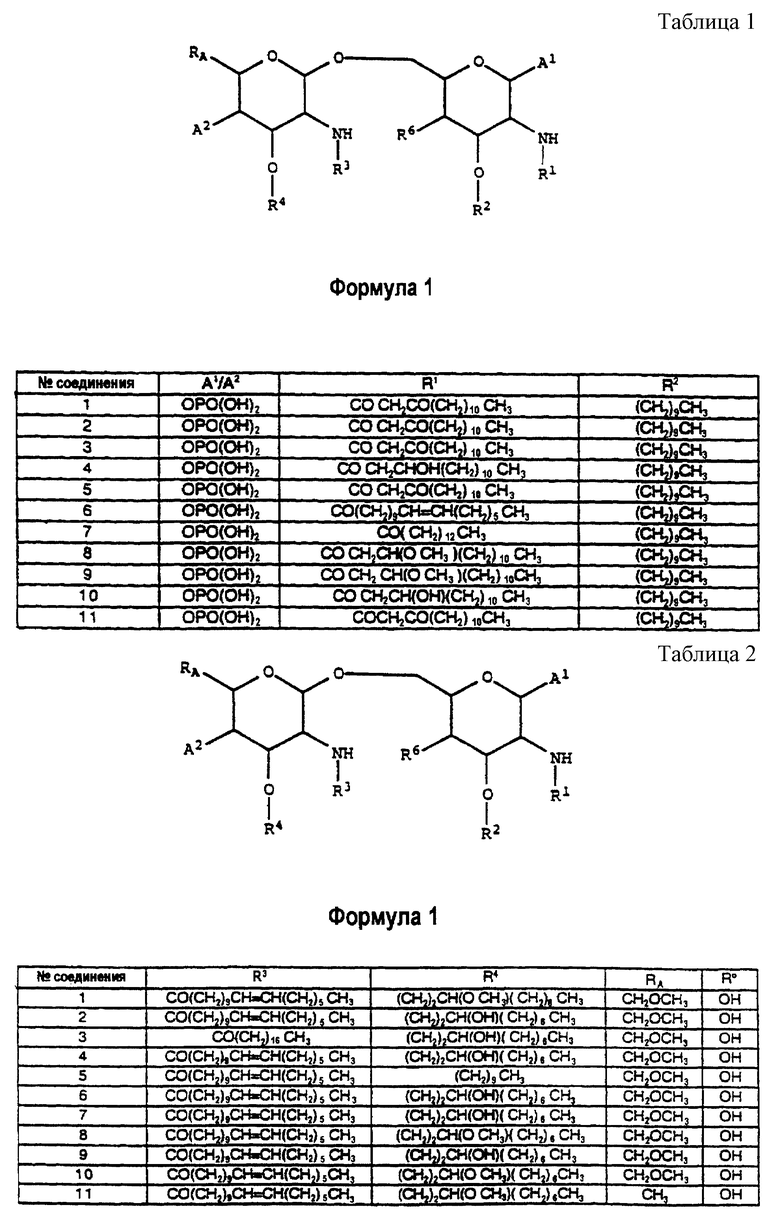
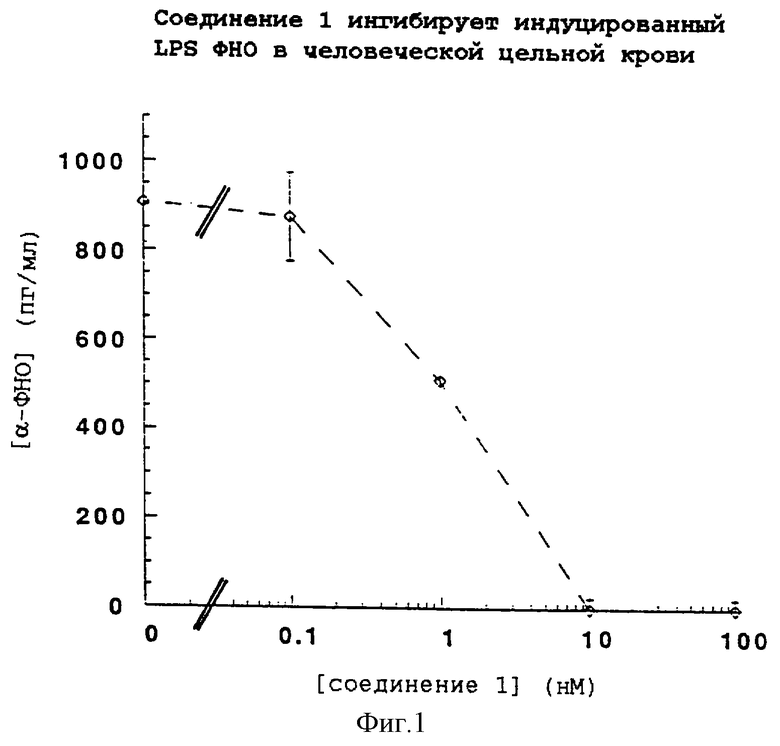
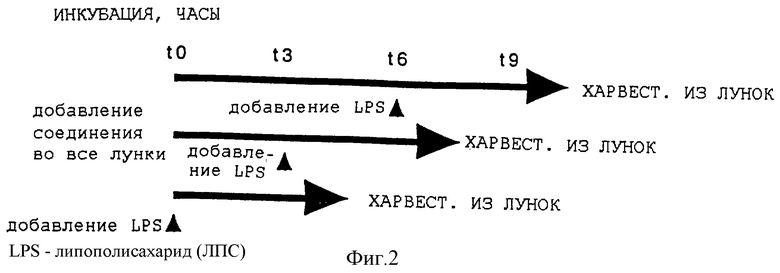
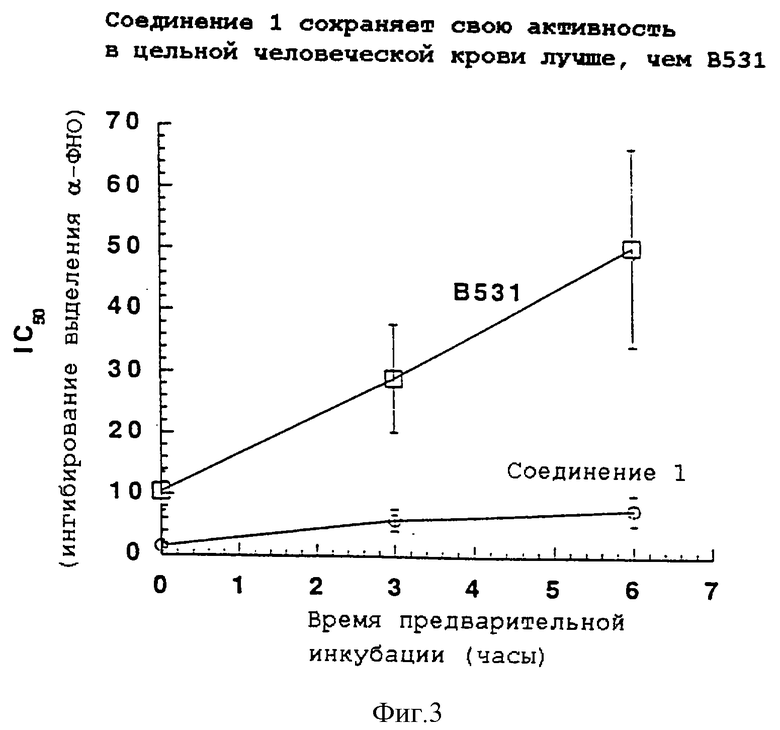
Описывается новый замещенный липосахарид формулы I, где R1 представляет собой группу (а), где J и K - линейный C1-C15алкил, R2 представляет собой линейный C5-C15алкил; R3 представляет собой группу (b), где A и B обозначают линейный C1-C15алкил, R4 представляет собой группу (с), где каждый U и V обозначает линейный C2-C15алкил и W обозначает линейный C1-C5алкил; RA представляет собой R5-O-CH2-, где R5 представляет собой линейный C1-C5алкил; R6 представляет собой гидрокси; A1 и A2 обозначают группу (d), или его фармацевтически приемлемая соль. Соединения могут использоваться при профилактике и при конструктивном лечении эндотоксинного воздействия, включая сепсис, септицемию, эндотоксикоз и различные формы септического шока. 13 з. п. ф-лы, 3 ил., 2 табл.
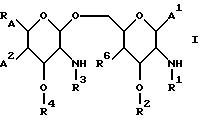
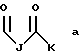
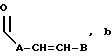
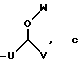
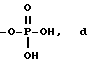
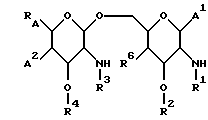
где R1 представляет собой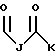
где J и K обозначают линейный C1-C15алкил;
R2 представляет собой линейный C5-C15алкил;
R3 представляет собой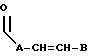
где A и B обозначают линейный C1-C15алкил;
R4 представляет собой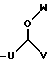
где каждый U и V обозначает линейный C2-C15алкил и W обозначает линейный C1-C5алкил;
RA представляет собой R5-O-CH2-, где R5 представляет собой линейный C1-C5алкил;
R6 представляет собой гидрокси;
A1 и A2 обозначают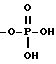
или его фармацевтически приемлемая соль.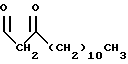
R2 представляет собой (CH2)9CH3;
R3 представляет собой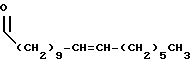
R4 представляет собой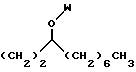
где W представляет собой C1-C5алкил;
R5 является -CH3.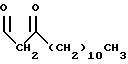
R2 представляет собой (CH2)9CH3;
R3 представляет собой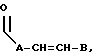
где A и B обозначает линейный C1-C15алкил;
R4 представляет собой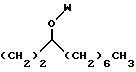
где W представляет собой C1-C5алкил;
R5 является -CH3.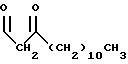
R2 представляет собой (CH2)9CH3;
R3 представляет собой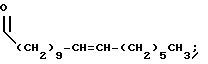
R4 представляет собой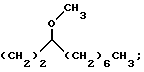
R5 является -CH3.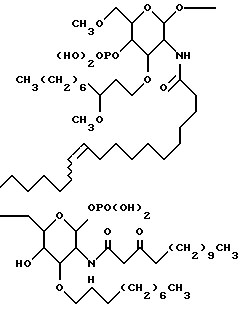
или его фармацевтически приемлемая соль.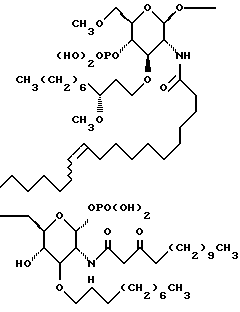
или его фармацевтически приемлемые соли.

