Изобретение относится к области органической химии и синтезу лекарственных веществ, конкретно к способу получения (1-фенил-2,3-диметил-4-иодпиразолона-5) (иодантипирина) формулы
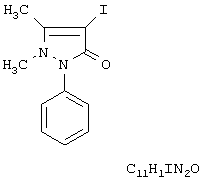
Иодантипирин - противовирусный препарат. Он обладает антипирическим, антиневрологическим действиями, проявляет высокую антивирусную активность против клещевого энцефалита. При изучении противовирусной активности иодантипирина было показано, что препарат индуцирует выработку β-интерферона, проявляет иммуномоделирующее действие в организме в отношении эритроцитарного и вирусного антигенов. Защитный эффект усиливается при комбинации с ремантадином и ацикловиром. Иодантипирин обладает более выраженным профилактическим действием по сравнению с иммуноглобулином, способствует более быстрому исчезновению вируса клещевого энцефалита из организма инфицированного. Сочетание противовирусной, интерфероногенной и иммуностимулирующей активности послужило причиной широкого распространения данного лекарственного средства в медицинской практике. [Яворовская В.Е., Саратиков А.С., Федоров Ю.В. Иодантипирин - средство для лечения и профилактики клещевого энцефалита // Эксперим. и клинич. Фармакол. - 1998. - Т.61, No 1. - 51-53 с.]. Иодантипирин представляет собой белый или почти белый кристаллический порошок горького вкуса, без запаха или со слабым специфическим запахом (ФСП 42-0311-1559-01), с температурой плавления 160-165°С и содержанием основного вещества (С11H11IN2О) не менее 99,0% в пересчете на сухое вещество.
Известен способ получения иодантипирина [CS, пат. 86123, 12 р 8/01; 1957 г.] окислительным иодированием антипирина иодом в воде в присутствие ацетата натрия. Данный способ получения иодантипирина заключается в том, что водный раствор антипирина и ацетата натрия доводят до кипения и добавляют одновременно при перемешивании смесь иода и иодида калия в воде и смесь соляной кислоты и перекиси водорода (вместо перекиси можно использовать озон, хлор). Затем рН реакционной массы доводят до 7-9, отфильтровывают выпавший осадок. Выход технического продукта составляет 98,5%. Недостатком способа являются сложность технологического процесса, использование токсичных реагентов. Кроме этого, вышеописанный способ не позволяет получить фармакопейный продукт, иодантипирин загрязнен ацетатом натрия и требует специальной очистки, которая приводит к высокой степени загрязнения сточных вод.
Известен способ получения иодантипирина [U.Vater Pharm. Praxis, Beilage Pharmazie, 1964, 2-3], заключающийся в обработке 1-фенил-2,3-диметилпиразолона-5 (антипирина) иодом в водной или водно-спиртовой среде в присутствии ацетата натрия. Недостатком указанного метода является повышенный расход иода, а главное присутствие в целевом продукте примесей (солей иодистоводородной кислоты), что не позволяет получить удовлетворяющий требованиям фармакопейной статьи продукт. Применение очистки технического продукта приводит к высокой степени загрязнения сточных вод и резкому снижению выхода фармакопейного иодантипирина. Осуществлению описанных выше способов получения иодантипирина в заводских условиях препятствует высокая степень загрязнения сточных вод.
Следующий способ получения иодантипирина [RU 2179551 С 07 D 231/22, публ. 2002] заключается в окислительном иодировании антипирина в нейтральной среде в изопропиловом спирте иодирующей смесью при температуре 80°С, которая достигается за счет самопроизвольного разогрева реакционной массы. Иодирующую смесь готовят из иодида калия, перекиси водорода и соляной кислоты (образуется KICl2).
Для получения иодантипирина в изопропиловый спирт помещают антипирин, перемешивают до его полного растворения и вносят стехиометрическое количество твердой щелочи (NaOH). Твердая щелочь практически не растворяется в изопропиловом спирте и находится в реакционной массе в виде суспензии. Затем в реакционную массу при интенсивном перемешивании прибавляют иодирующую смесь. При этом происходит иодирование антипирина до иодантипирина. Образующийся во время реакции хлористый водород нейтрализуется щелочью, что препятствует образованию побочных продуктов и обеспечивает высокую скорость иодирования антипирина. Поскольку окислительное иодирование антипирина происходит с выделением тепла, то температура реакционной массы повышается, при этом она зависит от скорости дозирования иодирующей смеси и ограничена температурой кипения реакционной массы (80°С).
После проведения иодирования реакционную массу охлаждают до 12-14°С, выпавший иодантипирин отфильтровывают, сушат. Выход иодантипирина количественный, Т.пл 162-164°С. Продукт полностью соответствует требованиям ФСП 42-0311-1559-01.
Недостатками описанного способа является использование изопропилового спирта, который относится к легко воспламеняющимся жидкостям и требует дополнительных технологических операций для утилизации; использование сильного основания (щелочи), что представляет определенную технологическую и экологическую опасность. Кроме того, описанная методика ограничена невозможностью использования более доступного сырья - бензолсульфокислого антипирина (БСКА), который является промышленным полупродуктом для получения антипирина, а также усложнением технологии, связанным с необходимостью точного дозирования подачи реагента.
Наиболее близким к заявленному изобретению являются следующий способ получения 1-фенил-2,3-диметил-4-иодпиразолона-5 (иодантипирина) [RU, пат. 21063444, С 07 D 231/16, 231.22, опубл. 1998 г.]. Иодантипирин получают из 1-фенил-2,3-диметилпиразолона (антипирина) или 1-фенил-2,3-диметилпиразолона бензолсульфокислого (бензолсульфокислого антипирина) окислительным иодированием в кислой среде, в воде, водно-органическом или органическом растворителе иодирующей смесью при температуре не выше 45°С (предпочтительно в интервале 2-10°С). Иодирующую смесь (KICl2) готовят из иодида калия, перекиси водорода и соляной кислоты.
Для получения иодантипирина раствор антипирина или БСКА в органическом или в водно-органическом растворителе обрабатывают по каплям иодирующей смесью при интенсивном перемешивании с такой скоростью, чтобы температура в конце прикапывания была не выше 45°С (предпочтительно 2-10°С). При этом образуется творожистый осадок.
Реакционную массу выдерживают при указанной температуре и перемешивании в течение 30 мин, затем к реакционной массе прибавляют 15-20% раствор гидроксида натрия, при перемешивании и температуре не выше 10°С выдерживают еще 30 минут. Белый осадок отфильтровывают, промывают холодной водой, высушивают, получают от 26 до 99.2% технического иодантипирина (в зависимости от использованного растворителя). Для получения иодантипирина, удовлетворяющего требованиям ВФС, технический иодантипирин перекристаллизовывают из метанола или этанола. Выход фармакопейного продукта не указан. После перекристаллизации из метанола или этанола получают иодантипирин с Т.пл. 160-161°С.
Недостатками способа по прототипу являются:
недостаточная чистота получаемого целевого продукта;
недостаточно высокий выход фармакопейного иодантипирина;
сложность технологического процесса, связанного с необходимостью поддержания оптимальной температуры реакции иодирования в пределах 2-10°С (повышение более 45°С приводит к резкому сокращению целевого продукта, см. пример 2 в описании к прототипу), что требует специального аппаратурного оформления;
использование органических растворителей (метанола или этанола), которые относятся к легко воспламеняющимся жидкостям и требуют дополнительных технологических операций для утилизации; использование сильного основания (щелочи), что представляет определенную технологическую и экологическую опасность.
Технической задачей, положенной в основу настоящего изобретения, является получение фармакопейного иодантипирина как из антипирина, так и бензолсульфокислого антипирина в условиях механической активации без использования органического растворителя.
Поставленная техническая задача решается тем, что в предлагаемом способе получения 1-фенил-2,3-диметил-4-иодпиразолона-5 (иодантипирина) реализуется прогрессивная методология твердофазного ведения процесса (отсутствие растворителя), при этом в качестве исходного субстрата наряду с антипирином может быть использован бензолсульфокислый антипирин.
В качестве иодирующих агентов согласно настоящему изобретению могут быть использованы известные реагенты, например иод или хлорид иода, а также комплексные аммонийные соли, например, Me4N+ICl2 и Et4N+ICl2. Сущность изобретения заключается в следующем.
Для получения иодантипирина по предлагаемому способу при комнатной температуре механически растирают исходный субстрат (антипирин или БСКА) и реагент (иод, хлорид иода, Me4N+ICl2 или Et4N+ICl2) в течение 15 минут. Процесс механического растирания реагирующих веществ может быть осуществлен известными способами, например, растиранием в ступке, шаровой мельнице, экструдере и др. После растирания реакционную массу выдерживают в течение 30 минут. Следует отметить, что процесс получения иодантипирина идет при комнатной температуре и не требует ее регулирования, что значительно упрощает аппаратурное оформление процесса. После выдержки реакционную массу обрабатывают определенным количеством водного раствора соды для нейтрализации кислотных компонентов, доводя рН среды до щелочного значения (преимущественно 10 ед. рН). Образующиеся в результате иодирования антипирина с помощью Me4N+ICl2 или Et4N+ICl2 алкиламмонийные соли, а также хлорид натрия и бензоат натрия легко растворяются в воде в ходе указанной обработки реакционной массы. Полученная водная суспензия иодантипирина кипятится 15 мин до полного растворения иодантипирина. После этого реакционную массу охлаждают до 20-22°С, и мало растворимый в воде иодантипирин выпадает в осадок в кристаллическом виде. Выпавший осадок отфильтровывают, промывают холодной водой от остатков солей и высушивают. Выход иодантипирина составляет 57-97% в зависимости от используемого субстрата и реагента, температура плавления - 160-161°С. Продукт полностью соответствует требованиям ФСП 42-0311-1559-01 (спектры УФ, ИК, содержание основного вещества).
Существенным отличием предлагаемого способа является проведение реакции в твердой фазе без растворителя. Использование твердофазной методологии позволило исключить органический растворитель не только в самом процессе иодирования, но и на стадии очистки технического иодантипирина. Кроме того, в данном способе возможно использование в качестве основания гидрокарбоната натрия вместо щелочи, что обеспечивает большую экологичность сточных вод. Безусловно, важным является возможность использования в качестве исходного субстрата бензолсульфокислого антипирина, что уменьшает стадийность процесса, поскольку антипирин в промышленности получают гидролизом указанной соли.
При использовании данной методики не наблюдается образование побочных продуктов, которые загрязняют целевой продукт. Иодантипирин получают с хорошими выходами, продукт соответствует требованиям ФСП 42-0311-1559-01.
Совокупность предлагаемых существенных отличий позволяет достичь следующего технического результата:
получить фармакопейный иодантипирин с высоким выходом;
исключить использование органических растворителей, повысив технологическую и экологическую безопасность, а также экономичность производства за счет проведения реакции в твердой фазе;
упростить процесс получения иодантипирина за счет проведения процесса при комнатной температуре без специального регулирования температуры;
позволяет получить иодантипирин как из антипирина, так и бензолсульфокислого антипирина, что приводит к сокращению промышленной стадии производства, заключающейся в получении антипирина из БСКА;
предлагаемый способ получения иодантипирина может быть реализован в заводских условиях.
Пример 1. Иодирование антипирина реагентом Ме4NICl2 в отсутствие растворителя в условиях механической активации.
В агатовой ступке в течение 15 минут растирают 5.6 г антипирина и 9.8 г Me4NICl2. Реакционную массу выдерживают в течение 30 минут, затем добавляют 500 см3 воды, 3.5 г гидрокарбоната натрия. Полученную суспензию кипятят в течение 15 минут до полного растворения твердой фазы. Охлаждают до температуры 20-22°С и добавляют 10 г хлорида натрия. Иодантипирин выпадает, кристаллы отфильтровывают, промывают на фильтре холодной водой и сушат. Выход чистого иодантипирина - 9 г (96%), белые кристаллы с температурой плавления 160-161°С.
Пример 2. Иодирование бензолсульфокислого антипирина реагентом Et4NICl2 в отсутствие растворителя в условиях механической активации.
В агатовой ступке в течение 15 минут растирают 12 г антипирина бензолсульфокислого и 11.6 г Et4NICl2. Выдерживают в течение 30 минут. Выделение и очистка целевого иодантипирина проводится по методике, описанной выше. Выход чистого иодантипирина - 8.2 г (88%), белые кристаллы с кремовым оттенком, температурой плавления 160-161°С.
Пример 3. Иодирование антипирина хлоридом иода в отсутствие растворителя в условиях механической активации.
В агатовой ступке растирают 5.6 г антипирина и 5.8 г хлорида иода в течение 15 минут. Реакционную массу выдерживают в течение 30 минут. Выделение и очистка целевого иодантипирина проводится по методике, описанной выше. Выход чистого иодантипирина - 5.9 г (63%), белые кристаллы, температура плавления 160-161°С.
Пример 4. Иодирование антипирина металлическим иодом в отсутствие растворителя в условиях механической активации.
В агатовой ступке растирают 5.6 г антипирина и 9 г иода в течение 15 минут. Выдерживают в течение 30 минут. Выделение и очистка целевого иодантипирина проводится по методике, описанной выше. Выход чистого иодантипирина - 5.4 г (57%), белые кристаллы с температурой плавления 160-161°С.
Все полученные продукты полностью соответствуют требованиям ФСП 42-0311-1559-01.
Промышленная применяемость.
Заявленный способ получения иодантипирина может быть использован в химико-фармацевтической промышленности и в промышленности тонкого органического синтеза.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ИОДПИРАЗОЛОНА-5 (ЙОДАНТИПИРИНА) | 2009 |
|
RU2401830C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ЙОДПИРАЗОЛОНА-5(ЙОДАНТИПИРИНА) (ВАРИАНТЫ) | 1996 |
|
RU2106344C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ЙОДПИРАЗОЛОНА-5(ЙОДАНТИПИРИНА) | 2000 |
|
RU2179551C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРАМИДОНА ИЗ АНТИПИРИНА | 1935 |
|
SU46924A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННОГО БЕНЗО[B]ТИОФЕНА (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-ФЕНИЛ-3-АРОИЛБЕНЗО[B]ТИОФЕНА (ВАРИАНТЫ), И ПРОИЗВОДНЫЕ БЕНЗО[B]ТИОФЕНА | 1996 |
|
RU2147582C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИОДПОЛИСТИРОЛА | 2011 |
|
RU2467018C1 |
| СПОСОБ ИОДИРОВАНИЯ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2506254C2 |
| ИНГИБИТОР КИСЛОТНОЙ КОРРОЗИИ | 2001 |
|
RU2194092C1 |
| Способ получения иодпроизводных аминонафто- или аминоантрахинонов | 1984 |
|
SU1182025A1 |
| СПОСОБ ВЫДЕЛЕНИЯ БЕНЗОЛСУЛЬФОКИСЛОГО АНТИПИРИНА | 1985 |
|
SU1325862A1 |
Изобретение относится к области органической химии, в частности к способу получения 1-фенил-2,3-диметил-4-иодпиразолона-5 (иодантипирина), являющегося лекарственным препаратом. Предлагается способ получения иодантипирина электрофильным иодированием антипирина или бензолсульфокислого антипирина в условиях механической активации без растворителя как на стадии синтеза, так и на стадии выделения. Использование данного способа позволяет получить фармакопейный иодантипирин с выходом 57-96% в зависимости от исходного субстрата и иодирующего реагента, значительно упростить процесс его получения и полностью исключить использование органического растворителя.
Способ получения 1-фенил-2,3-диметил-4-иодпиразолона-5 (иодантипирина) путем иодирования антипирина или бензолсульфокислого антипирина, отличающийся тем, что процесс проводят в твердой фазе в условиях механической активации компонентов реакции без использования органических растворителей как на стадии синтеза, так и на стадии выделения целевого продукта.
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ЙОДПИРАЗОЛОНА-5(ЙОДАНТИПИРИНА) (ВАРИАНТЫ) | 1996 |
|
RU2106344C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ЙОДПИРАЗОЛОНА-5(ЙОДАНТИПИРИНА) | 2000 |
|
RU2179551C1 |

