Область техники
Настоящее изобретение относится к новым производным ацилбензола, проявляющим гипогликемическое действие и/или обладающим защитным действием по отношению к β-клеткам или поджелудочной железе, или к их фармацевтически приемлемой соли и к фармацевтической композиции, содержащей указанные производные в качестве активных ингредиентов.
Уровень техники
Сахарный диабет является метаболическим заболеванием, характеризующимся, главным образом, хроническим гипергликемическим состоянием, обусловленным недостаточностью действия инсулина. Лечение диабета обычно осуществляется посредством лекарственной терапии совместно с диетотерапией и лечебной физкультурой. Примеры используемых пероральных гипогликемических средств, которые представляют собой класс терапевтических лекарственных средств от диабета, включают бигуанидные агенты и тиазолидиндионовые агенты, которые уменьшают инсулинорезистентность; агенты сульфонилмочевины и глиниды, которые стимулируют секрецию инсулина из панкреатических β-клеток; и ингибиторы α-глюкозидазы, которые тормозят поглощение сахара.
Однако было показано, что бигуанидные агенты обладают вредными побочными эффектами, такими как побочные реакции со стороны пищеварительной системы и лактацидоз; тиазолидиндионовые агенты проявляют вредные побочные эффекты, такие как прибавление в весе и отек; агенты сульфонилмочевины и глиниды проявляют вредные побочные эффекты, такие как гипогликемия или вторичная резистентность из-за длительного использования; и ингибиторы α-глюкозидазы проявляют вредные побочные эффекты, такие как диарея. Поэтому разработка перорального гипогликемического средства, которое сможет разрешить такие проблемы, является желательной.
Кроме того, в последние годы были разработаны пиперидиновые соединения в качестве пероральных гипогликемических средств с новыми структурами (см., например, патентную литературу 1-4). Также, были описаны оксадиазоловые соединения (см., например, патентную литературу 5 или 6).
Перечень ссылок
Патентная литература
Патентная литература 1: WO 07/116229.
Патентная литература 2: WO 07/003960.
Патентная литература 3: WO 07/003962.
Патентная литература 4: WO 05/061489.
Патентная литература 5: WO 11/016469.
Патентная литература 6: WO 11/016470.
Сущность изобретения
Проблемы, решаемые посредством изобретения
Однако с пиперидиновыми соединениями имеются трудности, которые заключаются в том, что достаточное гипогликемическое действие и защитное действие по отношению к β-клеткам или поджелудочной железе не достигается легко. Таким образом, целью настоящего изобретения является предоставление соединений с новой структурой, которые не были описаны и не рассматривались в указанной выше патентной литературе, и которые проявляют превосходный гипогликемический эффект и защитное действие по отношению к β-клеткам или поджелудочной железе, или их фармацевтически приемлемой соли; фармацевтической композиции, проявляющей превосходный терапевтический эффект и/или профилактический эффект против диабета 1 типа, диабета 2 типа и тому подобное, которые характеризуются повышением уровня сахара в крови вследствие нарушения метаболизма сахара; и фармацевтической композиции, проявляющей защитное действие по отношению к β-клеткам или поджелудочной железе.
Способы решения проблем
Настоящее изобретение предоставляет:
(1) Соединение, представленное общей формулой (I):
[Химическая формула 1]
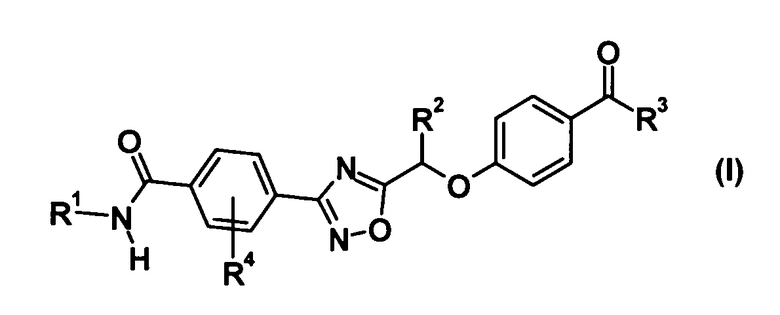
где R1 представляет собой гидрокси С1-С6 алкильную группу или гидрокси С3-С6 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α,
подгруппа заместителей α является группой, состоящей из гидроксильной группы и карбамоильной группы,
R2 представляет собой метильную группу или этильную группу,
R3 представляет собой С1-С6 алкильную группу или С3-С6 циклоалкильную группу, каждая из которой может быть замещена 1-3 атомами галогена, и
R4 представляет собой атом галогена;
или его фармацевтически приемлемую соль;
(2) соединение по пункту (1), где R1 представляет собой гидрокси С1-С3 алкильную группу или гидрокси С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α;
(3) соединение по пункту (1), где R1 представляет собой гидроксиэтильную группу, гидроксипропильную группу, гидроксиизопропильную группу или гидроксициклопентильную группу, каждая из которой может быть замещена одним заместителем, выбранным из подгруппы заместителей α;
(4) соединение по любому одному из пунктов (1)-(3), где R3 представляет собой С1-С4 алкильную группу или С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 атомами галогена;
(5) соединение по любому одному из пунктов (1)-(3), где R3 представляет собой изопропильную группу, трет-бутильную группу, циклопропильную группу или циклобутильную группу, каждая из которой может быть замещена одним атомом галогена;
(6) соединение по любому одному из пунктов (1)-(5), где R4 представляет собой атом фтора;
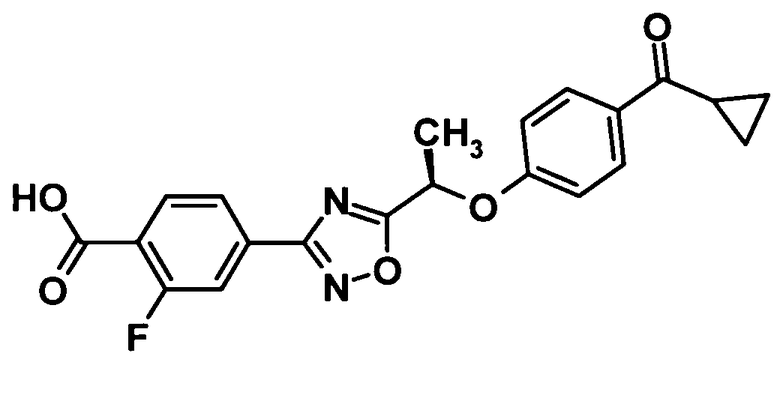
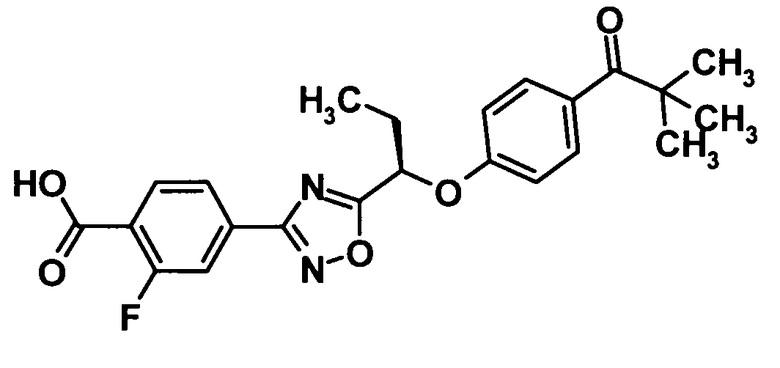
(7) соединение, выбранное из группы, состоящей из следующих соединений:
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2S)-2,3-дигидроксипропил]-2-фторбензамид,
N-[(1S)-2-амино-1-(гидроксиметил)-2-оксоэтил]-4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2R)-2,3-дигидроксипропил]-2-фторбензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R,2R)-2-гидроксициклопентил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид,
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S,2S)-2-гидроксициклопентил]бензамид,
4-(5-{(1R)-1-[4-(2,2-диметилпропаноил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид,
4-(5-{(1R)-1-[4-(циклобутилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид,
2-фтор-4-{5-[(1R)-1-{4-[(1-фторциклопропил)карбонил]фенокси}пропил]-1,2,4-оксадиазол-3-ил)-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид,
2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-{5-[(1R)-1-(4-изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамид, и
2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]-4-{5-[(1R)-1-(изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамид;
(8) фармацевтическую композицию, содержащую, в качестве активного ингредиента, соединение по любому одному из пунктов (1)-(7) или его фармацевтически приемлемую соль;
(9) фармацевтическую композицию по пункту (8) для лечения диабета 1 типа, диабета 2 типа или ожирения;
(10) фармацевтическую композицию по пункту (8) для защиты β-клеток или поджелудочной железы;
(11) применение соединения по любому одному из пунктов (1)-(7) или его фармацевтически приемлемой соли для приготовления фармацевтической композиции;
(12) способ лечения заболевания, включающий введение млекопитающему соединения по любому одному из пунктов (1)-(7) или его фармацевтически приемлемой соли; и
(13) способ по пункту (12), где млекопитающим является человек.
Полезные эффекты изобретения
Согласно настоящему изобретению предоставлено производное ацилбензола, проявляющее превосходный гипогликемический эффект и защитное действие относительно β-клеток или поджелудочной железы, или его фармацевтически приемлемая соль, фармацевтическая композиция, имеющая превосходный терапевтический эффект и/или профилактический эффект против диабета 1 типа, диабета 2 типа и тому подобное, который характеризуется повышением в крови уровня сахара, и фармацевтическая композиция, проявляющая защитное действие относительно β-клеток или поджелудочной железы.
Наилучшие способы осуществления изобретения
“С1-С6 алкильная группа”, как используют в описании настоящего изобретения, означает линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода. Конкретные примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, сек-бутильную группу, трет-бутильную группу, пентильную группу, 1,2-диметилпропильную группу, изопентильную группу, гексильную группу и изогексильную группу.
“С3-С6 циклоалкильная группа”, как используют в описании настоящего изобретения, означает насыщенную циклическую углеводородную группу, содержащую 3-6 атомов углерода, и примеры включают циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексильную группу.
“Гидрокси С1-С6 алкильная группа”, как используют в описании настоящего изобретения, означает группу, образованную замещением атома водорода “С1-С6 алкильной группы” гидроксильной группой. Конкретные примеры включают гидроксиметильную, 2-гидроксиэтильную, 4-гидроксибутильную, 5-гидроксипентильную и 6-гидроксигексильную группы.
“Гидрокси С3-С6 циклоалкильная группа”, как используют в описании настоящего изобретения, означает группу, образованную замещением атома водорода “С3-С6 циклоалкильной группы” гидроксильной группой. Конкретные примеры включают гидроксициклопропильную группу, гидроксициклобутильную группу, 2-гидроксициклопентильную группу и гидроксициклогексильную группу.
“Атом галогена”, как используют в описании настоящего изобретения, означает атом фтора, атом хлора, атом брома или атом йода.
“Фармацевтически приемлемая соль”, как используют в описании настоящего изобретения, означает соль, образованную при взаимодействии соединения согласно настоящему изобретению с кислотой или основанием.
Примеры соли включают соли с галогенводородными кислотами, такие как гидрофториды, гидрохлориды, гидробромиды и гидройодиды; соли с неорганическими кислотами, такие как гидрохлориды, нитраты, перхлораты, сульфаты и фосфаты; соли с низшими алкансульфокислотами, такие как метансульфонаты, трифторметансульфонаты и этансульфонаты; соли с арилсульфокислотами, такие как бензолсульфонаты и р-толуолсульфонаты; соли с органическими кислотами, такие как ацетаты, малаты, фумараты, сукцинаты, цитраты, аскорбаты, тартраты, оксалаты и малеаты; соли со щелочными металлами, такие как натриевые соли, калиевые соли и литиевые соли; соли с щелочноземельными металлами, такие как кальциевые соли и магниевые соли; соли металлов, такие как соли алюминия и соли железа; неорганические соли, такие как аммониевые соли; соли амина, включающие органические соли, такие как t-октиламинные соли, дибензиламинные соли, морфолиновые соли, глюкозаминные соли, соли фенилглициналкилового сложного эфира, этилендиаминные соли, соли N-метилглюкамина, гуанидиновые соли, диэтиламинные соли, триэтиламинные соли, дициклогексиламинные соли, N,N'-дибензилэтилендиаминные соли, хлорпрокаиновые соли, прокаиновые соли, диэтаноламинные соли, N-бензилфенэтиламинные соли, пиперазиновые соли, тетраметиламмониевые соли и трис(гидроксиметил)аминометановые соли; и соли аминокислот, такие как глициновые соли, лизиновые соли, аргининовые соли, орнитиновые соли, глутаматы и аспартаты.
Соединение согласно настоящему изобретению поглощает воду, например, при хранении в атмосфере, так что поглощенная вода может присоединиться к соединению и может образоваться гидрат. Поэтому такой гидрат также включен в представление о соли согласно настоящему изобретению.
Поскольку соединение согласно настоящему изобретению может иметь асимметрические атомы углерода в молекуле, соединение существует в виде оптических изомеров. Указанные изомеры и смеси указанных изомеров все представлены единой формулой, то есть общей формулой (I). Поэтому настоящее изобретение охватывает все оптические изомеры соединения, представленные общей формулой (I), и смеси указанных оптических изомеров при любых соотношениях. Такой оптический изомер может быть получен, например, при использовании сырых материалов с оптической активностью вместо сырых материалов, используемых в способах получения, ссылочных примерах и примерах, которые будут описаны ниже, или может быть получен путем оптического разделения, известного в данной области техники, соединения, полученного в способах получения, ссылочных примерах, примерах и тому подобном, которые будут описаны ниже, например, методом разделения диастереомеров, методом, основанном на ферментативной реакции, или методом оптического разделения, основанном на хроматографии.
Настоящее изобретение также может охватывать соединения, у которых один или более атомов, составляющих молекулу соединения, представленного общей формулой (I), были замещены изотопами атомов. Изотопы включают два класса радиоактивных изотопов и стабильных изотопов, и примеры изотопов включают, например, изотопы водорода (2Н и 3Н), изотопы углерода (11С, 13С и 14С), изотопы азота (13N и 15N), изотопы кислорода (15О, 17О и 18О) и изотопы фтора (18F). Композицию, содержащую меченое изотопом соединение, используют, например, в качестве терапевтического средства, профилактического средства, реагента для исследования, реагента для анализа, диагностического средства или in vivo средства для диагностической визуализации. Соединения, меченные изотопами, и смеси соединений, меченных изотопами при любых соотношениях, все включены в объем настоящего изобретения. Меченное изотопом соединение может быть получено способом, известным в данной области, например, при использовании сырых материалов, меченных изотопами, вместо сырых материалов, используемых в способах получения, которые будут описаны ниже.
Настоящее изобретение также может охватывать пролекарства соединения, представленного общей формулой (I). Пролекарство является производным соединения, представленного общей формулой (I), и означает соединение, которое посредством ферментативной реакции или химической реакции превращается в соединение согласно настоящему изобретению в живом организме.
Примеры пролекарства включают соединения, у которых аминогруппа в молекуле ацилирована, алкилирована или фосфорилирована; соединения, у которых карбонильная группа в молекуле этерифицирована или амидирована; и соединения, у которых гидроксильная группа в молекуле ацилирована, алкилирована или фосфорилирована (см., например, Povl Krogsgaard-Larsen, et al., “A Textbook of Drug Design and Development”, Second Edition, Harwood Academic Publishers, 1996, pp. 351-385). Такое пролекарство может быть получено из соединения, представленного общей формулой (I), способом, известным в данной области техники.
Группа R1 предпочтительно представляет собой гидрокси С1-С3 алкильную группу или С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α; более предпочтительно группа представляет собой гидроксиэтильную группу, гидроксипропильную группу, гидроксиизопропильную группу или гидроксициклопентильную группу, каждая из которых может быть замещена одним заместителем, выбранным из подгруппы заместителей α; и даже более предпочтительно группа представляет собой 2,3-дигидроксипропильную группу, 2-гидрокси-1-гидроксиметилэтильную группу, 1-карбамоил-2-гидроксиэтильную группу, 2-гидрокси-1-метилэтильную группу или 2-гидроксициклопентильную группу.
Группа R2 предпочтительно представляет собой этильную группу.
Группа R3 предпочтительно представляет собой С1-С4 алкильную группу или С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 атомами галогена; более предпочтительно группа представляет собой изопропильную группу, трет-бутильную группу, циклопропильную группу или циклобутильную группу, каждая из которых может быть замещена 1-3 атомами галогена; и даже более предпочтительно группа представляет собой изопропильную группу, трет-бутильную группу, циклопропильную группу, 1-фторциклопропильную группу или циклобутильную группу.
Группа R4 предпочтительно представляет собой атом фтора.
Предпочтительной комбинацией групп R1, R2, R3 и R4 в общей формуле (I) является комбинация, в которой R1 означает гидрокси С1-С3 алкильную группу или С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α; R2 означает этильную группу; R3 является С1-С4 алкильной группой или С3-С4 циклоалкильной группой, каждая из которой может быть замещена 1-3 атомами галогена, и R4 представляет собой атом фтора.
Более предпочтительной комбинацией является комбинация, в которой группа R1 представляет собой гидроксиэтильную группу, гидроксипропильную группу, гидроксиизопропильную группу или гидроксициклопентильную группу, каждая из которых может быть замещена одним заместителем, выбранным из подгруппы заместителей α; R2 является этильной группой; R3 является изопропильной группой, трет-бутильной группой, циклопропильной группой или циклобутильной группой, каждая из которых может быть замещена 1-3 атомами галогена, и R4 представляет собой атом фтора.
Даже более предпочтительной комбинацией является комбинация, в которой R1 представляет собой 2,3-дигидроксипропильную группу, 2-гидрокси-1-гидроксиметилэтильную группу, 1-карбамоил-2-гидроксиэтильную группу, 2-гидрокси-1-метилэтильную группу или 2-гидроксициклопентильную группу; R2 является этильной группой; R3 является изопропильной группой, трет-бутильной группой, циклопропильной группой, 1-фторциклопропильной группой или циклобутильной группой; и R4 представляет собой атом фтора.
Соединение согласно настоящему изобретению может быть получено, например, следующими способами.
В реакциях на различных стадиях способов, описанных ниже, когда соединение, служащее в качестве субстрата в реакции, содержит группу, которая ингибирует заданную реакцию (например, аминогруппа, гидроксильная группа или карбоксильная группа), введение защитной группы к такой группе и удаление введенной защитной группы может быть осуществлено при необходимости. Определенных ограничений на такие защитные группы не имеется, поскольку они являются обычно используемыми защитными группами, но примеры включают такие защитные группы, описанные в публикации T.H.Greene, P.G.Wuts, Protective Groups in Organic Synthesis. Third Edition, 1999, John Wiley & Sons, Inc., или тому подобное. Реакцию введения таких защитных групп и реакцию удаления защитных групп можно осуществлять обычными способами, такими как способы, описанные в упомянутой выше литературе.
[Химическая формула 2]
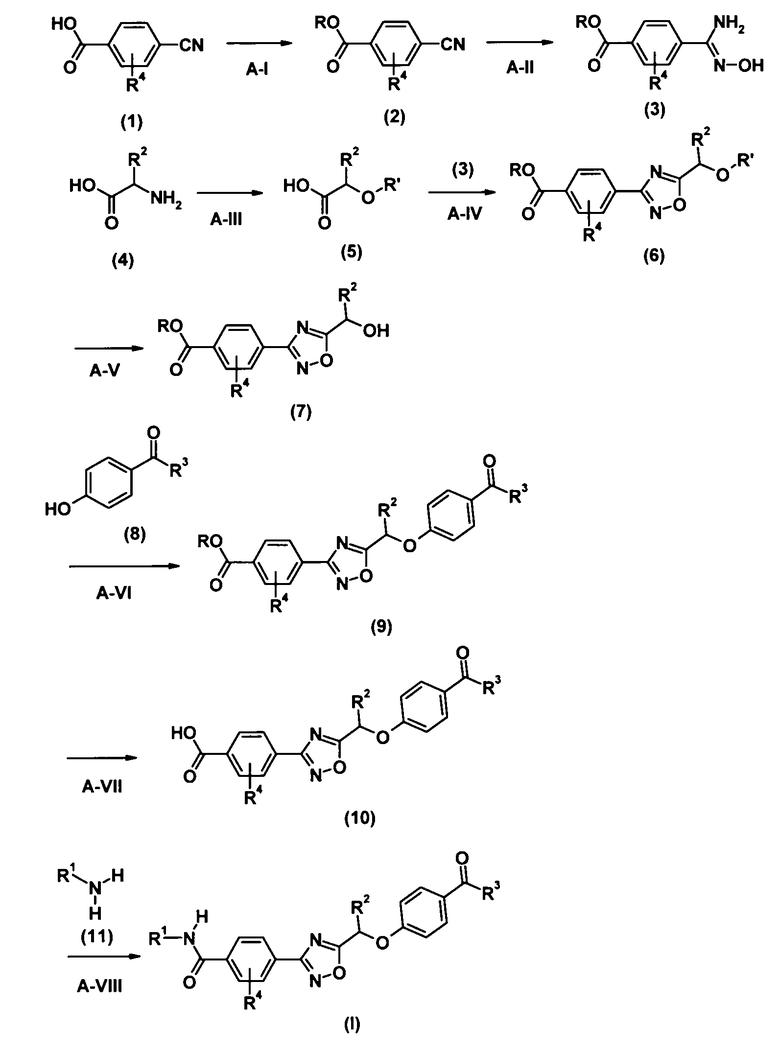
где R и R′ представляют собой защитную группу карбоксильной группы и защитную группу гидроксильной группы соответственно, и R1, R2, R3 и R4 соответственно имеют такие же значения, как определено выше.
Стадия A-I является стадией получения соединения (2) путем введения защитной группы R в карбоксильную группу соединения (1). Используемые в реакции растворитель, реагент, температура реакции и время реакции могут быть выбраны соответственно, например, опираясь на ссылку T.H.Greene, P.G.Wuts, Protective Groups in Organic Synthesis. Third Edition, 1999, John Wiley & Sons, Inc., или тому подобное.
Стадия A-II является стадией получения соединения (3) путем взаимодействия соединения (2) с гидроксиламином.
Примеры используемого в реакции растворителя включают метанол, этанол, бутанол, толуол, дихлорметан, диметилформамид (ДМФ), тетрагидрофуран (ТГФ) и диметилсульфоксид, и предпочтительным является этанол или ТГФ.
Примеры используемого в реакции гидроксиламина включают 50% масс./масс. водный раствор гидроксиламина и гидрохлорида гидроксиламина, и предпочтительным примером является 50% масс./масс. водный раствор гидроксиламина.
Температура реакции составляет от 0°С до 150°С и предпочтительно от 70°С до 90°С. Время реакции составляет от 30 минут до 24 часов и предпочтительно от 2 до 4 часов.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. Реакционную смесь охлаждают до комнатной температуры, далее растворитель подвергают дистилляции при пониженном давлении и образовавшийся остаток промывают водой.
Стадия A-III является стадией получения соединения (5) путем взаимодействия соединения (4) со сложным эфиром азотистой кислоты и карбоновой кислотой.
Примеры используемого в реакции эфира азотистой кислоты включают трет-бутилнитрит и н-бутилнитрит, и предпочтительным является трет-бутилнитрит.
Примеры используемой в реакции карбоновой кислоты включают, например, уксусную кислоту и пропионовую кислоту, и предпочтительной является уксусная кислота.
Температура реакции составляет от -20°С до 100°С и предпочтительно от 0°С до 70°С. Время реакции составляет от 1 до 24 часов и предпочтительно от 2 до 4 часов.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. Реакционную смесь охлаждают до комнатной температуры и затем растворитель подвергают дистилляции при пониженном давлении. Воду добавляют к образовавшемуся остатку и продукт экстрагируют органическим растворителем, таким как этилацетат. Полученный таким образом органический слой сушат над обезвоживающим веществом, таким как сульфат натрия. После удаления нерастворившегося вещества растворитель подвергают дистилляции при пониженном давлении.
Стадия A-IV является стадией получения соединения (6) путем взаимодействия соединения (3), полученного на стадии A-II, с соединением (5), полученным на стадии A-III, в присутствии конденсирующего реагента.
Примеры используемого в этой реакции растворителя включают, например, дихлорметан, тетрагидрофуран, 1,4-диоксан, ДМФ, диметилацетамид, предпочтительным растворителем является дихлорметан или ДМФ, и более предпочтительным является ДМФ.
Используемый в реакции конденсирующий реагент включает (i) карбодиимиды, такие как 1,3-дициклогексилкарбодиимид, 1,3-диизопропилкарбодиимид и 1-этил-3-(3-диметиламинопропил)карбодиимид (WSC) и комбинации указанных карбодиимидов и N-гидроксисоединений, таких как 1-гидроксибензотриазол, 1-гидрокси-7-азабензотриазол и N-гидроксисукцинимид, и (ii) имидазолы, такие как 1,1′-карбонилдиимидазол (CDI), и предпочтительной является комбинация WSC и 1-гидроксибензотриазола.
Температура реакции составляет от 0°С до 200°С и предпочтительно от 20°С до 120°С. Время реакции составляет от 30 минут до 24 часов и предпочтительно от 2 до 4 часов.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. Реакционную смесь охлаждают до комнатной температуры и затем растворитель подвергают дистилляции при пониженном давлении. Воду добавляют к образовавшемуся остатку и продукт экстрагируют органическим растворителем, таким как этилацетат. Полученный таким образом органический слой сушат над обезвоживающим веществом, таким как сульфат натрия. После удаления нерастворенного вещества растворитель подвергают дистилляции при пониженном давлении и остаток очищают хроматографией на силикагеле.
Стадия A-V является стадией получения соединения (7) путем удаления защитной группы R′ из соединения (6), полученного на стадии A-IV.
Примеры используемого в реакции растворителя включают метанол, этанол, 2-пропанол и n-бутанол, и предпочтительным растворителем является этанол.
Примеры используемого в реакции реагента включают карбонат калия, карбонат натрия, гидроксид калия и гидроксид натрия, и предпочтительным является карбонат калия.
Температура реакции составляет от -30°С до 100°С и предпочтительно от -20°С до 20°С. Время реакции составляет от 10 до 120 минут и предпочтительно от 20 до 50 минут.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. Кислоту, такую как хлористоводородная кислота, добавляют к реакционной смеси, чтобы она стала немного кислой или нейтральной, и продукт экстрагируют органическим растворителем, таким как этилацетат. Полученный таким образом органический слой сушат над обезвоживающим веществом, таким как сульфат натрия. После удаления нерастворенного вещества растворитель подвергают дистилляции при пониженном давлении и остаток очищают хроматографией на силикагеле.
Стадия A-VI является стадией получения соединения (9) путем взаимодействия соединения (7), полученного на стадии A-V, с соединением (8).
Примеры используемого в реакции растворителя включают ТГФ, дихлорметан, ацетонитрил и толуол, и предпочтительным растворителем является ТГФ.
Примеры используемого в реакции реагента включают (i) комбинации сложных эфиров азодикарбоновой кислоты, таких как диэтилазодикарбоксилат и ди-трет-бутилазодикарбоксилат, и фосфинов, таких как трифенилфосфин и трибутилфосфин, и (ii) (цианометилен)фосфораны, такие как (цианометилен)триметилфосфоран и (цианометилен)трибутилфосфоран, и предпочтительной комбинацией является комбинация ди-трет-бутилазодикарбоксилата и трифенилфосфина.
Температура реакции составляет от -20°С до 100°С и предпочтительно от 0°С до 40°С. Время реакции составляет от 10 минут до 6 часов и предпочтительно от 30 минут до 2 часов.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. После дистилляции растворителя при пониженном давлении воду добавляют к реакционной смеси и продукт экстрагируют органическим растворителем, таким как этилацетат. Полученный таким образом органический слой сушат над обезвоживающим веществом, таким как сульфат натрия. После удаления нерастворенного вещества растворитель подвергают дистилляции при пониженном давлении и остаток очищают хроматографией на силикагеле.
Стадией A-VII является стадия получения соединения (10) путем удаления защитной группы R из соединения (9), полученного на стадии A-VI.
Примеры используемого в реакции растворителя включают дихлорметан и этилацетат, и предпочтительным растворителем является дихлорметан.
Примеры используемого в реакции реагента включают хлористый водород и трифторуксусную кислоту, и предпочтительным реагентом является трифторуксусная кислота.
Температура реакции составляет от -20°С до 60°С и предпочтительно от 10°С до 30°С. Время реакции составляет от 10 минут до 6 часов и предпочтительно от 20 минут до 2 часов.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. Растворитель подвергают дистилляции при пониженном давлении и образовавшийся остаток промывают изопропиловым эфиром.
Стадией A-VIII является стадия получения соединения (I) согласно настоящему изобретению путем взаимодействия соединения (10), полученного на стадии A-VII, с аминосодержащим соединением (11), в присутствии конденсирующего реагента.
Примеры используемого в реакции растворителя включают дихлорметан, ТГФ, 1,4-диоксан, ДМФ и диметилацетамид, предпочтительным растворителем является дихлорметан или ДМФ, и более предпочтительным является ДМФ.
Используемый в реакции конденсирующий реагент является не очень ограниченным, поскольку он является средством, используемым в реакциях амидирования, и конденсирующие реагенты, описанные в публикации R.C. Larock, Comprehensive Organic Transformations, Second Edition, 1999, John Wiley & Sons, Inc., и тому подобное могут быть использованы. Конкретные примеры включают (i) сложные эфиры фосфорной кислоты, такие как диэтилфосфорилцианид; (ii) карбодиимиды, такие как 1,3-дициклогексилкарбодиимид, 1,3-диизопропилкарбодиимид и WSC и комбинации указанных карбодиимидов и N-гидроксисоединений, таких как 1-гидроксибензотриазол; (iii) имидазолы, такие как CDI; (iv) 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (DMT-MM); и (v) фосфаты, такие как О-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония гексафторфосфат (HATU) и О-бензотриазол-1-ил-N,N,N′,N′-тетраметилурония гексафторфосфат (HBTU). Предпочтительной является комбинация WSC и 1- гидроксибензотриазола.
Температура реакции составляет от 0°С до 100°С и предпочтительно от 0°С до 50°С. Время реакции составляет от 30 минут до 96 часов и предпочтительно от 1 до 12 часов.
Если требуется, например, последующая обработка, то ее можно осуществлять по следующей процедуре. Воду добавляют к реакционной смеси и продукт экстрагируют органическим растворителем, таким как этилацетат. Полученный таким образом органический слой промывают водой насыщенным раствором соли и тому подобное и сушат над обезвоживающим веществом, таким как сульфат натрия. Растворитель подвергают дистилляции при пониженном давлении и остаток очищают хроматографией на силикагеле.
Соединение согласно настоящему изобретению можно получать способами, описанными выше, и также можно легко получать из известных соединений согласно ссылочным примерам и примерам, которые будут описаны ниже.
Соединение согласно настоящему изобретению или его фармацевтически приемлемая соль, полученное описанными выше способами, имеет превосходный гипогликемический эффект, и поэтому оно может быть использовано в качестве активного ингредиента фармацевтической композиции, которую можно использовать в лечении и/или предотвращении диабета 1 типа, диабета 2 типа, гестационного диабета, гипергликемии, обусловленной другими факторами, нарушения толерантности к глюкозе (IGT), ожирения, ассоциированных с диабетом заболеваний (например, гиперлипидемия, гиперхолестеринемия, нарушение липидного метаболизма, гипертензия, жировой метаморфоз печени, метаболический синдром, отек, сердечная недостаточность, стенокардия, инфаркт миокарда, артериосклероз, гиперурикемия и подагра) или диабетических осложнений (например, ретиноз, почечная недостаточность, нейропатия, катаракта, гангрена нижней конечности, инфекции и кетоз).
Кроме того, соединение согласно настоящему изобретению или его фармацевтически приемлемая соль имеет превосходный защитный эффект по отношению к β-клеткам или поджелудочной железе и поэтому может быть использовано в качестве активного ингредиента фармацевтической композиции, которую можно использовать для защиты β-клеток или поджелудочной железы.
Соединение согласно настоящему изобретению также может быть использовано в комбинации с терапевтическим лекарственным средством от диабета, отличным от соединения согласно настоящему изобретению, терапевтическим лекарственным средством, применяемым против диабетических осложнений, терапевтическим лекарственным средством от гиперлипидемии, терапевтическим лекарственным средством от гипертензии и тому подобное.
Когда фармацевтическую композицию, содержащую соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, вводят млекопитающему (например, человеку, лошади, корове или свинье; предпочтительно человеку), фармацевтическую композицию можно вводить системно или местно и перорально или парентерально.
Соответствующие лекарственные формы фармацевтической композиции согласно настоящему изобретению могут быть выбраны согласно способу введения. Фармацевтическая композиция согласно настоящему изобретению может быть приготовлена способами, используемыми для приготовления различных обычно применяемых препаратов.
Примеры лекарственной формы фармацевтической композиции для перорального использования включают таблетки, пилюли, порошки, гранулы, капсулы, жидкости, суспензии, эмульсии, сиропы и эликсиры. Фармацевтические композиции таких лекарственных форм могут быть приготовлены обычными способами с использованием соответствующе выбранных, при необходимости, эксципиентов, связующих веществ, дезинтегрирующих средств, смазывающих средств, веществ, способствующих набуханию, набухающих добавок, покрывающих средств, пластификаторов, стабилизирующих средств, антисептиков, антиоксидантов, красителей, добавок, способствующих растворению, суспендирующих средств, эмульгаторов, подсластителей, консервантов, буферов, разбавителей, увлажняющих средств и тому подобное, которые обычно используют в качестве добавок.
Примеры лекарственных форм фармацевтической композиции для парентерального использования включают инъекции, мази, гели, кремы, горячие примочки, пластыри, аэрозоли, средства для ингаляции, спреи, глазные капли, капли в нос и суппозитории. Фармацевтические композиции таких лекарственных форм могут быть приготовлены обычными способами, соответствующе выбирая при необходимости стабилизирующие средства, антисептики, добавки, способствующие растворению, увлажняющие средства, консерванты, антиоксиданты, ароматизирующие вещества, гелеобразующие средства, нейтрализующие средства, буферы, изотоничные средства, поверхностно-активные вещества, красители, буферные вещества, загустители, смачивающие средства, наполнители, средства, стимулирующие всасывание, суспендирующие средства, связующие вещества и тому подобное, которые обычно применяют в качестве добавок.
Вводимое количество соединения согласно настоящему изобретению или его фармацевтически приемлемой соли может варьировать в зависимости от симптомов, возраста, массы тела или тому подобное. Однако в случае перорального введения соединение или его соль вводят один раз или несколько раз в день в количестве 1-2000 мг и предпочтительно 1-400 мг, относительно соединения, на дозу для взрослого; и в случае парентерального введения соединение или его соль вводят один раз или несколько раз в день в количестве 0,01-500 мг и предпочтительно 0,1-300 мг, относительно соединения, на дозу для взрослого.
В дальнейшем, настоящее изобретение будет описано более подробно посредством ссылочных примеров, примеров, примеров получения, примера препарата и тестовых примеров, но объем настоящего изобретения не предназначен ограничивать указанные примеры.
Примеры
(Ссылочный пример 1)
Трет-бутил 4-циано-2-фторбензоат
[Химическая формула 3]
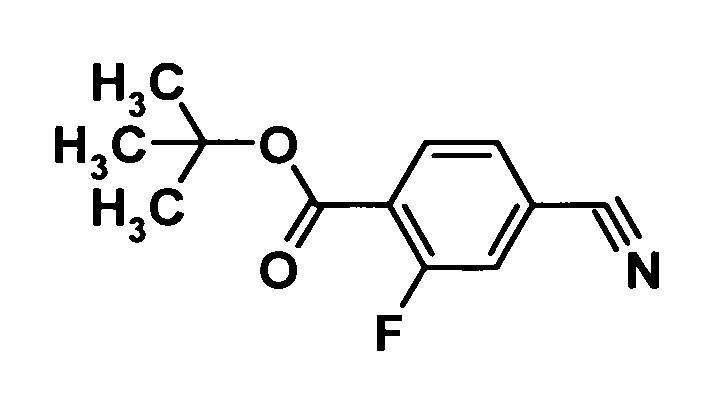
Ди-трет-бутил дикарбонат (145,4 г, 666 ммоль) и 4-диметиламинопиридин (7,40 г, 60,6 ммоль) добавляли к раствору 4-циано-2-фторбензоата в смеси трет-бутиловый спирт (1000 мл)-тетрагидрофуран (500 мл) и смесь перемешивали при 60°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и нерастворенный материал удаляли фильтрованием через целит. Растворитель отгоняли при пониженном давлении. Таким образом, был получен сырой продукт указанного в заголовке соединения.
(Ссылочный пример 2)
Трет-бутил 4-амино(гидроксиимино)метил-2-фторбензоат
[Химическая формула 4]
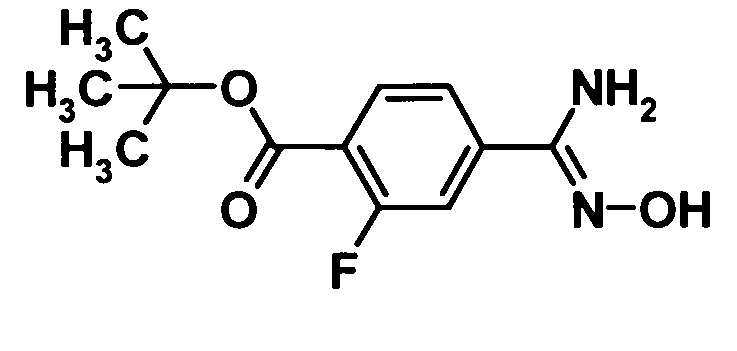
Водный 50%-ный раствор гидроксиламина (60 мл, 100 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 1 (11,0 г, 66,6 ммоль), в смеси этанол (100 мл)-тетрагидрофуран (50 мл) и смесь перемешивали при 80°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и затем растворитель отгоняли при пониженном давлении. Образовавшийся остаток промывали водой и сушили при 40°С в течение 2 дней при пониженном давлении. Таким образом, было получено указанное в заголовке соединение (150,0 г, выход: 98%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,89 (1H, т, J=8 Гц), 7,44 (2H, дд, J=8,2 Гц), 7,39 (2H, дд. J=11,2 Гц), 4,90 (2H, с), 1,60 (9H, с).
(Ссылочный пример 3)
Циклопропил (4-гидроксифенил)метанон
[Химическая формула 5]
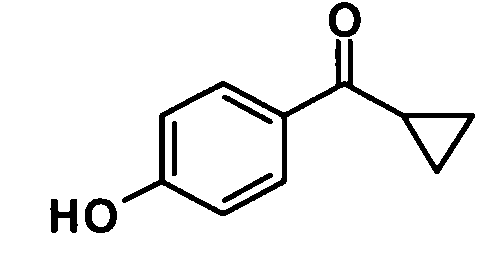
4-Хлорпропил (4-гидроксифенил)метанон (25,1 г, 127 ммоль) добавляли к 2 N водному раствору гидроксида натрия (283 мл, 566 ммоль) несколькими порциями при охлаждении льдом. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 6 часов, затем разбавленную серную кислоту (1,8 N) добавляли к реакционной смеси при охлаждении льдом до тех пор, пока рН не достигал величины 2. Реакционную смесь подвергали экстракции этилацетатом дважды. Полученный таким образом органический слой промывали водой и насыщенным раствором соли и затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 4:1→2:1, об./об.) с получением указанного в заголовке соединения (17,7 г, выход: 86%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,99-7,96 (2H, м), 6,93-6,89 (2H, м), 6,16 (1H, с), 2,67-2,61 (1H, м), 1,28-1,18 (2H, м), 1,09-0,97 (2H, м).
(Ссылочный пример 4)
(2S)-2-Ацетоксимасляная кислота
[Химическая формула 6]
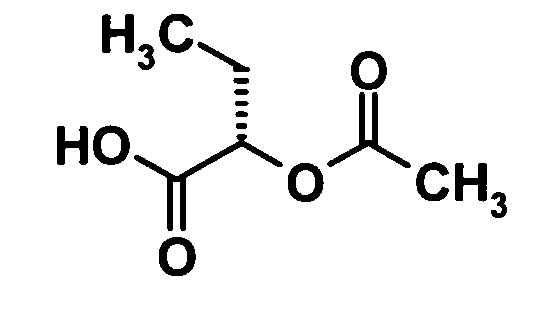
Ацетат натрия (11,9 г, 146 ммоль) и трет-бутилнитрит (15,0 г, 146 ммоль) добавляли к раствору (2S)-2-аминомасляной кислоты (10,0 г, 97,0 ммоль) в уксусной кислоте (300 мл) при охлаждении льдом и перемешивали при 60°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и затем растворитель отгоняли при пониженном давлении. Воду добавляли к остатку и смесь подвергали экстрагированию этилацетатом дважды. Полученный таким образом органический слой промывали водой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, затем остаток в виде азеотропной смеси с 1,4-диоксаном (50 мл) подвергали кипячению дважды. Таким образом, было получено указанное в заголовке соединение (8,4 г, выход: 60%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
5,00 (1H, м), 2,15 (3H, с), 1,94-1,90 (2H, м), 1,03 (3H, т. J=7 Гц);
МС (FAB+) m/z: 147 [M+H]+.
(Ссылочный пример 5)
Трет-бутил 4-{5-[(1S)-1-ацетоксипропил]-1,2,4-оксадиазол-3-ил}-2-фторбензоат
[Химическая формула 7]
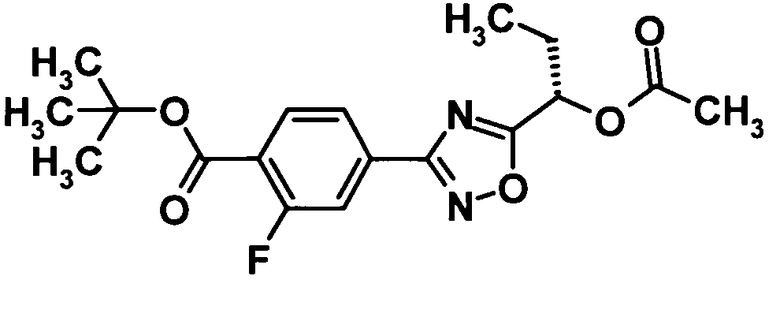
Моногидрат 1-гидроксибензотриазола (7,2 г, 53,0 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (20,3 г, 159 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 4 (7,8 г, 53,0 ммоль), в N,N-диметилформамиде (200 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Полученное в ссылочном примере 2 соединение (13,5 г, 53,0 ммоль) добавляли, смесь перемешивали в течение 30 минут и затем перемешивали при 100°С в течение 3 часов. Затем реакционную смесь охлаждали до комнатной температуры, воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали водой и 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→85:15, об./об.) с получением указанного в заголовке соединения (14,7 г, выход: 76%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,96 (1H, т, J=8 Гц), 7,90 (1H, дд. J=8, 2 Гц), 7,84 (1H, дд, J=ll,2 Гц), 5,92 (1H, т, J=7 Гц), 2,21 (3H, с), 2,16-2,08 (2H, м), 1,62 (9H, с), 1,05 (3H, т, J=7 Гц);
МС (FAB+) m/z: 365 [M+H]+.
(Ссылочный пример 6)
Трет-бутил 2-фтор-4-{5-[(1S)-1-гидроксипропил]-1,2,4-оксадиазол-3-ил}бензоат
[Химическая формула 8]
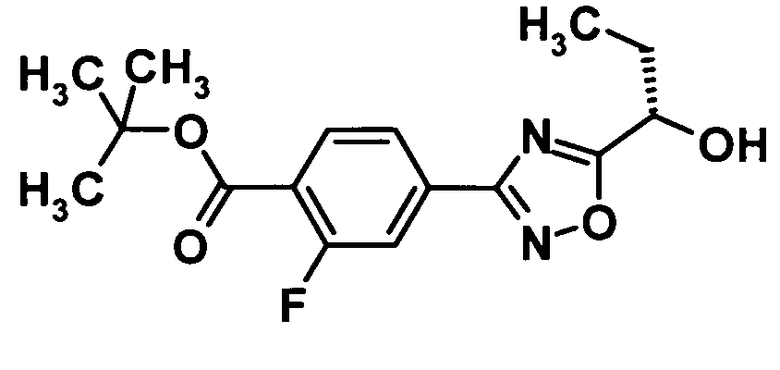
Карбонат калия (8,4 г, 61 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 5 (14,7 г, 40,3 ммоль), в метаноле (100 мл) при охлаждении льдом, и смесь перемешивали при той же температуре в течение 30 минут. К реакционной смеси добавляли 2 N хлористоводородную кислоту при той же температуре до тех пор, пока рН не достигал величины 6,0. Реакционную смесь подвергали экстрагированию этилацетатом дважды, полученный органический слой промывали водой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→80:20, об./об.) с получением указанного в заголовке соединения (12,9 г, выход: 84%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,97 (1H, т, J=8 Гц), 7,91 (1H, д, J=8 Гц), 7,85 (1H, д, J=11 Гц), 4,98 (1H, кв, J=6 Гц), 2,54 (1H, уш.с), 2,14-1,96 (2H, м), 1,62 (9H, с), 1,08 (3H, т, J=7 Гц);
МС (FAB+) m/z: 323 [M+H]+.
(Ссылочный пример 7)
Трет-бутил 4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоат
[Химическая формула 9]
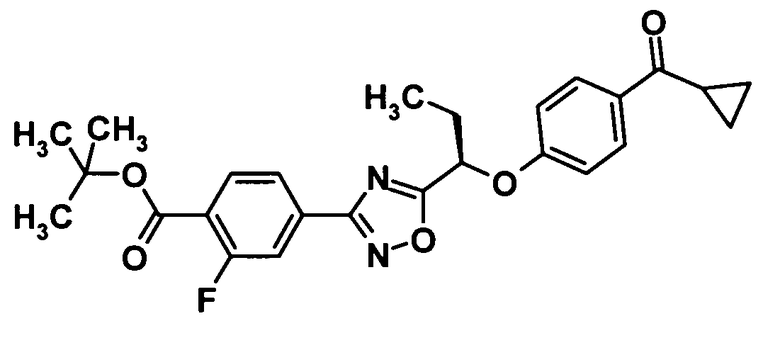
Ди-трет-бутилазодикарбоксилат (260 мг, 1,11 ммоль) и трифенилфосфин (300 мг, 1,11 мммоль) добавляли к раствору соединения, полученного в ссылочном примере 6 (300 мг, 0,931 ммоль), и соединения, полученного в ссылочном примере 3 (150 мг, 0,925 ммоль), в тетрагидрофуране (10 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→80:20, об./об.) с получением указанного в заголовке соединения (236 мг, выход: 55%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,00-7,94 (3H, м), 7,90-7,87 (1H, м), 7,84-7,81 (1H, м), 7,06-7,04 (2Н, м), 5,52 (1Н, дд, J=7, 6 Гц), 2,63-2,57 (1H, м), 2,34-2,25 (2H, м), 1,61 (9H, с), 1,21-1,18 (2H, м), 1,14 (3H, т, J=7 Гц), 1,01-0,98 (2H, м);
МС (FAB+) m/z: 466 [M+H]+.
(Ссылочный пример 8)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензойная кислота
[Химическая формула 10]
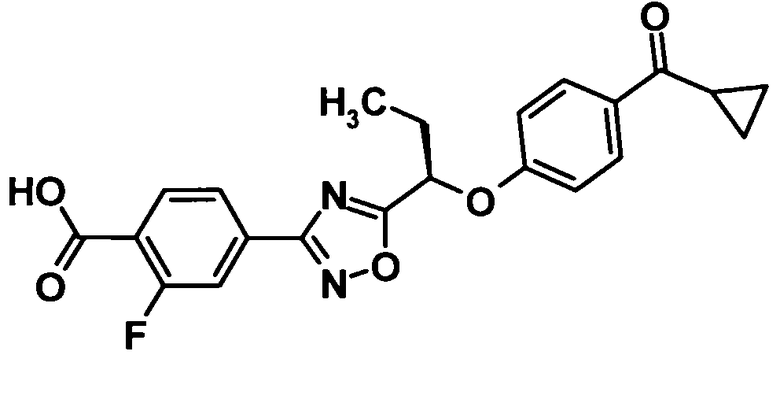
Трифторуксусную кислоту (10 мл) добавляли к раствору соединения, полученного в ссылочном примере 7 (236 мг, 0,506 ммоль), в дихлорметане (1 мл) при комнатной температуре и смесь перемешивали в течение 40 минут. Растворитель отгоняли при пониженном давлении и образовавшийся остаток промывали изопропиловым эфиром. Таким образом, было получено указанное в заголовке соединение (195 мг, выход: 94%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,14 (1H, т, J=8 Гц), 8,01-7,89 (4H, м), 7,04 (2H, дд. J=7, 2 Гц), 5,54 (1H, дд, J=7,6 Гц), 2,63-2,57 (1H, м), 2,35-2,21 (2H, м), 1,22-1,18 (2H, м), 1,15 (3H, кв, J=5 Гц), 1,02-0,99 (2H, м);
МС (FAB+) m/z: 411 [M+H]+.
(Ссылочный пример 9)
Трет-бутил 2-фтор-4-{5-[(1S)-1-гидроксиэтил]-1,2,4-оксадиазол-3-ил}бензоат
[Химическая формула 11]
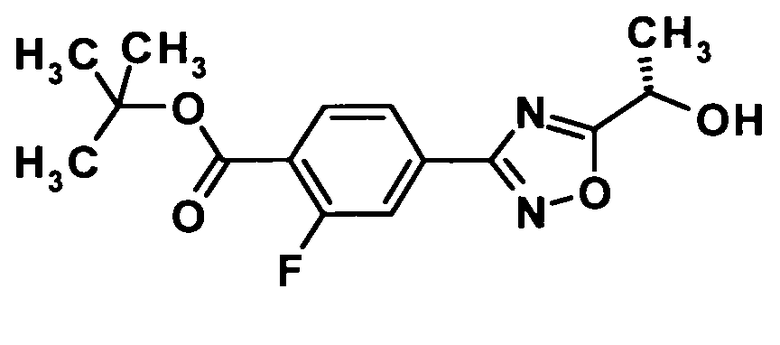
Моногидрат 1-гидроксибензотриазола (16,7 г, 109 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (41,8 г, 218 ммоль) добавляли к раствору (2S)-2-ацетоксипропионовой кислоты (14,4 г, 109 ммоль) в диметилформамиде (540 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение 30 минут. Соединение, полученное в ссылочном примере 2 (27,7 г, 109 ммоль), добавляли, смесь перемешивали в течение 10 минут и затем перемешивали при 90°С в течение 3 часов. Затем реакционную смесь охлаждали до комнатной температуры, к реакционной смеси добавляли воду и 10% водный раствор хлорида натрия и смесь подвергали экстрагированию этилацетатом дважды. Полученный таким образом органический слой промывали 10% водным раствором хлорида натрия и насыщенным водным раствором бикарбоната натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→85:15, об./об.).
Карбонат калия (12,7 г, 91,6 ммоль) добавляли к раствору полученного трет-бутил 4-{5-[(1S)-1-ацетоксиэтил]-1,2,4-оксадиазол-3-ил}-2-фторбензоата (32,1 г, 91,6 ммоль) в метаноле (360 мл) при охлаждении льдом и смесь перемешивали при той же температуре в течение 30 минут. К реакционной смеси добавляли 2 N хлористоводородную кислоту при той же температуре до тех пор, пока рН не достигал величины 6,0, и растворитель отгоняли при пониженном давлении. Воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом дважды. Полученный таким образом органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток отвердевал при использовании гексана. Таким образом, было получено указанное в заголовке соединение (26,4 г, выход: 93%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,97 (1H, т, J=8 Гц), 7,90 (1H, д, J=8 Гц), 7,84 (1H, д, J=5 Гц), 5,18 (1H, кв, J=7 Гц), 1,73 (4H, д, J=7 Гц), 1,60 (9H, с);
МС (FAB+) m/z: 309 [M+H]+.
(Ссылочный пример 10)
Трет-бутил 4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фторбензоат
[Химическая формула 12]
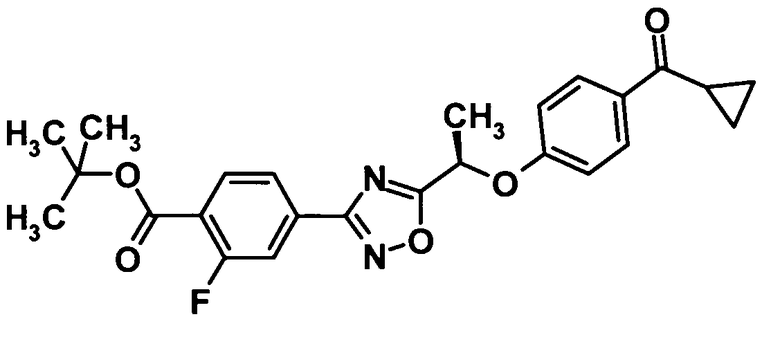
Трифенилфосфин (5,62 мг, 21,4 ммоль) и ди-трет-бутилазодикарбоксилат (4,93 г, 21,4 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 9 (6,00 г, 19,5 ммоль), и соединения, полученного в ссылочном примере 3 (3,47 г, 21,4 ммоль), в тетрагидрофуране (190 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение 40 минут. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→75:25, об./об.) с получением указанного в заголовке соединения 7,65 г, выход: 87%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,02-7,94 (3H, м), 7,89 (1H, д, J=8 Гц), 7,83 (1H, д, J=8 Гц), 7,05 (2H, д, J=8 Гц), 5,75 (1H, кв, J=7 Гц), 2,63-2,59 (1H, м), 1,92 (3H, д, J=4 Гц), 1,48 (9H, с), 1,20 (2H, м), 1,01-0,99 (2H, м).
(Ссылочный пример 11)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фторбензойная кислота
[Химическая формула 13]
Раствор трифторуксусной кислоты (20 мл) в дихлорметане (20 мл) добавляли к раствору соединения, полученного в ссылочном примере 10 (7,65 г, 16,9 ммоль), в дихлорметане (40 мл) при комнатной температуре, и смесь перемешивали в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток отвердевал при использовании смеси гексан:этилацетат (4:1, об./об.). Таким образом, было получено указанное в заголовке соединение (4,90 г, выход: 73%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,14 (1H, т, J=8 Гц), 8,01 (2H, д, J=9 Гц), 7,98-7,96 (1H, м), 7,93-7,90 (1H, м), 7,06 (2H, д, J=9 Гц), 5,76 (1H, кв, J=7 Гц), 2,64-2,57 (1H, м), 1,92 (3H, д, J=7 Гц), 1,23-1,18 (2H, м), 1,03-0,98 (2H, м);
МС (FAB+) m/z: 397 [M+H]+.
(Ссылочный пример 12)
1-[4-(Бензилокси)фенил]-2,2-диметилпропан-1-он
[Химическая формула 14]
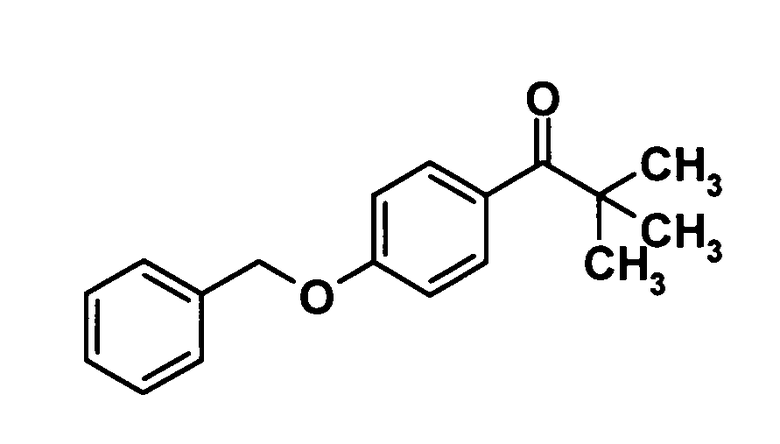
Магний (305 мг, 1,54 ммоль) добавляли к раствору 4-бензилоксибромбензола (3,00 г, 11,4 ммоль) в тетрагидрофуране (50 мл) при комнатной температуре и смесь перемешивали при 60°С в течение 30 минут. Реакционную смесь охлаждали до 0°С, 1 М раствор пивалонитрила в тетрагидрофуране (12 мл, 12 ммоль) добавляли и смесь перемешивали при комнатной температуре в течение 2 часов. Далее воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом один раз. Полученный таким образом органический слой промывали водой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 99:1→90:10, об./об.) с получением указанного в заголовке соединения 900 мг, выход: 29%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,86-7,83 (2H, м), 7,34-7,44 (5H, м), 6,96-6,98 (2H, м), 5,12 (2H, с), 1,37 (9H, с).
(Ссылочный пример 13)
1-(4-Гидроксиметил)-2,2-диметилпропан-1-он
[Химическая формула 15]
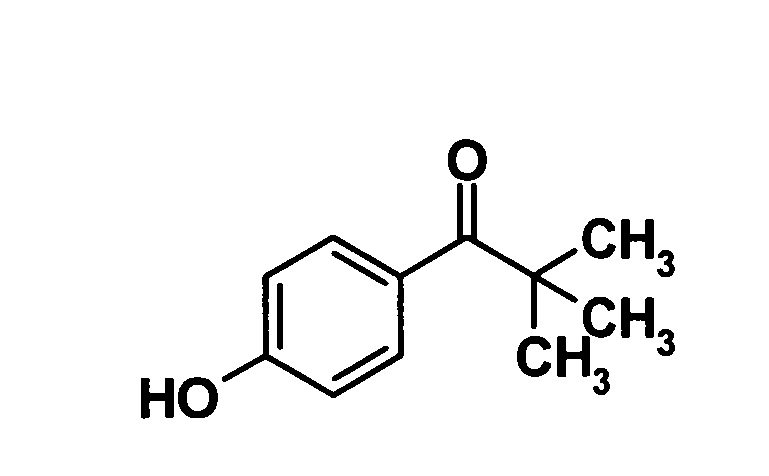
10% Палладий на угле (90 мг) добавляли к раствору соединения, полученного в ссылочном примере 12 (900 мг, 3,35 ммоль), в метаноле (30 мл) и смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере водорода. Далее реакционную смесь фильтровали через слой целита и растворитель отгоняли из маточного раствора при пониженном давлении. Образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 80:20→50:50, об./об.) с получением указанного в заголовке соединения (550 мг, выход: 92%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,82-7,80 (2H, м), 6,86-6,84 (2H, м), 5,77-5,61 (1H, уш.с), 1,37 (9H, с).
(Ссылочный пример 14)
Трет-бутил 4-(5-{(1R)-1-[4-(2,2-диметилпропаноил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоат
[Химическая формула 16]
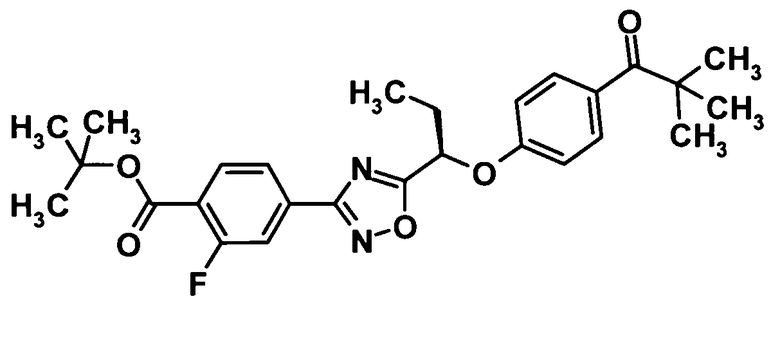
Ди-трет-бутилазодикарбоксилат (852 мг, 3,70 ммоль) и трифенилфосфин (971 мг, 3,70 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 6 (993 мг, 3,08 ммоль), и соединения, полученного в ссылочном примере 13 (550 мг, 3,08 ммоль), в тетрагидрофуране (30 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→80:20, об./об.) с получением сырого продукта, содержащего указанное в заголовке соединение (1,29 г).
(Ссылочный пример 15)
4-(5-{(1R)-1-[4-(2,2-Диметилпропаноил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензойная кислота
[Химическая формула 17]
Трифторуксусную кислоту (10 мл) добавляли к раствору соединения, полученного в ссылочном примере 14 (1,29 г), в дихлорметане (10 мл) при комнатной температуре и смесь перемешивали в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток промывали изопропиловым эфиром. Таким образом, был получен сырой продукт, содержащий указанное в заголовке соединение (900 мг).
(Ссылочный пример 16)
Циклобутил (4-гидроксифенил)метанон
[Химическая формула 18]
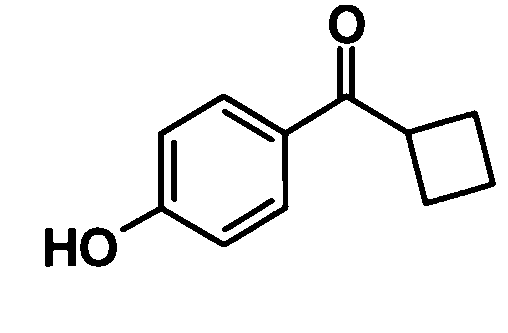
Хлорид алюминия (1,59 г, 11,9 ммоль) добавляли к раствору фенола (1,03 г, 10,9 ммоль) в дихлорметане (10 мл) при комнатной температуре и смесь перемешивали при этой температуре в течение 45 минут. Хлорангидрид циклобутанкарбоновой кислоты (1,33 мл, 11,7 ммоль) добавляли по каплям к полученной реакционной смеси при комнатной температуре и смесь перемешивали при этой же температуре в течение 4 часов. Далее, хлорид алюминия (1,58 г, 11,8 ммоль) добавляли при комнатной температуре и смесь перемешивали. После подтверждения полного растворения хлорида алюминия смесь оставляли еще в течение ночи. Реакционную смесь добавляли к хлористоводородной кислоте при охлаждении льдом и смесь подвергали экстрагированию этилацетатом один раз. Полученный таким образом органический слой промывали насыщенным раствором соли дважды и затем сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 90:10→80:20, об./об.) с получением указанного в заголовке соединения (993 мг, выход: 51%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
7,84 (2H, дт, J=9,2 Гц), 6,87 (2H, дт, J=9,2 Гц), 5,64 (1H, уш.с), 4,00-3,91 (1H, м), 2,44-2,39 (2H, м), 2,30-2,26 (2H, м), 2,13-2,02 (1H, м), 1,95-1,85 (1H, м)
(Ссылочный пример 17)
4-(5-{(1R)-1-[4-(Циклобутилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензойная кислота
[Химическая формула 19]
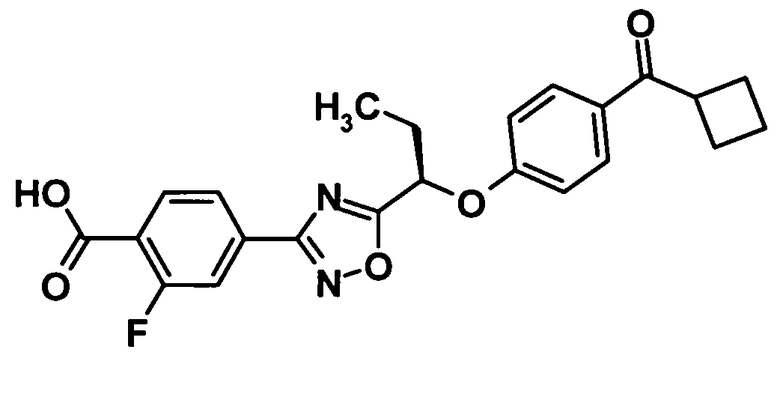
Трифенилфосфин (670 мг, 2,55 ммоль) и ди-трет-бутилазодикарбоксилат (588 мг-, 2,55 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 6 (716 мг, 2,32 ммоль), и соединения, полученного в ссылочном примере 16 (409 мг, 2,32 ммоль), в тетрагидрофуране (23 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение 40 минут. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 90:10, об./об.).
Трифторуксусную кислоту (5 мл) добавляли к раствору полученного трет-бутил 4-(5-{(1R)-1-[4-(циклобутилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензоата в дихлорметане (5 мл) при комнатной температуре и смесь перемешивали в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток отвердевал при использовании смеси гексан:этилацетат (5:1, об./об.). Таким образом, было получено указанное в заголовке соединение (531 мг, выход: 56%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,14 (1H, т, J=8 Гц), 7,97 (1H, д, J=8 Гц), 7,91 (1H, д, J=11 Гц), 7,87 (2H, д, J=9 Гц), 7,01 (2H, д, J=9 Гц), 5,51 (1H, т, J=6 Гц), 3,93 (1H, м), 2,45-2,20 (6H, м), 2,12-2,01 (1H, м), 1,95-1,85 (1H, м), 1,14 (3H, т, J=7 Гц);
МС (FAB+) m/z: 425 [M+H]+.
(Ссылочный пример 18)
4-Хлор-2-фтор-1-(4-фторфенил)бутан-1-он
[Химическая формула 20]
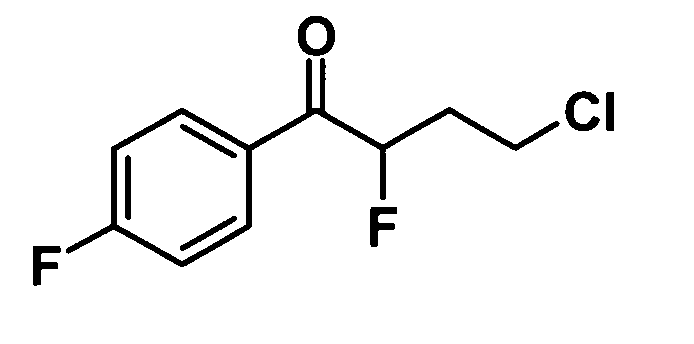
Раствор брома (8,76 г, 54,8 ммоль) в 1,4-диоксане (50 мл) добавляли к раствору 4-хлор-4′-фторбутирофенона (10,0 г, 49,8 ммоль) в 1,4-диоксане (50 мл) при комнатной температуре в течение 15 минут и смесь перемешивали при той же температуре в течение 10 минут. Воду добавляли к реакционной смеси и смесь подвергали экстрагированию гексаном дважды. Таким образом, полученный органический слой промывали водой, насыщенным водным раствором бикарбоната натрия, 1,5 М водным раствором сульфита натрия и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Часть (13,0 г) сырого продукта (14,3 г), полученного путем отгонки растворителя при пониженном давлении, растворяли в N,N-диметилформамиде (90 мл) и 18-кроун-6 (18,4 г, 69,8 ммоль) и фторид калия (4,05 г, 69,8 ммоль) добавляли к смеси при комнатной температуре. Смесь перемешивали при этой температуре в течение 2 с половиной часов, затем 18-кроун-6 (6,15 г, 23,3 ммоль) и фторид калия (1,35 г, 23,3 ммоль) добавляли к смеси и смесь еще перемешивали при этой же температуре в течение одного часа. Воду добавляли к реакционной смеси и смесь подвергали экстрагированию гексаном дважды. Таким образом, полученный органический слой промывали водой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 100:0→95:5, об./об.) с получением указанного в заголовке соединения (5,47 г, выход: 55%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,04 (2Н, дд, J=9,5 Гц), 7,18 (2H, дд, J=9,8 Гц), 5,84 (1H, ддд, J=49, 8, 4 Гц), 3,84-3,74 (2H, м), 2,46-2,35 (2H, м).
(Ссылочный пример 19)
(1-Фторциклопропил) (4-фторфенил)метанон
[Химическая формула 21]
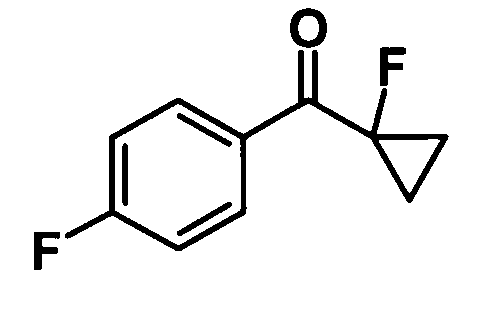
1,09 М раствор натрия бис(триметилсилил)амида в тетрагидрофуране (3,97 мл, 4,33 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 18 (861 мл, 3,94 ммоль), в ТГФ (8,0 мл) при 0°С в течение 30 минут и смесь перемешивали при комнатной температуре в течение 2,5 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и воду и смесь подвергали экстрагированию гексаном дважды. Таким образом, полученный органический слой промывали водой и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5, об./об.) с получением указанного в заголовке соединения (336 мг, выход: 47%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,10-8,05 (2H, м), 7,15 (2H, т, J=9 Гц), 1,61-1,54 (2H, м), 1,52-1,45 (2Н, м).
(Ссылочный пример 20)
(1-Фторциклопропил) {4-[(4-метоксибензил)окси]фенил}метанон
[Химическая формула 22]
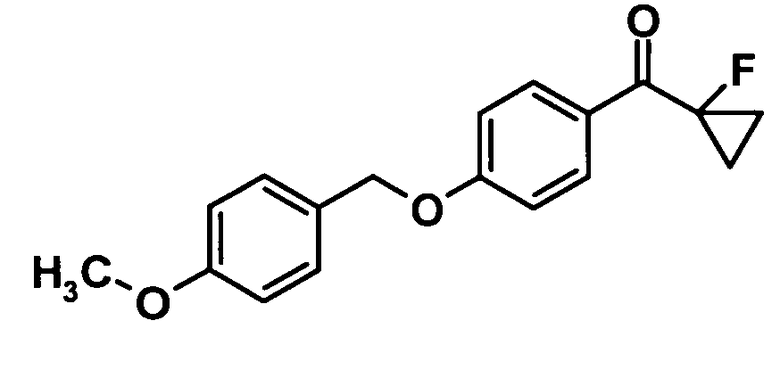
Трет-бутилат калия (225 мг, 2,00 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 19 (332 мг, 1,82 ммоль), и 4-метоксибензилового спирта (250 мкл, 2,00 ммоль) в N,N-диметилформамиде (9,0 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 1,5 часов. Насыщенный водный раствор хлорида аммония добавляли к реакционной смеси, смесь перемешивали при комнатной температуре в течение одного часа и затем выпавшее в осадок твердое вещество фильтровали и промывали водой и смесью 2-пропанол-вода (1:1). Таким образом, было получено указанное в заголовке соединение (396 мг, выход: 72%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,05 (2H, дд, J=9,1 Гц), 7,36 (2H, д, J=9 Гц), 7,02 (2H, д, J=9 Гц), 6,93 (2H, д, J=9 Гц), 5,07 (2H, с), 3,82 (3H, с), 1,58-1,51 (2H, м), 1,48-1,40 (2H, м).
(Ссылочный пример 21)
(1-Фторциклопропил) (4-гидроксифенил)метанон
[Химическая формула 23]
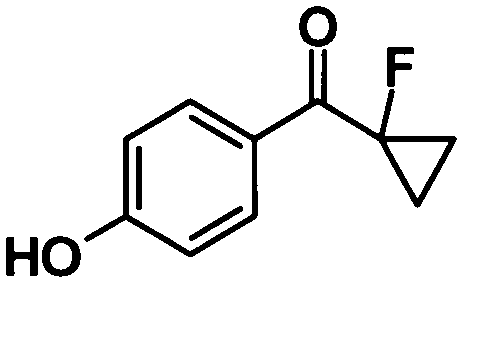
Метанол (0,4 мл) и концентрированную хлористоводородную кислоту (0,4 мл) добавляли к раствору соединения, полученного в ссылочном примере 20 (392 мг, 1,30 ммоль), в 1,4-диоксане (4,0 мл) при комнатной температуре, смесь перемешивали при этой температуре в течение 2 часов и затем нагревали при 60°С и еще перемешивали в течение 7 часов. Реакционную смесь охлаждали до комнатной температуры. Воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 90:10→75:25, об./об.) с получением указанного в заголовке соединения (217 мг, выход: 92%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,03 (2H, дд, J=9, 1 Гц), 6,89 (2H, д, J=9 Гц), 5,30 (1H, с), 1,58-1,51 (2H, м), 1,49-1,40 (2H, м).
(Ссылочный пример 22)
Трет-бутил 2-фтор-4-{5-[(1R)-1-{4-[(1-фторциклопропил)карбонил]фенокси}пропил]-1,2,4-оксадиазол-3-ил}бензоат
[Химическая формула 24]
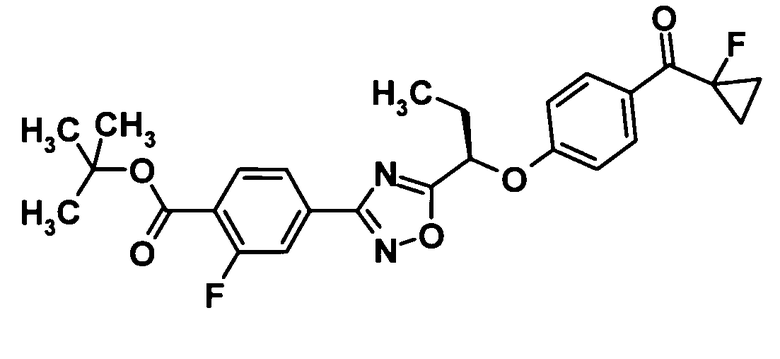
Трифенилфосфин (293 мг, 1,12 ммоль) и ди-трет-бутилазодикарбоксилат (253 мг, 1,12 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 6 (360 мг, 1,12 ммоль), и соединения, полученного в ссылочном примере 21 (183 мг, 1,02 ммоль), в тетрагидрофуране (5,0 мл) при 0°С, смесь перемешивали при этой же температуре в течение 5 минут и затем еще перемешивали при комнатной температуре в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 100:0→85:15, об./об.) с получением указанного в заголовке соединения (396 мг, выход: 80%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,04 (2H, дд, J=9,1 Гц), 7,97 (1H, дд, J=8,7 Гц), 7,89 (1H, дд, J=8,1 Гц), 7,83 (1H, дд, J=11,1 Гц), 7,04 (2H, д, J=9 Гц), 5,53 (1H, дд, J=7,6 Гц), 2,34-2,21 (2H, м), 1,61 (9H, с), 1,55-1,51 (2H, м), 1,48-1,40 (2H, м), 1,14 (3H, т, J=7 Гц).
(Ссылочный пример 23)
2-Фтор-4-{5-[(1R)-1-{4-[(1-фторциклопропил)карбонил]фенокси}пропил]-1,2,4-оксадиазол-3-ил}бензойная кислота
[Химическая формула 25]
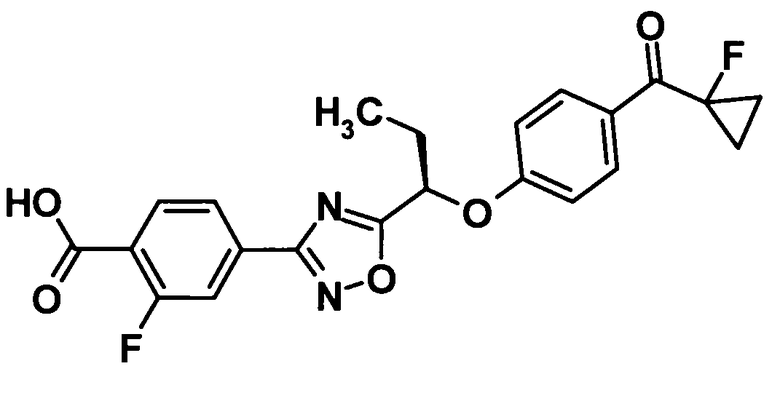
Трифторуксусную кислоту (2,0 мл) добавляли к раствору соединения, полученного в ссылочном примере 22 (390 мг, 0,805 ммоль), в дихлорметане (2,0 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток промывали смесью жидкостей гексан-этилацетат. Таким образом, было получено указанное в заголовке соединение (283 мг, выход: 82%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,14 (1H, дд, J=8,7 Гц), 8,04 (2H, дд, J=9,1 Гц), 7,97 (1H, дд, J=8,1 Гц), 7,91 (1H, дд, J=11,1 Гц), 7,04 (2H, д, J=9 Гц), 5,54 (1H, дд, J=7,6 Гц), 2,36-2,20 (2H, м), 1,57-1,51 (2H, м), 1,48-1,40 (2H, м), 1,15 (3H, т, J=7 Гц).
(Ссылочный пример 24)
Трет-бутил 2-фтор-4-{5-[(1R)-1-(4-изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензоат
[Химическая формула 26]
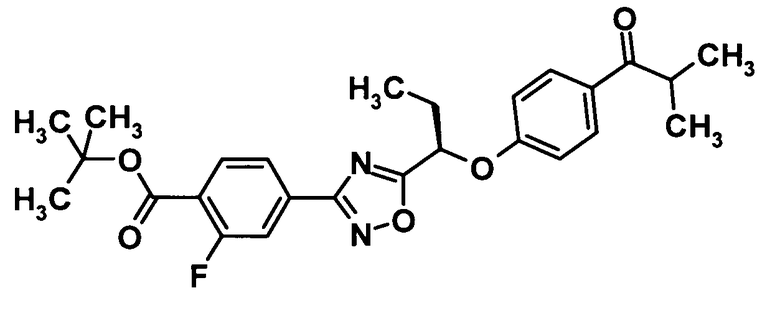
Трифенилфосфин (297 мг, 1,13 ммоль) и ди-трет-бутилазодикарбоксилат (261 мг, 1,13 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 6 (332 мг, 1,03 ммоль), и 4′-гидрокси-2-метилпропиофенона (186 мг, 1,13 ммоль) в тетрагидрофуране (5,0 мл) при 0°С, смесь перемешивали при этой же температуре в течение 5 минут и затем еще перемешивали при комнатной температуре в течение одного часа. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 95:5→85:15, об./об.) с получением указанного в заголовке соединения (268 мг, выход: 56%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
7,98-7,94 (1H, м), 7,93 (2H, д, J=9 Гц), 7,89 (1H, дд, J=8,2 Гц), 7,83 (1H, д, J=9 Гц), 7,03 (2H, дд, J=7,2 Гц), 5,51 (1H, дд, J=7,6 Гц), 3,54-3,42 (1H, м), 2,34-2,18 (2H, м), 1,55 (9H, с), 1,18 (6H, дд, J=7,2 Гц), 1,14 (3H, т, J=7 Гц).
(Ссылочный пример 25)
2-Фтор-4-{5-[(1R)-1-(4-изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензойная кислота
[Химическая формула 27]
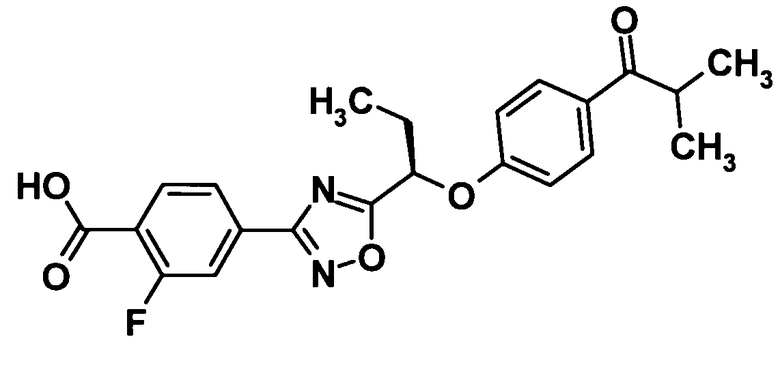
Трифторуксусную кислоту (1,5 мл) добавляли к раствору соединения, полученного в ссылочном примере 24 (268 мг, 0,572 ммоль), в дихлорметане (1,5 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 2 часов. Растворитель отгоняли при пониженном давлении и образовавшийся остаток промывали смесью жидкостей гексан-этилацетат. Таким образом, было получено указанное в заголовке соединение (223 мг, выход: 94%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,13 (1H, дд, J=8,7 Гц), 7,96 (1H, дд, J=8,2 Гц), 7,93 (2H, д, J=9 Гц), 7,91 (1H, дд, J=11,2 Гц), 7,03 (2H, д, J=9 Гц), 5,52 (1H, дд, J=7,6 Гц), 3,54-3,43 (1H, м), 2,35-2,19 (2H, м), 1,19 (6H, дд, J=7,2 Гц), 1,14 (3H, т, J=7 Гц).
(Ссылочный пример 26)
N-Циклопента-3-ен-1-ил-4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фторбензамид
[Химическая формула 28]
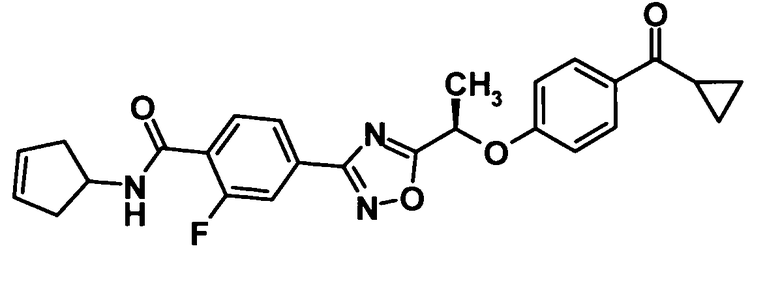
Моногидрат 1-гидроксибензотриазола (104 мг, 681 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (261 мг, 1,36 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 11 (270 мг, 681 ммоль), в N,N-диметилформамиде (3 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Далее 1-амино-3-циклопентена гидрохлорид (122 мг, 1,02 ммоль) и триэтиламин (142 мл, 1,02 ммоль) добавляли и смесь также перемешивали при той же температуре в течение 30 минут. Воду и 10% водный раствор хлорида натрия добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали водой и насыщенным водным раствором бикарбоната натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 100:0→70:30, об./об.) с получением указанного в заголовке соединения (212 мг, выход: 67%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,21 (1H, т, J=8 Гц), 8,01-7,96 (3H, м), 7,83 (1H, дд, J=12,1 Гц), 7,05 (2H, д, J=9 Гц), 6,94-6,87 (1H, м), 5,78-5,72 (3H, м), 4,84-4,75 (1H, м), 2,88 (2H, дд, J=15,8 Гц), 2,58-2,53 (1H, м), 2,34 (2H, дд, J=15,4 Гц), 1,91 (3H, д, J=7 Гц), 1,23-1,17 (2H, м), 1,03-0,97 (2H, м);
МС (FAB) m/z: 462 [M+H]+.
(Ссылочный пример 27)
(2R,3R)-3-Амино-1,2-бутандиола гидрохлорид
[Химическая формула 29]
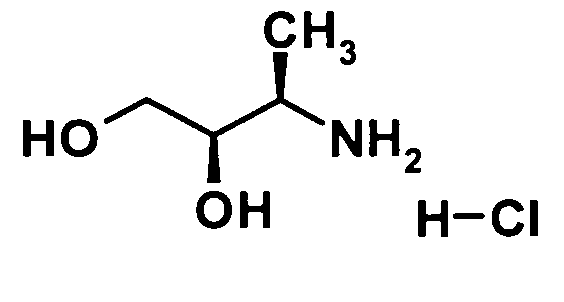
Гидроксид палладия на угле 20% (108 мг) добавляли к раствору (2R,3R,αR)-2-гидрокси-3-(N-бензил-N-α-метилбензиламино)бутанола, Tetrahedron: Asymmetry 2002, 13, 1555-1565, (1,08 г, 3,61 ммоль) в этаноле (20 мл) при комнатной температуре и смесь перемешивали при 60°С в течение 8 часов в токе водорода. Реакционную смесь охлаждали до комнатной температуры, и нерастворенный материал удаляли фильтрованием через целит. Далее часть (320 мг) образовавшегося остатка (466 мг), полученного путем отгонки растворителя при пониженном давлении, растворяли в этаноле (1,0 мл), 4 М раствор хлороводорода в 1,4-диоксане (1,13 мл, 4,54 ммоль) добавляли при комнатной температуре и смесь перемешивали при этой же температуре в течение 10 минут. Растворитель отгоняли при пониженном давлении. Таким образом, сырой продукт указанного в заголовке соединения (354 мг) был получен.
(Пример 1)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2S)-2,3-дигидроксипропил]-2-фторбензамид
[Химическая формула 30]
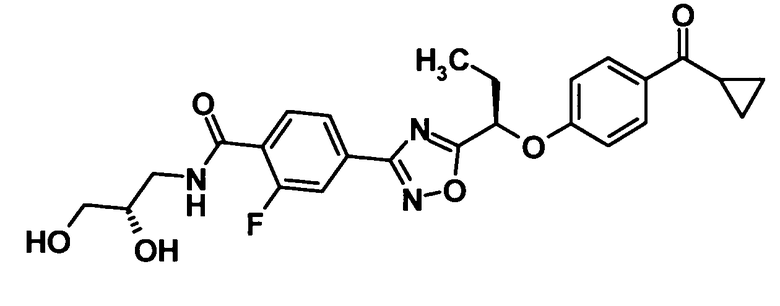
Моногидрат 1-гидроксибензотриазола (89,6 мг, 0,585 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (112 мг, 0,585 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 8 (200 мг, 0,487 ммоль), в N,N-диметилформамиде (1,4 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Далее раствор (S)-3-амино-1,2-пропандиола (133 мг, 1,46 ммоль) в N,N-диметилформамиде (1,0 мл) добавляли при 0°С и смесь также перемешивали при этой же температуре в течение одного часа. К реакционной смеси добавляли воду и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (дихлорметан:метанол = 100:0→90:10, об./об.) с получением указанного в заголовке соединения (199 мг, выход: 85%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,03-7,98 (3H, м), 7,87 (1H, дд, J=13,1 Гц), 7,28-7,16 (1H, м), 7,04 (2H, д, J=9 Гц), 5,53 (1H, т, J=6 Гц), 3,97-3,88 (1H, м), 3,76-3,58 (4H, м), 2,95 (1H, д, J=5 Гц), 2,81-2,72 (1H, м), 2,63-2,55 (1H, м), 2,35-2,18 (2H, м), 1,23-1,17 (2H, м), 1,15 (3H, т, J=7 Гц), 1,03-0,96 (2H, м);
МС (ES) m/z: 484 [M+H]+.
(Пример 2)
N-[(1S)-2-Амино-1-(гидроксиметил)-2-оксоэтил]-4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензамид
[Химическая формула 31]
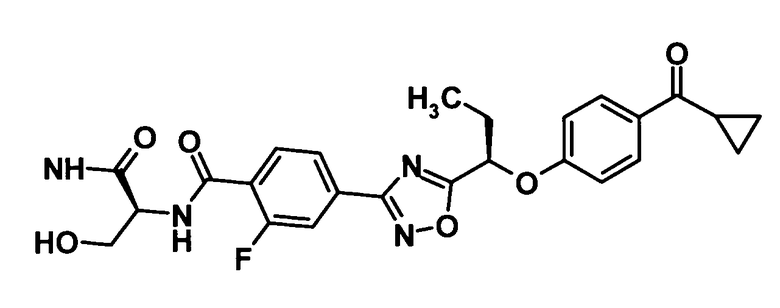
Моногидрат 1-гидроксибензотриазола (46,3 мг, 0,302 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (116 мг, 0,604 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 8 (124 мг, 0,302 ммоль), в диметилформамиде (1 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. L-Серинамида гидрохлорид (63,7 мг, 0,453 ммоль) и триэтиламин (147 мкл, 1,06 ммоль) добавляли и смесь также перемешивали при этой же температуре в течение 20 минут. Далее воду и 10% водный раствор хлорида натрия добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом три раза. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и 10% водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 70:30→этилацетат, об./об.) с получением указанного в заголовке соединения (69,5 мг, выход: 67%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,02-7,99 (3H, м), 7,92-7,89 (2H, м), 7,05 (2H, т, J=9 Гц), 6,77 (1H, с), 5,55-5,51 (2H, м), 4,72-4,68 (1Н, м), 4,44-4,36 (1H, м), 3,80-3,72 (1H, м), 3,07-3,01 (1H, м), 2,60 (1H, м), 2,29 (2H, дт, J=7,7 Гц), 1,23-1,14 (2H, м), 1,03-0,98 (2Н, м);
МС (FAB+) m/z: 497 [M+H]+.
(Пример 3)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид
[Химическая формула 32]
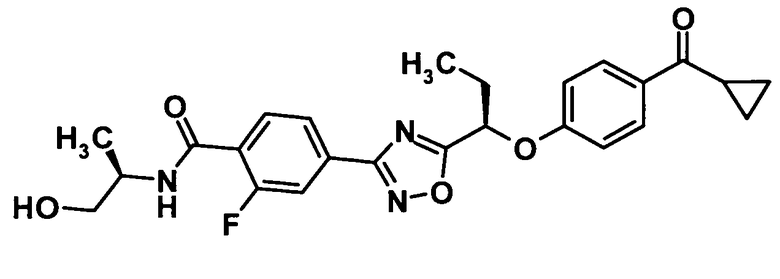
Моногидрат 1-гидроксибензотриазола (64,5 мг, 0,421 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (182 мг, 0,949 ммоль) и (R)-2-амино-1-пропанол (53,5 мг, 0,712 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 8 (195 мг, 0,475 ммоль), в N,N-диметилформамиде (5 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение одного часа. Далее воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом один раз. Таким образом, полученный органический слой промывали водой и 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 50:50→30:70, об./об.) с получением указанного в заголовке соединения (152,9 мг, выход: 69%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,01-7,97 (3H, м), 7,85 (1H, дд, J=12,1 Гц), 7,06-7,03 (2H, м), 6,90 (1H, дд, J=12,7 Гц), 5,52 (1H, т, J=6 Гц), 4,38-4,31 (1H, м), 3,81 (1H, дд, J=ll,4Гц), 3,68 (1H, дд, J=ll,6 Гц), 2,63-2,56 (1H, м), 2,44 (1H, уш.с), 2,34-2,20 (2H, м), 1,32 (3H, д, J=7 Гц), 1,22-1,18 (2H, м), 1,14 (3H, т, J=7 Гц), 1,02-0,97 (2H, м);
МС (FAB+) m/z: 468 [M+H]+.
(Пример 4)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид
[Химическая формула 33]
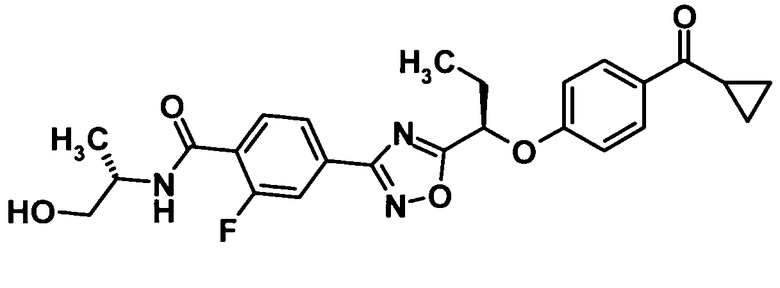
Используя соединение, полученное в ссылочном примере 8 (250 мг, 0,609 ммоль), N,N-диметилформамид (3,0 мл), 1-гидроксибензотриазола моногидрат (112 мг, 0,731 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (140 мг, 0,731 ммоль) и (S)-2-амино-1-пропанол (142 мкл, 1,83 ммоль), указанное в заголовке соединение (232 мг, выход: 81%) было получено способом, описанным в примере 1. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 70:30→0:100, об./об.).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,02-7,96 (3H, м), 7,85 (1H, дд, J=12,1 Гц), 7,04 (2H, д, J=9 Гц), 6,93-6,87 (1H, м), 5,52 (1H, дд, J=7,6 Гц), 4,39-4,31 (1H, м), 3,81 (1H, дд, J=11,4 Гц), 3,68 (1H, дд, J=11,6 Гц), 2,62-2,56 (1H, м), 2,47 (1H, с), 2,35-2,20 (2H, м), 1,32 (3H, д, J=7 Гц), 1,22-1,18 (2H, м), 1,14 (3H, т, J=7 Гц), 1,02-0,97 (2H, тт, J=8,2 Гц);
МС (ES) m/z: 468 [M+H]+.
(Пример 5)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2R)-2,3-дигидроксипропил]-2-фторбензамид
[Химическая формула 34]
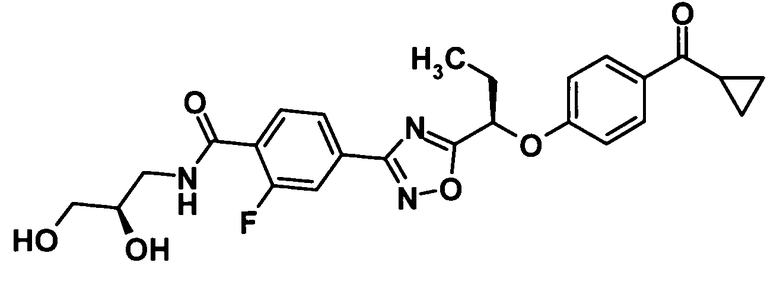
Используя раствор соединения, полученного в ссылочном примере 8 (200 мг, 0,487 ммоль), в N,N-диметилформамиде (1,0 мл), N,N-диметилформамида (1,4 мл), 1-гидроксибензотриазола моногидрат (89,6 мг, 0,585 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (112 мг, 0,585 ммоль) и (R)-3-амино-1,2-пропандиол (133 мг, 1,46 ммоль), указанное в заголовке соединение (203 мг, выход: 86%) было получено способом, описанным в примере 1. Также очистку осуществляли колоночной хроматографией на силикагеле (дихлорметан:метанол = 100:0→90:10, об./об.).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,03-7,98 (3H, м), 7,87 (1H, дд, J=13,1 Гц), 7,28-7,16 (1H, м), 7,04 (2H, д, J=9 Гц), 5,53 (1H, т, J=6 Гц), 3,97-3,88 (1H, м), 3,76-3,58 (4H, м), 2,95 (1H, д, J=5 Гц), 2,81-2,72 (1H, м), 2,63-2,55 (1H, м), 2,35-2,18 (2H, м), 1,23-1,17 (2H, м), 1,15 (3H, т, J=7 Гц), 1,03-0,96 (2H, м);
МС (ES) m/z: 484 [M+H]+.
(Пример 6)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид
[Химическая формула 35]
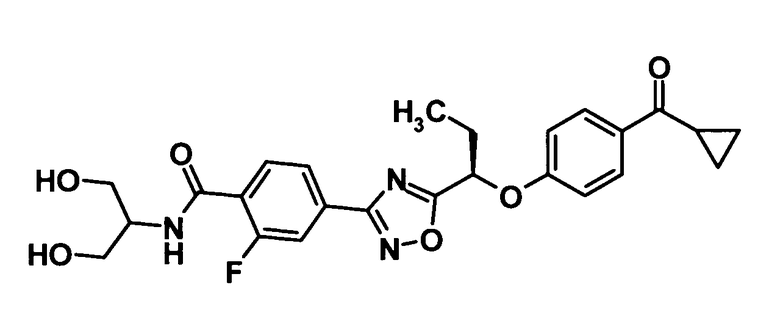
Используя соединение, полученное в ссылочном примере 8 (154 мг, 0,375 ммоль), диметилформамид (4 мл), 1-гидроксибензотриазола моногидрат (50,7 мг, 0,331 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (144 мг, 0,751 ммоль) и 2-амино-1,3-пропандиол (51,3 мкл, 0,563 ммоль), указанное в заголовке соединение (110 мг, выход: 61%) было получено способом, описанным в примере 3. Также очистку осуществляли колоночной хроматографией на силикагеле (дихлорметан:метанол = 99:1→90:10, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,19 (1H, т, J=8 Гц), 8,00-7,97 (3H, м), 7,86 (1H, дд, J=13,2 Гц), 7,52-7,47 (1H, м), 7,04 (2H, д, J=7 Гц), 5,53 (1H, т, J=6 Гц), 4,27-4,21 (1H, м), 4,02-3,93 (4H, м), 2,62-2,56 (1H, м), 2,51 (2H, дд, J=6,4Гц), 2,35-2,21 (2H, м), 1,21-1,18 (2H, м), 1,15 (3H, т, J=7 Гц), 1,02-0,97 (2H, м);
МС (FAB+) m/z: 484 [M+H]+.
(Пример 7)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид
[Химическая формула 36]
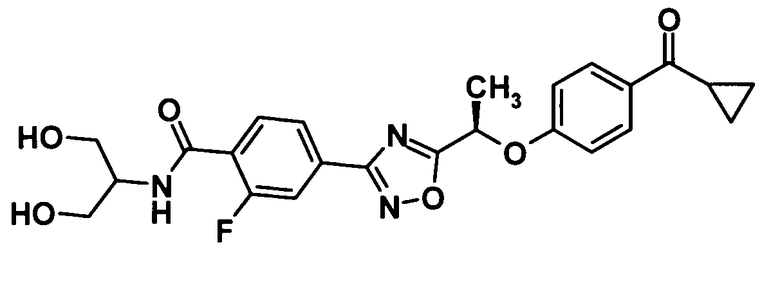
Моногидрат 1-гидроксибензотриазола (40,9 мг, 0,267 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (102 мг, 0,534 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 11 (106 мг, 0,267 ммоль), в диметилформамиде (1,5 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. 2-Амино-1,3-пропандиол (36,5 мг, 0,400 ммоль) добавляли и смесь перемешивали при этой же температуре в течение 20 минут. Далее воду и 10% водный раствор хлорида натрия добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом три раза. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 70:30→этилацетат, об./об.) с получением указанного в заголовке соединения (69,5 мг, выход: 55%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,17 (1H, т, J=8 Гц), 7,97-7,95 (3H, м), 7,83 (1H, д, J=12 Гц), 7,47-7,44 (1H, м), 7,01 (2H, т, J=9 Гц), 5,72 (1H, кв, J=7 Гц), 4,24-4,18 (1H, м), 4,00-3,94 (2H, м), 3,94-3,87 (2Н, м), 2,56 (1Н, дддд, J=8,8, 5,5 Гц), 2,37 (1H, м), 1,88 (3H, д, J=7 Гц), 1,19-1,15 (2H, м), 0,99-0,94 (2H, м);
МС (FAB+) m/z: 470 [M+H]+.
(Пример 8)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид
[Химическая формула 37]
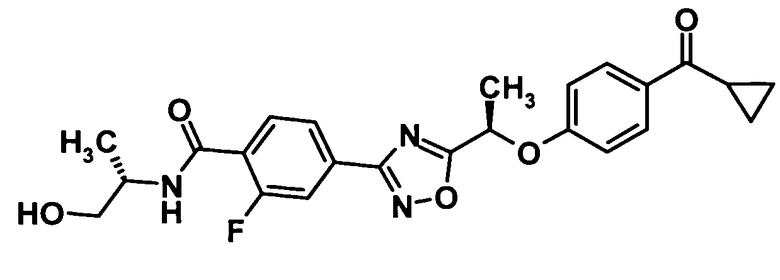
Используя соединение, полученное в ссылочном примере 11 (110 мг, 0,277 ммоль), диметилформамид (1,5 мл), 1-гидроксибензотриазола моногидрат (42,3 мг, 0,277 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (106 мг, 0,553 ммоль) и (S)-2-амино-1-пропанол (32,1 мкл, 0,414 ммоль), указанное в заголовке соединение (87,8 мг, выход: 70%) было получено способом, описанным в примере 7. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 80:20→30:70, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,21 (1H, т, J=8 Гц), 8,03-7,99 (3H, м), 7,87 (1H, д, J=12 Гц), 7,06 (2H, д, J=8 Гц), 6,95-6,89 (1H, м), 5,77 (1H, кв, J=7 Гц), 4,39-4,32 (1H, м), 3,82 (1H, дд, J=11,4 Гц), 3,70 (1H, дд, J=11,5 Гц), 2,62 (1H, дддд, J=8, 8, 5, 5 Гц), 1,93 (3H, д, J=7 Гц), 1,34 (3H, д, J=7 Гц), 1,24-1,20 (2H, м), 1,04-1,00 (2H, м);
МС (FAB+) m/z: 454 [M+H]+.
(Пример 9)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R,2R)-2-гидроксициклопентил]бензамид
[Химическая формула 38]
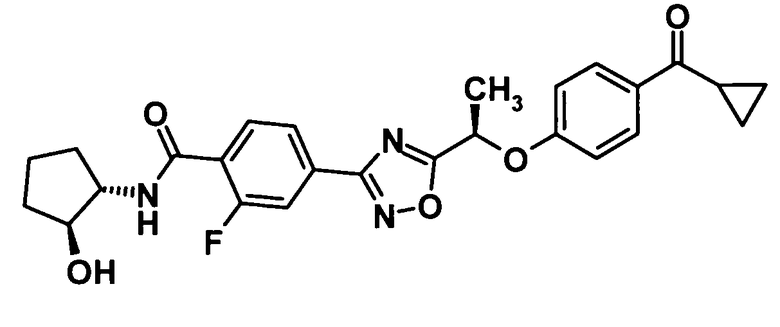
Используя соединение, полученное в ссылочном примере 11 (164 мг, 0,414 ммоль), диметилформамид (1,5 мл), 1-гидроксибензотриазола моногидрат (63,4 мг, 0,414 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (159 мг, 0,828 ммоль) и (1R,2R)-2-аминоциклопентанола гидрохлорид (85,5 мг, 0,621 ммоль), указанное в заголовке соединение (175 мг, выход: 87%) было получено способом, описанным в примере 7. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 80:20→30:70, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,22 (1H, т, J=8 Гц), 8,00 (3H, м), 7,86 (1H, д, J=12 Гц), 7,05 (2H, д, J=9 Гц), 6,93-6,86 (1H, м), 5,75 (1H, кв, J=7 Гц), 4,19 (1H, с), 4,16-4,05 (2H, м), 2,59 (1H, дддд, J=8, 8, 4, 4 Гц), 2,32-2,23 (1H, м), 2,15-2,05 (1H, м), 1,91 (3H, д, J=7 Гц), 1,91-1,72 (1H, м), 1,62-1,52 (2H, м), 1,23-1,18 (2H, м), 1,03-0,98 (2H, м);
МС (FAB+) m/z: 480 [M+H]+.
(Пример 10)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид
[Химическая формула 39]
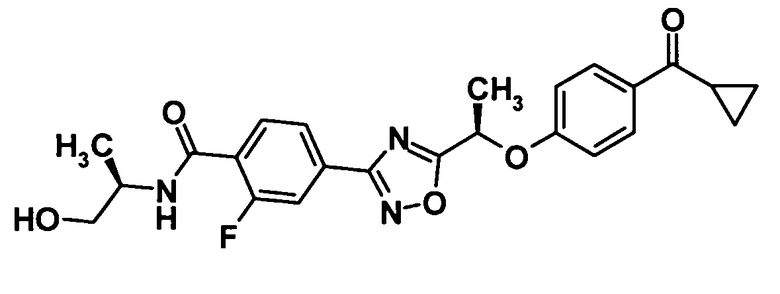
Используя соединение, полученное в ссылочном примере 11 (299 мг, 0,755 ммоль), диметилформамид (1,5 мл), 1-гидроксибензотриазола моногидрат (115 мг, 0,755 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (289 мг, 1,51 ммоль) и (R)-2-амино-1-пропанол (87,7 мкл, 1,13 ммоль), указанное в заголовке соединение (262 мг, выход: 77%) было получено способом, описанным в примере 7. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 80:20→30:70, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,02-7,97 (3H, м), 7,85 (1H, д, J=12 Гц), 7,05 (2H, д, J=12 Гц), 6,94-6,87 (1H, м), 5,75 (1H, кв, J=7 Гц), 4,38-4,31 (1H, м), 3,81 (1H, дд, J=11,4 Гц), 3,68 (1H, дд, J=11,6 Гц), 2,60 (1H, дддд, J=8, 8, 4, 4 Гц), 1,91 (3H, д, J=7 Гц), 1,32 (3H, д, J=7 Гц), 1,22-1,18 (2H, м), 1,02-0,98 (2H, м);
МС (FAB+) m/z: 454 [M+H]+.
(Пример 11)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S,2S)-2-гидроксициклопентил]бензамид
[Химическая формула 40]
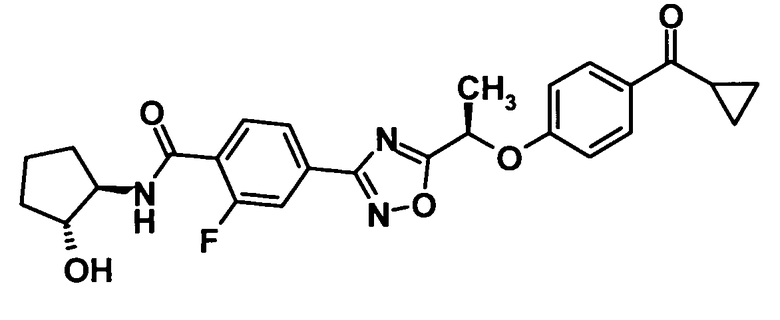
Используя соединение, полученное в ссылочном примере 11 (74,0 мг, 0,187 ммоль), диметилформамид (1 мл), 1-гидроксибензотриазола моногидрат (28,6 мг, 0,187 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (71,6 мг, 0,373 ммоль) и (1S,2S)-2-аминоциклопентанола гидрохлорид (38,5 мг, 0,280 ммоль), указанное в заголовке соединение (67,8 мг, выход: 76%) было получено способом, описанным в примере 7. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 80:20→30:70, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,22 (1H, т, J=8 Гц), 8,00 (3H, м), 7,86 (1H, д, J=12 Гц), 7,05 (2H, д, J=9 Гц), 6,93-6,86 (1H, м), 5,75 (1H, кв, J=7 Гц), 4,19 (1H, с), 4,16-4,05 (1H, м), 2,59 (1H, дддд, J=8, 8, 4, 4 Гц), 2,32-2,23 (1H, м), 2,15-2,05 (1H, м), 1,91 (3H, д, J=7 Гц), 1,91-1,72 (1H, м), 1,62-1,52 (2H, м), 1,23-1,18 (2H, м), 1,03-0,98 (2H, м);
МС (FAB+) m/z: 480 [M+H]+.
(Пример 12)
4-(5-{(1R)-1-[4-(2,2-Диметилпропаноил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид
[Химическая формула 41]
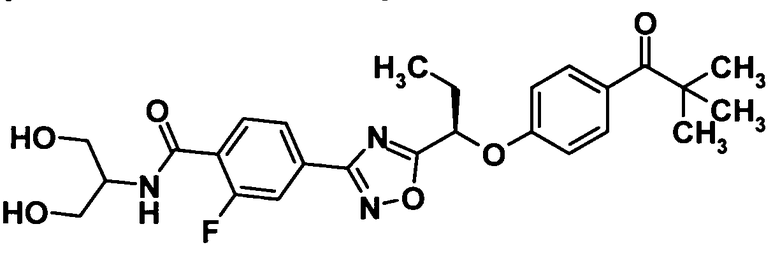
Используя соединение, полученное в ссылочном примере 15 (200 мг, 0,487 ммоль), диметилформамид (5 мл), 1-гидроксибензотриазола моногидрат (63,4 мг, 0,414 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (180,0 мг, 0,939 ммоль) и 2-амино-1,3-пропандиол (53,5 мкл, 0,587 ммоль), указанное в заголовке соединение (192 мг, выход: 82%) было получено способом, описанным в примере 3. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 50:50→30:70, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,18 (1H, т, J=8 Гц), 7,98-7,97 (1H, м), 7,86 (1H, д, J=12 Гц), 7,81-7,77 (2H, м), 7,54-7,50 (1H, м), 7,43-7,42 (1H, м), 6,98 (2H, д, J=9 Гц), 5,48 (1H, т, J=7 Гц), 4,24 (1H, с), 4,00 (2H, дд, J=ll,4 Гц), 3,93 (2H, дд, J=ll,4 Гц), 1,34 (9H, с), 1,14 (3H, т, J=7 Гц);
МС (FAB+) m/z: 500 [M+H]+.
(Пример 13)
4-(5-{(1R)-1-[4-(Циклобутилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид
[Химическая формула 42]
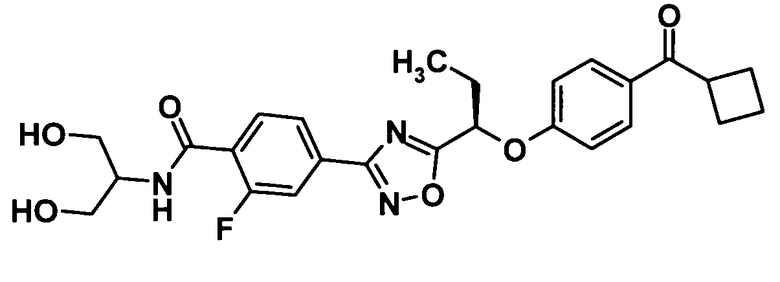
Используя соединение, полученное в ссылочном примере 17 (106 мг, 0,251 ммоль), диметилформамид (1,2 мл), 1-гидроксибензотриазола моногидрат (38,4 мг, 0,251 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (96,2 мг, 0,502 ммоль) и 2-амино-1,3-пропандиол (34,3 мкл, 0,376 ммоль), указанное в заголовке соединение (92,3 мг, выход: 74%) было получено способом, описанным в примере 7. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 70:30→этилацетат, об./об.).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,18 (1H, т, J=8 Гц), 7,97 (1H, д, J=8 Гц), 7,86-7,83 (3H, м), 7,52-7,46 (1H, м), 6,99 (2H, д, J=9 Гц), 5,48 (1H, т, J=6 Гц), 4,26-4,21 (1H, м), 4,01-3,87 (4H, м), 2,45-2,19 (6H, м), 2,10-2,05 (1H, м), 1,91-1,83 (1H, м), 1,12 (3H, т, J=8 Гц);
МС (FAB+) m/z: 498 [M+H]+.
(Пример 14)
2-Фтор-4-{5-[(1R)-1-{4-[(1-фторциклопропил)карбонил]фенокси}пропил]-1,2,4-оксадиазол-3-ил}-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид
[Химическая формула 43]
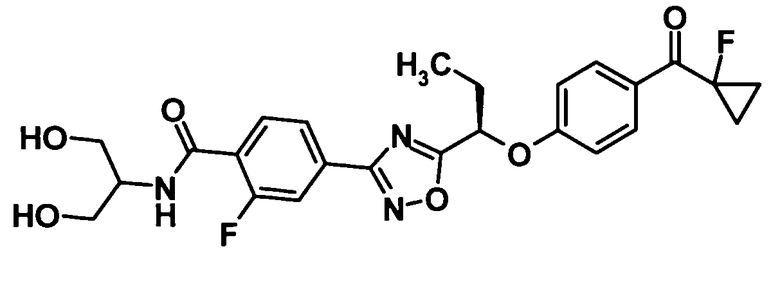
Используя раствор соединения, полученного в ссылочном примере 23 (67,0 мг, 0,156 ммоль), в N,N-диметилформамиде (1,0 мл), N,N-диметилформамид (1,0 мл), 1-гидроксибензотриазола моногидрат (28,7 мг, 0,188 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (36,0 мг, 0,188 ммоль) и 2-амино-1,3-пропандиол (57,0 мг, 0,626 ммоль), указанное в заголовке соединение (61,6 мг, выход: 79%) было получено способом, описанным в примере 1. Также очистку осуществляли колоночной хроматографией на силикагеле (дихлорметан:метанол = 100:0→90:10, об./об.).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,21 (1H, т, J=8 Гц), 8,04 (2H, дд, J=9,2 Гц), 7,99 (1H, дд, J=8,1 Гц), 7,87 (1H, дд, J=12,1 Гц), 7,54-7,47 (1H, м), 7,04 (2H, д, J=9 Гц), 5,54 (1H, дд, J=7,6 Гц), 4,28-4,21 (1H, м), 4,04-3,90 (4H, м), 2,40 (2H, дд, J=6,4 Гц), 2,36-2,20 (2H, м), 1,56-1,50 (2H, м), 1,49-1,39 (1H, м), 1,15 (3H, т, J=7 Гц);
МС (ES) m/z: 502 [M+H]+.
(Пример 15)
2-Фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-{5-[(1R)-1-(4-изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамид
[Химическая формула 44]
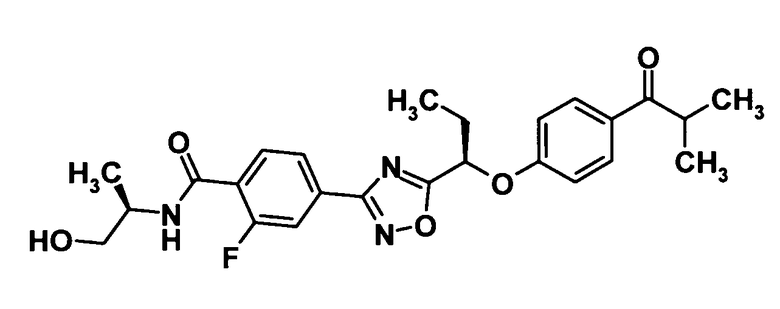
Используя соединение, полученное в ссылочном примере 25 (100 мг, 0,242 ммоль), N,N-диметилформамид (1,2 мл), 1-гидроксибензотриазола моногидрат (44,6 мг, 0,291 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (55,8 мг, 0,291 ммоль) и (R)-2-амино-1-пропанол (56,3 мкл, 0,727 ммоль), указанное в заголовке соединение (101 мг, выход: 89%) было получено способом, описанным в примере 1. Также очистку осуществляли колоночной хроматографией на силикагеле (гексан:этилацетат = 70:30→0:100, об./об.).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 7,98 (1H, дд, J=8,2 Гц), 7,93 (2H, д, J=9 Гц), 7,85 (1H, дд, J=13,2 Гц), 7,03 (2H, д, J=9 Гц), 6,95-6,85 (1H, м), 5,51 (1H, дд, J=7,6 Гц), 4,38-4,29 (1H, м), 3,84-3,77 (1H, м), 3,72-3,65 (1H, м), 3,53-3,43 (1H, м), 2,44 (1H, дд, J=6,5 Гц), 2,36-2,18 (2H, м), 1,32 (3H, д, J=7 Гц), 1,18 (6H, дд, J=7,2 Гц), 1,14 (3H, т, J=7 Гц);
МС (ES) m/z: 470 [M+H]+.
(Пример 16)
2-Фтор-N-[2-гидрокси-1-(гидроксиметил)этил]-4-{5-[(1R)-1-(изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамид
[Химическая формула 45]
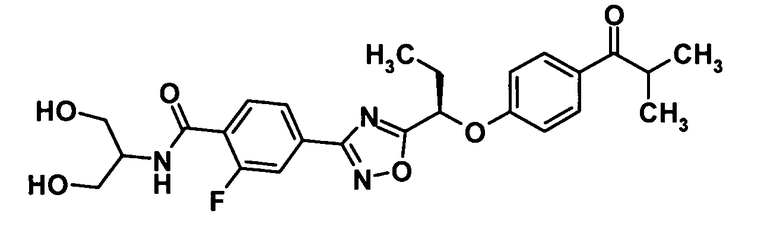
Используя раствор соединения, полученного в ссылочном примере 25 (118 мг, 0,286 ммоль), в N,N-диметилформамиде (1,0 мл), N,N-диметилформамид (1,4 мл), 1-гидроксибензотриазола моногидрат (52,6 мг, 0,343 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимид (65,8 мг, 0,343 ммоль) и 2-амино-1,3-пропандиол (78,2 мкл, 0,858 ммоль), указанное в заголовке соединение (121 мг, выход: 87%) было получено способом, описанным в примере 1. Также очистку осуществляли колоночной хроматографией на силикагеле (дихлорметан:метанол = 100:0→90:10, об./об.).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 7,98 (1H, дд, J=8,2 Гц), 7,93 (2H, д, J=9 Гц), 7,86 (1H, дд, J=12,2 Гц), 7,54-7,46 (1H, м), 7,03 (2H, д, J=9 Гц), 5,51 (1H, дд, J=7,6 Гц), 4,28-4,19 (1H, м), 4,04-3,87 (4H, м), 3,53-3,43 (1H, м), 2,50 (2H, с), 2,36-2,18 (2H, м), 1,18 (6H, дд, J=7,2 Гц), 1,14 (3H, т, J=7 Гц);
МС (ES) m/z: 486 [M+H]+.
(Пример 17)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-3-гидрокси-1-(гидроксиметил)пропил]бензамид
[Химическая формула 46]
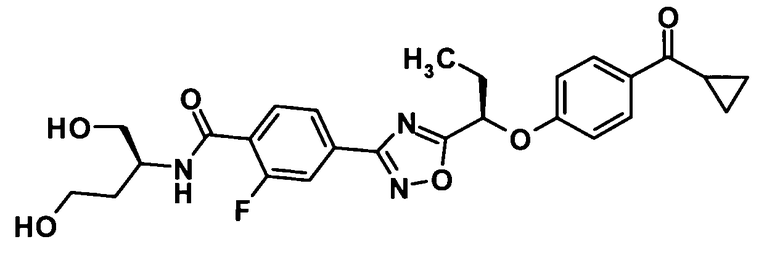
Моногидрат 1-гидроксибензотриазола (44,8 мг, 0,292 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (56,1 мг, 0,292 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 8 (100 мг, 0,244 ммоль), в диметилформамиде (1,2 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Далее раствор (2S)-2-амино-1,4-бутандиола (Bioorg. Med. Chem. Lett. 2007, 17, 2086-2090) (76,8 мг, 0,731 ммоль) в N,N-диметилформамиде (1,0 мл) добавляли при 0°С и смесь также перемешивали при этой же температуре в течение 30 минут. Далее воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (дихлорметан:метанол = 100:0→90:10, об./об.) с получением указанного в заголовке соединения (100 мг, выход: 83%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,19 (1H, т, J=8 Гц), 8,01-7,96 (3H, м), 7,85 (1H, дд, J=12,1 Гц), 7,43-7,36 (1H, м), 7,04 (2H, д, J=9 Гц), 5,53 (1H, дд, J=7,6 Гц), 4,43 (1H, с), 3,91-3,66 (4H, м), 3,20-3,13 (1H, м), 2,65-2,56 (2H, м), 2,36-2,19 (2H, м), 2,02-1,93 (1H, м), 1,87-1,79 (1H,м), 1,22-1,17 (2H, м), 1,15 (3H, т, J=7 Гц), 1,02-0,97 (2H, м).
МС (FAB+) m/z: 498 [M+H]+.
(Пример 18)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1r,3R,4S)-3,4-дигидроксициклопентил]-2-фторбензамид и
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1s,3R,4S)-3,4-дигидроксициклопентил]-2-фторбензамид
[Химическая формула 47]
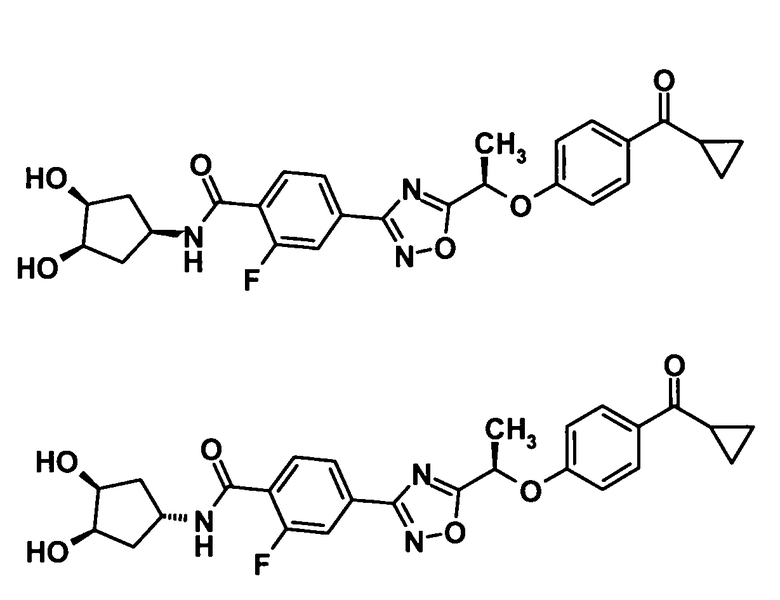
N-Метилморфолина оксид (84,3 мг, 698 ммоль) и тетроксид осмия (2,5% масс. трет-бутанольный раствор, 219 мкл, 17,5 мкмоль) добавляли к раствору соединения, полученного в ссылочном примере 26 (161 мг, 349 мкмоль), в смеси трет-бутанол/тетрагидрофуран/вода (2:2:1, об./об.) (5 мл) при комнатной температуре. Реакционную смесь перемешивали при этой же температуре в течение 20 минут. Далее насыщенный водный раствор тиосерной кислоты добавляли к реакционной смеси и смесь подвергали экстрагированию дихлорметаном три раза. Таким образом, полученный органический слой промывали водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 50:50→0:100, об./об.) с получением указанного в заголовке соединений (78,4 мг, выход: 45%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,19 (1H, т, J=8 Гц), 8,05-7,92 (2H, м), 7,84 (1H, д, J=13 Гц), 7,55-7,45 (1H, м), 7,05 (2H, д, J=8 Гц), 6,77-6,70 (1H, м), 5,75 (1H, кв, J=7 Гц), 4,77-4,67 (2/3H, м), 4,48-4,38 (1/3H, м), 4,35-4,27 (2/3H, м), 4,16-4,09 (1/3H, м), 2,89 (1/3H, д, J=4 Гц), 2,63-2,57 (1H, м), 2,33 (8/3H, м), 1,95-1,85 (5H, м), 1,22-1,19 (2H, м), 0,98-1,04 (2H, м);
МС (FAB+) m/z: 496 [M+H]+.
При разделении описанной выше смеси диастереомеров каждый диастереомер мог быть получен методом, известным в данной области техники, например, подвергая описанную выше смесь хиральной колоночной хроматографии.
(Пример 19)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(1R,2R)-2,3-дигидрокси-1-метилпропил]-2-фторбензамид
[Химическая формула 48]
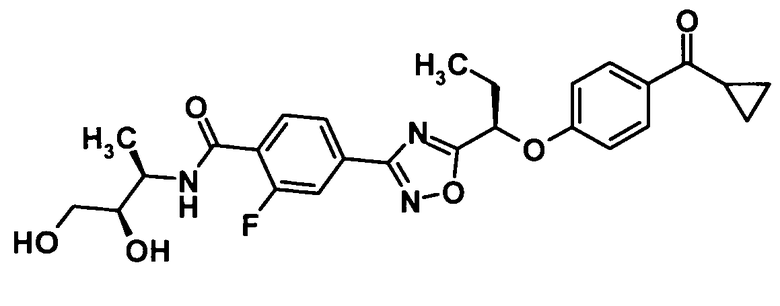
Моногидрат 1-гидроксибензотриазола (130 мг, 0,851 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (163 мг, 0,851 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 8 (291 мг, 0,709 ммоль), в N,N-диметилформамиде (2,0 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Далее триэтиламин (297 мкл, 2,13 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 27 (350 мг), в N,N-диметилформамиде (2,0 мл) при комнатной температуре, смесь перемешивали в течение 10 минут и затем реакционную смесь, полученную ранее, добавляли и общую смесь также перемешивали при этой же температуре в течение 30 минут. Далее воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (дихлорметан:этилацетат = 50:50→0:100, об./об.) с получением указанного в заголовке соединения (293 мг, выход: 83%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,19 (1H, т, J=8 Гц), 8,02-7,97 (3H, м), 7,87 (1H, дд, J=13,1 Гц), 7,04 (2H, д, J=9 Гц), 6,92-6,86 (1H, м), 5,53 (1H, дд, J=7,6 Гц), 4,28-4,19 (1H, м), 3,74-3,64 (2H,м), 3,53-3,46 (1H, м), 3,15 (1H, дд, J=9,5 Гц), 2,78 (1H, д, J=8 Гц), 2,63-2,57 (1H, м), 2,35-2,19 (2H, м), 1,41 (3H, д, J=7 Гц), 1,22-1,18 (2H, м), 1,15 (3H, т, J=7 Гц), 1,02-0,98 (2H, м).
МС (FAB+) m/z: 498 [M+H]+.
(Пример 20)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(1R,2S,3R)-2,3-дигидроксициклопентил]-2-фторбензамид и
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(1S,2R,3S)-2,3-дигидроксициклопентил]-2-фторбензамид
[Химическая формула 49]
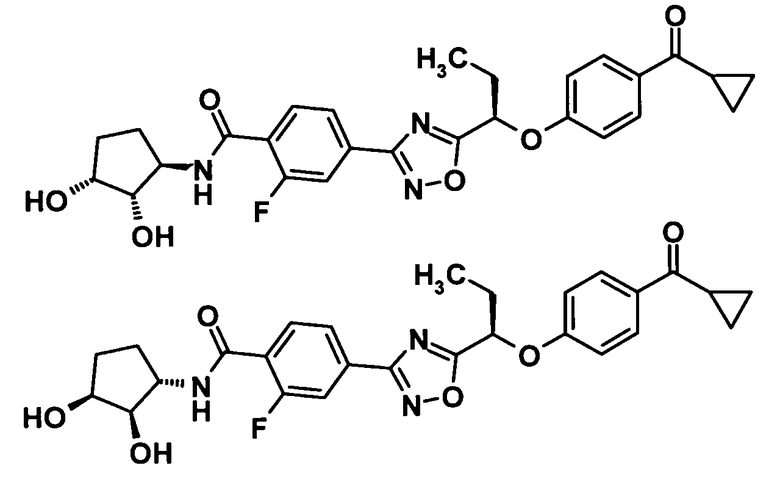
Триэтиламин (0,340 мл, 2,44 ммоль) добавляли к N,N-диметилформамидному (5,0 мл) раствору соединения, полученного в ссылочном примере 8 (250 мг, 0,609 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимида (234 мг, 1,22 ммоль), моногидрата 1-гидроксибензотриазола (93,3 мг, 0,609 ммоль) и (1RS,2SR,3RS)-3-аминоциклопентан-1,2-диола гидрохлорида (J. Org. Chem. 2009, 74, 6735-6748) (140 мг, 0,914 ммоль) при комнатной температуре и смесь перемешивали при этой же температуре в течение 71 часа. Воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом. Таким образом, полученный органический слой промывали водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали тонкослойной хроматографией на силикагеле (этилацетат) с получением смеси указанных в заголовке соединений (134 мг, выход: 43%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,22 (1H, т, J=8 Гц), 8,02-7,98 (3H, м), 7,88 (1H, дд, J=13,1 Гц), 7,06-7,02 (2H, м), 6,99-6,95 (1H, м), 5,55-5,51 (1H, м), 4,39-4,31 (1H, м), 4,20-4,18 (1H, м), 3,96-3,93 (1H, м), 2,63-2,56 (1H, м), 2,51-2,42 (1H, м), 2,35-2,21 (2H, м), 2,11-2,02 (1H, м), 1,96-1,88 (1H, м), 1,62-1,53 (1H, м), 1,22-1,18 (2H, м), 1,15 (3H, т, J=7 Гц), 1,02-0,98 (2H, м);
МС (ESI+) m/z: 510 [M+H]+.
При разделении описанной выше смеси диастереомеров каждый диастереомер мог быть получен методом, известным в данной области техники, например, подвергая описанную выше смесь хиральной колоночной хроматографии.
(Пример 21)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1R,2S,3R)-2,3-дигидроксициклопентил]-2-фторбензамид и
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1S,2R,3S)-2,3-дигидроксициклопентил]-2-фторбензамид
[Химическая формула 50]
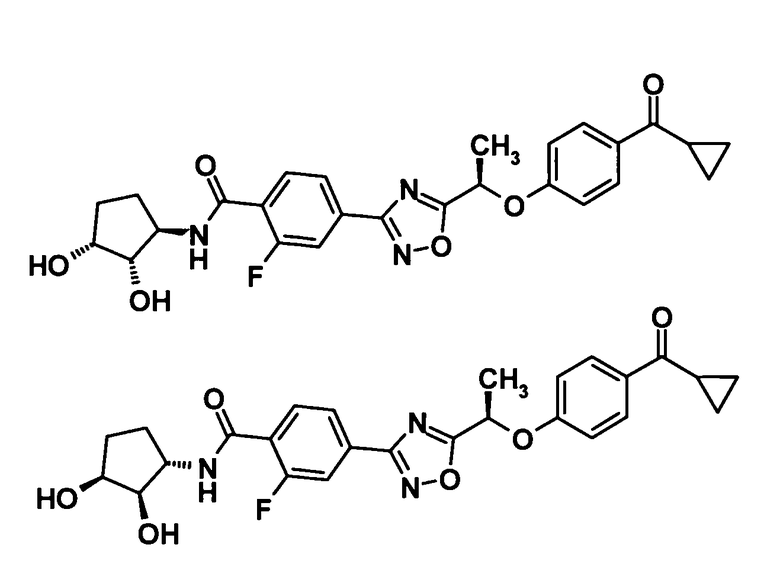
Триэтиламин (0,633 мл, 4,54 ммоль) добавляли к N,N-диметилформамидному (6,0 мл) раствору соединения, полученного в ссылочном примере 11 (300 мг, 0,757 ммоль), N-(3-диметиламинопропил)-N′-этилкарбодиимида (290 мг, 1,51 ммоль), моногидрата 1-гидроксибензотриазола (116 мг, 0,757 ммоль) и (1RS,2SR,3RS)-3-аминоциклопентан-1,2-диола гидрохлорида (J. Org. Chem. 2009, 74, 6735-6748) (174 мг, 1,14 ммоль) при комнатной температуре и смесь перемешивали при этой же температуре в течение 19 часов. Воду добавляли к реакционной смеси и смесь подвергали экстрагированию этилацетатом. Таким образом, полученный органический слой промывали водой и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали тонкослойной хроматографией на силикагеле (этилацетат) с получением смеси указанных в заголовке соединений (132 мг, выход: 35%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,22 (1H, т, J=8 Гц), 8,02-7,99 (3H, м), 7,88 (1H, дд, J=13,1 Гц), 7,07-7,03 (2H, м), 6,99-6,95 (1H, м), 5,76 (1H, кв, J=7 Гц), 4,39-4,31 (1H, м), 4,20-4,18 (1H, м), 3,96-3,93 (1H, м), 2,63-2,57 (1H, м), 2,52-2,42 (1H, м), 2,11-2,02 (1H, м), 1,96-1,88 (1H, м), 1,92 (3H, д, J=7 Гц), 1,62-1,53 (1H, м), 1,22-1,19 (2H, м), 1,03-0,98 (2H, м) ;
МС (ESI+) m/z: 496 [M+H]+.
При разделении описанной выше смеси диастереомеров каждый диастереомер мог быть получен методом, известным в данной области техники, например, подвергая описанную выше смесь хиральной колоночной хроматографии.
(Пример 22)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(3S,4S)-3,4-дигидроксициклопентил]-2-фторбензамид и
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(3R,4R)-3,4-дигидроксициклопентил]-2-фторбензамид
[Химическая формула 51]
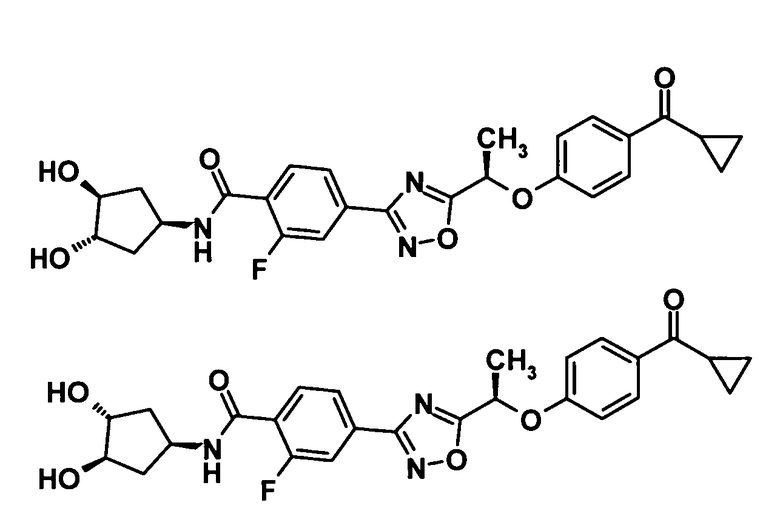
Моногидрат 1-гидроксибензотриазола (128 мг, 834 мкмоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (240 мг, 1,25 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 11 (330 мг, 834 мкмоль), в N,N-диметилформамиде (4 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Далее (1RS,2RS)-4-амино-3-циклопентан-1,2-диол (J. Med. Chem. 1992, 35, 2191-2195) (115 мг, 1,00 ммоль) добавляли и смесь также перемешивали при этой же температуре в течение 30 минут. Далее к реакционной смеси добавляли воду и смесь подвергали экстрагированию этилацетатом три раза. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат = 50:50→0:100, об./об.) с получением смеси указанных в заголовке соединений (218 мг, выход: 53%).
1H-ЯМР (400 МГц, CDCl3) δ м.д.:
8,18 (1H, т, J=8 Гц), 8,02-7,95 (3H, м), 7,83 (1H, д, J=12 Гц), 7,33-7,26 (1H, м), 7,04 (2H, д, J=9 Гц), 5,75 (1H, кв, J=7 Гц), 4, 69-4,59 (1H, м), 4,29 (1H, уш.с), 4,16 (1H, уш.с), 3,04 (1H, д, J=5 Гц), 2,63-2,51 (2H, м), 2,29 (1H, дт, J=14,6 Гц), 2,14 (1H, ддд, J=14, 8, 2 Гц), 1,91 (3H, д, J=7 Гц), 1,84-1,77 (1H, м), 1,74 (1H, д, J=2 Гц), 1,22-1,16 (2H, м), 1,04-0,98 (2H, м);
МС (FAB+) m/z: 496 [M+H]+.
При разделении описанной выше смеси диастереомеров каждый диастереомер мог быть получен методом, известным в данной области техники, например, подвергая описанную выше смесь хиральной колоночной хроматографии.
(Пример 23)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1R,2R)-2,3-дигидрокси-1-метилпропил]-2-фторбензамид
[Химическая формула 52]
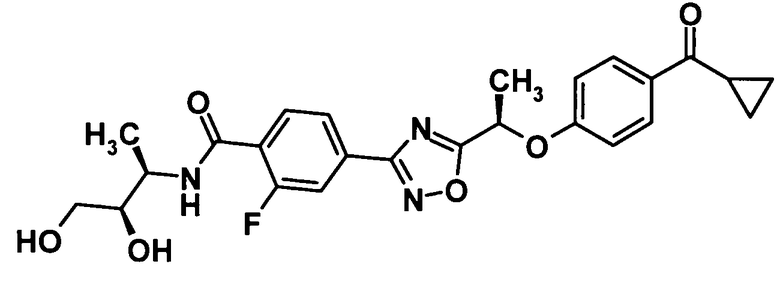
Моногидрат 1-гидроксибензотриазола (139 мг, 0,908 ммоль) и N-(3-диметиламинопропил)-N′-этилкарбодиимид (174 мг, 0,908 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 8 (300 мг, 0,757 ммоль), в N,N-диметилформамиде (2,0 мл) при комнатной температуре и смесь перемешивали при этой же температуре в течение 30 минут. Далее триэтиламин (316 мкл, 2,27 ммоль) добавляли к раствору соединения, полученного в ссылочном примере 27 (354 мг), в N,N-диметилформамиде (2,0 мл) при комнатной температуре и смесь перемешивали в течение 10 минут, затем реакционную смесь, полученную ранее, добавляли, и общую смесь также перемешивали при этой же температуре в течение 30 минут. Далее к реакционной смеси добавляли воду и смесь подвергали экстрагированию этилацетатом дважды. Таким образом, полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и затем сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и образовавшийся остаток очищали колоночной хроматографией на силикагеле (дихлорметан:этилацетат = 50:50→0:100, об./об.) с получением указанного в заголовке соединения (335 мг, выход: 92%).
1H-ЯМР (500 МГц, CDCl3) δ м.д.:
8,20 (1H, т, J=8 Гц), 8,01 (2H, д, J=9 Гц), 8,00 (1H, дд, J=8,1 Гц), 7,87 (1H, дд, J=12,1 Гц), 7,05 (2H, д, J=9 Гц), 6,93-6,8 6 (1H, м), 5,76 (1H, кв, J=7 Гц), 4,28-4,19 (1H, м), 3,74-3,64 (2H, м), 3,51-3,45 (1H, м), 3,17 (1H, дд, J=8,5 Гц), 2,80 (1H, дд, J=8,4 Гц), 2,63-2,58 (1H, м), 1,92 (3H, д, J=7 Гц), 1,41 (3H, д, J=6 Гц), 1,23-1,18 (2H, м), 1,03-0,98 (2H, м).
МС (FAB+) m/z: 484 [M+H]+.
(Типичный пример получения 1)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(1R,2R,3S)-2,3-дигидроксициклопентил]-2-фторбензамид и
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(1S,2S,3R)-2,3-дигидроксициклопентил]-2-фторбензамид
[Химическая формула 53]
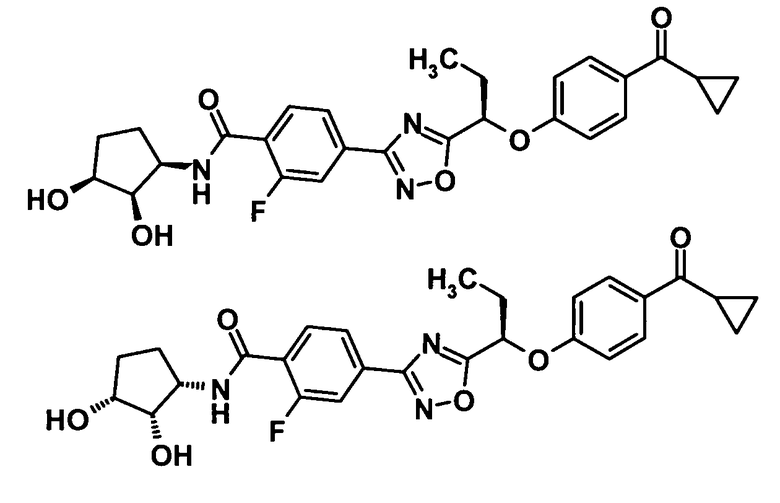
Используя соединение, полученное в ссылочном примере 8, и гидрохлорид (1RS,2SR,3SR)-3-аминоциклопентан-1,2-диола (J. Org. Chem. 2009, 74, 6735-6748), смесь указанных в заголовке соединений была получена таким же способом, как в примере 20.
При разделении описанной выше смеси диастереомеров каждый диастереомер мог быть получен методом, известным в данной области техники, например, подвергая описанную выше смесь хиральной колоночной хроматографии.
(Типичный пример получения 2)
4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1R,2R,3S)-2,3-дигидроксициклопентил]-2-фторбензамид и
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-N-[(1S,2S,3R)-2,3-дигидроксициклопентил]-2-фторбензамид
[Химическая формула 54]
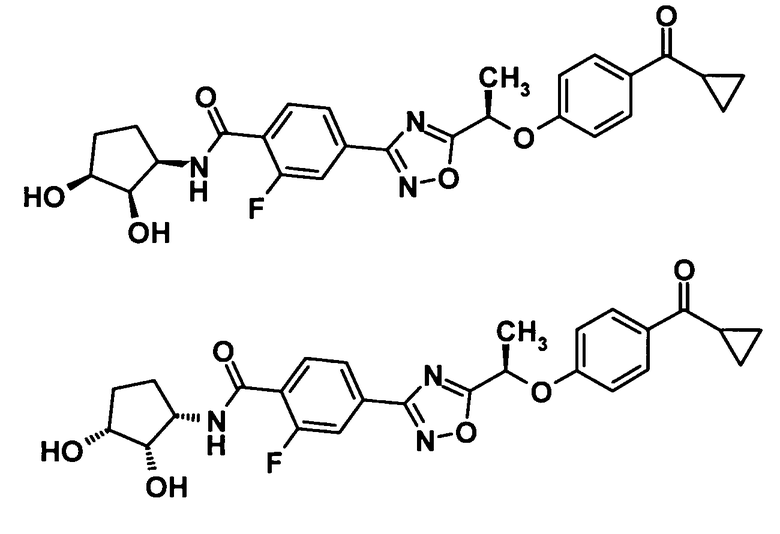
Используя соединение, полученное в ссылочном примере 11, и гидрохлорид (1RS,2SR,3SR)-3-аминоциклопентан-1,2-диола (J. Org. Chem. 2009, 74, 6735-6748), смесь указанных в заголовке соединений была получена способом, описанным в примере 21.
При разделении описанной выше смеси диастереомеров каждый диастереомер мог быть получен методом, известным в данной области техники, например, подвергая описанную выше смесь хиральной колоночной хроматографии.
(Пример препарата)
Каждое из соединений, полученных в примерах, в количестве 5 г, 90 г лактозы, 34 г кукурузного крахмала, 20 г кристаллической целлюлозы и 1 г стеарата магния смешивали в смесителе и затем смесь подвергали таблетированию в таблетировочной машине. Таким образом, таблетки были получены.
(Пример тестирования 1) Тест oGTT на мышах (пероральный тест на толерантность к глюкозе)
Дозируемый препарат (1 мг/мл каждого соединения) получали путем суспендирования в 0,5% масс./об. растворе метилцеллюлозы и последующего измельчения в агатовой ступке. Мышей самцов линии C57/BL6J (Charles River Laboratories Japan, Inc.) приобретали в возрасте 6-8 недель и затем использовали в возрасте 9-13 недель. Мышей не кормили от 17:00 до 18:00 часов один день до дня испытания, и испытание начинали после от 16 до 17 часов голодания. По пять мышей использовали в каждой группе. До введения соединения кровь собирали из хвостовой вены. После этого суспензию соединения вводили перорально при дозировке 10 мг/кг. Для отрицательной контрольной группы вводили 0,5% масс./об. раствор метилцеллюлозы. Через двадцать пять минут после дозирования соединения кровь собирали из хвостовой вены и затем через тридцать минут после дозирования соединения, 30% масс./об. раствор глюкозы вводили перорально в объеме 10 мл/кг. Кровь собирали из хвостовой вены через 15, 30, 60 и 120 минут после введения глюкозы. Каждый из образцов крови центрифугировали с получением плазмы и уровень глюкозы (мг/дл) в плазме измеряли посредством анализатора глюкозы (Glucoloader GXT, A&T Corp.). AUC глюкозы в плазме (мг/дл·мин) для каждой мыши рассчитывали, используя уровень глюкозы плазмы при 5 минутах до и через 15, 30, 60 и 120 минут после введения глюкозы. Арифметическое значение величины AUC рассчитывали для каждой группы и уменьшение AUC глюкозы плазмы, выраженное в процентах (%), по сравнению с отрицательной контрольной группой, рассчитывали как показатель эффективности.
В результате, соединения примеров 3-6, 8-19, 21 и 22 обнаружили от 5% до 20% уменьшения AUC (%) глюкозы плазмы, и соединения примеров 1, 2, 7, 20 и 23 обнаружили более чем 20% уменьшения AUC глюкозы плазмы.
(Пример тестирования 2) Тест oGTT на крысах и измерение концентрации соединения в плазме
Дозируемый препарат (1-10 мг/мл каждого соединения) можно было приготовить путем суспендирования в 0,5% масс./об. растворе метилцеллюлозы. Для оценки дозозависимого эффекта, суспензию соединения разбавляли 0,5% масс./об. раствором метилцеллюлозы последовательно. Самцы тучных крыс Цукера (Charles River Laboratories Japan, Inc.) могут быть использованы в возрасте 10- 18 недель. За два дня до теста oGTT, вес тела, уровни глюкозы в плазме и инсулина в плазме измеряли и крыс поровну распределяли в каждую группу (n=5-8) на основании указанных параметров. Крыс не кормили приблизительно от 15:00 в день до дня испытания oGTT. В день испытания oGTT дозируемый препарат вводили перорально крысам в объеме 1-5 мл/кг и через 30 минут после дозирования, от 25 до 50% масс./об. раствора глюкозы вводили перорально в объеме 4 мл/кг. Кровь собирали из хвостовой вены до дозирования соединения, за 5 минут до введения глюкозы, и через 30, 60, 120 и 180 минут после введения глюкозы. Полученную кровь центрифугировали для отделения плазмы и уровень глюкозы в плазме измеряли посредством анализатора глюкозы (Glucoloader GXT, A&T Corp.). Величины AUC глюкозы плазмы для каждой крысы рассчитывали после введения глюкозы. Уменьшение AUC, выраженное в процентах, глюкозы плазмы (%), по сравнению с контрольной группой, которой вводили растворитель, может быть выражено в виде эффективности соединения.
Образец плазмы, приготовленный описанным выше способом, может быть использован для измерения концентрации испытуемого соединения в плазме. Для того чтобы количественно оценить концентрацию испытуемого соединения в плазме, кровь собирали через 4-8 часов после введения и даже через 24 часа после введения. Из плазмы удаляли белок и затем подвергали жидкостной хроматографии/масс-спектрометрии для оценки концентрации испытуемого соединения в плазме.
(Пример тестирования 3) Оценка защитного эффекта в отношении панкреатических β-клеток
Защитное действие испытуемого соединения на панкреатические β-клетки можно подтвердить путем ссылки на метод, описанный в публикации Junko Ogawa, et al., Life Science, Vol. 65, No. 12, pp. 1287-1296 (1999).
Промышленное применение
Соединение согласно настоящему изобретению или его фармацевтически приемлемая соль проявляет активность в лечении и/или предотвращении диабета 1 типа, диабета 2 типа, гестационного диабета, гипергликемии вследствие других факторов, нарушенной толерантности к глюкозе, заболеваний, ассоциированных с диабетом, диабетических осложнений и тому подобное и, следовательно, является пригодным в качестве активного ингредиента фармацевтической композиции для защиты β-клеток или поджелудочной железы.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДНОЕ ПРОИЗВОДНОЕ | 2010 |
|
RU2536409C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ОКСАДИАЗОЛ, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2789456C2 |
| НОВЫЕ ЦИКЛИЧЕСКИЕ УГЛЕВОДОРОДНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2524949C2 |
| СУЛЬФОНАМИДНОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2017 |
|
RU2732572C2 |
| ПРОИЗВОДНЫЕ [1,8]НАФТИРИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕПЛИКАЦИИ ВИРУСА HCV | 2006 |
|
RU2467007C2 |
| ПРОТИВООПУХОЛЕВЫЙ АГЕНТ | 2018 |
|
RU2770824C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛКАРБАМОИЛБЕНЗОЛА | 2004 |
|
RU2330030C2 |
| СОЕДИНЕНИЯ ТРИАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ТРИАЗОЛА И СПОСОБ ЛЕЧЕНИЯ ГРИБКОВОЙ ИНФЕКЦИИ | 2002 |
|
RU2276670C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
Изобретение относится к соединению, представленному общей формулой (I), и его фармацевтически приемлемым солям
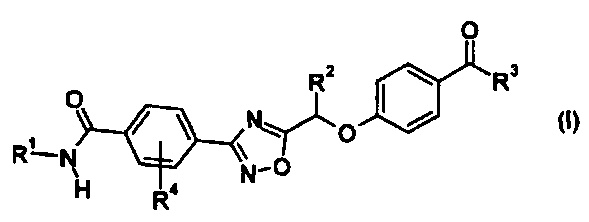
где R1 представляет собой гидрокси С1-С6 алкильную группу или гидрокси С3-С6 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α, подгруппа заместителей α является группой, состоящей из гидроксильной группы и карбамоильной группы, R2 представляет собой метильную группу или этильную группу, R3 представляет собой С1-С6 алкильную группу или С3-С6 циклоалкильную группу, каждая из которой может быть замещена 1-3 атомами галогена, и R4 представляет собой атом галогена. Изобретение также относится к индивидуальным соединениям; к фармацевтической композиции, обладающей гипогликемическим действием и/или защитным действием по отношению к β-клеткам или поджелудочной железе, содержащей, в качестве активного ингредиента, соединение изобретения; к применению соединений для приготовления фармацевтической композиции; к способу лечения диабета 1 типа или диабета 2 типа; и к способу для защиты β-клеток или поджелудочной железы. Технический результат: получены новые соединения общей формулы (I), которые обладают гипогликемическим действием и/или защитным действием по отношению к β-клеткам или поджелудочной железе. 22 н. и 13 з.п. ф-лы, 56 пр.
1. Соединение, представленное общей формулой (I):
где R1 представляет собой гидрокси С1-С6 алкильную группу или гидрокси С3-С6 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α,
подгруппа заместителей α является группой, состоящей из гидроксильной группы и карбамоильной группы,
R2 представляет собой метильную группу или этильную группу,
R3 представляет собой С1-С6 алкильную группу или С3-С6 циклоалкильную группу, каждая из которой может быть замещена 1-3 атомами галогена, и
R4 представляет собой атом галогена;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 представляет собой гидрокси С1-С3 алкильную группу или гидрокси С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 заместителями, выбранными из подгруппы заместителей α.
3. Соединение по п.1, где R1 представляет собой гидроксиэтильную группу, гидроксипропильную группу, гидроксиизопропильную группу или гидроксициклопентильную группу, каждая из которой может быть замещена одним заместителем, выбранным из подгруппы заместителей α.
4. Соединение по п.1, где R3 представляет собой С1-С4 алкильную группу или С3-С4 циклоалкильную группу, каждая из которой может быть замещена 1-3 атомами галогена.
5. Соединение по п.1, где R3 представляет собой изопропильную группу, трет-бутильную группу, циклопропильную группу или циклобутильную группу, каждая из которой может быть замещена одним атомом галогена.
6. Соединение по п.1, где R4 представляет собой атом фтора.
7. Соединение, выбранное из группы, состоящей из следующих соединений:
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2S)-2,3-дигидроксипропил]-2-фторбензамида;
N-[(1S)-2-амино-1-(гидроксиметил)-2-оксоэтил]-4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2R)-2,3-дигидроксипропил]-2-фторбензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R,2R)-2-гидроксициклопентил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамида;
4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S,2S)-2-гидроксициклопентил]бензамида;
4-(5-{(1R)-1-[4-(2,2-диметилпропаноил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамида;
4-(5-{(1R)-1-[4-(циклобутилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамида;
2-фтор-4-{5-[(1R)-1-{4-[(1-фторциклопропил)карбонил]фенокси}пропил]-1,2,4-оксадиазол-3-ил)-N-[2-гидрокси-1-(гидроксиметил)этил]бензамида;
2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-{5-[(1R)-1-(4-изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамида; и
2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]-4-{5-[(1R)-1-(изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамида.
8. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2S)-2,3-дигидроксипропил]-2-фторбензамид или его фармацевтически приемлемая соль.
9. N-[(1S)-2-Амино-1-(гидроксиметил)-2-оксоэтил]-4-(5-{(1R)-1-[4-(циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фторбензамид или его фармацевтически приемлемая соль.
10. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид или его фармацевтически приемлемая соль.
11. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид или его фармацевтически приемлемая соль.
12. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-N-[(2R)-2,3-дигидроксипропил]-2-фторбензамид или его фармацевтически приемлемая соль.
13. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид или его фармацевтически приемлемая соль.
14. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид или его фармацевтически приемлемая соль.
15. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S)-2-гидрокси-1-метилэтил]бензамид или его фармацевтически приемлемая соль.
16. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R,2R)-2-гидроксициклопентил]бензамид или его фармацевтически приемлемая соль.
17. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1R)-2-гидрокси-1-метилэтил]бензамид или его фармацевтически приемлемая соль.
18. 4-(5-{(1R)-1-[4-(Циклопропилкарбонил)фенокси]этил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[(1S,2S)-2-гидроксициклопентил]бензамид или его фармацевтически приемлемая соль.
19. 4-(5-{(1R)-1-[4-(2,2-Диметилпропаноил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид или его фармацевтически приемлемая соль.
20. 4-(5-{(1R)-1-[4-(2,2-Циклобутилкарбонил)фенокси]пропил}-1,2,4-оксадиазол-3-ил)-2-фтор-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид или его фармацевтически приемлемая соль.
21. 2-Фтор-4-{5-[(1R)-1-{4-[(1-фторциклопропил)карбонил]фенокси}пропил]-1,2,4-оксадиазол-3-ил}-N-[2-гидрокси-1-(гидроксиметил)этил]бензамид или его фармацевтически приемлемая соль.
22. 2-Фтор-N-[(1R)-2-гидрокси-1-метилэтил]-4-{5-[(1R)-1-(4-изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамид или его фармацевтически приемлемая соль.
23. 2-Фтор-N-[2-гидрокси-1-(гидроксиметил)этил]-4-{5-[(1R)-1-(изобутирилфенокси)пропил]-1,2,4-оксадиазол-3-ил}бензамид или его фармацевтически приемлемая соль.
24. Фармацевтическая композиция, обладающая гипогликемическим действием и/или защитным действием по отношению к β-клеткам или поджелудочной железе, содержащая, в качестве активного ингредиента, соединение по любому одному из пп.1-23 или его фармацевтически приемлемую соль.
25. Фармацевтическая композиция по п.24 для лечения диабета 1 типа или диабета 2 типа.
26. Фармацевтическая композиция по п.24 для защиты β-клеток или поджелудочной железы.
27. Применение соединения по любому одному из пп.1-23 или его фармацевтически приемлемой соли для приготовления фармацевтической композиции, обладающей гипогликемическим действием и/или защитным действием по отношению к β-клеткам или поджелудочной железе.
28. Применение по п.27, где фармацевтическая композиция представлена для лечения диабета 1 типа или диабета 2 типа.
29. Применение по п.27, где фармацевтическая композиция представлена для защиты β-клеток или поджелудочной железы.
30. Способ лечения диабета 1 типа или диабета 2 типа, включающий введение млекопитающему соединения по любому одному из пп.1-23 или его фармацевтически приемлемой соли.
31. Способ по п.30, где млекопитающее является человеком.
32. Способ для защиты β-клеток или поджелудочной железы, включающий введение млекопитающему соединения по любому одному из пп.1-23 или его фармацевтически приемлемой соли.
33. Способ по п.32, где млекопитающее является человеком.
34. Соединение по любому одному из пп.1-23 для применения в лечении диабета 1 типа или диабета 2 типа.
35. Соединение по любому одному из пп.1-23 для применения для защиты β-клеток или поджелудочной железы.
| ГИДРОХЛОРИДЫ ПРОИЗВОДНЫХ 5-ФЕНОКСИМЕТИЛ-1,2,4-ОКСАДИАЗОЛА, ОБЛАДАЮЩИЕ β И a -АДРЕНОЛИТИЧЕСКОЙ АКТИВНОСТЬЮ | 1982 |
|
SU1132505A1 |
| WO 9918091 A1, 15.04.1999 | |||
| WO 9519353 A1, 20.07.1995 | |||
| РАЗРАВНИВАТЕЛЬ-ИЗМЕЛЬЧИТЕЛЬ ПОЧВЫ | 0 |
|
SU246174A1 |
| WO 2004072050 A1, 26.08.2004 | |||
| WO 2007003960 A1, 11.01.2007 | |||
| WO 2010112461 A1, 07.10.2010. | |||

