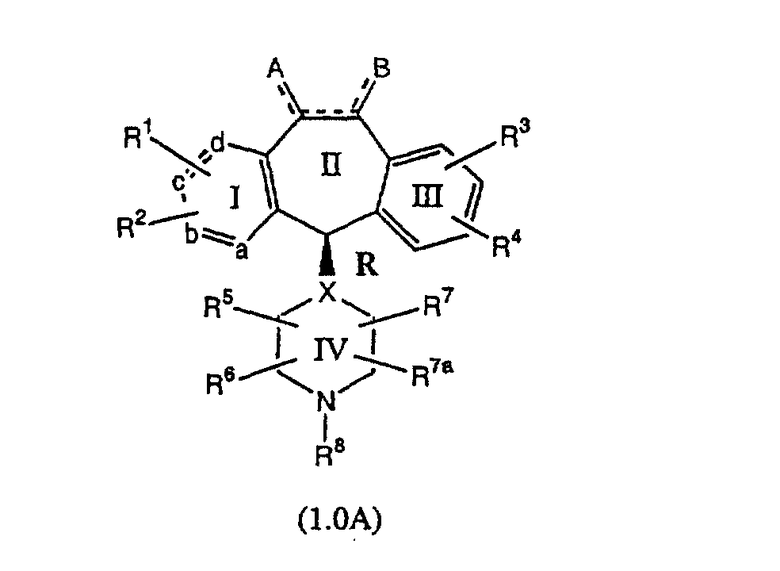
Настоящее изобретение относится к соединениям, пригодным для ингибирования фарнезилпротеинтрансферазы (ФПТ). Соединения, соответствующие настоящему изобретению, описываются формулой
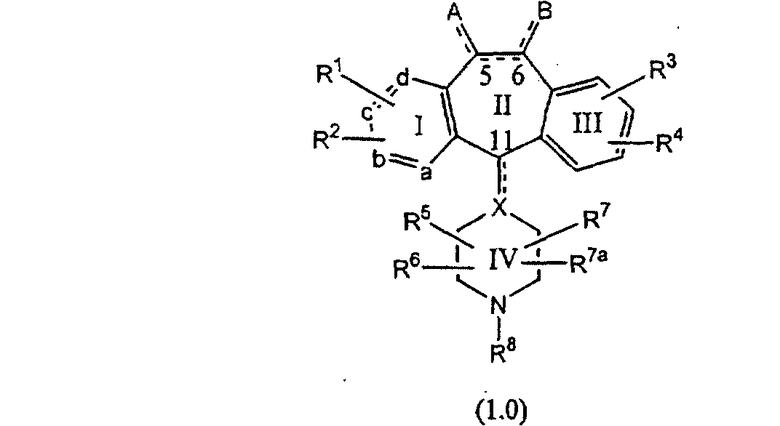
или являются его фармацевтически приемлемой солью или сольватом,
где один из а, b, c и d означает N или N+O-, а остальные а, b, с и d означают атом углерода, причем каждый атом углерода обладает группой R1 или R2, присоединенной к указанному атому углерода; или
все а, b, с и d означают атом углерода, причем каждый атом углерода обладает группой R1 или R2, присоединенной к указанному атому углерода;
штриховая линия (---) означает необязательные связи;
Х означает N или СН, когда необязательная связь (с атомом СН) отсутствует, и C, когда необязательная связь (с атомом СН) имеется;
когда имеется необязательная связь между атомом углерода 5 (т. е. С-5) и атомом углерода 6 (т. е. С-6) (т. е. имеется двойная связь между атомами С-5 и С-6), то имеется только один заместитель A, связанный с С-5, и имеется только один заместитель B, связанный с С-6, и A или B не означают Н;
если необязательная связь между атомом углерода 5 и атомом углерода 6 отсутствует (т.е. имеется ординарная связь между атомами С-5 и С-6), то имеются два заместителя A, связанные с С-5, причем каждый заместитель A выбран независимо, и два заместителя B, связанные с С-6, причем каждый заместитель B выбран независимо, т. е.
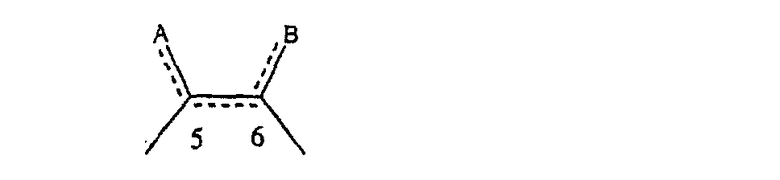
в формуле 1.0 означает
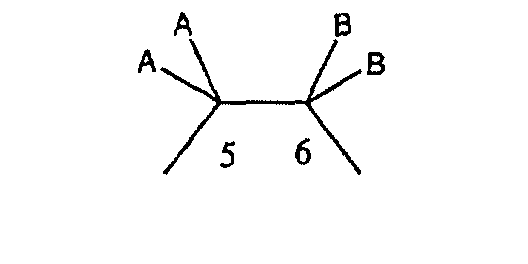
где имеется ординарная связь между атомами С-5 и С-6 и каждый A и каждый B выбраны независимо, и где хотя бы один из двух заместителей А или один из двух заместителей В означает Н и где хотя бы один из двух заместителей А или хотя бы один из двух заместителей В не означает Н (т. е. где имеется ординарная связь между атомами С-5 и С-6 и один из четырех заместителей (А, А, В и В) означает Н и один не означает Н);
А и B независимо выбраны из группы, включающей:
(1) -Н;
(2) -R9;
(3) -R9-C(O)R9;
(4) -R9-CO2-R9a;
(5) -(CH2)pR26;
(6) -C(O)N(R9), где R9 являются одинаковыми или разными;
(7) -C(O)NHR9;
(8) -C(O)NH-CH2-C(O)-NH2;
(9) -C(O)NHR26;
(10) -(CH2)pC(R9)-O-R9a;
(11) -(CH2)p(R9)2, где R9 являются одинаковыми или разными;
(12) -(CH2)pC(O)R9;
(13) -(CH2)pC(O)R278;
(14) -(CH2)pC(O)N(R9)2, где R9 являются одинаковыми или разными;
(15) -(CH2)pC(O)NH(R9);
(16) -(CH2)pC(O)N(R26)2, где R26 являются одинаковыми или разными;
(17) -(CH2)pN(R9)-R9a (например, -CH2-N(СН2-пиридин)-СН2-имидазол);
(18) -(CH2)pN(R26)2, где R26 являются одинаковыми или разными (например, -(СН2)рNH-СН2-СН3);
(19) -(CH2)pNHC(O)R50;
(20) -(CH2)pNHC(O)2R50;
(21) -(CH2)pN(C(O)R27a)2, где R27a являются одинаковыми или разными;
(22) -(CH2)pNR51C(O)R27 или R51 и R27 совместно с атомами, к которым они присоединены, образуют 5-или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(23) -(CH2)pNR51C(O)NR27 или R51 и R27 совместно с атомами, к которым они присоединены, образуют 5-или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(24) -(CH2)pNR51C(O)NR27a, где R273 являются одинаковыми или разными;
(25) -(CH2)pNHSOzN(R51), где R51 являются одинаковыми или разными;
(26) -(CH2)pNHCO2R50;
(27) -(CH2)pNC(O)NHR51;
(28) -(CH2)pCO2R51;
(29) -NHR9;
(30)
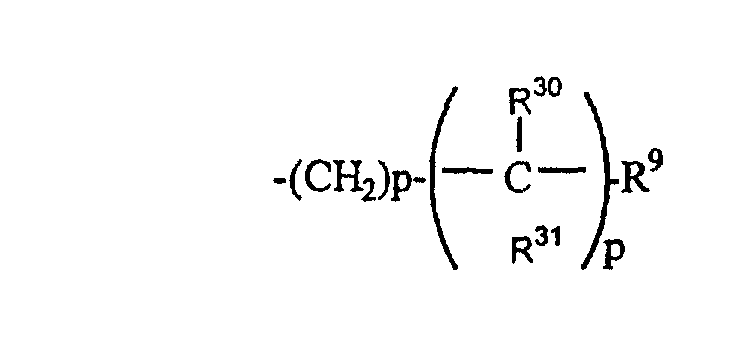
где R30 и R31 являются одинаковыми или разными;
(31)
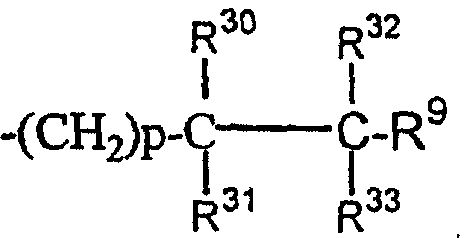
где R30, R31, R32 и R33 являются одинаковыми или разными;
(32) -алкенил-CO2R9a;
(33) -алкенил-С(O)R9a;
(34) -алкенил-CO2R51;
(35) -алкенил-С(O)-R27a;
(36) -(СН2)р-алкенил-CO2-R51;
(37) -(CH2)pC=NOR51 или
(38) -(СН2)р-фталимид;
р означает 0,1,2,3 или 4;
все R1 и R2 независимо выбраны из группы, включающей Н, галоген, -CF3, -OR10, -COR10, -SR10, -S(O)tR15, где t равно 0, 1 или 2, -N(R10)2, -NO2, -OC(O)R10, -CO2R10, -OCO2R15, -CN, -NR10COOR15, -SR15C(O)OR15, -SR15N(R13)2, при условии, что R15 в -SR15N(R13)2 не означает -CH2, и где все R13 независимо выбраны из группы, включающей Н или -C(O)OR15, бензотриазол-1-илоксил, тетразол-5-илтиоил или замещенный тетразол-5-илтиоил, алкинил, алкенил и алкил, указанная алкильная или алкенильная группа необязательно содержит в качестве заместителей галоген, -OR10 или -CO2R10;
R3 и R4 являются одинаковыми или разными и независимо выбраны из группы, включающей Н и любой из заместителей R1 и R2;
R5, R6, R7 и R73 независимо означают Н, -CF3, -COR10, алкил или арил, причем указанный алкил или арил необязательно содержит в качестве заместителей -OR10, -SR10, -S(O)tR15, -NR10COOR15, -N(R10)2, -NO2, -C(O)R10, -OCOR10, -OCO2R15, -CO2R10, -OPO3R10 или R5 объединен с R6 и означает =O или =S;
R8 выбран из группы, включающей:
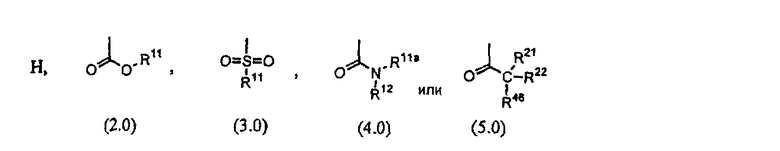
R9 выбран из группы, включающей:
(1) гетероарил;
(2) замещенный гетероарил;
(3) арилалкоксил;
(4) замещенный арилалкоксил;
(5) гетероциклоалкил;
(6) замещенный гетероциклоалкил;
(7) гетероциклоалкилалкил;
(8) замещенный гетероциклоалкилалкил;
(9) гетероарилалкил;
(10) замещенный гетероарилалкил;
(11) гетероарилалкенил;
(12) замещенный гетероарилалкенил;
(13) гетероарилалкинил и
(14) замещенный гетероарилалкинил;
где указанные замещенные группы R9 содержат один или большее количество (например, 1, 2 или 3) заместителей, выбранных из группы, включающей:
(1)-ОН;
(2) -CO2R14;
(3) -CH2OR14;
(4) галоген (например, Br, CI или F);
(5) алкил (например, метил, этил, пропил, бутил или трет-бутил);
(6) аминогруппу;
(7) тритил;
(8) гетероциклоалкил;
(9) циклоалкил (например, циклопропил или циклогексил);
(10) арилалкил;
(11) гетероарил;
(12) гетероарилалкил и
(13)
где R14 независимо выбран из группы, включающей: Н, алкил, арил, арилалкил, гетероарил и гетероарилалкил;
R93 выбран из группы, включающей: алкил и арилалкил;
R10 выбран из группы, включающей: Н, алкил, арил и арилалкил;
R11 выбран из группы, включающей:
(1) алкил;
(2) замещенный алкил;
(3) арил;
(4) замещенный арил;
(5) циклоалкил;
(6) замещенный циклоалкил;
(7) гетероарил;
(8) замещенный гетероарил;
(9) гетероциклоалкил;и
(10) замещенный гетероциклоалкил;
где указанные замещенные группы R11 содержат один или большее количество (например, 1, 2 или 3) заместителей, выбранных из группы, включающей:
(1)-ОН;
(2) галоген (например, Br, CI или F);
(3) алкил;
R1111 выбран из группы, включающей:
(1) Н;
(2) ОН;
(3) алкил;
(4) замещенный алкил;
(5) арил;
(6) замещенный арил;
(7) циклоалкил;
(8) замещенный циклоалкил;
(9) гетероарил;
(10) замещенный гетероарил;
(11) гетероциклоалкил; и
(12) замещенный гетероциклоалкил;
где указанные замещенные группы R113 содержат один или большее количество (например, 1, 2 или 3) заместителей, выбранных из группы, включающей:
(1) -ОН;
(2) -CN;
(3) -CF3;
(4) галоген (например, Br, CI или F);
(5) алкил;
(6) циклоалкил;
(7) гетероциклоалкил;
(8) арилалкил;
(9) гетероарилалкил;
(10) алкенил и (11) гетероалкенил;
R12 выбран из группы, включающей: Н и алкил;
R15 выбран из группы, включающей: алкил и арил;
R21, R22 и R46 независимо выбраны из группы, включающей:
(1) -Н;
(2) алкил (например, метил, этил, пропил, бутил или трет-бутил);
(3) арил (например, фенил);
(4) замещенный арил, необязательно содержащий один или большее количество заместителей, выбранных из группы, включающей: алкил, галоген, CF3 и ОН;
(5) циклоалкил (например, циклогексил);
(6) замещенный циклоалкил, необязательно содержащий один или большее количество заместителей, выбранных из группы, включающей: алкил, галоген, CF3 и ОН;
(7) гетероарил формулы:
(8) гетерциклоалкил формулы:

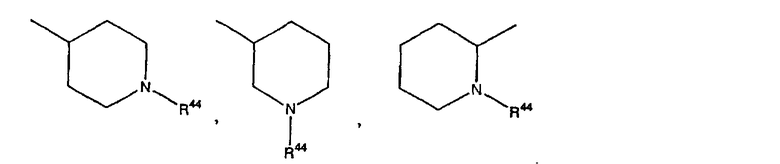
где R44 выбран из группы, включающей:
(1) -Н;
(2) алкил (например, метил, этил, пропил, бутил или трет-бутил);
(3) алкилкарбонил (например, СН3C(О)-);
(4) алкоксикарбонил (например, -C(O)O-трет-С4Н9, -С(O)O-С2Н5 и -С(О)ОСН3);
(5) галогеналкил (например, трифторметил) и
(6) -C(O)NH(R51);
где R21, R22 или R46 означают гетероциклоалкил приведенной выше формулы (т.е. кольцо V), кольцо V включает:
Примеры кольца V включают:
R26 выбран из группы, включающей:
(1) -Н;
(2) алкил (например, метил, этил, пропил, бутил или трет-бутил);
(3) алкоксил (например, метоксил, этоксил, пропоксил);
(4) -CH2-CN;
(5) R9;
(6) -CH2CO2H;
(7) -С(O)алкил и
(8) -СН2CO2алкил;
R27 выбран из группы, включающей:
(1) -Н;
(2) -ОН;
(3) алкил (например, метил, этил, пропил, или бутил) и
(4) алкоксил;
R27a выбран из группы, включающей:
(1) алкил (например, метил, этил, пропил, или бутил) и
(2) алкоксил;
R30, R31, R32 и R33 независимо выбраны из группы, включающей:
(1) -Н;
(2) -ОН;
(3) =O;
(4) алкил;
(5) арил (например, фенил) и
(6) арилалкил (например,бензил);
R50 выбран из группы, включающей:
(1) алкил;
(2) гетероарил;
(3) замещенный гетероарил и
(4) аминогруппу;
где указанные заместители в указанных замещенных группах R50 независимо выбраны из группы, включающей: алкил (например, метил, этил, пропил или бутил), галоген (например, Br, CI или F) и -OH;
R50a выбран из группы, включающей:
(1) гетероарил;
(2) замещенный гетероарил и
(3) аминогруппу;
R51 выбран из группы, включающей: -Н и алкил (например, метил, этил, пропил, бутил или трет-бутил).
Соединения, соответствующие настоящему изобретению: (i) in vitro активно ингибируют фарнезилпротеинтрансферазу, но не геранилгеранилпротеинтрансферазу I; (ii) блокируют фенотипическое изменение, индуцируемое формой трансформирующего Ras, которая является фарнезильным акцептором, но не формой трансформирующего Ras, которой придана способность являться геранилгеранильным акцептором; (III) блокируют внутриклеточный процессинг Ras, который является фарнезильным акцептором, но не Ras, которому придана способность являться геранилгеранильным акцептором, и (iv) блокируют аномальный рост клеток в культуре, индуцированный трансформирующим Ras.
Соединения, соответствующие настоящему изобретению, ингибируют фарнезилпротеинтрансферазу и фарнезилирование онкогенного белка Ras. Таким образом, настоящее изобретение дополнительно относится к способу ингибирования фарнезилпротеинтрансферазы (например, фарнезилпротеинтрансферазы ras) у млекопитающих, в частности у человека, путем введения эффективного количества (например, терапевтически эффективного количества) трициклических соединений, описанных выше. Введение пациентам соединений, соответствующих настоящему изобретению, для ингибирования фарнезилпротеинтрансферазы, применимо для лечения описанных ниже видов рака.
Настоящее изобретение относится к способу подавления или лечения аномального роста клеток, включая трансформированные клетки, путем введения эффективного количества (например, терапевтически эффективного количества) соединения, соответствующего настоящему изобретению. Аномальный рост клеток означает рост клеток, не зависящий от нормальных механизмов регуляции (например, утрату контактного ингибирования). К нему относятся аномальный рост: (1) опухолевых клеток (опухолей), экспрессирующих активированный онкоген Ras; (2) опухолевых клеток, в которых белок Ras активирован в результате онкогенной мутации в другом гене; и (3) доброкачественных и злокачественных клеток при других пролиферативных заболеваниях, при которых происходит аберрантная активация Ras.
Настоящее изобретение также относится к способу подавления или лечения роста опухоли путем введения эффективного количества (например, терапевтически эффективного количества) трициклических соединений, описанных в настоящем изобретении, млекопитающему (например, человеку), нуждающемуся в таком лечении. В частности, настоящее изобретение относится к способу подавления или лечения роста опухолей, экспрессирующих активированный онкоген Ras, путем введения эффективного количества (например, терапевтически эффективного количества) соединений, описанных выше.
Настоящее изобретение также относится к способу лечения пролиферативных заболеваний, в особенности разных видов рака (опухолей), включающему введение эффективного количества (например, терапевтически эффективного количества) соединения, соответствующего настоящему изобретению, млекопитающему (например, человеку), нуждающемуся в таком лечении, в сочетании с эффективным количеством хотя бы одного противоракового препарата, т. е. химиотерапевтического препарата, и/или проведением облучения.
Настоящее изобретение также относится к способу лечения пролиферативных заболеваний, в особенности разных видов рака (опухолей), включающему введение эффективного количества (например, терапевтически эффективного количества) соединения, соответствующего настоящему изобретению, млекопитающему (например, человеку), нуждающемуся в таком лечении, в сочетании с эффективным количеством хотя бы одного ингибитора трансдукции сигнала.
Примеры пролиферативных заболеваний (опухолей), для которых возможно подавление роста или лечение, включают (без наложения ограничений) рак легких (например, аденокарциному легких), разные виды рака поджелудочной железы (например, карциному поджелудочной железы, такую как карцинома поджелудочной железы неэндокринной этиологии), разные виды рака ободочной кишки (например, разные виды колоректального рака, такие как, например, аденокарцинома ободочной кишки и аденома ободочной кишки), миелоидную лейкемию (например, острую миелогенную лейкемию (ОМЛ)), фолликулярный рак щитовидной железы, миелодиспластический синдром (МДС), рак мочевого пузыря, плоскоклеточный рак, меланому, рак молочной железы и рак предстательной железы.
Предполагается, что настоящее изобретение также относится к способу подавления роста или лечения пролиферативных заболеваний, доброкачественных и злокачественных, при которых белки Ras аберрантно активируются вследствие онкогенной мутации в других генах - т.е. сам ген при мутации Ras не активируется в онкогенную форму, - причем указанное подавление роста или лечение осуществляется путем введения эффективного количества (например, эффективного с терапевтической точки зрения количества) трициклических соединений, описанных в настоящем изобретении, млекопитающему (например, человеку), нуждающемуся в таком лечении. В частности, для доброкачественного пролиферативного заболевания нейрофиброматоза, или опухоли, в которых Ras активируется вследствие мутации или сверхэкспрессии онкогенов тирозинкиназы (например, neu, scr, abl, Ick и fyn), подавление роста или лечение можно проводить с помощью трициклических соединений, описанных в настоящем изобретении.
Трициклические соединения, применимые в способах, соответствующих настоящему изобретению, подавляют или излечивают аномальный рост клеток. Не желая привязываться к какой-либо теории, можно предположить, что эти соединения могут действовать посредством ингибирования функции G-белка, такого как Ras р21, путем блокирования изопренилирования G-белка, что делает их пригодными для использования при лечении пролиферативных заболеваний, таких как рост опухоли и рак. Не желая привязываться к какой-либо теории, можно предположить, что эти соединения ингибируют фарнезилпротеинтрансферазу Ras и тем самым проявляют антипролиферативную активность по отношению к клеткам с трансформированным Ras.
Подробное описание изобретения
В настоящем изобретении следующие термины, если не указано иного, используются в приведенных ниже значениях:
ВОС означает трет-бутоксикарбонил;
CBZ означает C(O)ОСН2С6Н5 (т. е. бензилоксикарбонил);
CDI означает карбонил-бис(имидазол-1-ил);
СН2Cl2 означает дихлорметан;
dba означает дибензилиденацетон
DBU означает 1,8-диазабикло[5.4.0]ундец-7-ен;
DEA означает диэтаноламин;
DEAD означает диэтилазокарбоксилат;
DEC означает то же, что и EDCI, который означает 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид;
DIPEA означает N-диизопропилэтиламин;
DIBAL означает диизобутилалюминий;
DIBAL-H означает диизобутилалюминийгидрид;
DMF означает N,N-диметилформамид;
DPPA означает дифенилфосфоразидат;
Et означает этил;
EtOAc означает этилацетат;
EtOH означает этанол;
Et3N означает то же, что TEA, что означает триэтиламин;
Et2O означает диэтиловый эфир
НОВТ означает 1-гидроксибензотриазолгидрат;
IPA означает изопропанол;
iPrOH означает изопропанол;
LAH означает алюмогидрид лития;
LDA означает диизопропиламид лития;
Me означает метил;
МеОН означает метанол;
МН+ означает молекулярный ион с добавлением водорода, обнаруживающийся в масс-спектре молекулы;
NBS означает N-бромсукцинимид;
NMM означает N-метилморфолин;
PPh3 означает трифенилфосфин;
Ph означает фенил;
Pr означает пропил;
SEM означает 2,2-(триметилсилил)-этоксиметил;
TBDMS означает трет-бутилдиметилсилил;
t-BUTYL означает трет-бутил;
TFA означает трифторуксусную кислоту;
Tf означает SO2CF3;
THF означает тетрагидрофуран;
ББА означает то же, что и ББАМС, что означает масс-спектроскопию с бомбардировкой быстрыми атомами;
ВЭЖХ означает высокоэффективную жидкостную хроматографию;
ЖХМС означает жидкостную хроматографию - масс-спектроскопию;
МС означает масс-спектр;
МСВР означает масс-спектроскопию высокого разрешения;
МСХИ означает масс-спектроскопию с химической ионизацией;
МСЭР означает масс-спектроскопию с электрораспылением;
ТСХ означает тонкослойную хроматографию.
Обозначения в спектрах ЯМР:
s - синглет, d - дублет, t - триплет, q - квадруплет, m -мультиплет, b, br - широкий.
По крайней мере, один означает один или более (например, 1-6), более предпочтительно - от 1 до 4, а наиболее предпочтительно - 1, 2 или 3;
алкил означает линейные или разветвленные углеродные цепи и содержит от одного до двадцати атомов углерода, предпочтительно - от одного до шести атомов углерода, более предпочтительно - от одного до четырех атомов углерода, еще более предпочтительно - от одного до двух атомов углерода;
арилалкил означает алкильную группу, определенную выше, содержащую в качестве заместителя арильную группу, определенную ниже, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
алкоксил означает алкильный фрагмент, содержащий алкильную группу, определенную выше, ковалентно связанную с соседним структурным элементом через атом кислорода, например метоксил, этоксил, пропоксил, бутоксил и т.п.;
феноксил означает алкоксильный фрагмент, определенный выше, в котором ковалентно связанным фрагментом является арильная группа, определенная ниже, например, -O-фенил;
алкенил означает линейные или разветвленные углеродные цепи, содержащие не менее одной двойной углерод-углеродной связи и включающие от 2 до 12 атомов углерода, предпочтительно - от 2 до 6 атомов углерода, а наиболее предпочтительно - от 3 до 6 атомов углерода;
алкинил означает линейные или разветвленные углеродные цепи, содержащие не менее одной тройной углерод-углеродной связи и включающие от 2 до 12 атомов углерода, предпочтительно - от 2 до 6 атомов углерода, а наиболее предпочтительно - от 2 до 4 атомов углерода;
аминогруппа означает фрагмент -NH2;
арил (включая арильный фрагмент арилалкила и гетероарилалкила) означает кар-боциклическую группу, содержащую от 6 до 15 атомов углерода и включающую хотя бы одно ароматическое кольцо (например, арил представляет собой бензольное кольцо), причем все имеющиеся способные к замещению атомы углерода карбоциклической группы рассматриваются в качестве возможных положений замещения, и указанная карбоциклическая группа может необязательно содержать один или большее количество заместителей (например, от 1 до 3), выбранных из группы, включающей галоген, алкил, гидроксил, алкилоксил, феноксил, CF3, -C(O)N(R18)2, -SO2R18, -SO2(R18)2, амино-, алкиламино-, диалкиламиногруппу, -COOR23 и -NO2, где R18 означает Н, алкил, арил, арилалкил, гетероарил или циклоалкил и R23 означает алкил или арил;
циклоалкил означает насыщенные карбоциклические кольца, содержащие от 3 до 20 атомов углерода, предпочтительно - от 3 до 7 атомов углерода, причем указанное циклоалкильное кольцо необязательно содержит один или большее количество (например, 1, 2 или 3) одинаковых или разных алкильных групп (например, метильных или этильных);
циклоалкилалкил означает алкильную группу, определенную выше, содержащую в качестве заместителя циклическую группу, определенную выше, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
гетероциклоалкилалкил означает алкильную группу, определенную выше, содержащую в качестве заместителя гетероциклоалкильную группу, определенную ниже, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
галоген означает фтор, хлор, бром или йод;
галогеналкил означает алкильную группу, определенную выше, содержащую в качестве заместителя галогенидную группу, определенную выше, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
гетероарилалкил означает алкильную группу, определенную выше, содержащую в качестве заместителя гетероарильную группу, определенную ниже, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
гетероарилалкенил означает алкенильную группу, определенную выше, содержащую в качестве заместителя гетероарильную группу, определенную ниже, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
гетероалкил означает линейные или разветвленные углеродные цепи, содержащие от одного до двадцати атомов углерода, предпочтительно - от одного до шести атомов углерода, разделенные с помощью 1-3 гетероатомов, выбранных из группы, включающей -O-, -S- и -N-;
гетероалкенил означает линейные или разветвленные углеродные цепи, содержащие не менее одной двойной углерод-углеродной связи и включающие от одного до двадцати атомов углерода, предпочтительно - от одного до шести атомов углерода, разделенные с помощью 1-3 гетероатомов, выбранных из группы, включающей -O-, -S- и -N-;
гетероалкинил означает линейные или разветвленные углеродные цепи, содержащие не менее одной тройной углерод-углеродной связи и включающие от одного до двадцати атомов углерода, предпочтительно - от одного до шести атомов углерода, разделенные с помощью 1-3 гетероатомов, выбранных из группы, включающей -O-, -S- и -N-;
арилгетероалкил означает гетероалкильную группу, определенную выше, содержащую в качестве заместителя арильную группу, определенную выше, такую что связывание с другим заместителем осуществляется через алкильный фрагмент;
алкилкарбонил означает алкильную группу, определенную выше, ковалентно связанную с карбонильным фрагментом (-CO-), например -СОСН3;
алкилоксикарбонил означает алкильную группу, определенную выше, ковалентно связанную с карбонильным фрагментом (-CO-) через атом кислорода, например -C(O)-ОС2Н5;
гетероарил означает циклические группы, необязательно содержащие в качестве заместителей R3 и R4, включающие хотя бы один гетероатом, выбранный из группы, включающей O, S или N, причем указанный гетероатом включается в карбоциклическую кольцевую структуру и обладает количеством делокализованных пи-электронов, достаточным для придания ароматического характера, причем ароматические гетероциклические группы предпочтительно содержат от 2 до 14 атомов углерода и представляют собой, например, 2- или 3-фурил, 2- или 3-тиенил, 2-, 4- или 5-тиазолил, 2-, 4- или 5-имидазолил, 2-, 4- или 5-пиримидинил, 2-пиразинил, 3- или 4-пиридазинил, 3-, 5- или 6-[1,2,4-триазинил], 3-или 5-[1,2,4-тиадиазолил], 2-, 3-, 4-, 5-, 6- или 7-бензофуранил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 3-, 4- или 5-пиразолил, 2-, 4- или 5-оксазолил, триазолил, 2-, 3- или 4-пиридил или 2-, 3- или 4-пиридил-М-оксид, где пиридил-N-оксид можно представить в виде:
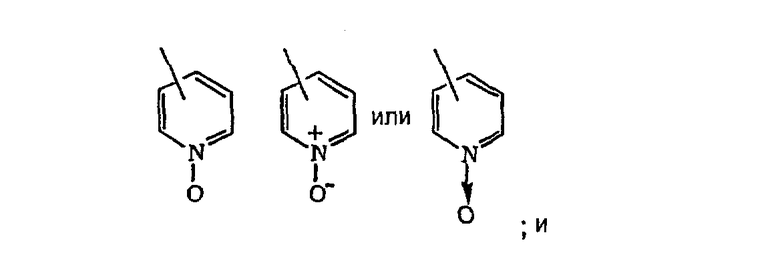
гетероциклоалкил означает насыщенное, разветвленное или неразветвленное карбоциклическое кольцо, содержащее от 3 до 15 атомов углерода, предпочтительно - от 4 до 6 атомов углерода, и в это карбоциклическое кольцо включено от 1 до 3 гетероатомных групп, выбранных из группы, включающей -O-, -S- и -NR24 (например, -NC(O)NH2), где R24 означает алкил, арил, -C(O)(NR18)2, где R18 такой, как определено выше, и подходящие гетероциклоалкильные группы включают 2- или 3-тетрагидрофуранил, 2- или 3-тетрагидротиенил, тетрагидропиракил, 2-, 3- или 4-пиперидинил, 2- или 3-пирролидинил, 1-, 2-, 3- или 4-пиперазинил, 2- или 4-диоксанил, морфолинил и
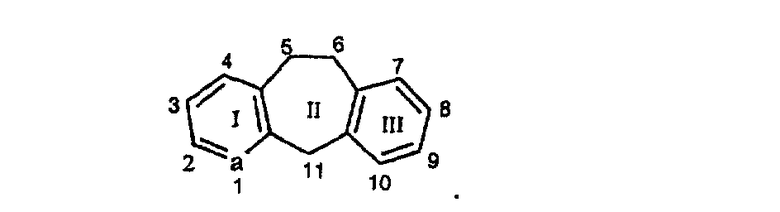
Нумерация положений трициклической кольцевой системы является следующей:
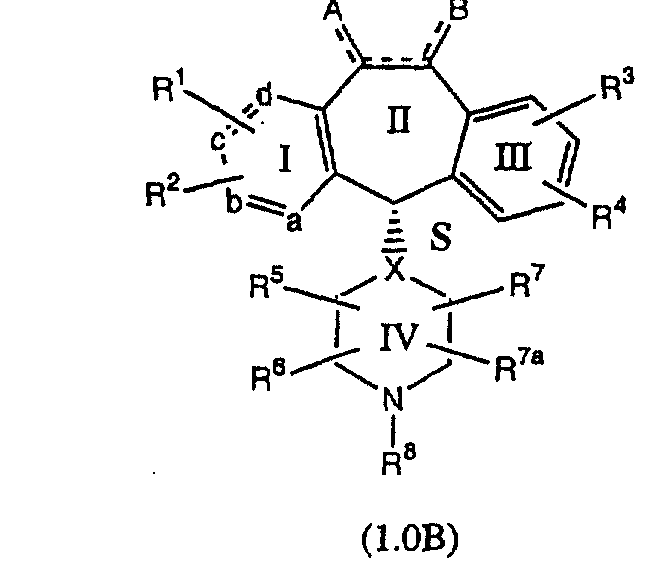
Соединения формулы 1.0 включают предпочтительный R-изомер:
Х = N или СН, а = N или C,
где необязательная связь между атомами С-5 и С-6 имеется, и B означает Н, или необязательная связь между атомами С-5 и С-6 отсутствует, и все В означают Н; и предпочтительный S-изомер:
X = N или СН, а = N или C,
где необязательная связь между атомами С-5 и С-6 имеется, и A означает Н, или необязательная связь между атомами С-5 и С-6 отсутствует, и все A означают Н.
Предпочтительно, чтобы R1, R2, R3 и R4 были независимо выбраны из группы, включающей Н и галоген, более предпочтительно, чтобы они были выбраны из группы, включающей Н, Br, F и CI, а еще более предпочтительно, чтобы они были выбраны из группы, включающей Н и CI. Типичные представители соединений формулы 1.0 включают дигалоген- (например, 3,8-дигалоген-) и моногалогензамещенные соединения (например, 8-галогензамещенные), такие как, например, (З-бром-8-хлор-), (3,8-дихлор-), (3-бром-) и (3-хлор-).
Предпочтительными заместителями а являются C и N, причем наиболее предпочтительным является N.
Предпочтительно, чтобы R8 был выбран из группы, включающей:
Более предпочтительно, чтобы R8 означал 2.0 или 4.0, а наиболее предпочтительно, чтобы R8 означал 4.0.
Предпочтительно, чтобы R11a был выбран из группы, включающей: алкил, замещенный алкил, арил, замещенный арил, гетероарил, замещенный гетероарил, циклоалкил и замещенный циклоалкил, где указанные группы R11a содержат заместители, независимо выбранные из группы, включающей: галоген (предпочтительно -F или CI), цианогруппу, -CF3 и алкил; и где указанные замещенные алкильные группы R11a содержат заместители, выбранные из группы, включающей галоген (предпочтительно -F или CI), цианогруппу и -CF3. Более предпочтительно, чтобы R11a был выбран из группы, включающей: алкил, арил, замещенный арил, циклоалкил или замещенный циклоалкил, где указанные замещенные арильные и замещенные циклоалкильные группы содержат заместители, независимо выбранные из группы, включающей:
галоген (предпочтительно -F или CI), CN и -CF3. Более предпочтительно, чтобы R11a был выбран из группы, включающей метил, трет-бутил, фенил, цианофенил, хлор-фенил, фторфенил и циклогексил. Еще более предпочтительно, чтобы R11a был выбран из группы, включающей: трет-бутил, цианофенил, хлорфенил, фторфенил и циклогексил. Еще более предпочтительно, чтобы R11a был выбран из группы цианофенилов, а еще более предпочтительным является п-цианофенил.
Предпочтительно, чтобы R11 был выбран из группы, включающей: алкил, циклоалкил и замещенный циклоалкил, где указанная замещенная циклоалкильная группа содержит 1, 2 или 3 заместителя, независимо выбранные из группы, включающей:
галоген (предпочтительно - хлор или фтор) и алкил (предпочтительно - метил или трет-бутил). Примеры групп R11 включают: метил, этил, пропил, трет-бутил, циклогексил и замещенный циклогексил. Более предпочтительно, чтобы R11 был выбран из группы, включающей метил, трет-бутил, циклогексил, хлорциклогексил (предпочтительно - п-хлорциклогексил) и фторциклогексил (предпочтительно - п-фторциклогексил). Наиболее предпочтительно, чтобы R11 был выбран из группы, включающей метил, трет-бутил и циклогексил, а еще более предпочтительными являются трет-бутил и циклогексил.
Предпочтительно, чтобы R12 был выбран из группы, включающей Н и метил. Наиболее предпочтительно, чтобы R12 означал Н.
Предпочтительно, чтобы R5, R6, R7 и R73 означали Н.
Предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) гетероарил;
(2) замещенный гетероарил;
(3) арилалкоксил;
(4) замещенный арилалкоксил;
(5) гетероциклоалкил;
(6) замещенный гетероциклоалкил;
(7) гетероциклоалкилалкил;
(8) замещенный гетероциклоалкилалкил;
(9) гетероарилалкил;
(10) замещенный гетероарилалкил;
(11) гетероарилалкенил и
(12) замещенный гетероарилалкенил;
где указанные замещенные группы R9 содержат один или большее количество (например, 1, 2 или 3) заместителей, независимо выбранных из группы, включающей:
(1) -ОН;
(2) -CO2R14; где R14 выбран из группы, включающей: Н и алкил (например, метил или этил), предпочтительно - алкил, наиболее предпочтительно - метил или этил;
(3) алкил, в качестве заместителей содержащий одну или большее количество групп -OH (например, 1, 2 или 3, предпочтительно - 1), например, -(CH2)qOH, где q равно 1-4, а предпочтительно, если q = 1;
(4) галоген (например, Br, F, I или Cl);
(5) алкил, обычно алкил с 1-6 атомами углерода, предпочтительно -алкил с 1-4 атомами углерода (например, метил, этил, пропил или бутил (предпочтительно - изопропил или трет-бутил));
(6) аминогруппу;
(7) тритил;
(8) гетероциклоалкил;
(9) арилалкил (например, бензил);
(10) гетероарил (например, пиридил) и
(11) гетероарилалкил (пиперидин-СН3).
Наиболее предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) гетероциклоалкил;
(2) замещенный гетероциклоалкил;
(3) гетероциклоалкилалкил;
(4) замещенный гетероциклоалкилалкил;
(5) гетероарилалкил;
(6) замещенный гетероарилалкил;
(7) гетероарилалкенил и
(8) замещенный гетероарилалкенил;
где указанные замещенные группы R9 содержат заместители, независимо выбранные из группы, включающей:
(1) -ОН;
(2) -CO2R14; где R14 выбран из группы, включающей: Н и алкил (например, метил или этил), предпочтительно - алкил и наиболее предпочтительно - метил или этил;
(3) алкил, в качестве заместителей содержащий одну или большее количество групп -OH (например, 1, 2 или 3, предпочтительно - 1), например, -(CH2)qOH, где q равно 1-4, а предпочтительно, если q = 1;
(4) галоген (например, Br или Cl);
(5) алкил, обычно алкил с 1-6 атомами углерода, предпочтительно - алкил с 1-4 атомами углерода (например, метил, этил, пропил, изопропил, бутил или трет-бутил, наиболее предпочтительно - трет-бутил);
(6) аминогруппу;
(7) тритил;
(8) гетероциклоалкил;
(9) арилалкил;
(10) гетероарил и
(11) гетероарилалкил.
Более предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) гетероциклоалкил;
(2) замещенный гетероциклоалкил;
(3) гетероциклоалкилалкил;
(4) замещенный гетероциклоалкилалкил;
(5) гетероарилалкил;
(6) замещенный гетероарилалкил;
(7) гетероарилалкенил и
(8) замещенный гетероарилалкенил;
где указанные замещенные группы R9 содержат заместители, независимо выбранные из группы, включающей:
(1) галоген (например, Br или Cl);
(2) алкил, обычно алкил с 1-6 атомами углерода, предпочтительно - алкил с 1-4 атомами углерода (например, метил, этил, пропил, изопропил, бутил или трет-бутил, наиболее предпочтительно -трет-бутил);
(3) алкил, в качестве заместителей содержащий одну или большее количество групп
-ОН (например, 1, 2 или 3, предпочтительно - 1), например, -(CH2)qOH, где q равно 1-4, а предпочтительно, если q = 1;
(4) аминогруппу;
(5) тритил;
(6) арилалкил и
(7) гетероарил.
Еще более предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) гетероциклоалкилалкил;
(2) замещенный гетероциклоалкилалкил;
(3) гетероарилалкил и
(4) замещенный гетероарилалкил;
где заместители для указанных замещенных групп R9 независимо выбраны из группы, включающей:
(1) галоген (например, Br или Cl);
(2) алкил, обычно алкил с 1-6 атомами углерода, предпочтительно - алкил с 1-4 атомами углерода (например, метил, этил, пропил, изопропил, бутил или трет-бутил, наиболее предпочтительно - трет-бутил);
(3) аминогруппу и
(4) тритил.
Еще более предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) гетероциклоалкилалкил;
(2) замещенный гетероциклоалкилалкил;
(3) гетероарилалкил и
(4) замещенный гетероарилалкил;
где заместители для указанных замещенных групп R9 независимо выбраны из группы, включающей:
(1) галоген (например, Br или Cl) и
(2) алкил, обычно алкил с 1-6 атомами углерода, предпочтительно - алкил с 1-4 атомами углерода (например, метил, этил, пропил, изопропил, бутил или трет-бутил, наиболее предпочтительно - трет-бутил),
Еще более предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) пиперидинил;
(2) пиперазинил;
(3) -(СН2)р-пиперидинил;
(4) -(СН2)р-пиперазинил;
(5) -(СН2)р-морфолинил и
(6) -(СН2)р-имидазолил;
где р равно от 0 до 1 и где циклический фрагмент каждой группы R9 необязательно содержит один, два или три заместителя, независимо выбранные из группы, включающей:
(1) галоген (например, Br или Cl) и
(2) алкил, обычно алкил с 1-6 атомами углерода, предпочтительно - алкил с 1-4 атомами углерода (например, метил, этил, пропил, изопропил, бутил или трет-бутил, наиболее предпочтительно - трет-бутил).
Еще более предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) пиперазинил;
(2) -(СН2)р-пиперидинил;
(3) -(СН2)р-имидазолил; и
(5) -(СН2)р-морфолинил,
где р равно от 1 до 4 и где циклический фрагмент каждой группы R9 необязательно содержит один, два или три заместителя, независимо выбранные из группы, включающей метил, этил и изопропил.
Еще более предпочтительно, чтобы R9 был выбран из группы -(СН2)-имидазолилов, где указанное имидазолильное кольцо необязательно содержит 1, 2 или 3 заместителя, предпочтительно - 1, выбранные из группы, включающей метил и этил.
Еще более предпочтительно, чтобы R9 был выбран из группы -(СН2)-(2-метил)-имидазолов.
Предпочтительно, чтобы хотя бы один из заместителей R21, R22 и R46 не означал Н и алкил. Более предпочтительно, чтобы R21 и R22 означали Н, а R46 не означал Н и алкил. Наиболее предпочтительно, чтобы R21 и R22 означали Н, а R46 был выбран из группы, включающей гетероарил и гетероциклоалкил.
Предпочтительно, чтобы указанные гетероарильные группы, которыми являются указанные R21, R22 и R46, представляли собой 3-пиридил, 4-пиридил, 3-пиридил-N-оксид или 4-пиридил-N-оксид; более предпочтительно - 4-пиридил или 4-пиридил-N-оксид; наиболее предпочтительно - 4-пиридил-N-оксид.
Предпочтительно, чтобы указанные гетероциклоалкильные группы, которыми являются указанные R21, R22 и R46, представляли собой пиперидиновое кольцо V:
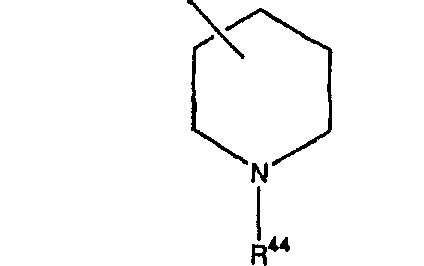
где R44 означает -C(O)NHR51, и предпочтительно, чтобы R51 означал -C(O)NH2. Более предпочтительно, чтобы пиперидиновое кольцо V означало:
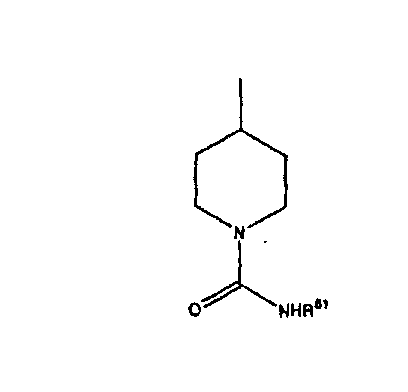
а наиболее предпочтительное кольцо V означает:
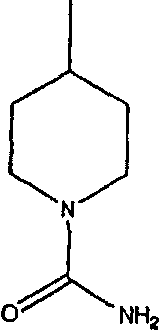
Таким образом, предпочтительно, чтобы R21, R22 и R46 были независимо выбраны из группы, включающей:
(1) Н;
(2) арил (наиболее предпочтительно - фенил);
(3) гетероарил и
(4) гетероциклоалкил (например, пиперидиновое кольцо V),
где хотя бы один из заместителей R21, R22 и R46 не означает Н, а наиболее предпочтительно, чтобы R21 и R22 означали Н, а R46 не означал Н, а более предпочтительно, чтобы R21 и R22 означали Н, а R46 был выбран из группы, включающей гетероарил и гетероциклоалкил, а еще более предпочтительно, чтобы R21 и R22 означали Н, а R46 означал пиперидиновое кольцо V; где предпочтительные значения гетероарила и пиперидинового кольца V являются такими, как указано выше.
Предпочтительно, чтобы A и В были независимо выбраны из группы, включающей:
(1) -Н;
(2) - R9;
(3) -R9-C(O)-R9;
(4) -R9-CO2-R98;
(5) -C(O)NHR9;
(6) -C(O)NH-CH2-C(O)-NH2;
(7) -C(O)NHR26;
(8) -(CH2)p(R9)2, где R9 являются одинаковыми или разными;
(9) -(CH2)pC(O)R9;
(10) -(CH2)pC(O)R27a;
(11) -(CH2)pC(O)N(R9)2, где R9 являются одинаковыми или разными;
(12) -(CH2)pC(O)NH(R9);
(13) -(CH2)pNHC(O)R50;
(15) -(CH2)pN(C(O)R27a)2, где R27a являются одинаковыми или разными;
(16) -(CH2)pNR51C(O)R27, причем необязательно R51 и R27 совместно с атомами, к которым они присоединены, образуют 5- или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(17) -(CH2)pNR51C(O)NR27, причем необязательно R51 и R27 совместно с атомами, к которым они присоединены, образуют 5- или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(18) -(CH2)pNR51C(O)N(R27a)2, где R27a являются одинаковыми или разными;
(19) -(CH2)pNHSO2N(R51), где R51 являются одинаковыми или разными;
(20) -(CH2)pNHCO2R50;
(21) -(CH2)pCO2R51;
(22) -NHR9;
(23)
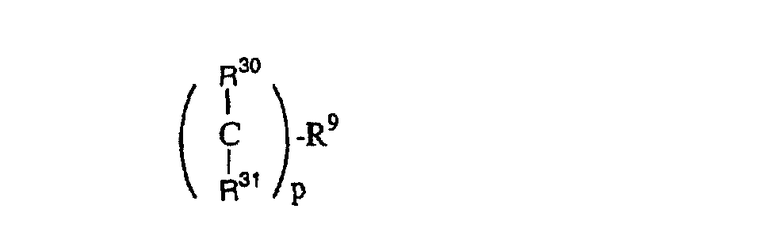
где R30 и R31 являются одинаковыми или разными и
(24)
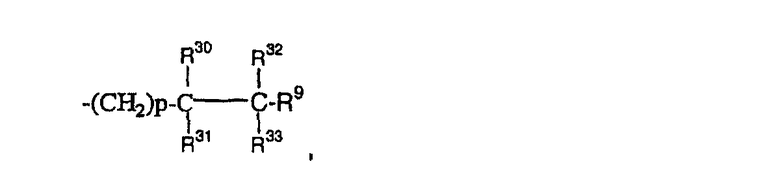
где R30, R31, R32 и R33 являются одинаковыми или разными.
Наиболее предпочтительно, чтобы А и В были независимо выбраны из группы, включающей:
(1) -Н;
(2) -R9;
(3) -R9-C(O)-R9;
(4) -R9-CO2-R9a;
(5) -C(O)NHR9;
(6) -(CH2)p(R9)2, где R9 являются одинаковыми или разными;
(7) -(CH2)pC(O)R9;
(8) -(CH2)pC(O)N(R9)2, где R9 являются одинаковыми или разными;
(9) -(CH2)pC(O)NH(R9);
(10) -(CH2)pNR51C(O)R27, причем необязательно R51 и R27 совместно с атомами, к которым они присоединены, образуют 5- или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(11) -(CH2)pNR51C(O)NR27, причем необязательно R51 и R27 совместно с атомами, к которым они присоединены, образуют 5- или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(13) -NHR9
Примеры A и В (без наложения ограничений) включают:
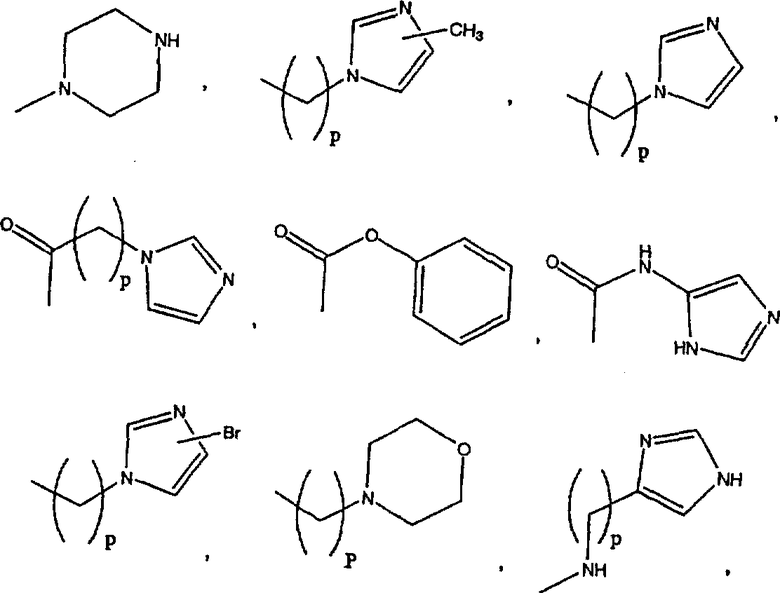
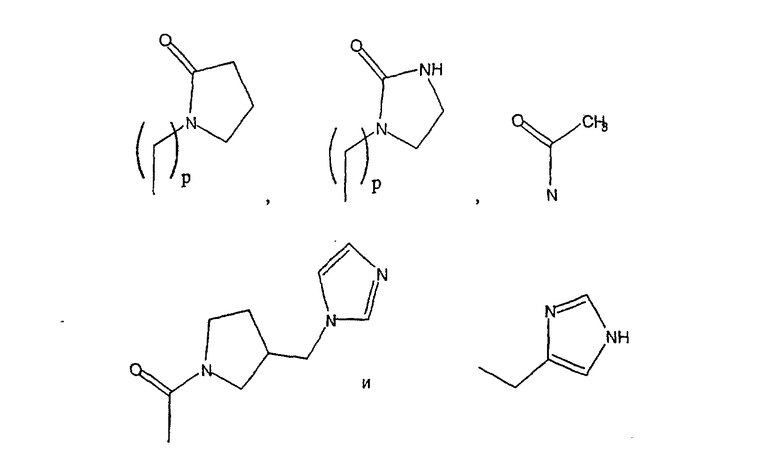
где р равно 0, 1, 2, 3 или 4.
Если имеется необязательная связь между атомами С-5 и С-6 (т. е. имеется двойная связь между атомами С-5 и С-6), то предпочтительно, чтобы одна из групп A и В означала Н, а другая означала R9, и предпочтительно, чтобы R9 был выбран из группы, включающей:
(1) гетероарил;
(2) замещенный гетероарил;
(3) арилалкил;
(4) замещенный арилалкил;
(5) арилалкоксил;
(6) замещенный арилалкоксил;
(7) гетероциклоалкил;
(8) замещенный гетероциклоалкил;
(9) гетероциклоалкилалкил;
(10) замещенный гетероциклоалкилалкил;
(11) гетероарилалкил;
(12) замещенный гетероарилалкил;
(13) алкенил;
(14) замещенный алкенил;
(15) гетероарилалкенил и
(16) замещенный гетероарилалкенил,
где заместители в указанных замещенных группах R9 независимо выбраны из группы, включающей:
(1) -ОН;
(2) -CO2R14;
(3) -CH2OR14;
(4) галоген;
(5) алкил (например, метил, этил, пропил, бутил или трет-бутил);
(6) аминогруппу;
(7) тритил;
(8) гетероциклоалкил;
(9) арилалкил;
(10) гетероарил и
(11) гетероарилалкил,
где R14 независимо выбран из группы, включающей: Н и алкил, предпочтительно - метил и этил.
Более предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа A означала Н, а группа B означала R9. Наиболее предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа A означала Н, а группа B означала R9, причем R9 был выбран из группы, включающей:
(1) арилалкил;
(2) замещенный арилалкил;
(3) арилалкоксил;
(4) замещенный арилалкоксил;
(5) гетероциклоалкил;
(6) замещенный гетероциклоалкил;
(7) гетероциклоалкилалкил;
(8) замещенный гетероциклоалкилалкил;
(9) гетероарилалкил;
(10) замещенный гетероарилалкил;
(11) алкенил;
(12) замещенный алкенил;
(13) гетероарилалкенил и
(14) замещенный гетероарилалкенил,
где заместители в указанных замещенных группах R9 независимо выбраны из группы, включающей:
(1) -ОН;
(2) галоген (предпочтительно Br);
(3) алкил (например, метил, этил, пропил, бутил или трет-бутил);
(4) аминогруппу и
(5) тритил.
Еще более предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа A означала Н, а группа B означала R9, причем R9 был выбран из группы, включающей:
(1) гетероциклоалкилалкил;
(2) замещенный гетероциклоалкилалкил;
(3) гетероарилалкил и
(4) замещенный гетероарилалкил,
где заместители в указанных замещенных группах R9 являются одинаковыми или разными алкильными группами (например, представляют собой (С1-С4)-алкил).
Еще более предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа A означала Н, а группа B означала R9, причем R9 был выбран из группы, включающей:
(1) гетероарил-(С1-С3)-алкил и
(2) замещенный гетероарил-(С1-С3)-алкил,
где заместители в указанных замещенных группах R9 являются такими, как указано выше.
Еще более предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа A означала Н, а группа В означала R9, причем R9 был выбран из группы, включающей:
(1) гетероарил-(С1-С3)-алкил, причем предпочтительным является гетероарил-СН3-, и
(2) (замещенный гетероарил)-(С1-С3)-алкил, причем предпочтительным является (замещенный гетероарил)-СН2-,
где в указанных замещенных группах R9 имеется один или большее количество (например, 1, 2 или 3) заместителей, причем предпочтительно, чтобы имелся один заместитель, которые представляют собой одинаковые или разные алкильные группы (например, -СН3, -С2Н5, -С3H4, а предпочтительной является -СН3).
Еще более предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа A означала Н, а группа В означала R9, причем R9 был выбран из группы, включающей:
(1) -СН2-имидазолил;
(2) (замещенный имидазолил)-СН2-;
(3) -(СН2)2-имидазолил;
(4) (замещенный имидазолил)-(СН2)2-;
(5) -(СН2)3-имидазолил;
(6) (замещенный имидазолил)-(СН2)3-;
(7) -СН2-пиперазинил;
(8) -СН2-морфолинил;
где в указанных замещенных группах R9 имеется один или большее количество (например, 1, 2 или 3) заместителей, причем предпочтительно, чтобы имелся один заместитель, которые представляют собой одинаковые или разные алкильные группы (например, -СН3, -С2Н5, -С3Н4), а предпочтительной является -СН3, и где предпочтительными являются замещенные имидазолильные группы вида:
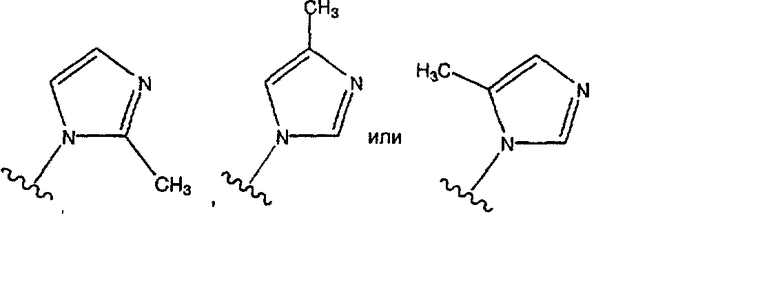
причем
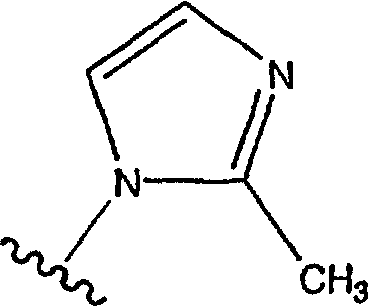
является наиболее предпочтительной.
Еще более предпочтительно, чтобы в случае, когда имеется двойная связь между атомами С-5 и С-6, группа А означает Н, а группа В означает R9, где R9 означает (замещенный имидазолил)-СН2-, и
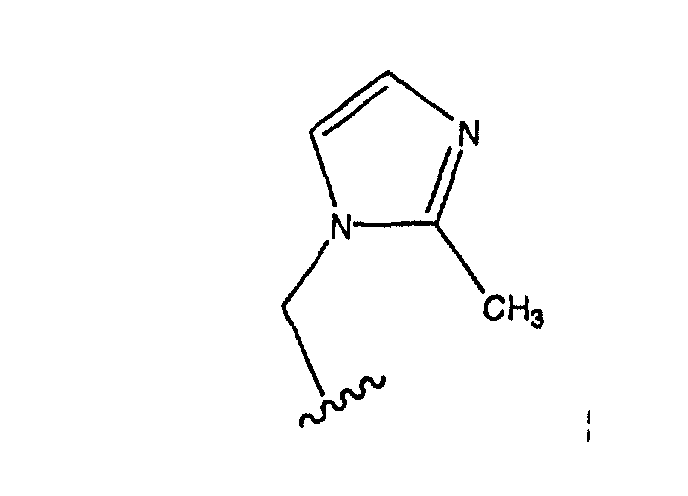
является предпочтительным.
Если B означает Н и A означает R9 и имеется двойная связь между атомами С-5 и С-6, то для А группы R9 являются такими, как указанные выше группы для В.
Если необязательная связь между атомами С-5 и С-6 отсутствует (т.е. если имеется ординарная связь между атомами С-5 и С-6), то все A и все В выбираются независимо, и определения для А и В являются такими, которые указаны выше для случая, когда имеется необязательная связь, при условии, что, если имеется ординарная связь между атомами С-5 и С-6, то один из двух заместителей A или один из двух заместителей В означает Н (т.е. если имеется ординарная связь между атомами С-5 и С-6, то один из четырех заместителей (А, А, В и В) должен означать Н).
Предпочтительно, чтобы между атомами С-5 и С-6 имелась двойная связь.
Соединения, соответствующие настоящему изобретению, обладающие R-и S-конфигурацией по атому С-11, включают:
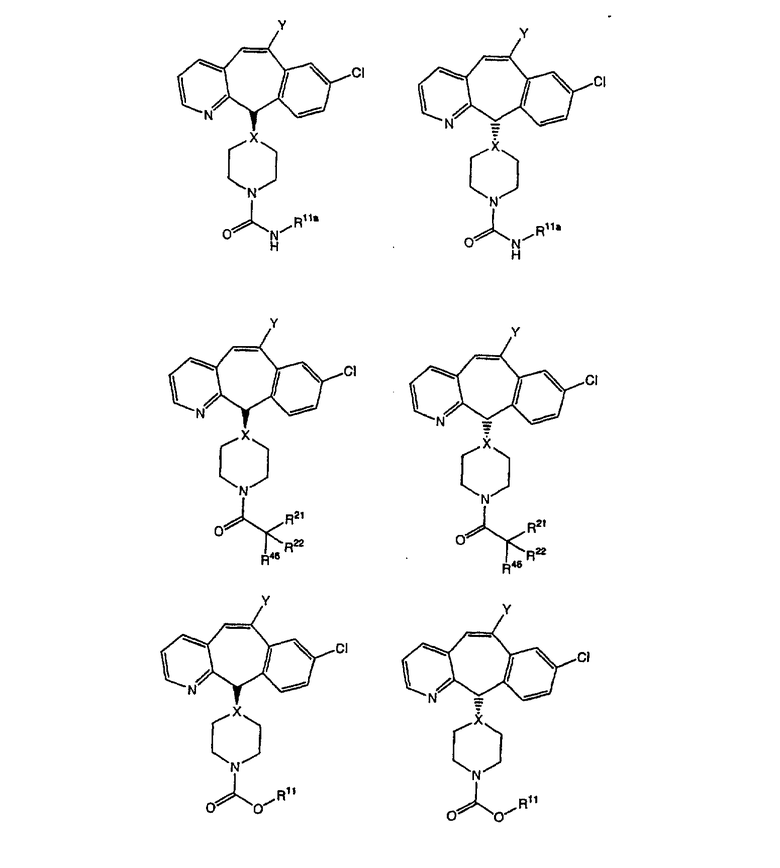
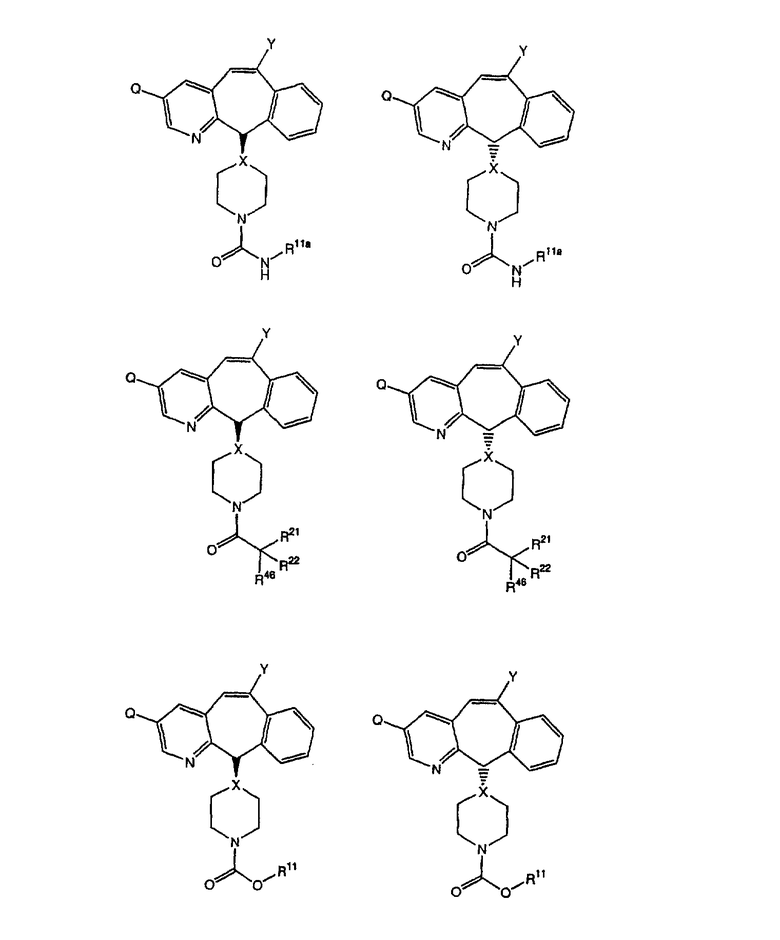
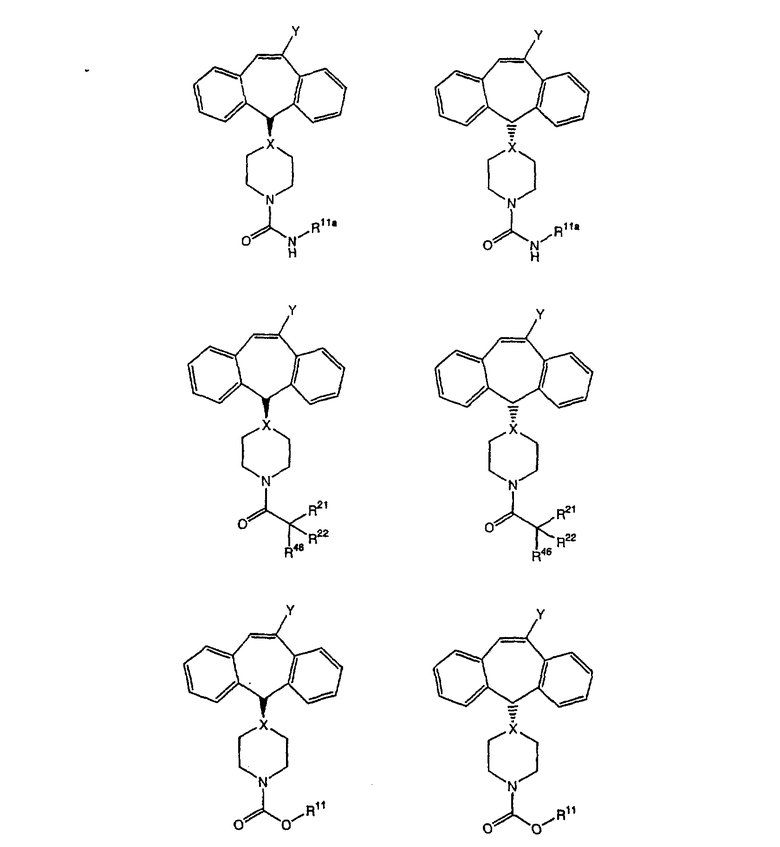
где X = N или C;
Q = Br или Cl;
Y = алкил, арилалкил или гетероарилалкил.
Предпочтительные соединения, соответствующие настоящему изобретению, перечислены ниже:
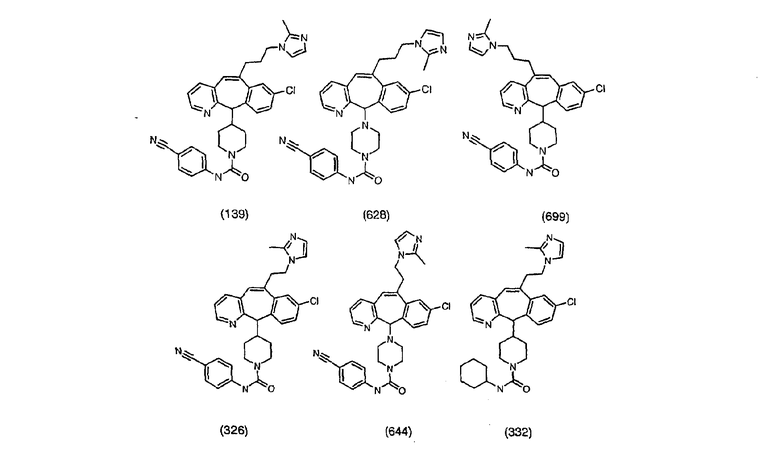
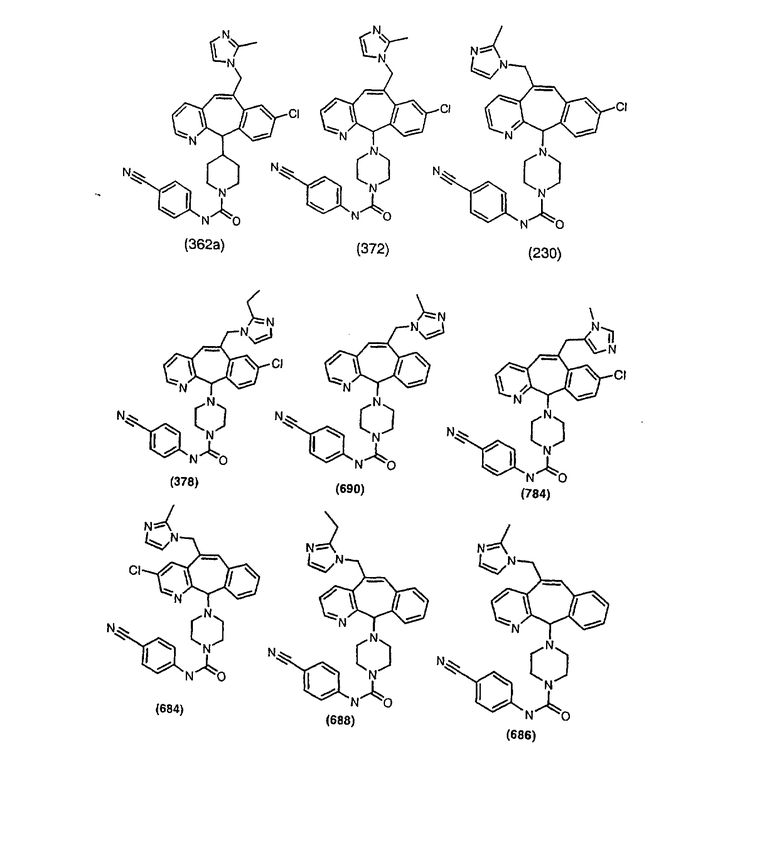
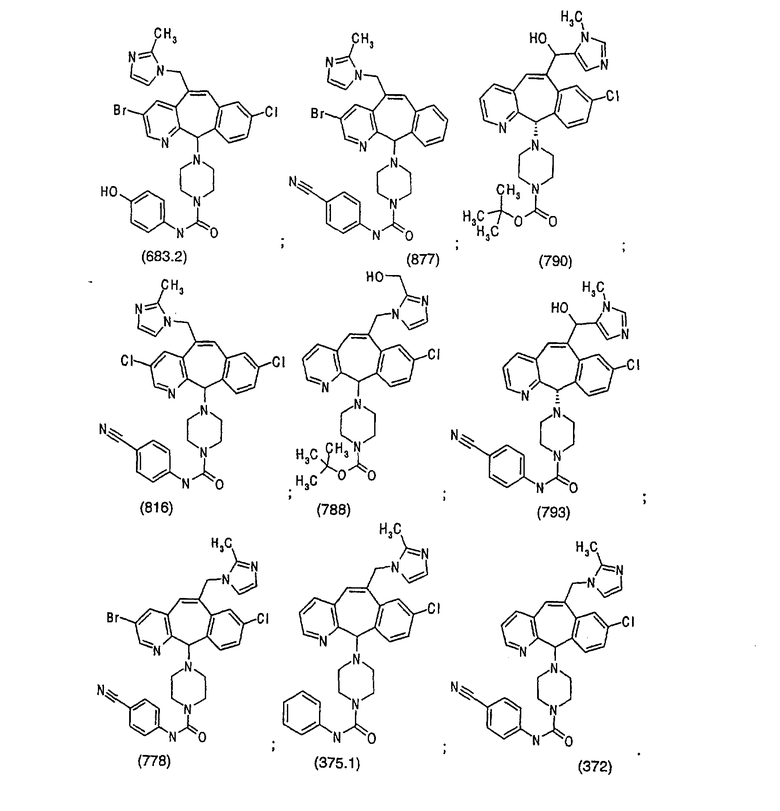
Более предпочтительные соединения, соответствующие настоящему изобретению, перечислены ниже:
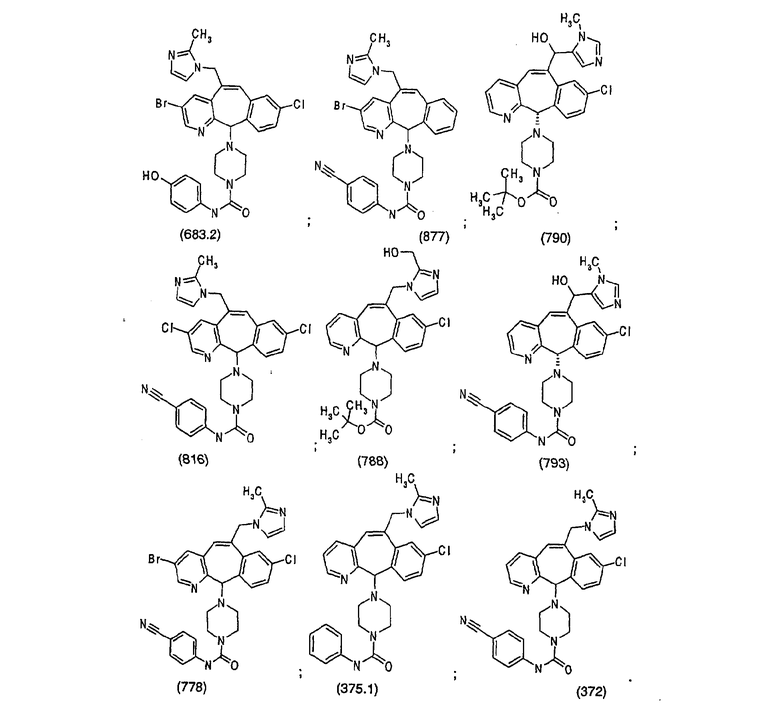
Наиболее предпочтительные соединения, соответствующие настоящему изобретению, перечислены ниже:
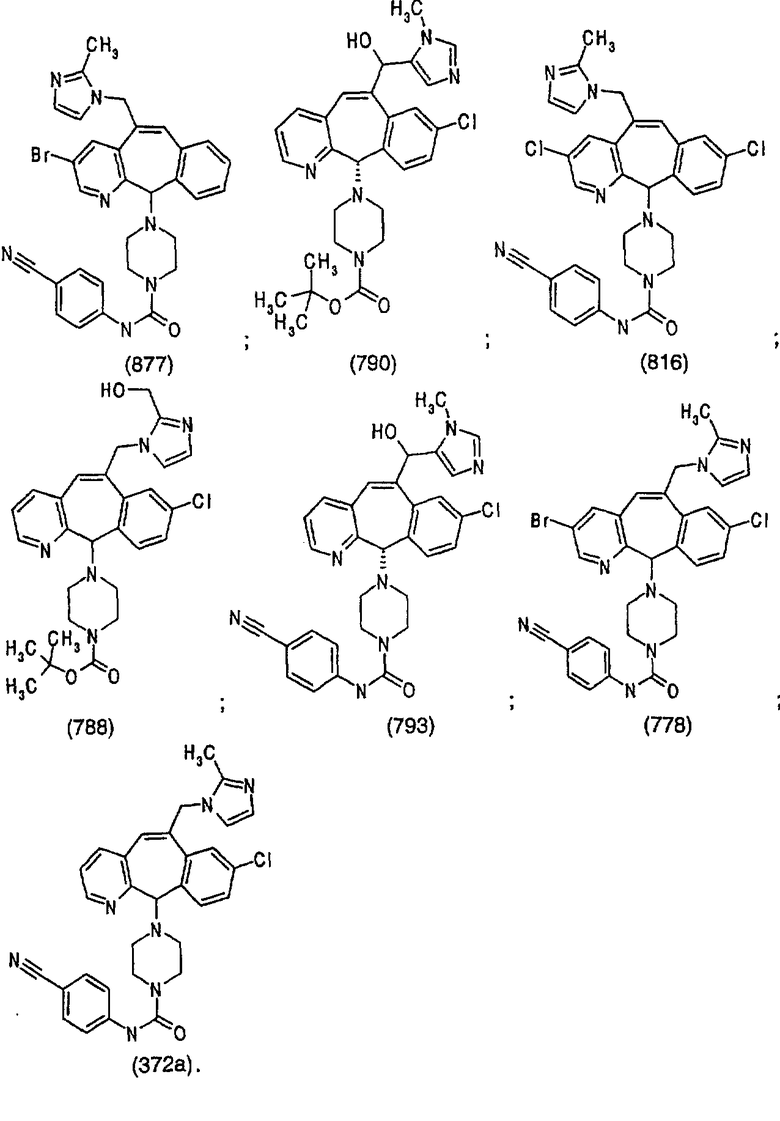
Отрезки, проведенные внутрь циклических систем, показывают, что указанная связь может быть образована с любыми способными к замещению атомами углерода цикла.
Некоторые соединения, соответствующие настоящему изобретению, могут находиться в различных изомерных (например, энантиомерных, диастереоизомерных, атропоизомерных) формах. Предполагается, что настоящее изобретение охватывает все такие изомеры, как чистые, так и смеси, включая рацемические смеси. Также охватываются енольные формы.
Некоторые трициклические соединения по своей природе являются кислотными, например соединения, в которых содержатся карбоксильная или фенольная гидроксильная группа. Эти соединения могут образовывать приемлемые с фармацевтической точки зрения соли. Примеры таких солей могут включать соли натрия, калия, кальция, алюминия, золота и серебра. Также предполагается, что настоящее изобретение охватывает соли, образованные с фармацевтически приемлемыми аминами, такими как аммиак, алкиламины, гидроксиалкиламины, М-метилглюкамин и т. п.
Некоторые основные трициклические соединения также образуют фармацевтически приемлемые соли, например молекулярные соли с кислотами. Например, атомы азота пиридинового типа могут образовывать соли с сильными кислотами, а соединения, в которых имеются основные заместители, такие как аминогруппы, также образуют соли и с более слабыми кислотами. Примерами кислот, подходящих для образования солей, являются хлористоводородная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие неорганические и карбоновые кислоты, хорошо известные специалистам в данной области техники. Соли получают путем взаимодействия соединения в виде свободного основания с достаточным количеством необходимой кислоты с образованием соли, происходящим обычным образом. Свободные основания можно выделить путем обработки соли подходящим разбавленным водным раствором основания, таким как разбавленный водный раствор NaOH, карбоната калия, аммиака и бикарбоната натрия. Свободные основания отличаются от соответствующих солей по некоторым физическим характеристикам, таким как растворимость в полярных растворителях, однако для задач настоящего изобретения по остальным характеристикам соли кислот и оснований эквивалентны соответствующим свободным основаниям.
В рамках объема настоящего изобретения все такие соли кислот и оснований считаются фармацевтически приемлемыми солями, и для задач настоящего изобретения все такие соли кислот и оснований считаются эквивалентными свободным основаниям соответствующих соединений.
Соединения формулы 1.0 могут находиться в несольватированных, а также в сольватированных формах, включая гидратированные формы, например полугидраты. Обычно сольватированные формы, образованные с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., для задач настоящего изобретения эквивалентны несольватированным формам.
Способы лечения пролиферативных заболеваний (рака), соответствующие настоящему изобретению, включают способ лечения (ингибирования) аномального роста клеток, включая трансформированные клетки, пациента, нуждающегося в таком лечении (например, млекопитающего, такого как человека), осуществляющийся путем введения, совместного или последовательного, эффективного количества соединения, соответствующего настоящему изобретению, и эффективного количества химиотерапевтического препарата и/или проведения облучения. Аномальный рост клеток означает рост клеток, не зависящий от нормальных механизмов регуляции (например, утрату контактного ингибирования), включая аномальный рост: (1) опухолевых клеток (опухолей), экспрессирующих активированный онкоген Ras; (2) опухолевых клеток, в которых белок Ras активирован в результате онкогенной мутации в другом гене; и (3) доброкачественных и злокачественных клеток при других пролиферативных заболеваниях.
В предпочтительных вариантах осуществления изобретения способы, соответствующие настоящему изобретению, включают способы лечения или подавления роста опухоли для пациента, нуждающегося в таком лечении (например, млекопитающего, такого как человека), осуществляющиеся путем введения, совместного или последовательного, (1) эффективного количества соединения, соответствующего настоящему изобретению, и (2) эффективного количества по крайней мере одного противоопухолевого препарата, препарата, воздействующего на микротрубочки, и/или проведения лучевой терапии. Примеры опухолей, которые можно подвергнуть лечению, включают (без наложения ограничений) разные виды эпителиального рака, например рак предстательной железы, рак легких (например, аденокарциному легких), разные виды рака поджелудочной железы (например, карциному поджелудочной железы, такую как карцинома поджелудочной железы неэндокринной этиологии), разные виды рака молочной железы, разные виды рака ободочной кишки (например, разные виды колоректального рака, такие как, например, аденокарцинома ободочной кишки и аденома ободочной кишки), рак яичника и рак мочевого пузыря. Другие виды рака, которые можно подвергнуть лечению, включают меланому, разные виды миелоидной лейкемии (например, острую миелогенную лейкемию), саркомы, фолликулярный рак щитовидной железы и миелодиспластический синдром. В частности, пролиферативное заболевание (опухоль), которое можно подвергнуть лечению, выбирается из группы, включающей рак легких, рак поджелудочной железы, рак предстательной железы и миелоидную лейкемию. Для способов, соответствующих настоящему изобретению, предпочтительно, чтобы заболевание (опухоль), которое можно подвергнуть лечению, было выбрано из группы, включающей рак легких и миелоидную лейкемию.
Способы лечения пролиферативных заболеваний, соответствующие настоящему изобретению, включают способ лечения (ингибирования) пролиферативных заболеваний, доброкачественных и злокачественных, при которых белки Ras аберрантно активируются вследствие онкогенной мутации в других генах - т.е. сам ген при мутации не активируется в онкогенную форму. Этот способ включает введение, совместное или последовательное, эффективного количества соединения, соответствующего настоящему изобретению, и эффективного количества противоопухолевого препарата и/или проведения лучевой терапии пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек). Примеры пролиферативных заболеваний, которые можно подвергнуть лечению, включают: доброкачественное пролиферативное заболевание нейрофиброматоз, или опухоли, при которых Ras активируется вследствие мутации или сверхэкспрессии онкогенов тирозинкиназы (например, neu, scr, abl, Ick, lyn, fyn).
Для лучевой терапии предпочтительно использовать γ-излучение.
Способы лечения пролиферативных заболеваний, соответствующие настоящему изобретению, включают способ лечения (ингибирования) аномального роста клеток, включая трансформированные клетки, пациента, нуждающегося в таком лечении (например, млекопитающего, такого как человека), осуществляющийся путем введения, совместного или последовательного, эффективного количества соединения, соответствующего настоящему изобретению, и эффективного количества по крайней мере одного ингибитора трансдукции сигнала.
Типичные ингибиторы трансдукции сигнала включают (без наложения ограничений):
(i) Ингибиторы Bcr/abl-киназы, такие как, например, STI 571 (Gleevec);
(ii) Ингибиторы рецептора эпидермального фактора роста (EFG), такие как, например, ингибиторы киназы (Iressa, OSI-774) и антитела (Imclone: C225 [Goldstein et al. (1955), Clin. Cancer Res. 1: 1311 -1318] и Abgenix: ABX-EGF), и
(iii) Ингибиторы рецептора Нег-2/neu, такие как, например, Herceptin® (трастузумаб).
При использовании в настоящем изобретении перечисленные ниже термины обладают приведенными ниже значениями (если не указано иного):
противоопухолевый препарат - химиотерапевтический препарат, действующий на раковую опухоль;
совместно - (1) одновременно по времени или (2) в разное время в течение обычного курса лечения; и
последовательно - (1) введение одного компонента, применяющегося в способе ((а) соединения, соответствующего настоящему изобретению, или (b) химиотерапевтического препарата, ингибитора трансдукции сигнала и/или проведения лучевой терапии) с последующим введением другого компонента или компонентов; после введения одного компонента следующий компонент можно ввести в основном сразу же после первого компонента или же следующий компонент можно ввести через достаточный для оказания воздействия период времени после введения первого компонента; достаточный для оказания воздействия период времени означает период времени, предоставленный для оказания максимального полезного воздействия, обусловленного введением первого компонента.
Термин "совместно с" при использовании в настоящем изобретении по отношении к комбинированной терапии означает, что препараты или компоненты вводятся совместно или последовательно в соответствии с приведенными выше определениями.
ХИМИОТЕРАПЕВТИЧЕСКИЕ ПРЕПАРАТЫ
Классы соединений, которые можно использовать в качестве химиотерапевтических препаратов (противоопухолевых препаратов/препаратов, воздействующих на микротрубочки), включают (без наложения ограничений): алкилирующие реагенты, антиметаболиты, природные вещества и их производные, гормоны и стероиды (включая синтетические аналоги) и синтетические вещества. Ниже приведены примеры соединений этих классов.
Алкилирующие реагенты (включая азотистый иприт и его производные, производные этиленимина, алкилсульфонаты, нитрозомочевины и триазены): урацилизотиоцианат, хлорметин, циклофосфамид (цитоксан®), ифосфамид, мелфалан, хлорамбуцил, пипоброман, триэтиленмеламин, триэтилентиофосфорамин, бусульфан, кармустин, ломустин, стрептозоцин, дакарбазин и темозоломид.
Антиметаболиты (включая антагонисты фолиевой кислоты, аналоги пиримидина, аналоги пурина и ингибиторы аденозиндезамидазы): метотрексат, 5-фторурацил, флоксуридин, цитарабин, 6-меркаптопурин, 6-тиогуанин, флударабинфосфат, пентостатин и гемцитабин.
Природные вещества и их производные (включая алкалоиды барвинка, противоопухолевые антибиотики, ферменты, лимфокины и эпиподофиллотоксины): винбластин, винкристин, виндезин, блеомицин, дактиномицин, даунорубицин, доксорубицин, эпирубицин, идарубицин, паклитаксел (паклитаксел продается под названием таксол® и более подробно описан ниже в подразделе под названием "Препараты, воздействующие на микротрубочки"), производные паклитаксела (например, таксотер), митрамицин, деоксикоформицин, митомицин-С, L-аспарагиназа, интерфероны (в особенности альфа-интерферон), этопозид и тенипозид.
Гормоны и стероиды (включая синтетические аналоги): 17α-этинилэстрадиол, диэтилстильбэстрол, тестостерон, преднизон, флуоксиместерон, дромостанолонпро-пионат, тестолактон, мегестролацетат, тамоксифен, метилпреднизолон, метилтестостерон, преднизолон, триамцинолон, хлоротрианизен, гидроксипрогестерон, аминоглутетимид, эстрамустин, медроксипрогестеронацетат, лейпролид, флутамид, торемифен, золадекс.
Синтетические вещества (включая неорганические комплексные соединения, такие как координационные комплексные соединения платины): цисплатин, карбоплатин, гидроксимочевина, амсакрин, прокарбазин, митотан, митоксантрон, левамизол и гексаметилмеламин.
Особенно предпочтительными являются противоопухолевые препараты, выбранные из группы, включающей циклофосфамид, 5-фторурацил, темозоломид, винкристин, цисплатин, карбоплатин и гемцитабин. Наиболее предпочтительно, если противоопухолевые препараты выбраны из группы, включающей гемцитабин, цисплатин и карбоплатин.
Способы безопасного и эффективного введения большинства этих химиотерапевтических препаратов известны специалистам в данной области техники. Кроме того, их введение описано в обычной литературе. Например, введение многих химиотерапевтических препаратов описано в книге "Physicians' Desk Reference" (PDR) ("Настольный справочник врача", например, в издании 1996 г. (Medical Economics Company, Montvale, NJ 07645-1742, USA), раскрытие которой включено в настоящее изобретение путем ссылки.
ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА МИКРОТРУБОЧКИ
Как показано выше, настоящее изобретение также относится к способам лечения пораженных клеток посредством взаимодействия этих клеток с соединением, ингибирующим ФПТ, соответствующим настоящему изобретению, и препаратом, воздействующим на микротрубочки (например, паклитакселом, производным паклитаксела или соединением типа паклитаксела). При использовании в настоящем изобретении препарат, воздействующий на микротрубочки, означает соединение, которое препятствует клеточному митозу, т.е. оказывает антимитотическое воздействие посредством влияния на образование и/или действие микротрубочек. Такими препаратами могут быть, например, препараты, стабилизирующие микротрубочки, или препараты, которые препятствуют образованию микротрубочек.
Препараты, воздействующие на микротрубочки, пригодные для использования в настоящем изобретении, хорошо известны специалистам в данной области техники, и включают (без наложения ограничений) аллоколхицин (NSC 406042), галихондрин В (NSC 609395), колхицин (NSC 757), производные колхицина (например, NSC 33410), доластатин 10 (NSC 376128), майтансин (NSC 153858), ризоксин (NSC 332598), паклитаксел (таксол®, NSC 125973), производные паклитаксела (например, таксотер, NSC 608832), тиоколхицин (NSC 361792), тритилцистеин (NSC 83265), винбластинсульфат (NSC 49842), винкристинсульфат (NSC 67574), эпотилон А, эпотилон и дискодермолид (см. Service, (1996) Science, 274: 2009), эстрамустин, нокодазол, МАР4 и т. п. Примеры таких препаратов также описаны в научной и патентной литературе, см., например, Bulinski (1997) J. Cell Sci. 110: 3055-3064; Panda (1997) Proc. Natl. Acad. Sci. USA 94: 10560-10564; Muhlradt (1997) Cancer Res. 57: 3344-3346; Nicolaou (1997) Nature 387: 268 -272; Vasquez (1997) Mol. Biol. Cell. 8: 973-985; Panda (1997); J. Biol. Chem. 271: 29807-29812.
Особенно предпочтительными препаратами являются соединения, обладающие активностью, сходной с активностью паклитаксела. К ним относятся (без наложения ограничений) паклитаксел и производные паклитаксела (соединения, сходные с паклитакселом) и аналоги. Паклитаксел и его производные (например, таксол и таксотер) имеются в продаже. Кроме того, способы получения паклитаксела и производных паклитаксела хорошо известны специалистам в данной области техники (см., например, патенты США: US 5569729, 5565478, 5530020, 5527924, 5508447, 5489589, 5488116, 5484809, 5478854, 5478736, 5475120, 5468769, 5461169, 5440057, 5422364, 5411984, 5405972 и 5296506).
Точнее, термин "паклитаксел" при использовании в настоящем изобретении относится к лекарственному препарату, продаваемому под названием таксол® (NSC №:125973). Таксол® ингибирует репликацию эукариотных клеток посредством усиления полимеризации тубулиновых фрагментов в стабилизированные пучки микротрубочек, которые неспособны перестроиться в структуры, необходимые для митоза. Из множества имеющихся хемотерапевтических лекарственных препаратов паклитаксел вызвал интерес вследствие обнаруженной при клинических испытаниях эффективности по отношению к стойким к воздействию лекарственных препаратов опухолям, включая опухоли яичника и молочной железы (Hawkins (1992) Oncology, 6: 17-23, Horwitz (1992) Trends Pharmacol. Sci. 13: 134-146, Rowinsky (1990) J. Natl. Cane. lnst. 82: 1247-1259).
Эффективность дополнительных препаратов, воздействующих на микротрубочки, можно оценить с помощью многих способов анализа, известных в данной области техники, например, с помощью полуавтоматических анализов, посредством которых определяется способность аналогов паклитаксела полимеризовать тубулин, совместно с клеточным анализом, предназначенным для определения способности этих соединений блокировать клетки при митозе (см. Lopes (1997) Cancer Chemother. Pharmacol. 41: 37-47).
Обычно активность исследуемого соединения определяется путем воздействия на клетки этого соединения и определения того, нарушается ли клеточный цикл, в частности, путем ингибирования акта митоза. Такое ингибирование может опосредоваться нарушением митотического аппарата, например нарушением нормального образования веретена. Клетки, митоз которых нарушен, могут характеризоваться измененной морфологией (например, сжатием микротрубочек, увеличением количества хромосом и т.п.).
В предпочтительном варианте осуществления соединения, предположительно обладающие способностью полимеризовать тубулин, исследуются in vitro. В предпочтительном варианте осуществления на культивированных клетках WR21 (полученных от мышей линии 69-2 wap-ras) исследуется способность соединений ингибировать пролиферацию и/или изменять морфологии клеток, в особенности, вызывать сжатие микротрубочек. После этого соединения, для которых получены положительные результаты, можно исследовать in vivo с использованием голых мышей, у которых имеются клетки опухоли WR21. Подробную схему этого способа исследования описал Porter (1995) Lab. Anim. Sd. (45(2): 145-150.
Другие способы исследования представляющей интерес активности соединений хорошо известны специалистам в данной области техники. Обычно такие исследования включают исследование ингибирования сборки и/или разборки микротрубочек. Исследование сборки микротрубочек описали, например, Gaskin et al. J. Molec. Biol., 89: 737-758. В патенте США US 5569720 также описаны проводимый in vitro и in vivo поиск соединений, обладающих активностью, сходной с активностью паклитаксела.
Способы безопасного и эффективного введения указанных выше препаратов, воздействующих на микротрубочки, хорошо известны специалистам в данной области техники. Кроме того, их введение описано в обычной литературе. В частности, введение многих химиотерапевтических препаратов описано в книге "Physicians' Desk Reference" (PDR), например, в издании 1996 г. (Medical Economics Company, Montvale, NJ 07645-1742, USA), раскрытие которой включено в настоящее изобретение путем ссылки.
Общие схемы синтеза
Для получения соединений, соответствующих настоящему изобретению, можно использовать следующие способы.
Трициклические пиридинсодержащие соединения
Специалисту в данной области техники должно быть понятно, что соединения, соответствующие настоящему изобретению и описываемые формулой 1, где одна из групп а, b, с и d означает N или N+O-, можно получить по следующим схемам:
Схема 1:
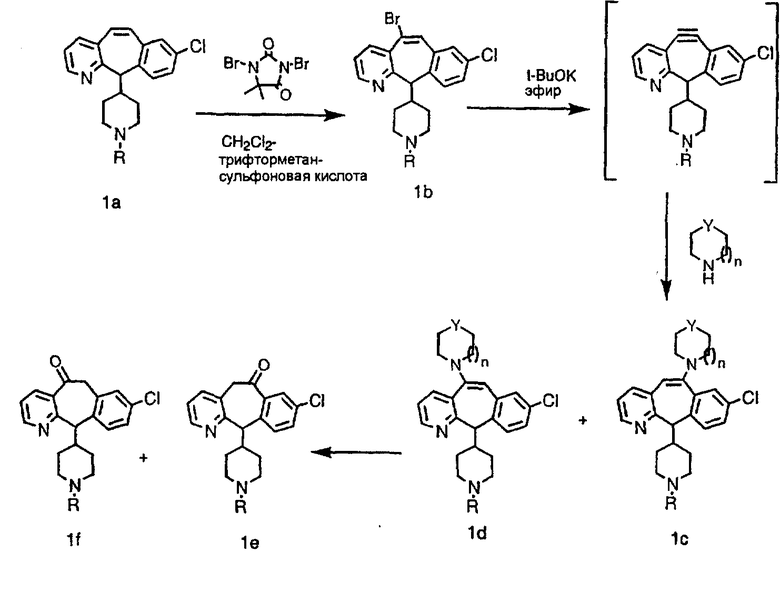
Синтез 5-бромированного трициклического соединения 1b начинают с мостикового олефина 1а (J. Med. Chem. (1998), 41, 1561-1567), который обрабатывают дибромдиметилгидантоином в среде, содержащей трифторметансульфоновую кислоту. Последующая обработка винилбромида трет-бутоксидом калия в присутствии соответствующего вторичного амина дает 5-и 6-замещенные енаминовые аддукты. Если Y означает NH (случай пиперазина), то ацилирование, сульфонирование и образование амида можно выполнить с помощью стандартных методик. Обработка этих аминных аддуктов водным раствором HCI, проводимая при соответствующих температурах, приводит к образованию 5-и 6-азакетонов, 1f и 1е соответственно.
Схема 2:
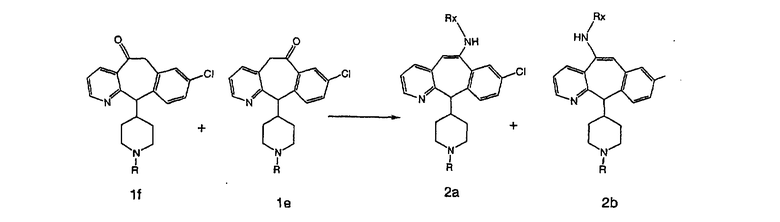
В случаях, когда требуются вторичные амины, синтез проводят, исходя из азакетонов 1f и 1е, как показано на схеме 2. Соответствующий кетон и амин кипятят с обратным холодильником в толуоле в присутствии п-толуолсульфоновой кислоты в аппарате Дина-Штарка.
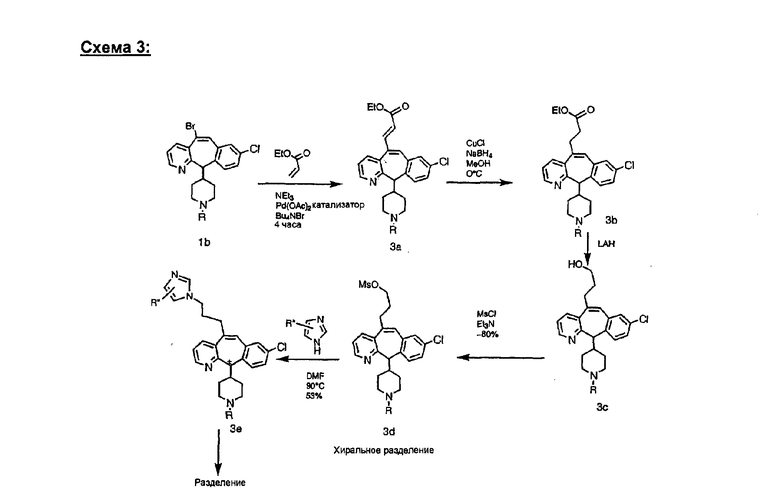
Синтез аналогов, содержащих разделительную группу из 3 атомов углерода, можно провести в соответствии со схемой 3. Таким образом, вводя трициклический винилбромид 1b в реакцию типа Хека с использованием этилакрилата и Pdo в качестве катализатора, получают α-β-ненасыщенный сложный эфир 3а. Восстановление сопряженных двойных связей проводят с помощью восстановительного реагента, хлорида двухвалентной меди - борогидрида натрия. Затем сложный эфир с помощью алюмогидрида лития восстанавливают в спирт. Обработка спирта метансульфонилхлоридом в подходящем апротонном растворителе с последующим вытеснением подходящей солью натрия дает искомые имидазолы. В большинстве случаев на этой стадии проводят разделение изомеров. Если группа R в 3е представляет собой ВОС, то ее удаление с помощью смеси HCI-диоксан дает гидрохлориды аминов. С помощью стандартных химических реакций эти амины превращают в мочевины, карбаматы, сульфонамиды и амиды.
Схема 4: ПОЛУЧЕНИЕ 6-ЗАМЕЩЕННЫХ УГЛЕРОДНЫХ АНАЛОГОВ
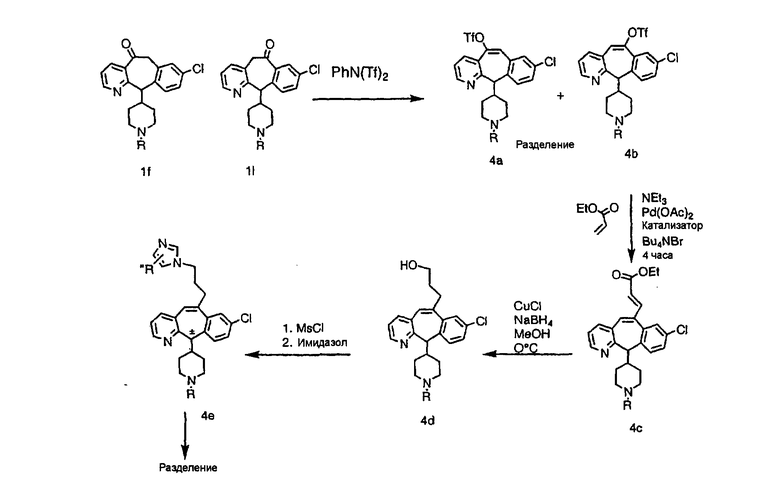
Получение 6-замещенных имидазолов с разделительными группами, содержащими 3 атома углерода, проводят так, как это показано на схеме 4. Смесь кетонов 1f и 1i обрабатывают N-фенилтрифторметансульфонимидом и получают разделяемую смесь 5-и 6-трифторметансульфонатзамещенных трициклических соединений. 6-Трифторметансульфонатзамещенный аддукт превращают в искомый аналог с разделительной группой, содержащей 3 атома углерода, с помощью последовательности реакций, аналогичной описанной на схеме 3 для 5-бромзамещенных трициклических соединений.
Схема 5: СИНТЕЗ АНАЛОГОВ, СОДЕРЖАЩИХ РАЗДЕЛИТЕЛЬНУЮ ГРУППУ ИЗ 2 АТОМОВ УГЛЕРОДА
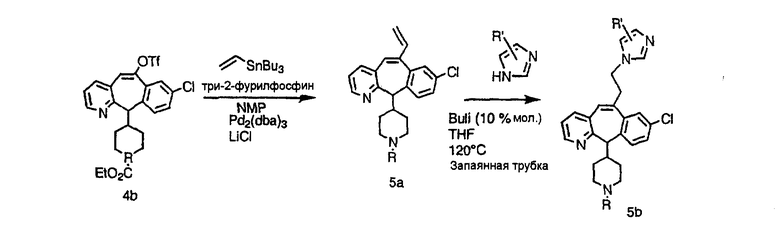
Аналоги с разделительными группами, содержащими два атома углерода, получают так, как это показано на схеме 5. Трифторметансульфонат 4b вводят в реакцию Стилле путем взаимодействия с трибутилвинилстаннатом использованием соответствующего производного Pdo в качестве катализатора и получают трициклическое винильное соединение 5b. Соединения с разделительными группами, содержащими два атома углерода, получают путем обработки трициклического соединения соответствующим имидазолом, который предварительно обрабатывают с помощью BuLi-THF в запаянной трубке и кипятят с обратным холодильником при 120°С. Последующее введение функциональных групп проводят так, как это описано выше. Аналогичным образом получают суберановое соединение.
Схема 6:
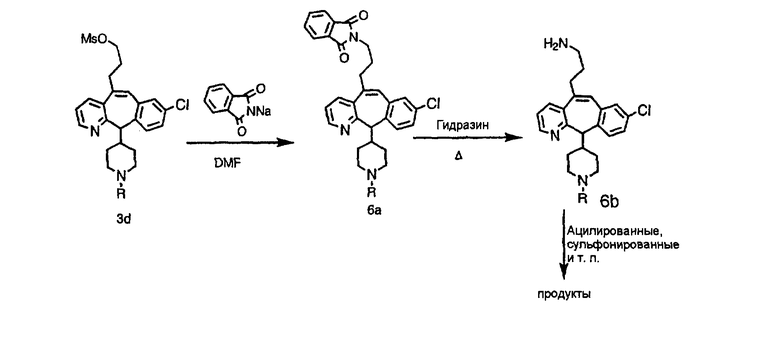
На схеме 6 показан способ получения амина 6b путем замещения мезилатной группы на фталимидную с последующим гидролизом фталимидного фрагмента в гидразин. Амин 6b можно превратить в искомые соединения, в которых имеются ацильная, сульфонильная, карбамоильная и мочевинная функциональные группы.
Схема 7:
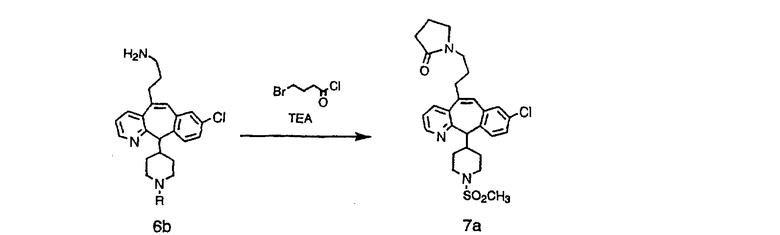
Лактамы 7а можно получить из амина 6b по их реакции с хлорангидридом броммасляной кислоты, как это показано на схеме 7.
Схема 8: Получение циклической мочевины
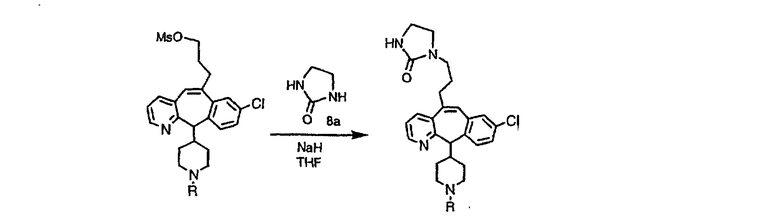
Циклическую мочевину можно получить из представленного выше мезилата путем его обработки солью циклической мочевины 8а, как это показано на схеме 8.
Схема 9: Получение 5-замещенных производных пропановой кислоты
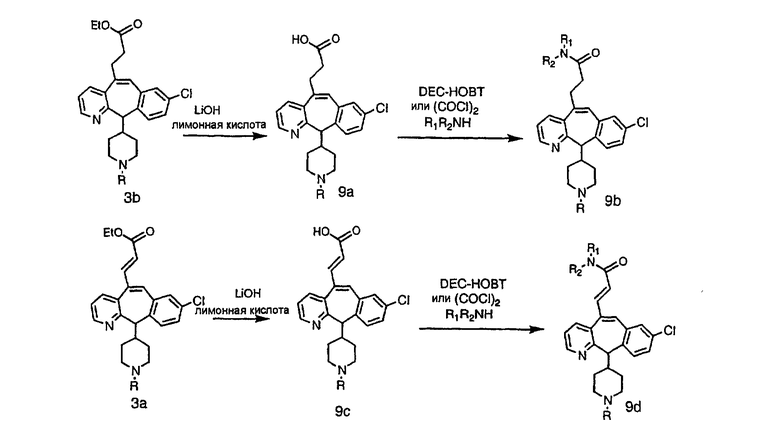
Из карбоновых кислот 9а и 9с с разделительными группами, содержащими 3 атома углерода, амиды получают по схеме 10 с использованием DEC-HOBT или из соответствующих хлорангидридов кислот.
Схема 10:
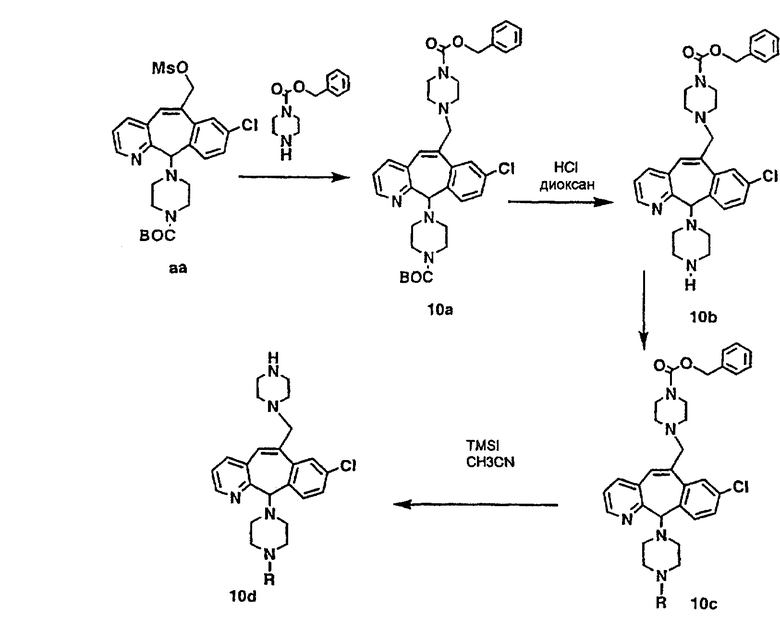
Получение соединений с пиперазиновыми группами, присоединенными через мостик, начинают с мезилата аа, который вводят в реакцию с пиперазином, у которого имеется защитная группа CBZ. Затем группу ВОС удаляют и в полученный амин 10с вводят необходимые функциональные группы. Отщепление группы CBZ от пиперазина проводят с помощью TMSI.
Схема 11: С-замещенные имидазол-3-метиленпиперидины
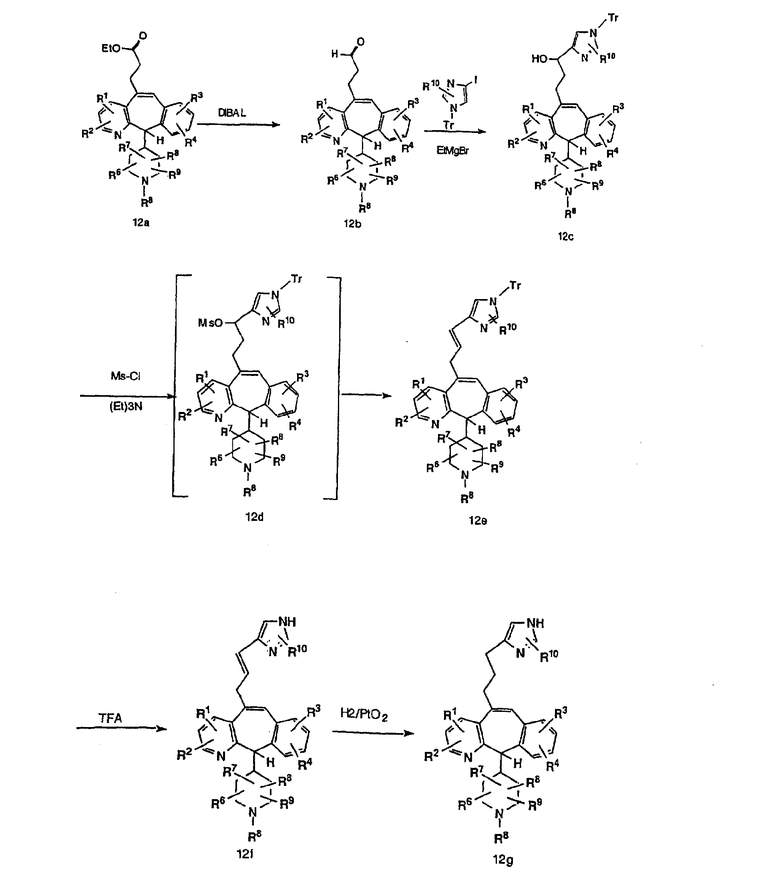
Соединение 12а восстанавливают с помощью DIBAL в инертном растворителе, таком как толуол или тетрагидрофуран, и после обработки кислотой получают 12b. Обработка 12b соответствующим образом замещенным и тритилированным имидазолйодидом в присутствии этилмагнийбромида в растворителях, таких как дихлорметан, при температуре окружающей среды дает аддукт 12с. Удаление гидроксильной группы путем превращения гидроксильной группы в соответствующую отщепляемую группу, такую как мезилатная, тозилатная или галогенидная, проводимое с помощью метансульфонилхлорида, п-толуолсульфонилхлорида или тионилхлорида, с последующим отщеплением, проводимым с помощью соответствующего основания, такого как триэтиламин, дает 12е. Удаление тритильной группы с помощью кислоты, такой как трифторуксусная кислота или хлористоводородная кислота, дает соединение 12f с двойной связью, которое затем гидрируют с использованием соответствующего катализатора, такого как оксид платины, при давлении водорода от 1 до 55 фунт-сила/дюйм2 в соответствующем растворителе, таком как этанол, и получают искомый продукт 12g.
Альтернативно, сложный эфир 12а можно омылить соответствующим основанием, таким как гидроксид лития, и получить кислоту 12h. Превращение кислоты 12h в "амид Вейнреба" с последующей реакцией с соответствующим образом замещенным и тритилированным имидазолйодидом в присутствии этилмагнийбромида в растворителях, таких как дихлорметан, при температуре окружающей среды дает аддукт 12с (показанный ниже на схеме 12).
Схема 12:
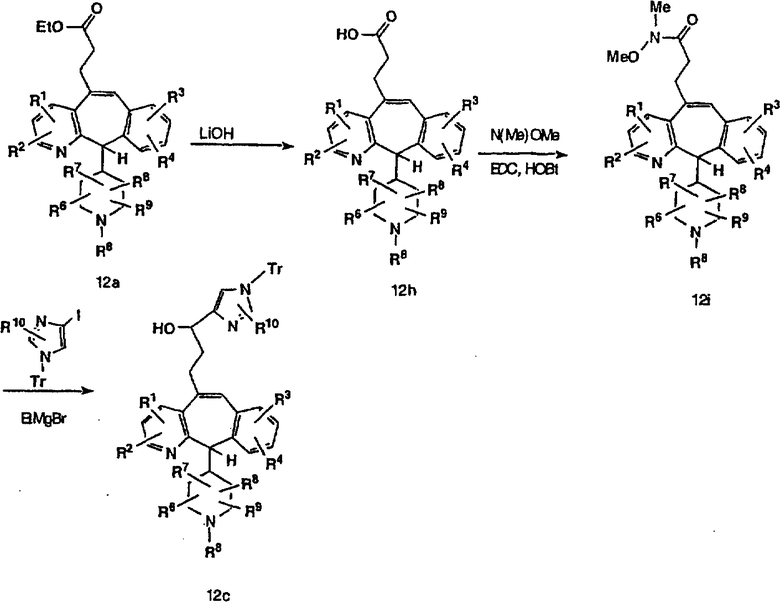
Схема 12а:
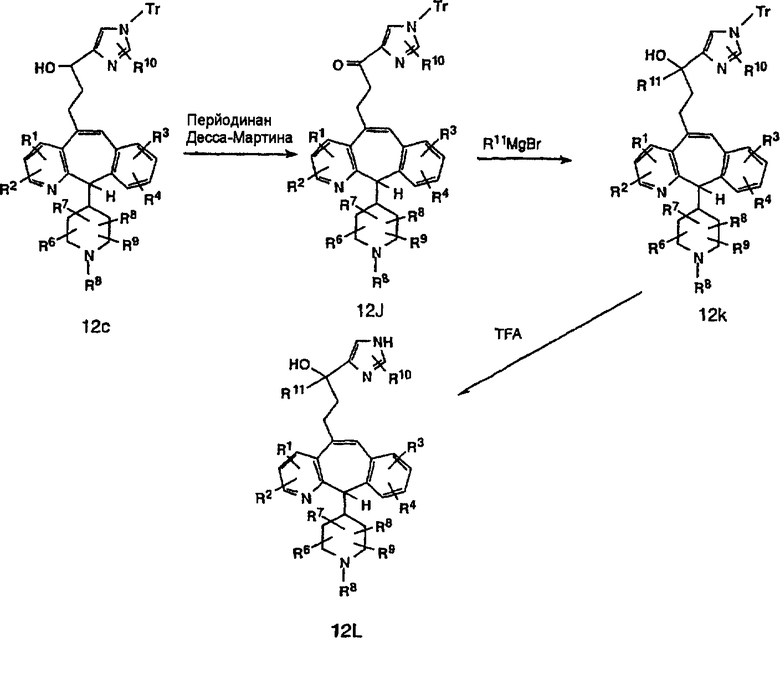
Соединения типа 12L получают так, как показано выше. Окисление гидроксильного соединения 12с можно провести с помощью перйодинана Десса-Мартина и получить 12j. Реакция с реагентом Гриньяра дает 12k. Тритильную группу удаляют при стандартных условиях, указанных выше, и получают искомое соединение 12L.
Схема 13: С-Замещенные производные имидазола с одним метиленовым мостиком
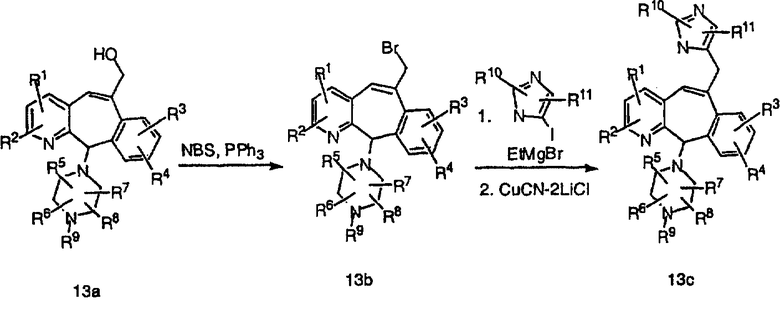
С-производные имидазола (13с) с одним метиленовым мостиком получают так, как показано выше. Сначала соединение 13а превращают в бромид 13b. Обработка соединения 13b купратами С-имидазола (полученными из соответствующего йодимидазола) дает аддукт 13с.
Схема 14: Получение пиперазинов с одной метиленовой группой
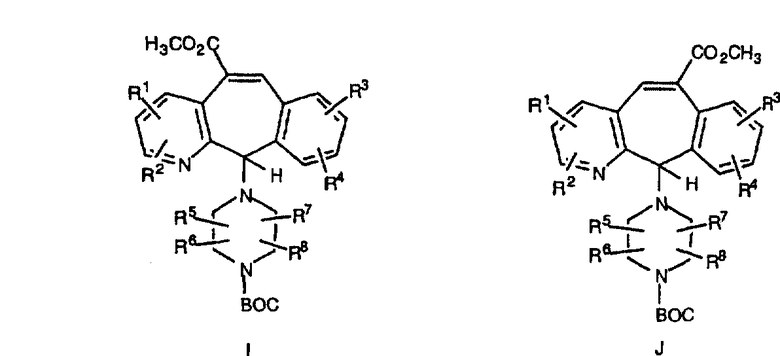
Кетон A бромируют бромирующими реагентами, такими как NBS, с использованием небольшого количества активатора, такого как бензоилпероксид, в растворителях, таких как дихлорметан, при повышенной температуре, такой как 80-100°С, и получают дибромированное соединение В.
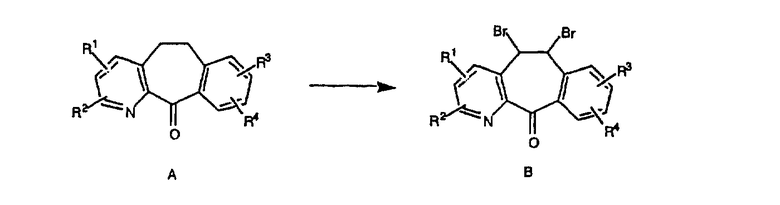
Дибромированное соединение B вводят в реакцию с основанием, таким как DBU, в растворителях, таких как дихлорметан, при температуре от 0°C до комнатной температуры и получают винилбромиды C и D. Эти винилбромиды разделяют с помощью хроматографии, такой как флэш-хроматография на силикагеле, с использованием смесей растворителей, таких как этилацетат и гексан. Альтернативно, винилбромиды C и D можно разделить путем кристаллизации из растворителей, таких как дихлорметан.
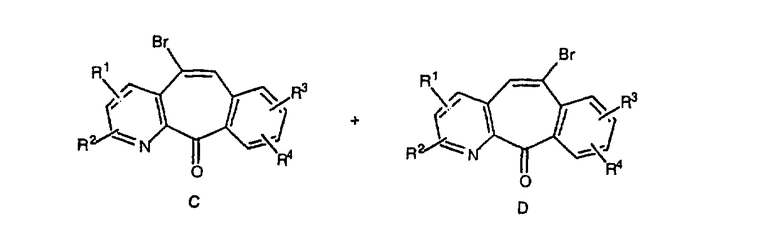
Кетогруппы разделенных винилбромидов C и D восстанавливают восстановительными реагентами, такими как NaBH4, в растворителях, таких как метанол или этанол, при температуре от 0°C до комнатной температуры, и получают соответствующие спирты Е и F.
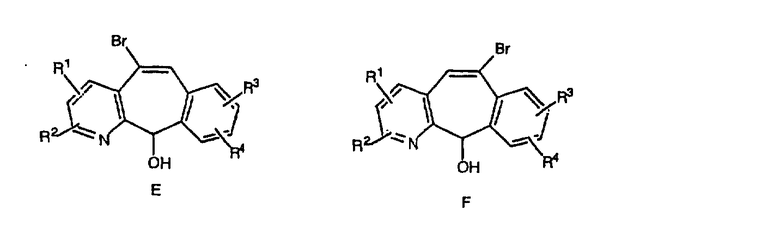
Гидроксигруппы полученных соединений Е и F превращают в отщепляемую группу, такую как галогенидная, с помощью реагентов, таких как SOCl2, в растворителях, таких как дихлорметан, содержащих основание, такое как 2,6-лутидин, проводя реакцию при температуре от 0°C до комнатной температуры. Полученные промежуточные галогениды без очистки вводят в реакцию с пиперазином или пиперазином, в котором имеются защитные группы, таким как ВОС-пиперазин, в растворителе, таком как дихлорметан, при комнатной температуре, и получают промежуточные продукты G и Н.
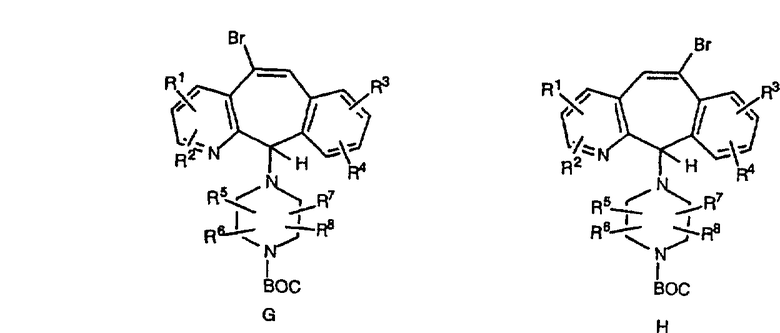
Промежуточные винилгалогениды карбонилируют с помощью газообразного СО при давлении, равном примерно 100 фунт-сила/дюйм2, и температуре от 80 до 100°C с использованием палладиевого катализатора, такого как PdCl2 и трифенилфосфина в толуоле и содержащего DBU и спирт, такой как метанол. Если используют метанол, то получают метиловые сложные эфиры I и J.
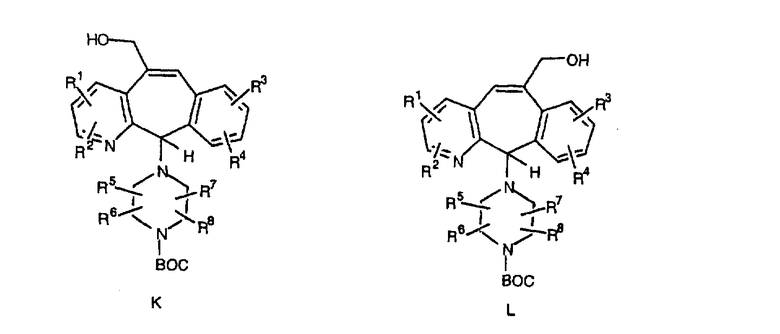
Сложноэфирные группы соединений I и J восстанавливают в гидроксиметильные с получением соединений K и L. Это можно сделать непосредственно путем удаления защитной группы ВОС с помощью TFA или смеси HCI-диоксан с последующим восстановлением с помощью восстановительного реагента, такого как DIBAL-H, и последующим повторным введением группы ВОС с помощью ди-трет-бутила и карбоната. Альтернативно, сложноэфирную группу можно подвергнуть гидролизу с помощью LiOH и воды с последующей нейтрализацией лимонной кислотой. Полученные карбоновые кислоты затем превращают в соединения с функциональными группами, которые легко восстанавливаются, такие как смешанный ангидрид или ацилимидазол. Это осуществляют путем введения полученных карбоновых кислот в реакцию с хлорформиатом для получения смешанного ангидрида или с карбонилдиимидазолом для получения ацилимидазола (Synlett. (1995), 839). Полученные активированные карбоновые кислоты восстанавливают с помощью NaBH4 в растворителях, таких как метанол, этанол или водный раствор THF.
Гидроксигруппы полученных соединений K и L превращают в отщепляемые группы, такие как метансульфонатная или арилсульфонатная, такая как тозилатная, по реакции с соответствующим сульфонилхлоридом в дихлорметане, содержащем основание, такое как триэтиламин. Отщепляемые сульфонатные группы можно заместить на нуклеофильные группы, такие как аминогруппы. Нуклеофильными группами также могут быть основные гетероциклы, такие как имидазол или замещенный имидазол. В случае имидазола сначала с помощью NaH в DMF получают анион имидазола, а затем его вводят в реакцию с указанным выше сульфонатом. Замещение сульфонатов на нуклеофильные группы дает соединения О и Р, которые можно превратить в соединения формулы 1.0, соответствующие настоящему изобретению, путем удаления защитной группы ВОС с последующим получением искомого амида, мочевины, карбамата или сульфонамида в полученном амине с помощью способов, хорошо известных в данной области техники.
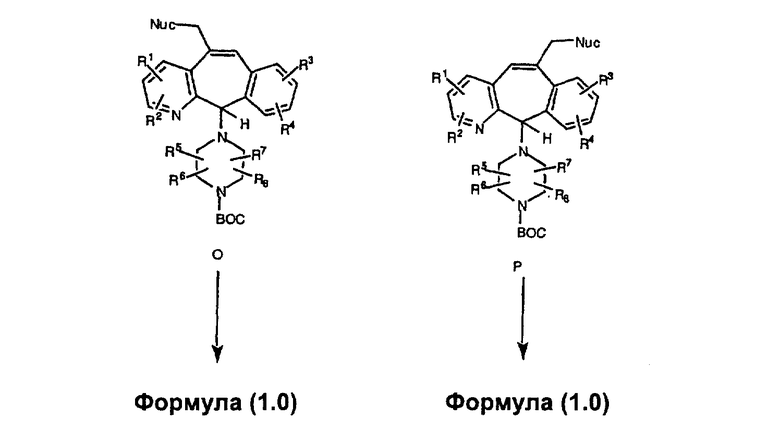
Схема 15: Получение пиперидинов с одной метиленовой группой
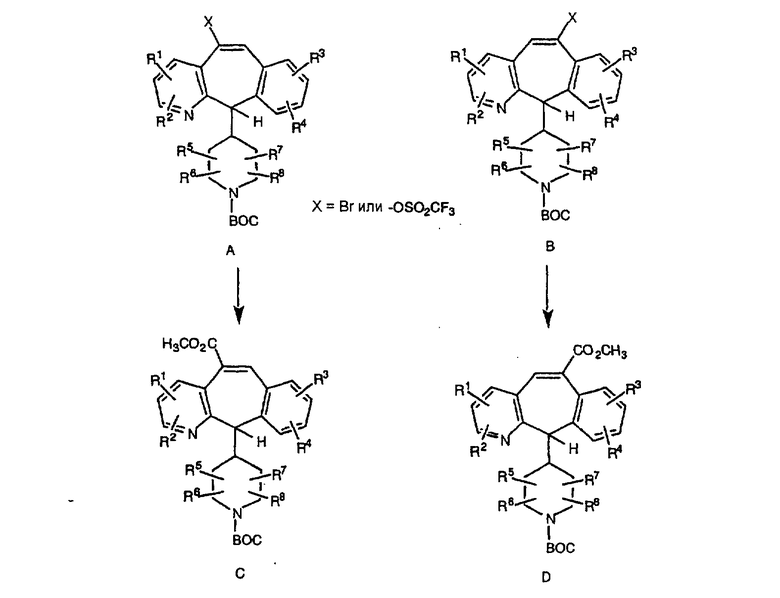
Промежуточные винилгалогенид или винилтрифторметансульфонат A и В (представленные на других общих схемах) карбонилируют с помощью газообразного СО при давлении, равном примерно 100 фунт-сила/дюйм2, и температуре от 80 до 100°С с использованием палладиевого катализатора, такого как PdCl2 и трифенилфосфина в толуоле, и содержащего DBU и спирт, такой как метанол. Если используют метанол, то получают метиловые сложные эфиры C и D. Промежуточные продукты C и D вводят в реакцию таким же образом, как и промежуточные продукты I и J, по общей схеме получения пиперазинов с одной метиленовой группой, и получают соединения формулы 1.0, соответствующие настоящему изобретению.
Схема 15а:
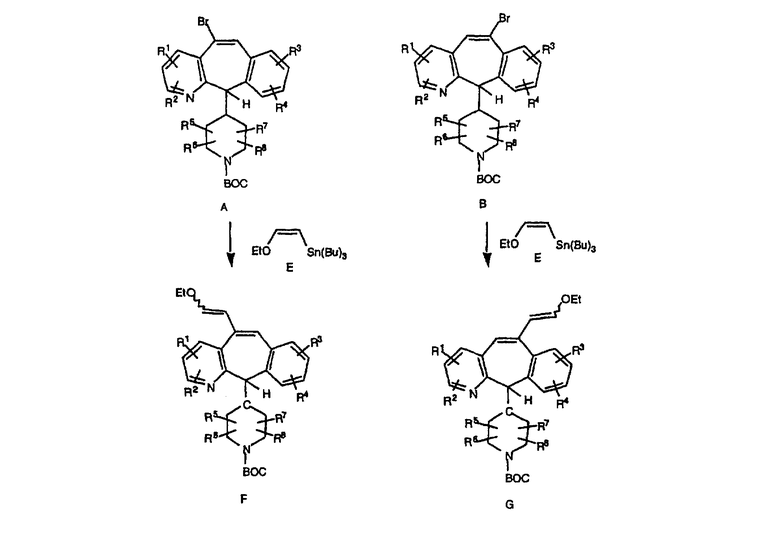
Альтернативно, промежуточные соединения A и В можно ввести в реакцию с виниловым эфиром Е в присутствии PdCl2 в соответствии с описанием в Tetrahedron, (1997), 47, 1877 и получить виниловые простые эфиры F и G (схема 15а). Выдерживание F и G, пока альдегид не перестанет обнаруживаться с помощью ЯМР (не менее двух недель), и последующие реакции с Н2(ОАс)2, KI, а затем с NaBH4 в соответствии с описанием в J. Chem Soc., Perkin Trans., (1984), 1069 и Tet. Lett., (1988), 6331 дает смеси Н, I и J, К. Промежуточные продукты Н и J разделяют и вводят в реакцию таким же образом, как и промежуточные продукты К и L, по общей схеме получения пиперазинов с одной метиленовой группой, и получают соединения формулы 1.0, соответствующие настоящему изобретению.
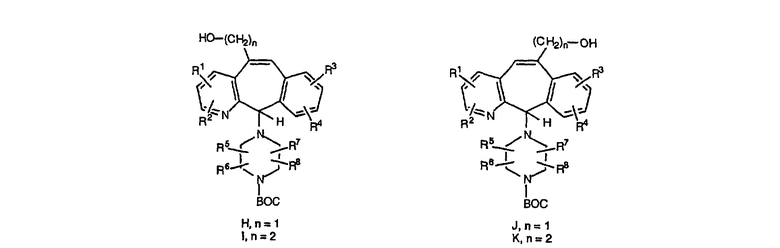
Схема 16: Разветвление метиленовой цепи
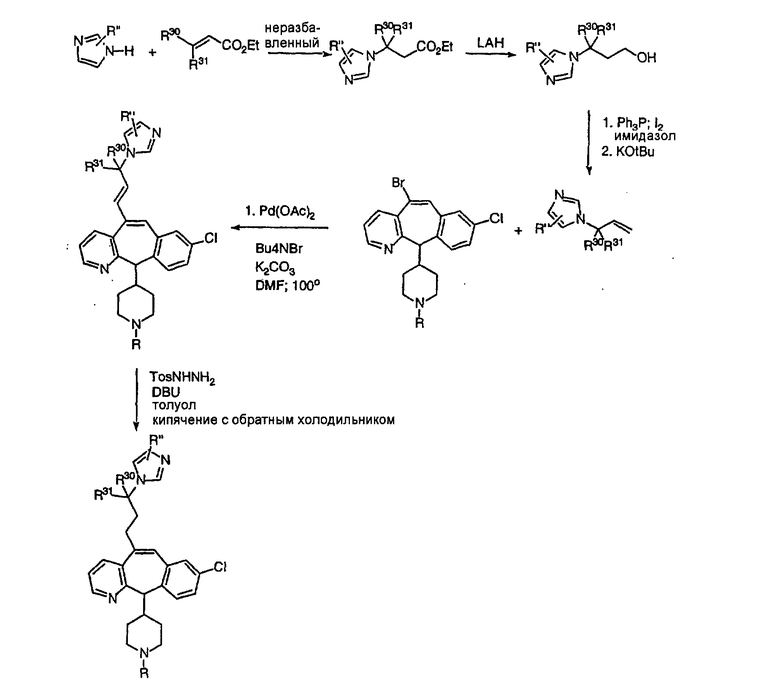
Соединения, содержащие заместители в цепи, можно синтезировать, исходя из замещенного этилакрилатного производного. Присоединение имидазола по двойной связи с последующим восстановлением дает концевой алкен, который можно присоединить к соответствующим образом замещенному винилбромиду в условиях реакции Хека. Селективное восстановление дизамещенного олефина дает насыщенное производное (схема 16).
Схема 17: Присоединенные по атому C имидазолы
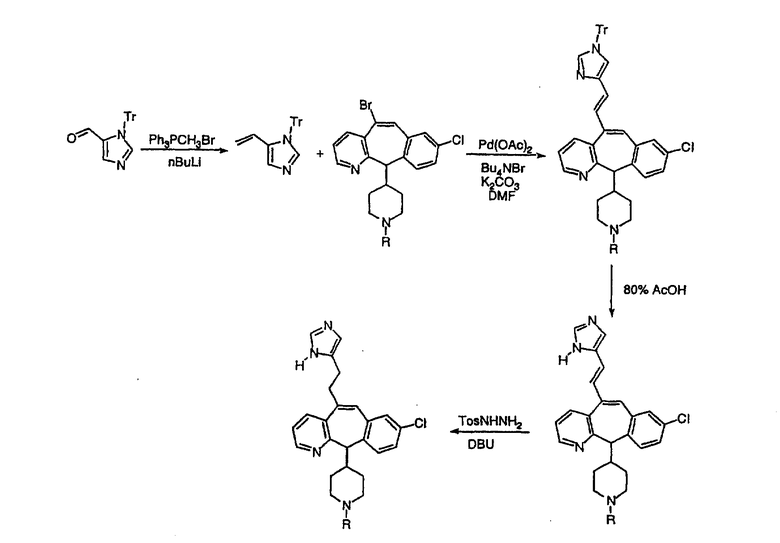
Синтез присоединенных по атому C имидазолов проводят по реакции Хека соответствующим образом замещенного винилимидазола с соответствующим винилбромидом. Селективное восстановление полученного дизамещенного олефина дает искомое соединение. Аналогичную процедуру можно провести для различных N-замещенных имидазолов и получить N-алкилпроизводные имидазола (схема 17).
Суберильные соединения
Специалисту в данной области техники должно быть понятно, что соединения, соответствующие настоящему изобретению и описываемые формулой 1.0, в которых а, b, с или d означают C, можно получить по следующим схемам.
Схема 18: Получение суберильных аналогов
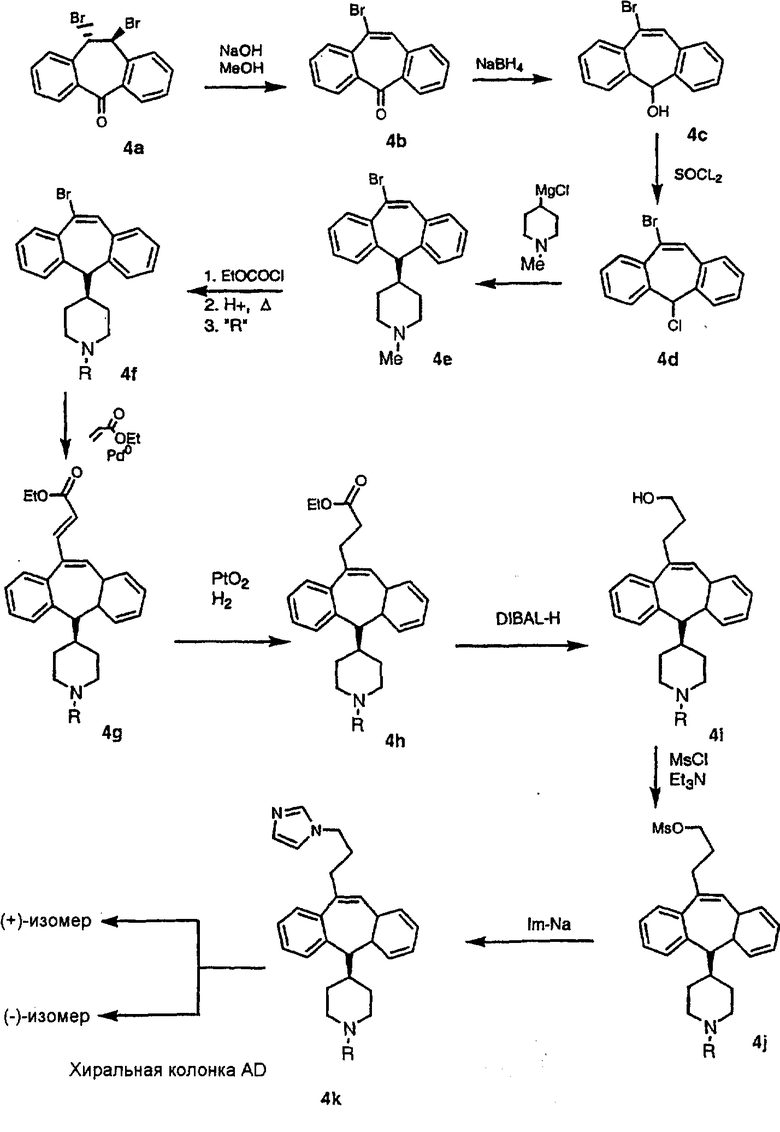
Трициклический винилбромидкетон 4b получают так, как это описано в работе Rupard et al. (J. Med. Chem. 1989, 32, 2261 -2268). Восстановление кетона в спирт 4с проводят с помощью NaBH4. Спирт превращают в хлорид 4d, а затем обрабатывают полученным из М-метилпиперидина реагентом Гриньяра и получают пиперидиновое производное 4е. Деметилирование проводят с помощью этилхлорформиата с проводимым после этого кислотным гидролизом и с последующим получением производного (т.е. сульфонилированием, ацилированием, карбамоилированием и т.п.). Получение соединений с имидазольными фрагментами, замещенными в положении 3, содержащимися в суберановых трициклических мостиковых соединениях, проводят аналогично тому, как это показано на схеме 3.
Получение промежуточных продуктов и Примеры
ПРИМЕР ПОЛУЧЕНИЯ 1
Стадия А Получение соединения (2).
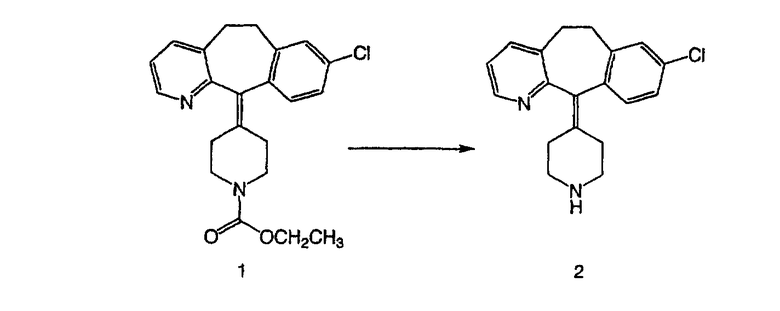
Лоратадин® (448 г, 1,17 моль) 12 ч кипятят с обратным холодильником в 2 л 70% водного раствора HCl (1,4 л концентрированной HCl в 600 мл Н2О). Затем реакционную смесь охлаждают и выливают на лед. После этого ее подщелачивают с помощью 950 мл 50% раствора NaOH, а затем экстрагируют с помощью CH2Cl2 (1 х 4 л и 2 х 2,5 л). Органическую фазу промывают рассолом, сушат над Na2SO4 и MgSO4 и затем фильтруют. После этого все летучие вещества удаляют и получают 368 г искомого соединения (2). МН+ =311.
Стадия В Получение соединения (3).
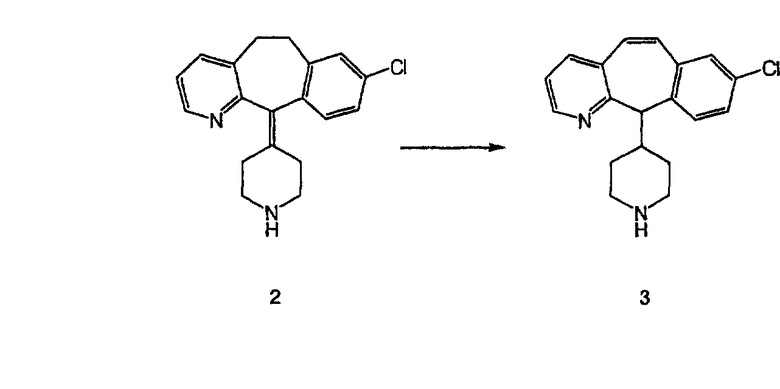
К соединению, полученному в Примере получения 1, стадия A (363 г, 1,17 моль), в атмосфере N2 прибавляют трифторметансульфоновую кислоту (1,8 кг). Реакционную смесь кипятят с обратным холодильником при 170°С. За протеканием реакции следят с помощью 1H ЯМР. Через 4 дня реакция проходит лишь на 63%. Через 8 дней с помощью 1H ЯМР обнаруживается, что реакция прошла на 80%; прибавляют еще 130 мл CF3SO3H и кипячение с обратным холодильником продолжают еще 24 ч. Затем реакционную смесь выливают на лед, подщелачивают с помощью 800 мл NaOH (50%) и дважды экстрагируют с помощью CH2Cl2 (1 х 8 л, затем 1 х 7 л). Органические фазы объединяют, промывают с помощью Н2О и фильтруют через целит. Затем их сушат над Na2SO4 и MgSO4 и повторно фильтруют через целит. Фильтрат концентрируют и получают черно-коричневое полужидкое вещество, которое предварительно адсорбируют на 600 г силикагеля, а затем хроматографируют на 2,3 кг силикагеля, элюируя сначала с помощью смеси 5% СН2ОН - CH2Cl2 (насыщенный аммиаком), а затем с помощью 10% СН3ОН - CH2Cl2 (насыщенный аммиаком), и получают 102 г искомого соединения (3) в виде твердого вещества. Т. пл. (температура плавления) = 73-75; МС (ББА) m/z 483 (МН+).
Стадия C Получение соединения (4).
К раствору соединения, полученного в Примере получения 1, стадия B (145 г), в 1 л CH2Cl2 при 0°С по каплям прибавляют этилхлорформиат (55 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем ее разбавляют с помощью 1 л CH2Cl2 и перемешивают с 2 л разбавленного NaHCO2, pH ˜ 7-8. Органический слой отделяют и сушат над Na2SO4 и MgSO4, фильтруют и концентрируют, получая 174 г коричнево-черной смолы. Неочищенное соединение очищают с помощью хроматографии на колонке с силикагелем, элюируя с помощью смеси 20-60% этилацетат - гексан, и получают искомое соединение (4). МС (ББА) m/z 383 (MH+).
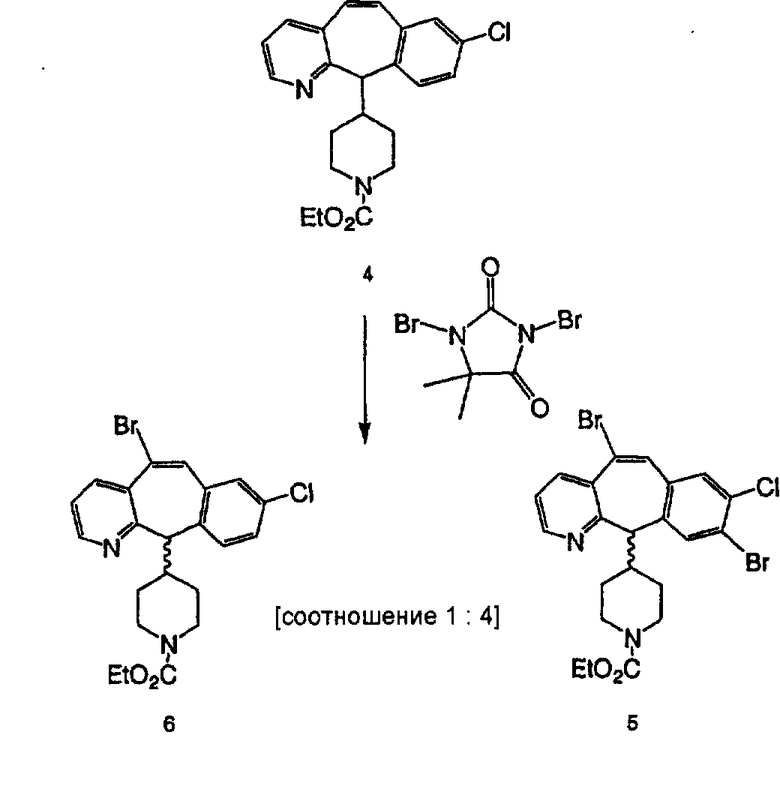
Соединение, полученное в Примере получения 1, стадия C (251 г, 0,65 моль), растворяют в 1,65 л CH2Cl2, а затем прибавляют дибромдиметилгидантоин (132 г, 0,462 моль). Раствор перемешивают, пока система не станет гомогенной. Раствор охлаждают до 0°C в атмосфере N2 и в течение 37 мин, поддерживая температуру равной от -1 до 1°C, прибавляют 174 мл CF3SO3Н. Реакционную смесь перемешивают 3 ч, охлаждают до -10°C и подщелачивают с помощью 50% NaOH (170 мл), поддерживая температуру ниже 1°С. Водную фазу экстрагируют с помощью CH2Cl2, а затем сушат над MgSO4 и концентрируют, получая 354 г желтого пенообразного вещества, которое хроматографируют на силикагеле, элюируя в градиентном режиме смесью 10-50% этилацетат-гексан, и получают 50 г соединения (5) (выход 14%) и 147 г искомого соединения (6) (выход 49%). Соединение (6): МС m/z (относительная интенсивность) 462 (МН+); соединение (5): МС m/z (относительная интенсивность) 542 (MH+).
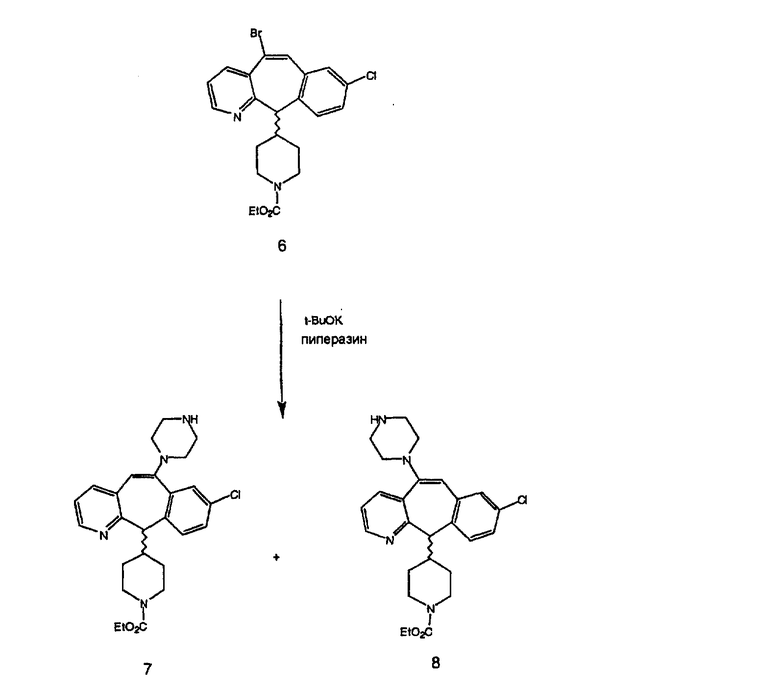
К раствору пиперазина (0,186 г, 2,2 ммоль, 5 экв.) в 5 мл THF прибавляют 0,20 г (0,4 ммоль) соединения 6 (полученного в Примере получения 1, стадия D). Реагенты перемешивают при комнатной температуре до полного растворения компонентов. К этой смеси одной порцией прибавляют трет-бутоксид калия (0,243 г, 2,1 ммоль, 5 экв.). Реакционную смесь 2 ч перемешивают при комнатной температуре. Весь THF удаляют на роторном испарителе, и полученный неочищенный продукт очищают с помощью флэш-хроматографии, элюируя смесью 3-4% (10% СН3ОН, насыщенного с помощью NH4OH)-CH2Cl2, и получают смесь искомых соединений (7) и (8). ББА m/z 467 (МН+).
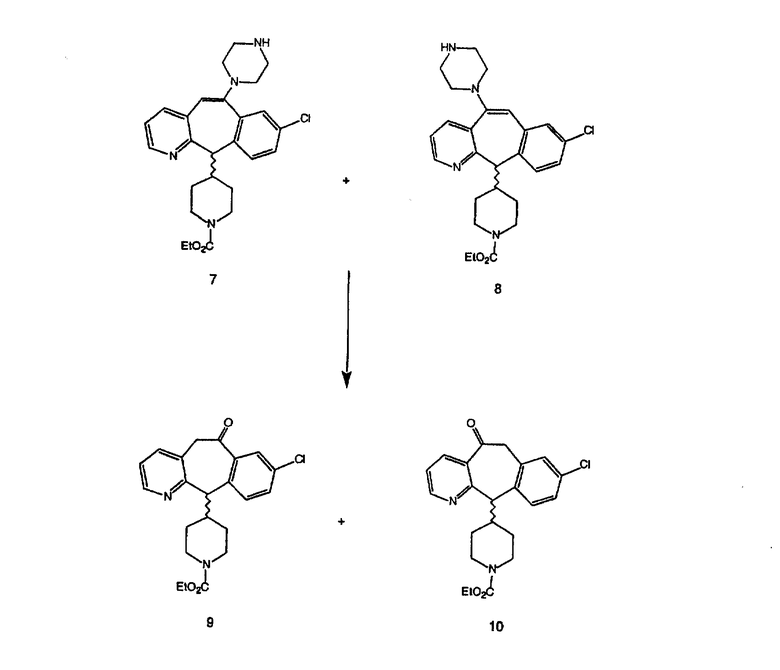
Смесь соединений, полученных в Примере получения 1, стадия Е (43,6 г), в 100 мл концентрированной HCI перемешивают при комнатной температуре в течение 16 ч. Реакционную смесь выливают на лед и подщелачивают концентрированным NH4OH, а затем экстрагируют с помощью CH2Cl2 и получают смесь соединений (9) и (10). МС (ББА) m/z 399 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 2
А Соединение (11).
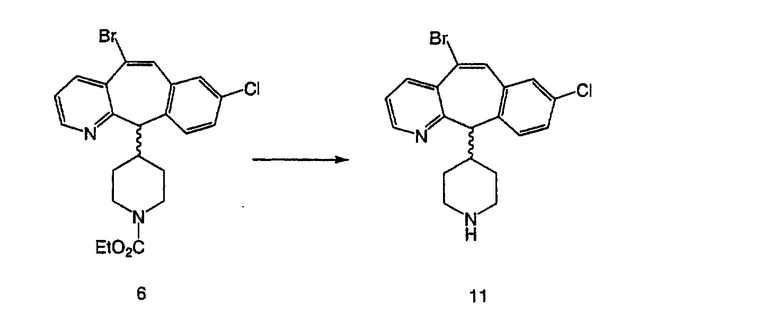
Соединение, полученное в Примере получения 1, стадия D (10 г, 21,7 ммоль), гидролизуют таким же способом, как это описано в Примере получения 1, стадия A, и получают искомое соединение (11). МН+ = 389.
В Соединение (12).
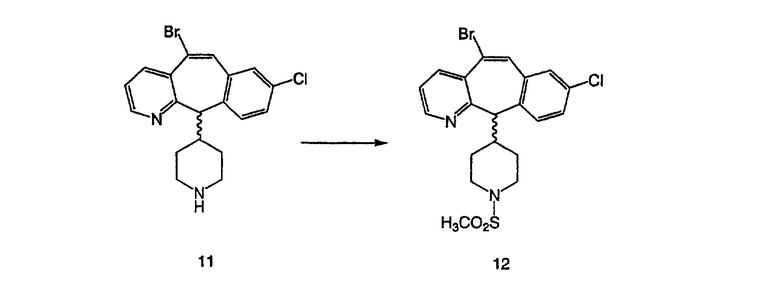
К амину, полученному в Примере получения 2, стадия A (20 г, 0,5 моль), и триэтиламину (10,4 г, 14,4 мл, 1,02 моль), растворенным в безводном дихлорметане (100 мл), прибавляют метансульфонилхлорид (8,8 г, 6 мл, 0,77 моль). После перемешивания при комнатной температуре в течение ночи раствор разбавляют дихлорметаном, промывают насыщенным раствором МаНСО3 и сушат над безводным сульфатом магния. Фильтрование и концентрирование в вакууме дает неочищенный продукт, который очищают с помощью флэш-хроматографии, элюируя смесью 1%СН3ОН (насыщенный аммиаком)-CH2Cl2, и получают искомое соединение (12). МС (ББА) m/z 469 (МН+).
Стадия C Получение соединений (13) и (14).
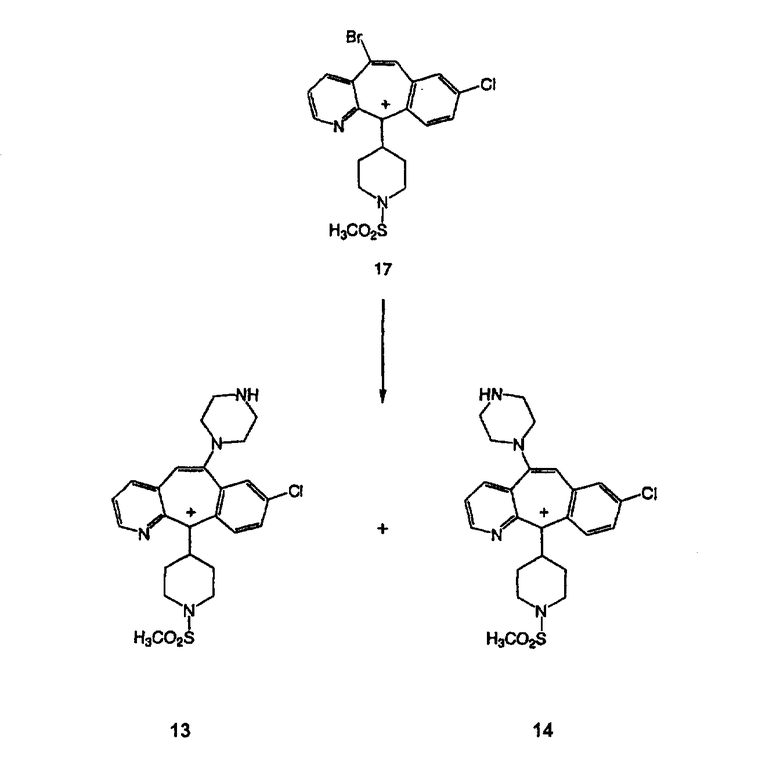
Продукт, полученный в Примере получения 2, стадия B (21,25 г, 45,3 ммоль), обрабатывают таким же способом, как это описано в Примере получения 1, стадия Е, и получают 22,2 г смеси соединений (13) и (14). МС (473) (MH+).
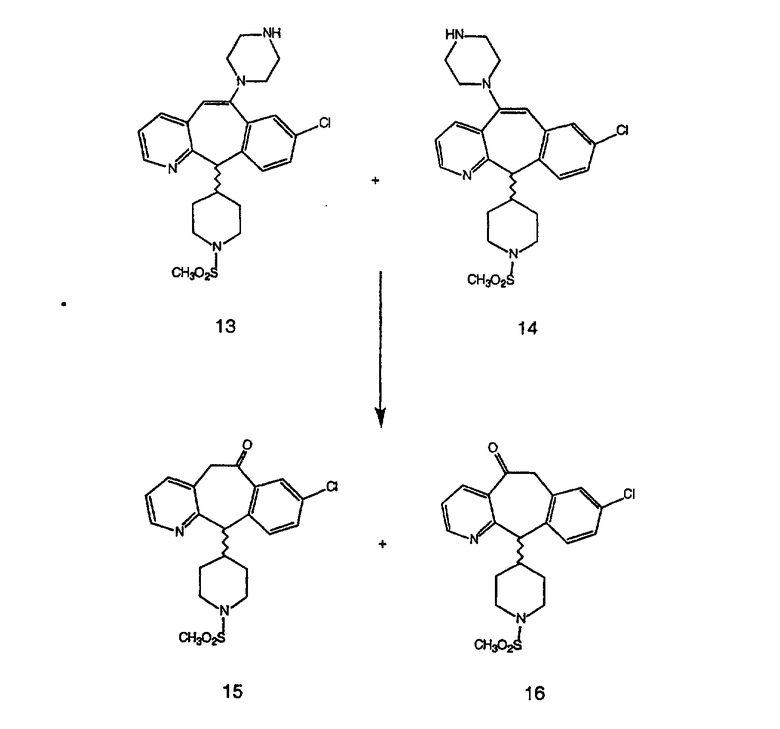
Продукт, полученный в Примере получения 2, стадия C (22,5 г), растворяют в 150 мл концентрированной HCl и перемешивают 16 ч. Реакционную смесь выливают на лед, подщелачивают концентрированным раствором NH4OH, а затем экстрагируют с помощью CH2Cl2, получая смесь соединений (15) и (16). МС (ББА) m/z 405 (МН+).
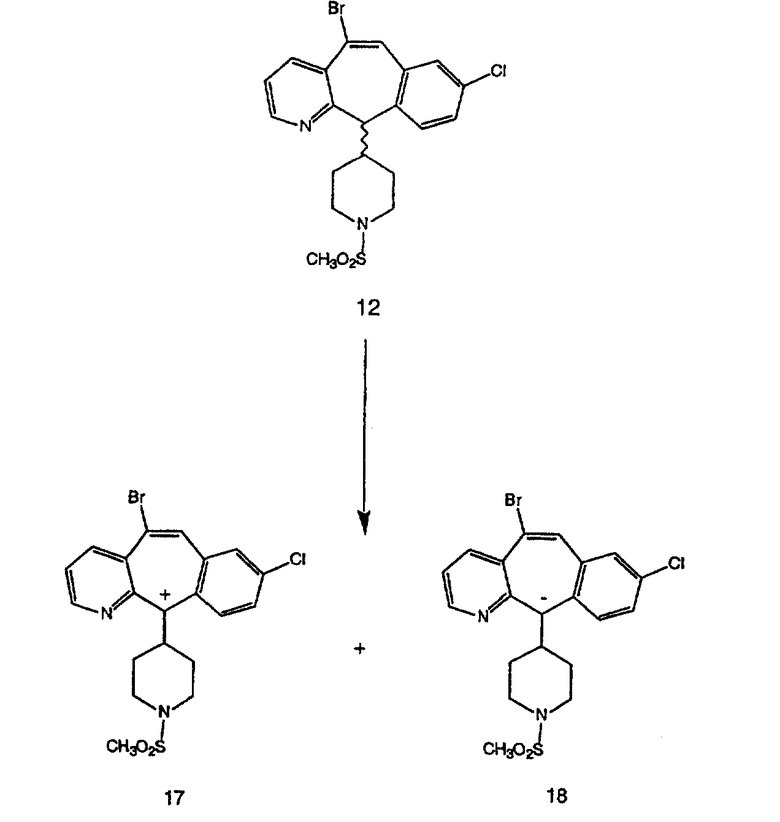
Разделение соединений, полученных в Примере получения 2, стадия C, с помощью ВЭЖХ с использованием колонки Chiralpack AD и с элюированием смесью 40-50% изопропанола: 60-50% гексана-0,2% диэтиламина дает энантиомерные амины (17) и (18).
Соединение (17): т. пл. = 118 -119; [α]D 22 = +136,9° (9,00 мг/2 мл МеОН); МС (ББА) m/z 469 (MH+).
Соединение (18): т. пл. = 119 -120; [α]D 22 = -178,2° (9,90 мг/2 мл МеОН); МС (ББА) m/z 469 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 3
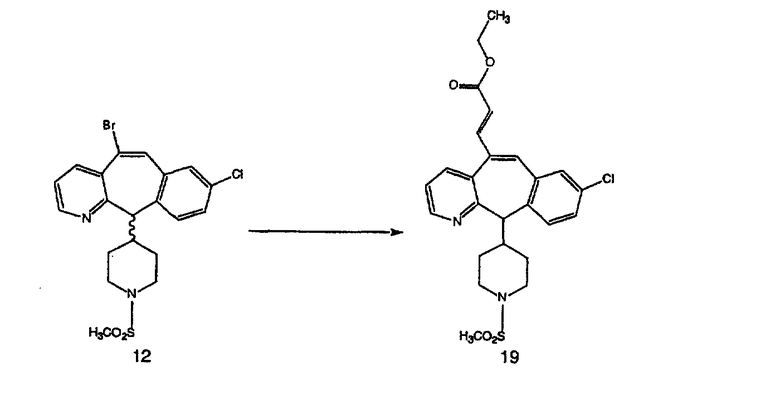
К раствору соединения, полученного в Примере получения 2, стадия B (2,0 г, 4,3 ммоль), в DMF (50 мл) в атмосфере азота прибавляют триэтиламин (17 мл), этиалкрилат (2,5 мл), карбонат калия (3 г, 21,4 ммоль), тетрабутиламмонийбромид (2,8 г, 8,6 ммоль) и ацетат палладия(II) (0,1255 г, 0,56 ммоль). Полученную смесь нагревают до 100°C и перемешивают 4 ч, а затем охлаждают до комнатной температуры и удаляют растворитель. К остатку прибавляют СН2Cl3 и воду, а затем смесь экстрагируют с помощью CH2Cl2. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха. Неочищенный продукт очищают с помощью флэш-хроматографии на колонке с диоксидом кремния, на котором предварительно адсорбируют продукт, элюируя в градиентном режиме смесью 30-50% этилацетат-гексан, и получают искомое соединение (19). МС 487 (МН+).
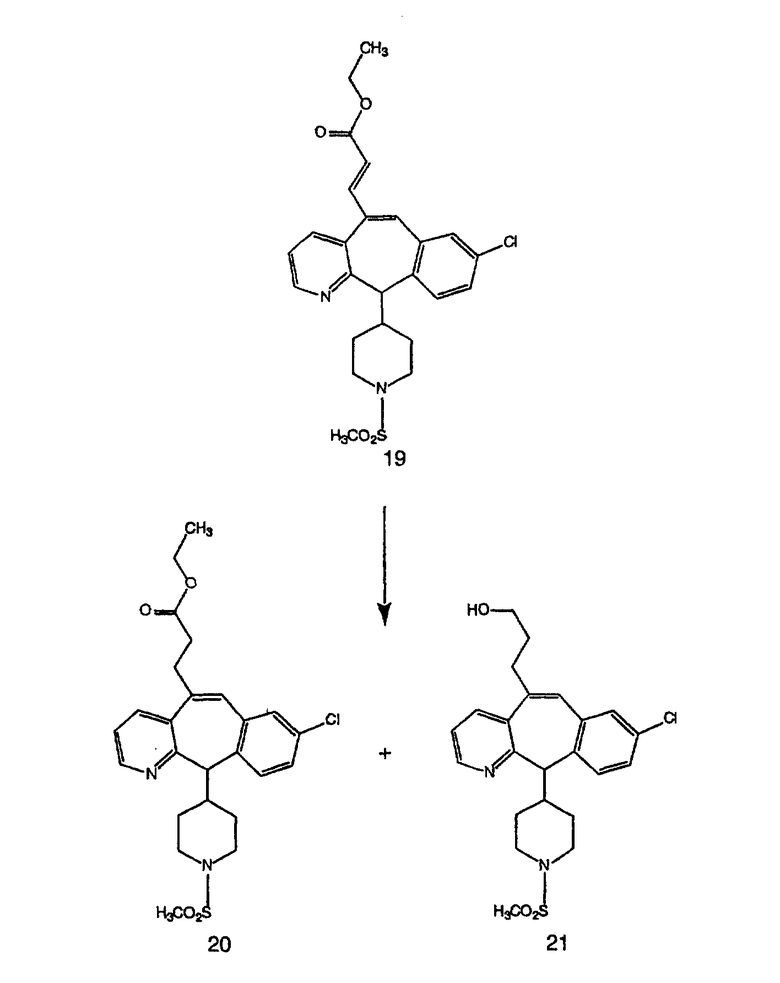
К раствору соединения, полученного в Примере получения 3, стадия A (6,4 г, 13 ммоль), в этаноле (50 мл) прибавляют хлорид двухвалентной меди (0,96 г, 9,7 ммоль). Реакционную смесь охлаждают до 0°С. Порциями прибавляют борогидрид натрия (4,97 г, 131 ммоль). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Прибавляют еще одну порцию борогидрида натрия (2,46 г, 65 ммоль) и реакционную смесь перемешивают еще 2 ч, а затем удаляют растворитель. К остатку прибавляют насыщенный раствор бикарбоната натрия и смесь экстрагируют с помощью СН2Cl2. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха, получая смесь искомых восстановленного сложного эфира (20) и спирта (21). Неочищенную смесь используют на следующей стадии без очистки.
Стадия С Получение соединения (22).
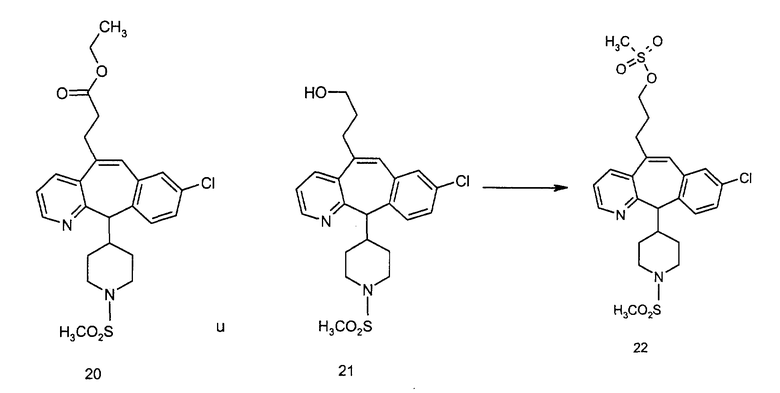
К раствору продукта, полученного в Примере получения 3, стадия B (5,74 г), в СН2Cl2 (100 мл) прибавляют триэтиламин (2,4 мл). Медленно прибавляют метансульфонилхлорид (0,8 мл) и смесь перемешивают в течение ночи при комнатной температуре. К реакционной смеси прибавляют насыщенный раствор бикарбоната натрия, а затем ее экстрагируют с помощью CH2Cl2. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха. Неочищенный продукт разделяют на колонке Biotage®, элюируя смесью 30% этилацетата-CHaClz, и получают искомое соединение (22). МС 525 (МН+). (извлеченный непрореагировавший сложный эфир (20)).
ПРИМЕР ПОЛУЧЕНИЯ 4
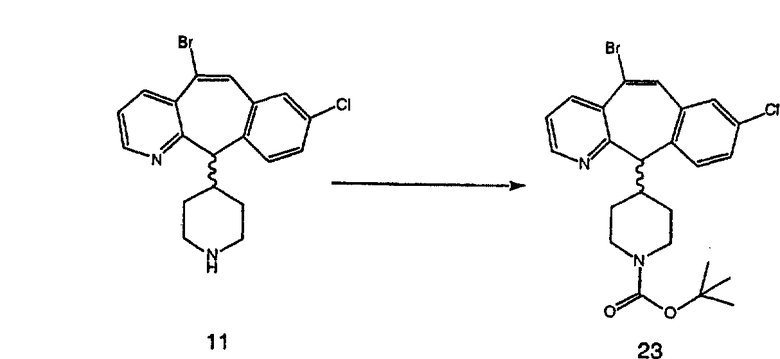
К раствору соединения (11), полученного в Примере получения 2, стадия A (20 г, 51,32 ммоль), в СН3ОН/Н2О (400 мл, 50: 1) прибавляют ди-трет-бутилдикарбонат (16,6 г, 77,0 ммоль). Значение рН доводят до 9 и смесь перемешивают 4 ч. Удаляют растворитель, а затем прибавляют воду. Смесь экстрагируют с помощью СН2Cl2. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха, получая искомое соединение (23). МС 491 (MH+).
В Соединение (24).
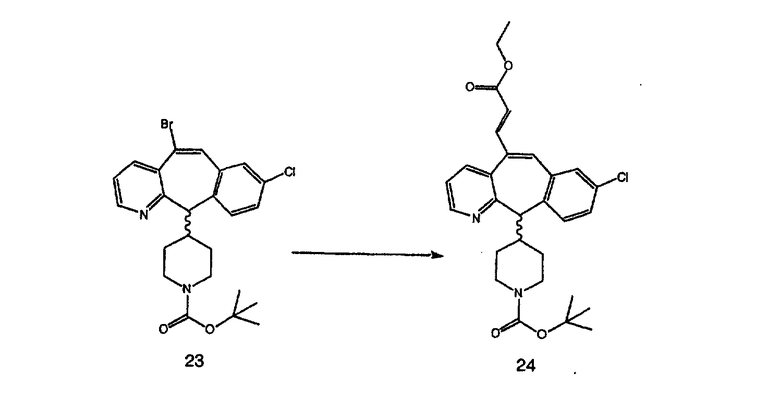
По методике, аналогичной использованной в Примере получения 3, стадия A, получают искомое соединение (24). МС 509 (МН+).
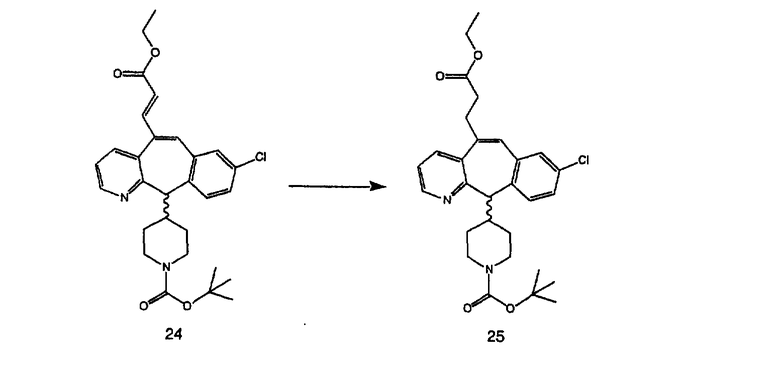
К раствору соединения, полученного в Примере получения 3, стадия В (19,62 г, 38,5 ммоль), в этаноле (150 мл) прибавляют оксид платины(IV) (1,962 г). Реакционную смесь перемешивают в течение ночи при комнатной температуре под давлением Н2, подаваемого из баллона. После прослеживания за протеканием реакции прибавляют еще 2% мас. оксида платины(IV) и реакционную смесь перемешивают в течение еще 6 ч под давлением Н2, подаваемого из баллона. Смесь фильтруют через целит и концентрируют досуха, получая искомое соединение (25) в виде белого твердого вещества. МС 511 (МН+).
Стадия D Получение соединения (26).
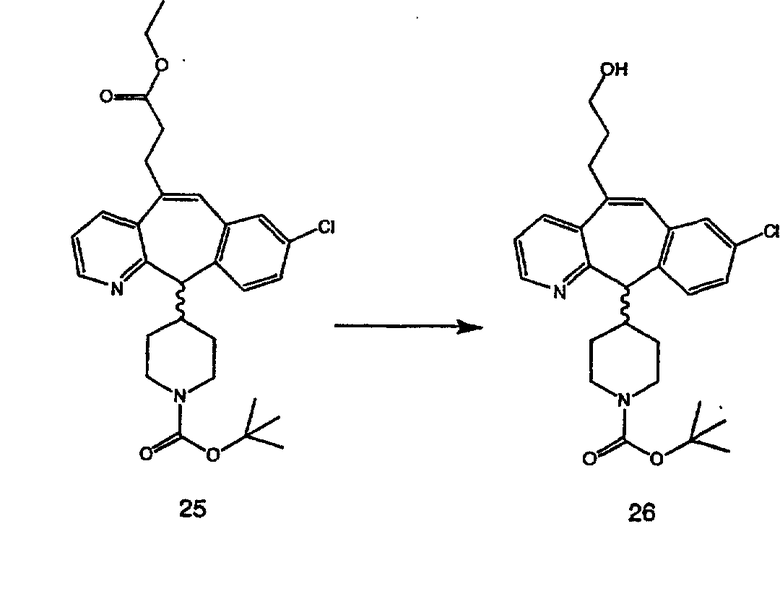
Продукт, полученный в Примере получения 3, стадия C (2,0 г, 3,9 ммоль), растворяют в THF (30 мл) и охлаждают до 0°C в бане со льдом. К этой реакционной смеси прибавляют диизобутилалюминийгидрид (7,8 мл, 7,8 ммоль). Реакционную смесь доводят до комнатной температуры и перемешивают в течение ночи. Реакция не доходит до конца. Смесь охлаждают в бане со льдом (0°С) и прибавляют свежий диизобутилалюминийгидрид/толуол (7,8 мл). После перемешивания реакционной смеси в течение еще 4 ч реакция еще не доходит до конца. Реакционную смесь охлаждают до 0°C и прибавляют еще 3,9 мл диизобутилалюминийгидрида. Реакционную смесь перемешивают еще 3 ч. Затем неочищенную реакционную смесь экстрагируют смесью этилацетат - 10% лимонная кислота и 1,0 н. NaOH. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха, получая искомое соединение (26). МС 471 (MH+).
Стадия Е Получение соединения (27).
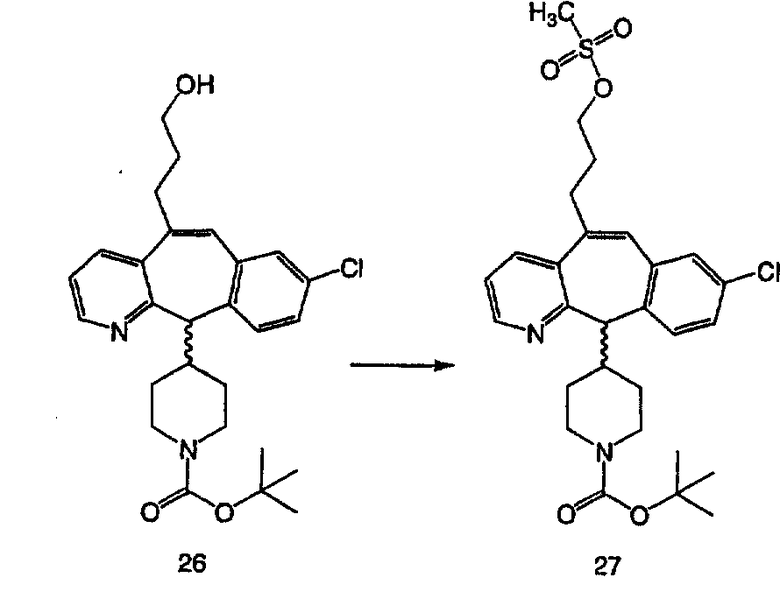
По методике, аналогичной использованной в Примере получения 3, стадия C, получают искомое соединение (27). МС 549 (МН+).
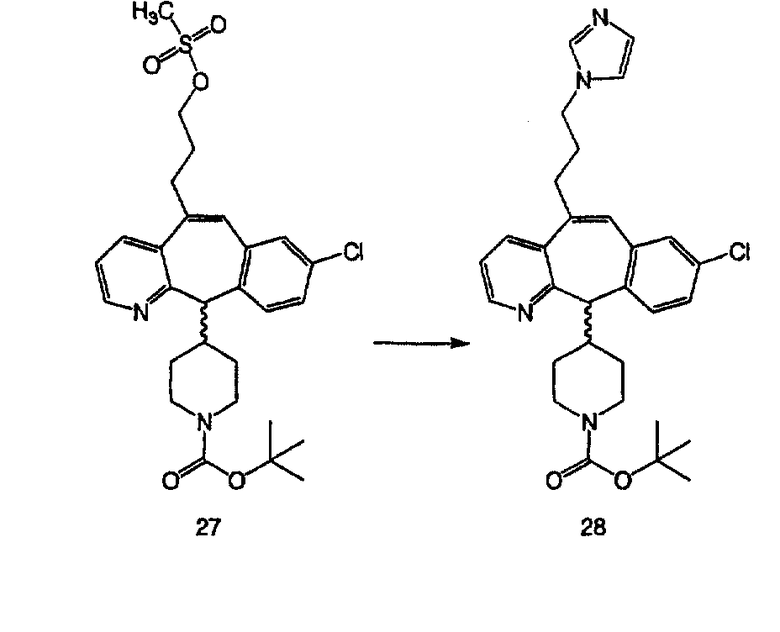
К раствору соединения, полученного в Примере получения 4, стадия Е (1,6 г, 3,01 ммоль), в DMF (50 мл) прибавляют имидазолилнатрий (Aldrich) (0,407 г, 4,52 ммоль). Реакционную смесь 2 ч нагревают при 90°С. Реакционную смесь охлаждают и DMF удаляют. Прибавляют насыщенный раствор бикарбоната натрия и смесь экстрагируют с помощью CH2Cl2. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха. Неочищенный продукт очищают с помощью хроматографии на колонке, элюируя с помощью смеси 2% СН3ОН, насыщенный аммиаком-CH2Cl2, и получают искомое соединение (28). МС 519 (MH+).
Стадия G Получение соединения (29).
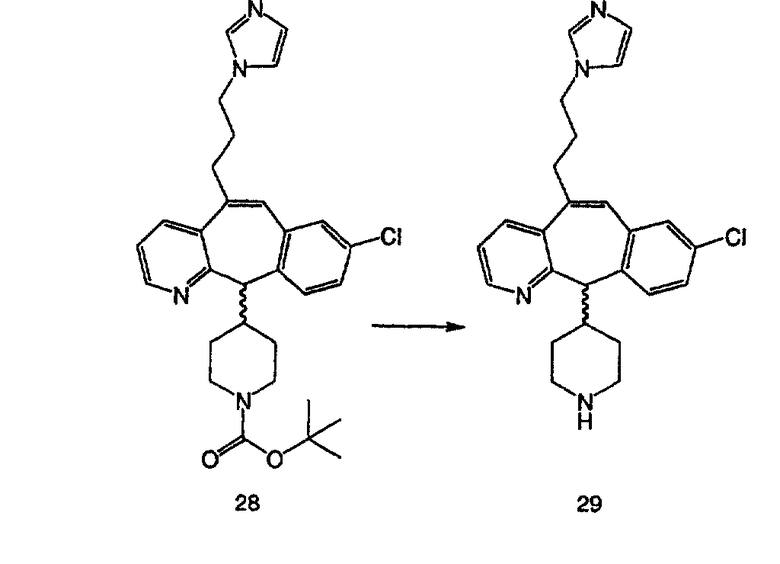
Продукт, полученный в Примере получения 4, стадия F (0,55 г, 1,08 ммоль), растворяют в 4 н. растворе диоксан/HCl (20 мл). Реакционную смесь 3 ч перемешивают при комнатной температуре, а затем концентрируют досуха, получая искомое соединение (29) в виде светло-желтого твердого вещества. МСВР 419 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 5
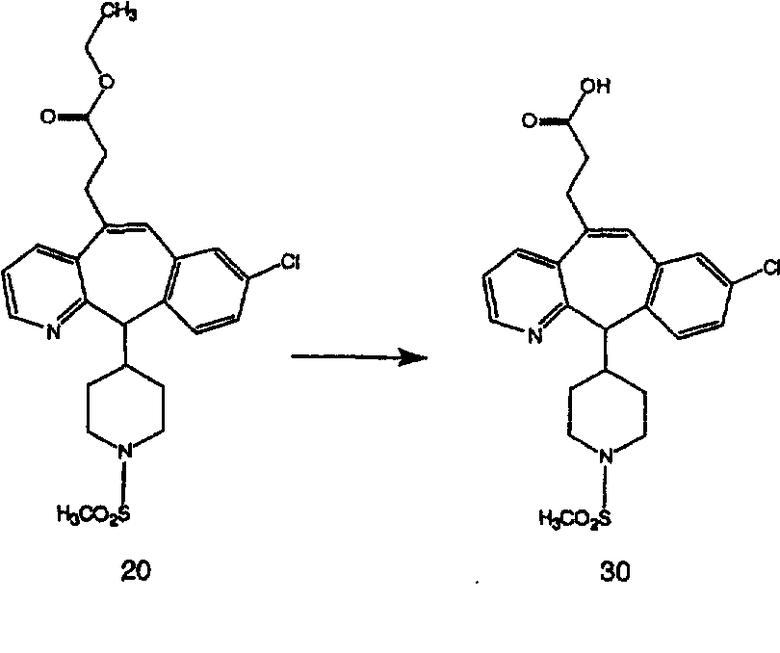
Соединение (20), полученное в Примере получения 3, стадия B (0,67 г, 1,37 ммоль), растворяют в THF (5 мл). К этой смеси прибавляют 1 н. раствор NaOH (6,9 мл) и полученный раствор перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, подкисляют 10% (мас./об.) лимонной кислотой и экстрагируют с помощью CH2Cl2. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха, получая искомое соединение (30) в виде желтого твердого вещества. Т.пл. 122,7 -123,4°С. МС 461 (МН+).
ПРИМЕР 1
Получение соединений (31) и (32).
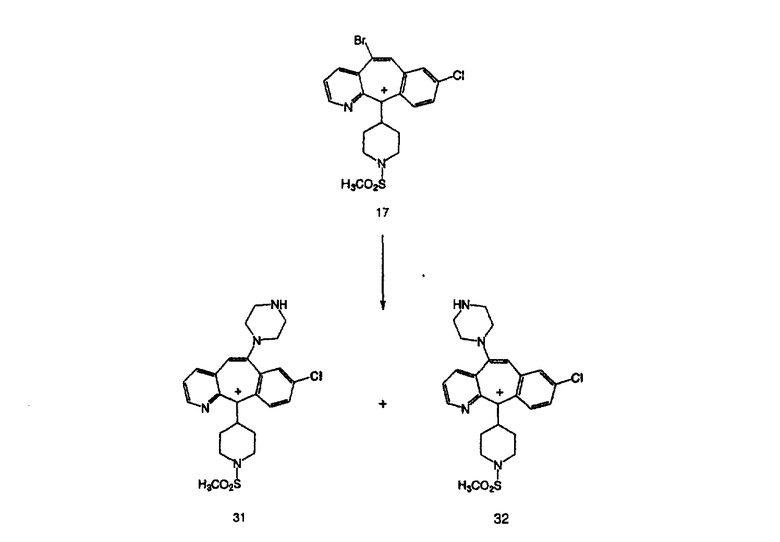
Соединение (17), полученное в Примере получения 2, стадия Е (0,31 г, 0,66 ммоль), обрабатывают таким же образом, как это описано в Примере получения 1, стадия Е, и получают смесь соединений (31) и (32), которую затем разделяют с помощью ВЭЖХ с использованием колонки Chiralpack AD, элюируя смесью 30% изопропанола-70% гексана-0,2% диэтиламина, и получают 0,04 г искомого соединения (31) и 0,07 г искомого соединения (32).
Соединение (31): т. пл. =174-175; [α]D 22 = +96,0° (3,6 мг/2 мл, CH2Cl2); МС (ББА) m/z 473 (MH+).
Соединение (32): т. пл. =173-174; [α]D 22 = +21,7° (8,4 мг/2 мл, CH2Cl2); МС (ББА) m/z 473 (MH+).
ПРИМЕР 2 Получение соединений (33) и (34).
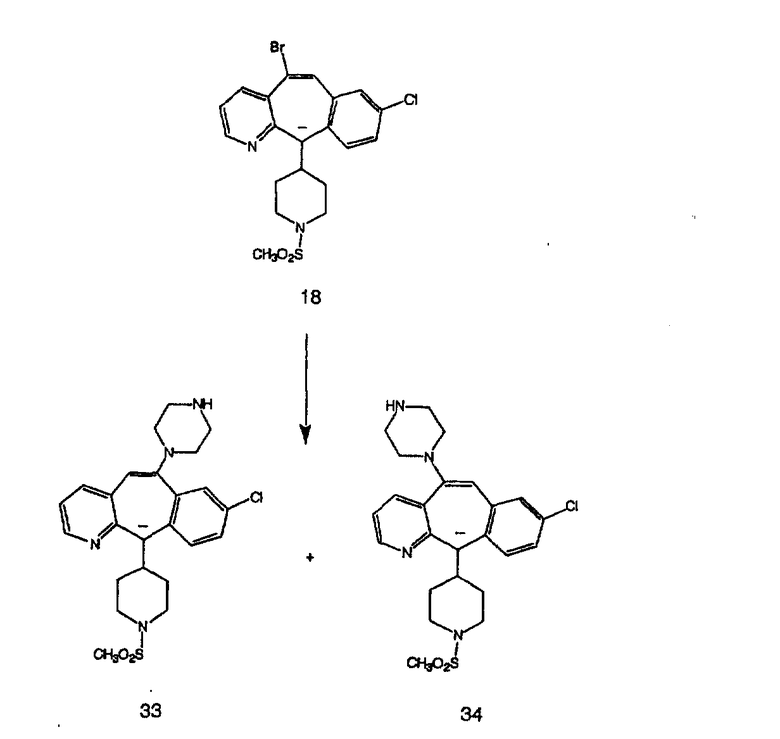
В соответствии с описанием, приведенным выше в Примере 1, 0,31 г соединения (18), полученного в Примере получения 2, стадия Е, превращают в смесь соединений (33) и (34), которую затем разделяют с помощью ВЭЖХ с использованием колонки Chiralpack AD, элюируя смесью 30% изопропанола-70% гексана-0,2% диэтиламина, и получают 0,12 г искомого соединения (33) и 0,04 г искомого соединения (34).
Соединение (33): т. пл. =178-179; [α]D 22 = -30,5° (9,5 мг/2 мл, CH2Cl2); МС (ББА) m/z 473 (MH+).
Соединение (34): т. пл. =172-173; [α]D 22 = -84° (3,5 мг/2 мл, СН2Cl2); МС (ББА) m/z 473 (MH+).
Получение соединений (35) и (36).
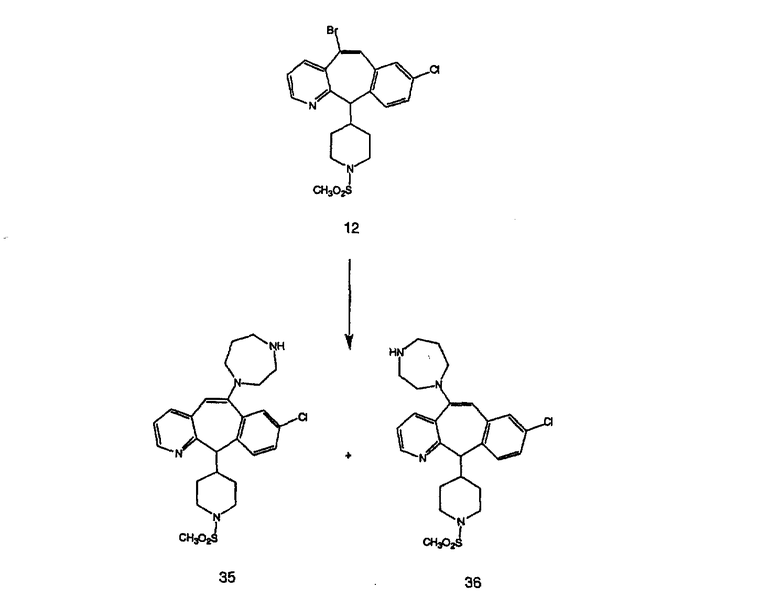
Продукт, полученный в Примере получения 2, стадия B (0,4 г, 0,86 ммоль), обрабатывают таким же образом, как это описано в Примере получения 1, стадия Е, используя гомопиперазин (Aldrich), и получают смесь соединений (35) и (36), которую затем разделяют с помощью флэш-хроматографии, элюируя смесью 30% СН3ОН, насыщенный с помощью NH3/CH2Cl2, и получают 0,13 г искомого соединения (35) и 0,17 г искомого соединения (36).
Соединение (35): т. пл. = 116 -117; МС (ББА) m/z 487 (MH+). Соединение (36): т. пл. = 111 -112; МС (ББА) m/z 487 (МН+).
ПРИМЕР 4 Получение соединений (37) и (38).
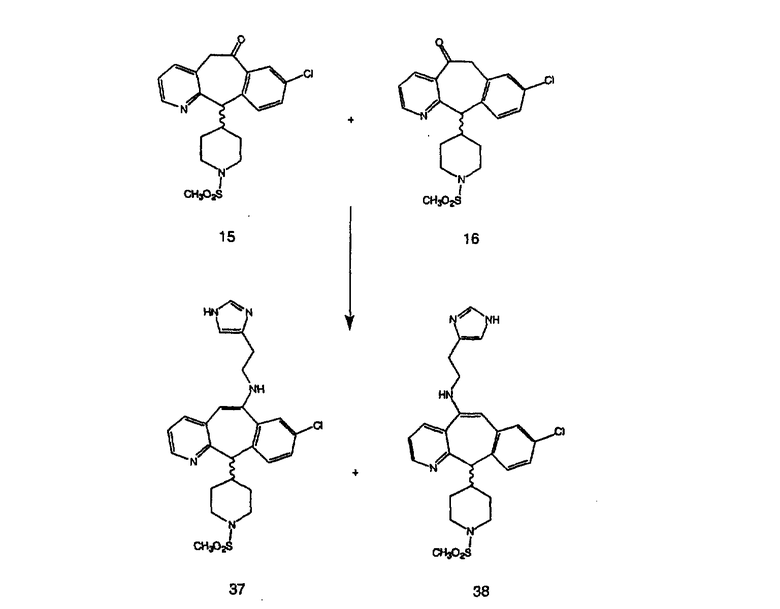
Кетоны, полученные в Примере получения 2, стадия D (0,50 г, 1,23 ммоль), Histamine® (0,21 г, 1,8 ммоль) и п-толуолсульфоновую кислоту (моногидрат) растворяют в безводном толуоле (40 мл) и 24 ч кипятят с обратным холодильником в аппарате Дина-Штарка с ловушкой. Затем реакционную смесь охлаждают, разбавляют этилацетатом и экстрагируют с помощью NaHCO3. После этого органический слой сушат над MgSO4 и концентрируют досуха. Очистка с помощью флэш-хроматографии на силикагеле с элюированием смесью 30% СН3ОН, насыщенный с помощью NH3/СН2Cl2, дает 0,17 г (выход 28%) 5-замещенного аддукта гистамина (38) в качестве первого элюирующегося продукта и 0,08 г (выход 13%) 6-замещенного аддукта гистамина (37) в качестве второго элюирующегося продукта.
Соединение (37): т. пл. =124-125; МС (ББА) m/z 498 (МН+). Соединение (38): т.пл. = 119 -120; МС (ББА) m/z 498 (MH+).
ПРИМЕРЫ 5 И 6
С использованием такой же методики, как и описанная выше, и с применением соответствующих аминов получают следующие смеси соединений:
ПРИМЕР 7
Получение соединений (43) и (44).
К раствору соединения (22), полученного в Примере получения 3, стадия C (1,0 г, 2,03 ммоль), в DMF (20 мл) прибавляют имидазолилнатрий (0,257 г, 2,85 ммоль). Реакционную смесь 2 ч нагревают при 90°С. Реакционную смесь охлаждают и DMF удаляют. Прибавляют насыщенный раствор бикарбоната натрия и экстрагируют с помощью CH2Cl2. Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage, элюируя с помощью смеси 3% СН3ОН (насыщенный аммиаком) - СН2Cl3, и получают искомое соединение в виде смеси энантиомеров. Смесь разделяют на чистые энантиомеры с помощью препаративной ВЭЖХ на колонке Chiral AD, элюируя с помощью смеси 35-40% изопропанол-гексан: 0,2% диэтиламин, и получают искомые соединения (43) и (44). МС 497 (MM*).
ПРИМЕР 8
Стадия A Получение соединения (45).
2-Метилимидазол растворяют в DMF (10 мл). К этому раствору прибавляют один эквивалент NaH и реакционную смесь 1 ч перемешивают при комнатной температуре.
Стадия B Получение соединения (46).
По методике, аналогичной использованной в Примере 7, заменяя имидазолилнатрий на 2-метилимидазолилнатрий (45), получают рацемическую смесь искомого соединения (46). МС 511 (МН+).
ПРИМЕР 9
СМЕСЬ СОЕДИНЕНИЙ (47) И (48)
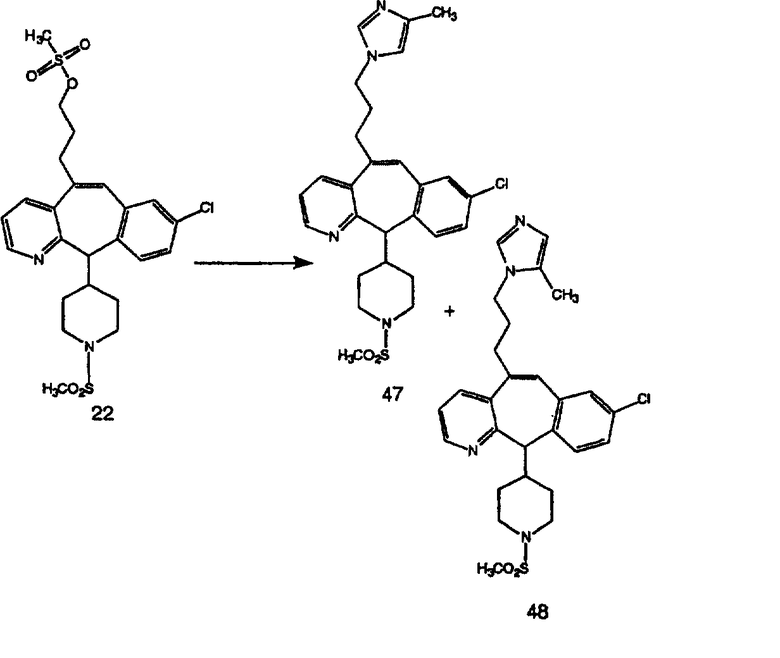
Соединение (22) вводят в реакцию таким же образом, как в Примере 8, используя 4-метилимидазол на стадии A, и получают смесь 4- и 5-метилзамещенных имидазолов (47) и (48).
ПРИМЕР 10
Стадия A Получение соединения (49).
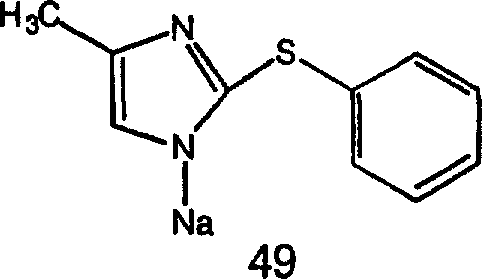
К метилимидазолу, содержащему защитную группу SEM (30 г, 0,141 моль), полученному по описанной в литературе методике, Whitten, J. P., J. Org. Chem. 1986, 51, 1891 -1894, в THF (250 мл) при -78°С в течение 1 ч прибавляют 2,5 М н-бутиллитий (74 мл, 0,184 моль). Раствор 1 ч перемешивают при -78°С, а затем в течение 1/2 ч прибавляют раствор дифенилдисульфида (34,27 г, 0,155 моль) в THF (125 мл). Смесь нагревают до комнатной температуры и перемешивают в течение ночи. Растворители удаляют и затем остаток разбавляют этилацетатом (250 мл) и промывают 1,0 М раствором NaOH (5 х 50 мл), а затем рассолом (50 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт (45,28 г, 0,141 моль) растворяют в этаноле (100 мл) и 5 М водном растворе HCI (100 мл) и 12 ч перемешивают при 60°С. Растворитель удаляют и остаток растворяют в дистиллированной H2O. Прибавляют 5 М водный раствор NaOH, пока рН не станет равным 8, а затем смесь экстрагируют этилацетатом. Объединенные органические слои промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют. Очистка с помощью флэш-хроматографии с элюированием с помощью смеси 70% гексан: ацетон дает продукт в виде белого твердого вещества. Затем в течение 1 ч проводят реакцию амина с NaH (1 эквивалент) в DMF и получают искомое соединение (49).
Стадия B Получение соединения (50).
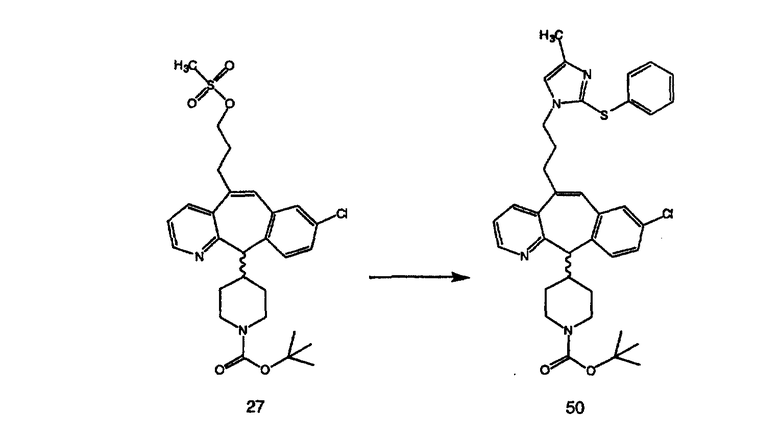
Соединение (27), полученное в Примере получения 4, стадия Е, вводят в реакцию таким же способом, как в Примере 8, используя 4-метил-2-фенилсульфанил-1Н-имидазолнатрий (49), и получают искомое соединение (50) в виде светло-желтого твердого вещества. МС 643 (МН+).
ПРИМЕР 11
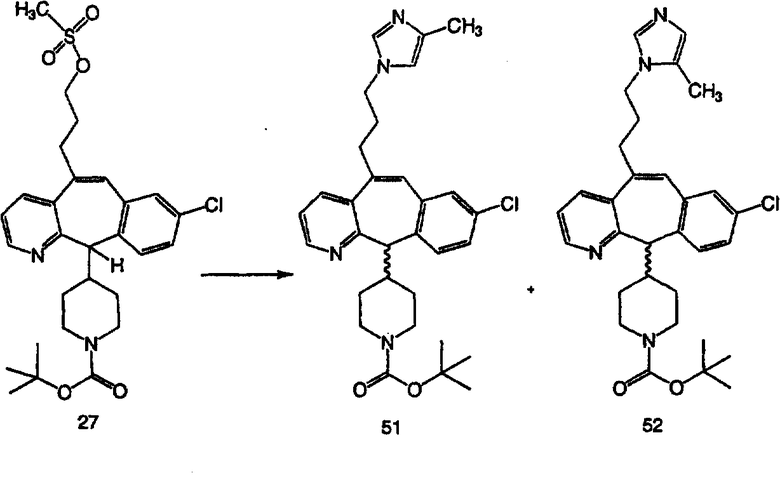
Соединение (27), полученное в Примере получения 4, стадия Е, обрабатывают таким же способом, как в Примере 9 выше, и получают смесь искомых 4- и 5-замещенных имидазолов (51) и (52).
Стадия В Получение чистых (+, -) соединений (53А) и (53В) и чистых (+, -) соединений (54А) и (54В).
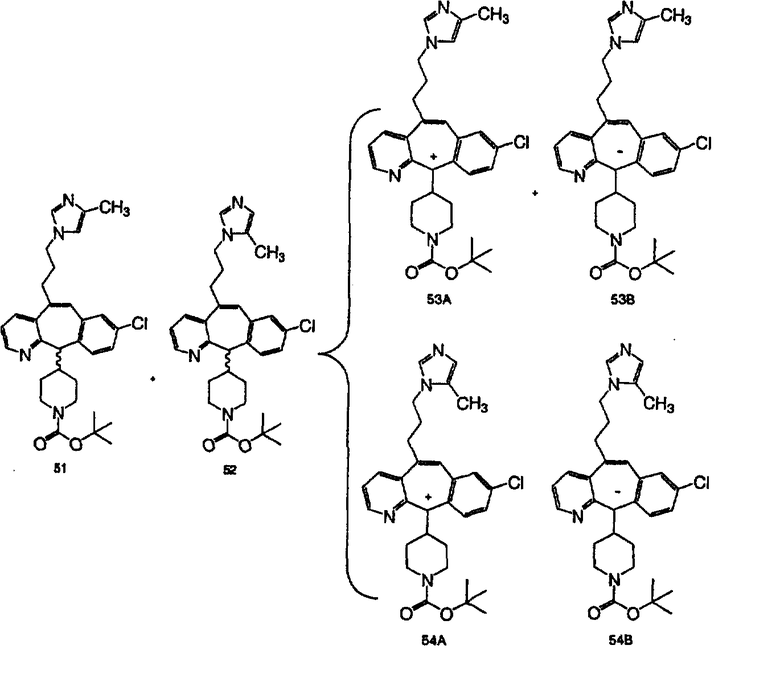
Соединения, полученные выше на стадии A, с помощью препаративной ВЭЖХ на колонке Chiral AD с элюированием смесью 20% изопропанол-гексан -0,2% диэтиламин дополнительно разделяют на смесь (4- и 5-)(+)-энантиомеров и (4- и 5-)(-)-энантиомеров. МС 532 (МН+). Затем пары чистых (+)- и (-)-энантиомеров вводят в реакцию с трифенилметилхлоридом (Aldrich) в CH2Cl2, начиная от 0°С и нагревая до комнатной температуры в течение 3 ч. Неочищенный продукт очищают с помощью хроматографии на колонке, элюируя смесью 50% этилацетат-ацетон, и получают чистые (+)- и (-)-4-метилзамещенные энантиомеры (53А) и (53В); МС 533 (MH+). Затем колонку промывают большим количеством 100% метанола, фракцию концентрируют и остаток обрабатывают метанолом, насыщенным аммиаком. Продукт очищают с помощью хроматографии на колонке, элюируя смесью 50% этилацетат-ацетон, и получают чистые (+)- и (-)-5-метилзамещенные энантиомеры (54А) И (54В); МС 533 (MH+).
ПРИМЕР 12
Получение соединений (55) и (56).
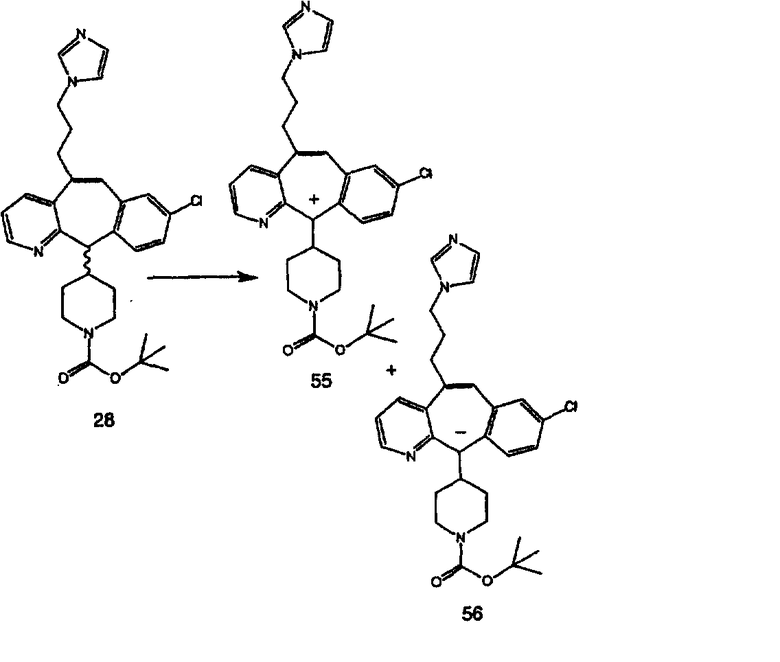
Соединение (28), полученное в Примере получения 4, стадия F, разделяют на чистые энантиомеры с помощью препаративной ВЭЖХ на колонке Chiral AD, элюируя с помощью смеси 20% изопропанол:гексан:0,2% диэтиламин, и получают чистые искомые соединения (55) и (56). МС 519 (MH+).
ПРИМЕР 13
Получение соединения (57).
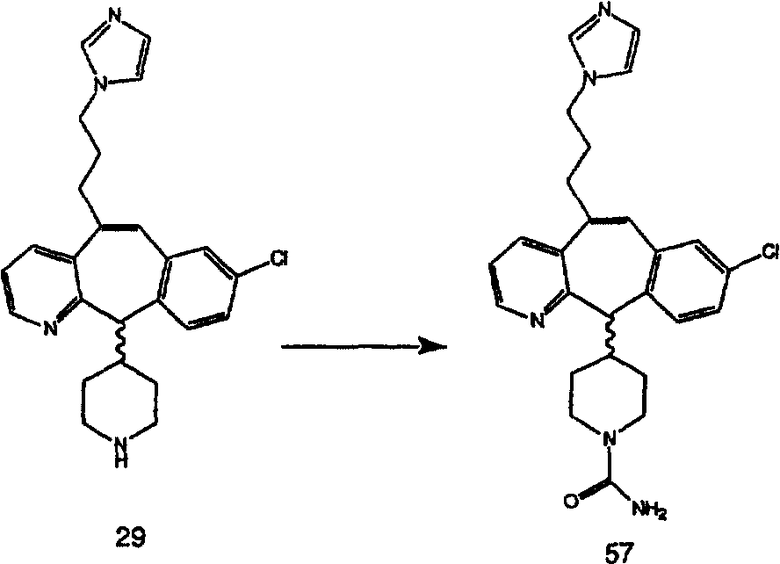
Соединение (28), полученное в Примере получения 4, стадия G (0,20 г, 0,48 ммоль), растворяют в CH2Cl2 (10 мл). Прибавляют триэтиламин (0,30 мл, 1,92 ммоль), а затем триметилсилилизоцианат (Aldrich) (1,3 мл, 9,6 ммоль) и перемешивают при комнатной температуре в течение ночи. Реакцию останавливают с помощью 1,0 н. раствора NaOH и экстрагируют с помощью CH2Cl2. Органический слой сушат над MgSO4, фильтруют и концентрируют. Очистка с помощью хроматографии на колонке с элюированием с помощью смеси 3-5% метанол, насыщенный аммиаком -CH2Cl2, дает искомое соединение (57) в виде белого твердого вещества. МС 464 (MH+).
ПРИМЕРЫ 14 И 15
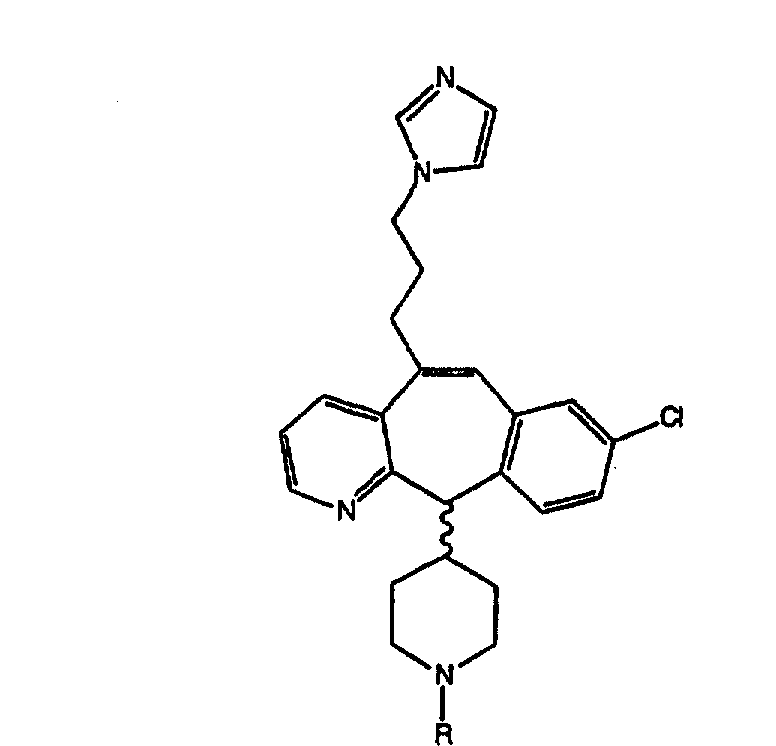
Используя соответствующие изоцианаты и применяя методику, описанную в Примере 13 выше, получают следующие соединения:
ПРИМЕР 16
Получение соединения (60).
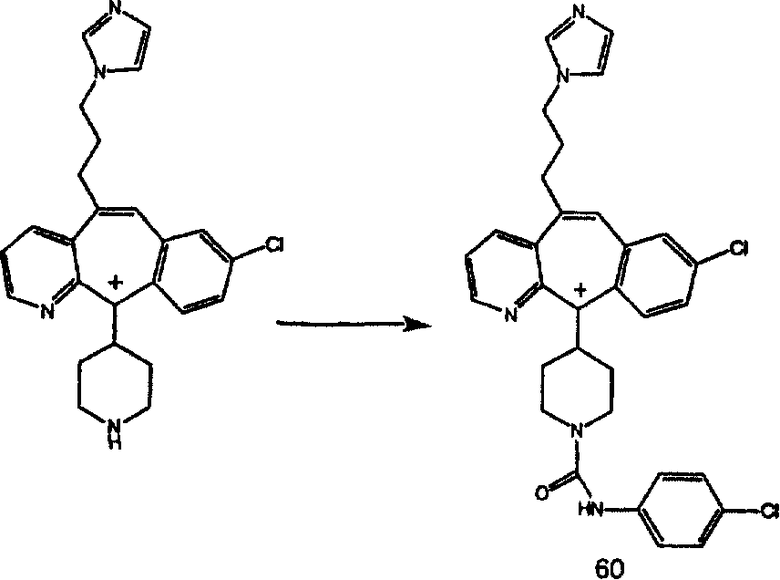
У соединения (55) защитную группу удаляют по методике, описанной в Примере получения 4, стадия G, и получают (+)-энантиомер исходного амина, который затем вводят в реакцию с 4-хлорфенилизоцианатом (Aldrich) (0,05 г, 0,34 ммоль) таким же образом, как в Примере 13 выше, и получают искомое соединение (60) в виде белого твердого вещества. МС 572 (MH+).
ПРИМЕР 17
Получение соединения (61).
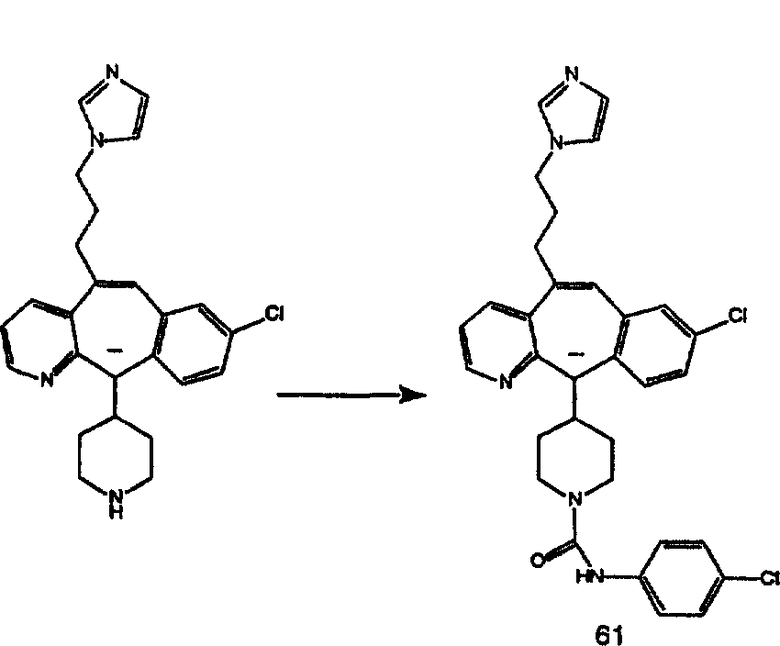
У соединения (56) защитную группу удаляют по методике, описанной в Примере получения 4, стадия G, и получают (-)-энантиомер исходного амина. Затем его вводят в реакцию таким же образом, как в Примере 16 выше, и получают искомое соединение (61) в виде белого твердого вещества. МС 572 (MH+).
ПРИМЕР 18
Получение соединения (62).
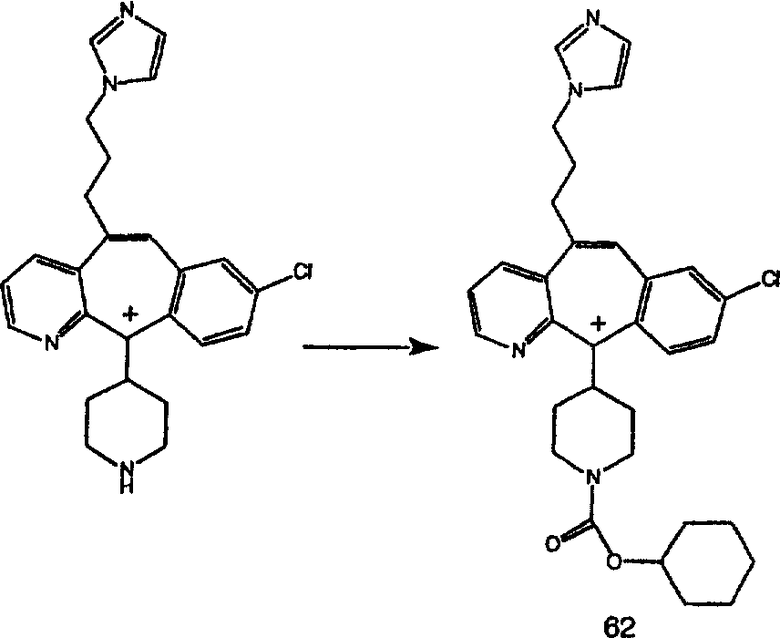
По методике, описанной в Примере 16, заменяя изоцианат на циклогексилхлорформиат (BASF), получают искомое соединение (62) в виде белого твердого вещества. МС 545 (MH+).
ПРИМЕР 19
Получение соединения (63).
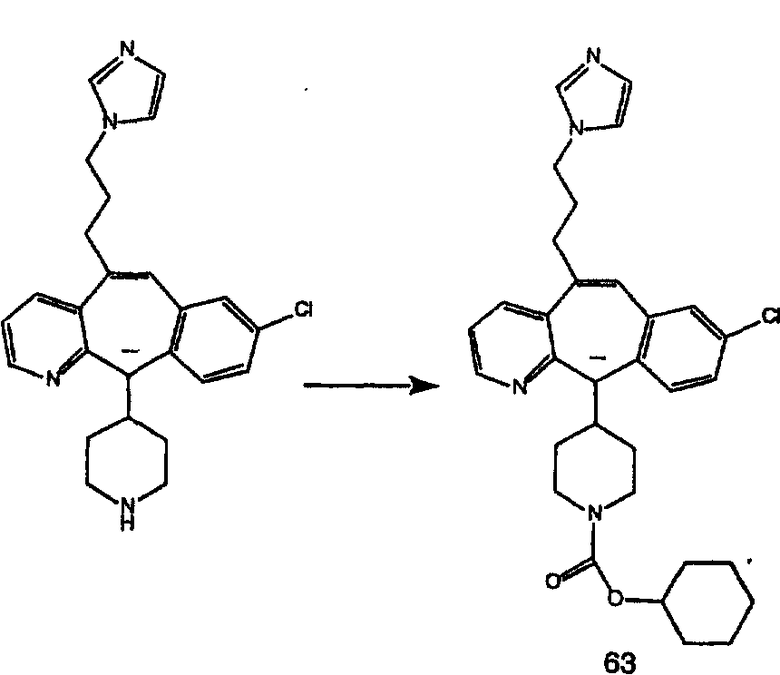
По методике, описанной в Примере 18 выше, используя (-)-энантиомер исходного амина, полученного в Примере 17, получают искомое соединение (63) в виде белого твердого вещества. МС 545 (МН+).
ПРИМЕР ПОЛУЧЕНИЯ 6
А Получение трибутил-(2-этоксивинил)-станнана (64).
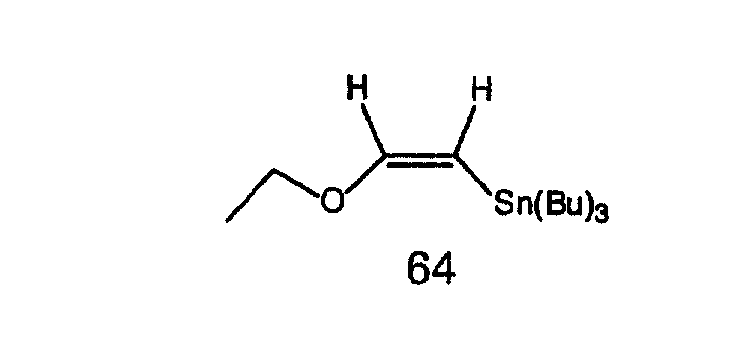
Этоксиэтин (Fluka), а затем гидрид трибутилолова (Aldrich) помещают в трубку, запаивают и в течение двух дней нагревают при 55°C. Затем реакционную смесь концентрируют в коричнево-красную жидкость. Очистка путем перегонки дает искомое соединение (64) в виде почти белой жидкости. Диапазон температур кипения 98-115°C (от 0,35 до 0,2 мм рт. ст.).
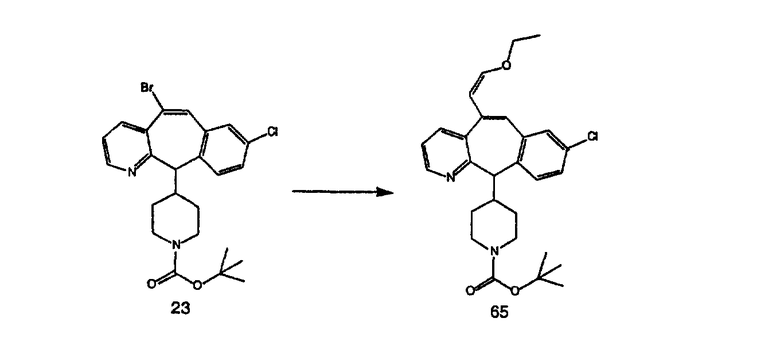
К раствору соединения (23), полученного в Примере получения 4, стадия А (6,51 г, 13,29 ммоль), дихлорбис(трифенилфосфин)палладия(И) (Aldrich) (0,373 г, 0,53 ммоль) и тетрабутиламмонийхлорида (Aldrich) (3,69 г, 13,29 ммоль) в DMF (50 мл) прибавляют соединение (64), полученное в Примере получения 6, стадия А. Реакционную смесь перемешивают в течение ночи в атмосфере азота при 75-80°С. Реакционную смесь охлаждают до комнатной температуры, а затем прибавляют раствор KF (0,93 г, 15,94 ммоль) в H2O (70 мл). При добавлении образуется осадок. Реакционную смесь перемешивают 15 мин, затем прибавляют CH2Cl2 и перемешивают еще 15 мин. Реакционную смесь экстрагируют с помощью CH2Cl2, органический слой сушат над сульфатом магния, фильтруют и концентрируют. Очистка путем хроматографии на колонке с силикагелем с элюированием смесями 1:3-1:1 этилацетат-гексан дает искомое соединение (65) в виде желтого твердого вещества, т. пл. 86-90°С.
Стадия C Получение соединения (66).
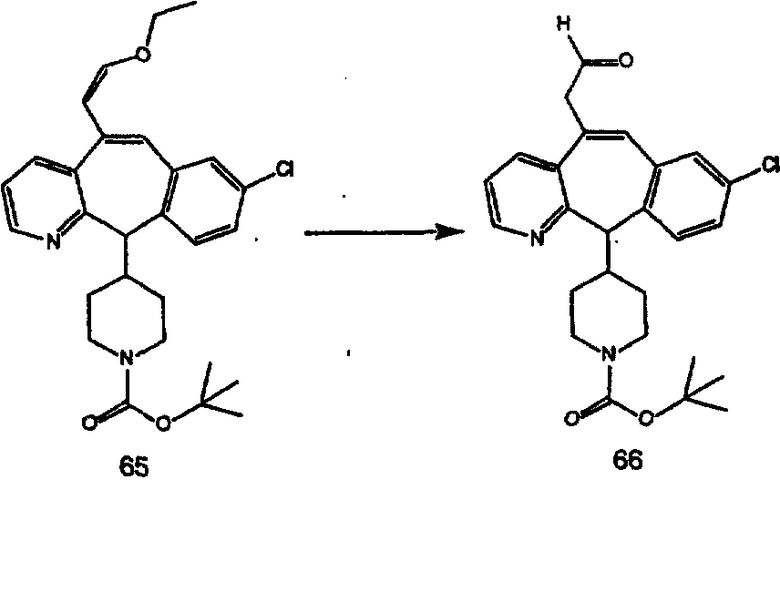
К раствору соединения (65), полученного в Примере получения 6, стадия B (3,25 г, 6,76 ммоль), в THF/H2O (33,7/7,3 мл) прибавляют ацетат ртути(II). Реакционную смесь 15 мин перемешивают при комнатной температуре, и за это время образуется осадок. Затем к смеси прибавляют насыщенный раствор KI (70-80 мл) и перемешивают 5 мин. Прибавляют CH2Cl2 и перемешивают 1 ч. Реакционную смесь экстрагируют с помощью СН2Cl2 (2 х 100 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют, получая искомое соединение (66) в виде светло-коричневого твердого вещества. МС 453 (MH+).
D Получение соединения (67).
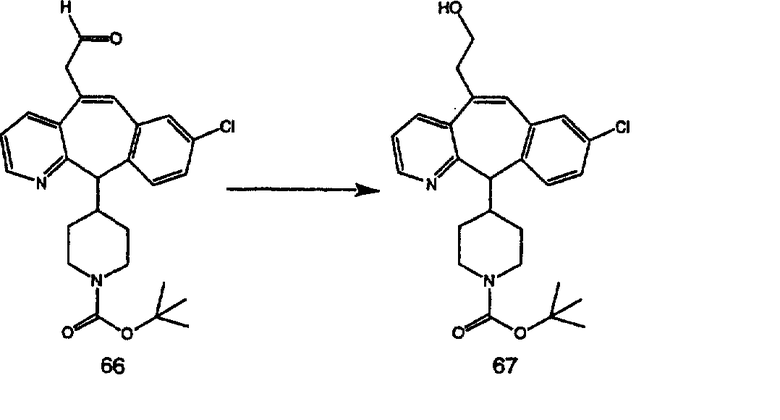
К раствору соединения (66), полученного в Примере получения 6, стадия С (3,06 г, 6,8 ммоль), в этаноле (40 мл) в течение 7 мин двумя порциями прибавляют борогидрид натрия (0,31 г, 8,1 ммоль). Реакционную смесь перемешивают 45 мин, а затем концентрируют, растворяют в этилацетате и промывают рассолом. Дополнительным количеством этилацетата повторно экстрагируют рассол и затем объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют. Дальнейшая очистка путем хроматографии на колонке с силикагелем с элюированием смесями 1:1-5:1 этилацетат-гексан дает искомое соединение (67) в виде белого твердого вещества. Диапазон температур кипения 120-130°С. МС 455 (МН+).
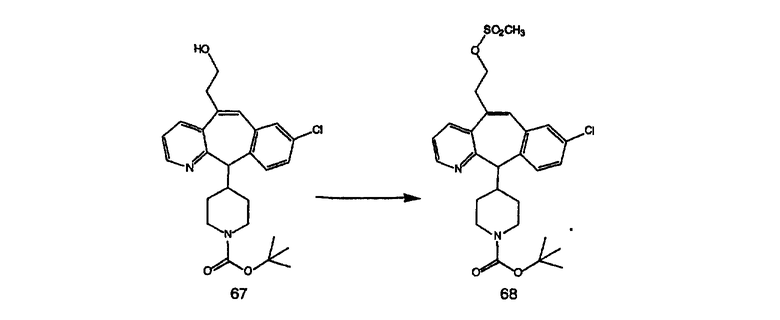
Соединение (67), полученное в Примере получения 6, стадия D, вводят в реакцию таким же образом, как в Примере получения 3, стадия C, и получают искомое соединение (68) в виде твердого вещества персикового цвета.
F Получение соединения (69).
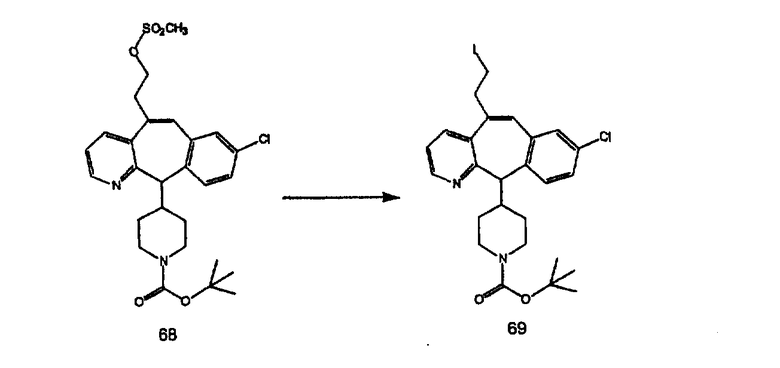
Соединение (68), полученное в Примере получения 6, стадия D (0,1 г, 0,19 ммоль), растворяют в THF (2,5 мл). К этой смеси прибавляют LiI (Aldrich) (0,064 г, 0,48 ммоль) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, растворяют в CH2Cl2 и промывают рассолом (25 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют и получают искомое соединение (69) в виде желто-коричневого твердого вещества.
ПРИМЕР 20
Получение соединения (70).
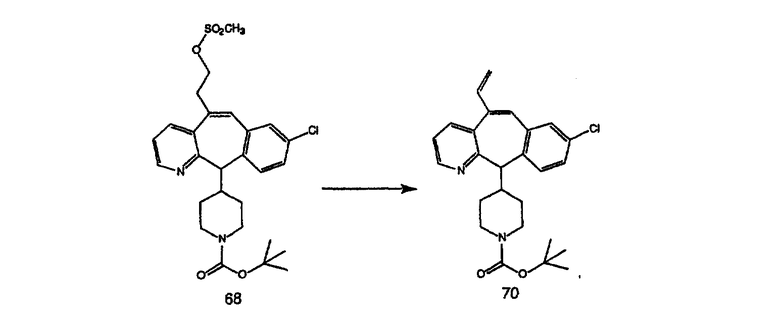
Соединение (68), полученное в Примере получения 6, стадия Е, вводят в реакцию таким же образом, как в Примере 8, стадия B, и получают искомое соединение (70) в виде белого твердого вещества. Т. пл. 94-101°С.
ПРИМЕР 21
Получение соединения (71).
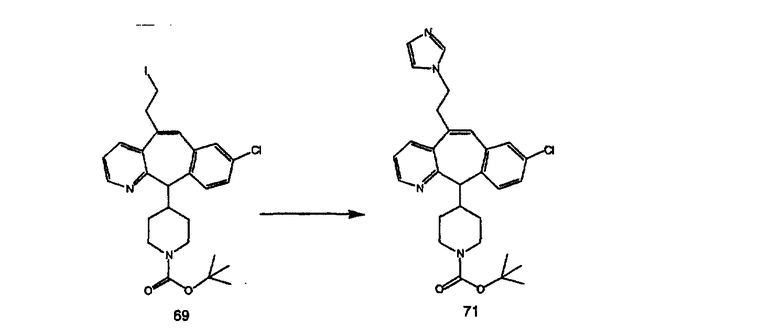
К соединению (69), полученному в Примере получения 6, стадия F (0,3 г, 0,05 ммоль), в СН3CN (1 мл) прибавляют имидазол (Aldrich) (0,014 г, 0,2 ммоль). Реакционную смесь нагревают до 52°C и перемешивают в течение ночи. Реакционную смесь охлаждают, концентрируют, а затем разбавляют этилацетатом и промывают рассолом. Органический слой сушат над сульфатом магния, фильтруют и концентрируют. Продукт очищают с помощью хроматографии на колонке с силикагелем, элюируя с помощью смеси 0-5% метанол (насыщенный аммиаком): СН2Cl2, и получают искомое соединение (71) в виде белого твердого вещества. МС (ББА) 505 (MH+).
ПРИМЕР 22
Получение соединения (72).
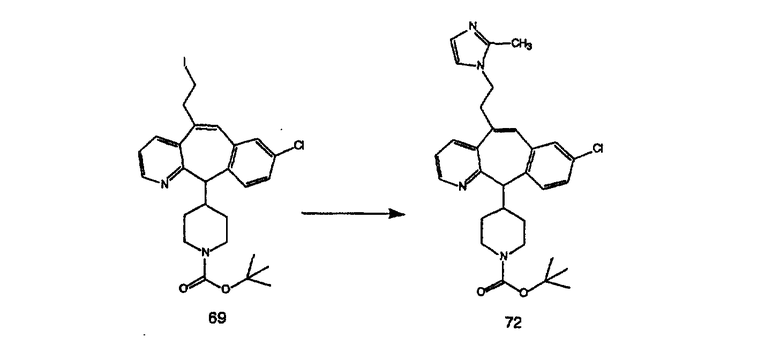
Заменяя имидазол на 2-метилимидазол и проводя реакции в основном таким же образом, как в Примере 21, получают искомое соединение (72) в виде светло-желтовато-коричневого твердого вещества, т. пл. 93-104°С.
ПРИМЕР 23
Получение соединения (73).
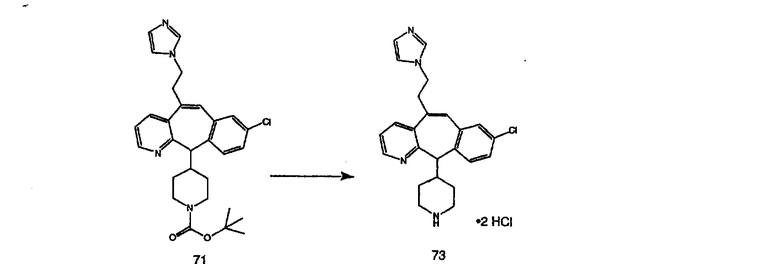
Соединение (71) (0,31 г, 0,06 ммоль), полученное в Примере 21, растворяют в 4 М растворе HCI/диоксан (0,5 мл) и перемешивают в течение 1 ч. Концентрирование реакционной смеси дает искомое соединение (73) в виде светло-желтого твердого вещества, т. пл. 195-205°С.
ПРИМЕР 24
Получение соединения (74).
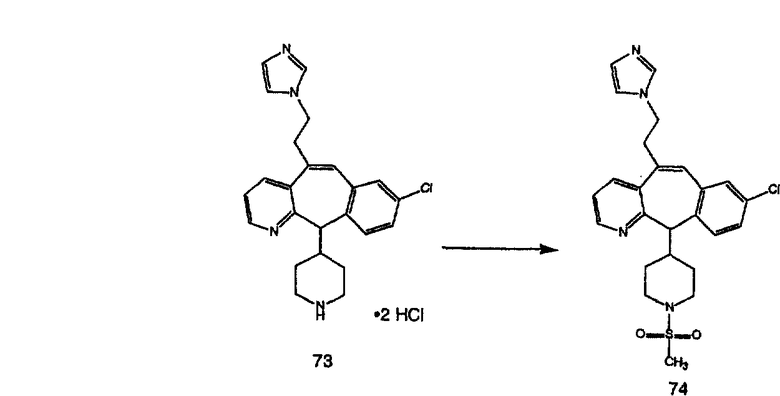
К раствору соединения (73), полученного в Примере 23 (0,026 г, 0,05 ммоль), в CH2Cl2 прибавляют триэтиламин (Aldrich) (0,046 мл, 0,33 ммоль), а затем метансульфонилхлорид (Aldrich) (0,01 мл, 0,1 ммоль). Реакционную смесь 36 ч перемешивают при комнатной температуре. Реакцию останавливают насыщенным раствором бикарбоната натрия (50 мл) и экстрагируют этилацетатом (2 х 75 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют. Продукт очищают с помощью препаративной тонкослойной хроматографии, элюируя с помощью смеси 90:10, CH2Cl2:метанол (насыщенный аммиаком), и получают искомое соединение (74), т.пл.105-116°С.
ПРИМЕР 25
Получение соединения (75).
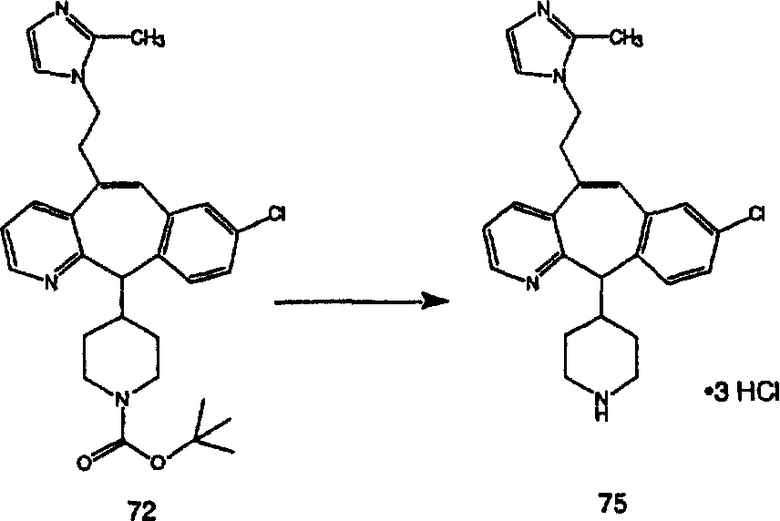
Соединение (72), полученное в Примере 22, 2 ч перемешивают с 4 М HCI/диоксан. Концентрированно реакционной смеси дает искомое соединение (75) в виде не совсем белого твердого вещества, т. пл. 185-203°С.
ПРИМЕРЫ 26 -29
Вводя в реакцию соединение (75), полученное в Примере 25, таким же образом, как описано в Примере 13, и используя соответствующий изоцианат, получают следующие соединения:
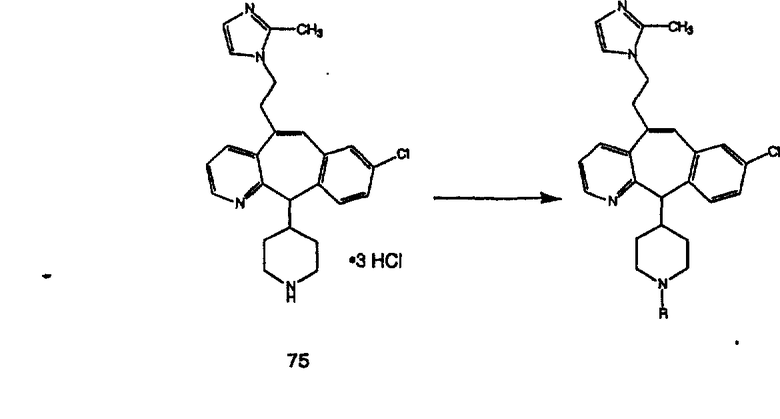
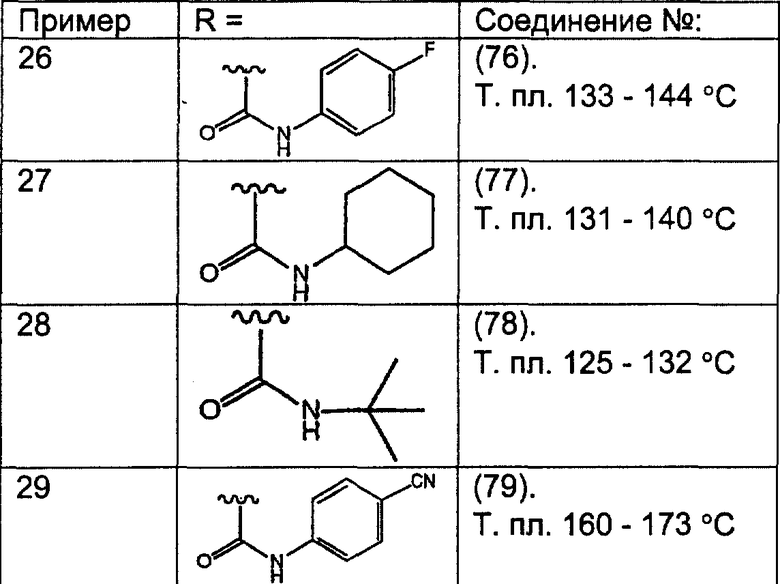
ПРИМЕР 30
А Получение циклогексилхлорформиата
Раствор циклогексанола (Aldrich) (25 мл, 0,2 моль) в CH2Cl2 (50 мл) при 0°C в течение 1 ч по каплям прибавляют к раствору фосгена в толуоле (262 мл 1,96 М раствора, 0,5 моль). Реакционную смесь в течение 3 ч нагревают до комнатной температуры и перемешивают в течение ночи. Летучие вещества удаляют и получают искомое соединение (80) в виде бесцветной жидкости.
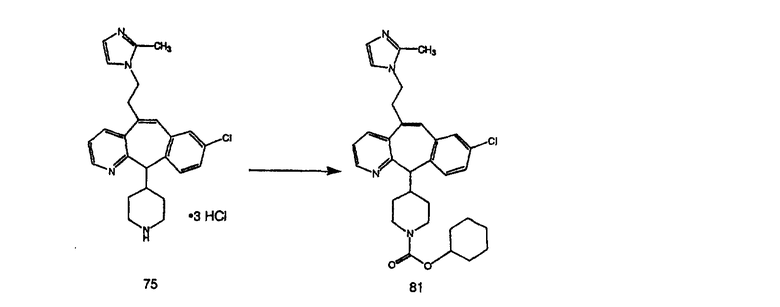
Вводя в реакцию соединение (75), полученное в Примере 25, таким же образом, как описано в Примере 13, но вместо изоцианата используя хлорангидрид кислоты (80), полученный в Примере 30, стадия A, получают искомое соединение (81) в виде не совсем белого полужидкого вещества, т. пл. 89-98°С.
ПРИМЕР 31
Получение соединения (82).
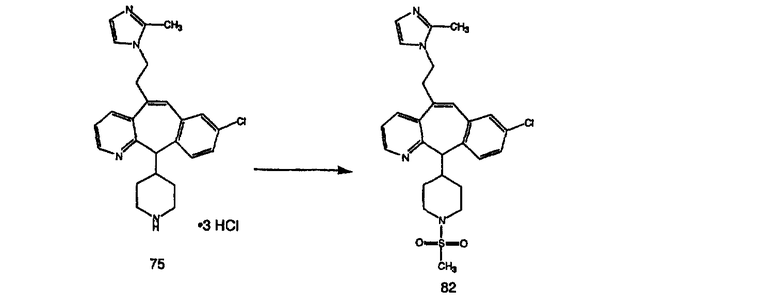
Вводя в реакцию соединение (75), полученное в Примере 25, таким же образом, как описано в Примере 13, но вместо изоцианата используя метансульфонилхлорид (80), получают искомое соединение (82) в виде желтовато-коричневого полужидкого вещества, т.пл. 120-129°С.
ПРИМЕР 32
Разделение соединения (75) на (+)- и (-)-энантиомеры (83) и (84).
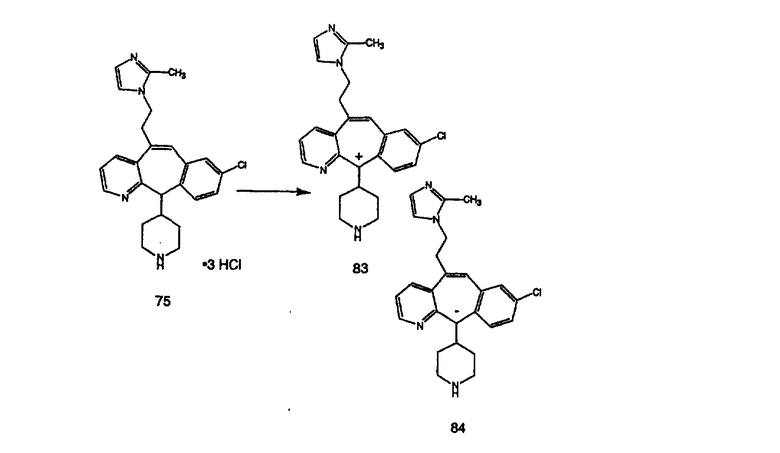
Соединение (75) разделяют на чистые (+)- и (-)-энантиомеры с помощью препаративной хроматографии на колонке Chiralpack-AD, элюируя с помощью смеси 85:15:0,2%, 2-пропанол:гексан/диэтиламин, и получают искомые соединения (83) и (84) соответственно.
ПРИМЕР 33
Получение соединения (85).
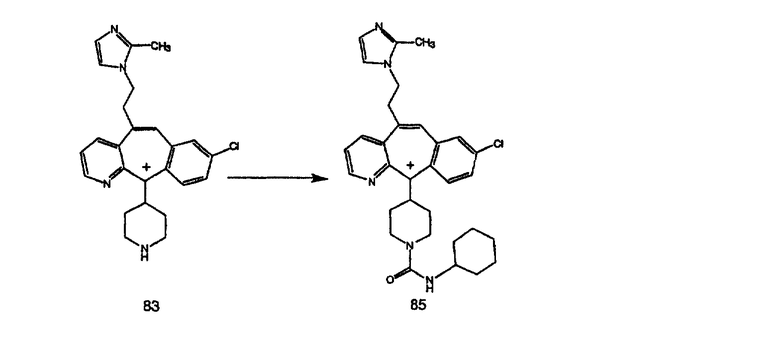
Соединение (83) вводят в реакцию таким же образом, как в Примере 27, и получают искомое соединение (85) в виде белого твердого вещества, т. пл. 122-129°С.
ПРИМЕР 34
Получение соединения (86).
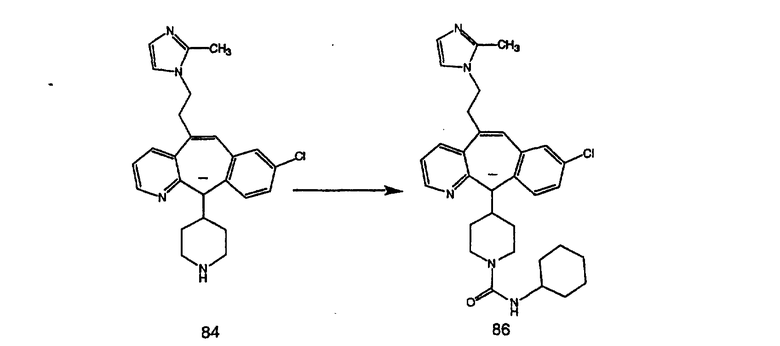
Соединение (84) вводят в реакцию таким же образом, как в Примере 27, и получают искомое соединение (86) в виде белого твердого вещества, т. пл. 118-133°С.
ПРИМЕР 35
Получение соединений (87) и (88).
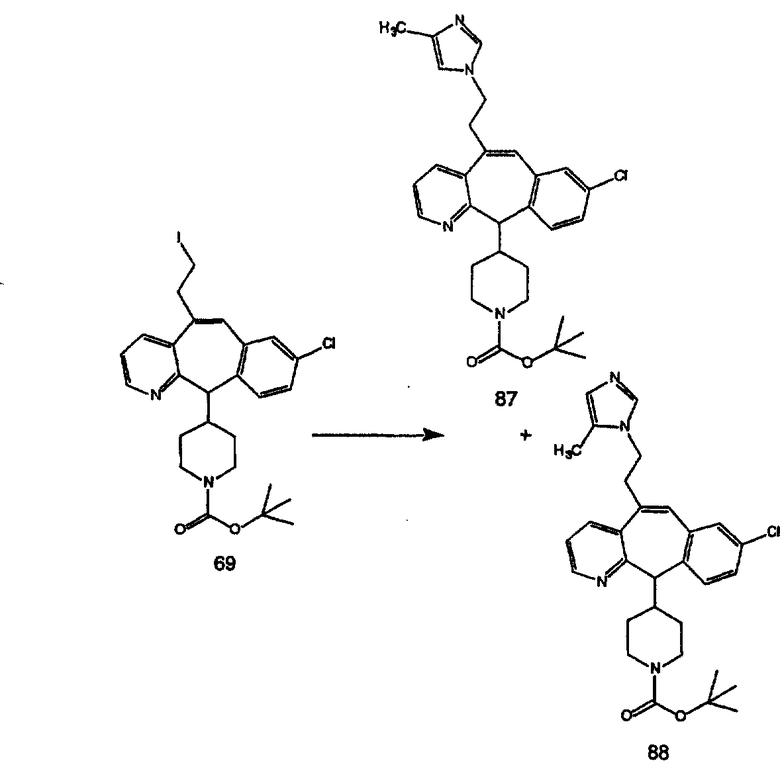
Соединение (69), полученное в Примере 19, вводят в реакцию таким же образом, как описано в Примере 21, заменяя имидазол на 4-метилимидазол, и получают смесь 4- и 5-замещенных имидазолов. После этого смесь (0,234 г, 0,45 ммоль) обрабатывают тритилхлоридом (Aldrich) (0,047 г, 0,17 ммоль) и разделяют с помощью препаративной тонкослойной хроматографии, элюируя с помощью смеси 1: 6 этилацетат-ацетон, и получают чистые изомеры (87) и (88), т. пл. (88) 97-107°C (белое твердое вещество).
ПРИМЕР 36
Получение соединения (89).
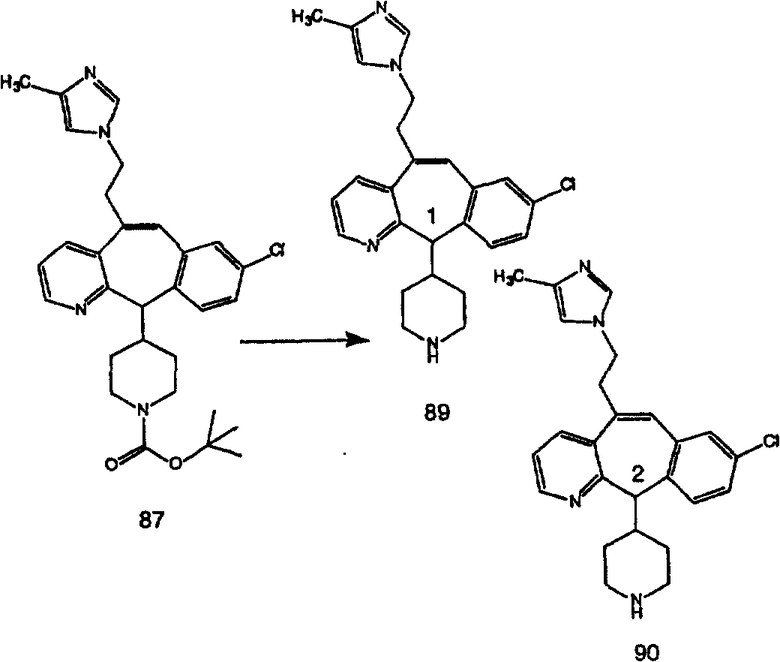
Соединение (87), полученное в Примере 35 (0,085 г, 0,16 ммоль), вводят в реакцию таким же образом, как описано в Примере 25. Полученную смесь энантиомеров затем разделяют с помощью препаративной хроматографии на колонке Chiralpack-AD, элюируя с помощью смеси 15-85% изопропанол - гексан с добавкой 0,2% диэтиламина, и получают энантиомеры 1 и 2 в виде не совсем белых твердых веществ.
ПРИМЕР 37
Получение соединения (91).
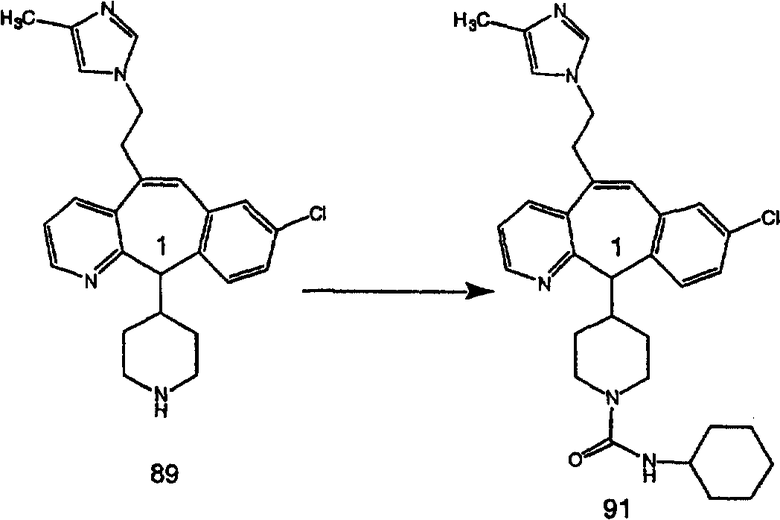
Этантиомерно чистое соединение (89), полученное в Примере 36 (0,02 г, 0,049 ммоль), вводят в реакцию способом, аналогичным использованному в Примере 27, и получают искомое соединение (91) в виде почти белого твердого вещества, т. пл. 130-142°С.
ПРИМЕР 38
Получение соединения (92).
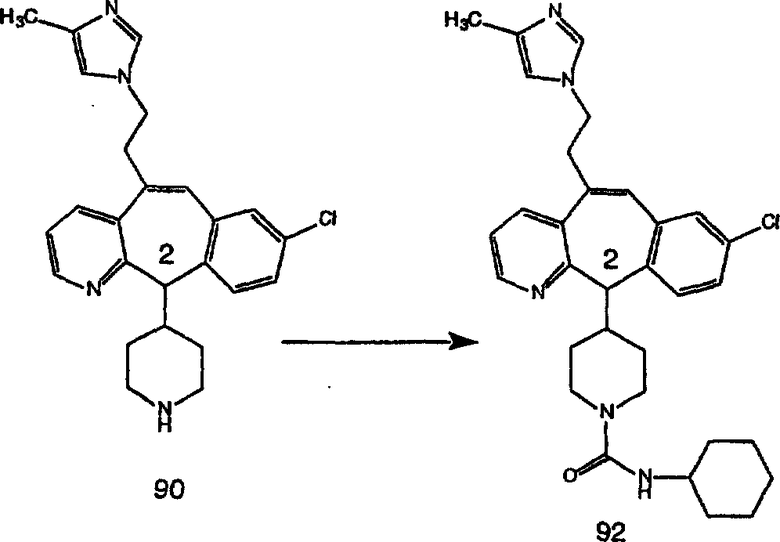
Этантиомерно чистое соединение (90), полученное в Примере 36 (0,023 г, 0,054 ммоль), вводят в реакцию способом, аналогичным использованному в Примере 27, и получают искомое соединение (92), т. пл. 125-135°С.
ПРИМЕР ПОЛУЧЕНИЯ 7
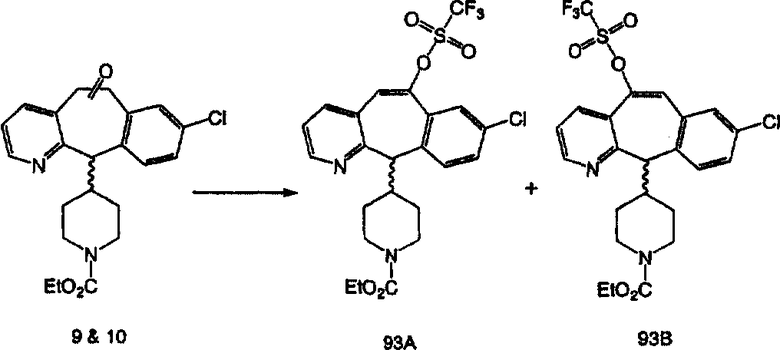
Смесь пиперазинилов (9) и (10), полученную в Примере получения 1, стадия F, в THF при -78°C вводят в реакцию с LDA (1,1 экв.) и перемешивают в течение 1,5 ч. Смесь нагревают до -20°С и затем прибавляют N-фенилтрифторметансульфонимид (1,1 экв.). Смесь перемешивают при комнатной температуре в течение ночи, а затем экстрагируют с помощью EtOAc и промывают с помощью H2O. Сушат над Na2SO4 и концентрируют. Очистка и разделение с помощью флэш-хроматографии на колонке с силикагелем дает чистые соединения (93А и 93В).
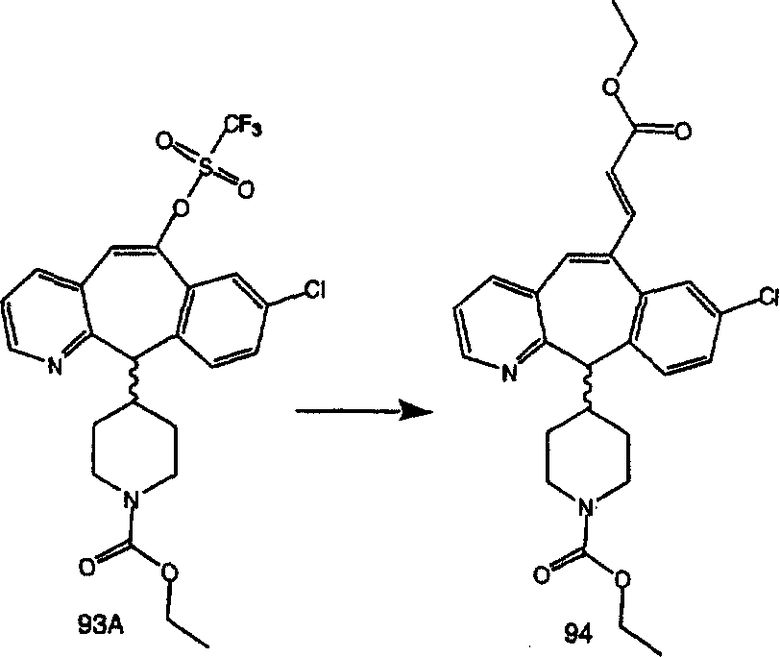
Полученное выше соединение (93А) растворяют в DMF. Последовательно прибавляют EtaN (29 экв.), этилацетат (5,4 экв.), K2СО3 (5 экв.), Bu4NBr (2 экв.) и ацетат палладия(II) (0,13 экв.). Смесь перемешивают и нагревают при 100°C в течение 4 ч. После охлаждения смесь концентрируют и остаток растворяют в CH2Cl2 и экстрагируют с помощью СН2Cl2/Н2О. Органический слой сушат над Na2SO4, а затем концентрируют и остаток очищают с помощью флэш-хроматографии на колонке с диоксидом кремния и получают искомое соединение (94).
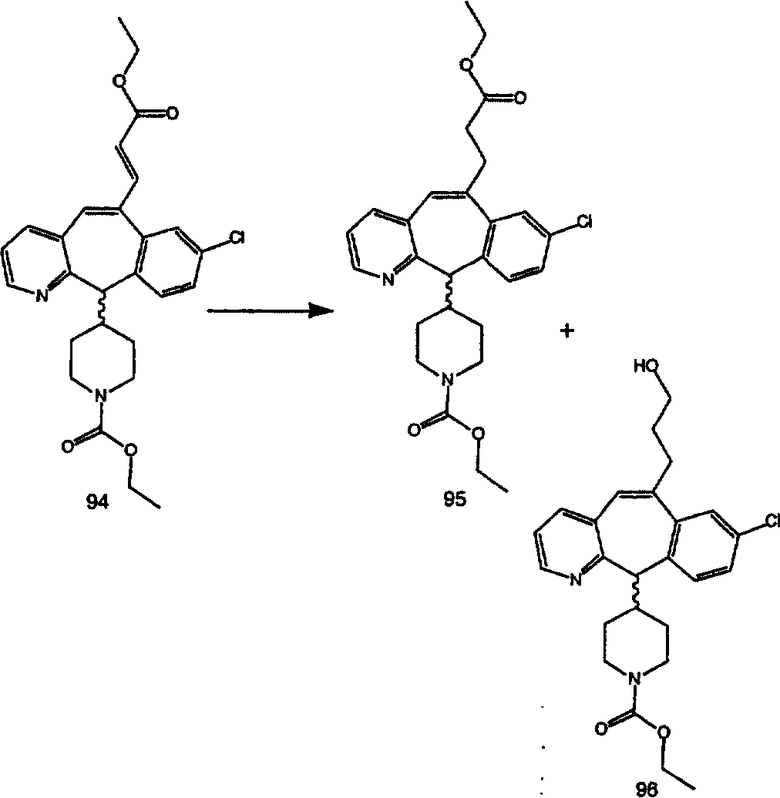
Соединение (94) растворяют в EtOH, охлаждают в бане со льдом и в течение 3 мин проводят реакцию с NaBH2 (15 экв.). Затем прибавляют CuCl (2 экв.) и 2 ч перемешивают при комнатной температуре. Затем смесь фильтруют, концентрируют и экстрагируют с помощью СН2Cl2. Промывают водой, затем рассолом, сушат над Na2SO4 и концентрируют в смесь искомого соединения (95) и гидроксисоединения (96).
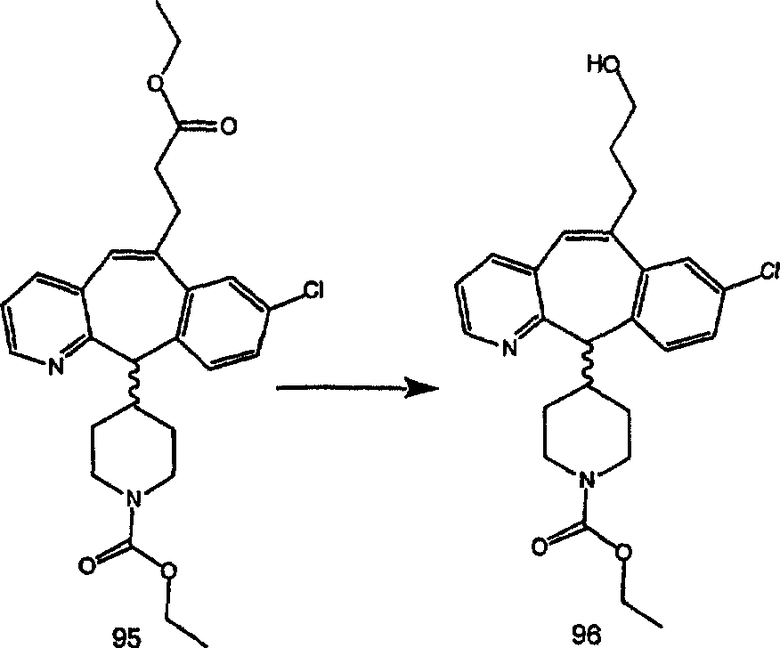
Соединение (95) вводят в реакцию с LiBH4 (3 экв.) в THF при кипячении с обратным холодильником в течении 4 ч. Прибавляют EtOAc и смесь промывают с помощью Na2CO3, затем сушат над Na2SO4 и концентрируют, получая искомое соединение (96).
Е Получение соединения (97).
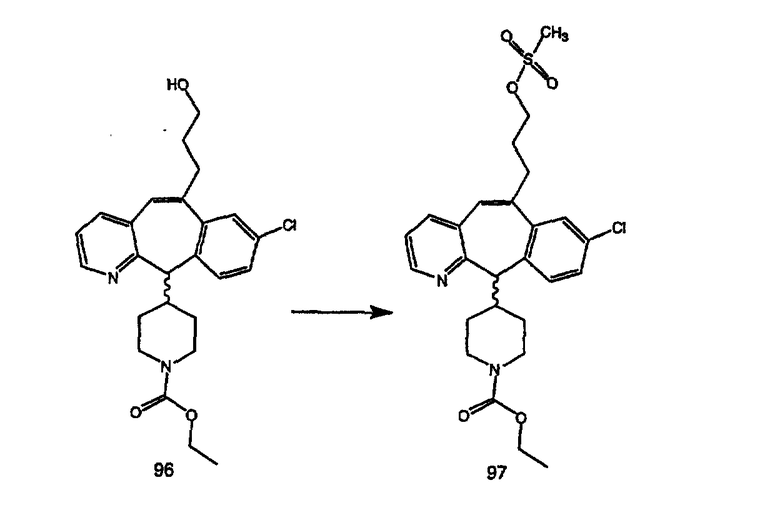
Соединение (96) растворяют в СН2Cl2, прибавляют Et3N (3 экв.), а затем метансульфонилхлорид (1,5 экв.). Смесь перемешивают при комнатной температуре в течение ночи, разбавляют с помощью СН2Cl2 и промывают с помощью Na2CO3. Сушат над Na2SO4 и концентрируют, получая искомое соединение (97).
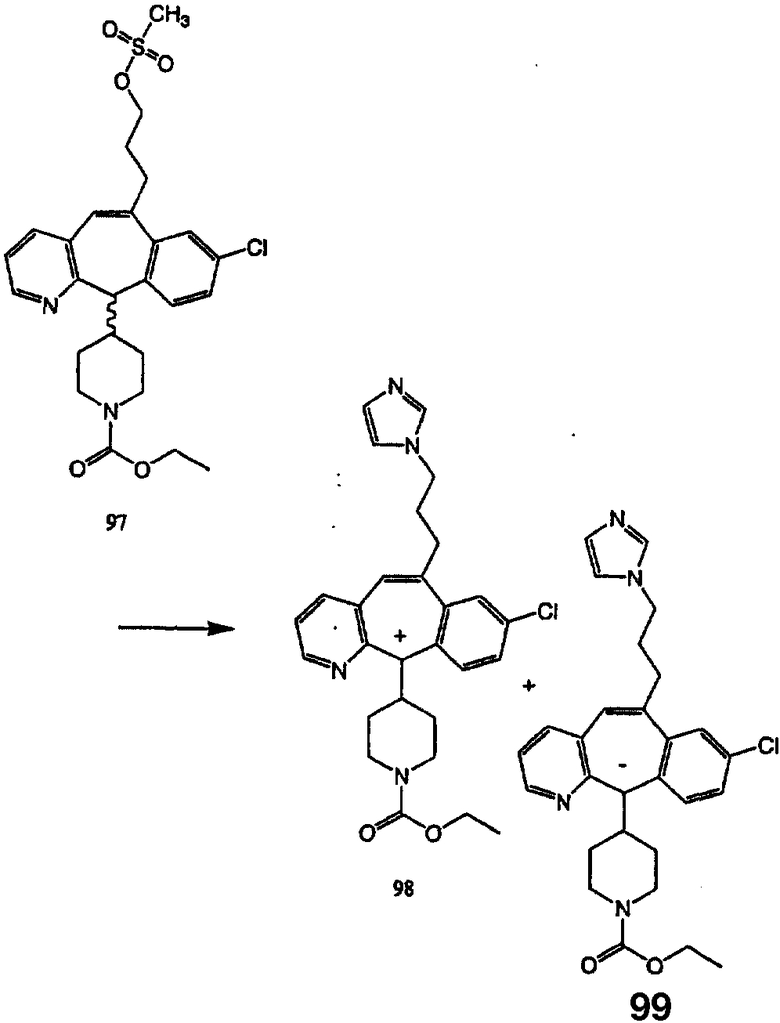
К раствору натриевой соли имидазола (Aldrich) в DMF прибавляют NaH (2 экв.). Перемешивают 15 мин, затем прибавляют соединение (97) (полученное выше) (1 экв.) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, а затем экстрагируют этилацетатом. Промывают с помощью Na2CO3, сушат над Na2SO4, фильтруют, а затем концентрируют. Неочищенный продукт очищают с помощью флэш-хроматографии на колонке с диоксидом кремния. Последующее разделение чистых (+)-энантиомеров и чистых (-)-энантиомеров проводят на колонке Chiracel AD и получают искомые соединения (98) и (99).
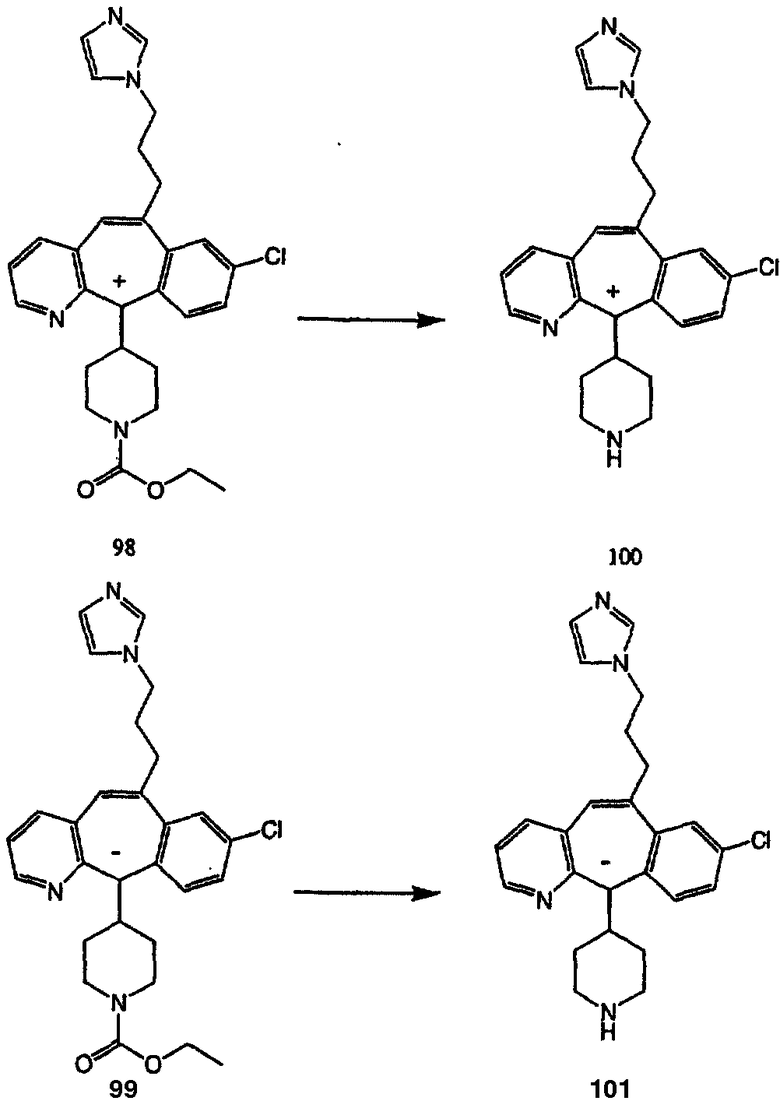
Соединения (98) и (99) по отдельности гидролизуют в их свободные амины путем проводимого в течение 5 ч кипячения с обратным холодильником в концентрированной HCl. Реакционные смеси по отдельности выливают на лед и подщелачивают с помощью NH4OH. Затем растворы экстрагируют с помощью СН2Cl2, сушат над Na2SO4, фильтруют и концентрируют, получая искомые соединения (100) и (101).
ПРИМЕР ПОЛУЧЕНИЯ 8
Получение соединений (102) и (103).
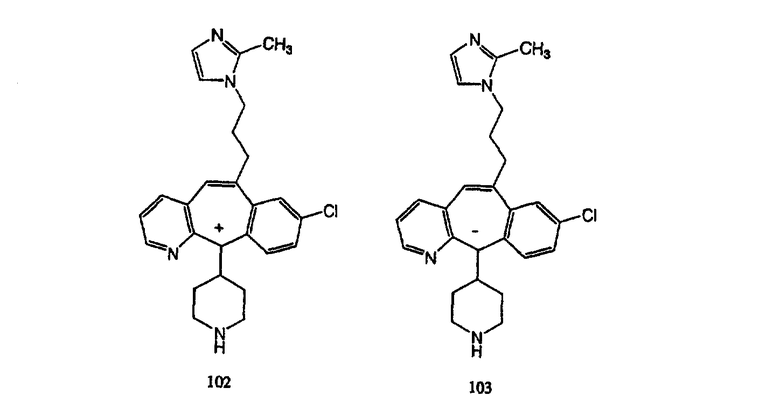
Способом, аналогичным описанному в Примере получения 7, стадии A - G, заменяя на стадии F натриевую соль имидазола на 2-метилимидазол, получают искомые соединения (102) и (103).
ПРИМЕР ПОЛУЧЕНИЯ 9
А Соединение (104).
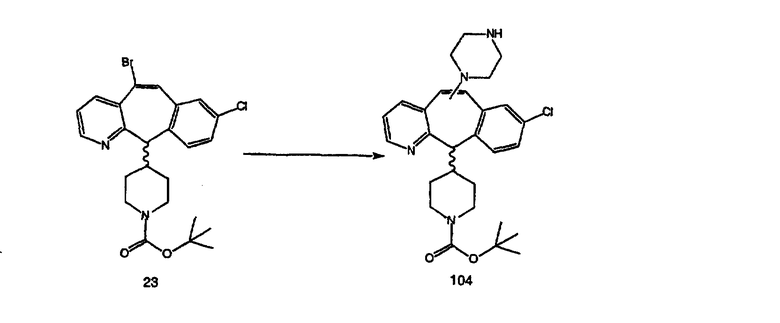
Соединение (23), полученное в Примере получения 4, вводят в реакцию с пиперазином таким же образом, как это описано в Примере получения 1, стадия Е, и получают искомое соединение (104).
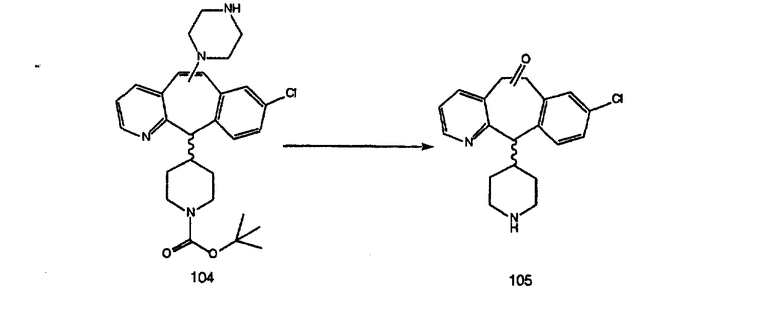
Полученное выше соединение (104) гидролизуют путем проводимого в течение ночи кипячения с обратным холодильником в 6 н. растворе HCl. Охлажденную реакционную смесь подщелачивают 50% мас./мас. раствором NaOH, а затем экстрагируют смесью 80% THF - EtOAc. Органический слой сушат над MgSO4, фильтруют и концентрируют досуха, получая искомое соединение (105).
С Получение соединений (106) и (107).
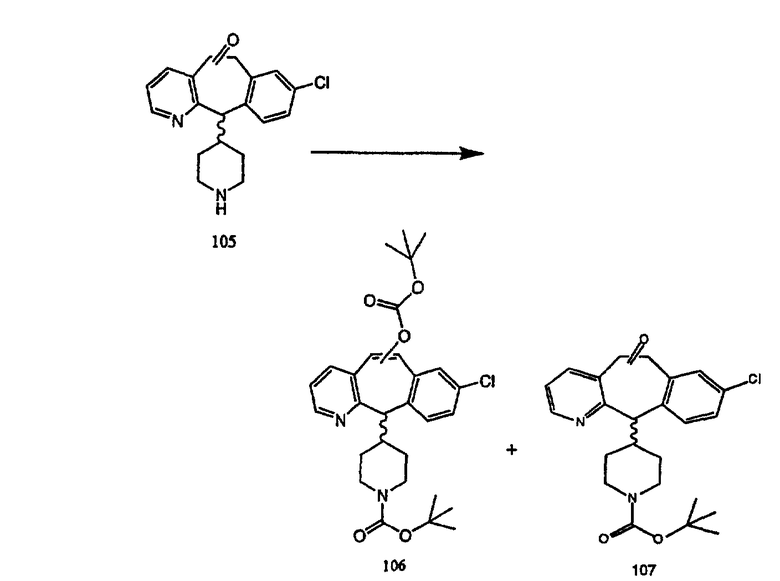
Соединение (105) растворяют в смеси МеОН:H2O состава 50:1, затем прибавляют ди-трет-бутилдикарбонат (2 экв.). Значение рН доводят до 9 и 4 ч перемешивают при комнатной температуре. Реакционную смесь концентрируют и экстрагируют с помощью CH2Cl2. Органический слой промывают с помощью Na2CO3, сушат, фильтруют и концентрируют досуха, получая смесь искомых соединений (106) и (107).
D Получение соединения (107).
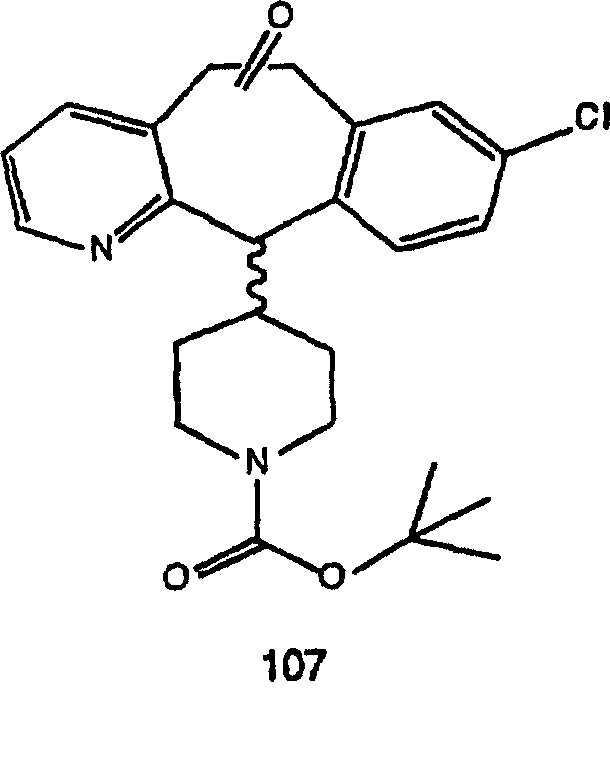
К смеси соединений (106) и (107), полученных выше на стадии C, в 80% MeOH/H2O при комнатной температуре прибавляют карбонат цезия (2 экв.). Реакционную смесь перемешивают в течение ночи. Затем смесь концентрируют, экстрагируют с помощью CH2Cl2, промывают с помощью Н2O, сушат над MgSO4, фильтруют и концентрируют досуха, получая искомое соединение (107).
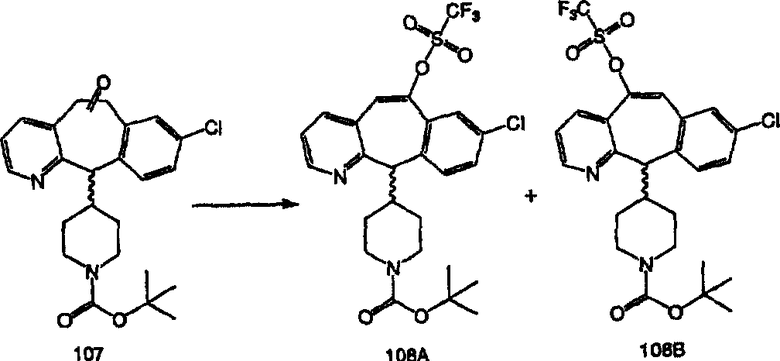
Соединение (107) вводят в реакцию с М-фенилтрифторметансульфонимидом способом, аналогичным описанному в Примере получения 7, стадия A, и получают искомые соединения (108А и 108В).
F Получение соединения (109).
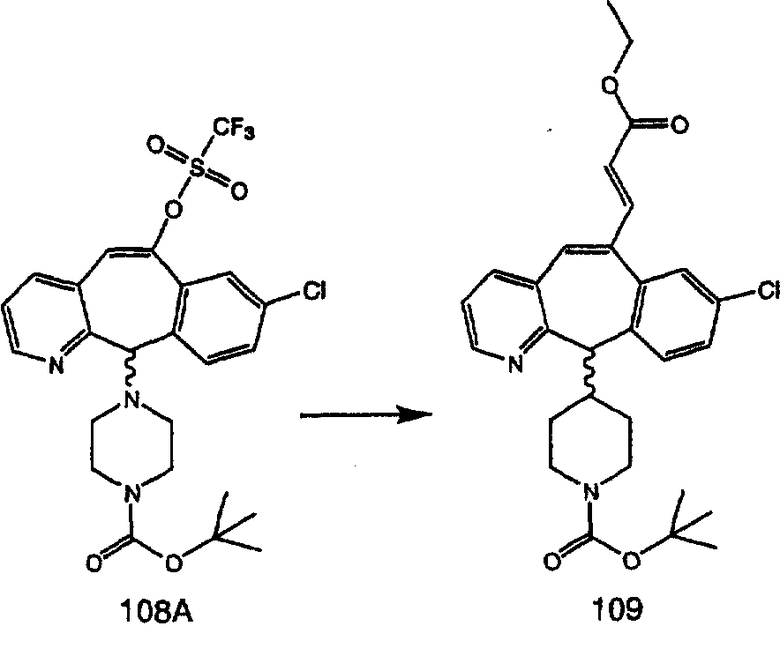
Соединение (108А) вводят в реакцию с этилакрилатом способом, аналогичным описанному в Примере получения 7, стадия B, и получают искомое соединение (109).
G Получение соединения (110).
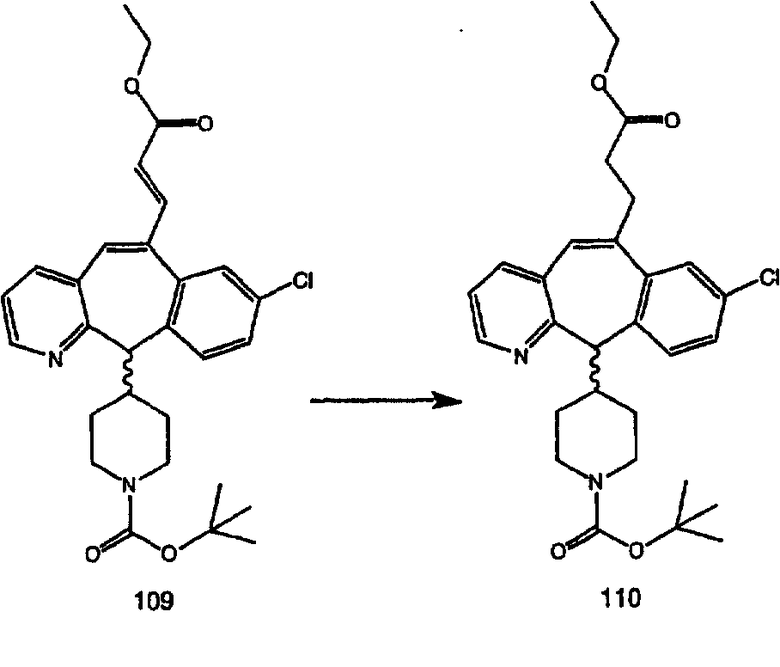
Соединение (109) вводят в реакцию с NaBH4 и CuCl способом, аналогичным описанному в Примере получения 7, стадия C, и получают искомое соединение (110).
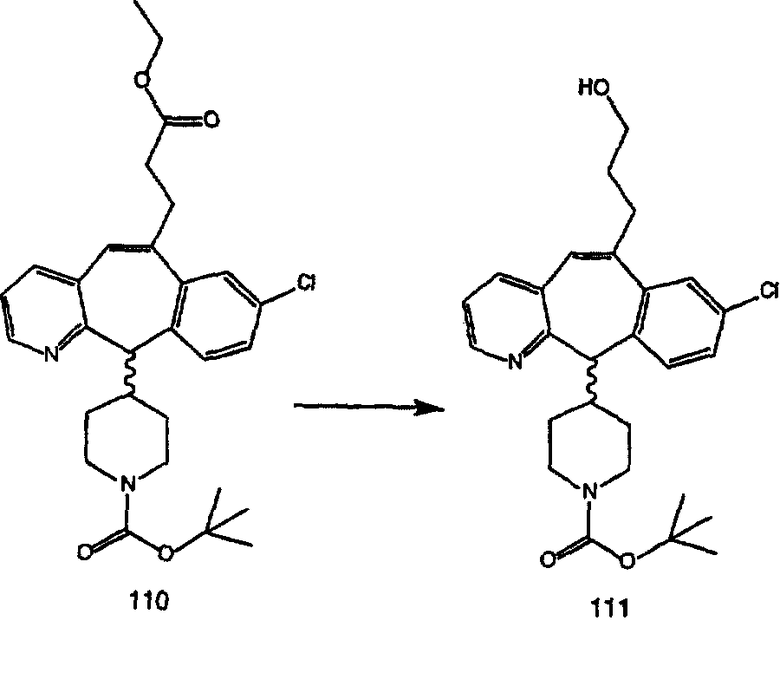
Соединение (110) растворяют в THF и затем прибавляют 1 М LiAlH4/THF (1 экв.) и 1,5 ч перемешивают при комнатной температуре. К смеси прибавляют H2O и 15% раствор NaOH, а затем экстрагируют с помощью EtOAc. Реакционную смесь промывают рассолом, сушат над MgSO4, фильтруют и концентрируют. Очистка с помощью флэш-хроматографии на колонке с диоксидом кремния с элюированием смесью 20% EtOAc/CH2Cl2 дает искомое гидроксисоединение (111).
I Получение соединения (112).
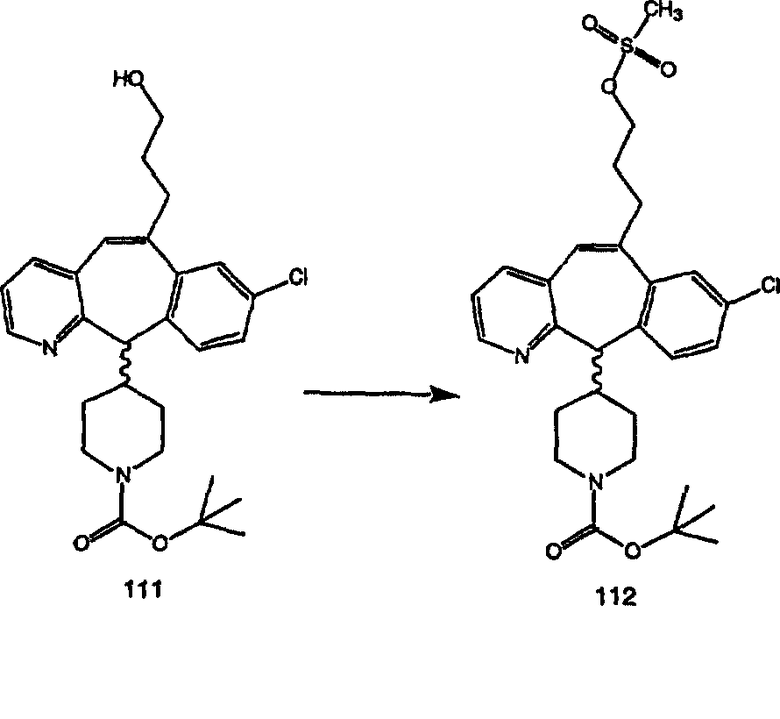
Соединение (111) вводят в реакцию с метансульфонилхлоридом способом, аналогичным описанному в Примере получения 7, стадия Е, и получают искомое соединение (112).
J Получение соединений (113), (114). (115) и (116).
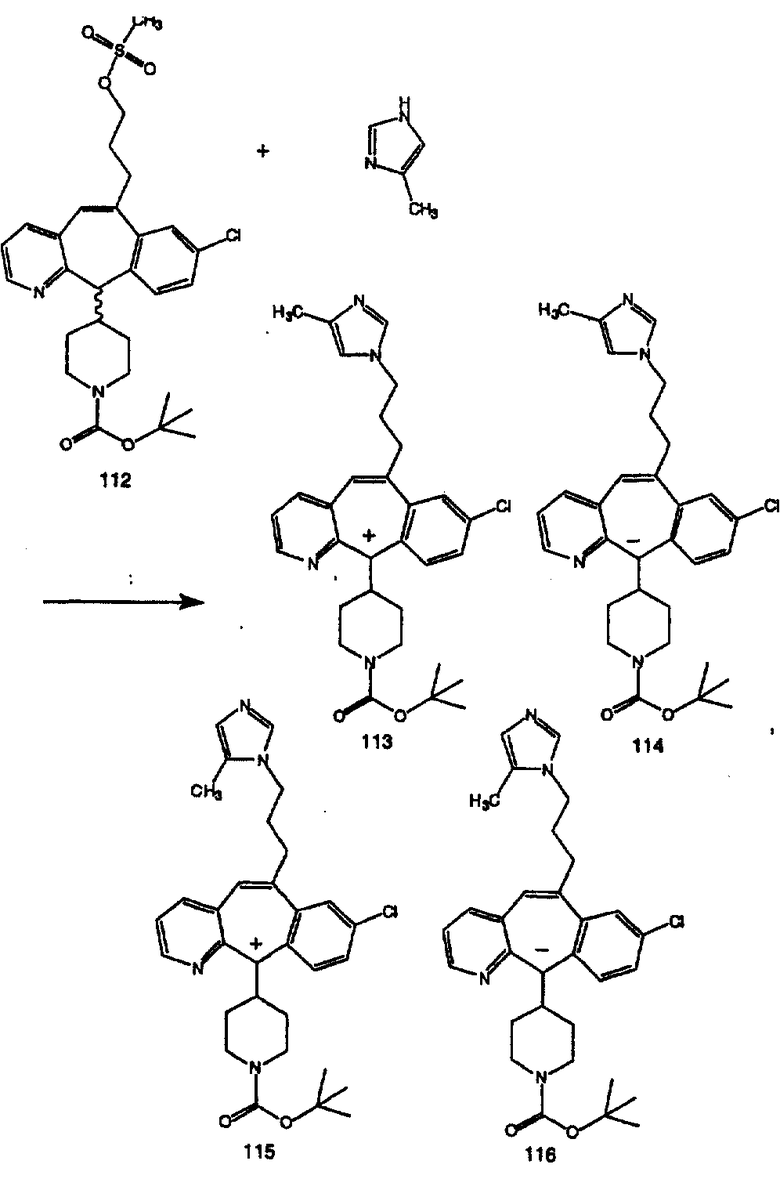
Соединение (112) вводят в реакцию способом, аналогичным описанному в Примере получения 7, стадия F, заменяя натриевую соль имидазола на 4-метилимидазол. Получают смесь (+,-)-4- и (+,-)-5-метилимидазолов. Смесь обрабатывают таким же образом, как описано в Примере 11, и получают чистые стереоизомеры (113), (114), (115) и (116).
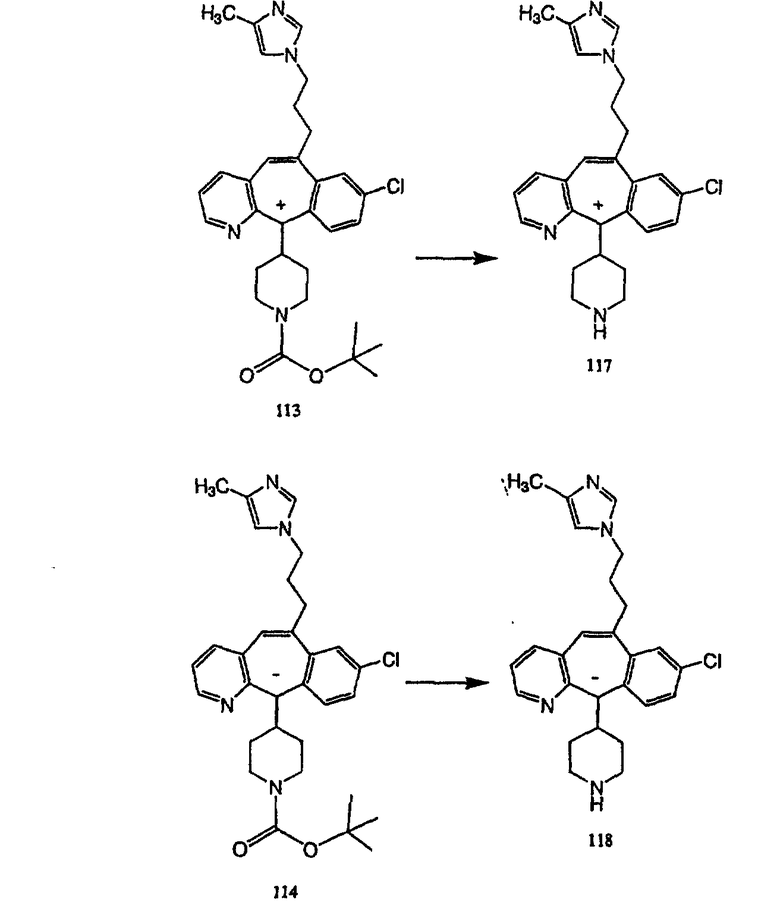
Соединения (113) и (114) гидролизуют в их свободные амины путем перемешивания в смеси HCI/диоксан в течение 4 ч. Затем смеси концентрируют досуха и получают искомые соединения (117) и (118).
ПРИМЕР ПОЛУЧЕНИЯ 10
Соединения (119) и (120).
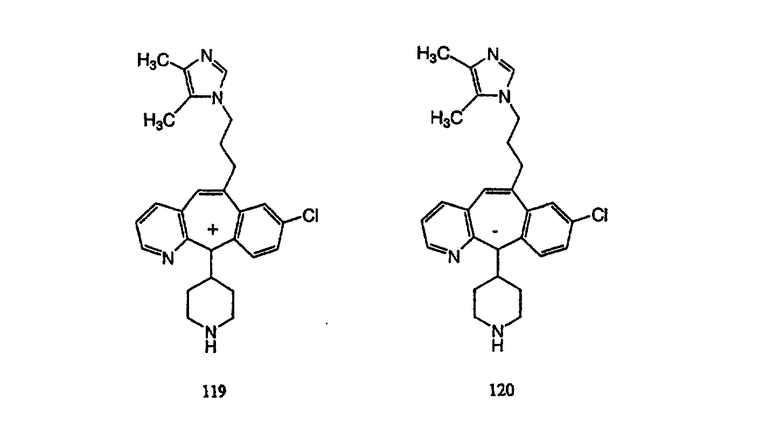
Способом, аналогичным описанному в Примере получения 9, стадии A - K, используя 4,5-диметилимидазол на стадии J, получают искомые соединения (119) и (120).
ПРИМЕРЫ 39-45
Соединения (100) или (101), полученные в Примере получения 7, вводят в реакцию таким же образом, как описано в Примере 13, используя соответствующий изоцианат или хлорформиат, и получают следующие соединения:
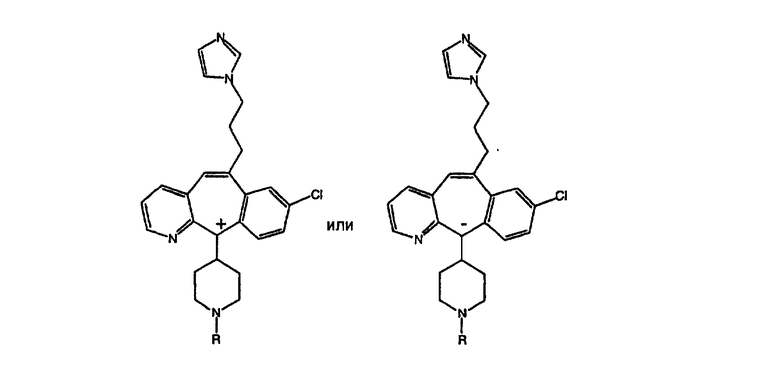
ПРИМЕРЫ 46 -51
Соединения (102) или (103), полученные в Примере получения 8, вводят в реакцию таким же образом, как описано в Примере 13, используя соответствующий изоцианат или хлорформиат, и получают следующие соединения:
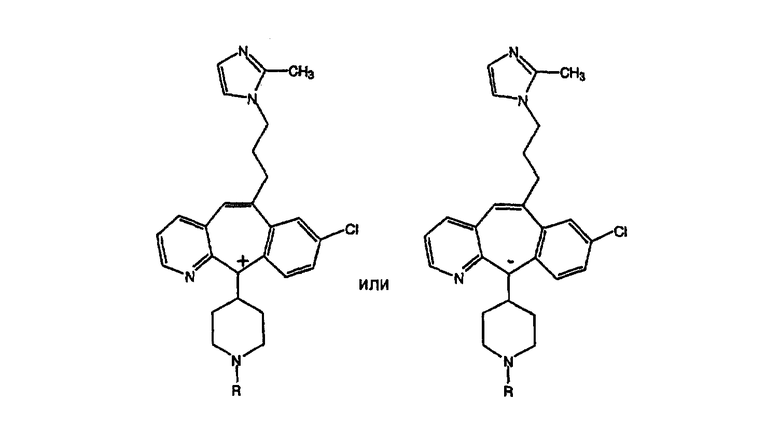
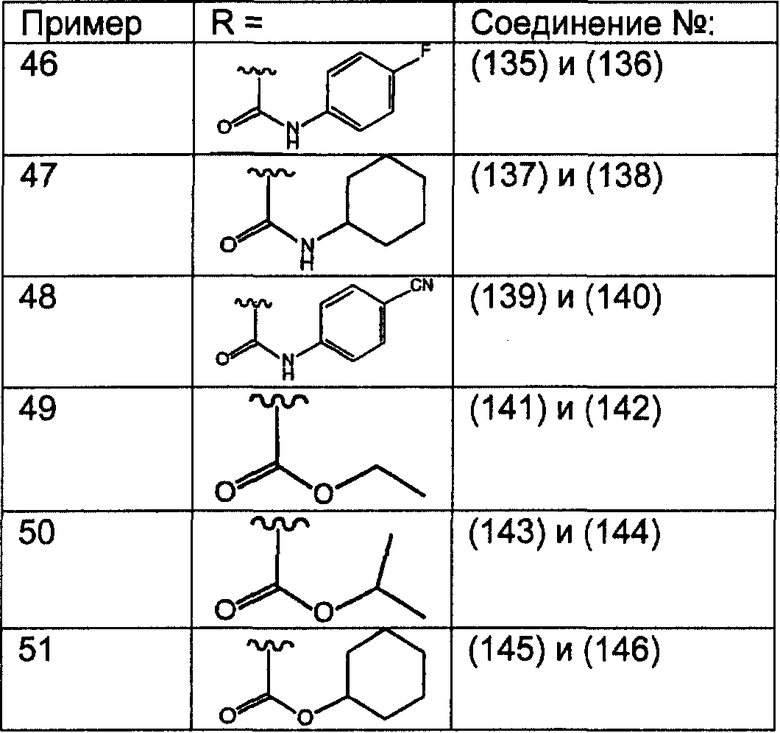
ПРИМЕРЫ 52 -59
Соединения (117) или (118), полученные в Примере получения 9, вводят в реакцию таким же образом, как описано в Примере 13, используя соответствующий изоцианат, хлорформиат или сульфонилхлорид, и получают следующие соединения:
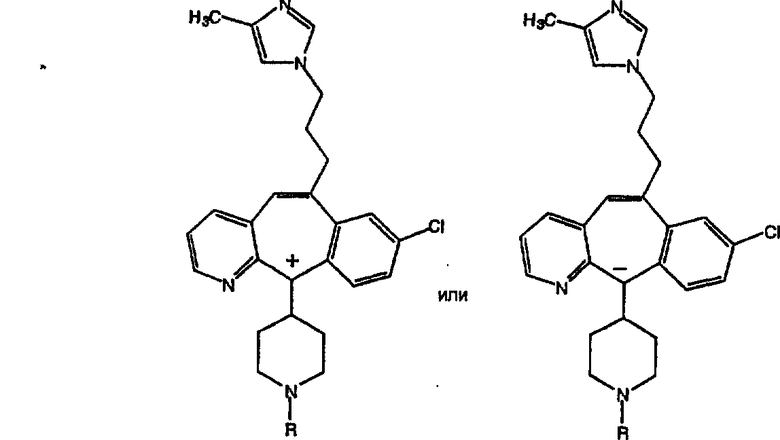
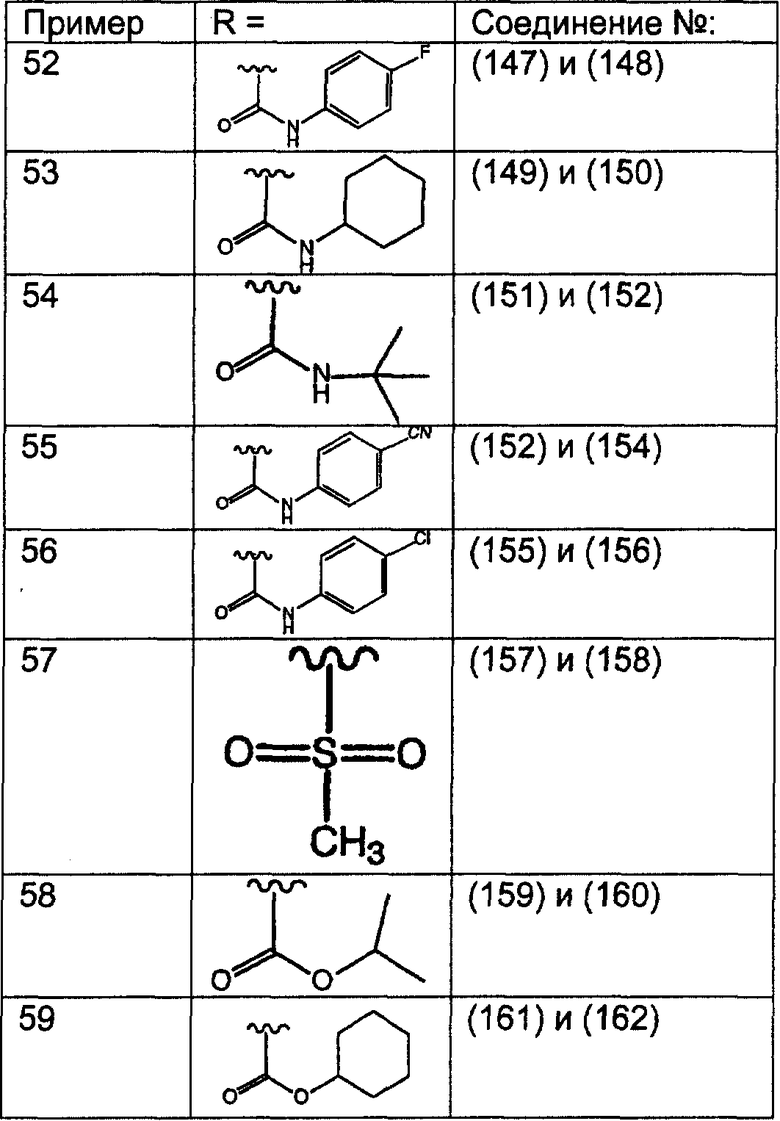
ПРИМЕРЫ 60-69
Соединения (119) или (120), полученные в Примере получения 10, вводят в реакцию таким же образом, как описано в Примере 13, используя соответствующий изоцианат, хлорформиат или сульфонилхлорид, и получают следующие соединения:
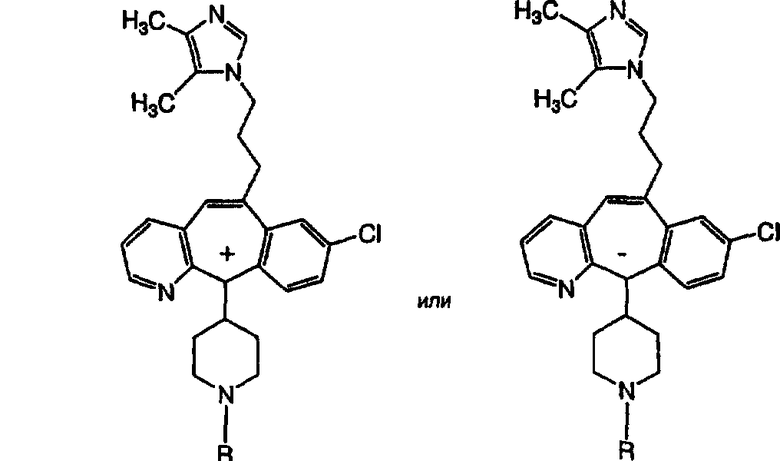
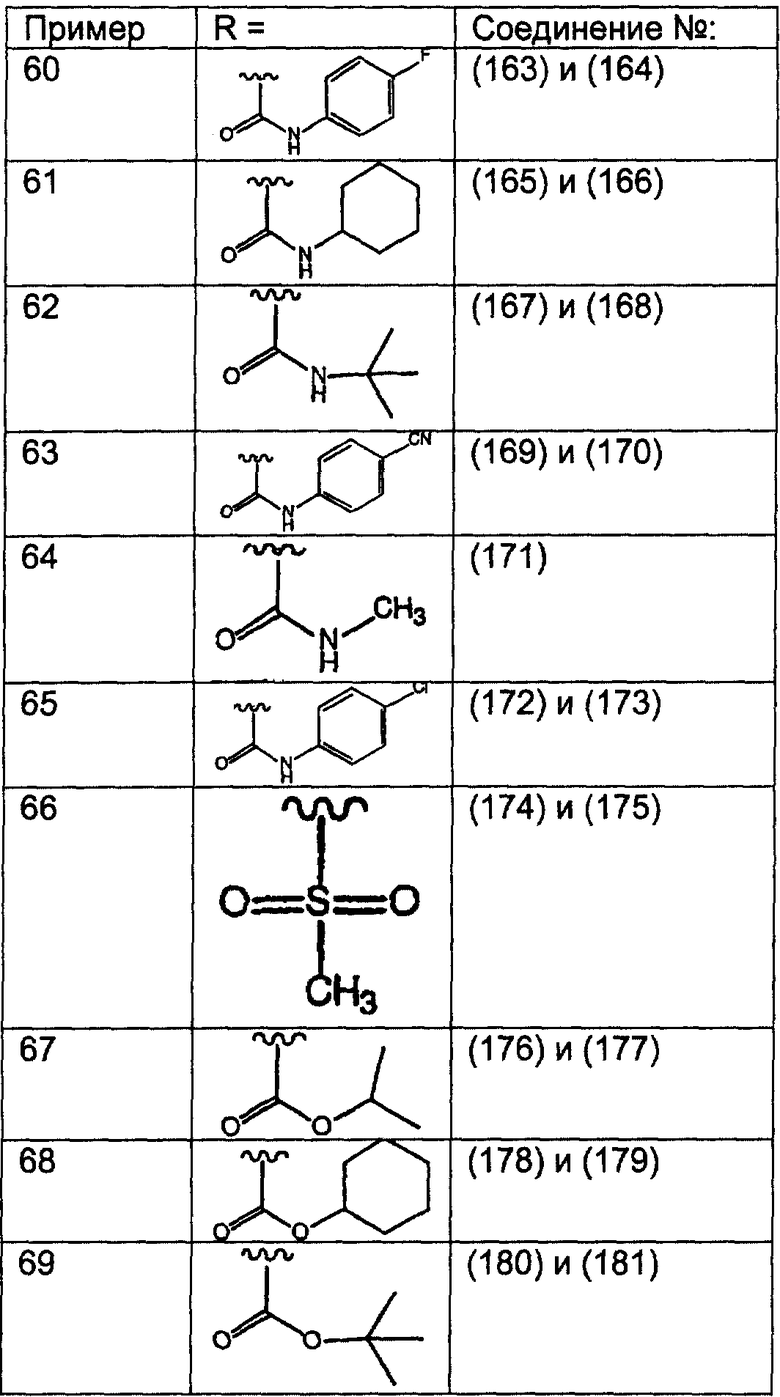
ПРИМЕР ПОЛУЧЕНИЯ 11
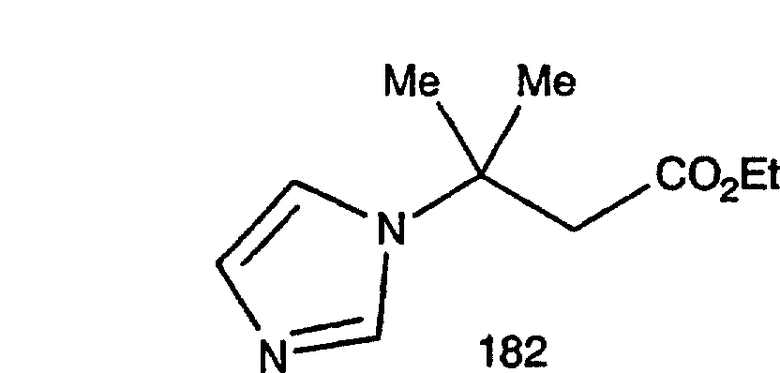
Этил-2,2-диметилакрилат (50,0 г, 2,0 экв.) при 90°C в течение 48 ч перемешивают с имидазолом (13,28 г, 200 ммоль). Полученный раствор охлаждают, разбавляйте помощью 300 мл смеси H2O - CH2Cl2 (1:1) и разделяют. Водный слой экстрагируют с помощью СН2Cl2 (2 х 75 мл) и объединенные органические слои сушат над Na2SO4 и концентрируют в вакууме. Неочищенную смесь очищают с помощью флэш-хроматографии с использованием 10% раствора МеОН в CH2Cl2 в качестве элюента и получают чистый продукт в виде прозрачного масла. МСХИ: MH+ = 197.
В Получение соединения (183).
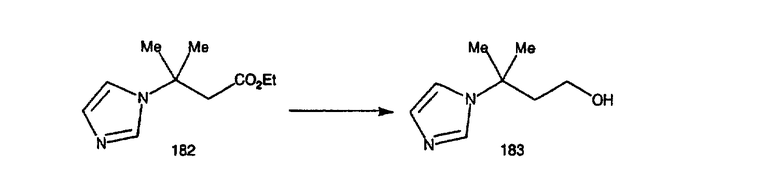
Раствор соединения, полученного в Примере получения 11, стадия A (10,0 г, 50,96 ммоль), обрабатывают с помощью LiAlH4 (51 мл, 1 М раствор в эфире, 1,0 экв.). Реакционную смесь перемешивают в течение 1 ч, а затем реакцию останавливают, по каплям прибавляя насыщенный раствор Na2SO4 (примерно 3,0 мл). Полученную взвесь сушат над Na2SO4 (твердым), разбавляют с помощью EtOAc (100 мл) и фильтруют через слой целита. Фильтрат концентрируют и получают неочищенный продукт, который используют без дополнительной очистки. МСХИ: MH+ = 155.
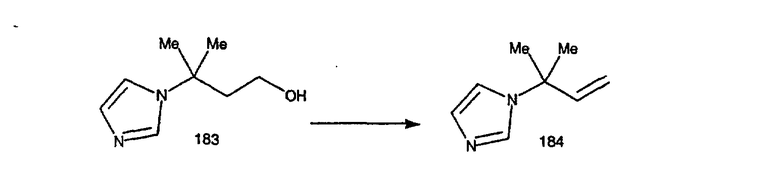
В течение 15 мин йод (3,83 г, 1,2 экв.) порциями прибавляют к раствору PH3Р (3,95 г, 1,2 экв.) и имидазола (1,02 г, 1,2 экв.) в CH2Cl2 (30 мл), а затем прибавляют раствор соединения, полученного в Примере получения 11, стадия B (3,83 г, 12,56 ммоль), в CH2Cl2 (10 мл). Полученный раствор перемешивают в течение 1 ч, а затем концентрируют в вакууме. Остаток растворяют в THF (100 мл), обрабатывают с помощью KOt-Bu (4,51 г, 3,2 экв.) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют водой (100 мл) и CH2Cl2 (100 мл), разделяют и водный слой экстрагируют с помощью CH2Cl2 (2 х 50 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Продукт очищают с помощью флэш-хроматографии с использованием в качестве элюентов неразбавленного EtOAc, затем 5% раствора МеОН в EtOAc и получают бледно-желтое масло (184). МСХИ: MH+=137.
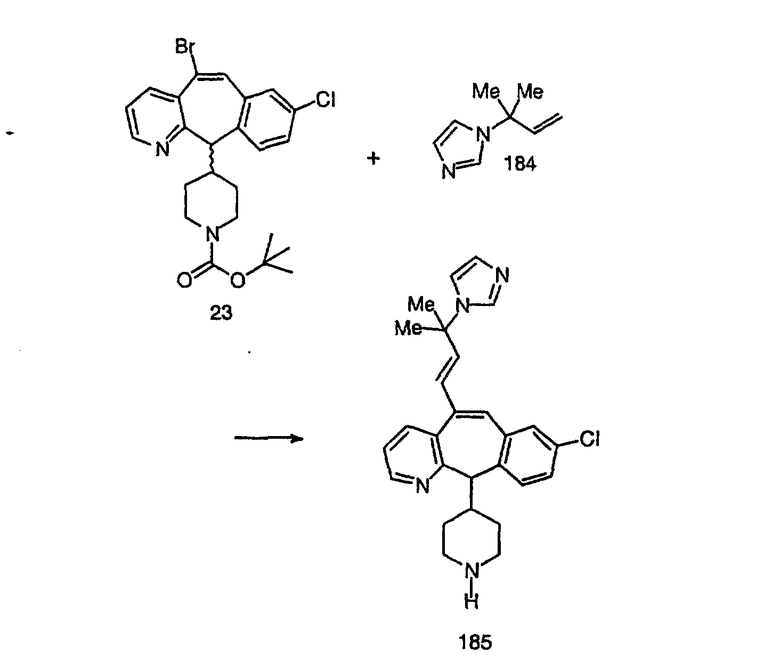
Pd(OAc)2 (0,023 г, 10 мол.%) прибавляют к раствору соединения (184), полученного в Примере получения 11, стадия С (0,30 г, 2,0 экв.), соединения (23) (0,50 г, 1,02 ммоль), Bu4NBr (0,66 г, 2,0 экв.), TEA (2,84 мл, 2,0 экв.) и K2СО3 (0,70 г, 5,0 экв.) в DMF (10 мл). Полученный раствор 48 ч нагревают при 100°C, охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток разбавляют водой (50 мл) и CH2Cl2 (50 мл), разделяют и водный слой экстрагируют с помощью CH2Cl2 (2 х 25 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают с помощью флэш-хроматографии на колонке с использованием 8% раствора МеОН в СН2Cl2 в качестве элюента и получают смесь состава 4:1 соединения (184) и продукта сочетания (185). Эту смесь (0,27 г) в течение 1,5 ч перемешивают в СН2Cl2:TFA (7,0 мл, 5:2). Неочищенный продукт концентрируют при пониженном давлении, нейтрализуют с помощью NaOH (1 н. раствор) и экстрагируют с помощью СН2Cl2 (3 х 20 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный остаток очищают с помощью флэш-хроматографии с использованием 15% (10% NH4OH в МеОН) раствора в СН2Cl2 в качестве элюента и получают искомое соединение (185) в виде желтовато-коричневого твердого вещества. МСХИ: МН+ = 445.
ПРИМЕР 70
Получение соединения (186).
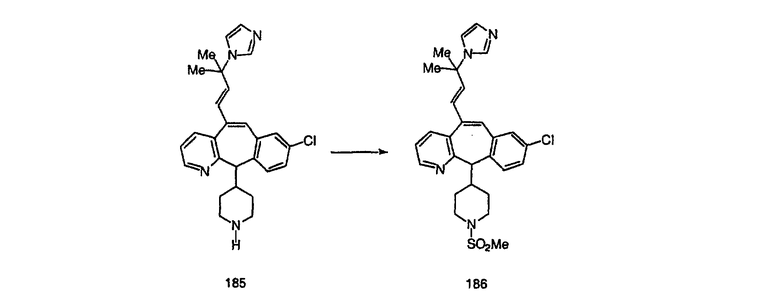
Метансульфонилхлорид (0,005 мл, 1,3 экв.) прибавляют к раствору соединения (185), полученного в Примере получения 11, стадия D (0,02 г, 0,045 ммоль), и TEA (0,010 мл, 1,5 экв.) в СН2Cl2 (1 мл). Полученный раствор 12 ч перемешивают при комнатной температуре и разбавляют насыщенным раствором NaHCO3 (5 мл), разделяют и водный слой экстрагируют с помощью СН2Cl2 (3х10 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют в вакууме. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 8% (10% NH4ОН в МеОН) раствора в CH2Cl2 в качестве элюента и получают искомое соединение (186) в виде желтовато-коричневого твердого вещества, т. пл. 124-129°С. ЖХМС: MH+ = 523.
ПРИМЕР 71
Получение соединения (187).
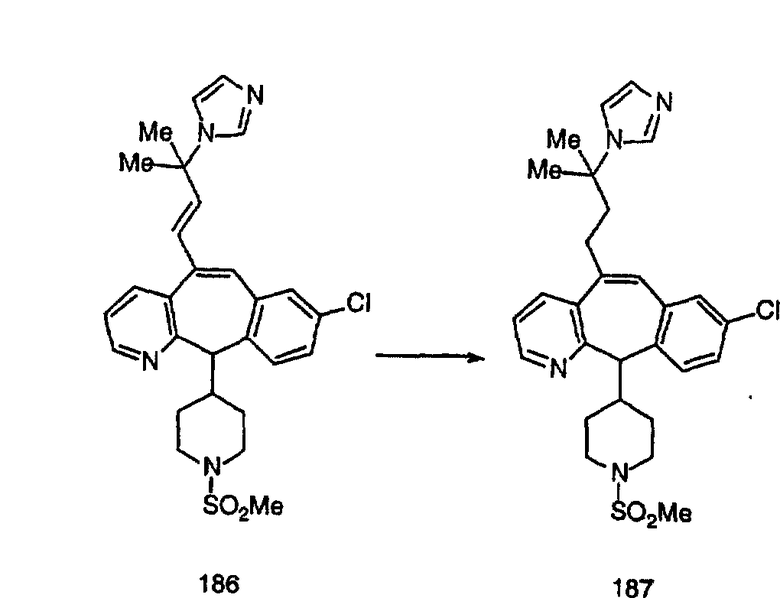
pTosNHNH2 (0,085 г, 3 экв.) прибавляют к раствору соединения (186), полученного в Примере 70 (0,08 г, 0,0153 ммоль), и DBU (0,11 мл, 5,0 экв.) в толуоле (5 мл) и полученный раствор кипятят с обратным холодильником. После этого каждые 2 ч в течение 6 ч раствор охлаждают и прибавляют дополнительное количество pTosNHNH2 (3 экв.) и раствор кипятят с обратным холодильником. После кипячения с обратным холодильником в течение 2 ч после прибавления последней порции раствор охлаждают, разбавляют с помощью CH2Cl2 (25 мл) и промывают насыщенным раствором NaHCO3 (3 х 20 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенную реакционную смесь очищают с помощью флэш-хроматографии с использованием 5% (10% NKtOH в МеОН) раствора в CH2Cl2 в качестве элюента и получают искомое соединение (187) в виде желтовато-коричневого твердого вещества, т. пл. 112-116°С. ЖХМС: MH+ = 525.
ПРИМЕР ПОЛУЧЕНИЯ 12
А Получение соединения (188).
Описанное в литературе соединение, 1Н-имидозол-4-карбальдегид, тритилируют в соответствии с описанной в литературе методикой: Kelley, et al.: J. Med. Chem. 20(5), (1977), 721, и получают искомое соединение (188).
В Получение соединения (189).
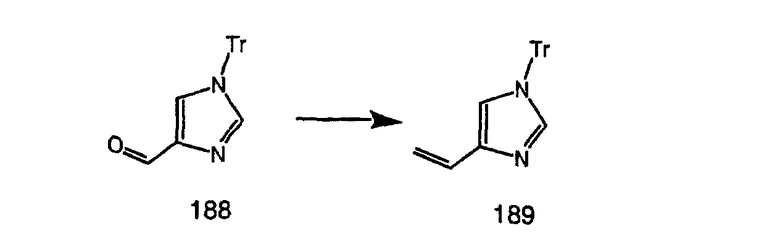
nBuLi (2,00 мл, 2,2 экв., 1,7 М раствор в гексанах) по каплям прибавляют к PH3РСН3Br (1,4 г, 2,3 экв.) в THF (10 мл). Полученный оранжевый раствор 30 мин перемешивают при комнатной температуре, а затем охлаждают до -78°С и прибавляют содержащий тритильную защитную группу 1(3)Н-имидазол-4-карбальдегид (0,50 г, 1,48 ммоль) в THF (7,0 мл). Полученный раствор медленно нагревают до комнатной температуры и перемешивают в течение ночи. Реакцию останавливают путем прибавления воды (20 мл) и экстрагируют с помощью CH2Cl2 (3 х 20 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют в вакууме. Неочищенный остаток очищают с помощью флэш-хроматографии с использованием 45% раствора гексанов в EtOAc в качестве элюента и получают искомое соединение (189) в виде белого твердого вещества.
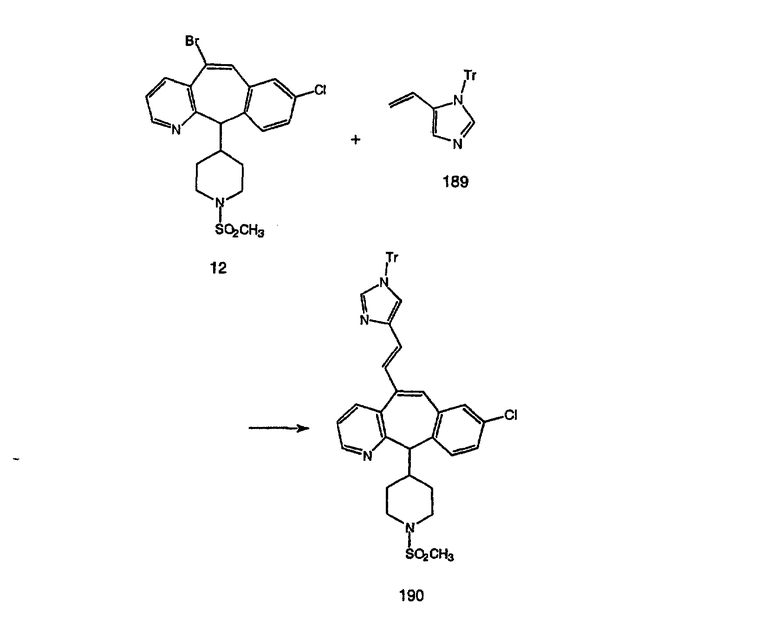
Pd(OAc)2 (0,021 г, 0,10 экв.) прибавляют к раствору соединения (12), полученного в Примере получения 2, стадия В (0,44 г, 0,95 ммоль), соединения (189), полученного в Примере получения 12, стадия В (0,32 г, 1,0 экв.), Bu4NBr (0,61 г, 2,0 экв.) и К2СО3 (0,66 г, 5,0 экв.) в DMF (8,0 мл). Полученный раствор нагревают при 100°C в течение ночи, охлаждают и концентрируют при пониженном давлении. Остаток разбавляют водой (50 мл) и CH2Cl2 (50 мл), разделяют и водный слой экстрагируют с помощью CH2Cl2 (2 х 50 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют в вакууме. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 100% СН2Cl2 в качестве элюента. ЖХМС: 723 (МН+).
ПРИМЕР 72
Получение соединения (191).
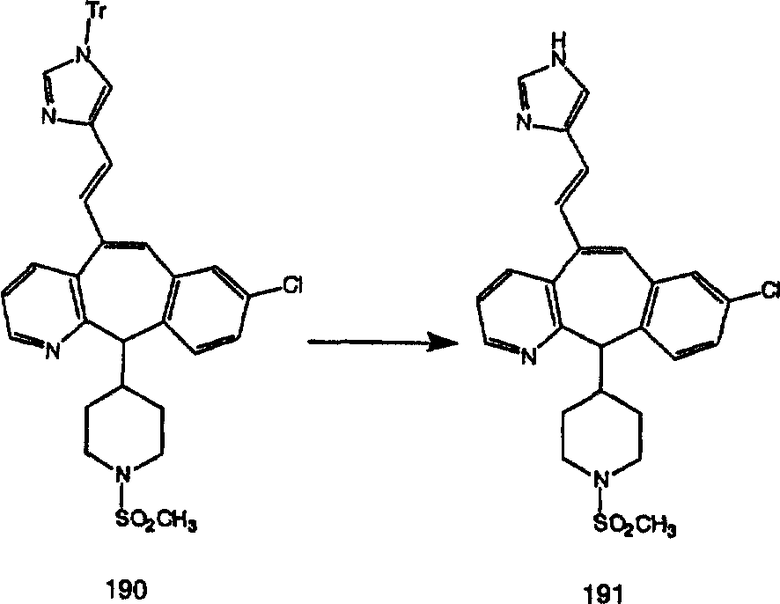
К раствору соединения, полученного в Примере получения 12, стадия C (1,43 г, 1,97 ммоль), в воде (70 мл) прибавляют АсОН (70 мл). Полученный раствор 2 ч кипятят с обратным холодильником, охлаждают до комнатной температуры и нейтрализуют, по каплям прибавляя 50% (мас./мас.) раствор NaOH. Затем раствор экстрагируют с помощью CH2Cl2 (3 х 200 мл) и объединенные органические слои сушат над Na2SO4 и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 10% (10% NH40H в МеОН) раствора в CH2Cl2 в качестве элюента, т.пл. 190°С. ЖХМС: МН+ = 483.
ПРИМЕР 73
Получение соединений (192) и (193).
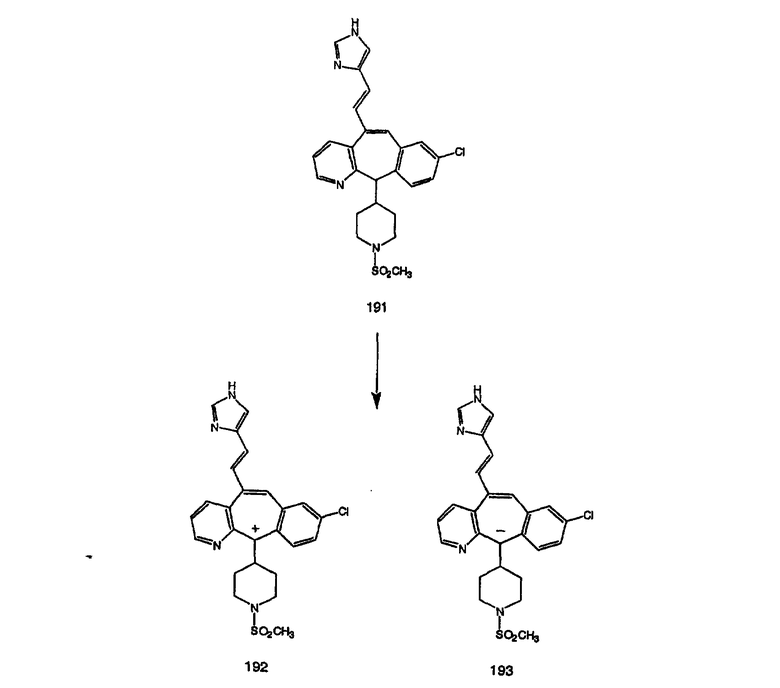
Соединение (191), полученное в Примере 72, разделяют на индивидуальные (+)- и (-)-энантиомеры с помощью препаративной ВЭЖХ с использованием колонки ChiralPak AD, элюируя с помощью смеси 70:30 гексаны:iPrOH, содержащей 0,2% диэтиламина.
Соединение (192): МС ББА: MH+=481; т. пл. = 109-112°c; [α]D 20 =+398° (2,0 мг в 2,0 мл МеОН).
Соединение (193): МС ББА: MH+ = 481; т. пл. =126-129°C; [α]D 20 = -367° (2,0 мг в 2,0 мл МеОН).
ПРИМЕР 74
Получение соединения (194).
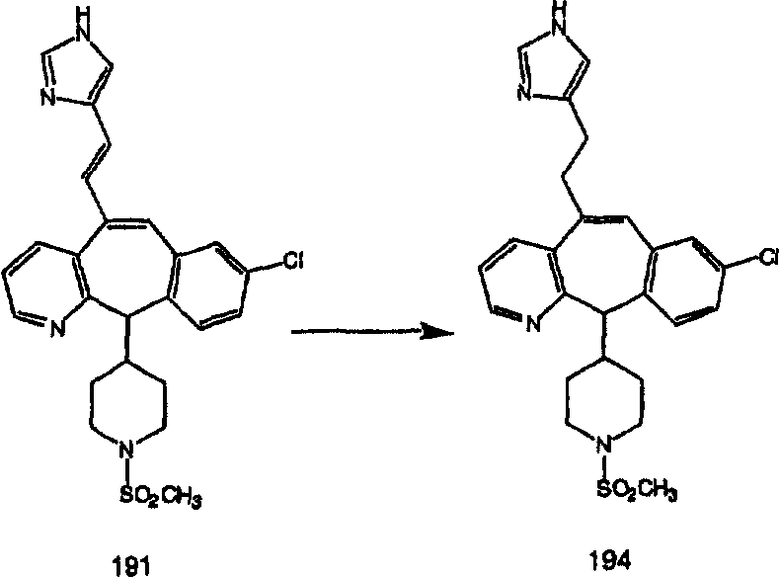
Соединение (191), полученное в Примере 72, растворяют в толуоле (50 мл) и прибавляют DBU (0,26 мл, 5,0 экв.) и pTosNHNH2 (0,33 г, 3,3 экв.). Полученный раствор 2,5 ч кипятят с обратным холодильником, а затем охлаждают до комнатной температуры и прибавляют дополнительное количество pTosNHNH2 (0,33 г, 3,3 экв.). Реакционную смесь кипятят с обратным холодильником еще 2 ч и охлаждают до комнатной температуры. Полученный раствор разбавляют насыщенным раствором NaHCO3 (100 мл) и экстрагируют с помощью CH2Cl2 (3 х 100 мл). Объединенные органические слои промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 5% (10% NH4OH в МеОН) раствора в CH2Cl2 в качестве элюента и получают чистый продукт (194), т. пл. 158-162°С. ЖХМС: MH+ = 483.
ПРИМЕР 75
Получение соединений (195) и (196).
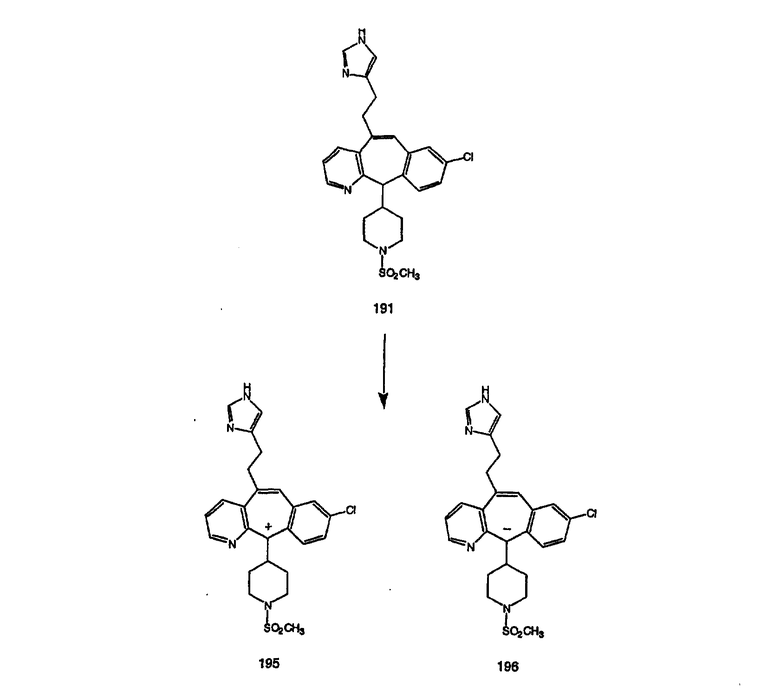
Способом, аналогичным описанному в Примере 73 выше, разделяют следующие энантиомеры:
Соединение (195): ЖХМС: МН+ = 483; т. пл. = 129-131°C; [α]D 20 = +134° (2,0 мг в 2,0 мл МеОН).
Соединение (196): ЖХМС: МН+ = 483; т. пл. =125-126°C; [α]D 20 = -105° (2,0 мг в 2,0 мл МеОН).
ПРИМЕР ПОЛУЧЕНИЯ 13
Получение соединения (197).
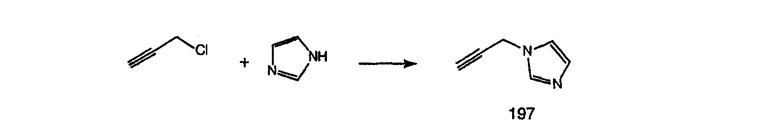
Имидазол (2,50 г, 36,72 ммоль) и основной оксид алюминия (15 г) смешивают и встряхивают 15 мин, а затем прибавляют пропаргилхлорид (2,66 мл, 1,0 экв.). Полученную смесь перемешивают 84 ч и суспендируют в EtOAc. Взвесь фильтруют и фильтрат промывают с помощью Н2O и рассола и сушат над Na2SO4. Раствор фильтруют и концентрируют при пониженном давлении, получая прозрачное масло.
ПРИМЕР 76
Получение соединения (198).
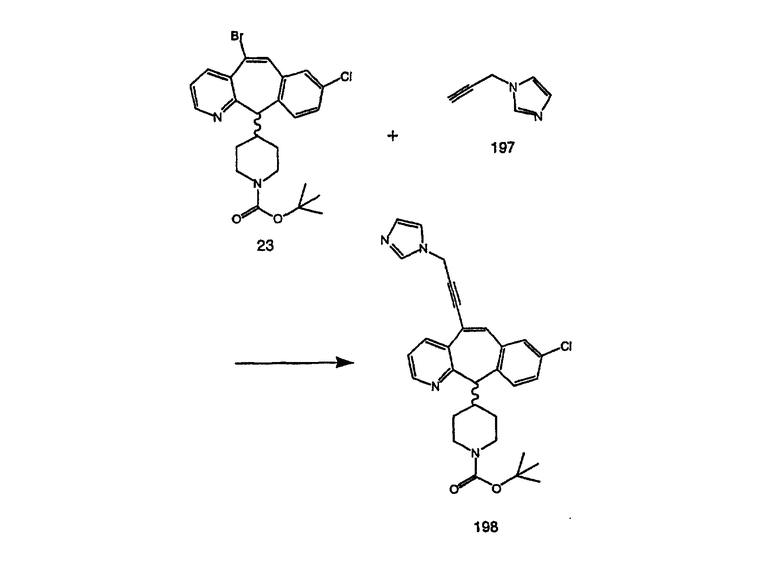
Раствор соединения (23) (0,50 г, 1,02 ммоль) и соединения (197), полученного в Примере получения 13 (0,22 г, 2,0 экв.), в TEA (3,0 мл) и пиридине (0,5 мл) дезоксигенируют 15 мин, а затем прибавляют PdCl2(PPh3)2 (0,018 г, 2,5 мол.%) и Cul (0,002 г, 1,0 мол.%). Полученный раствор нагревают в течение 48 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют с помощью Н2О и экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 8% раствора МеОН в СН2Cl2 в качестве элюента, т. пл. 109-112°С. ЖХМС: 515(MH+).
ПРИМЕР ПОЛУЧЕНИЯ 14
А Получение соединения (199).
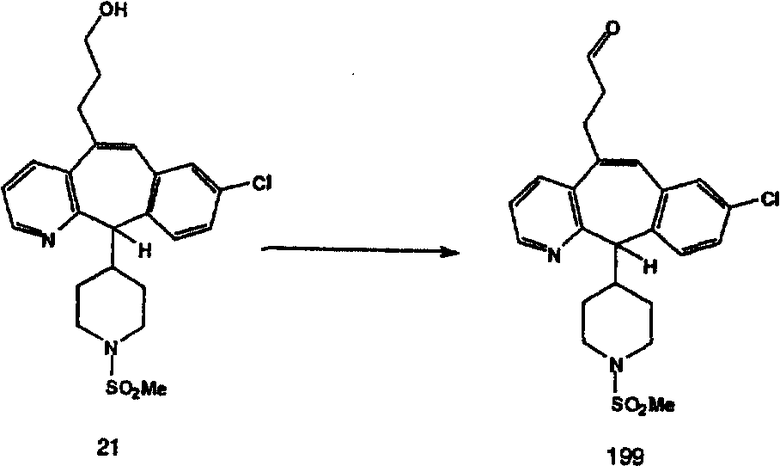
Соединение (21), полученное в Примере получения 3, стадия C (2,83 г, 6,37 ммоль), растворяют в 120 мл дихлорметана и 0,16 мл деионизированной воды. При температуре окружающей среды прибавляют перйодинан Десса-Мартина (3,85 г, 9 ммоль) в виде твердого вещества и реакционную смесь перемешивают 4 ч. Затем прибавляют 20% раствор Na2S2O3 (50 мл) и перемешивают 15 мин. Слои разделяют и дихлорметановый слой промывают насыщенным раствором NaHCO3, сушат над сульфатом магния, фильтруют и выпаривают, получая искомый продукт (199). МС ББА: 445 (MH+).
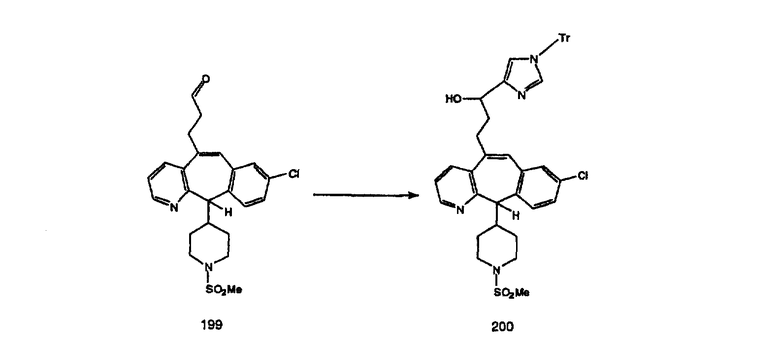
4-Йод-1-тритилимидазол (полученный по описанной в литературе методике: Kirk, Kenneth L. J. Heterocycl. Chem.; EN; 22; 1985; 57 -59) (0,48 г, 1,1 ммоль) растворяют в 5 мл дихлорметана в атмосфере сухого азота. Прибавляют этилмагнийбромид (0,36 мл) и реакционную смесь перемешивают. Через 30 мин соединение (199) (0,44 г, 1 ммоль) растворяют в 5 мл дихлорметана и при перемешивании прибавляют к реакционной смеси. После перемешивания в течение 4 ч при температуре окружающей среды смесь промывают насыщенным раствором хлорида аммония, сушат над сульфатом магния, фильтруют и выпаривают, получая твердый остаток. Продукт хроматографируют на флэш-колонке с силикагелем с использованием этилацетата в качестве элюента и получают искомое соединение (200). МС ББА: 756 (MH+).
ПРИМЕР 77
Получение соединения (201).
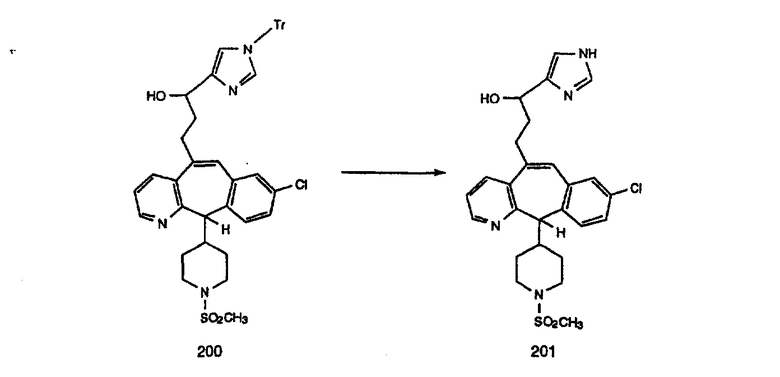
Соединение (200) (0,6 г) растворяют в 10 мл трифторуксусной кислоты и перемешивают при температуре окружающей среды. Через 7 ч реакционную смесь выпаривают досуха в вакууме и хроматографируют на силикагеле с использованием смеси 5% 2 н. раствор аммиака в метаноле/дихлорметан и получают искомое соединение (201). МС ББА: 514 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 15
А Получение соединения (202).
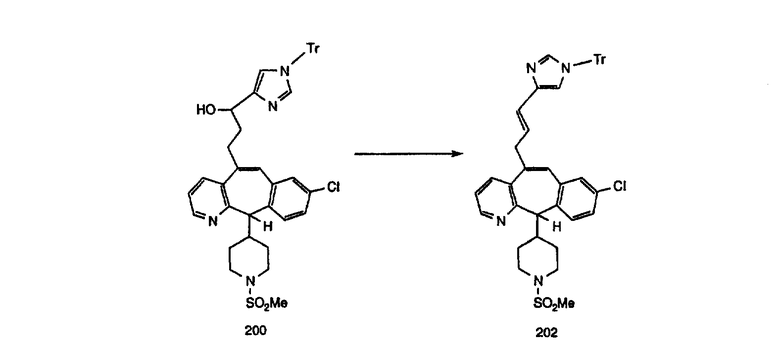
Соединение (200) (0,5 г, 0,66 ммоль) растворяют в 5 мл дихлорметана. Прибавляют триэтиламин (0,14 мл, 0,99 ммоль) и метансульфонилхлорид (0,062 мл, 0,79 ммоль) и реакционную смесь перемешивают 18 ч. Реакционную смесь прибавляют к рассолу и трижды экстрагируют дихлорметаном. Сушат над сульфатом магния, фильтруют и концентрируют досуха в вакууме, получая остаток, который хроматографируют на силикагеле с использованием этилацетата в качестве элюента и получают искомое соединение (202). МС ББА: 537 (МН+).
В Получение соединения (203).
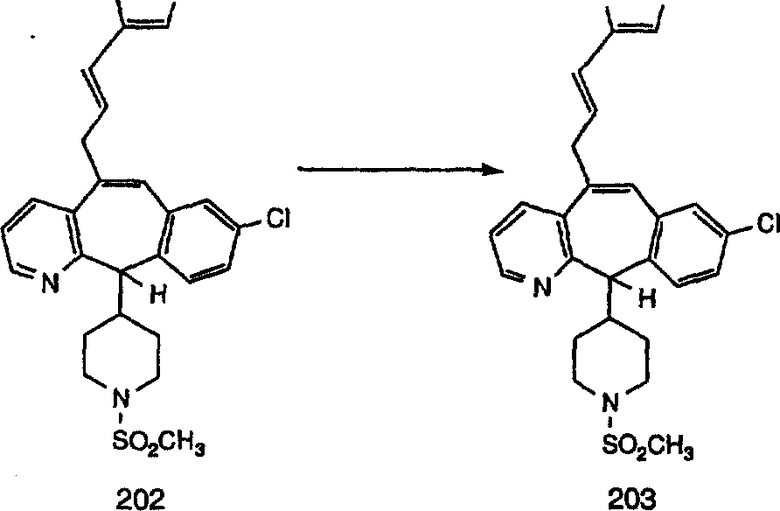
У соединения (202) тритильную группу удаляют таким же образом, как в Примере 77, и получают искомое соединение (203). МС ББА: 495 (МН+).
ПРИМЕР 78
Получение соединений (205. 206).
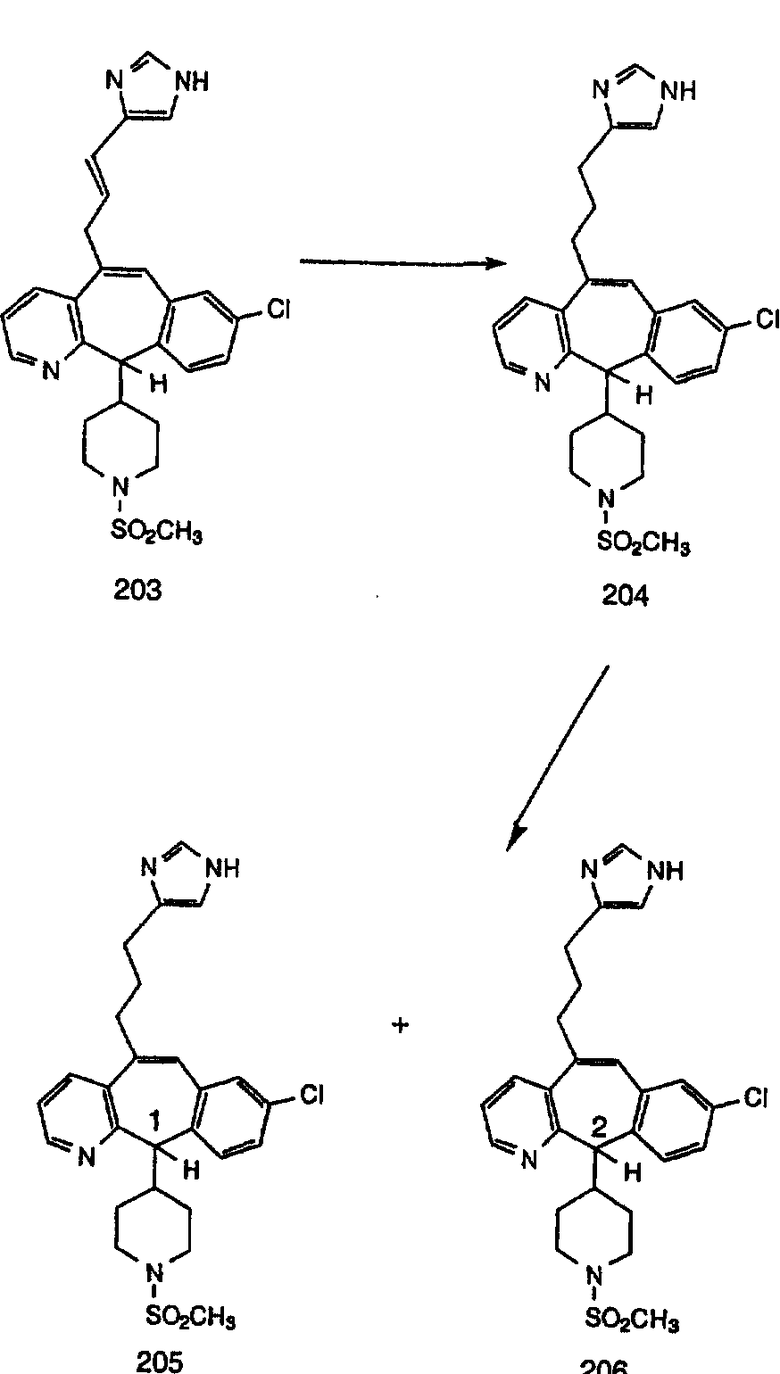
Соединение (203) (77 мг) в течение 24 ч гидрируют над PtO2 в этаноле в атмосфере водорода. После отфильтровывания катализатора и последующего выпаривания этанола и хроматографирования на колонке Chiral Technologies® AD для ВЭЖХ получают искомый продукт в виде двух чистых энантиомеров (205) и (206). МС ББА: 497 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 16
Получение соединения (207).
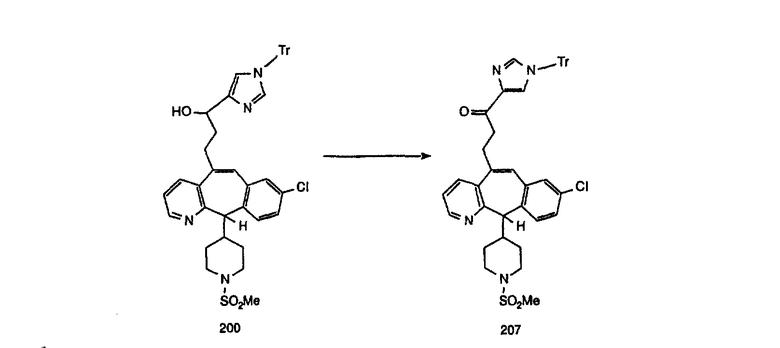
Соединение (200) (0,15 г, 0,198 ммоль) растворяют в 4 мл дихлорметана и 5 мкл деионизированной воды. Прибавляют перйодинан Десса-Мартина (0,12 г, 0,3 ммоль) и реакционную смесь перемешивают 4 ч. Прибавляют 5 мл 20% раствора Na2S2O3 и реакционную смесь перемешивают еще 15 мин. Слои разделяют и дихлорметановый слой промывают насыщенным раствором NaHCO3, сушат над сульфатом магния, фильтруют и выпаривают, получая искомое соединение (207). МС ББА: 753 (MH+).
ПРИМЕР 79
Получение соединения (208).
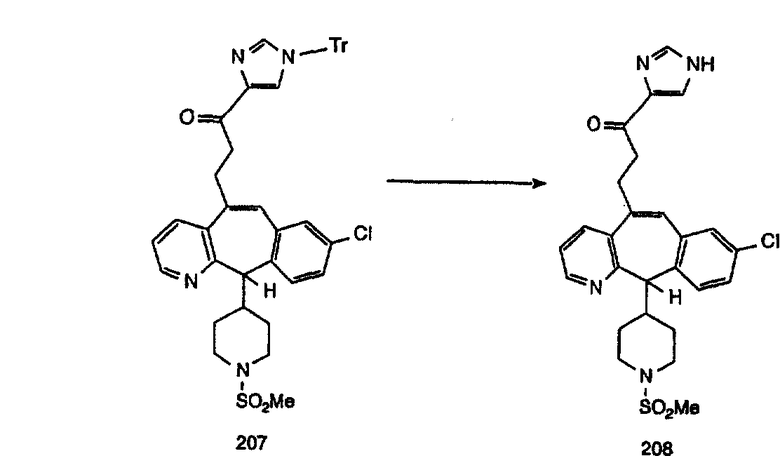
У соединения (207) тритильную группу удаляют таким же образом, как в Примере 77, и получают искомое соединение (208). МС ББА: 511 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 17
Получение соединения (209).
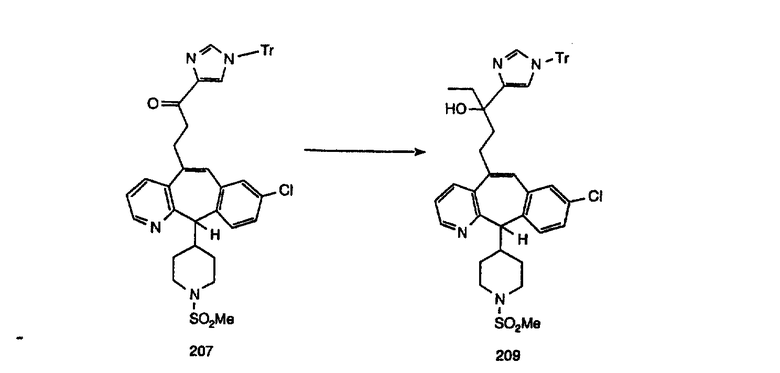
Соединение (207) (0,15 г, 0,2 ммоль) растворяют в 5 мл тетрагидрофурана. При температуре окружающей среды прибавляют этилмагнийбромид (0,1 мл, 3 М раствор в эфире) и перемешивают в атмосфере сухого азота. Через 2 ч прибавляют еще одну порцию этилмагнийбромида (0,1 мл, 3 М раствор в эфире). Через 4 ч реакционную смесь промывают насыщенным раствором хлорида аммония, сушат над сульфатом магния и выпаривают, получая искомое соединение (209). Продукт дополнительно очищают с помощью флэш-хроматографии на колонке с диоксидом кремния с элюированием смесью 50% этилацетат/гексаны. МС ББА: 783 (МН+).
ПРИМЕР 80
Получение соединения (210).
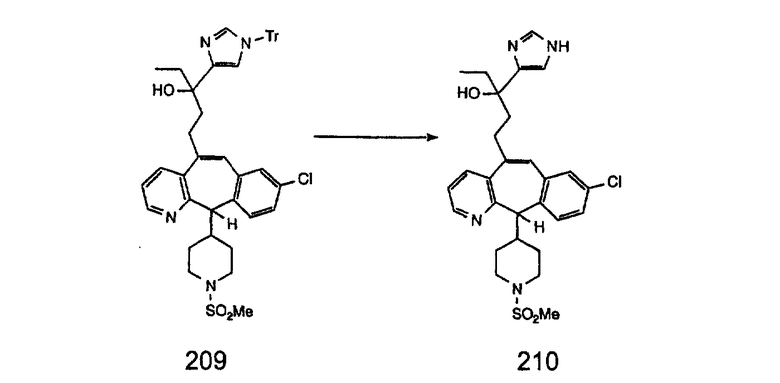
У соединения (209) тритильную группу удаляют таким же образом, как в Примере 77, и получают искомое соединение (210). МС ББА: 541 (МН+).
ПРИМЕР ПОЛУЧЕНИЯ 18
А Получение соединения (212).
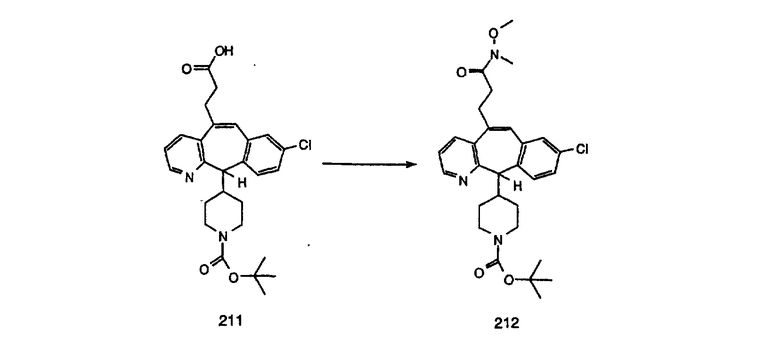
Соединение (211) (14 г, 29 ммоль), полученное гидролизом с помощью NaOH соединения (20), полученного в Примере получения 3, стадия B, растворяют в 400 мл DMF. Прибавляют 1-3(-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (8,3 г, 43 ммоль), 1-гидроксибензотриазол (5,9 г, 43 ммоль), триэтиламин (40 мл) и N,O-диметилгидроксиламингидрохлорид (3,8 г, 40 ммоль) и реакционную смесь перемешивают в атмосфере сухого азота при комнатной температуре. Через 24 часа реакционную смесь выливают в рассол и продукт дважды экстрагируют этилацетатом. После сушки над сульфатом магния, фильтрования и хроматографирования на силикагеле с использованием смеси 10% этилацетат/гексаны получают искомое соединение (212).
В Получение соединения (213).
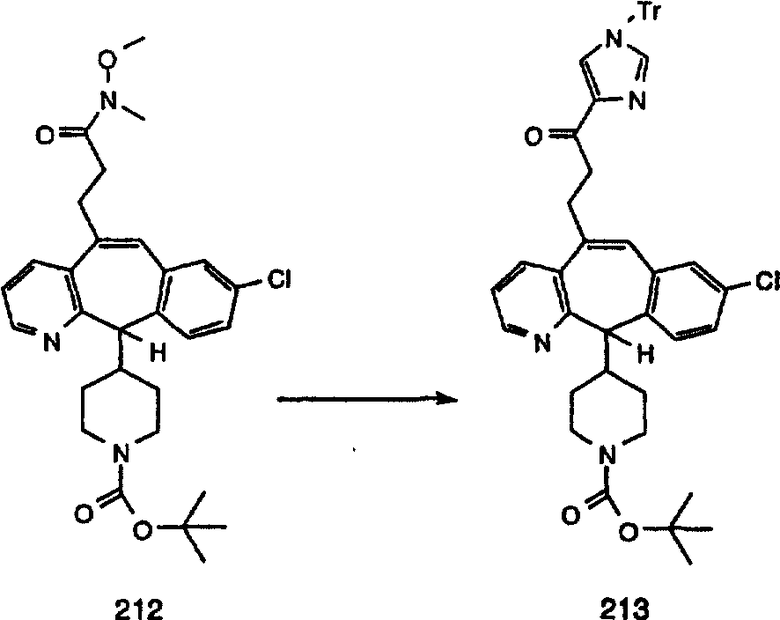
Соединение (212) (0,53 г, 1,01 ммоль) обрабатывают так, как в Примере получения 14, стадия B, и после хроматографирования на силикагеле получают искомое соединение (213).
ПРИМЕР 81
Получение соединений (214) и (215).
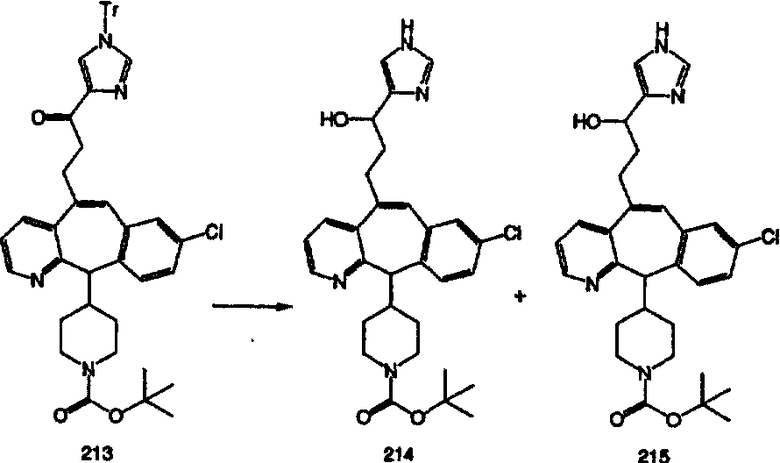
Соединение (213) (300 мг, 0,387 ммоль) растворяют в метаноле и при перемешивании порциями прибавляют борогидрид натрия (50 мг). Через 1 ч смесь прибавляют к 1 н. раствору HCl, а затем прибавляют 1 н. раствор NaOH и экстрагируют этилацетатом, получая неочищенный продукт, который в течение 5 ч обрабатывают неразбавленной трифторуксусной кислотой и выпаривают досуха. Смесь растворяют в метаноле и в течение 1 ч проводят реакцию с ди-трет-бутилдикарбонатом (0,2 г), поддерживая значение рН равным 10 с помощью 1 н. раствора NaOH. Затем в течение 15 мин смесь обрабатывают 2 н. метанольным раствором аммиака, а после этого выпаривают растворитель и проводят хроматографирование на силикагеле. Последующее разделение изомеров проводят с помощью ВЭЖХ на колонке Chiral Technologies® AD и получают чистые изомеры (214) и (215). МС ББА М + 1 = 535.
ПРИМЕР 82
Получение соединения (216).
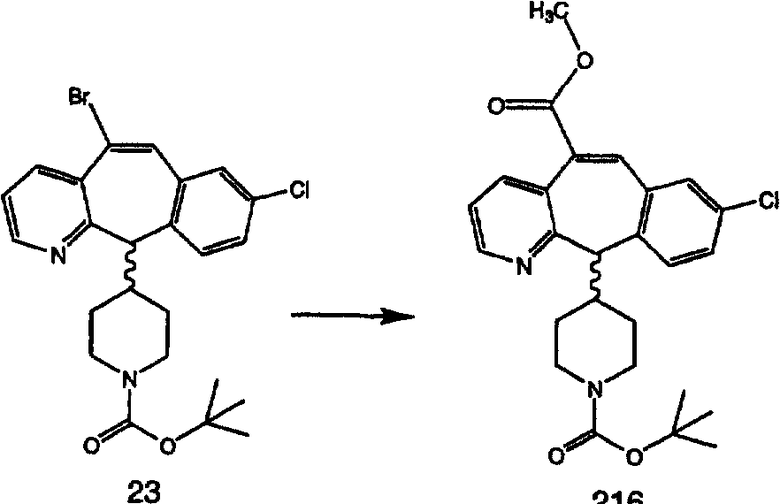
Соединение (23), полученное в Примере получения 4, стадия A (25,47 г, 52 ммоль), растворяют в 300 мл сухого толуола и 39,5 мл метанола. Прибавляют хлорид палладия (0,92 г), трифенилфосфин (6,887 г) и DBU (10,5 мл) и реакционную смесь переносят в автоклав. Автоклав продувают монооксидом углерода и создают в нем давление монооксида углерода, равное 100 фунтов/дюйм2, и смесь 5 ч перемешивают при 80°С. Реакционную смесь охлаждают в бане со льдом и 3-4 раза продувают азотом. Реакционную смесь переносят в делительную воронку и прибавляют 500 мл этилацетата. Смесь трижды промывают водой, сушат над сульфатом магния, фильтруют и выпаривают в вакууме досуха, получая темно-коричневую смолу. Смолу очищают с помощью хроматографии на силикагеле с использованием смеси 12,5-25% этилацетат/гексаны и получают 12,58 г чистого искомого продукта (216), МС ББА: 469 (MH+), и 9,16 г смеси двух соединений.
ПРИМЕР ПОЛУЧЕНИЯ 19
Получение соединения (217).
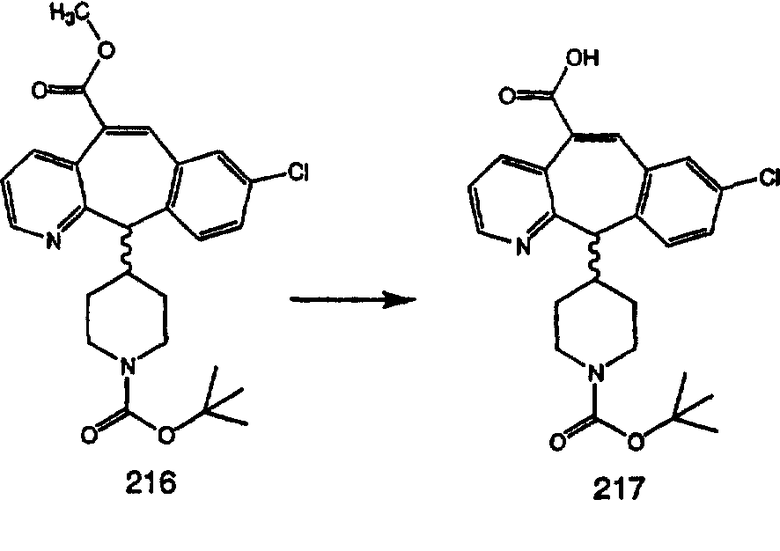
Соединение (216), полученное в Примере 82 (5,16 г, 11 ммоль), растворяют в метаноле (150 мл). Прибавляют 10% раствор гидроксида лития (2,9 мл) вместе с диоксаном (50 мл) и реакционную смесь перемешивают 4 ч. Прибавляют дополнительную порцию 10% раствора гидроксида лития (5,7 мл) и реакционную смесь перемешивают 18 ч. Реакционную смесь концентрируют до небольшого объема и разбавляют с помощью 50 мл воды. С помощью 10% раствора лимонной кислоты смесь подкисляют до рН = 3 и продукт экстрагируют дихлорметаном, получая искомое соединение (217). МС ББА: 455 (MH+).
ПРИМЕР ПОЛУЧЕНИЯ 20
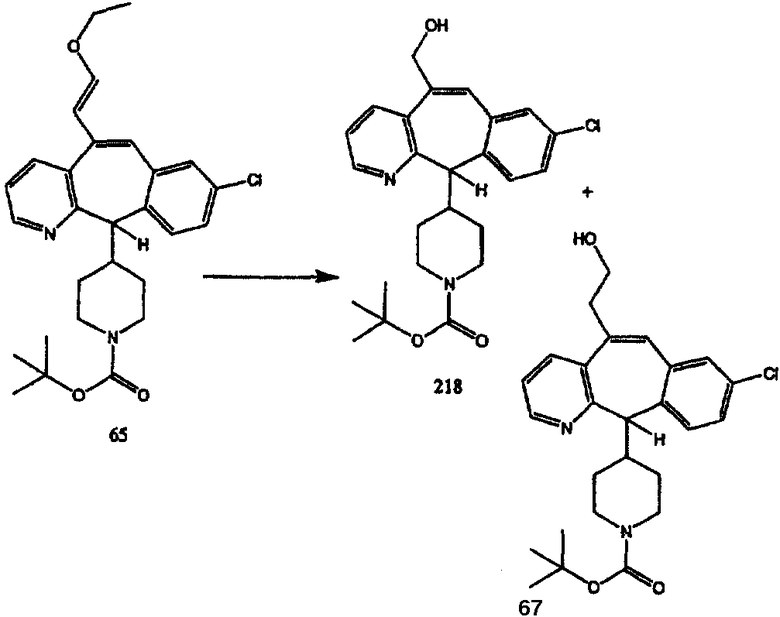
Соединение (65), полученное в Примере получения 6, стадия B, выдерживают примерно две недели при комнатной температуре, после чего с помощью ЯМР в неочищенном веществе обнаруживается некоторое количество альдегида. Затем это вещество обрабатывают так, как в Примере получения 6, стадии C и D, и получают смесь соединений (218) и (67). Неочищенную смесь разделяют с помощью флэш-хроматографии на колонке с диоксидом кремния с элюированием смесями 1:1-3:1 этилацетат:гексаны и получают искомое соединение (218).
В Получение соединения (219).
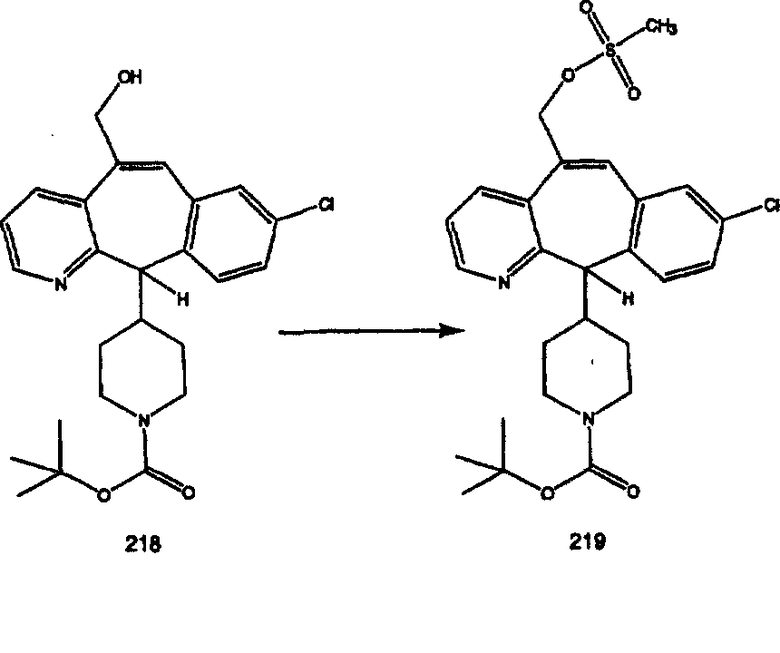
Соединение (218), полученное выше на стадии A, смешивают с триэтиламином (64,4 мл, 0,462 ммоль) в CH2Cl2 (4 мл), обрабатывают метилсульфонилхлоридом (17,93 мл, 0,231 ммоль) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют с помощью CH2Cl2 (70 мл), реакцию останавливают рассолом (25 мл) и экстрагируют. Органический слой сушат над MgSO4, фильтруют и концентрируют, получая не совсем белое твердое вещество (219) (93 мг, 100%).
С Получение соединения (220).
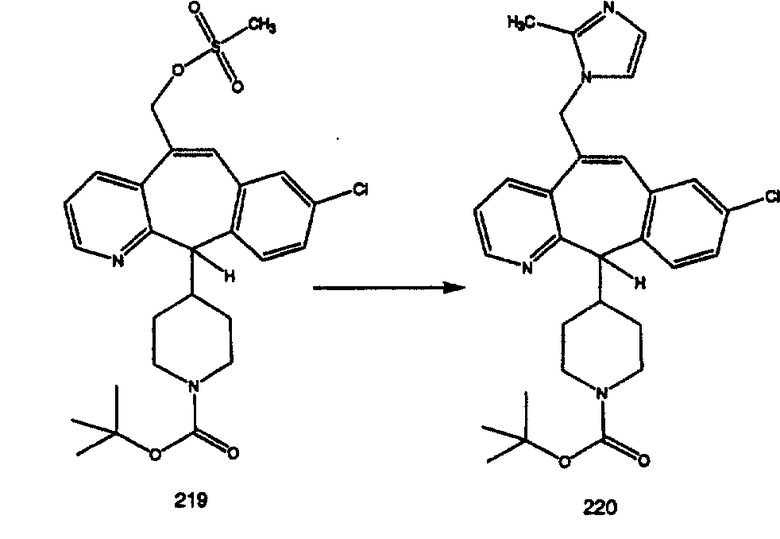
Соединение (219), полученное выше на стадии B, растворяют в DMF. К этому раствору прибавляют предварительно прореагировавший раствор 2-метилимидазола (145,27 мг, 1,734 ммоль) и NaH (60%) (69,4 мг, 1,734 ммоль) в DMF. Реакционную смесь 2 ч перемешивают при комнатной температуре. DMF удаляют и остаток растворяют в CH2Cl2, реакцию останавливают насыщенным водным раствором NaHCO3 и экстрагируют с помощью 2 х 100 мл CH2Cl2. Органические слои объединяют и очищают с помощью пластинок для препаративной ТСХ, получая не совсем белое твердое вещество (220).
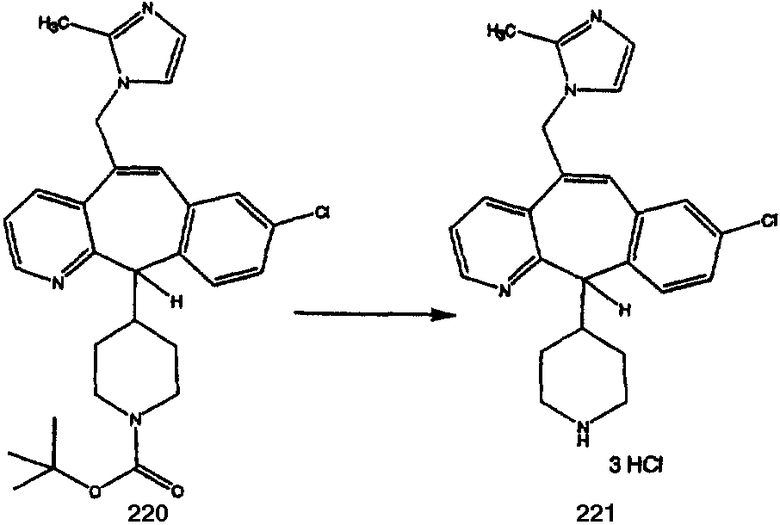
Соединение (220), полученное выше на стадии C, растворяют в 1,4-диоксане (3 мл). Затем к этому раствору прибавляют 4 М раствор HCI в диоксане (5 мл) и реакционную смесь 3 ч перемешивают при комнатной температуре. Затем смесь концентрируют и сушат в высоком вакууме, получая гидрохлорид (221) в виде не совсем белого твердого вещества.
ПРИМЕР 83
Получение соединения (222).
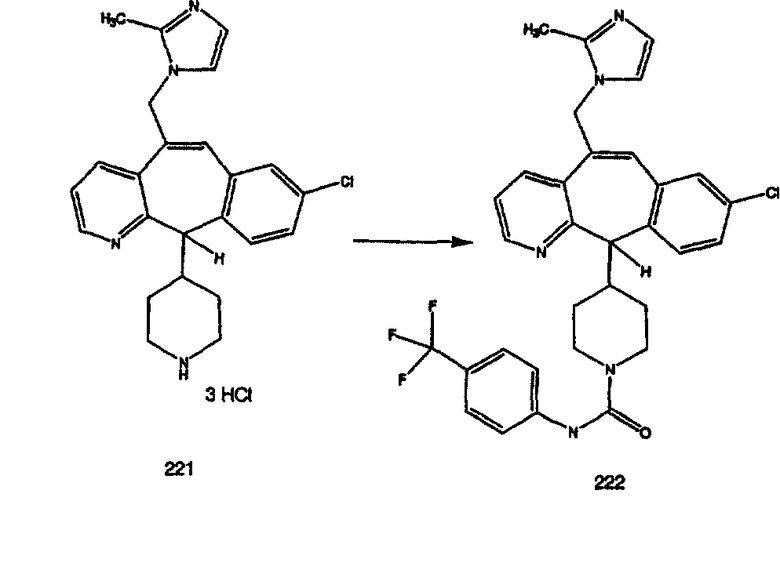
К раствору соединения (221), полученного в Примере получения 20, стадия D (51 мг, 0,126 ммоль), в триэтиламина (61,47 мл, 0,441 ммоль) в CH2Cl2 (2 мл) при 0°C прибавляют 4-трифторметилфенилизоцианат (20,26 мл, 0,139 ммоль). Реакционную смесь 2-3 ч перемешивают в атмосфере N2. CH2Cl2 и избыток триэтиламина удаляют в вакууме, и полученный продукт очищают с помощью препаративной тонкослойной хроматографии, элюируя смесью 98:2, СН2Cl2/(насыщенный раствор NH3 в МеОН), получая искомое соединение (222) в виде белого твердого вещества.
ПРИМЕР ПОЛУЧЕНИЯ 21
А Получение пиперидильного промежуточного продукта.
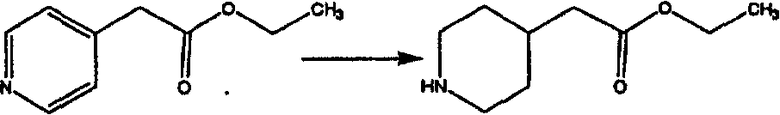
Коммерчески доступный этил-4-пиридилацетат (4,5 г, 27,2 ммоль), EtOH (70 мл) и 10% палладий на древесном угле (катализатор) при комнатной температуре в течение 94 ч встряхивают под давлением водорода, равном 55 фунтов/дюйм2. Смесь фильтруют через целит и остаток на фильтре промывают с помощью EtOH (4 х 40 мл). Фильтрат концентрируют и остаток очищают с помощью флэш-хроматографии на колонке с диоксидом кремния, элюируя смесью 3% (10% NH4OH: MeOH)/CH2Cl2.
В Получение этилового эфира (1-карбамоилпиперидин-4-ил)-уксусной кислоты.
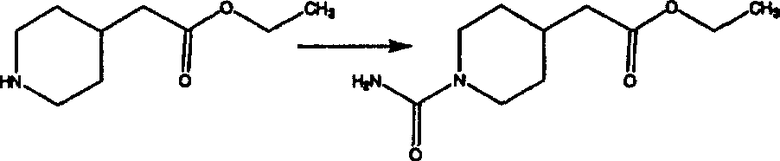
4-Пиридилуксусную кислоту (2,362 г), полученную выше на стадии A, растворяют в СН2Cl2 (118 мл). К этому раствору прибавляют триметилсилилизоцианат (27,87 мл). Реакционную смесь перемешивают 67 ч, а затем разбавляют с помощью CH2Cl2 (700 мл) и промывают насыщенным водным раствором NaHCO3 (150 мл). Водный раствор экстрагируют с помощью 2 х 200 мл СН2Cl2. Органические слои объединяют, сушат над MgSO4, фильтруют и концентрируют. Неочищенный продукт очищают с помощью флэш-хроматографии на колонке с диоксидом кремния, элюируя смесью 2% (10% NH4OH: MeOH)/CH2Cl2.
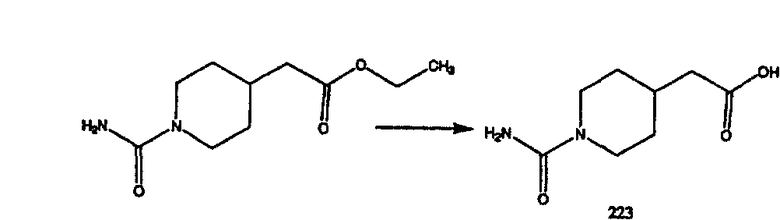
С Продукт, полученный выше на стадии B (40,63 мг, 0,1896 ммоль), растворяют в EtOH (2 мл) и CH2Cl2 (2 мл) и обрабатывают 1 М раствором LiOH (0,5 мл, 0,455 ммоль). Реакционную смесь нагревают до 50°C и перемешивают в течение 5 ч. Реакционную смесь охлаждают до комнатной температуры и обрабатывают с помощью 1 н. раствора HCI (0,57 мл, 0,531 ммоль) и 5 мин перемешивают. Полученную смесь концентрируют и сушат в высоком вакууме в течение 4 дней и получают искомое соединение (223) в виде белого твердого вещества.
ПРИМЕР 84
Получение соединения (224).
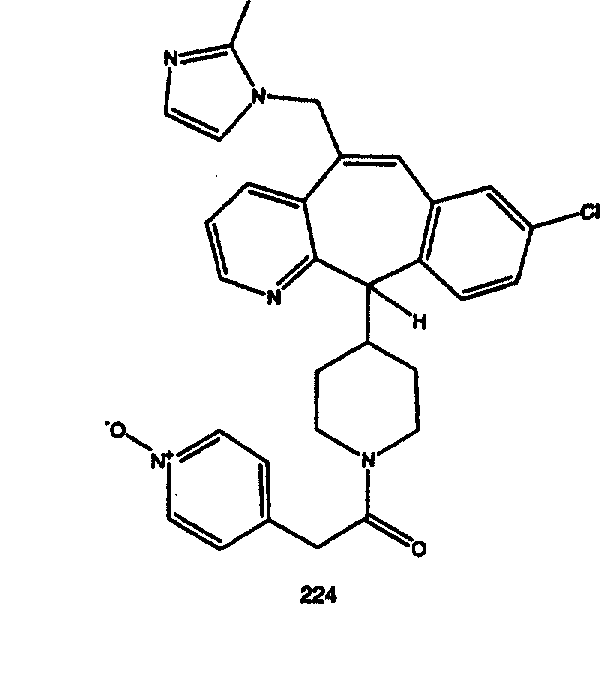
К раствору соединения (221), полученного в Примере получения 20, стадия D (51 мг, 0,126 ммоль), 4-метилморфолина (69,3 мл, 0,630 ммоль), DEC (31,44 мг, 0,164 ммоль) и НОВТ (22,2 мг, 0,164 ммоль) в DMF (2 мл) прибавляют l-N-оксид 4-пиридилуксусной кислоты (раскрыт в патенте США US 5719148; 2/17/98). Реакционную смесь 3 ч перемешивают при комнатной температуре. Реакционную смесь разбавляют с помощью CH2Cl2 и дважды промывают насыщенным водным раствором NaHCO3. Органические слои объединяют, концентрируют и очищают с помощью препаративной тонкослойной хроматографии, элюируя с помощью смеси 95:5, CH2Cl2:насыщенный раствор NH3 в МеОН, получая искомое соединение (224) в виде белого твердого вещества.
ПРИМЕР 85 Получение соединения (225).
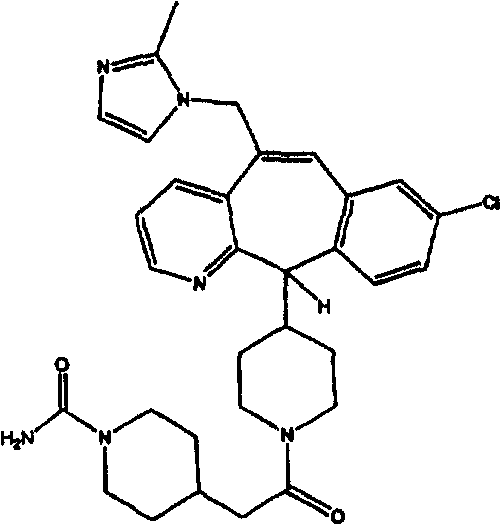
Соединение (221), полученное в Примере получения 20, стадия D (51 мг, 0,126 ммоль), смешивают с соединением (223), полученным в Примере получения 21, стадия C, и вводят в реакцию таким же образом, как в Примере 84, получая искомое соединение (225) в виде белого твердого вещества. (145-155°С, с разложением), MH+ 573.
ПРИМЕР 86
Получение соединения (226).
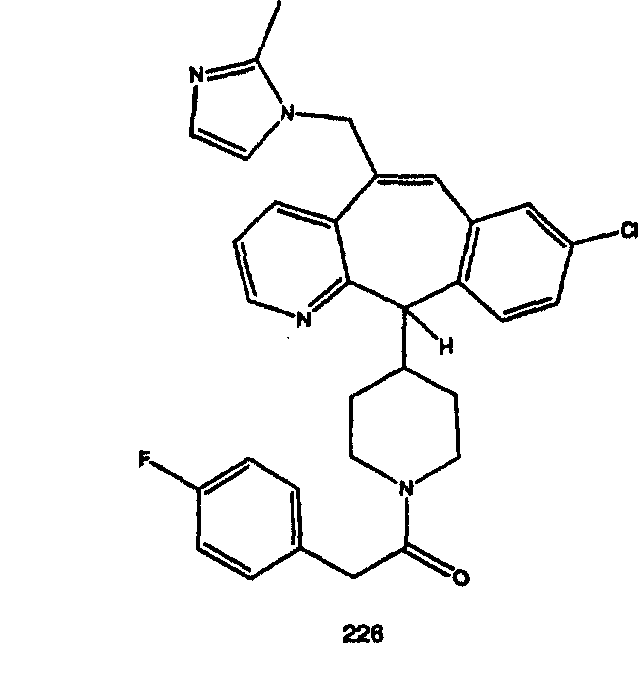
Соединение (221), полученное в Примере получения 20, стадия D (51 мг, 0,126 ммоль), смешивают с 4-фторсренилуксусной кислотой (Acros) (29,29 мг, 0,190 ммоль) и вводят в реакцию таким же образом, как в Примере 84, получая искомое соединение (226) в виде не совсем белого твердого вещества. (108-125°C, с разложением), MH+ 541.
ПРИМЕР ПОЛУЧЕНИЯ 22
Получение соединений (227 и 228).
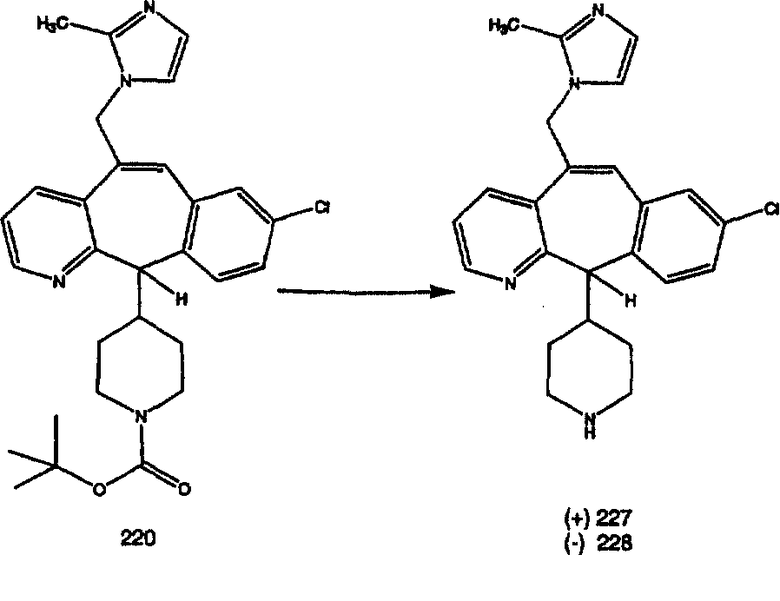
Соединение (220), полученное в Примере получения 20, стадия C (150 мг, 0,289 ммоль), обрабатывают 4 М раствором HCl в диоксане и 2 -3 ч перемешивают при комнатной температуре в атмосфере N2. Неочищенную смесь разделяют на чистый (+)-изомер (227) и (-)-изомер (228) с помощью препаративной хиральной ВЭЖХ с использованием колонки AD, элюируя смесью 85:15:2, гексаны:IPA:DEA.
ПРИМЕРЫ 87-90
Соответствующий (+)-изомер (соединение (227)) или (-)-изомер (соединение (228)), полученный выше в Примере получения 22, растворяют в СН2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии и получают следующие соединения (229-232):
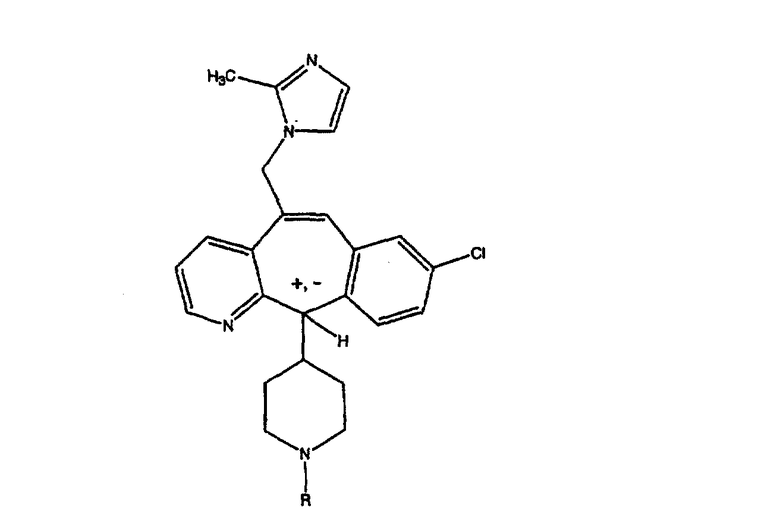
ПРИМЕР ПОЛУЧЕНИЯ 23
А Получение соединения (233).
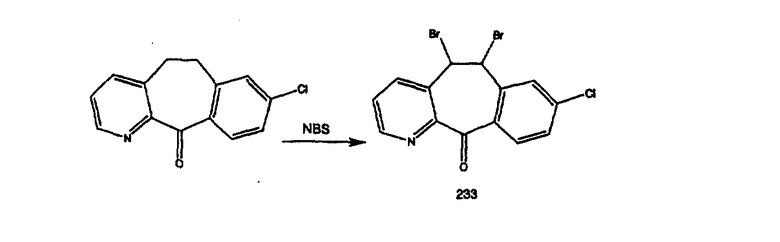
Трициклическое кетосоединение (раскрытое в патенте США US 5151423) (30,0 г, 123,2 ммоль) смешивают с NBS (48,2 г, 271,0 ммоль) и бензоилпероксидом (0,42 г) в ССЦ (210 мл). Реакционную смесь 10 ч нагревают при 80°С. Смесь охлаждают и 8 ч выдерживают. Образовавшийся осадок отфильтровывают. Прибавляют МеОН (200мл) и смесь 2 дня перемешивают. Твердое вещество отфильтровывают и сушат в вакууме до постоянной массы.
В Получение соединений (234а) и (234b).
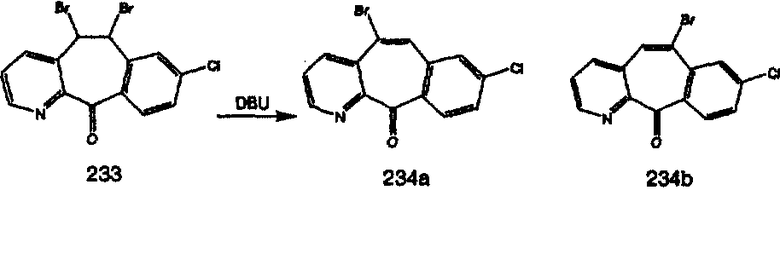
Дибромсоединение (233), полученное на стадии A (35,72 г, 88,97 ммоль), растворяют в CH2CI2 (1,5 л) и охлаждают до 0°С. По каплям прибавляют DBU (15,96 мл) и суспензию 3 ч перемешивают. Реакционную смесь концентрируют, повторно растворяют в CH2Cl2 (1,5 л), фильтруют через слой силикагеля и промывают смесью 5% EtOAc/CH2Cl2 (4 л). Объединенные промывочные растворы концентрируют и очищают с помощью флэш-хроматографии на колонке с силикагелем, элюируя с помощью смеси 10-30% EtOAc/гексан, а затем - 3% EtOAc/CH2Cl2, и получают чистые 5- и 6-монобромзамещенные соединения.
С Получение соединения (235).
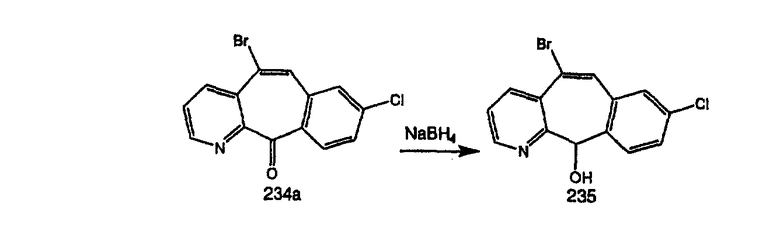
5-Бромзамещенное соединение (234а), полученное выше на стадии B (4,0 г, 12,45 ммоль), растворяют в МеОН и охлаждают до 0°С. Прибавляют NaBH4 (916,4 мг, 24,2 ммоль) и реакционную смесь перемешивают 5,5 ч. Растворитель удаляют и полученный остаток используют непосредственно.
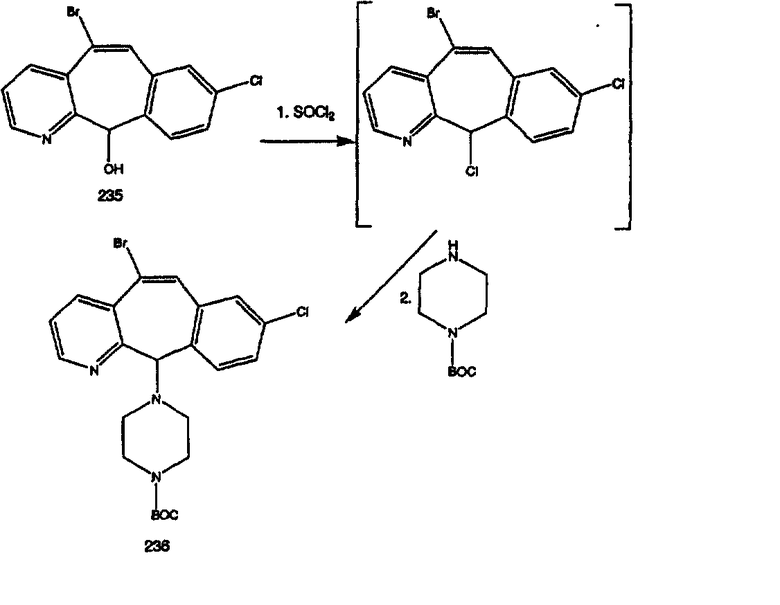
Спирт (235), полученный выше на стадии C (3,98 г, 12 ммоль), растворяют в СН2Cl2, охлаждают до 0°С и обрабатывают 2,6-лутидином (5,73 мл, 49 ммоль). Прибавляют SOCl2 (1,8 мл, 24,6 ммоль) и реакционную смесь перемешивают в течение 3 ч, доводя ее до комнатной температуры. Реакционную смесь выливают в 0,5 н. раствор NaOH (80 мл), экстрагируют и концентрируют в вакууме. Неочищенный продукт растворяют в СН3CN и обрабатывают 1,2,2,6,6-пентаметилпиперидином (4,45 мл, 24,6 ммоль) (Aldrich). Реакционную смесь нагревают до 60 -65°C, обрабатывают трет-бутил-1-пиперазинкарбоксилатом (2,32 г, 12 ммоль) (Aldrich) и перемешивают в течение ночи в атмосфере N2. Реакционную смесь концентрируют досуха, повторно растворяют в CH2CI2 и промывают насыщенным водным раствором МаНСО3. Органический слой сушат над Na2SO4, фильтруют и очищают с помощью флэш-хроматографии на колонке с силикагелем, элюируя с помощью смеси 1:4-1:2 EtOAc/гексаны, и получают продукт в виде белого твердого вещества.
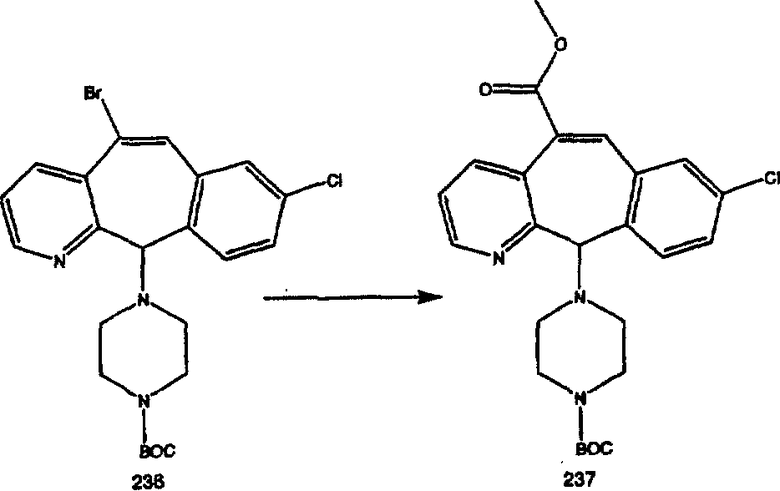
Бромированное соединение (236), содержащее защитную группу ВОС и полученное выше на стадии D (2 г, 4 ммоль), трифенилфосфин (0,54 г, 2 ммоль) и хлорид палладия (0,0723 г, 0,4 ммоль) смешивают с МеОН (10 мл) и толуолом (30 мл). К этой смеси прибавляют DBU (0,835 мл, 5,5 ммоль) и смесь герметизируют в бомбе Парра. Реакционную смесь в течение 5 ч перемешивают и обрабатывают с помощью СО при 80°C и давлении 90 фунтов/дюйм2. Реакционную смесь разбавляют с помощью EtOAc (200 мл) и промывают с помощью 2 х 80 мл H2O. Органический слой сушат над MgSO4, фильтруют и очищают с помощью флэш-хроматографии на колонке с диоксидом кремния, элюируя с помощью смеси 1:3 EtOAc/гексаны.
F Получение соединения (238).
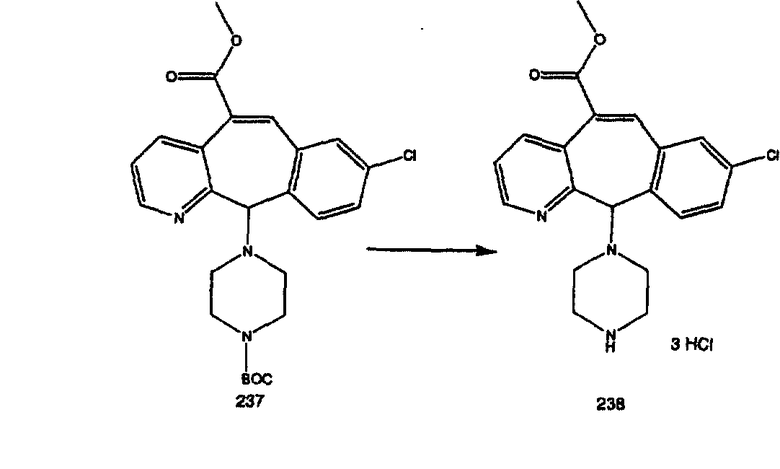
Соединение (237), полученное выше на стадии Е (1,73 г, 3,681 ммоль), обрабатывают 4 М раствором HCI в диоксане (35 мл) и 3 ч перемешивают при комнатной температуре. Реакционную смесь концентрируют в вакууме, и полученное желтовато-коричневое твердое вещество дополнительно сушат в высоком вакууме.
G Получение соединения (239).
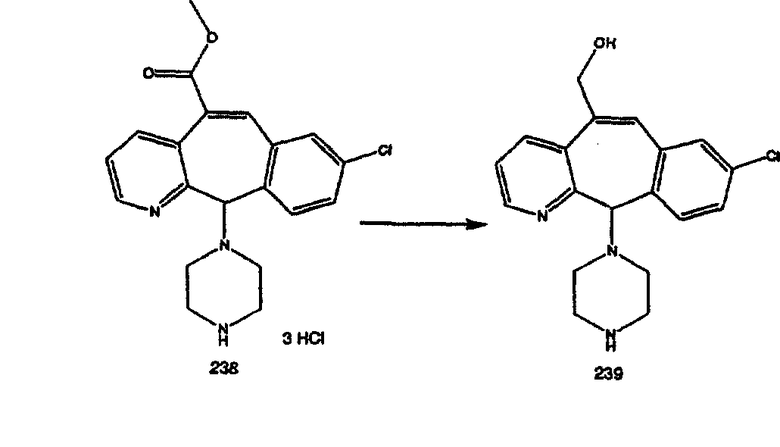
Соль HCI (238), полученную выше на стадии F (1,36 г, 3,68 ммоль), растворяют в THF, охлаждают до 0°C, обрабатывают 1 М раствором DIBAL в циклогексане (18,41 мл, 18 ммоль) и перемешивают при комнатной температуре в течение ночи. Смесь концентрируют досуха и непосредственно используют на следующей стадии.
Н Получение соединения (240).
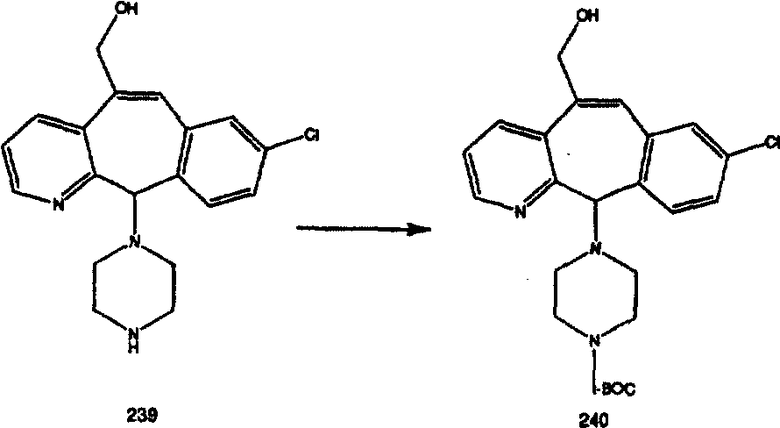
Спирт (239), полученный выше на стадии G, растворяют в МеОН (50 мл) и H2O (5 мл) и обрабатывают с помощью Вос-ангидрида (1,56 г, 7,14 ммоль). С помощью 1 н. раствора NaOH значение рН доводят примерно до 10. Реакционную смесь концентрируют, растворяют в CH2CI2 и промывают с помощью Н2O (2 х). Органический слой сушат над MgSO4, фильтруют и концентрируют, получая желтовато-коричневое твердое вещество, содержащее продукт и примеси.
Альтернативно, соединение (237) превращают в соединение (240) путем получения ацилимидазола с последующим восстановлением с помощью NaBH4, проводимым по следующей методике.
Соединение (237), полученное выше на стадии Е (7,0 ммоль), растворяют в смеси, состоящей из 15 мл метанола, 60 мл диоксана и 6 мл воды, содержащей 25 мл 10% водного раствора LiOH. Смесь 4 ч нагревают при 60°C, затем ее концентрируют в вакууме и с помощью 10% раствора лимонной кислоты значение рН доводят до 5,2. Остаток растворяют в СН2Cl2, промывают рассолом, сушат над MgSO4 и концентрируют в вакууме, получая карбоновую кислоту. Затем кислоту растворяют в 20 мл THF, содержащем 14 ммоль 1,1'-карбонилдиимидазола, и 18 ч нагревают при 38°C. Затем смесь концентрируют в вакууме, получая ацилимидазол. Остаток растворяют в смеси, состоящей из 21,2 мл THF и 5,3 мл воды, и охлаждают до 0°С. К этому раствору прибавляют 35 ммоль NaBH4 и его перемешивают в течение 1,5 ч. Затем прибавляют 5 мл рассола и 25 мл CH2CI2. Органический слой сушат над MgSO4, фильтруют и концентрируют, получая соединение (240) практически с количественным выходом.
I Получение соединения (241).
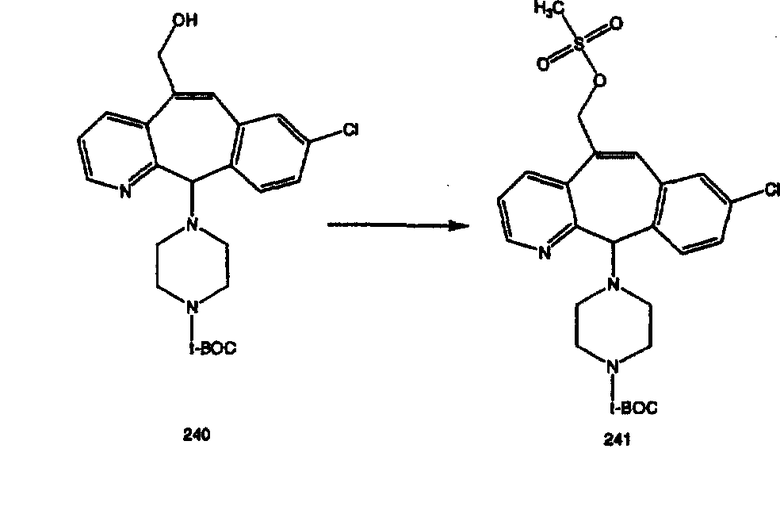
Неочищенный продукт (240), полученный выше на стадии Н (200 мг, 0,45 ммоль), растворяют в CH2Cl2 (2 мл) и обрабатывают триэтиламином (126 мл, 0,91 ммоль), а затем метансульфонилхлоридом (35 мл, 0,45 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют с помощью СН2Cl2 и реакцию останавливают насыщенным раствором NaCl. Органический слой сушат над MgSO4, фильтруют и концентрируют, получая соединение (241).
ПРИМЕР 91
Получение соединения (242).
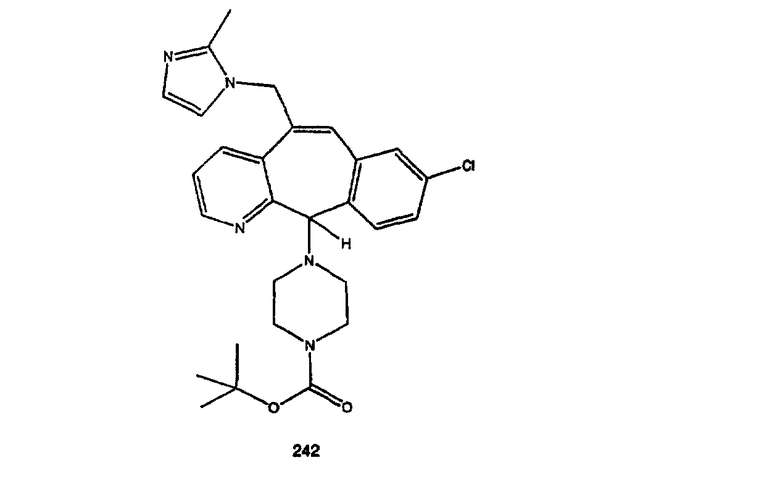
Мезилат (241), полученный выше в Примере получения 23, стадия I (230 мг, 0,442 ммоль), вводят в реакцию таким же образом, как и в Примере получения 20, стадия С. Очистку неочищенного продукта проводят на пластинах для препаративной ТСХ, элюируя смесью 95:5, СН2Cl2/МеОН(NH3), а затем 1:1, EtOAc:гексаны, и получают искомое соединение (242) в виде желтовато-коричневого твердого вещества, т. пл. = 105-116°C (с разложением). MH+ 506.
ПРИМЕР ПОЛУЧЕНИЯ 24
А Получение соединения (243).
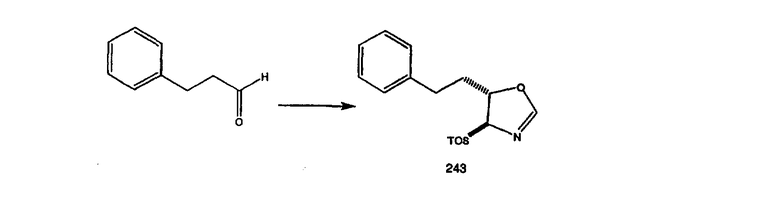
NaCI и 3-фенилпропионовый альдегид (ACROS) сушат в вакууме в течение ночи. Затем альдегид пропускают через активированный Al2О3. Тозилметилизоцианид (5 г, 25,6 ммоль) (ACROS) и сухой 3-фенилпропионовый альдегид (3,36 г, 25,1 ммоль) смешивают в EtOH (42 мл) и перемешивают 5 мин. К мутной смеси прибавляют сухой NaCN (1,23 г, 25,1 ммоль). Протекает экзотермическая реакция, и через 5 мин ТСХ указывает на то, что исходное вещество израсходовано. Реакционную смесь переносят в запаиваемую трубку и непосредственно используют в следующем эксперименте.
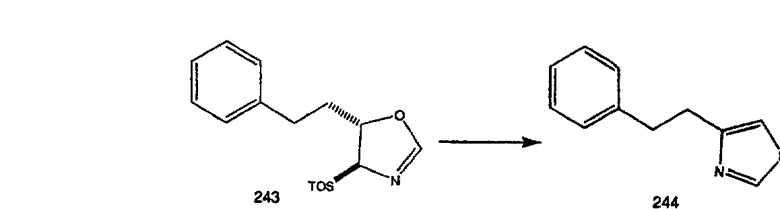
В Неочищенный продукт (243), полученный выше на стадии A (25 ммоль), с помощью EtOH разбавляют до полного объема, равного 65 мл. К этой смеси прибавляют 7 н. раствор NH3 в МеОН (100 мл) и реакционную смесь нагревают при 90°С в течение ночи (20 ч). Реакционную смесь охлаждают до комнатной температуры и перемешивают 2 ч, а затем концентрируют досуха. Неочищенный продукт очищают с помощью флэш-хроматографии на колонке с диоксидом кремния, элюируя в градиентном режиме с помощью смеси 1-5% МеОН (насыщенный аммиаком)/СН2Cl2 (244).
ПРИМЕР ПОЛУЧЕНИЯ 25
Получение соединения (245).
Пропионовый альдегид (1,5 г, 25,11 ммоль) (ACROS) и тозилметилизоцианид (5 г, 25,6 ммоль) вводят в реакцию таким же образом, как в Примере получения 24 выше, и получают искомое соединение (245).
ПРИМЕР ПОЛУЧЕНИЯ 26
Соединение (246) (+)-изомер
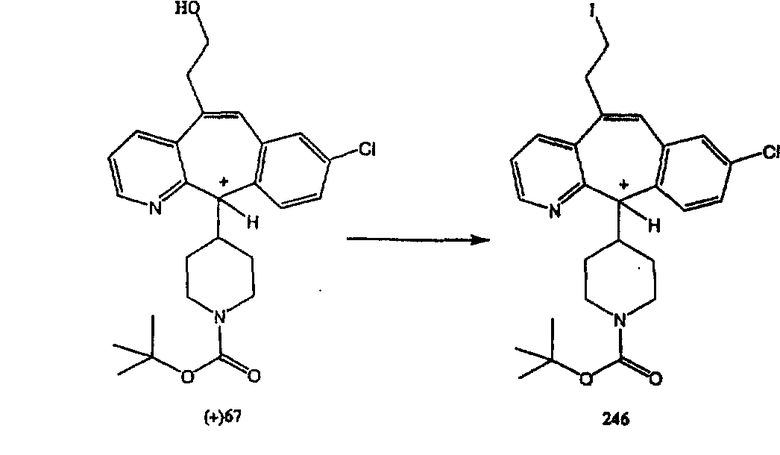
(+)-Изомер соединения (67), полученного в Примере получения 6, выделенный с помощью хроматографии на хиральной колонке AD, вводят в реакцию таким же образом, как в Примере получения 6, и получают соединение (246).
ПРИМЕРЫ 92 И 93
Получение соединений (247) и (248).
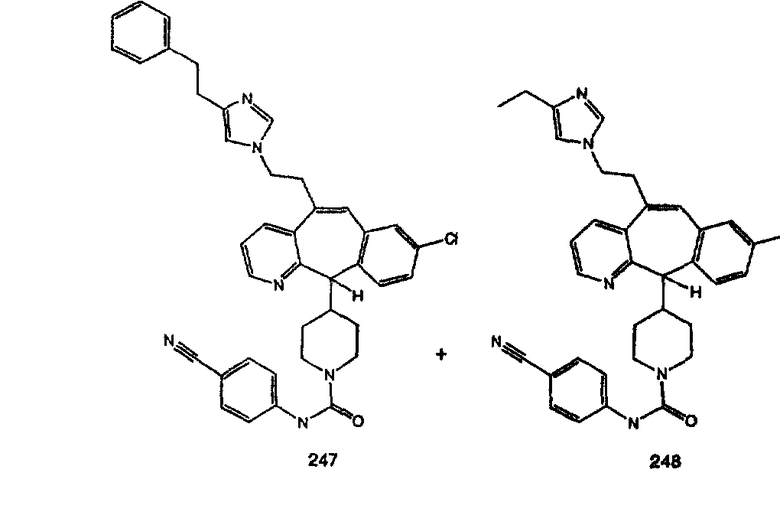
Соединение (246), полученное в Примере получения 26 выше, вводят в реакцию таким же образом, как в Примерах 22, 25 и 29, с использованием соответствующего имидазола или изоцианата соответственно и получают искомые соединения (247) и (248).
ПРИМЕРЫ 94-96
Получение соединений (249), (250) и (251).
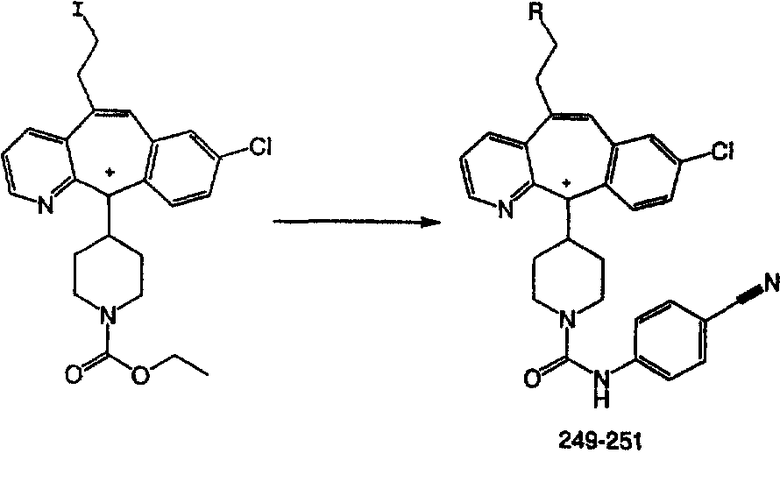
Способом, аналогичным использованному в Примере получения 26 выше, (+)-изомер карбамата получают и вводят в реакцию в основном таким же образом, как в Примерах 92 и 93, используя соответствующие имидазолы, и получают соединения (249)-(251), описанные в приведенной ниже таблице.
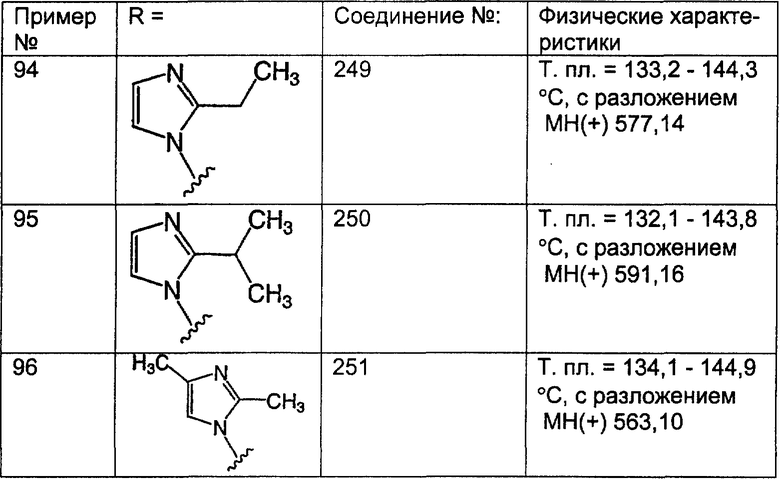
ПРИМЕРЫ 97-101
Получение соединений (252), (253), (254), (255) и (256).
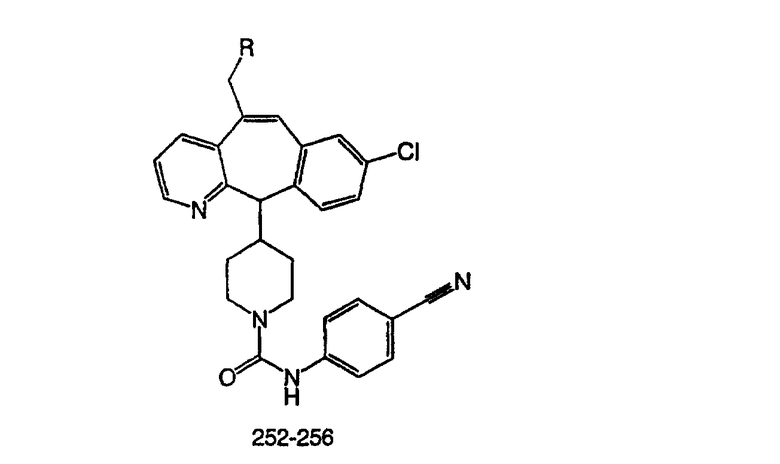
В основном таким же образом, как в Примере получения 20 и Примере 29, получены следующие соединения:
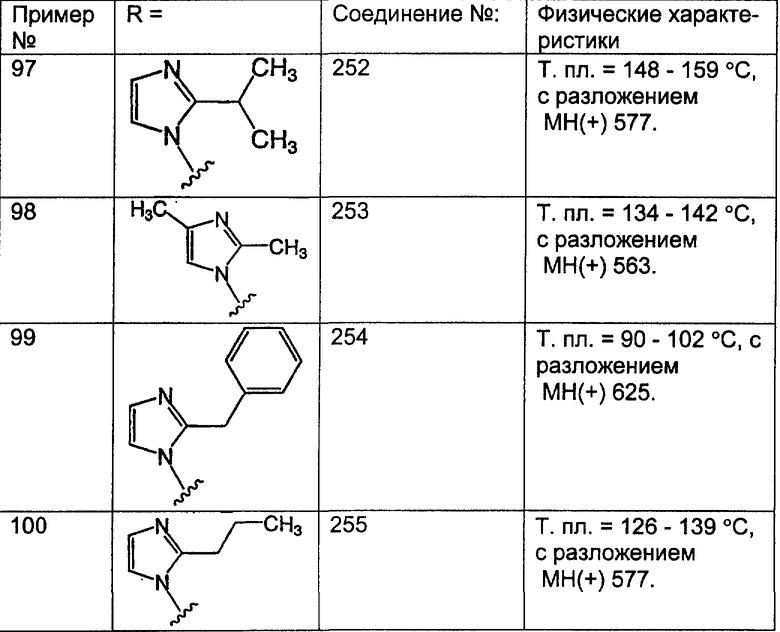
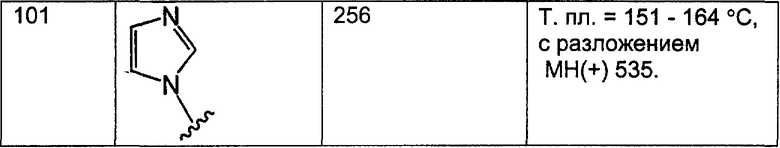
Получение соединения (257).
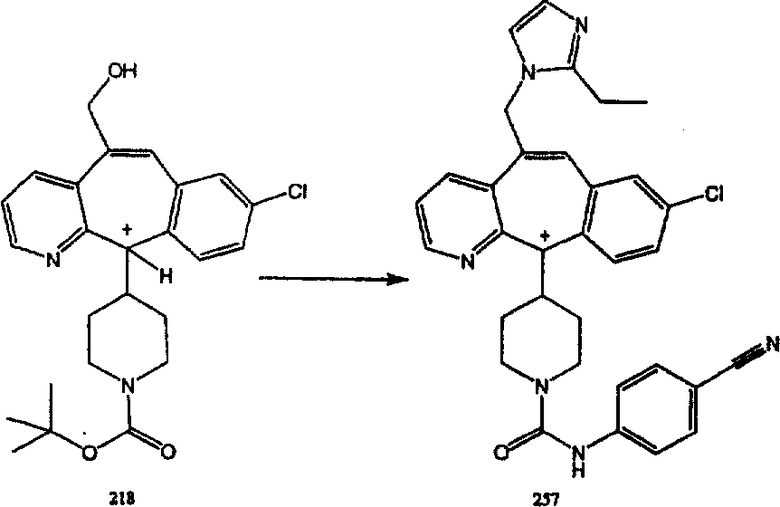
(+)-Изомер соединения (218), полученный в основном таким же образом, как в Примере получения 22, дополнительно вводят в реакцию таким же образом, как в Примере получения 6, стадии Е и F, Примерах 21, 23 и 29, используя 2-этилимидазол в Примере 21, и получают искомое соединение (257), (146 -157°C, с разложением), MH+ 564.
ПРИМЕР ПОЛУЧЕНИЯ 27
Соединения (258А) и (258В).
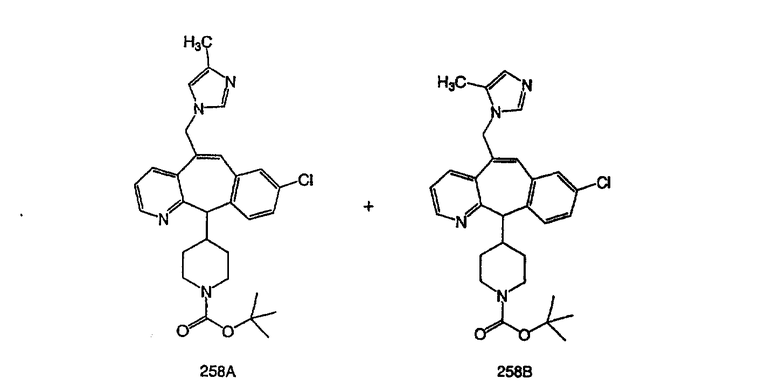
В основном таким же образом, как в Примере получения 20, с использованием 4-метилимидазола соединение (258) получают в виде смеси 4-и 5-замещенных производных имидазола. Эту смесь затем вводят в реакцию способом, аналогичным использованному в Примере 35, и получают разделенные изомеры (258А) и (259В).
ПРИМЕР 103
Получение соединения (259).
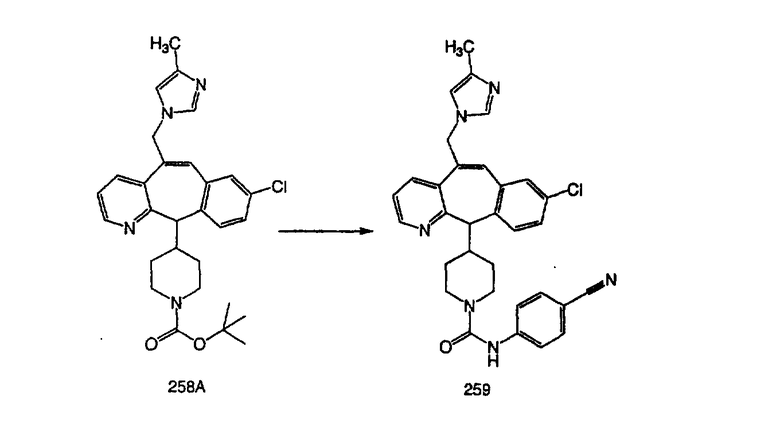
Чистый 4-метилимидазол (258А) вводят в реакцию, как в Примере получения 20, стадия D, и Примере 29, и получают искомое соединение (259) в виде белого твердого вещества, (128-138°C, с разложением), MH+ 549.
ПРИМЕР 104
Получение смеси соединений (260а) и (260b).
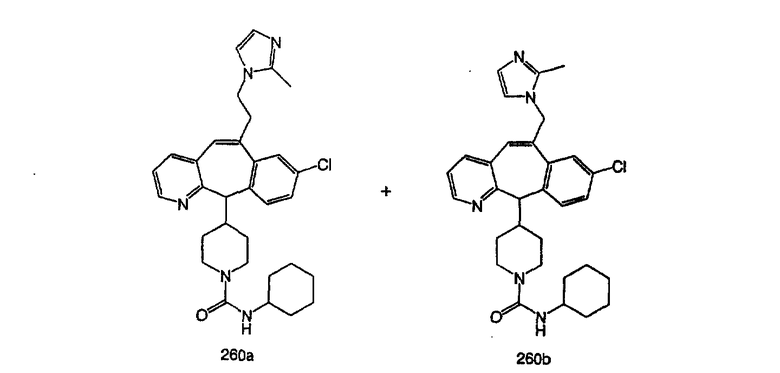
Стадия A
Соединение (108), полученное в Примере получения 9, стадия Е, вводят в реакцию с соединением (64), полученным в Примере получения 6, стадия А, в основном таким же образом, как в Примере получения 6, стадии В-F, и получают смесь йодсодержащих промежуточных продуктов, содержащих разделительные группы из одной и двух метиленовых групп.
Стадия B
Смесь двух промежуточных продуктов, полученных выше на стадии A, вводят в реакцию в основном таким же образом, как в Примере 22, и получают смесь производных имидазола, содержащих разделительные группы из одной и двух метиленовых групп.
Стадия C
Смесь, полученную выше на стадии B, вводят в реакцию таким же образом, как в Примере получения 20, стадия D, а затем вводят в реакцию с фенилизоцианатом, проводимую таким же образом, как в Примере 15, и получают искомое соединение в виде смеси (260а) и (260b) состава 1:1, (133-145°C, с разложением), MH+ 544.
ПРИМЕР ПОЛУЧЕНИЯ 28
Соединение (261)
Стадия A
Литература: Gazz. Chim. Ital. (1972) 102, 189-195; J. Org. Chem. (1991) 56,1166-1170.
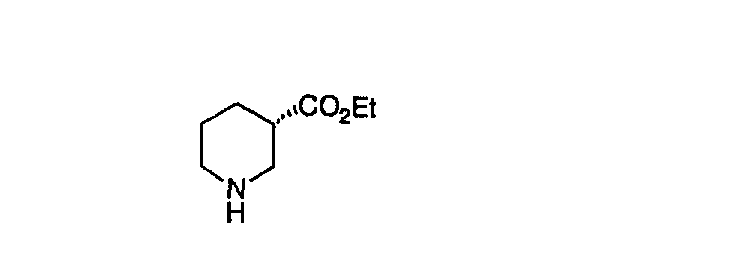
Этилнипекотат (70,16 г, 0,466 ммоль) и D-винную кислоту (67 г, 1,0 экв.) растворяют в горячем 95% EtOH (350 мл). Полученный раствор охлаждают до комнатной температуры и фильтруют и кристаллы промывают с помощью охлажденного льдом 95% EtOH. Затем кристаллы перекристаллизовывают из 95% EtOH (550 мл) и получают соль винной кислоты (38,5 г, выход 56%). Соль (38,5 г) растворяют в воде (300 мл) и охлаждают до 0°C, а затем нейтрализуют 3 М раствором NaOH. Раствор экстрагируют с помощью СН2Cl2 (5 х 100 мл) и объединенные органические экстракты сушат над Na2SO4 и концентрируют при пониженном давлении, получая прозрачное масло (19,0 г, выход 89%). МСХИ: MH+ = 158.
Стадия B
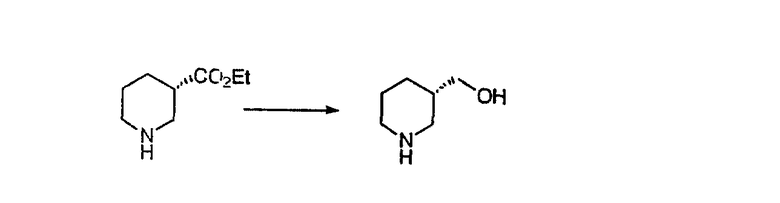
LAH (118 мл, 1,0 М раствор в Et2O, 1,0 экв.) в течение 20 мин при 0°С прибавляют к раствору продукта, полученного на стадии А (18,5 г, 0,125 ммоль), в THF (250 мл). Полученный раствор медленно нагревают до комнатной температуры, а затем 2 ч кипятят с обратным холодильником. Реакционную смесь охлаждают до комнатной температуры и реакцию останавливают путем медленного прибавления насыщенного раствора Na2SO4. Полученную взвесь сушат путем прибавления Na2SO4, фильтруют через целит и концентрируют, получая бесцветное масло (13,7 г, выход неочищенного продукта 98%). МСХИ: MH+ =116; [α]D 20 = -8,4° (5,0 мг в 2 мл МеОН).
Стадия C
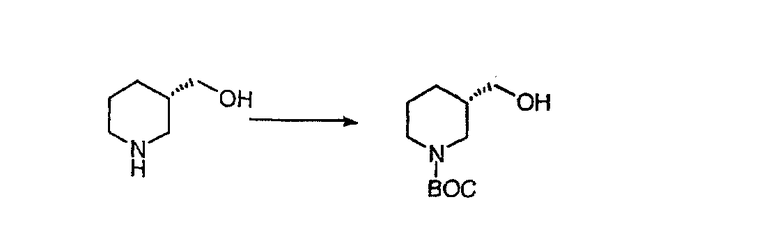
Продукт, полученный на стадии В (13,6 г, 0,104 ммоль), растворяют в МеОН (100 мл) и Н2O (100 мл), затем порциями прибавляют ди-трет-бутилдикарбонат (27,24 г, 1,2 экв.), поддерживания значение рН > 10,5 путем прибавления 50% раствора NaOH. Реакционную смесь еще 2,5 ч перемешивают при комнатной температуре и концентрируют в вакууме. Остаток разбавляют с помощью Н2O (350 мл) и экстрагируют с помощью CH2Cl2 (3 х 150 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 50% раствора EtOAc в гексане в качестве элюента и получают белое твердое вещество (12,13 г, выход 48%). МС ББА:MH2 = 216; [α]D 20 = +15,2° (5,0 мг в 2 мл МеОН).
Стадия D
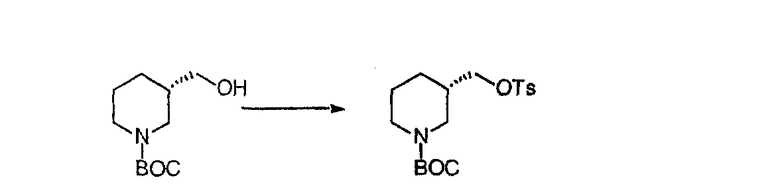
п-Толуолсульфонилхлорид (12,75 г, 1,2 экв.) при 0°C порциями прибавляют к раствору продукта, полученного на стадии C (12,00 г, 55,74 ммоль), в пиридине (120 мл). Полученный раствор перемешивают при 0°C в течение ночи. Реакционную смесь разбавляют с помощью EtOAc (300 мл) и промывают холодным 1 н. раствором HCI (5 х 300 мл), насыщенным раствором NaHCO3 (2 х 50 мл), Н2О (1 х 100 мл) и рассолом (1 х 100 мл), сушат над Na2SO4 и концентрируют в вакууме, получая бледно-желтое твердое вещество (21,0 г, выход неочищенного продукта 100%). МС ББА: МН+= 370.
Стадия Е
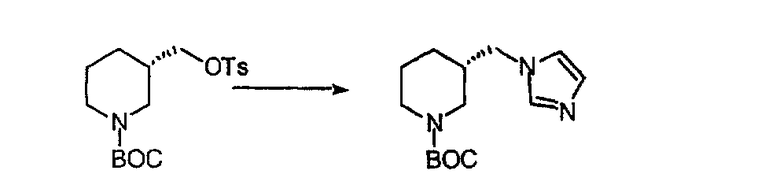
Продукт, полученный на стадии D (21,0 г, 55,74 ммоль), в DMF (300 мл) обрабатывают натриевой солью имидазола (8,37 г, 1,5 экв.) и полученный раствор 2 ч нагревают при 60°С. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток разбавляют с помощью H2O (300 мл) и экстрагируют с помощью СН2Cl2 (3 х 150 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают с помощью флэш-хроматографии с использованием 7% раствора МеОН в СН2Cl2 в качестве элюента и получают бледно-желтое твердое вещество (7,25 г, выход 49%). МС ББА: MH+= 266; [α]D 20 = +8,0° (5,0 мг в МеОН).
Стадия F
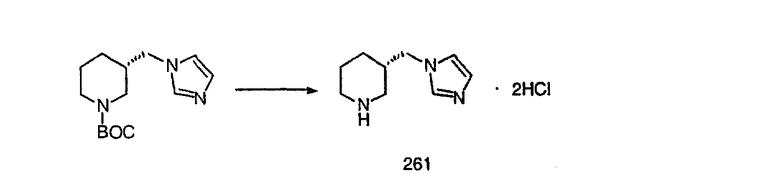
Продукт, полученные на стадии Е (5,50 г, 20,73 ммоль), в течение ночи перемешивают при комнатной температуре в 4 М растворе HCI в диоксане (50 мл). Полученный раствор концентрируют и остаток растирают с Et2O, получая соединение (261) в виде желтого твердого вещества (4,90 г, выход 99%). МСХИ: МН+ = 166.
ПРИМЕР ПОЛУЧЕНИЯ 29
Соединение (262)
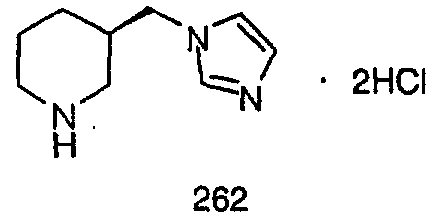
С помощью в основном такой же методики, как приведенная выше в Примере получения 28, с применением на стадии A L-винной кислоты вместо D-винной кислоты, получают соединение (262).
ПРИМЕР ПОЛУЧЕНИЯ 30
Получение соединений (263) и (264)
Стадия A
1N-трет-бутоксикарбонил-3(R)-и -3(S)-(1Н-имидазол-1-ил)-метилпирролидины
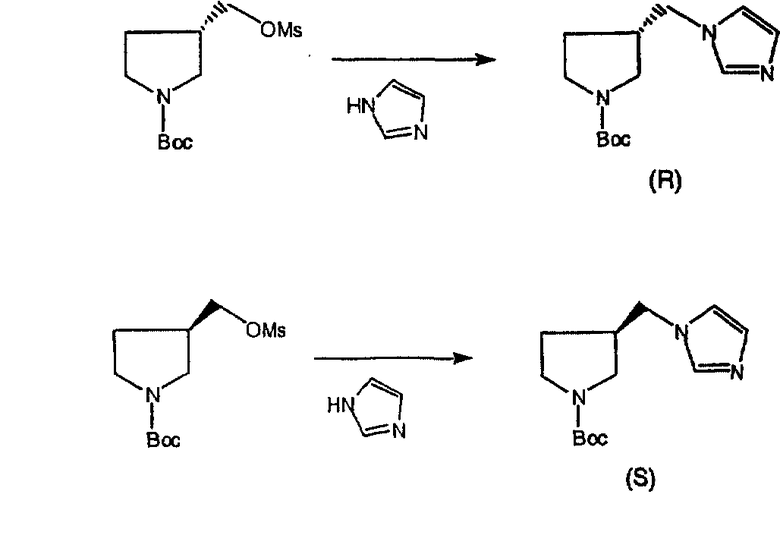
3(R)-(3-Метансульфонилоксиметил)-пирролидин (J. Med. Chem. 1990, 33, 77-77) (0,993 г, 3,56 ммоль) растворяют в безводном DMF (25 мл) и прибавляют натриевую соль имидазола (0,6 г, 10 ммоль). Смесь 2 ч нагревают при 60°C, а затем выпаривают досуха. Продукт экстрагируют с помощью CH2Cl2 и промывают рассолом. Экстракт в CH2Cl2 выпаривают досуха и получают искомое соединение (263) (1,1409 г, 100%). МСЭР: МС ББА (М + 1) = 252; 1H ЯМР (CDCl3) 1,45 (s, 9Н), 1,5-1,7 (m, 1H), 1,9 -2,1 (m, 1H), 2,5 -2,7 (m, 1H), 3,0 -3,2 (m, 1H), 3,3 -3,6 (m, 2H), 3,9 (dd, 2H), 6,9 (s, 1H), 7,1 (s,1H), 7,45 (s,1H).
Аналогичным образом из 3(8)-(3-метансульфонилоксиметил)-пирролидина (0,993 г, 3,56 ммоль) получают (S)-изомер искомого соединения (1,14 г, 100%).
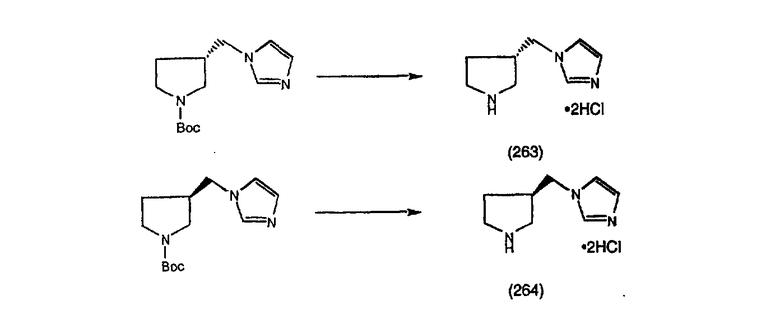
(R)-Продукт (0,48 г, 1,91 ммоль), полученный на стадии A, 2 ч перемешивают в 4 н. растворе HCI в диоксане (10 мл) и затем выпаривают досуха, получая искомое соединение (263) в виде соли с HCI.
Аналогичным образом получают (S)-изомер, соединение (264) в виде соли с HCl.
ПРИМЕР ПОЛУЧЕНИЯ 31
Соединения (265) и (266)
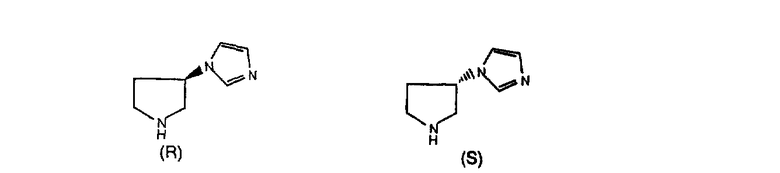
1N-Бензил-3(R)-гидроксипирролидин (5 г, 28,21 ммоль) и триэтиламин (7,86 мл, 56,35 ммоль) растворяют в CH2Cl2 (50 мл) и смесь перемешивают в атмосфере азота при 0°С. Прибавляют метансульфонилхлорид (2,62 мл, 33,87 ммоль) и раствор 2 ч перемешивают при комнатной температуре. Раствор разбавляют с помощью CH2Cl2 и промывают насыщенным раствором бикарбоната натрия, водой и сушат (MgSO4), фильтруют и выпаривают досуха, получая искомое (Р)-соединение (7,2 г, 96,4%). МС ББА (M+1) = 256; 1H ЯМР (CDCl3) 2,2 (m, 1H), 2,3(m, 1H), 2,52(m, 1H), 2,7-2,85 (т, 3Н), 2,95 (s, 3Н), 3,65 (q, 2Н), 5,16 (т, 1Н), 7,3 (s, 5H).
Аналогичным образом из 1N-бензил-3(3)-гидроксипирролидина (5 г, 28,21 ммоль) получают искомое (S)-соединение (7,15 г, 98%).
Стадия B
1N-Бензил-3(S)-и -(R)-(1 Н-имидазол-1 -ил)-пирролидины
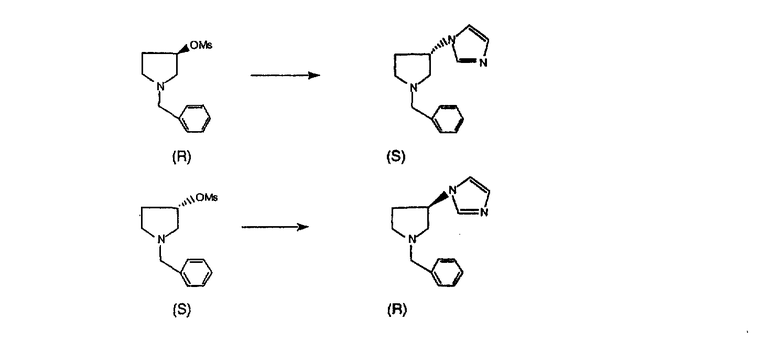
Раствор (R)-продукта, полученного на стадии A (2,0 г, 7,84 ммоль), при перемешивании прибавляют к раствору имидазола (1,1 г, 16,17 ммоль) в DMF (25 мл) в атмосфере азота. Смесь 16 ч перемешивают при 60°С. DMF выпаривают в вакууме. Оставшийся неочищенный продукт экстрагируют с помощью CH2Cl2 и экстракт последовательно промывают водой и рассолом и CH2Cl2 выпаривают, получая в остатке искомое вещество, которое хроматографируют на силикагеле, используя 3% (10% NH4OH в метаноле) - CH2Cl2 в качестве элюента, и получают искомое соединение (0,95 г, 50,56%). МС ББА (М + 1) = 228.
Аналогичным образом получают второй изомер.
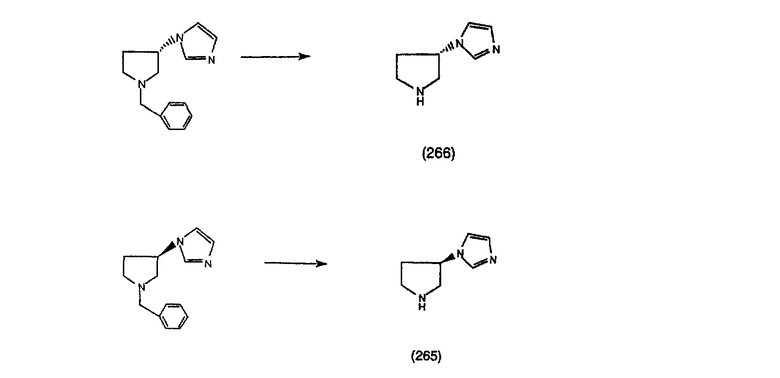
Смесь (З)-продукта (0,95 г), полученного на стадии B, и 10% Pd на угле (0,5 г) в EtOH (20 мл) в течение 24 ч встряхивают в атмосфере водорода при давлении 50 фунтов/дюйм2. Отфильтровывают катализатор и удаляют растворитель, получая искомое соединение (266) (0,522 г, 99,9%).
Аналогичным образом из 1,0 г исходного (R)-продукта, полученного на стадии В, и 10% Pd на угле (0,6 г) получают (Р)-изомер, соединение (265), с выходом 99%.
ПРИМЕР ПОЛУЧЕНИЯ 32
Соединения(267) и(268)
С помощью в основном такой же методики, как приведенная выше в Примере получения 31, с использованием L-или D-пролинола в качестве исходных соединений получают искомые соединения (267) и (268).
ПРИМЕР 105
Получение соединения (269).
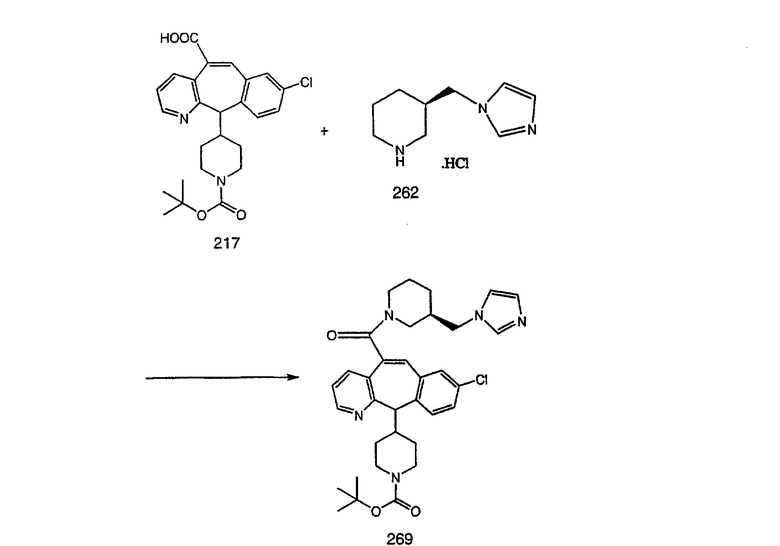
Соединение (217), полученное в Примере получения 19 (0,227 г, 0,499 ммоль), прибавляют к соединению (262), полученному в Примере получения 29 (0,131 г, 0,649 ммоль), DEC (0,249 г, 1,3 ммоль), HOST (0,175 г, 1,3 ммоль) и NMM (0,5 мл) в DMF (25 мл). Полученный раствор 24 ч перемешивают при комнатной температуре. Реакционную смесь разбавляют с помощью НаО, пока не перестанет происходить осаждение, и взвесь фильтруют. Осадок разбавляют с помощью CH2Cl2, промывают рассолом, сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают с помощью хроматографии, используя 5% (10% NH4OH в МеОН) раствор в CH2Cl2 в качестве элюента, и получают искомое соединение (269) (0,184 г, выход 62%).
ПРИМЕРЫ 106-111
Получение соединений (270) -(275).
С использованием подходящего амина из Примеров получения 28 -32 и в основном такой же методики, как и использованная выше в Примере 105, получают следующие соединения:
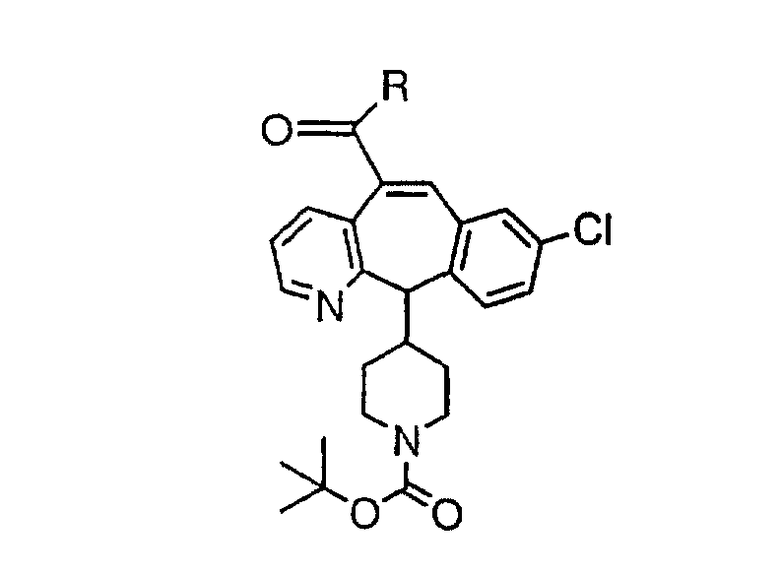
ПРИМЕР 112
Получение соединения (276).
Соединение (274), полученное выше в Примере 110 (0,125 г, 0,213 ммоль), в CH2Cl2 (50 мл) перемешивают с TFA (10 мл) при комнатной температуре в течение ночи. Реакционную смесь выпаривают, получая соль TFA (0,28 г), которую повторно растворяют в CH2Cl2 (50 мл) и охлаждают (на бане из воды со льдом). Прибавляют триэтиламин (0,1 мл), а затем сульфонилхлорид (0,038 г, 0,319 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь промывают бикарбонатом натрия и водой. Органический слой сушат над MgSO4 и выпаривают досуха, получая искомое соединение (276) (0,05 г, MH+ = 567).
ПРИМЕР 113
Получение соединения (277).
Используя в качестве исходного соединение (273), полученное выше в Примере 109, с помощью в основном такой же методики, как приведенная выше в Примере 112, получают соединение (277) (MH+ = 567).
ПРИМЕР ПОЛУЧЕНИЯ 33
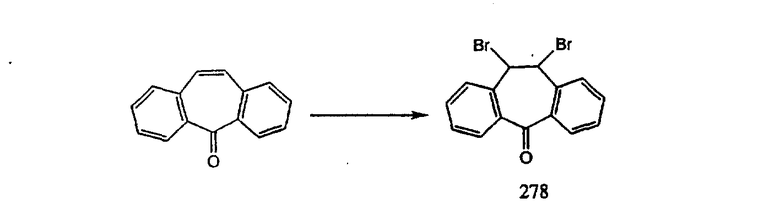
К раствору брома (33,0 г, 210 ммоль) в CCl4 (100 мл) при перемешивании при комнатной температуре прибавляют раствор дибензосуберенона (37,0 г, 179 ммоль) в CCl4 (200 мл). Полученный раствор 1,5 ч перемешивают при комнатной температуре. Белые кристаллы собирают фильтрованием и получают продукт (278) (60,12 г, выход 92%, МН+ = 367).
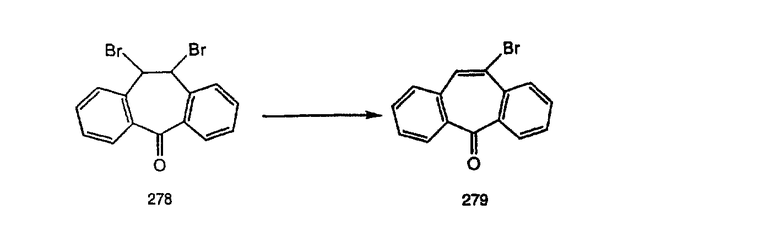
Раствор дибромсоединения (278), полученного на стадии A (60,0 г, 163 ммоль), и NaOH (20,0 г, 491 ммоль) в МеОН (500 мл) перемешивают и кипятят с обратным холодильником в течение 1,5 ч. Затем реакционную смесь охлаждают до комнатной температуры и перемешивают в течение ночи. Смесь выпаривают досуха и экстрагируют смесью СН2Cl2 - H2O. Объединенные органические слои сушат над MgSO4, фильтруют и выпаривают досуха, получая желтое твердое вещество (279) (46,34 г, выход 100%, М=285).
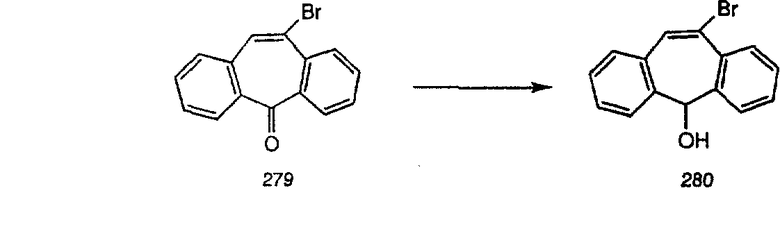
К раствору монобромсоединения (279), полученного на стадии B (10,0 г, 35,07 ммоль), в МеОН (200 мл) при перемешивании в атмосфере азота при 0°C прибавляют NaBH4 (1,94 г, 51,2 ммоль). Полученный раствор 1,5 ч перемешивают при 0°C, затем выпаривают, а после этого экстрагируют смесью CH2Cl2 - H2O. Объединенные органические слои сушат над MgSO4, фильтруют и выпаривают досуха, получая белое твердое вещество (280) (10,3 г, 100%, М = 287).
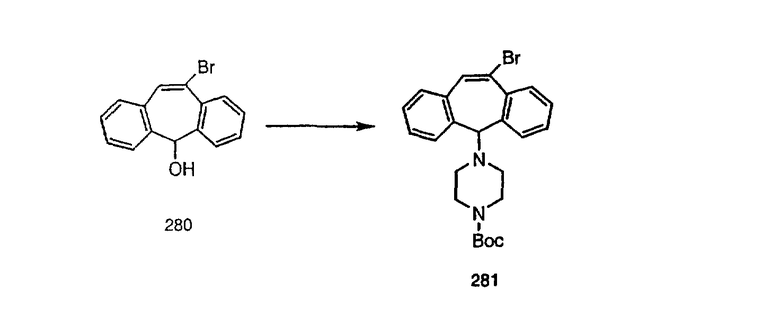
К раствору спирта (280), полученного на стадии C (10,0 г, 34,8 ммоль), в СН2Cl2 (200 мл) при перемешивании при 0°С прибавляют 2,6-лутидин (14,9 г, 139,3 ммоль) и тионилхлорид (8,28 г, 69,66 ммоль). Полученный раствор нагревают до комнатной температуры и перемешивают в течение ночи. Затем раствор выливают в 0,5 н. раствор NaOH, а после этого экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют досуха, получая неочищенное коричневое масло (15,5 г). К раствору этого коричневого масла (15,5 г) в ацетонитриле (200 мл) прибавляют 2,6-бис(диметил)-1-метилпиперидин (10,81 г, 69,66 ммоль) и М-Вос-пиперидин (6,49 г, 34,83 ммоль). Полученную смесь в течение ночи нагревают при 65°С. Смесь выпаривают досуха, а затем экстрагируют смесью СН2Cl2/насыщенный раствор NaHCO3. Объединенные органические слои сушат над Na2SO4, концентрируют и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 5% EtOAc/95% гексан, и получают содержащее защитную группу N-Boc соединение (281) (5,68 г, выход 36%, МН+ = 455).
Е Получение соединения (282).
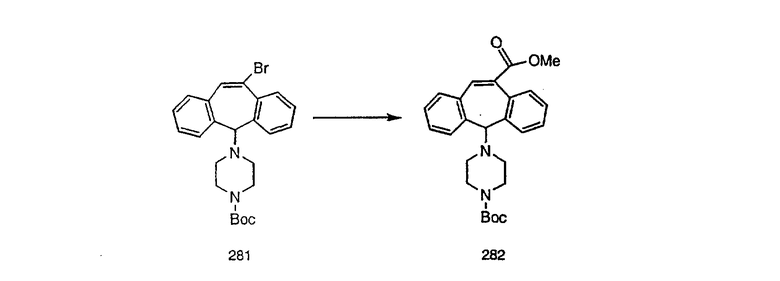
К раствору содержащего защитную группу N-Boc соединения (281), полученного на стадии D (4,0 г, 8,78 ммоль), в безводном толуоле (100 мл) и метаноле (20 мл) прибавляют трифенилфосфин (1,15 г, 4,39 ммоль), DBU (1,81 г, 11,9 ммоль) и хлорид палладия(II) (0,15 г, 0,88 ммоль). Полученную смесь продувают оксидом углерода при давлении от 80 до 100 фунтов/дюйм2 и в течение 5 ч нагревают при температуре 78-82°С, а затем перемешивают при комнатной температуре в течение ночи. Затем раствор экстрагируют с помощью EtOAc. Объединенные органические слои промывают водой, рассолом, сушат над Na2SO4, фильтруют, выпаривают и неочищенный продукт очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 10% EtOAc/90% гексан, и получают сложный эфир (282) (2,1 г, выход 55%, МН+ = 435).
F Получение соединения (283).
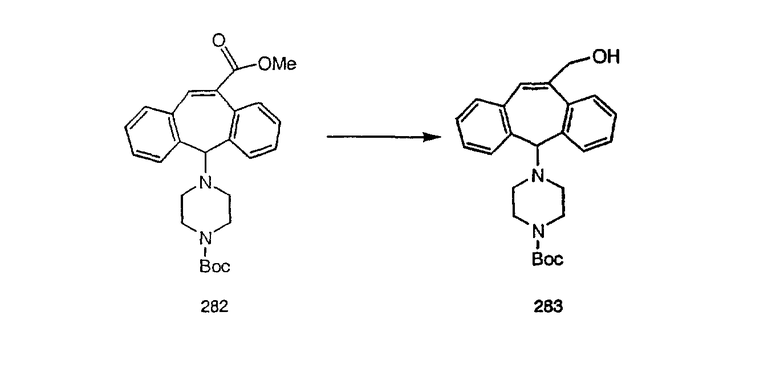
К раствору сложного эфира (282), полученного на стадии Е (1,2 г, 2,77 ммоль), в THF (15 мл) при 0°С прибавляют 1 М раствор DIBAL (16,62 мл, 16,62 ммоль). Полученный раствор 4 ч перемешивают при комнатной температуре. Затем к раствору прибавляют 10% раствор тартрата калия-натрия, а после этого экстрагируют с помощью EtOAc. Объединенные органические слои сушат над Na2SO4, фильтруют и выпаривают досуха, получая твердое вещество (283) (1,1 г, выход 100%, MH+ = 406).
G Получение соединения (284).
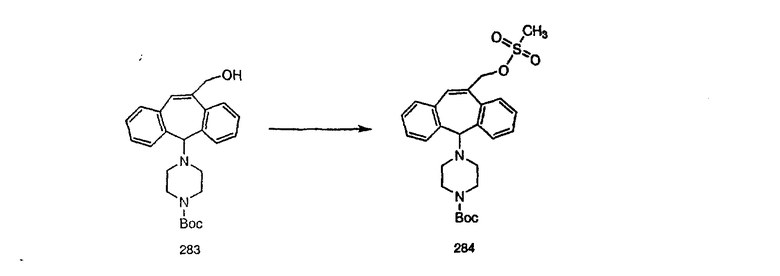
К раствору спирта (283), полученного на стадии F (0,62 г, 1,52 ммоль), в СН2Cl2 (15 мл) в атмосфере азота прибавляют триэтиламин (0,64 мл, 4,56 ммоль) и метансульфонилхлорид (0,26 г, 2,29 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи. Смесь промывают раствором NaHCO3, сушат над Na2SO4, фильтруют и концентрируют досуха, получая мезилат (284) (0,53 г, выход 76%, М-СН3SO3Н = 389,1).
Н Получение соединения (285).
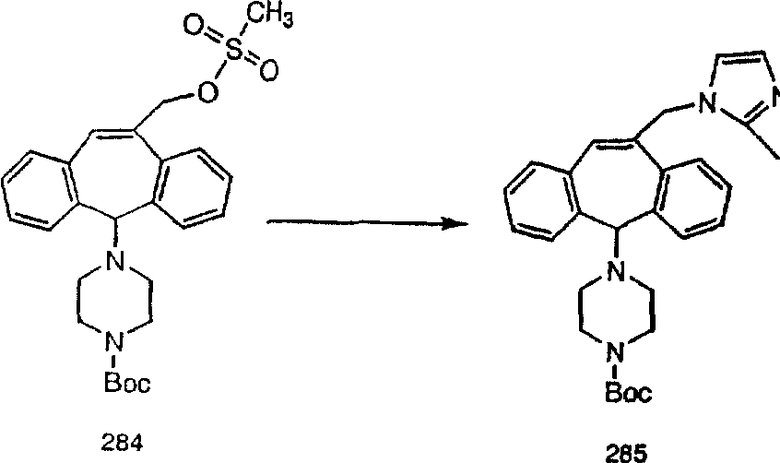
К раствору 1-метилимидазола (1,04 г, 12,7 ммоль) в DMF (10 мл) в атмосфере азота при перемешивании прибавляют NaH (0,305 г, 12,7 ммоль). Полученный раствор 15 мин перемешивают при комнатной температуре, а затем прибавляют мезилат (284), полученный на стадии G (2,05 г, 4,23 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем выпаривают досуха и экстрагируют раствором EtOAc-NaHCO3. Объединенные органические слои сушат над Na2SO4, концентрируют и неочищенный продукт очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 2% МеОН /98% NH3-CH2Cl2, и получают продукт (285) (0,775 г, выход 39%, МН+ = 471).
I Получение соединения (286).
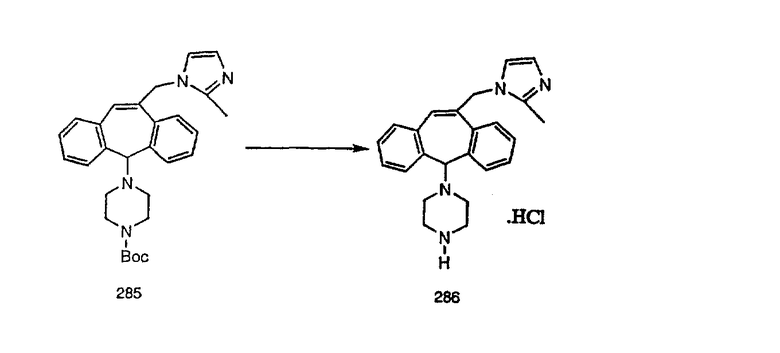
Раствор продукта (285), полученного на стадии Н (0,3 г, 0,64 ммоль), в 4 М растворе HCI в диоксане (40 мл) перемешивают при комнатной температуре в течение 3 ч, а затем концентрируют досуха, получая гидрохлорид искомого продукта (286) (0,42 г, выход 100%, MH+=371).
ПРИМЕРЫ 114 И 115
Соединения (287) и (288).
Рацемическую смесь, полученную выше в Примере получения 33, стадия Н, разделяют на чистые изомеры с помощью ВЭЖХ с использованием хиральной колонки AD, элюируя смесью 15% IPA/75% гексан/0,2% DEA, и получают соединения, приведенные в представленной ниже таблице:
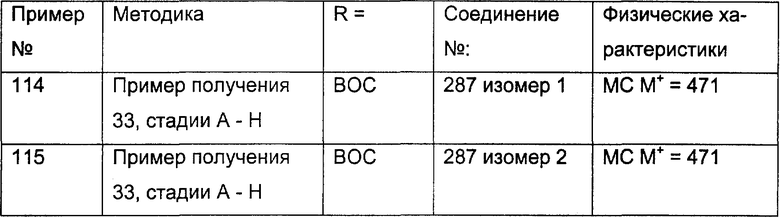
ПРИМЕРЫ 116-119
С использованием пиперазина (286), полученного в Примере получения 33, стадия I, в качестве исходного вещества и путем введения его в реакцию с соответствующим изоцианатом или сульфонилхлоридом с помощью в основном такой же методики, что и указанная в приведенной ниже таблице, получают следующие соединения:
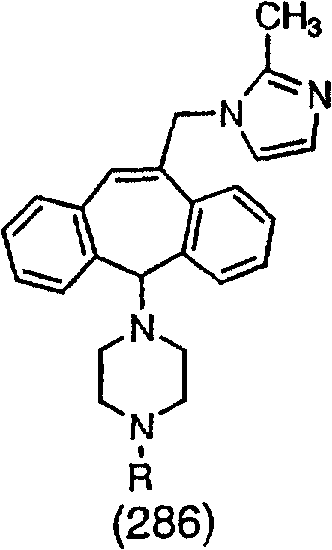
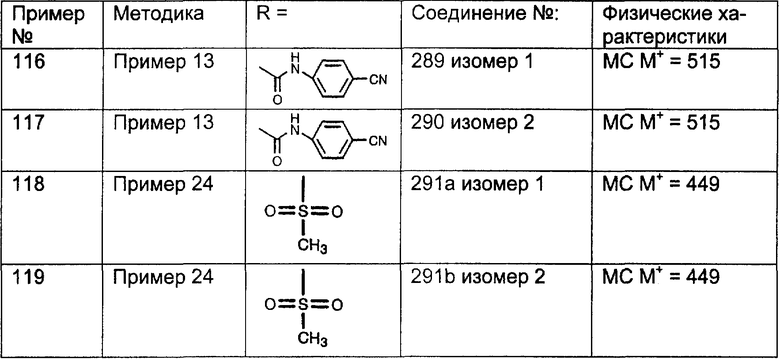
ПРИМЕР ПОЛУЧЕНИЯ 34
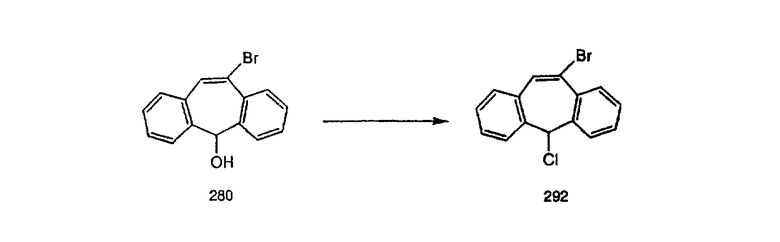
К раствору спирта (280), полученного в Примере получения 33, стадия C (30,0 г, 104,5 ммоль), в CH2Cl2 (500 мл) при перемешивании в атмосфере азота при -20°C прибавляют тионилхлорид (106,7 ил, 1,46 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, а затем выпаривают досуха. Неочищенную смесь разбавляют толуолом (50 мл), а затем при комнатной температуре дополнительно прибавляют SOCI2 (106,7 мл). Полученный раствор 2 ч кипятят с обратным холодильником, пока реакция не завершится. Затем реакционную смесь охлаждают до комнатной температуры и концентрируют досуха, получая светло-коричневое твердое вещество (292) (35,67 г, выход 100%, M-BrCI =191).
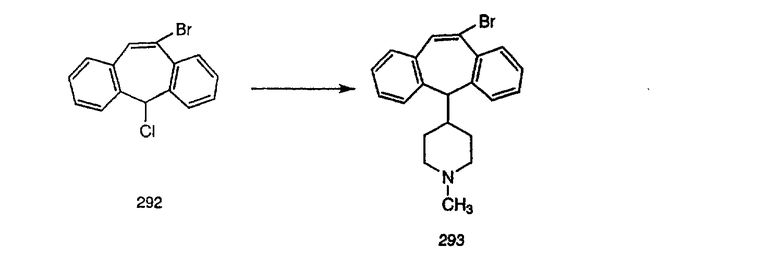
К суспензии Mg (3,63 г) в безводном THF (95 мл) в атмосфере азота при комнатной температуре прибавляют 4-хлор-1-метилпиперидин (3 мл, 10% от полного количества) и один небольшой кристалл йода. Полученный раствор кипятят с обратным холодильником, а затем прибавляют йодметан (0,5 мл) и оставшийся 4-хлор-1-метилпиперидин (27 мл). Реакционную смесь перемешивают 1 ч, а затем концентрируют досуха, получая неочищенный реагент Гриньяра (0,8 М).
К раствору хлорированного соединения (292), полученного в Примере получения 34, стадия A (35,67 г, 116,7 ммоль), в безводном THF (200 мл) в атмосфере азота при комнатной температуре по каплям прибавляют полученный выше реагент Гриньяра (0,8 М, 146 мл, 116,7 ммоль). Полученный раствор 3 ч перемешивают при комнатной температуре, а затем экстрагируют смесью EtOAc-Н2O. Объединенные органические слои сушат над MgSO4, фильтруют и выпаривают досуха, получая продукт (293) (49,25 г, выход 100%, MH+ = 368).
С Получение соединения (294).
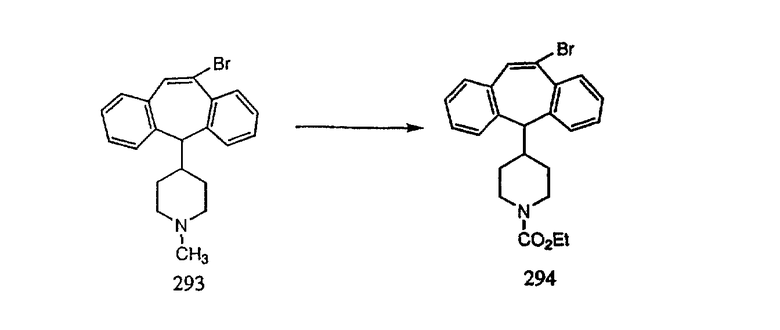
К раствору соединения (293), полученного выше на стадии В (42,9 г, 116,5 ммоль), в толуоле (400 мл) при перемешивании в атмосфере азота прибавляют триэтиламин (49 мл, 349,5 ммоль). Полученный раствор кипятят с обратным холодильником, а затем по каплям прибавляют этилхлорформиат (126 г, 1165 ммоль). Раствор продолжают кипятить с обратным холодильником в течение 2 ч. Затем реакционную смесь перемешивают при комнатной температуре в течение ночи, а после этого экстрагируют смесью EtOAc-1 н. раствор NaOH. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и неочищенный продукт очищают с помощью хроматографии на колонке с нормальной фазой силикагеля, элюируя смесью 30% EtOAc/70% гексан, и получают светло-желтое твердое вещество (294) (2,99 г, выход 12%, МН+ = 426,3).
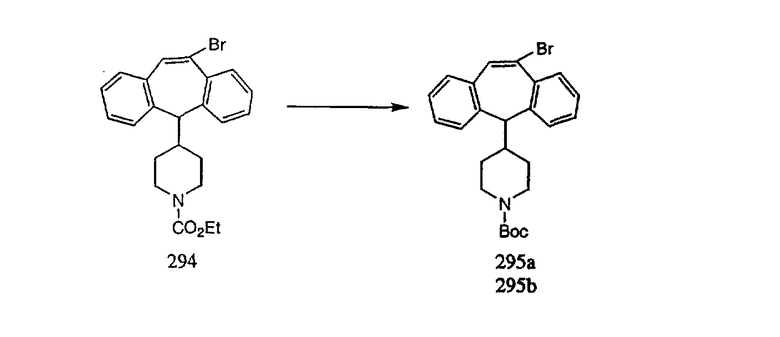
Раствор сложного эфира (294), полученного выше на стадии C (3,34 г, 7,83 ммоль), в 6 н. растворе HCl (20 мл) кипятят с обратным холодильником в течение ночи. Реакционную смесь охлаждают до комнатной температуры и подщелачивают раствором NH4OH, а затем экстрагируют с помощью CH2CI2. Объединенные органические слои сушат над MgSO4, фильтруют и выпаривают досуха, получая неочищенный свободный пиперидин (2,80 г, выход 100%, MH+ = 534).
К неочищенному материалу (полученному выше) (2,77 г, 7,82 ммоль) в 50% МеОН/1% Н2O (200 мл) прибавляют ди-трет-бутилдикарбонат (3,41 г, 15,64 ммоль). Значение рН реакционной смеси доводят до 9 и ее 4 ч перемешивают при комнатной температуре, выпаривают досуха, а затем экстрагируют с помощью CH2Cl2-Н2О. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью ВЭЖХ с использованием хиральной колонки AD, элюируя смесью 15% IPA/75% гексан/0,2% DEA, и получают чистые изомеры содержащих защитную группу N-Boc соединений (295а) и (295b) (3,42 г, выход 96%, MH+= 454).
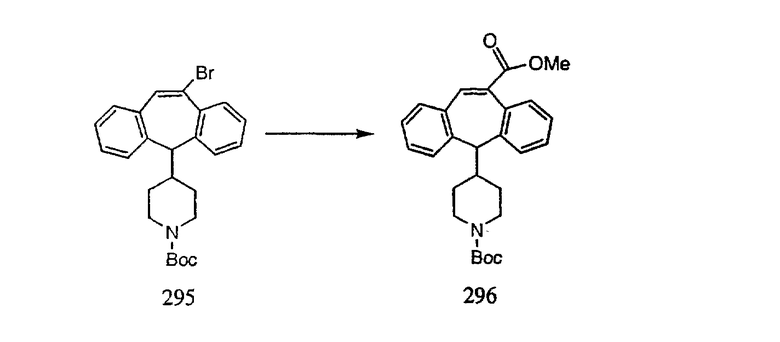
К раствору чистого (+)- или (-)-изомера, содержащего защитную группу N-Boc соединения, полученного выше на стадии D (4,0 г, 8,78 ммоль), в безводном толуоле (100 мл) и метаноле (20 мл) при перемешивании прибавляют трифенилфосфин (1,15 г, 4,39 ммоль), DBU (1,81 г, 11,9 ммоль) и хлорид палладия(II) (0,15 г, 0,88 ммоль). Полученную смесь продувают оксидом углерода при давлении от 80 до 100 фунт-сила/дюйм2 и в течение 5 ч нагревают при температуре 78-82°C, а затем перемешивают при комнатной температуре в течение ночи. После этого раствор экстрагируют с помощью EtOAc. Объединенные органические слои промывают водой, рассолом, сушат над Na2SO4, фильтруют, выпаривают и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 10% EtOAc/90% гексан, и получают сложный эфир (296а) или (296b) (2,1 г, выход 55%, MH+ = 435).
F Получение соединений (297а) и (297b).
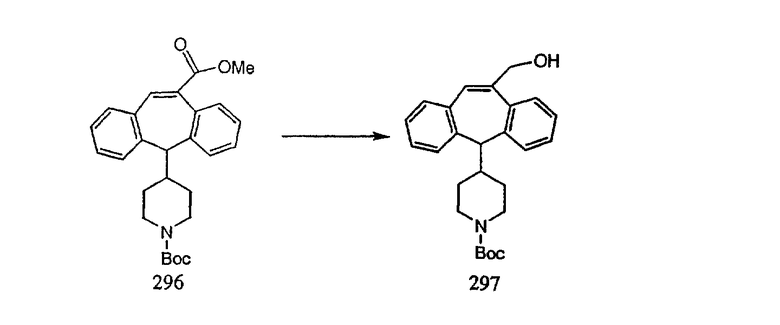
К раствору (+)- или (-)-изомера сложного эфира, полученного выше на стадии Е (1,2 г, 2,77 ммоль), в THF (15 мл) при перемешивании при 0°C прибавляют 1 М раствор DIBAL (16,62 мл, 16,62 ммоль). Полученный раствор 4 ч перемешивают при комнатной температуре. Затем к раствору прибавляют 10% раствор тартрата калия-натрия, а после этого экстрагируют с помощью EtOAc. Объединенные органические слои сушат над Na2SO4, фильтруют и выпаривают досуха, получая твердое вещество (297а) или (297b) (1,1 г, выход 100%, МН+ = 406).
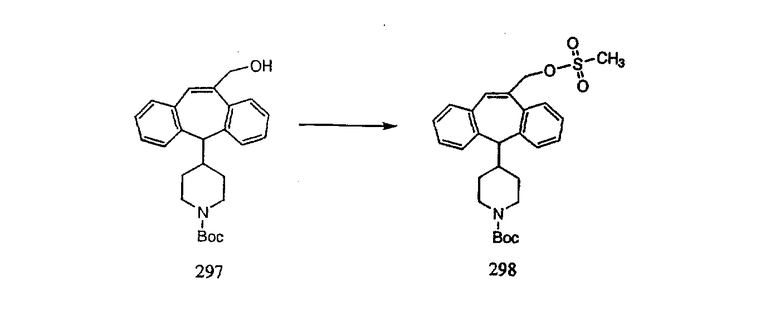
К раствору (+)- или (-)-изомера спирта, полученного выше на стадии F (0,62 г, 1,52 ммоль), в CH2Cl2 (15 мл) при перемешивании в атмосфере азота прибавляют триэтиламин (0,64 мл, 4,56 ммоль) и метансульфонилхлорид (0,26 г, 2,29 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи. Смесь промывают раствором NaHCO3, сушат над Na2SO4, фильтруют и концентрируют досуха, получая мезилат (298а) или (298b) (0,53 г, выход 76%, М-СН3SO3Н = 389,1).
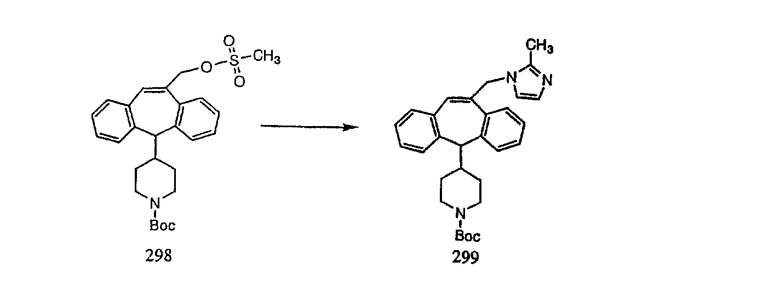
К раствору 1-метилимидазола (1,04 г, 12,7 ммоль) в DMF (10 мл) в атмосфере азота при перемешивании прибавляют NaH (0,305 г, 12,7 ммоль). Полученный раствор 15 мин перемешивают при комнатной температуре, а затем прибавляют (+)- или (-)-изомер мезилата (299), полученного выше на стадии G (2,05 г, 4,23 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем выпаривают досуха и экстрагируют раствором EtOAc-NaHCO3. Объединенные органические слои сушат над Na2SO4, концентрируют и неочищенный продукт очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 2% МеОН/98% NH3 - СН2Cl2, и получают продукт (299а) или (299b) (0,775 г, выход 39%, MH+ = 471).
I Получение соединений (300а) и (300b).
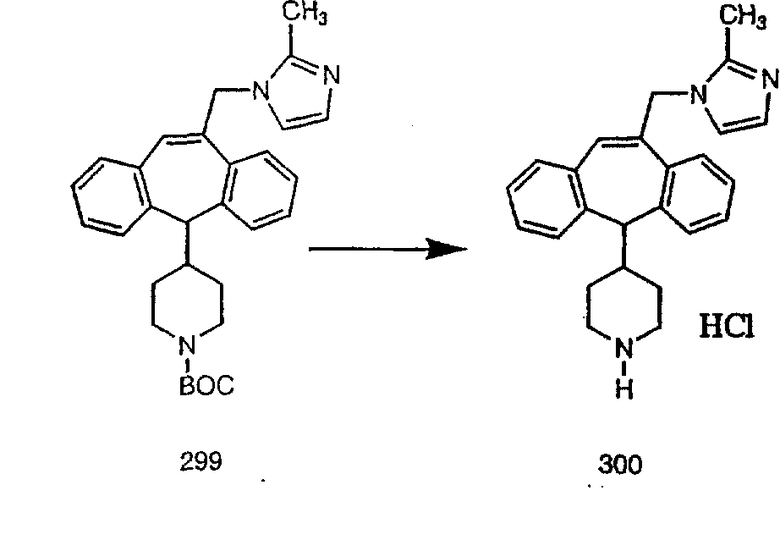
Раствор (+)- или (-)-изомера продукта, полученного выше на стадии Н (0,3 г, 0,64 ммоль), в 4 М растворе HCl в диоксане (40 мл) перемешивают при комнатной температуре в течение 3 ч, а затем концентрируют досуха, получая гидрохлорид продукта (300а) или (300b) (0,42 г, выход 100%, MH+ = 371).
ПРИМЕРЫ 120 И 121
С использованием соответствующего (+)- или (-)-изомера соединения (300) в качестве исходного вещества и путем введения его в реакцию аналогично тому, как это описано в Примере 13, с использованием соответствующего изоцианата получают следующие соединения:
ПРИМЕР ПОЛУЧЕНИЯ 35
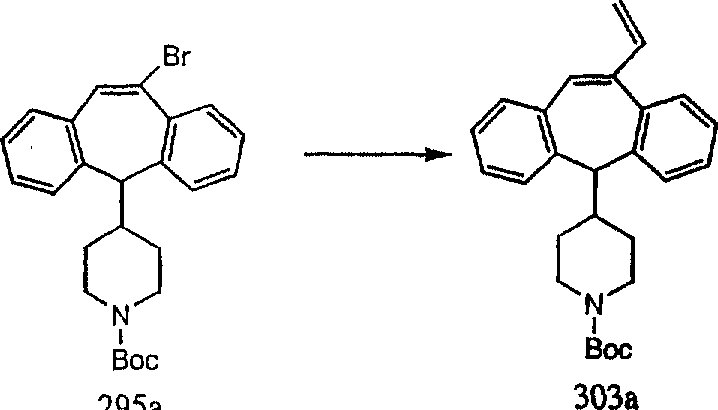
К раствору изомера 1 бромированного соединения (295а), полученного в Примере получения 34, стадия D (0,5 г, 1,10 ммоль), в 1-метил-2-пирролидиноне (4,3 мл) в атмосфере азота при перемешивании прибавляют хлорид лития (0,14 г, 3,3 ммоль), три-2-фурилфосфин (0,013 г, 0,04 ммоль) и трис(дибензилиденацетон)-дипалладий(0) (0,02 г, 0,02 ммоль). Полученный раствор 5 мин перемешивают при комнатной температуре, а затем прибавляют трибутилвинилолово (0,39 г, 1,24 ммоль). Затем реакционную смесь 2 ч нагревают при 85°C, а после этого экстрагируют с помощью EtOAc - Н2О. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с нормальной фазой силикагеля, элюируя смесью 10% МеОН/90% NH3 -CH2Cl2, и получают светло-желтое твердое вещество (303а) (0,06 г, выход 15%, MH+ = 390).
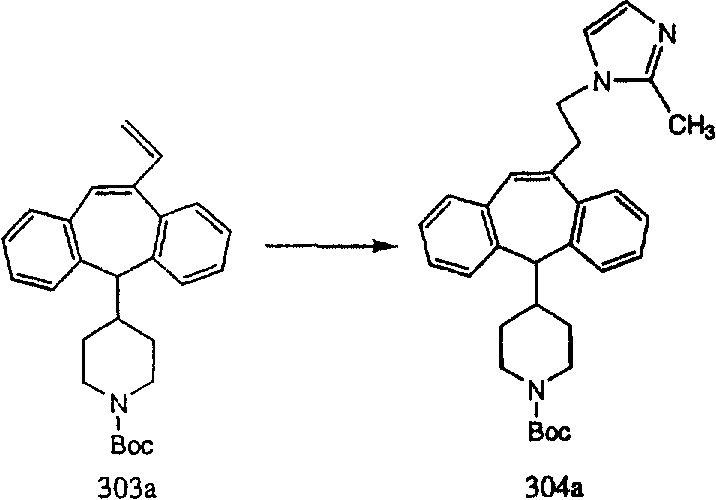
К раствору 1-метилимидазола (0,377 г, 4,6 ммоль) в безводном THF (4 мл) в атмосфере азота при -78°C и перемешивании прибавляют 2,5 М раствор n-BuLi/гексан (0,33 мл). Полученный раствор 30 мин перемешивают при -78°C, а затем дают ему нагреться до комнатной температуры. К этому раствору при перемешивании прибавляют алкен (303а), полученный выше на стадии A (0,78 г, 2,1 ммоль), в THF. Полученный раствор нагревают при 120°C в течение ночи, а затем охлаждают до комнатной температуры и экстрагируют с помощью EtOAc -H2O. Объединенные органические слои сушат над MgSO4, фильтруют, выпаривают и очищают с помощью хроматографии на колонке с нормальной фазой силикагеля, элюируя смесью 3% МеОН/97% NH3 - CH2CI2, и получают светло-желтое твердое вещество (304а) (0,09 г, выход 10%,MH+= 456,1).
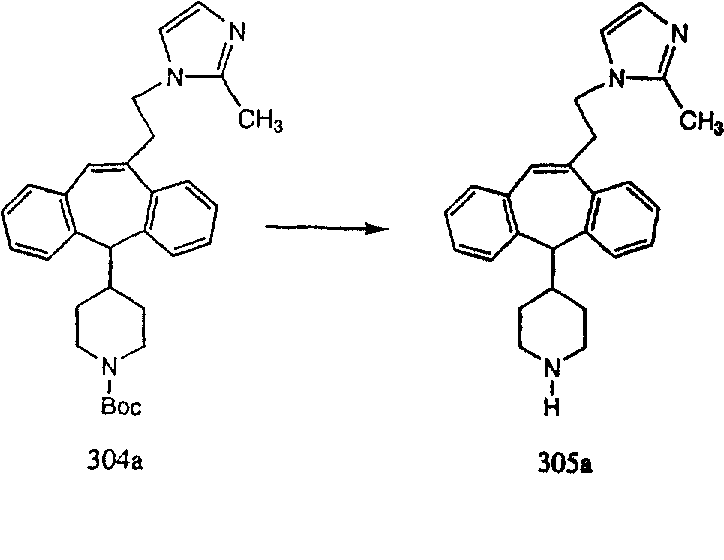
Раствор продукта, полученного выше на стадии B (0,18 г, 3,72 ммоль), в 4 М растворе HCl/диоксан (5 мл) перемешивают при комнатной температуре в течение 2 ч, а затем концентрируют досуха, получая неочищенное не совсем белое твердое вещество (305а) (0,22 г, выход 100%, MH+ = 384,2).
С использованием такой же методики, как и описанная выше в Примере получения 35, и изомера 2 содержащего защитную группу Boc бромированного соединения (258b) в качестве исходного вещества получают изомер 2 (305b) (MH+ = 384,2).
ПРИМЕРЫ 122-125
С использованием соответствующего (+)- или (-)-изомера соединения (305) в качестве исходного вещества и путем введения его в реакцию аналогично тому, как это описано в Примере 13, с использованием соответствующего изоцианата получают следующие соединения:
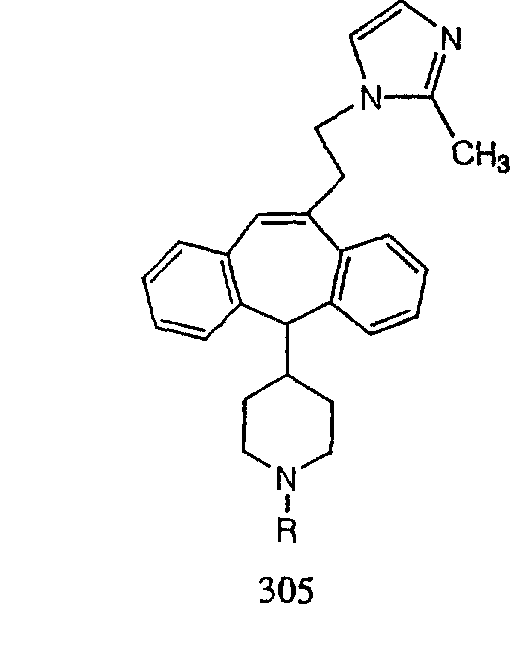
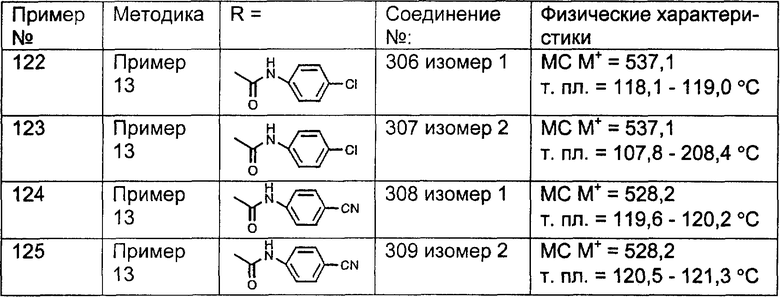
ПРИМЕР ПОЛУЧЕНИЯ 36
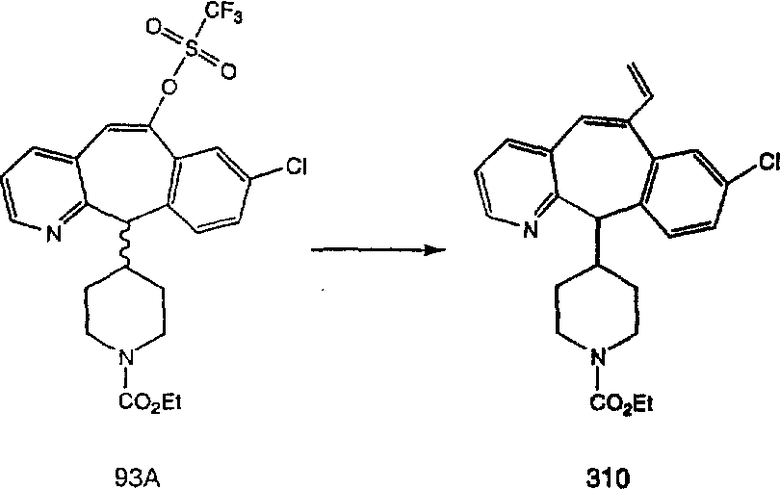
К раствору соединения (93А), полученного в Примере 7, стадия А (5,0 г, 10,02 ммоль), в 1-метил-2-пирролидоне (40 мл) в атмосфере азота при комнатной температуре прибавляют LiCl (1,27 г, 30,06 ммоль), три-2-фурилфосфин (0,093 г, 0,4 ммоль) и трис(дибензилиденацетон)дипалладий(0) (0,18 г, 0,2 ммоль). Полученный раствор 5 мин перемешивают при комнатной температуре, а затем прибавляют трибутилвинилолово (3,3 мл, 11,3 ммоль) и в течение ночи перемешивают при 80-85°С. Раствор охлаждают до комнатной температуры, а затем экстрагируют с помощью EtOAc - H2O. Органический слой сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 20% EtOAc/80% CH2Cl2, и получают продукт (310) (3,88 г, выход 95%, MH+ =409,1).
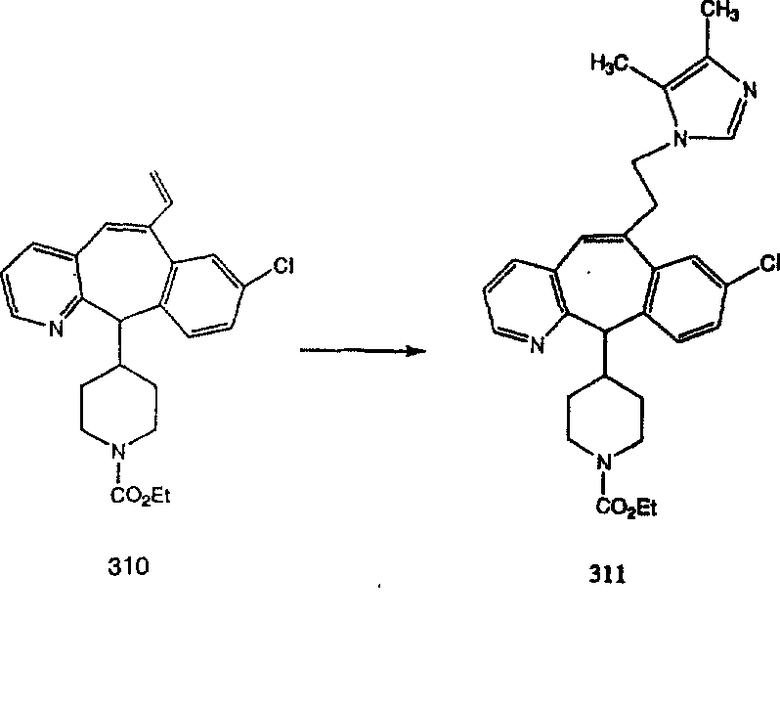
К раствору 4,5-диметилимидазола (25,8 мг, 0,268 ммоль) в безводном THF (0,2 мл) в атмосфере аргона при -78°C при перемешивании прибавляют 2,5 М раствор n-BuLi (0,032 мл, 0,08 ммоль). Полученный раствор нагревают до комнатной температуры, затем прибавляют алкен (310), полученный выше на стадии A (0,1 г, 0,24 ммоль), в безводном THF (0,2 мл). Затем раствор 25 ч нагревают на масляной бане при 120°C в течение 25 ч, а после этого экстрагируют с помощью CH2Cl2 - Н2О. Затем объединенные органические слои промывают рассолом, сушат над Na2SO4, фильтруют и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 5% МеОН/95% CH2CI2, и получают продукт (311) (0,046 г, выход 100%, MH+ = 505).
С Получение соединений (312а) и (312b).
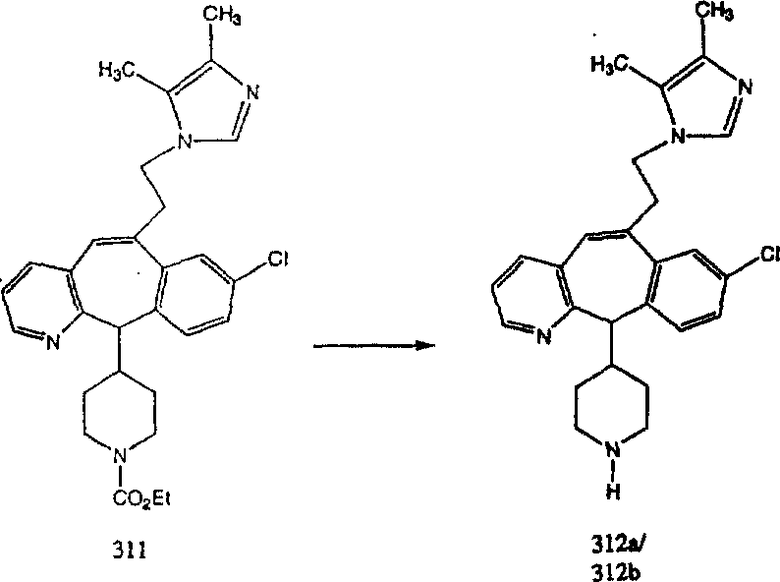
Раствор соединения (311), полученного выше на стадии B (0,57 г, 1,28 ммоль), в 6 н. растворе HCl (20 мл) 24 ч кипятят с обратным холодильником, а затем концентрируют досуха. Затем к остатку прибавляют насыщенный раствор NaHCO3 и NaCl. Раствор дважды экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над Na2SO4 и концентрируют досуха, получая неочищенный продукт (0,52 г, выход 93%). Затем неочищенный продукт растворяют в смеси 20% EtOH/80% гексан/0,2% DEA и очищают с помощью ВЭЖХ с использованием хиральной колонки AD, элюируя смесью 20-50% lPA/гексан/0,2% DEA (длина волны = 254 нм, ослабление = 1024, поглощение = 2), и получают чистые изомеры (312а) и (312b) (0,225 г, MH+=433).
ПРИМЕРЫ 126-133
С использованием соответствующего (+)- или (-)-изомера соединения (312) в качестве исходного вещества и путем введения его в реакцию аналогично тому, как это описано в Примере 13, с использованием соответствующего изоцианата или сульфонилхлорида получают следующие соединения:
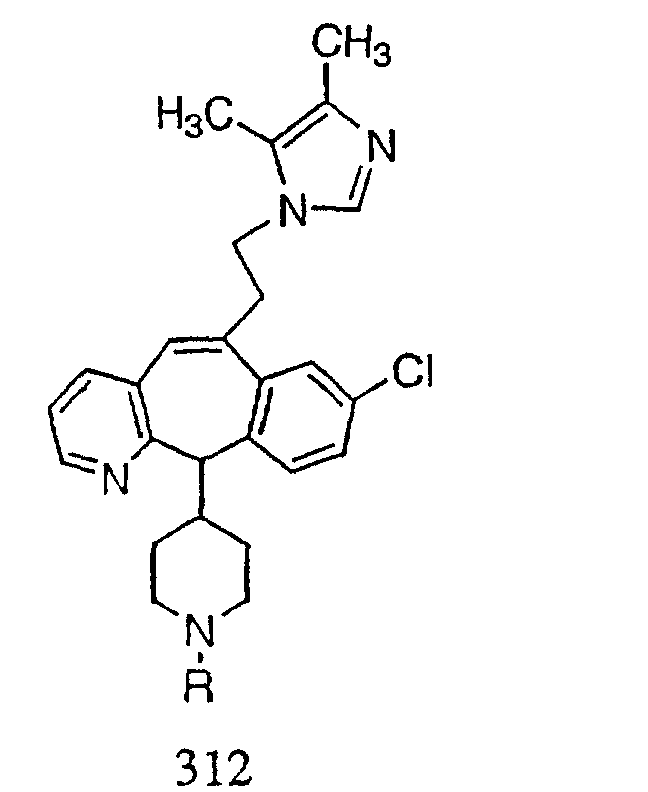
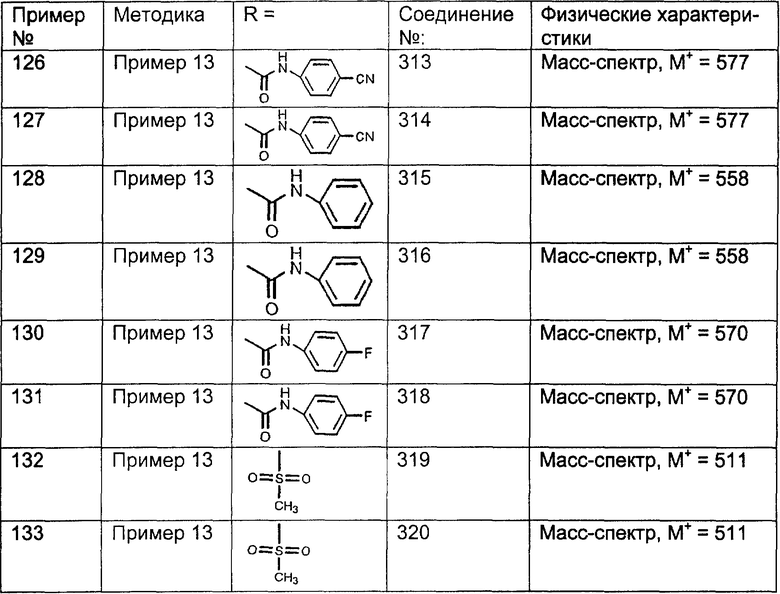
ПРИМЕР ПОЛУЧЕНИЯ 37
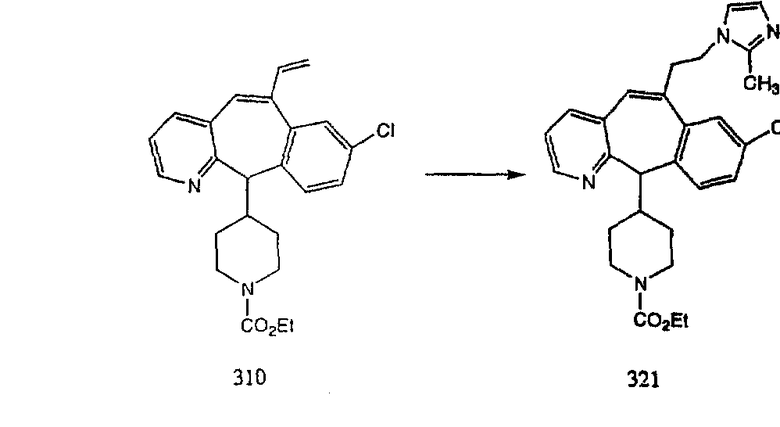
К раствору соединения (310), полученного в Примере получения 36, стадия A (0,66 г, 8,1 ммоль), в THF (4,0 мл) в атмосфере азота при -78°С по каплям прибавляют 2,5 М раствор H-BuLi/гексан (1,5 мл). Полученный раствор 30 мин перемешивают при -78°C, а затем прибавляют 1-метилимидазол (3,0 г, 7,3 ммоль) в THF (3,0 мл). Раствор затем нагревают при 120°C в течение выходных дней, а затем охлаждают до комнатной температуры и концентрируют досуха. Смесь экстрагируют с помощью EtOAc - Н2О, сушат над MgSO4, фильтруют и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% МеОН/97% NH3 - CH2Cl2, и получают продукт (321) (1,64 г, выход 46%, MH+ = 491,1).
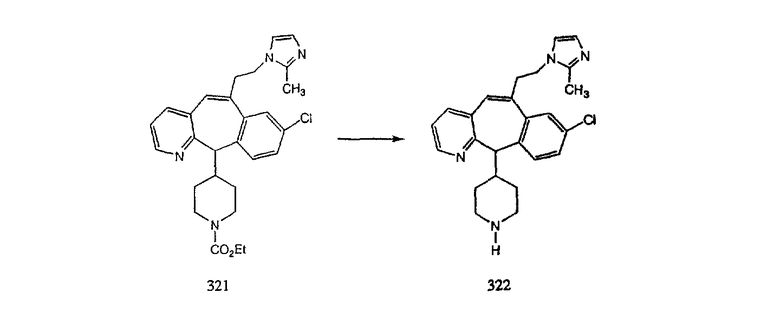
Раствор соединения (321), полученного выше в Примере получения 37, стадия A (0,6 г, 1,22 ммоль), в 12 н. растворе HCl (10 мл) кипятят с обратным холодильником в течение ночи, а затем концентрируют досуха, получая остаток в виде смолы. Этот остаток растворяют в насыщенном растворе NaHCO3, насыщают с помощью NaCl, a затем 10 мин перемешивают с CH2CI2. Твердое вещество отфильтровывают, а водный слой дважды экстрагируют с помощью СН2Cl2 и органический слой сушат над Na2SO4, фильтруют и концентрируют досуха, получая соединение (322) в виде светло-коричневого твердого вещества (566 мг, МН+ = 419,1).
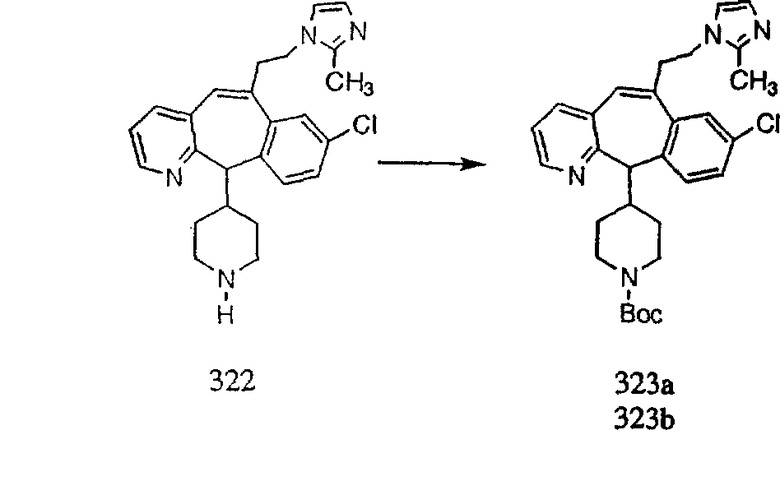
К раствору соединения (322), полученного выше на стадии B (0,566 г, 1,35 ммоль), в МеОН (20 мл) и hW (1 мл) при 0°С прибавляют Вос-ангидрид (0,44 г, 2,02 ммоль). Раствор подщелачивают с помощью 1 н. раствора NaOH, так чтобы рН = 8,5 -9,5, и концентрируют досуха, а затем экстрагируют с помощью CH2CI2 - Н2О, сушат над Na2SO4, фильтруют и концентрируют досуха, получая смесь изомеров 1 и 2 (0,63 г, выход 100%). Изомеры разделяют с помощью ВЭЖХ с использованием препаративной хиральной колонки AD, элюируя смесью 15% IPA/85% гексан/0,2% DEA (длина волны = 254 нм, ослабление = 64, поглощение = 1), и получают изомер 1 (323а) (0,28 г, MH+ = 519,2) и изомер 2 (323b) (0,28 г, MH+= 519,2).
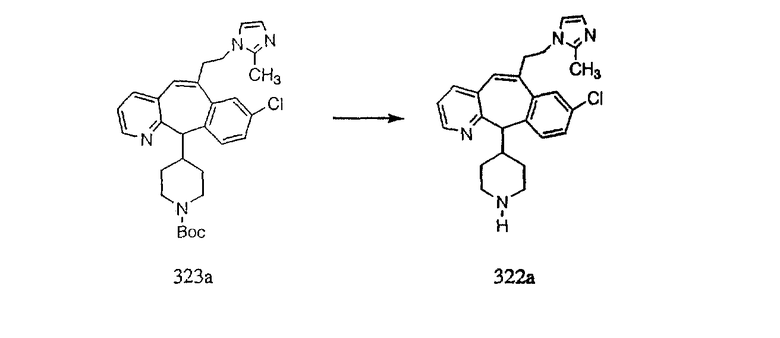
Раствор соединения (323а), изомер 1, полученного выше на стадии C (0,24 г, 0,46 ммоль), в растворе 4 н. HCl/диоксан (20 мл) перемешивают при комнатной температуре в течение 1 ч. К раствору прибавляют CH2Cl2 (7 мл) и реакционную смесь перемешивают 2 ч, а затем концентрируют досуха. Раствор 5 мин перемешивают с насыщенным раствором NaHCO3, насыщают с помощью NaCl, а затем трижды экстрагируют с помощью СН2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и выпаривают досуха, получая соединение (322а), изомер 1 (0,163 г, выход 84%, МН+= 419,2).
Соединение (322b) получают способом, аналогичным использованному на стадии D выше, применяя в качестве исходного соединение (323b), и получают второй изомер (0,193 г, выход 84%, МН+ = 419,2).
ПРИМЕРЫ 134-147
С использованием в качестве исходного соединение (322а) (изомер 1) или (322 (изомер 2) и проводя реакцию аналогично тому, как это описано в Примере 13, используя соответствующий изоцианат, хлорформиат или сульфонилхлорид (или, в случае карбоновой кислоты, используя сочетание с применением DEC), получают следующие соединения:
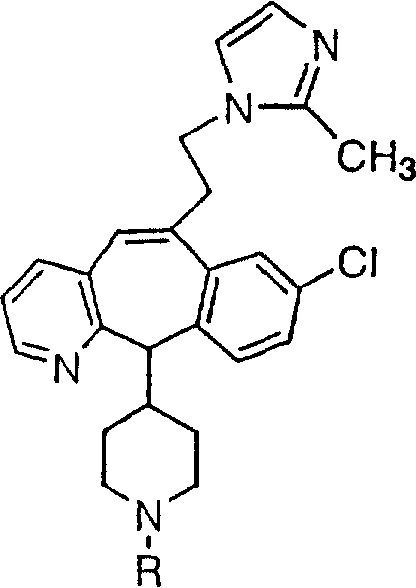
ПРИМЕР ПОЛУЧЕНИЯ 38
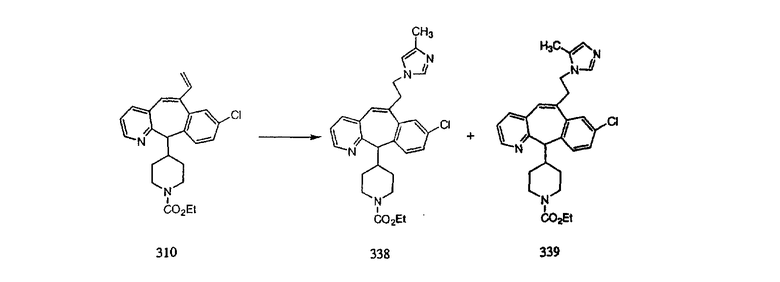
К раствору соединения (310), полученного в Примере получения 36, стадия A (3,0 г, 7,34 ммоль), в THF (8 мл) в атмосфере азота при -78°С по каплям прибавляют 2,5 Мраствор n-BuLi/гексан (0,65 мл, 8,07 ммоль). Полученный раствор 30 мин перемешивают при -78°C, а затем прибавляют 4-метилимидазол (0,66 г, 8,07 ммоль) в THF. Раствор в течение ночи нагревают при 120°C, охлаждают до комнатной температуры и концентрируют досуха. Реакционную смесь экстрагируют с помощью EtOAc-Н2О и органический слой сушат над MgSO4, фильтруют и концентрируют, получая смесь 4-метилзамещенного (338) и 5-метилзамещенного (339) продуктов (2,76 г, выход 76%, М+=491,1).
В Разделение соединений (338а/b) и (339а/b). Способом, аналогичным описанному в Примере 11, смесь продуктов, полученную выше на стадии А, с помощью ВЭЖХ на хиральной колонке сначала разделяют на чистые 4- и 5-замещенные (+)-энантиомеры и чистые 4- и 5-замещенные (-)-энантиомеры, а затем, после обработки трифенилметилхлоридом по методике, описанной в Примере 11, соединения далее разделяют на чистые изомеры 4-замещенного соединения (338а) (МС, M+ = 491; т. пл. = 72,1-73,0°С) и (338b) (МС, M+ = 491; т. пл. = 68,9 -69,0°С) и 5-замещенного соединения (339а) и (339b).
С Получение соединения (340а).
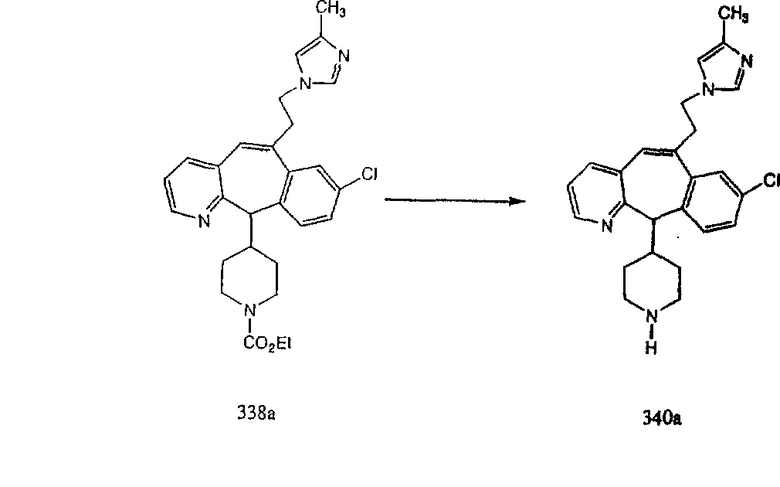
Раствор соединения (338а), полученного выше на стадии B (0,035 г, 0,071 ммоль), в 4 н. растворе HCl (2,0 мл) кипятят с обратным холодильником в течение ночи. Раствор охлаждают до комнатной температуры, подщелачивают раствором NH4OH и экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют, получая чистый изомер 1, соединение (340а) (0,0334 г, выход 100%, МН+ = 419,1; т. пл. = 60,3-61,0°С).
Способом, аналогичным использованному выше, применяя соединение (338b) (изомер 2), получают соединение (340b) (MH+ = 419,1).
ПРИМЕРЫ 148-156
Используя в качестве исходного (+)- или (-)-изомер соединения (340) и вводя его в реакцию аналогичным образом с использованием методики, указанной в приведенной ниже таблице, и соответствующего изоцианата, хлорформиата или сульфонилхлорида, получают следующие соединения:
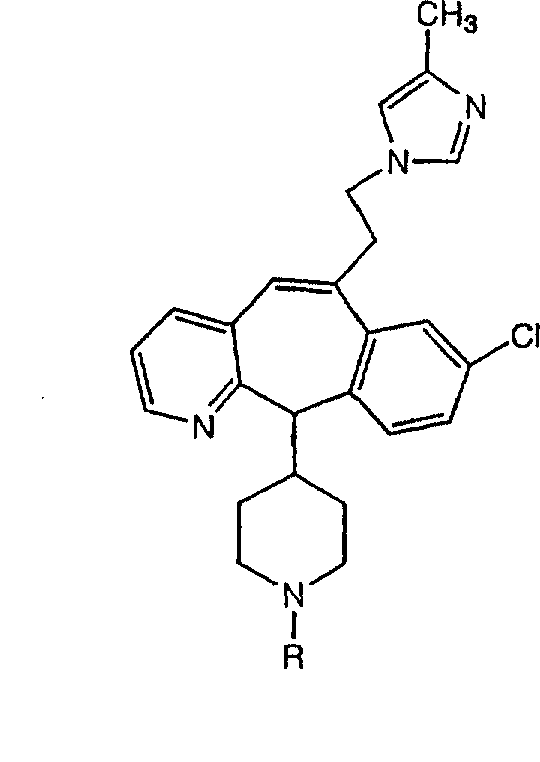
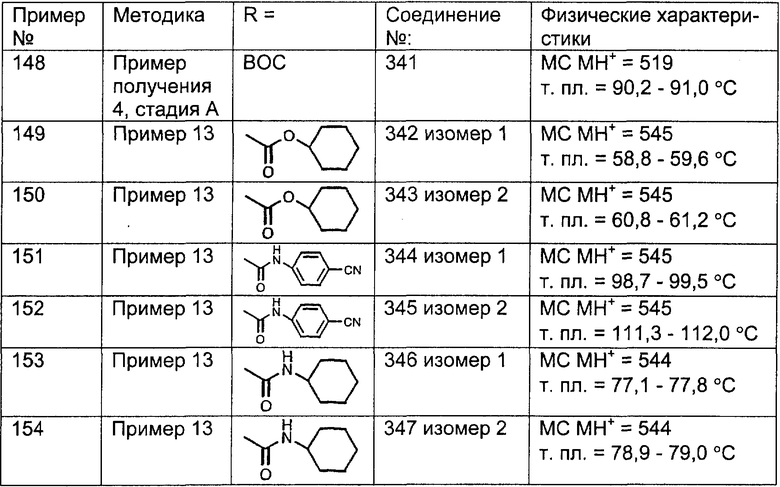
ПРИМЕР ПОЛУЧЕНИЯ 39
Получение соединения (350а).
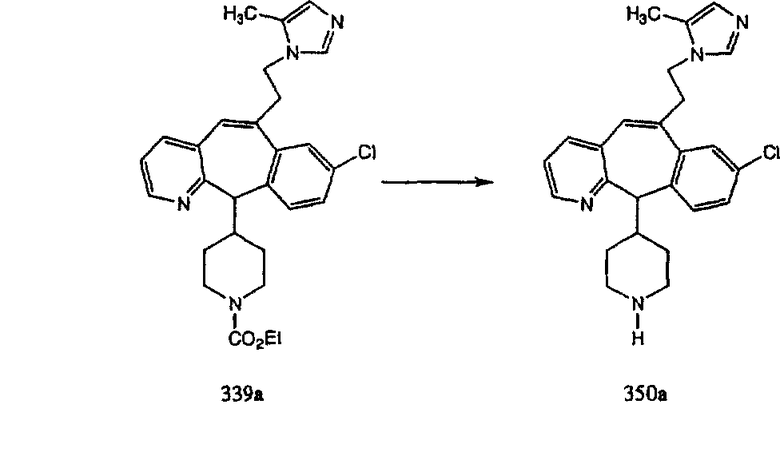
Соединение (339а) вводят в реакцию способом, аналогичным описанному в Примере получения 38, стадия C, и получают соединение (350а) (изомер 1) (0,13 г, выход 76%, MH+= 419,3).
Соединение (350b) (изомер 2) получают таким же образом, как и выше.
ПРИМЕРЫ 157-160
Используя в качестве исходного (+)- или (-)-изомер соединения (350) и вводя его в реакцию аналогичным образом с использованием методики, указанной в приведенной ниже таблице, и соответствующего Boc или изоцианата, получают следующие соединения:
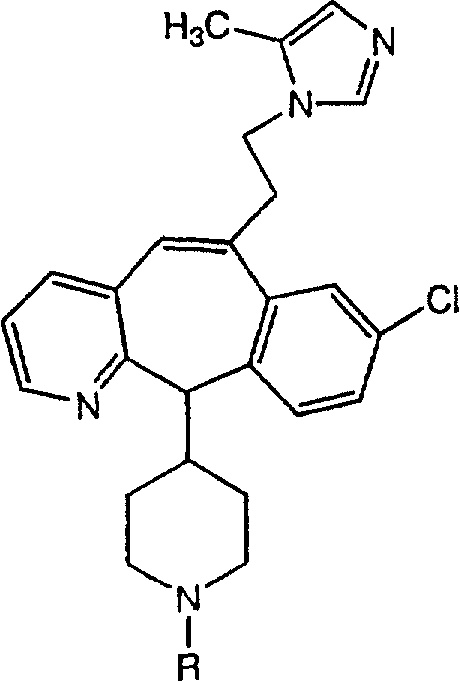
ПРИМЕР ПОЛУЧЕНИЯ 40
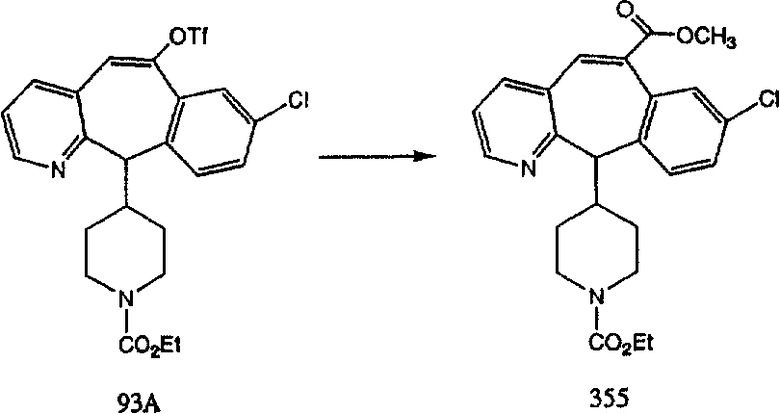
К раствору соединения (93А), полученного в Примере получения 7, стадия A (2,92 г, 5,5 моль), в безводном толуоле (70 мл) и МеОН (10 мл) прибавляют трифенилфосфин (0,72 г, 2,75 ммоль), DBU (1,11 мл, 7,42 ммоль) и PdCl2 (0,097 г, 0,55 ммоль). Полученный раствор продувают с помощью СО (100 фунтов/дюйм2), затем 5 ч нагревают при 80°С. Раствор охлаждают до комнатной температуры, продувают азотом и выпаривают досуха, получая коричневое масло. Продукт очищают с помощью хроматографии на колонке с силикагелем, элюируя с помощью смесей от 1% МеОН/99% CH2Cl2 до 4% МеОН/96% СН2Cl2, и получают соединение (355) (2,22 г, выход 92,5%, МН+ 441,1).
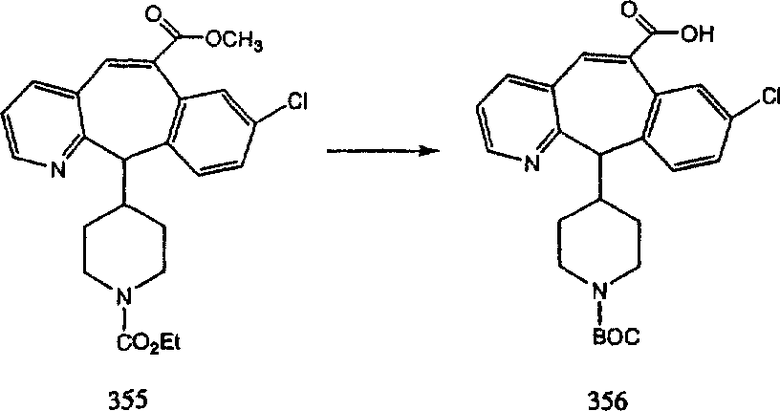
Раствор соединения (355), полученного в Примере получения 40, стадия A (2,2 г, 4,99 моль), в 6 н. растворе HCl (50 мл) в течение ночи нагревают при 100-110°С. Раствор охлаждают до комнатной температуры и выпаривают досуха, получая неочищенный продукт. К раствору неочищенного вещества в МеОН (50 мл) и Н2О (1 мл) при 0°C прибавляют Вос-ангидрид (1,63 г, 7,48 ммоль). Полученный раствор подщелачивают 1 н. раствором NaOH до рН = 8,5-9,5 и 2 ч перемешивают при 0°C, а затем выпаривают досуха и экстрагируют с помощью смеси EtOAc -5% раствор лимонной кислоты. Органический слой промывают с помощью Н2О, затем рассола, сушат над Na2SO4, фильтруют и концентрируют досуха, получая соединение (356) в виде желтого твердого вещества (2,29 г, выход 100%, МН+ = 455,1).
С Получение соединения (357).
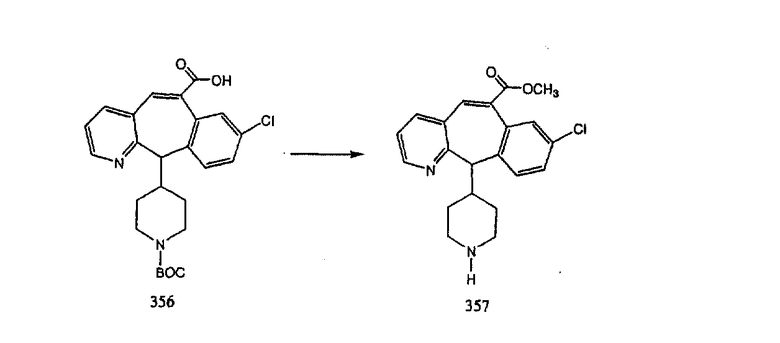
К раствору соединения (356), полученного выше в Примере получения 40, стадия В (2,26 г, 4,97 ммоль), в безводном бензоле (18,0 мл) и МеОН (2 мл) в течение 5 мин прибавляют (триметилсилил)диазометан (3 мл, 5,99 ммоль) в 2 М 1 н. гексане. Полученный раствор 1 ч перемешивают при комнатной температуре, затем выпаривают досуха, получая 2,33 г неочищенного вещества (MH+ = 369).
Раствор неочищенного вещества (полученного выше) в 4 н. растворе HCI в диоксане (25 мл) 1 ч перемешивают при комнатной температуре. Затем реакционную смесь выпаривают досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя с помощью смесей от 2% МеОН/98% СН2Cl2 до 6% МеОН/94% CH2CI2, а затем 50% (10% NH4OH/СН3ОН/50% CH2Cl2). Собранные фракции выпаривают досуха и разбавляют с помощью CH2Cl2. Затем органический раствор промывают насыщенным раствором NaHCO3 и рассолом, сушат над Na2SO4, фильтруют и выпаривают досуха, получая соединение (357) (1,26 г, выход 68,3%, MH+ = 369).
D Получение соединения (358).
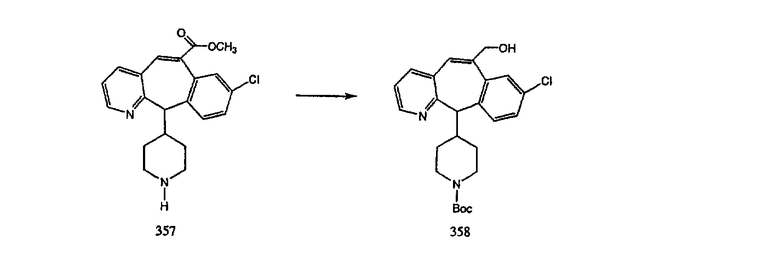
К раствору соединения (357), полученного выше в Примере получения 40, стадия C (0,6 г, 1,62 ммоль), в безводном THF (6 мл) при 0°C прибавляют DIBAL (1 М раствор в толуоле) (9,78 мл, 9,78 ммоль). Полученный раствор нагревают до комнатной температуры и перемешивают в течение ночи. Затем реакцию останавливают с помощью МеОН и реакционную смесь выпаривают досуха, получая неочищенный продукт.
К неочищенному веществу (полученному выше) в МеОН при 0°C прибавляют Вос-ангидрид (1,06 г, 4,9 ммоль). Полученный раствор подщелачивают 1 н. раствором NaOH до рН = 8,5 -9,5 и 1 ч перемешивают, а затем выпаривают досуха. Неочищенное вещество разбавляют с помощью CH2Cl2 с получением взвеси. Затем осадок фильтруют через целит и CH2Cl2 промывают с помощью H2O, затем рассола, сушат над Na2SO4, фильтруют и концентрируют досуха. Неочищенный спирт (358) (1,27 г, выход 100%) используют на следующей стадии без дополнительной очистки.
Е Получение соединения (359).
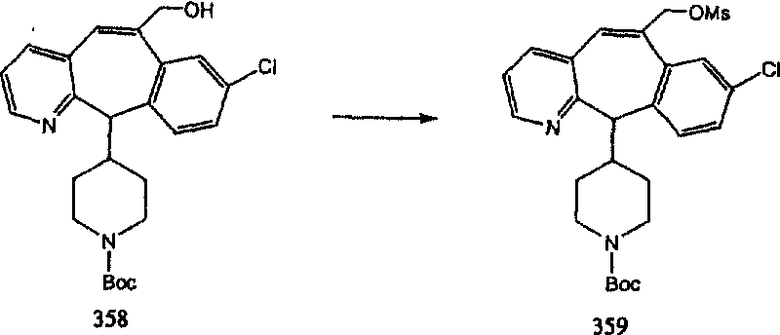
К охлажденному раствору спирта (358), полученному выше на стадии D (1,2 г, 2,73 ммоль), в безводном СН2Cl2 (12 мл) при 0°C прибавляют триэтиламин 1,14 мл, 8,18 ммоль) и метансульфонилхлорид (0,3 мл, 4,1 ммоль). Полученный раствор нагревают до комнатной температуры, перемешивают в течение ночи, затем реакцию останавливают с помощью H2О и перемешивают 10 мин. Реакционную смесь промывают водой, затем рассолом, фильтруют и выпаривают досуха, получая соединение (359) (1,22 г, выход 86%).
F Получение соединений (360а) и (360b).
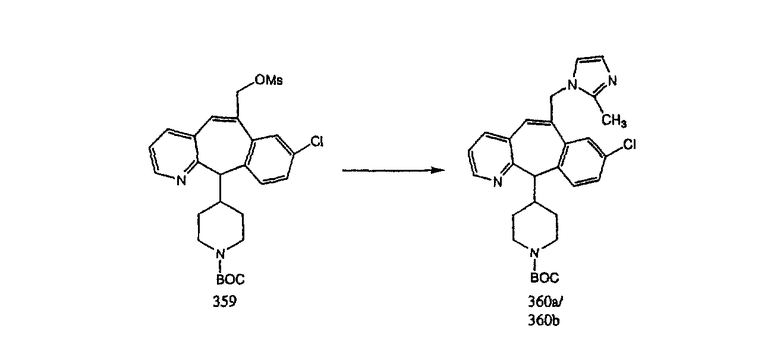
К раствору безводного DMF (5 мл) при 0°C прибавляют NaH (0,19 г, 8,18 ммоль) и 2-метилимидазол (0,67 г, 8,18 ммоль). Полученный раствор нагревают до комнатной температуры и перемешивают 20 мин. К этой реакционной смеси прибавляют раствор соединения (359), полученного выше на стадии Е (1,22 г, 2,3 ммоль), в безводном DMF (5 мл). Полученный раствор перемешивают в течение ночи при комнатной температуре, затем разбавляют с помощью EtOAc и промывают водой, затем рассолом. Органический слой сушат над Na2SO4, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя с помощью смесей от 1% МеОН/99% CH2Cl2 до 5% МеОН/95% СН2Cl2, и получают продукт в виде смеси изомеров (1,18 г, выход 100%, МН+ = 505,2). Разделение смеси продуктов с помощью ВЭЖХ с использованием препаративной колонки AD и с элюированием смесью 25% IPA/75% гексан/0,2% DEA (в изократическом режиме 60 мл/мин) дает чистый изомер 1 (360а) (0,251 г, MH+ = 505,1) и изомер 2 (360b) (0,251 г, MH+ = 505,1) в виде светло-розовых соединений.
G Получение соединений (361 а) и (361 b).
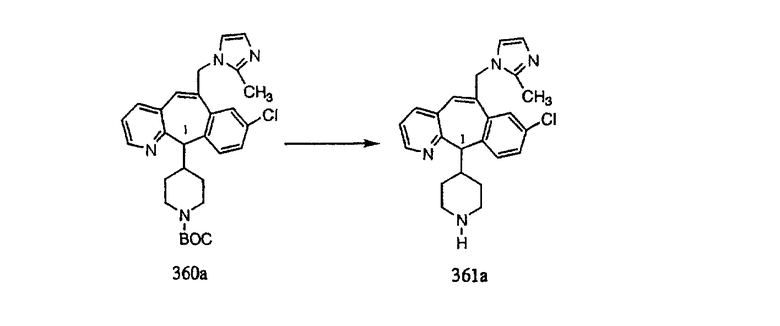
Раствор соединения (360а) (изомер 1), полученного выше на стадии F (0,2 г, 0,4 ммоль), в 4 н. растворе HCl в диоксане (10 мл) 2 ч перемешивают при комнатной температуре, затем выпаривают досуха и получают соединение (361 а) (0,292 г, выход 100%).
Соединение (361b) (изомер 2) получают способом, аналогичным использованному выше, с использованием в качестве исходного соединения (361 b), полученного в Примере получения 40, стадия F.
ПРИМЕРЫ 161-166
Используя в качестве исходного (+)- или (-)-изомер соединения (361) и вводя его в реакцию способом, сходным с описанным в Примере 13, и с использованием соответствующего изоцианата, указанного в приведенной ниже таблице, получают следующие соединения:
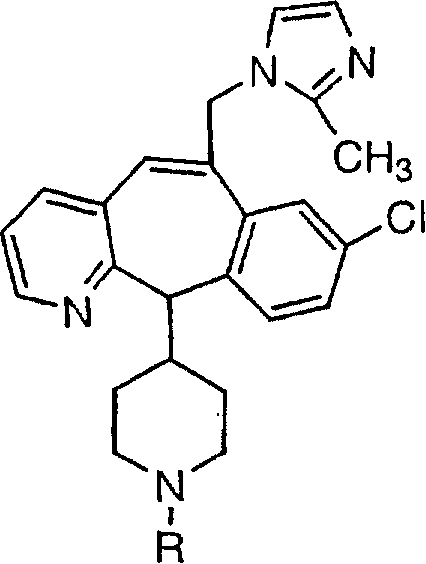
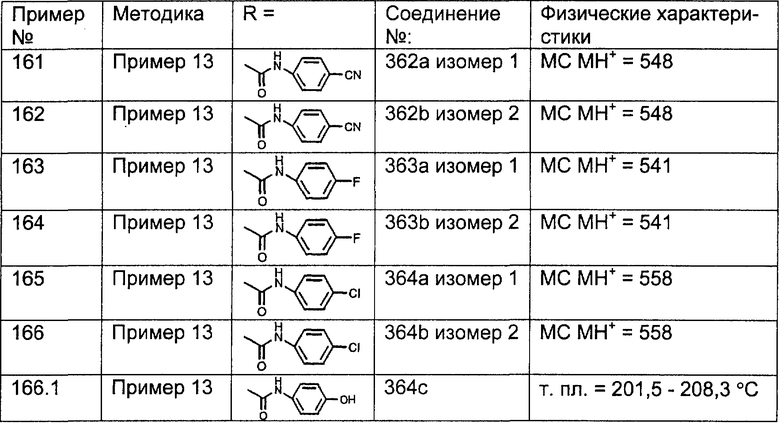
ПРИМЕР ПОЛУЧЕНИЯ 41
Соединение (365).
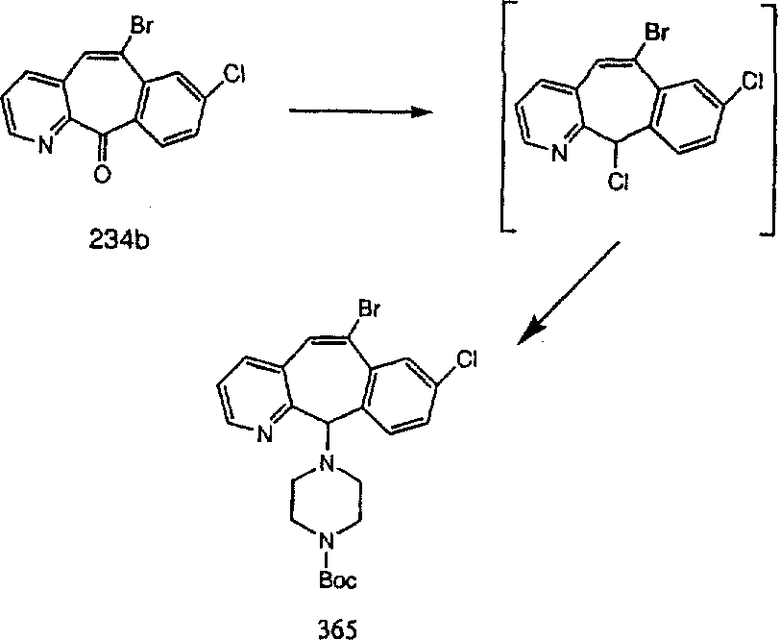
В основном таким же образом, как в Примере получения 23, стадии А-D, с использованием 6-бромзамещеного продукта, полученного на стадии B, т. е. соединения (234b), получают соединение (365) (76,6 г, выход 100%).
ПРИМЕР ПОЛУЧЕНИЯ 42
А Получение соединения (366).
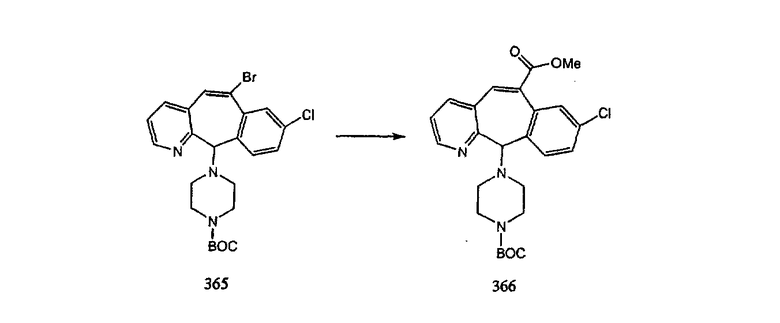
К раствору соединения (365), полученного в Примере получения 41 (4,0 г, 8,16 ммоль), в толуоле (75 мл) и МеОН (20 мл) прибавляют трифенилфосфин (1,099 г, 4,08 ммоль), DBU (1,7 г, 11,02 ммоль) и хлорид палладия (0,145 г, 0,82 ммоль). Полученный раствор в течение 5 ч обрабатывают с помощью СО при 78-82°C и давлении 100 фунтов/дюйм2, а затем экстрагируют с помощью EtOAc-H2O. Объединенные органические слои затем промывают рассолом, сушат над Na2SO4, концентрируют досуха и очищают с помощью хроматографии на колонке, элюируя смесью 30% EtOAc/70% гексан, и получают соединение (366) (3,12 г, выход 100%, МН+ = 470,1).
В Получение соединения (367).
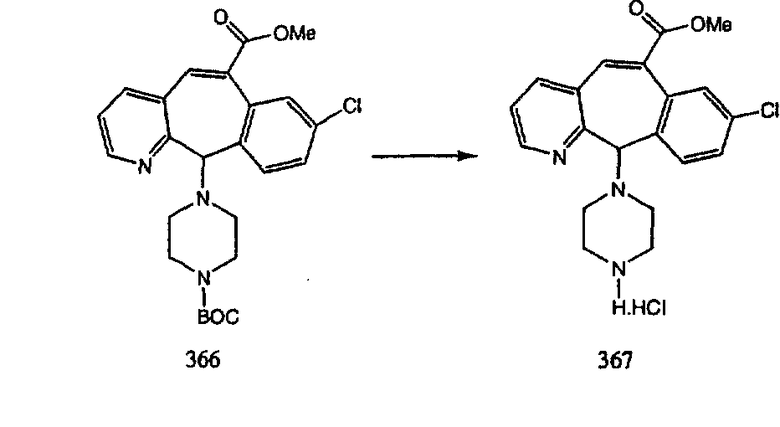
Раствор соединения (366), полученного выше на стадии A (3,1 г, 6,6 ммоль), в 4 М растворе HCl/диоксан (1,20 мл) перемешивают в течение 3 ч, а затем концентрируютдосуха и получают неочищенную соль - соединение (367) (3,89 г, выход 100%, МН+ = 370,2).
С Получение соединения (368).
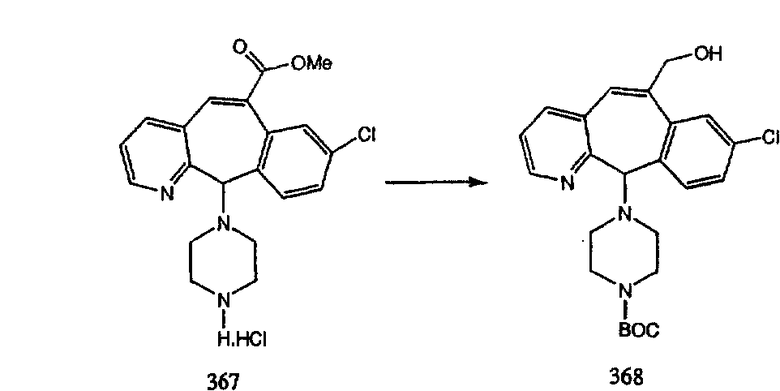
К раствору соединения (367), полученного выше на стадии B (3,43 г, 8,45 ммоль), в THF (60 мл) при 0°С прибавляют DIBAL (7,21 г, 50,7 ммоль). Полученный раствор нагревают до комнатной температуры, перемешивают в течение ночи, а затем концентрируют досуха, после чего прибавляют Вос-ангидрид (3,69 г, 16,9 ммоль). Затем реакционную смесь экстрагируют с помощью CH2Cl2 - Н2O, фильтруют, сушат над Na2SO4 и концентрируют досуха, получая соединение (368) (3,75 г, выход 100%, W = 442,4).
С.1 Альтернативный способ получения соединения (368).
Раствор соединения (366), полученного выше на стадии А (23,46 г, 50,98 ммоль), в CH2Cl2 - МеОН - Н2О (120 мл, 600 мл, 60 мл соответственно) смешивают с LiOH (12,0 г, 350,88 ммоль) и при 40°С нагревают с обратным холодильником в течение ночи. Из реакционной смеси удаляют растворитель и остаток разбавляют с помощью CH2Cl2 и подкисляют до рН = 6 с помощью 1 н. раствора HCl. Органический слой отделяют и промывают водой, сушат над Na2SO4 и концентрируют. Продукт растворяют в THF (285 мл) при 0°С. Прибавляют триэтиламин (6 мл, 42,97 ммоль) и этилхлорформиат (4,1 мл, 42,97 ммоль) и 1 ч перемешивают при 0°С. Реакционную смесь фильтруют и фильтрат охлаждают до -70°С. К этому фильтрату прибавляют NaBH4 (3,97 г, 104,94 ммоль) и 1 ч перемешивают при -70°С, после чего по каплям прибавляют 40 мл МеОН. Растворители удаляют и остаток растворяют в метиленхлориде, промывают насыщенным водным раствором NaHCO3, затем рассолом,сушат над Na2SO4 и концентрируют, получая соединение (368) в виде твердого вещества.
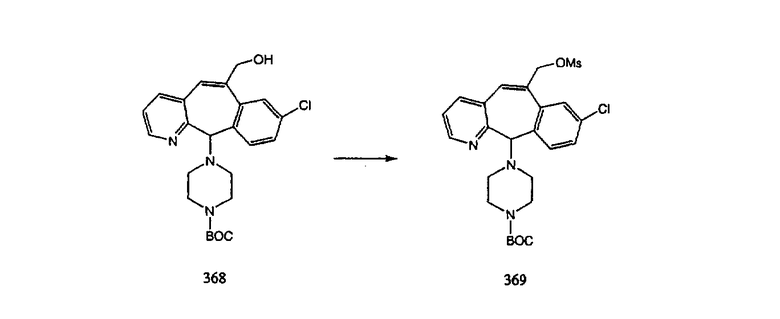
К раствору соединения (368), полученного выше на стадии С (3,74 г, 8,46 ммоль), в СН2Cl2 (100 мл) прибавляют триэтиламин (3,5 мл, 25,38 ммоль) и метансульфонилхлорид (1,45 г, 2,7 ммоль). Полученный раствор в течение ночи перемешивают при комнатной температуре в атмосфере азота, а после этого промывают насыщенным раствором NaHCO3, затем рассолом и сушат над Na2SO4, получая мезилат (369) (3,86 г, выход 88%).
Е Получение соединений (370а) и (370b).
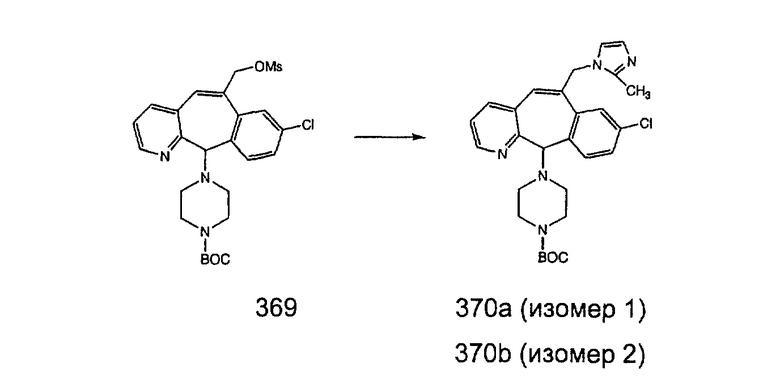
К раствору 2-метилимидазола (2,43 г, 29,68 ммоль) в DMF (30 мл) в атмосфере N2 прибавляют NaH (0,53 г, 22,3 ммоль) и 10 мин перемешивают, а затем прибавляют соединение (369), полученное выше на стадии D (3,86 г, 7,42 ммоль). Раствор перемешивают в течение ночи. Затем раствор концентрируют досуха и экстрагируют с помощью EtOAc-NaHCO3, сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке, элюируя с помощью 2% Ме-ОН-NH3/98% CH2Cl2, и получают смесь изомеров. Последующее разделение проводят с помощью препаративной хиральной ВЭЖХ с использованием колонки AD, элюируя смесью 25% IPA/75% гексан/0,2% DEA, и получают чистое соединение (370а) (изомер 1) (0,160 г) и соединение (370b) (изомер 2) (0,140 г) (МН+ = 506,1).
F Получение соединений (371а) и (371b).
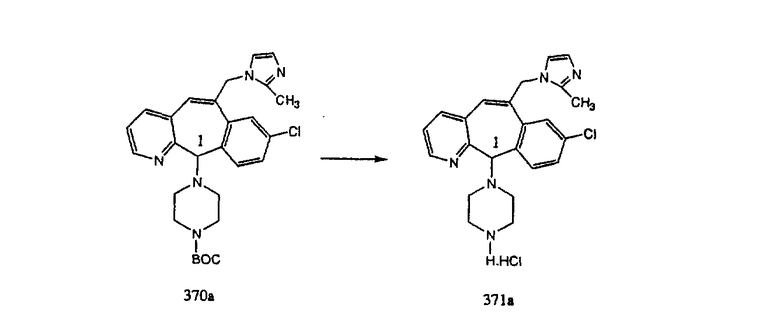
Раствор соединения (370а) (изомер 1), полученного выше на стадии Е (0,105 г, 0,21 ммоль), в 4М растворе HCl/диоксан (10 мл) 3 ч перемешивают при комнатной температуре и концентрируют досуха и получают соединение (371а) (0,147 г, выход 100%).
Соединение (370b) (изомер 2), полученное на стадии Е, обрабатывают таким же образом, как и изомер 1 выше, и получают соединение (371b) (изомер 2).
ПРИМЕР 167
Получение соединения (372).
К раствору соединения (371 а) (1,3 г, 2,94 ммоль) в CH2Cl2 (60 мл) прибавляют триэтиламин (1,3 мл, 9,4 ммоль) и п-цианофенилизоцианат (0,466 г, 3,24 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, затем экстрагируют с помощью CH2Cl2 и насыщенного раствора NaHCO3. Органический слой сушат над Na2SO4, выпаривают и остаток очищают с помощью хроматографии на колонке, элюируя смесью 1-2% МеОН - NH3/98% CH2Cl2, и получают соединение (372) (0,870 г, выход 48%), см. приведенную ниже таблицу.
ПРИМЕР 168
Получение соединения (373).
Соединение (371b) (изомер 2) вводят в реакцию аналогично тому, как это описано в Примере 13, с использованием п-цианофенилизоцианата и получают соединение (373), см. приведенную ниже таблицу.
ПРИМЕР 169
Получение соединения (374).
Соединение (371а) (изомер 1) вводят в реакцию аналогично тому, как это описано в Примере 13, с использованием п-цианофенилизоцианата и получают соединение (374), см. приведенную ниже таблицу.
ПРИМЕР 170
Получение соединения (375).
Соединение (371b) (изомер 2) вводят в реакцию аналогично тому, как это описано в Примере 13, с использованием п-хлорфенилизоцианата и получают соединение (375), см. приведенную ниже таблицу.
ПРИМЕРЫ 167-170
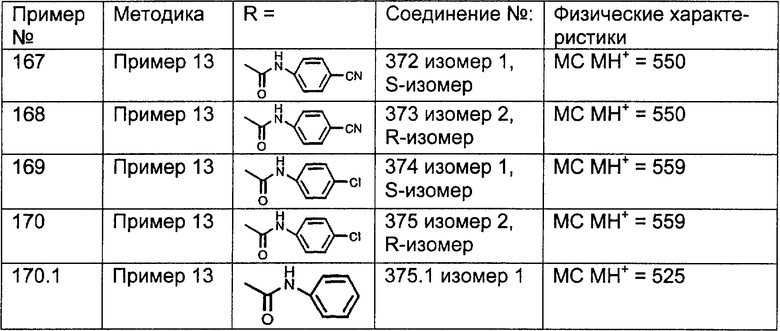
ПРИМЕР ПОЛУЧЕНИЯ 43
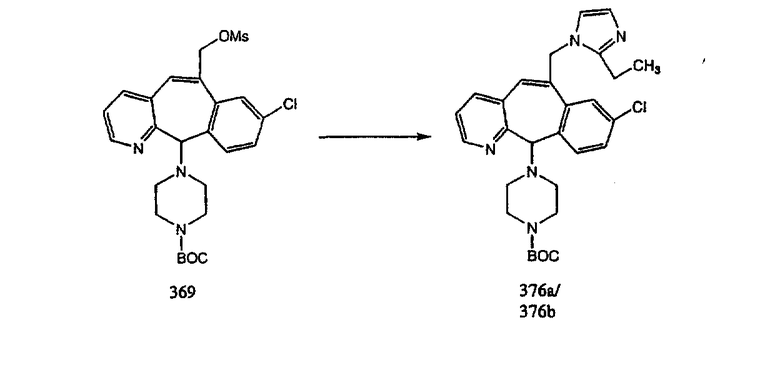
К раствору 1-этилимидазола (0,33 г, 3,46 ммоль) в DMF (5 мл) в атмосфере азота прибавляют NaH (0,083 г, 3,46 ммоль) и 10 мин перемешивают, а затем прибавляют соединение (369), полученное в Примере получения 42, стадия D (0,6 г, 1,15 ммоль) и перемешивают в течение ночи. Затем раствор выпаривают досуха, разбавляют этилацетатом, промывают бикарбонатом натрия, сушат над сульфатом натрия и концентрируют досуха. Реакционную смесь очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% МеОН/97% CH2Cl2, и получают смесь изомеров. Последующее разделение проводят с помощью препаративной ВЭЖХ с использованием хиральной колонки AD и получают чистое соединение (376а) (изомер 1) и соединение (376b) (изомер 2) (MH+ = 520,1).
В Получение соединений (377а) и (377b).
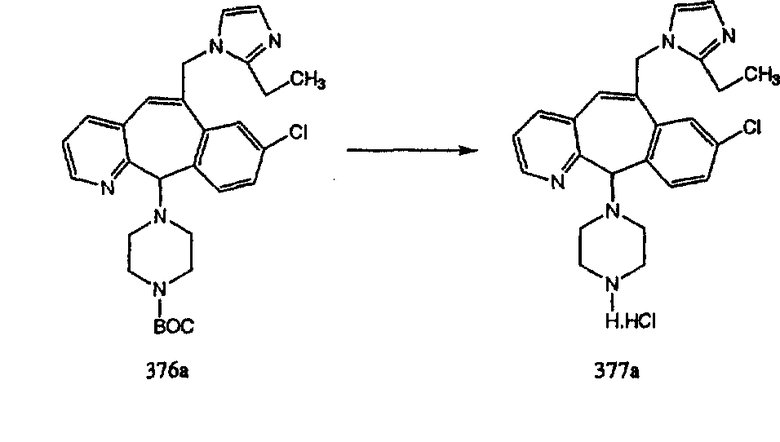
Раствор соединения (376а), полученного на стадии А (0,107 г, 0,2 ммоль), в 4 М растворе HCl в диоксане (10 мл) перемешивают при комнатной температуре в течение 2 ч, а затем концентрируют досуха, получая соединение (377а) (изомер 1) (0,13 г, выход 100%, MH+= 420,1).
Соединение (376b) вводят в реакцию способом, аналогичным использованному выше, и получают соединение (377b) (изомер 2) (МН+ = 420,1).
ПРИМЕРЫ 171-174
С использованием соответствующего (+)- или (-)-изомера соединения (377) в качестве исходного вещества и путем введения его в реакцию аналогично тому, как это описано в Примере 13, с использованием соответствующего изоцианата, указанного в приведенной ниже таблице, получают следующие соединения:
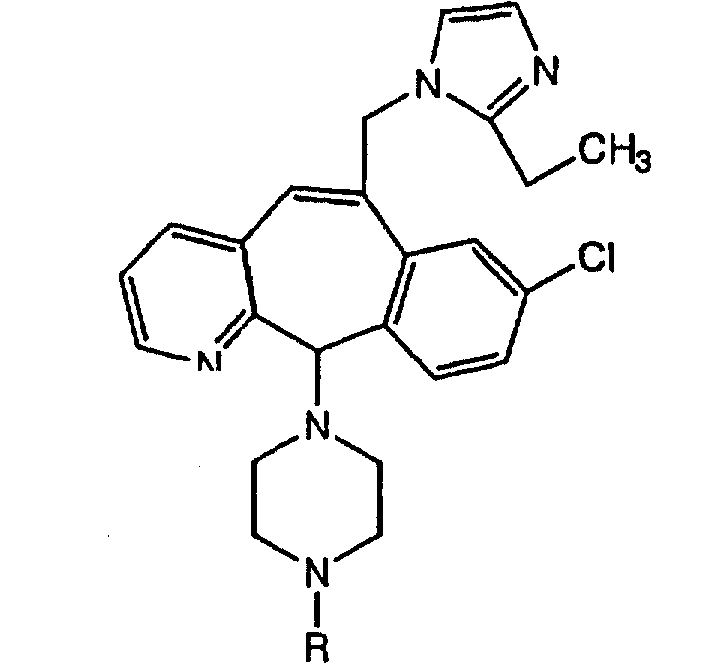
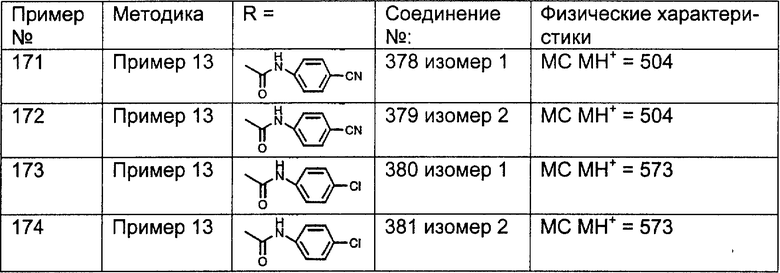
ПРИМЕР ПОЛУЧЕНИЯ 44
Соединения(382а)и (382b).
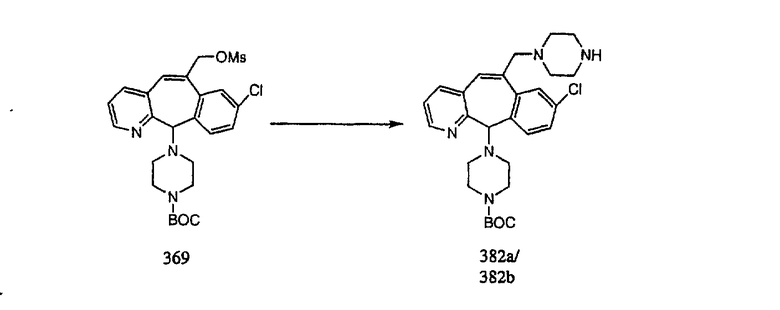
К раствору соединения (369), полученного в Примере получения 42, стадия D (0,5 г, 0,96 ммоль), в CH3CN (80 мл) прибавляют пиперазин (0,25 г, 2,88 ммоль) и 2,6-бис(диметил)-1-метилпиперидин (0,597 г, 3,84 ммоль). Полученный раствор 4 ч перемешивают при комнатной температуре, концентрируют досуха и экстрагируют с помощью CH2Cl2 - NaHCO3. Объединенные органические слои сушат над Na2SO4 и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% МеОН/97% CH2Cl2, и получают продукт, состоящий из 2 изомеров (0,28 г, выход 57%). Эти два изомера разделяют с помощью ВЭЖХ с использованием хиральной колонки adm получают чистое соединение (382а) (изомер 1) (0,136 г, MH+ = 510,3) и соединение (382b) (изомер 2) (0,14 г, MH+ = 510,3).
ПРИМЕР ПОЛУЧЕНИЯ 45
А Соединения (383а) и (383b).
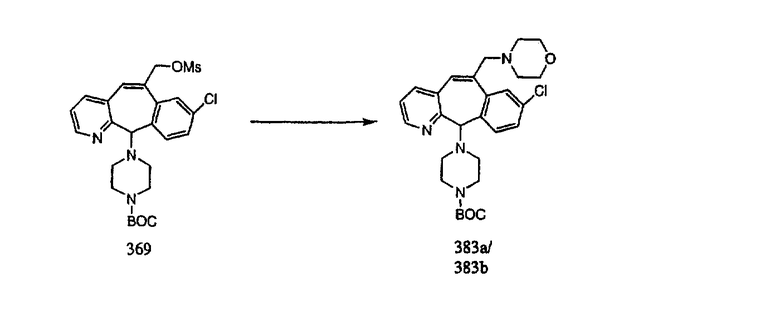
К раствору соединения (369), полученного в Примере получения 42, стадия D (1,2 г, 2,31 ммоль), в CH3CN (100 мл) прибавляют морфолин (0,8 г, 9,23 ммоль) и 2,6-бис(диметил)-1-метилпиперидин (1,9 г, 12,24 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи и концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - NaHCO3. Объединенные органические слои сушат над Na2SO4 и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 1% NH3 - МеОН/99% CH2Cl2, и получают продукт, состоящий из двух изомеров (1,1 г, выход 82%). Эти два изомера разделяют помощью ВЭЖХ с использованием хиральной колонки AD и получают чистое соединение (383а) (изомер 1) (0,24 г, MH+ = 425,1) и соединение (383b) (изомер 2) (0,112 г, MH+ = 425,1).
В Получение соединения (384а).
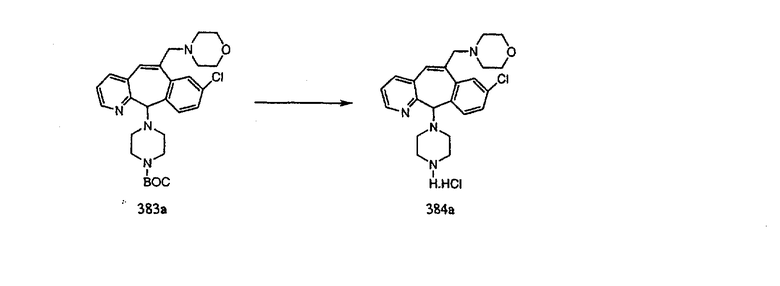
Раствор соединения (383а), полученного на стадии А (0,19 г, 0,37 ммоль), в 4 М растворе HCI/диоксан (25 мл) 2,5 ч перемешивают при комнатной температуре и концентрируют досуха, получая соединение (384а) (0,194 г, МН+ = 411,1).
Соединение (384b) получают способом, аналогичным использованному выше, с использованием в качестве исходного соединения (383b), полученного на стадии А.
ПРИМЕР 175
Получение соединений (385а) и (385b).
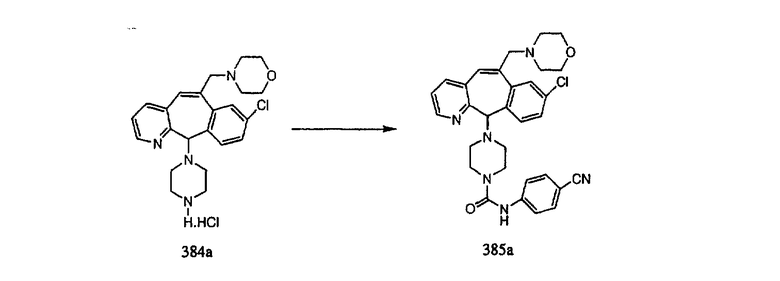
К раствору соединения (384а), полученного выше в Примере получения 45, стадия В (0,05 г, 0,11 ммоль), в безводном CH2Cl2 (5 мл) прибавляют триэтиламин (0,036 г, 0,36 ммоль) и 4-цианофенилизоцианат (0,018 г, 0,173 ммоль). Полученный раствор 4 ч перемешивают при комнатной температуре в атмосфере азота и концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - NaHCO3. Объединенные органические слои сушат над Na2SO4 и концентрируют досуха, получая соединение (385а) (изомер 1) (0,06 г, выход 100%, NH+ = 555,4).
С использованием в качестве исходного соединения (384b), полученного в Примере получения 45, стадия В, и проводя реакцию таким же образом, как и выше, получают соединение (385b) (изомер 2) (МН+ = 555,4).
ПРИМЕР ПОЛУЧЕНИЯ 46
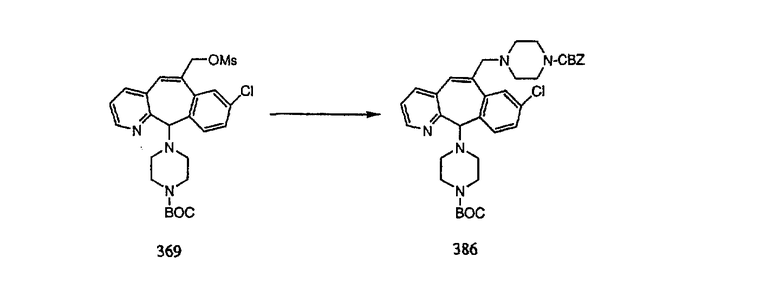
К раствору соединения (369), полученного в Примере получения 42, стадия D (3,0 г, 5,77 ммоль), в CH3CN (150 мл) прибавляют 2,6-бис(диметил)-1-метилпиперидин (7,16 г, 16,16 ммоль) и бензил-1-пиперазинкарбоксилат (7,61 г, 34,62 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - NaHCO3. Объединенные органические слои сушат над Na2SO4, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 1% NH3 - МеОН/99% CH2Cl2, а затем смесью 30% EtOAc/70% гексан, и получают искомое соединение (386) (1,24 г, выход 67%, MH+ = 644,2).
В Получение соединения (387).
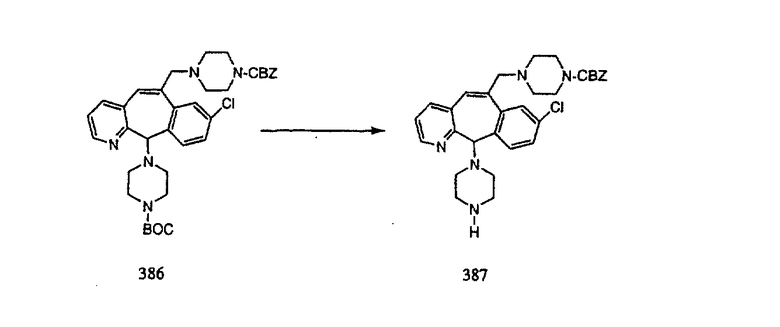
Раствор соединения (386), полученного выше на стадии А (0,5 г, 0,77 ммоль), в 4 М растворе HCl/диоксан (50 мл) 2 ч перемешивают при комнатной температуре. Затем раствор выливают на лед и подщелачивают 1 н. раствором NaOH, а после этого экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над Na2SO4и концентрируют досуха, получая соединение (387) (0,43 г, выход 100%, МН+ = 544,5).
С Получение соединений (388а) и (388b).
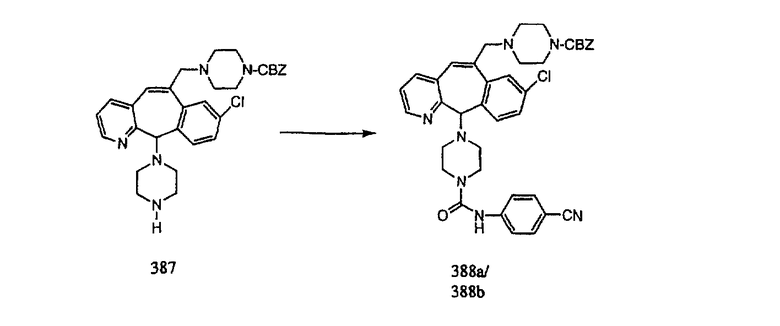
Соединение (387), полученное выше на стадии В, вводят в реакцию способом, аналогичным описанному в Примере 175, и получают смесь 2 изомеров (0,102 г, выход 55%). Последующее разделение изомеров с помощью ВЭЖХ на хиральной колонке AD дает чистое соединение (388а) (изомер 1) (0,05 г, МН+ = 688,2) и соединение (388b) (изомер 2) (0,048 г, MH+ = 688,2)
ПРИМЕРЫ 176 И 177
Соединение (387), полученное в Примере получения 46, стадия В, вводят в реакцию способом, аналогичным описанному в Примере 175, и с использованием соответствующего изоцианата, указанного в приведенной ниже таблице, и получают следующие соединения:
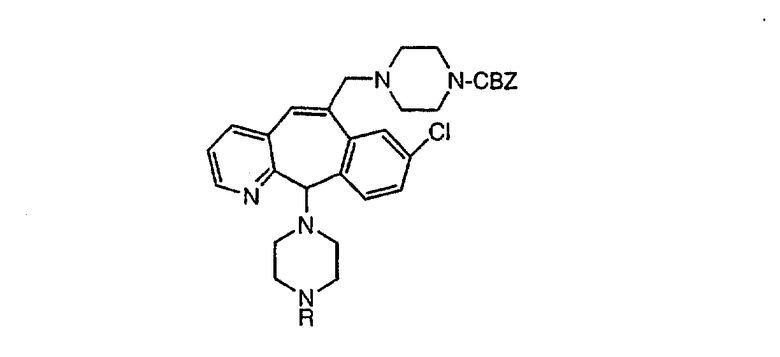
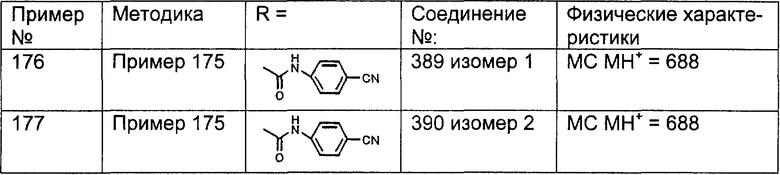
ПРИМЕР 178
Получение соединений (391а) и (391b).
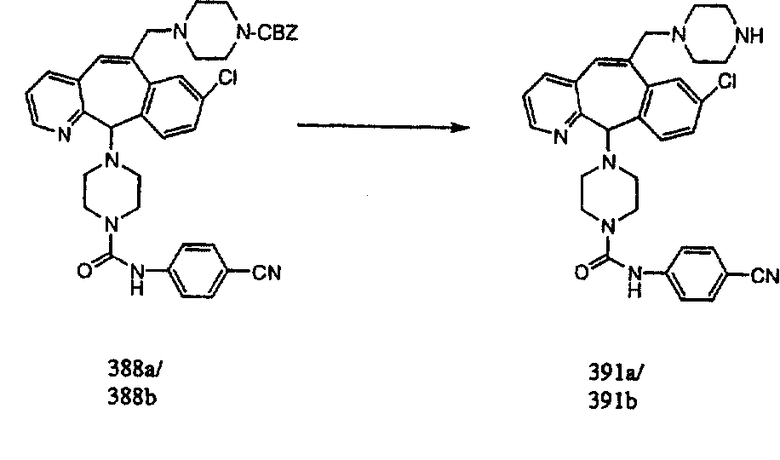
К раствору соединения (388а), полученного в Примере получения 46, стадия С (0,05 г, 0,086 ммоль), в CH3CN (1 мл) при 0°С прибавляют йодтриметилсилан (0,05 мл, 0,343 ммоль). Полученный раствор 1 ч перемешивают при 0°С и концентрируют досуха. Затем остаток выливают в 1 н. раствор HCl, а после этого экстрагируют эфиром. Затем водный слой подщелачивают 10% раствором NH4OH и после этого экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над Na2SO4 и концентрируют досуха, получая соединение (391а) (изомер 1) (0,02 г, выход 42,5%, MH+= 554,1).
С использованием в качестве исходного соединения (388b), полученного в Примере получения 46, стадия С, и проводя реакцию таким же образом, как и выше, получают соединение (391b) (изомер 2) (MH+ = 554,1).
ПРИМЕР ПОЛУЧЕНИЯ 47
А Соединение (394).
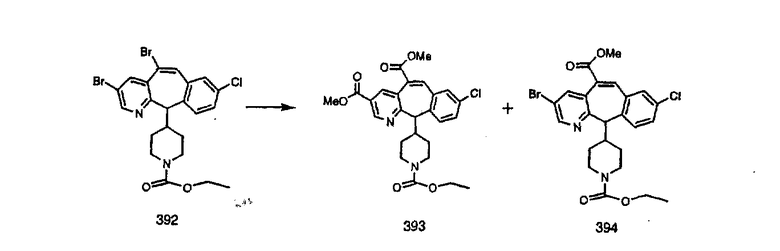
К раствору соединения (392) (5,0 г, 9,24 ммоль), полученного по методике, описанной в Journal of Medicinal ChemiStry (1988), 41(10), 1563, в МеОН (20 мл) и толуоле (50 мл) при комнатной температуре прибавляют трифенилфосфин (1,21 г, 4,62 ммоль), DBU (1,90 г, 12,48 ммоль) и хлорид палладия (0,16 г, 0,92 ммоль). Полученный раствор 6 ч перемешивают при 80°С, а затем в течение ночи при комнатной температуре. Затем раствор концентрируют досуха и получают два продукта. Искомый продукт очищают с помощью хроматографии на колонке с нормальной фазой силикагеля, элюируя смесью 30% EtOAc/70% гексан, и получают белое твердое соединение (394) (2,24 г, выход 47%, MH+ = 521,1).
В Получение соединения (395).
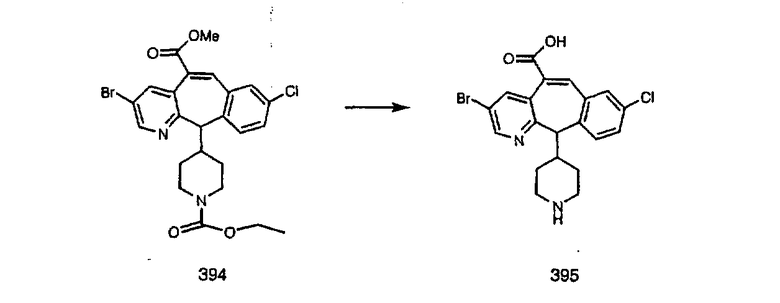
Раствор соединения (394), полученного выше на стадии А (2,38 г, 4,58 ммоль), в концентрированной HCl (40 мл) кипятят с обратным холодильником в течение ночи. Затем раствор охлаждают до комнатной температуры и подщелачивают раствором NH4OH, а после этого экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют досуха, получая белое твердое соединение (395) (1,03 г, выход 52%, MH+ = 435,1).
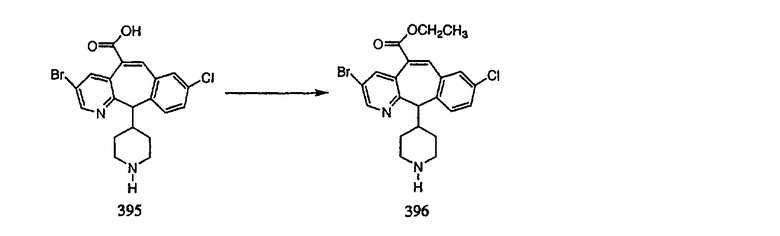
Через раствор соединения (395), полученного на стадии В (1,03 г, 2,37 ммоль), в чистом EtOH (50 мл) в течение 5 мин барботируют безводный газообразный СН2Cl2. Затем раствор 30 мин нагревают при 60°С, охлаждают до комнатной температуры и концентрируют досуха, получая соединение (396) (1,1 г, выход 100%, МН+ = 463,1).
D Получение соединения (397).
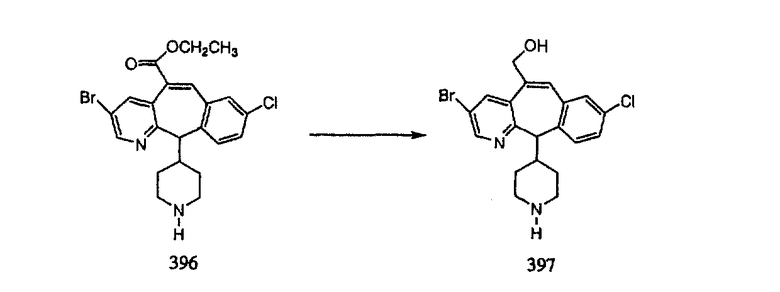
К раствору соединения (396), полученного на стадии С (1,09 г, 2,19 ммоль), в THF (10 мл) при 0°С по каплям прибавляют DIBAL/толуол (11,0 мл, 10,95 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, затем реакцию останавливают с помощью H2O и концентрируют досуха, получая светло-коричневое твердое соединение (397) (1,2 г, выход 100%, МН+ = 421,1).
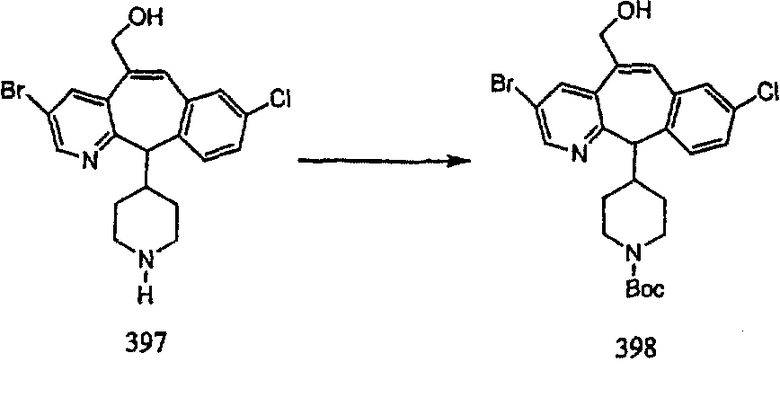
К раствору соединения (397), полученного на стадии D (0,92 г, 2,19 ммоль), в смеси 50% МеОН/1% H2O (50 мл) при комнатной температуре прибавляют Вос-ангидрид (0,95 г, 4,38 ммоль). Значение рН полученного раствора доводят до 9 и его 4 ч перемешивают при комнатной температуре, затем концентрируют досуха, после чего экстрагируют с помощью CH2Cl2 - Н2О. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют досуха, получая светло-коричневое твердое соединение (398) (0,91 г, выход 80%, MH+ = 521,1).
F Получение соединения (399).
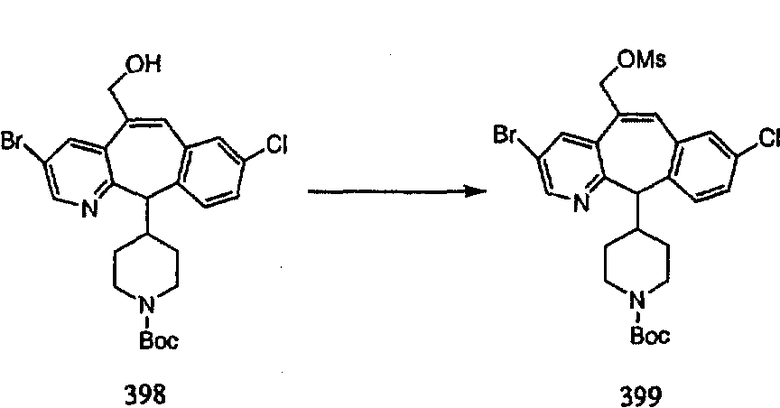
К раствору соединения (398), полученного на стадии Е (0,91 г, 1,75 ммоль), в CH2Cl2 (10 мл) прибавляют триэтиламин (0,73 мл, 5,25 ммоль) и метансульфонилхлорид (0,3 г, 2,62 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, а затем промывают раствором NaHCO3, сушат над Na2SO4, фильтруют и концентрируют досуха, получая мезилат в виде светло-желтого твердого соединения (399) (0,94 г, выход 90%).
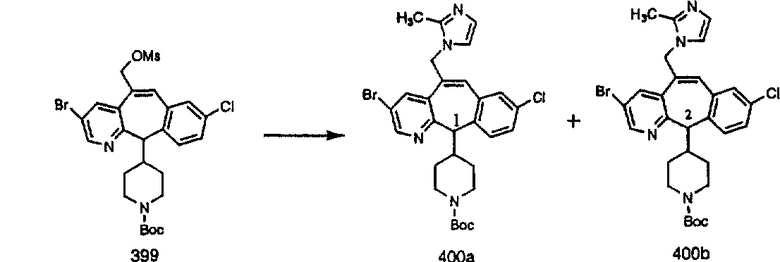
К раствору соединения (399), полученного на стадии F (0,93 г, 1,60 ммоль), в DMF (10 мл) в атмосфере азота прибавляют 2-метилимидазол (0,19 г, 2,3 ммоль) и NaH (0,037 г). Полученный раствор 15 мин перемешивают при комнатной температуре, а затем 3 ч при 90°С. Затем раствор охлаждают до комнатной температуры и концентрируют досуха, а после этого экстрагируют с помощью СН2Cl2 - NaHCO3. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют и очищают с помощью хроматографии на колонке с нормальной фазой силикагеля, элюируя смесью 5% МеОН - NH3/95% СН2Cl2, и получают смесь двух изомеров в виде светло-красного твердого вещества (0,39 г, выход 42%, MH+ = 585,1). Эти 2 изомера разделяют с помощью препаративной ВЭЖХ с использованием хиральной колонки AD, элюируя смесью 15% IPA/85% гексан/0,2% DEA, и получают соединение (400а) (изомер 1) в виде светло-коричневого твердого вещества (0,10 г, выход 11%) и соединение (400b) (изомер 2) в виде белого твердого вещества (0,10 г, выход 11%).
Н Получение соединения (401).
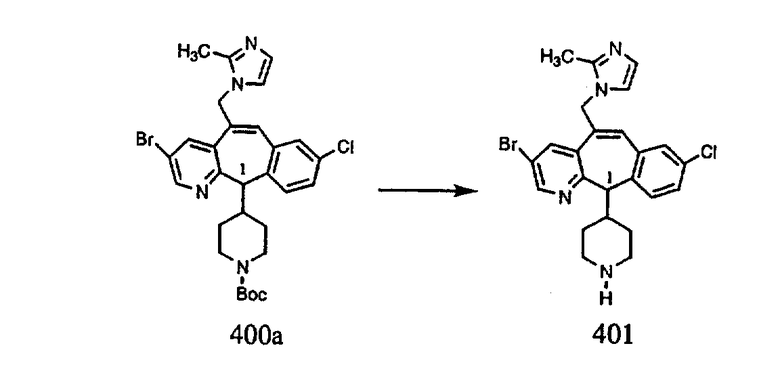
Раствор соединения (400а) (изомер 1), полученного выше на стадии G (0,07 г, 0,12 ммоль), в 4 М растворе HCl/диоксан (3 мл) 3 ч перемешивают при комнатной температуре, затем концентрируют досуха, получая белое твердое соединение (401) (0,06 г, выход 100%).
I Получение соединения (402).
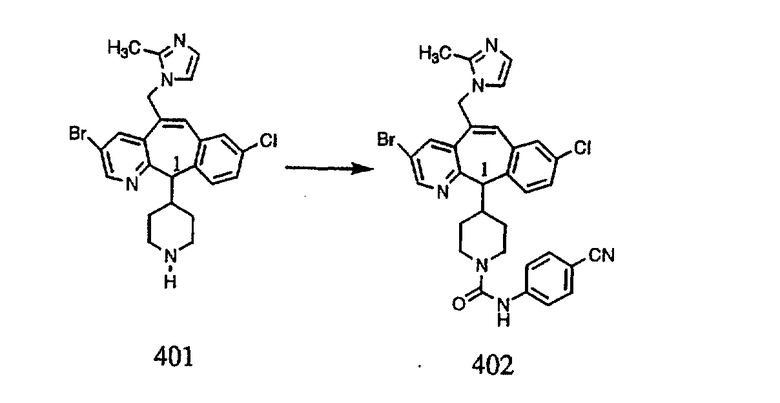
К раствору соединения (401), полученного выше на стадии Н (0,057 г, 0,12 ммоль), в СН2Cl2 (5 мл) в атмосфере азота прибавляют триэтиламин (0,026 г, 0,20 ммоль) и 4-цианофенилизоцианат (0,019 г, 0,13 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, а затем экстрагируют с помощью СН2Cl2 - NaHCO3. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют досуха, получая соединение (402) (изомер 1) в виде белого твердого вещества (0,053 г, выход 70%, MH+ = 629,3).
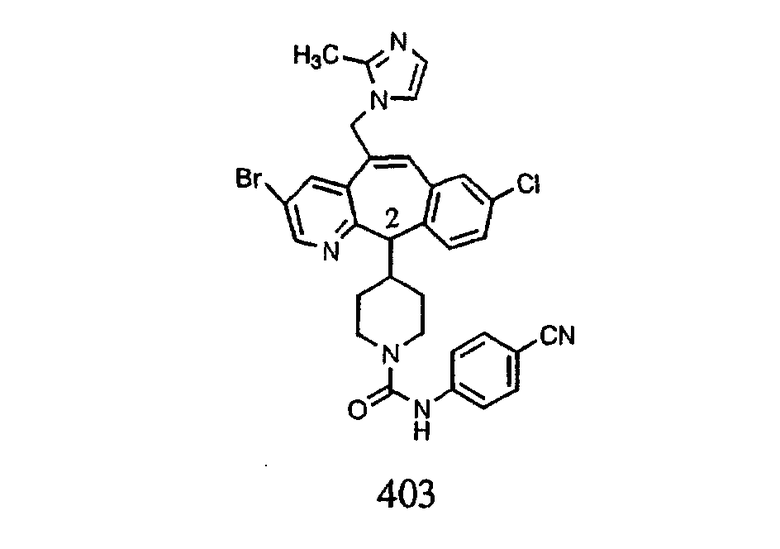
Соединение (400b) вводят в реакцию способом, аналогичным использованному выше на стадиях Н и I, и получают соединение (403) (изомер 2) (0,059 г, выход 79%, MH+ = 629,3).
ПРИМЕР ПОЛУЧЕНИЯ 48
Соединение (404).
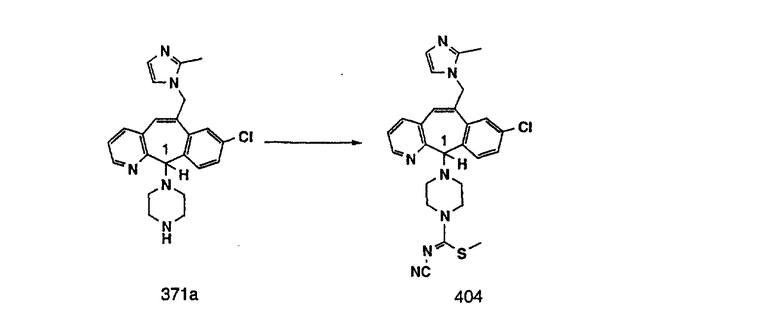
Соединение (371а) (изомер 1), полученное в Примере получения 42, стадия F (70 мг, 0,17 ммоль), растворяют в 1 мл этанола и 50 мкл триэтиламина. Прибавляют диметил-N-цианимидотиокарбонат (45 мг, 0,29 ммоль) и реакционную смесь 24 ч перемешивают при 85°С. Этанол выпаривают при пониженном давлении и продукт хроматографируют на силикагеле, используя смесь 5% раствор аммиака в метаноле - дихлорметан, и получают 47 мг искомого соединения (404) (МС ББА, М + 1 = 504).
ПРИМЕР 179
Получение соединения (405).
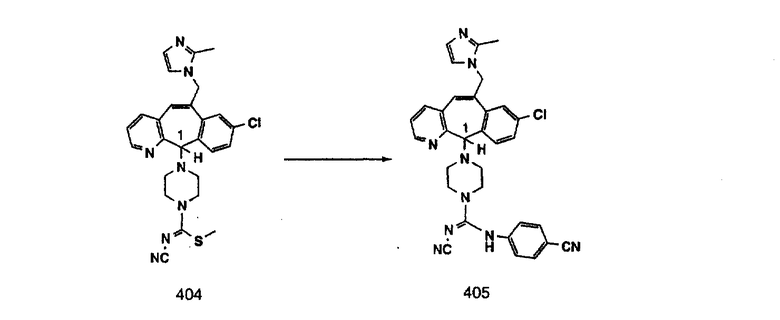
К раствору п-циананилина (53 мг, 0,45 ммоль) в 1 мл N,N-диметилформамида прибавляют гидрид натрия (18 мг, 0,45 ммоль). После перемешивания в атмосфере сухого азота в течение 0,5 ч, прибавляют соединение (404) (изомер 1), полученное выше в Примере получения 48 (40 мг, 0,08 ммоль), и реакционную смесь 4 ч перемешивают при 55°С. Реакционную смесь охлаждают до температуры окружающей среды и прибавляют к рассолу. Неочищенный продукт 3 раза экстрагируют дихлорметаном. Экстракты концентрируют и неочищенный продукт хроматографируют на силикагеле, используя смесь 5% раствор аммиака в метаноле/дихлорметан, и получают 17,6 мг искомого продукта (405). МС ББА, М + 1 = 574,1.
ПРИМЕРЫ 180 И 181
Получение соединений (407) и (408).
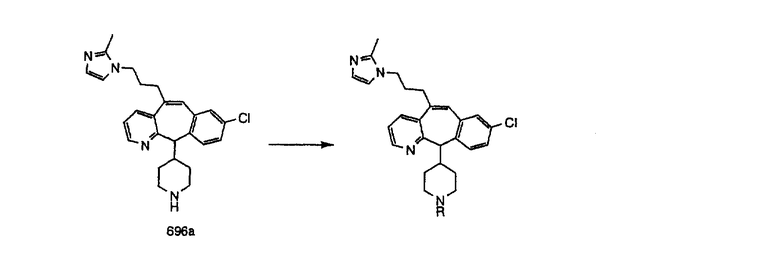
Соединение (696а), полученное в Примере получения 59, стадия В, вводят в реакцию таким же образом, как в Примере получения 48 и Примере 179, используя подходящий реагент R, и получают следующие соединения:

ПРИМЕР ПОЛУЧЕНИЯ 49
Соединения (51а) и (51b).
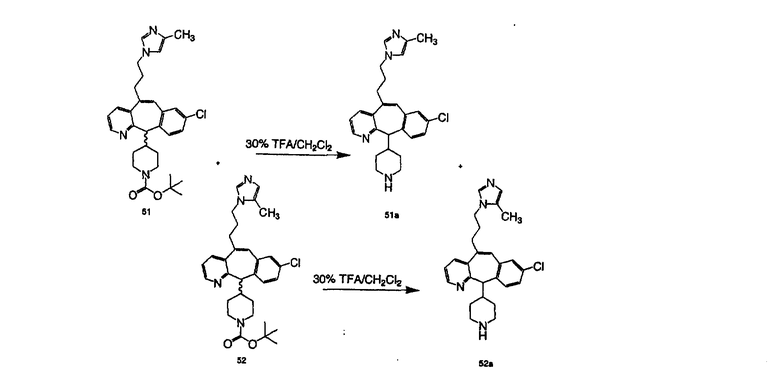
Соединения (51) и (52), полученные в Примере 11, стадия А, вводят в реакцию с TFA в СН2Cl2 и получают соединения (51а) и (52а).
Получение библиотеки
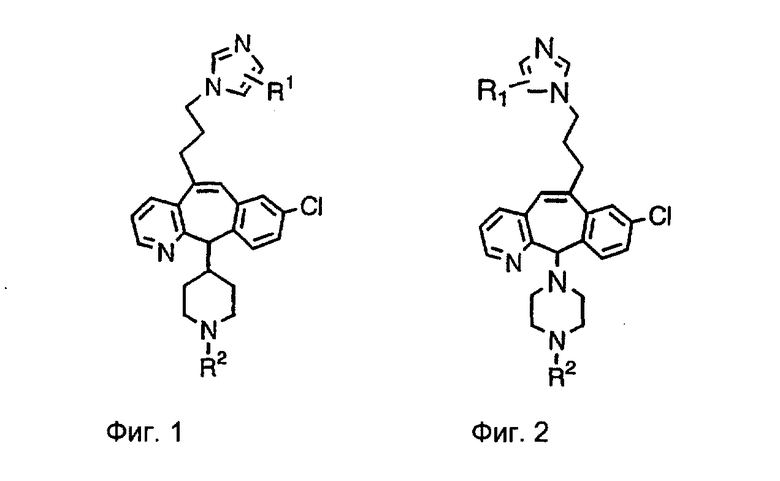
Библиотеку соединений получают с помощью параллельных синтезов в растворе. Общая структура этих соединений показана выше на фиг.1. Группа R1 имидазола может означать Н или СН3, группа R2 у атома N1 пиперидина в этой библиотеке соединений меняется.
Библиотеку соединений получают с использованием в качестве исходных соединения (29), полученного в Примере получения 4, или соединений (51а) и (52а), полученных выше в Примере получения 49, как это показано на схеме А. Синтез начинают в пробирках путем введения соединений (29), (51а) или (52а) с множеством эквивалентов различных изоцианатов, аминов, кислот, хлорангидридов кислот, сульфонилхлоридов и хлорформиатов в дихлорметане или хлороформе. Если необходимым продуктом является мочевина, то реакцию можно проводить непосредственно с использованием изоцианатов или, альтернативно, в течение нескольких часов обрабатывать амины с помощью CDI с последующей обработкой исходных соединений этим раствором в течение ночи. Если используют кислоты, то реакцию проводят в течение ночи в присутствии осуществляющего реакцию сочетания реагента, такого как PyBrop, и основания, такого как DIEA (диизопропилэтиламин). Если используют хлорангидриды кислот, сульфонилхлориды или хлорформиаты, то реакцию обычно проводят в присутствии триэтиламина. После завершения реакции в пробирки для проведения реакции прибавляют избыточное количество полистироламинометильной смолы и реакционную смесь выдерживают в течение ночи. После этого содержимое каждой пробирки профильтровывают в другую пробирку через хроматографическую колонку Bio-Rad Poly-Prep и смолу промывают дихлорметаном и МеОН. Объединенные фильтраты концентрируют на роторном испарителе. Затем остаток, содержащийся в каждой пробирке, растворяют в смеси H2O/CH3CN (50/50, содержащей 1% TFA) и для получения чистого продукта очищают с помощью системы Gilson 215 liquid Handling-ВЭЖХ. Продукт идентифицируют с помощью масс-спектроскопии. Соединения библиотеки, полученные таким образом, представлены в таблице 1 и таблице 2.
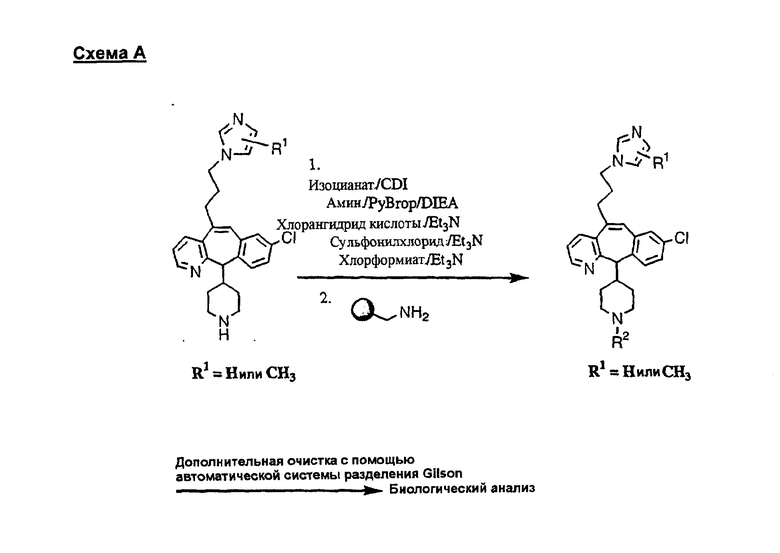
ПРИМЕРЫ 182-283
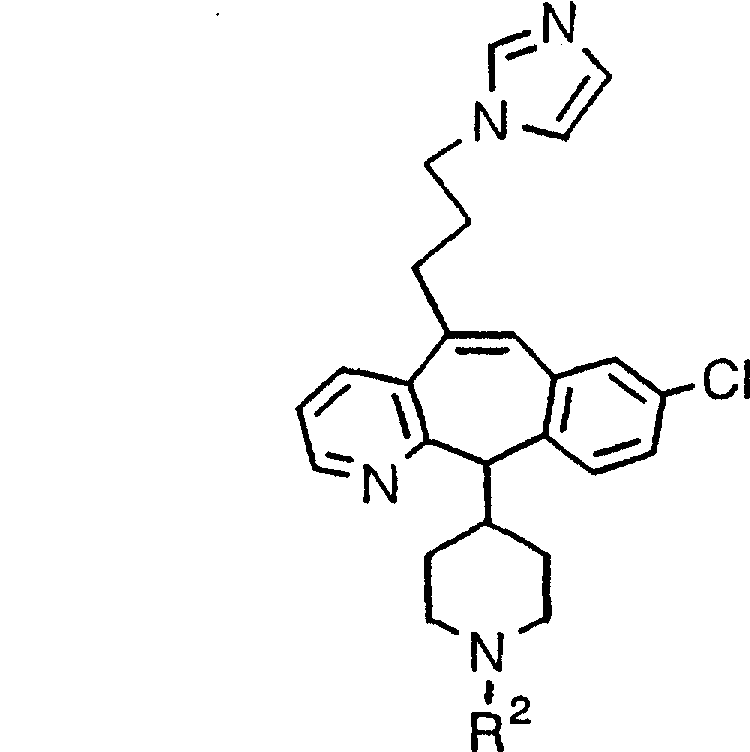
ТАБЛИЦА 1
ТАБЛИЦА 2
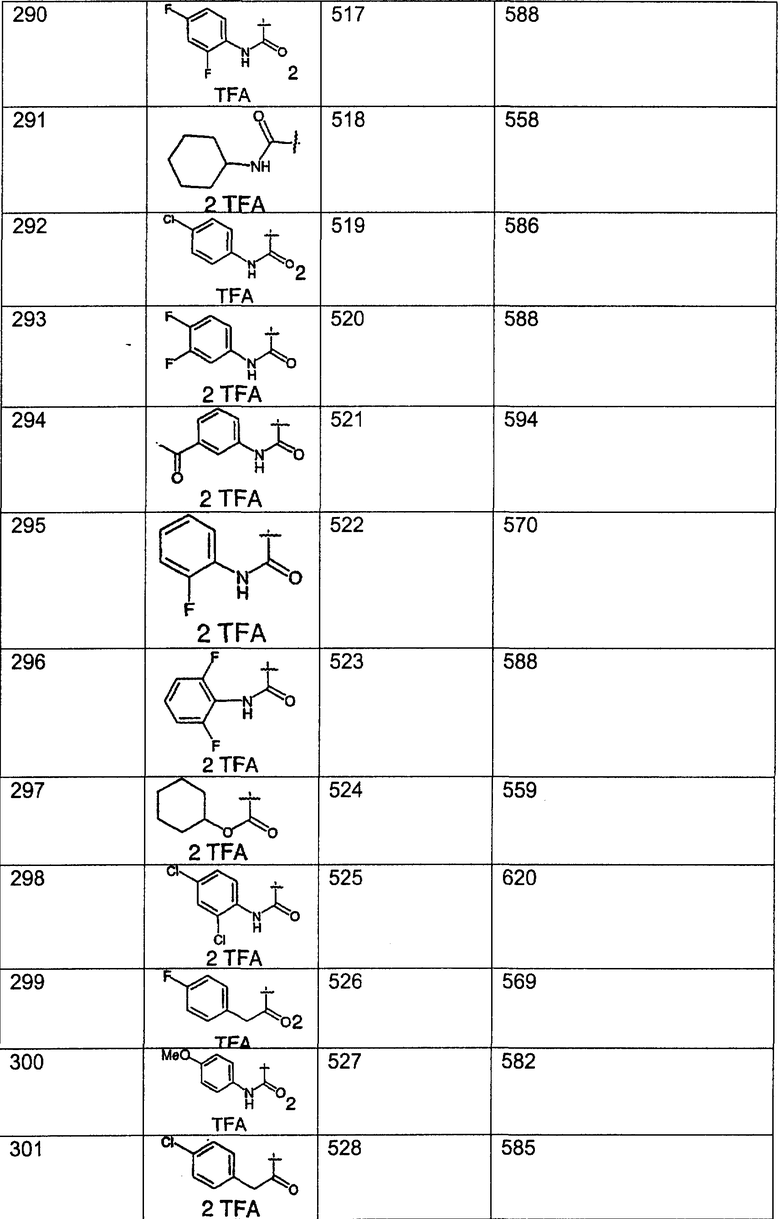
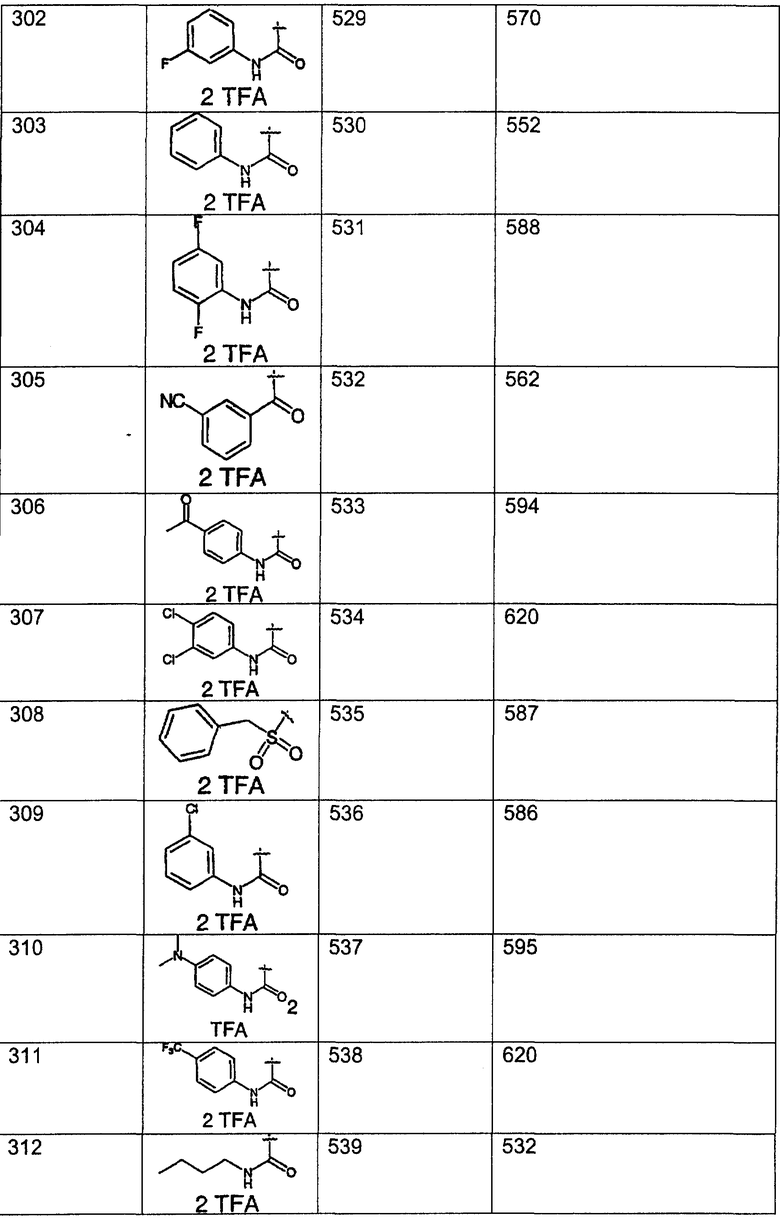
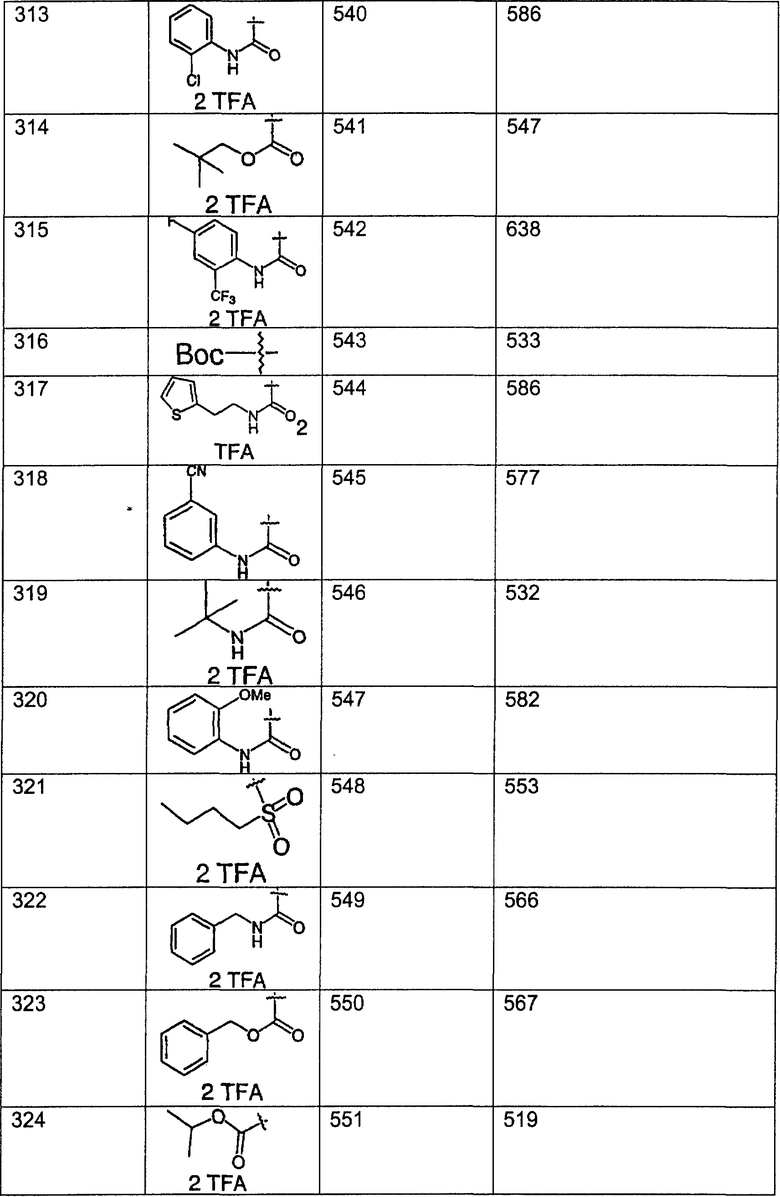
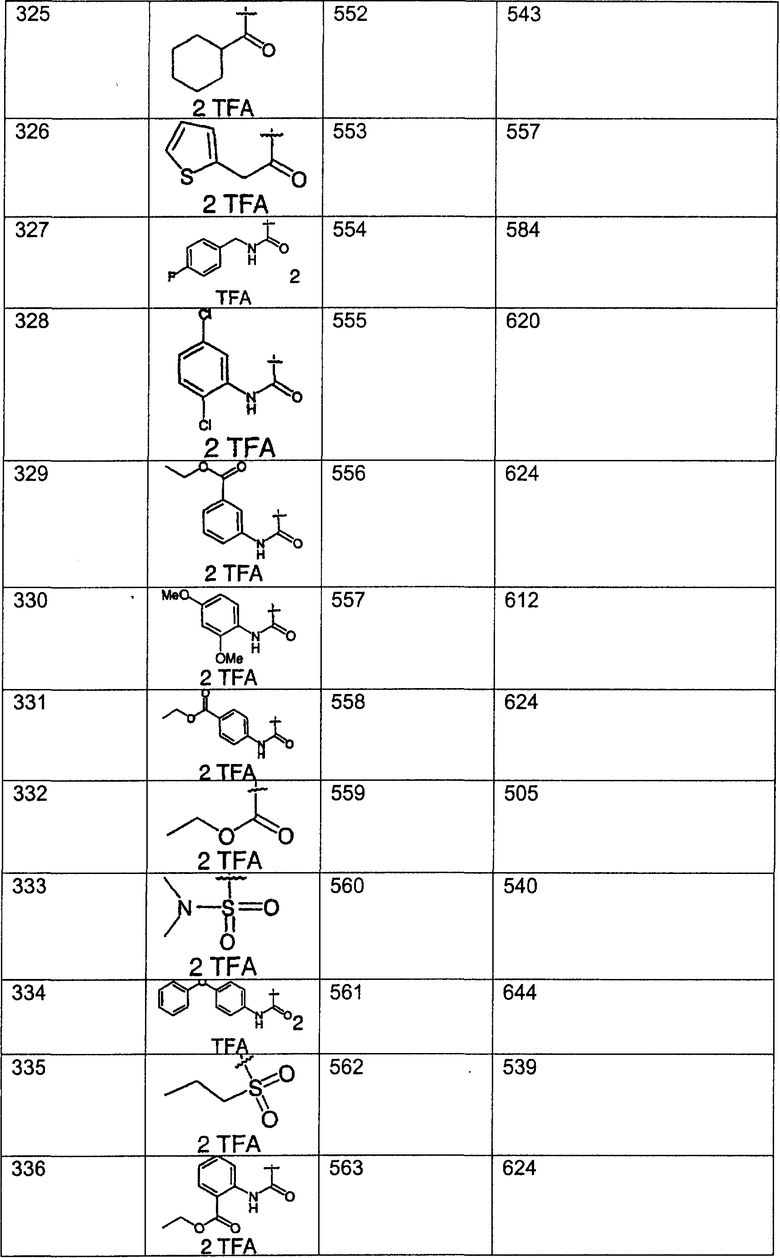
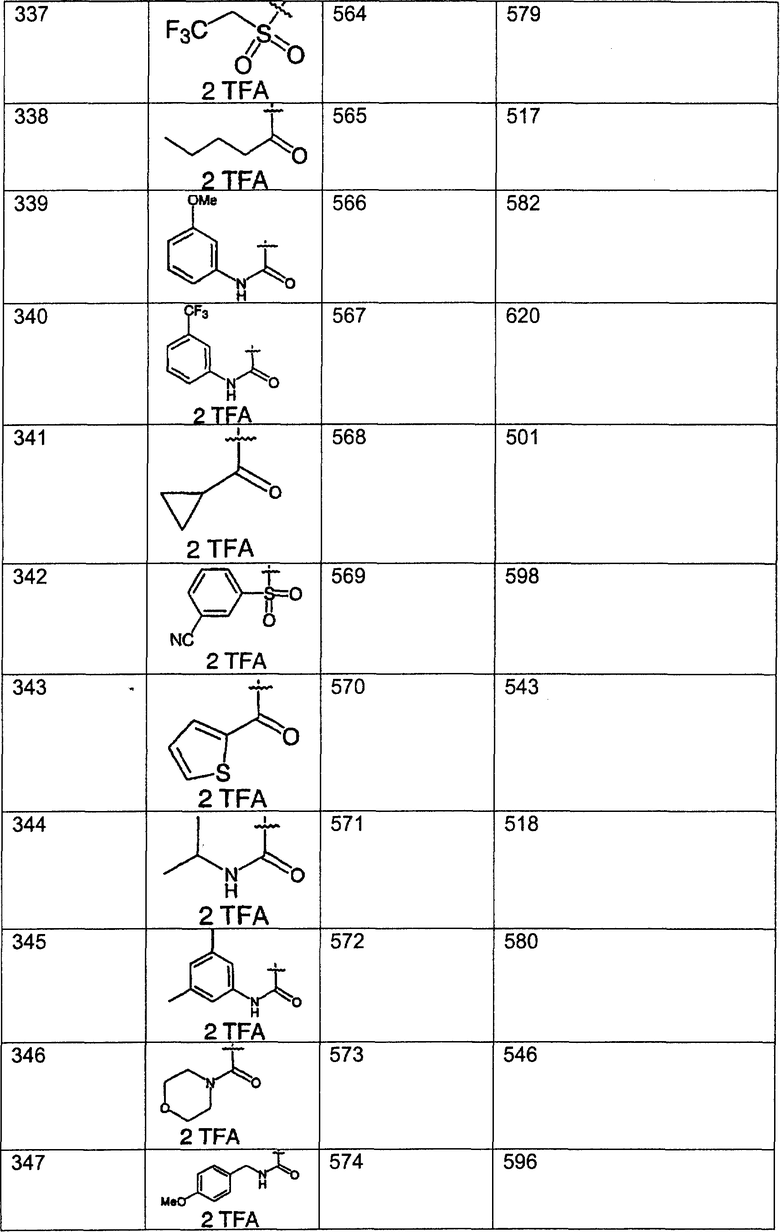
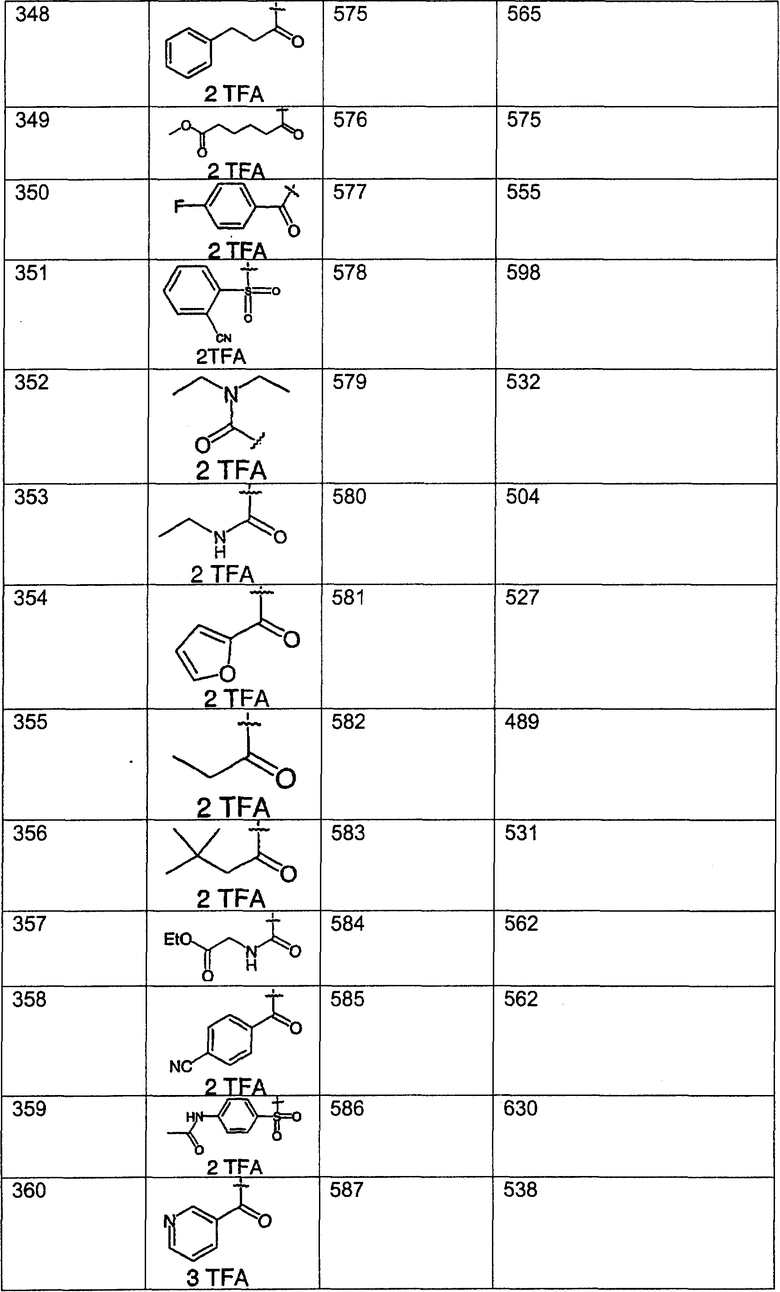
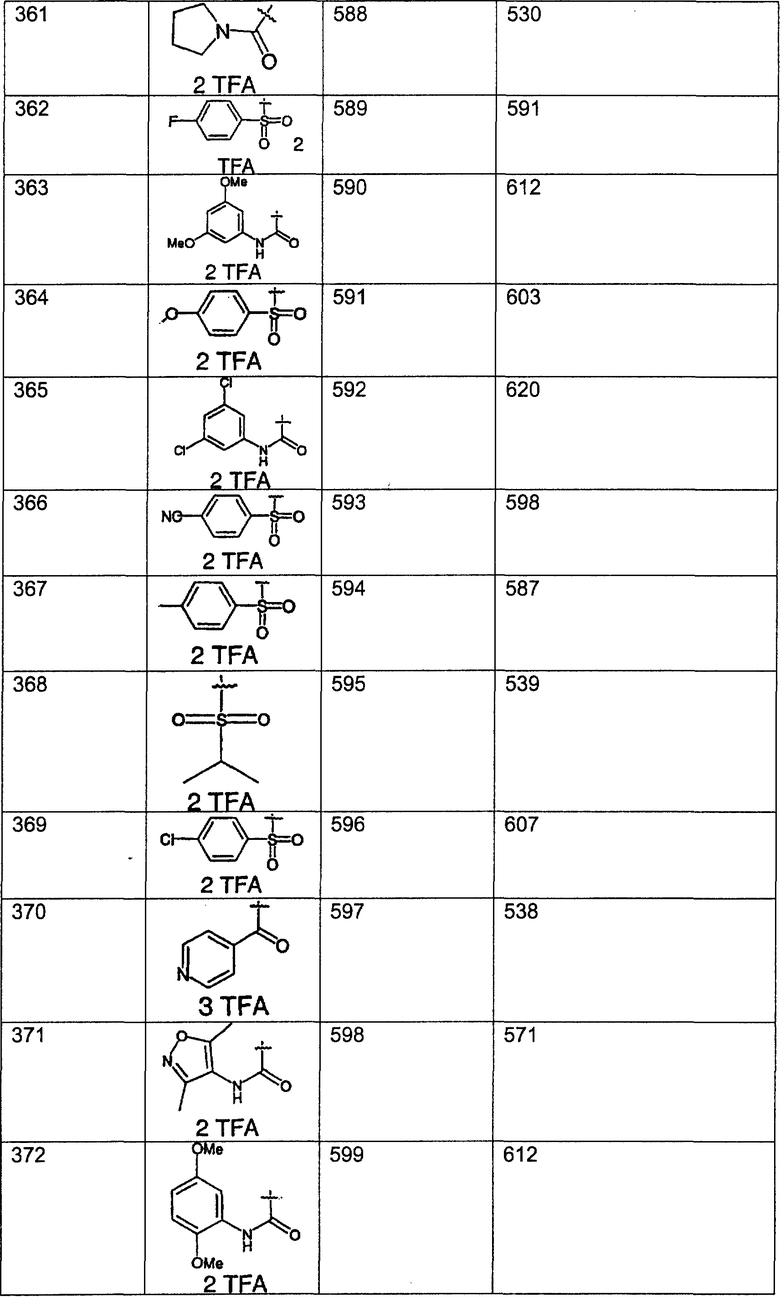
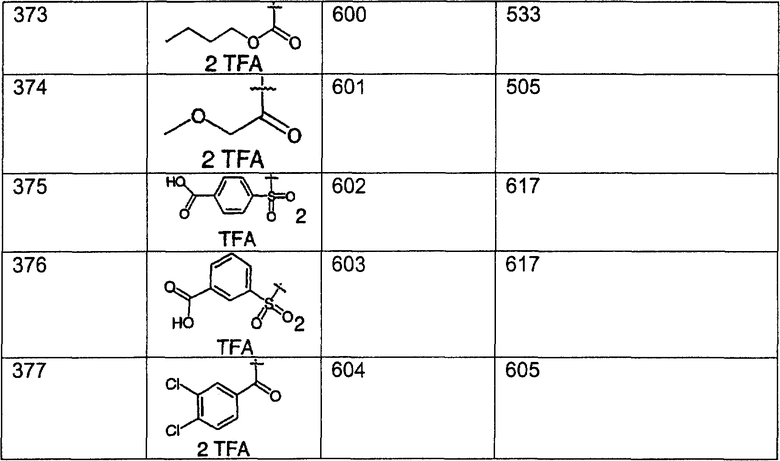
ПРИМЕР ПОЛУЧЕНИЯ 50
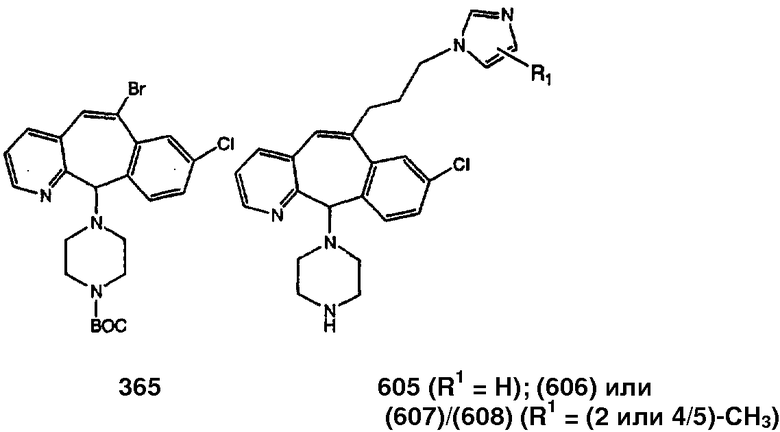
Соединение (365), полученное в Примере получения 41, вводят в реакцию в основном таким же образом, как в Примере получения 4, используя соответствующий имидазол, и получают соединение (605), где R1 = Н, или соединения (605) и (607)/(608), где R1 = (2 или 4/5)-СН3.
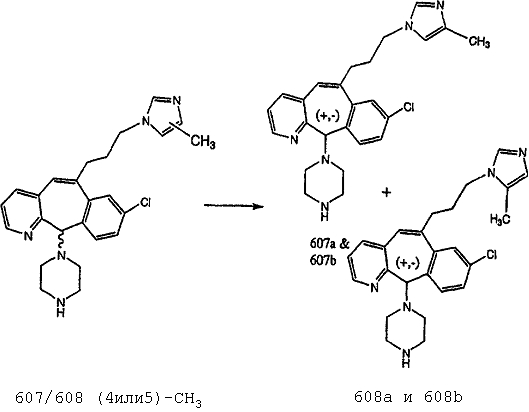
Соединения (607) и (608), полученные выше на стадии А, обрабатывают таким же образом, как описано в Примере 11, и получают чистые энанитомеры (+,-)-4-метилимидазола и чистые энанитомеры (+,-)-5-метилимидазола, соединения (607а), (607b) и (608а), (608b) соответственно.
Серию соединений получают способом, описанным выше, с использованием для схемы 2 в качестве исходных соединения (605), соединения (606), соединений (607)/(608), (607а), (607b) или соединений (608а), (608b). Общая структура этих соединений показана выше на фиг.2. Группа R1 у имидазольного кольца может означать Н или СНз, группа R2 у атома N1 пиперазина в этой серии соединений меняется. Серии соединений, полученных таким образом, представлены в таблице 3, таблице 4 и таблице 5.
ПРИМЕРЫ 378-396
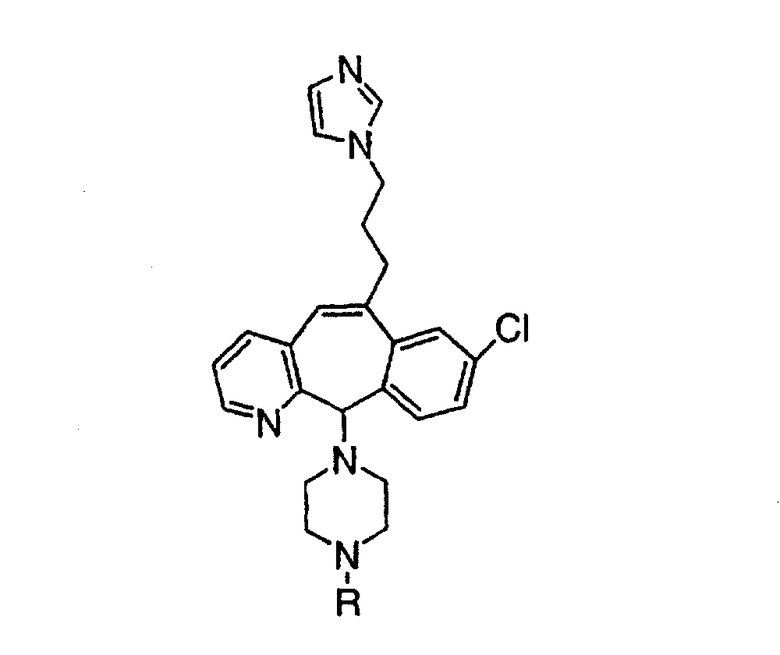
ТАБЛИЦА 3
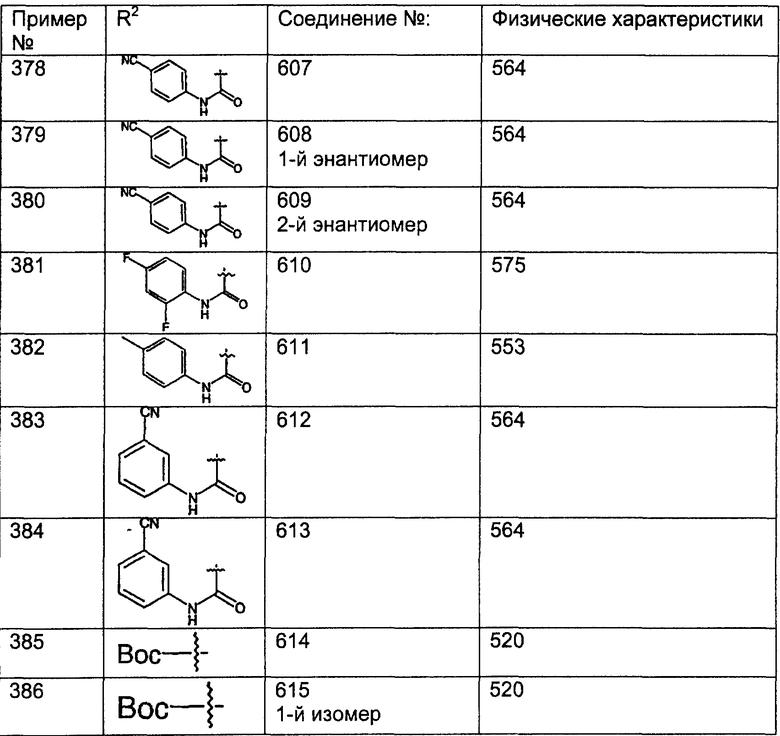
ПРИМЕРЫ 397-401
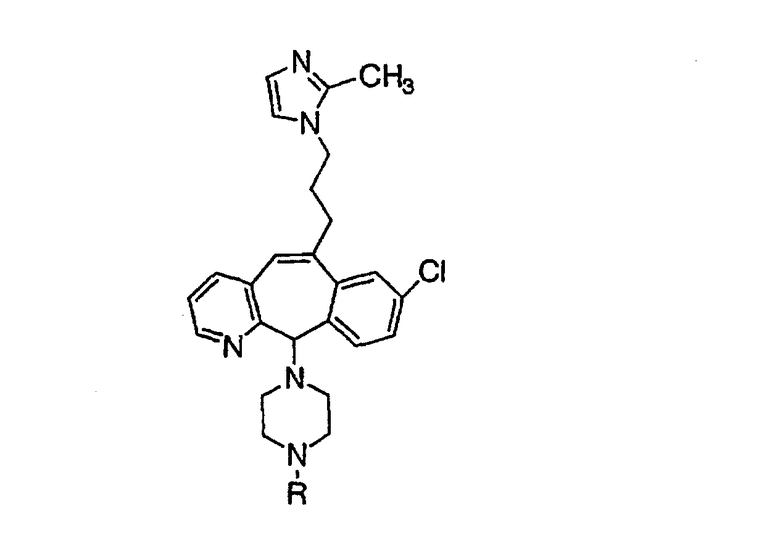
ТАБЛИЦА 4
ПРИМЕРЫ 402 -406
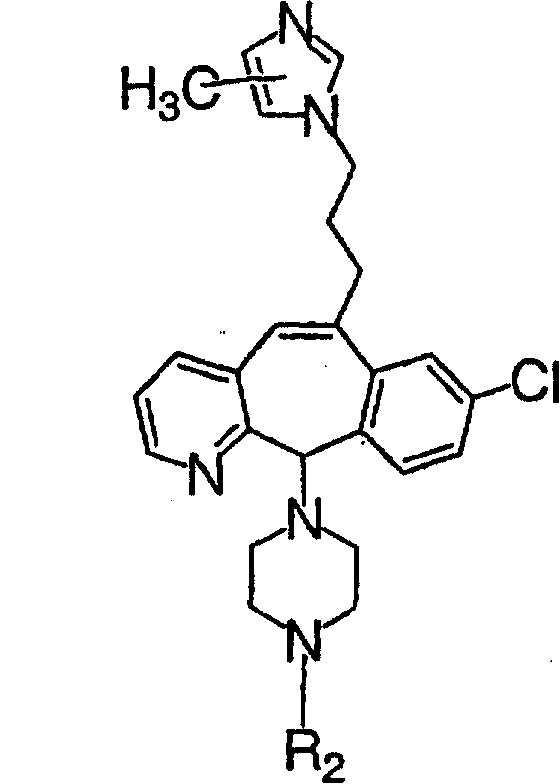
ТАБЛИЦА 5
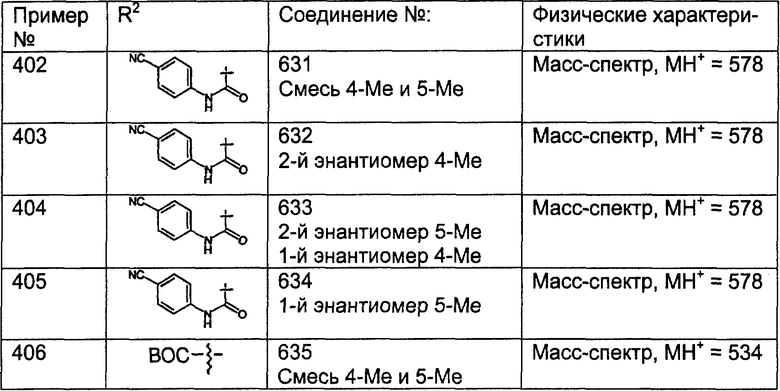
ПРИМЕР ПОЛУЧЕНИЯ 51
Получение соединения (636).
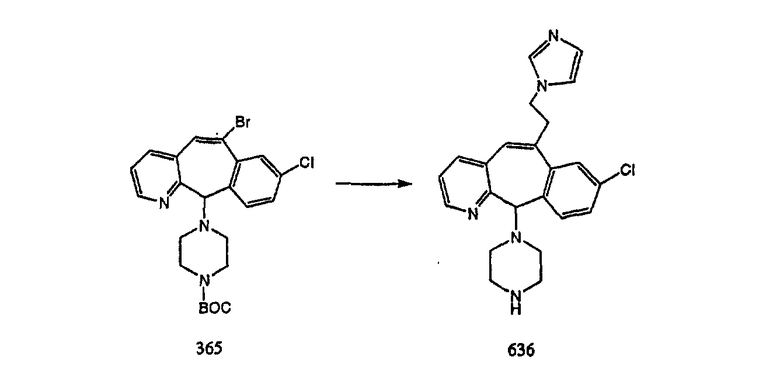
Соединение (365), полученное в Примере получения 41, вводят в реакцию в основном таким же образом, как в Примере получения 35, на стадии В заменяя 1-метилимидазол на имидазол, и получают соединение (636) (МН+ = 406). Затем соединение (636) вводят в реакцию получения библиотеки соединений, описанную выше, по методике, представленной на схеме 2, и получают соединения, представленные ниже в таблице 6:
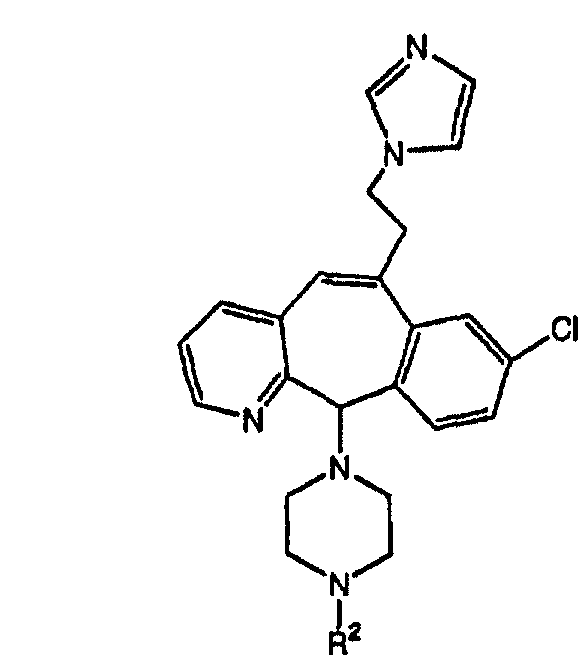
ТАБЛИЦА 6
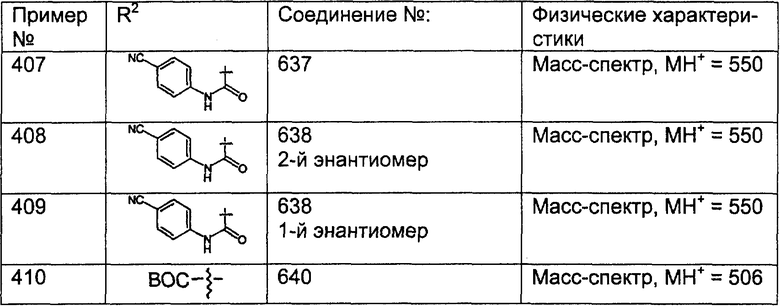
ПРИМЕР ПОЛУЧЕНИЯ 52
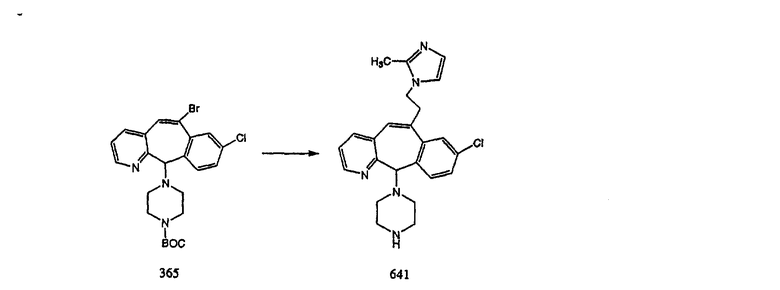
Соединение (365) вводят в реакцию таким же образом, как в Примере получения 51, заменяя имидазол на 1-метилимидазол, и получают соединение (641) (MH+ = 420). Затем соединение (641) дополнительно вводят в реакцию получения библиотеки соединений, описанную выше, по методике, представленной на схеме 2, и получают соединения, представленные ниже в таблице 7:
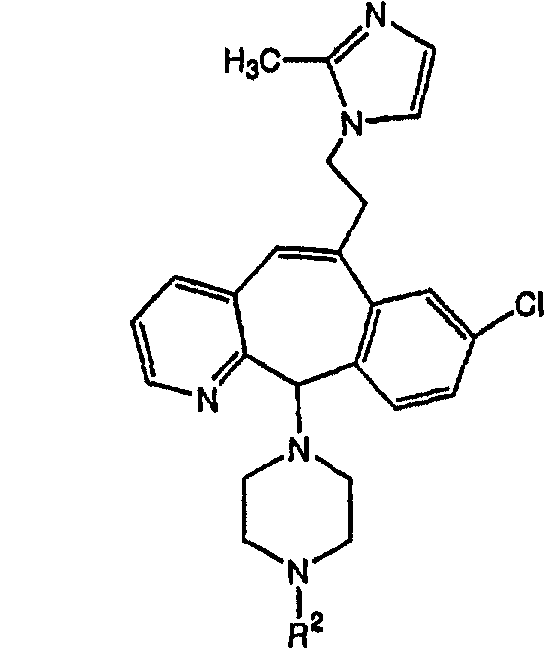
ТАБЛИЦА 7
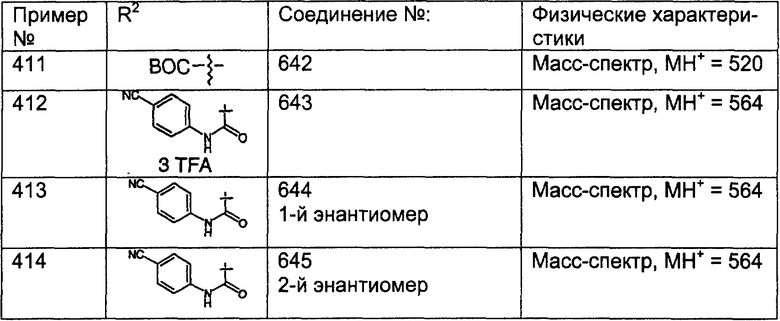
ПРИМЕР 415
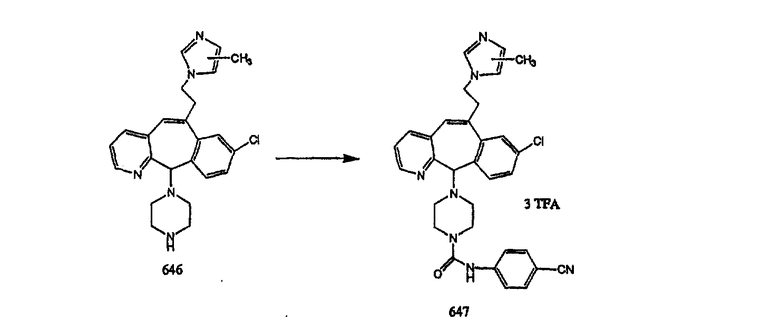
В основном таким же образом, как выше в Примере получения 52, заменяя 4-метилимидазол, и получают промежуточный исходный амин, соединение (646). Затем его вводят в реакцию в основном таким же образом, как выше в Примерах 411-414, и получают соединение (647) в виде смеси изомеров, содержащих 4- и 5-метилимидазол (масс-спектр, MH+ = 564).
ПРИМЕР ПОЛУЧЕНИЯ 53
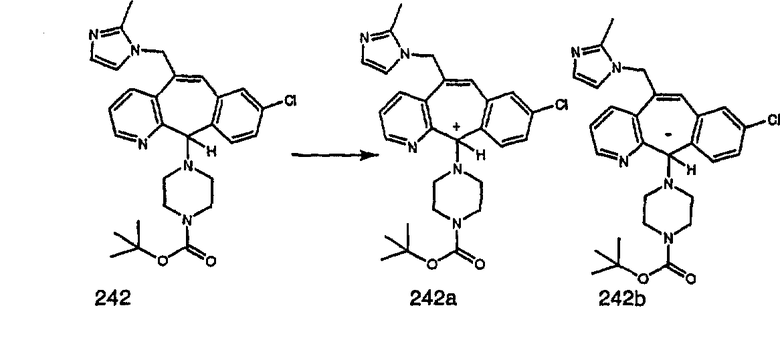
Рацемическое соединение (242), полученное в Примере 91, разделяют с помощью препаративной хиральной хроматографии (колонка Chiralpack AD, 5 см х 50 см, скорость потока 100 мл/мин, 20% 2-пропанол/гексан + 0,2% диэтиламин) и получают два энантиомера (242а) и (242b).
Соединение (242а), [α]D 25 = +144,8° (3,16 мг/2 мл МеОН). Соединение (242b), [α]D 25 = -144,8° (2,93 мг/2 мл МеОН).
ПРИМЕР ПОЛУЧЕНИЯ 54
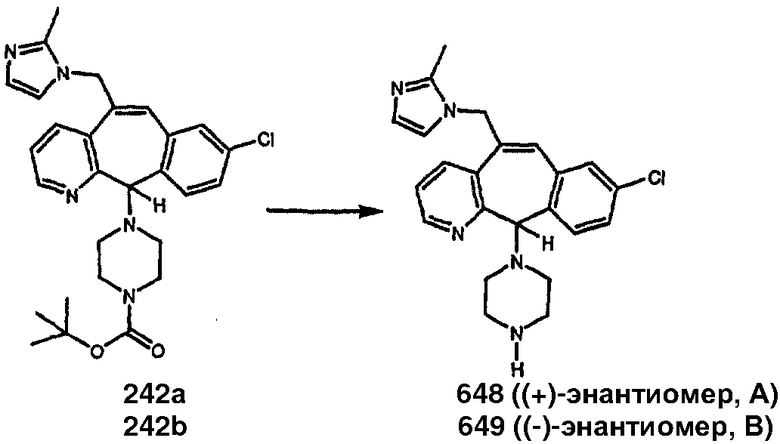
Соединения (242а) и (242b), полученные выше в Примере получения 53, по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают гидрохлориды, соединение (648) и соединение (649).
648 ((+)-энантиомер, изомер А), МН+ = 406,1793.
649 ((-)-энантиомер, изомер В), МН+ = 406,1789.
ПРИМЕР ПОЛУЧЕНИЯ 55
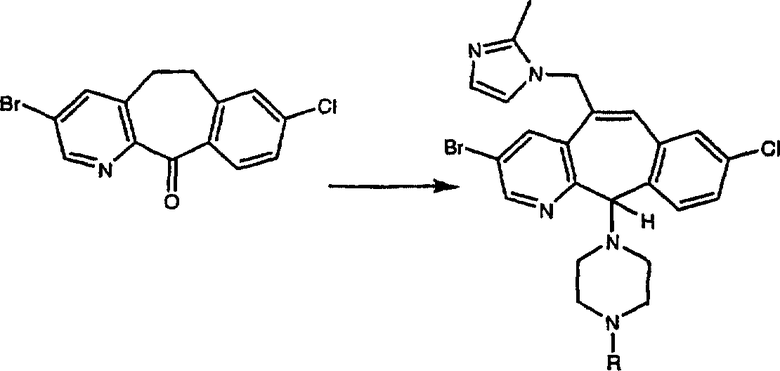
R=BOC
(650) ((+)-энантиомер, А)
(651) ((-)-энантиомер, В)
R=H
(652) ((+)-энантиомер, А)
(653) ((-)-энантиомер, В)
3-Бром-8-хлоразакетон (патент США US 5977128, Пример получения 11, стадия А (1999)) вводят в реакцию в основном таким же образом, как в Примере получения 23 и Примере 91, и получают М-ВОС-производные (650) и (651). Затем соединения (650) и (651) по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (652) ((+)-энантиомер, изомер А) и (653) ((-)-энантиомер, изомер В).
Соединение (650), ВОС-производное, [α]D 25 = +69,6° (2,5 мг/2 мл МеОН). Соединение (651), ВОС-производное, [α]D 25 = -90,0° (3,3 мг/2 мл МеОН).
Соединение (652) ((+)-энантиомер, изомер А), МН+ = 485. Соединение (653) ((-)-энантиомер, изомер В), МН+ = 485.
ПРИМЕР ПОЛУЧЕНИЯ 56
Стадия А
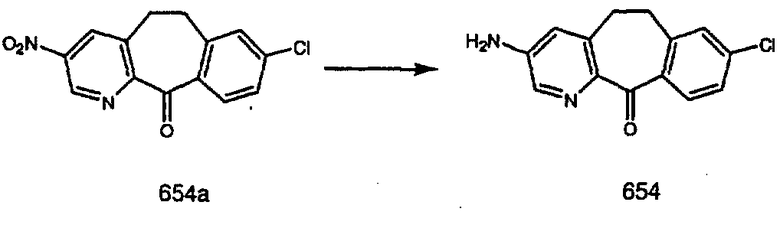
Соединение (654а) (202 г, 0,7 моль) (J.Org. Chem. 1998, 63, 445) растворяют в этаноле (5 л). К этой смеси прибавляют 12 н. раствор HCl (80 мл) и порошкообразное железо (180 г) и реакционную смесь кипятят с обратным холодильником в течение ночи. Для завершения реакции прибавляют дополнительное количество HCI и железа. Реакционную смесь фильтруют и осадок промывают горячим метанолом (1 л). Фильтрат концентрируют в вакууме до объема, равного примерно 600 мл, а затем подвергают распределению между 4 л CH2Cl2 и 1,3 л 1,3 н. раствора NaOH. Органический слой сушат над MgSO4 и фильтруют в горячем состоянии. Фильтрат концентрируют в вакууме и получают аминокетон, соединение (654) (184 г).
Стадия В
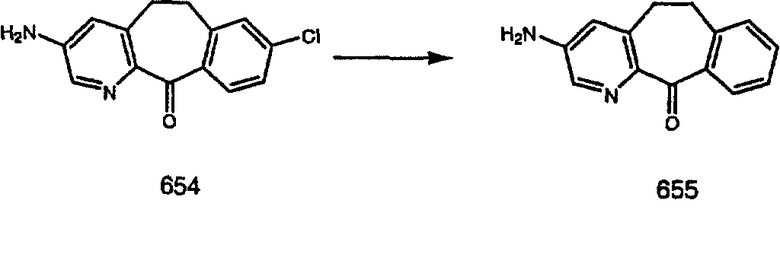
Соединение (654), полученное выше на стадии А (15 г, 57,98 ммоль), растворяют в 750 мл этанола, содержащего 3,75 г 5% Pd/C (50% взвесь в воде) и 37,69 г (579,82 ммоль) формиата аммония. Смесь 2,5 ч кипятят с обратным холодильником, а затем перемешивают при комнатной температуре в течение ночи. Реакционную смесь фильтруют, концентрируют в вакууме и хроматографируют на силикагеле с использованием смеси состава 95: 5, метиленхлорид (насыщенный аммиаком) - метанол и получают 6,15 г чистого соединения (655) в виде желтого твердого вещества.
Стадия С
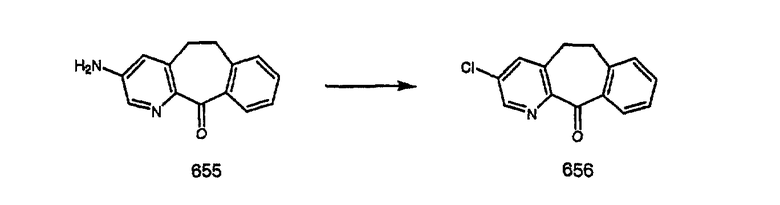
К взвеси соединения (655) (4,79 г, 21,37 ммоль), полученного выше на стадии А, в 75 мл ацетонитрила, охлажденной до 0°С, в атмосфере азота прибавляют трет-бутилнитрит (10,31 г, 32,05 ммоль) и CuCl2 (3,45 г, 24,64 ммоль). Смесь нагревают до комнатной температуры, перемешивают в течение ночи, а затем концентрируют в вакууме. Остаток превращают во взвесь в 30 мл 1 н. раствора HCl, затем нейтрализуют водным раствором NH4OH и экстрагируют с помощью 3 х 100 мл этилацетата. Органический слой сушат над Na2SO4, концентрируют в вакууме и хроматографируют на силикагеле с использованием смеси гексан:этилацетат (70:30) и получают чистое соединение (656).
Стадия D
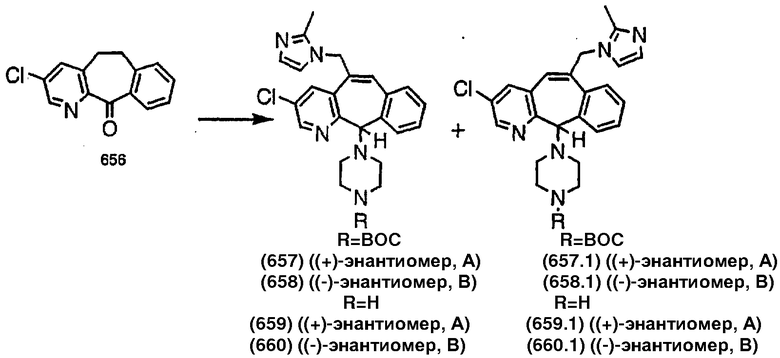
Соединение (656), полученное выше на стадии В, вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем, как в Примере 91, и получают N-BOC-производные (657), (658), (657.1) и (658.1). Соединения (657), (658), (657.1) и (658.1) затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (659) ((+)-энантиомер, изомер А), (659.1) ((+)-энантиомер, изомер А), (660) ((-)-энантиомер, изомер В) и (660.1) ((-)-энантиомер, изомер В).
Соединение (657), ВОС-производное, [α]D 25 = +59,5° (3,3 мг/2 мл МеОН). Соединение (658), ВОС-производное, [α]D 25 = -57,1° (3,3 мг/2 мл МеОН).
Соединение (659) ((+)-энантиомер, изомер А), МН+ = 406. Соединение (660) ((-)-энантиомер, изомер В), MH+ = 406.
Соединение (659.1) ((+)-энантиомер, изомер А), MH+ = 406. Соединение (660.1) ((-)-энантиомер, изомер В), MH+ = 406.
ПРИМЕР ПОЛУЧЕНИЯ 57
R=H
(666) ((+)-энантиомер, А) (668) ((-)-энантиомер, В)
(667) ((+)-энантиомер, А) (669) ((-)-энантиомер, В)
Соединение (661) вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем, как в Примере 91, и получают N-BOC-производные (662), (663), (664) и (665). Соединения (662), (663), (664) и (665) затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D,и получают энантиомеры (666) и (667) ((+)-энантиомер, изомер А), и (668) и (669) ((-)-энантиомер, изомер В). С5-и С6-винилбромидные промежуточные продукты разделяют с помощью хроматографии на силикагеле с использованием смеси гексан:этилацетат (80: 20) в основном таким же образом, как это описано в Примере получения 23,стадия В.
Соединение (662), ВОС-производное. Соединение (663), ВОС-производное.
Соединение (664), ВОС-производное. Соединение (665), ВОС-производное.
Соединение (666) ((+)-энантиомер, изомер А), MH+ = 372. Соединение (667) ((-)-энантиомер, изомер В), МН+ = 372.
Соединение (668) ((+)-энантиомер, изомер А), МН+ = 372. Соединение (669) ((-)-энантиомер, изомер В), МН+ = 372.
ПРИМЕР ПОЛУЧЕНИЯ 58
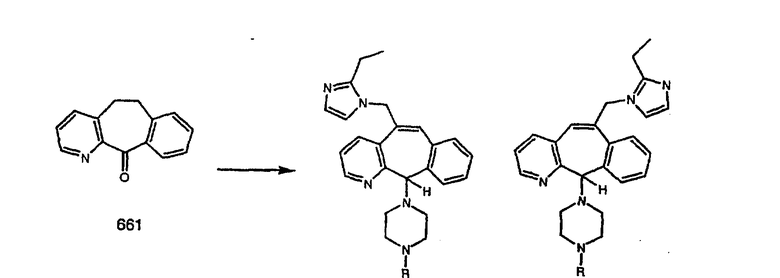
R=H
(674) ((+)-энантиомер, А) (676) ((-)-энантиомер, В)
(675) ((+)-энантиомер, А) (677) ((-)-энантиомер, В)
Соединение (661) вводят в реакцию в основном таким же образом, как в Примере получения 23 и Примере 91, заменяя 2-метилимидазол на 2-этилимидазол, и получают N-BOC-производные (670), (671), (672) и (673). Соединения (670), (671), (672) и (673) затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (674) и (675) ((+)-энантиомер, изомер А), и (676) и (677) ((-)-энантиомер, изомер В). С5-и С6-винилбромидные промежуточные продукты разделяют с помощью хроматографии на силикагеле с использованием смеси гексан:этилацетат (80:20) таким же образом, как это описано в Примере получения 23, стадия В.
Соединение (670), ВОС-производное ((+)-энантиомер, изомер А). Соединение (671), ВОС-производное ((+)-энантиомер, изомер А).
Соединение (672), ВОС-производное ((-)-энантиомер, изомер В). Соединение (673), ВОС-производное ((-)-энантиомер, изомер В).
Соединение (674) ((+)-энантиомер, изомер А), МН+ = 386. Соединение (675) ((+)-энантиомер, изомер А), MH+ = 386.
Соединение (676) ((-)-энантиомер, изомер В), MH+ = 386. Соединение (677) ((-)-энантиомер, изомер В), МН+ = 386.
ПРИМЕРЫ 416-419
Соответствующий (+)-энантиомер (648) или (-)-энантиомер (649), полученный выше в Примере получения 54, растворяют в CH2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице 8:
ТАБЛИЦА 8
-
ПРИМЕРЫ 420 И 421
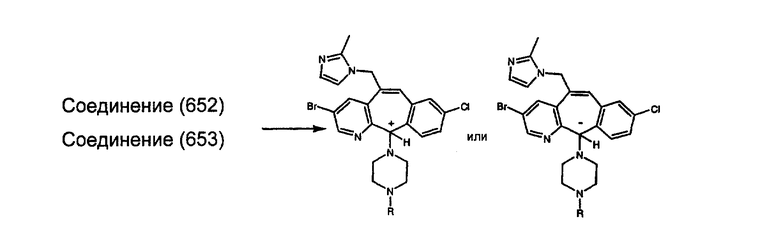
Соответствующий (+)-энантиомер (652) или (-)-энантиомер (653), полученный выше в Примере получения 55, растворяют в CH2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице 9:
ТАБЛИЦА 9
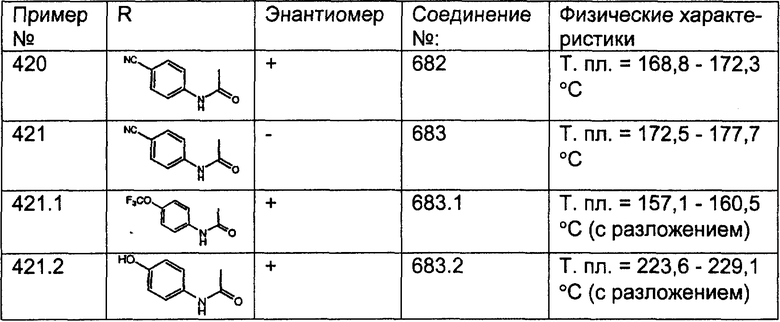
ПРИМЕРЫ 422 И 423
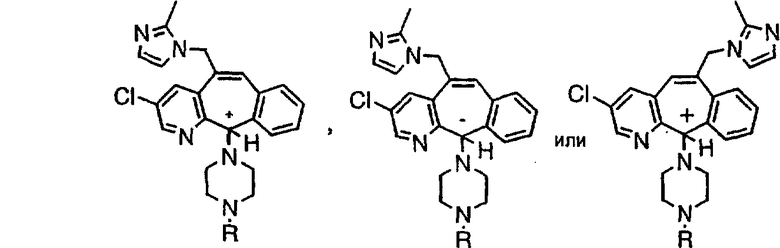
Соответствующий (+)-энантиомер (659) или (-)-энантиомер (660) или (+)-энантиомер (659А), полученный выше в Примере получения 56, растворяют в CH2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице 10:
ТАБЛИЦА 10
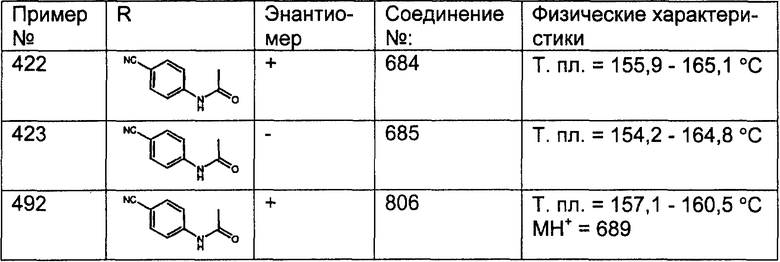
ПРИМЕРЫ 424 И 425
Соответствующий (+)-энантиомер (666) или (-)-энантиомер (668), полученный выше в Примере получения 57, растворяют в CH2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице 11:
ТАБЛИЦА 11
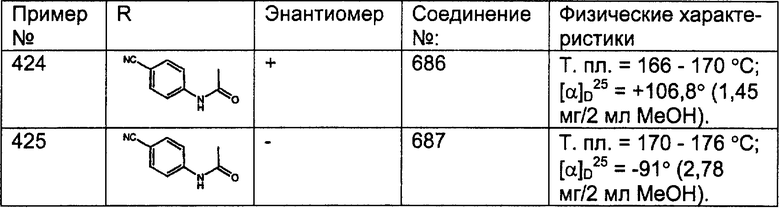
ПРИМЕРЫ 426 И 427
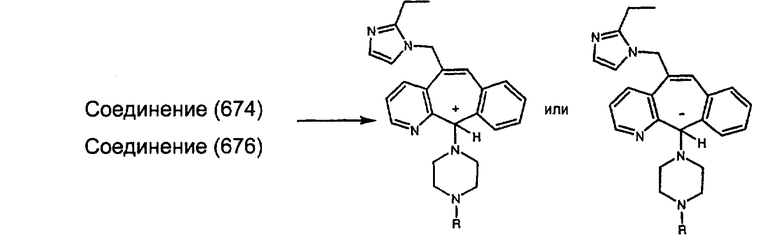
Соответствующий (+)-энантиомер (674) или (-)-энантиомер (676), полученный выше в Примере получения 58, растворяют в CH2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице 12:
ТАБЛИЦА 12
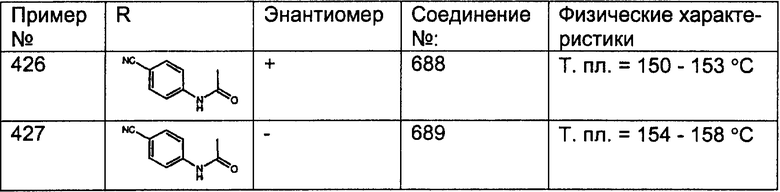
ПРИМЕРЫ 428 И 429
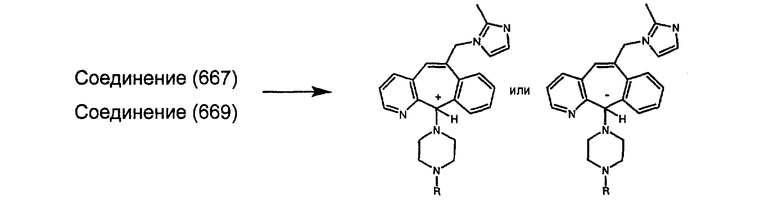
Соответствующий (+)-энантиомер (667) или (-)-энантиомер (669), полученный выше в Примере получения 57, растворяют в СН2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные в таблице ниже:
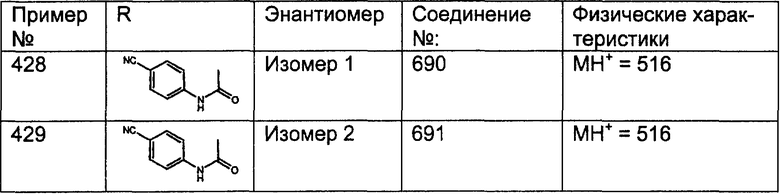
ПРИМЕРЫ 430 И 431
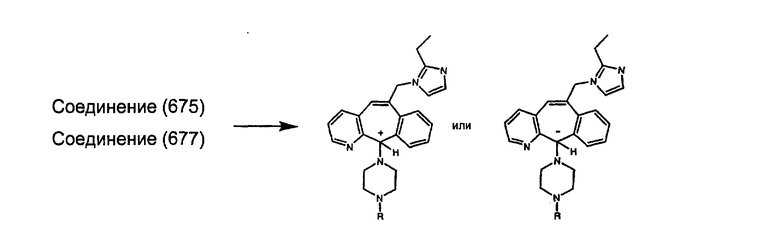
Соответствующий (+)-энантиомер (675) или (-)-энантиомер (677), полученный выше в Примере получения 58, растворяют в CH2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные в таблице ниже:
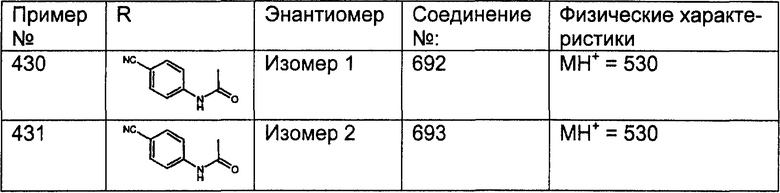
ПРИМЕР ПОЛУЧЕНИЯ 59
Соединения типа А (696а), (696b) и типа В (697а), (697b).
К раствору 2-метилимидазола (1,80 г, 21,97 ммоль) в безводном DMF (40 мл) при комнатной температуре и при перемешивании прибавляют NaH (5,3 г, 21,97 ммоль) и соединение (27), полученное в Примере получения 4, стадия Е (4,0 г, 7,33 ммоль). Полученный раствор 1 ч перемешивают при комнатной температуре и концентрируют досуха, а затем экстрагируют раствором EtOAc-NaHCO3. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют досуха, получая смесь соединений, содержащих ординарную и двойную связь. Эти соединения дополнительно очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 2% МеОН/NH3/98% CH2Cl2, и получают чистое соединение типа А (694) (0,450 г) (МН+ = 533) и смесь соединения типа А (694) и соединения типа В (695) (2,55 г) (NH2 = 535).
Соединения (694) и (695) дополнительно очищают с помощью препаративной ВЭЖХ, элюируя смесью 15% IPA/85% гексан/0,2% DEA, и получают соединение типа В (695а) (изомер 1, 0,58 г, МН+ = 535,4) и соединение типа А (694а) (изомер 1, 0,61 г, MH+ = 533) и смесь соединений (694b) и (695b) (изомеры 2, 0,84 г).
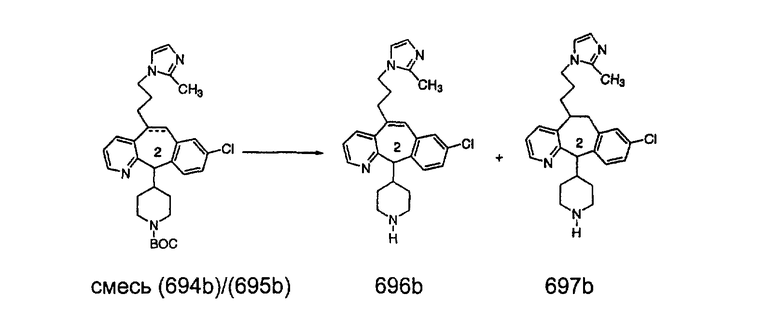
Смесь соединений (694b)/(695b), полученных выше на стадии А (0,8 г, 1,5 ммоль), в 4 н. растворе HCl/диоксан (40 мл) 3 ч перемешивают при комнатной температуре, затем концентрируют досуха, в качестве продукта получая смесь соединений, из которых удалены защитные группы. Продукт дополнительно очищают с помощью препаративной ВЭЖХ, элюируя смесью 15% IPA/85% гексан/0,2% DEA, и получают чистое соединение (696b) типа А (изомер 2, 0,29 г) и чистое соединение (697b) типа В (изомер 2, 0,19 г).
Стадия С Получение соединений (696а) и (697а).
У соединений (694а) и (695а) (чистый изомер 1) защитные группы удаляют по отдельности с использованием 4 н. раствора HCl/диоксан, что выполняют в основном по той же методике, что и при получении изомеров 2, описанной выше, и получают соответствующие N-Н-продукты (696а) типа А (изомер 1) и (697а) типа В (изомер 1).
ПРИМЕРЫ 432-437
Соединение (696а) (изомер 1) вводят в реакцию в основном таким же образом, как в Примере 13, с подходящим хлорформиатом или изоцианатом и получают следующие соединения, приведенные ниже в таблице 13.
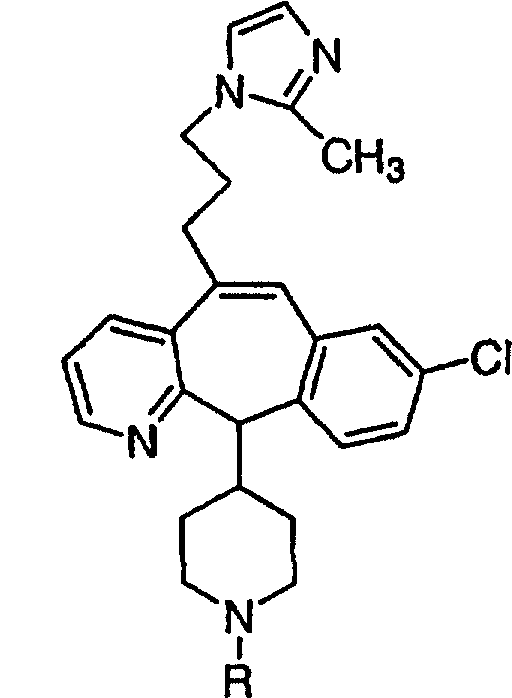
Таблица 13: 5-Замещенные аналоги 2-метилпропилимидазола с мостиковой двойной связью.
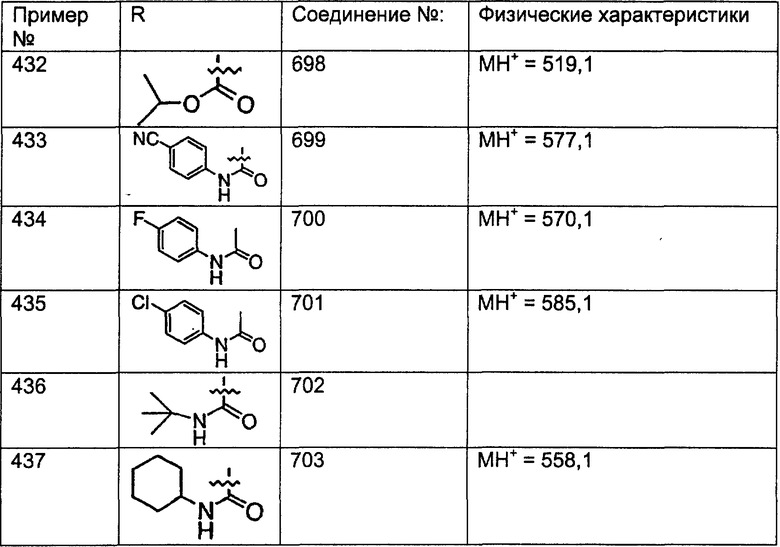
ПРИМЕРЫ 438-442
Соединение (697а) (изомер 1) вводят в реакцию в основном таким же образом, как в Примере 13, с подходящим хлорформиатом или изоцианатом и получают следующие соединения, приведенные ниже в таблице 14.
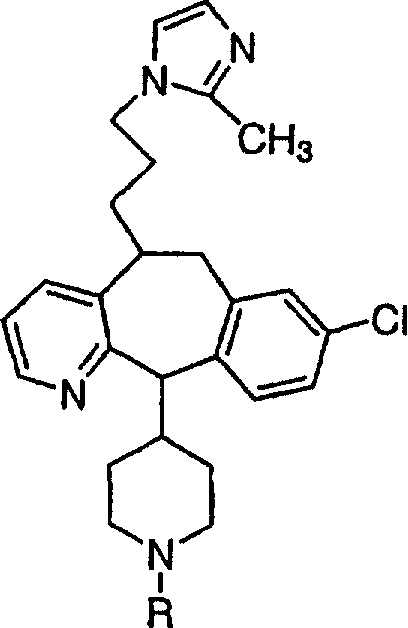
Таблица 14: 5-Замещенные аналоги 2-метилпропилимидазола с мостиковой ординарной связью.
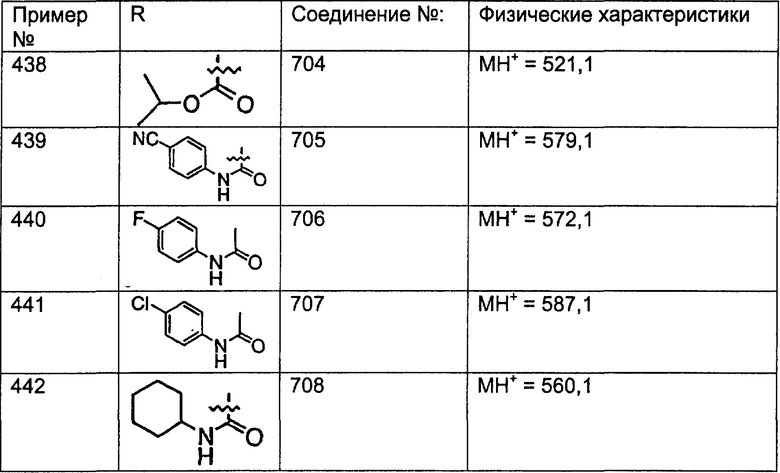
ПРИМЕР ПОЛУЧЕНИЯ 60
Соединения (711a) и (711b), (712а) и (712b).
Стадия А Получение соединений (709а), (709b), (710а), и (710b).
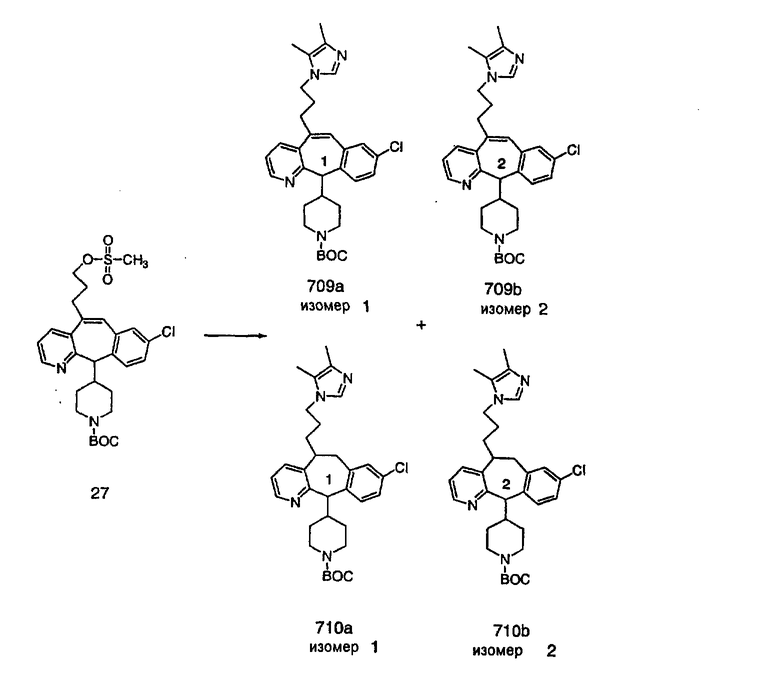
К раствору 4,5-диметилимидазола (1,08 г, 11,25 ммоль) в безводном DMF (35 мл) при перемешивании при комнатной температуре прибавляют NaH (0,27 г, 11,2 ммоль) и перемешивают 10 мин, а затем прибавляют соединение (27), полученное в Примере получения 4, стадия Е (4,0 г, 7,32 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи. К этому раствору прибавляют раствор 4,5-диметилимидазола (0,35 г, 3,65 ммоль) и NaH (0,088 г, 3,67 ммоль) в DMF (5 мл). Этот раствор 4 ч нагревают при 80-90°С, а затем охлаждают до комнатной температуры и после этого экстрагируют раствором EtOAc-Н2O. Объединенные органические слои промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 50% EtOAc/50% гексан и 5% MeOH/CH2Cl2, и получают смесь продуктов - соединение (709) типа А и соединение (710) типа В (1,2 г, МН+ = 547,3). Продукты дополнительно очищают с помощью препаративной ВЭЖХ с использованием хиральной колонки AD, элюируя смесью 15% IPA/85% гексан/0,2% DEA, и получают 4 отдельных соединения:
Соединение (709а) изомер 1, тип А (0,291 г, MH+ = 547,3), соединение (710а) изомер
1, тип В (0,305 г, МН+= 549,3) и
соединение (709b) изомер 2, тип А (0,280 г, МН+ = 547,3), соединение (710b) изомер
2, тип В (0,2 г, МН+ = 549,3),
Стадия В Получение соединений (711а), (711b), (712а), и (712b).
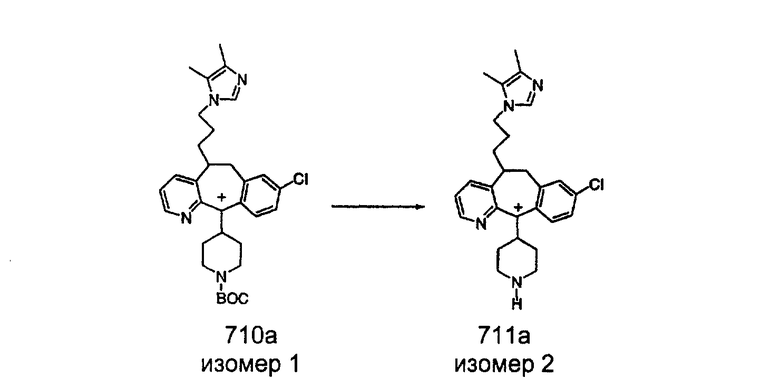
Раствор соединения (710а) (изомер 1, тип В) (0,245 г, 0,45 ммоль) в 4 н. растворе HCl/диоксан (2 мл) 3 ч перемешивают при комнатной температуре, затем концентрируют досуха и получают соединение (711 а) (изомер 1, тип В) (0,184 г, выход 98%) (MH+ = 455,1).
Соединения (711b) (изомер 2, тип В), (712а) (изомер 1, тип А) и (712b) (изомер 2, тип А) получают способом, аналогичным приведенному выше на стадии В способу получения соединения (711а) (изомер 1, тип В).
(711b) (0,085 г, выход 75%), (712а) (0,141 г, выход 75%), (712b) (0,106 г, выход 59%).
ПРИМЕРЫ 443-447
Соединения (711а) и (711b) по отдельности вводят в реакцию по методике, описанной в Примере 13, с подходящими хлорформиатами или изоцианатами и получают следующие соединения, приведенные ниже в таблице 15.
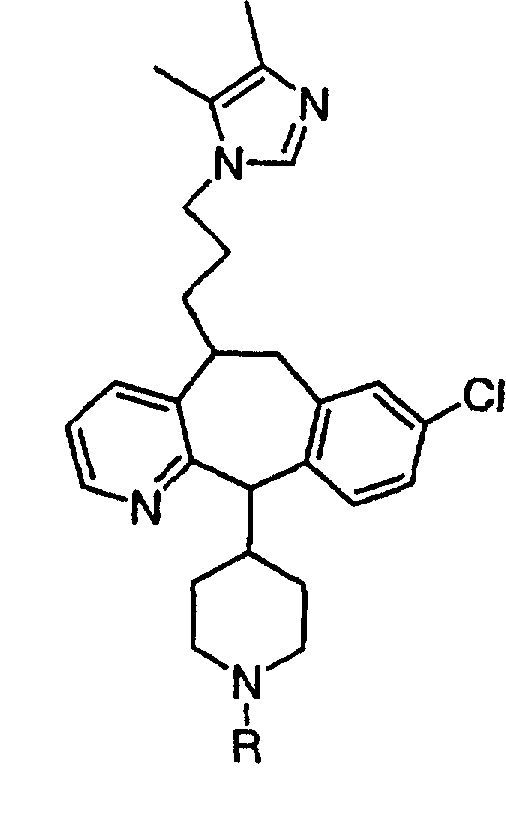
Таблица 15: 5-Замещенные аналоги 4,5-диметилпропилимидазола с мостиковой ординарной связью.
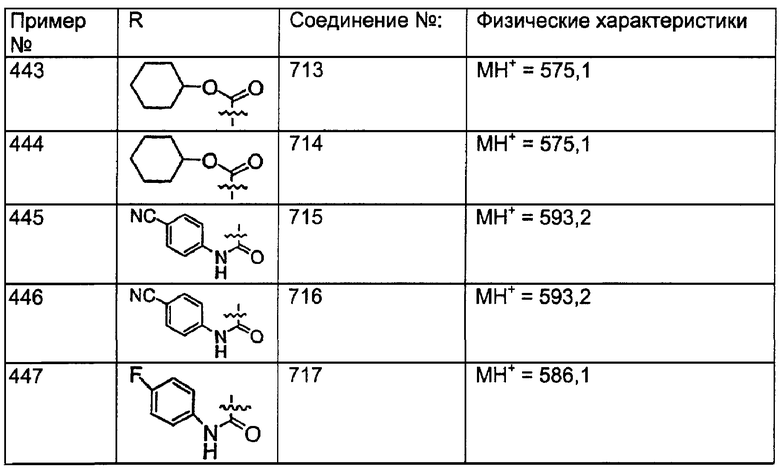
ПРИМЕРЫ 448-454
Соединения (712а) и (712b) по отдельности вводят в реакцию по методике, описанной в Примере 13, с подходящими хлорформиатами или изоцианатами и получают следующие соединения, приведенные ниже в таблице 16.
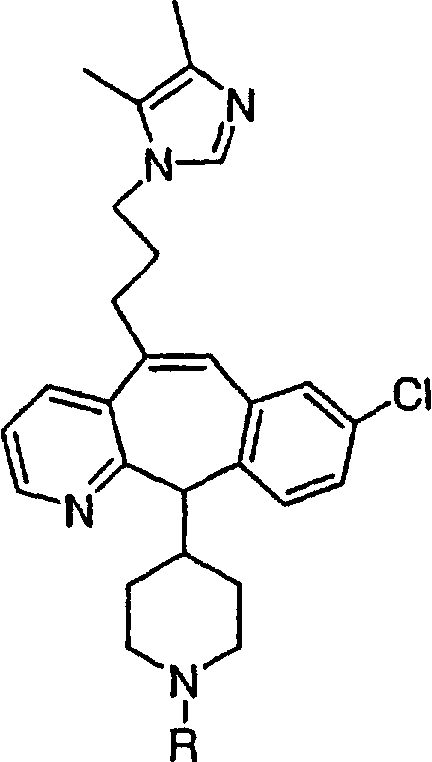
Таблица 16: 5-Замещенные аналоги 4,5-диметилпропилимидазола с мостиковой двойной связью.
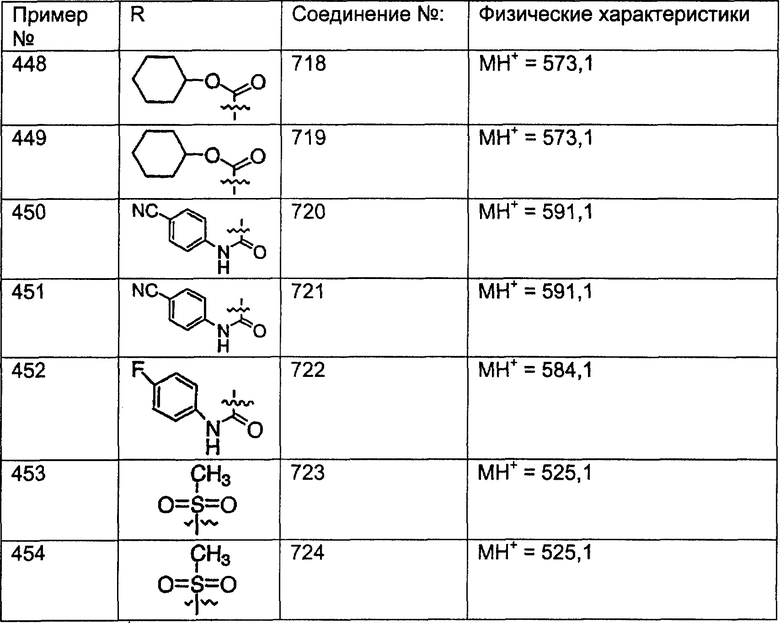
ПРИМЕР ПОЛУЧЕНИЯ 61
Соединения (727а), (727b), (728а) и (728b).
Стадия А Получение соединений (725а), (725b), (726а) и (726b).
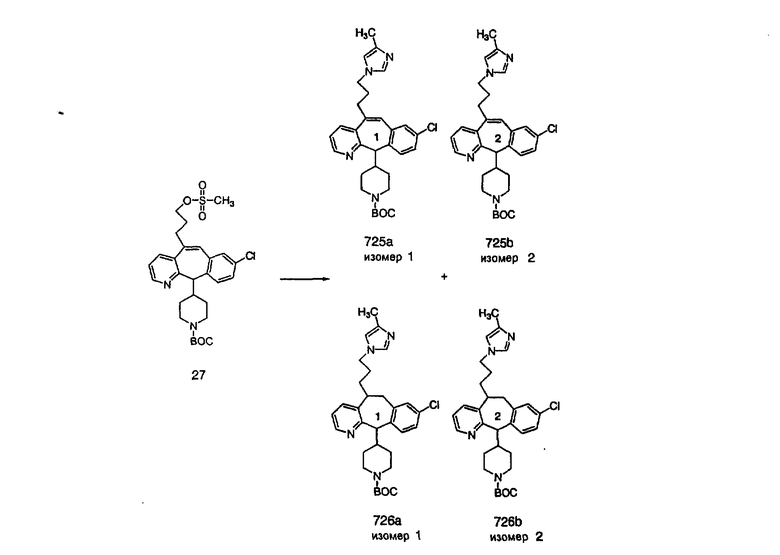
Соединение (27), полученное в Примере получения 4, стадия Е, вводят в реакцию в основном таким же способом, как описано выше в Примере получения 60, стадия А, заменяя 4,5-диметилимидазол на 4-метилимидазол, и в качестве продуктов получают четыре отдельных соединения.
ВОС-производные:
Соединение (725а) изомер 1, тип А (0,69 г, MH+ = 533,1). Соединение (725b) изомер 2, тип А (0,10 г, MH+ = 533,1). Соединение (726а) изомер 1, тип В (0,35 г, МН+ = 535,1). Соединение (726b) изомер 2, тип В (0,22 г, MH+ = 535,1).
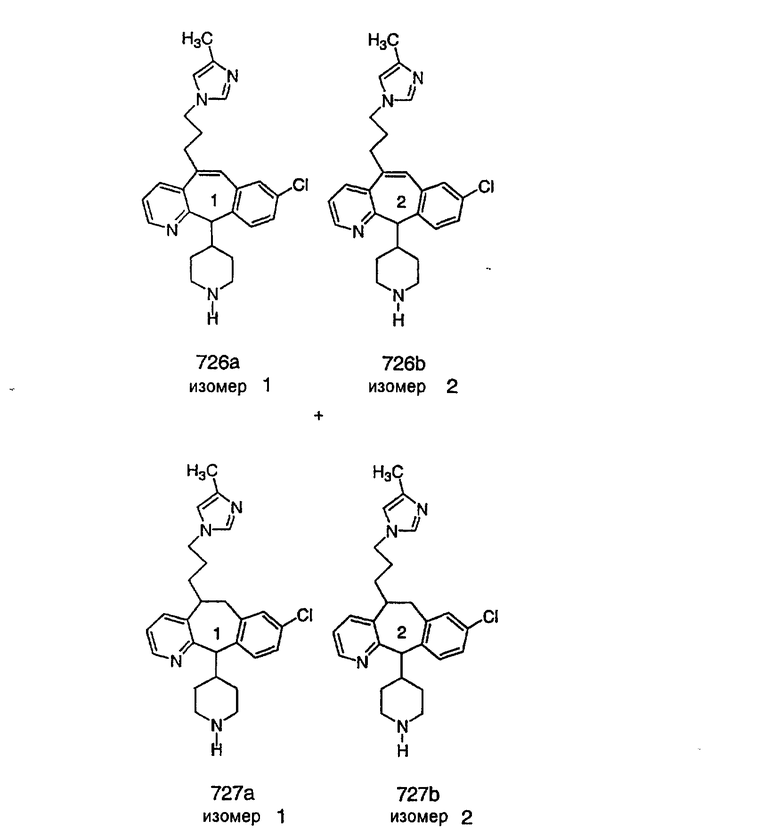
В основном таким же способом, как описано в Примере получения 60, стадия В, получают -NH-производные:
Соединения:
(727а) изомер 1 тип В (0,3 г, выход 100%, MH+ = 435,1), (727b) изомер 2 тип В, (728а) изомер 1 тип А и (728b) изомер 2 тип А.
ПРИМЕРЫ 455-459
Соединения (727а) и (727b) по отдельности вводят в реакцию по методике, описанной в Примере 13, с подходящими хлорформиатами или изоцианатами и получают следующие соединения, приведенные ниже в таблице 17.
Таблица 17: 5-Замещенные аналоги 4-метилпропилимидазола с мостиковой ординарной связью.
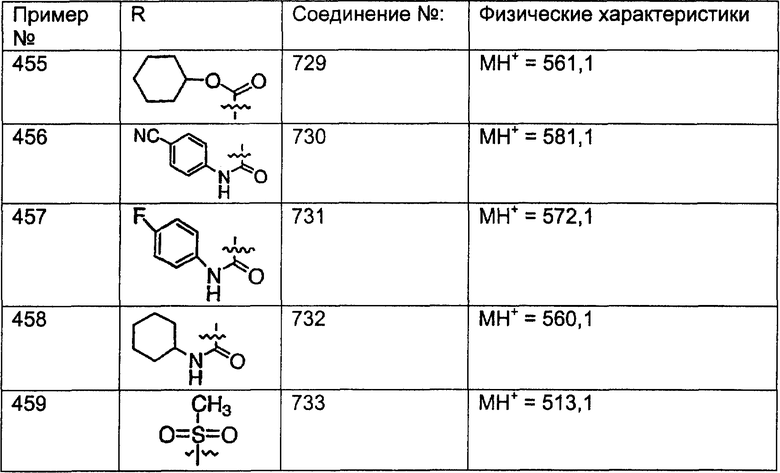
ПРИМЕРЫ 460 -469
Соединения (728а) и (728b) по отдельности вводят в реакцию по методике, описанной в Примере 13, с подходящими хлорформиатами или изоцианатами и получают следующие соединения, приведенные ниже в таблице 18.
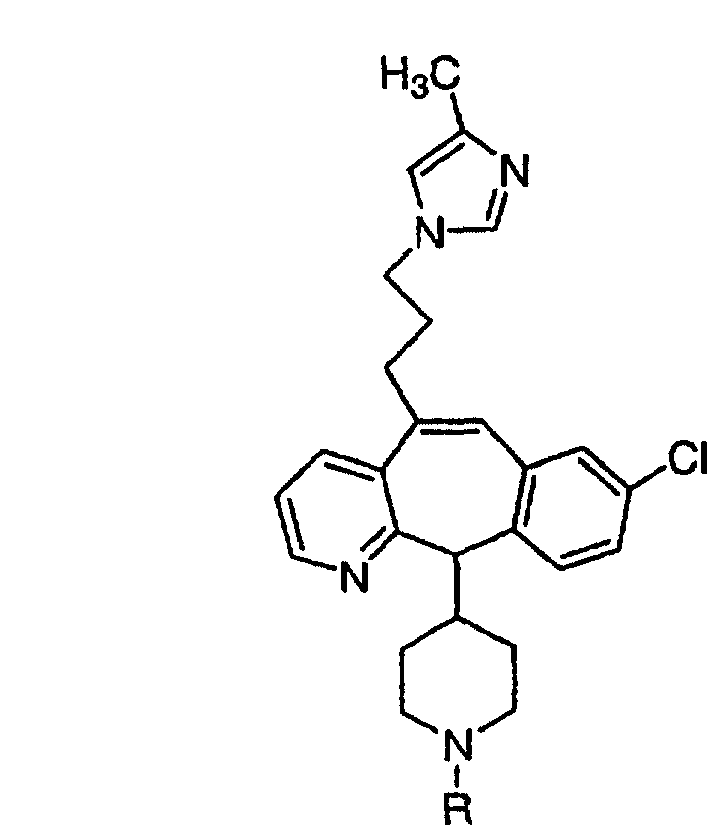
Таблица 18: 5-Замещенные аналоги 4-метилпропилимидазола с мостиковой двойной связью.
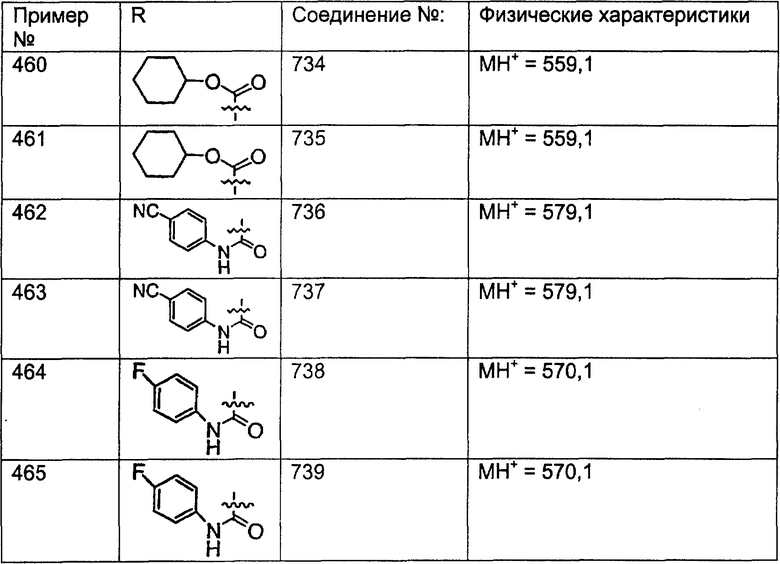
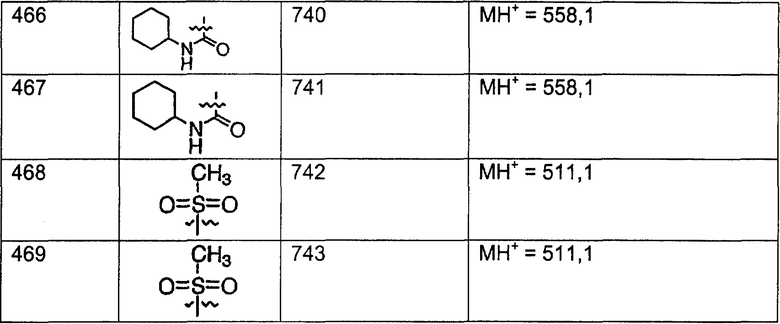
ПРИМЕР 470
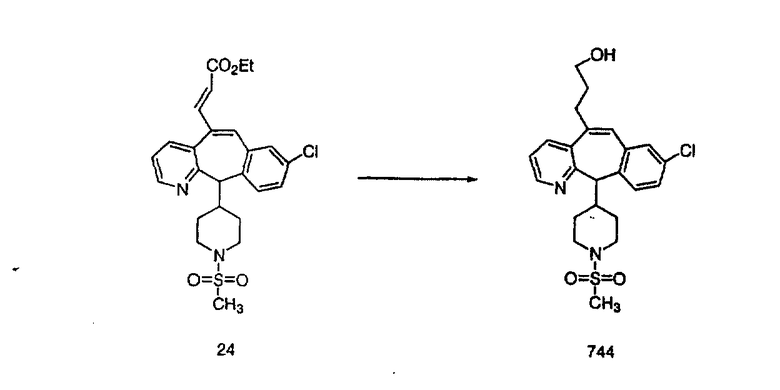
К раствору соединения (24), полученного в Примере получения 4, стадия D (4,0 г, 8,2 ммоль) при перемешивании в атмосфере азота прибавляют CuCI (0,7 г, 8,2 ммоль). Раствор охлаждают до 0°С, затем порциями прибавляют NaBH4 (4,66 г, 123,2 ммоль). Полученный раствор 6 ч перемешивают при 0°С, концентрируют досуха, а затем экстрагируют с помощью СН2Cl3 - насыщенный раствор NaHCO3. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют и очищают с помощью хроматографии на колонке с 200 мл нормальной фазы силикагеля, элюируя смесью 20% EtOAc/CH2Cl2, и получают соединение (744) (3,62 г, выход 99%, MH+ = 447).
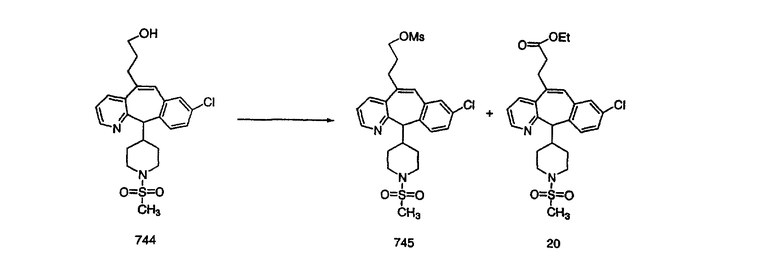
К раствору соединения (744), полученного выше на стадии А (3,0 г, 5,7 ммоль), в CH2Cl2 (100 мл) при комнатной температуре при перемешивании в атмосфере азота прибавляют триэтиламин (2,4 мл, 17,1 ммоль) и метансульфонилхлорид (0,98 г, 8,7 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, затем промывают насыщенным раствором NaHCO3. Объединенные органические слои сушат над Na2SO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке Biotage, элюируя смесью 30% EtOAc/70% CH2Cl2, и получают соединение (745) в виде белого твердого вещества (1,19 г, МН+ = 525,1) и соединение (20) (1,31 г, МН+ = 489,1).
Стадия С Получение соединения (746).
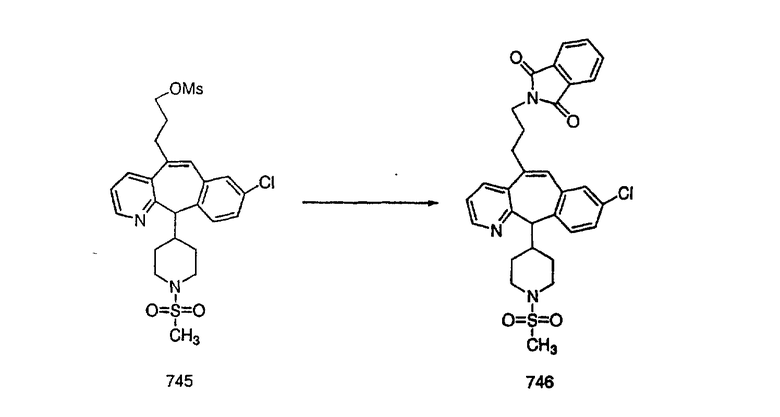
К раствору соединения (745), полученного выше на стадии В (2,17 г, 4,3 ммоль), в DMF (500 мл) при комнатной температуре при перемешивании в атмосфере азота прибавляют калиевое производное фталимида (1,20 г, 0,5 ммоль). Полученный раствор 4 ч нагревают при 90°С, охлаждают до комнатной температуры, концентрируют досуха и экстрагируют с помощью CH2Cl2 - насыщенный раствор NaHCO3. Объединенные органические слои сушат над Na2SO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 50-70% EtOAc/гексан, и получают соединение (746) в виде белого твердого вещества (1,76 г, выход 71%, MH+ = 577,0).
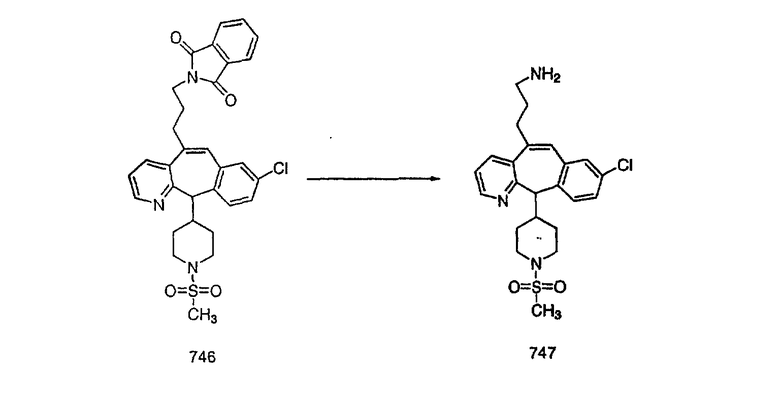
К раствору соединения (746), полученного выше на стадии С (1,67 г, 2,9 ммоль), в EtOH (50 мл) при комнатной температуре при перемешивании прибавляют моногидрат гидразина (0,29 г, 5,8 ммоль). Полученный раствор 4 ч кипятят с обратным холодильником, охлаждают до комнатной температуры, концентрируют досуха и экстрагируют с помощью СН2Cl2 - H2O. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и получают соединение (747) в виде белого твердого вещества (1,23 г, выход 95%, MH2 = 446,1).
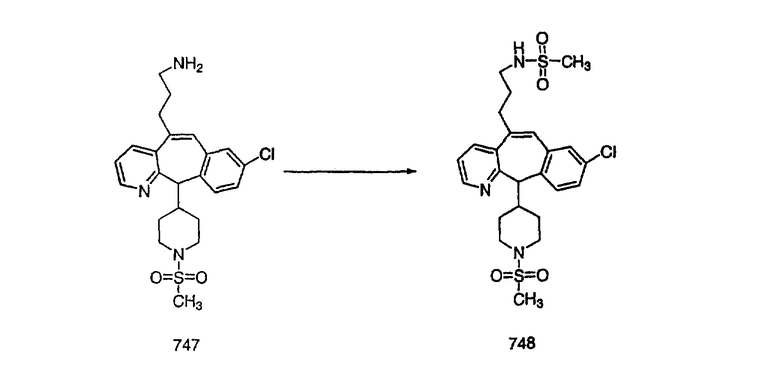
К раствору соединения (747), полученного на стадии D (0,1 г, 0,22 ммоль), в CH2Cl2 (5 мл) при комнатной температуре при перемешивании в атмосфере азота прибавляют TEA (0,06 мл, 0,45 ммоль) и метансульфонилхлорид (0,038 г, 0,34 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, затем промывают насыщенным раствором NaHCO3. Объединенные органические слои сушат над Na2SO4, фильтруют и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% MeOH-NH3/CH2Cl2, и получают соединение (748) в виде белого твердого вещества (0,087 г, выход 76%, МН+ = 524,0).
ПРИМЕР 471 Получение соединения (749).
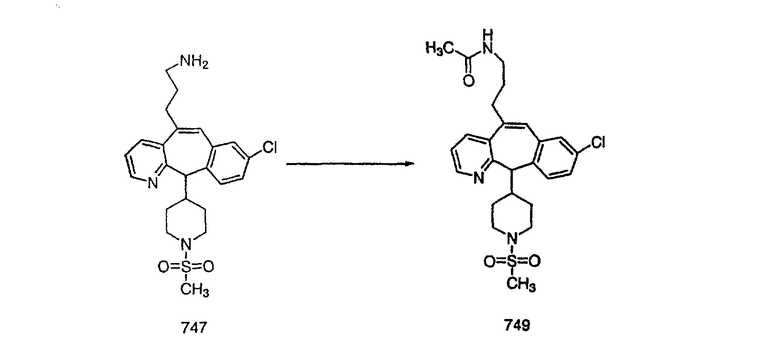
Соединение (747), полученное выше в Примере 470, стадия D, вводят в реакцию в основном таким же образом, как в Примере 470, стадия Е, используя ацетилхлорид, и получают соединение (749) (0,048 г, выход 45%, MH+ = 488,2).
ПРИМЕР 472
Стадия А Получение соединения (750).
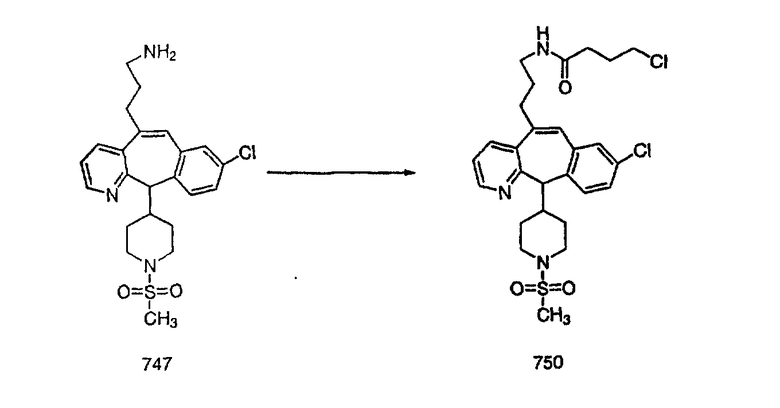
Соединение (747), полученное выше в Примере 470, стадия D, вводят в реакцию в основном таким же образом, как в Примере 470, стадия Е, используя 4-хлорбутилхлорид (ACROS), и получают соединение (750) (0,67 г, выход 100%, MH+ =514,1).
Стадия В Получение соединения (751).
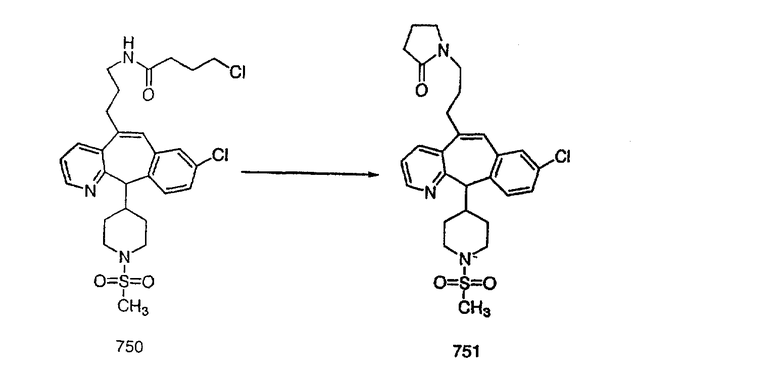
К раствору соединения (750), полученного на стадии А (0,575 г, 1,11 ммоль), в толуоле (15 мл) при комнатной температуре при перемешивании в атмосфере азота прибавляют КаСО3 (0,55 г, 4,01 ммоль). Полученный раствор перемешивают при комнатной температуре в течение выходных дней, а затем 7 ч нагревают при 55°С. После этого раствор охлаждают до комнатной температуры, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке, элюируя смесью 1,5% МеОН-NH3/98,5% СН2Cl2, и получают соединение (751) в виде белого твердого вещества (0,15 г, выход 26%, MH+ = 524,1).
ПРИМЕР 473
Стадия А Получение соединения (752).
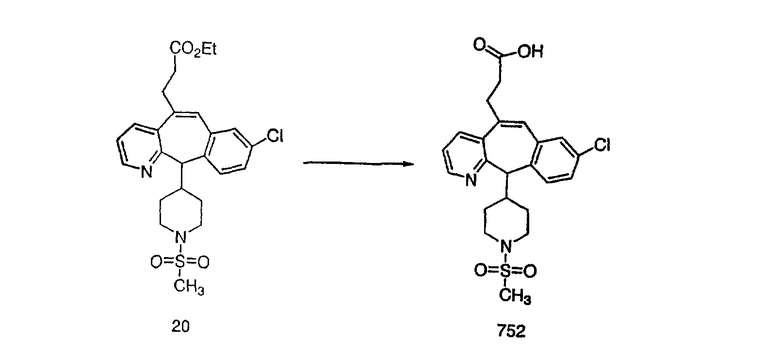
К раствору соединения (20), полученного в Примере 470, стадия В (0,67 г, 1,37 ммоль), в THF (5 мл) при перемешивании прибавляют 1 н. раствор NaOH (6,9 мл, 6,88 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи и концентрируют досуха. Затем раствор подкисляют 10% лимонной кислотой и после этого экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют досуха, получая соединение (752) в виде светло-желтого вещества (0,33 г, выход 52%, МН+ 461,1).
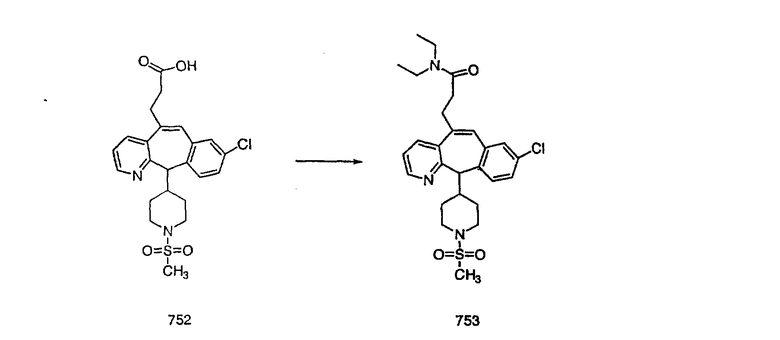
К раствору соединения (752), полученного выше на стадии А (0,1 г, 0,23 ммоль), в CH2Cl2 (5 мл) при комнатной температуре при перемешивании в атмосфере азота прибавляют оксалилхлорид (0,97 г, 7,62 ммоль) и диэтиламин (0,47 г, 6,43 ммоль). Полученный раствор 1 ч перемешивают при комнатной температуре и концентрируют досуха. Затем неочищенный продукт очищают с помощью хроматографии на колонке, элюируя смесью 2% МеОН-NH3/98% CH2Cl2, и получают соединение (753) в виде белого твердого вещества (0,051 г, выход 49,5%, МН+ = 516,1).
ПРИМЕР 474
Получение соединения (754).
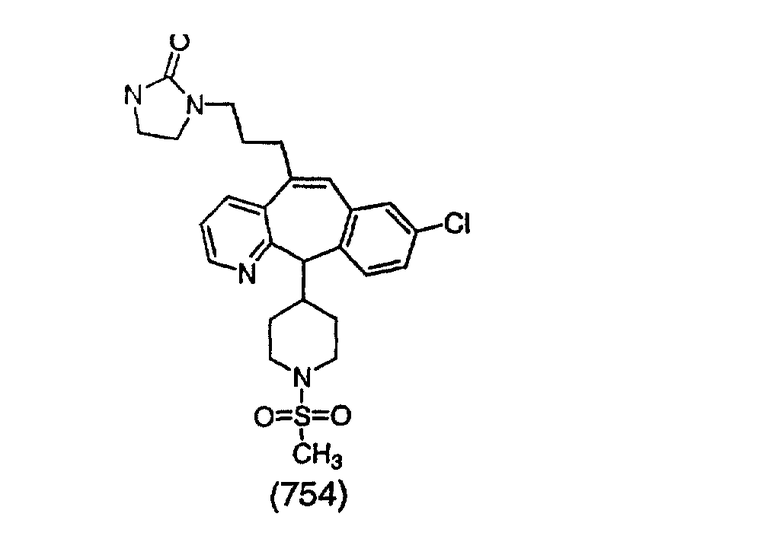
К раствору 2-имидазолидона (0,22 г, 2,0 ммоль) в DMF (10 мл) при перемешивании прибавляют NaH (0,28 г, 2,0 ммоль). Полученный раствор 1 ч перемешивают прикомнатной температуре. Затем при продувании азотом при комнатной температуре этот раствор прибавляют к раствору соединения (22), полученного в Примере получения 3, стадия С (0,67 г, 1,3 ммоль), в DMF (20 мл). Полученный раствор 2 ч нагревают при 90°С, концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - насыщенный раствор NaHCO3. Затем объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% МеОН-МН3/97% CH2Cl2, и получают светло-желтое твердое вещество (754) (0,17 г, выход 25%, MH+ = 515,1).
ПРИМЕР 475
Стадия А Получение соединения (755).
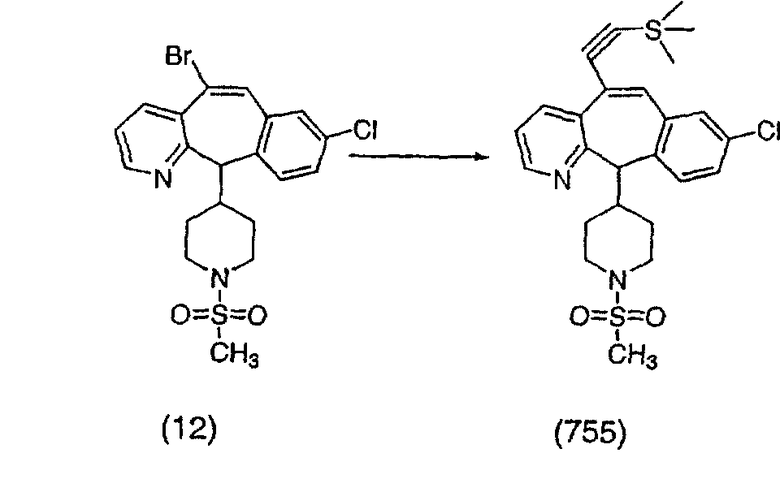
К раствору соединения (12), полученного в Примере получения 2, стадия В (15,75 г, 0,366 ммоль), в DMF (200 мл) при комнатной температуре при перемешивании при продувании азотом прибавляют триметилсилилацетален (12,14 г, 124 ммоль), дихлорид бис(трифенилфосфин)палладия(II) (0,47 г, 0,67 ммоль), EtaN (13,1 мл, 94 ммоль), CuI (0,89 г, 4,7 ммоль) и NaI (1,53 г, 10 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, концентрируют досуха и экстрагируют с помощью CH2Cl2 - Н2О. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 20% EtOAc/80% гексан, и получают продукт (755) (12,35 г, М = 485).
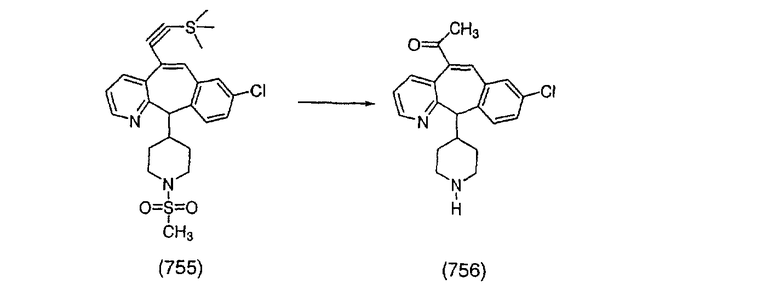
Раствор соединения (755), полученного выше на стадии А (4,48 г, 9,24 ммоль), в концентрированной HCl (100 мл) кипятят с обратным холодильником в течение ночи. Затем раствор охлаждают до комнатной температуры и подщелачивают 50% (мас./мас.) раствором NaOH и после этого экстрагируют с помощью CH2Cl2. Органический слой сушат над MgSO4, фильтруют и концентрируют досуха и получают не совсем белое твердое вещество (756) (4,40 г, выход 100%, MH+ = 353,1).
Стадия С Получение соединения (757).
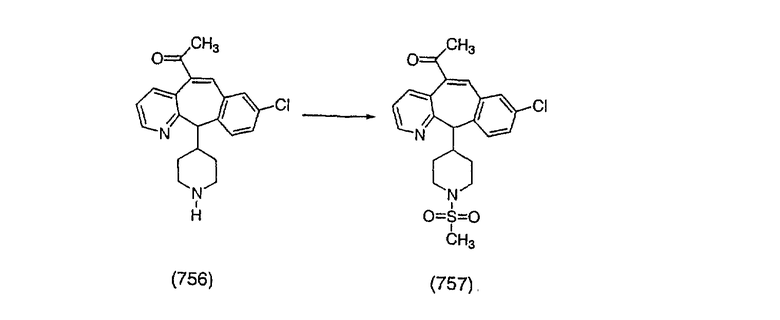
К раствору соединения (756), полученного на стадии В (3,15 г, 8,93 ммоль), в СН2Cl2 (100 мл) при перемешивании прибавляют Et3N (2,5 мл, 17,85 ммоль) и метансульфонилхлорид (0,51 г, 4,46 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи. Затем раствор промывают насыщенным раствором NaHCO3 и органический слой сушат над MgSO4, фильтруют, концентрируют досуха и получают неочищенный продукт (4,31 г, выход 100%, MH+ = 431,1).
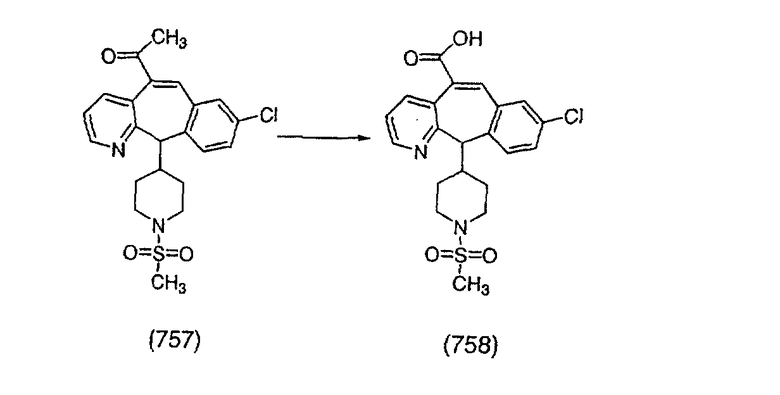
Раствор соединения (757), полученного на стадии С (3,84 г, 8,91 ммоль), в 4% NaCIO (150 мл) и 45% растворе NaOH (15 мл) 2 ч кипятят с обратным холодильником, затем охлаждают до комнатной температуры и прибавляют насыщенный раствор бисульфита натрия (150 мл). Затем значение рН раствора доводят до 6,5 и экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют досуха и получают светло-желтое твердое вещество (3,31 г, выход 86%, MH+ = 433,1).
Стадия Е Получение соединения (759).
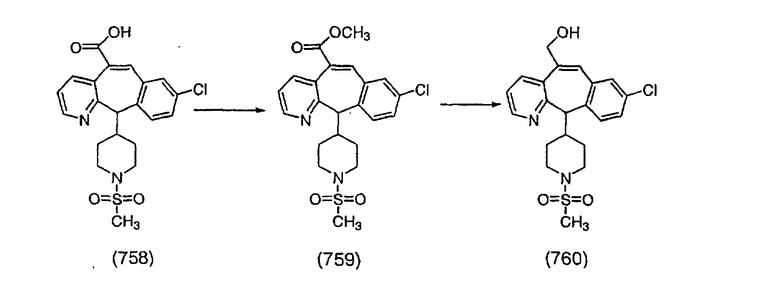
К раствору соединения (758), полученного на стадии D (3,31 г, 7,65 ммоль), в толуоле (80 мл) и МеОН (50 мл) при комнатной температуре при перемешивании в атмосфере азота прибавляют триметилсилилдиазометан (2,0 М раствор в гексане) (3,4 мл, 68,8 ммоль) при 0°С, пока бесцветный раствор не станет желтым. Полученный раствор полчаса перемешивают при 0°С и концентрируют досуха, получая неочищенный продукт (759).
К раствору полученного выше неочищенного продукта (759) в THF (30 мл) при 0°С прибавляют DIBAL (15,3 мл, 15,3 ммоль). Полученный раствор 2 ч перемешивают при 0°С, а затем экстрагируют 10% раствором лимонной кислоты и 1 н. раствором NaOH. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют досуха, получая светло-желтое твердое вещество (760) (2,90 г, выход 90%, MH+= 419,1).
Стадия F Получение соединения (761).
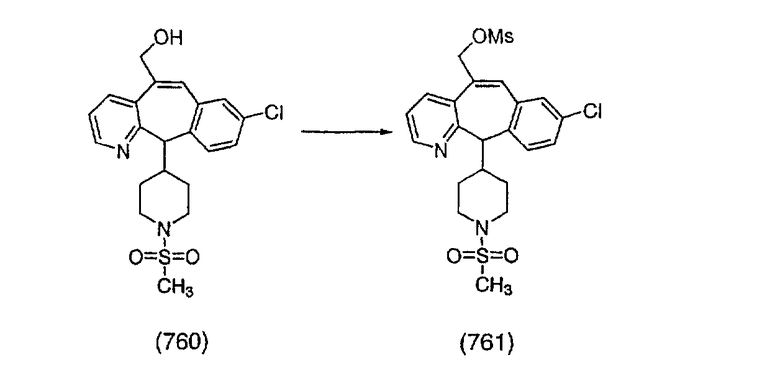
Соединение (760) вводят в реакцию в основном таким же образом, как выше на стадии С, и получают соединение (761).
Стадия G Получение соединения (762).
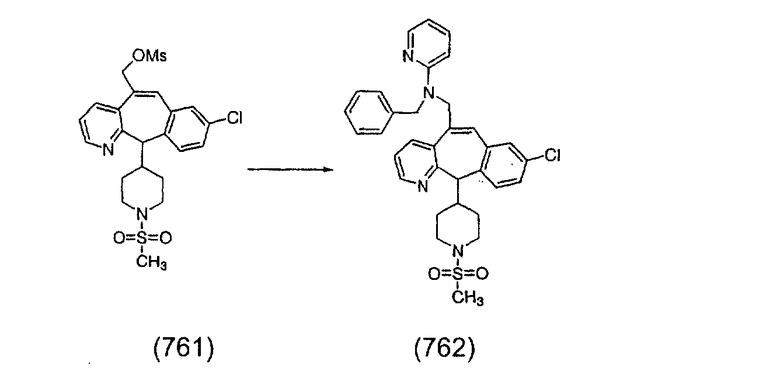
К раствору 2-бензиламинопиридина (0,115 г, 0,624 ммоль) в DMF (10 мл) при перемешивании при комнатной температуре прибавляют NaH (9,81 г, 0,41 ммоль) и раствор 0,5 ч перемешивают. К раствору мезилата, полученного на стадии F (0,2 г, 0,41 ммоль), в DMF (10 мл) при перемешивании и при продувании азотом прибавляютполученный выше раствор 2-бензиламинопиридина в DMF. Полученный раствор 3 ч нагревают при 90°С, концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - насыщенный раствор NaHCO3, а после этого сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 5% МеОН-NH3/CH2Cl2, и получают светло-желтое твердое вещество (762) (0,03 г, выход 13%, MH+ = 585,1).
ПРИМЕР 476
Получение соединения (768).
Стадия А Получение соединения (763).
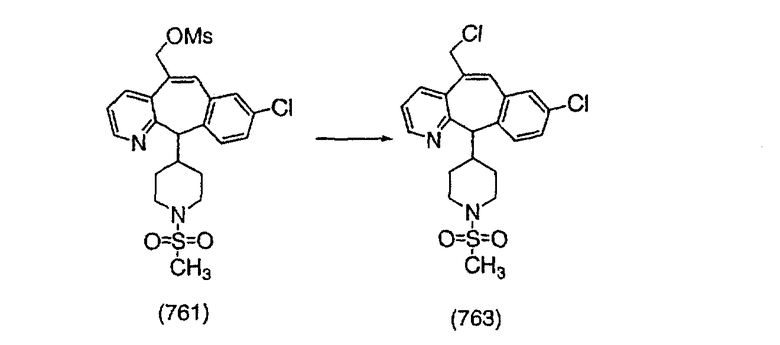
В основном таким же образом, как в Примере 475, стадия Е, получают соединение (763).
Стадия В Получение соединения (764).
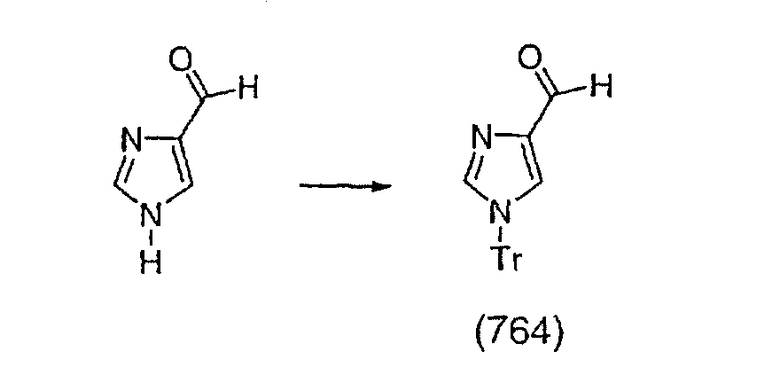
К раствору 4(5)-имидазолкарбоксальдегида (20,0 г, 0,208 ммоль) в CH2Cl2 (200 мл) при перемешивании прибавляют EtsN (29,0 мл, 0,208 ммоль). Затем раствор охлаждают до 0°С, а после этого при 0°С прибавляют трифенилметилхлорид (52,8 г, 0,18 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи и затем его промывают рассолом, водой и концентрируют досуха, получая белое твердое вещество (63,0 г, выход 98%, MH+ = 339,1).
Стадия С Получение соединения (765).
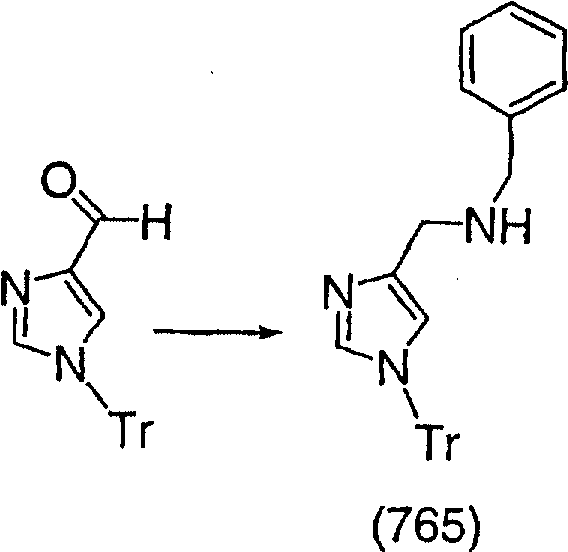
К раствору исходного вещества бензиламина (0,99 г, 8,87 ммоль) в МеОН (50 мл) при перемешивании при продувании азотом при комнатной температуре прибавляют ацетат натрия (0,73 г, 8,87 ммоль), молекулярные сита 3 А (3,0 г) и альдегид (3,0 г, 8,87 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи, а затем прибавляют NaBH4 (0,67 г, 17,74 ммоль) и после этого перемешивают 4 ч и концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - 1 н. раствор NaOH. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 2% MeOH-NH3/98% CH2Cl2, и получают светло-желтое масло (3,75 г, выход 98%, MH+ = 430,2).
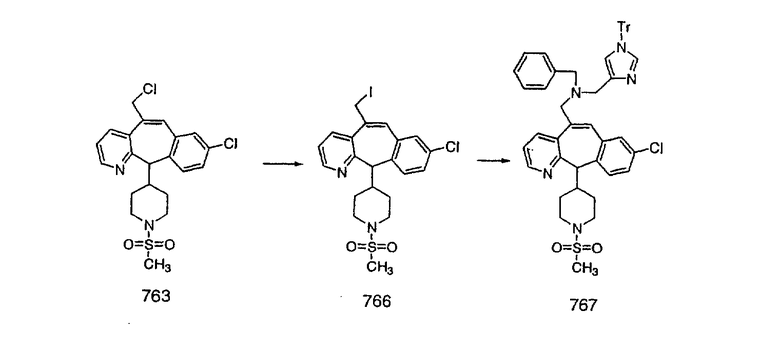
К раствору соединения (764), полученного на стадии B (0,41 г, 1,14 ммоль), в DMF (10 мл) при перемешивании при комнатной температуре в атмосфере азота прибавляют NaH (0,02 г, 0,84 ммоль). Полученный раствор 1 ч перемешивают при комнатной температуре.
К раствору соединения (763), полученного на стадии A (0,4 г, 0,84 ммоль), в ацетоне (30 мл) при перемешивании при комнатной температуре при продувании азотом прибавляют NaI (0,12 г, 0,84 ммоль). Полученный раствор 1 ч кипятят с обратным холодильником, а затем концентрируют досуха, получая соединение (766). К неочищенному соединению (766) прибавляют DMF (10 мл), полученный выше раствор соединения (764) и NaH (0,02 г, 0,84 ммоль). Полученный раствор в течение ночи нагревают при 90°C, затем концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 2% MeOH-NH3/98% CH2Cl2, и получают соединение (767) в виде светло-желтого твердого вещества (0,23 г, выход 33%, MH2 = 830,4).
Стадия Е Получение соединения (768).
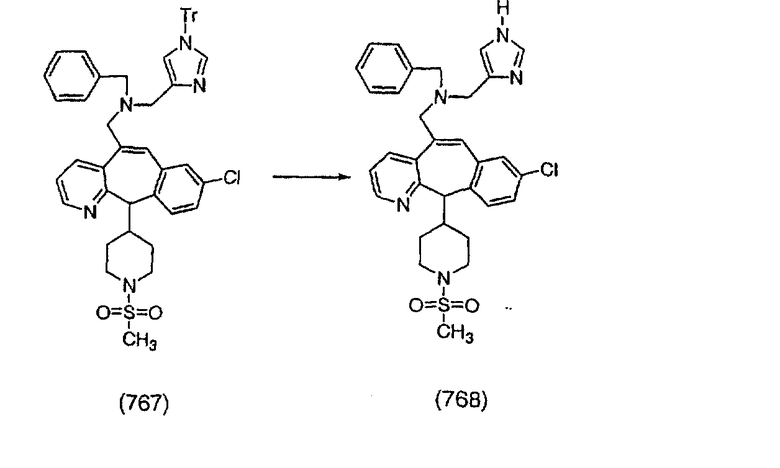
Раствор соединения (767), полученного на стадии С (0,238 г, 0,29 ммоль), в 80% уксусной кислоте в Н2O кипятят с обратным холодильником в течение 2 ч, а затем концентрируют досуха и после этого экстрагируют с помощью CH2Cl2 - 1 н. раствор NaOH. Объединенные органические слои сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% МеОН-NH3/97% CH2Cl2, и получают белое твердое вещество (0,10 г, выход 62%, MH+ = 588,2).
ПРИМЕР ПОЛУЧЕНИЯ 62
Стадия A 1N-трет-бутоксикарбонил-3(R)-и -3(S)-(1Н-имидазол-1-ил)-метилпирролидины
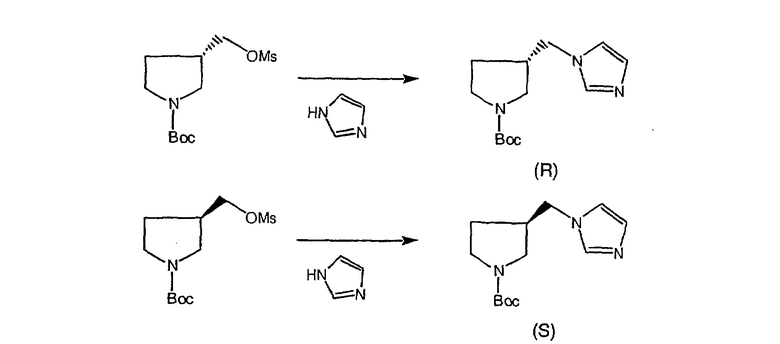
3(R)-(3-Метансульфонилоксиметил)-пирролидин (J. Med. Chem. 1990, 33, 77-77) (0,993 г, 3,56 ммоль) растворяют в безводном DMF (25 мл) и прибавляют натриевую соль имидазола (0,6 г, 10 ммоль). Смесь 2 ч нагревают при 60°С, а затем выпаривают досуха. Продукт экстрагируют с помощью CH2Cl2 и промывают рассолом. Экстракт в CH2Cl2 выпаривают досуха и получают искомое соединение (1,1409 г, 100%). МС ЭР: МС ББА(М+ 1)=252; 1H ЯМР (CDCl3) 1,45 (s,9H), 1,5-1,7 (m, 1H), 1,9-2,1 (m, 1H), 2,5 -2,7 (m, 1H), 3,0 -3,2 (m, 1H), 3,3 -3,6 (m, 2H), 3,9 (dd, 2H), 6,9 (s, 1H), 7,1 (s,1H), 7,45 (s,1H).
Аналогичным образом из 3(S)-(3-метансульфонилоксиметил)-пирролидина (0,993 г, 3,56 ммоль) получают (S)-изомер (1,1409 г, 100%).
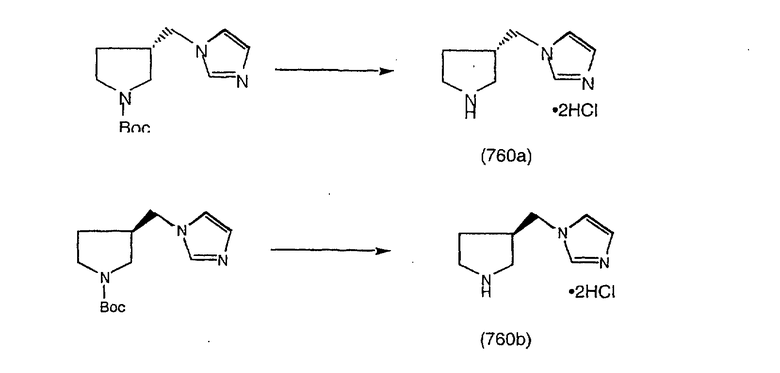
Соединение (0,48 г, 1,91 ммоль), полученное на стадии А, 2 ч перемешивают в 4 н. растворе HCl в диоксане (10 мл) и затем выпаривают досуха, получая искомое соединение, которое используют для сочетания с трициклической кислотой.
Аналогичным образом получают (S)-изомер.
ПРИМЕР 477
Получение соединения (771).
Стадия А Получение соединения (769).
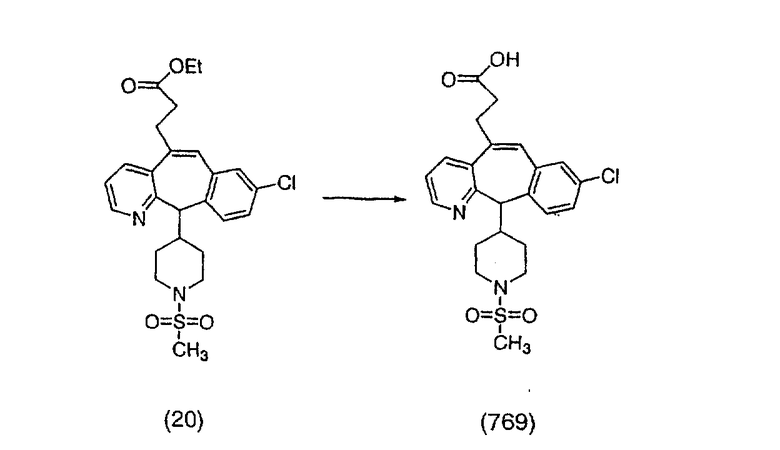
К раствору соединения (20), полученного в Примере получения 3, стадия В (4,86 г, 9,94 ммоль), в EtOH (100 мл) прибавляют 1 н. раствор LiOH (80 мл). Полученный раствор перемешивают при комнатной температуре в течение ночи и концентрируют досуха, а затем растворяют в CH2Cl2. Затем с помощью 1 н. раствора HCl значение рН доводят до 6,5-7,0. Затем водный слой отделяют и концентрируют досуха, а после этого растворяют в THF и получают литиевую соль (4,86 г, выход 100%, M+Li =467,1).
Стадия В Получение соединения (771).
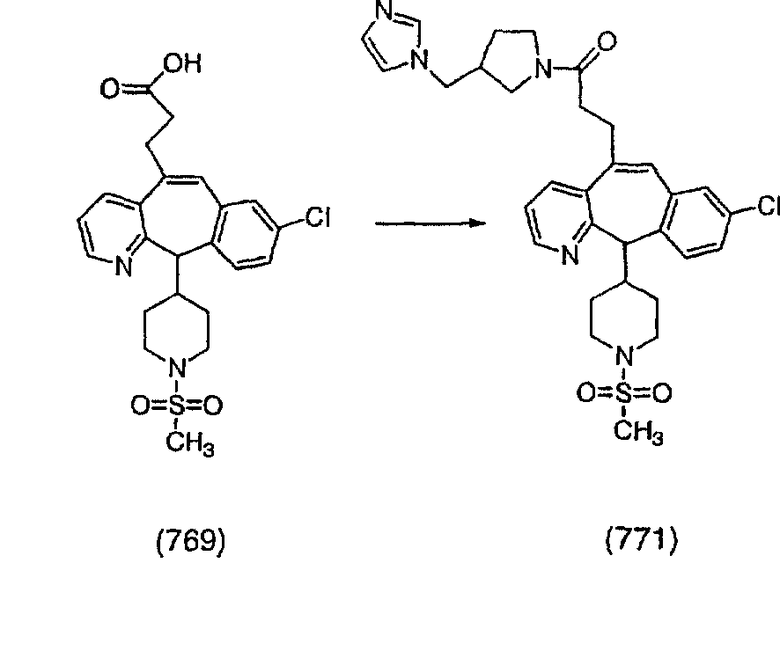
К раствору соединения (769), полученного выше на стадии А (0,38 г, 0,84 ммоль), в DMF (10 мл) при перемешивании при комнатной температуре при продувании азотом прибавляют соединение (770), полученное в Примере получения 62 (0,163 г, 1,09 ммоль), бензотриазил-М-окситрис(диметиламино)фосфонийгексафторфосфат (0,44 г, 1,01 ммоль) и EtsN (0,5 мл, 3,36 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи и концентрируют досуха, а затем экстрагируют с помощью смеси CH2Cl2 -10% раствор лимонной кислоты. Затем объединенные органические слои промывают насыщенным раствором NaHCO3, рассолом, сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% MeOH-NH3/CH2Cl2, и получают светло-желтое твердое вещество (0,12 г, М = 594,2).
ПРИМЕР ПОЛУЧЕНИЯ 63
Соединение (772)
Стадия А 1-N-трет-бутоксикарбонил-4-гидроксипиперидин.
К раствору 4-гидроксипиперидина (2 г, 19,78 ммоль) и триэтиламина (4,16 мл, 29,67 ммоль) в CH2CI2 (20 мл) прибавляют ди-трет-бутилдикарбонат (5,18 г, 23,72 ммоль) и 16 ч перемешивают при комнатной температуре. Раствор разбавляют с помощью CH2Cl2 и промывают водой, сушат (MgSO4), фильтруют и выпаривают, получая искомое соединение (3,95 г, 99%). МС ББА (М + 1) = 202.
Стадия B 1-N-трет-бутоксикарбонил-4-метансульфонилоксипиперидин.
Соединение, полученное выше на стадии А (3,5 г, 17,39 ммоль), и триэтиламин (4,85 мл, 34,79 ммоль) растворяют в СН2Cl2 (30 мл) и смесь перемешивают в атмосфере азота при 0°С. Прибавляют метансульфонилхлорид (1,62 мл, 20,88 ммоль) и раствор 2 ч перемешивают при комнатной температуре. Раствор разбавляют с помощью CH2Cl2 и промывают насыщенным водным раствором бикарбоната натрия, водой и сушат (MgSO4), фильтруют и выпаривают досуха, получая искомое соединение (4,68 г, 96,4%). МСЭР: m/z = 280 (MH+).
Стадия С 1-N-трет-бутоксикарбонил-4-(1Н-имидазол-1-ил)-пиперидин.
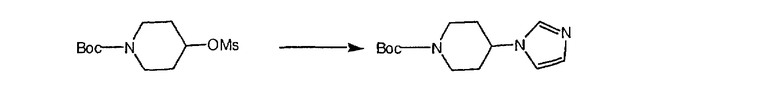
Раствор соединения, полученного на стадии В (4,0 г, 14,32 ммоль), в DMF (120 мл) при перемешивании в атмосфере азота прибавляют к раствору NaH (0,52 г, 21,66 ммоль) и имидазола (1,46 г, 21,47 ммоль) в DMF (20 мл). Смесь 16 ч перемешивают при 60°С. DMF выпаривают в вакууме. Полученный неочищенный продукт экстрагируют с помощью CH2Cl2 и экстракт последовательно промывают водой и рассолом и CH2Cl2 выпаривают, получая в остатке искомое вещество, которое хроматографируют на силикагеле, используя в качестве элюента смесь 3% (10% концентрированный NH4OH в метаноле) CH2Cl2, и получают искомое соединение (0,94 г, 26%). МС ББА (М + 1) = 252. 1H ЯМР (CDCl3) 1,4 (s, 9Н), 1,6-1,8 (m, 2H), 2,0 (dd, 2H), 2,8 (dt, 2H), 4,05 (m, 1Н), 4,2 (m, 2H), 6,9 (s, 1H), 7,0 (s, 1H), 7,65 (s, 1H).
Стадия D 4-(1 Н-имидазол-1-ил)-пиперидин.
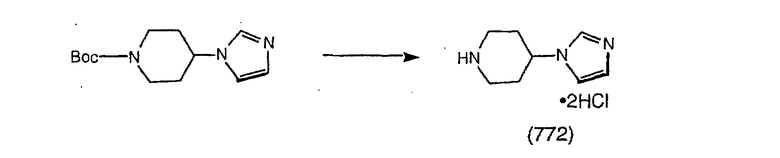
Соединение, полученное на стадии С (0,21 г, 0,836 ммоль), 2 ч перемешивают в 4 н. растворе HCI в диоксане (5 мл), а затем выпаривают досуха и получают искомое соединение (772), которое используют для сочетания с трициклической кислотой.
ПРИМЕР 478 Получение соединения (773).
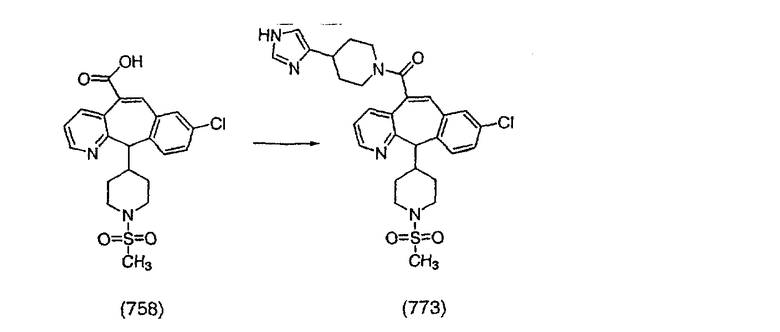
К раствору соединения (758), полученного в Примере 475, стадия D (0,2 г, 0,46 ммоль), в CH2Cl2 (5 мл) в атмосфере азота при перемешивании при комнатной температуре прибавляют соединение (772), полученное в Примере получения 63, стадия D (0,19 г, 0,55 ммоль), бензотриазоил-N-окси-трис-(диметиламино)фосфонийгексафторфосфат (0,25 г, 0,55 ммоль) и EtaN (0,3 мл, 1,85 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи и концентрируют досуха, а затем экстрагируют с помощью смеси CH2Cl2 - 10% раствор лимонной кислоты. Объединенные органические слои затем промывают насыщенным раствором МаНСО3, рассолом, сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью 3% МеОН-NH3/СН2Cl2, и получают белое твердое вещество (773) (0,013 г, выход 5%, М = 566,2).
ПРИМЕР 479
Получение соединений (774 -777).
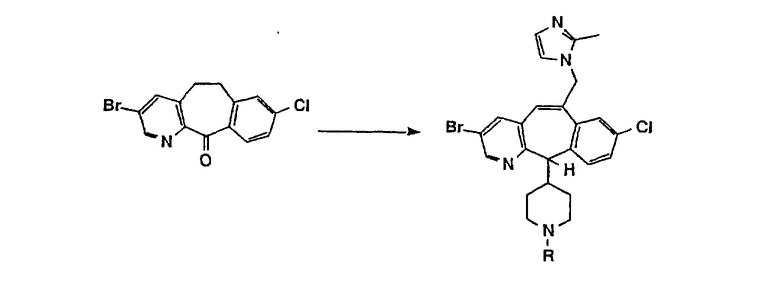
R = N-BOC
(774) (энантиомер 1), (М + 1 = 584).
(775) (энантиомер 2), (М + 1 = 584).
R=H
(776) (энантиомер 1).
(777) (энантиомер 2).
3-Бром-8-хлоразакетон (патент США US 5977128, Пример получения 11, стадия А (1999)) вводят в реакцию в основном таким же образом, как в Примере получения 23 и Примере 91, и получают М-ВОС-производные (774) и (775). Соединения (774) и (775) по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (776) и (777).
ПРИМЕР 480
Получение соединений (778) и (779).
В основном, таким же образом, как в Примерах 420 и 421, получают соединения (778) и (779).
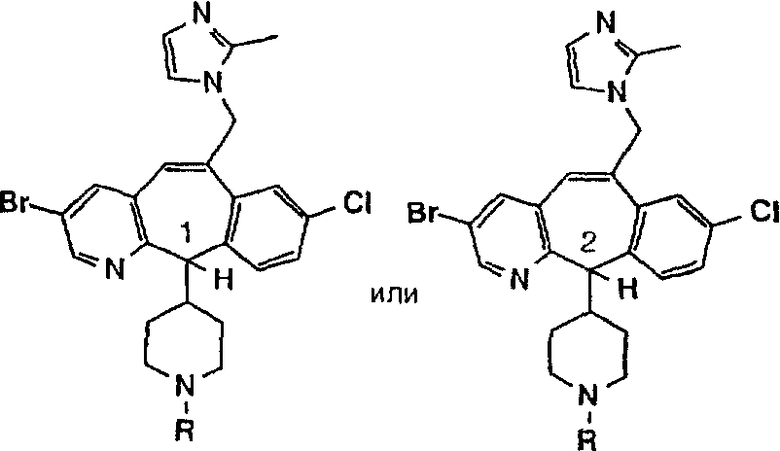
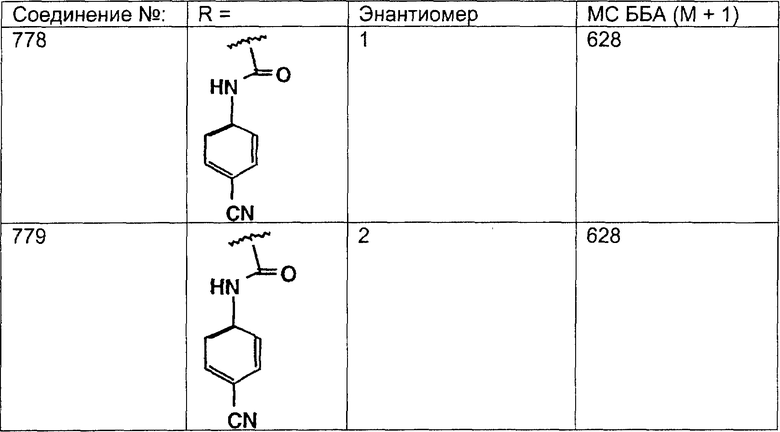
Физические характеристики:
(778): 1Н ЯМР (Varians 400 МГц, COCl3, ppm): 5 = 8,564 (1 Н, d, J = 2 Гц), 7,784 (1 Н, d, J = 2 Гц), 7,624 (1H, d, J = 2 Гц), 7,51 -7,37 (5Н, т), 7,305 (1Н, s), 7,267 (1Н, s), 6,870 (1Н, s), 6,867 (1H, s), 6,579 (1H, s), 5,282 (1H, d, J = 16 Гц), 5,031 (1H, d, J = 17 Гц), 4,576 (1H,s), 3,176 (4Н, br ddd, J=6, 14 и 58 Гц), 2,485 (3Н, s), 1,950 (4Н, dd, J = 6 и 9 Гц); МС (т/е) 630 (М + Н), 340, 327, 293, 263, 249; MCBP (Jeol JMS-HX110A) Рассчитано для C31H27BrCIN7O 628,1227 (М + 1), найдено 628,1229.
ПРИМЕР 481
Получение соединений (780) и (781).
В основном, таким же образом, как в Примере 70, получают соединения (780) и (781).
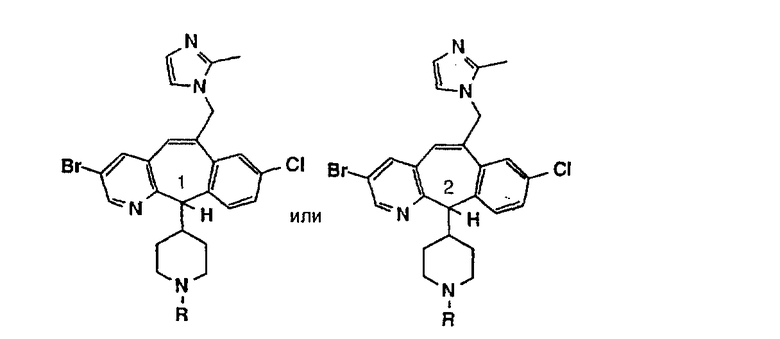
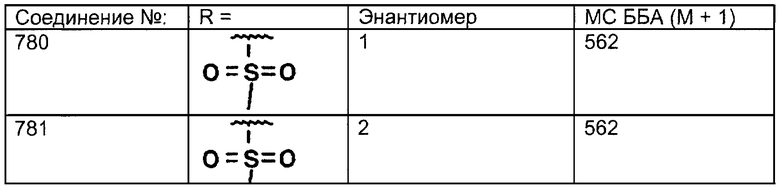
ПРИМЕР ПОЛУЧЕНИЯ 64
Стадия А Соединение (782).
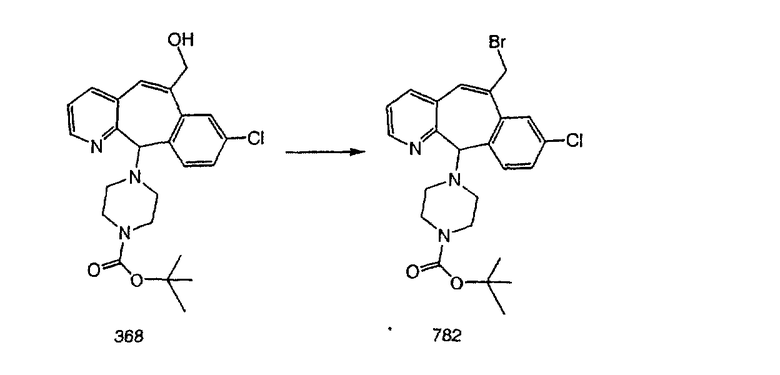
Соединение (368), полученное в Примере получения 42, стадия С (2,34 г, 5,29 ммоль), при 0°С растворяют в 25 мл CH2Cl2. Прибавляют PPh3 (1,66 г, 6,34 ммоль) и NBS (1,03 г, 5,82 ммоль). Через 90 мин реакционную смесь разбавляют с помощью СН2Cl2 (20 мл), промывают насыщенным раствором NaHCO3, рассолом и сушат над MgSO4. Неочищенный продукт очищают на колонке с силикагелем (гексаны/EtOAc от 4:1 до 2: 1) и получают 1,8 г соединения (782) в виде светло-желтого твердого вещества. МС, М + 1 = 504.
Стадия В Соединение (783).
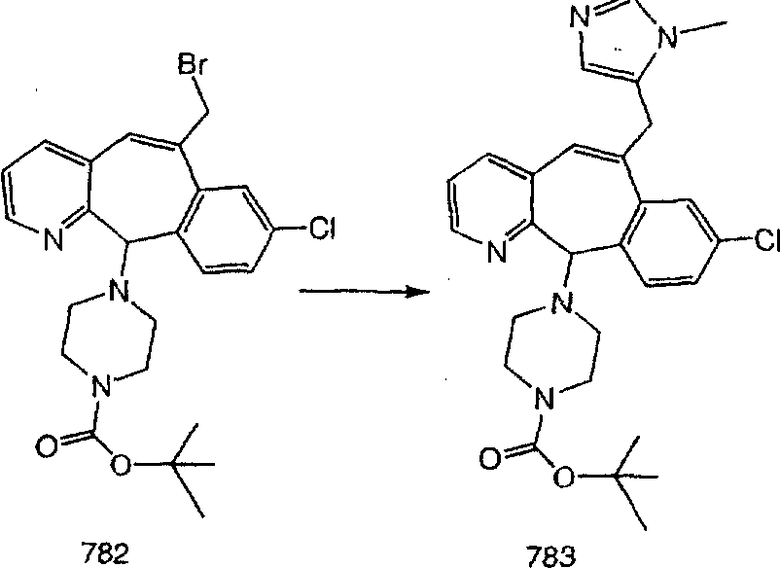
5-Йод-1N-метилимидазол (455 мг, 2,18 ммоль) при комнатной температуре растворяют в 10 мл THF. По каплям прибавляют EtMgBr (2,4 мл, 1,0 М раствор в THF). Через 30 мин реакционную смесь охлаждают до 0°С. Затем прибавляют 10 мл раствора CuCN (175 мг, 1,96 ммоль) в THF и LiCI (166 мг, 3,9 ммоль). Еще через 10 мин прибавляют соединение (782), полученное выше на стадии А (989 мг, 1,96 ммоль, в 10 мл THF). Реакционную смесь перемешивают в течение ночи. Для остановки реакции прибавляют насыщенный раствор NH4CI. Полученную эмульсию фильтруют через воронку с пористым фильтром и фильтрат дважды экстрагируют с помощью EtOAc. Органический слой промывают раствором NaHCO3 и рассолом, сушат над сульфатом магния, фильтруют и выпаривают в вакууме. Полученное неочищенное вещество хроматографируют на колонке с силикагелем (с использованием смеси гексаны/EtOAc -1:1, а затем CH2Cl2/MeOH -10: 1) и получают 330 мг искомого вещества. МС, М + 1 = 506. Энантиомеры разделяют на хиральной колонке AD.
ПРИМЕР 482
Получение соединения (784).
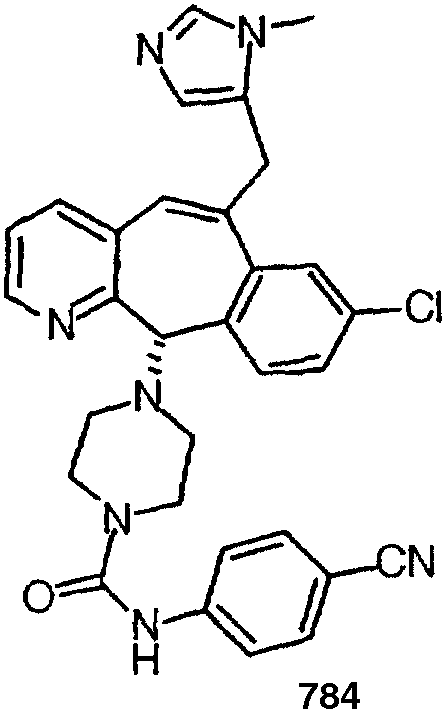
Соединение (783), полученное выше в Примере получения 64, стадия В (40 мг), при комнатной температуре растворяют в СН2Cl2 (5 мл), а затем прибавляют TFA (0,5 мл). Через 2 ч растворитель выпаривают в вакууме и дважды выпаривают совместно с PhCH3. Затем неочищенную смесь растворяют в СН2Cl2 (4 мл) и по каплям прибавляют EtaN, пока раствор не станет основным по индикаторной бумаге. Прибавляют 4-цианфенилизоцианат (14 мг). Через 5 мин реакционную смесь досуха выпаривают в вакууме. Неочищенное вещество очищают с помощью пластины для препаративной ТСХ (10: 1, CH2Cl2/MeOH) и получают 23 мг соединения (784) в виде белого твердого вещества. МС, М + 1 = 550.
ПРИМЕР 483
Получение соединения (785).
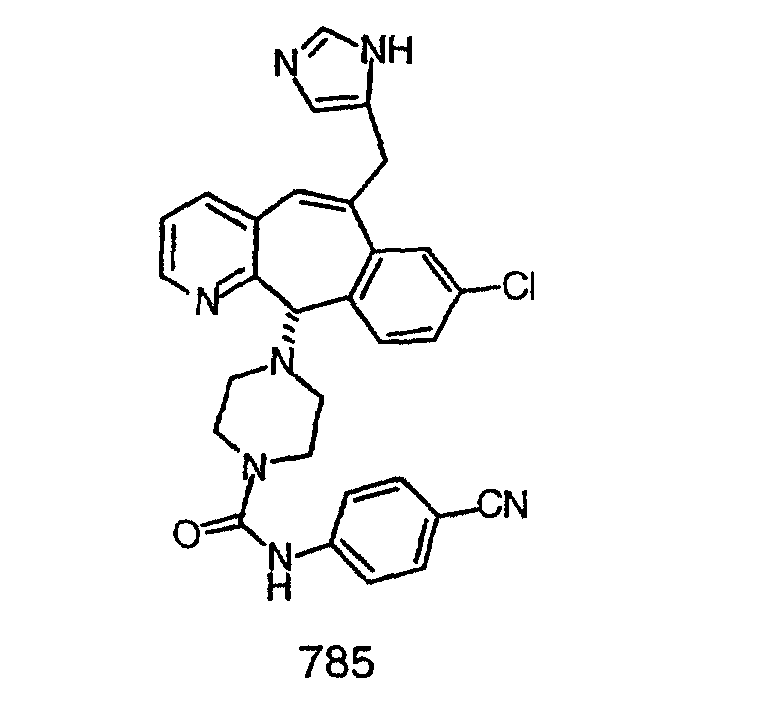
Соединение (785) получают с помощью в основном такой же методики, как приведенная в Примере получения 28 и Примере 482, заменяя 5-йод-1М-метилимидазол на 4-йод-1-тритилимидазол.
ПРИМЕР 484
Получение соединений (786) и (787).
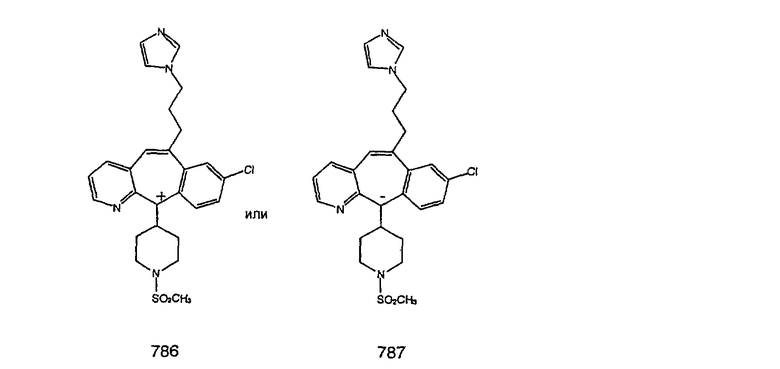
Соединения (786) и (787) получают с помощью в основном такой же методики, как приведенная в Примере получения 7, заменяя кетоны (9) и (10) на кетоны (15) и (16), полученные в Примере получения 2, стадия D.
Соединение (786) MH+ = 497; [α]D 20 = +15,3°;
соединение (787) МН+ = 497; [α]D 20 = -13,4°;
ПРИМЕР 485
Получение соединения (788).
С помощью в основном такой же методики, как приведенная в Примере получения 33, стадии Е-Н, но заменяя соединение (281) на соединение (365) и 1-метилимидазол на 2-гидроксиметилимидазол, получают соединение (788).
(788): 1Н ЯМР (Varians400 МГц, CDCI3, ppm): δ = 8,5 (1Н, dd), 7,34 (1Н, s), 7,59 (1Н, d), 7,4 (2Н, m), 7,25 (2Н, m), 7,04 (1Н, s), 6,9 (1Н, s), 6,6 (1Н, s), 5,37 (2Н, dd), 4,8 (2Н, dd), 4,6 (1 H, s), 3,2 (5Н, br s), 2,0 (2Н, br s), 1,9 (2Н, br s), 1,4 (9Н, s).
ПРИМЕР ПОЛУЧЕНИЯ 65
Стадия А Соединение (789).
К раствору спирта (3,8 г, 8,6 ммоль) в CH2Cl2 (100 мл) в атмосфере азота прибавляют MnO2 (40 г). Полученный раствор 4 дня перемешивают при комнатной температуре. Затем смесь фильтруют через слой целита, используя этилацетат (500 мл) в качестве элюента. Фильтрат концентрируют и получают желтую жидкость (4,0 г, МН+ 440,1). Неочищенное вещество разделяют на чистые изомеры с помощью ВЭЖХ, используя хиральную колонку AD и элюируя смесью 20% IPA/80% гексан/0,2% DEA (изомер 1, 810 мг, изомер 2, 806 мг),
Стадия В Соединение (790).
К раствору имидазолсодержащего реагента Гриньяра, полученного из 5-йод-1М-метилимидазола (312 мг, 1,5 ммоль, Пример получения 64, стадия В), прибавляют раствор альдегида (791) (380 мг, 0,86 ммоль) в CH2CI2 (10 мл). После перемешивания при комнатной температуре в течение ночи смесь 1 ч нагревают при 40°С. После повторного охлаждения до комнатной температуры для остановки реакции прибавляют насыщенный раствор NH4Cl. Органический слой сушат и растворитель выпаривают. Затем остаток очищают на колонке с силикагелем (от 2% до 10% МеОН в СН2Cl2) и получают продукт в виде коричневого масла (207 мг, выход 46%, MH4 = 522,1). После этого диастереоизомеры разделяют с помощью ВЭЖХ на хиральной колонке AD, элюируя смесью 20% IPA/80% гексаны/0,2% DEA.
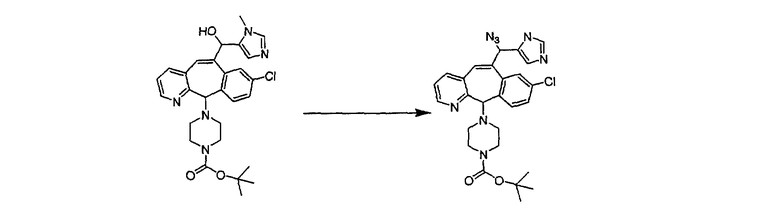
К раствору (790) (200 мг, 0,38 ммоль) в 5 мл THF при комнатной температуре прибавляют DPPA (210 мг, 0,76 ммоль), а затем прибавляют DBU (120 мг, 0,76 ммоль). Смесь перемешивают в течение ночи, а затем разбавляют этилацетатом (30 мл), два раза промывают водой и один раз рассолом. Органический слой сушат и растворитель выпаривают. Остаток очищают с помощью препаративной ТСХ (10% МеОН в СН2Cl2 с включением 0,2% МН3) и получают продукт (791) (102,8 мг, Mt-Г = 547,1). Также извлекают исходное вещество (790) (58 мг). Диастереоизомеры разделяют на хиральной колонке AD.
ПРИМЕР 486
Получение соединения (792).
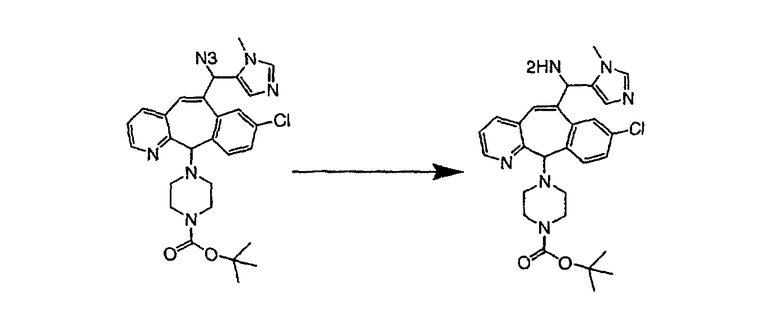
К влажному раствору (791) (48 мг, 0,09 ммоль) в THF (3 мл) при комнатной температуре прибавляют PPh3 (32 мг, 0,12 ммоль). После перемешивания в течение ночи реакционную смесь концентрируют и остаток очищают с помощью препаративной ТСХ (10% МеОН в CH2Cl2 с включением 0,2% NH3) и получают белое твердое вещество (24,3 мг). Затем белое твердое вещество повторно растворяют в THF/H2O (5 мл/0,5 мл) и смесь кипятят с обратным холодильником в течение ночи. Затем реакционную смесь подвергают распределению между этилацетатом и водой. Органический слой сушат и концентрируют. Остаток очищают с помощью препаративной ТСХ (5% МеОН в CH2Cl2 с включением 0,2% МН3) и получают желтое твердое вещество (792) (8,3 мг, МН+ = 521,1).
ПРИМЕР 487
Получение соединения (793).
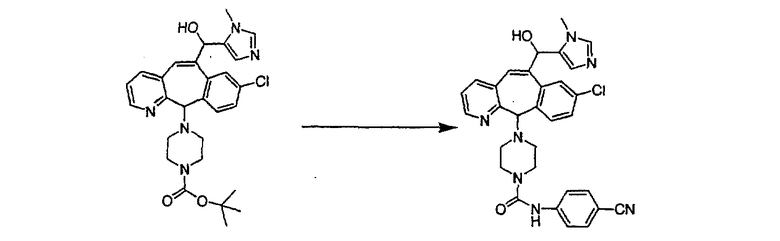
Соединение (790) превращают в соединение (793) с помощью в основном такой же методики, что и описанная в Примере 482. МС, M+ = 566,1).
ПРИМЕР 488
Получение соединения (794).
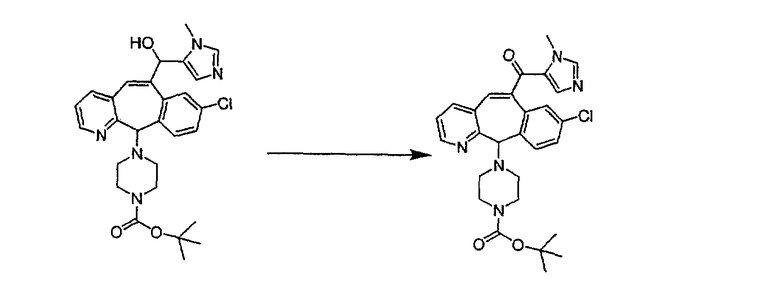
Соединение (790) превращают в соединение (794) с помощью в основном такой же методики, что и описанная в Примере получения 65, стадия А. МС, M+ = 520,1.
ПРИМЕР 489
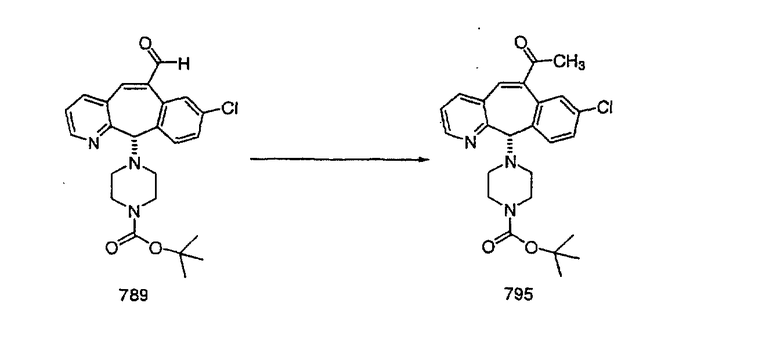
Альдегид (789), полученный в Примере получения 65, стадия А (150 мг, 0,34 ммоль), растворяют в THF (6 мл). К этому раствору по каплям прибавляют MeMgBr (0,3 мл, 3,0 М раствор в Et2O). После перемешивания при комнатной температуре в течение 4 ч реакцию останавливают насыщенным раствором NH4CI и экстрагируют этилацетатом. Органический слой промывают рассолом, сушат и концентрируют, получая желтое твердое вещество (150 мг). Затем неочищенный продукт растворяют в СН2Cl2 (5 мл). К этому раствору прибавляют перйодинан Десса-Мартина (210 мг) и каплю воды. Через 1 ч прибавляют водный раствор Na2S2O3 (4 мл, 10%). Смесь перемешивают 10 мин и экстрагируют с помощью CH2Cl2. Органический слой промывают с помощью NaHCO3, сушат и концентрируют. Неочищенное вещество очищают на пластинах для препаративной ТСХ (5% метанол в CH2Cl2) и получают метилкетон (795) в виде желтого твердого вещества (70 мг).
Стадия В Соединение (795.1).
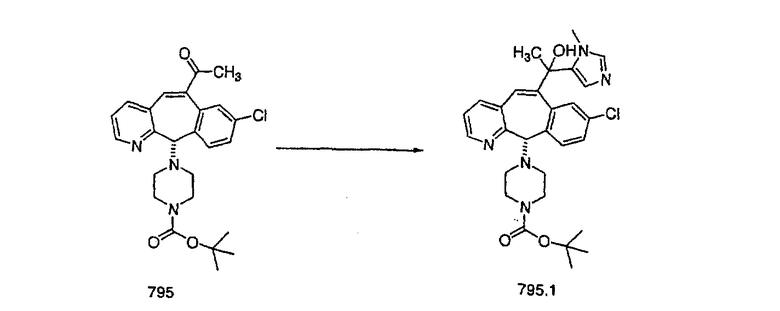
К раствору имидазолсодержащего реагента Гриньяра, полученного из 5-йод-1 N-метилимидазола (624 мг, 3 ммоль, см. Пример получения 64, стадия В, с использованием ClCH2СН2Cl вместо THF в качестве растворителя), прибавляют раствор метилэтилкетона (795) (272 мг, 0,06 ммоль) в ClCH2CH2CI (6 мл). Смесь 1,5 ч нагревают при 60°С. После охлаждения до комнатной температуры для остановки реакции прибавляют насыщенный раствор NH4CI. Органический слой сушат и затем выпаривают досуха. Затем остаток очищают на колонке с силикагелем (от 2% до 10% Ме-ОН в СН2Cl2) и получают продукт (795.1) в виде коричневого твердого вещества (63 мг, селективность по диастереоизомерам 10:1, МН+ = 536,1). Главный диастереоизомер: (CDCl3, 300 МГц) 8,47 (d, 1Н), 7,66 (d, 1Н), 7,57 (s, 1H), 7,54 (s, 1H), 7,34 (d, 1 Н), 7,25 -7,22 (m, 1 Н), 7,05 (s, 1 Н), 6,89 (s, 1 Н), 6,82 (s, 1 Н), 4,61 (s, 1 Н), 3,84 (s, ЗН), 3,24 (br s, 4H), 2,24 (m, 2H), 2,02 -2,00 (m, 2H), 1,88 (s, 3H), 1,41 (s, 9H).
Стадия С Соединение (795.2).
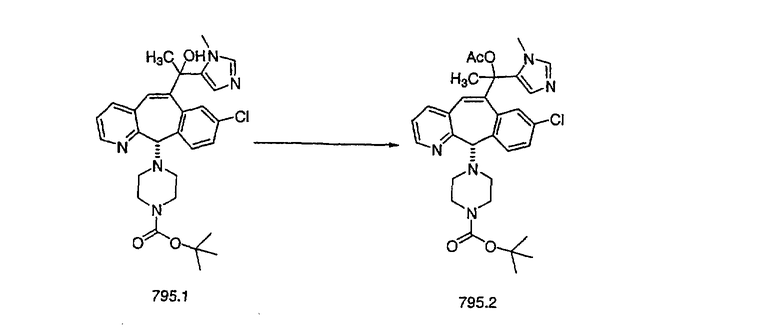
Соединение (795.1) можно превратить в ацетат (795.2) путем его введения в реакцию с 1 эквивалентом уксусного ангидрида и 2 эквивалентами пиридина.
Стадия D Соединение (795.3).
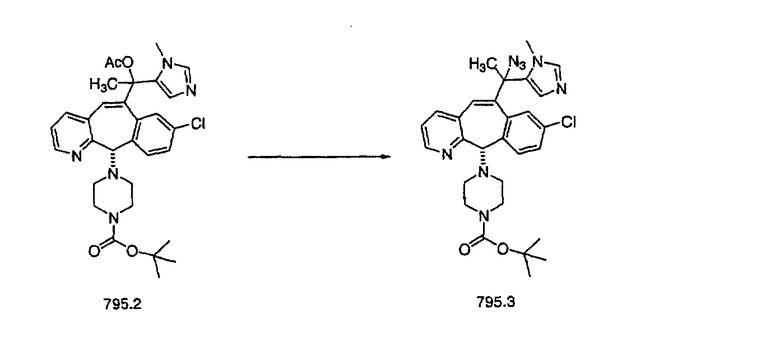
Соединение (795.2) можно превратить в соединение (795.3) путем его введения в реакцию с 1,5 эквивалентами NaN3, 15-краун-5 и каталитическим количеством Pd(dba)2/PPh3.
Альтернативно, (795.3) можно синтезировать путем обработки (795.1) с помощью NaNa, TFA, а затем (Вос)2O и триэтиламина.
Стадия Е Соединение (795.4).
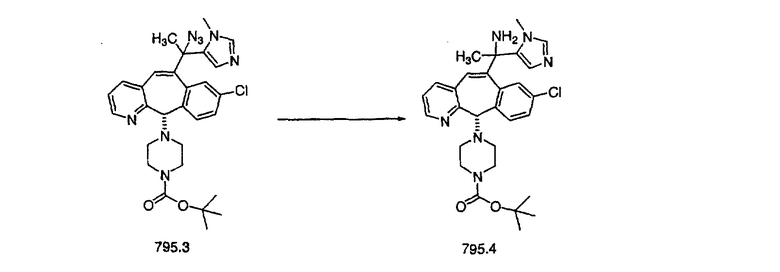
Соединение (795.4) можно получить по реакции (795.3) с P(СН3)3/Н2О.
ПРИМЕР ПОЛУЧЕНИЯ 66
Соединения (796)-(803).
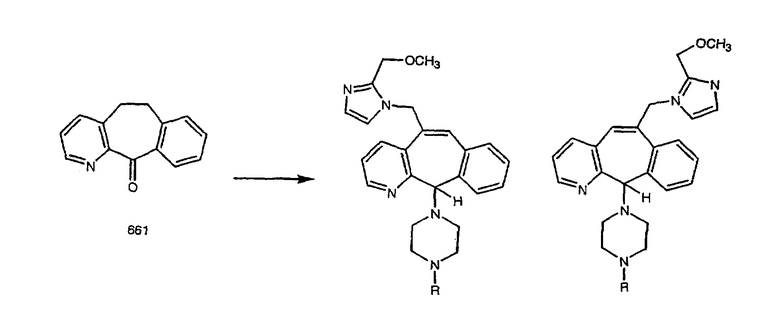
R=H
(800) ((+)-энантиомер, А) (802) ((-)-энантиомер, В)
R=H
(801) ((+)-энантиомер, А) (802) ((-)-энантиомер, В)
Соединение (661) вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем, как в Примере 91, и получают М-ВОС-производные (796), (797), (798) и (799). Соединения (796), (797), (798) и (799) затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (800), (801) ((+)-энантиомеры, изомер А) и (802), (803) ((-)-энантиомеры, изомер В). С5-и С6-винилбромидные промежуточные продукты разделяют с помощью хроматографии на силикагеле с использованием смеси гексан:этилацетат (80:20) таким же образом, как это описано в Примере получения 23, стадия В.
ПРИМЕРЫ 490-491
Получение соединений (804) и (805).
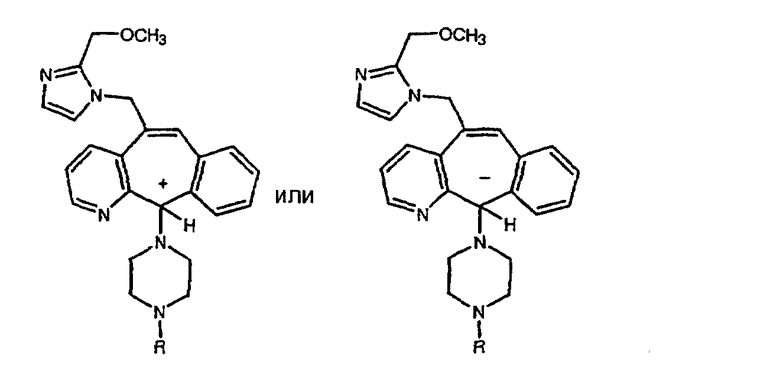
Соответствующий (+)-энантиомер (800) или (-)-энантиомер (802), полученный выше в Примере получения 66, растворяют в СН2Cl2, обрабатывают соответствующим изоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:

ПРИМЕР ПОЛУЧЕНИЯ 67
Стадия А Соединение (807).
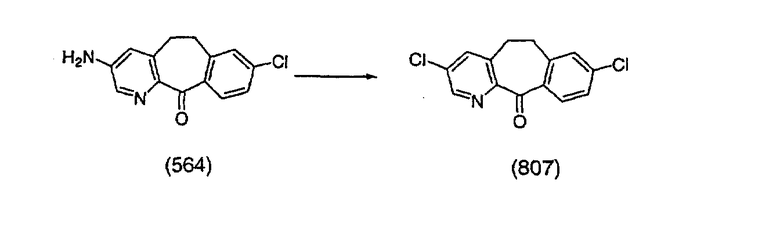
15,4 г (115 ммоль) CuCl2 и 17 мл (144 ммоль) трет-бутилнитрита прибавляют к 400 мл сухого СН3CN. Реакционную смесь охлаждают до 0°С и прибавляют 25 г кетона (564). Реакционную смесь нагревают до комнатной температуры и перемешивают 2 дня. Смесь концентрируют в вакууме. Затем к остатку прибавляют 1 н. раствор HCl, пока реакция не станет нейтральной, а после этого прибавляют NH4OH, пока реакция не станет щелочной. После экстракции этилацетатом органический слой сушат над MgSO4 и концентрируют в вакууме, получая соединение (807). Альтернативно, соответствующий спирт (564) можно ввести в реакцию, как это указано выше, а затем окислить с помощью MnO2 в CH2Cl2 и получить соединение (807).
Стадия В Соединения (808)-(815).
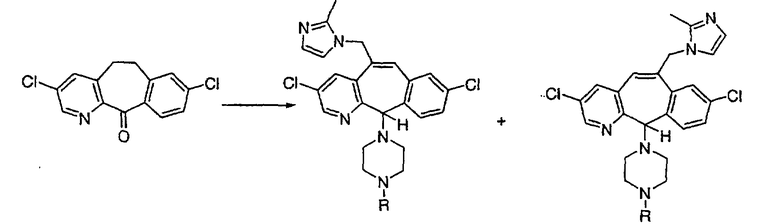
R=H
(812) (энантиомер 1) (814) (энантиомер 2)
R=H
(813) (энантиомер 1) (815) (энантиомер 2)
Соединение (807), полученное выше на стадии В, вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем как в Примере 91, и получают М-ВОС-производные (808), (809), (810) и (811). Их затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (812) и (814), а также энантиомеры (813) и (815). С5-и С6-винилбромидные промежуточные продукты разделяют с помощью хроматографии на силикагеле с использованием смеси гексан:этилацетат таким же образом, как это описано в Примере получения 23, стадия В.
ПРИМЕР 493
Получение соединений (816) и (817).
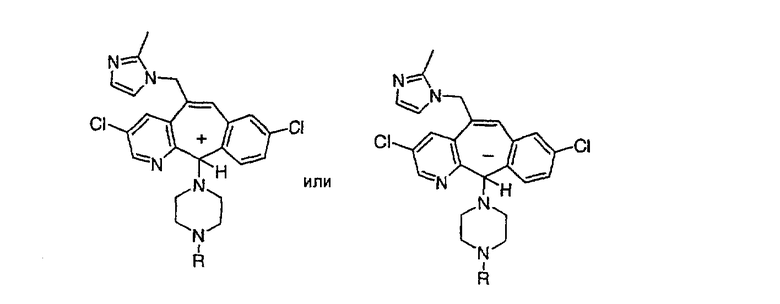
Соответствующий энантиомер (812) (энантиомер 1) или (814) (энантиомер 2), полученный выше в Примере получения 67, стадия В, растворяют в CH2CI2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
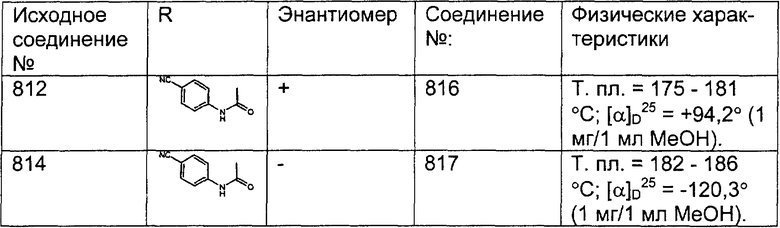
ПРИМЕР 494
Получение соединений (818) и (819).
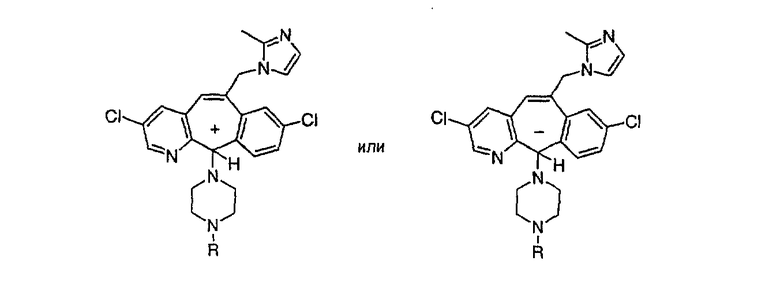
Соответствующий энантиомер (813) (энантиомер 1) или (815) (энантиомер 2), полученный выше в Примере получения 67, стадия В, растворяют в CH2Cl2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
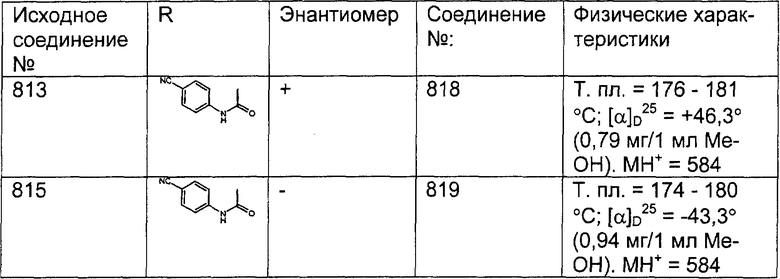
ПРИМЕР ПОЛУЧЕНИЯ 68
Соединения (820)-(827).
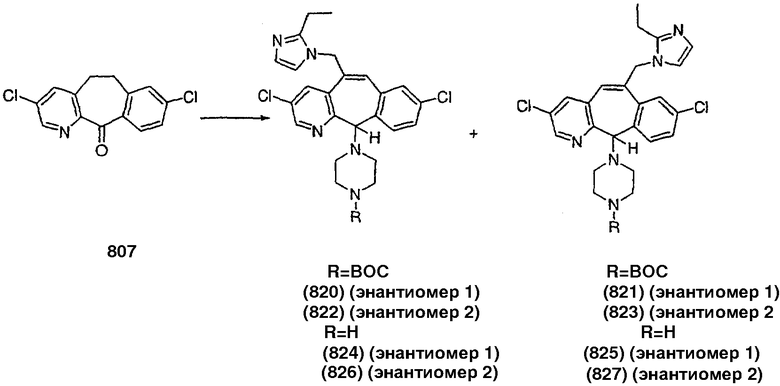
Соединение (807), полученное выше в Примере получения 67, стадия А, вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем, как в Примере 91, заменяя 2-метилимидазол на 2-этилимидазол, и получают N-BOC-производные (820), (821), (822) и (823). Их затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (824) и (826), а также энантиомеры (825) и (827). С5-и С6-винилбромидные промежуточные продукты разделяют с помощью хроматографии на силикагеле с использованием смеси гексан:этилацетат таким же образом, как это описано в Примере получения 23, стадия В.
ПРИМЕР 495
Получение соединений (828) и (829).
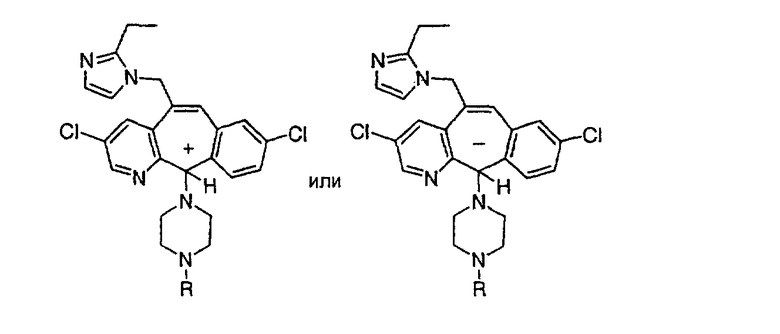
Соответствующий энантиомер (824) (энантиомер 1) или (826) (энантиомер 2), полученный выше в Примере получения 68, растворяют в CH2Cl2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
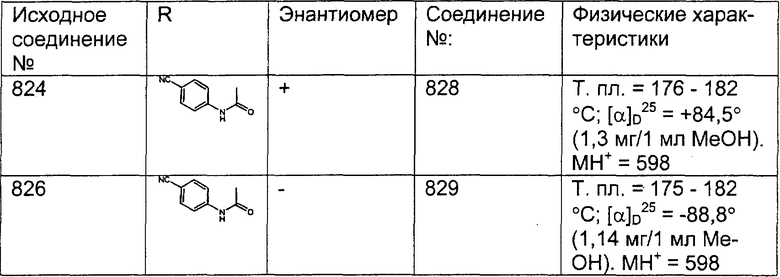
ПРИМЕР 496
Получение соединений (830) и (831).
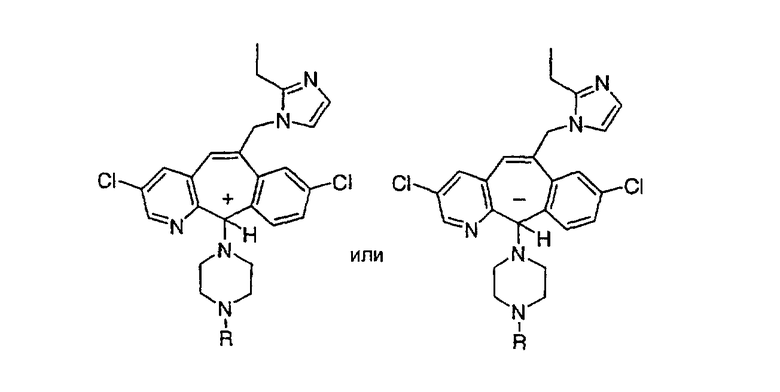
Соответствующий энантиомер (825) (энантиомер 1) или (827) (энантиомер 2), полученный выше в Примере получения 68, растворяют в CH2Cl2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
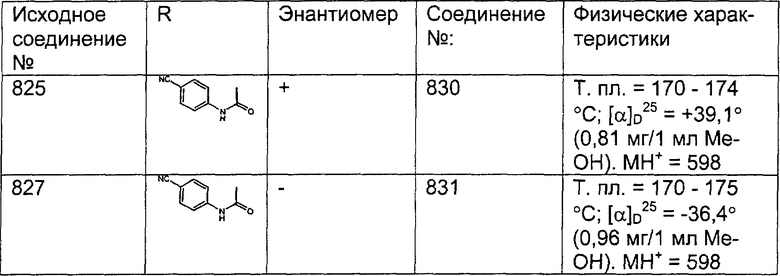
ПРИМЕР ПОЛУЧЕНИЯ 69
Соединения (832)-(835).
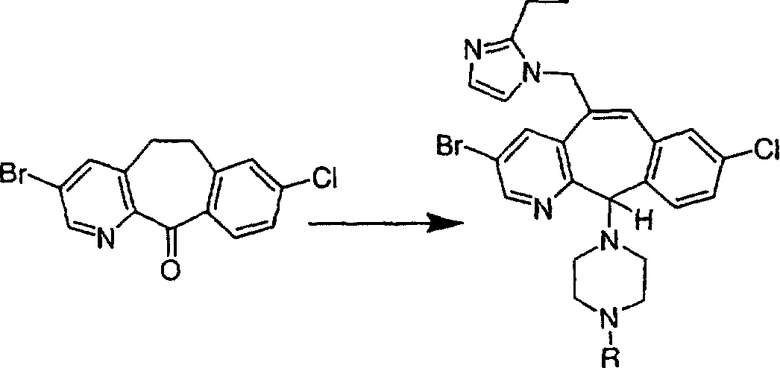
R=BOC
(832) (энантиомер А)
(833) (энантиомер В) R=H
(834) (энантиомер А)
(835) (энантиомер В)
3-Бром-8-хлоразакетон (патент США US 5977128, Пример получения 11, стадия А (1999)) вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем как в Примере 91, заменяя 2-метилимидазол на 2-этилимидазол, и получают М-ВОС-производные (832) и (833). Затем их по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (834) и (835).
ПРИМЕР 497
Получение соединений (836) и (837).
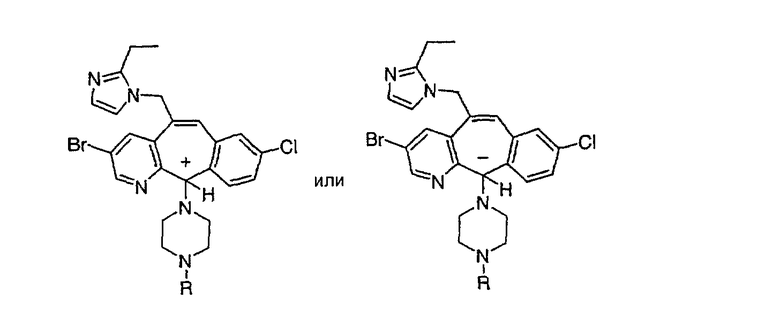
Соответствующий энантиомер (834) (энантиомер 1) или (835) (энантиомер 2), полученный выше в Примере получения 69, растворяют в CH2Cl2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
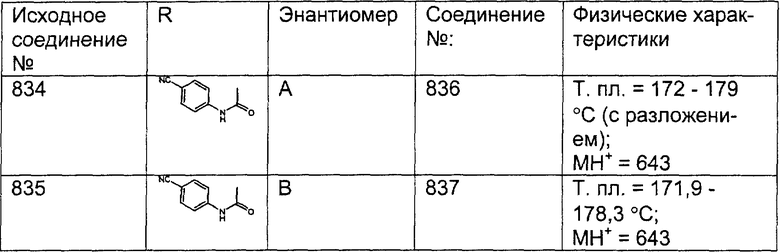
ПРИМЕР ПОЛУЧЕНИЯ 70
Соединения (838)-(841).
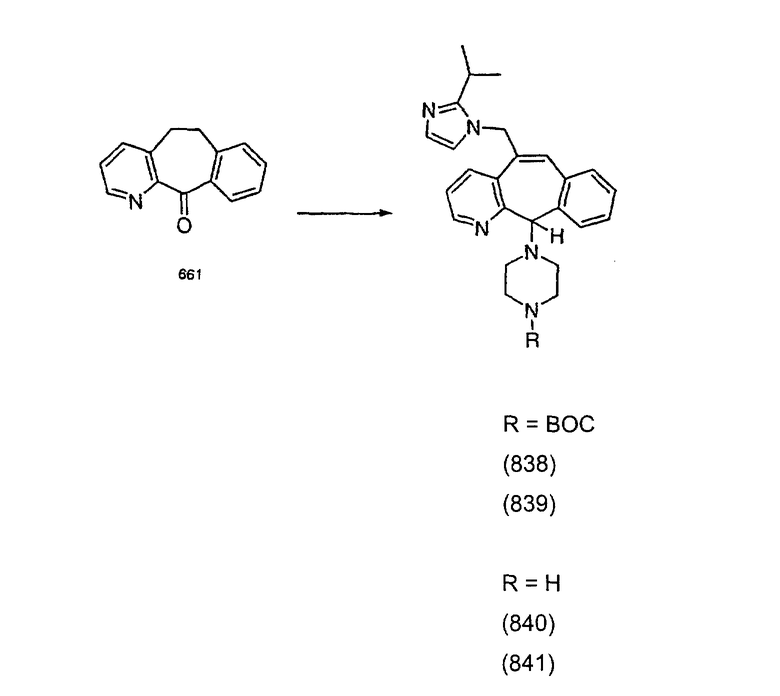
Соединение (661) вводят в реакцию в основном таким же образом, как в Примере получения 23, а затем как в Примере 91, заменяя 2-метилимидазол на 2-изопропилимидазол, и получают М-ВОС-производные (838) и (839). Их затем по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (840) и (841).
ПРИМЕР 498
Получение соединений (842) и (843).
Соответствующий энантиомер (840) (энантиомер 1) или (841) (энантиомер 2), полученный выше в Примере получения 70, растворяют в СН2Cl2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
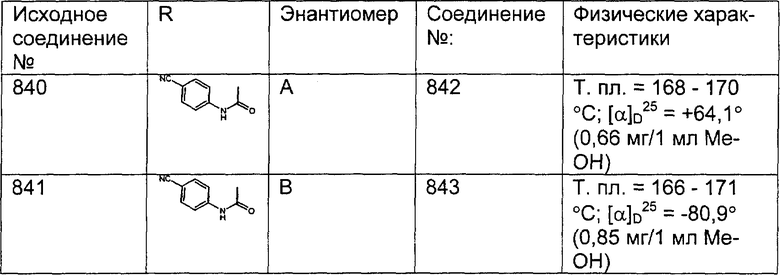
ПРИМЕР ПОЛУЧЕНИЯ 71
Соединения (844)-(847).
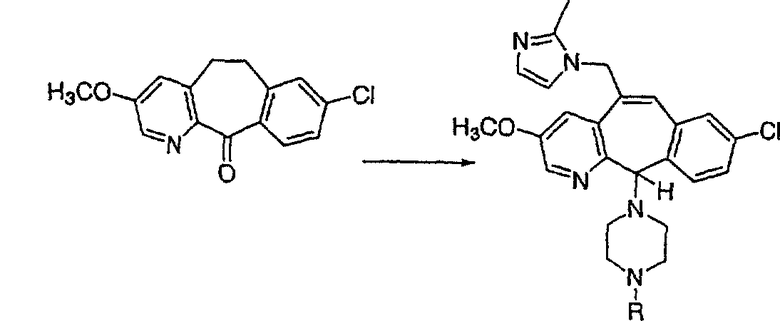
R=BOC
(844)
(845)
R=H
(846) (энантиомер А)
(847) (энантиомер В)
3-Метокси-8-хлоразакетон (патент США US 5977128 (1999), Пример 2, стадия D) вводят в реакцию в основном таким же образом, как в Примере получения 23 и Примере 91, и получают М-ВОС-производные (844) и (845). Затем эти соединения по отдельности вводят в реакцию в основном таким же образом, как в Примере получения 19, стадия D, и получают энантиомеры (846) (А) и (847) (В).
ПРИМЕР 499
Получение соединений (848) и (849).
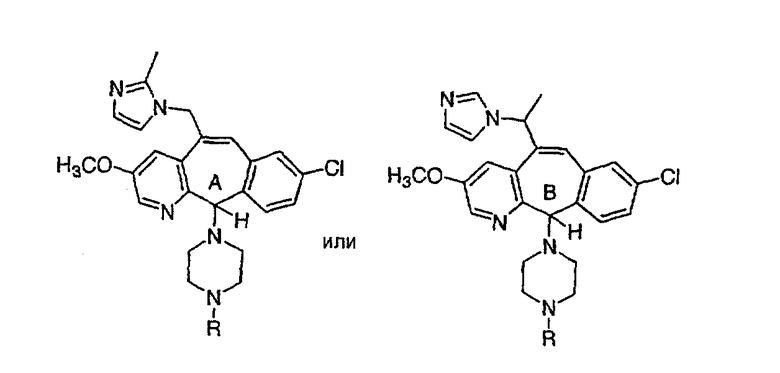
Соответствующий энантиомер (846) (энантиомер А) или (847) (энантиомер В), полученный выше в Примере получения 71, растворяют в CH2Cl2, обрабатывают 4-цианфенилизоцианатом и перемешивают при комнатной температуре в течение ночи. Неочищенный продукт очищают непосредственно с помощью препаративной тонкослойной хроматографии на силикагеле или хроматографии на колонке с силикагелем и получают следующие соединения, представленные ниже в таблице:
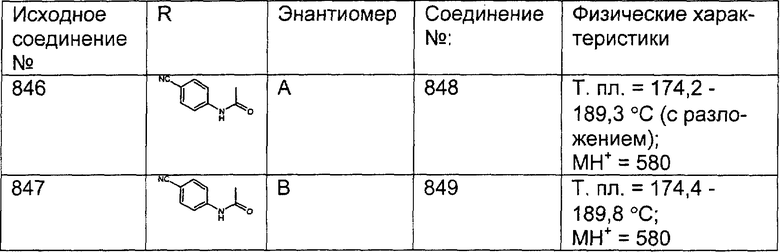
ПРИМЕР 500
Получение соединения (850).
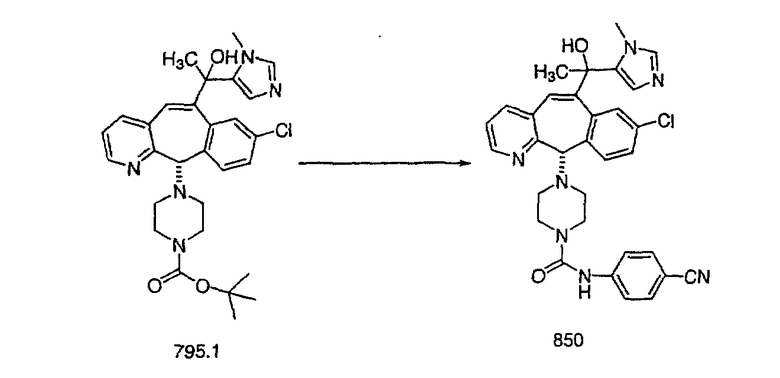
Соединение (850) можно получить с помощью в основном такой же методики, что и описанная в Примере 482.
ПРИМЕР 501
Получение соединения (851).
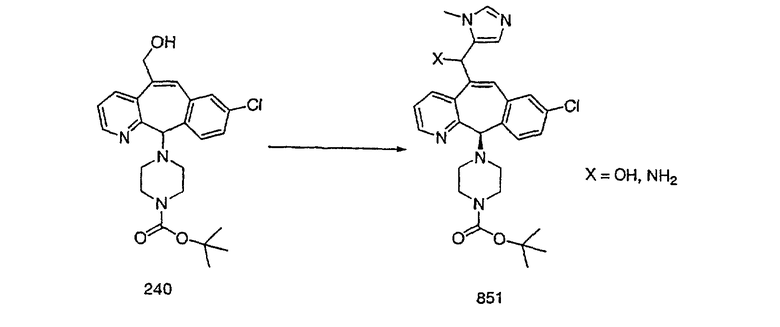
С использованием в качестве исходного соединения (240), полученного в Примере получения 23, стадия Н, соединение (851) можно получить с помощью в основном такой же методики, как приведенная в Примере получения 65, стадии А и В.
ПРИМЕР 502
Получение соединения (852).
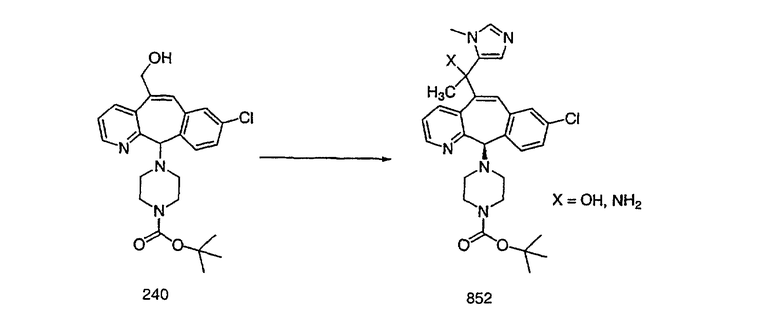
С использованием в качестве исходного соединения (240), полученного в Примере получения 23, стадия Н, соединение (852) можно получить с помощью в основном такой же методики, как приведенная в Примере получения 65, стадия А, и в Примере 489, стадии А-Е.
ПРИМЕР ПОЛУЧЕНИЯ 72
Стадия А Получение соединений (853) и (854).
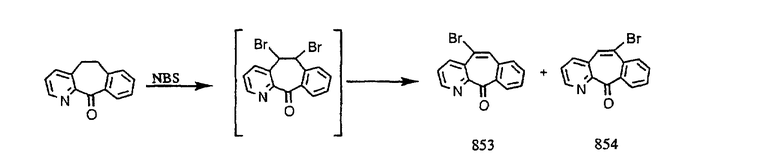
Исходное трициклическое кетосоединение (раскрытое в патенте США US №5151423) (56,5 г, 270 ммоль) вводят в CCl4 в реакцию с NBS (105 г, 590 ммоль) и бензоилпероксидом (0,92 г). Реакционную смесь 5 ч нагревают при 80°С. Смесь охлаждают и образовавшийся осадок отфильтровывают и обрабатывают с помощью DBU (25,59 мл) в THF (300 мл). Полученный раствор 24 ч перемешивают при комнатной температуре, затем выпаривают, а после этого экстрагируют с помощью СН2Cl3 - H2O. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая смесь двух соединений, которые разделяют на флэш-колонке с силикагелем, элюируя смесью гексан - 50% EtOAc, и получают искомое соединение (853): δн (CDCI3): 8,8 (dd, 1H), 8,45 (dd, 1H), 7,99 (m, 1H), 7,92 (s, 1H), 7,59-7,64 (m, 3H), 7,23 (dd, 1 Н) и (854): 8н (CDCIs): 8,19 (dd, 1 Н), 7,99 (dd, 1 Н), 7,82 (dd, 1 Н), 7,25 -7,65 (m, 4H), 7,22 (s, 1H).
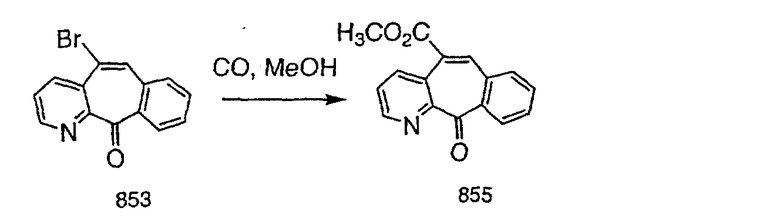
Соединение (853) (25 г), трифенилфосфин (13,75 г) и хлорид палладия (1,5 г) смешивают в МеОН (30 мл) и толуоле (200 мл). К этой смеси прибавляют DBU (18 мл) и смесь герметизируют в бомбе Парра. Смесь перемешивают и 5 ч обрабатывают с помощью СО при 80°С и давлении, равном 100 фунт-сила/дюйм2. Реакционную смесь разбавляют с помощью EtOAc и промывают водой. Органический слой сушат над MgSO4, фильтруют и очищают с помощью флэш-хроматографии, элюируя смесью CH2Cl2-10% EtOAc, и получают искомое соединение (855): δн (CDCl3): 8,8 (dd, 1 Н), 8,40 (dd, 1 Н), 8,2 (s, 1 Н), 8,04 (dd, 1 Н), 7,59 -7,64 (m, 4H), 3,95 (s, 3H).
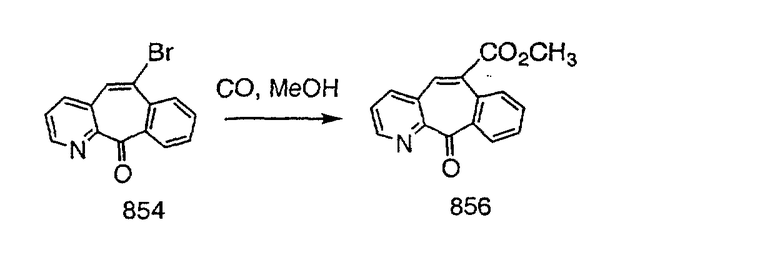
Введение соединения (854) в реакцию в основном таким же способом, как это описано выше на стадии В, дает искомое соединение (856). δн (CDCl3): 8,85 (dd, 1H), 7,5 -8,0 (m, 2H), 7,8 (s, 1H), 7,25 -7,31 (m, 4H).
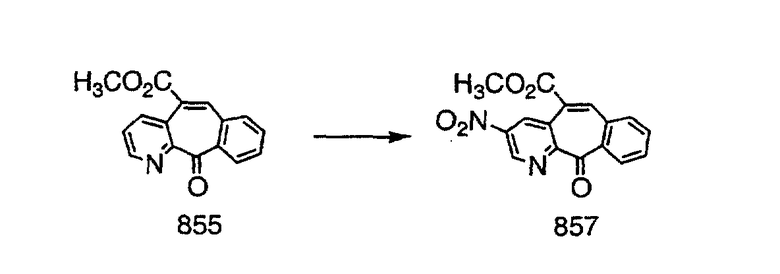
Соединение (855) (19,5 г, 73,5 ммоль) растворяют в CH2Cl2 (100 мл) и охлаждают до 0°С. Прибавляют тетрабутиламмонийнитрат (31,36 г, 103 ммоль) и трифторуксусный ангидрид (18,52 г, 88 ммоль) и смесь 5 ч перемешивают при комнатной температуре. Реакционную смесь концентрируют досуха, а затем экстрагируют с помощью CH2Cl2 - NaHCO3. Объединенные органические слои сушат над MgSO4 и концентрируют досуха, а остаток хроматографируют на силикагеле, используя СН2Cl2 - EtOAc (25%), и получают искомое соединение (857) (12,4 г), δн (CDCl3): 9,45 (dd, 1H), 9,05 (dd, 1H), 8,28 (s, 1H), 8,0 (dd, 1H), 7,65 (m, 3Н), 3,98 (s, 3Н).
Стадия Е Получение соединения (858).
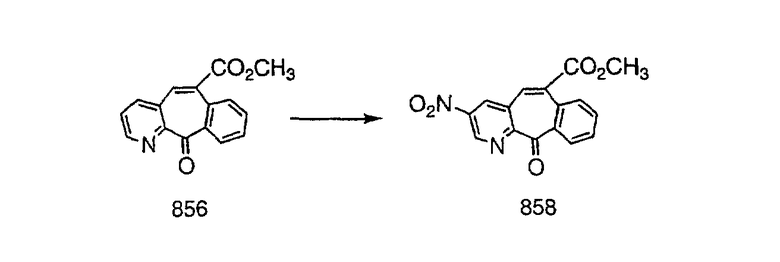
Введение соединения (856) в реакцию в основном таким же способом, как это описано выше на стадии D, дает искомое соединение (858). МН+ =311.
Стадия F Получение соединения (859).
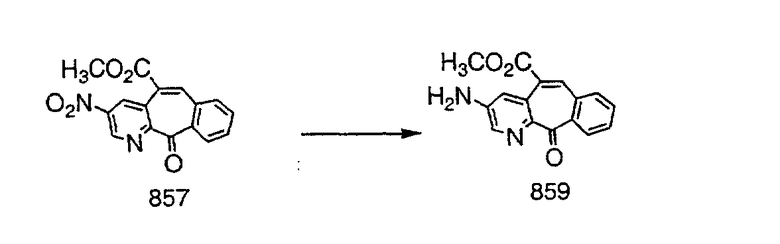
Соединение (857) (6 г) в течение ночи гидрируют в МеОН (100 мл) над никелем Ренея (4,2 г) при комнатной температуре, подавая водород из баллона. Катализатор отфильтровывают и фильтрат выпаривают досуха, получая искомое соединение (859) (4,66 г). MH+=281.
Стадия G Получение соединения (860).
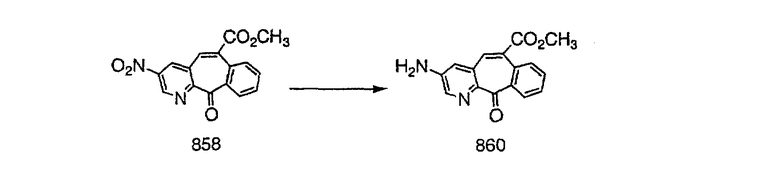
Введение соединения (858) в реакцию в основном таким же способом, как это описано выше на стадии F, дает искомое соединение (860). МН+ = 281.
Стадия Н Получение соединения (861).
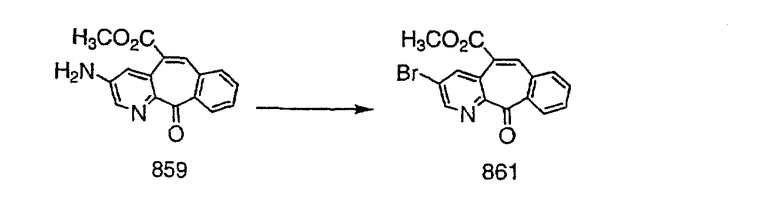
К суспензии соединения (859) (2,1 г) в 48% HBr при 0°C прибавляют нитрит натрия (1,55 г), а затем бром (2,11 мл). Смесь перемешивают при комнатной температуре в течение ночи. Затем по каплям прибавляют концентрированный раствор NH4OH, пока реакция не станет щелочной (по лакмусовой бумаге). Реакционную смесь экстрагируют с помощью СН2Cl2, промывают рассолом, сушат над MgSO4, фильтруют и растворитель выпаривают, получая искомое соединение (861) (1,75 г). МН+ = 345.
Стадия I Получение соединения (862).
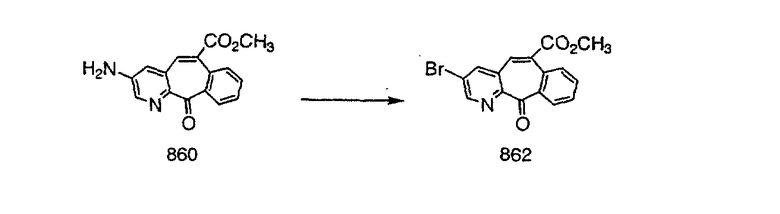
Введение соединения (861) в реакцию в основном таким же способом, как это описано выше на стадии Н, дает искомое соединение (862). MH+ = 345.
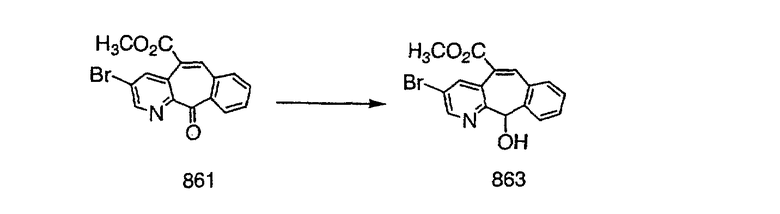
К раствору соединения (861) (1,6 г, 4,64 ммоль) в МеОН (30 мл) при перемешивании в атмосфере азота при 0°C прибавляют NaBH4 (0,3 г, 7,9 ммоль). Полученный раствор 24 ч перемешивают при комнатной температуре, затем выпаривают, а после этого экстрагируют с помощью CH2CI2 - Н2O. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая искомое соединение (863) (1,58 г). MH2 = 347.
Стадия K Получение соединения (864).
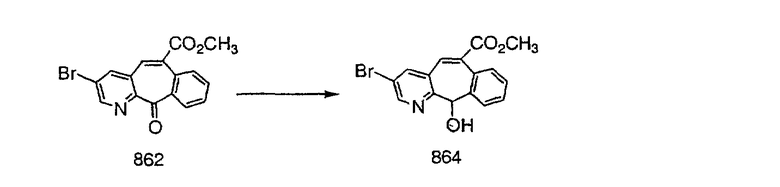
Введение соединения (862) в реакцию в основном таким же способом, как это описано выше на стадии J, дает искомое соединение (864). МН+ = 347.
Стадия L Получение соединения (865).
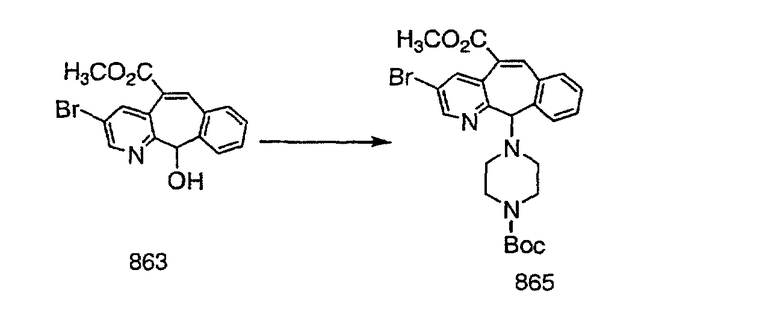
Соединение (863) (1,57 г) 4 ч перемешивают при комнатной температуре в тионилхлориде (10 мл), а затем выпаривают досуха. Полученное неочищенное масло растворяют в ацетонитриле (50 мл) и в течение ночи кипятят с обратным холодильником с N-Вос-пиперазином (1,41 г) и триэтиламином (3,91 г). Смесь выпаривают досуха, а затем экстрагируют с помощью СН2Cl2 - NaHCO3. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая коричневую смолу, которую очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью гексан - 20% EtOAc, и получают искомое соединение (865) (0,69 г). MH+ = 515.
Стадия М Получение соединения (866).
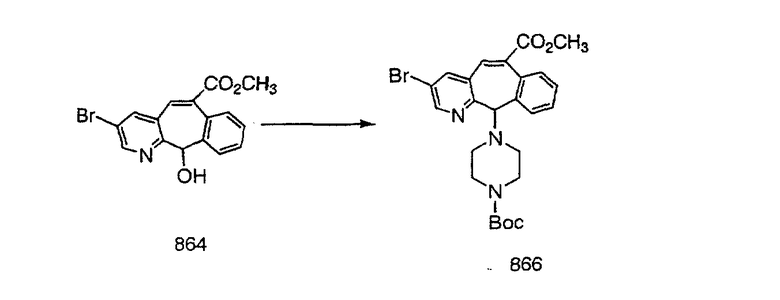
Введение соединения (864) в реакцию в основном таким же способом, как это описано выше на стадии L, дает искомое соединение (866). МН+ = 515.
Стадия N Получение соединения (867).
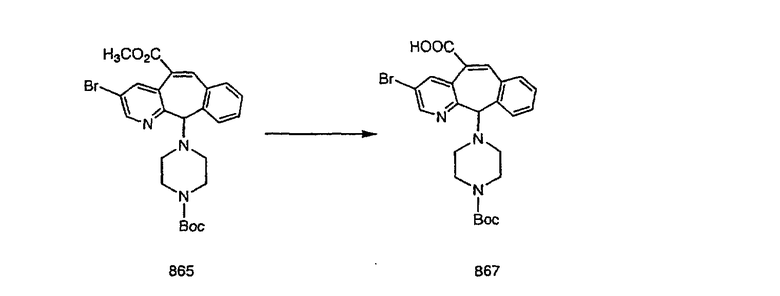
Соединение (865) (0,65 г, 1,26 ммоль) 2 ч кипятят с обратным холодильником с LiOH (0,45 г, 18,79 ммоль) в МеОН (15 мл) и воде (1 мл). Прибавляют 10% водный раствор лимонной кислоты, пока значение рН не станет равным 3,5, а затем экстрагируют с помощью CH2Cl2 - рассол. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая белое твердое вещество (867) (0,60 г). МН+ = 501.
Стадия О Получение соединения (868).
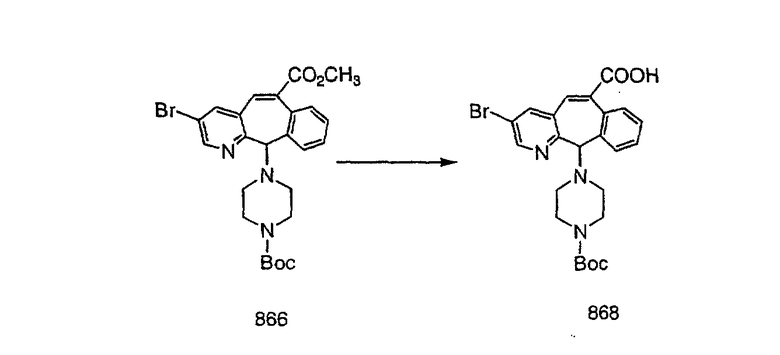
Введение соединения (866) в реакцию в основном таким же способом, как это описано выше на стадии N, дает искомое соединение (868). MH+ = 501.
Стадия P Получение соединения (869).
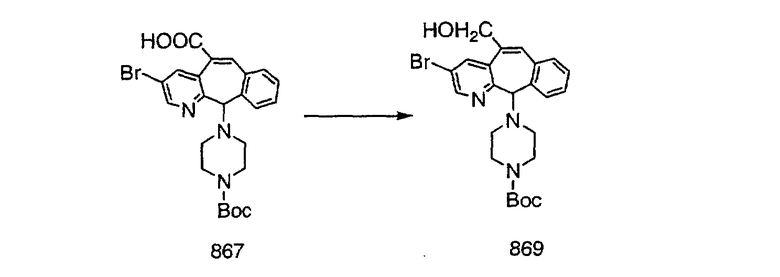
Соединение (867) (0,60 г, 1,21 ммоль) при 40°С в течение ночи перемешивают с карбонилдиимидазолом (0,59 г, 3,63 ммоль) в THF (15 мл). Реакционную смесь охлаждают в бане со льдом, затем прибавляют NaBH4 (0,28 г, 7,31 ммоль) и перемешивают при комнатной температуре в течение ночи. Смесь выпаривают досуха, а затем экстрагируют с помощью Ch2Cl2 - вода. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая коричневую смолу, которую очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью гексан - 50% EtOAc, и получают искомое соединение (869) (0,493 г). МН+ = 487.
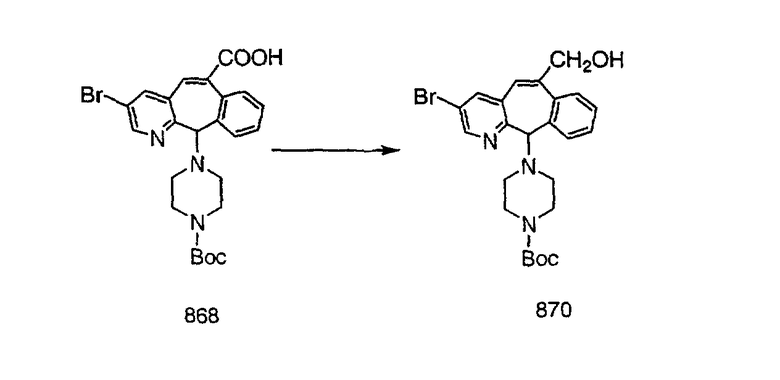
Введение соединения (868) в реакцию в основном таким же способом, как это описано выше на стадии P, дает искомое соединение (870). MH+ = 487.
Стадия R Получение соединения (871).
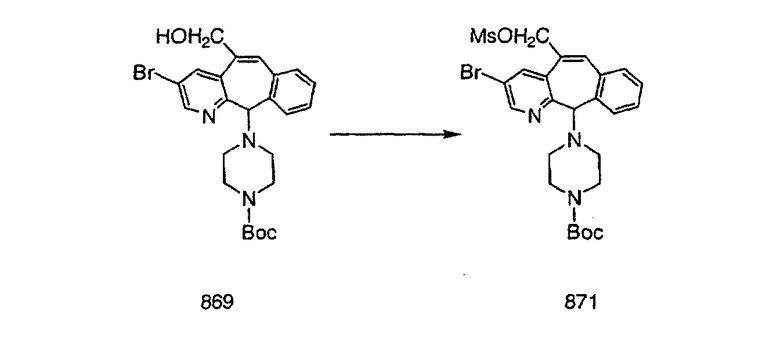
Соединение (869) (0,38 г, 0,78 ммоль) при комнатной температуре в течение ночи перемешивают с метансульфонилхлоридом (0,33 г, 1,296 ммоль) и триэтиламином (0,68 г, 6,72 ммоль) в THF (10 мл). Смесь выпаривают досуха, а затем экстрагируют с помощью CH2Cl2 - вода. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая искомое соединение (871) (0,369 г). MH+ = 565.
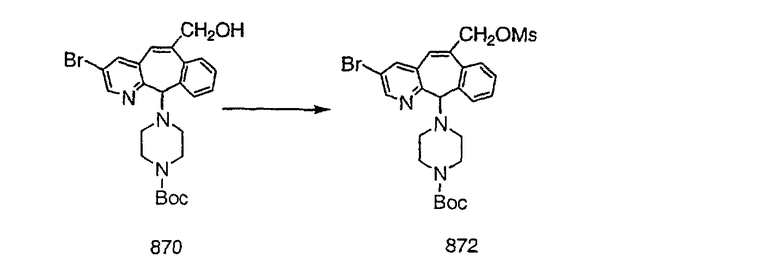
Введение соединения (870) в реакцию в основном таким же способом, как это описано выше на стадии R, дает искомое соединение (872). МН+ = 565.
Стадия Т Получение соединений (873) и (874).
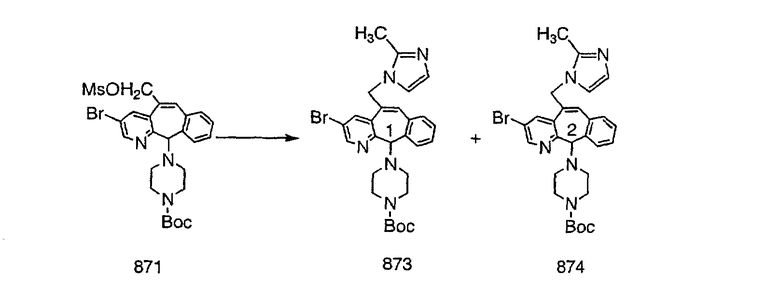
Соединение (871) (0,369 г, 0,653 ммоль) при комнатной температуре в течение ночи перемешивают с 2-метилимидазолом (0,188 г, 2,28 ммоль) в DMF (5 мл). Смесь выпаривают досуха, а затем экстрагируют с помощью CH2Cl2 - вода. Органический слой сушат над MgSO4, фильтруют, выпаривают досуха, а затем очищают с помощью препаративной тонкослойной хроматографии на пластине с силикагелем, элюируя смесью СН2Cl2 - 5% (МеОН -10% NH4OH), и получают продукт в виде смеси изомеров (1,126 г). MH+ = 551. Разделение смеси продуктов с помощью ВЭЖХ с использованием препаративной колонки AD и с элюированием смесью 20% IPA/80% гексан/0,2% DEA (в изократическом режиме 60 мл/мин) дает чистый изомер 1 (873) (0,06 г, MH+ = 551) и изомер 2 (874) (0,0061 г, MH+ = 551).
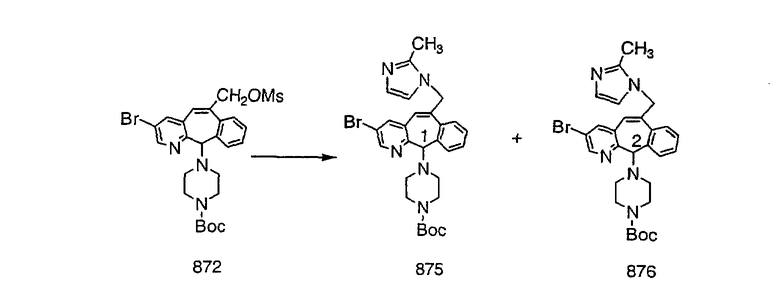
Введение соединения (872) в реакцию в основном таким же способом, как это описано выше на стадии Т, дает искомые соединения (875), MH+ = 551, и (876), МН+ = 551.
ПРИМЕР 503
Соединение (877).
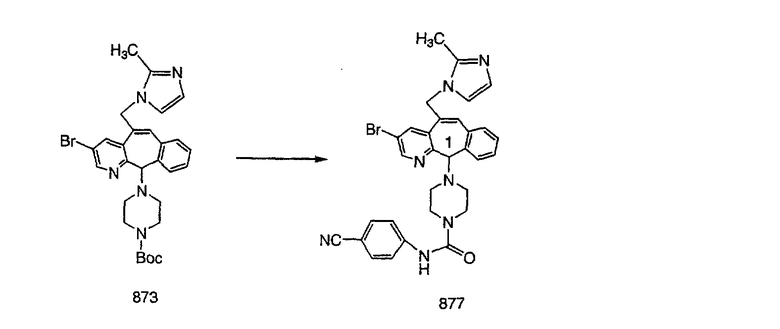
Соединение (873) (0,043 г, 0,078 ммоль) 4 ч перемешивают при комнатной температуре с TFA (5 мл) в СН2Cl2 (5 мл). Затем смесь выпаривают досуха. К остатку прибавляют п-цианофенилизоцианат (0,0123 г, 0,086 ммоль) и триэтиламин (0,5 мл) в СН2Cl2 (5 мл) и смесь 2 ч перемешивают при комнатной температуре. Смесь выпаривают досуха, а затем экстрагируют с помощью CH2Cl2 - рассол. Органический слой сушат над MgSO4, фильтруют и выпаривают досуха, получая коричневую смолу, которую очищают с помощью препаративной тонкослойной хроматографии на пластине с силикагелем, элюируя смесью CH2Cl2 - 5% (МеОН -10% NH4OH), и получают искомое соединение (877) (0,0394 г). MH+ = 595, δн (CDCI3): 8,6 (1Н), 8,05 (1Н), 7,22-7,5 (8Н), 6,99 (1Н), 6,95 (1Н), 6,93 (1Н), 4,99-5,25 (2Н), 4,6 (1Н), 3,1-3,25 (4Н), 2,25 (ЗН), 1,8-2,05 (4Н).
ПРИМЕР 504
Соединение(878).
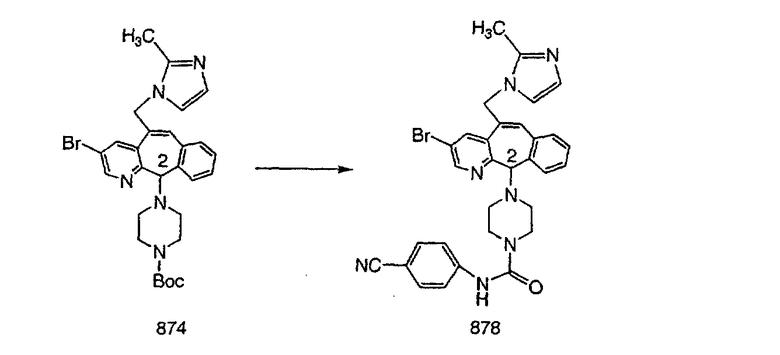
Введение соединения (874) в реакцию в основном таким же способом, как это описано выше в Примере 503, дает искомое соединение (878), МН+ = 595, δн (CDCl3): 8,6 (1Н), 8,05 (1Н), 7,22-7,5 (8Н), 6,99 (1Н), 6,95 (1Н), 6,93 (1Н), 4,99-5,25 (2Н), 4,6 (1Н), 3,1-3,25 (4Н), 2,25 (3Н), 1,8-2,05 (4Н).
ПРИМЕР 505
Соединение (879).
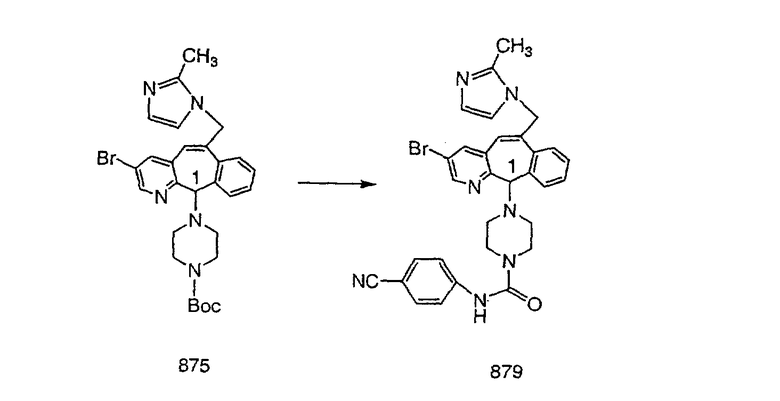
Введение соединения (875) в реакцию в основном таким же способом, как это описано выше в Примере 503, дает искомое соединение (879), MH+ = 595, δн (CDCl3): 8,55 (1Н), 7,78 (1Н), 7,65 (1Н), 7,4-7,51 (6Н), 6,98 (1Н), 6,9 (1Н), 6,85 (1Н), 5,05-5,3 (2Н), 4,6 (1 Н), 3,1-3,25 (4Н), 2,5 (3Н), 1,8-2,00 (4Н).
АНАЛИЗЫ
Активность ФПТ определяют путем измерения переноса [3H]-фарнезила от [3H]-фарнезилпирофосфата на биотинилированный пептид, полученный из С-конца Н-Ras (биотин-CVLS). Реакционная смесь содержит: 50 мМ ТРИС [трис(гидроксиметиламино)метан] с рН = 7,7, 5 мМ MgCl2, 5мкМ Zn++, 5 мМ DTT (ди-тиотреитол), 0,1% Triton-X, 0,05 мкМ пептида, 0,03 нМ очищенной фарнезилпроте-интрансферазы человека, 0,180 мкМ [3H]-фарнезилпирофосфата с добавлением трициклического соединения указанной концентрации или растворителя, выступающего в качестве контроля, до суммарного объема, равного 100 мкл. Реакционную смесь инкубируют в инкубаторе с устройством для встряхивания Vortemp при 37°С, 45 оборотах в минуту в течение 60 мин и реакцию останавливают с помощью 150 мкл ЭДТК (этилендиаминтетрауксусная кислота), содержащей 0,5% БСА (бычий сывороточный альбумин) и 1,3 мг/мл микрошариков Streptavidin SPA. Радиоактивность измеряют с помощью жидкостного сцинтилляционного счетчика Wallach 1450 Microbeta. Инкубирование в процентах рассчитывают по сравнению с растворителем, выступающим в качестве контроля.
COS Cell IC50 (анализ, основанный на использовании клеток) проводят с помощью методик анализа, описанных в WO 95/10516, опубликованной 20 апреля 1995 г. GGPT IC50 (ингибирование геранилгеранилпротеинтрансферазы, ферментный анализ in vitro), биохимический анализ Cell Mat и определение противоопухолевой активности (исследования противоопухолевой активности in vivd) можно было провести с помощью методик анализа, описанных в WO 95/10516. Раскрытие WO 95/10516 включено в настоящее изобретение путем ссылки.
Различные опухолевые клетки (от 5 х 105 до 8 х 106) инокулируют подкожно в бок 5-6-недельных бестимусных самок мышей с мутацией по гену nude. Использованы три модели опухолевых клеток: фибробласты мышей, трансформированные с помощью H-Ras; клетки немелкоклеточного рака легких человека НТВ-177 и клетки меланомы LOX человека. Животным вводят только растворитель бета-циклодекстран или соединения в растворителе два раза в день (BID) или один раз вдень (QD) в течение 7 дней в неделю в течение 1 (х1), 2 (х2) или 3 (х3) недель. Выраженное в процентах ингибирование роста опухоли по сравнению с растворителем, выступающим в качестве контроля, определяют путем измерения опухоли. Результаты представлены в приведенной ниже таблице.
(Например, режим ро, BID, x3 означает: перорально, два раза в день в течение 7 дней (14 раз в неделю) в течение 3 недель).
Анализ на мягком агаре:
Для линий опухолеродных клеток характерен свободный рост. Опухолевые клетки человека можно суспендировать в среде для выращивания, содержащей 0,3% агарозы и ингибитор фарнезилпротеинтрансферазы в заданной концентрации. Раствор можно нанести на среду для выращивания, отвержденную с помощью 0,6% агарозы и содержащую ингибитор фарнезилпротеинтрансферазы в такой же концентрации, как и верхний слой. После отверждения верхнего слоя чашки можно инкубировать в течение 10-16 дней при 37°C и содержании CO2, равном 5%, чтобы обеспечить рост колоний. После инкубирования колонии можно окрасить путем нанесения агара с раствором МТТ (3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолийбромид, тиазолил синий) (1 мг/мл в забуференном фосфатом физиологическом растворе). Можно подсчитать количество колоний и определить значения IC50:
Соединения, соответствующие настоящему изобретению, обладают значениями IC50 для ФПТ в диапазоне от 0,001 до 100 нМ и значениями IC50 в мягком агаре в диапазоне от 0,01 до 50 нМ.
Предпочтительные соединения, соответствующие настоящему изобретению, обладают значениями IC50 для ФПТ в диапазоне от менее 0,06 до 0,44 нМ и значениями IC50 в мягком агаре в диапазоне от менее 0,05 до 25 нМ.
Наиболее предпочтительные соединения, соответствующие настоящему изобретению, обладают значениями IC50 для ФПТ в диапазоне от менее 0,05 до 3,0 нМ и значениями IC50 в мягком агаре в диапазоне от 0,5 до 5 нМ.
При изготовлении фармацевтических композиций, описанных в настоящем изобретении, инертные, фармацевтически приемлемые носители могут быть твердыми или жидкими. К твердым формам препаратов относятся порошки, таблетки, диспергирующиеся гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут содержать от примерно 5 до примерно 95% активного компонента. Подходящие твердые носители известны в данной области техники и включают, например, карбонат магния, стеарат магния, тальк, сахар и лактозу. Таблетки, порошки, облатки и капсулы можно использовать в качестве твердых дозировочных форм, пригодных для перорального введения. Примеры фармацевтически приемлемых носителей и способов изготовления различных композиций приведены в работе A. Gennato (ed.), Remington's Pharmaceutical Sciences, 18th Edition, (1990), Mack Publishing Co., Easton, Pennsylvania.
Жидкие формы композиций включают растворы, суспензии и эмульсии. В качестве примера можно указать водные или водно-пропиленгликолевые растворы для парентеральных инъекций или прибавление подсластителей и замутнителей в пероральные растворы, суспензии и эмульсии. К жидким формам композиций также могут относиться растворы для внутриназального введения.
Аэрозольные композиции, пригодные для ингаляции, могут включать растворы и твердые вещества в порошкообразной форме, которые могут сочетаться с фармацевтически приемлемым носителем, таким как сжатый инертный газ, например азот.
В объем настоящего изобретения также включены твердые формы композиций, которые предназначены для превращения в жидкие формы композиций, предназначенных для перорального или парентерального введения, которое выполняется незадолго до использования. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения, соответствующие настоящему изобретению, также можно вводить чрескожно. Чрескожные композиции могут представлять собой кремы, лосьоны, аэрозоли и/или эмульсии, и они могут быть включены в чрескожный пластырь матричного или резервуарного типа, что обычно используется в данной области техники для такой цели.
Предпочтительно, чтобы фармацевтическая композиция содержалась в разовой дозировочной форме. В такой форме композиция разделяется на разовые дозы подходящей величины, содержащие соответствующие количества активных компонентов, т.е. количества, достаточные для достижения необходимой цели.
Количество соединений согласно настоящему изобретению в разовой дозе лекарственного препарата обычно может меняться или регулироваться в диапазоне от примерно 0,01 до примерно 1000 мг, предпочтительно - от примерно 0,01 до примерно 750 мг, более предпочтительно - от примерно 0,01 до примерно 500 мг, а наиболее предпочтительно - от примерно 0,01 до примерно 250 мг в соответствии с конкретным случаем применения.
Количество и частота введения соединений согласно настоящему изобретению и/или их фармацевтически приемлемых солей будет регулироваться в соответствии с решением лечащего врача, учитывающего такие факторы как возраст, состояние и массу пациента, а также тяжесть симптомов, подвергающихся лечению. Типичный рекомендованный суточный дозировочный режим при пероральном введении может включать введение от примерно 0,04 до примерно 4000 мг/сутки, назначаемых в виде одной или разделенных доз, предпочтительно - в виде двух-четырех разделенных доз.
Химиотерапевтические средства и/или лучевую терапию можно назначать совместно с соединениями, соответствующими настоящему изобретению, в дозировке и режиме, указанном в информационном листке для утвержденных к применению средств, в "Physicians' Desk Reference" (PDR), а также в лечебных протоколах, хорошо известных в данной области техники. В приведенной ниже таблице 1.0 представлены диапазоны доз и дозировочные режимы для некоторых типичных химиотерапевтических препаратов, применяющихся в способах, соответствующих настоящему изобретению. Специалистам в данной области техники должно быть понятно, что назначение химиотерапевтических средств и/или лучевой терапии может меняться в зависимости от подвергающегося лечению заболевания и известных результатов применения химиотерапевтического средства и/или лучевой терапии для лечения этого заболевания. Также по решению квалифицированного врача лечебные протоколы (т. е. дозировки и режим приема) могут меняться в соответствии с наблюдающимися результатами введения пациенту химиотерапевтических средств (т. е. противоопухолевого средства или облучения) и наблюдающейся реакции заболевания на введенные лечебные средства.
В предпочтительном примере комбинированной терапии при лечении рака поджелудочной железы соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с противоопухолевым препаратом, гемцитабином, который вводят в дозировке от 750 до 1350 мг/м2 ежедневно в течение трех из четырех недель курса лечения.
В предпочтительном примере комбинированной терапии при лечении рака легких соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с противоопухолевым препаратом, паклитакселом, который вводят в дозировке от 65 до 175 мг/м2 один раз каждые три недели.
В предпочтительном примере комбинированной терапии при лечении глиомы соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в видедвух разделенных доз совместно с противоопухолевым препаратом, темозоломидом, который вводят в дозировке от 100 до 250 мг/м2.
В другом примере комбинированной терапии соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с противоопухолевым препаратом, цисплатином, который вводят внутривенно в диапазоне от 50 до 100 мг/м2 один раз каждые четыре недели.
В другом примере комбинированной терапии соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с противоопухолевым препаратом, карбоплатином, который вводят внутривенно в диапазоне от 300 до 360 мг/м2 один раз каждые четыре недели.
В другом примере комбинированной терапии соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с химиотерапевтическим препаратом, карбоплатином, который вводят внутривенно в диапазоне от 300 до 360 мг/м2 один раз каждые четыре недели, и с химиотерапевтическим препаратом, паклитакселом, который вводят в дозировке от 65 до 175 мг/м2 один раз каждые три недели.
В еще одном примере комбинированной терапии соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с химиотерапевтическим препаратом, цисплатином, который вводят внутривенно в диапазоне от 50 до 100 мг/м2 один раз каждые четыре недели, и с химиотерапевтическим препаратом, гемцитабином, который вводят в дозировке от 65 до 175 мг/м2 один раз каждые три недели.
Терапию путем ингибирования трансдукции сигнала можно назначать в соответствии с дозировкой и режимом приема, указанными в информационном листке для утвержденных к применению средств, в "Physicians' Desk Reference" (PDR), а также в лечебных протоколах, хорошо известных в данной области техники. В приведенной ниже таблице 2.0 представлены диапазоны доз и дозировочные режимы для некоторых типичных ингибиторов трансдукции сигнала. Специалистам в данной области техники должно быть понятно, что введение ингибитора трансдукции сигнала может меняться в зависимости от подвергающегося лечению заболевания и известных результатов применения ингибитора трансдукции сигнала для лечения этого заболевания. Также по решению квалифицированного врача лечебные протоколы (т.е. дозировки и режим приема) могут меняться в соответствии с наблюдающимися результатами ведения пациенту ингибитора трансдукции сигнала и наблюдающейся реакции заболевания на назначенные лечебные средства.
В другом примере комбинированной терапии соединение формулы (I) вводят перорально в диапазоне от 50 до 400 мг/сутки в виде двух разделенных доз совместно с ингибитором трансдукции сигнала, ингибитором рецептора киназы EGF, Iressa (ZD1839), который вводят перорально в диапазоне от 150 до 700 мг/сутки.
Таблица 1.0
Типичные дозировки и дозировочные режимы для химиотерапевтических препаратов
Карбоплатин:
Таксотер:
300 -360 мг/м2 каждые четыре недели (ВВ)
60-100 мг/м2 каждые три недели (ВВ)
Таблица 2.0
Типичные дозировки и дозировочные режимы для ингибиторов трансдукции сигнала
В способах, соответствующих настоящему изобретению, соединение-ингибитор ФПТ формулы (I) назначают совместно или последовательно с другим терапевтическим препаратом (т.е. с химиотерапевтическим препаратом, ингибитором трансдукции сигнала и/или облучением). Таким образом, не обязательно, чтобы, например, терапевтический препарат и соединение-ингибитор ФПТ формулы (I) назначался одновременно, непосредственно один перед другим или один после другого.
Кроме того, в общем случае соединение-ингибитор ФПТ формулы (I), химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение не должны назначаться в одной фармацевтической композиции и вследствие различных физических и химических характеристик должны вводиться различными путями. Например, соединение-ингибитор ФПТ формулы (I) можно вводить перорально, чтобы создавать и поддерживать в крови требующиеся концентрации, тогда как химиотерапевтический препарат можно вводить внутривенно. Определение режима введения и целесообразности назначения, если это возможно, то в одной фармацевтической композиции, проводит клинический врач. Начальное назначение можно сделать в соответствии с установленными протоколами, известными в данной области техники, а затем, на основании полученных эффектов, клинический врач может изменить дозировку, способ введения и время введения.
Выбор конкретного соединения-ингибитора ФПТ формулы (I), химиотерапевтического препарата, ингибитора трансдукции сигнала и/или облучения будет зависеть от диагноза, поставленного лечащим врачом, и его заключения о состоянии пациента и соответствующем протоколе лечения.
Соединение-ингибитор ФПТ формулы (I), химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение могут назначаться совместно (т.е. одновременно, непосредственно один перед другим или один после другого, или в рамках одного протокола лечения) или последовательно в зависимости от природы пролиферативного заболевания, состояния пациента, выбора конкретного химиотерапевтического препарата, ингибитора трансдукции сигнала и/или облучения, назначаемых совместно (т.е. в рамках одного протокола лечения) с соединением-ингибитором ФПТ формулы (I).
Если соединение-ингибитор ФПТ формулы (I), химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение не назначаются одновременно, то начальный порядок введения соединения-ингибитора ФПТ формулы (I), химиотерапевтического препарата, ингибитора трансдукции сигнала и/или облучения может не быть существенным. Таким образом, соединение-ингибитор ФПТ формулы (I) можно вводить первым, после него вводить химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение; или химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение могут быть введены первыми, а затем введено соединение-ингибитор ФПТ формулы (I). Это чередующееся назначение можно повторять при одном протоколе лечения. Определение порядка назначения и количество повторных назначений каждого терапевтического препарата в протоколе лечения проводит квалифицированный врач после оценки подлежащего лечению заболевания и состояния пациента. Например, химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение могут быть введены первыми, в особенности если он представляет собой цитотоксичный препарат, а затем лечение будет продолжено путем введения соединения-ингибитора ФПТ формулы (I), а после этого, если это будет сочтено целесообразным, будет назначен химиотерапевтический препарат, ингибитор трансдукции сигнала и/или облучение и т.п., пока протокол лечения не будет завершен.
Таким образом, в соответствии с опытом и квалификацией лечащий врач при проведении лечения может в каждом протоколе изменить ведения компонента (терапевтического средства - т.е. ингибитора ФПТ формулы (I), химиотерапевтического препарата, ингибитора трансдукции сигнала и/или облучения), использующегося при лечении, в соответствии с индивидуальной потребностью пациента.
Лечащий врач при принятии решения о том, какое лечение является эффективным при назначенной дозе, будет рассматривать общее состояние пациента, а также более определенные признаки, такие как ослабление связанных с заболеванием симптомов, подавление роста опухоли, реальное уменьшение размера опухоли или подавление метастазов. Размер опухоли можно измерить с помощью стандартных способов, таких как радиологическое исследование, например сканирование с помощью компьютерной аксиальной томографии или магнитно-резонансной визуализации, и последовательные измерения можно использовать для установления того, заторможен ли или даже обращен рост опухоли. Для того чтобы помочь оценке эффективности лечения, можно использовать наличие ослабления связанных с заболеванием симптомов, таких как боль, и улучшения общего состояния.
Хотя настоящее изобретение описано вместе с конкретными вариантами осуществления, приведенными выше, для среднего специалиста в данной области техники будут ясными многие его альтернативы, модификации и изменения. Подразумевается, что все такие альтернативы, модификации и изменения включены в объем и сущность настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА, КАК ИНГИБИТОРЫ ЦИКЛИН-ЗАВИСИМОЙ КИНАЗЫ | 2005 |
|
RU2414472C9 |
| ПОЛИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ АНТАГОНИСТА РЕЦЕПТОРА H, И СПОСОБ ЛЕЧЕНИЯ С ЕГО ПРИМЕНЕНИЕМ | 2001 |
|
RU2301231C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ СОДЕРЖАЩИЕ, И ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ CCR5 | 2000 |
|
RU2299206C9 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА И ПИПЕРИДИНА | 2002 |
|
RU2326120C9 |
| ПЕПТИДИЛЬНЫЕ ГЕТЕРОЦИКЛЫ, ИСПОЛЬЗУЕМЫЕ ПРИ ЛЕЧЕНИИ ТРОМБИНАССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ | 1996 |
|
RU2181125C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО-ОБОГАЩЕННЫХ ТЕТРАГИДРОБЕНЗОТИЕПИНОКСИДОВ | 1998 |
|
RU2236406C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2408591C9 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2531588C2 |
| ТРИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ ТРОМБИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2329264C9 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2316546C2 |
Настоящее изобретение относится к новым трициклическим соединениям, их фармацевтически приемлемым солям и сольватам, пригодным для ингибирования фарнезилпротеинтрансферазы. Описывается соединение формулы
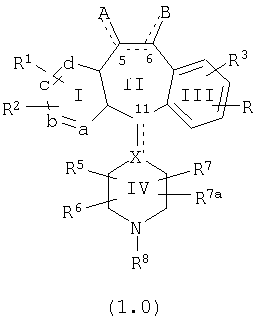
или его фармацевтически приемлемая соль или сольват, где один из а, b, с и d означает N или N+O-, а остальные а, b, с и d означают атом углерода, причем каждый атом углерода обладает группой R1 или R2, присоединенной к указанному атому углерода; или все а, b, с и d означают атом углерода, причем каждый атом углерода обладает группой R1 или R2, присоединенной к указанному атому углерода; штриховая линия ( ) означает необязательные связи; Х означает N или СН, когда необязательная связь отсутствует, и означает С, когда необязательная связь имеется; когда имеется необязательная связь между атомом углерода 5 и атомом углерода 6, то имеется только один заместитель А, связанный с атомом углерода 5, и имеется только один заместитель В, связанный с атомом углерода 6, и А или В отличны от Н; если необязательная связь между атомом углерода 5 и атомом углерода 6 отсутствует, то имеются два заместителя А, связанные с атомом углерода 5, и два заместителя В, связанные с атомом углерода 6, причем, по меньшей мере, один из двух заместителей А или один из двух заместителей В означает Н, и причем, по меньшей мере, один из двух заместителей А или один из двух заместителей В имеет значение, отличное от Н, остальные радикалы такие, как описано в формуле изобретения. Также раскрыты фармацевтическая композиция, включающая такие соединения, способ ингибирования аномального роста клеток и способы лечения пролиферативных заболеваний, таких как рак. 5 н. и 47 з.п. ф-лы, 20 табл.
) означает необязательные связи; Х означает N или СН, когда необязательная связь отсутствует, и означает С, когда необязательная связь имеется; когда имеется необязательная связь между атомом углерода 5 и атомом углерода 6, то имеется только один заместитель А, связанный с атомом углерода 5, и имеется только один заместитель В, связанный с атомом углерода 6, и А или В отличны от Н; если необязательная связь между атомом углерода 5 и атомом углерода 6 отсутствует, то имеются два заместителя А, связанные с атомом углерода 5, и два заместителя В, связанные с атомом углерода 6, причем, по меньшей мере, один из двух заместителей А или один из двух заместителей В означает Н, и причем, по меньшей мере, один из двух заместителей А или один из двух заместителей В имеет значение, отличное от Н, остальные радикалы такие, как описано в формуле изобретения. Также раскрыты фармацевтическая композиция, включающая такие соединения, способ ингибирования аномального роста клеток и способы лечения пролиферативных заболеваний, таких как рак. 5 н. и 47 з.п. ф-лы, 20 табл.
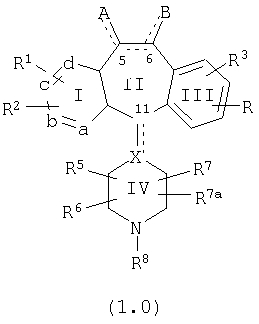
или его фармацевтически приемлемая соль или сольват, где
один из а, b, с и d означает N или N+O-, а остальные а, b, с и d означают атом углерода, причем каждый атом углерода обладает группой R1 или R2, присоединенной к указанному атому углерода; или
все а, b, с и d означают атом углерода, причем каждый атом углерода обладает группой R1 или R2, присоединенной к указанному атому углерода;
штриховая линия ( ) означает необязательные связи;
) означает необязательные связи;
Х означает N или СН, когда необязательная связь отсутствует, и означает С, когда необязательная связь имеется;
когда имеется необязательная связь между атомом углерода 5 и атомом углерода 6, то имеется только один заместитель А, связанный с атомом углерода 5, и имеется только один заместитель В, связанный с атомом углерода 6, и А или В отличны от Н;
если необязательная связь между атомом углерода 5 и атомом углерода 6 отсутствует, то имеются два заместителя А, связанные с атомом углерода 5, и два заместителя В, связанные с атомом углерода 6, причем, по меньшей мере, один из двух заместителей А или один из двух заместителей В означает Н, и причем, по меньшей мере один из двух заместителей А или один из двух заместителей В имеет значение, отличное от Н, причем каждый заместитель А и В выбран независимо из группы, включающей:
(1) -Н;
(2) -R9;
(3) -R9-C(O)-R9;
(4) -R9-CO2-R9a;
(7) -C(O)NHR9;
(8) -C(O)NH-CH2-C(O)-NH2;
(9) -C(O)NHR26;
(11) -(CH2)p(R9)2, где R9 являются одинаковыми или разными;
(12) -(CH2)pC(O)R9;
(13) -(CH2)pC(O)R27a;
(14) -(CH2)pC(O)n(R9)2, где R9 являются одинаковыми или разными;
(15) -(CH2)pС(O)NH(R9);
(19) -(CH2)pNHC(O)R50;
(20) -(CH2)pNHC(O)2R50;
(21) -(CH2)pN(C(O)R27a)2, где R27a являются одинаковыми или разными;
(22) -(CH2)pNR51C(O)R27 или R51 и R27 совместно с атомами, к которым они присоединены, образуют 5- или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(23) -(CH2)pNR51C(O)NR27 или R51 и R27 совместно с атомами, к которым они присоединены, образуют 5- или 6-членное гетероциклоалкильное кольцо при условии, что, если R51 и R27 образуют кольцо, то R51 не означает Н;
(24) -(CH2)pNR51C(O)N(R27a)2, где R27a являются одинаковыми или разными;
(25) -(CH2)pNHSO2N(R51)2, где R51 являются одинаковыми или разными;
(26) -(CH2)pNHCO2R50;
(28) -(CH2)pCO2R51; (29) -NHR9; (30)
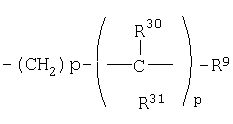
где R30 и R31 являются одинаковыми или разными; и
(31)
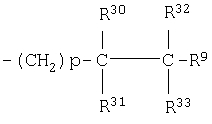
где R30, R31, R32 и R33 являются одинаковыми или разными;
р означает 0, 1, 2, 3 или 4;
все R1 и R2 независимо выбраны из группы, включающей Н, галоген, -CF3, -OR10, -COR10, -SR10, -S(O)tR15, где t равно 0, 1 или 2, -N(R10)2, -NO2, -OC(O)R10, -CO2R10, -OCO2R15, -CN, NR10COOR15, -SR15C(O)OR15, -SR15N(R13)2, при условии, что R15 в -SR15N(R13)2 не означает -СН2, и где все R13 независимо выбраны из группы, включающей Н или -C(O)OR15, бензотриазол-1-илоксил, тетразол-5-илтиоил или замещенный тетразол-5-илтиоил, алкинил, алкенил и алкил, причем указанная алкильная или алкенильная группа необязательно содержит в качестве заместителей галоген, -OR10 или -CO2R10;
R3 и R4 являются одинаковыми или разными и независимо выбраны из группы, включающей Н и любой из заместителей R1 и R2;
R5, R6, R7 и R7a независимо означают Н, -CF3, -COR10, алкил или арил, причем указанный алкил или арил необязательно содержит в качестве заместителей -OR10, -SR10, -S(O)tR15, -NR10COOR15, -N(R10)2, -NO2, -C(O)R10, -OCOR10, -OCO2R15, -CO2R10, -ОРО3R10, или R5 объединен с R6 и означает =O или =S;
R8 выбран из группы, включающей
H, 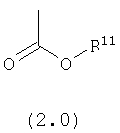 ,
,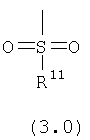 ,
,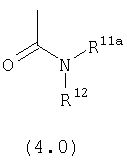 или
или 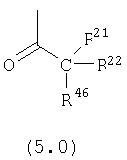
R9 выбран из группы, включающей (1) гетероарил; (2) замещенный гетероарил; (5) гетероциклоалкил; (6) замещенный гетероциклоалкил; (7) гетероциклоалкилалкил; (8) замещенный гетероциклоалкилалкил; (9) гетероарилалкил; (10) замещенный гетероарилалкил; (11) гетероарилалкенил; (12) замещенный гетероарилалкенил; (13) гетероарилалкинил; и (14) замещенный гетероарилалкинил;
где указанные замещенные группы R9 содержат один или большее количество заместителей, выбранных из группы, включающей
(1) -ОН; (2) -CO2R14; (3) -CH2OR14; (4) галоген; (5) алкил; (6) аминогруппу; (7) тритил; (8) гетероциклоалкил; (9) циклоалкил; (10) арилалкил; (11) гетероарил; (12) гетероарилалкил и (13)
где R14 независимо выбран из группы, включающей: Н, алкил, арил, арилалкил, гетероарил и гетероарилалкил;
R9a выбран из группы, включающей алкил и арилалкил;
R10 выбран из группы, включающей Н, алкил, арил и арилалкил;
R11 выбран из группы, включающей (1) алкил; (2) замещенный алкил; (3) арил; (4) замещенный арил; (5) циклоалкил; (6) замещенный циклоалкил; (7) гетероарил; (8) замещенный гетероарил; (9) гетероциклоалкил; и (10) замещенный гетероциклоалкил;
где указанные замещенные группы R11 содержат 1,2 или 3 заместителя, выбранных из группы, включающей (1) -ОН; (2) галоген и (3) алкил;
R11a выбран из группы, включающей (1) Н; (2) ОН; (3) алкил; (4) замещенный алкил; (5) арил; (6) замещенный арил; (7) циклоалкил; (8) замещенный циклоалкил; (9) гетероарил; (10) замещенный гетероарил; (11) гетероциклоалкил; и (12) замещенный гетероциклоалкил;
где указанные замещенные группы R11a содержат один или большее количество заместителей, выбранных из группы, включающей
(1) -ОН; (2) -CN; (3) -CF3; (4) галоген; (5) алкил; (6) циклоалкил; (7) гетероциклоалкил; (8) арилалкил; (9) гетероарилалкил; (10) алкенил и (11) гетероалкенил;
R12 выбран из группы, включающей Н и алкил;
R15 выбран из группы, включающей алкил и арил;
R21, R22 и R46 независимо выбраны из группы, включающей (1) -Н; (2) алкил; (3) арил; (4) замещенный арил, необязательно содержащий один или большее количество заместителей, выбранных из группы, включающей алкил, галоген, CF3 или ОН; (5) циклоалкил; (6) замещенный циклоалкил, необязательно содержащий один или большее количество заместителей, выбранных из группы, включающей алкил, галоген, CF3 или ОН;
(7) гетероарил формулы:
 и
и 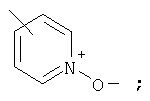
(8) гетероциклоалкил формулы:
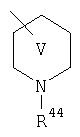
где R44 выбран из группы, включающей (1) -Н; (2) алкил; (3) алкилкарбонил; (4) алкоксикарбонил; (5) галогеналкил и (6) -C(O)NH(R51);
где R21, R22 или R46 означают гетероциклоалкил приведенной выше формулы, кольцо V означает
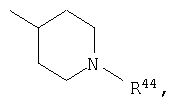
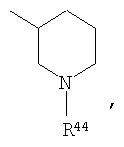
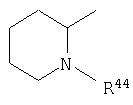
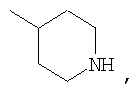
R26 выбран из группы, включающей
(1) -Н; (2) алкил; (3) алкоксил; (4) -CH2-CN; (5) R9; (6) -CH2CO2H; (7) -С(O)алкил и (8) -CH2СО2алкил;
R27 выбран из группы, включающей
(1) -Н; (2) -ОН; (3) алкил и (4) алкоксил;
R27a выбран из группы, включающей
(1) алкил и (2) алкоксил;
заместители от R30 до R33 независимо выбраны из группы, включающей
(1) -Н; (2) -ОН; (3) =O; (4) алкил; (5) арил и (6) арилалкил;
R50 выбран из группы, включающей
(1) алкил; (2) гетероарил; (3) замещенный гетероарил и (4) аминогруппу;
где указанные заместители в указанных замещенных группах R50 независимо выбраны из группы, включающей алкил, галоген и -ОН;
R50a выбран из группы, включающей
(1) гетероарил; (2) замещенный гетероарил и (3) аминогруппу;
R51 выбран из группы, включающей -Н и алкил.
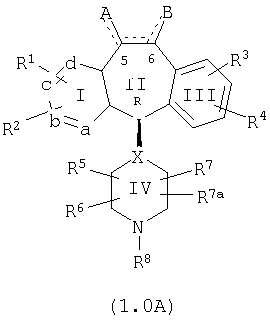
где Х=СН или N;
В означает Н, если необязательная связь между атомами С-5 и С-6 имеется, и если необязательная связь между атомами С-5 и С-6 отсутствует, то все В означают Н;
или обладающее структурой:
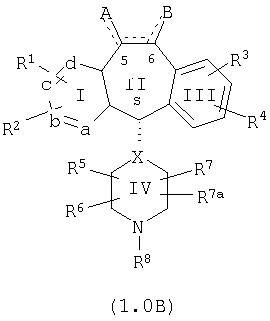
где Х=СН или N;
А означает Н, если необязательная связь между атомами С-5 и С-6 имеется, и если необязательная связь между атомами С-5 и С-6 отсутствует, то все А означают Н.
и 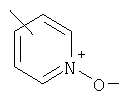 или
или
или гетероциклоалкил формулы:
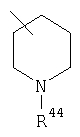
где указанные замещенные группы в качестве заместителей содержат алкил, алкилкарбонил или галогеналкил;
(5) R44 выбран из группы, включающей H и -C(O)NH2;
(6) R8 выбран из группы, включающей
(а) группу 2.0, в которой R11 выбран из группы, включающей трет-бутил или циклогексил;
(b) группу 3.0, в которой R11 выбран из группы, включающей метил или трет-бутил;
(c) группу 4.0, в которой R12 означает Н, a R11а выбран из группы, включающей трет-бутил, цианофенил, хлорфенил, фторфенил или циклогексил;
(d) группу 5.0, в которой R21 и R22 означают Н, а R46 выбран из группы, включающей
(1) гетероарил формулы:
и или
(2) гетероциклоалкил формулы:
и где R44 означает -С(O)NН2.
(1) гетероциклоалкилалкил формулы -(СН2)n-гетероциклоалкил;
(2) замещенный гетероциклоалкилалкил формулы -(СН2)n-замещенный гетероциклоалкил;
(3) гетероарилалкил формулы -(СН2)n-гетероарил;
(4) замещенный гетероарилалкил формулы -(CH2)n-замещенный гетероарил,
где n равно 1, 2 или 3 и заместители для указанных замещенных групп R9 независимо выбраны из группы, включающей
(1) -ОН; (2) -CO2R14; (3) -CH2OR14; (4) галоген; (5) алкил; (6) аминогруппу; (7) тритил; (8) гетероциклоалкил; (9) арилалкил; (10) гетероарил и (11) гетероарилалкил,
где R14 независимо выбран из группы, включающей Н и алкил.
(1) гетероарил; (2) замещенный гетероарил; (7) гетероциклоалкил; (8) замещенный гетероциклоалкил; (9) гетероциклоалкилалкил; (10) замещенный гетероциклоалкилалкил; (11) гетероарилалкил; (12) замещенный гетероарилалкил; (15) гетероарилалкенил и (16) замещенный гетероарилалкенил,
заместители для указанных замещенных групп R9 независимо выбраны из группы, включающей
(1) -ОН; (2) -CO2R14; (3) -CH2OR14; (4) галоген; (5) алкил; (6) аминогруппу; (7) тритил; (8) гетероциклоалкил; (9) арилалкил; (10) гетероарил и (12) гетероарилалкил.
где R14 независимо выбран из группы, включающей Н и алкил.
(1) гетероциклоалкилалкил формулы -(СН2)n-гетероциклоалкил;
(2) замещенный гетероциклоалкилалкил формулы -(CH2)n-замещенный гетероциклоалкил;
(3) гетероарилалкил формулы -(CH2)n-гетероарил;
(4) замещенный гетероарилалкил формулы -(CH2)n-замещенный гетероарил,
где заместители для указанных замещенных групп R9 независимо выбраны из группы, включающей
(1) -ОН; (2) -CO2R14; (3) -CH2OR14; (4) галоген; (5) алкил; (6) аминогруппу; (7) тритил; (8) гетероциклоалкил; (9) арилалкил; (10) гетероарил и (11) гетероарилалкил.
где указанные заместители в указанных замещенных группах выбраны из группы, включающей (1) галоген; (2) -CN и (3) -CF3.
где указанные заместители в указанных замещенных группах выбраны из группы, включающей (1) галоген; (2) цианогруппу и (3) -CF3.
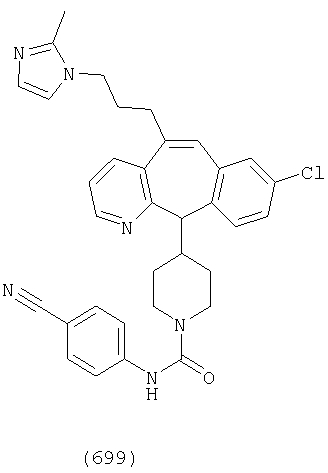
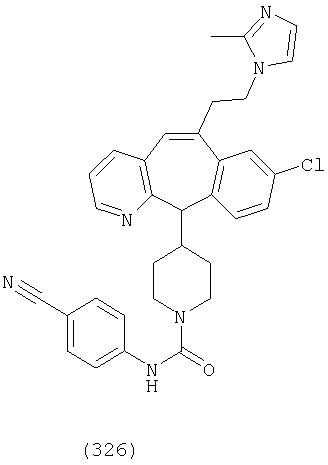
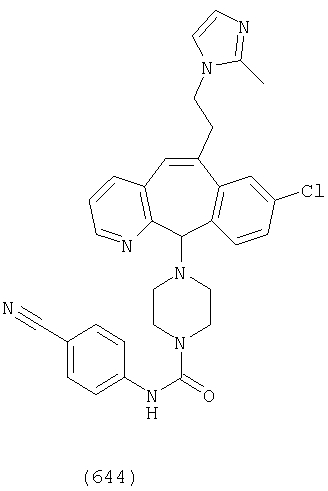
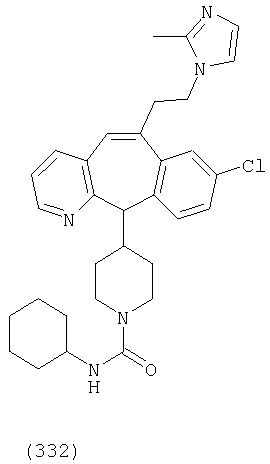
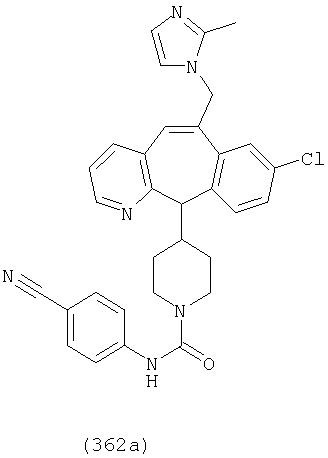
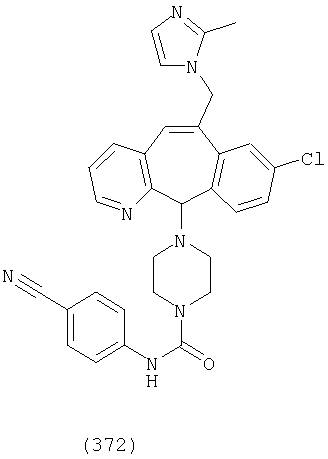
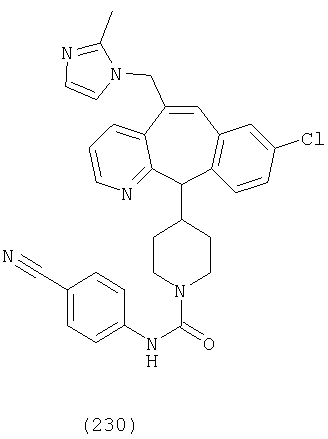
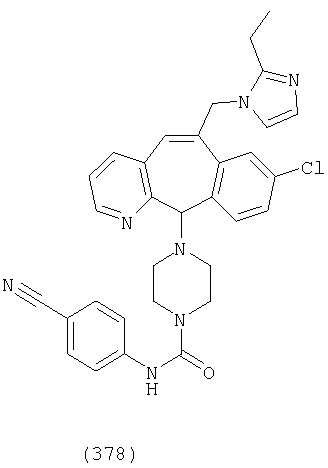
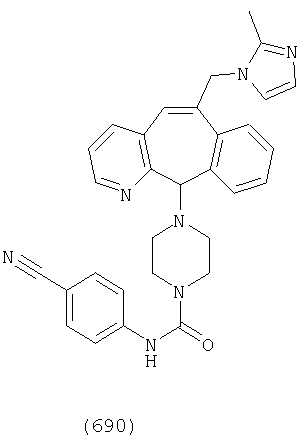
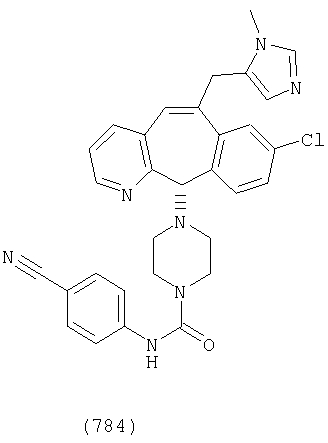

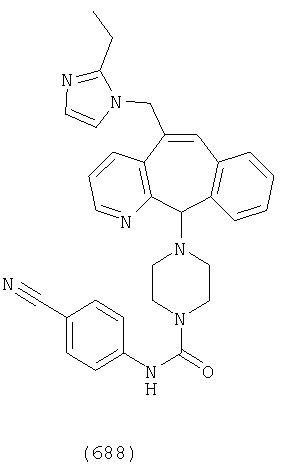
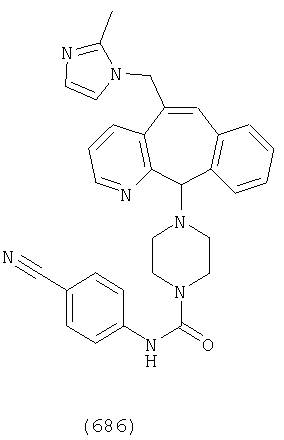
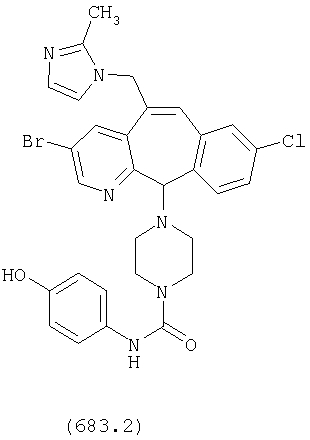
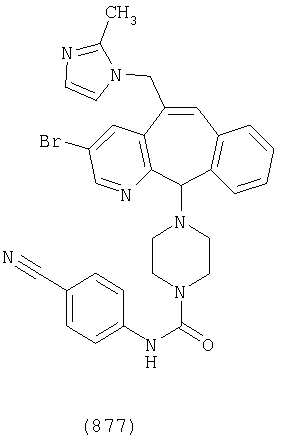
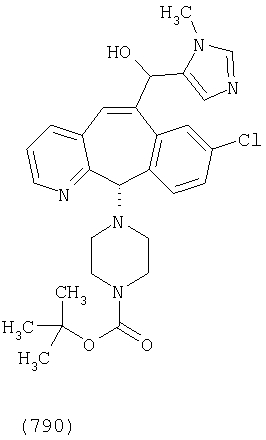
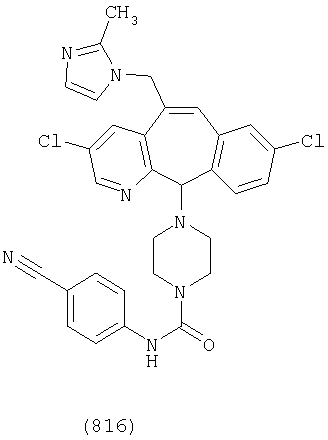
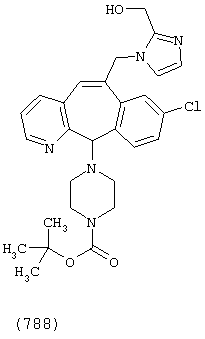
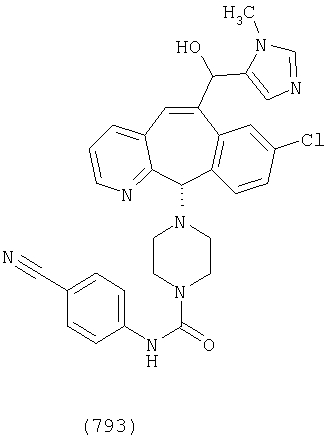
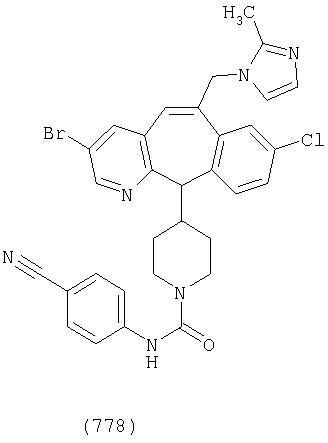
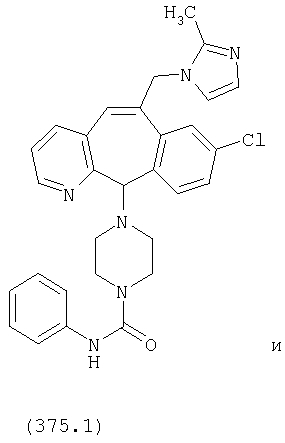
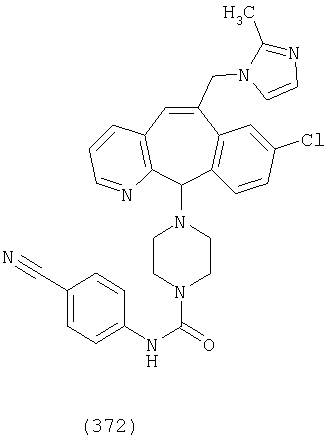
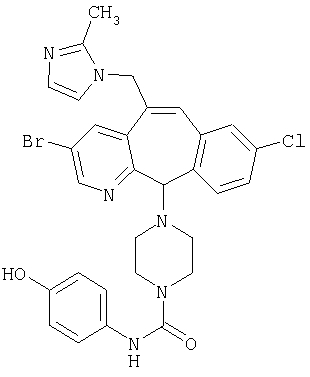
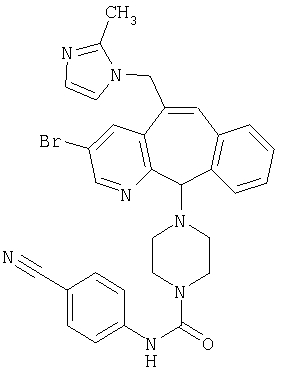
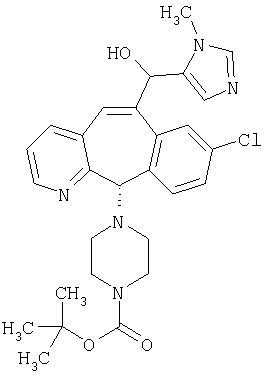
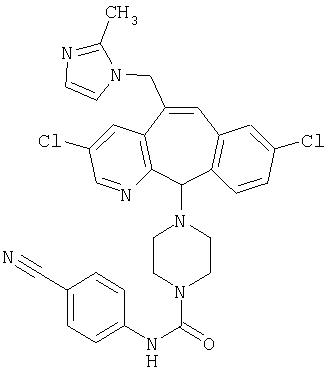
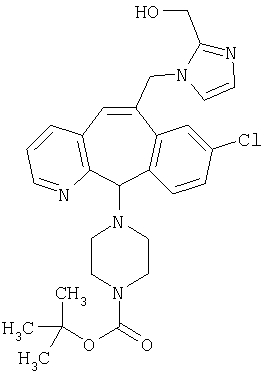
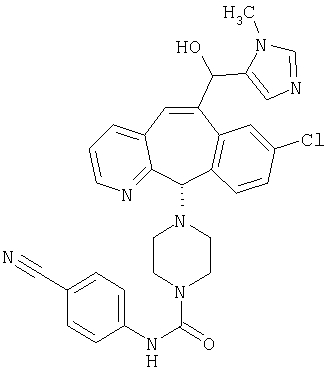
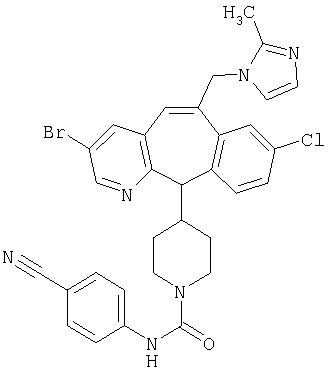
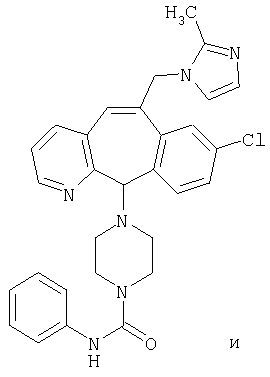
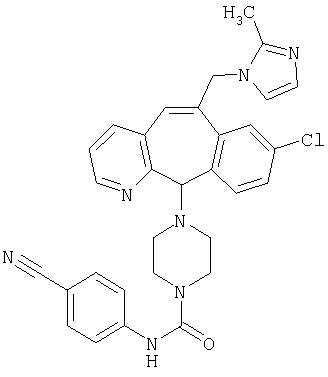
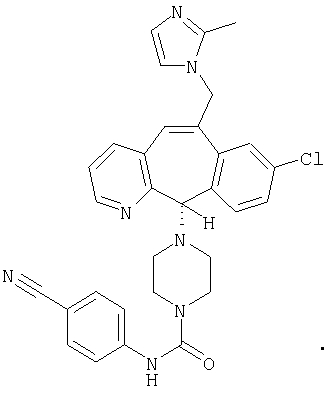
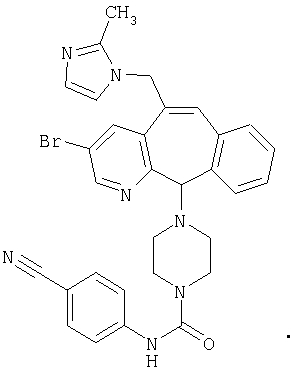
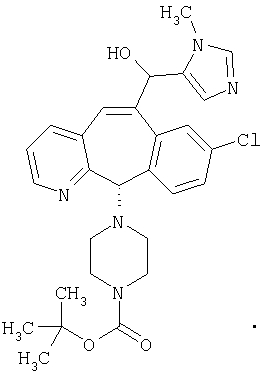
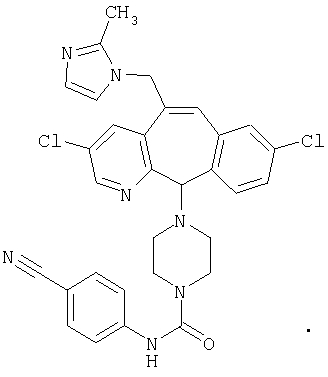
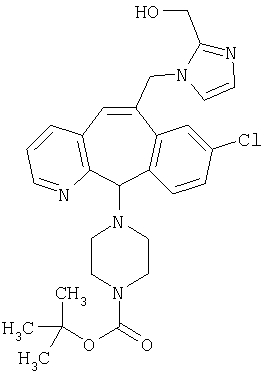
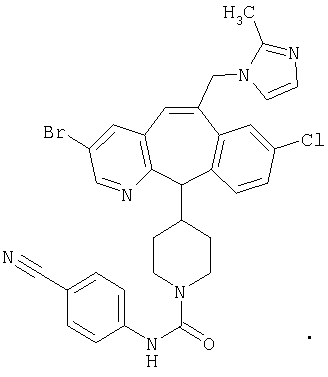
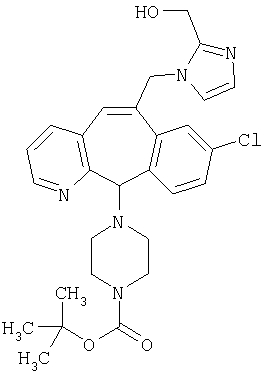
и
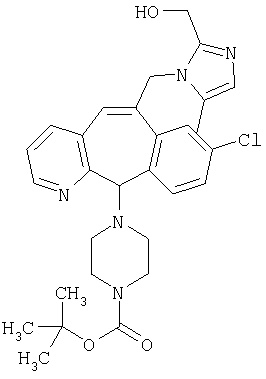
и
химиотерапевтический препарат представляет собой противоопухолевый препарат, выбранный из группы, включающей цисплатин, карбоплатин и гемцитабин, и/или препарат, воздействующий на микротрубочки, выбранный из группы, включающей таксол и таксотер, и соединение по п.1 выбрано из группы, включающей:
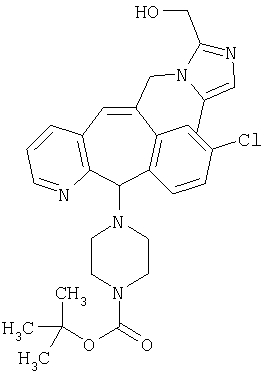
и
ингибитор трансдукции сигнала выбран из группы, включающей Gleevec, Iressa, OSI-774, Imclone C225, Abgenix ABX-EGF и герцептин, и соединение по п.1 выбрано из группы, включающей:
и
| US 6071907 A, 06.06.2000 | |||
| US 5089496 A, 18.02.1992 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 5925648 A, 20.07.1999 | |||
| ПРОИЗВОДНЫЕ БИС-БЕНЗО- ИЛИ БЕНЗОПИРИДО-ЦИКЛОГЕПТАПИПЕРИДИНА, ПИПЕРИДИЛИДЕНА И ПЕПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2086549C1 |

