Описание
Предшествующий уровень техники
Настоящее изобретение относится к производным пиперазина, применимым в качестве селективных антагонистов CCR5, фармацевтическим композициям, содержащим эти соединения, и способам лечения с помощью этих соединений. Настоящее изобретение также относится к использованию комбинации антагониста CCR5, соответствующего настоящему изобретению, и одного или большего количества противовирусных или других препаратов, используемых для лечения инфицирования вирусом иммунодефицита человека (ВИЧ). Настоящее изобретение также относится к использованию антагониста CCR5, соответствующего настоящему изобретению, в чистом виде или в комбинации с другим препаратом, при лечении отторжения трансплантанта цельного органа, болезни "трансплантант против хозяина", артрита, ревматоидного артрита, воспалительного заболевания кишечника, атопического дерматита, псориаза, астмы, аллергических заболеваний и рассеянного склероза.
Несомненным является глобальный кризис здравоохранения, вызванный ВИЧ, возбудителем синдрома приобретенного иммунодефицита (СПИД), и, хотя последние достижения лекарственной терапии привели к успеху в замедлении прогрессирования СПИД, все еще сохраняется необходимость в более безопасном, более эффективном, менее дорогостоящем способе борьбы с этим вирусом.
Сообщали, что ген CCR5 играет роль в сопротивлении инфицированию ВИЧ. Инфицирование ВИЧ начинается с присоединения вируса к мембране клетки-мишени путем взаимодействия с клеточным рецептором CD4 и молекулой сорецептора хемокина и продолжается путем репликации и распространения инфицированных клеток в крови и других тканях. Имеются различные хемокиновые рецепторы, но для макрофагового тропического ВИЧ, предположительно основного патогенного штамма, который реплицируется in vivo на ранних стадиях инфицирования, главным хемокиновым рецептором, необходимым для вхождения ВИЧ в клетку, является CCR5. Поэтому создание помех взаимодействию между вирусным рецептором CCR5 и ВИЧ может заблокировать вхождение ВИЧ в клетку. Настоящее изобретение относится к небольшим молекулам, которые являются антагонистами CCR5.
Сообщали, что рецепторы CCR5 опосредуют перенос клеток при воспалительных заболеваниях, таких как артрит, ревматоидный артрит, атопический дерматит, псориаз, астма и аллергические заболевания, и предполагается, что ингибиторы таких рецепторов применимы для использования при лечении таких заболеваний, а также при лечении других воспалительных заболеваний и патологических состояний, таких как воспалительное заболевание кишечника, рассеянный склероз, отторжение трансплантанта цельного органа и болезнь "трансплантант против хозяина".
Родственные производные пиперазина, являющиеся антагонистами мускарина, применимые для лечения нарушений познавательной способности, таких как болезнь Альцгеймера, раскрыты в патентах США 5883096, 6037352, 5889006.
А-М. Vandamme et al., Antiviral Chemistry & Chemotherapy, 9: 187 - 203 (1998), раскрыли применяющиеся в настоящее время способы клинического лечения ВИЧ-1-инфекций у людей, включающие комбинации хотя бы трех лекарственных препаратов, или так называемую антиретровирусную терапию ("ВААВТ"); ВААВТ включает различные комбинации нуклеозидных ингибиторов обратной транскриптазы ("НИОТ"), ненуклеозидных ингибиторов обратной транскриптазы ("ННИОТ") и ингибиторы протеазы ВИЧ ("ИП"). Для восприимчивых, не употребляющих наркотиков пациентов ВААВТ эффективно уменьшает смертность и перерастание ВИЧ-1 в СПИД. Однако эти полилекарственные способы лечения не приводят к уничтожению ВИЧ-1, а длительное лечение обычно приводит к возникновению полилекарственной резистентности. Поэтому приоритетной остается задача разработки новых способов лекарственного лечения, обеспечивающих более эффективное воздействие на ВИЧ-1.
Краткое описание изобретения
Настоящее изобретение относится к лечению ВИЧ-инфекции, включающей назначение млекопитающему, нуждающемуся в таком лечении, эффективного количества антагониста CCR5, описываемого структурной формулой I:
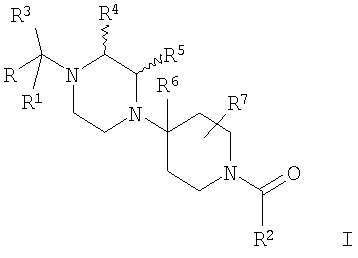
или его приемлемой с фармацевтической точки зрения соли, где
R означает R8-фенил, R8-пиридил, R8-тиофенил или R8-нафтил;
R1 означает водород или C1-С6-алкил;
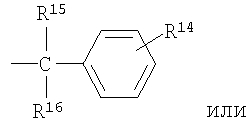
R2 означает R9-, R10-, R11-фенил; R9-, R10-, R11-замещенный 6-членный гетероарил; R9-, R10-, R11-замещенный 6-членный гетероарил-N-оксид; R12-, R13-замещенный 5-членный гетероарил; нафтил; флуоренил; дифенилметил
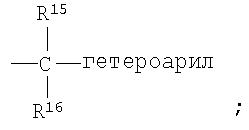
R3 означает водород, C1-С6-алкил, (С1-С6)-алкокси-(С1-С6)-алкил, С3-С10-циклоалкил, С3-С10-циклоалкил-(С1-С6)-алкил, R8-фенил, R8-фенил-(C1-C6)-алкил, R8-нафтил, R8-нафтил-(C1-C6)-алкил, R8-гетероарил или R8-гетероарил-(C1-C6)-алкил.
R4, R5, R7 и R13 независимо выбраны из группы, включающей водород и (C1-С6)-алкил;
R6 означает водород, C1-С6-алкил или С2-С6-алкенил;
R8 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, C1-С6-алкил, C1-С6-алкоксил, -CF3, CF3О-, СН3С(О)-, -CN, СН3SO2-, CF3SO2-, R14-фенил, R14-бензил, СН3С(=NOCH3), CH3C(=NOCH2CH3),

-NH2, -NHCOCF3, -NHCONH-(C1-C6)-алкил, -NHCO-(С1-С6)-алкил, -NHSO2-(C1-С6)-алкил, 5-членный гетероарил и
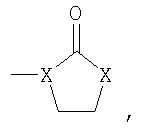
где Х означает -О-, -NH- или -N(СН3)-;
R9 и R10 независимо выбраны из группы, включающей (С1-С6)-алкил, галоген, -NR17R18, -ОН, -CF3, -ОСН3, -O-ацил, -OCF3 и -Si(СН3)3;
R11 означает R9, водород, фенил, -NO2, -CN, -CH2F, -CHF2, -СНО, CH=NOR17, пиридил, пиридил-N-оксид, пиримидил, пиразинил, -N(R17)CONR18R19, -NHCONH(хлор-(С1-С6)-алкил), -NHCONH((C3-C1)-циклоалкил-(C1-C6)-алкил), -NHCO(C1-C6)-алкил, -NHCOCF3, -NHSO2N((С1-С6)-алкил)2, -NHSO2(C1-C6)-алкил, -N(SO2CF3)2, -NHCO2(С1-С6)-алкил, С3-С10-циклоалкил, -SR20, -SOR20, -SO2R20, -SO2NH(C1-C6)-алкил, -OSO2(С1-С6)-алкил, -OSO2CF3, гидрокси-(С1-С6)-алкил), -CONR17R18, -CON(CH2CH2-O-СН3)2, -OCONH(C1-C6)-алкил, -CO2R17, -Si(СН3)3 или -В(ОС(СН3)2)2;
R12 означает (С1-С6)-алкил, -NH2 или R14-фенил;
R14 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, (С1-С6)-алкил, -CF3, -CO2R17, -CN, (С1-С6)-алкоксил и галоген;
R15 и R16 независимо выбраны из группы, включающей водород и C1-С6-алкил, или R15 и R16 совместно представляют собой С2-С5-алкиленовую группу и вместе с атомом углерода, к которому они присоединены, образуют спирановое кольцо, содержащее от 3 до 6 атомов углерода;
R17, R18 и R19 независимо выбраны из группы, включающей Н и C1-С6-алкил; и
R20 означает C1-С6-алкил или фенил.
Предпочтительными являются соединения формулы I, в которых R означает R8-фенил или R8-нафтил, в особенности, если R8 является единственным заместителем, и в особенности, если заместитель R8 находится в 4-положении. Для группы R8-фенил предпочтительными заместителями R8 являются -CF3, -OCF3, СН3SO2-, СН3СО-, CH3C(=NOCH3)-, Br и I. Для группы R8-нафтил предпочтительным заместителем R8 является C1-С6-алкоксил. Также предпочтительными являются соединения формулы I, в которых R3 означает водород, (С1-С6)-алкил, R8-фенил, R8-бензил или R8-пиридил; более предпочтительными заместителями R3 являются метил, этил, фенил, бензил и пиридил. Предпочтительно, чтобы R1 означал водород. Для соединений формулы I предпочтительно, чтобы R6 означал водород или метил, в особенности метил. Предпочтительно, чтобы R4 означал метил; предпочтительно, чтобы R5 и R7 означали водород.
В соединениях формулы I предпочтительно, чтобы R2 означал R9-, R10-, R11-фенил, R9-, R10-, R11-пиридил или его N-оксид, или R9-, R10-, R11-пиримидил. Если R2 означает пиридил, то предпочтительно, чтобы он представлял собой 3- или 4-пиридил, а если он означает пиримидил, то предпочтительно, чтобы он представлял собой 5-пиримидил. Предпочтительно, чтобы заместители R9 и R10 были присоединены к циклическим атомам углерода, соседним с атомом углерода, соединяющим цикл с остальной частью молекулы, а заместитель R11 может быть присоединен к любому остальных незамещенных циклических атомов углерода, например, как это показано на следующих структурах:
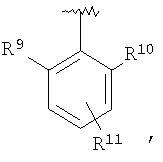
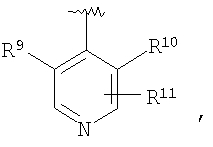
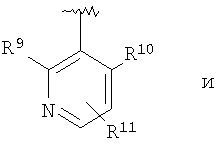
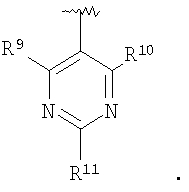
Предпочтительными заместителями R9 и R10 являются: (С1-С6)-алкил, в особенности метил; галоген, в особенности хлор или бром, -ОН и -NH2. Если R2 означает фенил, то предпочтительно, чтобы R11 означал водород или -ОН; если R11 означает пиридил, то предпочтительно, чтобы R11 означал водород, а если R2 означает пиримидил, то предпочтительно, чтобы R11 означал водород, метил или фенил. Примерами особенно предпочтительных групп R2 являются следующие:
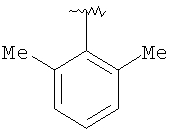

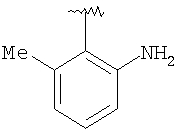
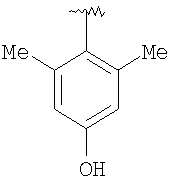
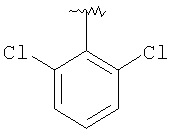

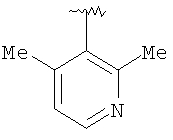
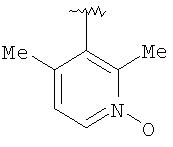


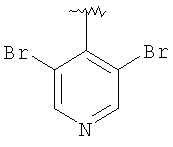
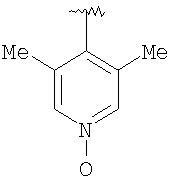


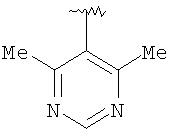
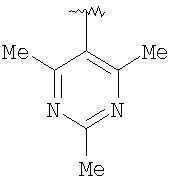
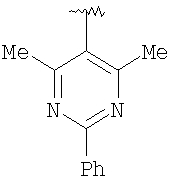
Также заявлены в качестве новых соединения, являющиеся антагонистами CCR5, описываемые структурной формулой II:

или его приемлемая с фармацевтической точки зрения соль, где
(1) Ra означает R8a-фенил, R8b-пиридил, R8b-тиофенил или R8-нафтил;
R1 означает водород или C1-С6-алкил;
R2 означает R9-, R10-, R11-фенил; R9-, R10-, R11-замещенный 6-членный гетероарил; R9-, R10-, R11-замещенный 6-членный гетероарил-N-оксид; R12-, R13-замещенный 5-членный гетероарил; нафтил; флуоренил; дифенилметил
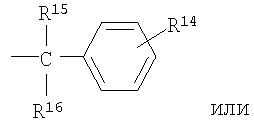

R3 означает водород, C1-С6-алкил, (С1-С6)-алкокси-(С1-С6)-алкил, С3-С10-циклоалкил, С3-С10-циклоалкил-(С1-С6)-алкил, R8-фенил, R8-фенил-(С1-С6)-алкил, R8-нафтил, R8-нафтил-(C1-C6)-алкил, R8-гетероарил или R8-гетероарил(С1-С6)-алкил.
R4, R5, R7 и R13 независимо выбраны из группы, включающей водород и (С1-С6)-алкил;
R6 означает водород, C1-С6-алкил или С2-С6-алкенил;
R8 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, C1-С6-алкил, C1-С6-алкоксил, -CF3, CF3О-, СН3С(О)-, -CN, СН3SO2-, CF3SO2-, R14-фенил, R14-бензил, СН3С(=NOCH3), CH3C(=NOCH2CH3),

-NH2, -NHCOCF3, -NHCONH-(C1 -C6)-алкил, -NHCO-(С1-С6)-алкил, -NHSO2-(C1-С6)-алкил, 5-членный гетероарил и

где Х означает -О-, -NH- или -N(СН3)-;
R8a означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, -CF3, CF3О-, -CN, CF3SO2-, R14-фенил, -NHCOCF3, 5-членный гетероарил и
где Х такой, как определено выше;
R8b означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, -CF3, CF3О-, СН3С(О)-, -CN, CF3SO2-, R14-бензил, CH3C(=NOCH3), CH3C(=NOCH2CH3),
-NHCOCF3, 5-членный гетероарил и
где Х такой, как определено выше;
R9 и R10 независимо выбраны из группы, включающей (С1-С6)-алкил, галоген, -NR17R18, -ОН, -CF3, -ОСН3, -O-ацил, -OCF3 и -Si(СН3)3;
R11 означает R9, водород, фенил, -NO2, -CN, -CH2F, -CHF2, -СНО, -CH=NOR17, пиридил, пиридил-N-оксид, пиримидил, пиразинил, -N(R17)CONR18R19, -NHCONH(хлор-(C1-C6)-алкил), -NHCONH((C3-C1)-циклоалкил-(С1-С6)-алкил), -NHCO(C1-C6)-алкил, -NHCOCF3, -NHSO2N((C1-C6)-алкил)2, -NHSO2(С1-С6)-алкил, -N(SO2CF3)2, -NHCO2(С1-С6)-алкил, С3-С10-циклоалкил, -SR20, -SOR20, -SO2R20, -SO2NH(C1-C6)-алкил, -OSO2(С1-С6)-алкил, -OSO2CF3, гидрокси-(С1-С6)-алкил), -CONR17R18, -CON(CH2CH2-O-CH3)2, -OCONH(C1-C6)-алкил, -CO2R17, -Si(СН3)3 или -В(ОС(СН3)2)2;
R12 означает (С1-С6)-алкил, -NH2 или R14-фенил;
R14 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, (C1-C6)-алкил, -CF3, -CO2R17, -CN, (C1-C6)-алкоксил и галоген; R15 и R16 независимо выбраны из группы, включающей водород и С1-С6-алкил, или R15 и R16 совместно представляю собой C2-C5-алкиленовую группу и вместе с атомом углерода, к которому они присоединены, образуют спирановое кольцо, содержащее от 3 до 6 атомов углерода;
R17,R18 и R19 независимо выбраны из группы, включающей H и С1-С6-алкил; и R20 означает С1-С6-алкил или фенил.
Предпочтительными являются соединения формулы I, в которых R означает R8-фенил или R8-нафтил, в особенности, если R8 является единственным заместителем, и в особенности, если заместитель R8 находится в 4-положении. Для группы R8-фенил предпочтительными заместителями R8 являются -CF3,-OCF3, CH3SO2-, CH3CO-, CH3C(=NOCH3)-, Br и I. Для группы R8-нафтил предпочтительным заместителем R8 является С1-С6-алкоксил. Также предпочтительными являются соединения формулы I, в которых R3 означает водород, (С1-С6)-алкил, R8-фенил, R8-бензил или R8-пиридил; более предпочтительными заместителями R3 являются метил, этил, фенил, бензил и пиридил. Предпочтительно, чтобы R1 означал водород. Для соединения формулы I предпочтительно, чтобы R6 означал водород или метил, в особенности метил. Предпочтительно, чтобы R4 означал метил; предпочтительно, чтобы R5 и R7 означали водород.
В соединениях формулы I предпочтительно, чтобы R2 означал R9-, R10-, R11-фенил, R9-, R10-, R11-пиридил или его N-оксид, или R9-, R10-, R11-пиримидил. Если R2 означает пиридил, то предпочтительно, чтобы он представлял собой 3- или 4-пиридил, а если он означает пиримидил, то предпочтительно, чтобы он представлял собой 5-пиримидил. Предпочтительно, чтобы заместители R9 и R10 были присоединены к циклическим атомам углерода, соседним с атомом углерода, соединяющим цикл с остальной частью молекулы, а заместитель R11 может быть присоединен к любому из остальных незамещенных циклических атомов углерода, например, как это показано на следующих структурах:
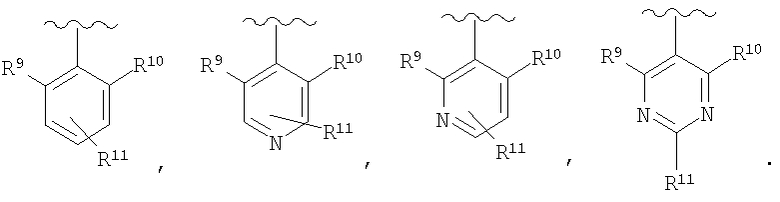
Предпочтительными заместителями R9 и R10 являются С1-С6-алкил, в особенности метил; галоген, в особенности хлор или бром, -OH и -NH2. Если R2 означает фенил, то предпочтительно, чтобы R11 означал водород или -OH; если R2 означает пиридил, что предпочтительно, чтобы R11 означал водород, а если R2 означает пиримидил, что предпочтительно, чтобы R11 означал водород, метил или фенил. Примерами особенно предпочтительных групп R2 являются следующие:
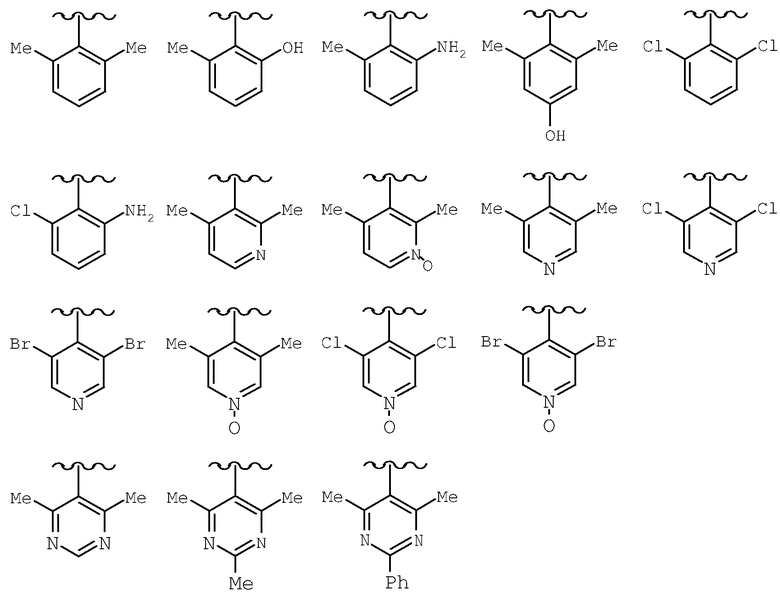
Также заявлены в качестве новых соединения, являющиеся антагонистами CCR5, описываемые структурной формулой II:

или его приемлемая с фармацевтической точки зрения соль, где
(1) Ra означает R8a-фенил, R8b-пиридил, R8b-тиофенил или R8-нафтил;
R1 означает водород или С1-С6-алкил;
R2 означает R9-, R10-, R11-фенил; R9-, R10-, R11-замещенный 6-членный гетероарил; R9-, R10-, R11-замещенный 6-членный гетероарил-N-оксид; R12-, R13-замещенный 5-членный гетероарил; нафтил; флуоренил; дифенилметил

R3 означает водород, С1-С6-алкил, (С1-С6)-алкокси-(С1-С6)-алкил, С3-С10-циклоалкил, С3-С10-циклоалкил-(С1-С6)-алкил, R8-фенил, R8-фенил-(С1-С6)-алкил, R8-нафтил, R8-нафтил-(С1-С6)-алкил, R8-гетероарил или R8-гетероарил-(С1-С6)-алкил.
R4, R5, R7 и R13 независимо выбраны из группы, включающей водород и (С1-С6)-алкил;
R6 означает водород, С1-С6-алкил или С2-С6-алкенил;
R8 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, С1-С6-алкил, С1-С6-алкоксил, -CF3, CF3О-, СН3С(О)-, -CN, CH3SO2-, CF3SO2-, R14-фенил, R14-бензил, СН3С(=NOCH3), СН3С(=NOCH2CH3),
-NH2, -NHCOCF3, -NHCONH-(С1-С6)-алкил, -NHCO-(С1-С6)-алкил, -NHSO2-(С1-С6)-алкил, 5-членный гетероарил и

где X означает -O-, -NH- или -N(CH3)-;
R8a означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, -CF3, CF3О-, -CN, CF3SO2-, R14-фенил, -NHCOCF3, 5-членный гетероарил и
где X такой, как определено выше;
R8b означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, -CF3, CF3О-, СН3С(О)-, -CN, CF3SO2-, R14-бензил, CH3C(=NOCH3), CH3C(=NOCH2CH3),
-NOCOCF3, 5-членный гетероарил и
где X такой, как определено выше;
R9 и R10 независимо выбраны из группы, включающей (С1-С6)-алкил, галоген, -NR17R18, -OH, -CF3, -OCH3, -O-ацил, -OCF3 и -Si(СН3)3;
R11 означает R9, водород, фенил, -NO2, -CN, -CH2F, -CHF2, -CHO, -CH=NOR17, пиридил, пиридил-N-оксид, пиримидил, пиразинил, -N(R17)CONR18R19, -NHCONH(хлор-(С1-С6)-алкил), -NHCONH((С3-С1)-циклоалкил-(С1-С6)-алкил), -NHCO(С1-С6)-алкил, -NHCOCF3, -NHSO2N((С1-С6)-алкил)2, -NHSO2(С1-С6)-алкил, -N(SO2CF3)2, -NHCO2(С1-С6)-алкил, C3-C10-циклоалкил, -SR20, -SOR20, -SO2R20, -SO2NH(С1-С6)-алкил, -OSO2(С1-С6)-алкил, -OSO2CF3, гидрокси-(С1-С6)-алкил), -CONR17R18, -CON(CH2CH2-O-CH3)2, -OCONH(С1-С6)-алкил, -CO2R17, -Si(CH3)3 или -B(OC(CH3)2)2;
R12 означает (С1-С6)-алкил, -NH2 или R14-фенил;
R14 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, (С1-С6)-алкил, -CF3, -CO2R17, -CN, (С1-С6)-алкоксил и галоген;
R15 и R16 независимо выбраны из группы, включающей водород и С1-С6-алкил, или R15 и R16 совместно представляет собой C2-C5-алкиленовую группу и вместе с атомом углерода, к которому они присоединены, образуют спирановое кольцо, содержащее от 3 до 6 атомов углерода;
R17, R18 и R19 независимо выбраны из группы, включающей H и С1-С6-алкил; и
R20 означает С1-С6-алкил или фенил; или
(2) Ra означает R8-фенил, R8-пиридил или R8b-тиофенил;
R2 означает флуоренил; дифенилметил
и R1, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18, R19 и R20 являются такими, как определено в (1).
Предпочтительными соединениями формулы II являются соединения, определенные в(1).
Более предпочтительными являются соединения формулы II(1), в которых Ra означает R8a-фенил или R8-нафтил, где R8a означает -CF3, -OCF3 или галоген, а R8 означает С1-С6-алкоксил. Предпочтительно, чтобы заместитель R8a или R8 представлял собой единственный заместитель; особенно предпочтительно, чтобы заместитель R8a или R8 находился в 4-положении. Также предпочтительными являются соединения формулы II(1), в которых R3 означает водород, (С1-С6)-алкил, R8-фенил, R8-бензил или R8-пиридил; более предпочтительными заместителями R3 являются метил, этил, фенил, бензил и пиридил. Предпочтительно, чтобы R1 означал водород. Для соединения формулы II(1) предпочтительно, чтобы R6 означал водород или метил, в особенности метил. Предпочтительно, чтобы R4 означал метил; предпочтительно, чтобы R5 и R7 означали водород.
Предпочтительно, чтобы в формуле II(1) R2 являлся таким, как определено для формулы I, т.е. представлял собой R9-, R10-, R11-фенил; R9-, R10-, R11-пиридил или его N-оксид; R9-, R10-, R11-замещенный пиримидил, где заместители R9, R10 и R11 являются такими, как определено выше для предпочтительных соединений формулы I.
Другим вариантом осуществления настоящего изобретения является фармацевтическая композиция для лечения ВИЧ-инфекции, включающая эффективное количество антагониста CCR5 формулы II в сочетании с приемлемым с фармацевтической точки зрения носителем. Другим вариантом осуществления настоящего изобретения является фармацевтическая композиция для лечения отторжения трансплантанта цельного органа, болезни "трансплантант против хозяина", артрита, ревматоидного артрита, воспалительного заболевания кишечника, атопического дерматита, псориаза, астмы, аллергических заболеваний и рассеянного склероза, содержащая эффективное количество антагониста CCR5 формулы II в сочетании с приемлемым с фармацевтической точки зрения носителем.
Еще одним вариантом осуществления настоящего изобретения является способ лечения ВИЧ-инфекции, включающий назначение человеку, нуждающемуся в таком лечении, эффективного количества антагониста CCR5 формулы II. Другим вариантом осуществления настоящего изобретения является способ лечения отторжения трансплантанта цельного органа, болезни "трансплантант против хозяина", артрита, ревматоидного артрита, воспалительного заболевания кишечника, атопического дерматита, псориаза, астмы, аллергических заболеваний и рассеянного склероза, включающий назначение человеку, нуждающемуся в таком лечении, эффективного количества соединения-антагониста CCR5 формулы I или II.
Еще одним вариантом осуществления настоящего изобретения является использование антагониста CCR5 формулы I или II, соответствующего настоящему изобретению, в комбинации с одним или большим количеством противовирусных или других препаратов, применимых для лечения инфицирования вирусом иммунодефицита человека при лечении СПИД. Еще одним воплощением настоящего изобретения является использование антагониста CCR5 формулы I или II, соответствующего настоящему изобретению, в комбинации с одним или большим количеством препаратов, применимых для лечения отторжения трансплантанта цельного органа, болезни "трансплантант против хозяина", воспалительного заболевания кишечника, ревматоидного артрита и рассеянного склероза. CCR5 и противовирусные и другие препараты, которые являются компонентами комбинации, можно назначать в единой дозировочной форме или их можно назначать по отдельности; предполагается возможность использования набора, включающего отдельные дозировочные формы активных веществ.
Подробное описание изобретения
Если не указано иного, то в настоящем изобретении следующие термины используются в соответствии с приведенными ниже описаниями.
Алкил представляет собой линейные и разветвленные углеродные цепи и содержит от одного до шести атомов углерода.
Алкенил представляет собой С2-С6-углеродные цепи, содержащие одну или две ненасыщенные связи, при условии, что две ненасыщенные связи не находятся рядом друг с другом.
Замещенный фенил означает, что фенильная группа может быть замещена в любом доступном положении фенильного кольца.
Ацил означает радикал карбоновой кислоты, имеющий формулу алкил-С(О)-, арил-С(О)-, арилалкил-С(О)-, (С3-С7)-циклоалкил-С(O)-, (С3-С7)-циклоалкил-(С1-С6)-алкил-С(О)- и гетероарил-С(О)-, где алкил и гетероарил являются такими, как это определено в настоящем изобретении; арил означает R14-фенил или R14-нафтил и арилалкил означает арил-(С1-С6)-алкил, где арил является таким как это определено выше.
Гетероарил представляет собой циклические ароматические группы, содержащие 5 или 6 атомов, или бициклические группы, содержащие от 11 до 12 атомов, включающие 1 или 2 гетероатома, независимо выбранные из группы, включающей О, S и N, причем указанный гетероатом (гетероатомы) входят в структуру карбоциклического кольца и обладают количеством делокализованных пи-электродов, достаточным для придания ароматического характера, при условии, что кольца не содержат соседних атомов кислорода и/или серы. В случае 6-членных гетероарильных циклов атомы углерода могут содержать замещающие группы R9, R10 или R11. По атомам азота могут образовываться N-оксиды. Предполагается, что могут использоваться все региоизомеры, например, 2-пиридил, 3-пиридил и 4-пиридил. Типичными 6-членными гетероарильными группами являются пиридильная, пиримидильная, пиразинильная, пиридазинильная и их N-оксиды. В случае 5-членных гетероарильных циклов атомы углерода могут содержать замещающие группы R12 и R13. Типичными 5-членными гетероарильными группами являются фурильная, тиенильная, пирролильная, тиазолильная, изотиазолильная, имидазолильная, пиразолильная и изоксазолильная. 5-Членные циклы с одним гетероатомом могут соединяться по 2- или 3-положениям; предпочтительно, чтобы 5-членные циклы с двумя гетероатомами соединялись по 4-положению. Типичные бициклические группы представляют собой сконденсированные с бензольным кольцом циклические системы, образовавшиеся из указанных выше гетероарильных групп, например, хинолильная, фталазинильная, хиназолинильная, бензофуранильная, бензотиенильная и индолильная.
Предпочтительные положения замещения для 6-членных гетероарильных циклов, представляющих собой заместители R2, описаны выше. Если R2 представляет собой 5-членную гетероарильную группу, то предпочтительно, чтобы заместители R12 и R13 были присоединены к циклическим атомам углерода, соседним с атомом углерода, соединяющим цикл с остальной частью молекулы, и предпочтительно, чтобы заместитель R12 означал алкил; однако, если рядом с атомом углерода, соединяющим цикл с остальной частью молекулы (например, как в 2-пирролильном фрагменте), находится гетероатом, то предпочтительно, чтобы R12 был присоединен к атому углерода, соседнему с атомом углерода, соединяющему цикл с остальной частью молекулы.
Галогены означают фтор, хлор, бром и йод.
Одно или большее количество, предпочтительно от одного до четырех, противовирусных препаратов, пригодных для использования в анти-ВИЧ-1 препаратах, можно использовать в комбинации с антагонистом CCR5, соответствующим настоящему изобретению. Противовирусный препарат или препараты можно сочетать с антагонистом CCR5 в одной дозировочной форме или антагонист CCR5 и противовирусный препарат или препараты можно назначать одновременно или последовательно в виде отдельных дозировочных форм. Противовирусные препараты, которые предполагается использовать в комбинации с соединениями, соответствующими настоящему изобретению, включают нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, ингибиторы протеазы и другие противовирусные лекарственные препараты, перечисленные ниже и не относящиеся к этим классам. В частности, комбинации, известные под названием ВААВТ (высокоактивная антиретровирусная терапия) предполагается использовать в комбинации с антагонистами CCR5, соответствующими настоящему изобретению.
Термин "нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы" ("НИОТ") при использовании в настоящем изобретении означает нуклеозиды, нуклеотиды и их аналоги, которые ингибируют активность обратной транскриптазы ВИЧ-1, фермента, которые катализирует превращение вирусной геномной РНК ВИЧ-1 в провирусную ДНК ВИЧ-1.
Типичные подходящие НИОТ включают зидовудин (AZT), выпускаемый под торговым названием RETROVIR компанией Glaxo-Wellcome Inc., Research Triangle, NC 27709; диданозин (ddl), выпускаемый под торговым названием VIDEX компанией Bristol-Myers Squibb Co., Princeton, NJ 08543; зальцитабин (ddC), выпускаемый под торговым названием HMD компанией Roche Pharmaceuticals, Nutley, NJ 07110; ставудин (d4T), выпускаемый под торговым названием ZERIT компанией Bristol-Myers Squibb Co., Princeton, NJ 08543; ламивудин (3ТС), выпускаемый под торговым названием EPIVIR компанией Glaxo-Wellcome Research Triangle, NC 27709; абакавир (1592U89), раскрытый в WO 96/30025 и выпускаемый под торговым названием ZIAGEN компанией Glaxo-Wellcome Research Triangle, NC 27709; адефовир дипивоксил [bis(POM)-PMEA], выпускаемый под торговым названием PREVON компанией Gilead Sciences, Foster City, CA 94404; лобукавир (BMS-180194), нуклеозидный ингибитор обратной транскриптазы, раскрытый в ЕР-0358154 и ЕР-0736533 и находящийся в процессе разработки компанией Bristol-Myers Squibb, Princeton, NJ 08543; ВСН-10652, ингибитор обратной транскриптазы (в виде рацемической смеси ВСН-10618 и ВСН-10619), находящийся в процессе разработки компанией Biochem Pharma, Laval, Quebec H7V, 4A7, Canada; эмитрицитабин [(-)-FTC], выпускаемый по лицензии Emory University по выданному Emory Univ. патенту США №5814639, и находящийся в процессе разработки компанией Triangle Pharmaceuticals, Durham, NC 27707; beta-L-FD4 (также известный под названием beta-L-D4C и химическим названием бета-L-2',3'-дидезокси-5-фторцитеден), выпускаемый по лицензии Yale University, выданной компании Vion Pharmaceuticals, New Haven CT 06511; DAPD, пуриновый нуклеозид, (-)-бета-D-2,6,-диапинопуриндиоксолан, раскрытый в ЕР 0656778, выпускаемый по лицензии Emory University и the University of Georgia, выданной компании Triangle Pharmaceuticals, Durham, NC 27707; лоденосин (FddA), 9-(2,3-дидезокси-2-фтор-b-D-трео-пентофуранозил)-аденин, устойчивый к действию кислот ингибитор обратной транскриптазы на основе пурина, предложенный в NIH (Национальный институт здравоохранения США) и находящийся в процессе разработки компанией U.S. Bioscience Inc., West Conshohoken, PA 19428.
Термин "ненуклеозидные ингибиторы обратной транскриптазы" ("ННИОТ") при использовании в настоящем изобретении означает ненуклеозиды, которые ингибируют активность обратной транскриптазы ВИЧ-1.
Типичные подходящие ННИОТ включают невирапин (BI-RG-587), выпускаемый под торговым названием VIRAMUNE компанией Boehringer Ingelheim, поставщиком для Roxane Laboratories, Columbus, ОН 43216; делавирадин (ВНАР, U-90152), выпускаемый под торговым названием RESCRIPTOR компанией Pharmacia & Upjohn Co., Bridgewater NJ 08807; эфавиренц (DMP-266), бензоксазин-2-он, раскрытый в WO 94/03440 и выпускаемый под торговым названием SUSTIVA выпускаемый компанией DuPont Pharmaceutical Co., Wilmington, DE 19880-0723; PNU-142721, фуропиридин-тио-пиримид, находящийся в процессе разработки компанией Pharmacia and Upjohn, Bridgewater NJ 08807; AG-1549 (ранее Shionogi # S-1153); 5-(3,5-дихлорфенил)-тио-4-изопропил-1-(4-пиридил)-метил-1Н-имидазол-2-илметилкарбонат, раскрытый в WO 96/10019 и находящийся в процессе клинической разработки компанией Agouron Pharmaceuticals, inc., LaJolla CA 92037-1020; МКС-442 (1-(этоксиметил)-5-(1-метилэтил)-6-(фенилметил)-(2,4(1Н,3Н)-пиримидиндион), раскрытый компанией Mitsubishi Chemical Co. и находящийся в процессе разработки компанией Triangle Pharmaceuticals, Durham, NC 27707; и (+)-каланолид A (NSC-675451) и В, производные кумарина, раскрытые в выданном NIH патенте США №5489697, лицензия на который выдана компании Med Chem Research и который представляет собой назначаемый перорально препарат (+) каланолид А, разработанный совместно с компанией Vita-Inves.
Термин "ингибитор протеазы" ("ИП") при использовании в настоящем изобретении означает ингибиторы протеазы ВИЧ-1, фермента, необходимого для протеолитического расщепления предшественников вирусного полипротеина (например, вирусных полипротеинов GAG и GAG Pol), на индивидуальные функциональные протеины, обнаруживаемые при ВИЧ-1-инфекции. К ингибиторам протеазы ВИЧ относятся соединения, обладающие пептидомиметической структурой, большой молекулярной массой (7600 Да) и по существу пептидным характером, например, CRIXIVAN (выпускаемый компанией Merck), а также непептидные ингибиторы протеазы, например, VIRACEPT (выпускаемый компанией Agouron).
Типичные подходящие ИП включают саквинавир (Ro 31-8959), выпускаемый в твердых желатиновых капсулах под торговым названием INVIRASE и в мягких желатиновых капсулах под торговым названием FORTOUASE компанией Roche Pharmaceuticals, Nutley, NJ 07110-1199; ритонавир (АВТ-538), выпускаемый под торговым названием NORVIR компанией Abbott Laboratories, Abbott Park, IL 60064; индинавир (МК-639) выпускаемый под торговым названием CRIXIVAN компанией Merck & Co., inc., West Point, PA 19486-0004; нелфинавир (AG-1343), выпускаемый под торговым названием VIRACEPT компанией Agouron Pharmaceuticals, Inc., LaJolla CA 92037-1020; ампренавир (141W94), торговое название AGENERASE, непептидный ингибитор протеазы, находящийся в процессе разработки компанией Vertex Pharmaceuticals, Inc., Cambridge, MA 02139-4211 и выпускаемый компанией Glaxo-Wellcome, Research Triangle, NC в соответствии с программой расширенного распространения; ласинавир (BMS-234475) выпускаемый компанией Bristol-Myers Squibb, Princeton, NJ 08543 (первоначально разработанный компанией by Novartis, Basel, Switzerland (CGP-61755); DMP-450, циклическая мочевина, предложенная компанией Dupont и находящаяся в процессе разработки компанией Triangle Pharmaceuticals; BMS-2322623, азапептид, находящийся в процессе разработки компанией Bristol-Myers Squibb, Princeton, NJ 08543, в качестве ИП 2-го поколения для HIV-1; АВТ-378 находящийся в процессе разработки компанией Abbott, Abbott Park, IL 60064; и AG-1549 активный при пероральном приеме имидазолкарбамат, предложенный компанией Shionogi (Shionogi #S-1153) и находящийся в процессе разработки компанией Agouron Pharmaceuticals, Inc., LaJolla CA 92037-1020.
Другие противовирусные препараты включают гидроксимочевину, рибавирин, IL-2, IL-12, пентафусид и Yissum Project No. 11607. Гидроксимочевина (Droxia), ингибитор рибонуклеозидтрифосфатредуктазы, фермента, участвующего в активации Т-клеток, предложенный компанией NCI, находится в процессе разработки компанией i Bristol-Myers Squibb; при доклинических испытаниях обнаружено, что он оказывает синергетическое воздействие на активность диданосина и исследован совместно со ставудином. IL-2 раскрыт в ЕР-0142268, выданном Ajinomoto, EP-0176299, выданном Takeda, и патентах США №№ RE 33653, 4530787, 4569790, 4604377, 4748234, 4752585 и 4949314, выданных Chiron, и выпускается под торговым названием PROLEUKIN (алдеслейкин) компанией Chiron Corp., Emeryville, CA 94608-2997 в виде лиофилизированного порошка для внутривенного вливания и подкожного введения после восстановления влагосодержания и разбавления водой; при подкожном введении предпочтительна доза, равная от примерно 1 до примерно 20 миллионов МЕ/сутки; при подкожном введении более предпочтительна доза, равная примерно 15 миллионам МЕ/сутки. IL-12 раскрыт в WO 96/25171 и выпускается компанией Roche Pharmaceuticals, Nutley, NJ 07110-1199 и компанией American Home Products, Madison, NJ 07940; при подкожном введении предпочтительна доза, равная от примерно 0,5 мкг/кг/сут до примерно 10 мкг/кг/сут. Пентафусид (DP-178, Т-20), синтетический пептид, состоящий из 36 аминокислот, раскрытый в патенте США №5464933, выпускаемый по лицензии Duke University, выданной компании Trimeris, которая разрабатывает пентафусид совместно с Duke University; пентафусид действует путем ингибирования слияния HIV-1 с мембранами-мишенями. Пентафусид (3-100 мг/сут) назначают в виде продолженного подкожного вливания или инъекции совместно с эфавиренцем и двумя ИП пациентам-носителям HIV-1, которые не поддаются лечению с помощью трехкомпонентных комбинированных препаратов; предпочтительно назначать 100 мг/сут. Yissum Project No. 11607, синтетический протеин на основе протеина HIV-1 Vif, находится в стадии доклинической разработки компанией Yissum Research Development Co., Jerusalem 91042, Israel. Рибавирин, 1-β-D-рибофуранозил-1Н-1,2,4-триазол-3-карбоксамид, выпускает компания ICN Pharmaceuticals, Inc., Costa Mesa, CA; его изготовление и рецептура описаны в патенте США №4211771.
При использовании в настоящем изобретении термин "анти-ВИЧ-1 препарат" означает любой анти-ВИЧ-1 лекарственный препарат, который оказался подходящим для лечения ВИЧ-1 инфекций у людей в чистом виде или в качестве части полилекарственных комбинированных препаратов, в особенности трехкомпонентных и четырехкомпонентных комбинированных препаратов ВААВТ. Обычно известные подходящие анти-ВИЧ-1 препараты включают (но не ограничиваются только ими) полилекарственные комбинированные препараты, такие как содержащие (i) хотя бы три анти-ВИЧ-1 препарата, выбранных из группы, включающей два НИОТ, один ИП, второй ИП и один ННИОТ, и (ii) не менее двух анти-ВИЧ-1 препаратов, выбранных из группы, включающей ННИОТ и ИП. Типичные подходящие ВААВТ - полилекарственные комбинированные препараты включают:
(а) трехкомпонентные комбинированные препараты, такие как два НИОТ и один ИП или (b) два НИОТ и один ННИОТ и (с) четырехкомпонентные комбинированные препараты, такие как два НИОТ, один ИП и второй ИП или один ННИОТ. При лечении пациентов, не употребляющих наркотиков, предпочтительно начинать анти-ВИЧ-1 лечение с назначения трехкомпонентных комбинированных препаратов; назначение двух НИОТ и одного ИП предпочтительно, если отсутствует непереносимость ИП. Необходимо согласие пациента на прием лекарственного препарата. Каждые 3-6 месяцев необходимо проверять уровни CD4+ и ВИЧ-1-РНК в плазме. При достижении плато вирусной нагрузки можно добавить четвертый лекарственный препарат, например, один ИП или один ННИОТ. В приведенной ниже таблице дополнительно описаны типичные препараты:
АНТИ-ВИЧ-1 ПОЛИЛЕКАРСТВЕННЫЕ КОМБИНИРОВАННЫЕ ПРЕПАРАТЫ
А. Трехкомпонентные комбинированные препараты
1. Два НИОТ1 + один ИП2
2. Два НИОТ1 + один ННИОТ2
В. Четырехкомпонентные комбинированные препараты4
Два НИОТ + один ИП + второй ИП или один ННИОТ
С. АЛЬТЕРНАТИВЫ:
Два НИОТ1
Один НИОТ5 + один ИП2
Два ИП6 ± один НИОТ7 или ННИОТ3
Один ИП2 + один НИОТ7 + один ННИОТ3
ПРИМЕЧАНИЯ К ТАБЛИЦЕ
1. Один из следующих вариантов: зидовудин + ламивудин; зидовудин + диданосин; ставудин + ламивудин; ставудин + диданосин; зидовудин + зальцитабин.
2. Мягкие желатиновые капсулы индинавира, нелфинавира, ритонавира или саквинавира.
3. Невирапин или делавирдин.
4. См. А-М.Vandamme et al., Antiviral Chemistry & Chemotherapy, 9: 187 p.193-197 и рис.1+2.
5. Альтернативные режимы для пациентов, которые не могут соблюдать рекомендованный режим из-за несогласия или токсичности, и для тех, для которых рекомендованный режим не привел к результатам или у которых наблюдается рецидив болезни. Комбинации, включающие два нуклеозида, для некоторых пациентов могут привести к резистентности ВИЧ и неблагоприятному исходу.
6. Большая часть данных получена при использовании саквинавира и ритонавира (по 400 мг два раза в день).
7. Зидовудин, ставудин или диданосин.
Препаратами, которые применяются для лечения ревматоидного артрита, отторжения трансплантанта, болезни "трансплантант против хозяина", воспалительного заболевания кишечника и рассеянного склероза и которые можно назначать в комбинации с антагонистами CCR5, соответствующими настоящему изобретению, являются следующие:
отторжение трансплантанта цельного органа и болезнь "трансплантант против хозяина: иммунодепрессанты, такие как циклоспорин и интерлейкин-10 (IL-10), такролимус, антилимфоцитарный глобулин, антитела ОКТ-3 и стероиды;
воспалительное заболевание кишечника: IL-10 (см. патент США 5368854) стероиды и азульдифин;
ревматоидный артрит: метотрексат, азатиоприн, циклофосфамид, стероиды и микофенолят мотефил;
рассеянный склероз: бета-интерферон, альфа-интерферон и стероиды.
Некоторые соединения, соответствующие настоящему изобретению, могут существовать в различных изомерных формах (например, энантиомерных, дистереоизомерных, атропоизомерных и ротамерных). Настоящее изобретение охватывает все такие изомеры, как в чистом виде, так и в смесях, включая рацемические смеси.
Некоторые соединения по своей природе являются кислотными, например соединения, в которых содержатся карбоксильная или фенольная гидроксильная группа. Эти соединения могут образовывать приемлемые с фармацевтической точки зрения соли. Примеры таких солей могут включать соли натрия, калия, кальция, алюминия, золота и серебра. Также предполагается, что настоящее изобретение охватывает соли, образованные с приемлемыми с фармацевтической точки зрения аминами, такими как аммиак, алкиламины, гидроксиалкиламины, N-метилглюкамин и т.п.
Некоторые основные соединения также образуют приемлемые с фармацевтической точки зрения соли, например молекулярные соли с кислотами. Например, атомы азота пиридинового типа могут образовывать соли с сильными кислотами, а соединения, в которых имеются основные заместители, такие как аминогруппы, также образуют соли и с более слабыми кислотами. Примерами кислот, подходящих для образования солей, являются хлористоводородная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие неорганические и карбоновые кислоты, хорошо известные специалистам в данной области техники. Соли получают путем взаимодействия соединения в виде свободного основания с достаточным количеством необходимой кислоты с образованием соли, происходящим обычным образом. Свободные основания можно выделить путем обработки соли подходящим разбавленным водным раствором основания, например, разбавленным водным раствором NaOH, карбоната калия, аммиака и бикарбоната натрия. Свободные основания отличаются от соответствующих солей по некоторым физическим характеристикам, таким как растворимость в полярных растворителях, однако для целей настоящего изобретения по остальным характеристикам соли кислот и оснований эквивалентны соответствующим свободным основаниям.
Для задач настоящего изобретения все такие соли кислот и оснований считаются приемлемыми с фармацевтической точки зрения солями и для целей настоящего изобретения все такие соли кислот и оснований считаются эквивалентными свободным основаниям соответствующих соединений.
Соединения, соответствующие настоящему изобретению, можно получить способами, известными в данной области техники, например, с помощью методик, описанных на приведенных ниже схемах реакций, способами, описанными в приведенных ниже примерах, и путем использования способов, описанных в WO 96/26196 и WO 98/05292.
Следующие растворители и реагенты в настоящем изобретении могут обозначаться указанными аббревиатурами: тетрагидрофуран (THF), этанол (EtOH), метанол (МеОН), уксусная кислота (НОАс или АсОН), этилацетат (EtOAc), N,N-диметилформамид (DMF), трифторуксусная кислота (TFA), 1-гидроксибензотриазол (НОВТ), м-хлорпербензойная кислота (МСРВА), триэтиламин (Et3N), диэтиловый эфир (Et2O), диметилсульфоксид (DMSO) и 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (DEC). RT означает комнатную температуру, а ТСХ означает тонкослойную хроматографию. Me означает метил, Et означает этил, Pr означает пропил, Bu и означает бутил, tButyl означает трет-бутил, Ph означает фенил и Ас означает ацетил.
Схема 1
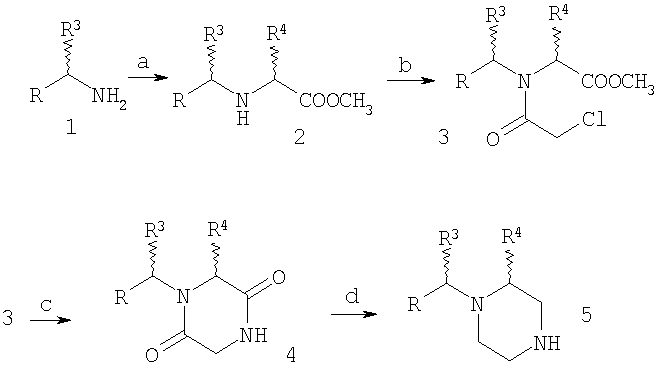
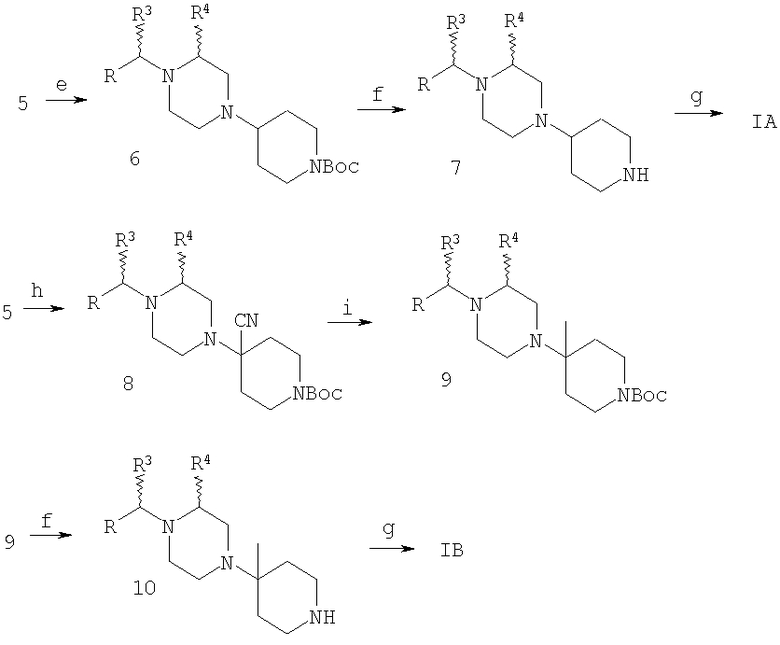
Реагенты и условия: а: R4CH(OSO2CF3)CO2CH3, основание (например, К2СО3); b: ClCH2COCl; с: NH3; d: NaBH4-BF3; e: N-Boc-4-пиперидон, NaBH(OAc)3; f: CF3СО2Н; g: ацилирование; h: N-Boc-4-пиперидон, Ti(OPr-i)4, Et2AlCN; i: CH3MgBr.
На Схеме 1 бензиламин (1), где R и R3 являются такими, как определено выше, и R1 означает водород, через (2) и (3) превращают в дикетопиперазин (4), где R4 является такими, как определено выше, который восстанавливают в пиперазин (5). В зависимости от необходимого заместителя R6 его обрабатывают двумя способами. Восстановительное аминирование дает (6), у которого можно удалить защитную группу с получением (7) и затем окончательно ацилировать с получением соединения формулы IA, где R5 и R6 означают H; альтернативно, использование (5) в модифицированной реакции Штрекера дает аминонитрил (8), который после обработки метилмагнийбромидом дает (9), удаление защитной группы дает (10), а конечное N-ацилирование дает соединение формулы IB, где R5 означает H, а R6 означает метил. Ацилирование (7) и (10) проводят в стандартных условиях, например, с помощью соединения R2COOH и реагентов, таких как DEC и НОВТ. Использование хирального соединения формулы 1, например, (S)-метил-4-замещенного бензиламина и хирального лактата на стадии а, например метил-(R)-лактаттрифторметансульфоната, приводит к получению хиральных соединений формул IA и IB.
Схема 2
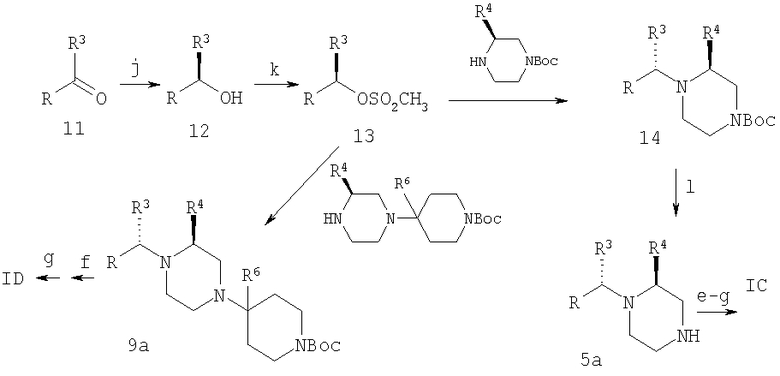
Реагенты: j: оксаборазолидин, ВН3; k: СН3SO2Cl, основание; l: CF3CO2H.
На Схеме 2 соединения получают с помощью процесса алкилирования с использованием предварительно полученного производного пиперазина. Например, предпочтительные соединения с S,S-конфигурацией по этой схеме можно получить путем хирального восстановления кетона (11) в спирт (12), его активирования путем превращения в метилсульфонилат и замещения с инверсией путем обработки подходящим пиперазином, который может содержать одну защитную группу, тогда окончательная обработка потребует удаления защитной группы с последующим проведением стадий (е)-(g), описанных на Схеме 1, с получением IC, или его можно обработать до проведения стадии замещения, тогда для получения ID конечными являются стадии (f) и (g) (удаление защитной группы и ацилирование), как на Схеме 1.
Схема 3
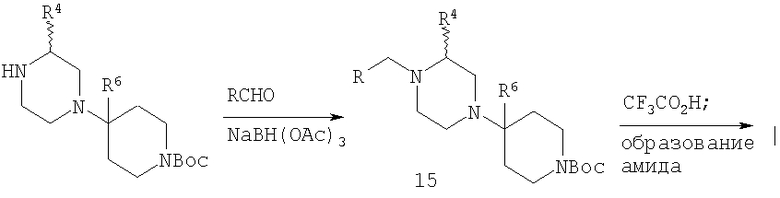
Для соединений, в которых R3 и R1 означают Н, можно использовать алкилирование, представленное на Схеме 2, или методику восстановительного аминирования, в качестве примера представленную на Схеме 3.
Схема 4
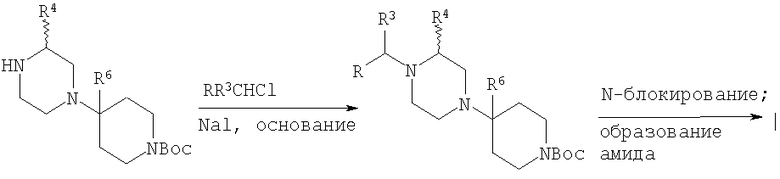
Для диарильных соединений, в которых R и R3 означают арил, предпочтительна методика алкилирования, в качестве примера представленная на Схеме 4.
Схема 5
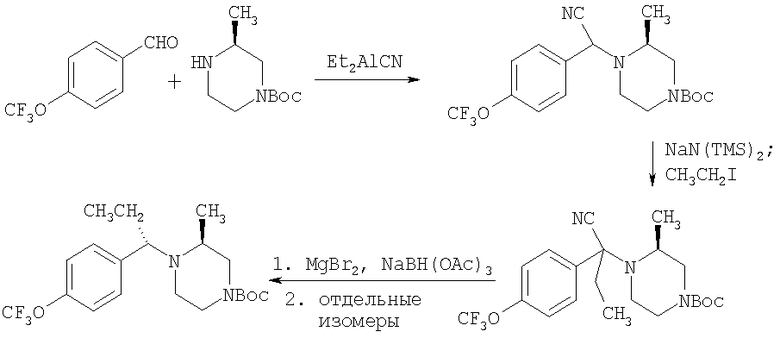
Пиперазины формулы 14, в частности, такие, в которых R3 означает (С1-С6)-алкил или бензил, также можно получить с помощью процесса, в котором фрагмент
вводят путем алкилирования-децианирования, как это показано выше. Реакция проиллюстрирована на примере соединений, в которых R означает CF3О-фенил, R1 означает водород, R3 означает этил и R4 означает метил, а с использованием соответствующих исходных соединений аналогичным образом можно получить другие соединения формулы 14.
Схема 6

Реагенты: m: ВОС2О, основание; n: R6MgBr; о: CCl3CO2Н, NaBH3CN; p: CF3CO2Н; q: NaBH4, BF3.
Как показано на Схеме 6, соединения, содержащие в пиперазиновом цикле дополнительную алкильную группу R5, можно получить из дикетопиперазиновых промежуточных продуктов (4), представленных на Схеме 1. Соединение (4) активируют путем превращения в N-(трет-бутоксикарбонильное) соединение (17); присоединение реагента Гриньяра и последовательное восстановление, удаление защитной группы и восстановление лактама дают (21), который можно использовать для получения соединений формулы I способом, описанным для промежуточного продукта (5) на Схеме 1.
Схема 7

Многие пиперазины, в которых R означает R8-фенил (или их Вос-производные), приведенные на Схеме 1, можно получить из общего промежуточного продукта, в котором R8 означает I. Несколько примеров показано на приведенной выше схеме, когда R8 превращают в Cl, CN, -C(O)NH2, H, Ph и п-ClC6H4CH2-. Подробные методики этих превращений представлены в приведенных ниже примерах. Затем полученный пиперазин или ВОС-пиперазин обрабатывают так, как это показано на Схеме 1.
Схема 8
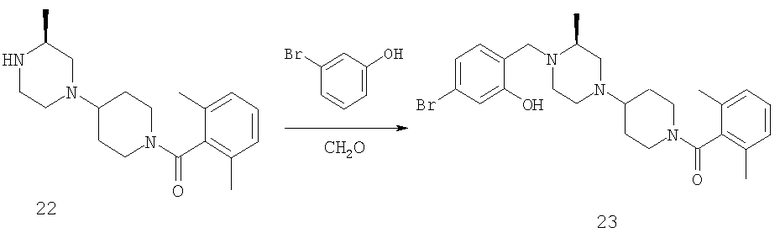
Некоторые соединения, соответствующие настоящему изобретению, можно получить по реакции Манниха, как это показано на примере, приведенном на Схеме 8.
Получение соединений, применимых в настоящем изобретении, пояснено с помощью последующих примеров получения, которые не следует считать ограничивающими объем настоящего изобретения. Для специалиста в данной области техники могут быть очевидны альтернативные пути и механизмы получения и аналогичные структуры, входящие в объем настоящего изобретения.
Пример 1
Стадия 1: Метил-R-лактат (5,0 г) перемешивают с CH2Cl2 (40 мл) при -70°C и прибавляют ангидрид трифторметансульфоновой кислоты (7,6 мл), а затем 2,6-лутидин (7,8 мл). Охлаждение прекращают, перемешивают 0,5 ч, промывают с помощью 2 н. HCl и раствор органических соединений прибавляют к (S)-метил-4-бромбензиламину (9,0 г) и K2СО3 (11,2 г) в воде (60 мл). Перемешивают 20 ч при комнатной температуре, органическую фазу сушат над K2СО3, выпаривают, хроматографируют на силикагеле с помощью Et2O-CH2Cl2 и получают искомый продукт (7,50 г) в виде густого масла.
Стадия 2: Продукт, полученный на стадии 1 (7,5 г) 5 ч кипятят с обратным холодильником в 1,2-дихлорэтане (40 мл) и ClCH2COCl (5,0 мл), затем выпаривают и полученный остаток без обработки используют на следующей стадии.
Стадия 3: Продукт, полученный на стадии 2, перемешивают с DMSO (80 мл), водой (10 мл) и NaI (8 г), охлаждают льдом, прибавляют концентрированный раствор NH4OH (15 мл) и 20 ч перемешивают при комнатной температуре. По каплям прибавляют воду (200 мл), собирают твердое вещество, тщательно промывают водой и сушат при 70°C/5 мм, получая дикетопиперазин, пригодный для использования на следующей стадии.
Стадия 4: Смесь продукта, полученного на стадии 3 (6,8 г), 1,2-диметоксиэтана (60 мл) и NaBH4 (3,4 г) перемешивают в атмосфере N2, по каплям прибавляют BF3·Et2O (6,8 мл), затем 10 ч нагревают при 100°С. Охлаждают и по каплям прибавляют СН3ОН (20 мл), а затем концентрированную HCl (30 мл). 1 ч нагревают при 100°С, охлаждают, подщелачивают избытком 2 н. NaOH и экстрагируют с помощью EtOAc. Сушат над K2СО3 и выпаривают, получая пиперазин (5,85 г), пригодный для использования на следующей стадии.
Стадия 5: Смесь продукта, полученного на стадии 4 (5,48 г), N-Boc-4-пиперидинона (4,32 г), НОАс (1,15 мл), CH2Cl2 (80 мл) и триацетоксиборогидрида натрия (NaBH(ОАс)3) (8,3 г) перемешивают при комнатной температуре в течение 20 ч. Медленно прибавляют избыток водного раствора Na2СО3, перемешивают 0,5 ч, разделяют и органическую фазу фильтруют через слой силикагеля, а для элюирования всего продукта промывают смесью CH2Cl2-Et2O состава 10:1. Выпаривают и остаток растворяют в Et2O (100 мл). Перемешивают и по каплям прибавляют 4 М раствор HCl в 1,4-диоксане (10 мл). Собирают твердое вещество, промывают с помощью Et2O и перемешивают с CH2Cl2 и избытком водного раствора NaOH. Органическую фазу сушат над K2СО3 и выпаривают, получая искомый продукт (5,45 г).
Стадия 6: Смесь продукта, полученного на стадии 5 (1,5 г), и TFA (4 мл) перемешивают при комнатной температуре в течение 2 ч. Выпаривают, растворяют в CH2Cl2 и промывают избытком 1 н. раствора NaOH. Сушат над K2СО3 и выпаривают, получая искомый продукт (1,15 г).
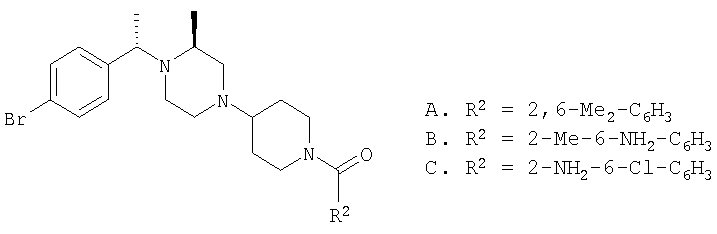
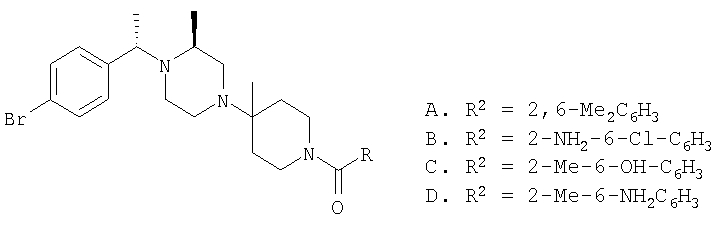
Соединение 1А: С помощью стандартной методики продукт, полученный на стадии 6, вводят в реакцию с 2,6-диметилбензоилхлоридом в CH2Cl2 и водным раствором NaOH и продукт превращают в гидрохлорид. Т.пл. (температура плавления): 185-192°С (с разложением). МСВР (масс-спектроскопия высокого разрешения): Найдено: 498,2130; МН+. Рассчитано: 498,2120.
Соединение 1В: С помощью стандартной методики продукт, полученный на стадии 6, вводят в реакцию сочетания с 2-амино-6-метилбензойной кислотой с использованием НОВТ и DEC с диизопропилэтиламином в DMF, очищают амид с помощью препаративной ТСХ и превращают в гидрохлорид. Т.пл.: 188-196°С (с разложением). МСВР: Найдено: 499,2069; МН+. Рассчитано: 499,2072.
Соединение 1С: С помощью описанной выше методики продукт, полученный на стадии 6, вводят в реакцию сочетания с 2-амино-6-хлорбензойной кислотой и после очистки превращают в гидрохлорид. Т.пл.: 192-200°С (с разложением). МСВР: Найдено: 519,1530; МН+. Рассчитано: 519,1526.
Пример 2
Стадия 1: Продукт, полученный в Примере 1, стадия 4 (1,00 г), N-трет-бутоксикарбонил-4-пиперидинон (0,77 г) и изопропоксид титана(IV) (Ti(OiPr)4) (1,00 г) в течение 20 ч перемешивают при комнатной температуре в CH2Cl2 (15 мл), 3 ч кипятят с обратным холодильником и охлаждают до комнатной температуры. Прибавляют диэтилалюминийцианид (Et2AlCN) (4,2 мл 1 М раствора в толуоле) и 5 суток перемешивают при комнатной температуре в атмосфере N2. Обрабатывают с помощью смеси CH2Cl2 - водный раствор NaOH, сушат и выпаривают органическую фазу и хроматографируют на силикагеле с использованием CH2Cl2-СН3ОН (100:1), получая искомый продукт (0,72 г).
Стадия 2: Продукт, полученный на стадии 1 (0,70 г), 20 ч перемешивают в сухом THF (15 мл) в атмосфере N2 с CH3MgBr (4 мл 3 М раствора в Et2O). Обрабатывают смесью EtOAc-вода и органическую фазу фильтруют через силикагель и промывают с помощью EtOAc. Выпаривают и получают искомый продукт (0,65 г).
Стадия 3: У продукта, полученного на стадии 2, с помощью TFA удаляют защитную группу в соответствии с методикой, описанной в Примере 1, стадия 6.
Соединение 2А: Продукт, полученный на стадии 3, вводят в реакцию с диметил-бензоилхлоридом, как это описано в Примере 1, и превращают в хлористоводородную соль. Т.пл.: 180-187°С (с разложением). МСВР: Найдено: 512,2272; МН+. Рассчитано: 512,2276.
Соединение 2В: Продукт, полученный на стадии 3, вводят в реакцию с 2-амино-6-хлорбензойной кислотой, как это описано в Примере 1, неочищенный продукт очищают с помощью препаративной ТСХ и превращают в хлористоводородную соль. Т.пл.: 195-200°С (с разложением). МСВР: Найдено: 535,1662; МН+. Рассчитано: 535,1652.
Соединение 2С: Продукт, полученный на стадии 3, вводят в реакцию с 2-гидрокси-6-метилбензойной кислотой, как это описано в Примере 1, неочищенный продукт очищают с помощью препаративной ТСХ и превращают в хлористоводородную соль. Т.пл.: 206-210°С (с разложением). МСВР: Найдено: 514,2067; МН+. Рассчитано: 514,2069.
Соединение 2D: Продукт, полученный на стадии 3, вводят в реакцию с 2-амино-6-метилбензойной кислотой по методике, аналогичной описанной в Примере 1, неочищенный продукт очищают с помощью препаративной ТСХ и превращают в хлористоводородную соль. Т.пл.: 202-209°С (с разложением). МСВР: Найдено: 513,2227; MH+. Рассчитано: 513,2229.
Пример 3
Стадия 1: Смесь гидрохлорида метилового эфира S-аланина (14 г), безводного Na2СО3 (60 г), сухого CH3CN (126 мл), хлордифенилметана (22,3 г) и NaI (6 г) в течение 6 ч при перемешивании кипятят с обратным холодильником. Охлаждают, прибавляют лед с водой и экстрагируют с помощью Et2O (350 мл, затем 50 мл). Экстракты в Et2O объединяют и промывают порциями 1 н. HCl: 200 мл, 100 мл, затем 4×10 мл. Водные кислотные экстракты объединяют, перемешивают и небольшими порциями прибавляют избыток Na2CO3, пока смесь не станет щелочной. Экстрагируют с помощью Et2O, сушат над MgSO4 и выпаривают, получая N-дифенилметильное соединение (23,2 г).
Стадия 2: Все полученное выше соединение 4 ч кипятят с обратным холодильником с ClCH2COCl (10 мл) в дихлорэтане (60 мл). Выпаривают и выпаривают совместно с толуолом (20 мл). Остаток растворяют в CH2Cl2 (200 мл), в течение 0,5 ч перемешивают с активированным углем (10 г), фильтруют и выпаривают. При охлаждении льдом остаток перемешивают с DMSO (200 мл) и постепенно прибавляют концентрированный водный раствор NH3 (100 мл), затем NaI (10 г). 20 ч перемешивают при комнатной температуре. Прибавляют воду, охлажденную льдом (500 мл), собирают твердое вещество, тщательно промывают водой, затем несколькими небольшими порциями смеси гексан-Et2O состава 10:1 и сушат в высоком вакууме при 50°С, получая твердый дикетопиперазин (15,5 г). Небольшую порцию перекристаллизовывают из смеси CH2Cl2-гексаны. Т.пл.: 186-188°С. [α]D 20=+272,6°.
Стадия 3: Продукт, полученный на стадии 2 (4,0 г), перемешивают в диметоксиэтане (1,6 г) и NaBH4 (1,6 г) в атмосфере N2 и медленно прибавляют BF3.Et2O (3,2 мл). 20 ч кипятят с обратным холодильником. Охлаждают и по каплям прибавляют СН3ОН (10 мл), а затем концентрированную HCl (15 мл). 2 ч кипятят с обратным холодильником, а затем обрабатывают избытком 2 н. водного раствора NaOH и экстрагируют с помощью CH2Cl2. Сушат над K2СО3 и выпаривают. Хроматографируют на силикагеле, элюируя смесями CH2Cl2-СН3ОН, а в заключение смесью CH2Cl2:СН3ОН:NH4OH состава 5:1:0,1 об./об./об. Экстракты объединяют и выпаривают фракции продукта, получая искомый продукт (1,95 г) в виде бледно-желтой смолы.
Стадия 4: Смесь продукта, полученного на стадии 3 (0,50 г), N-аллилоксикарбонил-4-пиперидона (0,40 г), CH2Cl2 (5 мл) и NaBH(OAc)3 (0,70 г) 20 ч перемешивают при комнатной температуре. Обрабатывают с помощью СН2Cl2 и избытка водного раствора NaOH, сушат над MgSO4, выпаривают и выделяют продукт с помощью препаративной ТСХ, элюируя с помощью 10% раствора Et2O в CH2Cl2, и получают искомое соединение (0,80 г) в виде масла, загрязненного небольшим количеством исходного кетона, но пригодного для использования на следующей стадии.
Стадия 5: Перемешивают смесь продукта, полученного на стадии 4 (0,80 г), СН3CN (20 мл), воды (5 мл) и пиперидина (1,5 мл). Прибавляют три-(4-сульфофенил)-фосфин (0,072 г) и палладий(II)ацетат (0,02 г) и 2 ч перемешивают при комнатной температуре в атмосфере N2. Обрабатывают водным раствором NaOH, экстрагируют смесью толуол: CH2Cl2 состава 5:1 об./об., сушат над K2СО3 и выпаривают, получая желтое масло, пригодное для ацилирования.
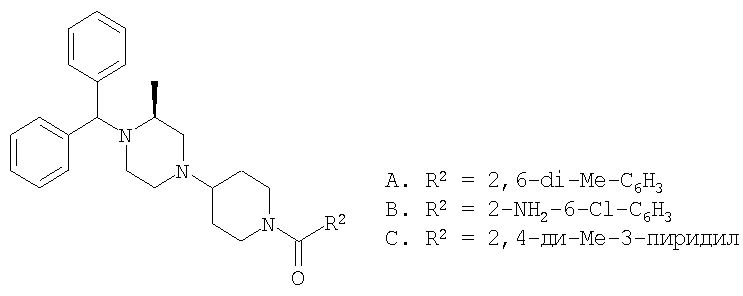
Соединение 3А: Смесь продукта, полученного на стадии 5 (0,10 г), N-(2,6-диметоксибензоил)-4-пиперидинона (0,10 г), CH2Cl2 (2 мл) и NaBH(ОАс)3 (0,15 г) в течение 2,5 ч при перемешивании кипятят с обратным холодильником, охлаждают и обрабатывают с помощью CH2Cl2 и водного раствора NaOH. Сушат над MgSO4, выпаривают и с помощью препаративной ТСХ выделяют основной продукт, элюируя смесью Et2O:СН2Cl2 состава 3:1 об./об. Осаждают гидрохлорид, получая искомое соединение в виде хлористоводородной соли (0,13 г). Т.пл.: 173-177°С (с разложением). МСВР: Найдено: 482,3175; МН+. Рассчитано: 482,3171.
Соединение 3В: Продукт, полученный на стадии 5, вводят в реакцию сочетания с 2-амино-6-метилбензойной кислотой с использованием DEC-HOBT, как это описано в Примере 1, выделяют продукт с помощью препаративной ТСХ и осаждают гидрохлорид, получая соединение 3В. Т.пл.: 188-195°С (с разложением). МСВР: Найдено: 503,2567; МН+. Рассчитано: 503,2578.
Соединение 3С: Продукт, полученный на стадии 5, вводят в реакцию сочетания с 2-амино-6-метилбензойной кислотой с использованием DEC-HOBT, как это описано выше, выделяют продукт с помощью препаративной ТСХ и осаждают гидрохлорид, получая соединение 3С. Т.пл.: 180-188°С (с разложением). МСВР: Найдено: 483,3114; МН+. Рассчитано: 483,3124.
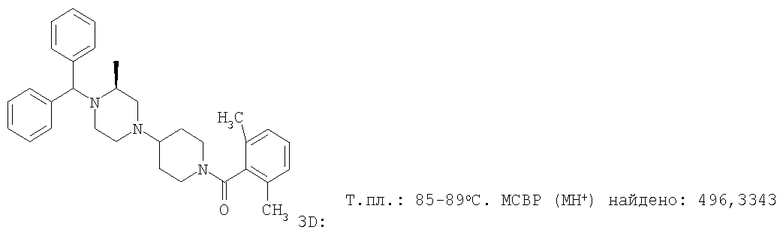
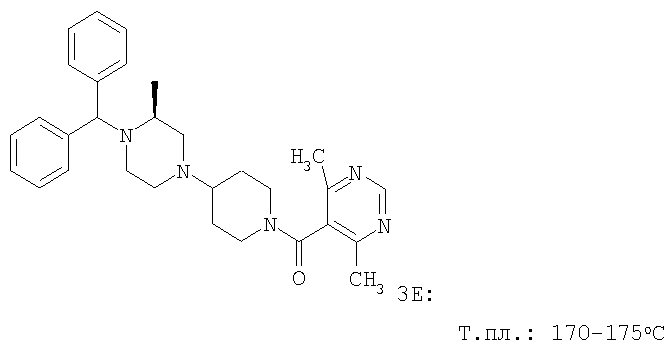
С помощью методик, аналогичных описанным выше, получают следующие соединения:
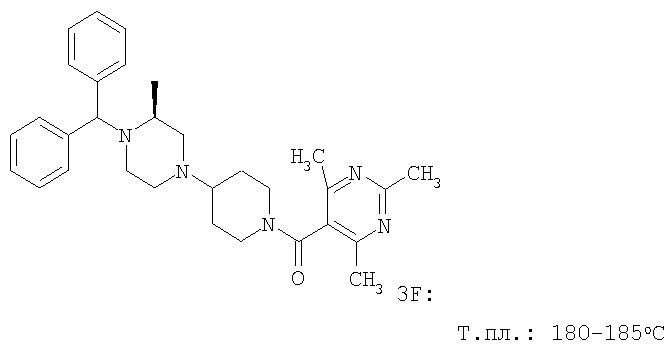
Пример 4
Стадия 1: Раствор 4-трифторметилацетофенона (1,88 г; 10 ммоль) в сухом THF (10 мл) охлаждают в бане со льдом и обрабатывают свежеприготовленным (S)-2-метилоксаборолидином (0,54 г; 2 ммоль). Через 10 мин в течение 5 мин по каплям прибавляют 2 М раствор комплекса боран-метилсульфид (3 мл; 6 ммоль) в THF. По истечении 30 мин ТСХ показывает, что исходное вещество превращено в более полярный продукт. Реакцию осторожно останавливают с помощью примерно 5 мл МеОН, прибавляя его до прекращения выделения пузырьков; летучие вещества удаляют в вакууме. Остаток растворяют в CH2Cl2 и промывают 1 н. HCl, водой, 10% раствором NaHCO3 и солевым раствором. Концентрирование в вакууме дает 2 г желтой смолы. С помощью флэш-хроматографии на силикагеле (ФХСГ) с использованием 10-20% раствора EtOAc в гексанах получают искомый хиральный спирт (1,6 г; 84%) в виде бесцветного масла. ТСХ, Rf=0,6 в 25% EtOAc: гексаны.
Стадия 2: К раствору продукта, полученного на стадии 1 (1,55 г; 8,16 ммоль), в 10 мл CH2Cl2, охлажденному в бане со льдом, прибавляют Et3N (2,3 мл; 16,32 ммоль) и CH3SO2Cl (0,87 мл; 10,6 ммоль) и получают белый мутный раствор. Реакцию останавливают водой и органическое вещество экстрагируют с помощью CH2Cl2, промывают водой, 1 н. HCl, 10% раствором NaHCO3 и солевым раствором. Концентрирование в вакууме дает хиральный метилсульфонилат (2,1 г; 96%) в виде бледно-желтого масла. ТСХ, Rf=0,6 в 25% EtOAc: гексаны.
Стадия 3: Раствор продукта, полученного на стадии 2 (2,1 г; 7,8 ммоль), 2(S)-метилпиперазина, в который с помощью N-BOC введена защитная группа [1,56 г; 7,8 ммоль - полученного по реакции продажного 2(S)-метилпиперазина с N-(трет-бутоксикарбонилокси)-фталимидом] и 2,2,6,6-тетраметилпиперидина (1,34 мл; 8 ммоль) в 14 мл сухого CH3CN кипятят с обратным холодильником, пока по данным ТСХ не произойдет полное исчезновение метилтолуолсульфоната (16 ч). Реакционную смесь охлаждают до комнатной температуры, разбавляют с помощью СН2Cl2 (50 мл) и промывают водой (3×100 мл) и солевым раствором. Органические экстракты сушат над твердым MgSO4, а затем концентрируют, получая 2,8 г желтой смолы. С помощью ФХСГ (20% раствор EtOAc в гексанах) выделяют искомый (S,S)-диастереоизомер (1,5 г; 52%) и его бензильный эпимер, (R,S)-диастереоизомер (0,5 г; 17%) с суммарным выходом 65%. ТСХ, Rf=0,75 (S,S) и 0,56 (R,S) в 25% EtOAc: гексаны.
Стадия 4: К раствору продукта, полученного на стадии 3, в 12 мл CH2Cl2 прибавляют TFA (6 мл) и полученный желто-оранжевый раствор 8 ч перемешивают при комнатной температуре. Реакцию останавливают путем прибавления 1 н. раствора NaOH, доводя значение рН до 10. Экстракция с помощью CH2Cl2 дает 1,1 г желтого сиропа. С помощью ФХСГ с использованием 10% раствора СН3ОН в CH2Cl2 удаляют менее полярную примесь, а для элюирования искомого свободного амина (S,S)-диастереоизомера необходимо градиентное элюирование с использованием 1% раствора Et3N в 10% СН3ОН: CH2Cl2. Выход =0,9 г (75%). ТСХ, Rf=0,5 в 10% СН3ОН: CH2Cl2.
Стадия 5: Бесцветный раствор продукта, полученного на стадии 1 (0,9 г; 3,3 ммоль), 4-пиперидинона (0,86 г; 4,3 ммоль), NaB(OAc)3H (1,05 г; 4,95 ммоль) и ледяной АсОН (80 мкл) в 8 мл CH2Cl2 один день перемешивают при комнатной температуре. ТСХ указывает на отсутствие исходного материала. Реакционную смесь разбавляют с помощью 50 мл СН2Cl2, промывают 1 н. раствором NaOH, водой (дважды) и солевым раствором. Экстракт в СН2Cl2 сушат над безводным MgSO4 и концентрируют, получая 1,7 г желтого масла. Для выделения чистого продукта (1,3 г; 86%) в виде белой пены используют ФХСГ (25% раствор ацетона в гексанах). ТСХ, Rf=0,6 в 25% ацетон/гексаны.
Стадия 6: К раствору продукта, полученного на стадии 5 (1,3 г; 2,87 ммоль), в CH2Cl2 (10 мл) прибавляют TFA (5 мл) и полученный желто-оранжевый раствор 7 ч перемешивают при комнатной температуре. Реакцию останавливают с помощью 1 н. раствора NaOH, доводя значение рН до 10. Органический продукт экстрагируют с помощью 50 мл CH2Cl2, промывают водой, затем солевым раствором и сушат над MgSO4. Концентрирование дает свободный амин (0,98 г; 98%) в виде желтого сиропа. ТСХ, Rf=0,1 в 25% ацетон/гексаны.
Стадия 7: Продукт, полученный на стадии 6 (0,78 г; 2,21 ммоль), DEC (0,65 г; 3,4 ммоль), НОВТ (0,46 г; 3,4 ммоль) и 2-амино-6-хлорбензойную кислоту (0,51 г; 2,9 ммоль) растворяют в 8 мл СН2Cl2, к которому прибавлен диизопропилэтиламин (0,7 мл), и смесь 16 ч перемешивают при комнатной температуре. Анализ с помощью ТСХ указывает на отсутствие исходного вещества и обнаруживает два перекрывающихся пятна соединений средней полярности (ротамеры пространственно затрудненного амида), являющихся основным продуктом. Неочищенный продукт (1,3 г) выделяют с помощью экстракции и очищают с помощью ФХСГ с использованием в качестве элюента 25% раствора ацетона в CH2Cl2, получая искомое соединение (0,88 г; 80%) в виде бледно-желтой пены. ТСХ, Rf=0,45 и 0,5 в 25% ацетон/гексаны.
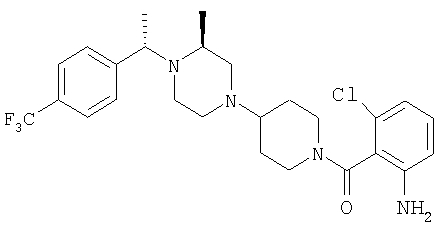
К раствору свободного основания искомого соединения (0,76 г; 1,54 ммоль) в CH2Cl2 (5 мл) прибавляют раствор хлористого водорода в Et2O (1 М; 3 мл) и мгновенно получают белый осадок. После перемешивания при комнатной температуре в течение 2 ч летучие вещества удаляют на роторном испарителе и белый осадок суспендируют в сухом толуоле (3×10 мл) и подвергают азеотропной перегонке. Полученное таким образом белое вещество суспендируют в сухом Et2O, содержащем 10% EtOAc, перемешивают 30 мин, фильтруют и промывают с помощью Et2O (100 мл). Хлористоводородную соль искомого соединения сушат в высоком вакууме и получают почти белое твердое вещество (0,88 г; 95%). Т.пл.: 205-210°С.
С использованием соответствующих карбоновых кислот продукт, полученный на стадии 6, превращают в другие амиды (4А-4Е), как это описано на стадии 7. Ниже приведены физико-химические характеристики соединений 4-4Е, обладающих следующей структурой:
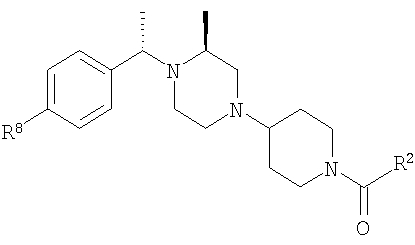
где R8 и R2 являются такими, как указано в таблице: 1

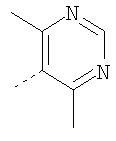
Пример 5
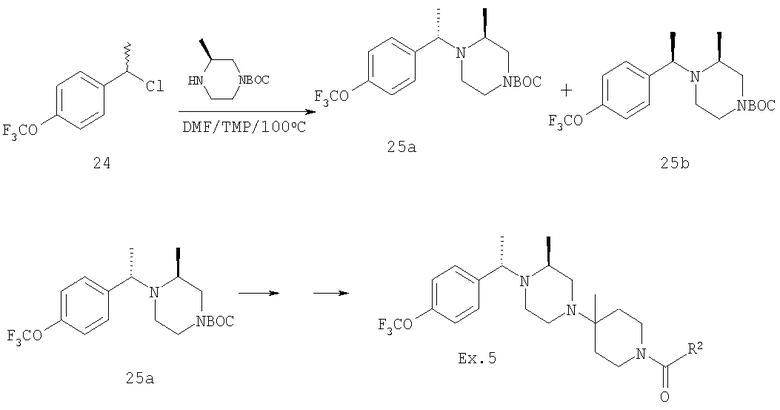
Раствор рацемического бензилхлорида 24 (1,26 г; 5,62 ммоль), который непосредственно перед использованием получают из соответствующего карбинола, 2(S)-метилпиперазин (1,12 г; 5,62 ммоль) и 2,2,6,6-тетраметилпиперидин (ТМР) (1,9 мл; 11,2 ммоль) растворяют в сухом DMF (2 мл) и 10 ч нагревают при 100-110°C (внутренняя температура). Анализ с помощью ТСХ указывает на отсутствие 24 и образование двух хорошо разделяющихся продуктов. Смесь разбавляют водой и органические вещества экстрагируют с помощью Et2O. Органические экстракты промывают насыщенным раствором NH4Cl и солевым раствором и концентрируют в вакууме, получая 2 г неочищенного продукта. Флэш-хроматография на силикагеле и элюирование сначала с помощью 25% Et2O-гексан, а затем с помощью 25% EtOAc-гексан дает ˜0,5 г 25а и ˜0,5 г 25b соответственно (суммарный выход ˜45%). ТСХ, Rf=0,6 (для 25а) и 0,4 (для 25b) в 25% EtOAc-гексаны. Очистку 25а проводят так, как это описано выше, и получают конечные продукты 5-5F, обладающие формулой
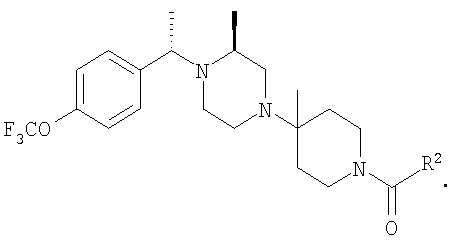
где R2 являются такими, как указано в таблице: 2

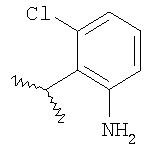
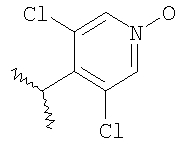
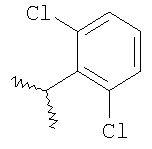
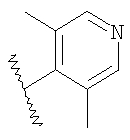

Пример 6
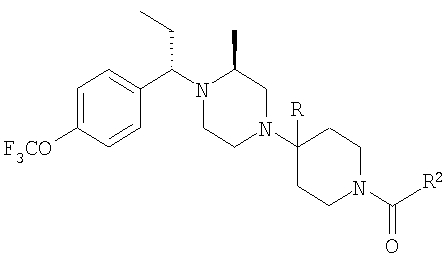
Стадия 1:
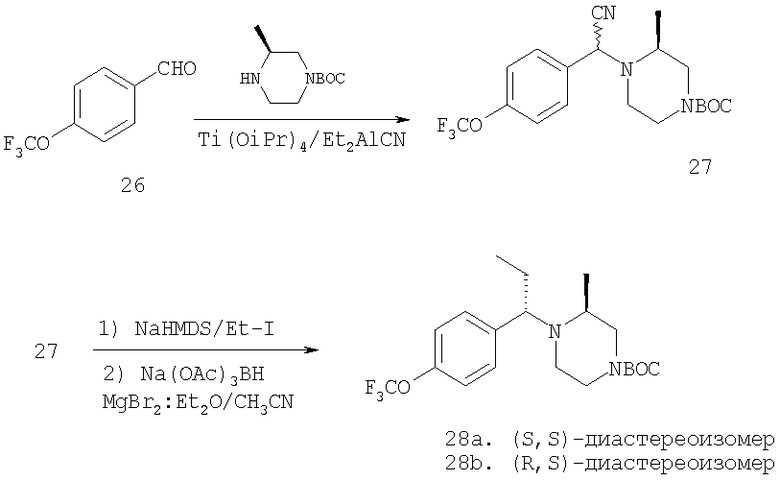
Смесь альдегида 26 (3,9 г; 20,5 ммоль), 2(S)-метил-N-ВОС-пиперазина (4,1 г; 20,5 ммоль) и Ti(OiPr)4 (6,1 мл; 20,5 ммоль) в 40 мл CH2Cl2 перемешивают в течение 24 ч при комнатной температуре. Прибавляют Et2AlCN и перемешивают еще день. Реакционную смесь обрабатывают так, как это описано выше, и после обработки с помощью ФХСГ получают 4,71 г (58%) цианамида 27 (ТСХ, Rf=0,45/0,5 для диастереоизомеров, получаемых с использованием 25% Et2O-гексаны в качестве растворителя).
Стадия 2: К раствору 27 (1 г; 2,5 ммоль) в сухом THF, охлаждаемую на бане твердая углекислота/ацетон, прибавляют гексаметилдисилазид натрия (1 М; 3,1 мл). Полученный ярко-желтый раствор обрабатывают с помощью СН3СН2I (7,5 ммоль; 0,6 мл). Баню с твердой углекислотой удаляют и реакционную смесь 15 мин перемешивают при комнатной температуре, а затем 30 мин осторожно нагревают на теплой водяной бане (40°С). С помощью ТСХ обнаруживаются два хорошо разделенных пятна. Стандартная экстракция и очистка с помощью ФХСГ дает два алкилированных соединения (суммарный выход 0,7 г; 70%). ТСХ, Rf=0,6 и 0,4 (25% EtOAc/гексаны).
Стадия 3: Продукт, полученный на стадии 2, в течение дня перемешивают с NaBH(ОАс)3 (2х) и MgBr2:Et2O (lx) в CH3CN. Реакцию останавливают водой, органические вещества экстрагируют с помощью EtOAc и обрабатывают, получая 0,8 г неочищенного продукта. ФХСГ (25% EtOAc-гексаны) дает ˜0,4 г каждого диастереоизомера (суммарный выход ˜100%). ТСХ, Rf=0,55 (28а) и 0,45 (28b) в 25% EtOAc-гексаны.
Стадия 4: Соединение 28а (S,S-диастереоизомер) обрабатывают с помощью обычной 5-стадийной схемы и завершают синтез соединений Примера 6, 6А и 6В с метильной группой в ипсо-положении, а также соединений 6С и 6D без метильной группы в ипсо-положении:
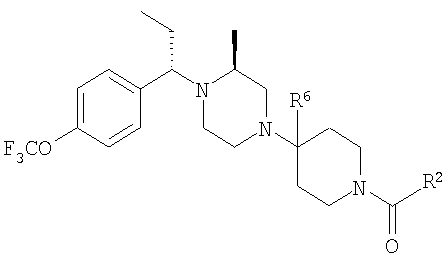
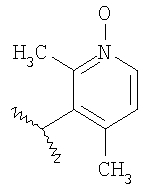

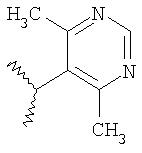
Пример 7
Синтез соединений с алкил- или арилсульфонильной группой R8 в параположении начинают с соответствующего паразамещенного ацетофенона, который обрабатывают, как в Примере 4, стадии 1-6, и получают содержащие сульфоновую группу соединения Примера 7, обладающие формулой:
где R8 и R2 являются такими, как указано в таблице: 4
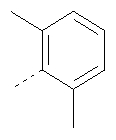
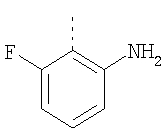
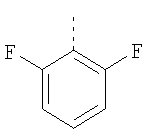
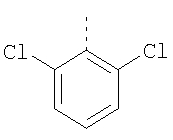
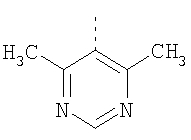
Пример 8
Стадия 1: Раствор продукта, полученного в Примере 4, стадия 4 (1,25 г; 4,6 ммоль), N-BOC-4-пиперидинона (0,91 г; 4,6 ммоль) и Ti(OiPr)4 (1,4 мл; 4,6 ммоль) в 10 мл CH2Cl2 перемешивают при температуре окружающей среды в течение 24 ч. Затем реакционную смесь обрабатывают с помощью Et2AlCN (5,5 мл; 1 М раствор в толуоле) и продолжают перемешивание в течение 20 ч. Реакционную смесь разбавляют с помощью EtOAc и перемешивают с насыщенным раствором NaHCO3 (10 мин) и слои разделяют как можно быстрее. Мутный (от наличия неотделенного водного слоя) органический слой обрабатывают избытком целита и фильтруют, промывая осадок на фильтре с помощью EtOAc. Профильтрованные слои разделяют и органический слой промывают водой и солевым раствором, сушат над безводным MgSO4 и концентрируют, получая 2,16 г (98%) смолы янтарного цвета.
Стадия 2: Полученный на стадии 1 амин Штрекера (2,16 г) растворяют в сухом THF, охлаждают в бане со льдом и обрабатывают с помощью CH3MgBr (7,5 мл 3 М раствора в Et2O). Через 1 ч баню со льдом удаляют и желтую гетерогенную реакционную смесь 18 ч перемешивают при комнатной температуре. Реакцию останавливают насыщенным раствором NH4Cl, реакционную смесь разбавляют водой и экстрагируют с помощью CH2Cl2. Концентрирование дает 2,2 г желтой смолы, которую очищают с помощью ФХСГ, отделяя основной продукт от более полярных примесей путем элюирования смесью CH2Cl2:EtOAc состава 1:1. ипсометильное соединение выделяют в виде желтой смолы (1,85 г; 88%). ТСХ, Rf=0,5 в смеси Et2O: гексаны состава 1:1.
Стадия 3: К раствору продукта, полученного на стадии 2 (1,5 г; 3,2 ммоль), в 10 мл CH2Cl2 прибавляют TFA (6 мл) и 2 ч перемешивают при 25°С. Реакцию останавливают с помощью 1 н. раствора NaOH, доводя значение рН до 9-10, и экстрагируют с помощью CH2Cl2, получая 1,2 г неочищенного продукта. С помощью ФХСГ с использованием смеси CH2Cl2:EtOAc состава 1:1 удаляют все менее полярные примеси, а посредством градиентного элюирования с помощью 10% раствора СН3ОН в CH2Cl2, а в конце - помощью 10% (примерно 7 н. NH3) раствора СН3ОН в CH2Cl2 выделяют свободный пиперидин в виде желтой смолы (1,07 г; 90%). ТСХ, Rf=0,2 в 10% СН3ОН: CH2Cl2.
Стадия 4: Раствор продукта, полученного на стадии 3 (1,03 г; 2,8 ммоль), 2,3-диметилникотиновой кислоты (0,42 г; 2,8 ммоль), DEC (0,8 г; 4,2 ммоль), НОВТ (0,57 г; 4,2 ммоль) и диизопропилэтиламина (1 мл; 5,6 ммоль) в СН2Cl2 (15 мл) перемешивают 24 ч при 25°С. Реакционную смесь разбавляют с помощью CH2Cl2 (25 мл), промывают водой, 10% раствором NaHCO3 и солевым раствором, а затем концентрируют, получая 1,6 г неочищенного масла. Очистка этого вещества посредством ФХСГ с использованием градиентного элюирования с помощью 10% ацетон - СН2Cl2, а затем с помощью 2-5% СН3ОН в CH2Cl2 дает искомое соединение (1,1 г; 80%) в виде белой пены. ТСХ, Rf=0,45 в 5% СН3ОН-СН2Cl2.
Выделенное выше свободное основание искомого соединения (1 г; 2 ммоль) растворяют в смеси EtOAc:Et2O состава 1:1 (8 мл) и прибавляют свежеприготовленный раствор хлористого водорода в Et2O (6,1 мл 1 М раствора), что немедленно приводит к образованию белого осадка. После перемешивания в течение 1 ч при 25°C летучие вещества удаляют в вакууме. Продукт суспендируют в Et2O и фильтруют, промывая фильтрат с помощью Et2O. Полученную таким образом соль хлористоводородной искомого соединения сушат в вакууме (1,1 г; Т.пл.: 213-215°С). МСВР (МН+): 503,2997.
Указанные ниже амиды 8А-8Е получены аналогичным образом из продукта, полученного на стадии 3, с использованием соответствующих кислот и аналогичным образом получены соединения 8F-8Н, в которых заместитель R8 означает п-метилсульфонильную группу.
где R8 и R2 являются такими, как указано в таблице: 5




С помощью методик, приведенных после таблицы, получают соединения 8S-8ЕЕ, обладающие структурой
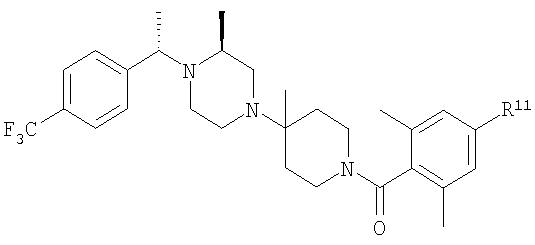
где R11 определен в таблице: 6

8S: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (75 мг, 0,16 ммоль), EDC (61 мг, 0,32 ммоль), НОВТ ((49 мг, 0,32 ммоль), iPr2NEt (0,16 мл, 0,96 ммоль) и 2,6-диметил-4-гидроксибензойную кислоту (53 мг, 0,32 ммоль) смешивают с CH2Cl2 и 20 ч перемешивают при 25°C. Раствор концентрируют. Очистка с помощью препаративной ТСХ (EtOAc, SiO2) дает искомое соединение в виде желтого масла. Т.пл. (2×HCl соль): 210-220°С. МСВР (МН+): Рассчитано для C29H39O2N3F3, 518,2994; Найдено, 518,2997.
8Т: 8S (100 мг, 0,19 ммоль), этилизоцианат (0,05 мл, 0,58 ммоль) и Et3N (0,13 мл, 0,95 ммоль) смешивают с СН2Cl2 и 16 ч перемешивают при 25°С. Раствор разбавляют с помощью CH2Cl2 и промывают с помощью 1 н. NaOH. Органический слой сушат (Na2SO4), фильтруют и концентрируют. Очистка с помощью препаративной ТСХ (2/1 EtOAc/гексаны, SiO2) дает искомое соединение в виде желтого масла.
8U: 8S (250 мг, 0,48 ммоль), метансульфонильный ангидрид (250 мг, 1,44 ммоль) и NaH (38 мг, 60 мас.% в масле) смешивают с THF и 20 ч перемешивают при 25°С. Раствор разбавляют с помощью EtOAc и промывают насыщенным раствором NaHCO3. Органический слой сушат (Na2SO4), фильтруют и концентрируют. Очистка с помощью препаративной ТСХ (1/1 EtOAc/гексаны, SiO2) дает искомое соединение в виде желтого масла (280 мг, 98%).
8V: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (50 мг, 0,1 ммоль), EDC (38 мг, 0,2 ммоль), НОВТ (27 мг, 0,2 ммоль), iPr2NEt (0,07 мл, 0,4 ммоль) и 2,6-диметил-4-(4-пиридил-N-оксид)-бензойную кислоту (73 мг, 0,3 ммоль) (описание методики получения см. ниже) смешивают с CH2Cl2 и 19 ч перемешивают при 25°С. Раствор концентрируют. Очистка с помощью препаративной ТСХ (2/1 EtOAc/гексаны, SiO2) дает 8V в виде желтого масла.
Получение 2,6-диметил-4-(4-пиридил-N-оксид)-бензойной кислоты
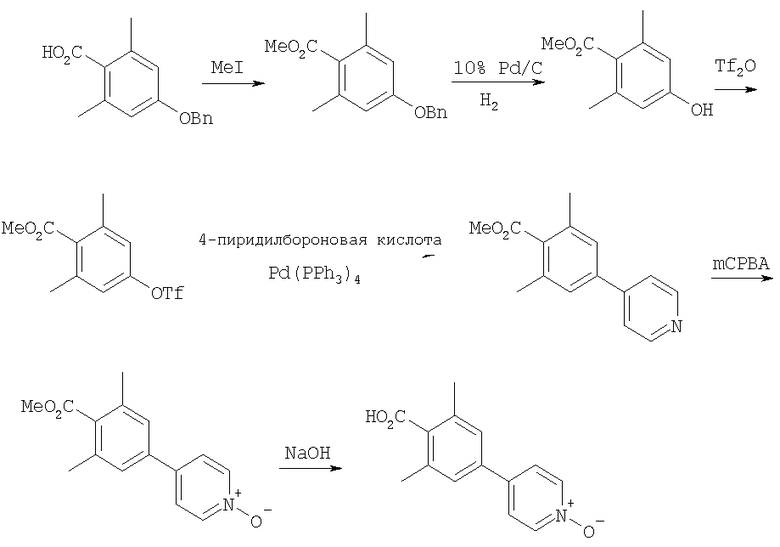
Стадия A: 4-Бензилокси-2,2-диметилбензойную кислоту (8,7 г, 34 ммоль; Thea, S. et al. Journal of the American Chemical Society 1985, 50, 1867), MeI (3,2 мл, 51 ммоль) и Cs2CO3 (17 г, 51 ммоль) в течение 17 ч перемешивают в DMF при 25°С. Раствор фильтруют и подвергают распределению между Et2O и водой. Водный слой экстрагируют с помощью Et2O. Объединенные слои, содержащие Et2O, промывают водой и солевым раствором. Органический слой сушат (MgSO4), фильтруют и концентрируют. Очистка с помощью флэш-хроматографии (10/1 гексаны/Et2O, SiO2) дает 8,6 г (94%) метилового эфира в виде бесцветного масла.
Стадия B: Защищенный бензильной группой фенол (8,5 г, 32 ммоль) и Pd/C (750 мг, 10 мас.% Pd) смешивают с СН3ОН. В раствор подают Н2 под давлением 50 фунт-сила/дюйм2 и при 25°C в течение 17 ч встряхивают в приборе Парра. Раствор фильтруют (через целит). Концентрированно дает 5,6 г (98%) фенола в виде белого твердого вещества.
Стадия C: Фенол (3,5 г, 19,4 ммоль) и iPr2NEt (3,76 г, 29,1 ммоль) растворяют в CH2Cl2 при 0°С. К этому раствору при 0°С по каплям прибавляют ангидрид трифторметансульфокислоты (Tf2O) (4,2 мл, 25,2 ммоль). Раствор нагревают до 25°C и при этой температуре перемешивают 4,5 ч. Раствор разбавляют с помощью CH2Cl2 и промывают насыщенным раствором NaHCO3. Водный слой экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат над Na2SO4. Фильтрование и концентрированно дает неочищенный арилтрифторметансульфонат. Очистка с помощью флэш-хроматографии (10/1 гексаны/Et2O, SiO2) дает 5,7 г (94%) трифторметансульфоната в виде желтого масла.
Стадия D: Трифторметансульфонат (1 г, 3,2 ммоль), 4-пиридинбороновую кислоту (1,2 г, 9,6 ммоль), Pd(PPh3)4 (370 мг, 0,32 ммоль) и Na2CO3 (1 г, 9,6 ммоль) смешивают с DME/H2O (4/1, 25 мл). Раствор 18 ч нагревают в атмосфере N2 при 90°С (масляная баня). Раствор подвергают распределению между EtOAc и Н2О. Водный слой экстрагируют с помощью EtOAc. Объединенные слои, содержащие EtOAc, сушат (Na2SO4). Фильтрование и концентрирование дает темно-коричневое масло. Очистка с помощью флэш-хроматографии (3/1 гексаны/Et2O, SiO2) дает 770 мг (100%) пиридильного производного в виде оранжевого масла.
Стадия Е: Пиридильное производное (390 мг, 1,6 ммоль) и mCPBA (550 мг, 3,2 ммоль) растворяют в CH2Cl2. Раствор 18 ч перемешивают при 25°С. Раствор разбавляют с помощью CH2Cl2 и промывают с помощью 1 н. NaOH. Органический слой сушат (Na2SO4). Фильтрование и концентрирование дает 400 мг (97%) N-оксида в виде оранжевого масла. МСВР (МН+): Рассчитано для С15Н16О3Н, 258,1130; Найдено, 258,1131.
Стадия F: Метиловый сложный эфир (400 мг, 1,6 ммоль) смешивают с 5 мл 3 н. NaOH и 2 мл EtOH. Раствор 20 ч кипятят с обратным холодильником. Раствор концентрируют. Остаток обрабатывают с помощью концентрированной HCl. Полученное твердое вещество отфильтровывают и промывают водой и солевым раствором. После сушки в высоком вакууме получают свободную кислоту (377 мг, 100%) в виде желтовато-коричневого твердого вещества. Т.пл.: >225°С (с разложением). Рассчитано для С14Н14О3N, 244,0974; Найдено, 244, 0981.
8W: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (1,34 г, 2,8 ммоль), 2,6-диметил-4-формилбензойную кислоту (500 мг, 2,8 ммоль) (описание методики получения см. ниже), EDC (1,1 г, 5,6 ммоль), НОВТ (760 мг, 5,6 ммоль) и iPr2NEt (2 мл, 11 ммоль) вводят в реакцию сочетания при стандартных условиях. Очистка с помощью флэш-хроматографии (2/1 гексаны/EtOAc, SiO2) дает 898 мг (61%) 8W в виде желтой пены.
Получение 2,6-диметил-4-формилбензойной кислоты
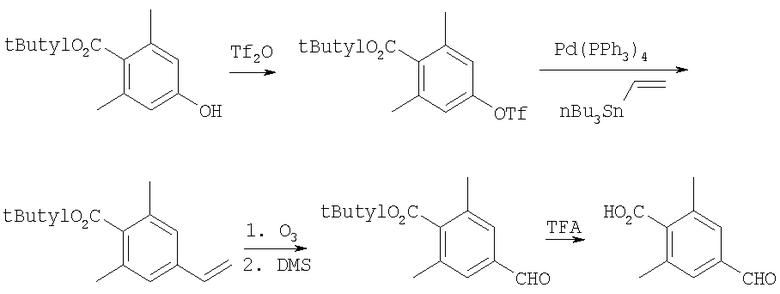
Стадия A: трет-Бутиловый эфир 4-гидрокси-2,6-диметилбензойной кислоты (6,4 г, 29 ммоль) и iPr2NEt (5,6 г, 43 ммоль) смешивают с СН2Cl2 и охлаждают до 0°С. К этому раствору при 0°С медленно прибавляют Tf2O (5,8 мл, 34 ммоль). Раствор 3 ч перемешивают при 0°С. Раствор подвергают распределению между насыщенным раствором NaHCO3 и СН2Cl2. Водный слой экстрагируют с помощью СН2Cl2. Объединенные органические слои сушат (Na2SO4). Фильтрование и концентрирование дает коричневое масло. Очистка с помощью флэш-хроматографии (20/1 гексаны/Et2О, SiO2) дает 7,99 г (82%) трифторметансульфоната виде желтого твердого вещества.
Стадия B: Трифторметансульфонат (5 г, 15 ммоль), LiCl (1,25 г, 30 ммоль), Pd(PPh3)4 (340 мг, 0,3 ммоль) и винилтрибутилолово (4,5 мл, 16 ммоль) смешивают с THF в атмосфере N2. Раствор 16 ч нагревают при 70°С. Раствор подвергают распределению между EtOAc и насыщенным раствором KF. Смесь фильтруют. Органический слой отделяют и водные слои экстрагируют с помощью EtOAc. Объединенные органические слои сушат (MgSO4). Фильтрование и концентрирование дает желтое масло. Очистка с помощью флэш-хроматографии (20/1 гексаны/Et2O, SiO2) дает 1,96 г (57%) олефина в виде желтого масла.
Стадия C: Олефин (0,6 г, 2,6 ммоль) смешивают с CH2Cl2/MeOH (1/1). Раствор охлаждают до -78°С. Через раствор барботируют озон, пока не сохранится темно-синяя окраска. Реакцию останавливают диметилсульфидом. Реакционную смесь концентрируют и получают альдегид в виде масла.
Стадия D: трет-Бутиловый сложный эфир (650 мг, 2,8 ммоль) и TFA (3 мл) смешивают с CH2Cl2 и 19 ч перемешивают при 25°С. Концентрированно раствора дает кислоту в виде бежевого твердого вещества.
8Х: 8W (100 мг, 0,19 ммоль), H2NOMe-HCl (28 мг, 0,34 ммоль), NaOAc (32 мг, 0,46 ммоль) смешивают в МеОН. Раствор 17 ч перемешивают при 25°С. Раствор концентрируют. Остаток подвергают распределению между СН2Cl2 и 1 н. NaOH. Водный слой экстрагируют с помощью СН2Cl2. Объединенные органические слои сушат (Na2SO4). Фильтрование и концентрирование дает неочищенный продукт. Очистка с помощью препаративной ТСХ (1/1 гексаны/EtOAc, SiO2) дает 85 мг 8Х (84%).
8Y: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (75 мг, 0,16 ммоль), и 4-дифторметил-2,6-диметилбензойную кислоту (32 мг, 0,16 ммоль) вводят в реакцию сочетания при стандартных условиях (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (2/1 гексаны/EtOAc, SiO2) дает 64 мг 8Y (73%).
Получение 4-дифторметил-2,6-диметилбензойной кислоты

Стадия A: Альдегид (400 мг, 1,7 ммоль), [бис-(2-метоксиэтил-амино]-трифторид серы (640 мг, 2,9 ммоль) и EtOH (0,02 мл, 0,34 ммоль) смешивают с 1,2-дилхорэтаном и перемешивают 6 ч при 65°С и 19 ч при 25°С. Реакцию останавливают с помощью насыщенного раствора NaHCO3. Водный слой экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат (Na2SO4). Фильтрование и концентрирование дает неочищенный продукт. Очистка с помощью препаративной ТСХ (10/1 гексаны/Et2O, SiO2) дает 210 мг (50%) дифторпроизводного.
Стадия B: трет-Бутиловый сложный эфир (210 мг, 0,82 ммоль) и HCl (2,1 мл 4 М раствора в диоксане, 8,2 ммоль) смешивают с МеОН. Раствор 20 ч перемешивают при 45°С. Раствор концентрируют и получают кислоту в виде белого твердого вещества.
8Z: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (811 мг, 1,7 ммоль), и 4-[(этиламино)-карбониламино]-2,6-диметилбензойную кислоту (400 мг, 1,7 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания при стандартных условиях (EDC/HOBT/iPr2NEt). Очистка с помощью флэш-хроматографии (1/1 гексаны/ацетон, SiO2) дает 803 мг (81%) 8Z в виде пены.
Получение 4-[(этиламино)-карбониламино]-2,6-диметилбензойной кислоты
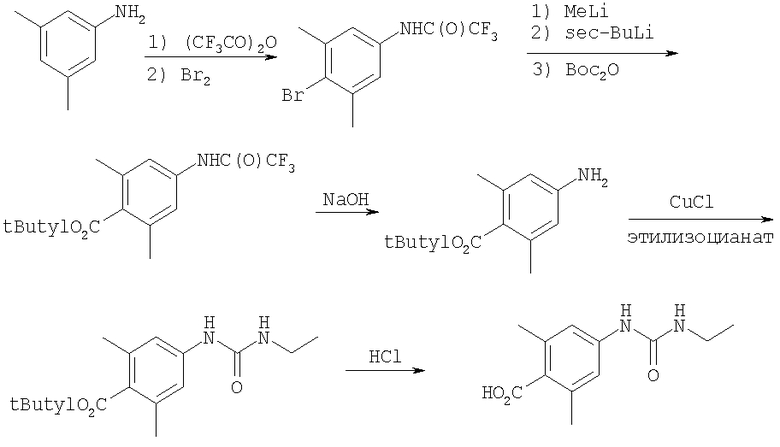
Стадия A: 3,5-Диметиланилин (18,5 мл, 149 ммоль) смешивают с CH2Cl2. Раствор охлаждают на водяной бане. К раствору медленно прибавляют трифторуксусный ангидрид (29,5 мл, 209 ммоль). После прибавления раствор 15 мин перемешивают при 25°С. При поддержании водяной бани при комнатной температуре к раствору медленно прибавляют бром (7,3 мл, 142 ммоль). Раствор 3,5 ч перемешивают при 25°С. Реакцию останавливают с помощью 10% раствора Na2S2O3. Водный слой экстрагируют с помощью СН2Cl2. Объединенные органические слои сушат (MgSO4), обрабатывают активированным углем и фильтруют. Концентрирование дает оранжевое твердое вещество. Очистка с помощью перекристаллизации (гексаны/Et2O) дает две порции (всего 34 г, 77%) бромированного производного в виде белого твердого вещества.
Стадия B: Арилбромид (17 г, 57 ммоль) смешивают с THF и в атмосфере N2 охлаждают до -78°С. При -78°C к этому раствору медленно прибавляют метиллитий/LiBr (54 мл 1,5 М раствора в Et2O, 80 ммоль). Через 5 мин перемешивания к раствору реакционной смеси при -78°C медленно прибавляют втор-BuLi (62 мл 1,3 М раствора в циклогексане, 80 ммоль). Через 5 мин к этому раствору при -78°C прибавляют ди-трет-бутилдикарбонат (22,5 г, 103 ммоль) в THF. Раствор нагревают до 25°С. Через 30 мин проводят распределение реакционной смеси между водой и СН2Cl2. Водный слой экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат (MgSO4). Фильтрование и концентрирование дает желтое твердое вещество. Очистка с помощью флэш-хроматографии (от 1/1 до 1/4 гексаны/СН2Cl2, SiO2) дает 13,1 г (72%) трет-бутилового сложного эфира в виде почти белого твердого вещества.
Стадия C: Трифторацетамид (10 г, 31 ммоль) и NaOH (2,5 г, 62 ммоль) смешивают с MeOH/H2O (3/1) и 3 ч нагревают при 60°С. Раствор подвергают распределению между водой и CH2Cl2. Водный слой экстрагируют с помощью СН2Cl2. Объединенные органические слои промывают водой и сушат (Na2SO4). Фильтрование и концентрирование дает 6,4 г (93%) анилина в виде оранжевого твердого вещества.
Стадия D: Анилин (1 г, 4,5 ммоль), этилизоцианат (0,4 мл, 5 ммоль) и CuCl (90 мг, 0,9 ммоль) смешивают с DMF при 0°С. Раствор нагревают до 25°C и при этой температуре перемешивают 2 ч. Раствор подвергают распределению между EtOAc и 10% NH4OH. Водный слой экстрагируют с помощью EtOAc. Объединенные слои промывают солевым раствором и сушат (MgSO4). Фильтрование и концентрирование дает желтое твердое вещество. Очистка с помощью флэш-хроматографии (от 3/1 до 1/1 гексаны/EtOAc, SiO2) дает 904 мг (69%) мочевины в виде желтого твердого вещества.
Стадия Е: трет-Бутиловый сложный эфир (900 мг, 3,1 ммоль) и 4 М раствор HCl в диоксане (3 мл) смешивают с iPrOH и нагревают 3,5 ч при 45°C и 16,5 ч при 25°С. Раствор концентрируют при пониженном давлении. Остаток подвергают распределению между Et2O и 1 н. NaOH. Щелочной водный раствор экстрагируют с помощью Et2O. Водный слой охлаждают до 0°C и подкисляют концентрированной HCl (рН 1-2). Водный слой экстрагируют с помощью EtOAc. Объединенные слои, содержащие EtOAc, сушат (Na2SO4). Фильтрование и концентрирование дает 400 мг (55%) кислоты в виде белого твердого вещества.
8АА: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (2 г, 4,3 ммоль), и 4-амино-2,6-диметилбензойную кислоту (описание методики получения см. ниже) вводят в реакцию сочетания при стандартных условиях (EDC/HOBT/iPr2NEt). Очистка с помощью флэш-хроматографии (2/1 гексаны/ацетон, SiO2) дает 1,16 г (52%) 8АА в виде желтой пены.
Получение 4-амино-2,6-диметилбензойной кислоты

трет-Бутиловый сложный эфир (950 мг, 4,3 ммоль) и HCl (11 мл, 4 М раствор в диоксане) смешивают с МеОН и 20 ч нагревают при 45°С. Раствор концентрируют и с количественным выходом получают кислоту (710 мг).
8ВВ: 8АА (100 мг, 0,19 ммоль) и этансульфонилхлорид (0,02 мл, 0,21 ммоль) смешивают с пиридином и 19 ч перемешивают при 25°С. Раствор концентрируют. Остаток подвергают распределению между 1 н. NaOH и СН2Cl2. Водный слой экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат (Na2SO4). Фильтрование и концентрированно дает коричневое масло. Очистка с помощью препаративной ТСХ (2/1 гексаны/ацетон, SiO2) дает 100 мг (86%) 8ВВ в виде бесцветного масла.
8СС: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (127 мг, 0,27 ммоль), и 4-фтор-2,6-диметилбензойнуюй кислоту (58 мг, 0,35 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания в соответствии с общей методикой (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (2/1 гексаны/EtOAc, SiO2) дает 8СС в виде бесцветного масла (87 мг бис-HCl соли, 54%).
Получение 4-фтор-2,6-диметилбензойной кислоты
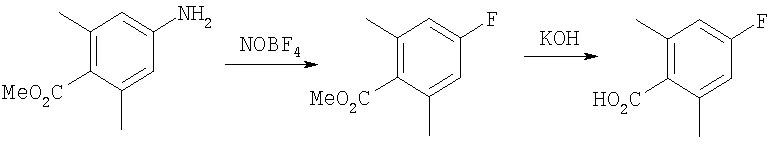
4-Амино-2,6-диметилбензойную кислоту (200 мг, 1,1 ммоль) и NOBF4 (196 мг, 1,7 ммоль) 30 мин нагревают в 1,2-дихлорбензоле при 100°С. Раствор охлаждают и разбавляют с помощью МеОН и воды. Прибавляют несколько гранул (2-3) КОН и раствор 16 ч кипятят с обратным холодильником. Раствор концентрируют. Остаток подвергают распределению между Et2O и 1 н. NaOH. Водный слой экстрагируют с помощью Et2O. Водный слой охлаждают до 0°С и подкисляют концентрированной HCl (рН=1-2). Водный слой экстрагируют с помощью CH2Cl2. Органические слои сушат (Na2SO4). Фильтрование и концентрированно дает 58 мг (31%) кислоты в виде желтовато-коричневого твердого вещества.
8DD: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (150 мг, 0,31 ммоль), и 4-хлор-2,6-диметилбензойную кислоту (76 мг, 0,41 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания в соответствии с общей методикой (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (4/1 гексаны/ацетон, SiO2) дает 8DD в виде бесцветного масла.
Получение 4-хлор-2,6-диметилбензойной кислоты
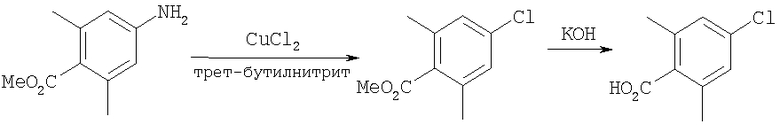
4-Амино-2,6-диметилбензойную кислоту (172 мг, 0,96 ммоль) и CuCl2 (155 мг, 1,15 ммоль) смешивают с CH3CN при 0°С. При 0°C к этому раствору прибавляют трет-бутилнитрит (0,17 мл, 1,4 ммоль). Раствор нагревают до 25°c, а затем 45 мин нагревают при 65°С. Раствор подвергают распределению между Et2O и водой. Водный слой экстрагируют с помощью Et2O. Объединенные органические слои промывают солевым раствором и сушат (MgSO4). Фильтрование и концентрированно дает метиловый сложный эфир. Метиловый эфир гидролизуют, как это описано выше для фторпроизводного (КОН). После экстракции получают 4-хлор-2,6-диметилбензойную кислоту (158 мг, 89%) в виде желтого твердого вещества.
8ЕЕ: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (180 мг, 0,38 ммоль), и 4-бром-2,6-диметилбензойную кислоту (95 мг, 0,41 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания в соответствии с общей методикой (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (4/1 гексаны/ацетон, SiO2) дает 8ЕЕ в виде бесцветного масла (140 мг бис-HCl соли, 56%).
Получение 4-бром-2,6-диметилбензойной кислоты
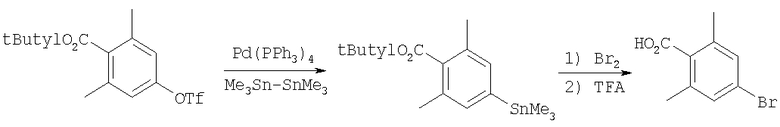
Стадия A: Трифторметансульфонат (500 мг, 1,48 ммоль), гексаметилолово (1,48 ммоль), LiCl (377 мг, 8,9 ммоль) и Pd(PPh3)4 (171 мг, 0,15 ммоль) 21 ч нагревают в THF (70°С) в атмосфере N2. Раствор подвергают распределению между Et2O и буферным раствором с рН 7 (NH4OAc). Водный слой экстрагируют с помощью Et2O. Объединенные слои, содержащие Et2O, промывают солевым раствором и сушат (Na2SO4). Фильтрование и концентрирование дает неочищенный арилстаннат в виде желтого полужидкого вещества.
Стадия B: Арилстаннат (0,74 ммоль) смешивают с CH2Cl2 при 0°С. К этому раствору прибавляют бром (0,7 мл 1 М раствора Br2 в CH2Cl2. Раствор 30 мин перемешивают при 0°С. Раствор разбавляют с помощью CH2Cl2 и промывают 10% раствором Na2S2O3. Водный слой экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат (Na2SO4). Раствор фильтруют. К этому раствору прибавляют TFA (2 мл) и раствор 17 ч перемешивают при 25°С. Раствор концентрируют. Остаток подвергают распределению между Et2O и 1 н. NaOH. Водный слой экстрагируют с помощью Et2O. Водный слой охлаждают до 0°C и подкисляют концентрированной HCl (рН 1-2). Водный слой экстрагируют с помощью Et2O. Объединенные слои сушат (Na2SO4). Фильтрование и концентрирование дает 100 мг (59%) кислоты в виде белого твердого вещества.
С помощью методик, приведенных после таблицы, получают соединения 8FF-8НН, обладающие структурой
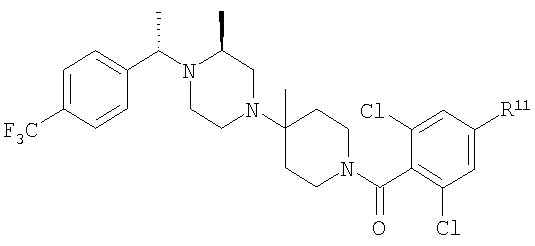
где R11 определен в таблице: 7
8FF: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (100 мг, 0,21 ммоль), и 2,6-дихлор-4-метоксибензойную кислоту (140 мг, 0,63 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания в соответствии с общей методикой (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (3/1 гексаны/EtOAc, SiO2) дает 8FF в виде бесцветного масла (27 мг, 23%).
8GG: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (330 мг, 0,7 ммоль), и 2,6-дихлор-4-гидроксибензойную кислоту (290 мг, 1,4 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания в соответствии с общей методикой (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (1/1 гексаны/EtOAc, SiO2) дает 8GG в виде бесцветного масла (75 мг, 19%).
Получение 2,6-дихлор-4-гидроксибензойной кислоты

2,6-Дихлор-4-метоксибензойную кислоту (500 мг, 2,3 ммоль) смешивают с CH2Cl2 и охлаждают до -78°С. При -78°C к этому раствору прибавляют BBr3 (6,9 мл 1 М раствора в CH2Cl2). Раствор нагревают до 25°С и при этой температуре перемешивают 16 ч. Реакцию останавливают с помощью 3 н. раствора NaOH. Водный слой экстрагируют с помощью CH2Cl2. Водный слой охлаждают (0°С) и подкисляют концентрированной HCl (рН 1-2). Водный слой экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат (Na2SO4). Фильтрование и концентрирование дает неочищенный фенол, который используют без дополнительной очистки.
8НН: Тригидрохлорид продукта, полученного в Примере 8, стадия 3 (96 мг, 0,2 ммоль), и 2,6-дихлор-4-(4-пиридил-N-оксид)-бензойную кислоту (55 мг, 0,2 ммоль) (описание методики получения см. ниже) вводят в реакцию сочетания в соответствии с общей методикой (EDC/HOBT/iPr2NEt). Очистка с помощью препаративной ТСХ (1/5 гексаны/ацетон, SiO2) дает 8НН в виде бесцветного масла (54 мг, 43%).
Получение 2,6-дихлор-4-(4-пиридил-N-оксид)-бензойной кислоты
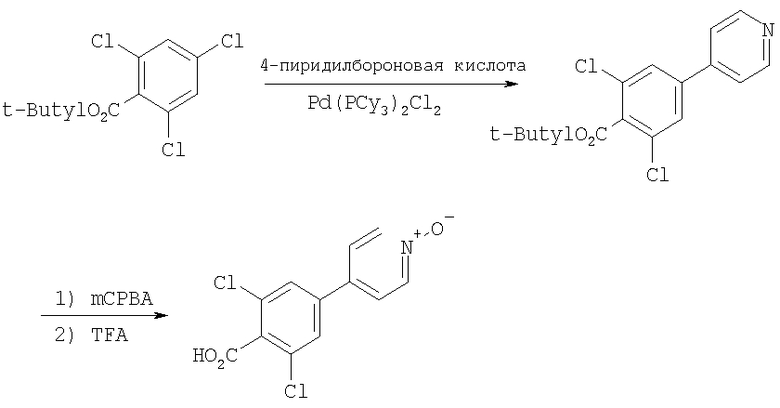
трет-Бутиловый эфир 2,4,6-трихлорбензойной кислоты (500 мг, 1,8 ммоль), 4-пиридилбороновую кислоту (270 мг, 2,16 ммоль), Pd(PCy3)2Cl2 (130 мг, 0,18 ммоль) и CsF (540 мг, 3,6 ммоль) смешивают с NMP и нагревают при 100°C в атмосфере N2 (16 ч). Раствор подвергают распределению между EtOAc и водой. Водный слой экстрагируют с помощью EtOAc. Объединенные органические слои промывают водой и олевым раствором и сушат (Na2SO4). Фильтрование и концентрирование дает неочищенный продукт. Очистка с помощью препаративной ТСХ (1/1 гексаны/EtOAc, SiO2) дает 68 мг пиридильного эфира. трет-Бутиловый эфир превращают в кислоту, как это сделано выше для диметильного производного (a. mCBA /b. TFA).
С помощью подходящих исходных веществ и методик, описанных для примеров 8S-8НН, получают соединения, обладающие следующей структурой:
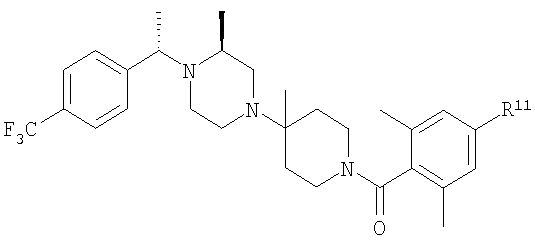
где R11 определен в таблице: 8

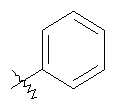
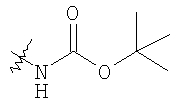


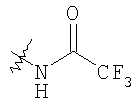
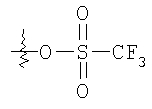
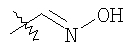
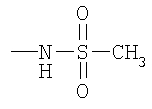
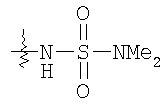
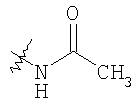
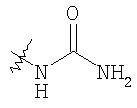

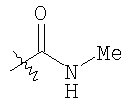
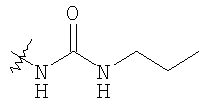
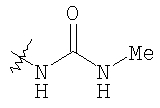

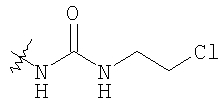
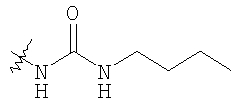
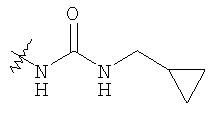
Все температуры плавления определены для бисгидрохлоридов (2×HCl) и только в случае 8РР - для свободного основания.
С использованием производных трифторметансульфонатного промежуточного продукта, описанного в 8Z, с помощью методик, аналогичных описанным выше, и данных приведенной ниже таблицы для 8АО-8AQ получают соединения, обладающие следующей структурой:
где R11 определен в таблице: 9
8АО:
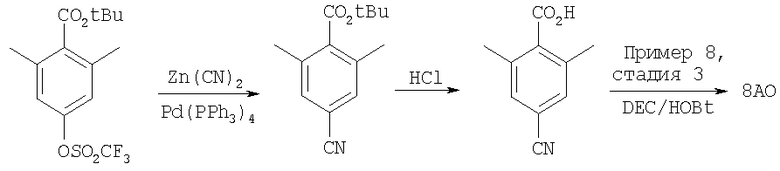
Стадия 1: Трифторметансульфонатный промежуточный продукт (см. 8W) (0,4 г), Zn(CN)2 (0,2 г), Pd(PPh3)4 (0,3 г) и DMF (1,5 мл) нагревают при 80°C в течение 17 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют с помощью EtOAc и насыщенного водного раствора NaHCO3. Слой, содержащий EtOAc, отделяют, промывают водой и солевым раствором, сушат и выпаривают с получением неочищенного масла, которое очищают с помощью препаративной тонкослойной хроматографии (пластины, покрытые 2000 мкМ диоксида кремния; элюент - смесь гексаны: EtOAc состава 8:1) и после отделения соответствующей зоны с выходом 77% получают цианосодержащий промежуточный продукт (0,2 г).
Стадия 2: Продукт, полученный на стадии 1 (0,2 г), растворяют в МеОН (1,5 мл) и прибавляют HCl (4 М раствор в 1,4-диоксане; 2 мл). Полученный раствор 3 ч перемешивают при 50°C и выпаривают. Этот неочищенный промежуточный продукт (0,038 г) и продукт, полученный в Примере 8, стадия 3 (65 мг; в виде тригидрохлорида), обрабатывают таким же образом, что и в Примере 8, стадия 4, с использованием DMF (2 мл), НОВТ (45 мг), DEC (60 мг) и диизопропилэтиламина (0,1 мл) и после выделения и очистки получают свободное основание 8АО, которое с выходом в 95% превращают в хлористоводородную соль (45 мг).
8АР:
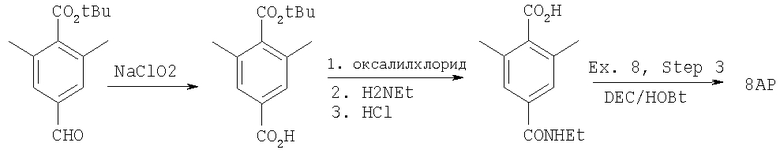
Стадия 1: 2,6-Диметил-4-формилбензойную кислоту (1,96 г) (см. 8W) растворяют в трет-бутаноле (94 мл) и 2-метил-2-бутене (24 мл). К этому раствору по каплям прибавляют раствор NaClO2 (6,89 г) и моногидрата NaH2PO4 (8,17 г) в воде (45 мл). После завершения прибавления устанавливают значение рН, равное 3, и образуются два слоя. Органический слой отделяют и выпаривают, получая промежуточный продукт - кислоту (1,80 г) в виде белого кристаллического вещества, которое используют без дополнительной очистки.
Стадия 2: К раствору продукта, полученного на стадии 1 (0,62 г), CH2Cl2 (5 мл) и DMF (1 капля) прибавляют оксалилхлорид (0,31 мл) и полученный раствор перемешивают 10 мин, после чего прибавляют вторую порцию оксалилхлорида (0,30 мл). Реакционную смесь перемешивают 10 мин, прибавляют толуол и реакционную смесь выпаривают досуха. Прибавляют СН2Cl2 (10 мл) и EtNH2 (1 мл) и реакционную смесь перемешивают двое суток, а затем ее подвергают распределению между солевым раствором и СН2Cl2. Слой, содержащий СН2Cl2, выпаривают и прибавляют HCl (4 мл 4 М раствора в 1,4-диоксане). Полученный раствор перемешивают 3 ч и выпаривают, получая твердое вещество, которое промывают с помощью Et2O и собирают, получая амидный промежуточный продукт (0,13 г) с выходом 24%.
Стадия 3: Продукт, полученный в Примере 8, стадия 3 (60 мг, в виде тригидрохлорида), и продукт, полученный на стадии 2 (35 мг), обрабатывают таким же образом, как и в Примере 8, стадия 4, и после обработки и очистки получают 8АР в виде свободного основания, которое с выходом 62% превращают в хлористоводородную соль (50 мг).
8AQ:
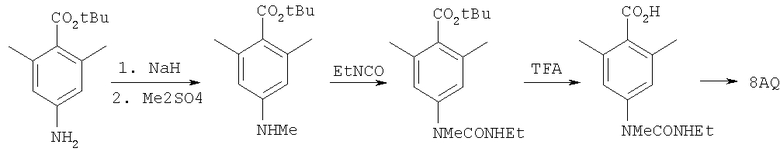
Стадия 1: К раствору аминного промежуточного продукта (2 г) (см. 8Z) прибавляют NaH (0,4 г 60% дисперсии в масле). Полученную суспензию перемешивают 15 мин и прибавляют Me2SO4. После кипячения с обратным холодильником в течение 1,5 ч реакционную смесь охлаждают до комнатной температуры, выливают в насыщенный водный раствор NH4Cl и экстрагируют с помощью Et2O. После выпаривания неочищенную реакционную смесь хроматографируют на силикагеле, элюируя смесью гексаны: EtOAc состава 4:1, и после выпаривания соответствующих фракций получают промежуточный продукт - метиламин (0,8 г) с выходом 38%.
Стадия 2: Продукт, полученный на стадии 1 (0,12 г), THF (5 мл) и EtNCO (54 мг) кипятят с обратным холодильником в течение 17 ч. Прибавляют EtNCO (54 мг) и 1,4-диоксан (2 мл) и полученный раствор 17 ч нагревают в запаянной трубке при 65°С. Раствор охлаждают, выпаривают и очищают с помощью препаративной тонкослойной хроматографии (силикагель; 25% EtOAc:CH2Cl2) и с выходом 64% получают искомый продукт (0,1 г) в виде кристаллического твердого вещества.
Стадия 3: Продукт, полученный на стадии 2 (0,1 г), обрабатывают таким же образом, как и в Примере 8, стадия 3, и получают искомый промежуточный продукт (0,08 г), который без дополнительной обработки используют на следующей стадии.
Стадия 4: Продукт, полученный в Примере 8, стадия 3 (75 мг; в виде тригидрохлорида), и продукт, полученный на стадии 3 (0,04 г), обрабатывают таким же образом, как и в Примере 8, стадия 4, и после обработки и очистки получают 8AQ в виде свободного основания, которое с выходом 62% превращают в хлористоводородную соль (65 мг).
С помощью приведенных выше методик и коммерчески доступных кислот получают соединения 8AY-8ВТ, обладающие структурой
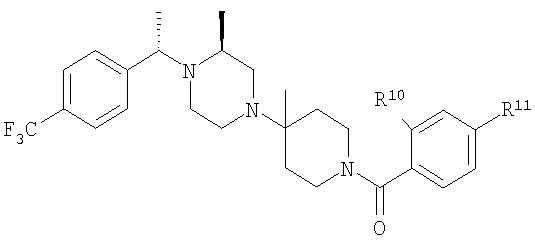
где R10 и R11 определены в таблице: 10

С использованием методик, аналогичных описанным выше, получают следующие соединения

где R8, R3, R6 и R2 являются такими, как указано в таблице: 11


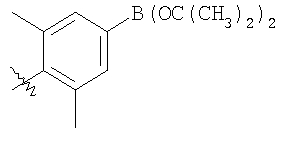
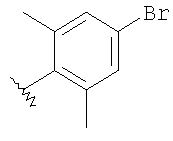
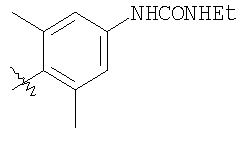
Пример 9
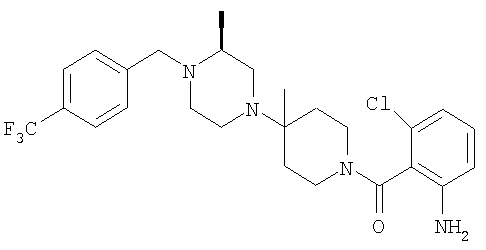
Стадия 1: Раствор 4-N-ВОС-2(S)-метилпиперазина (1,5 г; 7,5 ммоль), 4-метокси-бензилхлорида (1,1 мл; 8,1 ммоль) и диизопропилэтиламина (1,5 мл) в сухом CH3CN кипятят с обратным холодильником в течение 5 ч. Реакционную смесь охлаждают до комнатной температуры и летучие вещества удаляют в вакууме. Остаток растворяют в CH2Cl2 (30 мл) и промывают водой и солевым раствором.
Концентрирование дает неочищенный продукт, который очищают с помощью ФХСГ (10% EtOAc-гексаны) и получают 2,1 г (88%) продукта в виде бледно-желтой жидкости.
К раствору полученного соединения (2,1 г; 6,56 ммоль) в 12 мл CH2Cl2 прибавляют TFA (6 мл) и смесь 1,5 ч перемешивают при 25°С. Реакцию останавливают с помощью 1 н. раствора NaOH и доводят значение рН до 10. Экстрагирование с помощью CH2Cl2 дает искомый продукт (1,4 г; 97%) в виде бесцветной смолы.
Стадия 2: Смесь продукта, полученного на стадии 1 (1,4 г; 6,36 ммоль), N-BOC-4-пиперидинона (1,27 г; 6,4 ммоль) и Ti(OiPr)4 (1,9 мл; 6,4 ммоль) перемешивают 24 ч при 25°С. К реакционной смеси прибавляют 1 М раствор Et2AlCN в толуоле (7,6 мл) и смесь еще один день перемешивают при температуре окружающей среды. Полученный таким образом амин Штрекера обрабатывают и выделяют (2,7 г; 100%) так, как это описано в Примере 8, стадия 2. ТСХ, Rf=0,3 в 25% EtOAc-CH2Cl2.
Амин Штрекера (2,7 г; 6,3 ммоль) растворяют в 15 мл сухого THF при 0°C и к нему прибавляют CH3MgBr (3 М в Et2O; 10,5 мл). Через 1 ч баню со льдом удаляют, и реакции в течение 15 ч дают возможность протекать при комнатной температуре. Анализ гетерогенной реакционной смеси с помощью ТСХ не обнаруживает изменение исходного вещества; смесь 5 ч нагревают при 60°C и ТСХ не обнаруживает изменений. Реакцию останавливают с помощью насыщенного раствора NH4Cl и органические продукты экстрагируют с помощью СН2Cl2. Обработка неочищенного продукта (2,7 г) с помощью ФХСГ с использованием в качестве элюента смеси 15% ацетона-гексанов дает искомое ипсо-метильное соединение в виде бесцветной смолы (2,3 г; 87%).
Стадия 3: Продукт, полученный на стадии 2 (1,7 г; 4,08 ммоль), формиат аммония (1,4 г; 22 ммоль) и 10% палладий на угле (0,4 г) смешивают в 20 мл СН3ОН и 5 ч кипятят с обратным холодильником. Реакционную смесь фильтруют через целит и удаляют летучие вещества. Остаток растворяют в CH2Cl2 и промывают 10% раствором NaOH, водой и солевым раствором. Концентрирование в вакууме дает 1,1 г (92%) бледно-желтой смолы.
Стадия 4: Раствор продукта, полученного на стадии 3 (0,12 г; 0,4 ммоль), п-трифторметилбензилбромида (0,1 г; 0,4 ммоль) и диизопропилэтиламина (0,1 мл) в сухом СН3CN осторожно нагревают (60-70°С) в течение 16 ч. Смесь охлаждают и органический продукт выделяют путем экстракции с помощью CH2Cl2. Обработка с помощью ФХСГ (10-30% Et2O-CH2Cl2; Rf=0,4) дает основной продукт в виде бесцветной пленки (0,12 г; 68%).
Обработка указанного выше продукта (в CH2Cl2) с помощью TFA (1 мл) в течение 1 ч с последующим подщелачиванием и стандартной обработкой дает искомое соединение (0,09 г; 96%) в виде бесцветной пленки.
Стадия 5: Продукт, полученный на стадии 4 (0,045 г; 0,13 ммоль), и 6-хлорантраниловую кислоту (0,022 г; 0,13 ммоль) вводят в реакцию сочетания, как это описано в Примере 1, и после обработки и ФХСГ (5% раствор СН3ОН в CH2Cl2) искомое соединение выделяют в виде бесцветной пленки (0,058 г; 90%).
Хлористоводородную соль искомого соединения получают обычным образом с помощью реакции свободного основания с 1 М HCl-Et2O и обработки осадка с получением бежевого твердого вещества (0,066 г).
С использованием аналогичной методики продукт, полученный на стадии 3, превращают в другие соединения путем алкилирования пиперазинового атома азота с помощью соответствующего галогенида с последующим удалением защитной группы и сочетания пиперидинильного фрагмента с соответствующей кислотой с получением амидов общей формулы:
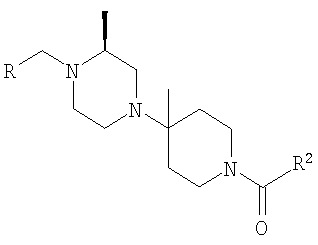
где R и R2 являются такими, как указано в таблице: 12

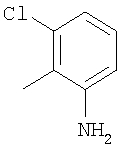





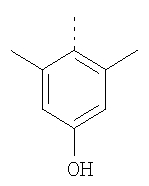
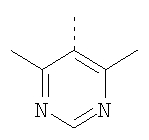
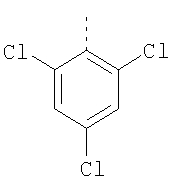
С помощью аналогичной методики, описанной ниже, также получают соединения, в которых R означает 4-этоксинафтил:
Стадии 1-3: См. Пример 9.
Стадия 4А: 4-Гидроксинафтальдегид (0,86 г) и K2СО3 (1,38 г, 2 экв.) в CH3CN (35 мл) обрабатывают с помощью СН3CH2I (0,80 мл, 2 экв.) и полученную смесь 20 ч перемешивают при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают с помощью EtOAc и смесь фильтруют. Фильтрат подвергают распределению с Н2O. Высушенный (MgSO4) раствор в EtOAc концентрируют в вакууме и получают оранжево-коричневый остаток (0,89 г). Этот остаток помещают на пластины для препаративной тонкослойной хроматографии (10, 1000 мкм) и элюируют с помощью CH2Cl2, получая искомое соединение (0,82 г).
Стадия 4: Продукты, полученные на стадии 3 (0,270 г; 0,95 ммоль) и стадии 4А (0,571 г; 2,9 ммоль) в CH2Cl2 (25 мл), 30 мин перемешивают при комнатной температуре в атмосфере аргона. Прибавляют Na(OAc)3BH (0,506 г; 3,4 ммоль). Через 19 ч реакцию останавливают с помощью разбавленного раствора NaOH. Водный слой промывают с помощью CH2Cl2 (3х). Объединенные растворы в СН2Cl2 промывают водой (3х), а затем солевым раствором. Высушенный (MgSO4) раствор концентрируют СН2Cl2 до ˜50 мл. Прибавляют Amberlyst 15 (4,5 мэкв./г: 2,4 г; 11,025 мэкв.). Через 19 ч прибавляют дополнительную порцию Amberlyst 15 (2,3 г). Через 7 ч смолу промывают с помощью CH2Cl2 (5x), THF (5x), THF:Н2О (5х), Н2O (5x), СН3ОН (5x) и CH2Cl2 (5x). Смолу элюируют с помощью 2 М раствора NH3 в СН3ОН (300 мл) (3х), а затем концентрируют в вакууме, получая масло янтарного цвета (0,215 г). Неочищенный материал помещают на пластины для препаративной тонкослойной хроматографии (4, 1000 мкм) и элюируют с помощью CH2Cl2: 2 М раствор NH3 в СН3ОН (9:1), получая масло янтарного цвета (0,125 г, 36%).
Стадия 5: С помощью соответствующей карбоновой кислоты по методике, описанной в Примере 9, стадия 5, получают следующие соединения:

ЖХМС (жидкостная хроматография - масс-спектроскопия) найдено М+H=531;
ВЭЖХ* (высокоэффективная жидкостная хроматография): Время удерживания равно 5,52 мин.
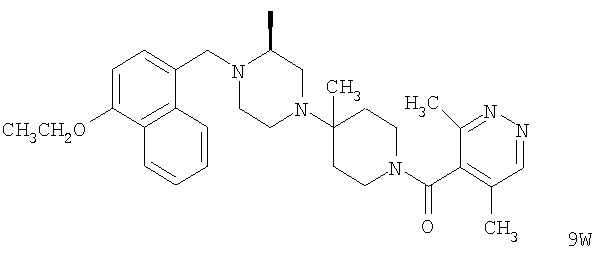
ЖХМС найдено М+Н=516; ВЭЖХ*: Время удерживания равно 5,66 мин.
*ВЭЖХ: Колонка VYDAC 218TP5505; градиент 5-95% В в течение 5 мин, выдерживание 2 мин; Раствор А: 0,1% TFA/H2О, Раствор В: 0,1% TFA/CH3CN; при 245 нм.
С использованием аналогичной методики в случае, когда исходный пиперазин не содержит метильного заместителя, получают следующие соединения

Пример 10
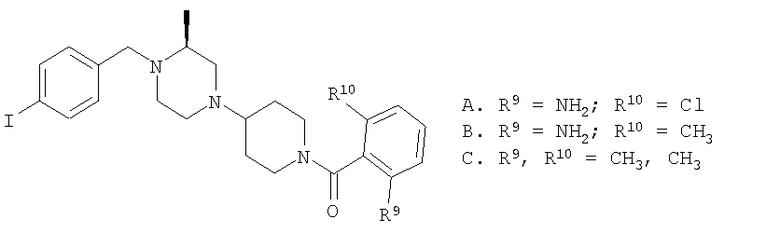
Стадия 1: Раствор 4-N-ВОС-2(S)-метилпиперазина (0,4 г; 2 ммоль), п-йодбензальдегида (0,46 г; 2 ммоль) и NaBH(OAc)3 (0,65 г; 3 ммоль) в 6 мл CH2Cl2 осторожно кипятят с обратным холодильником в течение 14 ч. Содержимое охлаждают, разбавляют с помощью 30 мл СН2Cl2 и промывают 1 н. раствором NaOH, водой и солевым раствором, получая желтое масло (0,8 г). Обработка с помощью ФХСГ (25% EtOAc-гексан) дает искомый продукт (0,66 г; 79%) в виде бесцветной пленки. ТСХ, Rf=0,6 в 25% EtOAc-гексан.
Защитную группу ВОС удаляют из продукта (0,66 г; 1,58 ммоль) путем обработки с помощью TFA (1 мл) в CH2Cl2 (2 мл). После стандартной обработки получают моноалкилированный пиперазин (0,5 г; 100%) в виде бесцветной смолы.
Стадия 2: К раствору продукта, полученного на стадии 1 (0,5 г; 1,58 ммоль) и N-ВОС-пиперидинона (0,6 г; 3 ммоль) в 5 мл СН2Cl2 прибавляют NaBH(OAc)3 (0,63 г; 3 ммоль) и две капли АсОН и полученный раствор 16 ч перемешивают при температуре окружающей среды. После обычной обработки и ФХСГ получают искомый продукт (0,6 г; 76%) в виде бесцветного масла. ТСХ, Rf=0,4 в 25% ацетон-CH2Cl2.
Свободный пиперидин (0,38 г; 79%) получают из соединения с защитной группой N-BOC (0,6 г; 1,2 ммоль), путем обработки с помощью TFA (2 мл) в СН2Cl2 (5 мл).
Соединение 10А: Сочетание 6-хлорантраниловой кислоты (0,065 г; 0,38 ммоль) с продуктом, полученным на стадии 2 (0,127 г; 0,32 ммоль) в присутствии DEC (0,092 г; 0,48 ммоль), НОВТ (0,065 г; 0,48 ммоль) и диизопропилэтиламина (0,1 мл) с последующим выделением продукта проводят так, как это описано выше. Эта методика дает соединение 10А (0,13 г; 73%) в виде бесцветной пленки. ТСХ, Rf=0,5/0,45 для пары поворотных изомеров в 2% СН3ОН-СН2Cl2.
Хлористоводородную соль искомого соединения получают обычным способом. Т.пл.: 198-202°С. МСВР (МН+)=553,1231.
Соединение 10В: Сочетание продукта, полученного на стадии 2, с 6-метилантраниловой кислотой дает соединение 10В (хлористоводородную соль) с выходом 73%. Т.пл.: 197-200°С. МСВР (МН+)=533,1774.
Соединение 10С: 2,6-Диметилбензойную кислоту вводят в реакцию сочетания с продуктом, полученным на стадии 2, и получают амид 10С (хлористоводородную соль) с выходом 50%. Т.пл.: 202-205°С. МСВР (МН+)=532,1826.
Пример 11
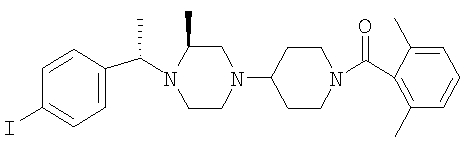
Стадия 1: (S)-Метилбензиламин (27 мл; 0,2 моль) в CH2Cl2 (50 мл) в течение 15 мин по каплям прибавляют к охлажденному льдом трифторуксусному ангидриду (40 мл) в CH2Cl2 (200 мл). Смесь 1 ч перемешивают при комнатной температуре, после этого при охлаждении на водяной бане со льдом прибавляют йод (27 г; 0,106 моль), затем [бис-(трифторацетокси)-йод]-бензол (25 г; 0,058 моль). После перемешивания в темноте при комнатной температуре в течение ночи прибавляют дополнительное количество [бис-(трифторацетокси)-йод]-бензола (24 г; 0,056 моль) и смесь перемешивают при комнатной температуре в течение еще одного дня. Смесь разбавляют с помощью CH2Cl2 (500 мл) и охлажденного льдом Na2SO3 (10% водный раствор, 500 мл) и перемешивают 0,5 ч. Органический слой отделяют и промывают с помощью NaHCO3, фильтруют через короткую колонку с силикагелем и промывают с помощью CH2Cl2 (500 мл). После выпаривания СН2Cl2 прибавляют Et2O (125 мл) и смесь перемешивают 10 мин. К раствору в Et2O постепенно прибавляют гексаны (600 мл) и смесь перемешивают 0,5 ч. Осадок собирают и промывают гексанами. Белое твердое вещество сушат при комнатной температуре и получают йодпроизводное (36,5 г, выход 53%, Rf=0,7, EtOAc/гексаны, 1:3).
Стадия 2: Продукт, полученный на стадии 1 (11,2 г, 0,033 ммоль), растворяют в СН3ОН (200 мл) и по каплям прибавляют раствор NaOH (15 г, 0,375 моль) в воде (100 мл). Смесь 2,5 ч перемешивают при комнатной температуре. После выпаривания СН3ОН водный слой экстрагируют с помощью Et2O (3×100 мл) и объединенные органические порции промывают солевым раствором, сушат над Na2SO4, фильтруют и концентрируют, получая свободный амин.
Метил-R-лактат (4,08 г, 0,039 моль) растворяют в CH2Cl2 (40 мл) и смесь перемешивают и в атмосфере N2 охлаждают до -78°C в смеси ацетон-CO2. Прибавляют трифторметансульфоновый ангидрид (10,2 г, 0,036 ммоль), а затем 2,6-лутидин (6,27 г, 0,059 моль) и смесь 5 мин перемешивают при -78°С. Смесь нагревают до комнатной температуры и перемешивают 30 мин. Прибавляют дополнительное количество CH2Cl2 и раствор промывают с помощью 2 н. HCl. К раствору трифторметансульфоната прибавляют свежеприготовленный амин, а затем раствор K2СО3 (18 г, 0,132 моль) в воде (20 мл). Смесь перемешивают при комнатной температуре в течение ночи. Экстракция с помощью CH2Cl2 с последующей хроматографией на колонке с силикагелем дает вторичный амин (8,27 г, выход 75%, Rf=0,65, гексаны/EtOAc, 3:1) в виде желтого сиропа.
Стадия 3: Амин, полученный на стадии 2 (17,3 г, 0,052 моль), растворяют в дихлорэтане (100 мл) и ClCH2COCl (117,2 г, 82 мл, 1,04 моль). Смесь 3 ч кипятят с обратным холодильником при перемешивании. Растворитель и ClCH2COCl удаляют в вакууме. Оставшийся желтый сироп растворяют в DMSO (40 мл) при 0°C и прибавляют NaI (5,2 г, 0,035 моль) и NH4OH (56 мл, 1,04 моль). Реакционную смесь 30 мин перемешивают при 0°C, нагревают до комнатной температуры и перемешивают в течение ночи. К смеси прибавляют воду (100 мл) и осадок отфильтровывают и промывают водой. Полученное белое твердое вещество сушат на воздухе и получают дикетопиперазин (14,3 г, выход 77%, Rf=0,56, гексаны/EtOAc, 3:1).
Стадия 4: Дикетопиперазин, полученный на стадии 3 (14,3 г, 0,04 моль), растворяют в диметоксиэтане (200 мл) и к раствору прибавляют NaBH4 (15,1 г, 0,4 моль) и BF3.Et2O (34 г, 29,5 мл, 0,24 моль). Смесь 3 ч кипятят с обратным холодильником и затем охлаждают примерно до 0°C в бане со льдом. К смеси медленно прибавляют СН3ОН (500 мл) и концентрированную HCl (300 мл). Смесь перемешивают 20 мин при комнатной температуре, а затем 45 мин при кипячении с обратным холодильником. Смесь концентрируют и прибавляют NaOH, пока значение рН не станет равным более 10. Экстракция с помощью EtOAc дает искомый пиперазин в виде желтого сиропа (12,9 г, выход 98%).
Стадия 5: Продукт, полученный на стадии 4 (1,9 г, 5,79 ммоль), N-BOC-4-пиперидон (5,73 г, 28,8 ммоль), NaBH(ОАс)3 (6,1 г, 28,8 ммоль) и 2 М АсОН (5,76 мл, 11,52 ммоль) смешивают в CH2Cl2 (150 мл) и смесь перемешивают в течение ночи. После удаления растворителя прибавляют раствор NaOH (3 н.), и экстракция с помощью EtOAc с последующей хроматографией на силикагеле дает чистый пиперазинопиперидин (2,21 г, выход 75%, Rf=0,18, гексаны/EtOAc, 1:1) в виде сиропа.
Стадия 6: Продукт, полученный на стадии 5 (1,9 г, 3,7 ммоль), растворяют в CH2Cl2 (10 мл) и прибавляют TFA (10 мл). Смесь 2 ч перемешивают при комнатной температуре. После удаления растворителя и TFA при пониженном давлении к оставшемуся сиропу прибавляют раствор NaOH (3 н.), и экстракция с помощью EtOAc дает свободный пиперазинопиперидин (1,3 г, выход 85%) в виде желтого сиропа. К раствору свободного пиперазинопиперидина (200 мг, 0,484 ммоль) в CH2Cl2 (2 мл) прибавляют 2,6-диметилбензойную кислоту (150 мг, 0,99 ммоль), DEC (191 мг, 0,99 ммоль) и НОВТ (135 мг, 0,99 ммоль). Смесь перемешивают при комнатной температуре в течение ночи и затем растворитель удаляют при пониженном давлении. К оставшемуся сиропу прибавляют раствор NaOH (3 н.), и экстракция с помощью EtOAc с последующей хроматографией на колонке дает искомое соединение (210 мг, выход 80%, Rf=0,37, CH2Cl2/СН3ОН, 20:1). МСВР (в виде хлористоводородной соли): рассчитано для C27H37N3OI (М+Н+), 546,1981; найдено, 546,1965. Т.пл.: 190°С (с разложением).
С использованием аналогичной методики получают соединения формулы
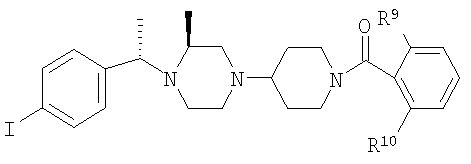
где R9 и R10 являются такими, как указано в таблице: 13
Пример 12
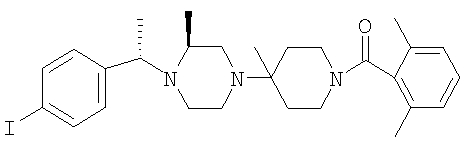
Стадия 1: К раствору продукта, полученного в Примере 11, стадия 4 (1,4 г, 4,2 ммоль), и 1-трет-бутоксикарбонил-4-пиперидона (0,93 г, 4,67 ммоль) в СН2Cl2 прибавляют Ti(OiPr)4 (1,19 г, 4,2 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 1 М Et2AlCN (5,04 мл, 5,04 ммоль), смесь перемешивают при комнатной температуре в течение ночи и растворитель выпаривают. К остатку прибавляют насыщенный раствор NaHCO3, и экстракция с помощью EtOAc дает амин Штрекера в виде желтого сиропа. Сироп растворяют в THF (40 мл) и к раствору прибавляют 3 М CH3MgBr (7 мл, 21 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, затем охлаждают до 0°C и прибавляют насыщенный раствор NH4Cl. Экстракция с помощью EtOAc с последующей хроматографией на силикагеле дает пиперазинопиперидин (1,78 г, выход 81%, Rf=0,52, гексаны/EtOAc, 2:1).
Стадия 2: Продукт, полученный на стадии 1, обрабатывают способом, описанным в Примере 11, стадия 6, и получают искомое соединение. Т.пл.: 190°С (с разложением). МСВР (в виде хлористоводородной соли): найдено, 560,2145.
С использованием аналогичной методики получают соединения формулы
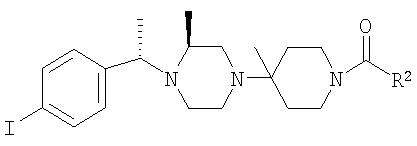
где R2 являются такими, как указано в таблице: 14

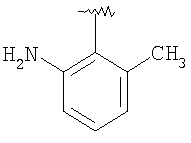


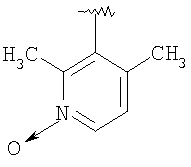
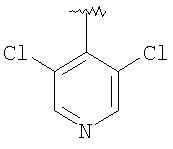
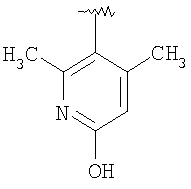
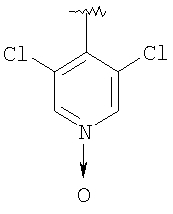
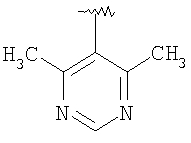
Пример 13

Стадия 1: К раствору защищенного с помощью N-BOC продукта, полученного в Примере 11, стадия 4 (250 мг, 0,581 ммоль), в DMF (2,5 мл) прибавляют CuCl (1 г, 10,1 ммоль). Суспензию 24 ч перемешивают при 110°C в атмосфере N2. После охлаждения смеси до комнатной температуры прибавляют NH4OH и раствор постепенно становится ярко-синим. Экстракция с помощью EtOAc дает смесь хлорзамещенного пиперазина и его ВОС-производного. После обработки смеси с помощью TFA (5 мл) в СН2Cl2 (2 мл) в течение 2 ч растворитель выпаривают и прибавляют NaOH (3 н.). Экстракция с помощью EtOAc дает чистый пиперазин (110 мг, 79%) в виде желтого сиропа.
Стадия 2: Продукт, полученный на стадии 1, обрабатывают способом, описанным в Примере 11, стадии 5 и 6, и получают искомое соединение. Т.пл.: 180°С (с разложением). МСВР (в виде хлористоводородной соли): найдено, 454,2617.
С использованием аналогичной методики получают соединения формулы
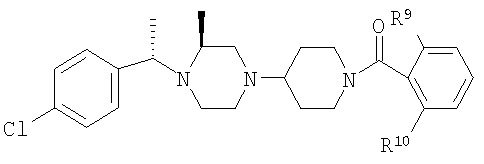
где R9 и R10 являются такими, как указано в таблице: 15
С использованием продукта, полученного на стадии 1, в методике Примера 12 получают соединения формулы
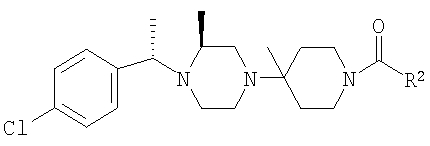
где R2 являются такими, как указано в таблице: 16
Пример 14
Стадия 1: К раствору защищенного с помощью N-BOC продукта, полученного в Примере 11, стадия 4 (5 г, 0,012 ммоль) в DMF (20 мл) прибавляют CuCN (20,8 г, 0,23 моль). Суспензию 22 ч перемешивают при 110°C в атмосфере N2. После охлаждения смеси до комнатной температуры прибавляют NH4OH, и раствор постепенно становится ярко-синим. Экстракция с помощью EtOAc с последующей хроматографией на колонке с силикагелем дает цианпроизводное (2,29 г, выход 60%, Rf=0,5, гексаны/EtOAc, 4:1), карбоксамидное производное (0,95 г, выход 23,6%, Rf=0,2, CH2Cl2/СН3ОН, 10:1) и незамещенное производное (85 мг, выход 2,4%, Rf=0,75, гексаны/EtOAc, 2:1).
Стадия 2: У циансоединения, полученного на стадии 1, в кислой среде удаляют группу ВОС и полученный амин превращают в искомое соединение по методике, описанной в Примере 11, стадии 5 и 6. МСВР (в виде хлористоводородной соли): найдено, 445,4970.
Пример 15
Стадия 1: К раствору защищенного с помощью N-BOC продукта, полученного в Примере 11, стадия 4 (1,4 г, 3,26 ммоль) и CuCl (1,61 г, 16,3 ммоль) в СН3ОН при 0°C медленно прибавляют NaBH4 (3,69 г, 97,6 ммоль). Образуется черный осадок. Смесь нагревают до комнатной температуры и перемешивают в течение ночи. Осадок удаляют фильтрованием через целит, а СН3ОН удаляют в вакууме. Экстракция с помощью EtOAc дает искомое соединение (1 г, выход 100%, Rf=0,55, гексаны/EtOAc, 5:1) в виде сиропа.
Стадия 2: У продукта, полученного на стадии 1, в кислой среде удаляют группу ВОС и полученный амин превращают в искомое соединение по методике, описанной в Примере 11, стадии 5 и 6. Т.пл.: 195°С. МСВР (в виде хлористоводородной соли): найдено, 420,3016.
С помощью аналогичной методики получают следующее соединение:
МСВР (в виде хлористоводородной соли): найдено, 441,2426.
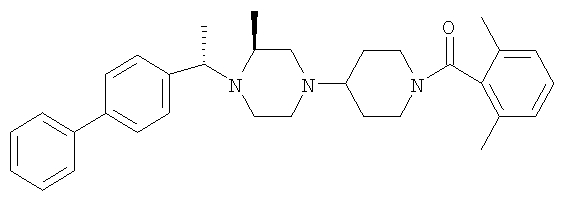
Пример 16
Стадия 1: К раствору защищенного с помощью N-BOC продукта, полученного в Примере 11, стадия 4 (2,5 г, 5,8 ммоль) в бензоле прибавляют фенилборную кислоту (1,68 г, 13,8 ммоль), 2 М Na2CO3 (14 мл) и тетракис-(трифенилфосфин)-палладий (0,67 г, 0,58 ммоль). Смесь кипятят с обратным холодильником при перемешивании в течение ночи. Экстракция с помощью EtOAc с последующей хроматографией на колонке с силикагелем дает фенилпроизводное (1,37 г, выход 62%, Rf=0,5, гексаны/EtOAc, 5:1) в виде сиропа.
Стадия 2: У продукта, полученного на стадии 1, в кислой среде удаляют группу ВОС и полученный амин превращают в искомое соединение по методике, описанной в Примере 11, стадии 5 и 6. Т.пл.: 190°С. МСВР (в виде хлористоводородной соли): найдено, 496,3319.
С использованием аналогичной методики получают соединения формулы
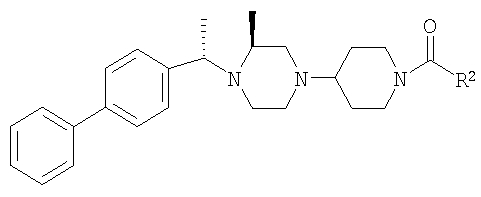
где R2 являются такими, как указано в таблице: 17
Пример 17
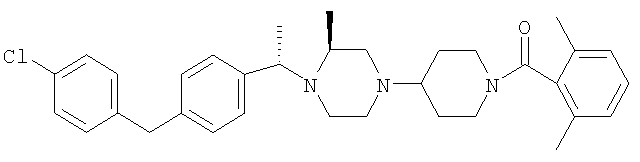
Стадия 1: Защищенный с помощью N-BOC продукт, полученный в Примере 11, стадия 4 (800 мг, 1,88 ммоль), растворяют в сухом THF и в атмосфере N2 температуру доводят до -78°С. Прибавляют бутиллитий (2,5 М раствор, 0,832 мл, 2 ммоль) и смесь 10 мин перемешивают при -78°С. Раствор по каплям прибавляют к п-хлорбензилальдегиду (234 мг, 2,07 ммоль) в THF при -78°С. Смесь 30 мин перемешивают при -78°C, а затем постепенно нагревают до комнатной температуры. К смеси прибавляют насыщенный раствор NH4Cl, и экстракция с помощью EtOAc с последующей хроматографией на колонке с силикагелем дает искомый спирт (30 мг, выход 3,6%, Rf=0,5, гексаны/EtOAc, 2:1) в виде желтого сиропа.
Стадия 2: Раствор спирта, полученного на стадии 1 (40 мг, 0,090 ммоль), триэтил-силана (52 мг, 0,45 ммоль) и TFA (5 мл) в CH2Cl2 (5 мл) 2 ч кипятят с обратным холодильником при перемешивании. После удаления CH2Cl2, триэтилсилана и TFA при пониженном давлении к оставшемуся сиропу прибавляют раствор NaOH (3 н.). Экстракция с помощью EtOAc дает хлорбензильное производное (20 мг, выход 68%) в виде желтого сиропа.
Стадия 3: Продукт, полученный на стадии 2, превращают в искомое соединение с помощью методики, описанной в Примере 11, стадии 5 и 6. Т.пл.: 170°С (с разложением). МСВР (в виде хлористоводородной соли): найдено, 544,3101.
Пример 18
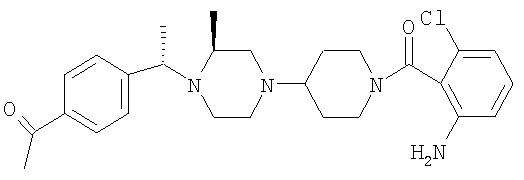
Стадия 1: К раствору защищенного с помощью N-BOC 4-пиперидинильного производного циансоединения, полученного в Примере 14, стадия 1 (510 мг, 1,24 ммоль), в Et2O (4 мл) по каплям прибавляют 3 М CH3MgBr (4 мл). Смесь кипятят с обратным холодильником при перемешивании в течение ночи. После охлаждения раствора в бане со льдом прибавляют 12 н. HCl (4 мл) и смесь 2 ч перемешивают на паровой бане. Раствор охлаждают до комнатной температуры и прибавляют гранулы твердого NaOH, пока значение рН не станет равным более 10. Экстракция с помощью EtOAc/СН3ОН (3:1) дает искомый метилкетон (249 мг, выход 61%) в виде сиропа.
Стадия 2: Продукт, полученный на стадии 1, обрабатывают в соответствии со стандартной методикой получения пептида по реакции сочетания с использованием DEC, описанной в Примере 11, стадия 6, и получают искомое соединение. Т.пл.: 210°С. МСВР (в виде хлористоводородной соли): найдено, 483,2522.
С использованием аналогичной методики получают следующее соединение:

Т.пл.: 210°С (с разложением). МСВР (в виде хлористоводородной соли): найдено, 463,3088.
Пример 19
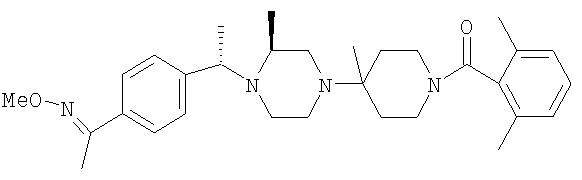
Стадия 1: К раствору продукта, полученного в Примере 22 (140 мг, 0,29 ммоль), в СН3ОН (10 мл) и EtOH (1 мл) прибавляют NH2OCH3·HCl (738 мг, 8.84 ммоль) и NaOAc (725 мг, 8,84 ммоль). Суспензию перемешивают при 40°C в течение ночи, растворители выпаривают и к остатку прибавляют воду. Экстракция с помощью EtOAc с последующей хроматографией на силикагеле дает искомое соединение (99 мг, выход 68%, Rf=0,38. CH2Cl2/СН3ОН, 20:1). МСВР (в виде тартрата): Рассчитано для C31H45N4O2 (M+H+), 505,3543; Найдено, 505,3542.
С использованием аналогичной методики получают соединения формулы:
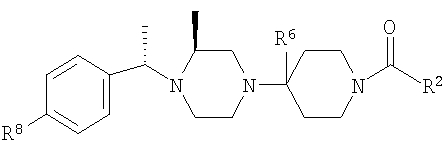
где R8, R6 и R2 являются такими, как указано в таблице: 18

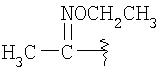
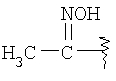
Пример 20
Свободный пиперазино-пиперидин, полученный в Примере 11, стадия 6 (1,7 г, 3,3 ммоль), растворяют в CHCl3 (30 мл; = основной раствор А). 250 мкл основного раствора А (0,027 ммоль) прибавляют к взвеси 0,15 г (˜0,14 ммоль) карбодиимида, связанного со смолой (получают путем реакции смолы Argopore-Cl с 1-(3-диметиламинопропил)-3-этилкарбодиимидом в DMF), при 100°С в DMF (1,5 мл) в полиэтиленовом патроне SPE. К этой смеси прибавляют 75 мкл 1 М раствора 5-метил-3-фенилизоксазол-4-карбоновой кислоты в DMF (0,075 ммоль) и НОВТ (24 мкл 1 М раствора в DMF). Эту смесь встряхивают 14 ч, фильтруют и к фильтрату прибавляют 0,1 г смолы Amberlyst-15 (0,47 ммоль). Встряхивают 1-2 ч, фильтруют и смолу дважды промывают каждым из следующих растворителей: THF, CH2Cl2 и СН3ОН, а затем промывают с помощью THF и CH2Cl2. Смолу обрабатывают с помощью 2 М раствора NH3 в СН3ОН (1 раз в течение 30 мин и 1 раз в течение 5 мин). Фильтраты объединяют и концентрируют при пониженном давлении, получая искомое соединение. ЖХМС, найдено, MH+=599,1 (рассчитанная молекулярная масса, 598); ТСХ, Rf=0,74 (CH2Cl2/СН3ОН/NH4ОН (95/5/0,5)).
С использованием описанной выше методики и соответствующих карбоновых кислот получают следующие соединения
где R2 являются такими, как указано в таблице: 19
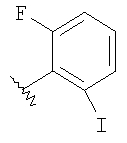

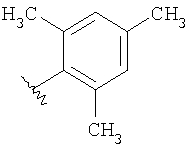
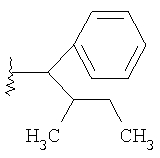
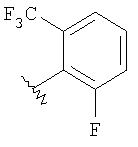
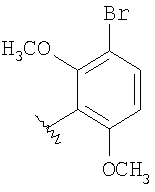

Пример 21
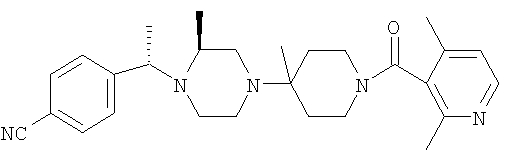
Стадия 1: У циансоединения, полученного в Примере 14, стадия 1, в кислой среде удаляют группу ВОС и полученный амин (1,59 г, 6,96 ммоль), 1-трет-бутоксикарбонил-4-пиперидон (1,66 г, 8,35 ммоль) и Ti(OiPr)4 (2,18 г, 7,66 ммоль) в CH2Cl2 перемешивают при комнатной температуре в течение ночи. Прибавляют 1 М Et2AlCN (8,35 мл, 8,35 ммоль), смесь перемешивают при комнатной температуре в течение ночи и выпаривают растворитель. К остатку прибавляют насыщенный раствор NaHCO3, и экстракция с помощью EtOAc с последующей хроматографией на колонке дает амин Штрекера в виде желтого сиропа (1,76 г, выход 58%, Rf=0,70, гексаны/EtOAc, 2:1).
Стадия 2: Амин, полученный на стадии 1 (200 мг, 0,46 ммоль), растворяют в безводном THF (2 мл) и по каплям прибавляют CH3MgBr (0,76 мл, 2,29 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, затем охлаждают до 0°С. Прибавляют насыщенный раствор NH4Cl (10 мл), и образуется осадок. Прибавляют воду (40 мл), и осадок исчезает. Экстракция с помощью EtOAc с последующей хроматографией на колонке дает искомое ипсо-метилпроизводное (169 мг, выход 86%, Rf=0,53, гексаны/EtOAc, 2:1).
Стадия 3: Продукт, полученный на стадии 2, обрабатывают способом, описанным в Примере 11, стадия 6, и получают искомое соединение. Разложение 198°С. МСВР (в виде хлористоводородной соли): найдено, 460,3079.
С использованием аналогичной методики получают соединения формулы
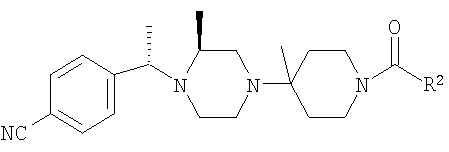
где R2 являются такими, как указано в таблице: 20
Пример 22
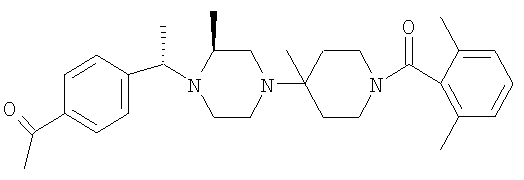
Стадия 1: Амин Штрекера, полученный в Примере 21, стадия 1 (380 мг, 0,87 ммоль), при кипячении с обратным холодильником в течение ночи обрабатывают с помощью CH3MgBr (2,9 мл, 8,7 ммоль) в Et2O (5 мл). Смесь охлаждают в бане со льдом и по каплям прибавляют воду (5 мл). Прибавляют 12 н. HCl (6 мл) и смесь 2 ч перемешивают на паровой бане. После охлаждения смеси на льду прибавляют NaOH, пока значение рН раствора не станет равным более 10. Экстракция с помощью EtOAc дает свободный амин в виде сиропа (370 мг, выход 100%).
Стадия 2: Продукт, полученный на стадии 1, превращают в искомое соединение в соответствии с методикой получения пептида по реакции сочетания, описанной в Примере 11, стадия 6. Т.пл.: 80-85°С. МСВР: найдено, 476,3271.
С использованием аналогичной методики получают соединения формулы
где R2 являются такими, как указано в таблице: 21
Пример 23

Стадии 1-3:

Стадия 1: Этилдиацетоацетат (93,4 г), CS2СО3 (185 г) и CH3CN (550 мл) смешивают друг с другом с помощью верхней механической мешалки. Прибавляют СН3CN (50 мл) и полученную смесь охлаждают до 0°С. По каплям прибавляют метилтрифторметансульфонат (88,6 г) и после завершения прибавления охлаждающую баню удаляют. Смесь перемешивают 1 ч при комнатной температуре, фильтруют и соль промывают с помощью Et2O (2×50 мл). Органический экстракты объединяют и прибавляют Et2O (300 мл). Полученную смесь фильтруют, осадок на фильтре промывают с помощью Et2O (2×100 мл), экстракты, содержащие Et2O, объединяют и упаривают до половинного объема. Раствор охлаждают на бане со льдом и один раз промывают охлажденным (0°С) 2 н. раствором NaOH (рН 11). Слой, содержащий Et2O, сушат над MgSO4, фильтруют и выпаривают, получая с выходом 65% искомый продукт в виде желтой жидкости (64,7 г), который используют на следующей стадии без дополнительной обработки.
Стадия 2: При комнатной температуре смешивают продукт, полученный на стадии 1 (64,2 г), раствор этоксида натрия в этаноле (коммерчески доступный раствор; 21 мас.%; 113 г), этанол (587 мл) и формамидинацетат (36,2 г). После кипячения с обратным холодильником в течение 4 ч смесь охлаждают до комнатной температуры, образующийся осадок отфильтровывают и этанол удаляют в вакууме. Полученную жидкость подвергают распределению между водой и CH2Cl2 и водный слой экстрагируют с помощью СН2Cl2 (3×150 мл). Экстракты в CH2Cl2 в сушат над MgSO4, фильтруют и выпаривают, получая темную неочищенную жидкость (50,7 г), которую очищают с помощью хроматографии на силикагеле (980 г, элюент - 4:1, гексаны: EtOAc). После выпаривания соответствующих фракций искомый продукт (28,5 г) выделяют с выходом 46% и используют на следующей стадии без дополнительной обработки.
Стадия 3: Продукт, полученный на стадии 2 (28,1 г), NaOH (6,72 г), воду (65 мл) и EtOH (130 мл) смешивают при комнатной температуре и 1 ч кипятят с обратным холодильником. Полученный раствор охлаждают до комнатной температуры и летучие вещества удаляют в вакууме до образования густой пасты. Прибавляют воду (20 мл), смесь охлаждают до 0°C и при перемешивании по каплям прибавляют концентрированную HCl (14,3 мл). Образовавшийся белый осадок собирают с помощью фильтрования, промывают ледяной водой (2×10 мл) и в течение 30 мин сушат на воздухе с отсасыванием. Полученное белое твердое вещество обрабатывают толуолом (2×20 мл), растворитель удаляют в вакууме при 50°С и затем 18 ч сушат в вакууме (1 мм рт.ст.). Искомый продукт выделяют в виде белого твердого вещества с выходом 63%. Т.пл.: 176-178°С. Элементный анализ C7H8N2O2: рассчитано: С, 55,26%; Н, 5,30%; N, 18,41%; найдено: С, 55,13%; Н, 5,44%; N, 18,18%.
Вторую порцию продукта выделяют путем выпаривания досуха полученного выше водного фильтрата и прибавления воды (20 мл). Полученную смесь 5 мин перемешивают при комнатной температуре, охлаждают на бане со льдом и образовавшийся осадок собирают путем фильтрования. Полученное твердое вещество промывают ледяной водой (2×5 мл) и сушат, как описано выше, получая продукт (4,68 г) в виде твердого вещества кремового цвета, что приводит к суммарному выходу, равному 83%.
Стадия 4: Продукт, полученный в Примере 4, стадия 6 (в виде тригидрохлорида; 5,4 г), DMF (11,3 мл), НОВТ (3,07 г), диизопропилэтиламин (12,3 мл) и продукт, полученный на стадии 3 (3,45 г), смешивают и в течение 15 мин порциями прибавляют DEC (4,35 г). Полученную смесь 18 ч нагревают при 45°С, охлаждают до комнатной температуры, разбавляют с помощью EtOAc (80 мл) и промывают 2 н. раствором NaOH (25 мл). Водный слой экстрагируют с помощью EtOAc (3×25 мл), органические экстракты объединяют, промывают солевым раствором, сушат над Na2SO4, фильтруют и выпаривают. Полученное неочищенное масло очищают с помощью хроматографии на силикагеле (170 г; элюент - 76:19:5, гексаны: EtOAc: Et3N). После выпаривания соответствующих фракций свободное основание искомого соединения (5,21 г) выделяют в виде слабоокрашенной пены с выходом 91%.
Стадия 5: К охлажденному (0°С) раствору свободного основания, полученного на стадии 4 (2,00 г), и EtOAc (20 мл) прибавляют HCl (3,0 мл 4,0 М раствора в 1,4-диоксане). Полученную смесь нагревают до комнатной температуры, разбавляют с помощью Et2O (20 мл), фильтруют, промывают с помощью Et2O (2×20 мл), 10 мин сушат на воздухе с отсасыванием и затем 5 ч сушат в вакууме (1 мм рт.ст.) при 90°С, получая искомое соединение (2,30 г) в виде белого твердого вещества с выходом 97%. Т.пл.: 159-162°С. Элементный анализ C27H35N5OF3·2HCl·0,5H2O: рассчитано: С, 55,38%; Н, 6,71%; N, 11,96%; Cl, 12,11%; найдено: С, 55,19%; Н, 6,69%; N, 11,75%; Cl, 11,45%.
По аналогичным методикам получают дополнительные производные пиримидина:
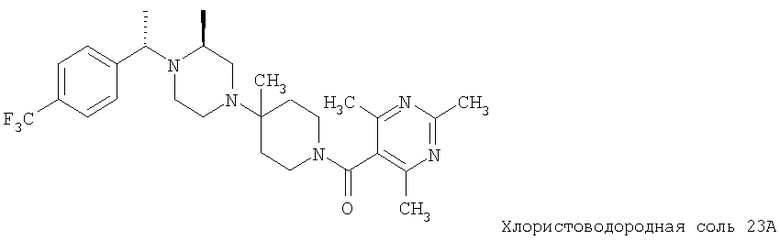
Стадии 1-2:
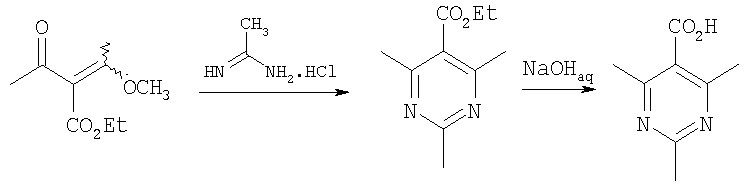
Стадия 1: Продукт, полученный в Примере 23, стадия 1, обрабатывают таким же способом, как и в Примере 23, стадия 2, заменяя формамидинацетат на ацетамидингидрохлорид (2,03 г). Используют следующие количества реагентов: продукт, полученный в Примере 23, стадия 1 (4,0 г), этанол (20 мл) и раствор этоксида натрия в этаноле (коммерчески доступный раствор; 21 мас.%; 8,03 г). После экстрагирования и очистки, выполняемых так, как это описано выше, продукт выделяют в виде бесцветной жидкости (1,7 г) с выходом 41% и используют на следующей стадии без дополнительной обработки.
Стадия 2: Продукт, полученный на стадии 1 (1,7 г), обрабатывают таким же способом, как и в Примере 23, стадия 3, с использованием этанола (5 мл), воды (5 мл) и NaOH (1,0 г). После экстракции и очистки, проводимой так, как это описано выше, продукт выделяют (0,12 г) в виде белого твердого вещества с выходом 8% и используют на следующей стадии без дополнительной обработки.
Стадия 3: Продукт, полученный в Примере 4, стадия 6 (0,5 г), и продукт, полученный на стадии 2 (предыдущая стадия) (0,028 г), вводят в реакцию при тех же условиях, что и в Примере 23, стадия 4, с использованием НОВТ (20 мг), DEC (45 мг), диизопропилэтиламина (40 мг) и DMF (1,5 мл). После экстракции и очистки, проводимой так, как это описано выше, продукт выделяют в виде хлористоводородной соли с использованием методики, описанной в Примере 23, стадия 5, и получают искомое соединение (77 мг) в виде белого твердого вещества с суммарным выходом для двух стадий, равным 97%. Т.пл.: 185-190°С.
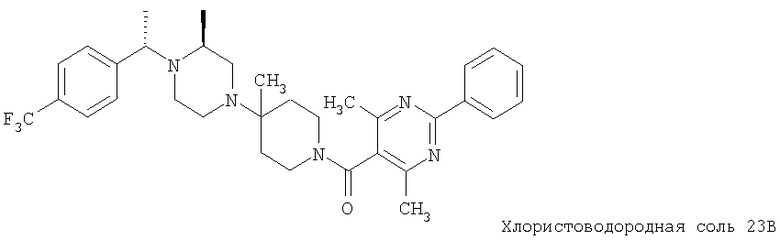
Стадии 1-2:
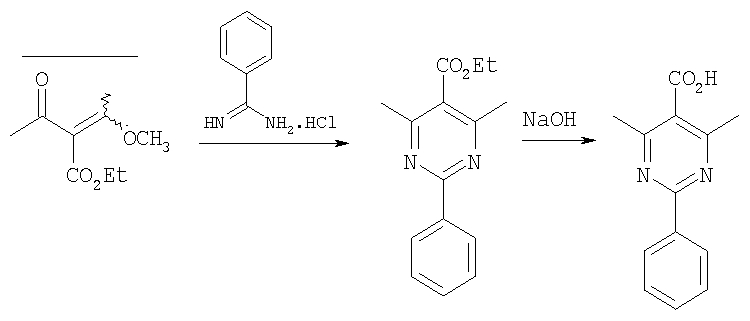
Стадия 1: Продукт, полученный в Примере 23, стадия 1, обрабатывают таким же способом, как и в Примере 23, стадия 2, заменяя формамидинацетат на бензамидингидрохлорид (3,35 г). Используют следующие количества реагентов: продукт, полученный в Примере 23, стадия 1 (4,0 г), этанол (20 мл) и раствор этоксида натрия в этаноле (коммерчески доступный раствор; 21 мас.%; 8,03 г). После экстрагирования и очистки, выполняемых так, как это описано выше, продукт выделяют в виде бесцветной жидкости (4,5 г) с выходом 82% и используют на следующей стадии без дополнительной обработки.
Стадия 2: Продукт, полученный на стадии 1 (4,5 г), обрабатывают таким же способом, как и в Примере 23, стадия 3, с использованием этанола (10 мл), воды (10 мл) и NaOH (2,0 г). После экстракции и очистки, проводимой так, как это описано выше, продукт выделяют (3,0 г) в виде белого твердого вещества с выходом 77% и используют непосредственно на следующей стадии.
Стадия 3: Продукт, полученный в Примере 4, стадия 6 (75 мг) и продукт, полученный на стадии 2 (предыдущая стадия) (39 мг), вводят в реакцию при тех же условиях, что и в Примере 23, стадия 4, с использованием НОВТ (35 мг), DEC (53 мг), диизопропилэтиламина (100 мг) и DMF (2 мл). После экстракции и очистки, проводимой так, как это описано выше, продукт выделяют в виде хлористоводородной соли с использованием методики, описанной в Примере 23, стадия 5, и получают искомое соединение (98 мг) в виде белого твердого вещества с суммарным выходом для двух стадий, равным 96%. Т.пл.: 250-253°С.
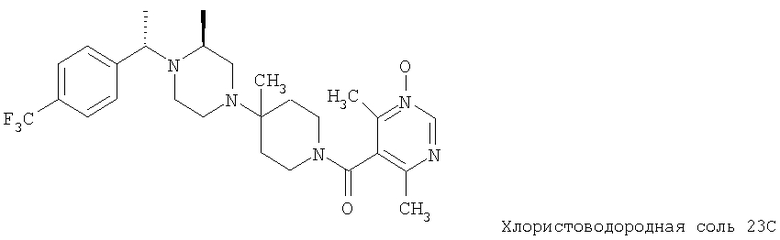
Стадии 1-2:
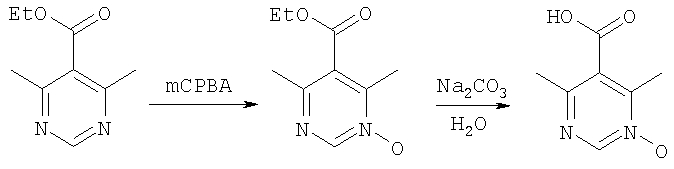
Стадия 1: Продукт, полученный в Примере 23, стадия 2 (528 мг), растворяют в CH2Cl2 (5,0 мл) и при комнатной температуре тремя порциями прибавляют мета-хлорбензойную кислоту (mCPBA) (600 мг). Полученную смесь 24 ч перемешивают при комнатной температуре и прибавляют СН2Cl2 (2 мл) и mCPBA (200 мг). Через 3 ч смесь выливают в колонку с силикагелем (40 г) и элюируют смесью гексаны: EtOAc, 1:1, а затем смесью СН2Cl2:СН3ОН, 10:1. После выпаривания соответствующих фракций продукт выделяют (512 мг) в виде воскообразного белого твердого вещества с выходом 89% и используют на следующей стадии без дополнительной обработки.
Стадия 2: Продукт, полученный на стадии 1, растворяют в СН3ОН (1,8 мл) и прибавляют 1,0 М раствор Na2CO3 (1,5 мл). После перемешивания при комнатной температуре в течение 36 ч полученную смесь выпаривают досуха, прибавляют толуол (2 мл) и смесь выпаривают досуха. Полученное неочищенное твердое вещество (153 мг) используют на следующей стадии без дополнительной очистки.
Стадия 3: Продукт, полученный в Примере 4, стадия 6 (94 мг), и продукт, полученный на стадии 2 (предыдущая стадия) (76 мг), вводят в реакцию при тех же условиях, что и в Примере 23, стадия 4, с использованием НОВТ (92 мг), DEC (130 мг), диизопропилэтиламина (0,14 мл) и DMF (0,25 мл). После экстракции и очистки с помощью тонкослойной хроматографии (пластина с диоксидом кремния 1000 мкм; элюент - 95:5, EtOAc:Et3N) свободное основание искомого соединения (52 мг) выделяют в виде пены с выходом 40%. МСВР: Рассчитано: МН+: C27H37N5O2F3: 520,2899; Найдено: 520,2908.
Стадия 4: Продукт, полученный на стадии 3 (52 мг), вводят в реакцию при тех же условиях, что и в Примере 23, стадия 5, с использованием EtOAc (1,0 мл) и HCl (4,0 М раствор в 1,4-диоксане; 75 мкл) и после обработки получают искомое соединение (44,5 мг) в виде белого твердого вещества с выходом 76%. Т.пл.: разложение при температуре выше 161°С.
С использованием аналогичной методики получают соединения формулы
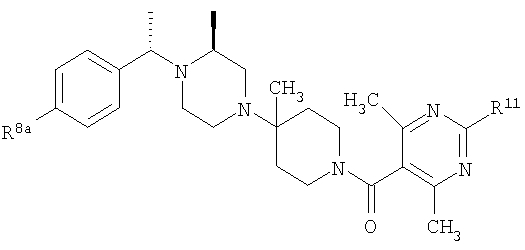
где R8a и R11 являются такими, как указано в таблице: 22

Пример 24
Арилциклопропиламиды
Методика A:
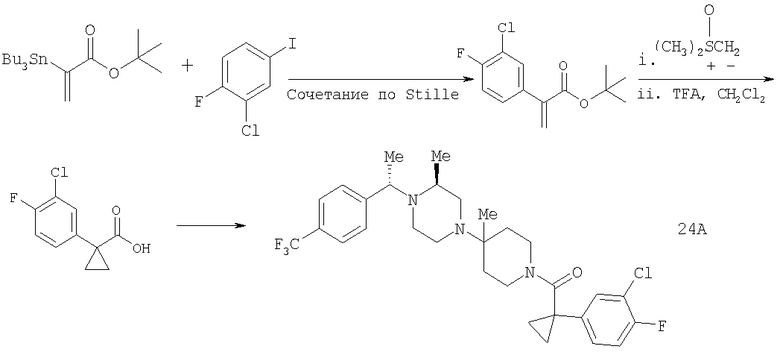
Стадия 1: К станнану (0,39 г, 0,95 ммоль) в DMF (10 мл) прибавляют 2-хлор-4-фторйодбензол (0,73 г, 2,86 ммоль), CuI (0,19 г, 1,05 ммоль) и тетракис-(трифенилфосфин)-палладий(0) (0,11 г, 0,095 ммоль). Реакционную смесь 21 ч перемешивают в атмосфере N2 при комнатной температуре. Реакционную смесь прибавляют к Et2O и гетерогенный раствор фильтруют через слой целита, промывая с помощью EtOAc. Фильтрат промывают водой и солевым раствором и сушат (MgSO4). Фильтрование и выпаривание растворителя в вакууме дает остаток, который предварительно адсорбируют на силикагеле. Очистка с помощью хроматографии на силикагеле (4% EtOAc/гексан) дает арилакрилат (0,19 г, 78%), который используют на следующей стадии без дополнительной обработки.
Стадия 2: К триметилсульфоксониййодиду (0,18 г, 0,81 ммоль) в DMSO (1,6 мл) прибавляют трет-бутоксид калия (0,09 г, 0,81 ммоль). Реакционную смесь 1 ч перемешивают при комнатной температуре, а затем прибавляют арилакрилат (0,19 г, 0,74 ммоль) в DMSO (1,6 мл). Реакционную смесь 5 ч перемешивают при комнатной температуре и прибавляют воду. Смесь экстрагируют с помощью EtOAc. Объединенные органические слои промывают водой и солевым раствором и сушат (MgSO4). Фильтрование и выпаривание растворителя в вакууме дает арилциклопропиловый сложный эфир, который вводят в реакцию непосредственно, смешивая с CH2Cl2 (3 мл) и прибавляя TFA (0,5 мл). Реакционную смесь 15 ч перемешивают при комнатной температуре и затем концентрируют в вакууме, получая арилциклопропилкарбоновую кислоту (0,14 г, 91% - 2 стадии). Без дополнительной очистки карбоновую кислоту вводят в реакцию сочетания с продуктом, полученным в Примере 8, стадия 3, с помощью методики, описанной в Примере 8, стадия 4, и получают 24А в виде хлористоводородной соли. МСВР (М+Н): найдено, 566,2561.
Методика B:
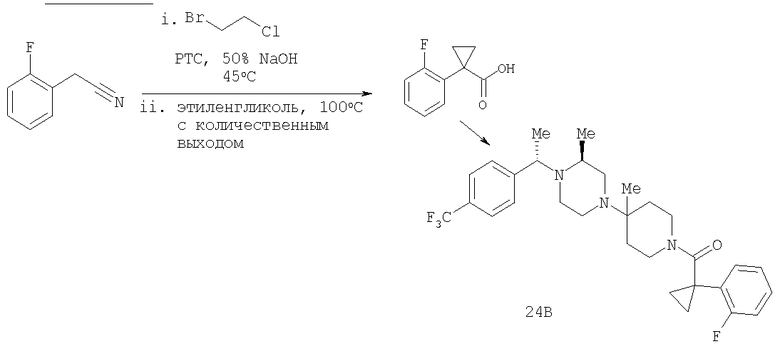
К 2-фторфенилацетонитрилу (0,80 г, 5,92 ммоль), бензилтриэтиламмонийхлориду (0,03 г, 0,12 ммоль) и 1-бром-2-хлорэтану (1,70 г, 11,9 ммоль) прибавляют 50% водный раствор NaOH (3,5 мл). Реакционную смесь 21 ч перемешивают при 45°C и прибавляют этиленгликоль (3 мл). Реакционную смесь нагревают до 100°C и перемешивают 7 ч. После охлаждения до комнатной температуры реакционную смесь разбавляют водой и промывают с помощью EtOAc. С помощью 6 н. водного раствора HCl водный слой подкисляют до значения рН, равного 2-3. Подкисленный раствор экстрагируют с помощью Et2O. Объединенные экстракты в Et2O промывают водой и солевым раствором и сушат (MgSO4). Фильтрование и выпаривание растворителя в вакууме дает бледно-желтое твердое вещество (1,06 г, 99%). Арилциклопропиловую кислоту вводят в реакцию сочетания с продуктом, полученным в Примере 8, стадия 3, с использованием методики, описанной в Примере 8, стадия 4, и получают 24 В в виде хлористоводородной соли. МСВР (М+Н): найдено, 532,2949.
С использованием аналогичной методики получают соединения формулы
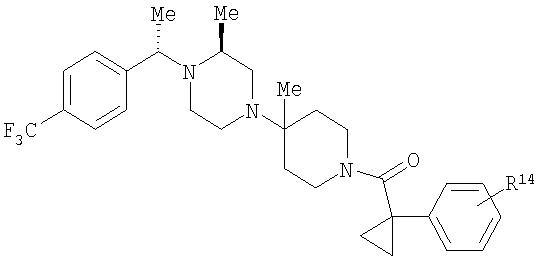
где
являются такими, как указано в таблице: 23




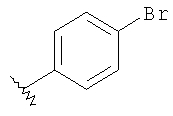
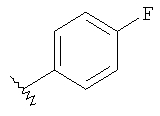
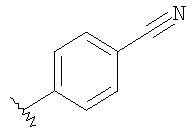

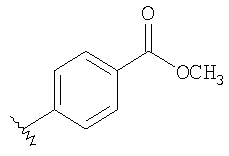
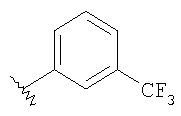


Пример 25
Стадия 1:
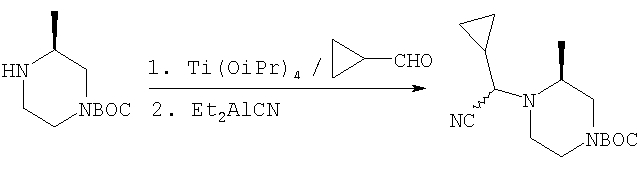
Циклопропилкарбоксальдегид (3,4 мл) S-метил-N-ВОС-пиперазин (8,28 г), CH2Cl2 (82 мл) и Ti(OiPr)4 (15,80 мл) смешивают и 23 ч перемешивают при комнатной температуре, а затем полученный раствор охлаждают до 0°C и прибавляют Et2AlCN (1,0 М в толуоле; 62,1 мл). Раствор 5 ч перемешивают при комнатной температуре. Прибавляют смесь KF (20 г) и целита (10 г), а затем осторожно прибавляют EtOAc (120 мл) и воду (120 мл). Полученную взвесь перемешивают 15 мин, фильтруют, промывают с помощью EtOAc (3×35 мл) и слой, содержащий EtOAc, отделяют, промывают солевым раствором, сушат над Na2SO4, фильтруют и выпаривают, получая искомый промежуточный продукт (12,0 г), который используют непосредственно на следующей стадии.
Стадия 2:
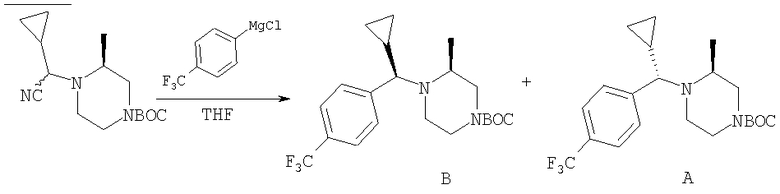
К раствору 4-йодбензотрифторида (40 г) и THF (52 мл) при 0°C прибавляют изопропилмагнийхлорид (2,0 М в Et2O; 74 мл). Полученный раствор 1 ч перемешивают при комнатной температуре и затем в течение 10 мин прибавляют к охлажденному до 0°C раствору продукта, полученного на стадии 1 (10,0 г) и THF (26 мл). Реакционную смесь нагревают до комнатной температуры, перемешивают в течение ночи и прибавляют EtOAc (50 мл). После перемешивания в течение 10 мин прибавляют 2 н. раствор NaOH (50 мл) и полученную смесь перемешивают 30 мин, фильтруют и соли промывают с помощью EtOAc (3×20 мл). Объединенные экстракты в EtOAc промывают солевым раствором, сушат над Na2SO4, фильтруют и выпаривают, получая неочищенный продукт (28 г) в виде масла золотистого цвета, которое хроматографируют на силикагеле (1 кг), элюируя с помощью смеси гексаны: EtOAc (8:1). Два диастереоизомерных продукта собирают в виде одной фракции (15,9 г) и дополнительно очищают с помощью хроматографии на колонке, как это описано выше, и получают промежуточный продукт A (Rf=0,47 в смеси гексаны: EtOAc, 4:1; 5,34 г), который загрязнен неидентифицированной примесью. (Также собирают второй диастереоизомер В (Rf=0,29 в смеси гексаны: EtOAc, 4:1)).
Стадия 3:
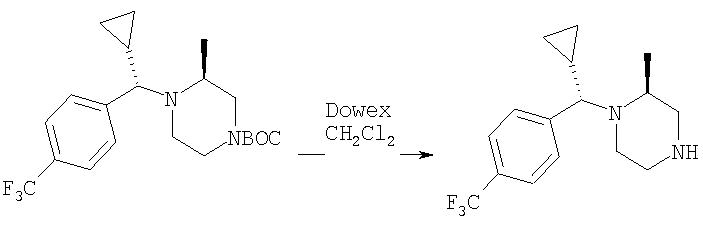
К раствору A, полученного на стадии 2 (3,96 г), в CH2Cl2 (120 мл) прибавляют ионообменную смолу DOWEX 50Х2-100 (15 г) и полученную смесь 2,5 ч встряхивают при комнатной температуре. Смолу отфильтровывают и промывают с помощью CH2Cl2 (2×40 мл). Смолу обрабатывают с помощью 7 н. раствора NH3 в СН3ОН (30 мл), смолу отфильтровывают и эту процедуру повторяют дважды. Экстракты в СН3ОН объединяют и выпаривают. Полученное масло обрабатывают смесью толуол: CH2Cl2 (1:1, 15 мл) и выпаривают с получением пиперазинового промежуточного продукта (0,80 г) в виде прозрачного масла. МСВР: Рассчитано: МН+: C16H21N2F3: 299,1735; найдено: 299,1748.
Стадия 4:

Продукт, полученный на стадии 3 (0,57 г), обрабатывают таким же способом, что и в Примере 8, стадия 3, используя N-BOC-4-пиперидон (0,42 г), CH2Cl2 (3,84 мл), Ti(OiPr)4 (3,39 мл), Et2AlCN (2,88 мл) и CH3MgBr (3,0 М раствор в Et2O; 3,2 мл) с получением искомого продукта (0,78 г) в виде прозрачного масла с выходом 82%.
Стадия 5: Продукт, полученный на стадии 4 (0,12 г), обрабатывают с помощью смеси АсОН-Ch2Cl2 (3:1, об./об.; 1,4 мл), а затем BF3Et2O (0,14 мл). После перемешивания в течение 1 ч полученный раствор разбавляют с помощью CH2Cl2 (10 мл), охлаждают до 0°C и с помощью твердого NaOH доводят значение рН до 10. Прибавляют воду (10 мл) и отделяют слой, содержащий CH2Cl2. После дополнительной экстракции с помощью CH2Cl2 (2×10 мл) органический слой промывают водой, солевым раствором, сушат над Na2SO4, фильтруют и выпаривают, получая свободный пиперидин (80 мг) с выходом 81%.
Стадия 6: Продукт, полученный на стадии 5 (57 мг), обрабатывают таким же способом, что и в Примере 8, стадия 4, с использованием DMF (0,30 мл), НОВТ (41 мг), DEC (57 мг), диизопропилэтиламина (0,08 мл) и 4,6-диметил-5-пиримидинкарбоновой кислоты (43 мг); реакционную смесь 5 ч перемешивают при 45°С. Очистку неочищенного масла проводят с помощью препаративной тонкослойной хроматографии (адсорбент - диоксид кремния: 2000 мкм; элюент - 76:19:5, EtOAc: гексаны: Et3N) и после элюирования необходимой зоны (1:1, CH2Cl2:МеОН) и концентрирования растворителя получают искомое соединение (70 мг) в виде прозрачного масла с выходом 93%. Хлористоводородную соль получают так, как это описано в Примере 8, стадия 4 (78 мг) с выходом 100%. Т.пл.: 147-149°С.
С помощью аналогичной методики получают следующее соединение:
Т.пл.: >188°C (с разложением).
Пример 26

Стадия 1:
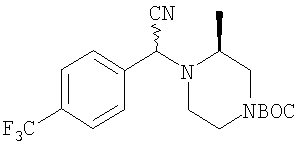
Искомое соединение получают способом, сходным с описанным в Примере 25, стадия 1, с использованием п-трифторметилбензальдегида (20 г) вместо цикло-пропилкарбоксальдегида и после обработки получают смесь диастереоизомеров (22,7 г) с выходом 59%.
Стадия 2:
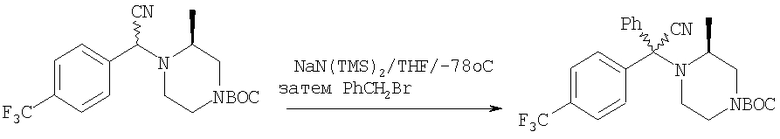
К охлажденному до -70°C раствору продукта, полученного на стадии 1 (1,9 г), в THF (15 мл) прибавляют NaHMDS (1,0 М раствор в THF; 7,5 мл), а затем бензил-бромид (2 мл). Охлаждающую баню удаляют и полученный раствор перемешивают 45 мин. Прибавляют концентрированный NH4OH (10 мл) и реакционную смесь перемешивают 30 мин. Полученную смесь подвергают распределению между водой и CH2Cl2, экстракты в CH2Cl2 отделяют и выпаривают и неочищенное масло очищают с помощью хроматографии на колонке (силикагель; элюенты-гексаны: CH2Cl2, 2:1; гексаны: EtOAc, от 10:1 до 7:1) и после выпаривания соответствующих фракций получают смесь промежуточных продуктов (1,92 г) в виде желтой пены.
Стадия 3:
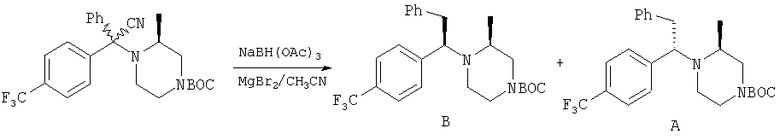
Смесь, полученную на стадии 2 (1,91 г), СН3СН (35 мл), триацетоксиборогидрид натрия (4,0 г) и магнийбромидэфират (2,25 г) смешивают и 70 ч перемешивают при комнатной температуре. Прибавляют воду (25 мл) и затем постепенно прибавляют раствор Na2CO3 (10 г) в воде (50 мл). После экстракции с помощью EtOAc (2×50 мл), сушки и упаривания органического слоя полученное масло очищают с помощью препаративной тонкослойной хроматографии (пластины с диоксидом кремния 5×2000 мМ; элюент - 6:1 гексаны: EtOAc). Менее полярную зону отбирают, обрабатывают с помощью смеси метанол: СН2Cl2 состава 1:1, фильтруют и выпаривают, получая промежуточный продукт A (0,84 г) в виде белой пены. МСВР: Рассчитано: МН+: C25H29O2N2F3: 449,2407; найдено: 449,2416.
Стадия 4: Продукт, полученный на стадии 3 (0,81 г), обрабатывают таким же способом, что и в Примере 8, стадия 3, используя TFA (5 мл) и CH2Cl2 (10 мл), и после обработки получают свободный пиперазин (0,60 г) в виде прозрачной смолы. МСВР: Рассчитано: МН+: C20H23N2F3: 349,1892; найдено: 349,1894.
Стадия 5: Продукт, полученный на стадии 4 (0,39 г), обрабатывают таким же способом, что и в Примере 8, стадия 1, используя N-BOC-4-пиперидон (0,25 г), CH2Cl2 (8 мл), Ti(OiPr)4 (0,40 мг), Et2AlCN (2 мл) и CH3MgBr (3,0 М раствор в Et2O; 1,5 мл) с получением искомого пиперидинильного промежуточного продукта с защитной группой ВОС (0,44 г) в виде прозрачного масла с выходом 72%. МСВР: Рассчитано: MH+: С31H42О2N3F3: 546,3307; найдено: 546,3315.
Стадия 6: Продукт, полученный на стадии 5 (0,43 г), обрабатывают таким же способом, что и в Примере 8, стадия 3, используя TFA (3 мл), CH2Cl2 (2 мл) и воду (0,2 мл), и после обработки получают свободный пиперидинильный промежуточный продукт (0,37 г) в виде прозрачного масла.
Стадия 7: Продукт, полученный на стадии 6 (50 мг), обрабатывают таким же способом, что и в Примере 8, стадия 4, используя CH2Cl2 (3 мл), НОВТ (28 мг), DEC (40 мг), диизопропилэтиламин (42 мг) и 4,6-диметил-5-пиримидинкарбоновую кислоту (24 мг); реакционную смесь 2 дня перемешивают при комнатной температуре. С использованием методики, описанной в Примере 8, стадия 4, гидрохлорид искомого соединения (59 мг) получают с выходом 91% (из продукта стадии 5). Т.пл.: 187-196°С. МСВР: Рассчитано: МН+. С33Н40ON5F3: 580,3263; Найдено: 580,3263.
С использованием аналогичной методики получают соединения формулы
где R8a, R3 и R2 являются такими, как указано в таблице: 24
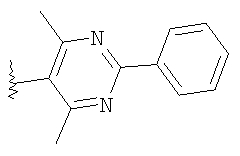

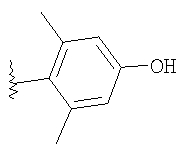
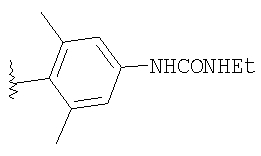
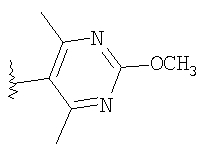
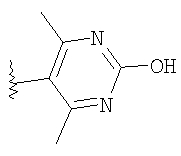
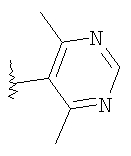


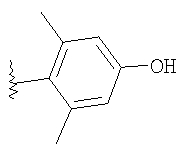
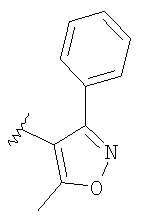
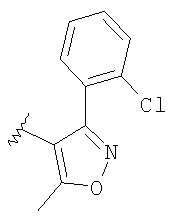

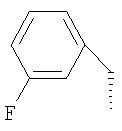
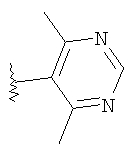

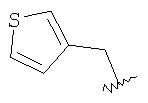
Пример 27
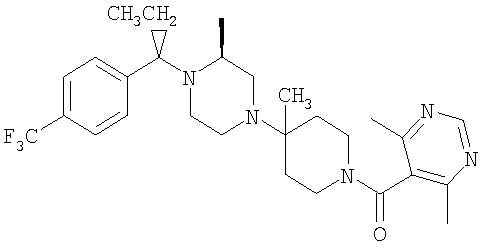
Стадия 1:

4'-(Трифторметил)-пропиофенон (2,02 г, 0,01 моль) и (S)-2-метил-CBS-оксазаборолидин (1 М раствор в THF) (2,0 мл, 0,002 моль) в THF (10 мл) охлаждают на бане со льдом и к смеси по каплям прибавляют комплекс боран-метил-сульфид (2 М раствор в THF) (3 мл, 0,006 моль). Смесь 30 мин перемешивают при 0°C и медленно прибавляют СН3ОН, пока не перестанут выделяться пузырьки.
Растворители удаляют при пониженном давлении и к смеси прибавляют раствор HCl (1 н.). Экстракция с помощью EtOAc с последующей хроматографией на силикагеле дает спирт (1,47 г) с выходом 72%.
Стадия 2: Раствор продукта, полученного на стадии 1 (4,32 г, 0,021 моль), и Et3N (5,9 мл, 0,042 моль) в СН2Cl2 (20 мл) охлаждают до 0°C в бане со льдом и по каплям прибавляют СН3SO2Cl (2,13 мл, 0,028 моль). Смесь 1 ч перемешивают при 0°C и баню со льдом удаляют. К смеси прибавляют воду, и экстракция с помощью СН2Cl2 дает метилсульфонат (5,99 г) с количественным выходом.
Стадия 3: Продукт, полученный на стадии 2 (5,93 г, 0,021 моль), и 1-трет-бутоксикарбонил-3S-метилпиперазин (4,2 г, 0,021 моль) растворяют в безводном CH3CN (20 мл) и к раствору прибавляют высушенный в сушильном шкафу K2СО3 (4,35 г, 0,032 моль). Смесь 2 дня кипятят с обратным холодильником при перемешивании, затем разбавляют водой. Экстракция с помощью EtOAc с последующей хроматографией на силикагеле дает искомый продукт (3,16 г) с выходом 39%.
Стадия 4: К раствору продукта, полученного на стадии 3 (1,15 г, 2,59 ммоль), в CH2Cl2 (5 мл) прибавляют TFA (10 мл) и смесь 2 ч перемешивают при комнатной температуре, а затем концентрируют при пониженном давлении. К остатку прибавляют NaOH (3 н.), и, экстракция с помощью EtOAc дает искомый амин с количественным выходом.
Стадия 5: Продукт, полученный на стадии 4, и 1-трет-бутоксикарбонил-4-пиперидрн (0,94 г, 4,74 ммоль) обрабатывают с помощью Ti(OiPr)4, Et2AlCN и CH3MgBr способом, сходным с описанным в Примере 8, стадия 1, и получают искомый продукт (1,09 г) с выходом 87% (из амина стадии 4).
Стадия 6: К раствору продукта, полученного на стадии 5 (0,76 мг, 1,57 ммоль), в CH2Cl2 (2 мл) прибавляют TFA (4 мл) и смесь 2 ч перемешивают при комнатной температуре, а затем концентрируют при пониженном давлении. К остатку прибавляют NaOH (3 н.), и экстракция с помощью EtOAc дает искомый амин с количественным выходом.
Стадия 7: Амин, полученный на стадии 6, и 4,6-диметилпиримидин-5-карбоновую кислоту вводят в реакцию сочетания, как это описано в Примере 8, стадия 4, и получают искомое соединение (0,58 г) с выходом 72%. Т.пл.: 160°С. МСВР: Рассчитано: МН+: Найдено: 518,3123.
С использованием аналогичной методики получают соединения формулы

где Z, R3, R6 и R2 являются такими, как указано в приведенной ниже таблице: 25

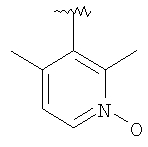
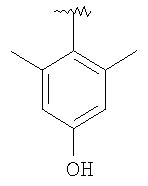
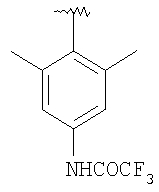
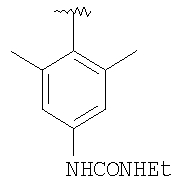
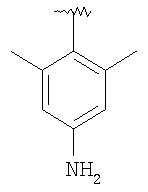
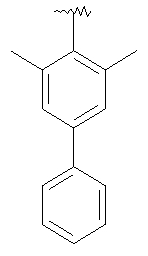
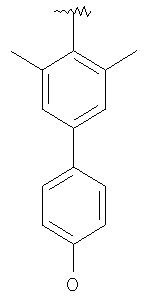
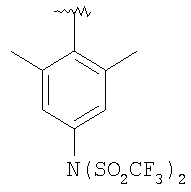


С использованием аналогичных методик также получают следующие соединения:

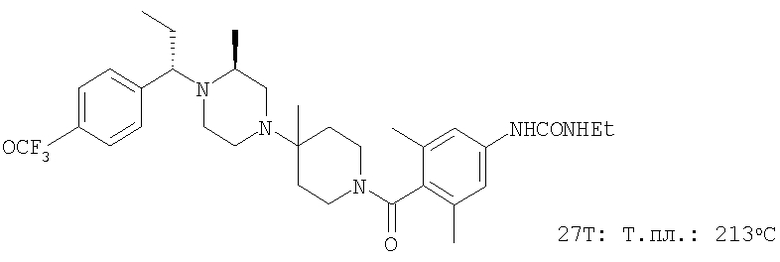
Пример 28
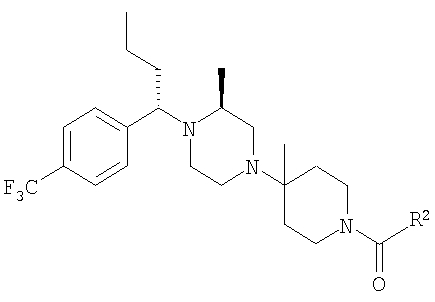
Стадии 1-4:
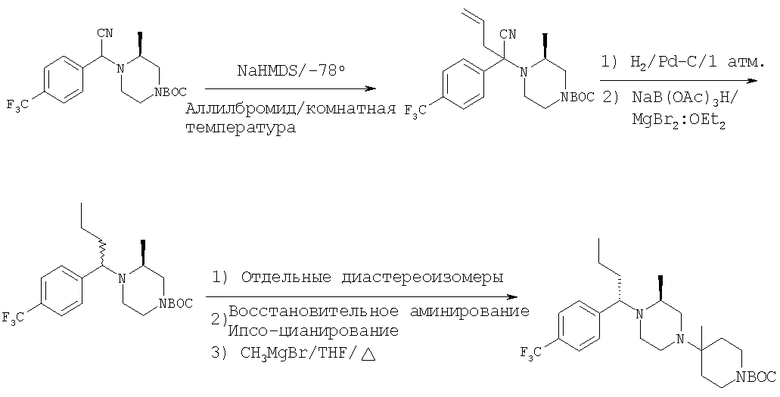
Стадия 1: Цианамин получают из п-трифторметилбензальдегида и 2(S)-метил-4-(трет-бутоксикарбонил)-пиперазина точно по методике, описанной в Примере 6, стадия 1.
Стадия 2: Раствор цианамина 2 (2,5 г; 6,53 ммоль) в 30 мл сухого THF помещают в сосуд в атмосфере N2 и охлаждают до -78°С. Этот раствор обрабатывают раствором гексаметилдисилазида натрия в THF (1 М; 26 мл), а через 5 мин - неразбавленным аллилбромидом (6 мл). После удаления бани смеси дают нагреться до комнатной температуры (˜1 ч) и желтая окраска раствора переходит в темно-красновато-коричневую. Реакцию останавливают насыщенным раствором NH4Cl и продукт экстрагируют с помощью EtOAc, промывают водой, солевым раствором и сушат. Концентрирование в вакууме дает коричневое полужидкое вещество. ФХСГ этого материала с использованием в качестве элюента 25% раствора Et2O в гексане дает 2,5 г (92%) искомого продукта в виде смолы янтарного цвета (ТСХ, Rf=0,65, 0,6 для двух перекрывающихся зон).
Стадия 3: Раствор продукта, полученного на стадии 2 (2,4 г), в СН3ОН обрабатывают с помощью 10% Pd/C (0,2 г) и в сосуд подают Н2 из баллона. После перемешивания в течение 4 ч при комнатной температуре катализатор отделяют путем фильтрования через целит. Концентрирование фильтрата дает смолу янтарного цвета.
Полученный выше α-пропилнитрат растворяют в CH3CN (12 мл), прибавляют эфират бромида магния (2,1 г; 8,14 ммоль) и триацетоксиборгидрид натрия (3,44 г; 16,2 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакцию останавливают водой и подщелачивают с помощью насыщенного раствора NaHCO3. Органические продукты экстрагируют с помощью EtOAc и обрабатывают, получая ˜2 г неочищенного вещества. С помощью ФХСГ (10-25% раствор Et2O в гексане) выделяют два диастереоизомерных продукта (суммарный выход 1,7 г; 79% для двух стадий):
(S,S)-Диастереоизомер (А): ТСХ, Rf=0,6 (25% Et2O-гексан). 0,9 г бесцветной смолы.
(R,S)-Диастереоизомер (В): ТСХ, Rf=0,5 (25% Et2O-гексан). 0,8 г бесцветной смолы.
Стадия 4: Удаление защитной группы ВОС от промежуточного продукта A проводят путем обработки с помощью TFA в CH2Cl2. Выделенный свободный пиперазин (0,68 г; 2,3 ммоль), N-(трет-бутоксикарбонил)-4-пиперидинон (0,45 г; 2,3 ммоль) и Ti(OiPr)4 (0,7 мл; 2,5 ммоль) растворяют в 10 мл CH2Cl2 и перемешивают в течение ночи. В реакционную смесь вводят Et2AlCN (1 M раствор в толуоле; 2,7 мл) и полученный раствор перемешивают один день. Реакционную смесь разбавляют с помощью EtOAc и реакцию останавливают водой. Для облегчения фильтрования солей титана и алюминия прибавляют целит. Двухфазный фильтрат промывают водой, солевым раствором и сушат. Концентрированно в вакууме дает 1,1 г желтой смолы (ТСХ, Rf=0,55 в 25% Et2O-гексан).
Полученное ипсо-циансоединение растворяют в сухом THF (8 мл) и обрабатывают раствором CH3MgBr (3 М в Et2O; 6 мл) и перемешивают в течение ночи при комнатной температуре. Колбу с реакционной смесью помещают в баню с холодной водой и реакцию осторожно останавливают насыщенным раствором NH4Cl. Органический продукт экстрагируют с помощью EtOAc и промывают водой и солевым раствором. Концентрированно продукта, очищенного с помощью ускоренной ФХСГ (10-25% раствор EtOAc в гексане) дает ВОС-пиперидинильное соединение в виде бледно-желтой смолы (1,1 г; 100%). ТСХ, Rf=0,6 в 25% Et2O-гексан.
Стадия 5: Защитную группу ВОС от пиперидинового атома азота продукта, полученного на стадии 4, удаляют путем обработки с помощью TFA в CH2Cl2. Подщелачивание с помощью 1 М раствора NaOH и обработка с помощью СН2Cl2 дают пиперидин без защитной группы с выходом 90%. Этот промежуточный продукт вводят в реакцию сочетания (EDCI, НОВТ) с арил- и гетероарилкарбоновыми кислотами и получают амиды, примеры которых приведены в следующей таблице:
где R2 являются такими, как указано в таблице: 26




С использованием аналогичной методики получают следующие соединения:
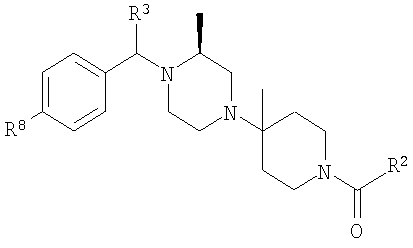
где R8, R3 и R2 являются такими, как указано в таблице: 27
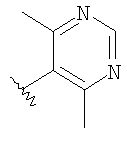
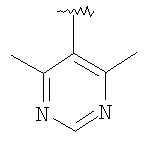
С использованием 3-фторбензилбромида или -хлорида вместо бензилбромида в методике, описанной в Примере 28, стадии 1-4 (изомер B обрабатывается на стадии 3), с последующим использованием методики, описанной в Примере 1, стадия 5, и с последующим использованием методики, описанной в Примере 26, стадии 6-7, получают следующее соединение (хлористоводородную соль):
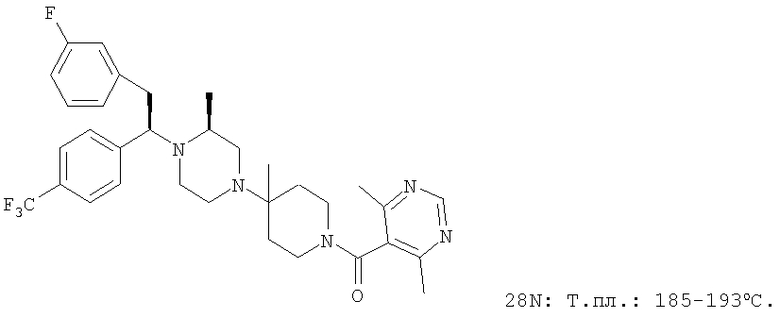
Пример 29
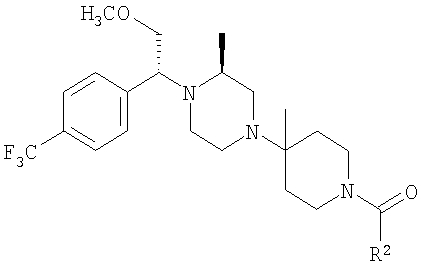
Стадии 1-3:
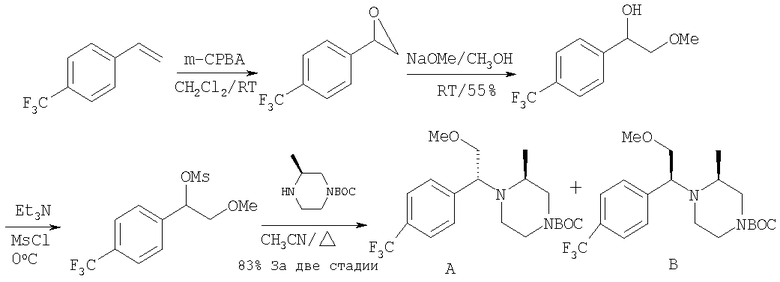
Стадия 1: Натриевую соль m-СРВА прибавляют к раствору п-трифторметилстирола (3 г; 17,4 ммоль) в 30 мл СН2Cl2 и 20 ч перемешивают при комнатной температуре. Прибавляют примерно 20 мл насыщенного раствора NaHCO3 и 2 ч перемешивают при комнатной температуре. Смесь разбавляют с помощью 20 мл CH2Cl2 и органический продукт экстрагируют в слой СН2Cl2. Органический экстракт обрабатывают и получают неочищенный продукт. ФХСГ дает 3 г (90%) искомого эпоксида в виде бесцветного масла. (ТСХ, Rf=0,8 (25% раствор EtOAc в гексане).
Стадия 2: К раствору продукта, полученного на стадии 1 (2 г; 10,6 ммоль), в 20 мл безводного СН3ОН прибавляют свежеприготовленный NaOCH3 (0,6 г; 10,6 ммоль). После перемешивания при комнатной температуре в течение одного дня СН3ОН удаляют в вакууме. Остаток растворяют в CH2Cl2 и промывают водой и солевым раствором. Концентрирование с последующей обработкой с помощью ФХСГ дает 1,3 г (55%) карбинола в виде бесцветного масла (Rf=0,30, 50% раствор Et2O в гексане).
Стадия 3: Карбинол, полученный на стадии 2 (1,3 г; 5,9 ммоль), растворяют в СН2Cl2 и охлаждают в бане со льдом. Последовательная обработка с помощью Et3N (1,7 мл; 12 ммоль) и СН3SO2Cl (0,6 мл; 7,7 ммоль) и перемешивание в течение 30 мин дает метилсульфонат. Продукт экстрагируют стандартным способом (выход =100%).
Метилсульфонат (1,76 г; 5,9 ммоль) и 2(S)-метил-4-(трет-бутоксикарбонил)-пиперазин (2,4 г; 12 ммоль) растворяют в 5 мл СН3СН и 19 ч кипятят с обратным холодильником. Реакционную смесь охлаждают до комнатной температуры и непосредственно обрабатывают с помощью флэш-хроматографии на силикагеле. Элюирование с помощью 25%, а затем 50% раствора Et2O в гексане позволяет выделить диастереоизомерные продукты А и В (суммарный выход =86%).
A: Rf=0,5 (50% Et2O в гексане). Светло-желтая смола (0,9 г; 42%).
В: Rf=0,4 (50% Et2O в гексане). Смола янтарного цвета (1,13 г; 44%).
Стадия 4: Восстановительное аминирование свободного пиперазина, полученного из A (0,9 г; 2,2 ммоль), с помощью N-ВОС-пиперидин-4-она с введением ипсо-метильной группы проводят так, как это описано в Примере 1, стадия 4, и получают пиперидинильное соединение с защитной группой ВОС (0,87 г; 92%). Rf=0,3 (50% Et2O в гексане).
Стадия 5: От пиперидинильного атома азота защитную группу ВОС удаляют с помощью TFA и полученное соединение вводят в реакцию сочетания с кислотами с использованием методики на основе EDCI/HOBT, описанной в Примере 8, стадия 4, и получают соединения, представленные в приведенной ниже таблице:
где R2 являются такими, как указано в таблице: 28
Пример 30
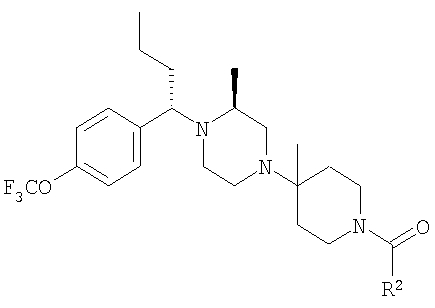
Стадия 1:

Раствор п-трифторметоксибензальдегида (0,48 мл, 3,36 ммоль), пиперидинопиперазина (1,00 г, 3,36 ммоль) и бензотриазола (0,48 г, 4,00 ммоль) в сухом толуоле 6 ч кипятят с обратным холодильником. Реакционную смесь охлаждают до комнатной температуры и растворитель удаляют в вакууме. После подтверждения образования продукта, выполненного с помощью ЯМР, продукт используют на следующей стадии без дополнительной очистки.
Стадия 2:
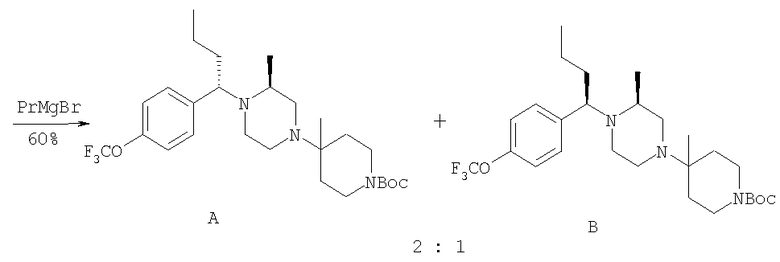
К раствору продукта, полученного на стадии 1 (1,16 г, 1,97 ммоль), в 20 мл толуола прибавляют раствор н-пропилмагнийбромида (2 М в Et2O, 1,1 мл) и смесь 15 ч перемешивают при комнатной температуре. Реакцию останавливают, выливая реакционную смесь в насыщенный водный раствор NH4Cl со льдом. Водный слой экстрагируют с помощью EtOAc, промывают 1 М раствором NaOH, водой и солевым раствором. Концентрированно и очистка с помощью ФХСГ (20% EtOAc-гексан) дает искомый продукт А. Дополнительное элюирование с помощью 30% раствора EtOAc в гексане дает (R,S)-диастереоизомер В.
Стадия 3: Для удаления защитной группы ВОС амин А обрабатывают с помощью TFA в CH2Cl2. Сочетание свободного пиперидина с кислотой с использованием EDCI/HOBT дает соединения 30-30В, представленные в приведенной ниже таблице; аналогичные методики используют для получения соединений 30С-30I.
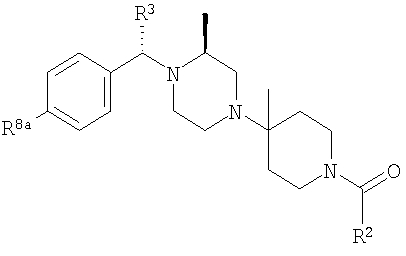

Пример 31

Раствор продукта, полученного в Примере 12, стадия 2 (150 мг, 0,27 ммоль), имидазола (27,4 мг, 0,403 ммоль), 1,10-фенантролина (48 мг, 0,27 ммоль), транс, транс-дибензилиденацетона (6,28 мг, 0,027 ммоль), комплекса меди(II) с бензолтриф-торметансульфонатом (15 мг, 0,027 ммоль) и Cs2СО3 (96,1 мг, 0,30 ммоль) в ксилоле (2 мл) перемешивают при 110°C в течение 5 ч. Реакционную смесь охлаждают до комнатной температуры и прибавляют насыщенный раствор NaHCO3. Экстракция с помощью EtOAc с последующей хроматографией на силикагеле дает искомое соединение (70 мг, выход 52%). Разложение 215°C (хлористоводородная соль). МСВР: Рассчитано для С29Н39ClN3OS (М+Н+), 500,3389; Найдено, 500,3396.
Для определения антагонистической активности соединений, соответствующих настоящему изобретению, по отношению к CCR5, можно использовать следующие методики анализа.
Анализ связывания с CCR5 мембраны.
При высокопроизводительном скрининге, в котором используется анализ связывания с CCR5 мембраны, определяются ингибиторы связывания RANTES. В этом анализе используют мембраны, приготовленные из клеток NIH 3Т3, экспрессирующих хемокиновый рецептор CCR5 человека, который обладает способностью связываться с RANTES, природным лигандом этого рецептора. С использованием планшета с 96 лунками препараты мембран 1 ч инкубируют с помощью 125I-RANTES в присутствии или в отсутствии соединения. Соединения последовательно разбавляют в диапазоне от 0,001 мкг/мл до 1 мкг/мл и исследуют трижды. Реакционные смеси фильтруют через фильтры из стекловолокна и тщательно промывают. Результаты подсчетов для групп анализов усредняют и данные приводят в виде концентраций, необходимых для ингибирования 50% полного связывания 125I-RANTES. Соединения, для которых при анализе связывания с мембраной обнаружена высокая активность, дополнительно характеризуют путем анализа вторичного проникновения ВИЧ-1 в клетки и анализа репликации.
Анализ проникновения ВИЧ-1:
Репортерные вирионы с нарушенной репликацией получают путем сотрансфекции плазмиды, кодирующей штамм NL4-3 ВИЧ-1 (который модифицирован путем мутации гена оболочки и введения репортерной плазмиды с геном люциферазы), и плазмиды, кодирующей один из нескольких генов оболочки ВИЧ-1, в соответствии с описанием в работе Connor et al., Virology, 206 (1995), р.935-944. После трансфекции этих двух плазмид с помощью осаждения фосфатом кальция на 3-й день собирают вирусную надосадочную жидкость и определяют функциональный вирусный титр. Эти основные растворы затем используют для инфицирования клеток U87, стабильно экспрессирующих CD4 и хемокиновый рецептор CCR5, которые предварительно инкубируют в присутствии или в отсутствии исследуемого соединения. Инфицирование проводят в течение 2 ч при 37°С, клетки промывают и среду заменяют на свежую среду, содержащую соединение. Клетки инкубируют в течение 3 суток, подвергают лизису и определяют люциферазную активность. Результаты приводят в виде концентраций, необходимых для ингибирования 50% люциферазной активности в контрольных культурах.
Анализ репликации ВИЧ-1:
В этом анализе с целью определения эффективности анти-ССК5 соединений для блокирования инфицирования первичными штаммами ВИЧ-1 используют первичные моноядерные клетки периферической крови или стабильные клетки линии U87-CCR5. Первичные лимфоциты берут у нормальных здоровых доноров, очищают и стимулируют in vitro с помощью РНА и IL-2 за трое суток до инфицирования. С использованием планшета с 96 лунками клетки подвергают предварительной обработке лекарственным препаратом в течение 1 ч при 37°C и затем инфицируют культурами макрофагового тропического ВИЧ-1. После инфицирования клетки промывают для удаления остаточного инокулята и в течение 4 суток культивируют в присутствии соединений. Собирают надосадочную жидкость культур и вирусную репликацию измеряют путем определения концентрации вирусного антигена р24.
Анализ потока кальция.
Клетки, экспрессирующие сорецептор CCR5 ВИЧ, обрабатывают красителем, чувствительным по отношению к кальцию, а затем добавляют соединение или природный лиганд CCR5. Соединения, обладающие антагонистической способностью, будут индуцировать в клетке сигнал кальциевого потока, а антагонисты CCR5 определяются, как соединения, которые сами по себе не индуцируют сигнал, но могут блокировать сигнал природных лигандов CCR5.
Анализ связывания GTPγS:
При анализе связывания GTPγS определяется активация рецептора лигандами CCR5. В этом анализе измеряют связывание GTP, меченного 35S, с G-белками, связанными с рецептором, которое происходит вследствие активации рецептора соответствующим лигандом. В этом анализе лиганд CCR5, RANTES, инкубируют с мембранами клеток, экспрессирующих CCR5, и связь с активацией рецептора (или связывание) устанавливают путем количественного определения связанной метки 35S. Обладает ли соединение агонистической способностью, в этом анализе количественно определяют по индуцированию активации рецептора, или, альтернативно, обладает ли оно антагонистической способностью, - путем измерения ингибирования связывания RANTES по конкурентному или не конкурентному механизму.
Анализ хемотаксиса:
Анализ хемотаксиса - это функциональный анализ, который сравнительным образом характеризует агонистические и антагонистические характеристики исследуемого соединения. В анализе измеряется способность неприлипающей линии клеток мышей, экспрессирующих CCR5 человека (BaF-550), мигрировать через мембрану под влиянием исследуемых соединений и природных лигандов (т.е. RANTES, MIP-1β). Клетки мигрируют через проницаемую мембрану по направлению к соединениям, обладающим агонистической активностью. Соединения, являющиеся антагонистами, не только не могут вызывать хемотаксис, но и способны ингибировать миграцию клеток под влиянием известных лигандов CCR5.
Роль рецепторов хемокинов СС, таких как рецепторы CCR5, при воспалительных заболеваниях рассмотрена в таких публикациях, как Immunology Letters, 57, (1997), 117-120 (артрит); Clinical & Experimental Rheumatology, 17, (4) (1999), р.419-425 (ревматоидный артрит); Clinical & Experimental Immunology, 117 (2) (1999), р.237-243 (атопический дерматит); International Journal of Immunopharmacology, 20 (11) (1998), р.661-7 (псориаз); Journal of Allergy & Clinical Immunology, 100 (6, Pt 2) (1997), p.S52-5 (астма); и Journal of Immunology, 159 (6) (1997), р.2962-72 (аллергические заболевания).
При анализах по определению ингибирования связывания RANTES соединения, соответствующие настоящему изобретению, обладают активностью в диапазоне значений Ki от примерно 0,5 до примерно 1500 нМ, а предпочтительные соединения обладают активностью в диапазоне от примерно 0,5 до примерно 750, более предпочтительные - от примерно 0,5 до 300 нМ, а наиболее предпочтительные - от примерно 0,5 до 50 нМ. Результаты для предпочтительных и репрезентативных соединений формул I и II, полученные при исследованиях по определению ингибирования связывания RANTES, представлены в приведенной ниже таблице.
Для приготовления фармацевтических композиций, включающих соединения-антагонисты CCR5, соответствующие настоящему изобретению, используемые инертные, приемлемые с фармацевтической точки зрения носители могут быть твердыми или жидкими. К твердым формам препаратов относятся порошки, таблетки, диспергируемые гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут включать от примерно 5 до примерно 95% активного компонента. Подходящими твердыми носителями, известными в данной области техники, являются, например, карбонат магния, стеарат магния, тальк, сахар и лактоза. Таблетки, порошки, облатки и капсулы можно использовать в виде твердых дозировочных форм, пригодных для перорального приема. Примеры приемлемых с фармацевтической точки зрения носителей и способов изготовления различных композиций приведены в работе A. Gennaro (ed.), Remington's Pharmaceutical Sciences, 18th Edition, (1990), Mack Publishing Co., Easton, Pennsylvania.
К жидким формам препаратов относятся растворы, суспензии и эмульсии. В качестве примеров можно привести водные и водно-пропиленгликолевые растворы для парентеральных инъекций или прибавление подсластителей и замутнителей в растворы, суспензий и эмульсий, предназначенные для перорального приема. К жидким формам препаратов также могут относиться растворы для внутриносового введения.
Аэрозольные препараты, пригодные для ингаляции, могут представлять собой растворы и твердые вещества в порошкообразной форме, которые могут сочетаться с приемлемыми с фармацевтической точки зрения носителями, такими как сжатый инертный газ, например, азот.
Также включены твердые формы препаратов, которые предназначены для проводимого непосредственно перед употреблением превращения в жидкие формы препаратов, предназначенные для перорального или парентерального введения. К таким жидким формам относятся растворы, суспензии и эмульсии.
Соединения-антагонисты CCR5, соответствующие настоящему изобретению, также могут вводиться чрескожно. Чрескожные композиции могут представлять собой кремы, лосьоны, аэрозоли и/или эмульсии и могут включаться в предназначенные для чрескожного введения пластыри матричного и резервуарного типа, которые принято использовать для этой цели в данной области техники.
Предпочтительно вводить соединения-антагонисты CCR5 перорально.
Предпочтительно, чтобы фармацевтический препарат представлял собой разовую дозировочную форму. В такой форме препарат можно разделить на подходящие разовые дозы, содержащие соответствующие количества активного компонента, например, в количестве, эффективном для достижения необходимой цели.
Количество активного компонента в разовой дозе препарата может меняться или подбираться в диапазоне от примерно 10 до примерно 500 мг, предпочтительно - от примерно 25 до примерно 300 мг, более предпочтительно - от примерно 50 до примерно 250 мг, а наиболее предпочтительно - от примерно 55 до примерно 200 мг в соответствии с конкретным случаем применения.
Реальная применяющаяся дозировка может меняться в зависимости от потребности пациента и тяжести патологического состояния, подвергающегося лечению. Определение необходимого дозировочного режима в конкретном случае делает специалист в данной области техники. Для удобства полную суточную дозу можно разделить и в соответствии с необходимостью принимать отдельными порциями в течение суток.
Количество и частоту приема соединений-антагонистов CCR5, соответствующих настоящему изобретению, и/или их приемлемых с фармацевтической точки зрения солей определяется решением лечащего врача, учитывающего такие факторы, как возраст, состояние и массу пациента, а также тяжесть подвергающегося лечению патологического состояния. Типичный рекомендованный суточный дозировочный режим при пероральном приеме может меняться от примерно 100 до примерно 300 мг/сут, предпочтительно - от примерно 150 до примерно 250 мг/сут, а более предпочтительно - примерно 200 мг/сут, назначаемых в виде двух-четырех разделенных доз.
Дозы и дозировочные режимы приема НИОТ, ННИОТ, ИП и других препаратов должен определять лечащий врач с учетом доз и дозировочных режимов, указанных на листке-вкладыше или в соответствии с указаниями в описании препаратов с учетом возраста, пола и состояния пациента и тяжести инфицирования с помощью ВИЧ-1.
Хотя настоящее изобретение описано с помощью представленных выше конкретных вариантов осуществления, специалисту с общей подготовкой в данной области техники должны быть понятны их многочисленные альтернативы, модификации и вариации. Предполагается, что все такие альтернативы, модификации и вариации соответствуют объему и сущности настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА И ПИПЕРИДИНА | 2002 |
|
RU2326120C9 |
| СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2237060C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНИЛПИПЕРАЗИНА, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2006 |
|
RU2411241C2 |
| ТРИЦИКЛИЧЕСКИЕ ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2293734C9 |
| ПОЛИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ АНТАГОНИСТА РЕЦЕПТОРА H, И СПОСОБ ЛЕЧЕНИЯ С ЕГО ПРИМЕНЕНИЕМ | 2001 |
|
RU2301231C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И СПОСОБ ИДЕНТИФИКАЦИИ АГЕНТА НА ЕГО ОСНОВЕ | 1999 |
|
RU2245335C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИЙ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2290179C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| Микроциклические аналоги и способы их применения и получения | 1999 |
|
RU2822750C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2076100C1 |
Описываются новые производные пиперазина общей формулы

или их фармацевтически приемлемые соли, где
Ra-R8a-фенил, R8b-пиридил или R8-нафтил; R1-водород; R2-R9-, R10-, R11 - замещенный фенил; R9-, R10-, R11 - замещенный пиридил или пиримидил; R9-, R10-, R11 - замещенный пиридил-N-оксид или пиримидил-N-оксид; R12-, R13 - замещенный оксазолил; нафтил; флуоренил;
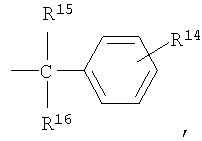

R3-водород, C1-С6-алкил, (С1-С6)-алкокси-(С1-С6)-алкил, С3-С10-циклоалкил, С3-С10-циклоалкил-(С1-С6)-алкил, R8-фенил, R8-фенил-(С1-С6)-алкил или R8 - тиенил-(С1-С6)-алкил; R4, R5, R7 и R13 - независимо означают водород или (С1-С6)-алкил; R6-водород или C1-С6-алкил; R18 - 1-3 заместителя, независимо означают водород, галоген, C1-С6-алкоксил и -CF3; R8a - 1-3 заместителя, независимо означают водород, галоген, -CF3, CF3О-, -CN, R14-фенил, -NHCOCF3 и имидазолил; R8b - 1-3 заместителя, независимо означают водород или галоген; R9 и R10 - независимо означают (С1-С6)-алкил, галоген, -NR17R18, -ОН, -CF3 и -ОСН3; R11 означает R9, водород, фенил, -NO2, -CN, -СН2F, -CHF2, -СНО, -CH=NOR17, пиридил, пиридил-N-оксид, пиримидинил, пиразинил, N(R17)CONR18R19, -NHCONH(хлор-(С1-С6)-алкил), -NHCONH((С3-С10)-циклоалкил-(С1-С6)-алкил), -NHCO(С1-С6)-алкил, -NHCOCF3, -NHSO2N((C1-C6)-алкил)2, -NHSO2(C1-C6)-алкил, -N(SO2CF3)2, -NHCO2(C1-С6)-алкил, С3-С10-циклоалкил, -SR20, -OSO2(С1-С6)-алкил, -OSO2CF3, гидрокси-(С1-С6)-алкил, -CONR17R18, -CON(СН2СН2-O-СН3)2, -OCONH(С1-С6)-алкил, -Si(CH3)3 или - В(ОС(СН3)2)2; R12 - (С1-С6)-алкил или R14-фенил; R14 - 1-3 заместителя, независимо означают водород, (С1-С6)-алкил, -CF3, -CO2R17, -CN, (С1-С6)-алкоксил и галоген; R15 и R16 - независимо означают водород и C1-С6-алкил, или R15 и R16 совместно означают С2-С5-алкиленовую группу и вместе с атомом углерода, к которому они присоединены, образуют С3-6 спирановое кольцо. R17, R18 и R19 - независимо означают водород или C1-С6-алкил; и R20 - C1-С6-алкил, фармацевтические композиции, их содержащие, и применение новых соединений в качестве антагонистов CCR5 для лечения ВИЧ-инфекции, артрита, астмы, рассеянного склероза и др. 5 н. и 24 з.п.ф-лы, 30 табл.

или их фармацевтически приемлемые соли, где
Ra означает R8a-фенил, R8b-пиридил или R8-нафтил;
R1 означает водород;
R2 означает R9-, R10-, R11- замещенный фенил; R9-, R10-, R11 - замещенный пиридил или пиримидил; R9-, R10-, R11- замещенный пиридил-N-оксид или пиримидил-N-оксид; R12-, R13- замещенный оксазолил; нафтил; флуоренил;
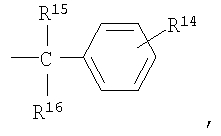

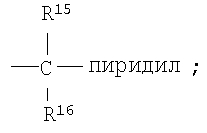
R3 означает водород, C1-С6-алкил, (С1-С6)-алкокси-(С1-С6)-алкил, С3-С10-циклоалкил, С3-С10-циклоалкил-(С1-С6)-алкил, R8-фенил, R8-фенил-(С1-С6)-алкил или R8-тиенил-(С1-С6)-алкил;
R4, R5, R7 и R13 независимо выбраны из группы, включающей водород и (С1-С6)-алкил;
R6 означает водород или C1-С6-алкил;
R8 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, C1-С6-алкоксил и -CF3;
R8a означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, галоген, -CF3, CF3О-, -CN, R14-фенил, -NHCOCF3 и имидазолил;
R8b означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород или галоген;
R9 и R10 независимо выбраны из группы, включающей (С1-С6)-алкил, галоген, -NR17R18, -ОН, -CF3 и -ОСН3;
R11 означает R9, водород, фенил, -NO2, -CN, -CH2F, -CHF2, -СНО, -CH=NOR17, -пиридил, пиридил-N-оксид, пиримидинил, пиразинил, N(R17)CONR18R19, -NHCONH(хлор-(C1-C6)-алкил), -NHCONH((C3-C10)-циклоалкил-(С1-С6)-алкил), -NHCO(C1-C6)-алкил, -NHCOCF3, -NHSO2N((C1-С6)-алкил)2, -NHSO2(C1-C6)-алкил, -N(SO2CF3)2, -NHCO2(C1-C6)-алкил, С3-С10-циклоалкил, -SR20, -OSO2(С1-С6)-алкил, -OSO2CF3, гидрокси-(С1-С6)-алкил, -CONR17R18, -CON(СН2СН2-O-СН3)2, -OCONH(C1-C6)-алкил, -Si(CH3)3 или -В(ОС(CH3)2)2;
R12 означает (С1-С6)-алкил или R14-фенил;
R14 означает от 1 до 3 заместителей, независимо выбранных из группы, включающей водород, (С1-С6)-алкил, -CF3, -CO2R17, -CN, (С1-С6)-алкоксил и галоген;
R15 и R16 независимо выбраны из группы, включающей водород и C1-С6-алкил, или R15 и R16 совместно представляют собой С2-C5-алкиленовую группу и вместе с атомом углерода, к которому они присоединены, образуют спирановое кольцо, содержащее от 3 до 6 атомов углерода;
R17, R18 и R19 независимо выбраны из группы, включающей водород и C1-С6-алкил; и
R20 означает C1-С6-алкил.

где R8a означает -CF3, CF3О- или галоген; или Ra означает
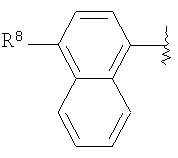
где R8 означает C1-С6-алкоксил.

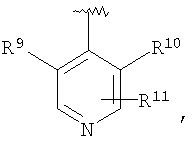
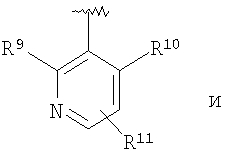

где R9 и R10 выбраны из группы, включающей (С1-С6)-алкил, галоген, ОН и -NH2.
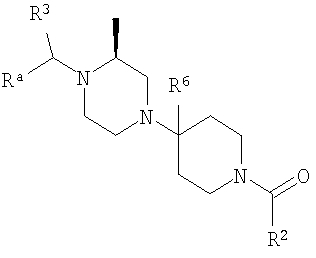
где Ra, R3, R6 и R2 имеют указанные в следующей таблице значения:

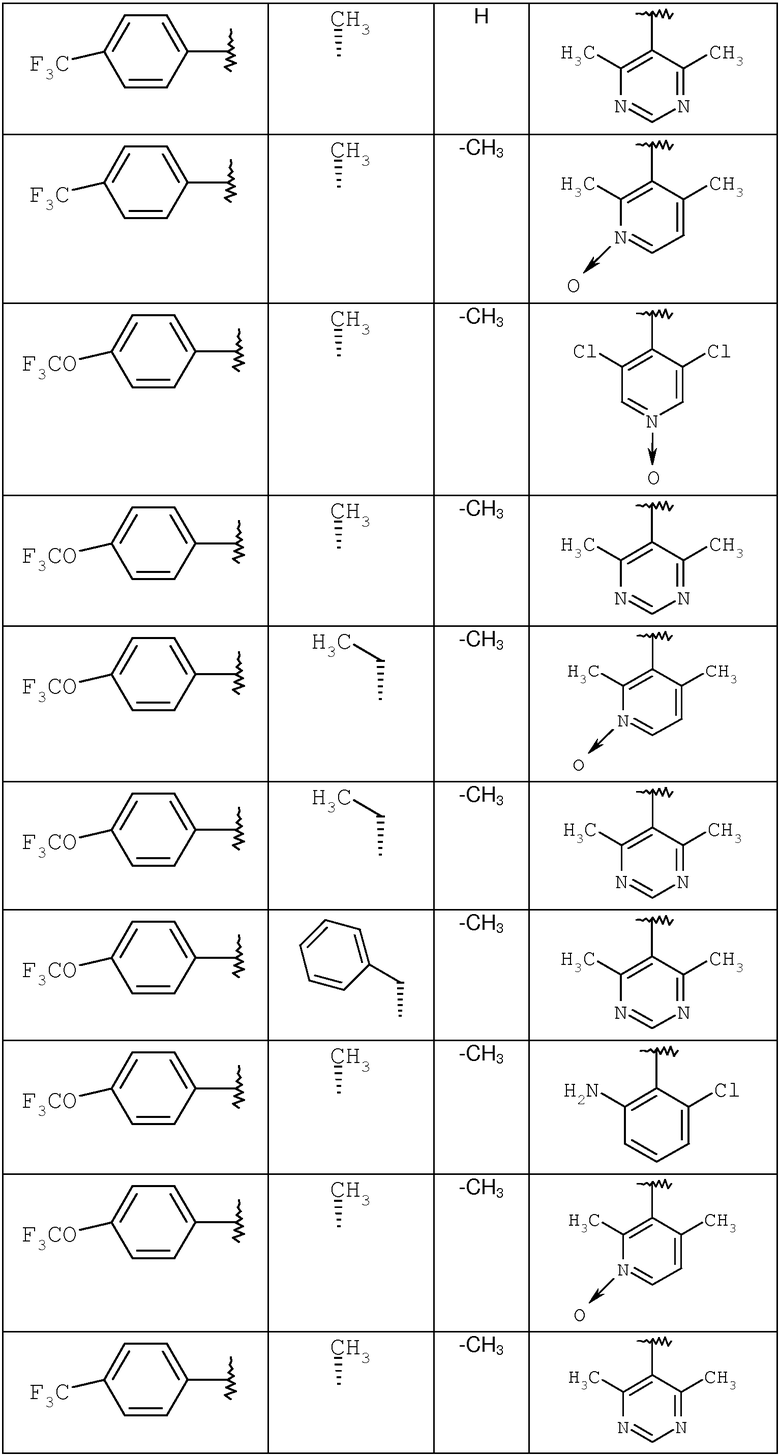

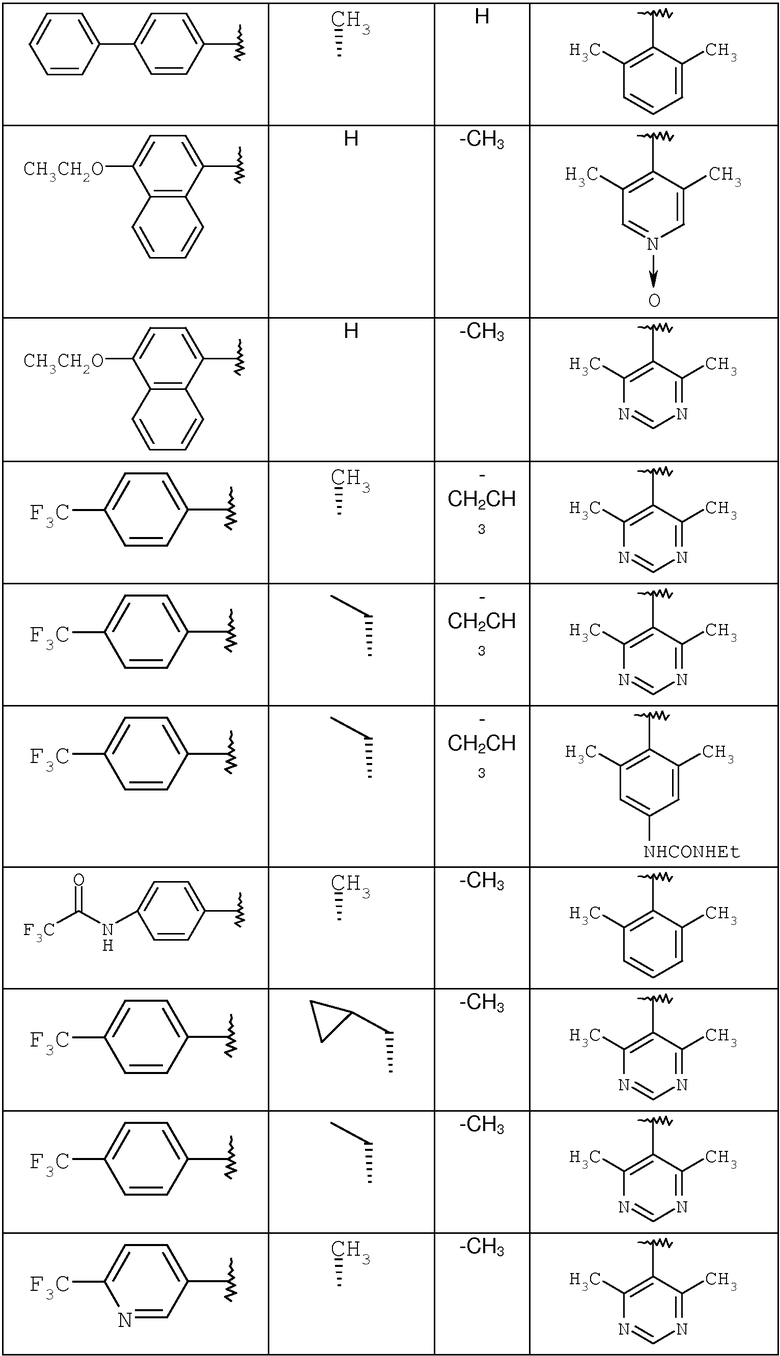
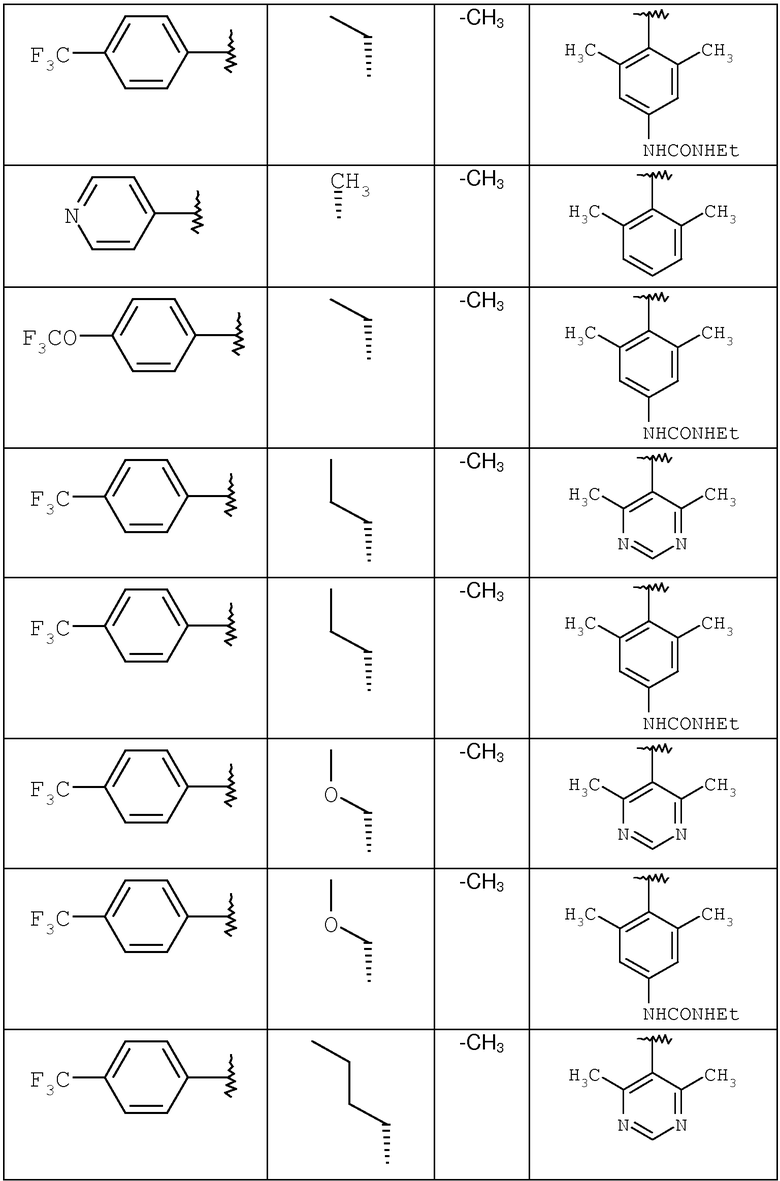
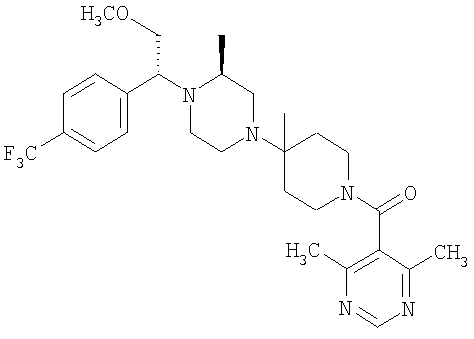


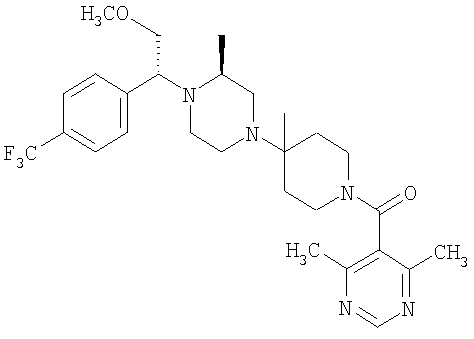
| WO 9805292 А, 12.02.1998 | |||
| WO 9806697 A, 19.02.1998 | |||
| WO 9801425 A, 15.01.1998 | |||
| ПИРИДИЛ- ИЛИ ПИРИМИДИЛСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ПИПЕРАЗИНА ИЛИ 1,4-ДИАЗАЦИКЛОГЕПТАНА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ АКТИВНЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПСИХОТРОПНЫМ ДЕЙСТВИЕМ | 1989 |
|
RU2021269C1 |
| ЭФИРЫ БИС-ФЕНИЛПИПЕРАЗИННИКОТИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1993 |
|
RU2127732C1 |

