Предшествующий уровень техники
В WO 95/14007, опубликованной 26 мая 1995 г., описаны антагонисты типа имидазола для рецептора Н3.
В WO 99/24405, опубликованной 20 мая 1999 г., описаны лиганды типа имидазола для рецептора Н3.
В патенте США US 5869479, выданном 9 февраля 1999 г., описаны композиции для симптоматического лечения аллергического ринита с использованием комбинации хотя бы одного антагониста гистаминового рецептора H1 и хотя бы одного антагониста гистаминового рецептора Н3.
Вследствие проявляющегося в данной области техники интереса к соединениям, которые воздействуют на рецепторы Н3, новые соединения, такие как антагонисты рецептора Н3, внесли бы ценный вклад в данную область техники. Настоящее изобретение вносит именно такой вклад.
Краткое описание изобретения
Настоящее изобретение относится к новым соединениям структуры
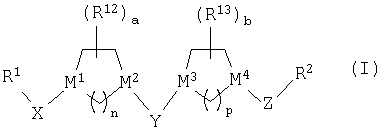
или к их фармацевтически приемлемой соли или сольвату, в которых
(1) R1 выбран из группы, включающей:
(a) арил;
(b) гетероарил;
(c) гетероциклоалкил;
(d) алкил;Ш
(e) циклоалкил и
(f) алкиларил,
где указанные группы R1 необязательно содержат от 1 до 4 заместителей, независимо выбранных из группы, включающей:
(1) галоген (например, Br, F или Cl, предпочтительно F или Cl);
(2) гидроксил (т.е. -ОН);
(3) низший алкоксил (например, (С1-С6)-алкоксил, предпочтительно (C1-C4)-алкоксил, более предпочтительно (С1-С2)-алкоксил, более предпочтительно метоксил);
(4) -CF3;
(5) CF3O-;
(6) -NR4R5;
(7) фенил;
(8) -NO2;
(9) -CO2R4;
(10) -CON(R4)2, где все R4 являются одинаковыми или разными;
(11) -S(O)mN(R20)2, где все R20 являются одинаковыми или разными и означают Н или алкильную группу, предпочтительно (С1-С4)-алкил, более предпочтительно (С1-С2)-алкил, а более предпочтительно метил;
(12) -CN и
(13) алкил или
(2) R1 и Х совместно образуют группу, выбранную из:

(3) Х выбран из =С(O), =C(NOR3), =C(NNR4R5),

(4) М1 означает атом углерода;
(5) М2 выбран из группы, включающей С и N;
(6) М3 и М4 независимо выбраны из группы, включающей С и N;
(7) Y выбран из группы, включающей -СН2-, =С(O), =C(NOR20) (где R20 является таким, как определено выше) и =C(S);
(8) Z означает (С1-С6)-алкильную группу;
(9) R2 означает пяти- или шестичленный гетероарильный цикл, указанный шестичленный гетероарильный цикл содержит 1 или 2 атома азота, а остальные атомы цикла являются атомами углерода, и указанный пятичленный гетероарильный цикл содержит 1 или 2 гетероатома, выбранных из группы, включающей азот, кислород и серу, а остальные атомы цикла являются атомами углерода; указанный пяти- или шестичленный гетероарильный цикл необязательно содержит от 1 до 3 заместителей, независимо выбранных из группы, включающей галоген, гидроксил, низший алкил, низший алкоксил, -CF3, CF3О-, -NR4R5, фенил, -NO2, -CO2R4, -CON(R4)2, где все R4 являются одинаковыми или разными, -CH2NR4R5, -(N)C(NR4R5)2 или -CN;
(10) R3 выбран из группы, включающей:
(a) водород;
(b) (С1-С6)-алкил;
(c) арил;
(d) гетероарил;
(e) гетероциклоалкил;
(f) арилалкил (например, арил-(С1-С4)-алкил, например, -(CH2)w-арил, где w равно от 1 до 4, предпочтительно от 1 до 2, наиболее предпочтительно 1, такой как, например, -СН2-фенил или -СН2-(замещенный фенил));
(g) -(CH2)e-C(O)N(R4)2, где все R4 являются одинаковыми или разными;
(h) -(CH2)e-C(O)OR4;
(i) -(CH2)e-C(O)R30, где R30 означает гетероциклоалкильную группу, такую как, например, морфолинильную, пиперидинильную, пиперазинильную или пирролидинильную, включая
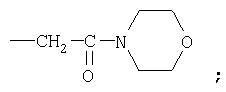
(j) -CF3 и
(k) -СН2CF3,
где указанные арил, гетероарил, гетероциклалкил и арильный фрагмент указанного арилалкила необязательно содержат от 1 до 3 (предпочтительно 1) заместителей, независимо выбранных из группы, включающей галоген (например, F или Cl), -ОН, -OCF3, -CF3, -CN, -N(R45)2, -CO2R45 и -C(O)N(R45)2, где все R45 независимо выбраны из группы, включающей H, алкил, алкиларил или алкиларил, где указанный арильный фрагмент содержит от 1 до 3 заместителей, независимо выбранных из группы, включающей -CF3, -ОН, галоген, алкил, -NO2 или -CN;
(11) R4 выбран из группы, включающей водород, (С1-С6)-алкил, арил, алкиларил, указанные арильные и арилалкильные группы необязательно содержат от 1 до 3 заместителей, выбранных из группы, включающей галоген, -CF3, -OCF3, -ОН, -N(R45)2, -CO2R45 или -C(O)N(R45)2 или -CN, где R45 являются такими, как определено выше;
(12) R5 выбран из группы, включающей водород, (С1-С6)-алкил, -C(O)R4, -C(O)2R4 и -C(O)N(R4)2, где все R4 выбраны независимо и R4 являются таким, как определено выше;
(13) или R4 и R5 вместе с атомом азота, с которым они связаны, образуют пяти- или шестичленное гетероциклоалкильное кольцо (например, морфолин);
(14) R6 выбран из группы, включающей алкил, арил, алкиларил, галоген, гидроксил, низший алкоксил, -CF3, CF3О-, -NR4R5, фенил, -NO2, -CO2R4, -CON(R4)2, где все R4 являются одинаковыми или разными, или -CN;
(15) R12 выбран из группы, включающей алкил, гидроксил, алкоксил или фтор;
(16) R13 выбран из группы, включающей алкил, гидроксил, алкоксил или фтор;
(17) а (нижний индекс у R12) равно от 0 до 2;
(18) b (нижний индекс у R13) равно от 0 до 2;
(19) с (нижний индекс у R6) равно от 0 до 2;
(20) e равно от 0 до 5;
(21) m равно от 1 до 2;
(22) n равно 1, 2 или 3 и
(23) p равно 1, 2 или 3 при условии, что, если М3 и М4 означают азот, то p равно 2 или 3 (т. е. если М3 и М4 означают азот, то р не равно 1).
Настоящее изобретение также относится к фармацевтической композиции, содержащей эффективное количество соединения формулы I и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей (например, верхних дыхательных путей), застоя (например, отека слизистой оболочки носа), гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна (например, гиперсомнии, сонливости и нарколепсии), расстройств центральной нервной системы, дефицита внимания с гиперактивностью (ДВГА), гипо- и гиперактивности центральной нервной системы (например, ажитации и депрессии) и других заболеваний центральной нервной системы (таких как болезнь Альцгеймера, шизофрения и мигрень), включающему назначение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I.
Настоящее изобретение также относится к способу лечения аллергии, включающему введение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I.
Настоящее изобретение также относится к способу лечения вызванных аллергией поражений дыхательных путей (например, верхних дыхательных путей), включающему введение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I.
Настоящее изобретение также относится к способу лечения застоя (например, отека слизистой оболочки носа), включающему введение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I.
Настоящее изобретение также относится к фармацевтической композиции, содержащей эффективное количество соединения формулы I и эффективное количество антагониста рецептора H1 в комбинации с фармацевтически приемлемым носителем.
Настоящее изобретение также относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей (например, верхних дыхательных путей) и застоя (например, отека слизистой оболочки носа), включающему введение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I в комбинации с эффективным количеством антагониста рецептора H1.
Настоящее изобретение также относится к способу лечения аллергии, включающему введение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I в комбинации с эффективным количеством антагониста рецептора H1.
Настоящее изобретение также относится к способу лечения вызванных аллергией поражений дыхательных путей (например, верхних дыхательных путей), включающему ведение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I в комбинации с эффективным количеством антагониста рецептора H1.
Настоящее изобретение также относится к способу лечения застоя (например, отека слизистой оболочки носа), включающему введение пациенту, нуждающемуся в таком лечении (например, млекопитающему, такому как человек), эффективного количества соединения формулы I в комбинации с эффективным количеством антагониста рецептора H1.
Подробное описание изобретения
В настоящем изобретении следующие термины, если не указано иного, используются в приведенных ниже значениях:
алкил (включая алкильные фрагменты алкоксила и алкиларила) означает линейные или разветвленные углеродные цепи и содержит от одного до двадцати атомов углерода, предпочтительно от одного до шести атомов углерода;
алкиларил означает алкильную группу, определенную выше, связанную с арильной группой, определенной ниже, такой что указанная арильная группа связана с остальной частью молекулы;
арил (включая арильный фрагмент алкиларила) означает карбоциклическую группу, содержащую от 6 до 15 атомов углерода и включающую хотя бы одно ароматическое кольцо (например, арил представляет собой бензольное кольцо), причем все имеющиеся способные к замещению атомы углерода карбоциклической группы рассматриваются в качестве возможных положений присоединения;
арилалкил означает арильную группу, определенную выше, связанную с алкильной группой, определенной выше, такой что указанная алкильная группа связана с остальной частью молекулы;
циклоалкил означает насыщенные карбоциклические кольца, содержащие от 3 до 20 атомов углерода, предпочтительно от 3 до 7 атомов углерода;
галоген означает фтор, хлор бром или йод;
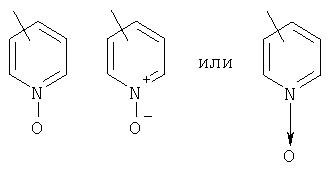
гетероарил означает циклические группы, содержащие хотя бы один гетероатом, выбранный из группы, включающей О, S или N, входящий в карбоциклическую кольцевую структуру и обладающий количеством делокализованных пи-электронов, достаточным для придания ароматического характера, причем ароматические гетероциклические группы предпочтительно содержат от 2 до 14 атомов углерода; примеры включают (без наложения ограничений) изотиазолил, изоксазолил, фуразанил, триазолил, тиазолил, тиенил, фуранил (фурил), пирролил, пиразолил, пиранил, пиримидинил, пиразинил, пиридазинил, пиридил (например, 2-, 3- или 4-пиридил), пиридил-N-оксид (например, 2-, 3- или 4-пиридил-N-оксид), триазинил, птеридинил, индолил (бензопирролил), пиридопиразинил, изохинолинил, хинолинил, хиноксолинил, нафтиридинил, где указанный пиридил-N-оксид можно представить в виде
гетероциклоалкил означает насыщенное карбоциклическое кольцо, содержащее от 3 до 15 атомов углерода, предпочтительно от 4 до 6 атомов углерода, и в это карбоциклическое кольцо включено от 1 до 3 гетероатомных групп, выбранных из группы, включающей -О-, -S- и -NR40, где R40 означает (С1-С6)-алкил, алкиларил, -C(O)R4, -C(O)OR4 или -C(O)N(R45)2, (где R45 является таким, как определено выше, и все R45 выбраны независимо); примеры включают (без наложения ограничений) 2- или 3-тетрагидрофуранил, 2- или 3-тетрагидротиенил, 2-, 3- или 4-пиперидинил, 2- или 3-пирролидинил, 2- или 3-пиперазинил, 2- или 4-диоксанил, 1,3-диоксоланил, 1,3,5-тритианил, пентаметиленсульфид, пергидроизохинолинил, декагидрохинолинил, триметиленоксид, азетидинил, 1-азациклопентанил, 1,3-дитианил, 1,3,5-триоксанил, морфолинил, тиоморфолинил, 1,4-тиоксанил и 1,3,5-гексагидротриазинил, тиазолидинил, тетрагидропиранил;
низший алкил означает алкильную группу, определенную выше, которая содержит от 1 до 6 атомов углерода, предпочтительно 1-4 атома углерода;
низший алкоксил означает алкоксильную группу, алкильный фрагмент которой содержит от 1 до 6 углерода, предпочтительно 1-4 атома углерода;
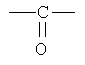
=С(O) означает
=C(NOR3) означает
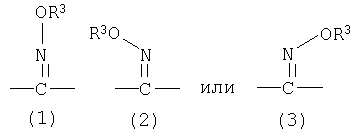
где (1) означает смесь изомеров оксима; (2) означает один геометрический изомер оксима, в котором группа -OR3 расположена с той же стороны от двойной связи, что и группа, расположенная слева от атома углерода; (3) означает один геометрический изомер оксима, в котором группа -OR3 расположена с той же стороны от двойной связи, что и группа, расположенная справа от атома углерода; (1) также можно представить в виде
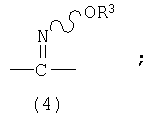
=C(NNR4R5) означает

и представляет собой смесь изомеров

-(N)C(NR4R5)2 означает
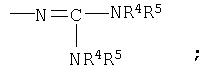
 в структуре
в структуре
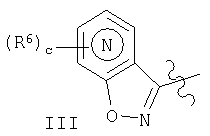
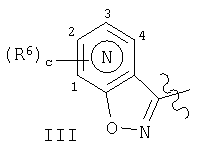
означает атом азота, который расположен в одном из 4 положений, не входящих в участок конденсирования цикла, т.е. в положениях 1, 2, 3 или 4, указанных ниже:
АсОН означает уксусную кислоту;
BOC означает трет-бутоксикарбонил;
CBZ означает карбонилбензилоксил (-С(O)ОСН2С6Н5);
CSA означает камфорсульфоновую кислоту;
DBU означает 1,8-диазабикло[5.4.0]ундец-7-ен;
DBN означает 1,5-диазабикло[4.3.0]нон-5-ен;
DCC означает дициклогексилкарбодиимид;
DEC означает 2-диэтиламиноэтилхлоридгидрохлорид;
DIBAL-H означает диизобутилалюминийгидрид;
DIPEA означает N,N-диизопропилэтиламин;
DIBAL означает диизобутилалюминий;
DMAP означает 4-(диметиламино)пиридин;
DMF означает диметилформамид;
DMSO означает диметилсульфоксид;
EDCI означает 1-(3-диметиламинопропил)-3-этилкарбодиимид;
EtOA- означает этилацетат;
EtOH означает этанол;
FMOC означает 9-флуоренметоксикарбонил;
НОВТ означает 1-гидроксибензотриазол;
LAH означает алюмогидрид лития;
LDA означает диизопропиламид лития;
m-CPBA означает м-хлорпербезнойную кислоту;
МеОН означает метанол;
NaBH(ОАс)3 означает триацетоксиборогидрид натрия;
NaBH4 означает борогидрид натрия;
NaBH3CN означает цианоборогидрид натрия;
NaHDMS означает гексаметилдисилилазид натрия;
NBS означает N-бромсукцинимид;
РСС означает пиридинийхлорхромат;
PG означает защитную группу;
РуВОР означает бензотриазол-1-илокситриспирролидинофосфонийгексафторфосфат;
t-BOC означает трет-бутоксикарбонил;
TEMPO означает 2,2,6,6-тетраметил-1-пиперидиноксильный свободный радикал;
TFA означает трифторуксусную кислоту;
THF означает тетрагидрофуран;
TMAD означает N,N,N'N'-тетраметилазодикарбоксамид;
TMEDA означает тетраметилэтилендиамин;
Tr трифенилметил;
Tris - трис(гидроксиметил)аминометан;
p-TsOH - п-толуолсульфоновая кислота;
Ки/ммоль означает кюри/ммоль (мера удельной радиоактивности);
Ki означает константу ингибирования для комплекса субстрат/рецептор;
рА2 означает -logEC50 в соответствии с определением, приведенным в J. Hey, Eur. J. Pharmacol., (1995), Vol.294, 329-335;
ББА означает масс-спектроскопию с бомбардировкой быстрыми атомами;
ВЭЖХ означает высокоэффективную жидкостную хроматографию;
ЖХМС означает жидкостную хроматографию - масс-спектроскопию;
МС означает масс-спектр;
МСВР означает масс-спектроскопию высокого разрешения;
МСНР означает масс-спектроскопию низкого разрешения;
ТСХ означает тонкослойную хроматографию.
Кроме того, при использовании в настоящем изобретении "верхние дыхательные пути" обычно означают верхнюю часть дыхательной системы, т.е. нос, горло и связанные с ними структуры.
Кроме того, при использовании в настоящем изобретении "эффективное количество" обычно означает эффективное с терапевтической точки зрения количество.
Отрезки, проведенные внутрь циклических систем, показывают, что указанная связь может быть образована с любыми способными к замещению атомами углерода цикла.
Некоторые соединения, соответствующие настоящему изобретению, могут находиться в различных изомерных (например, энантиомерных, диастереоизомерных и геометрических) формах. Предполагается, что настоящее изобретение охватывает все такие изомеры, как чистые, так и смеси, включая рацемические смеси. Также охватываются енольные формы.
Соединения, соответствующие настоящему изобретению, являются лигандами для гистаминового рецептора Н3. Соединения, соответствующие настоящему изобретению, также можно описать, как антагонисты рецептора Н3 или как антагонисты Н3.
Соединения, соответствующие настоящему изобретению, являются основными и образуют приемлемые с фармацевтической точки зрения соли с органическими и неорганическими кислотами. Примерами кислот, подходящих для образования таких солей, являются хлористоводородная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая и другие неорганические и карбоновые кислоты, хорошо известные специалистам в данной области техники. Соли получают путем взаимодействия свободного основания с достаточным количеством необходимой кислоты с образованием соли, происходящим обычным образом. Свободные основания можно выделить путем обработки соли подходящим разбавленным водным раствором основания, таким как разбавленный водный раствор гидроксида натрия, карбоната калия, аммиака и бикарбоната натрия. Свободные основания отличаются от соответствующих солей по некоторым физическим характеристикам, таким как растворимость в полярных растворителях, однако для задач настоящего изобретения по остальным характеристикам соли кислот и оснований эквивалентны соответствующим свободным основаниям.
Соединения формулы I могут находиться в несольватированных, а также в сольватированных формах, включая гидратированные формы, например полугидраты. Обычно сольватированные формы, образованные с приемлемыми с фармацевтической точки зрения растворителями, такими как вода, этанол и т.п., для задач настоящего изобретения эквивалентны несольватированным формам.
Соединения, соответствующие настоящему изобретению, можно комбинировать с антагонистом рецептора H1 (т.е. соединения, соответствующие настоящему изобретению, можно комбинировать с антагонистом рецептора H1 в фармацевтической композиции или соединения, соответствующие настоящему изобретению, можно назначать с антагонистом рецептора H1).
Известно, что многочисленные химические вещества обладают антагонистической активностью по отношению к рецептору H1. Многие применимые соединения можно отнести к классу этаноламинов, этилендиаминов, алкиламинов, фенотиазинов или пиперидинов. Примеры антагонистов рецептора H1 включают (без наложения ограничений): астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин (также известный как SCH-34117), дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин. Многие соединения можно без труда исследовать для определения активности по отношению к рецепторам Hi с помощью известных способов, включая специфическое блокирование сократительной реакции в ответ на воздействие гистамина для изолированной подвздошной кишки морской свинки. См., например, WO 98/06395, опубликованный 19 февраля 1998 г.
Таким образом, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 выбирают из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Кроме того, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 выбирают из группы, включающей астемизол, азатадин, азеластин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, каребастин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, эбастин, фексофенадин, лоратадин, левокарбастин, мизоластин, норастемизол и терфенадин.
Кроме того, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 выбирают из группы, включающей: азатадин, бромфенирамин, цетиризин, хлорфенирамин, каребастин, дезкарбэтоксилоратадин (также известный под обозначением SCH-34117), дифенгидрамин, эбастин, фексофенадин, лоратадин и норастемизол.
Кроме того, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 представляет собой лоратадин.
Кроме того, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 представляет собой дезкарбэтоксилоратадин.
Кроме того, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 представляет собой фексофенадин.
Кроме того, в способах, соответствующих настоящему изобретению, при которых соединение формулы I комбинируют с эффективным количеством антагониста рецептора H1, указанный антагонист рецептора H1 представляет собой цетиризин.
Предпочтительно, чтобы при использовании указанных выше способов подвергали лечению вызванные аллергией поражения дыхательных путей.
Также предпочтительно, чтобы при использовании указанных выше способов подвергали лечению аллергию.
Также предпочтительно, чтобы при использовании указанных выше способов подвергали лечению отек слизистой оболочки носа.
Предпочтительно, чтобы в указанных выше способах при использовании комбинации соединения формулы I (антагониста H3) и антагониста H1 антагонист H1 выбирали из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин. Более предпочтительно, чтобы антагонистом H1 являлся лоратадин или дезкарбэтоксилоратадин.
В способах, соответствующих настоящему изобретению, при которых антагонист Н3, соответствующий настоящему изобретению (соединение формулы I), вводят в комбинации с антагонистом H1, эти антагонисты можно вводить одновременно, последовательно (т.е. один после другого через относительно короткий промежуток времени) и поочередно (сначала один, а затем другой, через некоторый период времени). Обычно, когда антагонисты вводят последовательно или поочередно, антагонист Н3, соответствующий настоящему изобретению (соединение формулы I), назначают первым.
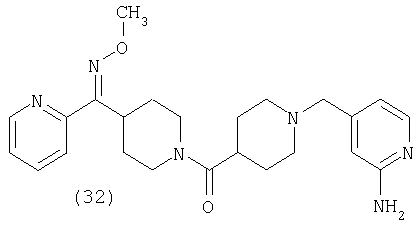
Таким образом, один вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 32 и фармацевтически приемлемый носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 54 и фармацевтически приемлемый носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 55 и фармацевтически приемлемый носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 253А и фармацевтически приемлемый носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 287 и фармацевтически приемлемый носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 320 и фармацевтически приемлемый носитель.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей, застоя, гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна, расстройств центральной нервной системы, дефицита внимания с гиперактивностью, гипо- и гиперактивности центральной нервной системы, болезни Альцгеймера, шизофрении и мигрени, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей, застоя, гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна, расстройств центральной нервной системы, дефицита внимания с гиперактивностью, гипо- и гиперактивности центральной нервной системы, болезни Альцгеймера, шизофрении и мигрени, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей, застоя, гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна, расстройств центральной нервной системы, дефицита внимания с гиперактивностью, гипо- и гиперактивности центральной нервной системы, болезни Альцгеймера, шизофрении и мигрени, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей, застоя, гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна, расстройств центральной нервной системы, дефицита внимания с гиперактивностью, гипо- и гиперактивности центральной нервной системы, болезни Альцгеймера, шизофрении и мигрени, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей, застоя, гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна, расстройств центральной нервной системы, дефицита внимания с гиперактивностью, гипо- и гиперактивности центральной нервной системы, болезни Альцгеймера, шизофрении и мигрени, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей, застоя, гипотензии, сердечно-сосудистого заболевания, заболеваний желудочно-кишечного тракта, гипер- и гипокинезии и секреции кислоты в желудочно-кишечном тракте, ожирения, расстройств сна, расстройств центральной нервной системы, дефицита внимания с гиперактивностью, гипо- и гиперактивности центральной нервной системы, болезни Альцгеймера, шизофрении и мигрени, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320.
Другой вариант осуществления настоящего изобретения относится к способу лечения вызванных аллергией поражений дыхательных путей, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32.
Другой вариант осуществления настоящего изобретения относится к способу лечения вызванных аллергией поражений дыхательных путей, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54.
Другой вариант осуществления настоящего изобретения относится к способу лечения вызванных аллергией поражений дыхательных путей, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55.
Другой вариант осуществления настоящего изобретения относится к способу лечения вызванных аллергией поражений дыхательных путей, включающему назначение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А.
Другой вариант осуществления настоящего изобретения относится к способу лечения вызванных аллергией поражений дыхательных путей, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287.
Другой вариант осуществления настоящего изобретения относится к способу лечения вызванных аллергией поражений дыхательных путей, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии или отека слизистой оболочки носа, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии или отека слизистой оболочки носа, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии или отека слизистой оболочки носа, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии или отека слизистой оболочки носа, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии или отека слизистой оболочки носа, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии или отека слизистой оболочки носа, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 32 и эффективное количество антагониста рецептора H1 и фармацевтически эффективный носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 54 и эффективное количество антагониста рецептора H1 и фармацевтически эффективный носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 55 и эффективное количество антагониста рецептора H1 и фармацевтически эффективный носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 253А и эффективное количество антагониста рецептора H1 и фармацевтически эффективный носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 287 и эффективное количество антагониста рецептора H1 и фармацевтически эффективный носитель.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, содержащей эффективное количество соединения 320 и эффективное количество антагониста рецептора Н1 и фармацевтически эффективный носитель.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32 в комбинации с эффективным количеством антагониста рецептора H1.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54 в комбинации с эффективным количеством антагониста рецептора H1.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55 в комбинации с эффективным количеством антагониста рецептора H1.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А в комбинации с эффективным количеством антагониста рецептора H1.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287 в комбинации с эффективным количеством антагониста рецептора H1.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320 в комбинации с эффективным количеством антагониста рецептора H1.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, хлорфенирамин, клемастин, циклизин, каребастин, ципрогептадин, карбиноксамин, дезкарбэтоксилоратадин, дифенгидрамин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, левокарбастин, меклизин, мизоластин, меквитазин, миансерин, ноберастин, норастемизол, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32 в комбинации с эффективным количеством антагониста рецептора Н1, выбранного из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему назначение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему назначение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин, дезкарбэтоксилоратадин, фексофенадин и цетиризин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 32 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин и дезкарбэтоксилоратадин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 54 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин и дезкарбэтоксилоратадин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему назначение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 55 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин и дезкарбэтоксилоратадин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему назначение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 253А в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин и дезкарбэтоксилоратадин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 287 в комбинации с эффективным количеством антагониста рецептора Н1, выбранного из группы, включающей лоратадин и дезкарбэтоксилоратадин.
Другой вариант осуществления настоящего изобретения относится к способу лечения аллергии, вызванных аллергией поражений дыхательных путей и застоя, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения 320 в комбинации с эффективным количеством антагониста рецептора H1, выбранного из группы, включающей лоратадин и дезкарбэтоксилоратадин.
Предпочтительно, чтобы R1 был выбран из группы, включающей
(A) арил (наиболее предпочтительно фенил);
(B) замещенный арил (например, замещенный фенил) и наиболее предпочтительно, чтобы заместители для указанного замещенного арила были выбраны из группы, включающей (1) галоген (например, моногалоген или дигалоген), более предпочтительно хлор или фтор, еще более предпочтительно монохлор, дихлор, монофтор или дифтор, и (2) алкил, более предпочтительно неразветвленный алкил (т.е. с линейной цепью, например, метил), еще более предпочтительно замещенный алкил, еще более предпочтительно алкил, содержащий в качестве заместителей галоген (например, 1, 2 или 3 атома галогена, такие как Cl или F), еще более предпочтительно алкил, содержащий в качестве заместителей атомы фтора, а еще более предпочтительно трифторметил;
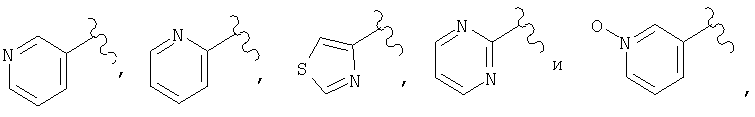
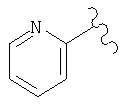
(C) гетероарил, наиболее предпочтительно пяти- или шестичленный гетероарильный цикл, более предпочтительно шестичленный гетероарильный цикл, а еще более предпочтительно пиридил, примеры гетероарильных циклов включают пиридил, тиенил, пиримидил, тиазолил и пиридил-N-оксид, а примерами наиболее предпочтительных гетероарильных колец являются
где более предпочтительным является
(D) замещенный гетероарил, наиболее предпочтительно галоген или алкилзамещенный гетероарил (например, галогенпиридил (например, фторпиридил) и алкилтиазолил), более предпочтительно замещенный гетероарил, в котором заместители независимо выбраны из группы, включающей одинаковые или разные алкильные группы (еще более предпочтительной является алкильная группа с линейной цепью, например, метил), еще более предпочтительно алкилзамещенный тиазолил, а еще более предпочтительно
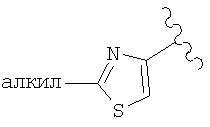
и еще более предпочтительно
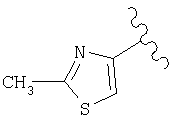
(Е) если R1 и Х образуют группировку совместно, то фрагмент имеет вид
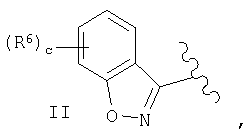
где наиболее предпочтительно, чтобы с равнялось 0 или 1 и, если с равно 1, то наиболее предпочтительно, чтобы R6 означал галоген и, если с равно 1, то наиболее предпочтительно, чтобы R6 означал фтор.
Предпочтительно, чтобы Х означал =C(NOR3), где наиболее предпочтительно, чтобы R3 был выбран из группы, включающей Н, алкил и галогензамещенный алкил (например, фторзамещенный алкил, такой как -CH2CF3), наиболее предпочтительно алкил, более предпочтительно метил и этил, а еще более предпочтительно метил.
Предпочтительно, чтобы М2 означал азот.
Предпочтительно, чтобы n равнялось 2.
Предпочтительно, чтобы а равнялось 0 или 1, а наиболее предпочтительно 0.
Предпочтительно, чтобы b равнялось 0 или 1, а наиболее предпочтительно 0.
Предпочтительно, чтобы с равнялось 0 или 1, а наиболее предпочтительно 0 и, если с равно 1, то предпочтительно, чтобы R6 означал галоген, и, если с равно 1, то наиболее предпочтительно, чтобы R6 означал фтор.
Предпочтительно, чтобы e равнялось 1-5.
Предпочтительно, чтобы Y означал =С(O) (т.е. =С=O).
Предпочтительно, чтобы М3 и М4 были выбраны такими, чтобы (1) один из них означал углерод, а другой - азот или (2) оба означали азот, причем наиболее предпочтительно, чтобы М3 означал углерод.
Предпочтительно, чтобы р равнялось 2. Предпочтительно, чтобы Z означал (С1-С3)-алкил, а наиболее предпочтительно
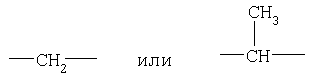
Предпочтительно, чтобы R2 означал шестичленный гетероарильный цикл, наиболее предпочтительно пиридил, замещенный пиридил, пиримидил или замещенный пиримидил, более предпочтительно пиридил, пиридил, в качестве заместителя содержащий -NR4R5, пиримидил и пиримидил, в качестве заместителя содержащий -NR4R5, еще более предпочтительно пиридил, пиридил, в качестве заместителя содержащий -NH2 (т.е. R4 и R5 означают Н), пиримидил и пиримидил, в качестве заместителя содержащий -NH2 (т.е. R4 и R5 означают Н), а еще более предпочтительно
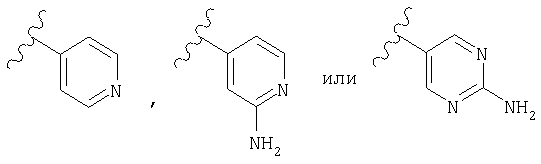
а еще более предпочтительно
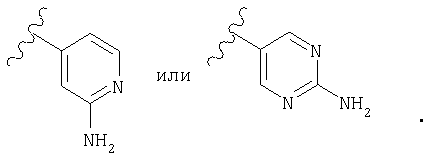
Предпочтительно, чтобы R3 означал Н или алкил, более предпочтительно Н или метил.
Предпочтительно, чтобы R4 означал Н или низший алкил, более предпочтительно Н или метил, а более предпочтительно - Н.
Предпочтительно, чтобы R5 означал Н, (С1-С6)-алкил или -C(O)R4, наиболее предпочтительно Н или метил, а более предпочтительно Н.
Предпочтительно, чтобы R12 означал алкил, гидроксил или фтор, а наиболее предпочтительно Н.
Предпочтительно, чтобы R13 означал алкил, гидроксил или фтор, а наиболее предпочтительно Н.
Типичные соединения, соответствующие настоящему изобретению, включают (без наложения ограничений) соединения 23, 30, 31, 32, 33, 41, 44, 45, 49, 50, 52, 53, 54, 55, 56, 57А, 59, 65, 75, 76, 80, 82, 83, 88, 92, 99, 104, 105, 110, 111, 117, 121, 123, 127, 128, 200-241, 244-273, 275 и 278-282, 287, 296, 301-439 и 446.
Таким образом, типичные соединения, соответствующие настоящему изобретению, включают (без наложения ограничений) соединения 23, 30, 31, 32, 33, 44, 45, 49, 50, 53, 54, 55, 59, 75, 76, 83, 88, 92, 99, 104, 110, 117, 128, 200, 201, 203-215, 217-241, 244-246, 246А, 247-253, 253А, 254-273, 275, 278 и 280-282, 317, 334 и 403.
Предпочтительные соединения, соответствующие настоящему изобретению, выбраны из группы, включающей соединения 23, 30, 31, 32, 33, 50, 53, 54, 55, 56, 57А, 59, 92, 212, 215, 218, 219, 220, 224, 225, 226, 227, 229, 233, 235, 237, 238, 246, 246А, 247, 248, 251, 253, 253А, 268-273, 275, 278-281, 287, 296, 301, 304-307, 309, 312, 314-318, 320-356 и 358-376.
Наиболее предпочтительные соединения, соответствующие настоящему изобретению, выбраны из группы, включающей соединения 30, 31, 32, 33, 54, 55, 56, 57А, 225, 237, 246А, 253А, 273, 280, 287, 296, 301, 304-307, 309, 312, 314-318, 320-348, 350-356, 359-372 и 374-376.
Таким образом, один вариант осуществления настоящего изобретения относится к соединению 32.
Другой вариант осуществления настоящего изобретения относится к соединению 54. Другой вариант осуществления настоящего изобретения относится к соединению 55. Другой вариант осуществления настоящего изобретения относится к соединению 253А. Другой вариант осуществления настоящего изобретения относится к соединению 287. Другой вариант осуществления настоящего изобретения относится к соединению 320.
Структуры указанных выше соединений представлены в приведенных ниже примерах и в приведенных ниже таблицах 1-3.
Наиболее предпочтительным соединением, соответствующим настоящему изобретению, является соединение формулы
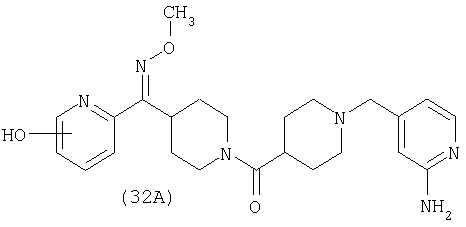
Настоящее изобретение также относится к соединению формулы
Настоящее изобретение также относится к соединению формулы
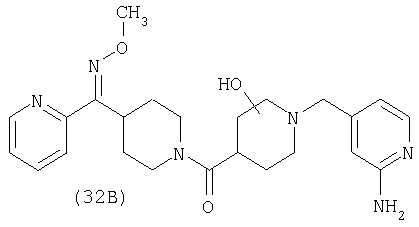
Соединения 32А и 32В также можно использовать в фармацевтических композициях и способах, соответствующих настоящему изобретению.
Для получения соединений, соответствующих настоящему изобретению, можно использовать описанные ниже способы.
Один путь синтеза включает линейную последовательность реакций получения искомых соединений, т.е.
А+В→АВ+С→АВС+D→ABCD.
Эта линейная последовательность реакций синтеза соединений, соответствующих настоящему изобретению, пояснена ниже. В представленной методике R1 означает арил, гетероарил или алкил; Х= кетон, оксим или замещенный оксим; М1=М3= углерод; М2=М4= азот; Y означает С=O; Z=CHR; R2 означает гетероарил и n и m равны 2 (по этой методике также можно получить соединения с n и m, равными 1).
Стадия 1: синтез кетона 8
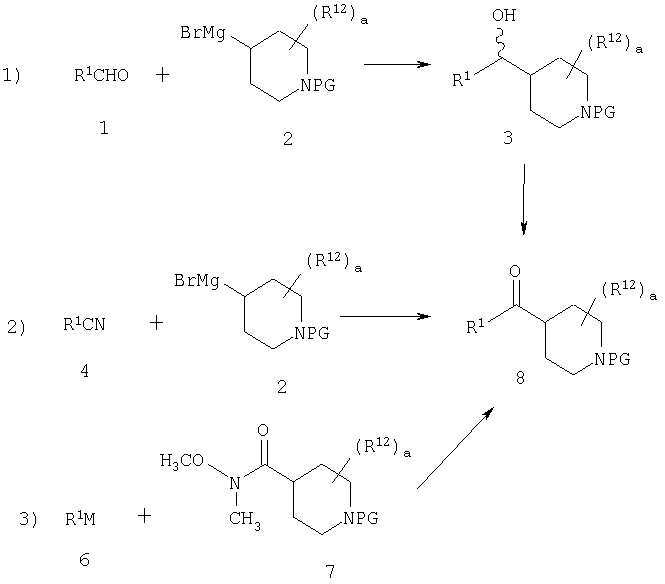
В приведенных выше уравнениях PG означает защитную группу, а М означает Li или MgX1 (где X1 означает Cl, Br или I).
В уравнениях (1) и (2) реагент Гриньяра 2 вводят в реакцию с электрофилом, таким как альдегид 1 или нитрил 4, в подходящем апротонном растворителе, таком как THF или эфир. PG означает защитную группу. Подходящие защитные группы, например, включают метил и бензил. В случае нитрила 4 обработка кислотой дает непосредственно кетон 8. Спирт 3 с помощью ряда различных реагентов можно окислить и получить 8. Альтернативно, амид 7 можно ввести в реакцию с металлоорганическим соединением и непосредственно получить кетон 8. Подходящие для этой стадии защитные группы включают карбаматную, амидную и т.п. Таким образом, примеры защитных групп для уравнения (3) включают t-Boc, CBZ и FMOC.
Стадия 2: отщепление защитной группы от 8
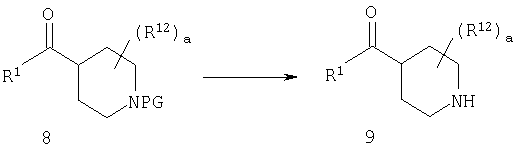
Если защитной группой PG является метильная группа, то указанную метильную группу можно удалить с помощью реагента, такого как хлорформиат; если PG означает карбаматную группу, такую как группа t-Boc, то ее можно удалить с помощью разбавленной кислоты, такой как, например, HCl.
Стадия 3: синтез 11
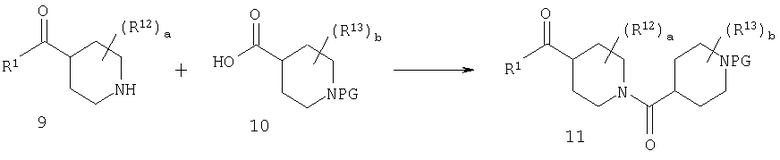
Амин 9 можно ввести в реакцию сочетания с кислотой 10 с помощью ряда способов, хорошо известных в данной области техники, таких как способы с использованием DCC или PyBOP. Альтернативно, кислоту 10 можно активировать путем превращения в хлорангидрид или смешанный ангидрид кислоты и затем ввести его в реакцию с амином 9 и получить 11. Подходящие защитные группы для 10 включают, например, t-Boc.
Стадия 4: синтез амина 12
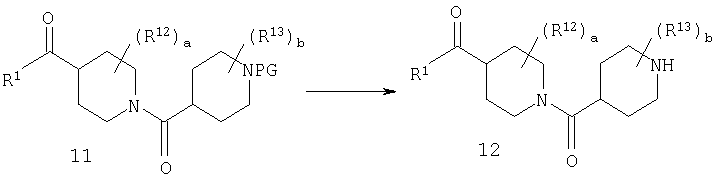
У соединения 11, в котором защитной группой является t-Boc, защитную группу можно удалить с помощью кислоты, такой как HCl, в диоксане или TFA в СН2Cl2 и получить амин 12.
Стадия 5: синтез соединения 14
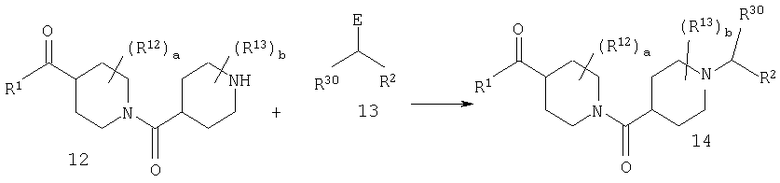
В 13 R3 означает алкильную группу. Е означает отщепляющуюся группу, галоген или Е означает карбонильную группу.
Соединение 14 можно получить по реакции амина 12 с 13. Если Е означает карбонильную группу (С=O), то 12 и 13 вводят в реакцию в растворителе, таком как CH2Cl2, в присутствии молекулярных сит. После завершения реакции (например, через промежуток времени от 1 до 10 ч) прибавляют восстановительный реагент, такой как NaBH(ОАс)3. Альтернативно, если Е означает атом галогена, такой как Cl или Br, то 12 и 13 вводят в реакцию в растворителе, таком как DMF, в присутствии третичного амина и получают продукт 14. Подходящие защитные группы включают, например, t-Boc, фталоил.
Стадия 6: синтез соединения 16
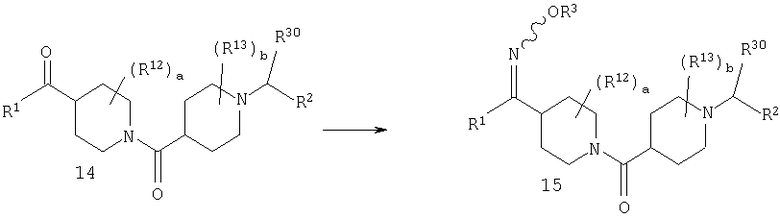
Соединение 14 можно превратить в оксим 15 путем введения 14 в реакцию с H2NOR3·HCl в пиридине при температуре, равной 40-60°С. Альтернативно, 14 можно ввести в реакцию с H2NOR3·HCl в спирте в качестве растворителя в присутствии основания, такого как NaOAc, и получить 15.
Альтернативный подход к синтезу соединений формулы I включает синтез двух половин молекулы с последующим сочетанием этих двух фрагментов, т.е.
А+В→АВ
С+D→CD
AB+CD→ABCD.
В этом случае синтез фрагмента АВ проводят так же, как это описано выше. Синтез фрагмента CD описан ниже.
Стадия 1: синтез соединения 17
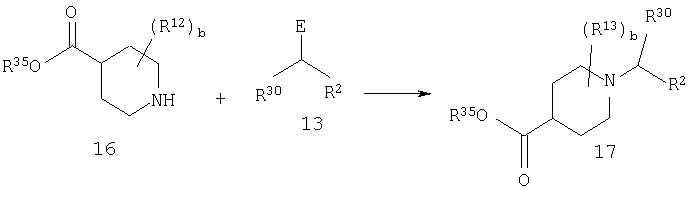
R30 является таким, как определено выше (т.е. алкилом). R35 означает метил или этил.
Соединение 17 синтезируют таким же образом, как это описано для синтеза соединения 14.
Стадия 2: синтез соединения 18
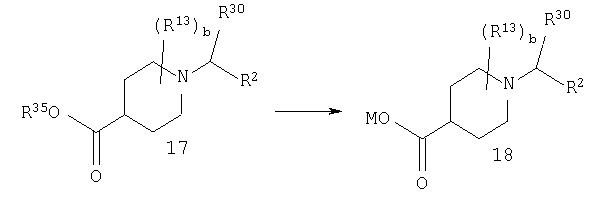
М означает Li, Na или К.
Соединение 17 омыляют в смеси растворителей, такой как, например: (1) EtOH или МеОН и вода, или (2) THF, вода и МеОН, с использованием основания щелочного металла, такого как LiOH или NaOH, при температуре от 50 до 100°С и получают соль 18.
Соединение 18 можно ввести в реакцию с соединением 9, как это описано выше, и получить 14. Остальные стадии являются такими же.
Соединения, использующиеся в настоящем изобретении, представлены в качестве примеров в приведенных ниже примерах, которые не следует рассматривать, как ограничивающие настоящее изобретение. Для специалиста в данной области техники должны быть понятными альтернативные пути синтеза и аналогичные структуры, входящие в объем настоящего изобретения.
Пример 1
Стадия 1
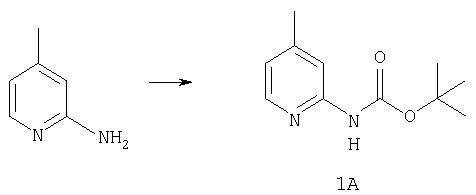
К раствору 10,81 г (100 ммоль) 2-амино-4-метилпиридина в 250 мл трет-бутанола прибавляют 26,19 г (120 ммоль) ВОС-ангидрида. Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют, подают на силикагель и подвергают флэш-хроматографии (от 30% гексаны/СН2Cl2 до 0-2% ацетон/CH2Cl2) и получают 15,25 г (73,32 ммоль; 73%) соединения 1А в виде белого твердого вещества.
Стадия 2
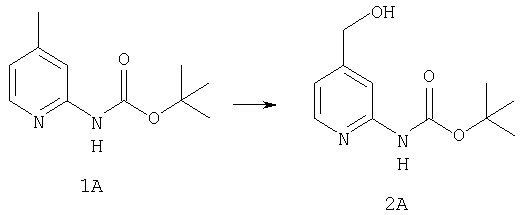
При -78°С к раствору 1А (35,96 г, 173 ммоль) в THF (1,4 л) в течение 30 мин порциями прибавляют 1,4 М раствор BuLi (272 мл, 381 ммоль) в гексанах. Реакционной смеси дают нагреться и 2 ч перемешивают при комнатной температуре, что приводит к образованию оранжевого осадка. Смесь повторно охлаждают до -78°С и в течение 6 ч, поддерживая температуру равной -78°С, через суспензию барботируют предварительно высушенный кислород (пропущенный через колонку Drierite). За это время окраска реакционной смеси переходит в оранжевую. При -78°С реакцию останавливают путем прибавления 51,4 мл (700 ммоль) Me2S, а затем 22 мл (384 ммоль) АсОН. Реакционной смеси дают нагреться и 48 ч перемешивают при комнатной температуре. Разбавляют водой и экстрагируют с помощью СН2Cl2, а затем концентрируют и подвергают флэш-хроматографии (0-15% ацетон/СН2Cl2) и получают 20,15 г (90 ммоль; 52%) спирта 2А в виде бледно-желтого твердого вещества.
Стадия 3
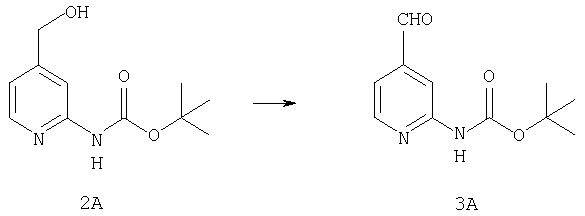
К раствору 19,15 г (85,5 ммоль) спирта 2А в 640 мл CH2Cl2 прибавляют насыщенный водный раствор 8,62 г (103 ммоль) NaHCO3 и 444 мг (4,3 ммоль) NaBr. Реакционную смесь охлаждают до 0°С и прибавляют 140 мг (0,90 ммоль) TEMPO. При энергичном перемешивании порциями в течение 40 мин прибавляют 122 мл 0,7 М (85,4 ммоль) продажного отбеливающего раствора (содержащего 5,25% NaOCl). Еще через 20 мин выдерживания при 0°С реакцию останавливают с помощью насыщенного водного раствора Na2S2O3 и реакционной смеси дают нагреться до комнатной температуры. Разбавляют водой и экстрагируют с помощью СН2Cl2, а затем концентрируют и подвергают флэш-хроматографии (от 30% гексаны/СН2Cl2 до 0-2% ацетон/СН2Cl2) и получают 15,97 г (71,9 ммоль; 84%) альдегида 3А в виде почти белого твердого вещества.
Стадия 4
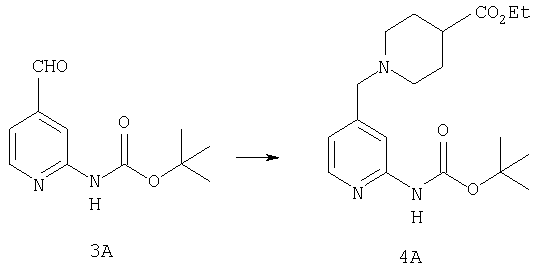
К раствору 11,87 г (53,5 ммоль) альдегида 3А в 370 мл CH2Cl2 прибавляют 9,07 мл (58,8 ммоль) этилизонипекотата, а затем четыре капли АсОН. Затем реакционную смесь 40 мин перемешивают при комнатной температуре, после чего прибавляют 22,68 г (107 ммоль) NaBH(ОАс)3. Реакционную смесь перемешивают при комнатной температуре в течение ночи, нейтрализуют насыщенным водным раствором NaHCO3, разбавляют водой и экстрагируют с помощью СН2Cl2. Концентрирование и флэш-хроматография (0-4% насыщенный раствор NH3 в MeOH/CH2Cl2 дает 19,09 г (52,6 ммоль, 98%) соединения 4А в виде почти белого твердого вещества.
Стадия 5
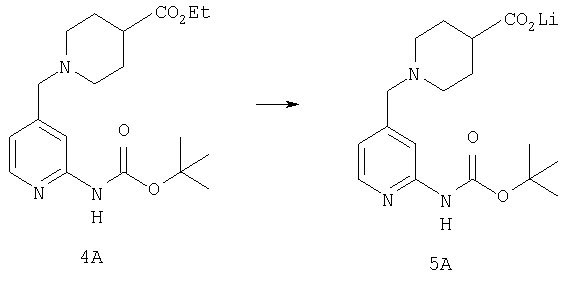
К раствору 1,57 г (4,33 ммоль) сложного эфира 4А в 10 мл смеси THF - вода - метанол состава 3:1:1 прибавляют 0,125 г (5,21 ммоль) LiOH. Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют и подвергают сушке в высоком вакууме, получая 1,59 г неочищенной кислоты 5А в виде желтоватого твердого вещества, которое используют без очистки.
Пример 2
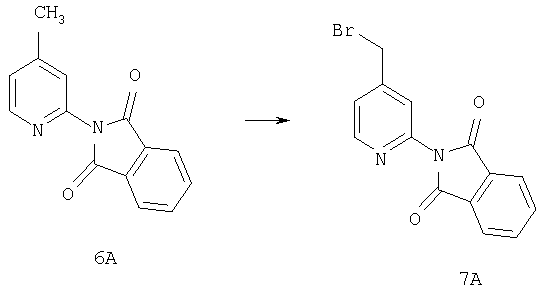
Раствор соединения 6А (42 ммоль), NBS (126 ммоль) и Bz2O2 (4,2 ммоль) в CCl4 (400 мл) 5 ч кипятят с обратным холодильником при 80°С, охлаждают и перемешивают при комнатной температуре в течение ночи. Реакционную смесь фильтруют, и концентрируют, остаток очищают на флэш-колонке (30% EtOAc/гексан) и получают искомое соединение 7А (3,1 г, 23%).
Пример 3
Стадия 1
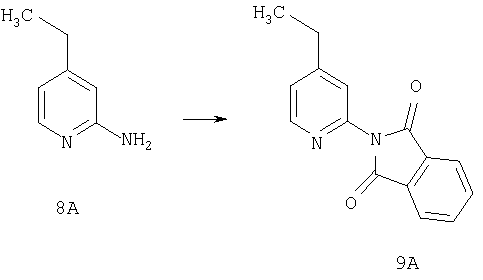
К раствору 8А (10 г, 79,4 ммоль) и DMAP (0,029 г, 0,24 ммоль) в метиленхлориде (150 мл) при 0°С по каплям прибавляют фталоилдихлорид (16,1 г, 79,4 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. После перемешивания в течение ночи реакционную смесь промывают насыщенным водным раствором NaHCO3, водой, сушат и концентрируют, получая соединение 9А в виде желтого твердого вещества (20 г, 99,8%), которое используют без дополнительной очистки.
Стадия 2
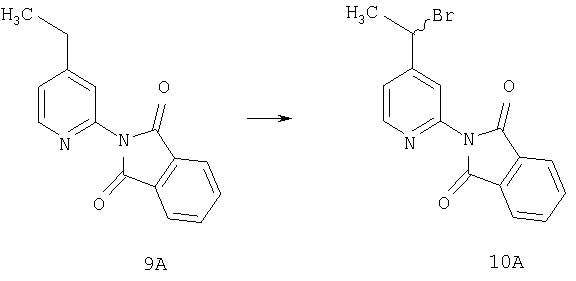
Способом, сходным с описанным в Примере 2, соединение 9А (20 г, 79,3 ммоль) превращают в соединение 10А.
Стадия 3
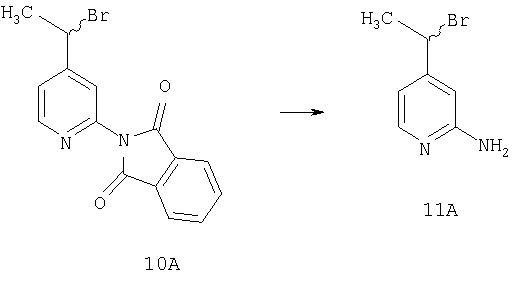
Соединение 10А (0,5 г, 1,5 ммоль) и гидразин (0,5 М раствор в этаноле, 5 мл, 2,5 ммоль) смешивают и перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют водой и экстрагируют метиленхлоридом. Органический слой сушат, концентрируют и остаток очищают на флэш-колонке (3% раствор метанола в этилацетате) и получают соединение 11А (0,2 г, 66%).
Пример 4
Стадия 1
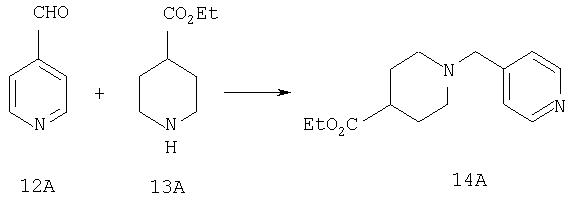
Соединения 12А (2 г, 18,3 ммоль) и 13А (3,5 г, 22 ммоль) растворяют в метиленхлориде и 1 ч перемешивают при комнатной температуре. Прибавляют NaBH(ОАс)3 (5,4 г, 25,6 ммоль) и реакционную смесь 5 ч перемешивают при комнатной температуре. Реакционную смесь промывают насыщенным водным раствором NaHCO3, водой, сушат, концентрируют и остаток очищают на флэш-колонке (2% раствор метанола в этилацетате). Получают соединение 14А (4,5 г, 99%).
Стадия 2
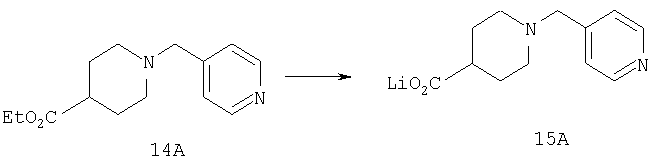
Способом, сходным с описанным в Примере 1, стадия 5, соединение 14А (0,35 г, 1,4 ммоль) превращают в соединение 15А (0,31 г, 100%).
Пример 5
Стадия 1
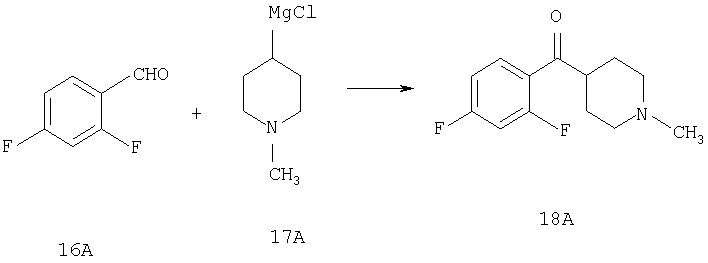
К раствору 2,4-дифторбензилальдегида (16А, 28,1 ммоль) в THF (10 мл) прибавляют реагент Гриньяра 17А (1,33 М раствор в THF, 30 мл) и смесь перемешивают при комнатной температуре в течение ночи. Реакцию останавливают насыщенным раствором NH4Cl (150 мл), три раза экстрагируют с помощью EtOAc (100 мл), сушат, фильтруют и концентрируют. Флэш-хроматография (20% МеОН/EtOAc) дает искомое соединение 18А (1,8 г,27%).
Стадия 2
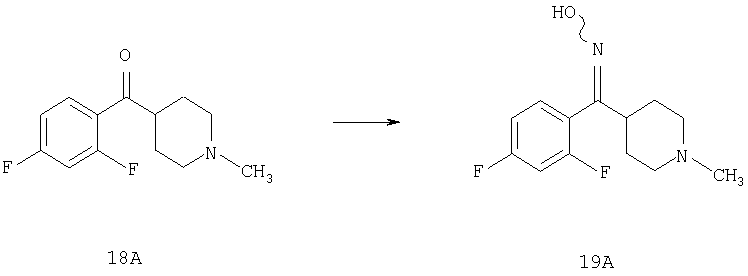
Соединение 18А (1,6 г, 6,7 ммоль), H2NHOH·HCl (0,95 г, 6,7 ммоль) и пиридин (7 мл) смешивают и в течение ночи нагревают при 60°С. Пиридин удаляют в вакууме и остаток обрабатывают метиленхлоридом и насыщенным водным раствором NaHCO3. Органический слой отделяют, сушат и, концентрируют и остаток очищают с помощью флэш-хроматографии, получая соединение 19А (1,4 г, 82%).
Стадия 3
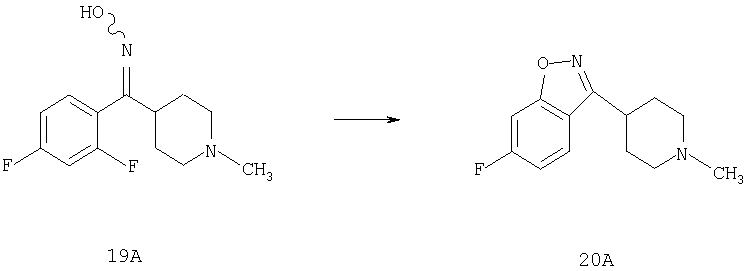
К суспензии NaH (0,41 г, 10,2 ммоль) в THF (10 мл) по каплям медленно прибавляют раствор 19А (1,3 г, 5,11 ммоль) в DMF (5 мл) и реакционную смесь перемешивают в течение ночи при 70-75°С. Смесь дважды экстрагируют с помощью EtOAc и трижды с помощью Н2O (30 мл), сушат над MgSO4 и концентрируют, получая неочищенное соединение 20А, которое используют без дополнительной очистки (1,04 г, 87%).
Стадия 4
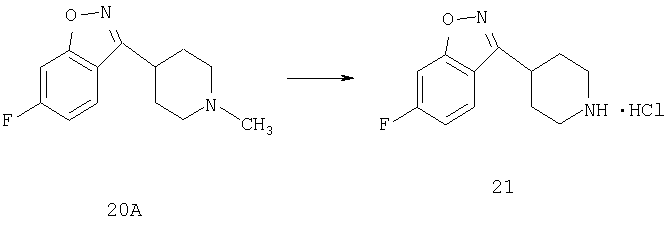
К раствору соединения 20А (4,3 ммоль) в дихлорметане (20 мл) при 0°С прибавляют 2-хлорэтилхлорформиат (6,2 ммоль) и триэтиламин (7,2 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают, к остатку прибавляют Et2O и непрореагировавшее исходное вещество удаляют фильтрованием. Фильтрат концентрируют и остаток повторно растворяют в МеОН и 30 мин кипятят с обратным холодильником. Удаление метанола дает продукт 21 (0,3 г), который используют без дополнительной очистки.
Стадия 5
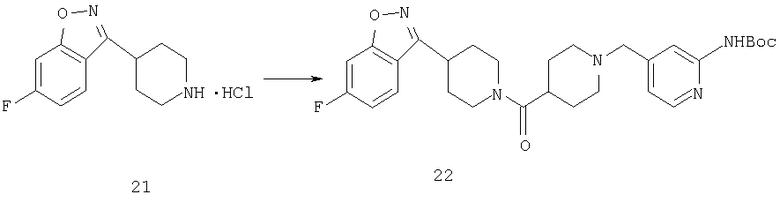
К смеси соединения 21 (1,64 ммоль), соединения 5А (1,64 ммоль) и PyBOP (1,64 ммоль) прибавляют DIPEA (4,92 ммоль) и СН2Cl2 (10 мл) и реакционную смесь перемешивают при комнатной температуре в течение выходных дней. Прибавляют насыщенный раствор NaHCO3 (100 мл) и реакционную смесь дважды экстрагируют с помощью CH2Cl2 (100 мл), сушат над твердым MgSO4, концентрируют, подвергают флэш-хроматографии (70% EtOAc/гексан) и получают соединение 22 (1,04 ммоль, 64%).
Стадия 6
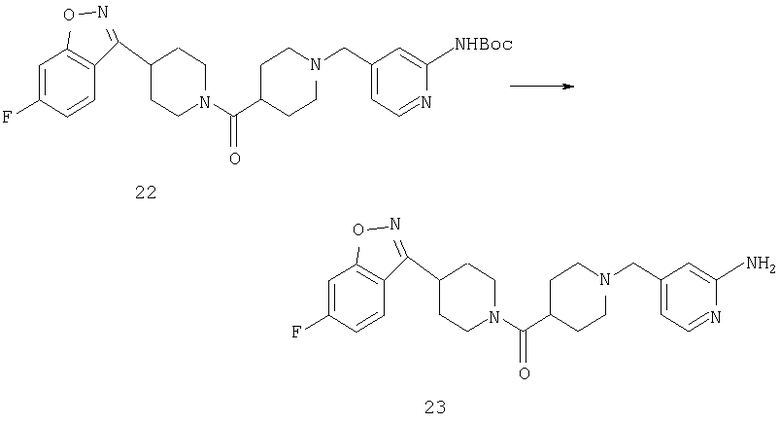
Соединение 22 (0,2 г, 0,37 ммоль) растворяют в CF3CO2Н (3 мл) и метиленхлориде (3 мл) и перемешивают при комнатной температуре в течение ночи. Растворитель удаляют выпариванием, прибавляют насыщенный раствор NaHCO3 и смесь экстрагируют метиленхлоридом. Органический слой сушат (MgSO4), фильтруют и концентрируют, остаток очищают с помощью флэш-хроматографии, получая соединение 23 (0,11 г, 68%).
Пример 6
Стадия 1
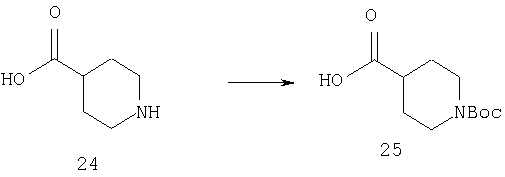
Раствор 24 (50 г, 387 ммоль) и триэтиламина (110 мл) в диоксане (400 мл) и воде (400 мл) при 4°С обрабатывают с помощью Boc2O (9,3 г, 426 ммоль). Охлаждающую баню удаляют и раствору дают нагреться до комнатной температуры. Через 21 ч с помощью вакуума объем уменьшают на две трети. Остаток выливают в этилацетат (250 мл) и воду (250 мл). Прибавляют насыщенный водный раствор NaHCO3 и органическую фазу отделяют и отбрасывают. Водную фазу подкисляют с помощью 10% раствора HCl и экстрагируют этилацетатом. Объединенные органические фазы промывают водой, рассолом, сушат (Na2SO4) и концентрируют, получая 25 в виде белого порошка (82 г, 94%).
Стадия 2
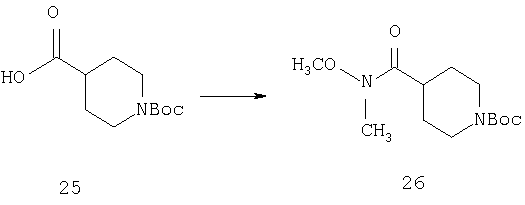
К раствору соединения 25 (40 г, 175 ммоль) в DMF (250 мл) при 4°С прибавляют N,O-диметилгидроксиламингидрохлорид (34 г), EDCl (44 г, 0,228 моль), НОВТ (2,4 г) и DIPEA (120 мл). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Затем реакционную смесь концентрируют в вакууме до половины объема и выливают в смесь этилацетат: вода состава 1:1. Органический слой отделяют и водный слой экстрагируют дополнительным количеством этилацетата. Объединенные органические фазы промывают насыщенным водным раствором NH4Cl, насыщенным водным раствором NaHCO3, водой и рассолом и сушат. Концентрирование дает 26 в виде светло-желтого масла (46,7 г, 99%).
Стадия 3
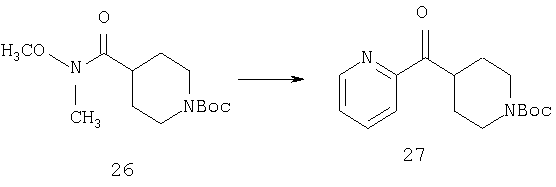
К раствору 2-бромпиридина (17,6 мл, 0,184 моль) в THF (600 мл) при -78°С в течение 15 мин по каплям прибавляют n-BuLi (115 мл 1,6 М раствора в гексанах, 0,184 моль). После перемешивания при этой температуре в течение еще 30 мин по каплям в течение 15 мин прибавляют раствор 26 (25 мг, 91,9 ммоль) в THF (500 мл). Реакционный сосуд снимают с охлаждающей бани, помещают на масляную баню и 1,5 ч нагревают при 60°С. Затем реакционную смесь охлаждают до 4°С, разбавляют эфиром (500 мл) и обрабатывают насыщенным водным раствором Na2SO4, (примерно 5 мл). Смесь переносят в коническую колбу и разбавляют дополнительным количеством эфира (700 мл). Прибавляют дополнительное количество насыщенного водного раствора Na2SO4, a затем твердый Na2SO4. Смесь фильтруют через слой твердого Na2SO4 и концентрируют в вакууме. Хроматографирование на флэш-колонке (0-20% раствор этилацетата в гексанах) дает соединение 27 в виде желтого масла (16,85 г, 63%).
Стадия 4
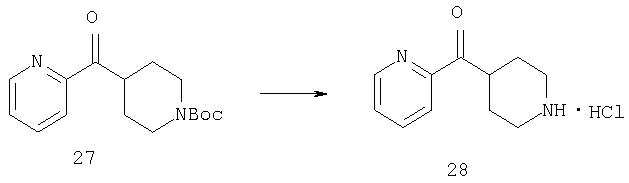
Раствор 27 (3,3 г, 11,4 ммоль) в метаноле (50 мл) обрабатывают 4 М раствором HCl в диоксане (50 мл) и 1,5 ч перемешивают при комнатной температуре. Удаление растворителя в вакууме дает 28 в виде коричневато-желтого порошка (3 г, 100%).
Стадия 5
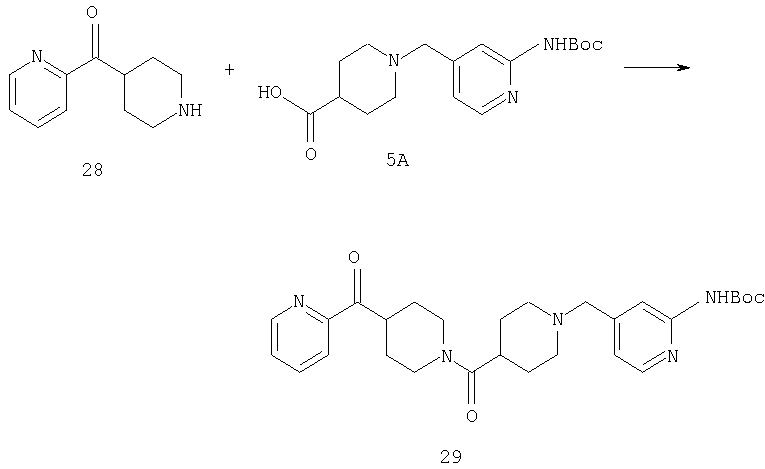
К суспензии соединения 5А (17,4 г, 50 ммоль), соединения 28 (11 г, 42 ммоль) и диизопропилэтиламина (34,6 мл, 199 ммоль) в DMF (125 мл) прибавляют НОВТ (7,83 г, 58 ммоль), EDC (18,54 г, 96,7 ммоль) и молекулярные сита с размером пор, равным 4 Å. Смесь 40 ч перемешивают при комнатной температуре, разбавляют метиленхлоридом (600 мл) и 0,5 н. раствором NaOH (400 мл) и фильтруют. Осадок тщательно промывают дополнительным количеством 0,5 н. раствора NaOH и метиленхлорида. Объединенные органические фазы концентрируют и дважды хроматографируют на силикагеле (от смеси гексан : метиленхлорид состава 1:1 до 6% раствора насыщенного раствора NH3 в метаноле в метиленхлориде), получают 29 в виде желтовато-коричневого твердого вещества (22,3 г), которое используют на следующей стадии без дополнительной обработки.
Стадия 6
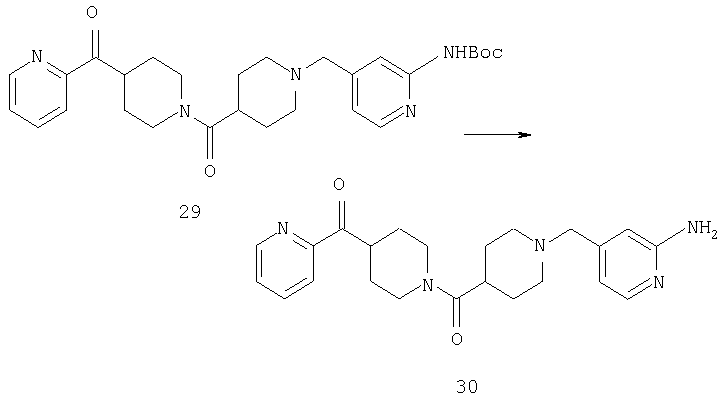
Раствор 29 (22,3 г, 44 ммоль) в метиленхлориде (120 мл) и трифторуксусной кислоте (60 мл) перемешивают при комнатной температуре в течение 7 ч. Реакционную смесь концентрируют, 3 ч подвергают воздействию высокого вакуума, растворяют в толуоле, концентрируют и повторно подвергают воздействию высокого вакуума. Полученное таким образом неочищенное коричневое масло используют на следующей стадии без дополнительной очистки.
Стадия 7
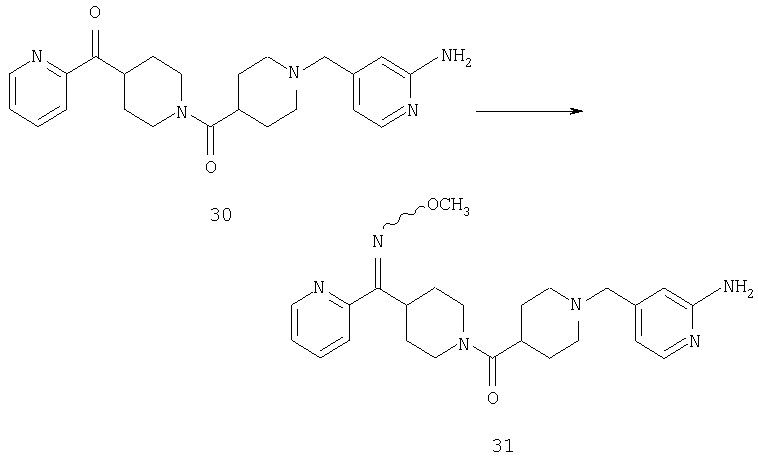
Соединение 30 (примерно 17,9 г, 44 ммоль) растворяют в пиридине (420 мл), обрабатывают с помощью Н2NOCH3·HCl (21,78 г, 264 ммоль) и 14 ч нагревают при 90°С. Затем реакционную смесь концентрируют и остаток растворяют в смеси метиленхлорида (500 мл) с 2 н. раствором NaOH (500 мл). Органическую фазу отделяют и водную фазу экстрагируют дополнительным количеством метиленхлорида (300 мл). Органические фазы сушат, концентрируют, остаток хроматографируют на SiO2 (0-13% NH3/MeOH в CH2Cl2) и получают желтое твердое вещество (9,26 г). Смешанные фракции, полученные на колонке, хроматографируют повторно и получают еще 3,23 г искомого вещества. Суммарный выход 12,49 г (общий выход для двух последних стадий равен 65%).
Стадия 8
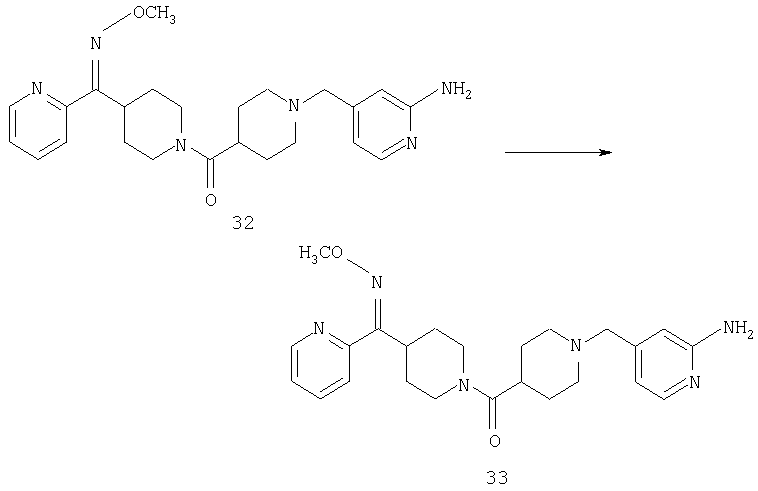
Соединение 31 (1 г) в этаноле (15 мл) разделяют на два чистых изомера с помощью колонки Chiralcel AD (20 мм × 500 мм) (элюент: 75:25, гексан : изопропанол с прибавлением 0,5% N,N-диэтиламина; скорость потока 50 мл/мин; УФ детектирование при длине волны 254 нм) и получают соединение 32 (0,6 г) и соединение 33 (0,4 г). [M+H]+ 437 для 32 и 33.
Альтернативно, соединение 32 можно получить из соединения 5А способом, аналогичным описанному для получения соединения 287 на стадии 3 Примера 28.
Пример 7
Стадия 1
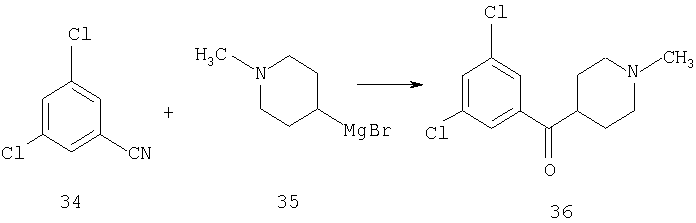
К раствору 34 (2,4 г, 13,5 ммоль) в THF (15 мл) прибавляют соединение 35 (26 мл 1,3 М раствора) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 2 н. раствор HCl, пока значение рН не станет меньше 2, и THF удаляют при пониженном давлении. Реакционную смесь нейтрализуют путем прибавления 1 н. раствора NaOH и водную фазу экстрагируют с помощью 5% раствора МеОН в EtOAc. Органическую фазу сушат, концентрируют и остаток хроматографируют (20% раствор МеОН в EtOAc), получая 36 (1,03 г, 28%).
Стадия 2
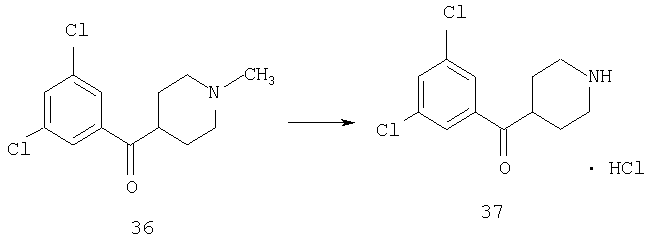
К раствору 36 (1,03 г, 3,78 ммоль) в 1,2-дихлорэтане (30 мл) прибавляют 1-хлорэтилхлорформиат (0,76 мл, 7,6 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и остаток промывают эфиром. Твердый остаток удаляют фильтрованием, эфир удаляют путем выпаривания и получают масло, которое растворяют в МеОН (15 мл) и 2 ч кипятят с обратным холодильником. Удаление растворителя дает 37, которое используют на следующей стадии без дополнительной очистки (1,4 г).
Стадия 3
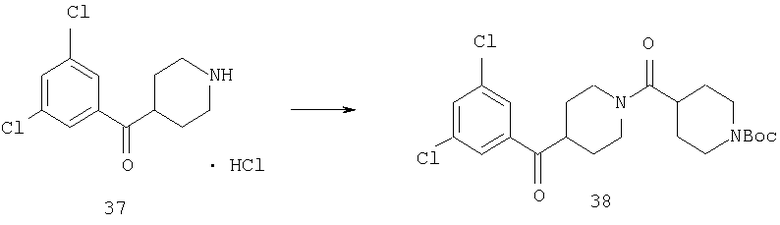
Соединение 37 (0,98 г, 3,78 ммоль), N-Вос-изонипекотиновую кислоту (0,87 г, 3,78 ммоль), DEC (1,11 г, 5,7 ммоль), НОВТ (0,68 г, 4,91 ммоль) и DIPEA (3 мл) смешивают в CH2Cl2 (40 мл) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют с помощью CH2Cl2 и промывают насыщенным водным раствором NaHCO3. Органический слой сушат, концентрируют и остаток хроматографируют (10% раствор гексана в EtOAc), получая 38 (1,61 г, 91%).
Стадия 4
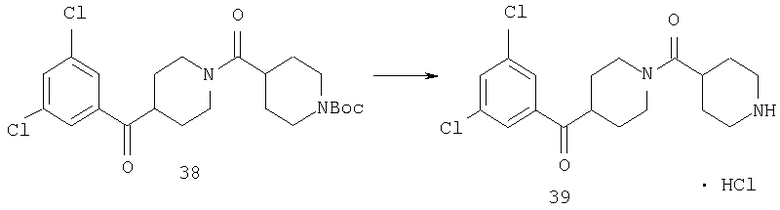
Соединение 38 (1,61 г, 3,43 ммоль) в CH2Cl2 (15 мл) обрабатывают 1 н. раствором HCl в диоксане (5,2 мл) и перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и получают 39 (1,65 г), который используют без дополнительной очистки.
Стадия 5
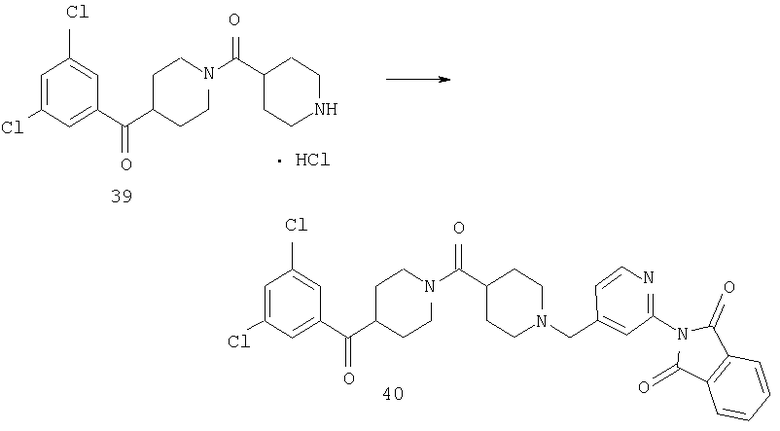
Соединения 39 (1,65 г, 4,01 ммоль), 7 (1,29 г, 3,07 ммоль) и Et3N (1,7 мл) смешивают в DMF (40 мл) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь растворяют в EtOAc и 4 раза промывают водой. Органический слой сушат и концентрируют, остаток очищают с помощью хроматографии (5% раствор МеОН в EtOAc), получая 40 (0,6 г, 47%).
Стадия 6
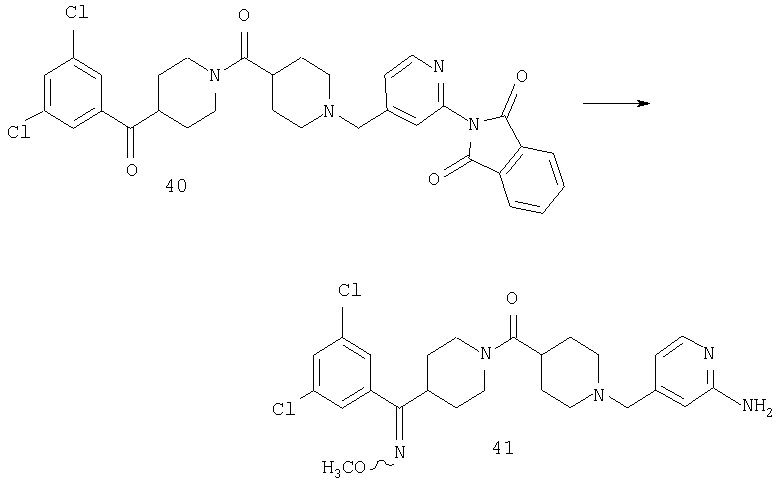
Раствор 40 (0,31 г, 0,51 ммоль) в пиридине (5 мл) обрабатывают с помощью H2NOMe·HCl (0,092 г, 1,08 ммоль) и нагревают при 60°С в течение ночи. Реакционную смесь разбавляют 10% раствором МеОН в CH2Cl2, промывают насыщенным водным раствором NaHCO3, сушат и концентрируют, остаток очищают с помощью хроматографии (10-15% раствор МеОН в EtOAc), получая 41 (0,09 г).
Пример 8
Стадия 1
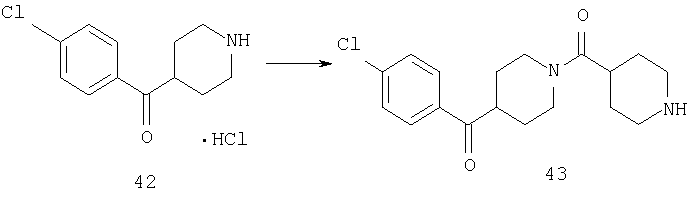
Способом, сходным с описанным в Примере 7, стадии 3-4, соединение 42 превращают в соединение 43.
Стадия 2
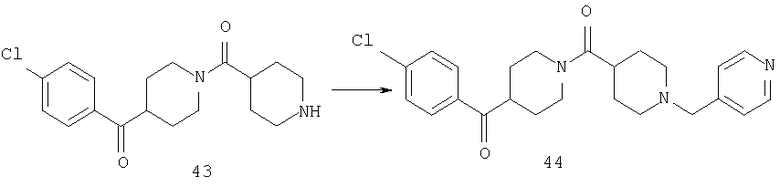
К раствору 43 (2,3 г, 6,3 ммоль) в CH2Cl2 (60 мл) прибавляют молекулярные сита с размером пор, равным 4Å, и 4-формилпиридин (0,68 мл, 6,9 ммоль) и смесь 3 ч перемешивают при комнатной температуре. Затем прибавляют NaBH(OAc)3 (2,7 г, 12,7 ммоль) и реакционную смесь перемешивают 1 ч. Реакцию останавливают путем прибавления NH4Cl, а затем прибавляют насыщенный водный раствор NaHCO3. Затем реакционную смесь экстрагируют с помощью EtOAc и объединенные органические слои сушат и концентрируют, получая остаток, который хроматографируют (20% раствор МеОН в EtOAc). Получают соединение 44 (2,3 г, 87%).
Стадия 3
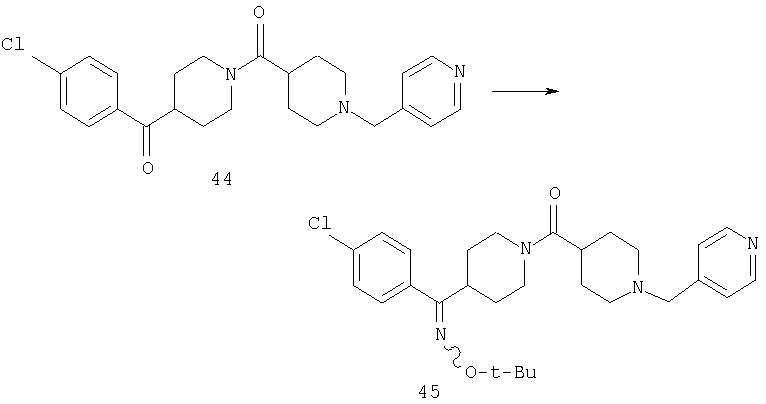
Способом, сходным с описанным в Примере 7, стадия 6, соединение 44 превращают в соединение 45.
Пример 9
Стадия 1
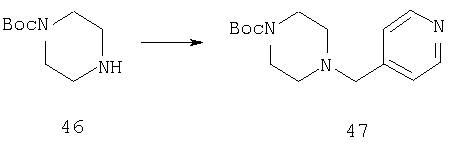
Способом, сходным с описанным в Примере 8, стадия 2, соединение 46 (1,13 г, 6 ммоль) превращают в соединение 47 (1,7 г, 100%).
Стадия 2
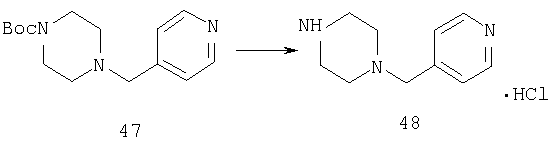
Способом, сходным с описанным в Примере 7, стадия 4, соединение 47 (1,7 г, 6,13 ммоль) превращают в соединение 48 (1,9 г, 100%).
Стадия 3
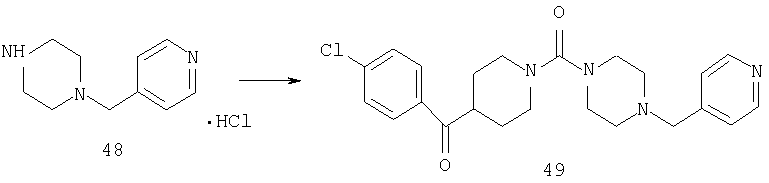
К смеси соединения 48 (0,57 г, 2 ммоль) и соединения 42 (0,52 г, 2 ммоль) в CH2Cl2 (20 мл) прибавляют Et3N (1,95 мл) и реакционную смесь охлаждают до -40°С. Прибавляют трифосген (0,2 г) и реакционную смесь перемешивают 2 ч при -40°С и 48 ч при комнатной температуре. Затем реакционную смесь промывают 1 н. раствором NaOH, рассолом и органический слой сушат. Концентрирование дает остаток, который очищают с помощью хроматографии на колонке (10% раствор МеОН в EtOAc.) и получают 49 (0,14 г, 55%).
Стадия 4
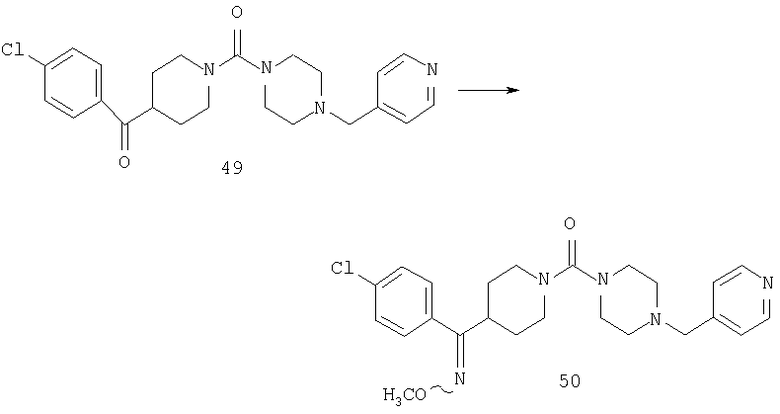
Способом, сходным с описанным в Примере 7, стадия 6, соединение 49 (0,09 г, 0,21 ммоль) превращают в соединение 50.
Пример 10
Стадия 1
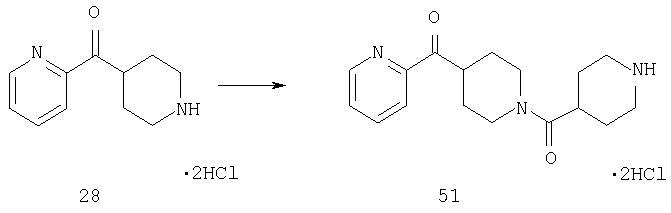
Способом, сходным с описанным в Примере 7, стадии 3-4, соединение 28 (2,6 г, 9,9 ммоль) превращают в соединение 51 (1,1 г).
Стадия 2
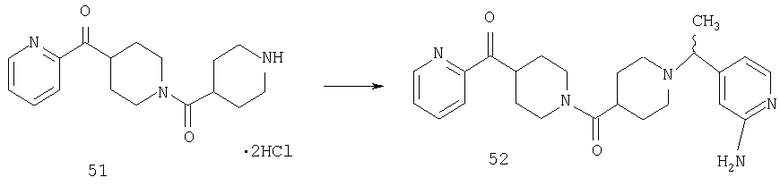
Способом, сходным с описанным в Примере 7, стадия 5, соединение 51 (1,1 г, 2,94 ммоль) превращают в соединение 11 (0,59 г, 2,94 ммоль) и получают соединение 52 (0,53 г).
Стадия 3
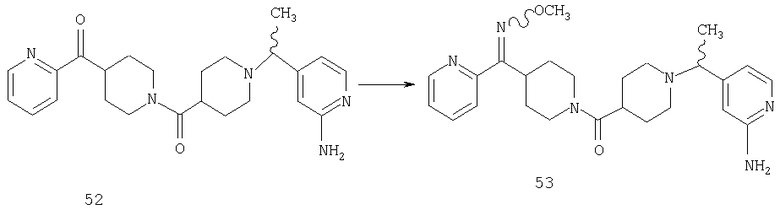
Способом, сходным с описанным в Примере 6, стадия 7, соединение 52 (0,53 г, 1,26 ммоль) превращают в соединение 53 (0,48 г).
Стадия 4
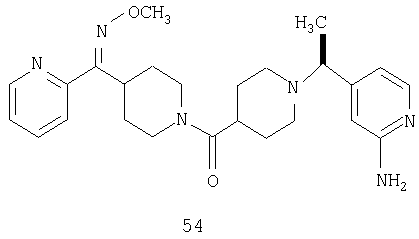
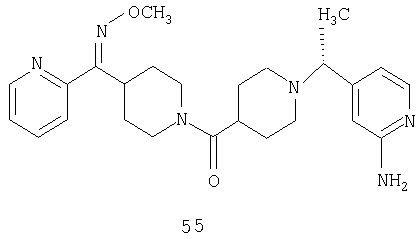
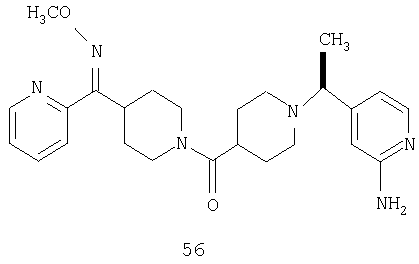
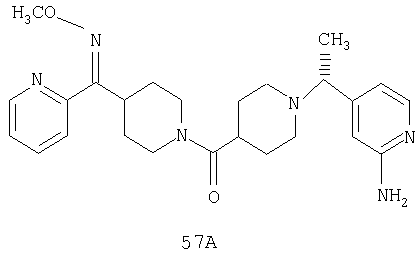
Способом, сходным с описанным в Примере 6, стадия 8, 4 диастереоизомера соединения 53 можно получить с использованием колонки Chiralcel AD (75:25, гексан : EtOAc с добавкой 0,5% Et2NH). Два более быстро элюирующиеся соединения (54 и 55) представляют собой Е-изомеры оксима, а медленнее элюирующиеся соединения (56 и 57А) представляют собой Z-изомеры оксима.
Пример 11
Стадия 1
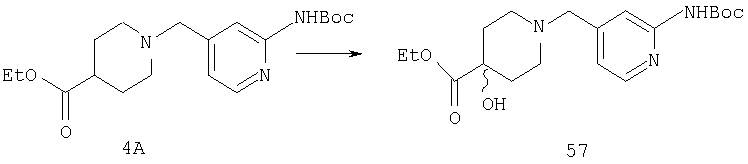
Раствор n-BuLi (4,2 мл 1,6 М раствора в гексане) в THF (25 мл) при -25°С обрабатывают с помощью (i-Pr)2NH (0,69 г, 6,8 ммоль). Реакционную смесь 1 ч перемешивают при 0°С, а затем охлаждают до -70°С. По каплям прибавляют соединение 4А (0,82 г, 2,26 ммоль) в THF (5 мл) и реакционную смесь перемешивают 2 ч при -70°С и 2 ч при -50°С. Реакционную смесь повторно охлаждают до -70°С и прибавляют (1S)-(+)-(10-камфорсульфонил)оксазиридин (1,04 г, 4,52 ммоль) в THF (5 мл). Реакционную смесь 2 ч перемешивают при -70°С и в течение ночи медленно нагревают до комнатной температуры. Реакцию останавливают насыщенным раствором NH4Cl и экстрагируют с помощью EtOAc. Органический слой сушат и концентрируют, остаток очищают с помощью хроматографии на колонке (1:1, гексан : EtOAc) и получают 57 (0,44 г, 51%).
Стадия 2
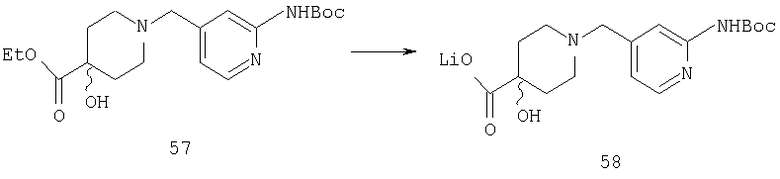
Способом, сходным с описанным в Примере 1, стадия 5, соединение 57 (0,42 г, 1,1 ммоль) превращают в соединение 58 (0,4 г).
Стадия 3
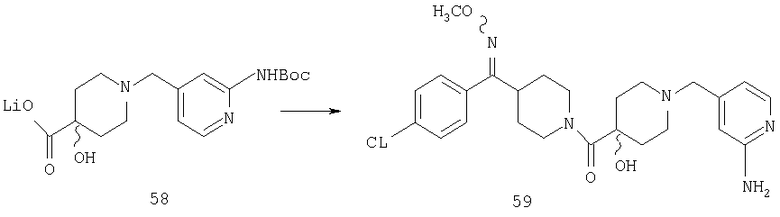
Способом, сходным с описанным в Примере 6, стадии 5-8, соединение 58 (0,25 г, 0,7 ммоль) превращают в соединение 59 (0,1 г).
Пример 12
Стадия 1
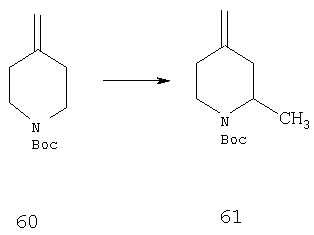
Раствор соединения 60 (10 г, 50,7 ммоль) в эфире (150 мл) при -78°С последовательно обрабатывают с помощью TMEDA (11,8 г, 101,4 ммоль) и n-BuLi (58,5 мл 1,3 М раствора в гексанах, 76 ммоль) и при этой температуре реакционную смесь перемешивают в течение 6 ч. Затем прибавляют неразбавленный СН3SO4СН3 (12,8 г, 101,4 ммоль) и реакционную смесь в течение ночи медленно охлаждают до комнатной температуры. Прибавляют насыщенный водный раствор NaCl и органический слой отделяют. Водный слой трижды экстрагируют эфиром и объединенные органические слои сушат, концентрируют и остаток хроматографируют (5% раствор EtOAc в гексане) и получают 61 (8,0 г, 75%).
Стадия 2
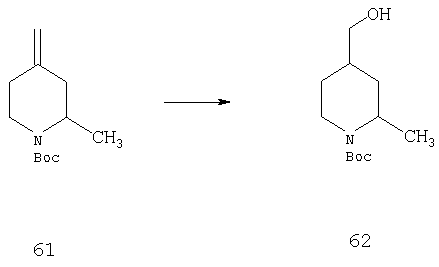
Раствор 61 (8 г, 37,9 ммоль) в THF (40 мл) при 0°С по каплям обрабатывают раствором ВН3·THF (45,4 мл 1,0 М раствора в THF, 45,4 ммоль) и реакционной смеси в течение ночи дают медленно нагреться до комнатной температуры. Реакционную смесь повторно охлаждают до 0°С и прибавляют EtOH (17 мл), буферный раствор с рН=7 (25 мл) и H2O2 (25 мл) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем растворитель удаляют в вакууме и остаток выливают в воду и CH2Cl2. Прибавляют 10% водный раствор NaOH (10 мл) и органический слой отделяют. Водный слой экстрагируют дополнительным количеством CH2Cl2 и объединенные органические слои сушат и концентрируют. Остаток хроматографируют (40% раствор EtOAc в гексане) и получают 62 (3 г).
Стадия 3
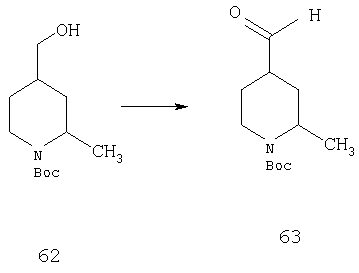
Раствор 62 (2,8 г, 12,2 ммоль) в EtOAc (30 мл) и NaBr (1,26 г, 0,12 ммоль) в насыщенном водном растворе NaHCO3 (30 мл) охлаждают до 0°С и обрабатывают с помощью TEMPO (0,02 г, 0,12 ммоль). Через 15 мин прибавляют NaOCl (17,44 мл) и смесь 3 ч перемешивают. Прибавляют насыщенный водный раствор Na2S2O2 и путем прибавления 1 н. раствора HCl значение рН доводят до 5-6. Смесь экстрагируют с помощью EtOAc и органические слои сушат и концентрируют. Остаток хроматографируют (10-20% раствор EtOAc в гексане) и получают соединение 63 (2,1 г, 76%).
Стадия 4
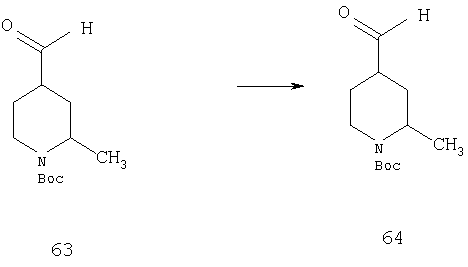
К суспензии РСС (0,95 г, 4,4 ммоль) в CH2Cl2 (5 мл) при охлаждении (0°С) по каплям прибавляют раствор 63 (0,5 г, 2,2 ммоль). Смесь перемешивают при комнатной температуре в течение ночи. Прибавляют дополнительное количество РСС (1 экв.) и смесь 2 ч кипятят с обратным холодильником. Реакционную смесь охлаждают, фильтруют через целит и концентрируют, получая неочищенное соединение 64 (1,5 г), которое используют без дополнительной очистки.
Стадия 5
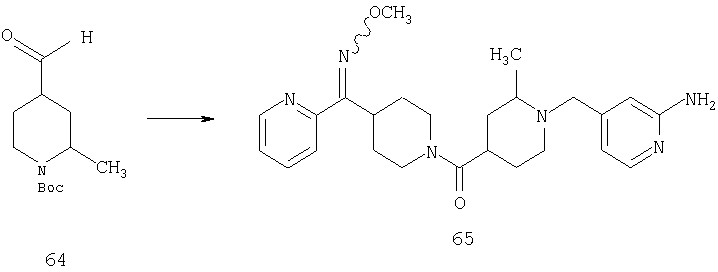
Способом, сходным с описанным в Примере 5, стадия 5, Примере 7, стадия 4, Примере 1, стадия 4, и Примере 6, стадии 6 и 7, 64 (0,73 г, 3 ммоль) превращают в 65 (0,1 г).
Пример 13
Стадия 1
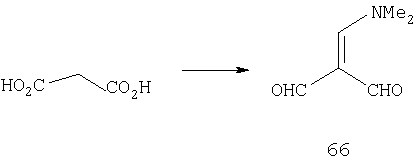
При 0°С к раствору соли Вильсмейера, полученной проводимым в течение 15 мин по каплям прибавлением оксихлорида фосфора (150,0 мл, 1,61 моль) к DMF (310,4 мл, 4,01 моль) с последующим охлаждением в бане со льдом, порциями в течение 45 мин прибавляют малоновую кислоту (40,1 г, 0,39 моль). Затем реакционную смесь нагревают до 100°С и перемешивание продолжают в течение 48 ч. После этого реакционной смеси дают охладиться до комнатной температуры и реакцию останавливают, медленно выливая реакционную смесь в суспензию NaHCO3 (808 г, 9,62 моль) в воде. Раствор сливают с избытка NaHCO3 и концентрируют досуха в вакууме. После сушки в высоком вакууме в течение 2 дней твердый остаток многократно промывают с помощью CH2Cl2, пока ТСХ не подтвердит полное удаление продукта. Объединенные органические экстракты концентрируют в вакууме и получают 41,0 г темно-коричневого масла, которое непосредственно используют на следующей стадии.
Стадия 2
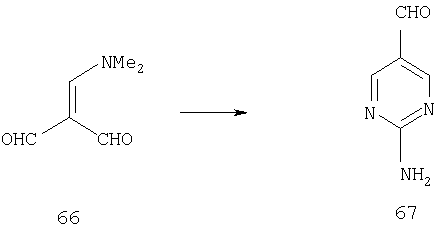
К раствору 32,5 г (256 ммоль) неочищенного малонового диальдегида 66 в 650 мл абсолютного этанола прибавляют 24,5 г (256 ммоль) гуанидингидрохлорида и 17,4 г (256 ммоль) этоксида натрия. Реакционную смесь 4 ч кипятят с обратным холодильником, охлаждают до комнатной температуры, концентрируют и в сухом виде в вакууме вносят в силикагель. Флэш-хроматография (0-10% МеОН/20% ацетона/CH2Cl2) дает 11,0 г (89,4 ммоль; 23% в расчете на малоновую кислоту (2 стадии)) пиримидина 67 в виде светло-желтого твердого вещества.
Стадия 3
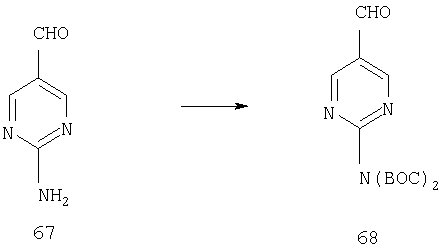
К смеси 166 мг (1,35 ммоль) аминопиридина 67, 17 мг (0,14 ммоль) DMAP и 418 мкл (3,00 ммоль) Et3N в 10 мл THF прибавляют 589 мг (2,7 ммоль) (ВОС)2О. Смесь 5 ч перемешивают при комнатной температуре, концентрируют досуха, вносят в силикагель и подвергают флэш-хроматографии (1-3% ацетон/СН2Cl2) и получают 117 мг (0,36 ммоль, 27%) соединения 68 в виде прозрачного масла.
Стадия 4
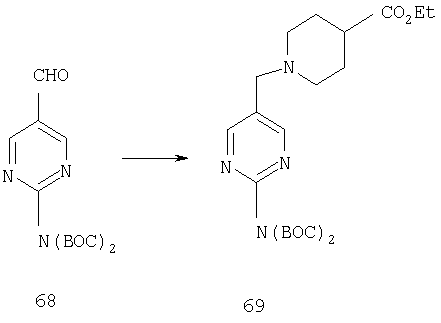
К раствору 117 мг (0,36 ммоль) альдегида 68 в 7 мл CH2Cl2 прибавляют 67 мкл (0,43 ммоль) этилизонипекотата и 5 мкл уксусной кислоты. Через 30 мин прибавляют 153 мг (0,72 ммоль) NaBH(ОАс)3. Смесь перемешивают при комнатной температуре в течение ночи, разбавляют с помощью CH2Cl2, промывают водным раствором NaHCO3, сушат и концентрируют, неочищенный остаток подвергают флэш-хроматографии (1-4% насыщенный раствор NH3 в MeOH/CH2Cl2) и получают 133 мг (0,29 ммоль, 81%) соединения 69 в виде прозрачной пленки.
Стадия 5
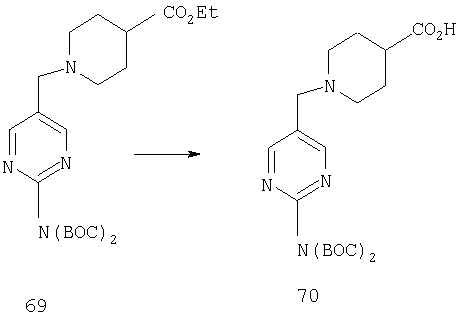
К раствору сложного эфира 69 в 5 мл смеси THF-вода-метанол состава 3:1:1 прибавляют 11 мг (0,44 ммоль) LiOH. Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют досуха и сушат в высоком вакууме, получая 134 мг неочищенной кислоты 70 в виде желтоватого твердого вещества, которое используют без очистки.
Пример 14
Стадия 1
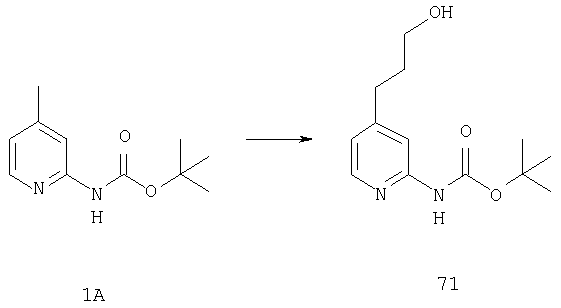
При -78°С к раствору 2,36 г (11,4 ммоль) пиколина 1А в 70 мл THF в течение 10 мин порциями прибавляют 16,3 мл 1,4 М раствора BuLi (22,8 ммоль) в гексанах. Реакционной смеси дают нагреться и ее 2 ч перемешивают при комнатной температуре, что приводит к образованию оранжевого осадка. Смесь повторно охлаждают до -78°С и в течение 1 мин через раствор барботируют оксид этилена, а затем перемешивают в течение 5 мин. Эту двустадийную последовательность повторяют восемь раз. Затем смеси дают нагреться до -50°С, перемешивают при этой температуре в течение 40 мин, реакцию останавливают с помощью 1,34 мл (23 ммоль) АсОН и реакционной смеси дают нагреться до комнатной температуры. Разбавление водой с последующей экстракцией с помощью АсОН, концентрирование органической фазы и флэш-хроматография неочищенного остатка (10-15% ацетон/CH2Cl2 дают 1,50 г (5,95 ммоль, 53%) соединения 71 в виде белого твердого вещества.
Стадия 2
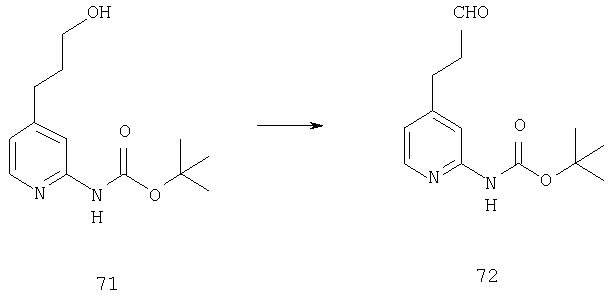
При -60°С к раствору 628 мкл (7,2 ммоль) оксалилхлорида в 20 мл CH2Cl2 по каплям прибавляют 1,03 мл (14,5 ммоль) DMSO. После перемешивания смеси в течение 15 мин при -55°С в течение 15 мин прибавляют раствор 1,50 г (5,95 ммоль) спирта 71 в 20 мл CH2Cl2. После завершения прибавления смесь 30 мин перемешивают при -55°С, а затем прибавляют 4,18 мл (30,0 ммоль) Et3N и перемешивают еще 15 мин. Затем реакционную смесь нагревают до комнатной температуры и разбавляют водой. Экстракция с помощью АсОН, концентрирование органической фазы и флэш-хроматография (1-15% ацетон/CH2Cl2) дают 1,00 г (4,00 ммоль, 67%) соединения 72 в виде почти белого твердого вещества.
Стадия 3
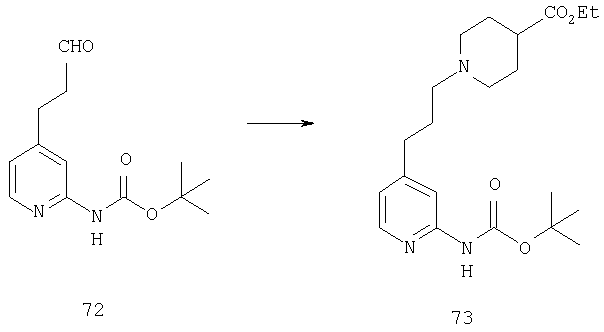
К раствору 1,00 г (4,0 ммоль) альдегида 72 в 25 мл CH2Cl2 прибавляют 617 мкл (4,8 ммоль) этилизонипекотата, а затем одну каплю АсОН. Затем реакционную смесь 40 мин перемешивают при комнатной температуре, после чего прибавляют 1,70 г (8,0 ммоль) NaBH(ОАс)3. Реакционную смесь перемешивают при комнатной температуре в течение ночи, нейтрализуют насыщенным водным раствором NaHCO3, разбавляют водой и экстрагируют с помощью CH2Cl2. Концентрирование и флэш-хроматография (0-4% насыщенный раствор NH3 в MeOH/CH2Cl2) дают 1,41 г (3,6 ммоль, 90%) соединения 73 в виде белого твердого вещества.
Стадия 4
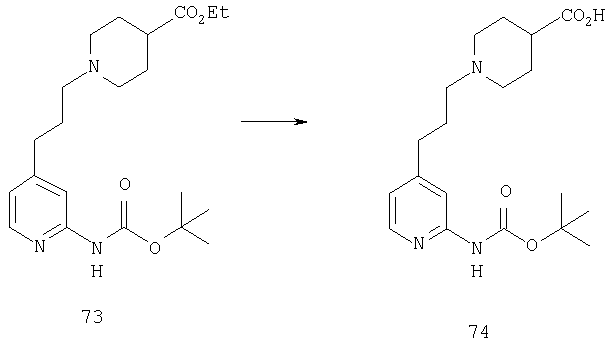
К раствору 534 мг (1,47 ммоль) сложного эфира 73 в 4 мл смеси THF - вода - метанол состава 3:1:1 прибавляют 60 мг (2,50 ммоль) LiOH. Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют досуха и сушат в высоком вакууме, получая 540 мг неочищенной кислоты 74 в виде белого твердого вещества, которое используют без очистки.
Пример 15
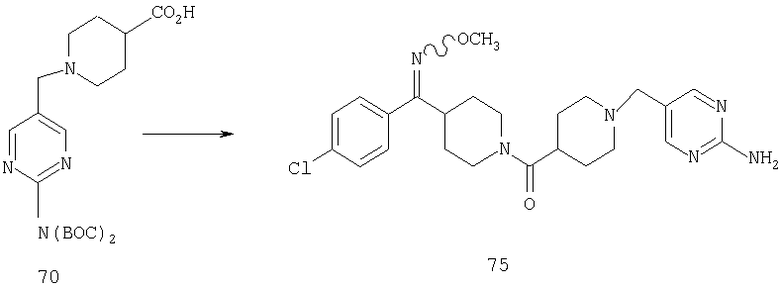
Способом, сходным с описанным в Примере 6, стадии 5, 6 и 7, 70 превращают в 75.
Пример 16
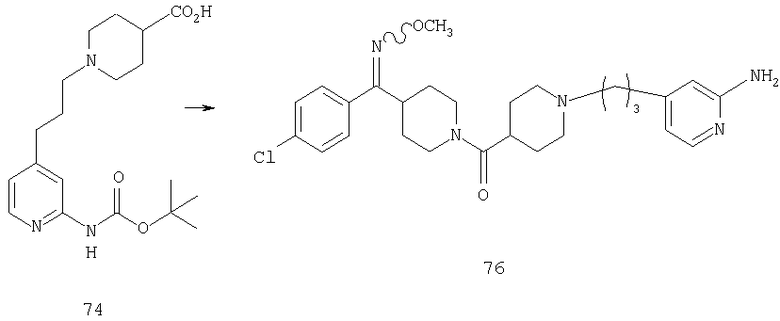
Способом, сходным с описанным в Примере 6, стадии 5, 6 и 7, 74 превращают в 76.
Пример 17
Стадия 1
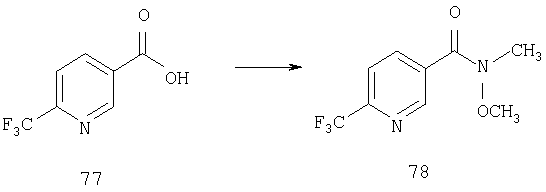
К раствору 77 (0,73 г, 3,82 ммоль) в CH2Cl2 (10 мл) прибавляют (COCl)2 (0,41 мл, 4,58 ммоль), а затем DMF (0,1 мл) и реакционную смесь 3 ч выдерживают при 40°С. Реакционную смесь концентрируют и получают коричневое твердое вещество, которое растворяют в CH2Cl2 (10 мл). Прибавляют N,O-диметилгидроксиламингидрохлорид (0,56 г, 5,73 ммоль) и DIPEA (1,33 мл) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакцию останавливают путем прибавления насыщенного водного раствора NaHCO3 и экстрагируют с помощью EtOAc. Объединенные органические слои сушат и концентрируют, остаток очищают с помощью хроматографии и получают 78 (3,2 г, 84%).
Стадия 2
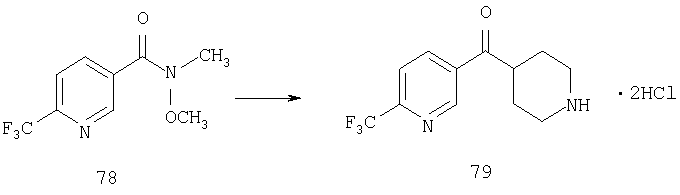
Способом, сходным с описанным в Примере 5, стадии 1 и 4, 78 (0,57 г, 2,41 ммоль) превращают в 79 (0,59 г).
Стадия 3
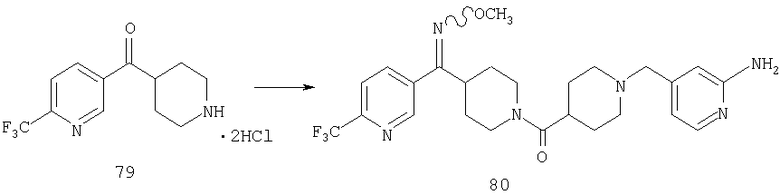
Способом, сходным с описанным в Примере 6, стадии 5, 6 и 7, 79 (0,38 г, 1,49 ммоль) превращают в 80 (0,24 г).
Пример 18
Стадия 1
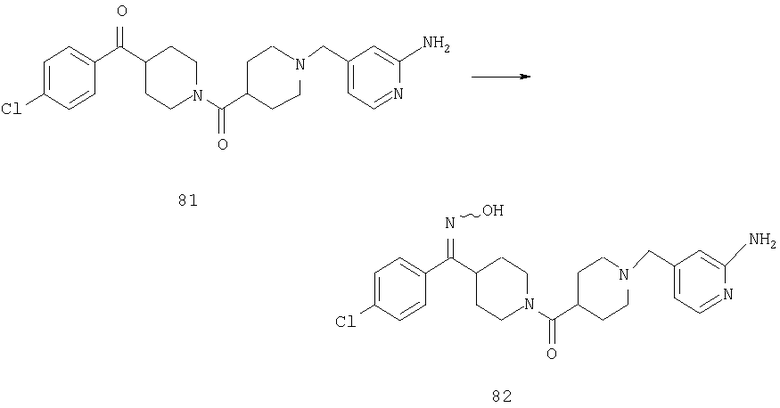
Способом, сходным с описанным в Примере 6, стадия 7, 81 (0,36 г, 0,53 ммоль; синтезировано таким же способом, как соединение 30) превращают в 82 (0,34 г).
Стадия 2
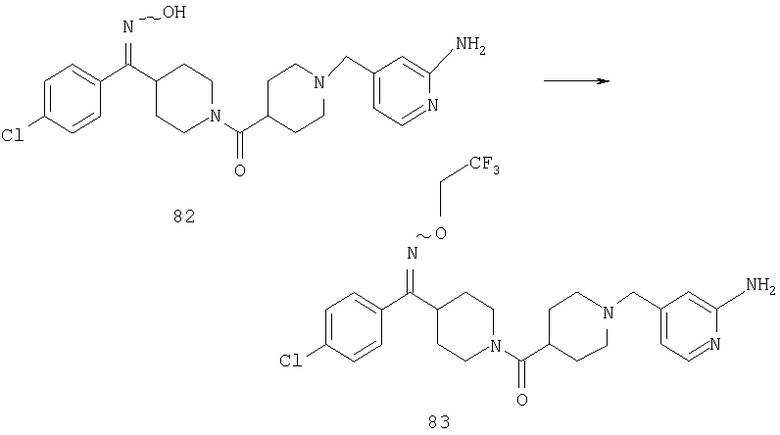
К раствору 82 (0,115 г, 0,25 ммоль) в DMF (4 мл) прибавляют NaH (60% дисперсия в минеральном масле, 0,03 г, 0,76 ммоль). Через 5 мин при комнатной температуре прибавляют CF3СН2OSO2CF3 (0,069 г, 0,3 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют с помощью EtOAc и для удаления DMF трижды экстрагируют водой. Органический слой сушат и концентрируют, получая остаток, который очищают с помощью хроматографии (10% МеОН/NH3 в EtOAc) и получают 83 (0,08 г, 30%).
Пример 19
Стадия 1
К раствору 17 (0,21 моль, 100 мл THF, -10°С) в течение 15 мин прибавляют 84 (0,14 моль) и реакционная смесь становится очень вязкой. Прибавляют дополнительное количество THF (100 мл) и в течение примерно 2,5 ч желтую суспензию нагревают от -10 до 10°С. Реакцию останавливают путем прибавления 100 мл насыщенного раствора NH4Cl и 100 мл H2O. Экстрагируют один раз с помощью EtOAc (300 мл) и восемь раз с помощью CH2Cl2 (150 мл). Сушат над твердым MgSO4 и фильтруют. Концентрируют и очищают с помощью флэш-хроматографии на силикагеле (от 3 до 10% МеОН (NH3)/СН2Cl2 и получают 85 (11 г, выход 38%).
Стадия 2
К смеси 85 (9,2 г) и MnO2 (42 г) прибавляют 200 мл CH2Cl2 и смесь перемешивают при комнатной температуре в течение ночи. Прибавляют дополнительное количество MnO2 (20 г) и реакционную смесь перемешивают еще 24 ч. MnO2 отфильтровывают, реакционную смесь концентрируют и очищают с помощью флэш-хроматографии на силикагеле (5 и 10% МеОН (NH3)/CH2Cl2, получают 86 (3,1 г, выход 33%).
Стадия 3
Способом, сходным с описанным в Примере 7, стадия 2, 86 (3,1 г) превращают в 87 (2,0 г, выход 68%).
Стадия 4
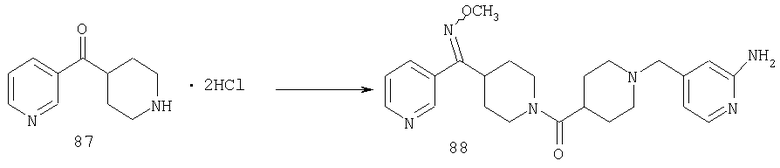
Способом, сходным с описанным в Примере 7, стадии 3, 4, 5 и 6, 87 превращают в 88.
Пример 20
Стадия 1
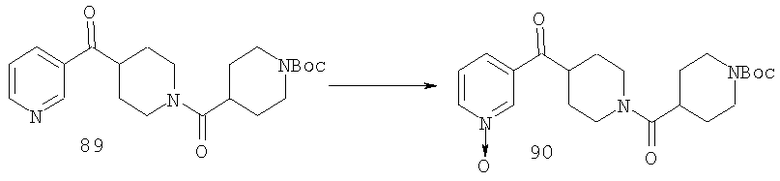
К раствору соединения 89 в CH2Cl2 (20 мл) при 0°С прибавляют m-СРВА (0,54 г) и реакционную смесь 25 мин перемешивают при 0°С, а затем 2 ч при комнатной температуре. Прибавляют 40% NH4OH (12 мл) и смесь перемешивают в течение 30 мин. Водный слой отделяют и экстрагируют с помощью СН2Cl2 (10 мл). Сушат (MgSO4), фильтруют и концентрируют в вакууме. Флэш-хроматография (5% МеОН (NH3)/CH2Cl2) дает 90 (0,67 г, 80%).
Стадия 2
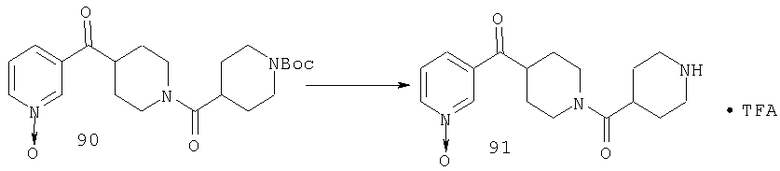
К раствору соединения 90 (0,65 г) в CH2Cl2 (6 мл) при -10°С прибавляют TFA (6 мл) и реакционную смесь в течение 1 ч перемешивают при температуре от -10 до 0°С. Концентрируют и дважды подвергают азеотропной перегонке с толуолом (20 мл), концентрируют досуха, получая 91 в виде смолообразного масла, которое используют без дополнительной обработки.
Стадия 3
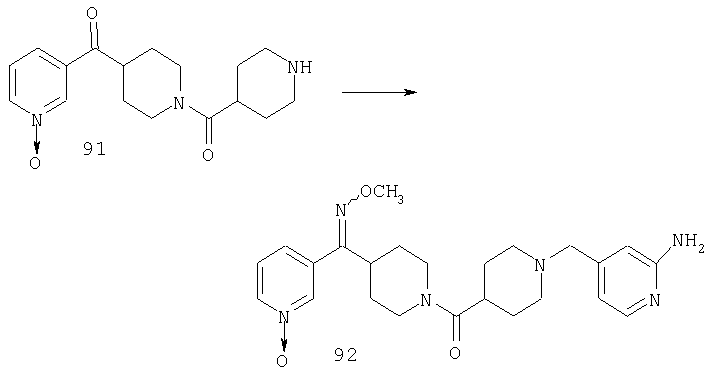
Способом, сходным с описанным в Примере 7, стадии 5 и 6, 91 превращают в 92.
Пример 21
Стадия 1
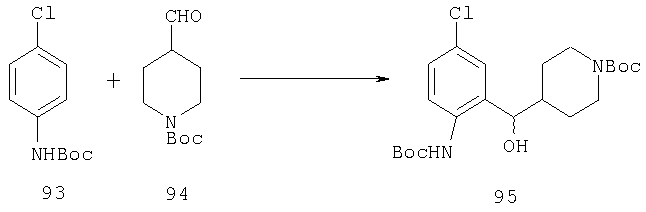
К раствору 93 (5,17 г, 22,7 ммоль) в THF (100 мл) при -50°С по каплям прибавляют n-BuLi (38,4 мл 1,3 М раствора в гексане. После выдерживания при -40°С в течение часа реакционную смесь повторно охлаждают до -50°С и прибавляют 95 (4,84 г, 22,7 ммоль) в THF (20 мл). После выдерживания при -50°С в течение 2,75 ч прибавляют ледяную уксусную кислоту, а затем насыщенный водный раствор NH4Cl. Смесь нагревают до комнатной температуры и слои разделяют. Водный слой экстрагируют с помощью EtOAc. Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют, получая остаток, который очищают с помощью флэш-хроматографии (от 1 до 3% МеОН/NH3 в CH2Cl2), получая 95 (6,35 г, 63%).
Стадия 2
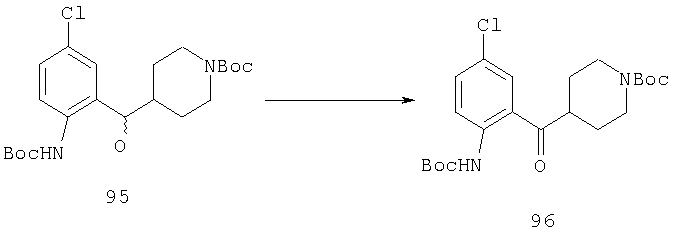
Способом, сходным с описанным в Примере 12, стадия 3, 95 (5,34 г, 12,11 ммоль) превращают в 96 (4,71 г, 75%).
Стадия 3
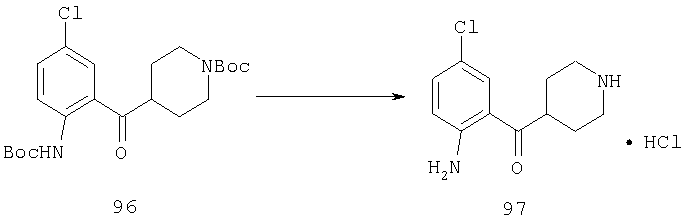
Способом, сходным с описанным в Примере 6, стадия 4, 96 (3,7 г, 8,43 ммоль) превращают в 97 (3,08 г, > 100%), которое используют на следующей стадии без дополнительной обработки.
Стадия 4
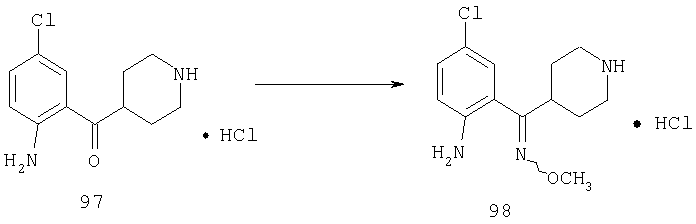
Соединение 97 (0,7 г, 2,25 ммоль), Н2NOCH3·HCl (0,94 г, 11,23 ммоль) и NaOAc (1,47 г, 17,97 ммоль) смешивают в 1-пентаноле (20 мл) и воде (2 мл) и 2 дня кипятят с обратным холодильником. Реакционную смесь охлаждают до комнатной температуры и прибавляют 0,5 н. раствор NaOH. EtOH удаляют в вакууме, прибавляют дополнительное количество воды (15 мл) и реакционную смесь экстрагируют с помощью 10% раствора EtOH в CH2Cl2 (полный объем 180 мл). Объединенные органические экстракты сушат и концентрируют, получая 98 (0,55 г, 92%).
Стадия 5
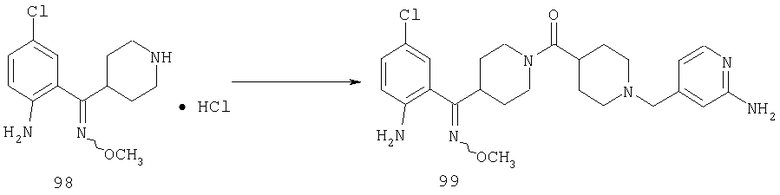
Способом, сходным с описанным в Примере 6, стадии 5, 6 и 7, 98 превращают в 99.
Пример 22
Стадия 1
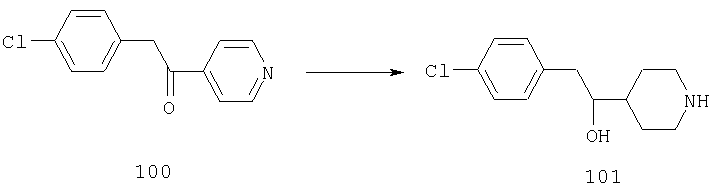
Получено по методике, описанной в J. Org. Chem., 1968, 33 (6), 2388.
Раствор 2,2 г (9,5 ммоль) 100 в 75 мл ледяной уксусной кислоты в течение 5 ч гидрируют в присутствии 0,5 г 10 мас.% платины-на-древесном угле. Реакционную смесь фильтруют для удаления катализатора и фильтрат концентрируют путем выпаривания при пониженном давлении, получая твердый остаток, который подщелачивают с помощью 0,5 н. раствора NaOH и экстрагируют метиленхлоридом (CH2Cl2). Экстракты в метиленхлориде сушат над безводным MgSO4 и концентрируют. Остаток очищают с помощью флэш-хроматографии, элюируя смесью 10-30% 7 н. NH3 - МеОН в CH2Cl2, и получают 0,82 г соединения 101 (температура плавления 158-163°С). ЖХМС m/z 240 (МН+).
Стадия 2
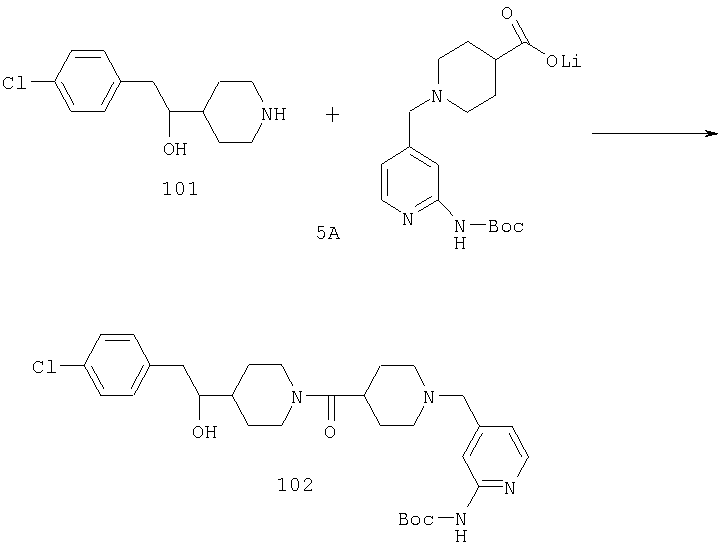
Смесь 0,12 г (0,52 ммоль) 101, 0,2 г (0,52 ммоль) 5А, 0,67 г (0,5 ммоль) 1-гидроксибензотриазолгидрата (НОВТ) и 0,11 г (0,57 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида (DEC) в 7 мл безводного диметилформамида (DMF) перемешивают при температуре окружающей среды в течение 18 ч. Смесь разбавляют водой и образовавшийся осадок отфильтровывают, получая 0,26 г 102, в виде белого твердого вещества (температура плавления 110-115°С). ЖХМС m/z 557 (МН+).
Стадия 3
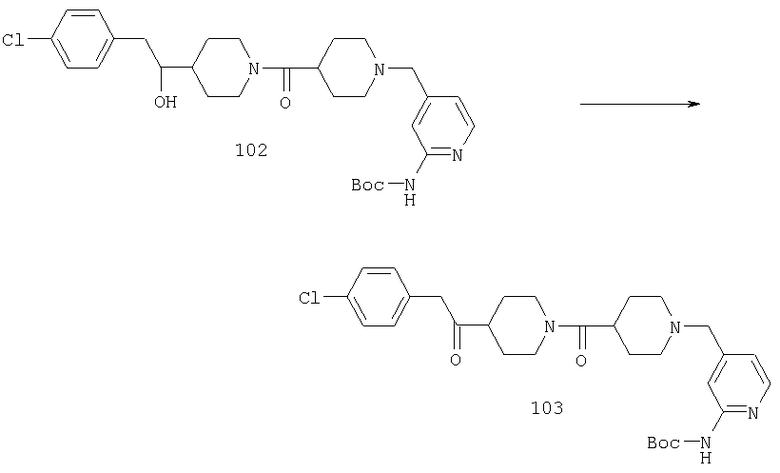
К раствору 0,34 г (2,7 ммоль) оксалилхлорида в 3 мл безводного CH2Cl2 при -70°С и при перемешивании прибавляют 0,44 г (5,7 ммоль) безводного метилсульфоксида в 2 мл CH2Cl2. После перемешивания при -70°С в течение 10 мин к реакционной смеси прибавляют 1,2 г (2,15 ммоль) 102 в 10 мл CH2Cl2. Перемешиваемую смесь выдерживают при -70°С в течение 0,5 ч, смешивают с 1,8 мл (13 ммоль) триэтиламина и затем смеси дают нагреться до комнатной температуры. Смесь разбавляют водой и экстрагируют с помощью CH2Cl2. Органические экстракты промывают рассолом, сушат над безводным MgSO4 и концентрируют, получая 1,18 г 103 в виде стекла. ЖХМС m/z 555 (МН+).
Стадия 4
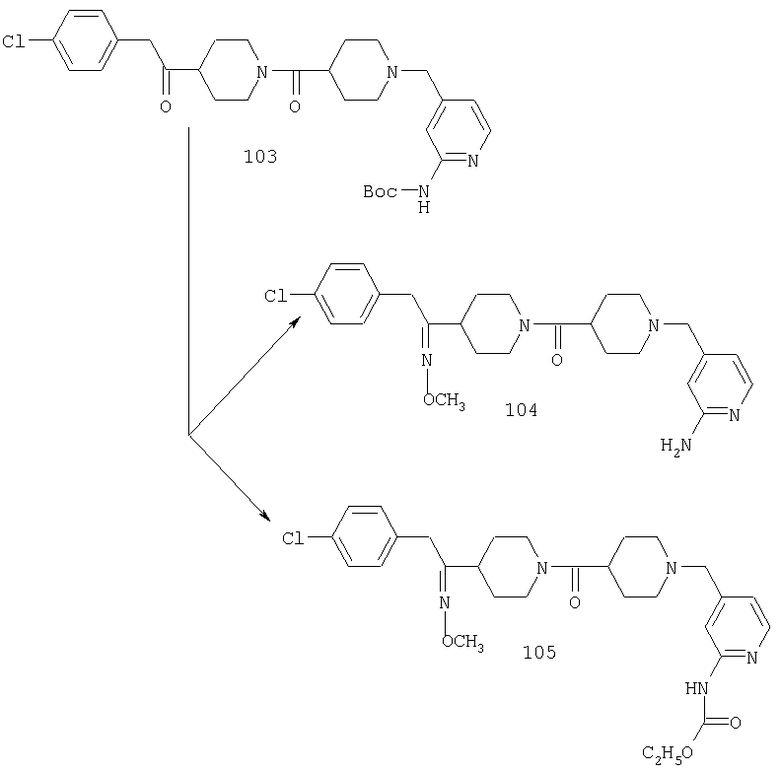
Раствор 0,8 г (1,44 ммоль) 103 и 0,6 г (7,2 ммоль) метоксиламингидрохлорида в 40 мл этанола и 40 мл пиридина 18 ч кипятят с обратным холодильником. Смесь концентрируют и остаток растворяют в смеси этилацетат/эфир и промывают водой. Органический раствор сушат над безводным MgSO4 и концентрируют, получая 0,65 г вязкого остатка, который растворяют в 8 мл трифторуксусной кислоты и 8 мл CH2Cl2 и 18 ч перемешивают при температуре окружающей среды. Раствор концентрируют, остаток подщелачивают 1 н. раствором NaHCO3 и экстрагируют этилацетатом. Органические экстракты промывают рассолом, сушат над безводным MgSO4 и концентрируют, получая смолистый остаток. Очистка этого остатка с помощью флэш-хроматографии с использованием 5-8% 7 н. NH3 - МеОН в CH2Cl2 дает 0,151 г 104 в виде смолы, ЖХМС m/z 484 (МН+), и 0,146 г 105 в виде стекла, ЖХМС m/z 556 (МН+).
Смешивание раствора 0,056 г свободного основания 104 в этилацетате с раствором 0,04 г малеиновой кислоты в этилацетате приводит к образованию осадка, который выделяют фильтрованием и получают 0,06 г дималеата 104 (температура плавления 155-160°С).
Пример 23
Стадия 1
2,4 г (10 ммоль) 106 восстанавливают способом, аналогичным описанному в Примере 22, стадия 1, и получают 1,5 г 107 в виде полужидкого вещества. ЖХМС m/z 240 (МН+).
Стадия 2
1,5 г (6,31 ммоль) 107 вводят в реакцию сочетания с соединением 3 способом, аналогичным описанному в Примере 22, стадия 2, и получают 3 г 108 в виде твердого вещества (температура плавления 104-106°С). ЖХМС m/z 557 (МН+).
Стадия 3
1,17 г (2,1 ммоль) 108 окисляют способом, аналогичным описанному в Примере 22, стадия 3, и получают 0,7 г 109 в виде стекла, ЖХМС m/z 557 (МН+).
Стадия 4
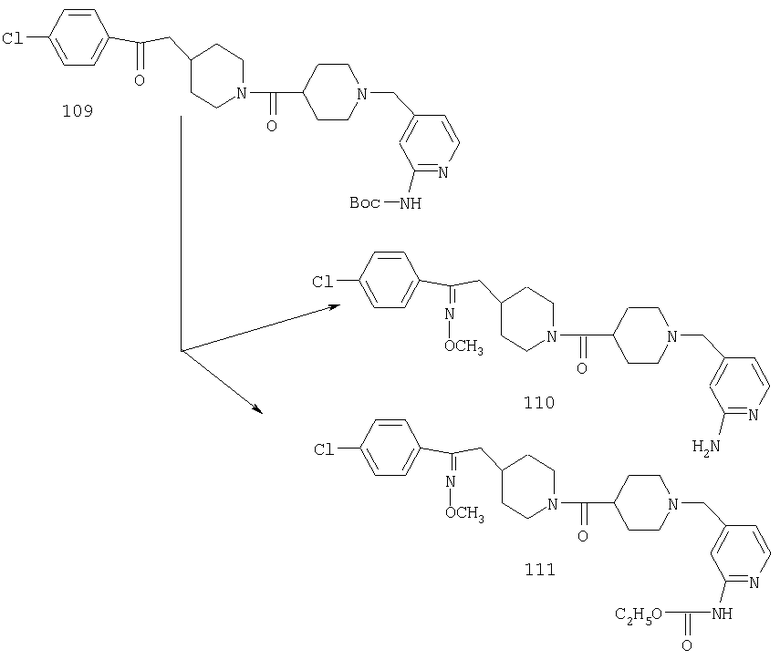
0,32 г (0,58 ммоль) 109 вводят в реакцию с 0,6 г (7,2 ммоль) метоксиламингидрохлорида таким же способом, который описан в Примере 22, стадия 4, и получают 0,065 г 110 в виде смолы, ЖХМС m/z 484 (МН+), и 0,12 г 111 в виде стекла, ЖХМС m/z 556 (МН+).
Пример 24
Стадия 1
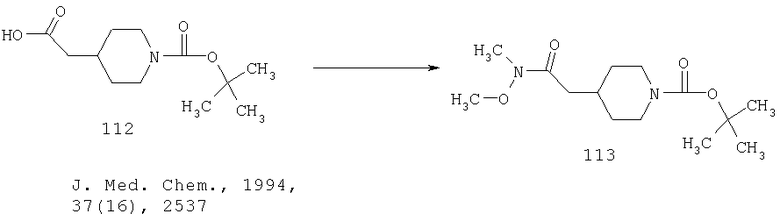
Смесь 18 г (74 ммоль) 112, 7,2 г (74 ммоль) N,О-диметилгидроксиламингидрохлорида, 19,4 г (15 ммоль N,N-диизопропилэтиламина, 1,1 г (8 ммоль) НОВТ и 14,2 г (74 ммоль) DEC в 80 мл безводного DMF в течение 18 ч перемешивают при температуре окружающей среды. Смесь разбавляют водой и экстрагируют с помощью этилацетата. Органические экстракты промывают 1% раствором NaHCO3 и рассолом, сушат над MgSO4 и концентрируют, получая 15,5 г 113 в виде масла. ЖХМС m/z 287 (МН+).
Стадия 2

К раствору 2,9 г (18 ммоль) 2-бромпиридина в 30 мл безводного THF при -78°С и при перемешивании в течение 0,5 ч по каплям прибавляют 7,5 мл 2,5 М раствора n-BuLi в гексане. После перемешивания при -78°С в течение 1 ч к реакционной смеси прибавляют раствор 5,1 г (17,8 ммоль) 113 в 15 мл THF. Смесь 48 ч перемешивают при температуре окружающей среды, смешивают с насыщенным водным раствором NH4Cl и экстрагируют эфиром. Органические экстракты промывают рассолом, сушат над безводным MgSO4 и концентрируют, получая 5,7 г 114 в виде масла. ЖХМС m/z 305 (МН+).
Стадия 3
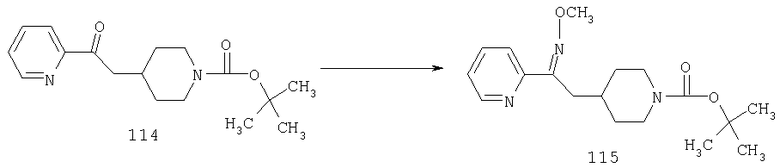
Раствор 3,15 г (10,4 ммоль) 114 и 3,47 г (41,6 ммоль) метоксиламингидрохлорида в 30 мл этанола и 30 мл пиридина 18 ч кипятят с обратным холодильником. Смесь концентрируют и остаток растворяют в эфире и промывают водой. Органический раствор сушат над безводным MgSO4 и концентрируют, получая 2,5 г 115 в виде масла. ЖХМС m/z 334 (МН+).
Стадия 4
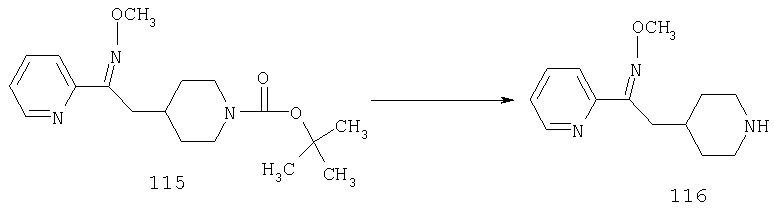
Раствор 2,4 г (7,2 ммоль) 22 в 20 мл CH2Cl2 и 20 мл трифторуксусной кислоты 1 ч перемешивают при температуре окружающей среды. Раствор концентрируют. Остаток подщелачивают насыщенным водным раствором NaHCO3 и экстрагируют с помощью CH2Cl2. Органические экстракты промывают рассолом, сушат над безводным MgSO4 и концентрируют, получая 1,41 г 23 в виде стекла. ЖХМС m/z 234 (МН+).
Стадия 5
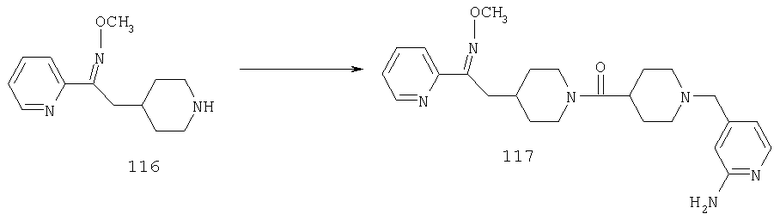
Смесь 0,466 г (2 ммоль) 116, 0,517 г (2,2 ммоль) 5А, 0,276 г (2 ммоль) НОВТ и 0,46 г (2,4 ммоль) DEC в 20 мл безводного DMF в течение 18 ч перемешивают при температуре окружающей среды. Смесь концентрируют путем выпаривания при пониженном давлении на бане с температурой 25-45°С и остаток хроматографируют с использованием 4% (7 н. NH3/СН3ОН) в CH2Cl2, получая 0,48 г сиропа, который растворяют в 15 мл EtOAc-EtOH (3:1, об.) и смешивают с раствором 0,26 г малеиновой кислоты в 10 мл EtOAc-EtOH (1:1). Полученный осадок отфильтровывают и получают 0,35 г малеата 117 (температура плавления 160-163°С). ЖХМС m/z 451 (МН+).
Пример 25
Стадия 1
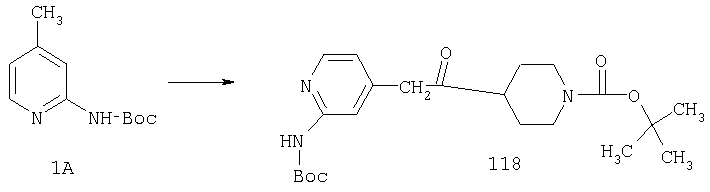
К раствору 4,16 г (20 ммоль) 1А в 80 мл безводного THF при -78°С в течение 25 мин при перемешивании по каплям прибавляют 2,5 М раствор n-BuLi в гексане. После перемешивания в течение 1 ч при температуре от -78°С до комнатной к реакционной смеси прибавляют раствор 6 г (22 ммоль) 26 в 100 мл безводного THF и 18 ч выдерживают при комнатной температуре. Смесь смешивают с насыщенным водным раствором NH4Cl и экстрагируют с помощью EtOAc. Органические экстракты промывают рассолом, сушат над безводным MgSO4 и концентрируют, получая 6,1 г 118 (температура плавления 146-149°С). ЖХМС m/z 420 (МН+).
Стадия 2
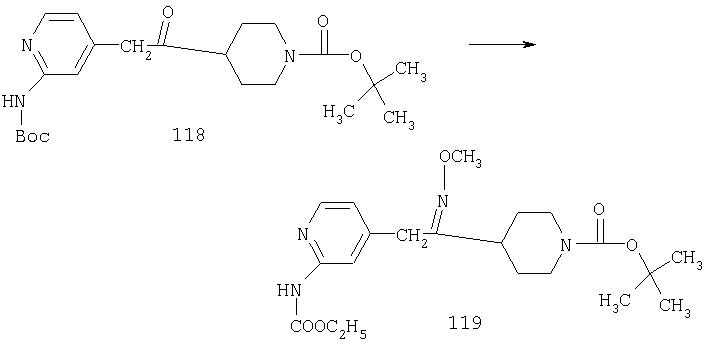
Раствор 3,71 г (8,8 ммоль) 118 и 3,7 г (44 ммоль) метоксиламингидрохлорида в 40 мл пиридина и 40 мл этанола 2 дня кипятят с обратным холодильником. Смесь концентрируют и остаток растворяют в CH2Cl2 и промывают насыщенным водным раствором NaCl. Органический раствор сушат над безводным MgSO4 и концентрируют, получая 2,6 г 119 в виде стекла. ЖХМС m/z 421 (МН+).
Стадия 3
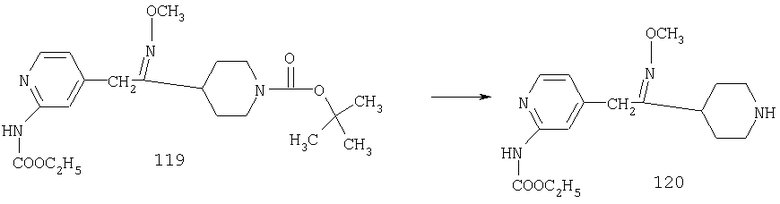
Раствор 0,9 г (2,14 ммоль) 119 в 10 мл CH2Cl2 и 10 мл трифторуксусной кислоты 2 ч перемешивают при температуре окружающей среды. Раствор концентрируют. Остаток растворяют в CH2Cl2, промывают насыщенным водным раствором NaHCO3 и рассолом, сушат над безводным MgSO4 и концентрируют, получая твердый остаток, который растирают с CH3CN и фильтруют, получая 0,29 г 120 (температура плавления 200-205°С). ЖХМС m/z 321 (МН+).
Стадия 4
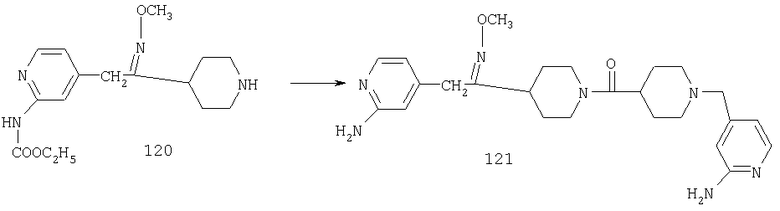
0,1 г (0,31 ммоль) 120 и 0,83 г (0,35 ммоль) 5А вводят в реакцию сочетания таким же способом, который описан в Примере 24, стадия 5, и получают 0,12 г малеата 121 (температура плавления 170-173°С). ЖХМС m/z 538 (МН+).
Пример 26
Стадия 1
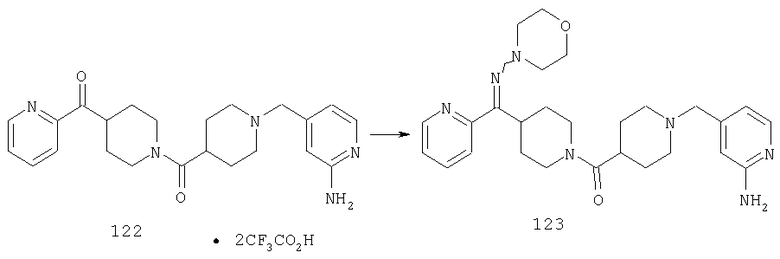
Способом, сходным с описанным в Примере 6, стадия 7, 122 (0,26 г, 0,41 ммоль) превращают в 123 (0,08 г, 40%).
Пример 27
Стадия 1
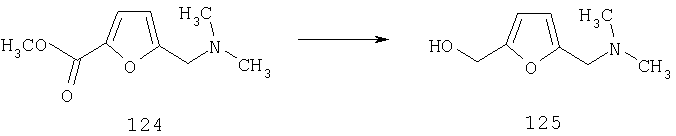
К суспензии LAH (0,83 г, 22 ммоль) в эфире (20 мл) при 0°С по каплям прибавляют 124 (3,2 г, 17,5 ммоль) в THF (15 мл). Реакционную смесь 1,5 ч перемешивают при 0°С и реакцию останавливают путем прибавления воды (0,8 мл), 20% водного раствора NaOH (0,8 мл) и воды (2,4 мл). Смесь перемешивают 15 мин, фильтруют и осадок на фильтре промывают с помощью CH2Cl2. Фильтрат концентрируют и получают масло, которое растворяют в эфире (30 мл), промывают рассолом и сушат (MgSO4). Фильтрование и концентрирование в вакууме дает 125 (2,5 г), который используют без дополнительной очистки.
Стадия 2
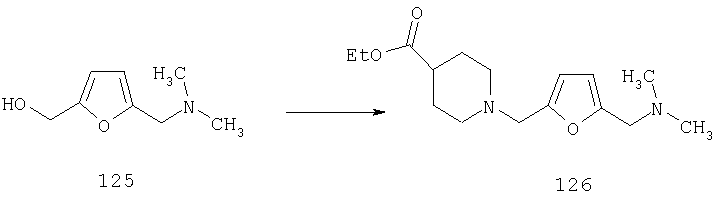
Стадия 3
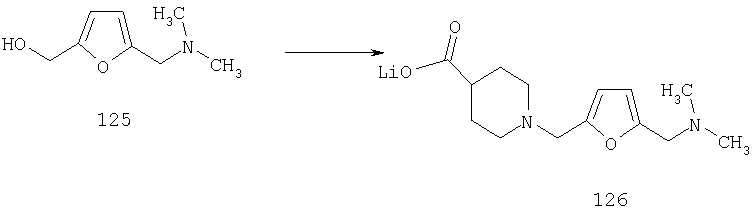
Аналогично примеру 1, стадиям 4 и 5, 125 превращают в 126.
Стадия 4
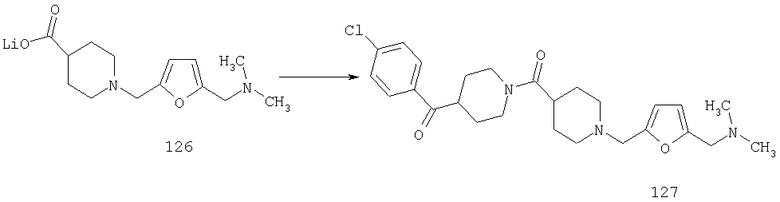
Способом, сходным с описанным в Примере 6, стадия 5, 126 превращают в 127.
Стадия 5
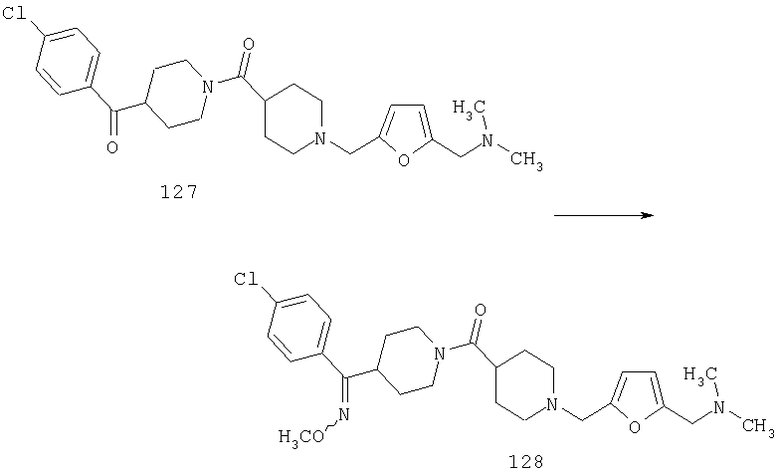
Способом, сходным с описанным в Примере 6, стадия 7, 127 превращают в 128.
Соединения, указанные в таблице 1 (первый столбец), получают из соединений, указанных в последнем столбце таблицы 1, с помощью по существу таких же методик, что и использованные в описанных выше примерах.
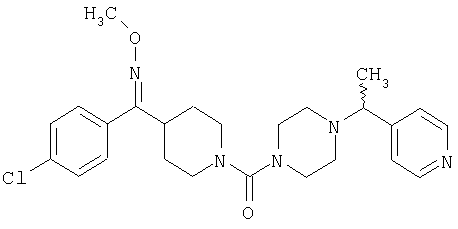
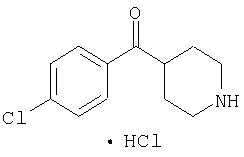
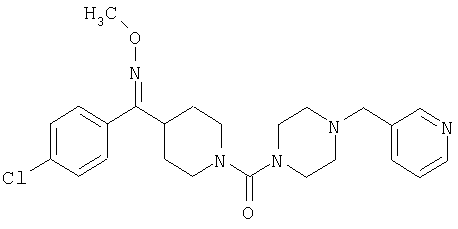
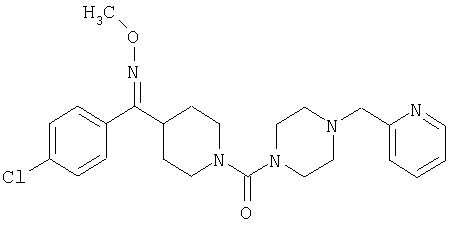
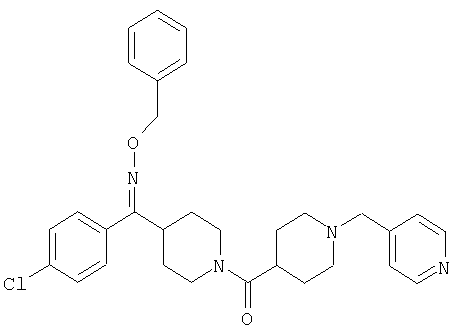
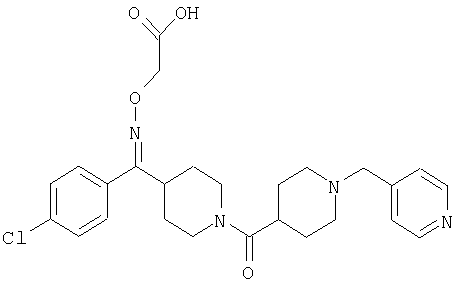
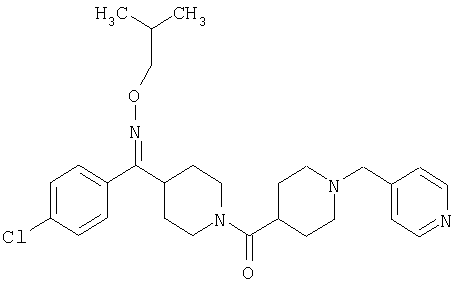
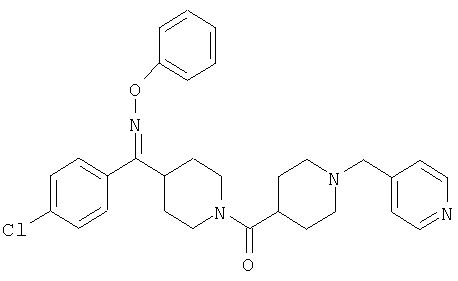
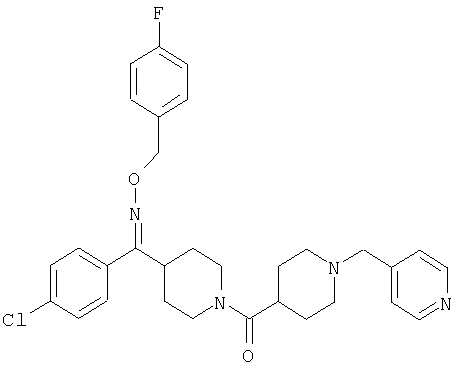
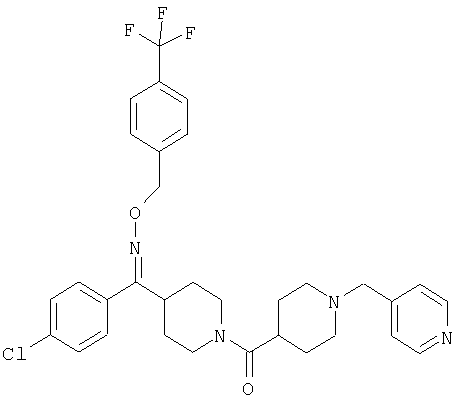
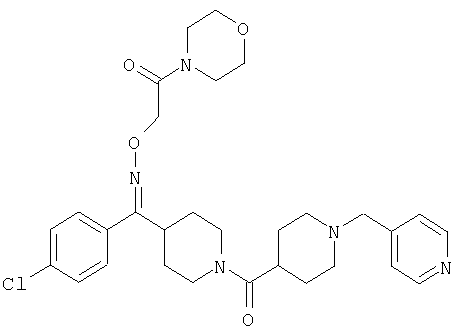
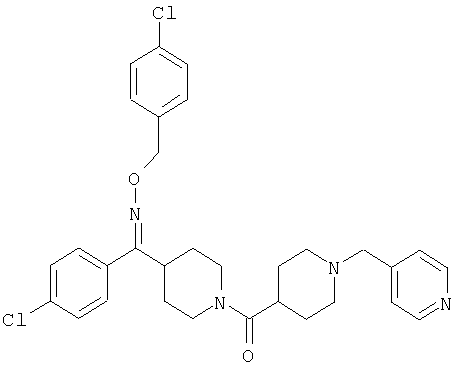
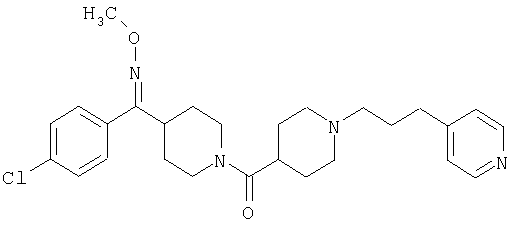
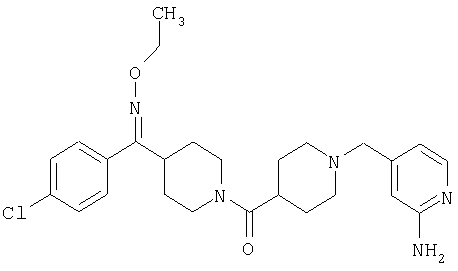
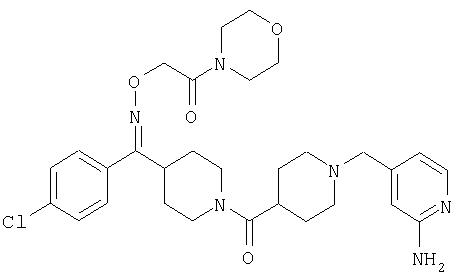
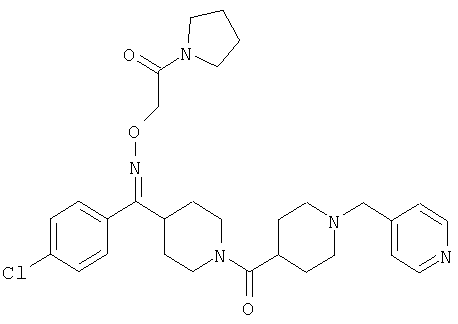
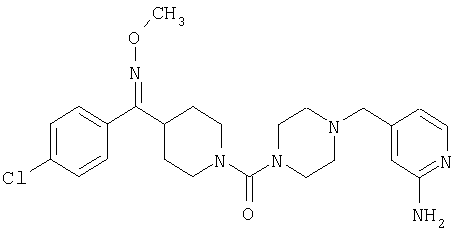
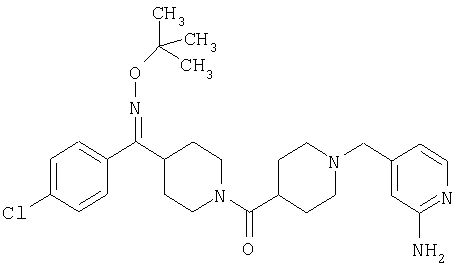
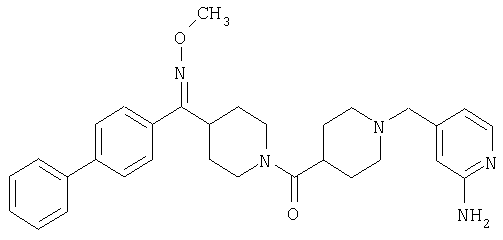
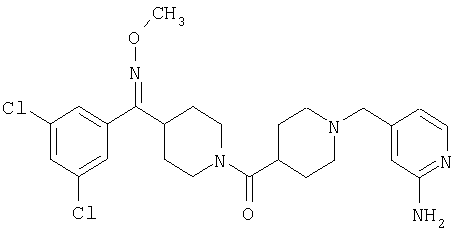
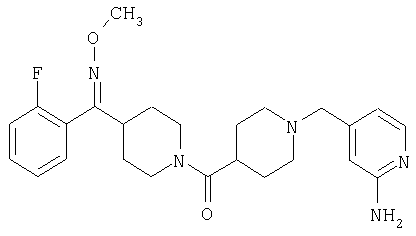
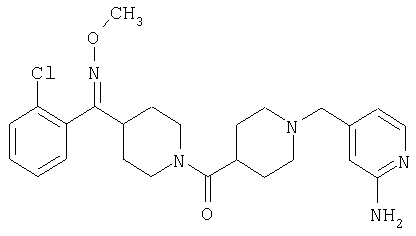
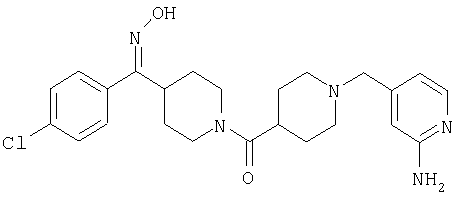
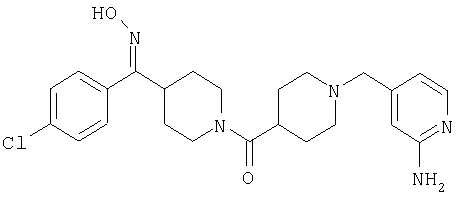
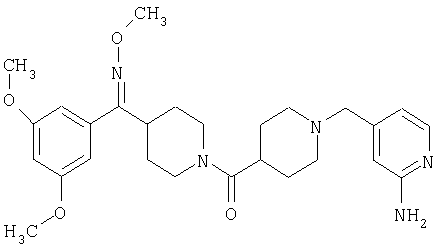
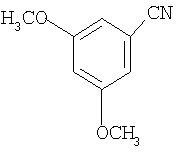
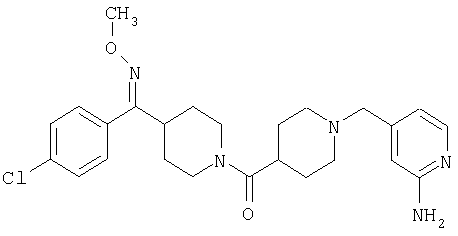
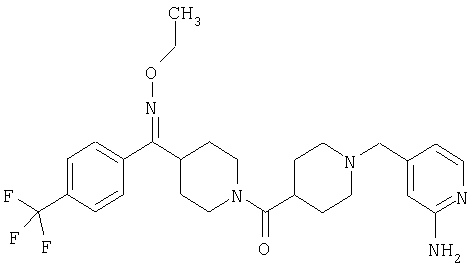
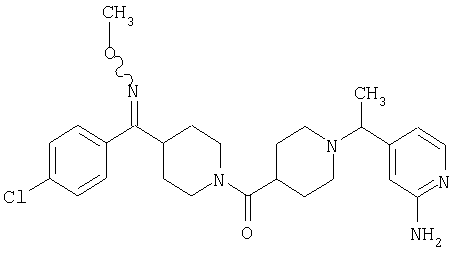
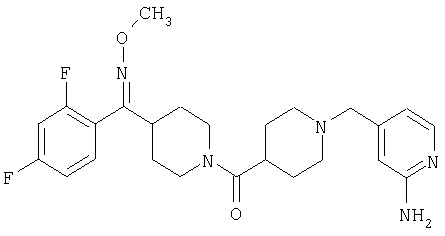
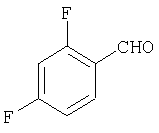
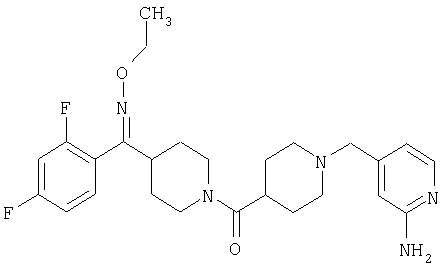
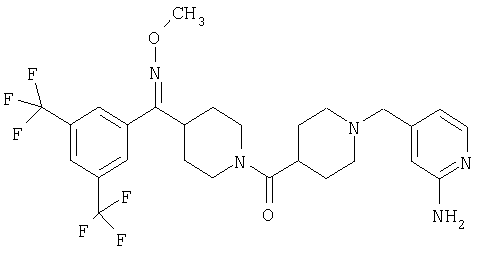
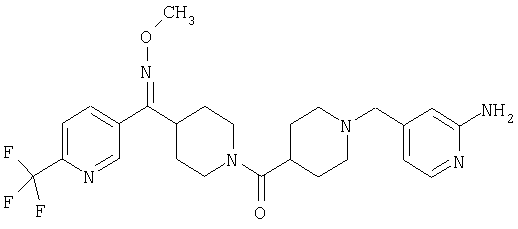
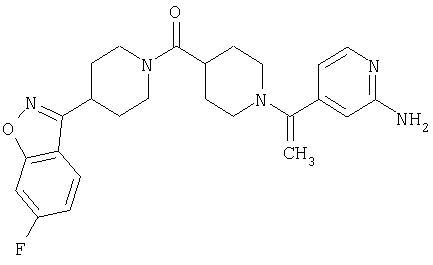
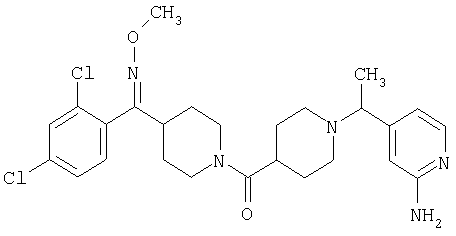
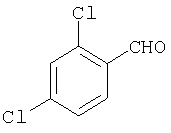
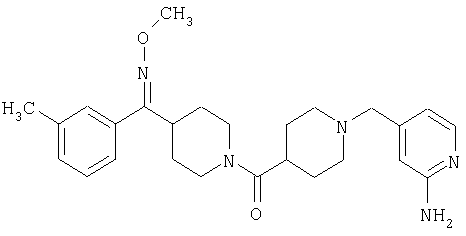
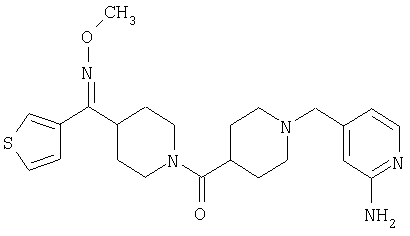
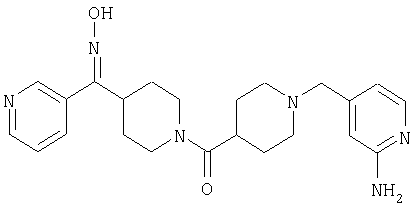
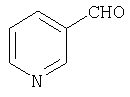
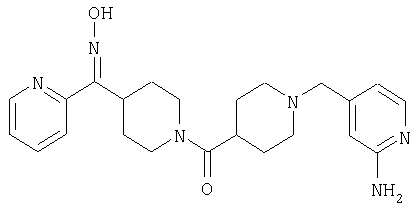
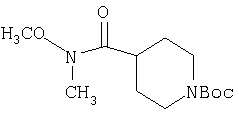
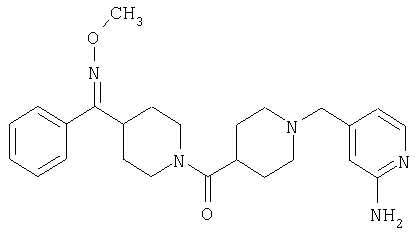
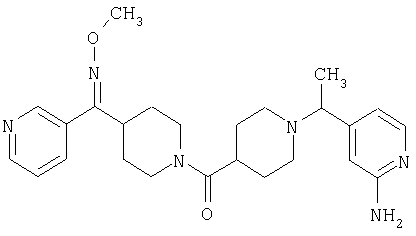
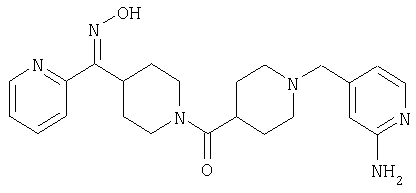
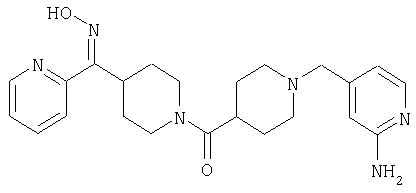
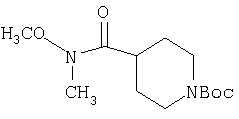
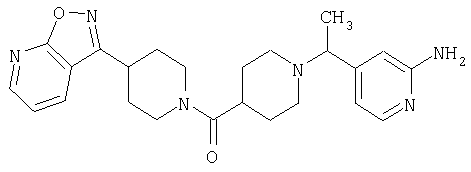
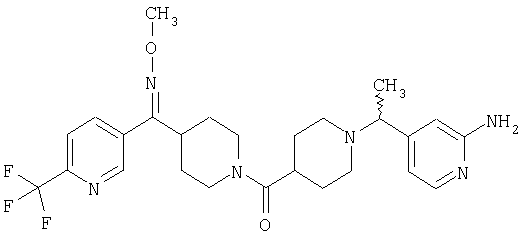
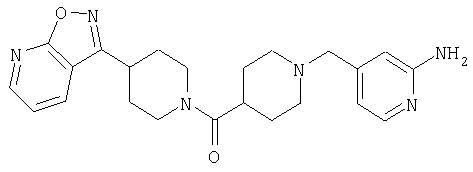
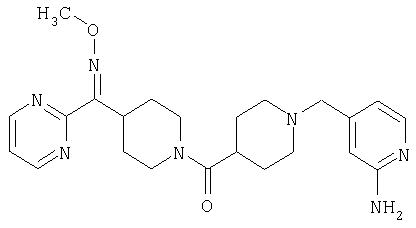
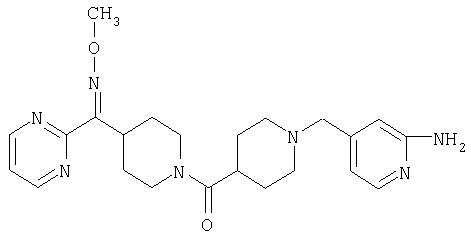
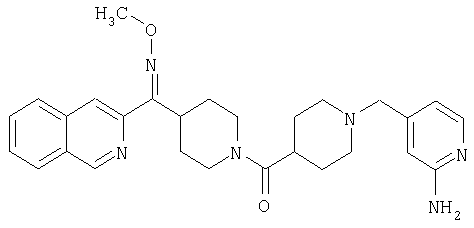
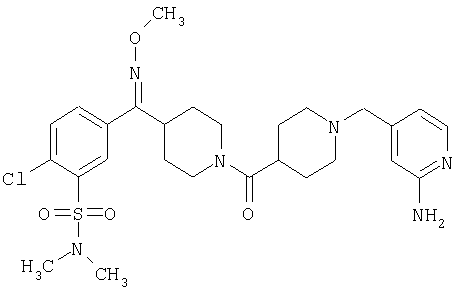
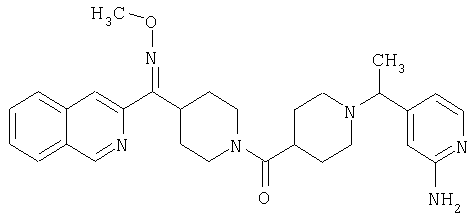
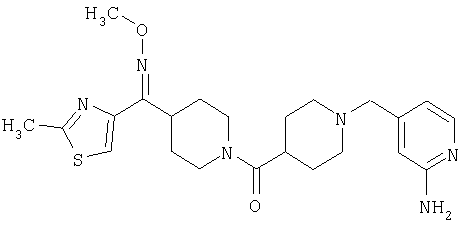
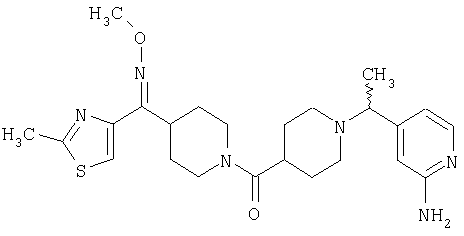
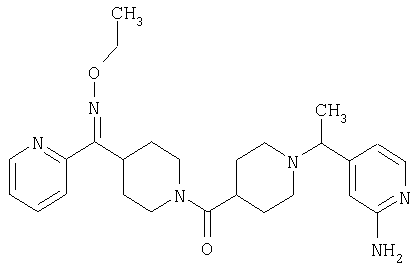
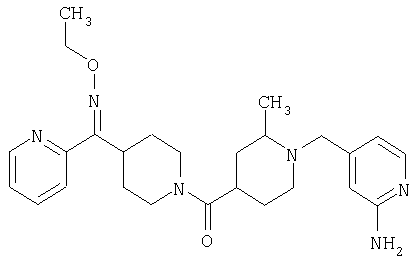
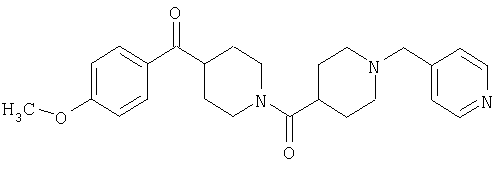
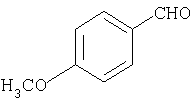
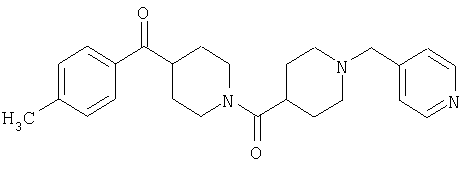
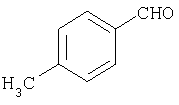
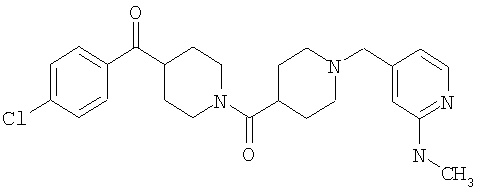
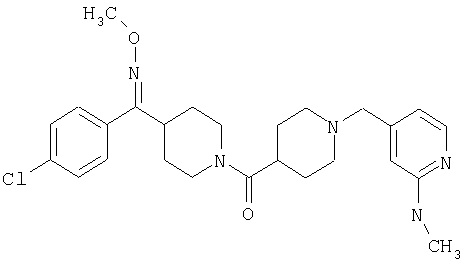
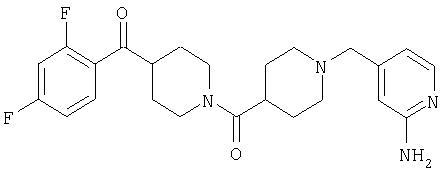
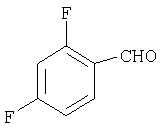
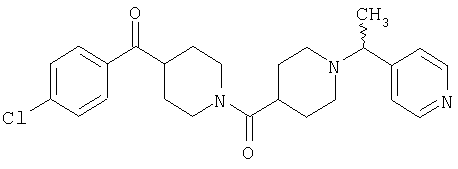
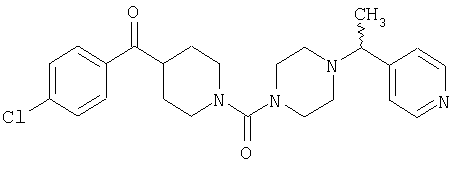
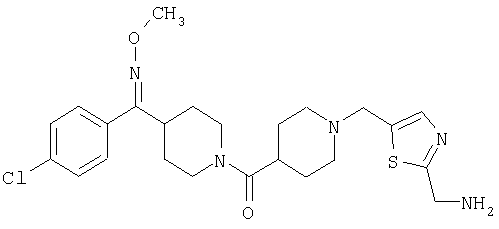
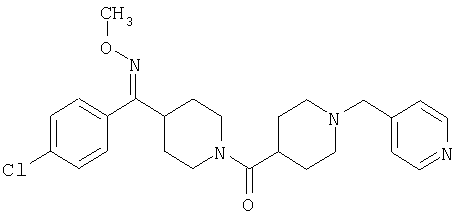
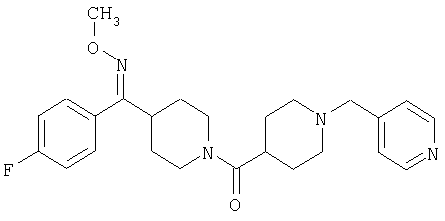
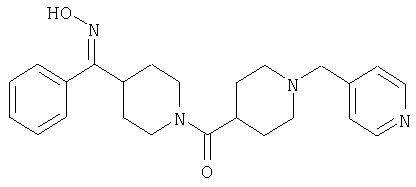
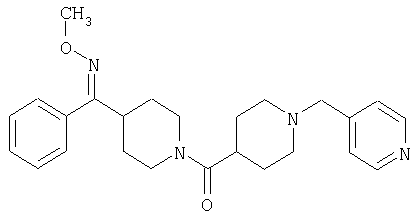
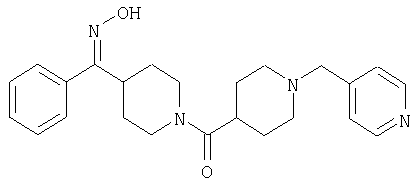
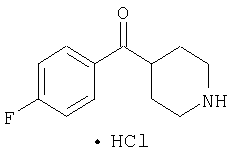
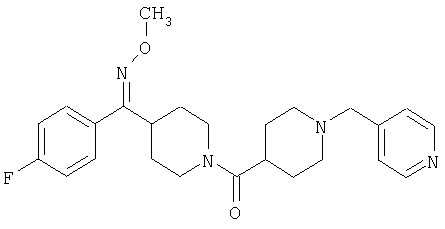
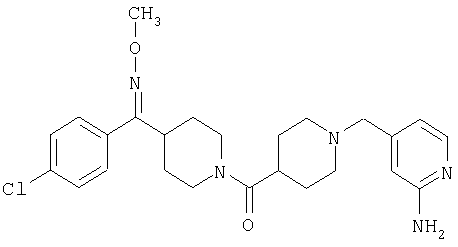
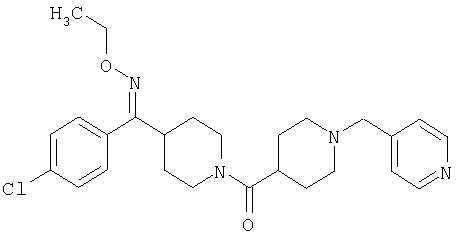
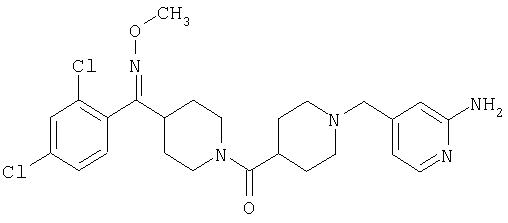
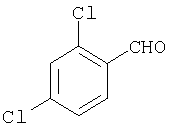
Приведенные ниже изомеры 246А и 253А можно отделить от приведенных выше 246 и 253 соответственно с помощью методик, хорошо известных специалистам в данной области техники.
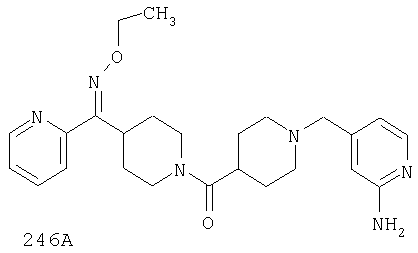
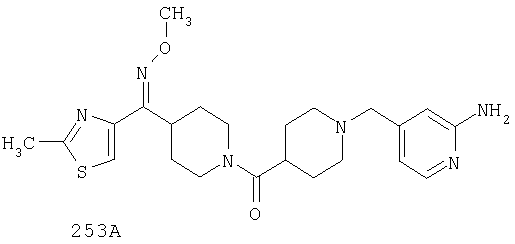
Пример 28
Стадия 1
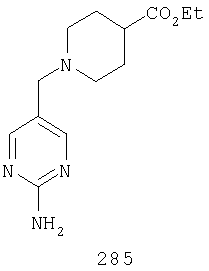
К раствору 1,00 г (8,13 ммоль) пиримидинового альдегида 67 (стадия 2 Примера 13) в 40 мл CH2Cl2 прибавляют 1,36 мл (10,58 ммоль) этилизонипекотата и 2 капли уксусной кислоты. Смесь 40 мин перемешивают при комнатной температуре, а затем прибавляют 2,58 г (12,17 ммоль) NaBH(ОАс)3. Затем смесь 20 ч перемешивают при комнатной температуре, разбавляют водным раствором NaOH (значение рН доводят до 11) и экстрагируют с помощью CH2Cl2. Органическую фазу сушат и концентрируют, остаток подвергают флэш-хроматографии (4-8% примерно 3 н. раствор NH3 в MeOH/CH2Cl2) и получают 1,55 г (5,87 ммоль, 72%) амина 285 в виде желтоватого твердого вещества.
Стадия 2
К раствору 3,83 г (14,51 ммоль) сложного эфира 285 в 60 мл смеси THF - H2O - МеОН состава 3:1:1 прибавляют 1,22 г (29,02 ммоль) моногидрата LiOH. Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют и подвергают сушке в высоком вакууме, получая 3,84 г неочищенной литиевой соли кислоты 286 в виде желтого твердого вещества. Вещество можно использовать непосредственно или можно очистить, пропуская через слой силикагеля и элюируя с помощью примерно 3 н. раствора NH3 в МеОН.
Стадия 3
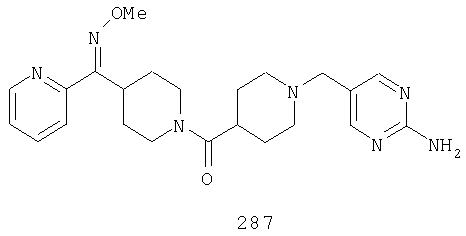
К смеси 3,32 г (14,05 ммоль) кислоты 286 и 4,07 г (14,05 ммоль) 4-[(Е)-(метоксиимино)-2-пиридинилметил]пиперидиндигидрохлорида (см. ниже соединение 447) в 40 мл DMF прибавляют 8,94 мл (70,25 ммоль) 4-этилморфолина и 14,0 мл (23,52 ммоль) раствора циклического ангидрида 1-пропанфосфоновой кислоты в этилацетате концентрации 50 мас.%. Реакционную смесь 4,5 ч перемешивают при 50°С, а затем 14 ч при комнатной температуре. Смесь концентрируют, а затем в течение 24 ч сушат в высоком вакууме для удаления оставшегося DMF. Остаток подвергают распределению между водным раствором NaOH и CH2Cl2, органическую фазу отделяют, сушат и концентрируют, остаток подвергают флэш-хроматографии (5-15% примерно 3 н. раствор NH3 в MeOH/CH2Cl2) и получают 4,60 г (10,51 ммоль, 75%) амида 287 в виде светло-желтовато-коричневого пенообразного вещества. МС 438 (М+1).
Пример 29
Стадия 1
Литература: J. Heterocyclic Chem., 1966. 3, 252.
3,4-Пиридиндикарбоксимид 288 (10,0 г, 67,5 ммоль) растворяют в 162 г 10% водного раствора NaOH и в бане со льдом с солью раствор охлаждают до температуры 7°С. По каплям прибавляют бром (3,6 мл, 70 ммоль). После прибавления раствор 45 мин нагревают на бане с температурой, равной 80-85°С. Затем желтый раствор охлаждают до температуры, равной 37°С, после этого по каплям прибавляют 17 мл ледяной уксусной кислоты, пока значение рН не станет равным 5,5. Полученную смесь в течение ночи хранят в холодильнике. Образовавшееся твердое вещество отфильтровывают и промывают с помощью 5 мл воды и 5 мл метанола. Реакция дает 6,35 г продукта 289, который плавится при 280-285°С (с разложением).
Стадия 2
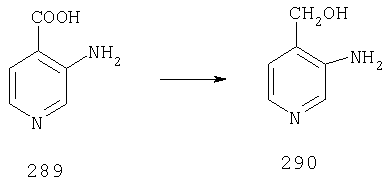
Твердое соединение 289 (9,5 г, 69 ммоль) осторожно прибавляют к трем аликвотам взвеси алюмогидрида лития (9,5 г, 250 ммоль) в 200 мл сухого тетрагидрофурана. Полученную горячую смесь два дня перемешивают при комнатной температуре. После охлаждения в бане со льдом реакцию останавливают, последовательно осторожно по каплям прибавляя 10 мл воды, затем 10 мл 15% водного раствора NaOH, затем 30 мл воды. Образовавшееся твердое вещество отфильтровывают через слой целита и несколько раз промывают с помощью THF. Масло, полученное после выпаривания растворителя, при выдерживании затвердевает. Реакционную смесь очищают с помощью флэш-хроматографии на силикагеле, используя в качестве элюента смесь 5% МеОН (NH3)/EtOAc, и получают 6,21 г (72%) соединения 290. ЖХМС: m/z 125 (М+1).
Стадия 3
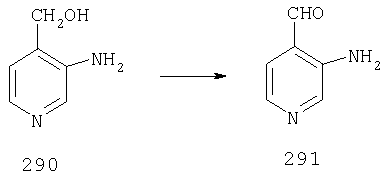
Диоксид марганца (29 г, 334 ммоль) при комнатной температуре и эффективном перемешивании одной порцией прибавляют к суспензии 3-амино-4-гидроксиметилпиридина 290 (5,0 г, 40,3 ммоль) в 500 мл хлороформа. Через два дня твердое вещество отфильтровывают через слой целита и промывают хлороформом. Удаление растворителя при пониженном давлении дает 4,2 г (85%) соединения 291 в виде желтого твердого вещества.
Стадия 4
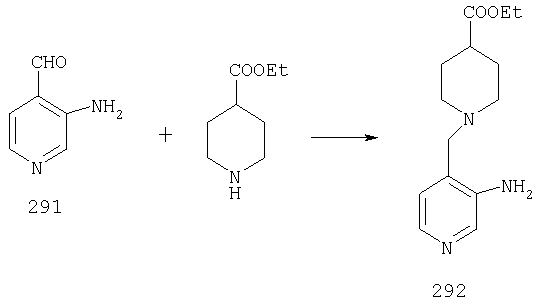
Раствор этилизонипекотата (12,5 г, 79,5 ммоль) и 3-аминопиридин-4-карбоксальдегида 291 (3,33 г, 27,3 ммоль) в сухом дихлорметане (400 мл) 1 ч перемешивают при комнатной температуре, затем прибавляют 60 г активированного молекулярного сита с размером пор, равным 3 Å. Смесь перемешивают еще 90 мин, затем при комнатной температуре одной порцией прибавляют 20 г (96,4 ммоль) триацетоксиборогидрида натрия. После перемешивания в течение трех дней твердое вещество отфильтровывают через слой целита и промывают дихлорметаном. Раствор 15 мин перемешивают со 100 мл насыщенного водного раствора бикарбоната натрия, затем органический слой отделяют от водного. Органический слой еще два раза промывают насыщенным водным раствором бикарбоната натрия, затем рассолом и сушат над безводным сульфатом натрия. После выпаривания растворителя полученное масло очищают с помощью флэш-хроматографии на силикагеле, используя в качестве элюента смесь EtOAc : гексаны : МеОН (NH3). Эта процедура дает 6,8 г (94%) соединения 292. МС-ББА: m/z=264 (М+1).
Стадия 5
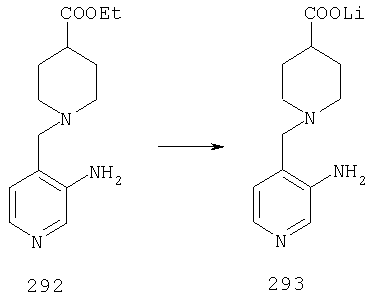
Этил-1-[(3-амино-4-пиридинил)метил]-4-пиперидинкарбоксилат 292 (4,75 г, 18,04 ммоль) 24 ч перемешивают при комнатной температуре с 1,51 г (36 ммоль) моногидрата гидроксида лития в 75 мл метанола. Удаление растворителя при пониженном давлении дает соединение 293 в виде белого твердого вещества.
Стадия 6
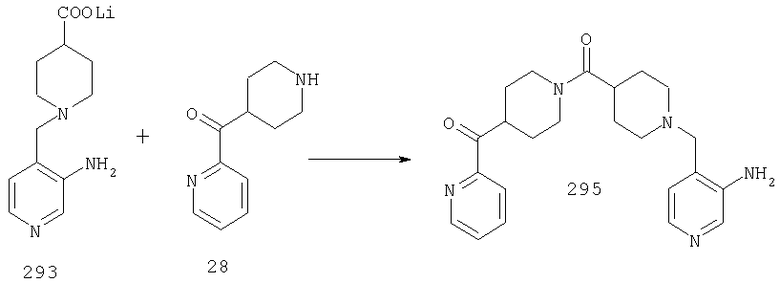
4-(2-Пиридинилкарбонил)пиперидин 28 (стадия 4 в Примере 6) (0,3 г, 1,58 ммоль), литий-1-[(3-амино-4-пиридинил)метил]-4-пиперидинкарбоксилат 293 (0,34 г, 1,4 ммоль), DEC (0,38 г, 2,0 ммоль) и НОВТ (0,27 г, 2,0 ммоль) при комнатной температуре два дня перемешивают в 10 мл сухого DMF. Реакцию останавливают с помощью 50 мл 0,5 н. водного раствора NaOH, а затем раствор экстрагируют дихлорметаном. Объединенные экстракты промывают рассолом и сушат над безводным сульфатом натрия. Продукт 295 выделяют с помощью флэш-хроматографии на силикагеле, используя в качестве элюента смесь EtOAc : гексаны : МеОН (NH3) (50:45:5). Выход 0,27 г (47%). МС-ББА: m/z=408 (M+1).
Стадия 7
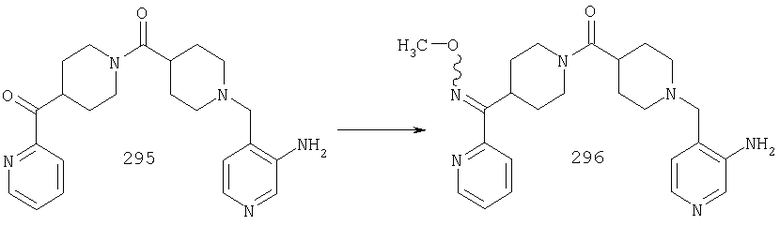
1-[[[1-[(3-Амино-4-пиридинил)метил]-4-пиперидинил]карбонил]-4-(2-пиридинилкарбонил)]пиперидин 295 (0,196 г, 0,48 ммоль) и метоксиамингидрохлорид (0,401 г, 4,8 ммоль) в 6,0 мл сухого пиридина в течение 24 ч в атмосфере N2 нагревают на бане с температурой 70°С. После удаления пиридина с использованием пониженного давления остаток обрабатывают насыщенным водным раствором бикарбоната натрия. Полученную смесь несколько раз экстрагируют дихлорметаном. Объединенные экстракты промывают рассолом и сушат над безводным сульфатом натрия. Реакционную смесь очищают с помощью препаративной тонкослойной хроматографии на силикагеле. Пластины элюируют смесью EtOAc : гексаны : МеОН (NH3) (60:35:5) и продукт 296 экстрагируют смесью 10% МеОН (NH3)/EtOAc. Выход 0,15 г (71%). МС-ББА: m/z=437 (М+1).
Пример 30
Стадия 1
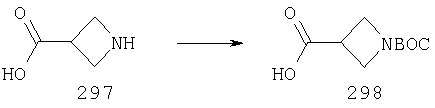
Смесь 297 (1 г, 10 ммоль) с раствором вода - диоксан состава 1:1 (50 мл) обрабатывают с помощью Et3N (4 мл, 13 ммоль) и BOC2O (2,8 г, 13 ммоль) при 4°С и дают нагреваться до 20°С в течение одного дня. Затем растворитель удаляют в вакууме. Остаток обрабатывают смесью вода-этилацетат состава 1:1 и органический слой отбрасывают. Водный слой подкисляют 1 н. водным раствором HCl и три раза экстрагируют этилацетатом. Объединенные органические фазы промывают водой и рассолом, сушат (Na2SO4) и концентрируют, получая 298 в виде белого твердого вещества (1,8 г, 90%).
Стадия 2
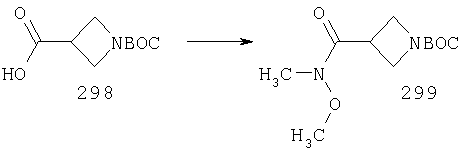
Смесь 298 (1,8 г, 9 ммоль), N,O-диметилгидроксиламингидрохлорида (2,6 г, 27 ммоль), EDCl (5 г, 27 ммоль), НОВТ (0,1 г, 1 ммоль) и DIPEA (12,5 мл, 72 ммоль) в DMF (30 мл) перемешивают в течение ночи при 20°С. Затем реакционную смесь концентрируют в вакууме до половины объема, выливают в воду и три раза экстрагируют этилацетатом. Объединенные органические фазы промывают насыщенным водным раствором NH4Cl, насыщенным водным раствором NaHCO3, водой и рассолом, сушат (Na2SO4) и концентрируют, получая 299 в виде прозрачного масла (2,1 г, 98%).
Стадия 3
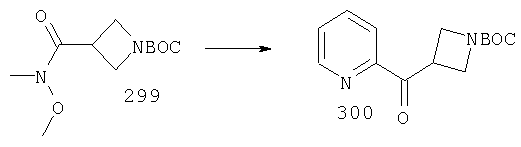
К раствору 2-бромпиридина (1,2 мл, 12 ммоль) в THF (60 мл) при -78°С в течение 15 мин по каплям прибавляют n-BuLi (8 мл 1,6 М раствора в гексанах, 12 ммоль). После перемешивания при этой температуре в течение еще 30 мин по каплям в течение 15 мин прибавляют раствор 299 (1 г, 4 ммоль) в THF (20 мл). Затем реакционную смесь 1 ч нагревают при 60°С. После охлаждения до 20°С реакционную смесь разбавляют эфиром, реакцию останавливают насыщенным водным раствором Na2SO4 и сушат над твердым Na2SO4. Затем смесь фильтруют через слой твердого Na2SO4 и концентрируют в вакууме. Хроматографирование на флэш-колонке (0-20% этилацетат-гексаны) дает 300 в виде желтого масла (0,12 г, 11%).
Стадия 4
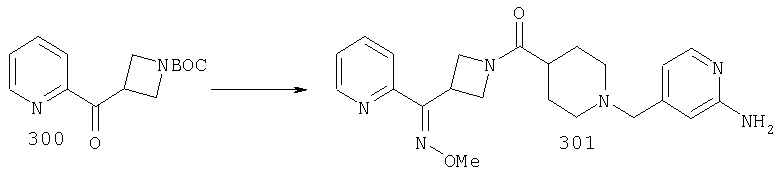
По методике, сходной с использованной на стадиях от 4 до 7 Примера 6, получают соединение 301. МС 409 (М+1).
По методикам, сходным с описанными в приведенных выше примерах, получают соединения, приведенные в таблице 2.
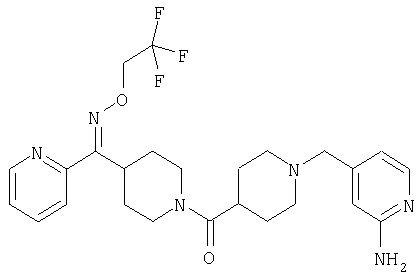
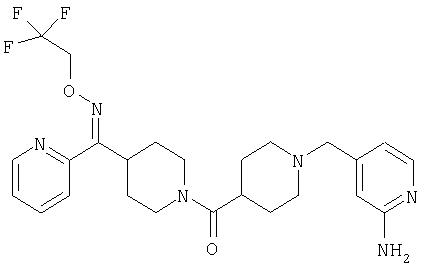
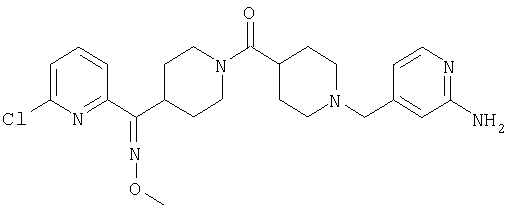
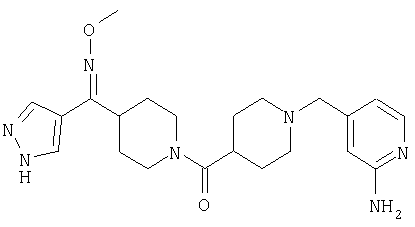
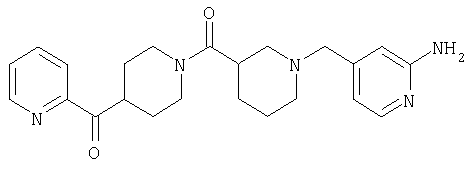
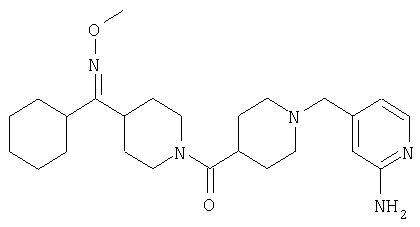
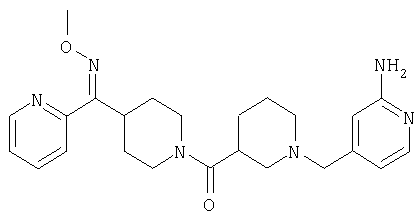
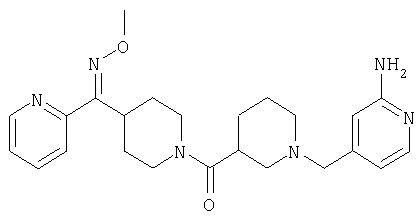
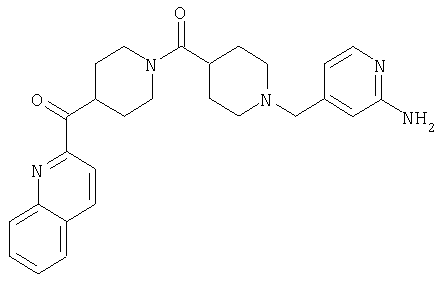
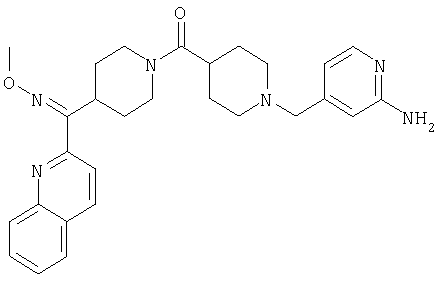
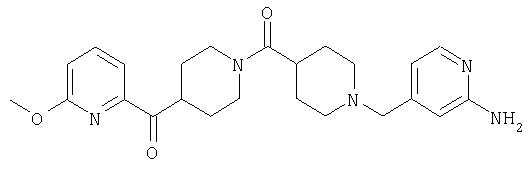
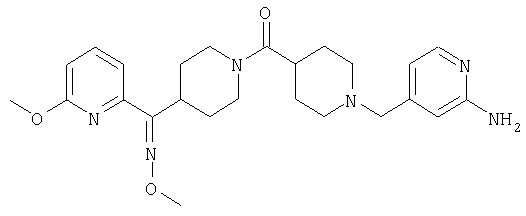
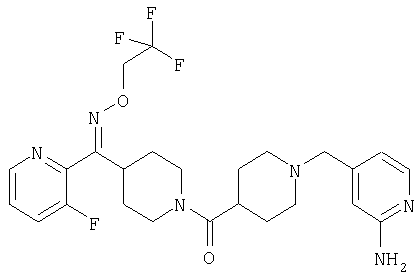
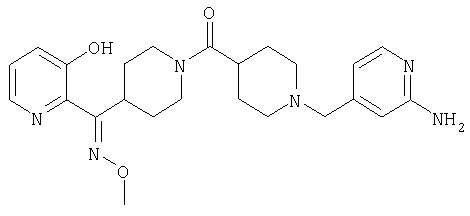
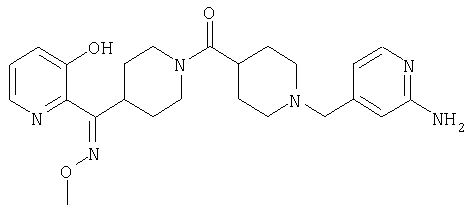
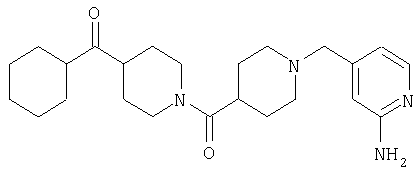
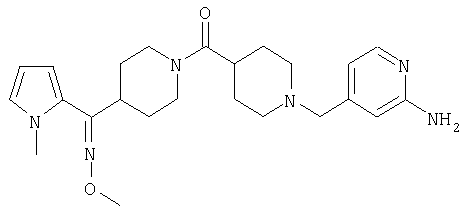
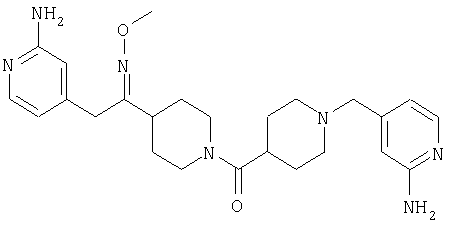
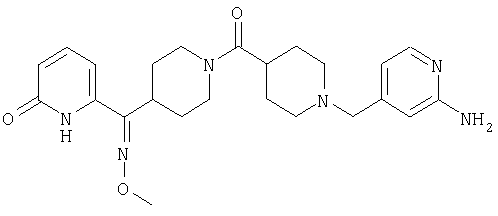
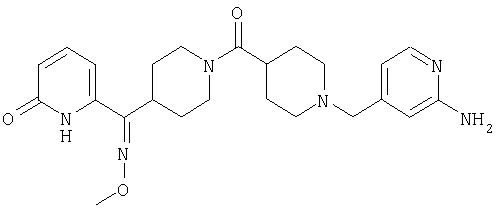
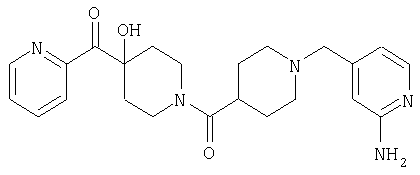
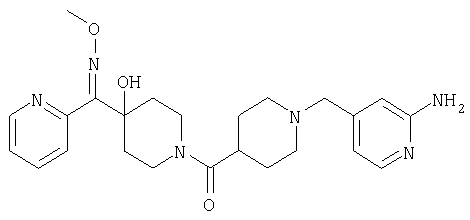
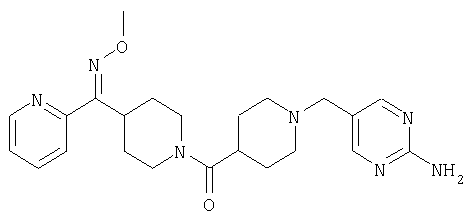
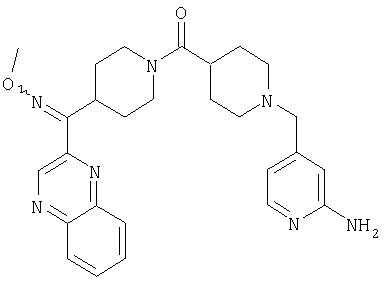
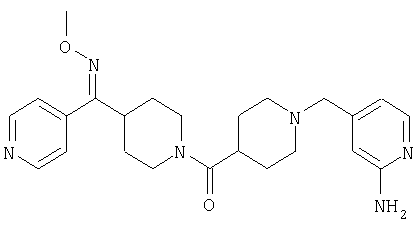
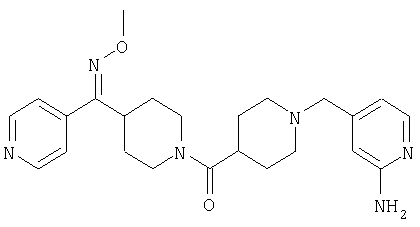
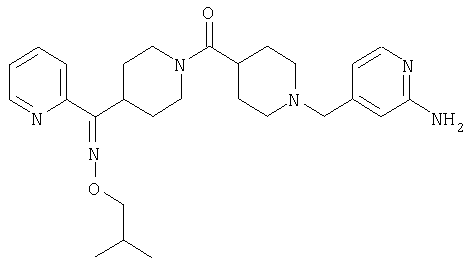
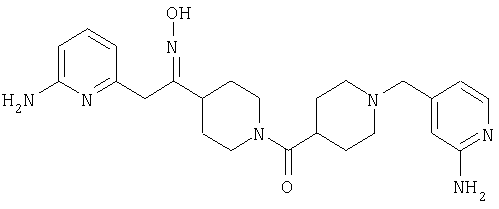
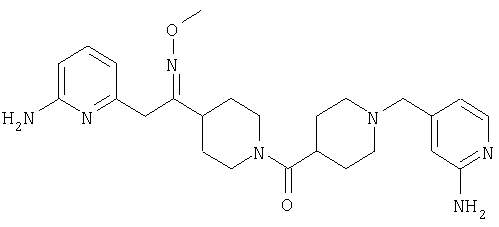
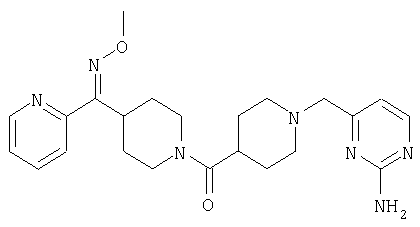
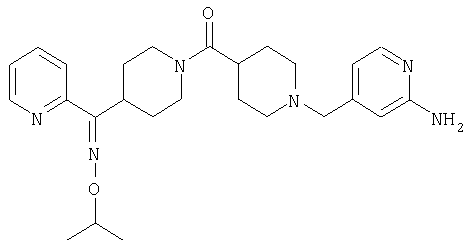
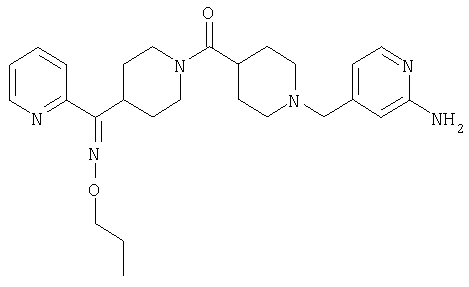
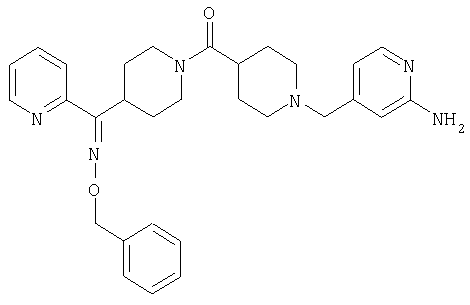
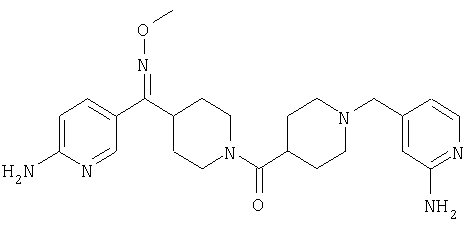
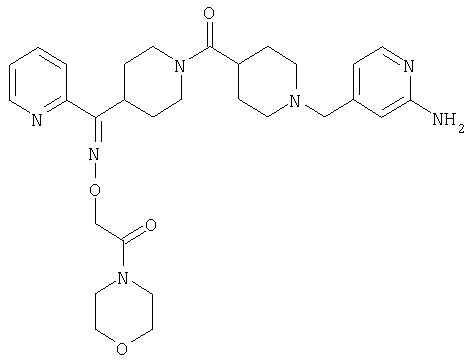
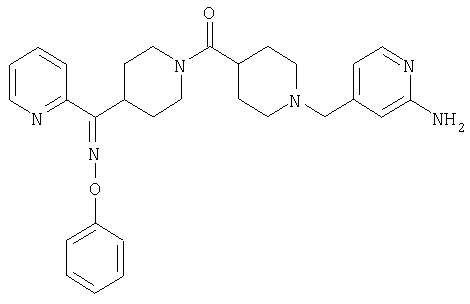
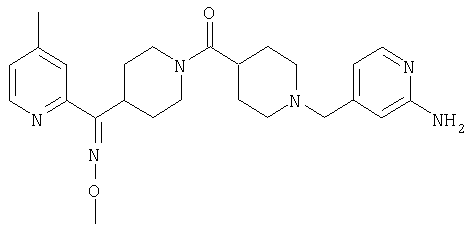
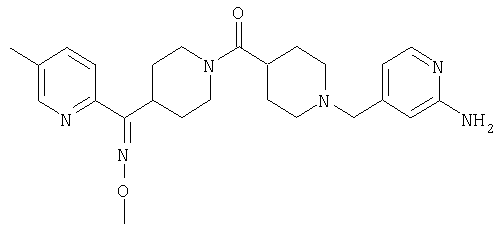
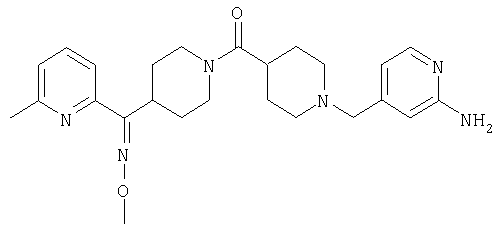
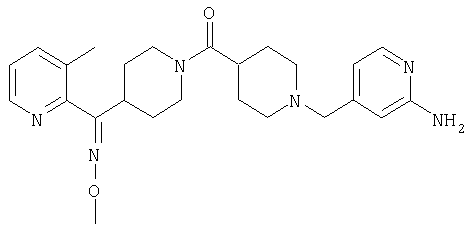
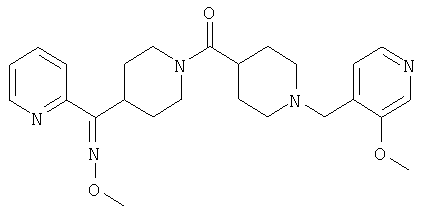
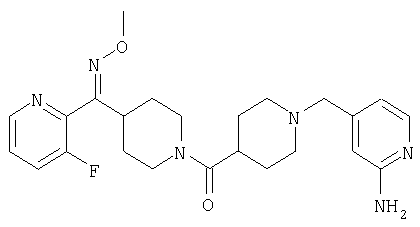
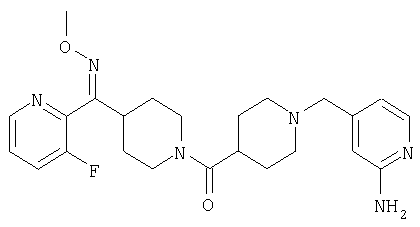
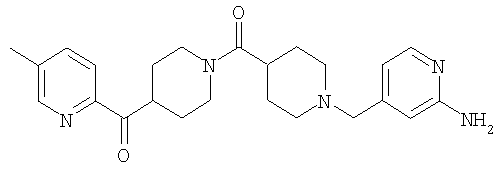
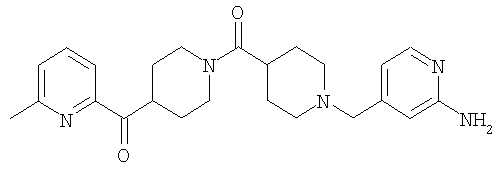
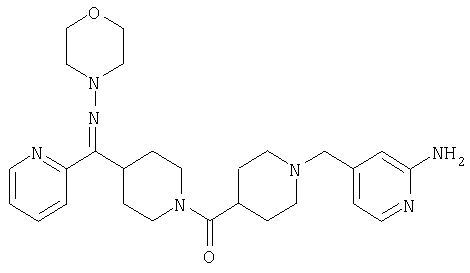
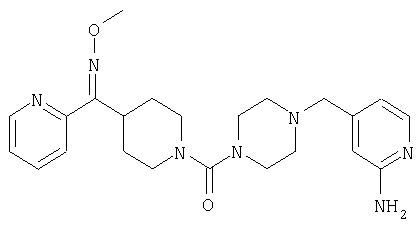
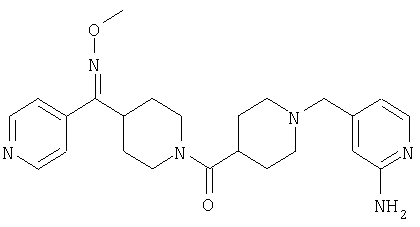
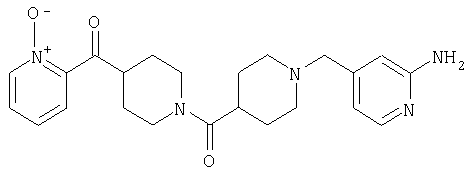
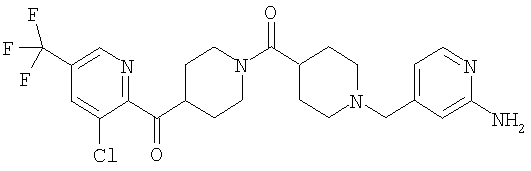
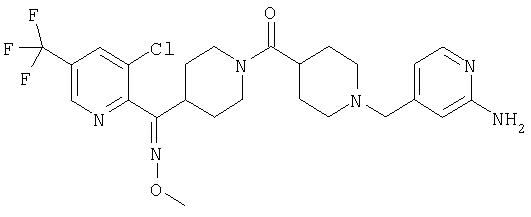
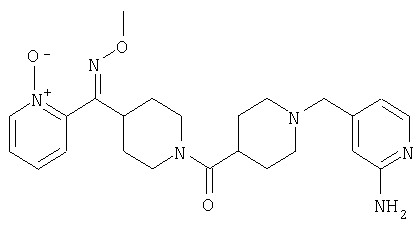
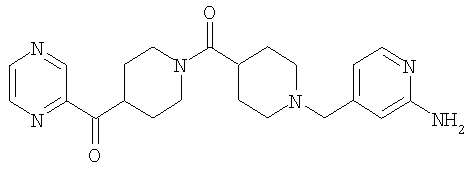
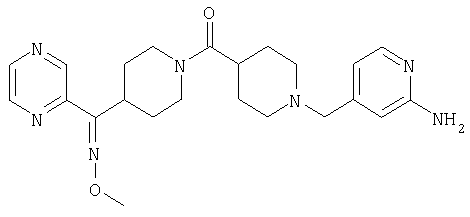
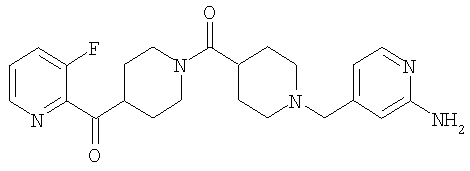
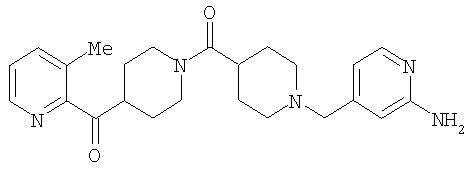
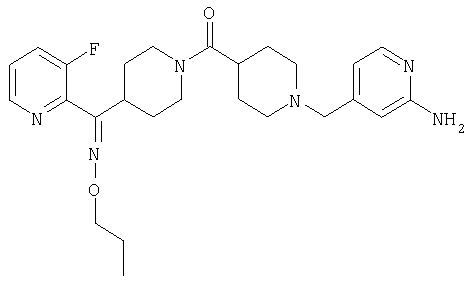
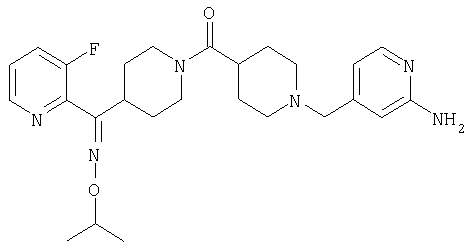
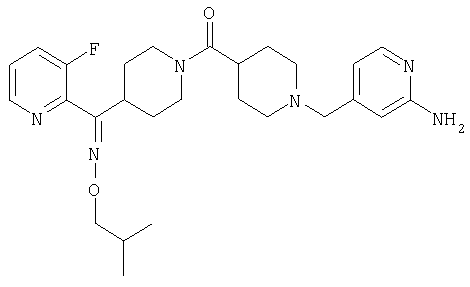
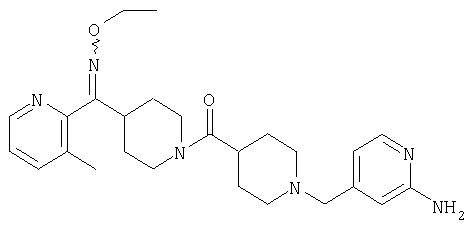
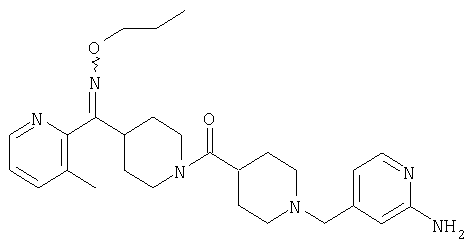
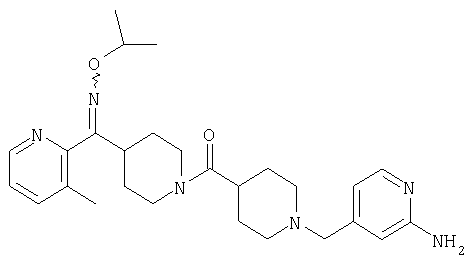
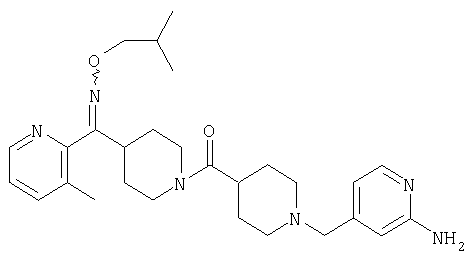
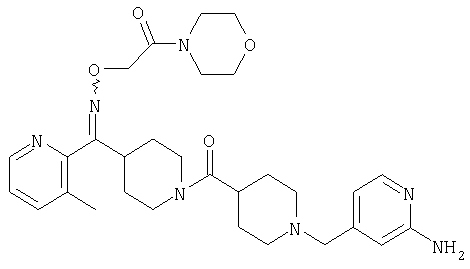
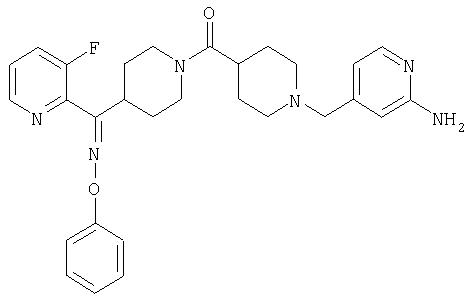
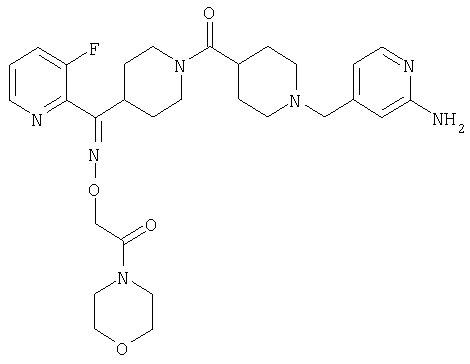
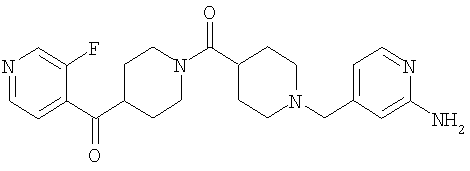
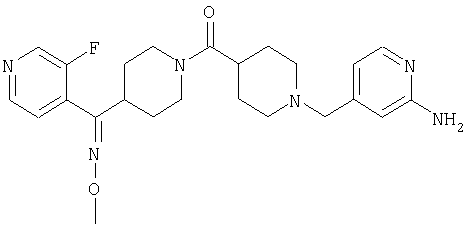
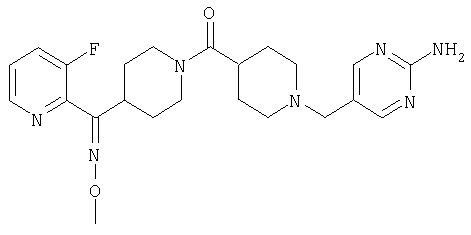
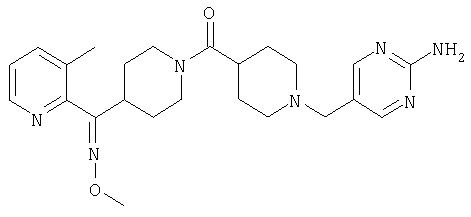
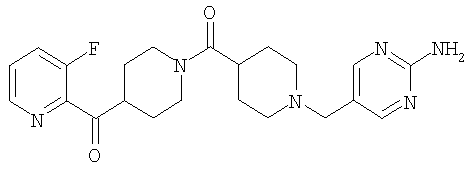
При использовании методик, сходных с описанными в приведенных выше примерах, с использованием исходных соединений, указанных в таблице 3, получают соединения, указанные в столбце "Структура" таблицы 3. Каждое соединение, указанное в таблице 3, является смесью изомеров оксимов, что отмечено с помощью символа  связи между атомом азота оксима и фрагментом ОН или OCH3.
связи между атомом азота оксима и фрагментом ОН или OCH3.
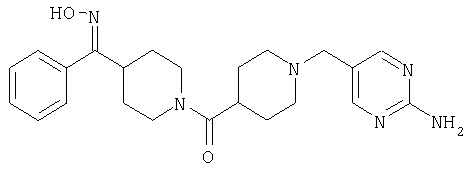
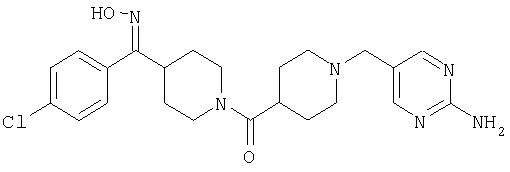
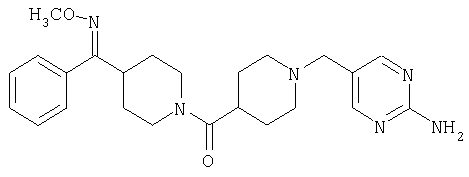
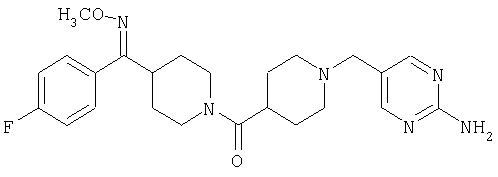
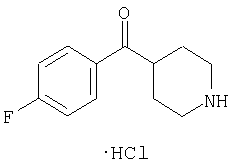
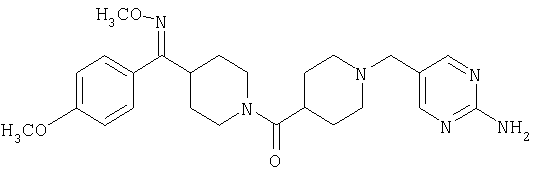
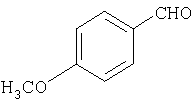
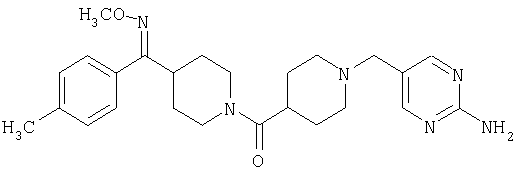
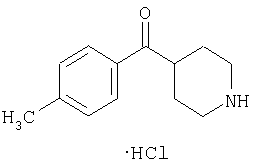
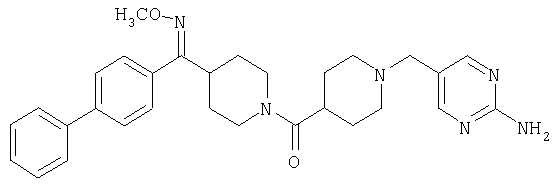
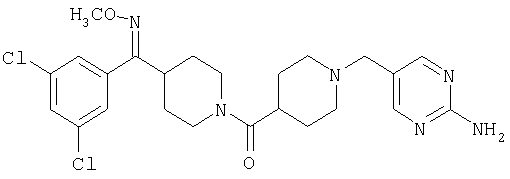
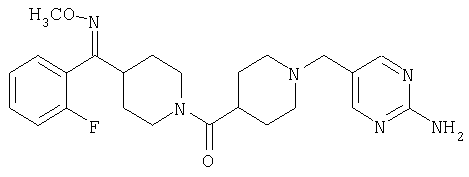
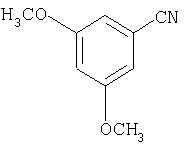
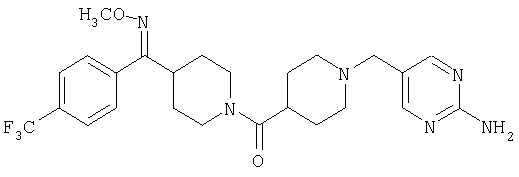
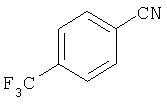
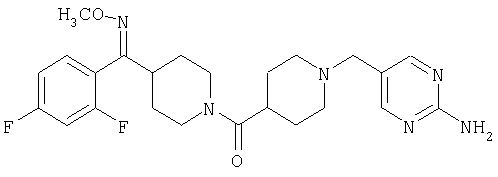
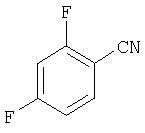
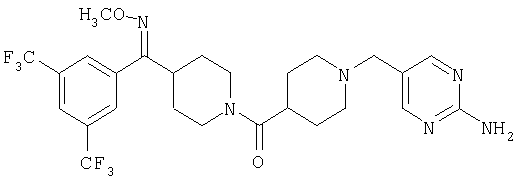
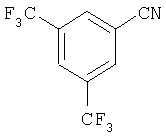
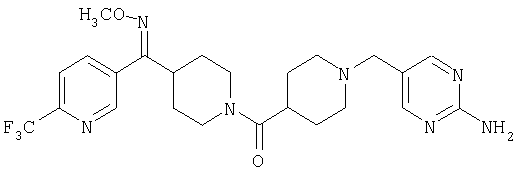
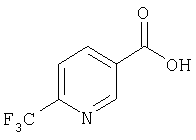
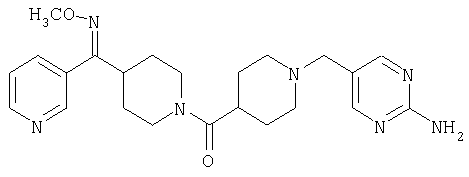
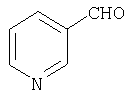
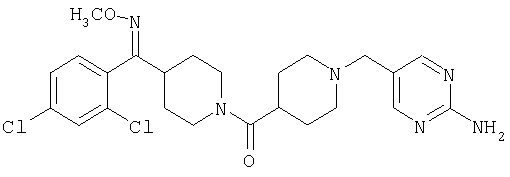
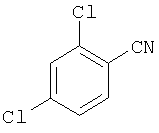
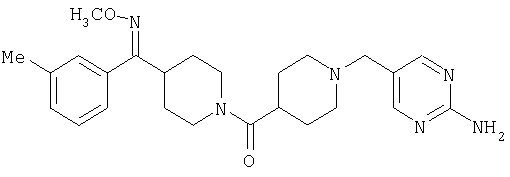
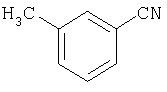
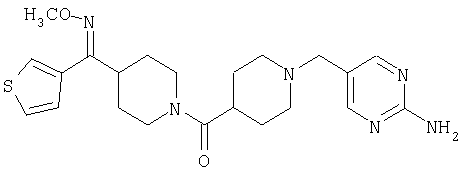
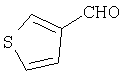
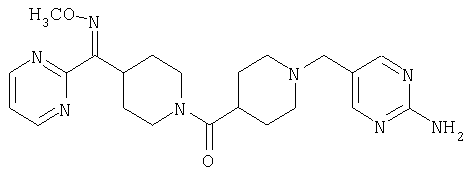
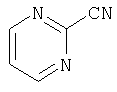
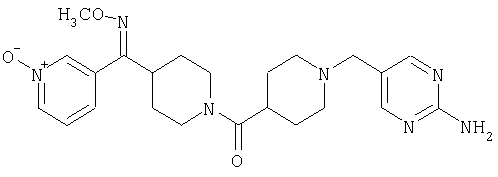
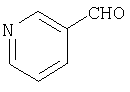
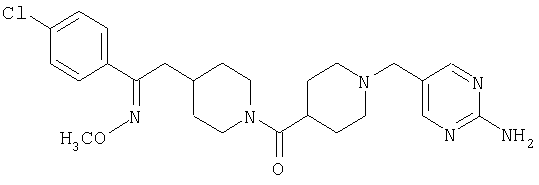
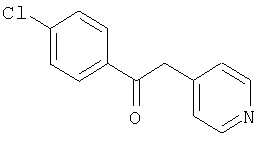
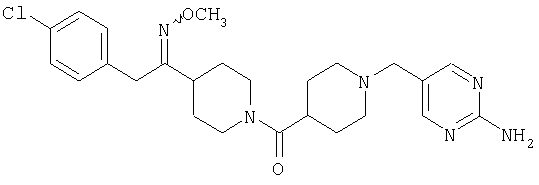
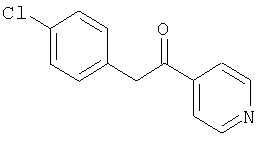
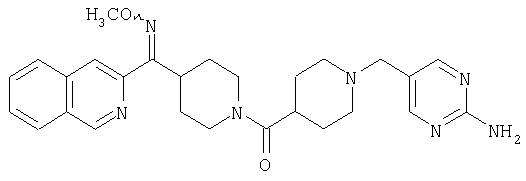
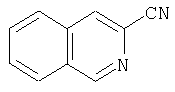
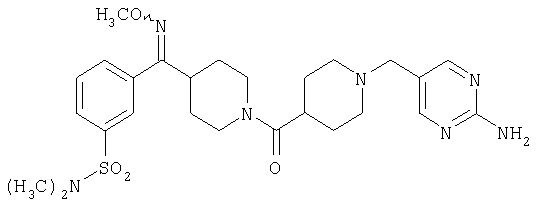
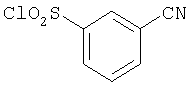
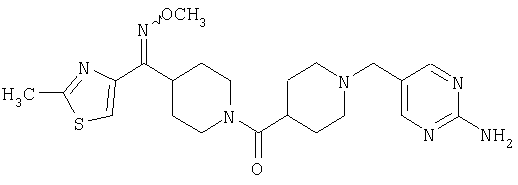
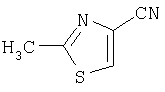
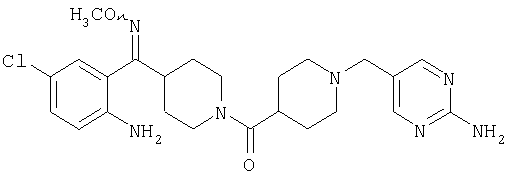
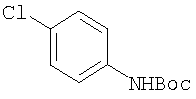
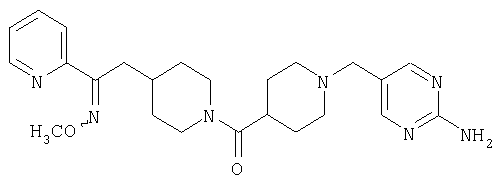
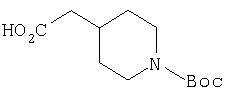
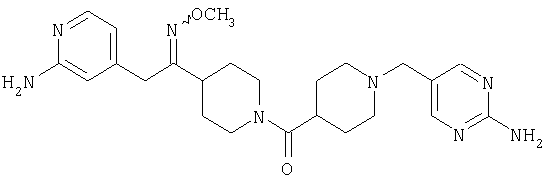
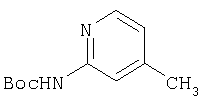
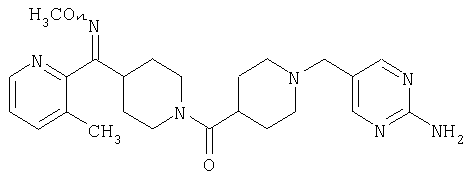
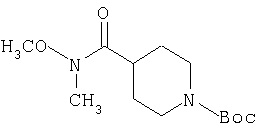
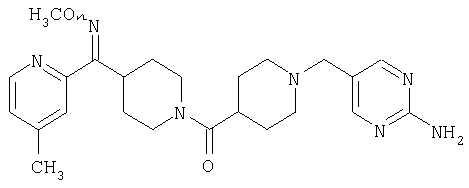
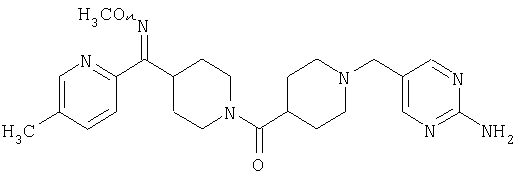
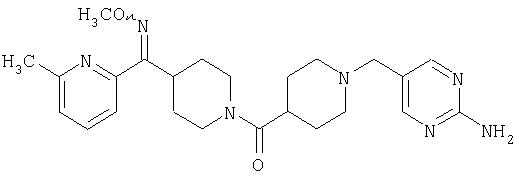
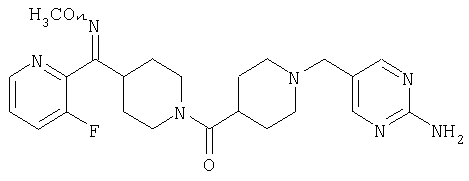
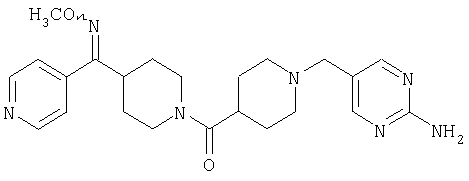
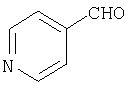
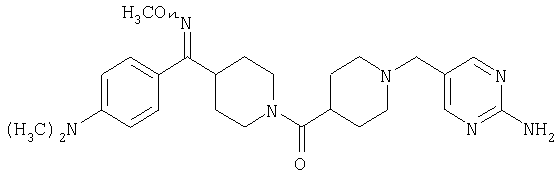
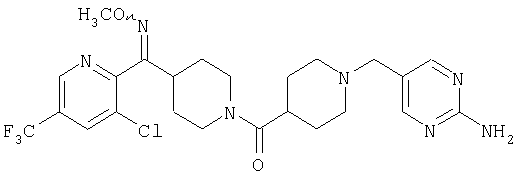
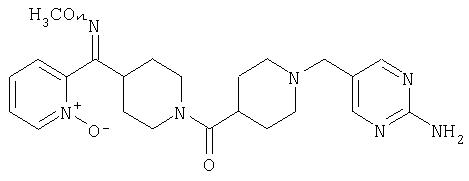
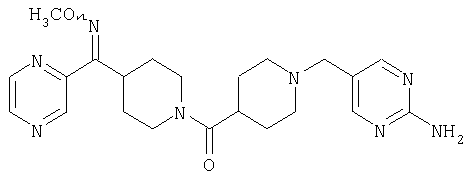
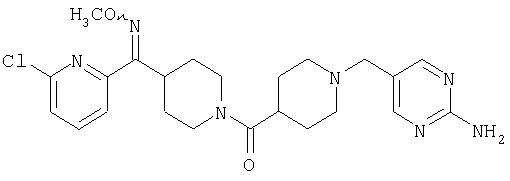
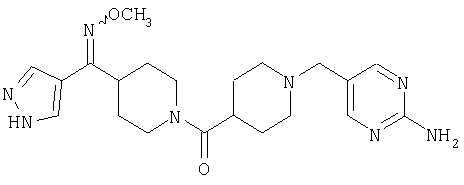
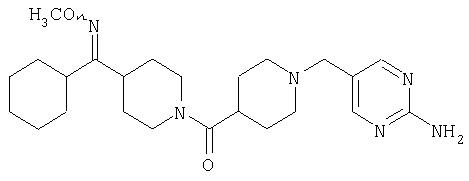
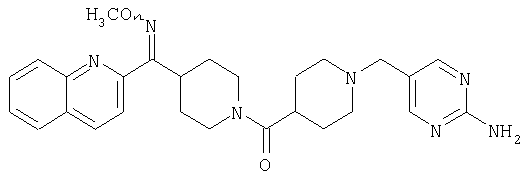
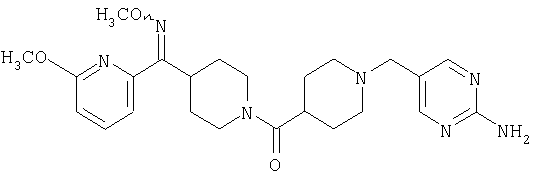
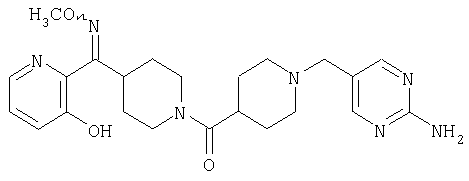
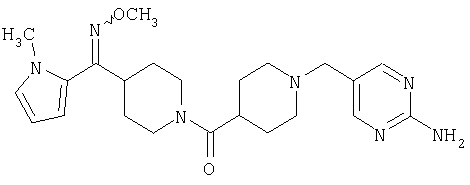
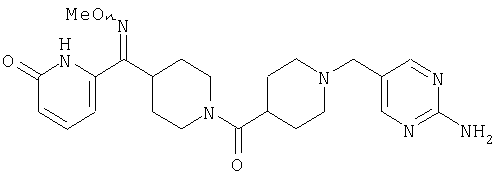
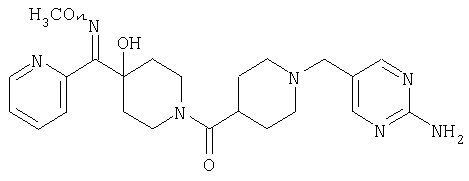
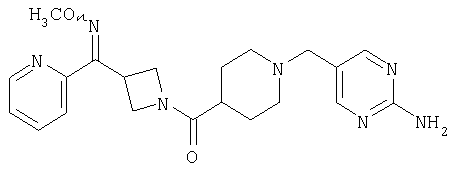
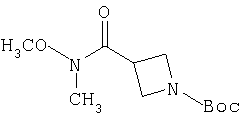
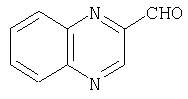
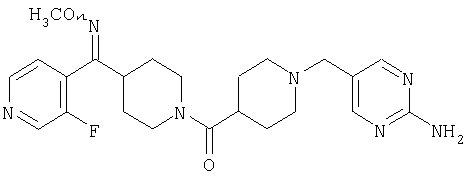
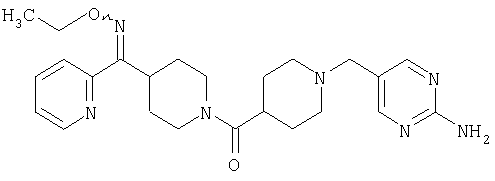
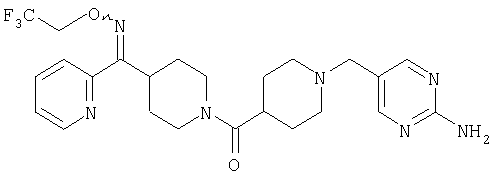
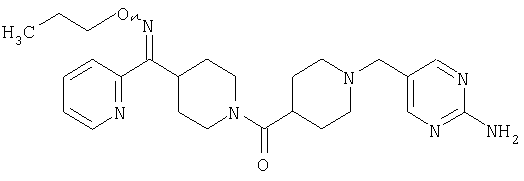
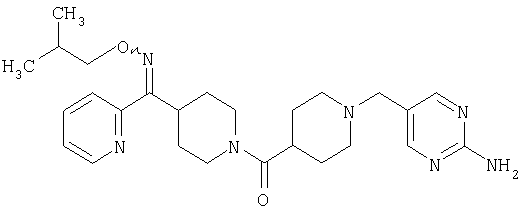
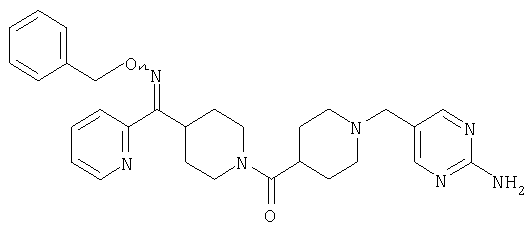
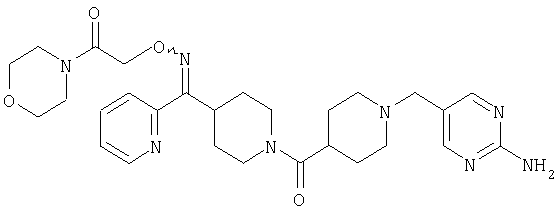
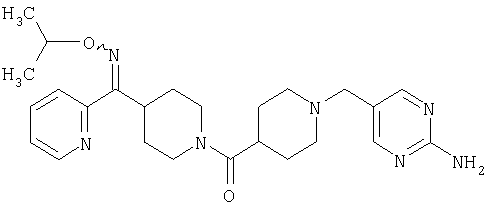
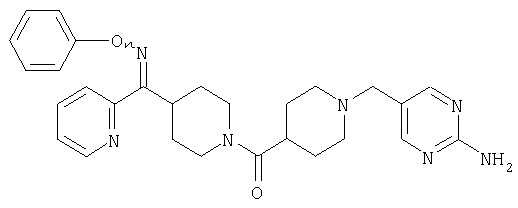
Пример 31
Стадия 1
К раствору LDA (233 мл, 2,0 М раствор в смеси THF/гептан/этилбензол, 0,466 моль) в THF (300 мл) при 0°С в течение 1,0 ч по каплям прибавляют раствор соединения 440 (100 г, 0,389 моль) в THF (примерно 400 мл). Красно-оранжевый раствор 30 мин перемешивают при 0°С, а затем с помощью канюли переносят в предварительно охлажденный (0°С) раствор N-фторбензолсульфонимида (153 г, 0,485 моль) в сухом THF (примерно 600 мл). Реакционную смесь 30 мин перемешивают при 0°С, а затем 18 ч при комнатной температуре. Общий объем растворителя уменьшают примерно до одной трети и прибавляют EtOAc (примерно 1 л). Раствор последовательно промывают водой, 0,1 н. раствором HCl, насыщенным водным раствором NaHCO3 и рассолом. Органический слой сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, получая неочищенную жидкость. Разделение с помощью флэш-хроматографии (смесь состава 6:1 гексаны - EtOAc) дает соединение 441 (93,5 г, 87%).
Стадия 2
Способом, сходным с описанным в Примере 6, стадия 4, соединение 441 превращают в соединение 442.
Стадия 3
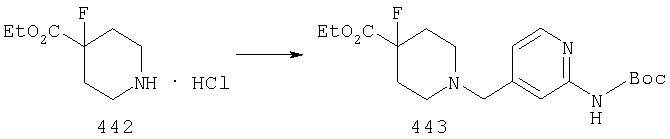
Способом, сходным с описанным в Примере 1, стадия 4, соединение 442 превращают в соединение 443.
Стадия 4
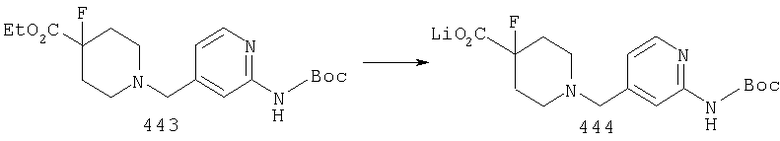
Способом, сходным с описанным в Примере 1, стадия 5, соединение 443 превращают в соединение 444.
Стадия 5
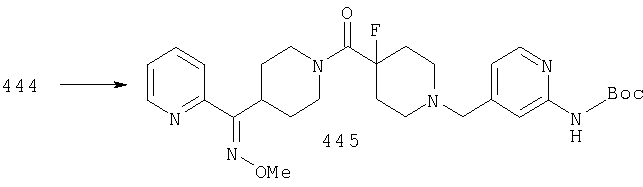
Способом, сходным с описанным в Примере 6, стадия 5, соединение 5 превращают в соединение 445.
Стадия 6
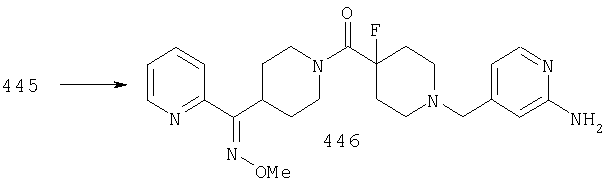
Способом, сходным с описанным в Примере 6, стадия 6, соединение 445 превращают в соединение 446.
В приведенных выше примерах соединение 4-[(Е)-(метоксиимино)-2-пиридинилметил]пиперидиндигидрохлорид
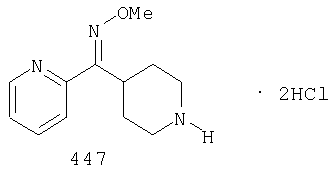
можно использовать для получения соединений, соответствующих настоящему изобретению, см., например, Примеры 6 и 28. Соединение 447 предпочтительно получать из соединения формулы
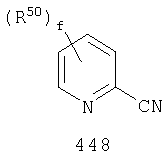
и из соединения формулы 449
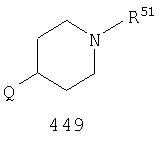
R50 означает алкильную или арильную группу, f равно от 0 до 4, R51 означает алкильную группу и Q означает галогенидную группу, где указанные алкильная, арильная и галогенидная группы являются такими, как определено выше.
Соединение 447 можно получить из 448 и 449 с помощью
(а) превращения соединения формулы 449 в соответствующий реагент Гриньяра (449А)
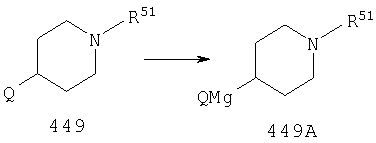
(b) введения соединения формулы 448 в реакцию с соединением формулы 449А с получением соединения формулы 450
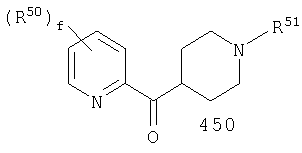
(с) введения соединения формулы 450 в реакцию с подходящим алкилхлорформиатом формулы 451
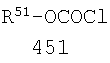
с получением соединения формулы 452
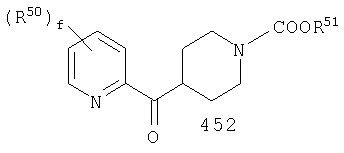
(d) получения соли (формулы 453)
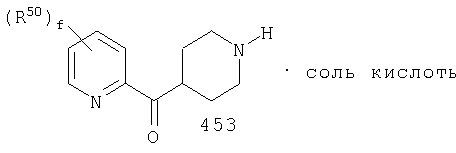
(е) введения соединения формулы 453 в реакцию с алкоксиамином (NH2OR51) или с его гидрохлоридом с получением оксима формулы 454
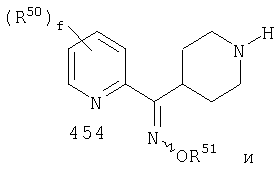
(f) изомеризации соединения формулы 454 с помощью обработки сильной кислотой и превращения в искомую соль кислоты формулы 454 с повышенным содержанием Е-изомера, когда содержание Е-изомера превышает содержание Z-изомера в соответствии с соотношением 90:10. Если f=0, то R51 означает метил, и кислота, использующаяся при изомеризации соединения 454, представляет собой HCl и конечным продуктом является соединение формулы 447.
Таким образом, получение можно представить следующим образом:
По описанной выше методике соединение 447 можно получить следующим образом:
Превращение соединения 461 в соединение 447 приводит в основном к Е-изомеру соединения 447 с высокой стереохимической чистотой и высоким выходом. Изомеризация смеси фенольных соединений с помощью кислотных катализаторов обсуждена в работе Т.Zsuzsanna et al., Hung. Magy. Km. Foly., 74 (3) (1968), 116-119.
Описанный выше способ начинают с соединения 449. На стадии 1 4-галоген-1-алкилпиперидин (или 4-галоген-1-арилпиперидин) по реакции с магнием превращают в соответствующий реагент Гриньяра (449А). Реакцию обычно проводят при температурах, равных примерно от -10°С до температуры кипения. Обычно для этой реакции являются подходящими углеводородный растворитель, такой как, например, толуол, ксилол, хлорбензол, дихлорбензол и т.п., или смеси перечисленных выше углеводородов с простым эфиром, таким как, например, (C5-C12)-алкильный простой эфир, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим (диэтиловый эфир диметиленгликоля), 1,4-диоксан, тетрагидрофуран и т.п. Раствор охлаждают до температуры от примерно -10°С до примерно 10°С и затем проводят реакцию с подходящим 2-цианопиридином (448) в течение примерно 10-120 мин. Примерами подходящих 2-цианопиридинов являются 2-цианопиридин, 4-метил-2-цианопиридин, 4-этил-2-цианопиридин, 4-фенил-2-цианопиридин и т.п. Предпочтительными являются 2-цианопиридин и 4-метил-2-цианопиридин. Реагент Гриньяра обычно используют в количестве, равном 1-4 молярным эквивалентам по отношению к соединению формулы 448, предпочтительно примерно 1-3 молярных эквивалента, а обычно примерно 1,5-2,5 молярных эквивалента. Продукт формулы 450 можно выделить с помощью методик, хорошо известных в данной области техники, таких как, например, обработка кислотой (например, HCl), предпочтительно в подходящем растворителе (например, тетрагидрофуране или этилацетате).
Затем продукт формулы 450 на следующей стадии можно ввести в реакцию с алкилхлорформиатом. Подходящими алкилхлорформиатами являются, например, метилхлорформиат, этилхлорформиат, пропилхлорформиат и т.п., а предпочтительными являются метилхлорформиат и этилхлорформиат. Обычно для этой реакции являются подходящими углеводородный растворитель, такой как, например, толуол, ксилол, хлорбензол, дихлорбензол и т.п., или смеси перечисленных выше углеводородов с простым эфиром, таким как, например, (C5-C12)-алкильный простой эфир, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим, 1,4-диоксан, тетрагидрофуран и т.п. Реакцию обычно проводят при температурах, равных примерно 25-100°С, предпочтительно примерно 40-90°С, а обычно примерно 50-80°С в течение примерно 1-5 ч.
После завершения реакции образовавшуюся кислоту обычно смывают и продукт формулы 452 можно выделить путем экстракции органическими растворителями.
Соединение формулы 452 затем можно превратить в соль путем обработки кислотой, такой как, например, серная кислота, хлористоводородная кислота, трифторуксусная кислота и т.п., обычно в растворителе при температурах от температуры окружающей среды до температуры кипения растворителя. Подходящие растворители включают углеводороды, такие как, например, толуол, ксилол, хлорбензол, дихлорбензол и т.п. Поскольку в соединении формулы 452 имеются два атома азота, то соль обычно содержит 2 моля кислоты на 1 моль соединения 452.
Соединение формулы 453 затем можно превратить в алкилоксим формулы 454 путем его введения в реакцию с алкоксиламином (или его гидрохлоридом) обычно в виде водного раствора. Подходящими алкоксиаминами являются, например, метоксиамин, этоксиамин и т.п. Предпочтительным является метоксиамин. Алкоксиамин (или его гидрохлорид) обычно используют в количестве, равном от примерно 1 до примерно 4 молярных эквивалентов, предпочтительно от примерно 1 до примерно 3 молярных эквивалентов, а обычно от примерно 1 до примерно 2 молярных эквивалентов. Обычно реакцию катализируют слабой кислотой, такой как, например, уксусная кислота, муравьиная кислота и т.п. или их смеси. Можно прибавить вспомогательный растворитель, такой как например, метанол, этанол, изопропанол, н-бутанол и т.п. или их смеси. Продукт формулы 454 после обработки является смесью Z- и Е-изомеров, соотношение между которыми, необходимое для их стереохимической обработки, можно установить с помощью способов, хорошо известных в данной области техники, таких как, например, ВЭЖХ.
Обработка соединения формулы 454 сильной кислотой в условиях проведения реакции, описанных ниже, приводит к изомеризации смеси Z- и Е-изомеров с образованием преимущественно Е-изомера. Обычно соединение формулы 454 можно растворить в растворителе, таком как, например, метанол, этанол, изопропанол, н-бутанол и т.п., в эфире, таком как метил-трет-бутиловый эфир, тетрагидрофуран и т.п., углеводороде, таком как, например, гептан, гексан, толуол и т.п., нитриле, таком как, например, ацетонитрил, бензонитрил и т.п. или в смесях таких растворителей. Растворенное соединение затем обрабатывают сильной кислотой, такой как, например, HCl, HBr, H2SO4 и т.п., при температурах в диапазонах от 20 до 100°С в течение примерно 1-20 ч. Кислоту обычно используют в количестве, равном от примерно 1 до примерно 8 молярных эквивалентов, предпочтительно от примерно 1 до примерно 6 молярных эквивалентов, а обычно от примерно 2 до примерно 4 молярных эквивалентов. Обработка обычно приводит к образованию соли преимущественно Е-изомера соединения формулы 454, которое в действительности представляет собой соединение формулы 447, если в 454 R51 - метил, n=0 и кислотой является HCl.
Как хорошо известно специалистам в данной области техники, продукты, получаемые на различных стадиях описанного выше способа, можно выделять и очищать с помощью обычных способов, таких как, например, фильтрование, перекристаллизация, жидкостная экстракция, дистилляция, осаждение, возгонка и т.п. Продукты можно анализировать или проверять их чистоту с помощью обычных способов, таких как, например, тонкослойная хроматография, ЯМР, ВЭЖХ, определение температуры плавления, масс-спектральный анализ, элементный анализ и т.п., хорошо известных специалистам в данной области техники.
Анализ связывания H3-рецептора
В этом эксперименте источником рецепторов H3 является головной мозг морской свинки. Животные имеют массу, равную 400-600 г. Мозговую ткань гомогенизируют с раствором 50 мМ Tris, pH=7,5. Конечная концентрация ткани в гомогенизированном буферном растворе составляет 10% (мас./об.). Для удаления комков ткани и дебриса гомогенизаты центрифугируют при 1000 g в течение 10 мин. Затем полученную надосадочную жидкость центрифугируют при 50000 g в течение 20 мин для осаждения мембран, которые затем три раза промывают в гомогенизирующем буферном растворе (каждый раз 50000 g в течение 20 мин). Мембраны замораживают и до использования хранят при -70°С.
Все исследуемые соединения растворяют в DMSO и затем разбавляют в связывающем буферном растворе (50 мМ Tris, pH=7,5) так, чтобы конечная концентрация составляла 2 мкг/мл при 0,1% DMSO. Затем мембраны (400 мкг белка) помещают в пробирки для проведения реакции. Реакцию начинают путем прибавления 3 нМ [3H]R-α-метилгистамина (8,8 Ки/ммоль) или 3 нМ [3H]Nα-метилгистамина (80 Ки/ммоль) и продолжают инкубацию при 30°С в течение 30 мин. Связанный лиганд отделяют от несвязанного лиганда фильтрованием и количество радиоактивного лиганда, связанного с мембранами, определяют с помощью жидкостной сцинтилляционной спектрометрии. Инкубацию всегда проводят дважды и стандартная погрешность всегда составляет менее 10%. Соединения, которые ингибируют более 70% специфического связывания радиоактивного лиганда с рецептором, для определения значения Ki (нМ) подвергают серийному разведению.
Соединения 23, 30, 31, 32, 33, 44, 45, 49, 50, 53, 54, 55, 56, 57А, 59, 75, 76, 83, 88, 92, 99, 104, 110, 117, 128, 200, 201, 203-215, 217-241, 244-246, 246А, 247-253, 253А, 254-273, 275, 278, 280-282, 287, 296, 301-310 и 312-379 обладают значениями Ki, находящимся в диапазоне от примерно 0,25 до примерно 370 нМ.
Предпочтительные соединения 23, 30, 31, 32, 33, 50, 53, 54, 55, 56, 57А, 59, 92, 212, 215, 218, 219, 220, 224, 225, 226, 227, 229, 233, 235, 237, 238, 246, 246А, 247, 248, 251, 253, 253А, 268-273, 275, 278-281, 287, 296, 301, 304-307, 309, 312, 314, 318, 320-356 и 358-376 обладают значениями Ki, находящимся в диапазоне от примерно 0,25 до примерно 33 нМ.
Наиболее предпочтительные соединения 30, 31, 32, 33, 54, 55, 56, 56А, 225, 237, 246А, 253А, 273, 280, 287, 296, 301, 304-307, 309, 312, 314-318, 320-348, 350-356, 359-372 и 374-376 обладают значениями Ki, находящимся в диапазоне от примерно 0,25 до примерно 16 нМ.
Более предпочтительное соединение 32 обладает значением Ki, равным 0,83 нМ.
Более предпочтительные соединения 54, 55, 253А, 287, 320 обладают значениями Ki, находящимся в диапазоне от примерно 1,05 до примерно 9,75 нМ.
При изготовлении фармацевтических композиций, описанных в настоящем изобретении, инертные, фармацевтически приемлемые носители могут быть твердыми или жидкими. К твердым формам препаратов относятся порошки, таблетки, диспергирующиеся гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут содержать от примерно 5 до примерно 95% активного компонента. Подходящие твердые носители известны в данной области техники и включают, например, карбонат магния, стеарат магния, тальк, сахар и лактозу. Таблетки, порошки, облатки и капсулы можно использовать в качестве твердых дозировочных форм, пригодных для перорального введения. Примеры фармацевтически приемлемых носителей и способов изготовления различных композиций приведены в работе A.Gennato (ed.), Remington's Pharmaceutical Sciences, 18th Edition, (1990), Mack Publishing Co., Easton, Pennsylvania.
Жидкие формы композиций включают растворы, суспензии и эмульсии. В качестве примера можно указать водные или водно-пропиленгликолевые растворы для парентеральных инъекций или прибавление подсластителей и замутнителей в пероральные растворы, суспензии и эмульсии. К жидким формам композиций также могут относиться растворы для внутриназального введения.
Аэрозольные композиции, пригодные для ингаляции, могут включать растворы и твердые вещества в порошкообразной форме, которые могут сочетаться с фармацевтически приемлемым носителем, таким как сжатый инертный газ, например азот.
В объем настоящего изобретения также включены твердые формы композиций, которые предназначены для превращения в жидкие формы композиций, предназначенных для перорального или парентерального введения, которое выполняется незадолго до использования. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения, соответствующие настоящему изобретению, также можно вводить чрескожно. Чрескожные композиции могут представлять собой кремы, лосьоны, аэрозоли и/или эмульсии и они могут быть включены в матрицу пластыря чрескожного воздействия или пластыря резервуарного типа, что обычно используется в данной области техники для такой цели.
Предпочтительно вводить соединение перорально.
Предпочтительно, чтобы фармацевтическая композиция содержалась в разовой дозировочной форме. В такой форме композиция разделяется на разовые дозы подходящей величины, содержащие соответствующие количества активных компонентов, например количества, достаточные для достижения необходимой цели.
Количество активного соединения, содержащегося в разовой дозе лекарственного препарата, в соответствии с конкретным случаем применения обычно может меняться или регулироваться в диапазоне от примерно 1 до примерно 150 мг, предпочтительно от примерно 1 до примерно 75 мг, более предпочтительно от примерно 1 до примерно 50 мг.
Реальная использующаяся доза может меняться в зависимости от требований пациента и тяжести подвергающегося лечению патологического состояния. Определение надлежащего дозировочного режима для конкретного случая проводит специалист в данной области техники. Для удобства полную суточную дозу можно разделять и назначать порциями в течение дня в соответствии с необходимостью.
Количество и частота введения соединений, соответствующих настоящему изобретению, и/или их фармацевтически приемлемых солей будет регулироваться в соответствии с решением лечащего врача, учитывающего такие факторы, как возраст, состояние и массу пациента, а также тяжесть симптомов, подвергающихся лечению. Типичный рекомендованный суточный дозировочный режим при пероральном введении может включать введение от примерно 1 до примерно 300 мг/сутки, предпочтительно от 1 до 75 мг/сутки, назначаемых в виде двух - четырех разделенных доз.
Описанные выше способы, соответствующие настоящему изобретению, в которых используется соединение формулы I, также включают использование одного или большего количества соединении формулы I, и описанные выше способы, соответствующие настоящему изобретению, включающие использование соединений формулы I в комбинации с антагонистом рецептора H1, также включают использование одного или большего количества соединении формулы I в комбинации с одним или большим количеством антагонистов рецептора H1.
Хотя настоящее изобретение описано вместе с конкретными вариантами осуществления, приведенными выше, для специалиста с общей подготовкой в данной области техники будут ясными многие его альтернативы, модификации и изменения. Подразумевается, что все такие альтернативы, модификации и изменения включены в объем и сущность настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОАРИЛЬНЫЕ ПРОИЗВОДНЫЕ, КАК ПРЕВОСХОДНЫЕ ЛИГАНДЫ ДЛЯ РЕЦЕПТОРА НОЦИЦЕПТИНА ORL-1 | 2002 |
|
RU2327698C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА И ПИПЕРИДИНА | 2002 |
|
RU2326120C9 |
| ТРИЦИКЛИЧЕСКИЕ ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2293734C9 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ СОДЕРЖАЩИЕ, И ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ CCR5 | 2000 |
|
RU2299206C9 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2408591C9 |
| СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2237060C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2563634C2 |
| ТРИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ ТРОМБИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2329264C9 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2007 |
|
RU2431631C2 |
| ПИРИДИЛПИПЕРИДИНОВЫЕ АНТАГОНИСТЫ РЕЦЕПТОРА ОРЕКСИНОВ | 2008 |
|
RU2470021C2 |
Изобретение относится к новым полициклическим соединениям формулы (I), в которой радикалы и символы имеют значения, определенные в формуле изобретения. Соединения формулы (I) обладают свойствами антагониста рецептора Н3. Объектами изобретения также являются фармацевтическая композиция, содержащая соединения формулы (I), и способ лечения болезни из группы, включающей заложенность носа, ожирение, сонливость, нарколепсию, дефицит внимания с гиперактивностью, болезнь Альцгеймера и шизофрению, с использованием соединений формулы (I) необязательно в комбинации с антагонистом рецептора H1. 3 н. и 36 з.п. ф-лы, 3 табл.
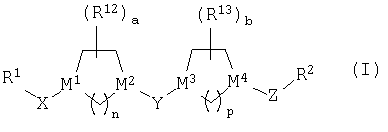
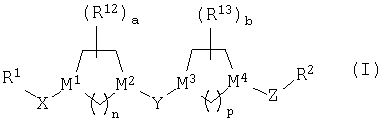
или его фармацевтически приемлемые соль или сольват, в котором
(1) R1 выбран из группы, включающей
(a) фенил;
(b) гетероарил, выбранный из группы, включающей пиридил, тиенил, пиримидил, хинолил, тиазолил, пиразолил, пиролил, хиназолил, пиридил-N-оксид и пиразинил; и
(е) (С3-С7)-циклоалкил;
где указанные группы R1 необязательно содержат от 1 до 4 заместителей, независимо выбранных из группы, включающей
(1) галоген;
(2) гидроксил;
(3) (С1-С6)-алкоксил;
(4) -CF3;
(6) -NR4R5;
(7) фенил;
(11) -S(O)mN(R20)2, где все R20 являются одинаковыми или разными и означают Н или C1-С6-алкильную группу; и
(13) (С1-С6)-алкил, незамещенный или замещенный галогеном; или
(2) R1 и Х совместно образуют группу, выбранную из
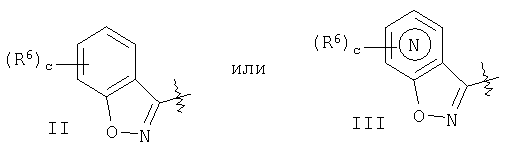
(3) Х выбран из =С(O), =C(NOR3), =C(NNR4R5),
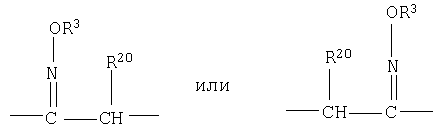
(4) M1 означает атом углерода;
(5) M2 означает N;
(6) M3 означает С или N и M4 означает N;
(7) Y означает =С(O);
(8) Z означает (С1-С6)-алкильную группу;
(9) R2 означает гетероарильный цикл, выбранный из группы, включающей пиридил, пиримидил, пиридил-N-оксид, фуранил, тиазолил, пиридазинил, пиразолил, оксазолил, тиенил, пиролил, тиадиазолил, бензотиенил и оксадиазолил, причем указанный гетероарильный цикл необязательно содержит от 1 до 3 заместителей, независимо выбранных из группы, включающей галоген, (С1-С6)-алкил, (С1-С6)-алкоксил, -NR4R5, -CO2R4, -CH2NR4R5 или -(N)C(NR4R5)2;
(10) R3 выбран из группы, включающей
(a) водород;
(b) (С1-С6)-алкил;
(c) фенил;
(f) фенил-(С1-С6)-алкил;
(h) -(CH2)e-C(O)OR4;
(i) -(CH2)e-C(O)R30, где R30 означает гетероциклоалкильную группу, выбранную из группы, включающей морфолинил и пирролидинил; или
(k) -СН2CF3;
где указанные арил, гетероарил, гетероциклалкил и арильный фрагмент указанного арилалкила необязательно содержат от 1 до 3 заместителей, независимо выбранных из группы, включающей галоген и -CF3;
(11) R4 выбран из группы, включающей водород и (С1-С6)-алкил;
(12) R5 выбран из группы, включающей водород, (С1-С6)-алкил и -С(O)2R4;
(13) или R4 и R5 вместе с атомом азота, с которым они связаны, образуют морфолинильное кольцо;
(14) R6 означает галоген;
(15) R12 выбран из группы, включающей гидроксил и фтор;
(16) R13 выбран из группы, включающей (С1-С6)-алкил, гидроксил и фтор;
(17) а равно 0 или 1;
(18) b равно 0 или 1;
(19) с равно 0 или 1;
(20) е равно 1;
(21) m равно 2;
(22) n равно 2 и
(23) p равно 1, 2 или 3 при условии что если М3 и М4 означают азот, то p равно 2 или 3.
(A) фенил;
(B) замещенный фенил, в котором заместители для указанного замещенного арила выбраны из группы, включающей (1) галоген, (2) C1-С6-алкил и (3) замещенный галогеном C1-C6-алкил;
(C) гетероарил, выбранный из группы, включающей пиридил, тиенил, пиримидил, хинолил, тиазолил, пиразолил, пиролил, хиназолил, пиридил-N-оксид и пиразинил;
(D) определенный в п.1 гетероарил, замещенный как указано в п.1; или
(Е) если R1 и Х образуют группировку совместно, то фрагмент имеет вид
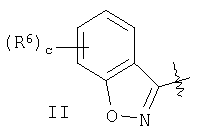
(A) фенил;
(B) замещенный фенил, в котором заместители для указанного замещенного фенила выбраны из группы, включающей (1) галоген, (2) C1-С6-алкил и (3) замещенный галогеном C1-С6-алкил;
(C) гетероарил, выбранный из группы, включающей пиридил, тиенил, пиримидил, тиазолил и пиридил-N-оксид;
(D) замещенный (С1-С6)-алкилом тиазолил или
(Е) если R1 и Х образуют группировку совместно, то фрагмент имеет вид
где с равно 0 или 1, и если с равно 1, то R6 означает галоген.
(A) фенил;
(B) замещенный фенил, в котором заместители для указанного замещенного фенила выбраны из группы, включающей хлор, фтор и трифторметил;
(C) гетероарил, выбранный из группы, включающей
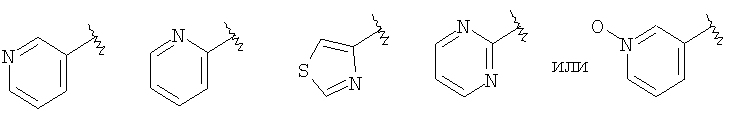
или
(D) замещенный гетероарил формулы
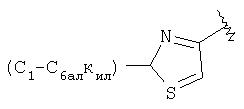
или
(Е) если R1 и Х образуют группировку совместно, то фрагмент имеет вид
где с равно 0 или 1, и если с равно 1, то R6 означает фтор.
(A) фенил;
(B) замещенный фенил, в котором заместители для указанного замещенного фенила выбраны из группы, включающей хлор, фтор и трифторметил;
(C) пиридил или
(D) замещенный гетероарил формулы
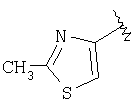
или
(Е) если R1 и Х образуют группировку совместно, то фрагмент имеет вид
где с равно 0 или 1, и если с равно 1, то R6 означает фтор.
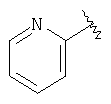
М3 означает углерод и М4 означает азот;
n равно 2;
а равно 0 или 1;
b равно 0 или 1;
с равно 0 или 1, и если с равно 1, то R6 означает галоген;
e равно 1 и
p равно 2;
Y означает =С(O) и
Z означает (С1-С3)-алкил.
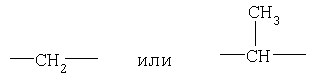
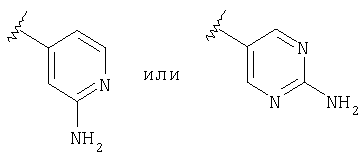
(1) R1 выбран из группы, включающей
(A) фенил;
(B) замещенный фенил, в котором заместители для указанного замещенного арила выбраны из группы, включающей(1) галоген, (2) C1-С6-алкил и (3) замещенный галогеном C1-С6-алкил;
(C) гетероарил, выбранный из группы, включающей пиридил, тиенил, пиримидил, хинолил, тиазолил, пиразолил, пиролил, хиназолил, пиридил-N-оксид и пиразинил;
(D) определенный в п.1 гетероарил, замещенный как указано в п.1; или (Е), если R1 и Х образуют группировку совместно, то фрагмент имеет вид
(2) Х означает =C(NOR3);
(3) R3 выбран из группы, включающей Н и (С1-С6)-алкил;
(4) М2 означает азот;
(5) Y означает =С(O);
(6) М3 означает С или N и М4 означает N;
(7) Z означает (С1-С3)-алкил и
(8) R2 означает шестичленный гетероарильный цикл, выбранный из группы, включающей пиридил, пиримидил, пиридил-N-оксид и пиридазинил.
(1) R1 выбран из группы, включающей
(A) фенил;
(B) замещенный фенил, в котором заместители для указанного замещенного фенила выбраны из группы, включающей (1) галоген, (2) C1-C6-алкил и (3) замещенный галогеном C1-C6-алкил;
(C) гетероарил, выбранный из группы, включающей пиридил, тиенил, пиримидил, тиазолил и пиридил-N-оксид; или
(D) замещенный (С1-С6)-алкилом тиазолил; или
(Е) если R1 и Х образуют группировку совместно, то фрагмент имеет вид
где с равно 0 или 1, и если с равно 1, то R6 означает галоген.
(2) R3 выбран из группы, включающей Н, метил и этил;
(3) n равно 2;
(4) а равно 0 или 1;
(5) b равно 0 или 1;
(6) с равно 0 или 1, и если с равно 1, то R6 означает галоген;
(7) e равно 1 и
(8) p равно 2;
(9) R4 означает Н или (С1-С6)-алкил;
(10) R5 означает Н или (С1-С6)-алкил;
(11) R12 означает гидроксил или фтор и
(12) R13 означает (С1-С6)-алкил, гидроксил или фтор.
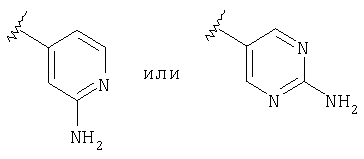
R1 означает
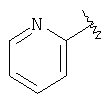
М2 означает азот, М3 означает углерод и М4 означает азот.
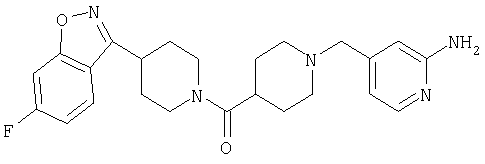
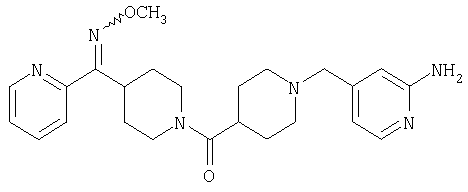
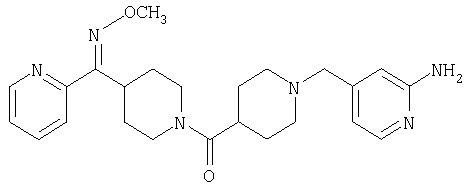
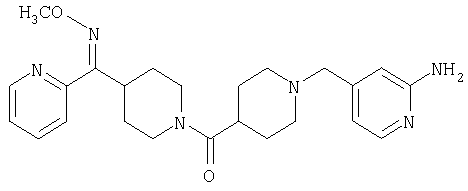
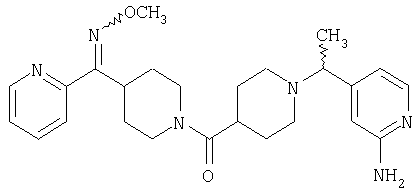
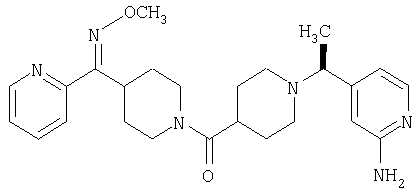
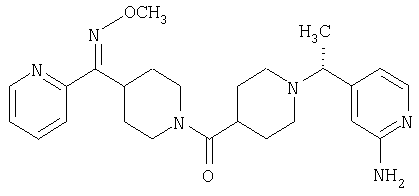
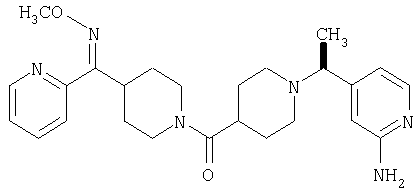
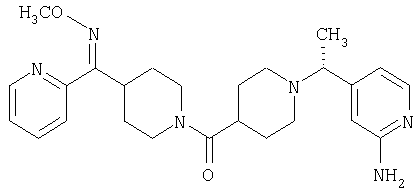
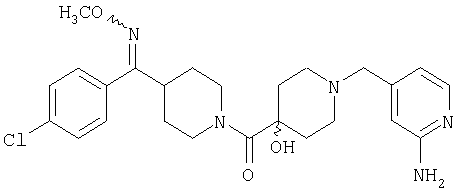
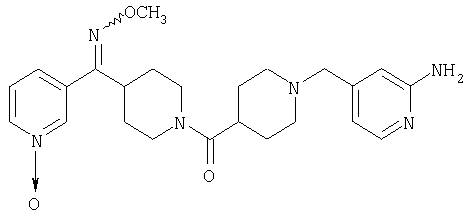
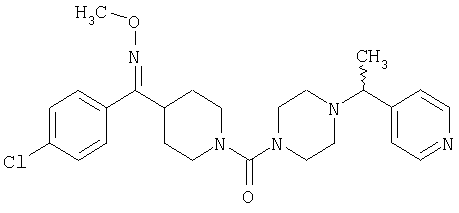
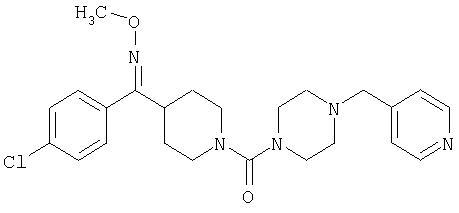
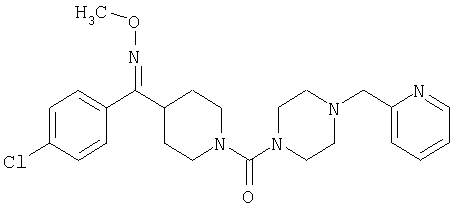
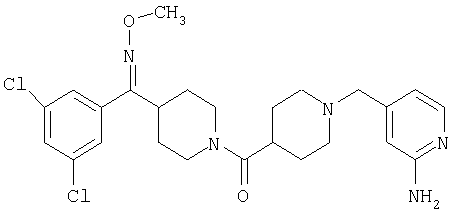
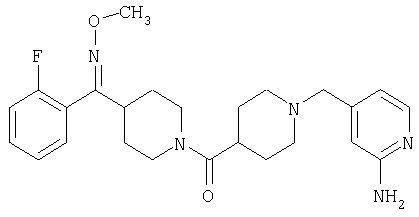
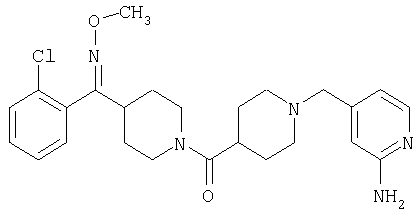
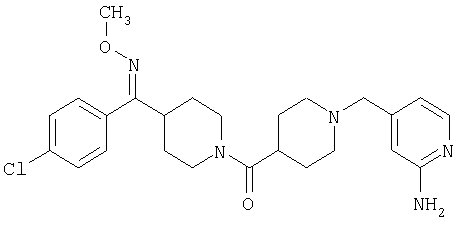
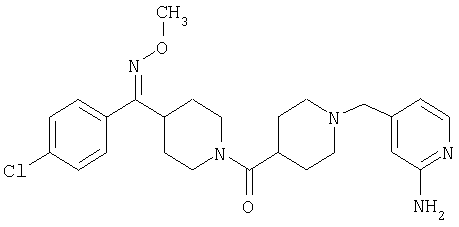
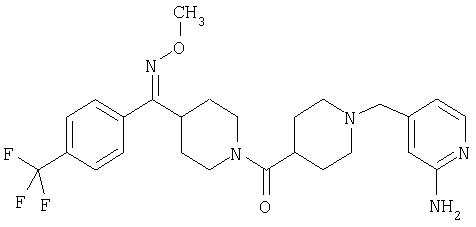
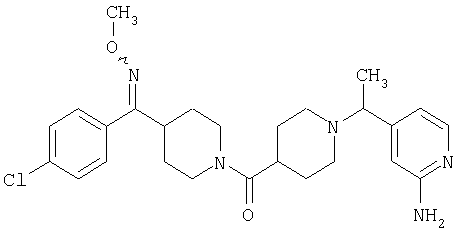
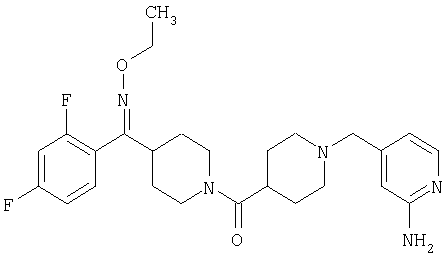
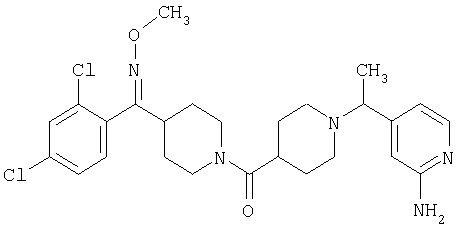
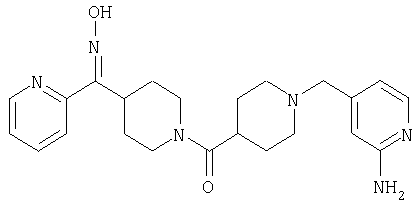
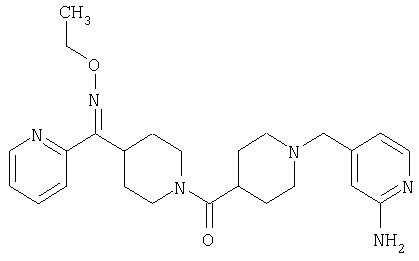
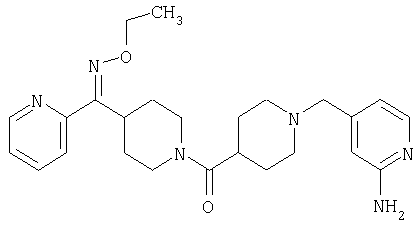
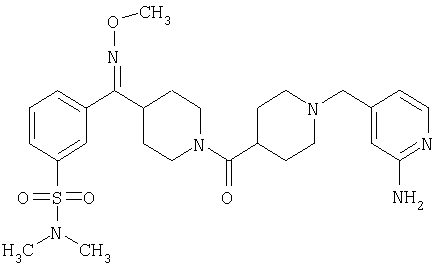
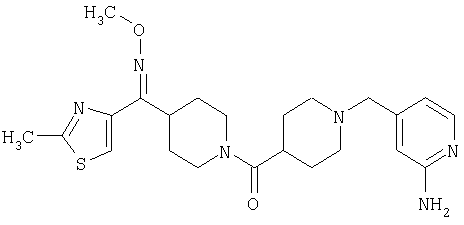
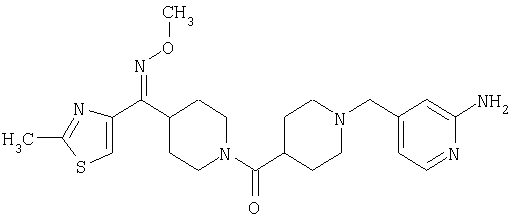
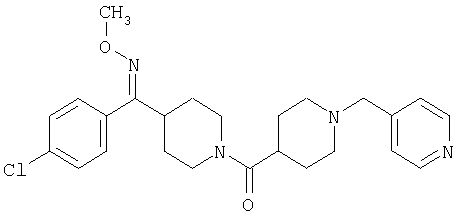
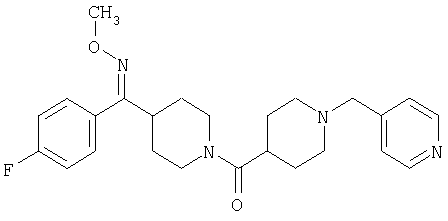
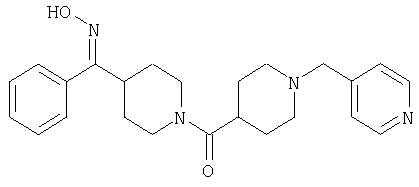
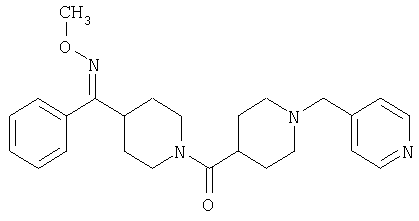
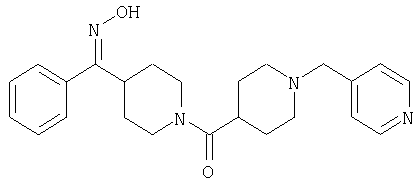
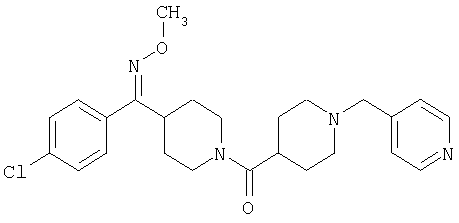
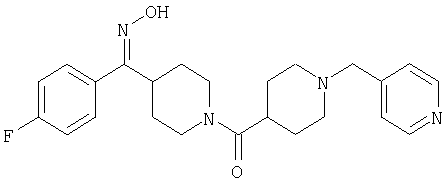
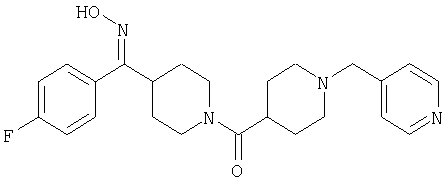
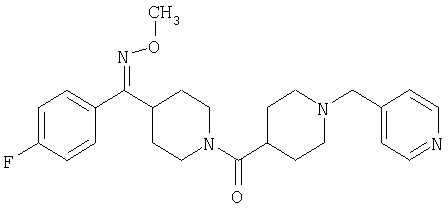
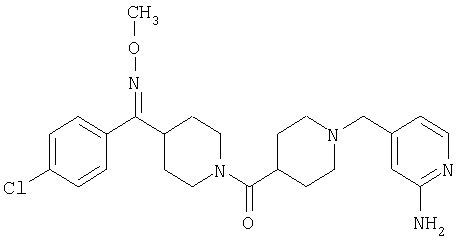
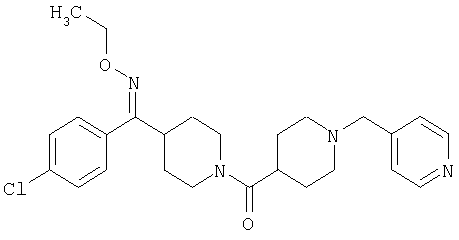
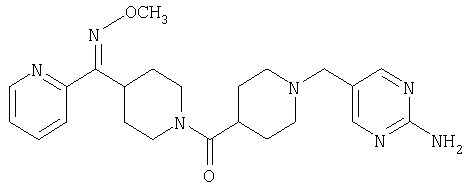
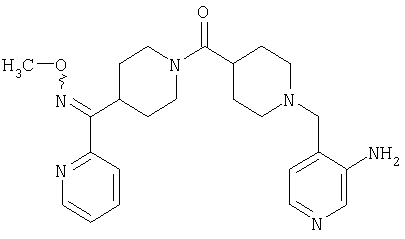
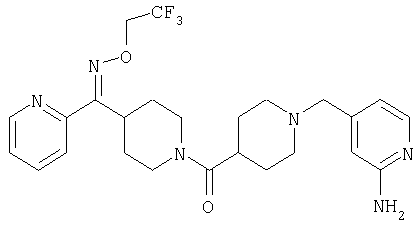
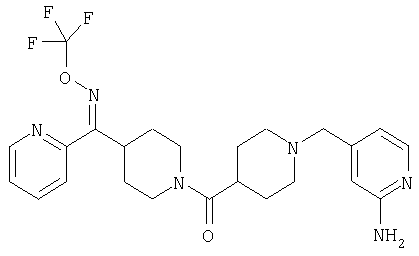
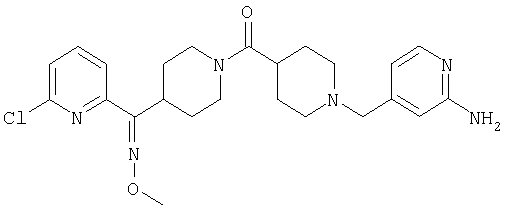
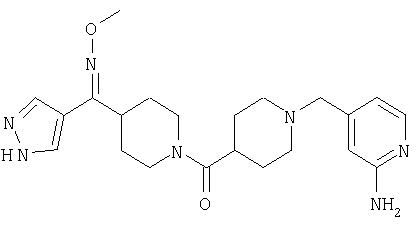
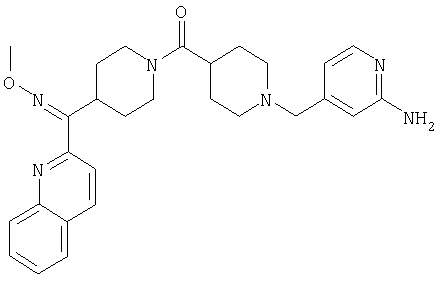
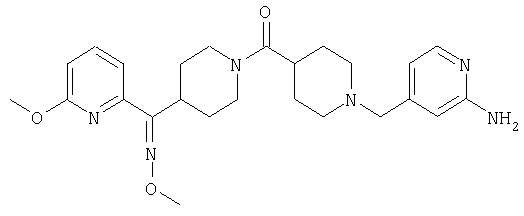
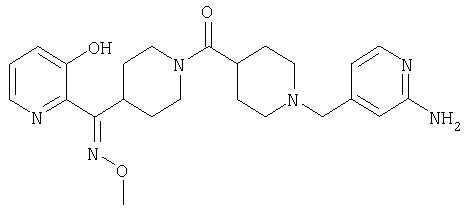
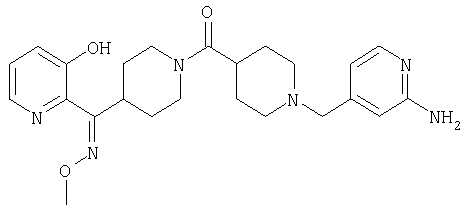
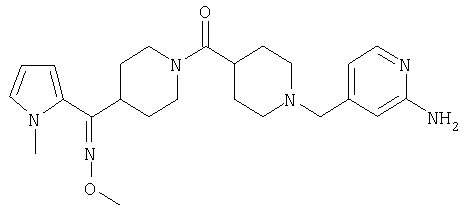
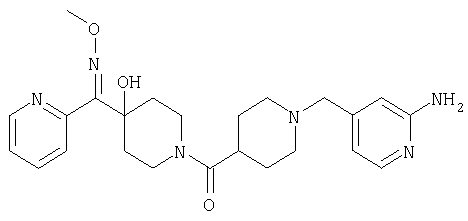
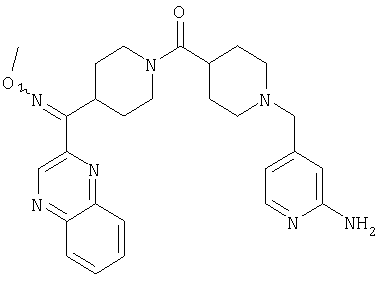
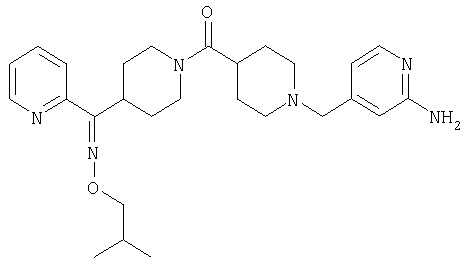
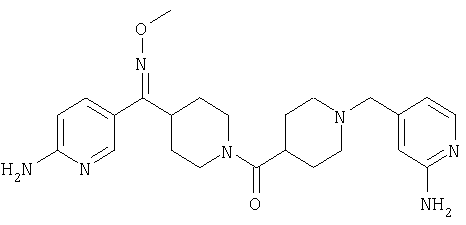
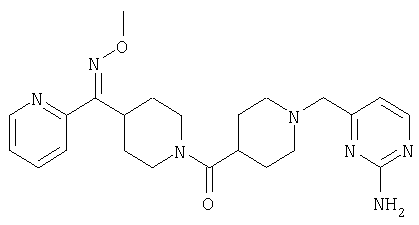
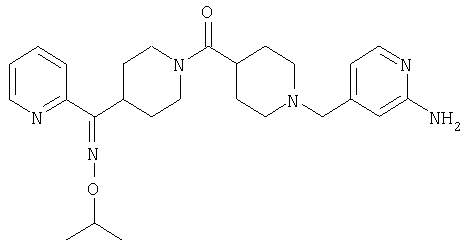
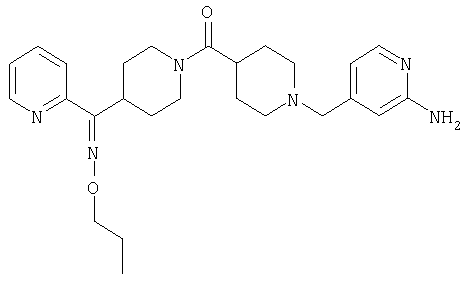
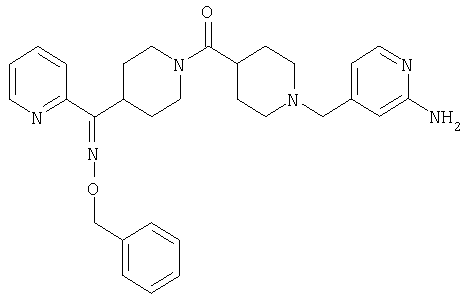
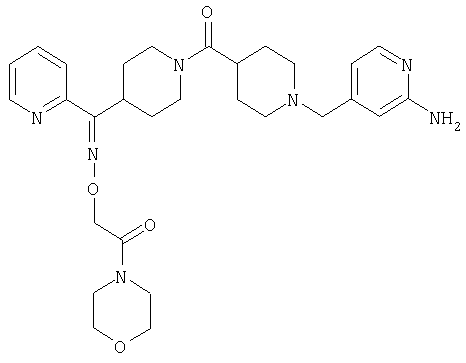
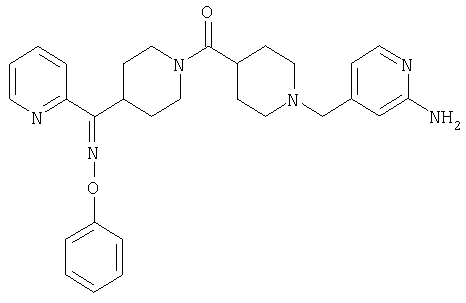
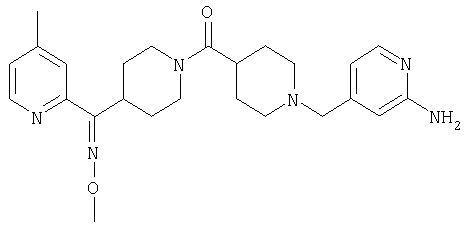
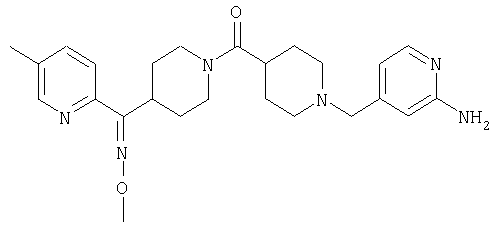
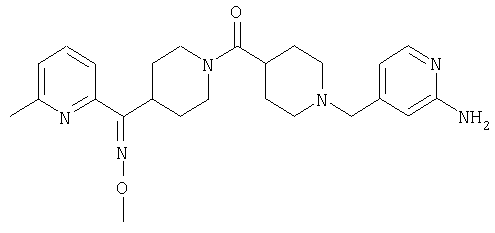
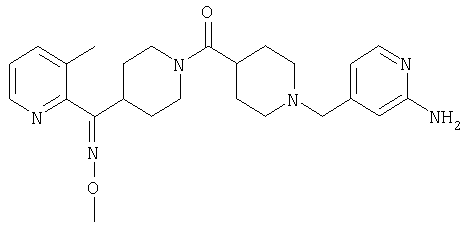
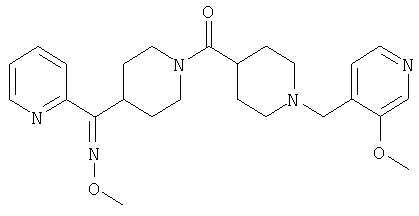
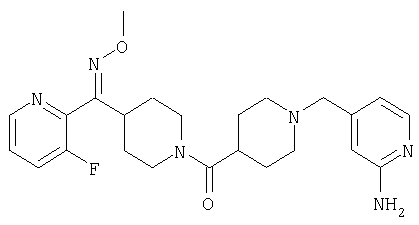
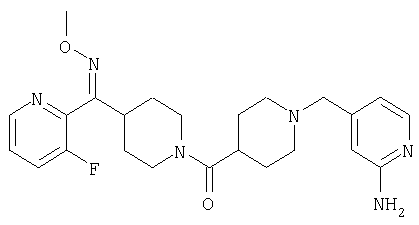
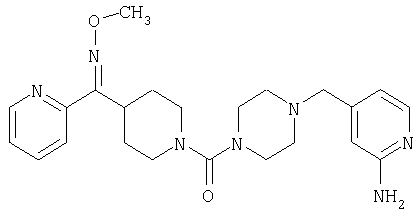
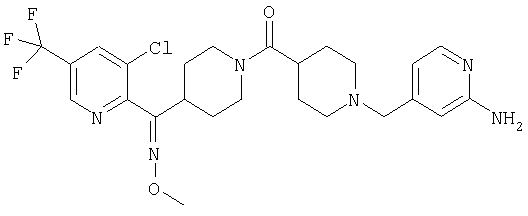
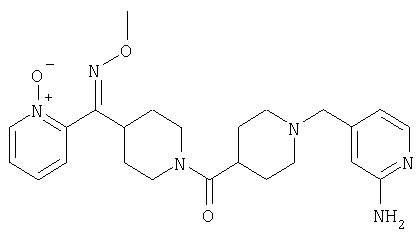
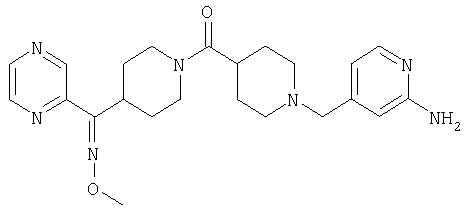
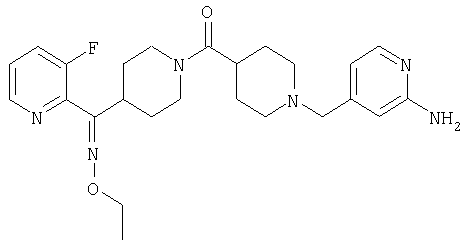
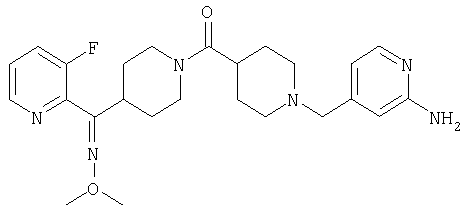
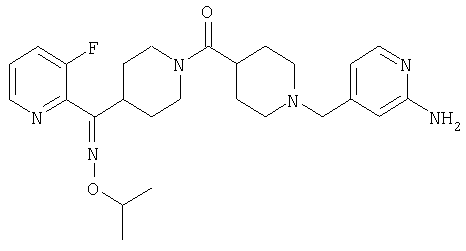
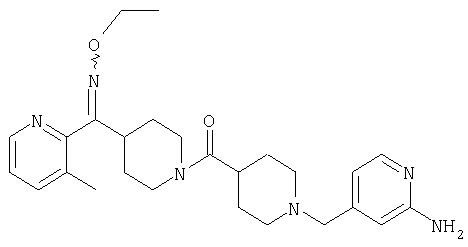
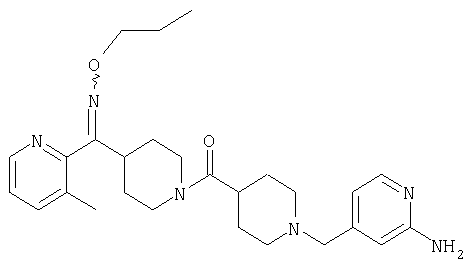
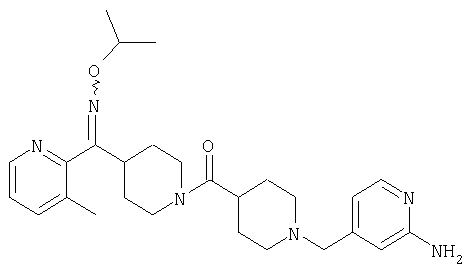
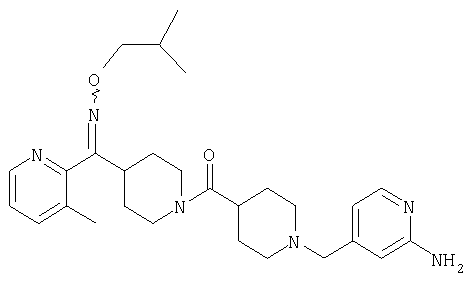
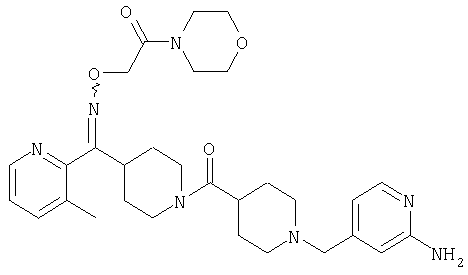
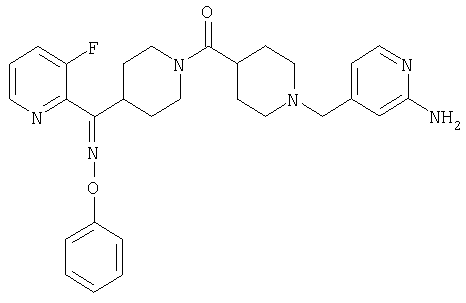
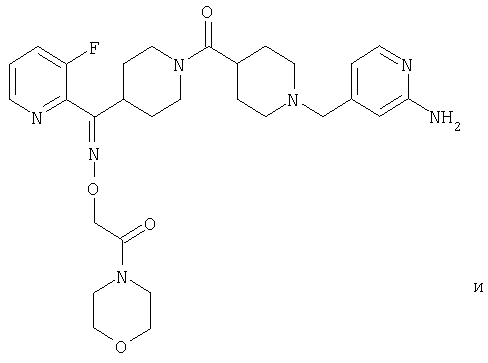
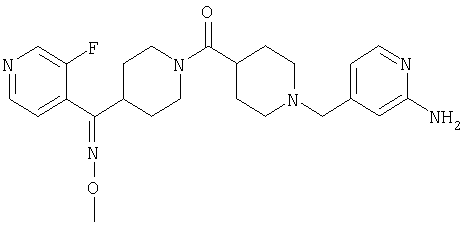
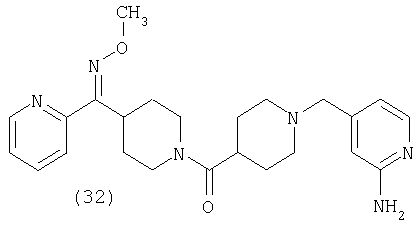
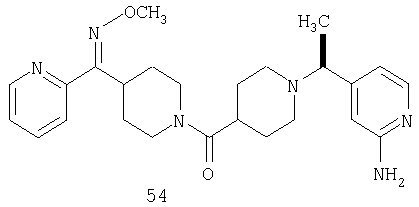
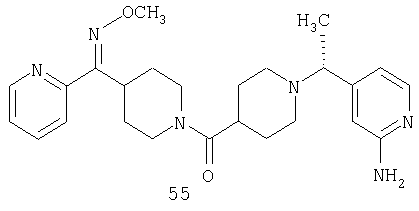
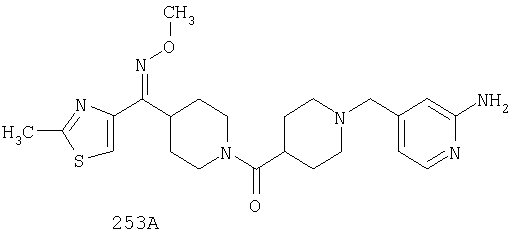
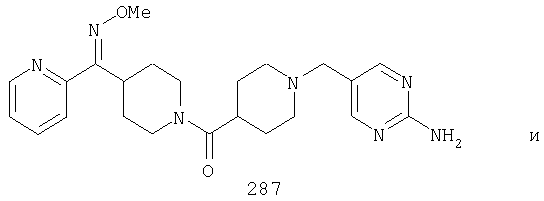
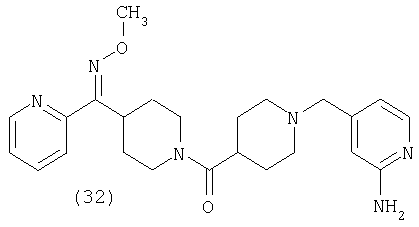
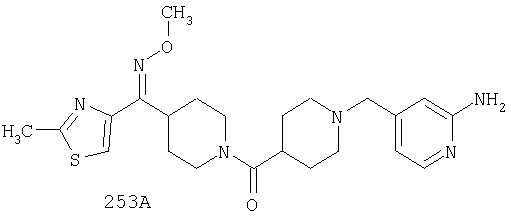
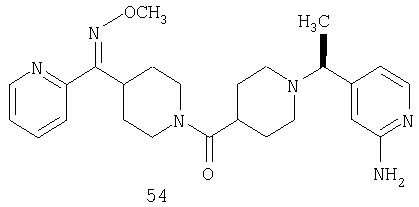
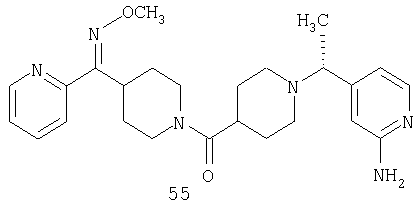
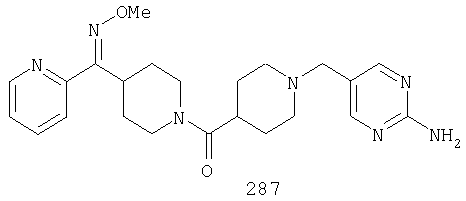
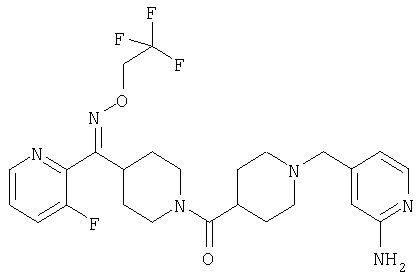
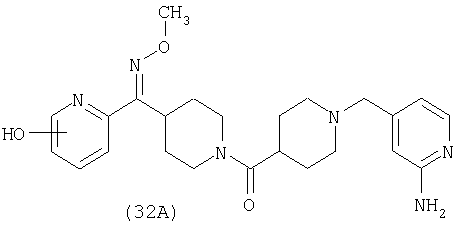
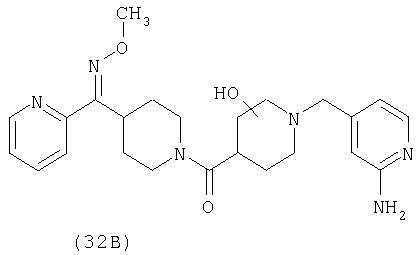
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| ПЕРЕРАБОТКА ПРИРОДНОГО ГАЗА С ПОЛУЧЕНИЕМ МЕТАНОЛА | 1998 |
|
RU2135454C1 |

