Предшествующий уровень техники
Область изобретения
Настоящее изобретение относится к антагонисту рецептора нейропептида нейрокинина-1 (NK1 или NK-1).
Описание предшествующего уровня техники
Тахикинины являются пептидными лигандами нейрокининовых рецепторов. Нейрокининовые рецепторы, такие как NK1, NK2 и NK3, участвуют во множестве биологических процессов. Они могут быть обнаружены в нервной системе и системе кровообращения млекопитающих, а также в периферических тканях. В связи с этим модуляция этих типов рецепторов была исследована для изучения возможности лечения или предупреждения заболеваний млекопитающих. Например, показано, что рецепторы NK1 влияют на проницаемость капилляров и выработку слизи. Типичные примеры антагонистов нейрокининовых рецепторов и их применения описаны в патенте США №5760018 (1998) (боль, воспаление, мигрень и рвота), патенте США №5620989 (1997) (боль, передача болевых ощущений и воспаление), WO 95/19344 (1995) (то же), WO 94/13639 (1994) (то же) и WO 94/10165 (1994) (то же). Другие типы антагонистов рецептора NK1 описаны в работе Wu et al., Tetrahedron 56, 3043-3051 (2000); Rombouts et al., Tetrahedron Letters 42, 7397-7399 (2001), и Rogiers et al., Tetrahedron 57, 8971-8981 (2001).
Было бы полезно получить антагонист NK1, который являлся бы активным, селективным и обладал полезными терапевтическими и фармакологическими характеристиками и хорошей стабильностью при метаболизме. Также было бы полезно получить антагонист NK1, который являлся бы эффективным для лечения различных физиологических нарушений, устранения симптомов и лечения заболеваний при сведении к минимуму побочных эффектов. В настоящем изобретении предпринята попытка обеспечить эти и другие преимущества, которые станут ясными из последующего описания.
Краткое содержание изобретения
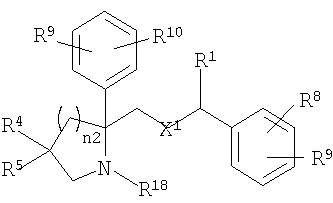
Настоящее изобретение относится к производным пирролидина и пиперидина, формулы (I)
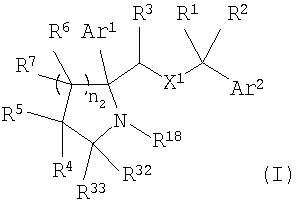
или к их фармацевтически приемлемой соли,
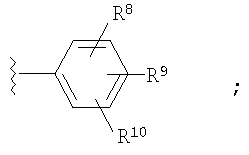
где Ar1 и Ar2 означают группу
X1 означает -О-;
R1 и R2 независимо выбраны из группы, включающей Н, C1-C6 алкил;
R3 выбран из Н;
каждый R6 выбран из Н;
каждый R7 выбран из Н;
n2 равно от 1 до 4;
R4 и R5 независимо выбраны из группы, включающей 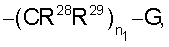
где n1 равно от 0 до 5 и
G означает Н, -ОН, -O-(C1-C6 алкил), -NR13R14, -NR12SO2R13, -NR12C(O)R14, -NR12(C(O)NR13R14), -C(O)NR13R14, C(O)OR13, -OC(O)R14, -OC(O)NR13R14, -C(=NOR14)(R13), 5-членный гетероциклоалкенил, содержащий N в качестве гетероатомов и необязательно содержащий от 1 до 4 заместителей, независимо выбранных из группы, включающей R30 и R31,
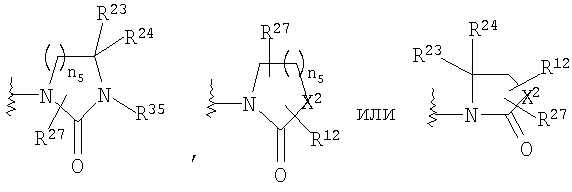
или
R4 и R5 совместно означают =NOR12, или
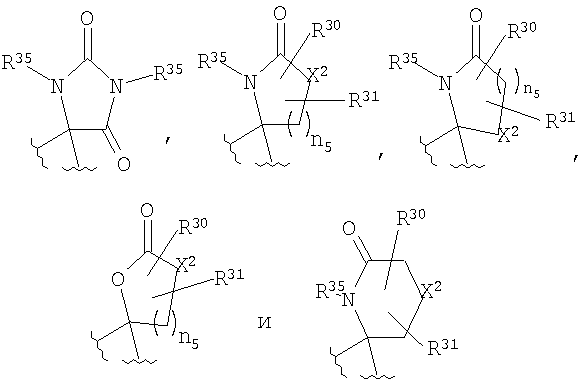
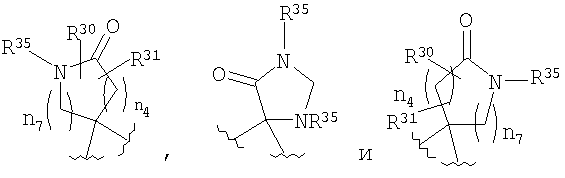
R4 и R5, совместно с атомом углерода, к которому они оба присоединены, образуют 4- - 6-членный гетероциклоалкильный или гетероциклоалкенильный цикл, содержащий от 1 до 3 групп, независимо выбранных из группы, включающей X2, при условии, что хотя бы один X2 означает -NR35-, -О-, или -SO2-, цикл необязательно содержит от 1 до 6 заместителей, независимо выбранных из группы, включающей R30 и R31;
при условии, что R4 и R5 оба не выбраны из группы, включающей Н, алкил и циклоалкил;
также при условии, что, когда один из R4 и R5 означает -ОН, то другой из R4 и R5 не означает алкил;
R8, R9 и R10 независимо выбраны из группы, включающей Н, -CF3;
R12 означает Н, C1-C6 алкил;
R13 и R14 независимо выбраны из группы, включающей Н, С1-С6 алкил, С3-С8 циклоалкил, -СН2CF3 и 5- или 6-членный гетероарил, содержащий атомы N и/или О в качестве гетероатомов, необязательно замещенный CN-группой, или
R13 и R14, совместно с атомом азота, к которому оба они присоединены, образуют 4- - 6-членный незамещенный насыщенный цикл, где один из атомов углерода цикла необязательно заменен на O в качестве гетероатома;
R15 означает C1-C6 алкил;
R18 означает Н;
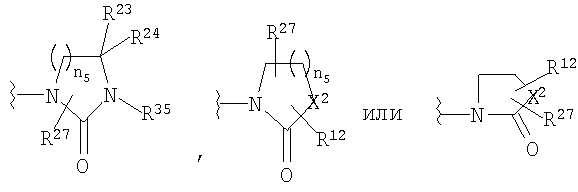
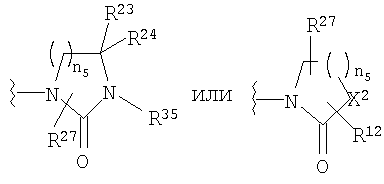
R23 и R24 означает Н;
R27 означает Н или C1-C6 алкил;
R28 и R29 независимо выбраны из группы, включающей Н и С1-С2 алкил;
R30 и R31 независимо выбраны из группы, включающей Н, C1-C6 алкил,
или R30 и R31, совместно с атомом углерода, к которому оба они присоединены, образуют =O;
R32 и R33 означают Н;
R35 означает Н, (С3-С8)циклоалкил(С1-С6)алкил, -Р(O)(ОН)2, аллил, гидрокси(С2-С6)алкил, (С1-С6)алкокси(С1-С6)алкил или
-(CH2)2-N(R12)-SO2-R15, где R12 и R15 имеют вышеуказанное значение;
X2 означает -NR35-, -О-, -СН2, -SO2-;
n5 равно от 1 до 3;
или к их диастереоизомеру, энантиомеру, стереоизомеру, региостереоизомеру, поворотному изомеру или таутомеру.
Соединения формулы (I) могут быть использованы для лечения различных заболеваний, устранения симптомов и физиологических нарушений, таких как рвота, депрессия, беспокойство и кашель.
Подробное описание изобретения
Приведенные ниже определения и термины используются в настоящем изобретении или иным образом известны специалисту в данной области техники. Если не указано иного, то приведенные ниже определения применимы ко всему описанию и формуле изобретения. Если не указано иного, то эти определения применимы независимо от того, используется ли термин по отдельности или в комбинации с другими терминами. Следовательно, определение "алкила" относится к "алкилу", а также к "алкильным" фрагментам "гидроксиалкила," "алкоксила" и т.п.
При использовании в настоящем изобретении термин "алкил" означает линейную или разветвленную углеводородную цепь, содержащую указанное количество атомов углерода. Если количество атомов углерода не указано, то углеродная цепь содержит от 1 до 24 атомов углерода, более предпочтительно от 1 до 12 атомов углерода, а наиболее предпочтительно от 1 до 6 атомов углерода.
При использовании в настоящем изобретении термин "циклоалкил" означает насыщенный, стабильный, неароматический карбоциклический цикл, содержащий от 3 до 8 атомов углерода. Циклоалкил может быть присоединен к любому эндоциклическому атому водорода с образованием стабильной структуры. Предпочтительные карбоциклические циклы содержат от 3 до 8 атомов углерода. Примеры циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и т.п.
При использовании в настоящем изобретении термин "4-6-членный гетероциклоалкил" означает насыщенное циклическое кольцо, содержащее от 3 до 5 атомов углерода и от 1 до 3 гетероатомов, независимо выбранных из группы, включающей -О-, -SO2- и -NR35-. Типичными гетероциклоалкильными группами являются пирролидинильная, имидазолидинильная, пиперидинильная, пиперазинильная, морфолинильная и т.п. Гетероциклоалкильное кольцо может быть присоединено к остальной части структуры через способный к замещению циклический атом углерода или способный к замещению циклический атом азота.
При использовании в настоящем изобретении термин "4-6-членный гетероциклоалкенил" означает циклическое кольцо, содержащее от 3 до 5 атомов углерода и от 1 до 3 гетероатомов, независимо выбранных из группы, включающей -О-, -SO2- и -NR35-, и содержащее в цикле хотя бы одну двойную связь, но не обладающее ароматическими характеристиками. Примерами таких колец являются
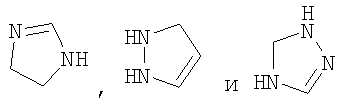
где кольцо может быть присоединено к остальной части структуры через способный к замещению циклический атом углерода или способный к замещению циклический атом азота (например, в R4, если G означает гетероциклоалкенил, он может быть соединен с группой 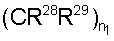 через способный к замещению циклический атом углерода или способный к замещению циклический атом азота).
через способный к замещению циклический атом углерода или способный к замещению циклический атом азота).
Если R4 и R5 образуют цикл с 1, 2 или 3 группами, независимо выбранными из группы, включающей X2, и 1 или 2 из X2 означают углерод, то переменный размер цикла можно обозначить с помощью n4 и n7, значения которых независимо выбраны из диапазона 0-3, при условии, что сумма n4 и n7 равна от 1 до 3. Типичная структура, в которой гетероатом означает -NR35-, X2 означает -CH2- и R30 и R31 совместно образуют карбонильную группу, представляется формулой
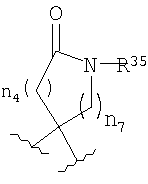 .
.
Если R4 и R5, совместно с атомом углерода, к которому они присоединены, образуют гетероциклоалкенильный цикл, примерами таких циклов являются
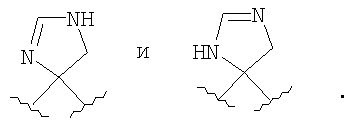
При использовании в настоящем изобретении термин "алкоксил" означает атом кислорода, связанный с углеродной цепью, такой как алкильная или алкенильная группа (например, -O-алкил или -O-алкенил). Типичные алкоксильные группы включают метоксильную, этоксильную и изопропоксильную группы.
При использовании в настоящем изобретении термин "гидроксиалкил" означает замещенную углеводородную цепь, предпочтительно алкильную группу, обладающую хотя бы одним гидроксильным заместителем (т.е. -ОН). Предпочтительные гидроксиалкильные группы включают гидроксиметильную, гидроксиэтильную и гидроксипропильную группы.
Если не известно, не утверждается или не показано противоположного, то участком присоединения составного заместителя (состоящего из элементов, обозначающих единый фрагмент) к описываемой структуре считается последний термин составного заместителя. Например, циклоалкильный заместитель присоединяется к описываемой структуре через последний "алкильный" фрагмент заместителя (например, структура - алкил-циклоалкил).
Если переменная, например, R8 появляется в структурной формуле более одного раза, то ее значение в каждом положении не зависит от ее значения в любом другом положении.
Соединения согласно изобретению могут также иметься в виде "пролекарственной формы", что означает, что соединения являются предшественниками лекарственных препаратов и после назначения пациенту выделяют лекарственный препарат in vivo посредством химических или физиологических процессов (например, пролекарственная форма, оказавшаяся в среде с физиологическим значением рН, или вследствие воздействия фермента превращается в необходимую форму лекарственного препарата). Обсуждение пролекарственных форм приведено в работах Т.Higuchi and V.Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of A.C.S. Symposium Series (1987), и Bioreversible Carriers in Drug Design, E.B.Roche, ed., American Pharmaceutical Association and Pergamon Press (1987), которые во всей своей полноте включены в настоящее изобретение путем ссылки.
За исключением указанного в приведенных примерах или иных указаний все числа, использованные в описании и формуле изобретения, означающие количества компонентов, условия протекания реакций и т.п., следует понимать, как во всех случаях содержащие слово "около".
В первую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, у которых по меньшей мере два из R8, R9 и R10 радикала Ar2 означают -CF3.
Во вторую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, у которых R8, R9 и R10 радикала Ar2 означают Н.
В третью группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, представляющиеся формулой
в которой X1 означает -О-, R8 и R9 означают -CF3, R9 и R10 означают Н, n2 равно 1 или 2 и R1, R4, R5 и R18 имеют вышеуказанное значение или же один из R4 и R5 означает Н и другой означает где n1 равно 0, 1 или 2 и G имеет вышеуказанное значение, или один из R4 и R5 означает Н и другой означает где n1 означает 0, а G выбран из группы, включающей -NR13R14, -NR12C(O)R14, -C(O)NR13R14, -OC(O)R14, -OC(O)NR13R14, -C(O)OR13, -NR12(C(O)NR13R14), -NR12SO2R13,
где R12-R14, R23, R24, R27, R35, X2 и n имеют вышеуказанное значение.
В четвертую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, представляющиеся формулой
в которой X1 означает -О-, R8 и R9 означают -CF3, R9 и R10 означают Н, n2 равно 1 или 2, R1, R4, R5 и R18 имеют вышеуказанное значение, R4 означает группу 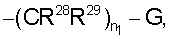 где n1 означает О, a G означает -ОН, -O(С1-С6)алкил, -NR13R14, -NR12C(O)R14, -NR12(C(O)NR13R14), -OC(O)R14, -OC(O)NR13R14, -NR12SO2R13,
где n1 означает О, a G означает -ОН, -O(С1-С6)алкил, -NR13R14, -NR12C(O)R14, -NR12(C(O)NR13R14), -OC(O)R14, -OC(O)NR13R14, -NR12SO2R13,
где R12-R14, R23, R24, R27, R35 и n5 имеют вышеуказанное значение, X2 означает -О-, -СН2- или -NR35-, где R35 имеет вышеуказанное значение, R5 означает группу где n1 означает О и G означает-C(O)OR13 или -C(O)NR13R14, или R12 и R27 независимо выбраны из группы, включающей Н и -СН3; n5 равно 1 или 2 или же R12 и R27 означают Н и n5 равно 1 или 2.
В пятую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, представляющиеся формулой
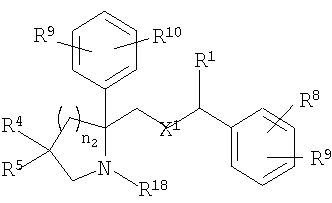
в которой X1 означает -О-, R8 и R9 означают -CF3, R9 и R10 означают Н, n2 равно 1 или 2, R1, R4 и R18 имеют вышеуказанное значение, а R4 и R5, совместно с атомом углерода, к которому оба они присоединены, образуют 4 - 6-членный гетероциклоалкильный или гетероциклоалкенильный цикл, содержащий от 1 до 3 групп, независимо выбранных из группы, включающей X2, при условии, что хотя бы один X2 означает -NR35-, -О- или -SO2-, при этом цикл необязательно содержит от 1 до 6 заместителей, независимо выбранных из группы, включающей R30 и R31, при этом 4 - 6-членный цикл более предпочтительно выбран из группы, включающей
где R35 означает Н, (С3-С8)циклоалкил(С1-С6)алкил или гидрокси(С1-С6)алкил; n5 равно 1, 2 или 3; X2 означает -NR35-, -CH2- или -О-, R30 означает Н или C1-C6 алкил и R31 означает Н или C1-C6 алкил, в частности 4 - 6-членный цикл выбран из группы, включающей
где R30 означает Н или C1-C6 алкил; R31 означает Н или C1-C6 алкил; каждый R35 независимо выбран из группы, включающей Н, (С3-С8)циклоалкил(С1-С6)алкил и гидрокси(С1-С6)алкил; n4 и n7 независимо равны 0-3, при условии, что сумма n4 и n7 равна 1-3.
В шестую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, представляющиеся формулой
в которой X1 означает -О-, R8 и R9 означают -CF3, R9 и R10 означают Н, n2 равно 1 или 2, R1, R4 и R18 имеют вышеуказанное значение, а R4 и R5, совместно с атомом углерода, к которому они присоединены, образуют 4 - 6-членный цикл, выбранный из группы, включающей
В седьмую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, выбранные из группы, включающей соединения Примеров 3, 9, 12а, 13, 14, 15, 20, 23, 29, 36, 40, 43b, 44b, 45, 50, 53, 56b, 57, 60а, 61, 62, 63, 72a, 73b, 74a, 75b, 76a, 82a, 82b, 90, 96, 105, 106b, 109, 110а, 111а, 112 и 113 и их стереоизомеры.
В восьмую группу предпочтительных соединений вышеприведенной формулы (I) входят соединения, выбранные из группы, включающей соединения формул
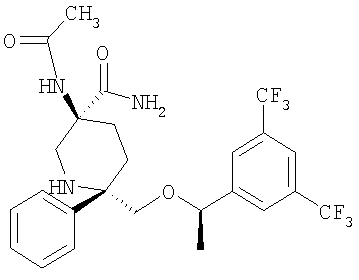
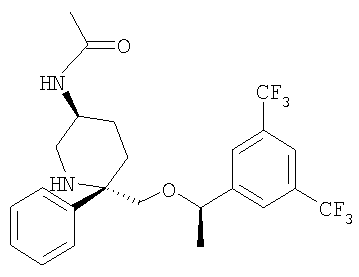
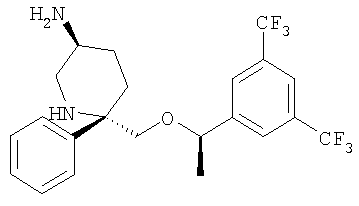
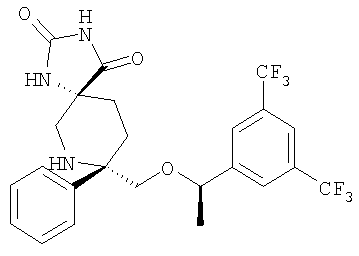
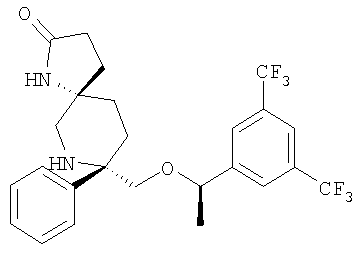
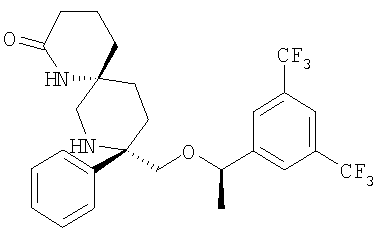
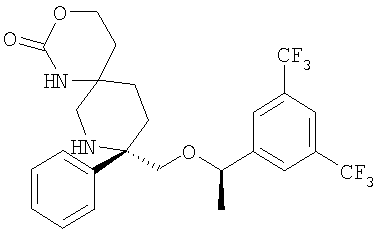
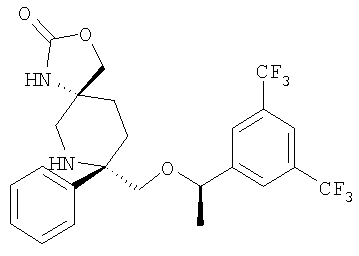 и
и
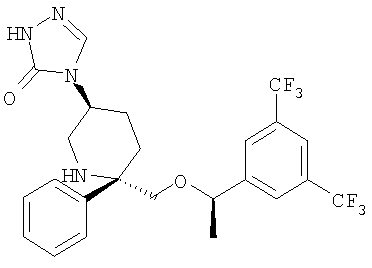
Соединения, обладающие формулой (I), могут являться эффективными антагонистами рецептора NK1 и оказывать влияние на его эндогенный агонист, Соединение Р, на клеточном рецепторе NK1 и поэтому могут быть полезны при лечении патологических состояний, вызванных или осложненных активностью указанного рецептора. Активность соединений, обладающих формулой (I), по отношению к NK1, NK2 и NK3 in vitro и in vivo можно определить с помощью различных методик, известных в данной области техники, таких как тестирование их способности ингибировать активность агониста NK1 - Соединения Р. Выраженное в процентах ингибирование активности агониста нейрокинина представляет собой разность между выраженным в процентах максимальным специфическим связыванием (МСС) и 100%. Выраженное в процентах МСС определяется по следующему уравнению, в котором РВМ означает "количество распадов в минуту":
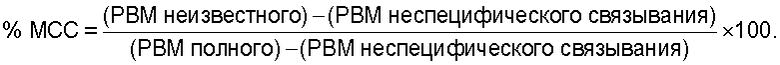
Концентрация, при которой соединение обеспечивает 50% ингибирование связывания, затем используется для определения константы ингибирования (Ki) с помощью уравнения Чанга-Прусова.
Активность in vivo можно измерить по ингибированию вызванной агонистом реакции лап песчанок при постукивании в соответствии с описанием, приведенным в работе Science, 281, 1640-1695 (1998), которая во всей своей полноте включена в настоящее изобретение для ссылки. Следует понимать, что соединения, обладающие формулой (I), могут ингибировать активность антагониста NK1 в различной степени. Например, некоторые соединения могут ингибировать активность антагониста NK1 сильнее, чем другие.
Соединения, соответствующие настоящему изобретению, обладают высоким сродством по отношению к рецептору NK1, о чем свидетельствуют результаты измерения значений Ki (в нМ). Активность соединений, соответствующих настоящему изобретению, определяют путем измерения их значений Ki. Чем меньше значение Ki, тем более активным является антагонистическое воздействие соединения на рецептор NK1. Соединения, соответствующие настоящему изобретению, обладают широким диапазоном активности. Средние значения Ki по отношению к NK1 для соединений, обладающих формулой (I), обычно составляют от 0,01 до около 1000 нМ, предпочтительно от около 0,01 до около 500 нМ, а наиболее предпочтительными являются значения от около 0,01 до около 100 нМ. Еще более предпочтительными являются соединения, обладающие средними значениями Ki для рецептора NK1, равными от 0,01 до около 10 нМ. Наиболее предпочтительные соединения обладают средними значениями Ki для NK1, равными от 0,01 до около 3 нМ. Отмеченные выше предпочтительные соединения обладают следующими значениями Ki: соединения Примеров 43b: 0,77 нМ; 72а: 0,66 нМ; 73b: 0,2 нМ; 109: 0,1 нМ; 110а: 0,41 нМ и 111а: 0,38 нМ.
Соединения, соответствующие настоящему изобретению, также оказывают весьма селективное антагонистическое воздействие на рецептор NK1 по сравнению с антагонистическим воздействием на рецепторы (i) NK2 и/или (ii) NK3. Если отношение селективности соединения больше примерно 100 для значения Ki для рецептора NK1 по сравнению со значением Ki для рецептора NK2 и/или независимо для значения Ki для рецептора NK3, то в настоящем изобретении это соединение определяется как эффективный антагонист рецептора NK1 по сравнению с рецепторами NK2 и/или NK3 соответственно.
Соединения, обладающие формулой (I), могут содержать хотя бы один асимметрический атом углерода. Подразумевается, что частью настоящего изобретения являются все изомеры, включая стереоизомеры, диастереоизомеры, энантиомеры, региоизомеры, таутомеры и поворотные изомеры. В объем настоящего изобретения также входят соли, сольваты, сложные эфиры и т.п., полученные из соединений, обладающих формулой (I), и их прекурсоры. Настоящее изобретение включает d- и I-изомеры как в чистом виде, так и в смеси, включая рацемические смеси. Изомеры можно получить с помощью обычных методик по реакции оптически чистых или оптически обогащенных исходных веществ или путем разделения изомеров соединения, обладающего формулой (I). Специалисты в данной области техники поймут, что для некоторых соединений, обладающих формулой (I), конкретные изомеры могут обладать большей фармакологической активностью, чем другие изомеры.
Имеется много способов применения соединений, обладающих формулой (I). Например, соединения, обладающие формулой (I), можно использовать в качестве антагонистов нейрокининовых рецепторов, в частности рецепторов NK1 у млекопитающих, таких как человек. Как таковые, они могут использоваться при лечении и предотвращении одного или большего количества болезненных состояний млекопитающего (человека и животного) (физиологических нарушений, симптомов и заболеваний), например, респираторных заболеваний (например, хронического заболевания легких, бронхита, пневмонии, астмы, аллергии, кашля и бронхоспазма), воспалительных заболеваний (например, артрита и псориаза), заболеваний кожи (например, атопического дерматита и контактного дерматита) офтальмологических заболеваний (например, ретинита, внутриглазной гипертензии и катаракты), патологических состояний центральной нервной системы, таких как депрессия (например, невротическая депрессия), тревоги (например, общей тревоги, социальной тревоги и панической тревоги), фобий (например, социальной фобии) и биполярных нарушений, зависимостей (например, алкогольной зависимости и злоупотребления психотропными веществами), эпилепсии, ноцицепции, психоза, шизофрении, болезни Альцгеймера, сопутствующего СПИДу слабоумия, болезни Тауна, заболеваний, связанных со стрессом (например, посттравматического стресса), навязчивых, компульсивных нарушений, нарушений питания (например, булимии, нервно-психической анорексии и потребления чрезмерного количества пищи), мании, предменструального синдрома, расстройств желудочно-кишечного тракта (например, синдрома раздраженной толстой кишки, болезни Крона, колита и рвоты), атеросклероза, фиброзных заболеваний (например, фиброза легких), ожирения, диабета типа II, сопутствующих боли нарушений (например, головной боли, такой как мигрень, нейропатической боли, послеоперационной боли и хронических болевых синдромов), заболеваний мочевого пузыря и мочеполовой системы (например, интерстициального цистита и недержания мочи) и тошноты. В частности, соединения, обладающие формулой (I), применимы для лечения болезненных состояний, связанных с проницаемостью капилляров и выработкой слизи. Поэтому соединения, соответствующие настоящему изобретению, особенно полезны для лечения и предупреждения астмы, рвоты, тошноты, депрессии, тревоги, кашля и связанных с болью заболеваний.
В еще одном варианте осуществления настоящего изобретения предоставлен способ устранения воздействия Соединения Р на участок рецептора нейрокинина-1 или блокирования одного или большего количества рецепторов нейрокинина-1 у млекопитающего, нуждающегося в таком лечении, включающий введение млекопитающему эффективного количества по меньшей мере одного соединения, обладающего формулой (I).
В другом варианте осуществления настоящего изобретения для лечения депрессии или тревоги эффективное количество одного или большего количества антагонистов рецептора NK1, соответствующих настоящему изобретению, можно скомбинировать с эффективным количеством одного или большего количества селективных ингибиторов повторного поглощения серотонина (ИСПС). ИСПС меняют синаптическую доступность серотонина посредством ингибирования пресинаптического повторного накопления выделенного нейронами серотонина. В патенте США №6162805, который во всей своей полноте включен в настоящее изобретение для ссылки, раскрыт способ лечения ожирения с помощью комбинированной терапии с использованием антагониста рецептора NK1 и ИСПС. Соединение (соединения), соответствующее настоящему изобретению, обладающее формулой (I), можно скомбинировать с одним или несколькими ИСПС в одной фармацевтической композиции или его можно назначать совместно, одновременно или последовательно с ИСПС.
Известны многочисленные химические соединения, меняющие синаптическую доступность серотонина посредством ингибирования пресинаптического повторного накопления выделенного нейронами серотонина. Типичные ИСПС включают (без наложения ограничений) следующие: флуоксетин, флувоксамин, пароксетин, серталин и их фармацевтически приемлемые соли. Другие соединения легко изучить и определить их способность к селективному ингибированию повторного накопления серотонина. Таким образом, настоящее изобретение относится к фармацевтической композиции, включающей по меньшей мере один антагонист рецептора NK1, обладающий формулой (I), и по меньшей мере один ИСПС, и способ лечения указанных выше болезненных состояний млекопитающего, при этом способ включает введение млекопитающему, нуждающемуся в таком лечении, эффективного количества фармацевтической композиции, включающей по меньшей мере один антагонист рецептора NK1, обладающий формулой (I), в комбинации с хотя бы одним ИСПС, таким как один из указанных выше.
В другом варианте настоящее изобретение относится к способу лечения рвоты, включающему введение пациенту, нуждающемуся в таком лечении, эффективного количества по меньшей мере одного антагониста рецептора NK1, обладающего формулой (I). Соединения, соответствующие настоящему изобретению, особенно полезны для лечения задержанного начала рвоты, такого как проявляющегося в период от 24 часов до нескольких дней после осуществления химиотерапии. См. работу Gonzales et al., Oncology Special Edition, Vol.5 (2002), p.53-58. Комбинации, по меньшей мере, одного антагониста рецептора NK1 и по меньшей мере одного другого противорвотного агента, такого как антагонист рецептора серотонина 5-НТ3, кортикостероида или замещенного бензамида можно использовать для лечения других форм рвоты, например, острой рвоты, вызванной химиотерапией, радиацией, движением и алкоголем (например, этанолом) и послеоперативной тошноты и рвоты. Примерами антагонистов рецептора серотонина 5-НТ3 являются палонсетрон, доласетрон, ондасетрон и гранисетрон или их фармацевтически приемлемые соли. Примером подходящего кортикостероида является дексаметасон. Примером замещенного бензамида является метоклопрамид.
Предпочтительные комбинации для лечения рвоты включают соединение формулы I и антагонист рецептора серотонина 5-НТ3; соединение формулы I и кортикостероид; соединение формулы I и замещенный бензамид; соединение формулы I, антагонист рецептора серотонина 5-НТ3 и кортикостероид; и соединение формулы I, замещенный бензамид и кортикостероид.
Когда антагонист рецептора NK1, соответствующий настоящему изобретению, комбинируется с ИСПС, антагонистом рецептора серотонина 5-НТ3, кортикостероидом или замещенным бензамидом для введения пациенту, нуждающемуся в таком лечении, два или большее количество активных компонентов можно вводить одновременно, совместно (один после другого через относительно короткий промежуток времени) или последовательно (сначала один, а затем другой через некоторый период времени).
Таким образом, соединения, соответствующие настоящему изобретению, можно использовать по отдельности или в комбинации с другими агентами. В дополнении к описанной выше комбинированной терапии с использованием антагониста рецептора NK1/ИСПС или антагониста рецептора серотонина 5-НТ3, соединения, обладающие формулой (I), можно комбинировать с другими активными агентами, такими как другие типы антагонистов рецептора NK1, простаноиды, антагонисты рецептора H1, агонисты α-адренергического рецептора, антагонисты эндотелинового рецептора, ингибиторы эндотелин-конвертирующего фермента, антагонисты ангиотензинового II рецептора, ингибиторы ангиотензинконвертирующего фермента, ингибиторы нейтральной металлоэндопептидазы, антагонисты ЕТA, ингибиторы ренина, агонисты рецептора серотонина 5-НТ2с, антагонисты ноцицептинового рецептора, ингибиторы rho-киназы, модуляторы калиевых каналов и/или ингибиторы белка 5, связанного с полилекарственной резистентностью (multidrug resistance protein 5). Предпочтительными терапевтическими агентами для комбинированной терапии совместно с соединениями, соответствующими настоящему изобретению, являются следующие: простаноиды, такие как простагландин E1; α-адренергические агонисты, такие как фонтоламинмезилат; агонисты допаминового рецептора, такие как апоморфин; антагонисты ангиотензина II, такие как лосартан, ирбесартан, валсартан и кандесартан; антагонисты ЕТA, такие как босентан и АВТ-627. Дозировочные диапазоны для этого другого препарата можно определить из литературы.
При изготовлении фармацевтических композиций из соединений, описанных в настоящем изобретении, инертные фармацевтически приемлемые носители могут быть твердыми или жидкими. К твердым формам препаратов относятся порошки, таблетки, диспергирующиеся гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут содержать от около 5 до около 95% активного компонента. Подходящие твердые носители известны в данной области техники и включают, например, карбонат магния, стеарат магния, тальк, сахар и лактозу. Таблетки, порошки, облатки и капсулы можно использовать в качестве твердых дозировочных форм, пригодных для перорального введения. Примеры фармацевтически приемлемых носителей и способов изготовления различных композиций приведены в работе A.Gennaro (ed.), Remington: The Science and Practice of Pharmacy, 20th Edition (2000), Lippincott Williams & Wilkins, Baltimore, MD.
Жидкие формы композиций включают растворы, суспензии и эмульсии. В качестве примера можно указать водные или водно-пропиленгликолевые растворы для парентеральных инъекций или прибавление подсластителей и замутнителей в растворы, суспензии и эмульсии для перорального введения. К жидким формам композиций также могут относиться растворы для внутриназального введения.
Аэрозольные композиции, пригодные для ингаляции, могут включать растворы и твердые вещества в порошкообразной форме, которые могут сочетаться с фармацевтически приемлемым носителем, таким как сжатый инертный газ, например азот.
В объем настоящего изобретения также включены твердые формы композиций, которые предназначены для превращения в жидкие формы композиций, предназначенных для перорального или парентерального введения, которое выполняется незадолго до использования. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения, соответствующие настоящему изобретению, также можно вводить чрескожно. Чрескожные композиции могут представлять собой кремы, лосьоны, аэрозоли и/или эмульсии и они могут быть включены в матрицу пластыря чрескожного воздействия или пластыря резервуарного типа, что обычно используется в данной области техники для такой цели.
Предпочтительно вводить соединение перорально.
Предпочтительно, чтобы фармацевтическая композиция содержалась в разовой дозировочной форме. В такой форме композиция разделяется на разовые дозы подходящей величины, содержащие соответствующие количества активных компонентов, т.е. количества, достаточные для достижения необходимой цели.
Количество активного соединения, содержащегося в разовой дозе лекарственного препарата, в соответствии с конкретным случаем применения обычно может меняться или регулироваться в диапазоне от около 0,01 до около 4000 мг, предпочтительно от около 0,02 до около 1000 мг, более предпочтительно от около 0,03 до около 500 мг, а наиболее предпочтительно от около 0,04 до около 250 мг.
Реальная использующаяся доза может меняться в зависимости от требований пациента и тяжести подвергающегося лечению патологического состояния. Определение надлежащего дозировочного режима для конкретного случая проводит специалист в данной области техники. Для удобства полную суточную дозу можно разделять и вводить порциями в течение дня в соответствии с необходимостью.
Количество и частота введения соединений формулы, соответствующих настоящему изобретению, и/или их фармацевтически приемлемых солей будет регулироваться в соответствии с решением лечащего врача, учитывающего такие факторы, как возраст, состояние и массу пациента, а также тяжесть заболевания, подвергающегося лечению. Типичный рекомендованный дозировочный режим для перорального введения может составлять от около 0,02 до около 2000 мг/сутки в виде двух - четырех разделенных доз.
Количество антагониста рецептора NK1 в комбинации с ИСПС или антагонистом рецептора серотонина 5-НТ3 (5-НТ3) в разовой дозе препарата можно менять или подбирать в диапазоне от около 10 до около 300 мг антагониста рецептора NK1 в комбинации с количеством ИСПС или 5-НТ3, составляющим от примерно 10 до примерно 100 мг. Кроме того, количество антагониста рецептора NK1 в комбинации с ИСПС или 5-НТ3 в разовой дозе препарата можно менять или подбирать в диапазоне от около 50 до около 300 мг антагониста рецептора NK1 в комбинации с количеством ИСПС или 5-НТ3, составляющим от около 10 до около 100 мг. В дополнение к этому, количество антагониста рецептора NK1 в комбинации с ИСПС или 5-НТ3 в разовой дозе препарата можно менять или подбирать в диапазоне от около 50 до около 300 мг антагониста рецептора NK1 в комбинации с количеством ИСПС или 5-НТ3, составляющим от около 20 до около 50 мг в зависимости от конкретного случая применения. Дозировочные диапазоны для кортикостероидов и замещенных бензамидов можно определить из литературы.
Альтернативно, для удобства пациента отдельные дозировочные формы соединений формулы I и других препаратов можно приготовить в одной упаковке в виде набора. Это особенно удобно, когда отдельные компоненты необходимо вводить в разных дозировочных формах (например, в виде таблетки и капсулы) или в разных режимах приема.
После улучшения состояния пациента при необходимости может вводиться поддерживающая доза соединения, композиции или комбинации, соответствующей настоящему изобретению. После этого доза или частота приема, или оба этих фактора в соответствии с симптомами могут быть уменьшены до уровня, при котором сохраняется улучшенное состояние. Если симптомы ослаблены до необходимого уровня, лечение следует прекратить. Однако после возврата каких-либо симптомов заболевания пациенту может потребоваться длительное периодическое лечение.
Соединения, соответствующие настоящему изобретению, могут находиться в сольватированных, а также в несольватированных формах. Обычно сольватированные формы с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., для задач настоящего изобретения эквивалентны несольватированным формам.
Соединения, соответствующие настоящему изобретению, могут образовывать фармацевтически приемлемые соли с органическими и неорганическими кислотами. Примерами кислот, пригодных для образования солей, являются хлористоводородная, серная, фосфорная, уксусная, лимонная, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, малеиновая, метансульфоновая кислоты и другие неорганические и карбоновые кислоты, хорошо известные специалистам в данной области техники. Соли получают путем взаимодействия свободных оснований с количеством необходимой кислоты, достаточным для образования соли обычным образом. Свободные основания могут быть регенерированы путем обработки соли разбавленным водным раствором подходящего основания, такого как разбавленный водный раствор гидроксида натрия, карбоната калия, аммиака или бикарбоната натрия. Свободные основания по некоторым физическим характеристикам, таким как растворимость в полярных растворителях, могут немного отличаться от соответствующих солей, но в остальном для задач настоящего изобретения соли эквивалентны своим соответствующим свободным основаниям.
Кислотные соединения, соответствующие настоящему изобретению (например, такие соединения, которые содержат карбоксильную группу), образуют фармацевтически приемлемые соли с неорганическими и органическими основаниями. Типичными примерами солей такого типа являются соли натрия, калия, кальция, алюминия, золота и серебра. В объем настоящего изобретения также включены соли, образованные с фармацевтически приемлемыми аминами, такими как аммиак, алкиламины, гидроксиалкиламины, N-метилглюкамин и т.п.
Ниже приведены общие и специальные методики получения соединений, обладающих формулой (I). При использовании в настоящем изобретении приведенные ниже аббревиатуры определяются следующим образом:
КДК означает круглодонную колбу;
Me означает метил;
Bu означает бутил;
Ас означает ацетил;
Et означает этил;
Ph означает фенил;
THF означает тетрагидрофуран;
ОАс означает ацетат;
(Вос)2O означает ди-трет-бутилдикарбонат;
(Boc) означает трет-бутоксикарбонил;
ТСХ означает тонкослойную хроматографию;
LAH означает алюмогидрид лития;
LDA означает диизопропиламид лития;
CDI означает 1,1-карбонилдиимидазол;
НОВТ означает гидроксибензотриазол;
DEC означает 1-[3-(диметиламино)пропил]-3-этилкарбодиимидгидрохлорид;
TFA означает трифторуксусную кислоту;
МТВЕ означает трет-бутилметиловый эфир;
DIEA или i-Pr2EtN означает диизопропилэтиламин;
DMF означает диметилформамид;
DMPU означает 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон;
TEMPO означает свободный радикал 2,2,6,6-тетраметил-1-пиперидинилоксил;
BuLi означает бутиллитий;
KHMDS означает бис(триметилолсилил)амид калия и
DBU означает 1,8-диазабицикло[5.4.0]ундец-7-ен.
Соединения, обладающие формулой (I), можно получить по методикам, известным специалистам в данной области техники. Типичные методики изложены ниже, хотя специалист в данной области техники должен понимать, что можно использовать другие методики и что для получения других соединений, входящих в объем настоящего изобретения, методики могут быть соответствующим образом модифицированы.
Общие методики синтеза
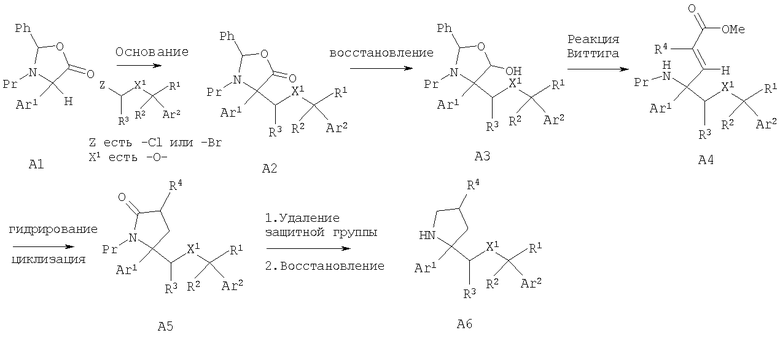
Соединения, обладающие формулой (I), обычно можно получить из соответствующего содержащего защитную группу производного оксалидинона А1, как это показано при следующих условиях, когда Ar1 и Ar2 являются такими, как определено в кратком содержании изобретения; X1 означает -О-; группы от R1 до R33, независимо одна от другой, определены так, как указано в кратком содержании изобретения, и n2 равно 1.
Стереоселективное алкилирование содержащего защитную группу оксалидинона А1 дает содержащий защитную группу оксалидинон А2. Частичное восстановление с помощью восстановительного реагента, такого как LAH, дает лактол A3. Реакция Виттига дает соответствующий олефин А4. Гидрирование олефина А4 и циклизация дает пактам А5. Если защитной группой (Pr) у атома азота является Cbz, то ее можно отщепить при условиях гидрирования. Проводимое при необходимости удаление защитной группы от атома азота лактама А5 с последующим восстановлением лактама с помощью восстановительного реагента, такого как LAH или LAH/AlCl3, предпочтительно LAH/AlCl3, дает замещенные пирролидины А6.
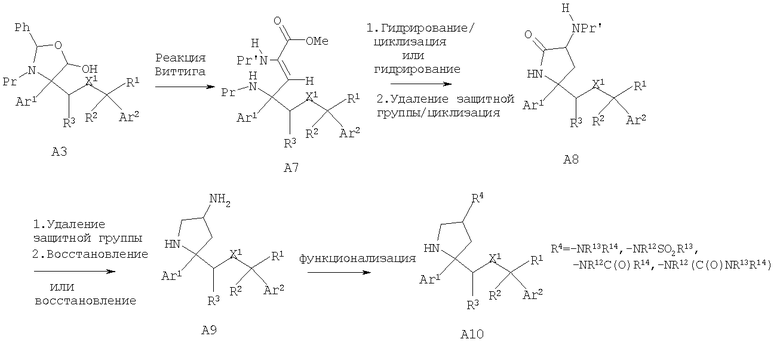
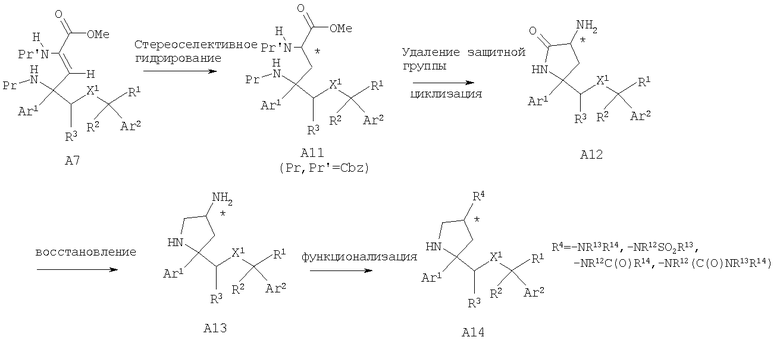
Соединения, обладающие формулой I, в которой n2 равно 1 и R4 означает -NR13R14, -NR12SO2R13, -NR12C(O)R14 или -NR12(C(O)NR13R14), также можно получить превращением лактола A3 в олефин А7 по реакции Виттига с использованием содержащего защитную группу у атома азота (N-Pr') реагента Виттига - сложного эфира глицина, где Pr' может означать защитную группу Boc или Cbz и Pr предпочтительно означает защитную группу Cbz. Гидрирование с использованием палладиевого катализатора и удаление защитной группы (если Pr означает группу Cbz) у олефина А7 с последующей самопроизвольной циклизацией дает лактам А8. Если Pr не означает Cbz или защитная группа легко отщепляется при стандартных условиях гидрирования, то после гидрирования олефина А7 проводят удаление защитной группы -NHPr и последующую циклизацию с получением лактама А8. Проводимое при необходимости удаление защитной группы для N-Pr' с последующим восстановлением лактама с помощью восстановительных реагентов, таких как LAH или LAH/AlCl3, предпочтительно LAH/AlCl3, дает аминопирролидины А9, в которые при стандартных условиях можно дополнительно ввести функциональные группы и получить N-замещенные пирролидины А10.
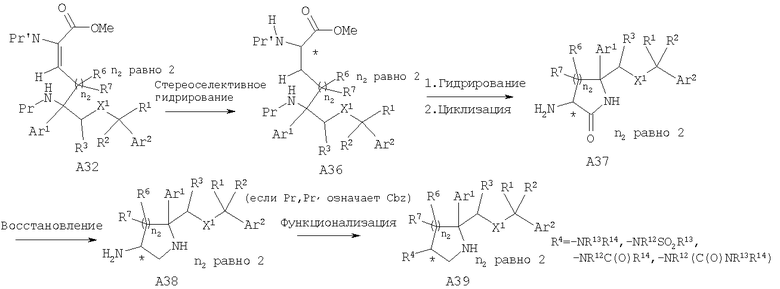
Специалисты в данной области техники должны понять, что стереоселективное гидрирование двойной связи олефина А7 также можно осуществить с помощью хирального катализатора гидрирования, такого как хиральный родиевый катализатор, что может дать хиральный сложный эфир А11. Удаление защитной группы (если Pr, Pr' означают группы Cbz) при стандартных условиях гидрирования с последующей самопроизвольной циклизацией даст хиральный аминолактам А12. Восстановление хирального аминолактама А12 с помощью восстановительных реагентов, таких как LAH или LAH/AlCl3, предпочтительно LAH/AlCl3, дает хиральные аминопирролидины А13, в которые при стандартных условиях можно дополнительно ввести функциональные группы и получить N-замещенные пирролидины А14.
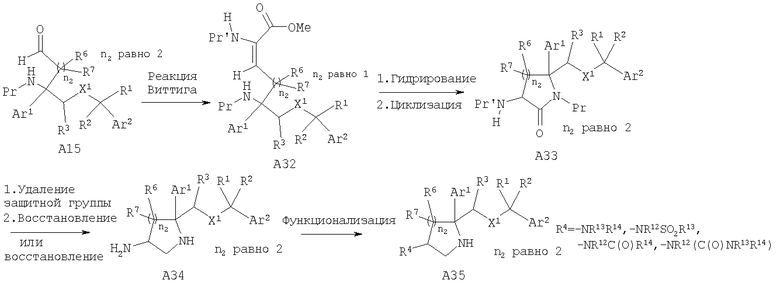
Соединения, обладающие формулой I, в которой n2 равно 2, 3 или 4, можно получить с помощью превращения лактола A3 в гомологизированные по атому углерода производные А15 (n равно 1, 2 или 3) с помощью стандартных химических реакций, известных специалистам в данной области техники. Особенно полезными для гомологизации этой углеродной цепи являются следующие: реакция Виттига с использованием метоксиметилтрифенилфосфонийбромида или аналогичного реагента, цианометилтрифенилфосфонийбромида, и реакция Хорнера-Эммонса и альдольная реакция. Гидрирование и циклизация в 6-, 7- и 8-членные лактамы А17 соответственно и удаление защитной группы и восстановление в 6-, 7- и 8-членные замещенные восстановленные лактамы А18, выполняются аналогично описанным выше методикам.
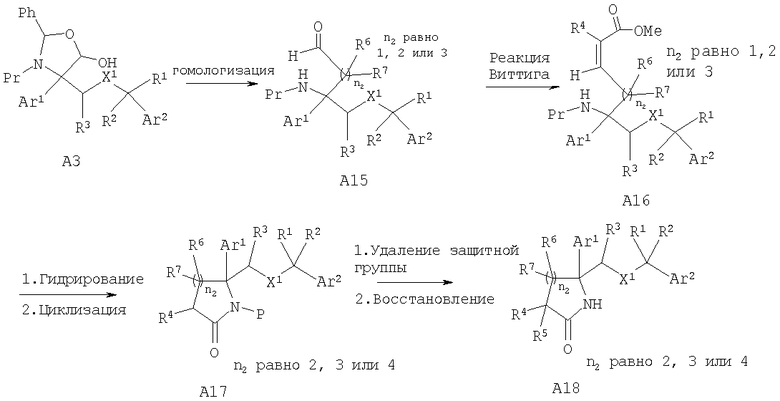
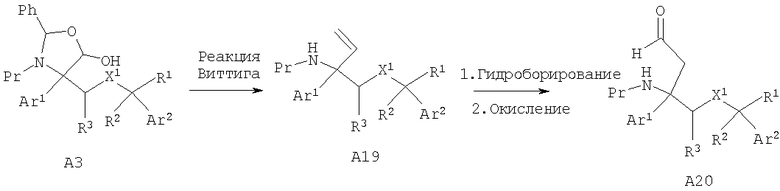
Другой способ получения альдегида А15, в котором R6, R7=Н, включает гомологизацию по Виттигу лактола A3 в производное этилена А19, которое после гидроборирования, предпочтительно с помощью 9-BBN, и последующего окисления дает альдегид А20.
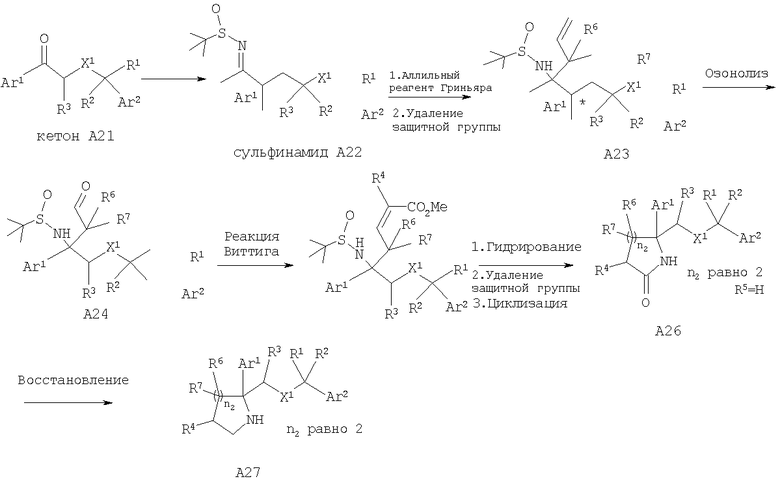
Альтернативно, соединения, обладающие формулой I, в которой n2 равно 2 и X1 означает -О-, можно получить с помощью превращения кетона А21 в сульфинамид с использованием подходящего сульфинамида (рацемического или хирального) и изопропоксида титана в соответствии с методикой, описанной в работе Cogan et al., Tetrahedron, 55, 8883 (1999). Сульфинамид А22 затем обрабатывают с помощью подходящего аллильного реагента Гриньяра, а затем подвергают озонолизу и получают альдегид А24. Специалисты в данной области техники должны понять, что добавление аллильного реагента Гриньяра дает А23, в котором R6, R7=Н, который далее можно модифицировать в аллильном положении и включить функциональные группы, указанные в определении R6 и R7, с помощью стандартных реакций, таких как алкилирование и гидроксилирование. Реакция Виттига с альдегидом А24 с последующим гидрированием, удалением защитной группы и циклизацией дает лактам А26. Стандартное восстановление лактама А26 дает замещенные пиперидины А27, в которых n2 равно 2.
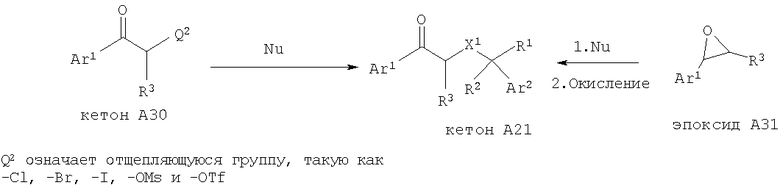
Если X1 является таким, как указано в кратком содержании изобретения, то кетон А21, в котором X1 означает эфирную, тиоэфирную или иминогруппу, можно получить с помощью нескольких различных методик с использованием имеющихся в продаже веществ. Кетон А28 можно подвергнуть ацилированию (Q1 означает -NH2, -ОН или -SH), восстановительному аминированию (Q1 означает -NH2), получить из него эфир (Q1 означает -ОН) с помощью стандартных методик алкилирования, получить из него тиоэфир (Q1 означает -SH) с помощью стандартных методик алкилирования или этерифицировать (Q1 означает -ОН или -SH). Альтернативно, соответствующий спирт А29 можно окислить в альдегид и обработать арильным или гетероарильным металлоорганическим реагентом с последующим окислением и получить кетон А21.
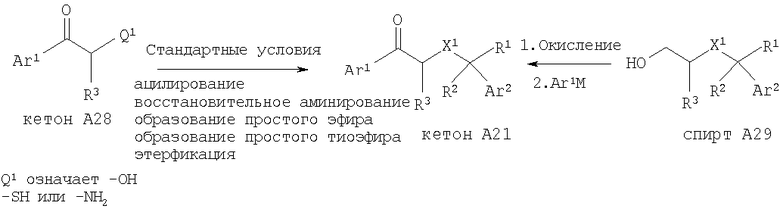
Другой способ получения кетона А21 включает нуклеофильное замещение отщепляющейся группы, такой как -Cl, -Br, -I, -OMs и -OTf, соседней к арильной или гетероарильной кетогруппе, см., например, WO 01/44200 (2001), которая во всей своей полноте включена в настоящее изобретение путем ссылки. В соответствии с этим подходящий замещенный стирол или гетероарилэпоксид можно раскрыть с помощью подходящего нуклеофильного реагента и получить искомый X1 по следующей схеме:
Соединения, обладающие формулой I, в которой n2 равно 2 и R4 или R5 означает -NR13R14, -NR12SO2R13, -NR12C(O)R14 или -NR12(C(O)NR13R14), также можно получить из альдегида А15 с использованием химических реакций, описанных выше для пирролидиновых соединений (n2=1).
Специалисты в данной области техники должны понять, что стереоселективное гидрирование двойной связи олефина А32 также можно осуществить с помощью хирального катализатора гидрирования, такого как хиральный родиевый катализатор, что может дать хиральный сложный эфир А36. Хиральный сложный эфир А36 можно превратить в хиральное функционализированное аминопиперидиновое соединение А39 с использованием химических реакций, описанных выше для хиральных функционализированных аминопирролидиновых соединений (n2=1).
Специалисты в данной области техники должны понять, что гомологизация альдегида А15 с последующими реакциями синтеза, описанными выше, приведет к циклическим восстановленным лактамам, в которых n2 равно 3 или 4.
Другой способ получения соединений, обладающих формулой I, в которой n2 равно 2 и X1 означает -О-, включает алкилирование аминопроизводного А40 с помощью подходящего замещенного аллилгалогенида, предпочтительно 2-замещенного аллилбромида, в бисолефин А41. Обработка бисолефина А41 катализатором Граббса или Шрока при использовании стандартных условий проведения метатезиса олефина дает ненасыщенное производное пиперидина А42. Удаление защитной группы от атома азота и гидрирование дает шестичленные циклические восстановленные лактамы или замещенные пиперидины А43. Если защитной группой (Pr) у атома азота является Cbz, то ее можно отщепить в условиях гидрирования. Специалисты в данной области техники должны понять, что алкилирование амина А40 с помощью подходящего замещенного алкилгалогенида с 4-5 атомами углерода в цепочке, содержащей концевой олефин, с последующими реакциями синтеза, описанными выше, приведет к замещенным циклическим восстановленным лактамам, в которых n2 равно 3 или 4.
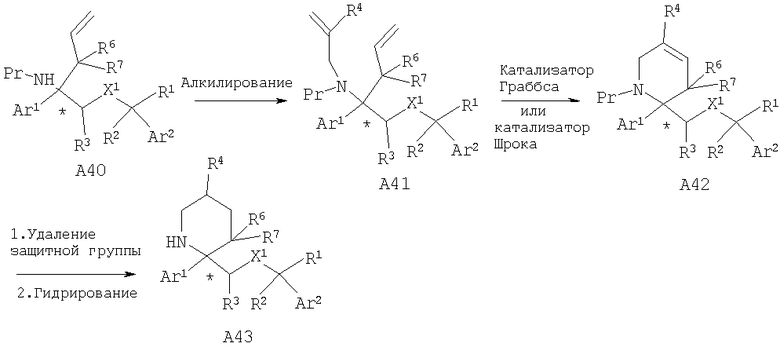
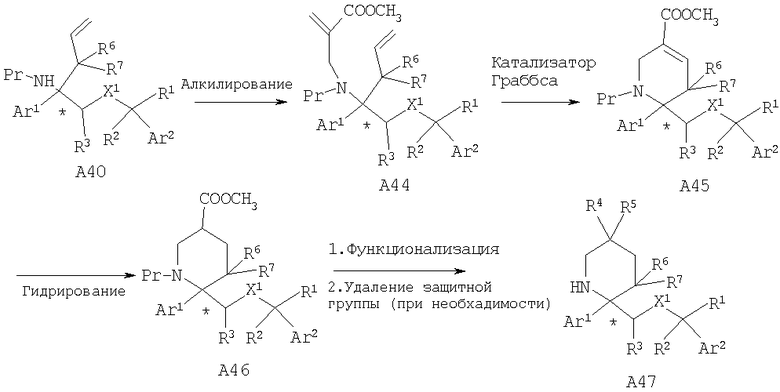
Если R4=СООСН3, то описанные выше химические реакции дают А46, в котором с использованием стандартных химических реакций сложноэфирную группу далее можно превратить в другие функциональные группы, такие как амидная (R4=CONR13R14) и спиртовая (R4=CH2ОН). Кроме того, пиперидин А46 можно дополнительно функционализировать с использованием таких химических реакций, как алкилирование, с последующим удалением защитной группы от атома азота, если это необходимо, и получить замещенные пиперидины А47.
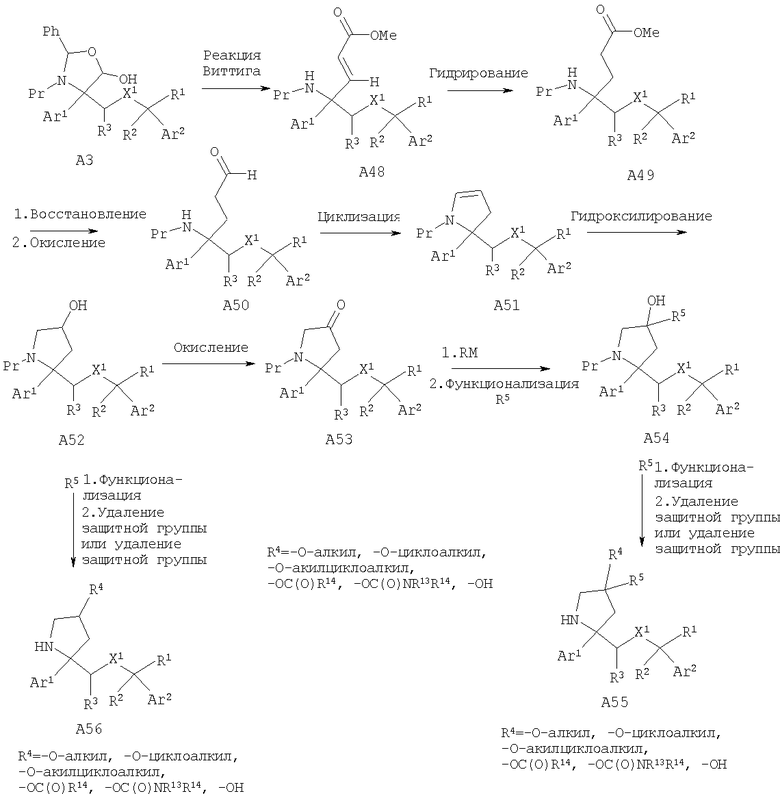
Другой способ получения соединений, обладающих формулой (I), в которой n2 равно 1, X1 означает -О- и R4 означает -ОН, -O-(C1-C6 алкил), -O-(С3-С8 циклоалкил), -O-(C1-C6 алкил)-(С3-С8 циклоалкил), -OC(O)R14 или -OCONR13R14, из лактола A3 включает реакцию Виттига с получением соответствующего сложного эфира олефина А48. Гидрирование сложного эфира олефина А48 с последующим восстановлением в спирт с помощью восстанавливающих реагентов - гидридов металлов, предпочтительно LiBH4, и последующее окисление, такое как окисление по Шверну, или с помощью хлорной извести дает альдегид А50. Циклизация альдегида А50 дает енамид А51, который после гидроксилирования, предпочтительно с использованием борана, дает спирт А52. Спирт А52 можно окислить при стандартных условиях окисления, таких как окисление по Шверну, и получить кетон А53. Обработка кетона подходящим металлоорганическим реагентом дает третичный спирт А54. Например, если необходимый заместитель R5 невозможно непосредственно ввести в металлоорганический реагент, может потребоваться дополнительная функционализация в положении R5. Гидроксильную группу спирта А54 можно дополнительно функционализировать с помощью стандартных химических реакций с последующим удалением защитной группы и получить дизамещенные пирролидины А55. Альтернативно, дополнительное модифицирование вторичного спирта А52 при стандартных условиях и удаление защитной группы у атома азота дает монозамещенные пирролидины А56.
Соединения, обладающие формулой I, в которой n2 равно 2, X1 означает -O- и R4 означает -ОН, -O-(C1-C6 алкил), -O-(С3-С8 циклоалкил), -O-(C1-C6 алкил)-(С3-С8 циклоалкил), -ОС(O)R14 или -OCONR13R14, можно получить из лактола A3. Реакция Виттига с последующим гидрированием и циклизацией в слабокислой среде, такой как п-толуолсульфоновая кислота, дает енамид А59. С использованием реакций синтеза, описанных выше для енамида А51, енамид А59 приведет к дизамещенным пиперидинам А63 и монозамещенным пиперидинам А64.
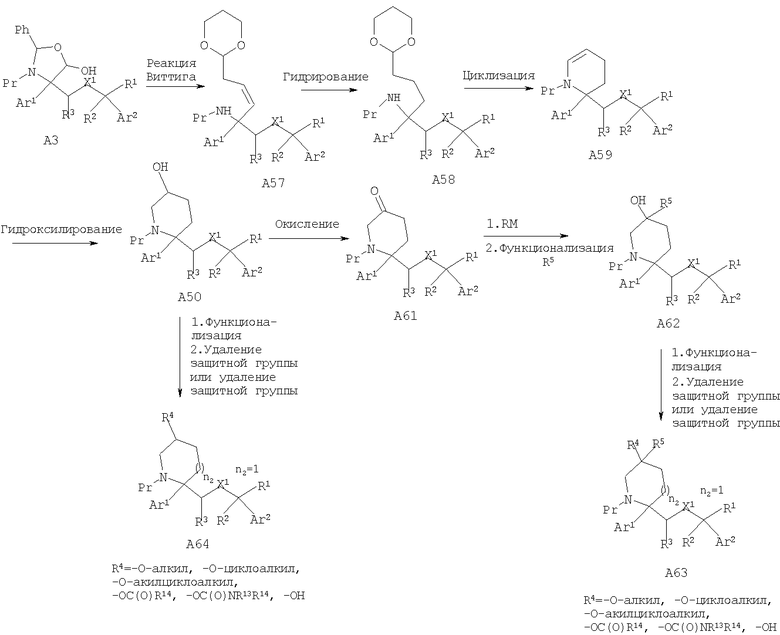
Специалисты в данной области техники должны понять, что гомологизация лактола A3 в альдегид А15, в котором n2 равно 2 или 3, с последующими реакциями синтеза, описанными выше, приведет к монозамещенным циклическим аминам А64 или дизамещенным циклическим аминам А63, в которых n2 равно 2 или 3.
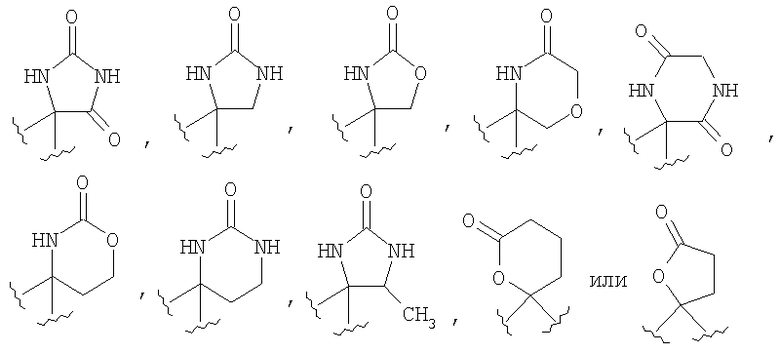
Соединения, обладающие формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О- и R4 и R5, совместно с атомом углерода, к которому они оба присоединены, образуют 5-членный цикл, можно получить из соответствующих кетонов. Кетон А65 превращается в соответствующий гидантоин А66 путем нагревания с KCN/карбонат аммония в смеси этанол/вода или путем использования альтернативных стандартных условий, известных специалистам в данной области техники. У амина удаляют защитную группу и получают гидантоин А67, который можно превратить в соответствующие аналоги мочевины А68 путем восстановления, предпочтительно с помощью LAH/AlCl3. Альтернативно, гидантоин А66 можно расщепить до аминокислоты А69 с использованием методики, описанной в работе Kubik, S.; Meissner, R.S.; Rebek, J. Tetrahedron Lett. 35, 6635 (1994). После стандартного введения защитной группы в аминокислоту А69 с образованием карбаматного производного (Pr') проводят активацию карбоновой кислоты. Одним таким способом активации является обработка фосгеном или эквивалентом фосгена, предпочтительно трифосгеном. Восстановление NBoc-UNCA A71 с помощью восстановительных реагентов, предпочтительно с помощью борогидрида лития, дает спирт А72, который можно превратить в пятичленное циклическое соединение, такое как карбамат А73, путем внутримолекулярной циклизации (если Pr' означает карбаматную защитную группу, такую как Вое) с использованием основания, предпочтительно NaH, с последующим удалением защитной группы. Альтернативно, спирт А72 можно окислить в NBoc-альдегид А74 при стандартных условиях окисления, таких как окисление по Шверну, и с помощью стандартных химических реакций, известных специалистам в данной области техники. NBoc-альдегид А74 можно превратить в циклические аналоги, такие как γ-лактам А75.
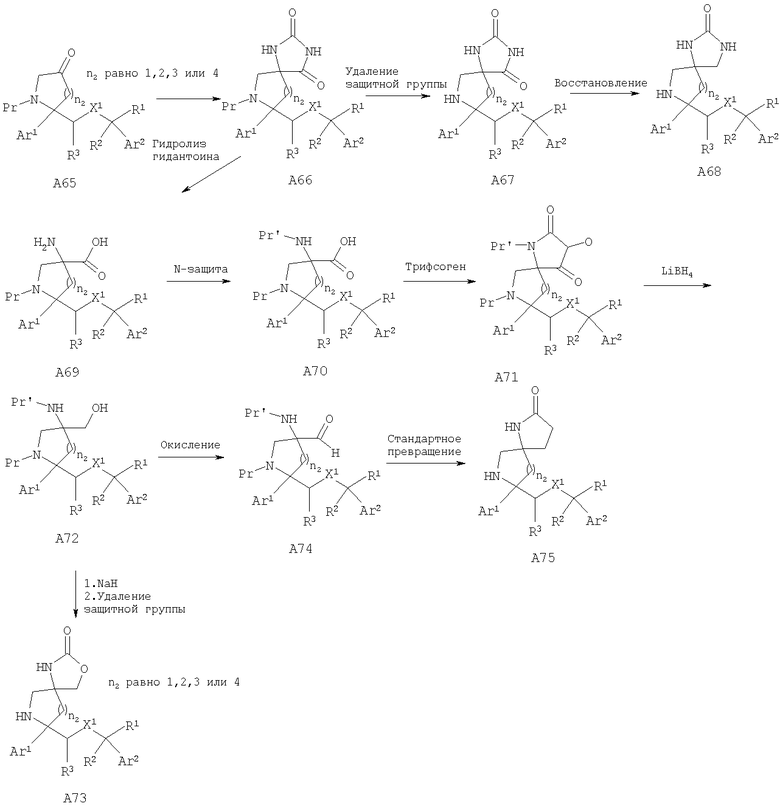
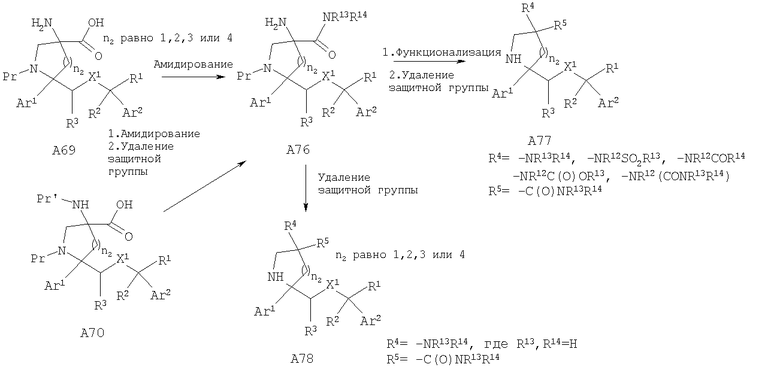
Соединения, обладающие формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О- и R4 означает -NR13R14, -NR12SO2R13, -NR12COR14, -NR12C(O)OR13 или -NR12(CONR13R14) и R5 означает -C(O)NR13R14, можно получить путем амидирования аминокислоты A69 с получением аминоамида А76 с последующей функционализацией аминогруппы и удалением защитной группы с получением дизамещенных аналогов А77. Альтернативно, NBoc-аминокислоту A70 можно ввести в реакцию с амином с последующим удалением защитной группы от группы N-Pr' и получить аминоамид А76. У аминоамида А76 также можно удалить защитную группу и получить аналоги А78, в которых R4 означает -NR13R14 и R13, R14=Н.
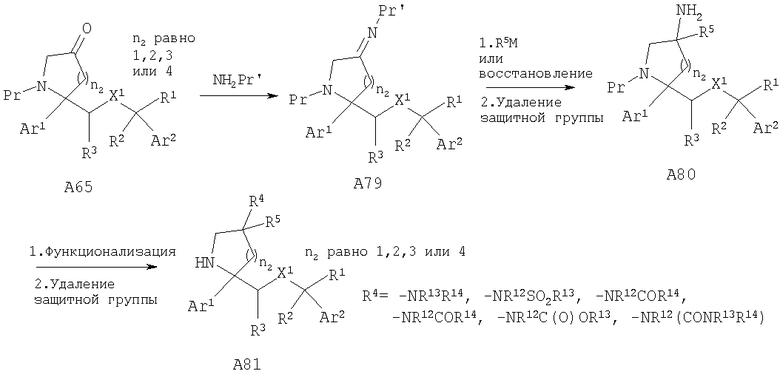
Другой способ получения соединений, обладающих формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О- и R4 означает -NR13R14, -NR12SO2R13, -NR12COR14, -NR12C(O)OR13 или -NR12(CONR13R14), включает обработку кетона А65 с содержащей защитную группу аминогруппой при подходящих условиях с получением имина А79. Нуклеофильное присоединение совместимых металлоорганических реагентов, таких как реагент Гриньяра, или восстановление (если R5=Н) имина А79 с последующим удалением защитной группы от атома азота (N-Pr') дает амин А80. Функционализация амина А80 при стандартных условиях и удаление защитной группы от атома азота дает замещенные пирролидины А81.
Соединения, обладающие формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О-, R5 означает Н и R4 означает гетероциклическую или гетероарильную группу, можно получить превращением кетона А65 в нитрил А87, альдегид А82 и карбоновую кислоту А85 через альдегид А82 с использованием стандартных условий окисления. Специалисты в данной области техники должны понять, что цианосоединение, альдегид и карбоновая кислота с помощью стандартных химических реакций могут дать соответствующую гетероциклическую или гетероарильную группу.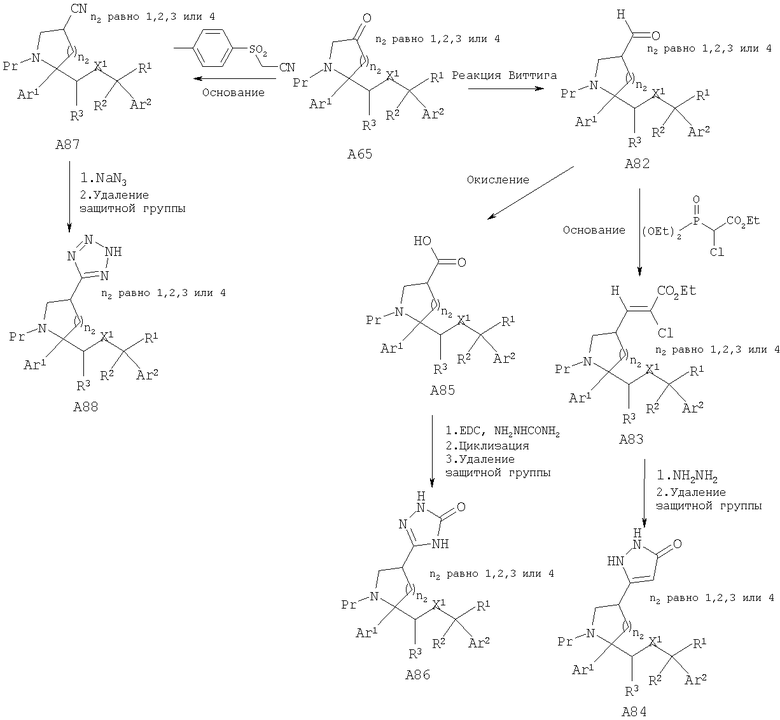
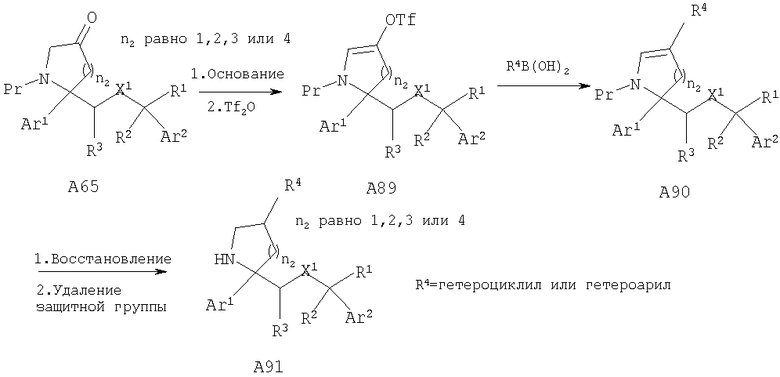
Другой способ получения соединений, обладающих формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О-, R5 означает Н и R4 означает гетероциклическую или гетероарильную группу, включает превращение кетона А65 в винилтрифторметансульфонат А89 с использованием основания, такого как LDA, и трифторметансульфонового ангидрида в качестве электрофильного реагента. Трифторметансульфонат А89 можно ввести в реакцию сочетания с подходящими металлоорганическими реагентами, предпочтительно с бороновой кислотой, и получить гетероциклическое или гетероарильное ненасыщенное соединение А90. Восстановление двойной связи с последующим удалением защитной группы амина (при необходимости) дает гетероциклические или гетероарильные замещенные циклические амины А91.
Соединения, обладающие формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О- и R4 означает -C(OR12)(R13)(R14), где R14 означает Н или -C(=NOR14)(R13), можно получить путем превращения альдегида А82 в спирт А92 путем присоединения металлоорганического реагента. Спирт А92 можно превратить в аналоги, такие как А93, или его можно окислить в кетон А94, из которого можно получить аналоги, такие как оксим А95, с использованием стандартных условий.
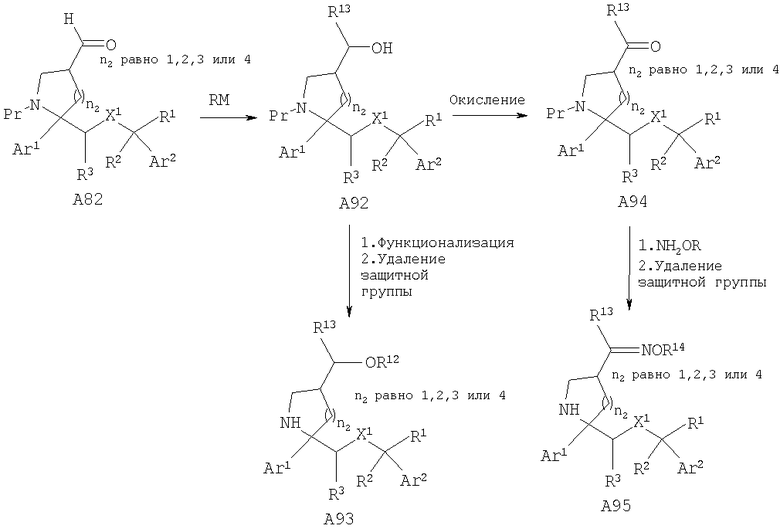
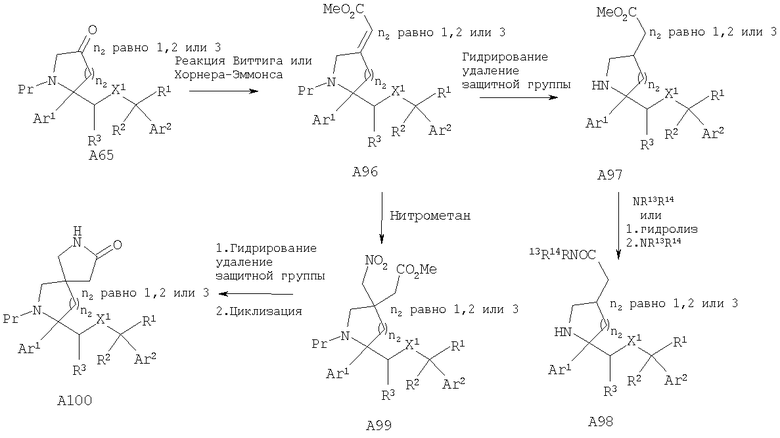
Соединения, обладающие формулой I, в которой n2 равно 1, 2, 3 или 4, X1 означает -О-, R5 означает Н и R4 означает -C(R28R29)CONR13R14, где R28, R29=Н или метил, можно получить путем превращения кетона А65 в ненасыщенный сложный эфир А96 с помощью реакции Виттига. Гидрирование двойной связи с последующим удалением защитной группы, если это необходимо, дает сложный эфир А97. Превращение сложного эфира в амиды А98 можно осуществить путем обработки аминами или превращения в кислоту и последующего введения в реакцию сочетания с аминами с помощью стандартных методик. Кроме того, ненасыщенный сложный эфир А96 также может привести к получению соединений, в которых R4 и R5 совместно с атомом углерода, к которому они присоединены, образуют пятичленный цикл, такой как лактам А100.
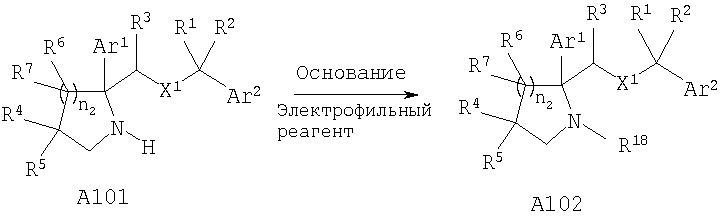
Специалисты в данной области техники должны понять, что функционализацию атома азота цикла, который образован из R4 и R5, если R4 и R5 совместно с атомом углерода, к которому они присоединены, образуют циклы, такие как гидантоин А67, мочевина А68 и лактам А100, можно осуществить на подходящей стадии синтеза путем депротонирования с помощью подходящего основания и реакции с необходимым электрофильным реагентом с получением заместителей, определенных для R35. Специалисты в данной области техники должны понять, что замещенный алкилгалогенид приведет к получению соответствующей замещенной C1-C6 алкильной группы и обработка тетрабензилпирофосфатом с последующим гидрированием приведет к получению R35= -Р(O)(ОН)2.
Функционализацию атома азота восстановленного лактама можно осуществить на подходящей стадии синтеза путем депротонирования с помощью подходящего основания и реакции с необходимым электрофильным реагентом с получением заместителей, определенных для R18. Специалисты в данной области техники должны понять, что замещенный алкилгалогенид приведет к получению соответствующей замещенной C1-C6 алкильной группы и обработка тетрабензилпирофосфатом с последующим гидрированием приведет к получению R18=-Р(O)(ОН)2.
Специалисты в данной области техники должны понять, что для образования различных функциональных групп могут потребоваться некоторые дополнительные стадии введения и удаления защитных групп. В соответствии с этим для обеспечения совместимости функциональных групп на разных стадиях синтеза порядок стадий синтеза может быть иным.
Конкретные способы получения
Примеры
Стадия 1:
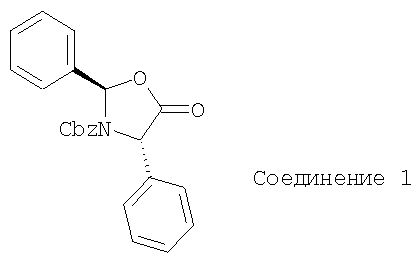
Соединение 1 получают с использованием методики синтеза, описанной М.J.O'Donnell, Z.Fang, X.Ма and J.С.Huffman, J. Am. Chem. Soc., 1997, 46, 617.
Стадия 2:
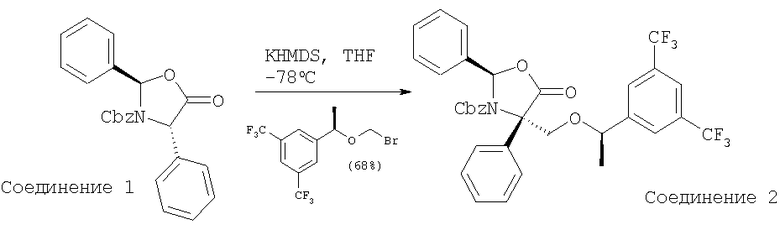
К продуваемому током азота раствору оксазолидинона - Соединения 1 (10,0 г, 0,027 моль, 1 экв.) в THF (500 мл) при -78°С прибавляют раствор KHMDS (0,5 М в толуоле, 64 мл, 0,032 моль, 1,18 экв.). После перемешивания при -78°С в течение 30 мин через канюлю в реакционную смесь прибавляют раствор бромметилового эфира (11,3 г, 0,032 моль, 1,18 экв.) в THF (100 мл) при -78°С. Раствор перемешивают при -78°С в течение 1 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl при -78°С. Реакционную смесь нагревают до комнатной температуры и прибавляют воду и EtOAc. Водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4), фильтруют и растворители, содержащиеся в фильтрате, удаляют в вакууме. Очистка с помощью хроматографии на колонке [гексан-толуол, 1:1 (об./об.)] дает Соединение 2 (11,7 г, 68%) в виде бесцветного масла.
МС с электрораспылением [М+1]+ 644,1.
Стадия 3:
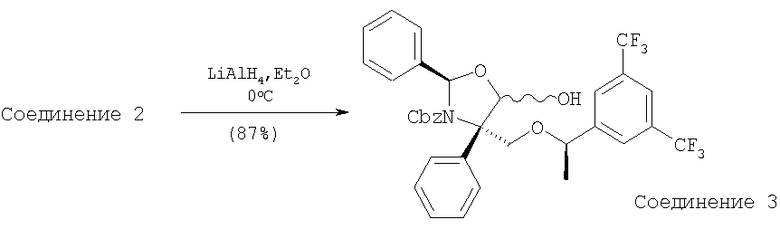
К раствору лактона - Соединения 2 (35,2 г, 0,055 моль, 1 экв.) в Et2O при 0°С прибавляют 1 М раствор LAH (17,8 мл, 0,018 моль, 0,32 экв.) в Et2O. Реакционную смесь перемешивают при 0°С в течение 30 мин, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. Прибавляют воду и полученные слои разделяют. Отделенный водный слой экстрагируют с помощью EtOAc (300 мл×2), объединенные органические слои сушат (MgSO4) и фильтруют. Растворители, содержащиеся в фильтрате, удаляют в вакууме и получают бесцветное масло. Масло растворяют в НОАс (240 мл) при комнатной температуре и прибавляют воду (60 мл). После перемешивания при комнатной температуре в течение 1 ч белое твердое вещество фильтруют, промывают водой и сушат в высоком вакууме. Перекристаллизация [гексан-толуол] дает Соединение 3 (23 г) в виде белого порошка. Все фильтраты объединяют, растворители удаляют в вакууме и получают желтое масло. Описанную выше процедуру [НОАс-Н2O, с последующей перекристаллизацией] повторяют и получают вторую порцию лактола - Соединения 3 (3 г). Растворители, содержащиеся в фильтрате, удаляют в вакууме и полученное масло подвергают хроматографированию на колонке [гексан-EtOAc, 6:1 (об./об.)] и получают третью порцию (4 г). Суммарный выход для Соединения 3 составляет 30 г, 87%.
МС с электрораспылением [М+1]+ 646,2.
Стадия 4:
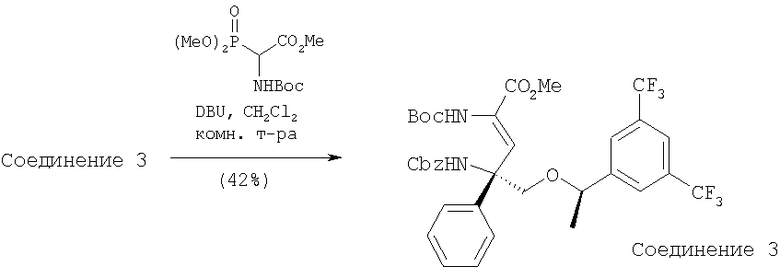
К раствору Соединения 3 (0,98 г, 1,52 ммоль, 1 экв.) и триметилового эфира NBoc-O-фосфоноглицина (1,26 г, 3,80 ммоль, 2,5 экв.) в CH2Cl2 (5 мл) при 23°С по каплям прибавляют DBU (0,57 мл, 3,80 ммоль, 2,5 экв.). Смесь перемешивают при 23°С в течение 4 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. Прибавляют Et2O и слои разделяют. Отделенный водный слой экстрагируют с помощью Et2O (250 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме с последующей очисткой с помощью хроматографии [гексан : эфир, 3:1 (об./об.)] дает Соединение 4 (587 мг, 52%) в виде белого вспененного вещества. МС с электрораспылением [М+1]+ 745,1.
Стадия 5:
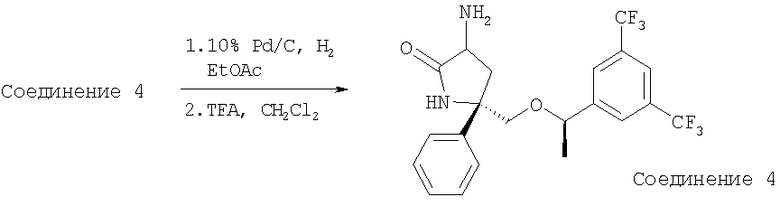
Раствор Соединения 4 (1,4 г, 1,88 ммоль, 1,0 экв.) в EtOAc (30 мл) продувают током N2. После прибавления палладия на угле (10%, 2 г) к колбе с реакционной смесью присоединяют баллон с Н2. Реакционную смесь перемешивают в течение 18 ч при 23°С в атмосфере Н2 и затем фильтруют и концентрируют. Остаток растворяют в безводном CH2Cl2 (45 мл), охлаждают до 0°С, затем обрабатывают с помощью раствора TFA (4,5 мл, 0,059 ммоль, 30,0 экв.). Реакционную смесь перемешивают при 0°С в течение 30 мин и затем при 23°С в течение еще 4 ч. Реакционную смесь разбавляют с помощью CH2Cl2 (300 мл), промывают насыщенным раствором NaHCO3 (100 мл). Органический слой сушат (Na2SO4), фильтруют, концентрируют и получают Соединение 5 (0,8 г, 95%).
Стадия 6:
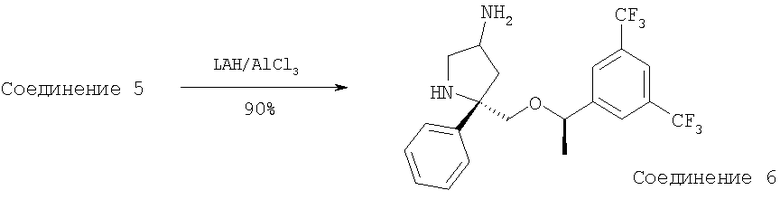
В высушенную на огне КДК объемом 25 мл помещают AlCl3 (0,089 г, 0,67 ммоль, 1,5 экв.). Колбу с реакционной смесью охлаждают до 0°С и осторожно прибавляют 1 М раствор LAH в Et2O (2 мл, 1,98 ммоль, 4,5 экв.). Реакционную смесь охлаждают до -78°С и медленно прибавляют раствор Соединения 5 (0,2 г, 0,44 ммоль, 1,0 экв.) в сухом THF (4 мл). Реакционную смесь перемешивают при -78°С в течение 2 ч, затем медленно нагревают до 23°С и перемешивают в течение 18 ч. Затем реакционную смесь охлаждают до 0°С и реакцию осторожно останавливают с помощью насыщенного водного раствора тартрата натрия-калия. Реакционную смесь растворяют в EtOAc (200 мл) и экстрагируют с помощью насыщенного водного раствора NaHCO3 (100 мл). Водный слой экстрагируют с помощью EtOAc (150 мл). Объединенные органические слои сушат над Na2SO4, фильтруют, концентрируют и получают Соединение 6 (180 мг, 95%). МС с электрораспылением [M+1]+ 433,1.
Стадия 7:
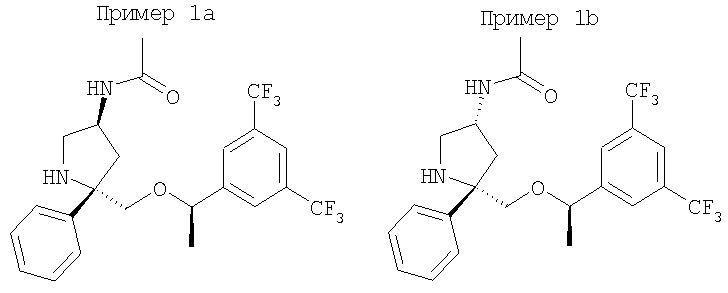
К раствору Соединения 6 (0,21 г, 0,486 ммоль, 1,0 экв.) в МеОН (3 мл) при 0°С прибавляют 2-трифторметил-N,N-диацетиланилин (0,131 г, 0,535 ммоль, 1,1 экв.). Смесь перемешивают при 0°С в течение 1 ч, затем нагревают до 23°С и перемешивают в течение 18 ч. Затем реакционную смесь концентрируют и очищают с помощью колонки Gilson, элюируя смесью вода/СН3CN, и получают смесь двух соединений (0,16 г). Очистка смеси с помощью ВЭЖХ с использованием колонки ChiralPak (98:2, гексан : IPA) дает менее полярный изомер - соединение Примера 1а (0,050 г, 22%), МС с электрораспылением [М+1]+ 475,1, и более полярный изомер - соединение Примера 1b (0,015 г, 7%), МС с электрораспылением [M+1]+ 475,1.
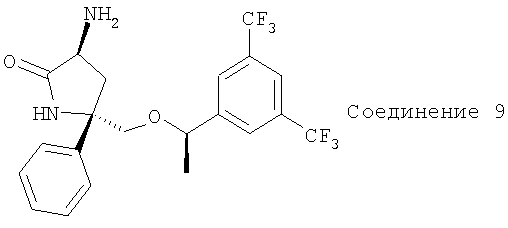
Получение соединения 9
Стадия 1:
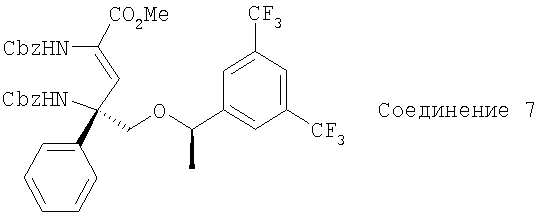
Соединение 7 получают с помощью методики, аналогичной использованной для получения Соединения 4, с использованием Соединения 3 и PO(OEt)2CH(NHCbz)CO2Me вместо PO(OMe)2CH(NHBoc)CO2Me. MC с электрораспылением [М+1]+ 745,1.
Стадия 2:
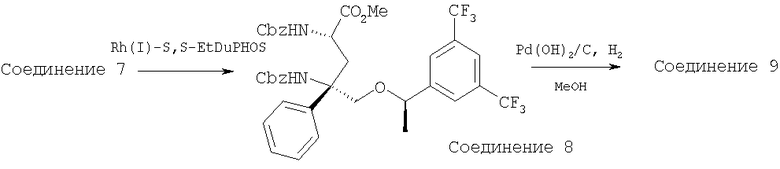
Соединение 7 (3,0 г, 4,03 ммоль, 1,0 экв.) растворяют в МеОН (30 мл) в реакционном сосуде Парра. Реакционный сосуд дегазируют с помощью N2 в течение 15 мин. К реакционной смеси в перчаточном ящике прибавляют (+)-1,2-бис-((2S,5S)-2,5-диэтилфосфолано)бензол(циклооктадиен)родий(I)трифторметансульфонат (0,12 г, 0,16 ммоль, 0,04 экв.) и встряхивают в атмосфере H2 при 60 фунт-сила/дюйм2 в течение 96 ч. Реакционную смесь переносят в КДК объемом 200 мл. К реакционной смеси прибавляют 20% Pd(OH)2/C (1 г) и ее перемешивают в атмосфере H2 при 23°С в течение 18 ч. За протеканием реакции следят с помощью ТСХ 9/1 EtOAc/СН3ОН. После завершения реакции реакционную смесь фильтруют через целит и концентрируют. Очистку выполняют с использованием слоя диоксида кремния с помощью 9:1 EtOAc/МеОН(NH3) и получают Соединение 9 (1,3 г, 72%). МС с электрораспылением [M+1]+ 447,1.
Получение соединения 10
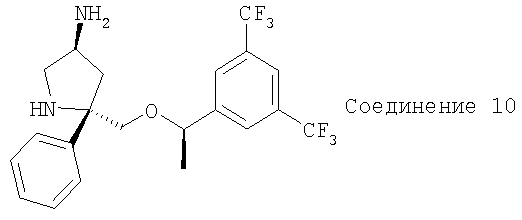
Соединение 10 получают с помощью методики, аналогичной использованной для получения Соединения 6, с использованием Соединения 9 вместо Соединения 5.
Пример 2
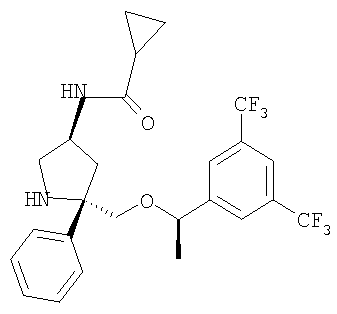
К раствору Соединения 10 (0,05 г, 0,116 ммоль, 1,0 экв.) в МеОН (2 мл) при -78°С прибавляют циклопропанкарбонилхлорид (12 мкл, 0,127 ммоль, 1,1 экв.). Смесь перемешивают при -78°С в течение 5 мин, затем нагревают до 23°С и перемешивают в течение 18 ч. Затем реакционную смесь концентрируют и растворяют в EtOAc (200 мл) и промывают с помощью насыщенного водного раствора NaHCO3 (1×100 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Очистка полученной смеси на колонке Biotage с использованием 5% МеОН/EtOAc дает соединение Примера 2 (0,04 г, 69%). МС с электрораспылением [М+1]+ 501.
Пример 3
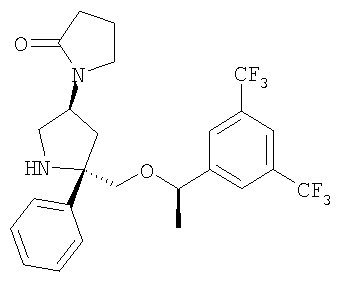
Стадия 1:
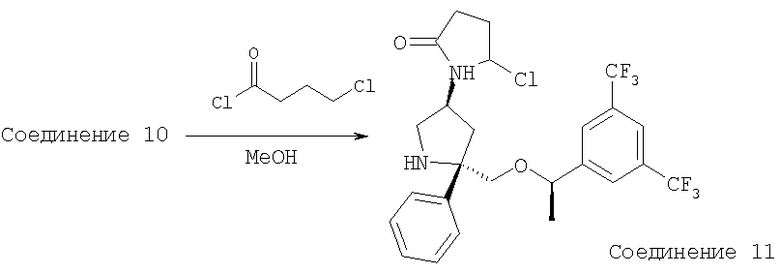
К раствору Соединения 10 (0,05 г, 0,116 ммоль, 1,0 экв.) в МеОН (2 мл) при -78°С прибавляют 4-хлорбутирилхлорид (14 мкл, 0,127 ммоль, 1,1 экв.). Смесь перемешивают при -78°С в течение 5 мин, затем нагревают до 23°С и перемешивают в течение 18 ч. Затем реакционную смесь концентрируют и растворяют в EtOAc (200 мл) и промывают с помощью насыщенного водного раствора NaHCO3 (1×100 мл). Органический слой сушат над Na2SO4, фильтруют, концентрируют и получают неочищенное Соединение 11, которое используют в следующей реакции без дополнительной очистки.
Стадия 2:
К раствору неочищенного Соединения 11 в сухом THF (2 мл) прибавляют NaH (60% дисперсия в минеральном масле, 0,014 г, 0,347 ммоль, 3 экв.) при 0°С и перемешивают в течение 5 мин, затем нагревают при 60°С в течение 2 ч. Реакционную смесь охлаждают до 0°С и реакцию осторожно останавливают водой (3 мл). Смесь выливают в EtOAc (100 мл) и промывают насыщенным водным раствором NaHCO3 (100 мл). Органический слой сушат над безводным Na2SO4, фильтруют и концентрируют. Очистка полученной смеси на колонке Biotage с использованием 5% МеОН/EtOAc дает соединение Примера 3 (0,20 г, 34%). МС с электрораспылением [М+1]+ 501,1.
Пример 4
Соединение Примера 4 (суммарный выход 53%) получают из Соединения 10 способом, аналогичным использованному для получения соединения Примера 3, но с использованием 5-хлорвалерилхлорида вместо 4-хлорбутирилхлорида. МС с электрораспылением [M+1]+ 515,1.
Пример 5
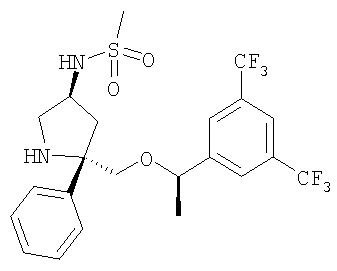
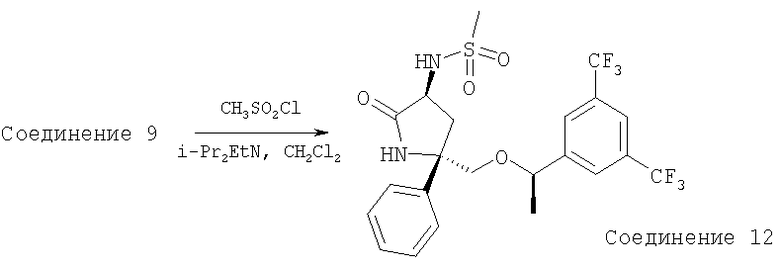
К раствору Соединения 9 (0,13 г, 0,29 ммоль, 1,0 экв.) в CH2Cl2 (3 мл) при 0°С прибавляют DIEA (0,11 мл, 0,61 ммоль, 2,1 экв.) и СН3SO2Cl (34 мкл, 0,435 ммоль, 1,5 экв.). Смесь перемешивают при 0°С в течение 30 мин, затем выливают в EtOAc (150 мл) и промывают насыщенным водным раствором NaHCO3 (100 мл). Органический слой сушат над безводным Na2SO4, фильтруют, концентрируют и получают неочищенное Соединение 12, которое используют в следующей реакции без дополнительной очистки.
Неочищенное Соединение 12 превращают в соединение Примера 5 (80 мг, выход 54%, за две стадии из Соединения 9) с помощью методики, аналогичной использованной для получения Соединения 6 из Соединения 5. МС с электрораспылением [М+1]+ 511,1.
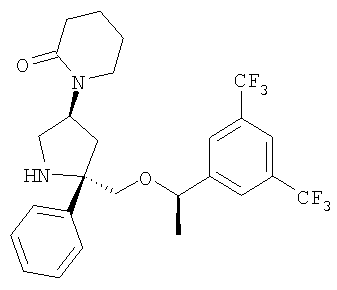
Пример 6а и Пример 6b
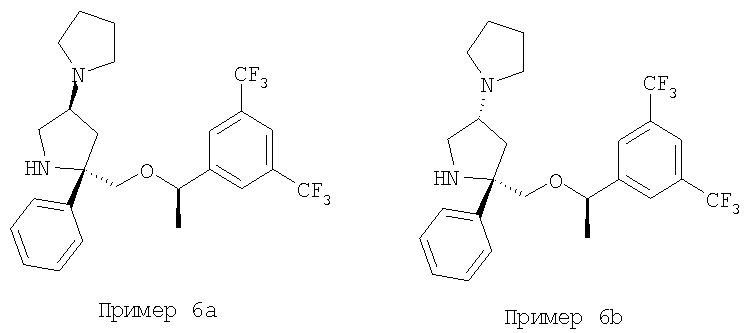
Стадия 1:
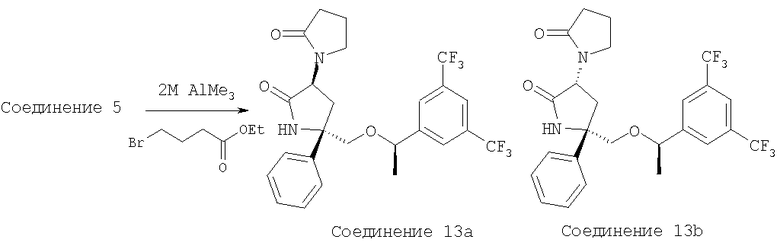
К раствору аминолактама - Соединения 5 (0,100 г, 0,224 ммоль, 1 экв.) в толуоле (7 мл) при 0°С, прибавляют 2 М раствор AlMe3 в толуоле (0,14 мл, 0,28 ммоль, 1,25 экв.). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 15 мин. Прибавляют этил-4-бромбутират и полученную смесь нагревают при 100°С в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры, выливают в EtOAc (20 мл) и последовательно промывают насыщенным водным раствором NaHCO3 (100 мл) и насыщенным водным раствором NaCl (100 мл). Органический слой сушат над безводным Na2SO4, фильтруют и концентрируют. Разделение с помощью ВЭЖХ на колонке ChiralCel OD с использованием (90/10) смеси гексан/IPA дает Соединение 13а (40 мг, 35%) и Соединение 13b (20 мг, 18%).
МС с электрораспылением [M+1]+ 515,1 для Соединения 13а.
МС с электрораспылением [М+1]+ 515,1 для Соединения 13b.
Соединения Примера 6а и Примера 6b получают с помощью методики, аналогичной использованной для получения Соединения 6, с использованием Соединений 13а и 13b вместо Соединения 5.
МС с электрораспылением [M+1]+ 487,11 для соединения Примера 6а.
МС с электрораспылением [M+1]+ 487,11 для соединения Примера 6b.
Пример 7
Соединение Примера 7 (суммарный выход 74%) получают из Соединения 10 способом, аналогичным использованному для получения соединения Примера 29 из соединения Примера 13. МС с электрораспылением [М+1]+ 476,1.
Пример 8
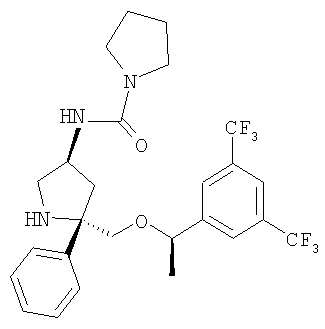
Соединение Примера 8 (суммарный выход 94%) получают из Соединения 10 способом, аналогичным использованному для получения соединения Примера 33 из соединения Примера 13. МС с электрораспылением [M+1]+ 430,1.
Пример 9
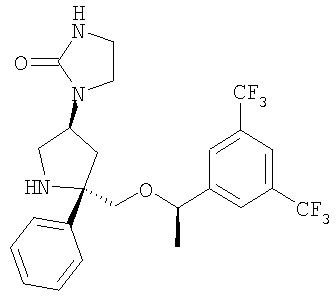
Соединение Примера 9 (суммарный выход 50%) получают из Соединения 10 способом, аналогичным использованному для получения соединения Примера 36 из соединения Примера 13. МС с электрораспылением [М+1]+ 502,1.
Пример 10
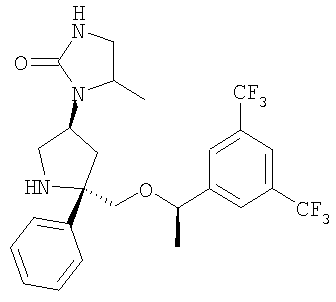
К раствору Соединения 10 (0,15 г, 0,3 ммоль, 1 экв.) в CH2Cl2 (2 мл) прибавляют метиллевулинат (0,041 мл, 0,33 ммоль, 1,1 экв.), а затем триацетоксиборогидрид натрия (0,127 г, 0,6 ммоль, 2 экв.) и реакционную смесь перемешивают при 23°С в течение 72 ч. Реакцию останавливают насыщенным водным раствором NaHCO3 (100 мл) и экстрагируют с помощью EtOAc (200 мл). Органический слой отделяют, сушат (Na2SO4), фильтруют и концентрируют. Смесь очищают с помощью хроматографии на колонке Gilson (1:9, вода : СН3CN) и получают искомое соединение (0,070 г, 47%). МС с электрораспылением [M+1]+ 515,1.
Пример 11
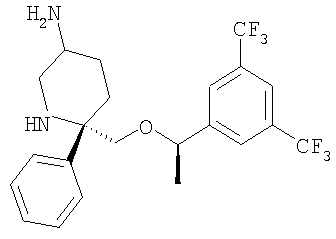
Стадия 1:
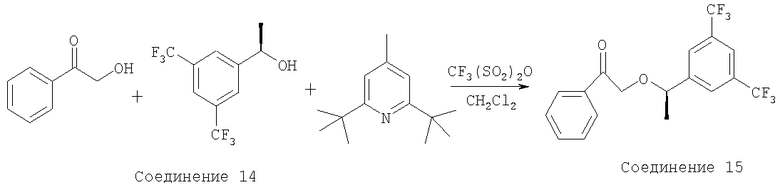
Методики получения Соединения 14 и Соединения 15 приведены в WO 01/44200.
Стадия 2:
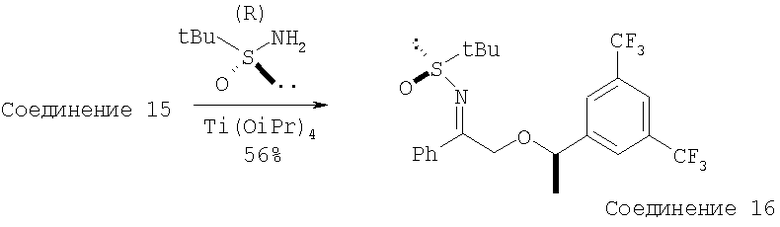
Колбу, содержащую кетон - Соединение 15 (1,05 г, 2,8 ммоль, 1 экв.) и (R)-трет-бутилсульфинамид (0,4 г, 3,3 ммоль, 1,8 экв.), подсоединяют к вакуумной линии на 5 мин. Затем колбу заполняют с помощью N2. К реакционной смеси с помощью шприца по каплям прибавляют Ti(OiPr)4 (1 мл). Реакционную смесь перемешивают при 23°С в течение 36 ч. Затем реакционную смесь выливают в рассол (10 мл) и EtOAc (20 мл) и энергично перемешивают в течение 10 мин. Полученную суспензию пропускают через слой целита 545. Слой целита несколько раз промывают с помощью EtOAc. Объединенные органические растворы сушат и концентрируют при пониженном давлении. Флэш-хроматография на колонке дает Соединение 16 (0,75 г, 56%).
Стадия 3:
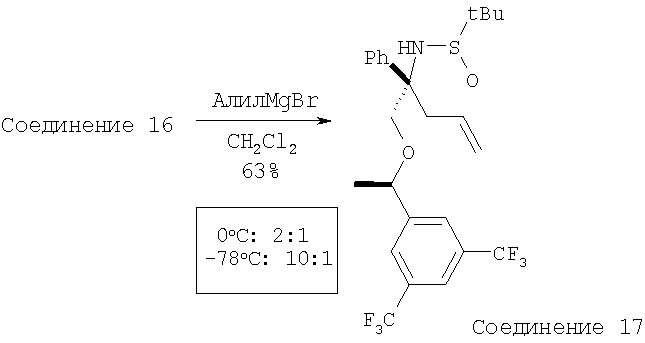
К раствору сульфинимина - Соединения 16 (2,44 г, 5,1 ммоль, 1 экв.) в CH2Cl2 при -78°С с помощью шприца по каплям прибавляют аллилмагнийбромид (6,1 мл, 6,1 ммоль, 1,2 экв., 1 М в Et2O). После перемешивания в течение 3 ч при -78°С реакцию останавливают с помощью насыщенного водного раствора NH4Cl и реакционной смеси дают нагреться до 23°С. Слои разделяют и водный слой экстрагируют с помощью EtOAc. Объединенные органические слои сушат и концентрируют. Флэш-хроматография на колонке дает Соединение 17 (1,672 г, 63%).
Стадия 4:
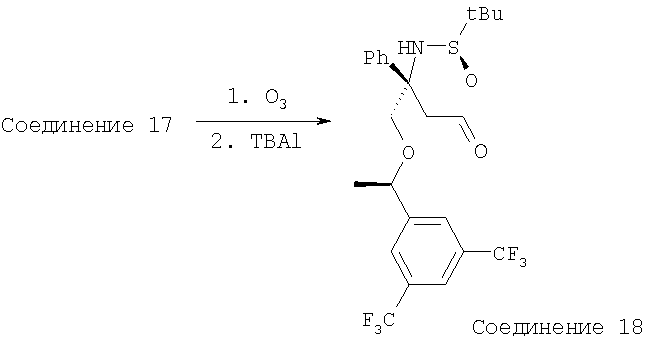
В КДК объемом 15 мл помещают Соединение 17 (245 мг, 0,47 ммоль, 1,0 экв.) и CH2Cl2 (2 мл). Этот бледно-оранжевый раствор охлаждают до -78°С и затем через него пропускают О3 со скоростью 1,0 мл/мин. После того как раствор станет бледно-синим, реакционную смесь перемешивают при -78°С в течение 10 мин. Затем через него пропускают N2 для удаления О3. Для разрушения комплекса прибавляют тетрабутиламмониййодид (177 мг, 0,47 ммоль, 1,0 экв.). Затем реакцию останавливают с помощью насыщенного раствора Na2S2O3 и экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат, фильтруют, концентрируют, затем повторно растворяют в Et2O и фильтруют. Остаток на фильтре растворяют в воде и экстрагируют с помощью Et2O. Объединенные слои, содержащие Et2O, сушат, фильтруют, концентрируют и получают Соединение 18 (243,5 мг, 99%). МС с электрораспылением [M+1]+ 524,1.
Стадия 5:
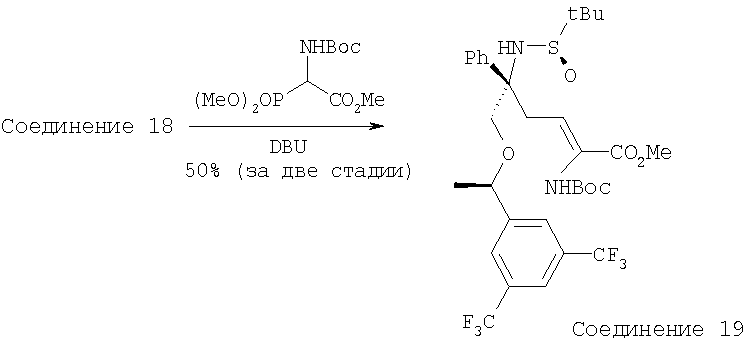
К раствору Соединения 18 (1,2 г, 2,29 ммоль, 1,0 экв.) и Вос-фосфоната (818 мг, 2,75 ммоль, 1,2 экв.) в DMF (20 мл) прибавляют Cs2СО3 (2,24 г, 6,87 ммоль, 3,0 экв.). После перемешивания при комнатной температуре в течение 3 ч смесь разбавляют с помощью Et2O и промывают водой (100 мл 2×) и рассолом. Объединенные водные слои дополнительно экстрагируют с помощью Et2O. Объединенные органические слои сушат, фильтруют, концентрируют и получают неочищенное коричневатое масло, которое очищают на колонке и получают Соединение 19 (830 мг, 55%). МС с электрораспылением [M+1]+ 695,2.
Стадия 6:
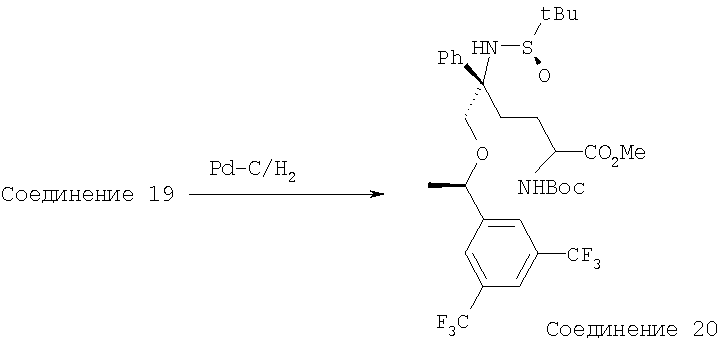
Раствор Соединения 19 (830 мг, 1,19 ммоль, 1,0 экв.) в EtOH (20 мл) продувают током N2. После прибавления палладия на угле (10%, 1,27 г, 1,19 ммоль, 1,0 экв.) к колбе с реакционной смесью присоединяют баллон с Н2. Реакционную смесь перемешивают в течение почти 24 ч, пока ТСХ не покажет завершение реакции. Смесь фильтруют, концентрируют и получают Соединение 20 в виде белого твердого вещества (790 мг, 95%). МС с электрораспылением [М+1]+ 697,2.
Стадия 7:
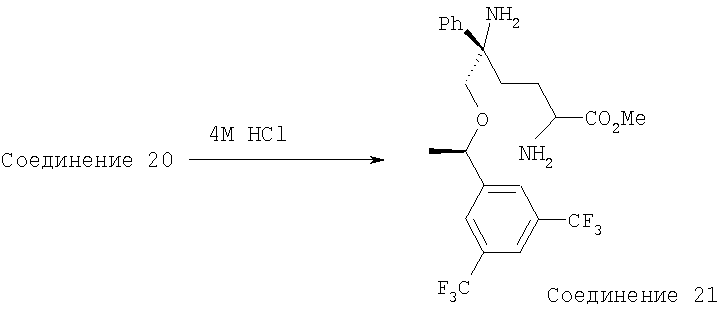
Раствор Соединения 20 (400 мг, 0,57 ммоль, 1,0 экв.) в безводном МеОН (4 мл) охлаждают до 0°С, затем обрабатывают с помощью 4 М раствора HCl в 1,4-диоксане (16 мл). Через 30 мин выдерживания при 0°С ее перемешивают при комнатной температуре в течение еще 3 ч. Растворитель выпаривают в вакууме и получают Соединение 21 в виде бледно-коричневого твердого вещества. МС с электрораспылением [М+1]+ 493,1.
Стадия 8:
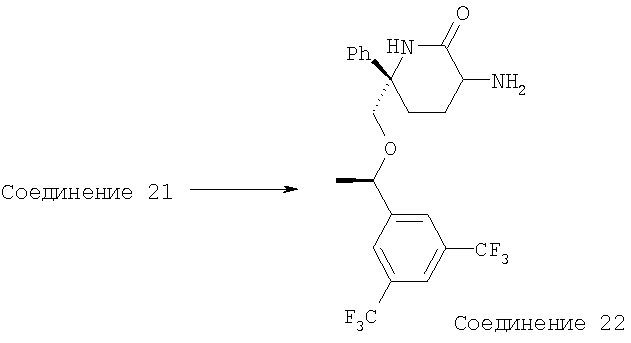
К раствору Соединения 21 в МеОН (50 мл) прибавляют K2СО3 (4,5 г). Смесь перемешивают в течение 30 мин, затем фильтруют, концентрируют и получают Соединение 22 (199 мг, 76%). МС с электрораспылением [М+1]+ 461,1.
Стадия 9:
В высушенную на огне КДК объемом 500 мл помещают AlCl3 (37,4 мг, 0,28 ммоль, 1,5 экв.). Колбу с реакционной смесью охлаждают до 0°С и с помощью шприца прибавляют безводный THF (1 мл). После перемешивания в течение 5 мин через канюлю прибавляют 1 М раствор LAH в Et2O (0,84 мл, 0,84 ммоль, 4,5 экв.). Баню со льдом удаляют и раствор перемешивают при комнатной температуре в течение 30 мин. Затем реакционную смесь охлаждают до -78°С и медленно прибавляют раствор Соединения 22 (50 мг, 0,187 ммоль, 1,0 экв.) в сухом THF (1 мл). Реакционную смесь перемешивают при -78°С и дают нагреться до комнатной температуры в течение ночи. После того как ТСХ (MeOH/CH2Cl2=1/9) показывает, что реакция завершена, реакционную смесь охлаждают до 0°С, разбавляют с помощью EtOAc и реакцию осторожно останавливают с помощью насыщенного водного раствора тартрата натрия-калия. Ее перемешивают при комнатной температуре в течение 30 мин до разделения на два слоя. Водный слой дополнительно экстрагируют с помощью EtOAc. Объединенные органические слои сушат над Na2SO4, фильтруют, концентрируют и получают соединение Примера 11 (34 мг, 41%). МС с электрораспылением [М+1]+ 447,1.
Пример 12а и Пример 12b
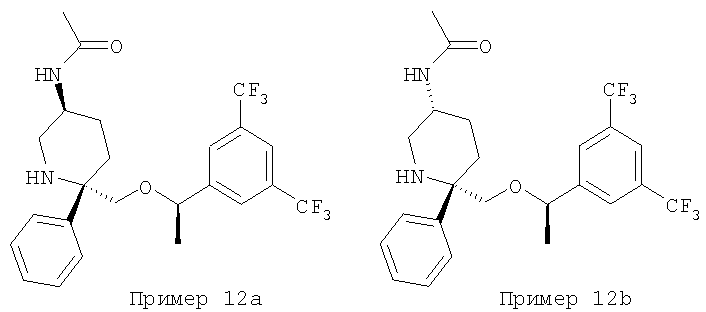
Стадия 1:
К раствору соединения Примера 11 (30 мг, 0,067 ммоль, 1,0 экв.) в CH2Cl2 (10 мл) при 0°С прибавляют DIEA (17,5 мкл, 0,10 ммоль, 1,5 экв.) и АС2O (6,3 мкл, 0,067 ммоль, 1,0 экв.). Смесь перемешивают при 0°С в течение 30 мин. Реакцию останавливают с помощью насыщенного водного раствора NaHCO3 (4 мл) и экстрагируют с помощью CH2Cl2. Объединенные органические слои сушат, фильтруют, концентрируют и получают неочищенный продукт (39 мг). Очистка смеси с помощью ВЭЖХ с использованием колонки ChiralPak AD (2:98, IPA : гексан) дает более полярный изомер - соединение Примера 12а, МС с электрораспылением [М+1]+ 489,1, менее полярный изомер - соединение Примера 12b, MC с электрораспылением [М+1]+ 489,1.
Пример 13
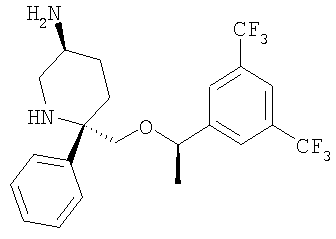
Стадия 1:
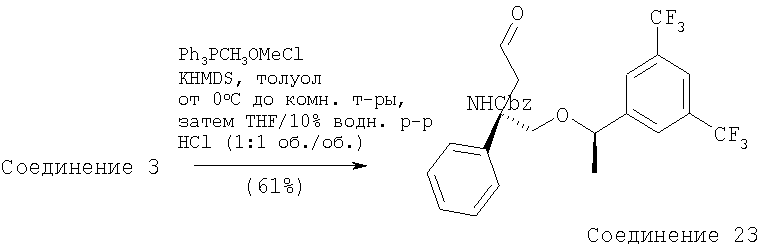
К суспензии (метоксиметил)трифенилфсофонийхлорида (21,3 г, 0,062 ммоль, 2,95 экв.) в толуоле (300 мл) при 0°С в атмосфере N2 прибавляют раствор бис(триметилсилил)амида калия (125 мл, 0,062 ммоль, 2,95 экв.). После перемешивания при 0°С в течение 1 ч прибавляют раствор Соединения 3 (13,4 г, 0,021 ммоль, 1 экв.) в толуоле (100 мл). Смесь перемешивают при нагревании от 0 до 23°С в течение 1 ч и затем реакцию останавливают с помощью насыщенного раствора NH4Cl. Прибавляют Et2O и слои разделяют. Отделенный водный слой экстрагируют с помощью Et2O (400 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и получают неочищенный эфир енола в виде желтого масла.
Неочищенный эфир енола растворяют в THF (100 мл) при 23°С и прибавляют водный раствор HCl (100 мл, 10% в воде). Смесь перемешивают в течение ночи и реакцию останавливают с помощью насыщенного раствора KHCO3. Прибавляют Et2O и слои разделяют. Отделенный водный слой экстрагируют с помощью Et2O (300 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме с последующей очисткой с помощью хроматографии [гексан : EtOAc, 4:1 (об./об.)] дает Соединение 23 (6,97 г, 61%) в виде желтого масла.
Стадия 2:
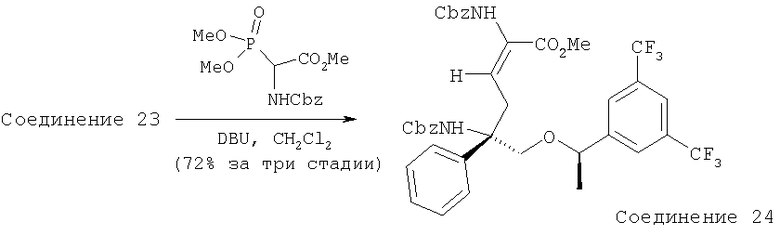
Соединение 24 получают из Соединения 23 с помощью методики, аналогичной использованной для получения Соединения 4 из Соединения 3 и с использованием PO(OEt)2CH(NHCbz)CO2Me вместо PO(OMe)2CH(NHBoc)CO2Me.
Стадия 3:
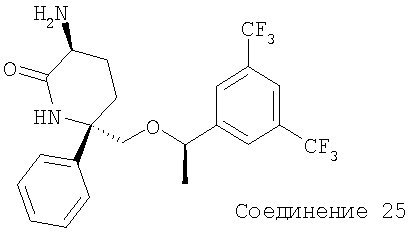
Соединение 25 получают с помощью методики, аналогичной использованной для получения Соединения 9 с использованием Соединения 24 вместо Соединения 7. МС с электрораспылением [М+1]+ 461,1.
Стадия 4:
Соединение Примера 13 (6,84 г, 73%) получают с помощью методики, аналогичной использованной для получения Соединения 6 с использованием Соединения 25 вместо Соединения 5. МС с электрораспылением [М+1]+ 447,1.
Пример 14
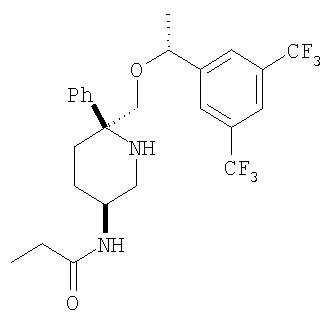
К раствору соединения Примера 13 (275 мг, 0,60 ммоль, 1,0 экв.) в безводном СН2Cl2 (10 мл) при -78°С прибавляют пропионилхлорид (52 мкл, 0,60 ммоль, 1,0 экв.). Реакция завершается за 30 мин. Реакцию останавливают с помощью 7 н. аммиака в МеОН (0,5 мл), затем реакционную смесь загружают непосредственно в колонку с диоксидом кремния, очищают и получают соединение Примера 14 (241,3 мг, 80%). МС с электрораспылением [М+1]+ 503,1.
Пример 15
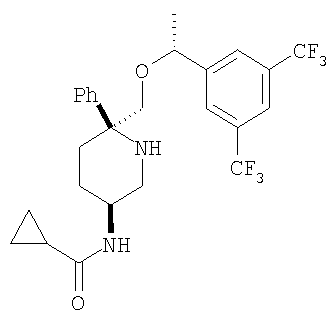
Соединение Примера 15 (выход 89%) получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием циклопропанкарбонилхлорида вместо пропионилхлорида. МС с электрораспылением [М+1]+ 515,1.
Пример 16
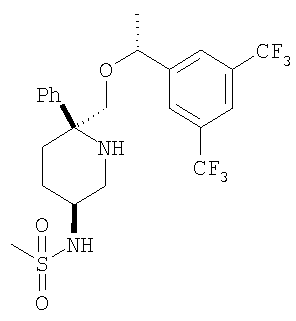
Соединение Примера 16 (выход 89%) получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием соединения Примера 13 и СН3SO2Cl вместо пропионилхлорида. МС с электрораспылением [М+1]+ 52,1.
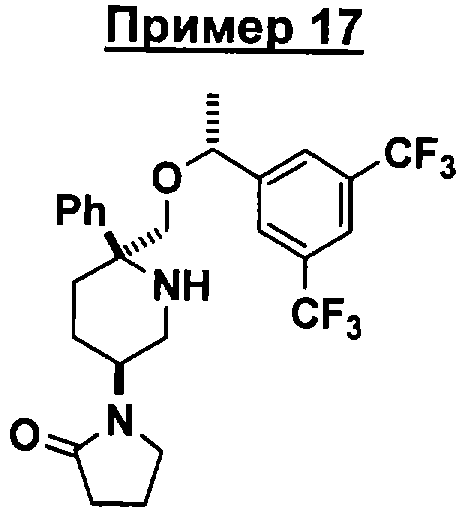
Соединение Примера 17 (суммарный выход 23%) получают с помощью методики, аналогичной использованной для получения соединения Примера 3 с использованием соединения Примера 13 вместо Соединения 10. МС с электрораспылением [М+1]+ 515,1.
Пример 18
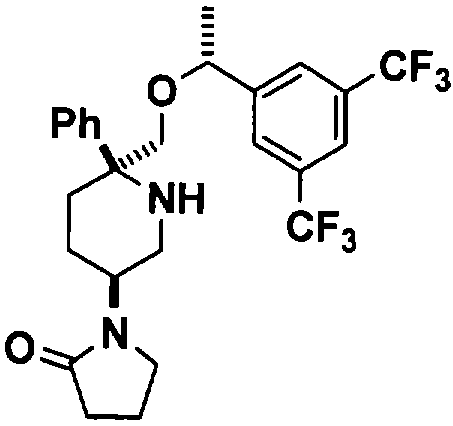
Соединение Примера 18 (суммарный выход 42%) получают с помощью методики, аналогичной использованной для получения соединения Примера 4 с использованием соединения Примера 13 вместо Соединения 10. МС с электрораспылением [М+1]+ 529,1.
Получение соединений 26, 27, 28 и 29:
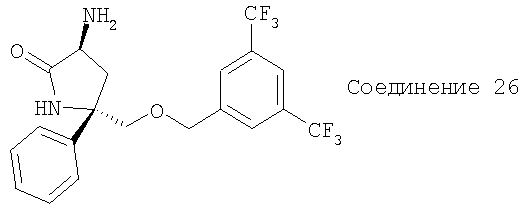
Соединение 26 получают из Соединения 1 с помощью методики, аналогичной использованной для получения Соединения 9.
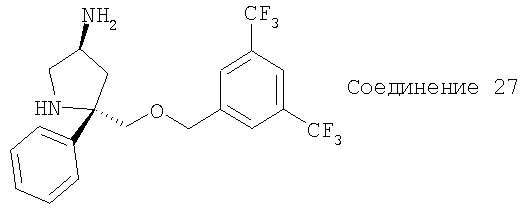
Соединение 27 получают с помощью методики, аналогичной использованной для получения Соединения 10.
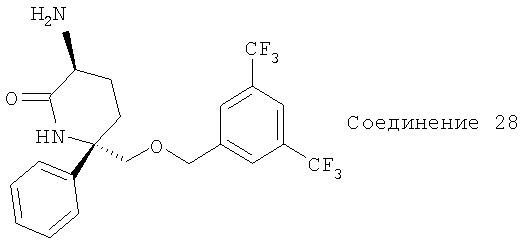
Соединение 28 (выход 90%) получают с помощью методики, аналогичной использованной для получения Соединения 25. МС с электрораспылением [M+1]+ 447,1.
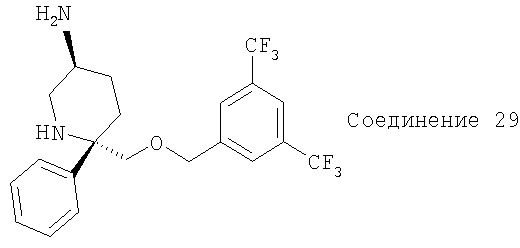
Соединение 29 получают с помощью методики, аналогичной использованной для получения соединения Примера 13. МС с электрораспылением [М+1]+ 433,1.
Пример 19
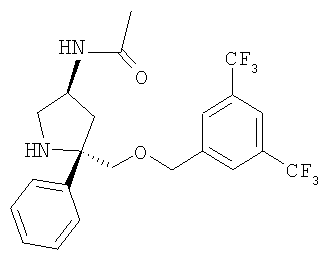
Соединение Примера 19 (40 мг, выход 70%) получают с помощью методики, аналогичной использованной для соединения Примера 1а с использованием Соединения 27 вместо Соединения 6. МС с электрораспылением [М+1]+ 461,1.
Пример 20
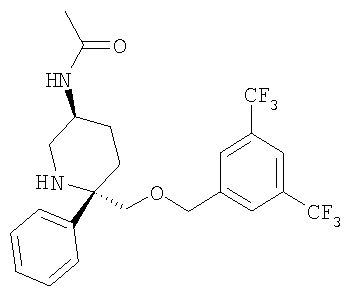
Соединение Примера 20 (99 мг, 72%) получают с помощью методики, аналогичной использованной для получения соединения Примера 1а с использованием Соединения 29 вместо Соединения 6. МС с электрораспылением [М+1]+ 475,1.
Пример 21
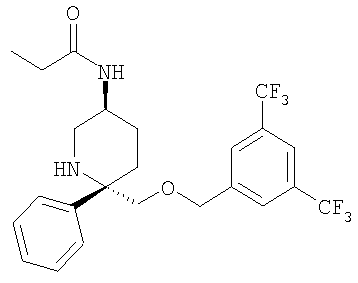
Соединение Примера 21 (74 мг, 66%) получают из Соединения 29 с помощью методики, аналогичной использованной для получения соединения Примера 2 из Соединения 10, с использованием пропионового ангидрида вместо циклопропанкарбонилхлорида. МС с электрораспылением [М+1]+ 489,1.
Пример 22
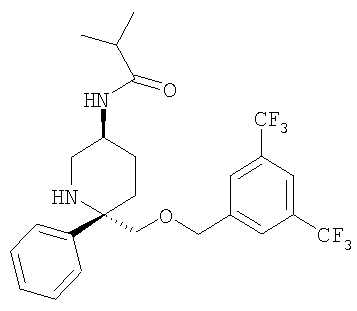
Соединение Примера 22 (75 мг, 78%) получают из Соединения 29 с помощью методики, аналогичной использованной для получения Соединения Примера 2 из Соединения 10, с использованием изобутирилхлорида вместо циклопропанкарбонилхлорида. МС с электрораспылением [М+1]+ 503,1.
Пример 23
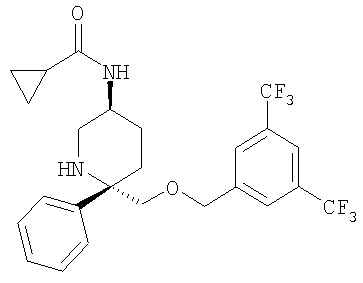
Соединение Примера 23 (9 мг, 35%) получают из Соединения 29 с помощью методики, аналогичной использованной для получения соединения Примера 2 из Соединения 10. МС с электрораспылением [M+1]+ 501,1.
Пример 24
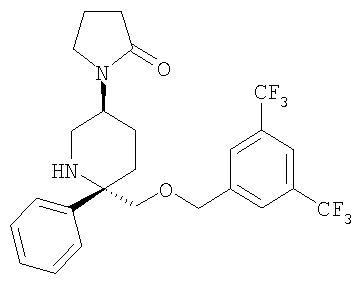
Соединение Примера 24 (31 мг, 71%) получают из Соединения 29 с помощью методики, аналогичной использованной для получения соединения Примера 3 из Соединения 10. МС с электрораспылением [М+1]+ 501,1.
Пример 25
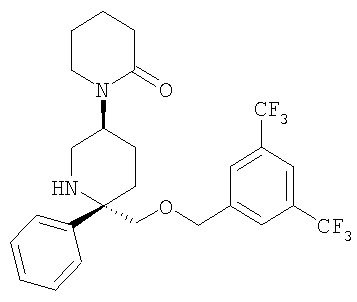
Соединение Примера 25 (68 мг, 68%) получают из Соединения 29 с помощью методики, аналогичной использованной для получения соединения Примера 4 из Соединения 10. МС с электрораспылением [М+1]+ 515,1.
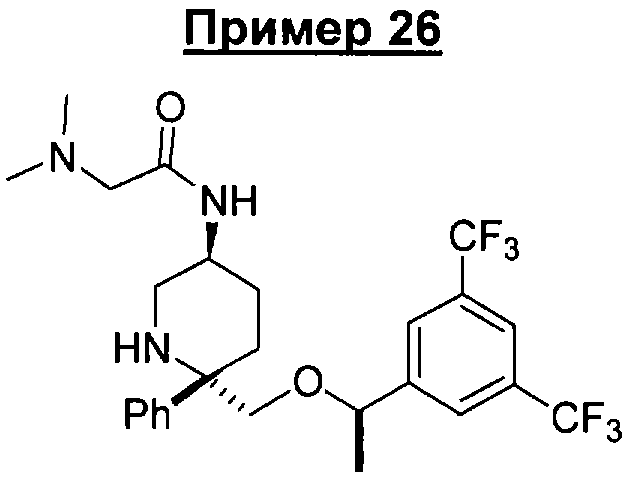
К раствору соединения Примера 13 (0,14 г, 0,314 ммоль, 1 экв.) в безводном DMF (1,6 мл) при 23°С прибавляют N,N-диметилглицин (33,95 мг, 0,329 ммоль, 1,05 экв.), а затем EDC.HCl (66,13 мг, 0,345 ммоль, 1,1 экв.) и реакционную смесь перемешивают при 23°С в течение 18 ч. Реакционную смесь разбавляют с помощью DMF (2,4 мл), очищают с помощью колонки Gilson и получают соединение Примера 26 (66 мг, 40%). МС с электрораспылением [М+1]+ 532,1.
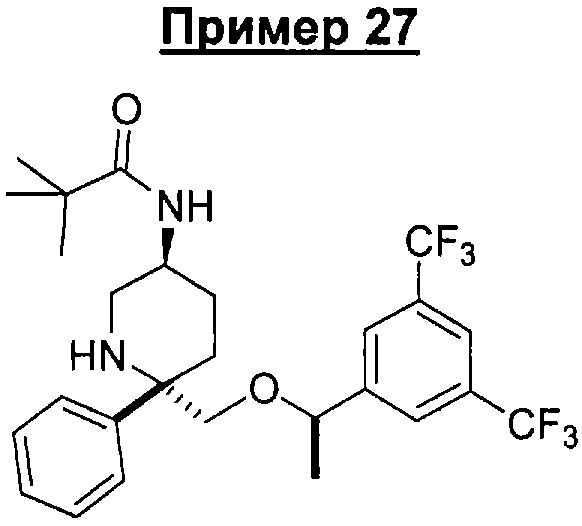
Соединение Примера 27 (выход 62%) получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием триметилацетилхлорида вместо пропионилхлорида. МС с электрораспылением [M+1]+ 531,1.
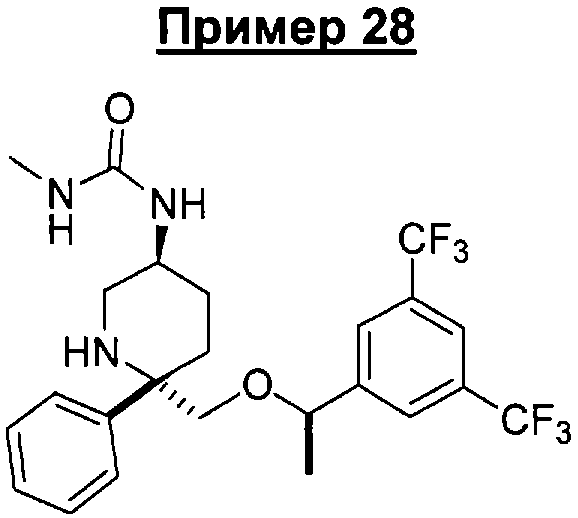 Соединение Примера 28 (105 мг, 74%) получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием метилизоцианата вместо пропионилхлорида. МС с электрораспылением [М+1]+ 504,1.
Соединение Примера 28 (105 мг, 74%) получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием метилизоцианата вместо пропионилхлорида. МС с электрораспылением [М+1]+ 504,1.
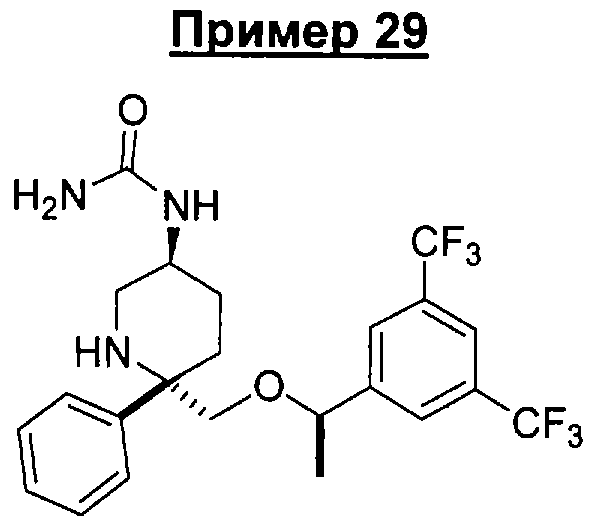
Соединение Примера 29 (146 мг, 754%) получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием триметилсилилизоцианата вместо пропионилхлорида. МС с электрораспылением [М+1]+ 490,1.
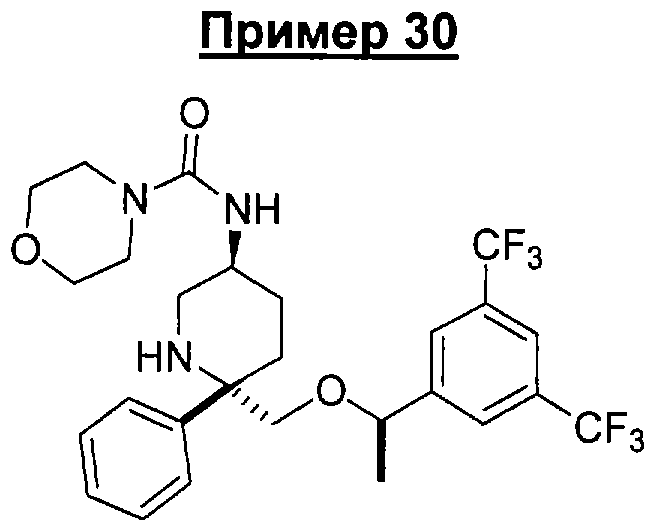
К раствору соединения Примера 13 (100 мг, 0,224 ммоль, 1 экв.) в безводном CH2Cl2 (2 мл) прибавляют 4-морфолинилкарбонилхлорид (28,7 мкл, 0,246 ммоль, 1,1 экв.) и DIEA (39 мкл, 0,223 ммоль, 1 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворение в воде и очистка с использованием колонки с диоксидом кремния дает соединение Примера 30 (53 мг, 42%). МС с электрораспылением [М+1]+ 560,1.
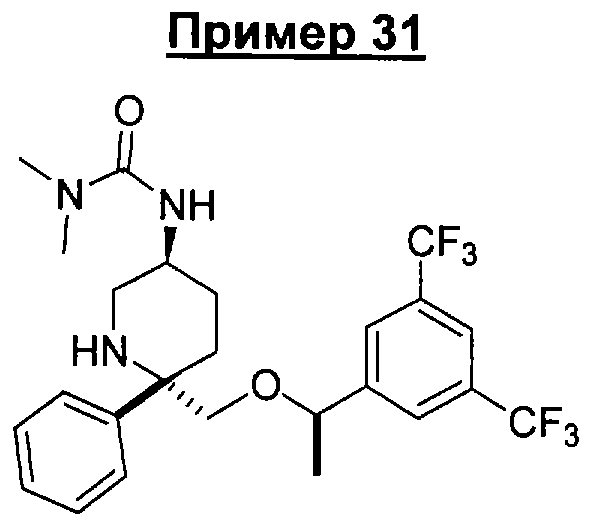
Соединение Примера 31 (выход 40%) получают с помощью методики, аналогичной использованной для получения соединения Примера 30, с использованием диметилкарбамилхлорида вместо 4-морфолинилкарбонилхлорида. МС с электрораспылением [М+1]+ 518,1.
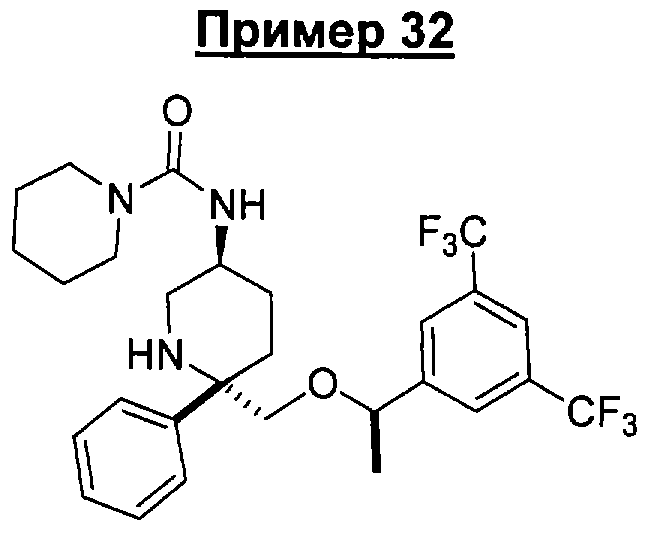
Соединение Примера 32 (выход 42%) получают с помощью методики, аналогичной использованной для получения соединения Примера 30, с использованием 1-пиперидинкарбонилхлорида вместо 4-морфолинилкарбонилхлорида. МС с электрораспылением [М+1]+ 558,1.
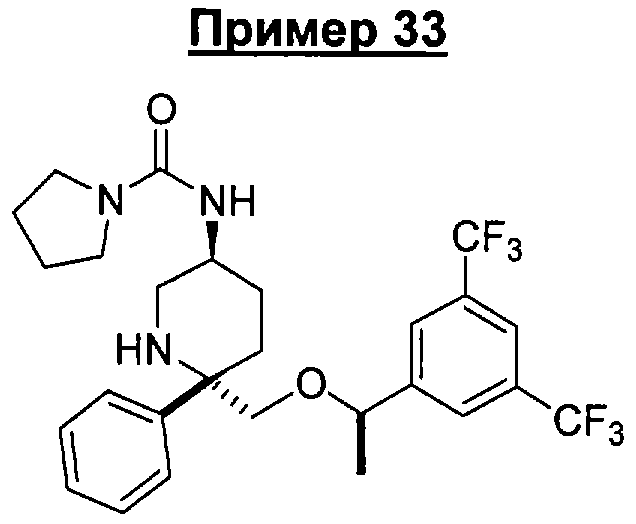
Соединение Примера 33 (выход 40%) получают с помощью методики, аналогичной использованной для получения соединения Примера 30, с использованием 1-пирролинидкарбонилхлорида вместо 4-морфолинилкарбонилхлорида. МС с электрораспылением [М+1]+ 544,1
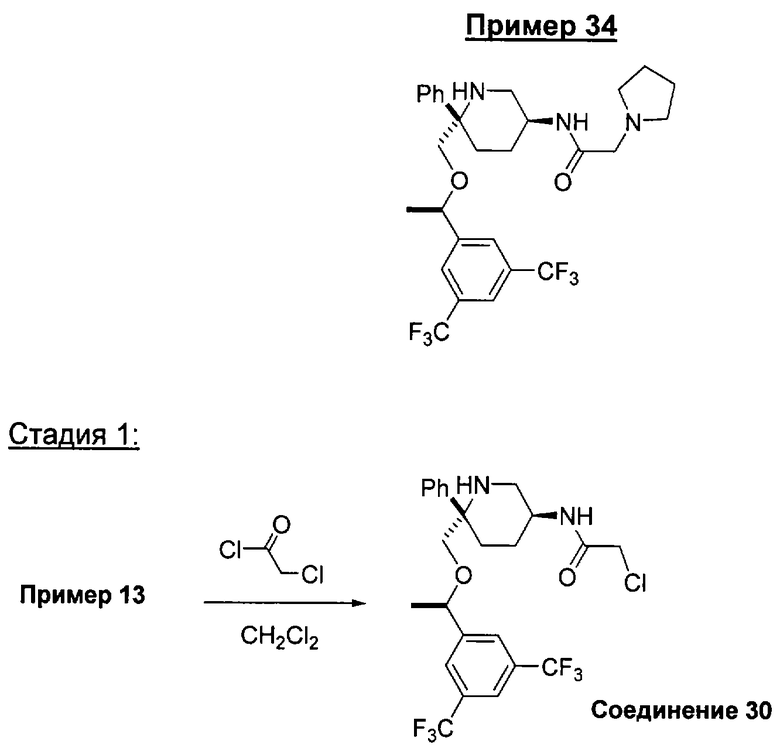
Соединение 30 (выход 43%) получают с помощью методики, аналогичной использованной для получения соединения Примера 10, с использованием хлорацетилхлорида вместо пропионилхлорида.
Стадия 2:
К раствору Соединения 30 (90 мг, 0,17 ммоль, 1 экв.) в безводном CH2Cl2 (0,5 мл) прибавляют пирролидин (17,2 мкл, 0,206 ммоль, 1,2 экв.) и DIEA (30 мкл, 0,17 ммоль, 1 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворение в воде и очистка с использованием колонки с диоксидом кремния дает соединение Примера 34 (45 мг, 47%). МС с электрораспылением [М+1]+ 558,1.
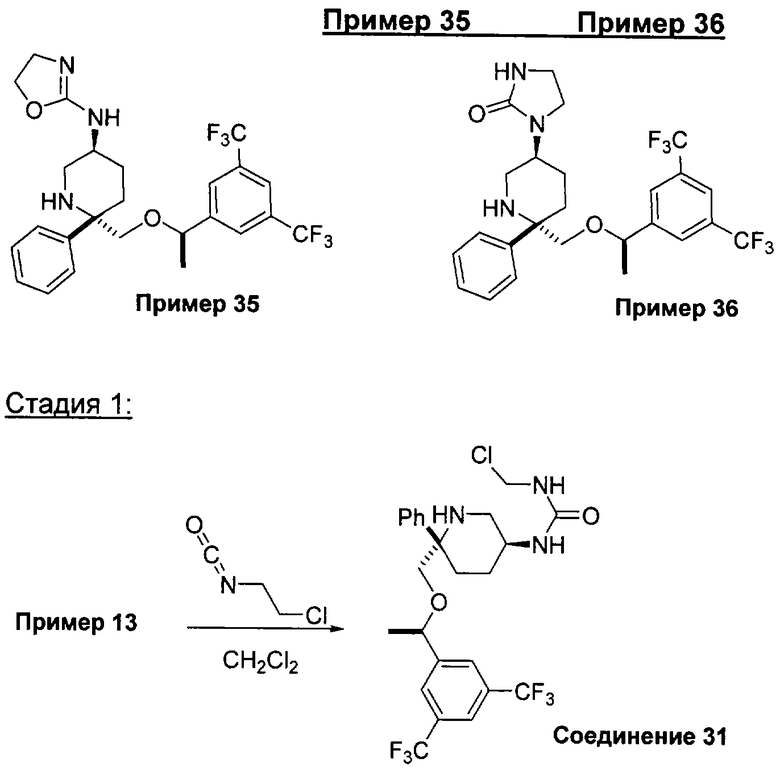
Соединение 31 получают с помощью методики, аналогичной использованной для получения соединения Примера 14, с использованием 2-хлорэтилизоцианата вместо пропионилхлорида.
Стадия 2:
К раствору Соединения 31 в безводном THF (7 мл) прибавляют NaH (25 мг, 0,625 ммоль, 1,7 экв., 60% дисперсия в минеральном масле) при 0°С. Полученный мутный раствор нагревают при 60°С в течение 2 ч. Растворение в воде дает неочищенный продукт, который очищают на колонке с силикагелем и получают менее полярное искомое соединение Примера 35 (10 мг, 5,4%), МС с электрораспылением [М+1]+ 516,1 и более полярное искомое соединение Примера 36 (122 мг, 66%), МС с электрораспылением [М+1]+ 516.1.
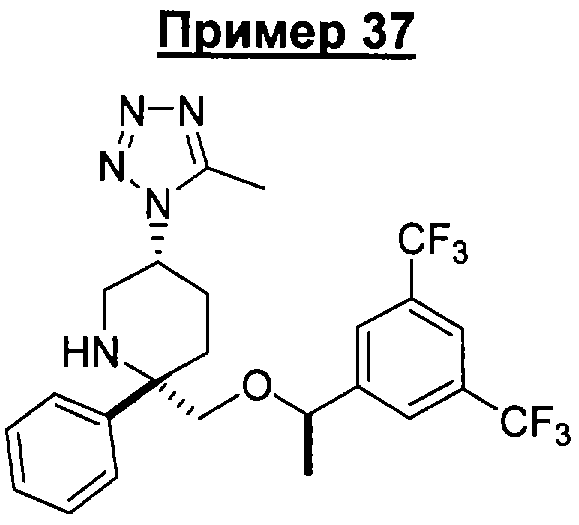
К раствору соединения Примера 12b (200 мг, 0,41 ммоль, 1 экв.) в безводном СН2Cl2 (1 мл) при 0°С прибавляют трифторметансульфоновый ангидрид (69 мкл, 0,41 ммоль, 1 экв.). Реакционную смесь перемешивают в течение 40 мин, а затем прибавляют NaN3 (26,6 мг, 0,41 ммоль, 1 экв.). Смесь нагревают до комнатной температуры в течение 2 ч. Растворитель удаляют в вакууме. Остаток очищают с помощью препаративной ТСХ (диоксид кремния) и получают соединение Примера 37 (4,5 мг, 2%). МС с электрораспылением [М+1]+ 514,1.
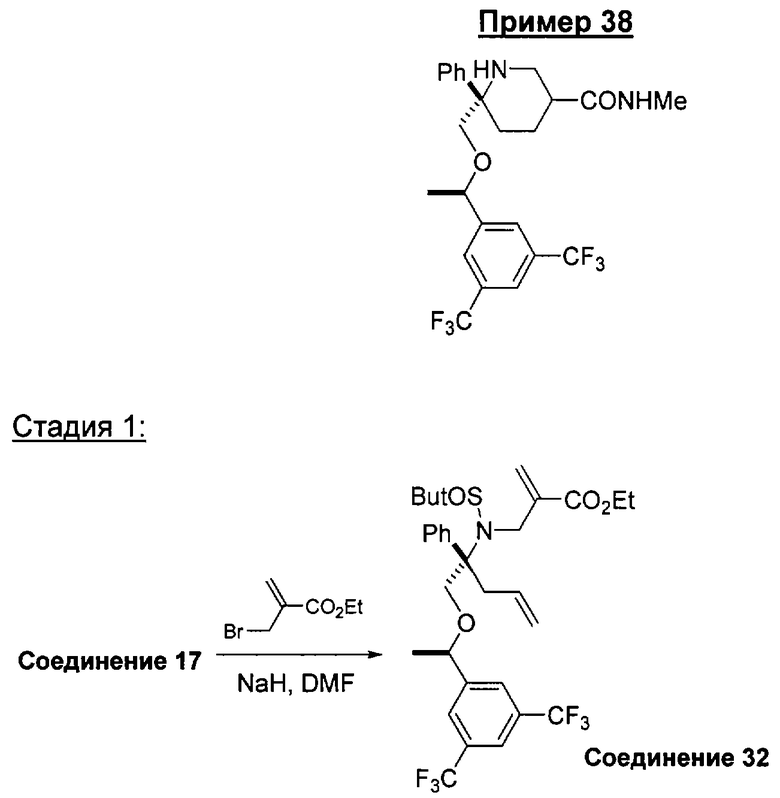
К Соединению 17 (0,3 г, 0,575 ммоль, 1 экв.) в атмосфере N2 в безводном DMF (3 мл) при 0°С прибавляют NaH (27,6 мг, 0,69 ммоль, 1,2 экв., 60% в минеральном масле) и реакционную смесь перемешивают в течение 1 ч. К полученной суспензии при энергичном перемешивании по каплям прибавляют этил-2-бромметилакрилат (0,088 мл, 0,629 ммоль, 1,1 экв.). Реакционной смеси дают нагреться до 23°С и перемешивают в течение 18 ч. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl и экстрагируют с помощью Et2O. Объединенные органические слои промывают водой, рассолом, сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают с использованием флэш-колонки с силикагелем и получают искомое Соединение 32 (0,199 г, 55%).
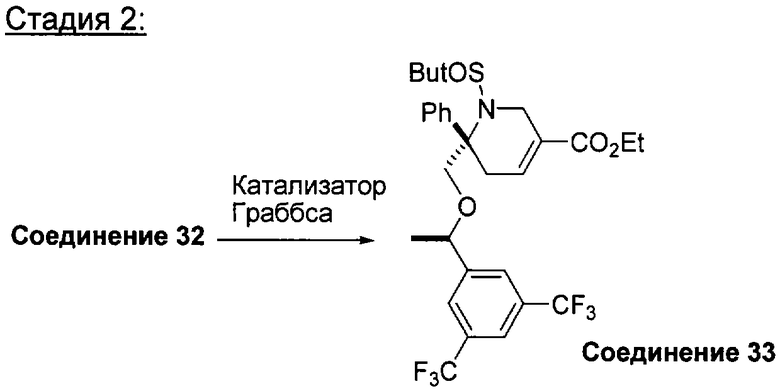
К раствору Соединения 32 (50 мг, 0,078 ммоль, 1 экв.) в безводном СН2Cl2 (0,8 мл) в атмосфере N2 прибавляют катализатор Граббса, трициклогексилфосфин-[1,3-бис(2,4,6-триметилфенил)-4,5-дигидроимидазол-2-илиден][бензилидин]рутений(IV)дихлорид (6,7 мг, 0,0079 ммоль, 0,1 экв.). Полученный коричневый раствор нагревают при 40-45°С в течение 2 ч. Затем растворитель удаляют, остаток очищают на колонке с силикагелем и получают искомое Соединение 33 (60 мг, 63%). МС с электрораспылением [M+1]+ 502,1.
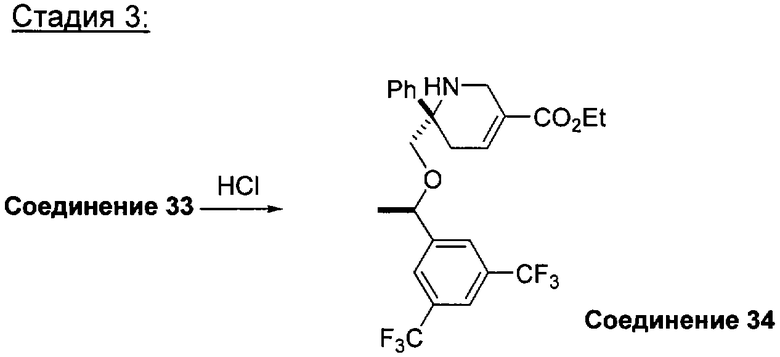
К раствору Соединения 33 (30 мг, 0,05 ммоль, 1 экв.) в абсолютном МеОН (0,5 мл) при 0°С прибавляют 4 н. раствор HCl в диоксане (0,5 мл). Полученный раствор перемешивают при 0°С в течение 4 ч. Затем растворитель удаляют и остаток растворяют в CH2Cl2 и пропускают через короткую колонку, заполненную K2СО3. Остаток Соединения 34 непосредственно используют на следующей стадии без дополнительной очистки.
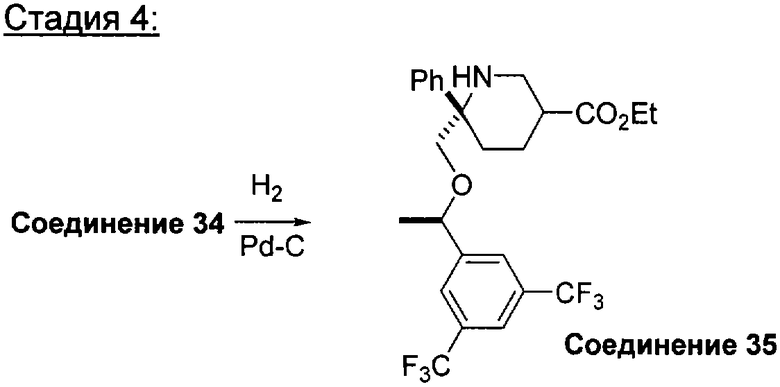
Раствор Соединения 34 (30 мг, 0,06 ммоль) в EtOH (5 мл) обрабатывают с помощью 10% Pd-C (32 мг, 0,03 ммоль) и гидрируют при 60 фунт-сила/дюйм2 ман. (psig) в течение 18 ч. Катализатор отфильтровывают и промывают с помощью EtOAc. Фильтрат концентрируют и полученный остаток Соединения 35 непосредственно используют на следующей стадии без дополнительной очистки.
Стадия 5:
К смеси HCl соли метиламина (52 мг, 0,77 ммоль, 12,8 экв.) в толуоле (0,2 мл) прибавляют Ме3Al (2 М в толуоле, 0,36 мл, 0,72 ммоль) и полученную смесь перемешивают в течение 30 мин. Раствор Соединения 35 (30 мг, 0,06 ммоль) в толуоле (0,5 мл) прибавляют к реакционной смеси с помощью шприца. Полученный раствор нагревают при 100°С в течение 18 ч. Затем реакционную смесь выливают в насыщенный водный раствор тартрата Na/K (10 мл), перемешивают в течение 10 мин и экстрагируют с помощью EtOAc (4×10 мл). Объединенные органические слои промывают рассолом и концентрируют. Остаток обрабатывают с помощью препаративной ТСХ и получают менее полярный изомер, соединение Примера 38а, МС с электрораспылением [М+1]+ 489,1, и более полярный изомер, соединение Примера 38b, MC с электрораспылением [M+1]+ 489,1.
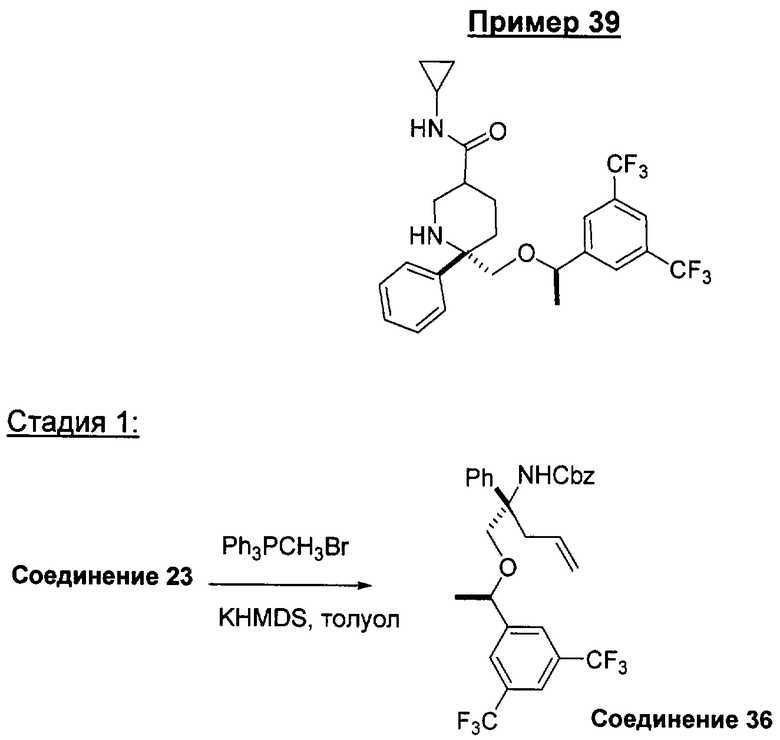
Соединение 36 (выход 63%) получают из Соединения 23 с помощью методики, аналогичной использованной для получения Соединения 23 из Соединения 3, и с использованием метилтрифенилфосфонийбромида вместо (метоксиметил)трифенилфосфонийхлорида.
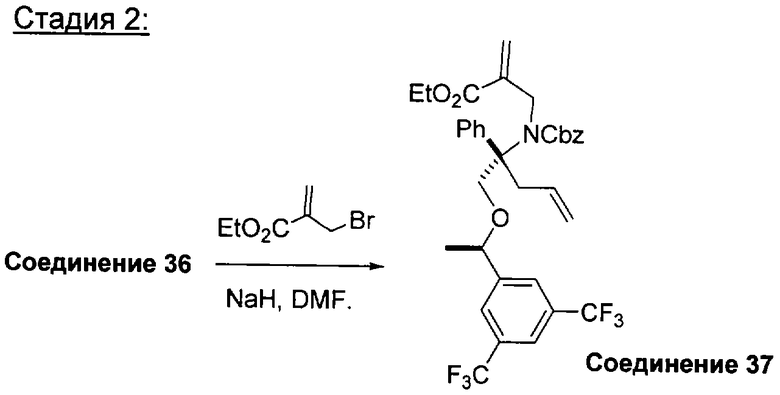
Соединение 37 (выход 50%) получают с помощью методики, аналогичной использованной для получения Соединения 32, с использованием Соединения 36 вместо Соединения 17.
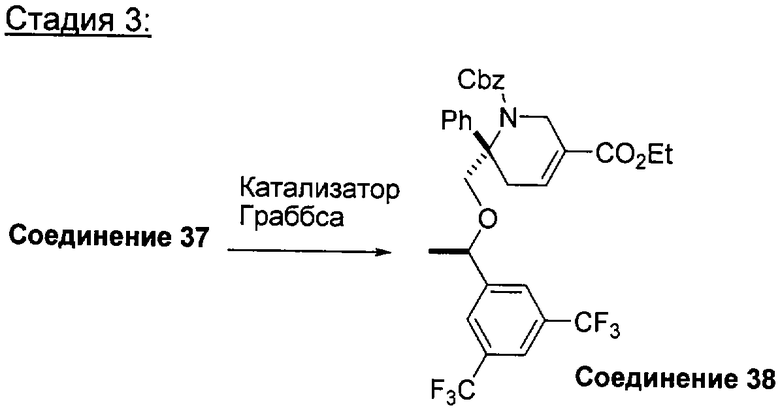
К раствору Соединения 37 (2,46 г, 3,71 ммоль, 1 экв.) в безводном CH2Cl2 (50 мл) в атмосфере N2 прибавляют катализатор Граббса (327 мг, 0,385 ммоль, 0,1 экв.). Полученный коричневый раствор нагревают при 40-45°С в течение ночи. Затем растворитель удаляют, остаток очищают на колонке с силикагелем и получают Соединение 38 (2,1 г, 89%).
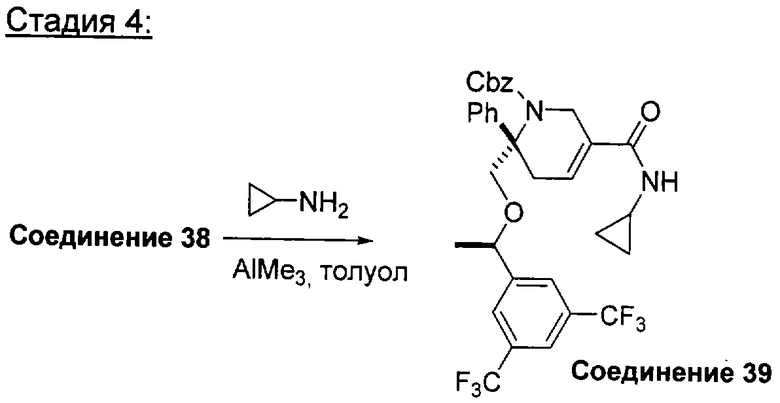 К смеси циклопропиламина (0,24 мл, 3,45 ммоль, 4,2 экв.) в толуоле (1,0 мл) прибавляют Ме3Al (2 М в толуоле, 1,71 мл, 3,41 ммоль, 4,2 экв.) и полученную смесь перемешивают в течение 30 мин. Раствор Соединения 38 (516 мг, 0,82 ммоль, 1 экв.) в толуоле (2,5 мл) прибавляют к реакционной смеси с помощью шприца. Полученный раствор нагревают при 60°С в течение 18 ч. Затем реакционную смеси выливают в насыщенный водный раствор тартрата Na/K, перемешивают в течение 10 мин и экстрагируют с помощью EtOAc (10 мл×4). Объединенные органические слои промывают рассолом и концентрируют. Остаток очищают на колонке с диоксидом кремния и получают Соединение 39 (360 мг, 68%).
К смеси циклопропиламина (0,24 мл, 3,45 ммоль, 4,2 экв.) в толуоле (1,0 мл) прибавляют Ме3Al (2 М в толуоле, 1,71 мл, 3,41 ммоль, 4,2 экв.) и полученную смесь перемешивают в течение 30 мин. Раствор Соединения 38 (516 мг, 0,82 ммоль, 1 экв.) в толуоле (2,5 мл) прибавляют к реакционной смеси с помощью шприца. Полученный раствор нагревают при 60°С в течение 18 ч. Затем реакционную смеси выливают в насыщенный водный раствор тартрата Na/K, перемешивают в течение 10 мин и экстрагируют с помощью EtOAc (10 мл×4). Объединенные органические слои промывают рассолом и концентрируют. Остаток очищают на колонке с диоксидом кремния и получают Соединение 39 (360 мг, 68%).
Стадия 5:
Раствор Соединения 39 (360 мг, 0,556 ммоль, 1 экв.) в EtOH (25 мл) обрабатывают с помощью 10% Pd-C (641 мг, 0,613 ммоль, 1,1 экв.) и гидрируют при 50 фунт-сила/дюйм2 в течение 6 ч. Катализатор отфильтровывают и промывают с помощью EtOAc. Остаток очищают на колонке с силикагелем и получают менее полярный изомер, соединение Примера 39а (54 мг, 19%), МС с электрораспылением [М+1]+ 515,1, и более полярный изомер, соединение Примера 39b (22 мг, 8%), МС с электрораспылением [М+1]+ 515,1.
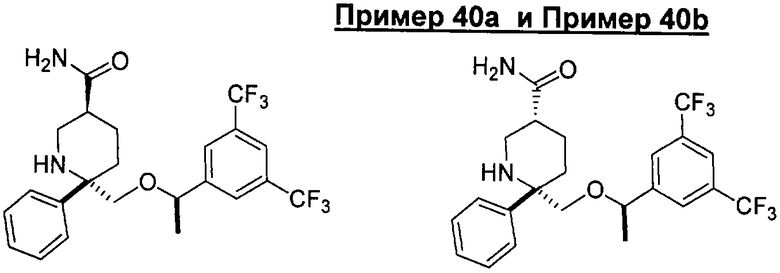
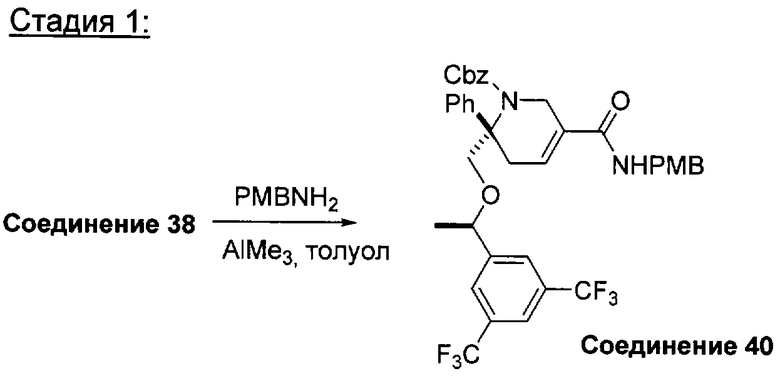
Соединение 40 (выход 55%) получают с помощью методики, аналогичной использованной для получения Соединения 39 с использованием п-метоксибензиламина вместо циклопропиламина.
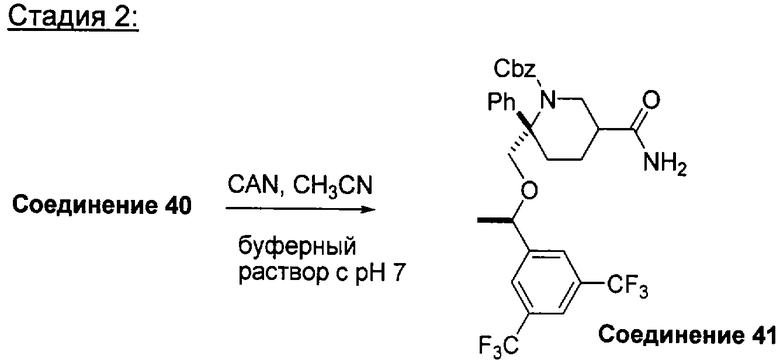
Раствор Соединения 40 (1 г, 1,38 ммоль, 1 экв.) в CH3CN (10 мл) и буферный раствор с рН 7 (3 мл) обрабатывают с помощью нитрата аммония-церия(IV) (2,17 г, 3,96 ммоль, 2,9 экв.) при комнатной температуре в течение 2 ч. Растворение в воде дает неочищенный продукт, который очищают на колонке с силикагелем и получают Соединение 41 (760 мг, 91%).
Стадия 3:
Соединение Примера 40а и соединение Примера 40b получают с помощью методики, аналогичной использованной для получения соединения Примера 39а и соединения Примера 39b, с использованием Соединения 41 вместо Соединения 39.
МС с электрораспылением [М+1]+ 475,1 для соединения Примера 40а (менее полярный изомер);
МС с электрораспылением [M+1]+ 475,1 для соединения Примера 40b (более полярный изомер).
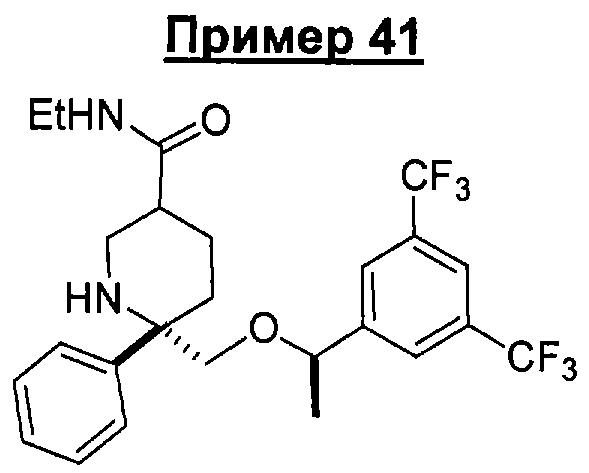
Соединение Примера 41а и соединение Примера 41b получают с помощью методики, аналогичной использованной для получения соединения Примера 38а и соединения Примера 38b, с использованием этиламина вместо метиламина.
МС с электрораспылением [М+1]+ 503,1 для соединения Примера 41а (менее полярный изомер);
МС с электрораспылением [М+1]+ 503,1 для соединения Примера 41b (более полярный изомер).
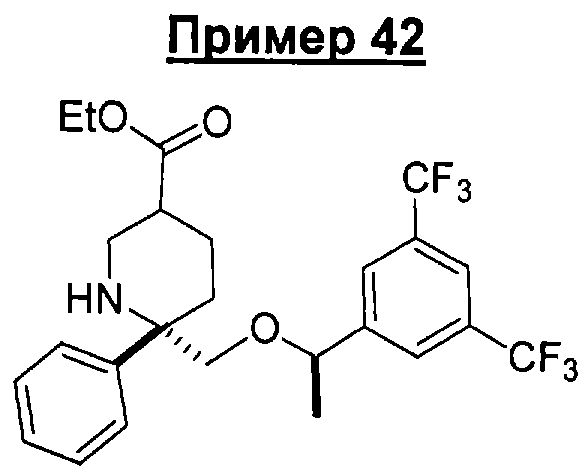
Смесь двух изомеров Соединения 35 разделяют с помощью хроматографии на колонке и получают чистое соединение Примера 42а и соединение Примера 42b.
МС с электрораспылением [М+1]+ 504,1 для соединения Примера 42а (менее полярный изомер);
МС с электрораспылением [М+1]+ 504,1 для соединения Примера 42b (более полярный изомер).
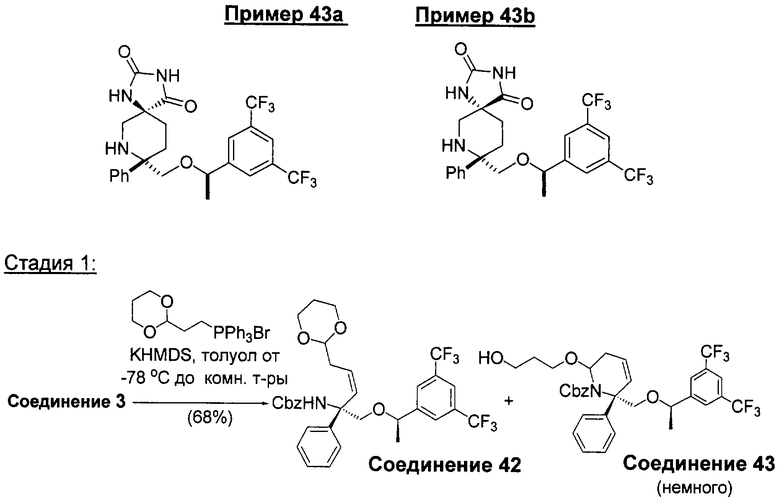
К суспензии лактола - Соединения 3 (60 г, 93,0 ммоль, 1 экв.) и реагента Виттига (93,5 г, 200,0 ммоль, 2,15 экв.) в толуоле (800 мл) при перемешивании и -78°С в атмосфере N2 по каплям прибавляют раствор KHMDS (0,5 М в толуоле, 558 мл, 280,0 ммоль, 3 экв.) при -78°С. Охлаждающую баню удаляют и желтую смесь нагревают до комнатной температуры с получением красного раствора. Смесь перемешивают при 23°С в течение еще 1 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. Прибавляют EtOAc и слои разделяют. Отделенный водный слой экстрагируют с помощью EtOAc (2×500 мл). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме с последующей хроматографией на колонке Biotage [от 5% EtOAc-гексан до 10% EtOAc-гексан] дает алкен - Соединение 42 в виде белого твердого вещества (40,5 г, 68%), МС с электрораспылением [М+1]+ 638,1. Продолжение элюирования дает неочищенный продукт циклизации - Соединение 43.
Стадия 2:
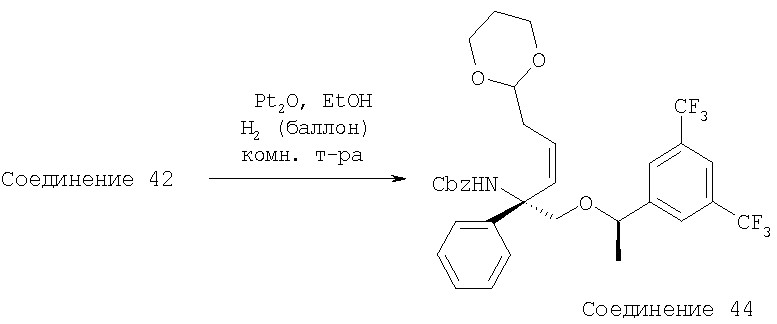
Суспензию алкена - Соединения 42 (40,5 г, 64 ммоль, 1 экв.) и PtO2 (1,44 г, 6,4 ммоль, 0,1 экв.) в EtOH (400 мл) перемешивают в атмосфере подаваемого из баллона Н2 при 23°С в течение 24 ч. Прибавляют еще одну порцию PtO2 (1,44 г, 6,4 ммоль, 0,1 экв.) и смесь перемешивают в течение еще 24 ч при 23°С. Катализатор отфильтровывают через слой целита. Этот раствор алкана - Соединения 44 используют на следующей стадии без дополнительной очистки.
Стадия 3:
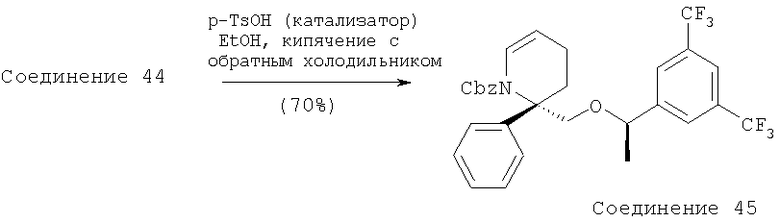
p-TsOH·H2O (2,42 г, 13,0 ммоль) прибавляют к этанольному раствору алкана - Соединения 44, полученного выше, и раствор кипятят с обратным холодильником в течение 4 ч. Раствор охлаждают до комнатной температуры и нейтрализуют с помощью Et3N. Растворители удаляют в вакууме и прибавляют EtOAc. Прибавляют насыщенный раствор NaHCO3 и слои разделяют. Отделенный водный слой экстрагируют с помощью EtOAc (300 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме с последующей хроматографией на колонке Biotage [10% эфир-гексан] дает енамид - Соединение 45 (первая порция) в виде желтого масла. Некоторое количество промежуточного продукта и исходное вещество регенерируют в виде желтого масла путем последующего элюирования с помощью [50% EtOAc-гексан]. Желтое масло растворяют в толуоле и прибавляют 10 мол.% p-TsOH. Смесь кипятят с обратным холодильником в течение 2 ч и охлаждают до комнатной температуры. Обрабатывают так, как указано выше, и получают енамид - Соединение 45 (25 г, 70%), МС с электрораспылением [М+1]+ 564,1, в виде желтого масла.
Стадия 4:
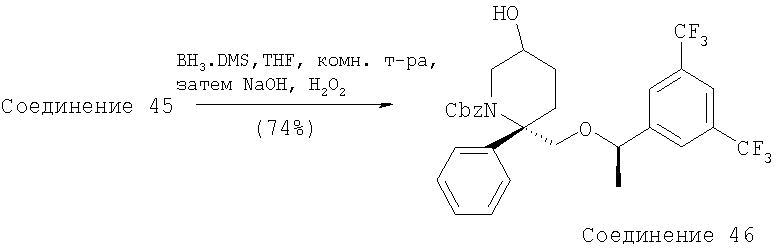
ВН3.Ме2S (13,6 мл, 133 ммоль, 3,02 экв.) прибавляют к раствору енамида - Соединения 45 (25 г, 44,0 ммоль, 1 экв.) в THF при 23°С в атмосфере N2. Смесь перемешивают при 23°С в течение 18 ч, затем охлаждают на бане из воды со льдом. Медленно прибавляют раствор NaOH (500 мл, 2 н.), а затем раствор Н2O2 (500 мл, 30% водный). Смесь перемешивают, нагревая от 0 до 23°С в течение 18 ч. Слои разделяют и отделенный водный слой экстрагируют с помощью Et2O (500 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме с последующей хроматографией на колонке Biotage [гексан-EtOAc, 3:1 (об./об.)] дает спирт - Соединение 46 в виде бесцветного масла (19 г, 74%), МС с электрораспылением [М+1]+ 582,1.
Стадия 5:
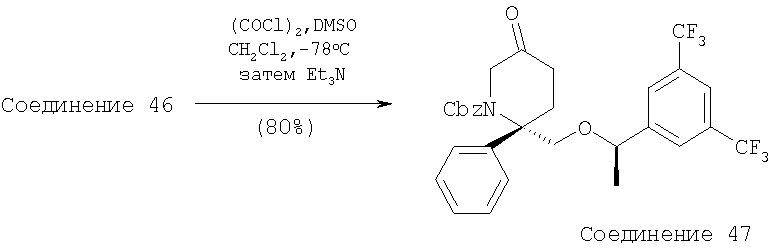
Оксалилхлорид (5,7 мл, 65,3 ммоль, 2 экв.) прибавляют к раствору DMSO (9,3 мл, 131,0 ммоль, 4 экв.) в CH2Cl2 (300 мл) при -78°С в атмосфере N2. Смесь перемешивают при -78°С в течение 15 мин, а затем прибавляют раствор спирта - Соединения 46 (19 г, 32,7 ммоль, 1 экв.) в CH2Cl2 (50 мл). Смесь перемешивают при -78°С в течение еще 1 ч и прибавляют Et3N (32 мл, 228,9 ммоль, 7 экв.). Охлаждающую баню удаляют и смесь нагревают до комнатной температуры, а затем реакцию останавливают с помощью насыщенного раствора NaHCO3. Слои разделяют и водный слой экстрагируют с помощью CH2Cl2 (300 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме с последующей хроматографией на колонке Biotage [гексан-эфир, 4:1 (об./об.)] дает кетон - Соединение 47 в виде бесцветного масла (15 г, 80%), МС с электрораспылением [M+1]+ 580,1.
Стадия 6:
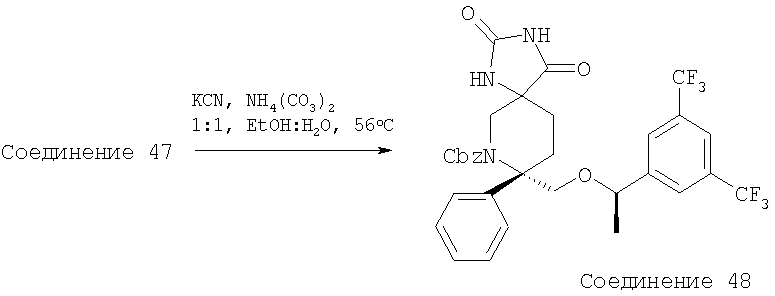
EtOH (150 мл) прибавляют к Cbz-кетону - Соединению 47 (15 г, 25,88 ммоль, 1 экв.), а затем прибавляют NH4(CO3)2 (9,95 г, 103,5 ммоль, 4 экв.) и раствор KCN (3,4 г, 51,77 ммоль, 2 экв.). Полученную смесь нагревают при 58°С в атмосфере N2 в течение 72 ч. ТСХ (1:1, EtOA : гексан) подтверждает полное использование исходного вещества. Реакционную смесь охлаждают до комнатной температуры и выливают в насыщенный водный раствор NaHCO3 (200 мл) и экстрагируют с помощью EtOAc (3×200 мл). Объединенные органические слои сушат над MgSO4 и концентрируют в вакууме и получают неочищенный Cbz-гидантоин - Соединение 48 (16,5 г, 98%), МС с электрораспылением [М+1]+ 650,1. Неочищенное вещество используют в следующей реакции без дополнительной очистки.
Стадия 7:
Неочищенный Cbz-гидантоин - Соединение 48 (16,5 г, 25,4 ммоль, 1 экв.) растворяют в МеОН (220 мл) и прибавляют 20% Pd(OH)2-C (3,6 г). Реакционную смесь встряхивают в приборе для гидрирования Парра в атмосфере Н2 при 40 фунт-сила/дюйм2 в течение 18 ч. ТСХ (1:1, EtOAc : гексан) подтверждает полное использование исходного вещества. Реакционную смесь фильтруют через слой целита и целит промывают с помощью МеОН. Полученный раствор концентрируют в вакууме. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (3:2, EtOAc : гексан). Собирают две главные фракции. Менее полярная фракция соответствует изомеру - соединению Примера 43а (3 г, суммарный выход для двух фракций 20%), МС с электрораспылением [М+1]+ 516,1. Более полярная фракция соответствует изомеру - соединению Примера 43b (4,5 г, суммарный выход для двух фракций 30%), МС с электрораспылением [M+1]+ 516,1.
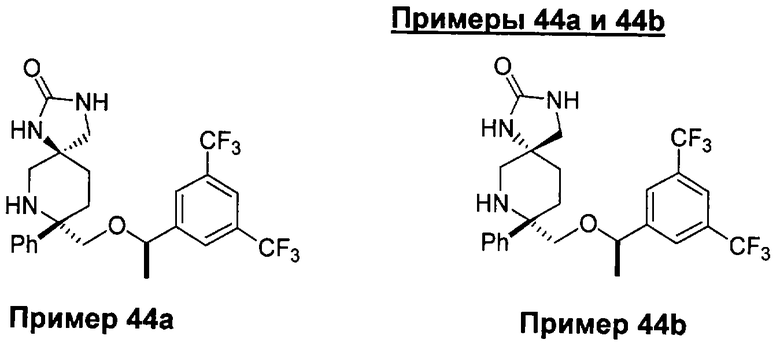
В высушенную на огне КДК объемом 25 мл помещают AlCl3 (0,01 г, 0,776 ммоль, 4 экв.). Колбу с реакционной смесью охлаждают до 0°С и прибавляют 1 М раствор LAH в Et2O (0,58 мл, 0,58 ммоль, 3 экв.). Смесь перемешивают при 0°С в течение 10 мин и затем через канюлю медленно прибавляют раствор соединения Примера 43b (0,1 г, 0,194 ммоль, 1 экв.) в сухом THF (3 мл). Реакционную смесь перемешивают при 0°С в течение 1 ч и затем дают нагреться до комнатной температуры и перемешивают в течение 18 ч. Затем реакционную смесь охлаждают до 0°С и реакцию осторожно останавливают с помощью насыщенного водного раствора тартрата натрия-калия. Затем ее перемешивают при 0°С в течение 30 мин. Смесь экстрагируют с помощью EtOAc (2×200 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (1:9, МеОН : EtOAc) и получают соединение Примера 44b (0,066 г, 68%), МС с электрораспылением [М+1]+ 502,1.
Соединение Примера 44а получают из соединения Примера 43а с помощью методики, аналогичной использованной для получения соединения Примера 44b из соединения Примера 43b. МС с электрораспылением [M+1]+ 502,1 для соединения Примера 44а.
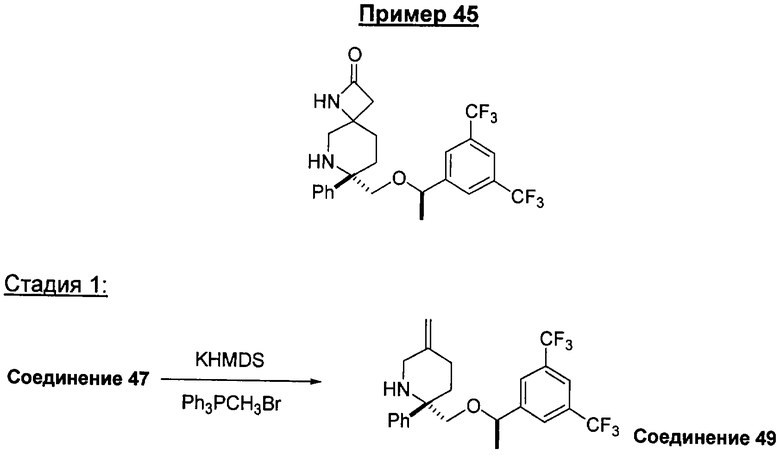
К суспензии (метил)трифенилфосфонийбромида (0,37 г, 1,04 ммоль, 3 экв.) в толуоле (5 мл) при 0°С в атмосфере N2 прибавляют раствор KHMDS (1,73 мл, 0,863 ммоль, 2,5 экв.). После перемешивания при 0°С в течение 1 ч прибавляют раствор Соединения 47 (0,2 г, 0,35 ммоль, 1 экв.) в толуоле (7 мл). Смесь перемешивают при 0°С в течение 1,5 ч и затем реакцию останавливают с помощью насыщенного раствора NaHCO3 (150 мл). Смесь экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (4:1, гексан : EtOAc) и получают Соединение 49 (0,196 г, 98%).
Стадия 2:
К раствору Соединения 49 (0,196 г, 0,34 ммоль, 1 экв.) в сухом Et2O (3 мл) при 0°С прибавляют хлорсульфонилизоцианат (0,045 мл, 0,51 ммоль, 1,5 экв.). Реакционную смесь перемешивают при 0°С в течение 1 ч и затем нагревают до 23°С. Прибавляют еще один эквивалент хлорсульфонилизоцианата и смесь перемешивают при 23°С в течение 18 ч. Реакционную смесь разбавляют с помощью Et2O (12 мл), прибавляют 10% водный раствор Na2SO3 и рН реакционной смеси доводят до 8 с помощью 2 М водного раствора КОН. Смесь перемешивают в течение 1,5 ч и затем промывают рассолом. Органический слой сушат (MgSO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (2:1, гексан : EtOAc) и получают неочищенный продукт - NCbz-лактам (20 мг), который превращают в смесь искомых продуктов - соединений Примеров 45а и 45b с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48. Смесь двух продуктов разделяют с помощью препаративной тонкослойной хроматографии (5:95, МеОН : EtOAc) и получают менее полярный изомер, соединение Примера 45а (0,006 г, 3,5% за четыре стадии), МС с электрораспылением [М+1]+ 487,1, и более полярный изомер, соединение Примера 45b (0,003 г, 1,79% за четыре стадии), МС с электрораспылением [М+1]+ 487,1.
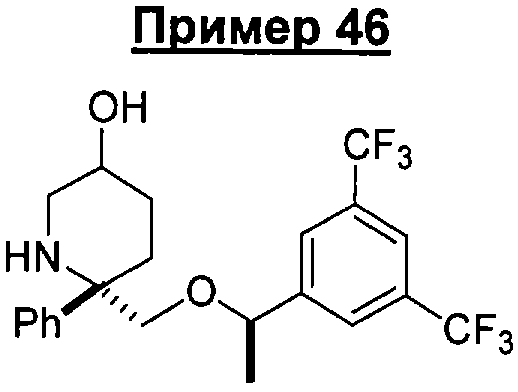
Соединение Примера 46а и соединение Примера 46b получают из Соединения 46 с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48.
МС с электрораспылением [М+1]+ 448,1 для соединения Примера 46а (менее полярный изомер);
МС с электрораспылением [М+1]+ 448,1 для соединения Примера 46b (более полярный изомер).
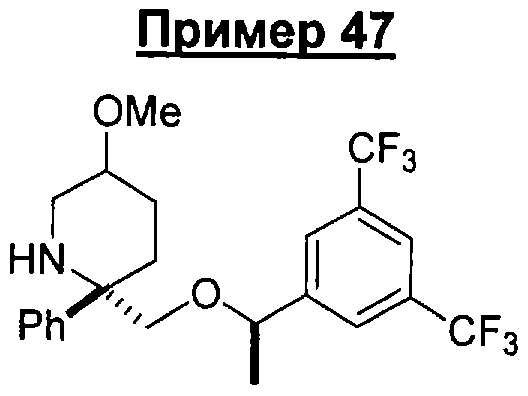
К раствору NCbz-спирта - соединения Примера 46 (0,125 г, 0,215 ммоль, 1 экв.) в сухом DMF (3 мл) при 0°С прибавляют NaH (60% в минеральном масле, 0,017 г, 0,43 ммоль, 2 экв.). Реакционную смесь перемешивают при 0°С в течение 20 мин и затем прибавляют СН3I (0,04 мл, 0,645 ммоль, 3 экв.) и смесь перемешивают при 23°С в течение 18 ч. Неочищенное вещество выливают в CH2Cl2 (100 мл) и промывают рассолом (100 мл×2). Органический слой сушат (MgSO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (4:1, гексан : EtOAc) и получают неочищенный продукт - NCbz-метиловый эфир (69 мг), который гидрируют в смесь искомых продуктов - соединения Примера 47а и 47b с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48. Смесь двух продуктов очищают с помощью хроматографии на колонке Biotage (1:4, гексан : EtOAc) и получают менее полярный изомер, соединение Примера 47а, МС с электрораспылением [М+1]+ 462,1 и более полярный изомер, соединение Примера 47b, МС с электрораспылением [М+1]+ 462,1.
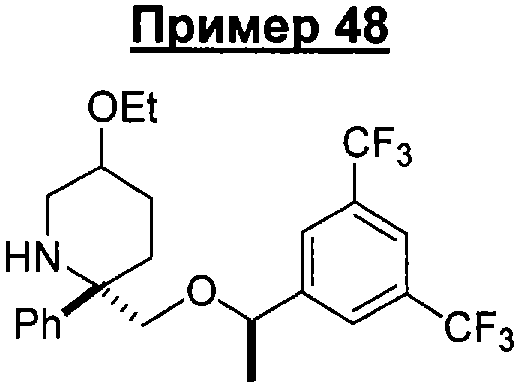
Соединение Примера 48а и соединение Примера 48b получают из Соединения 46 с помощью методики, аналогичной использованной для получения соединения Примера 47а и соединения Примера 47b, и с использованием этилйодида вместо метилйодида.
МС с электрораспылением [M+1]+ 476,1 для соединения Примера 48а (менее полярный изомер);
МС с электрораспылением [М+1]+ 476,1 для соединения Примера 48b (более полярный изомер).
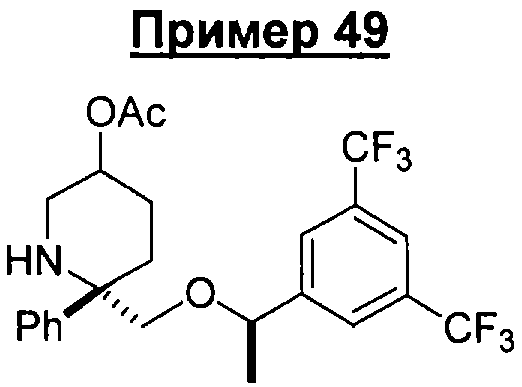
К раствору NCbz-спирта - Соединения 46 (0,118 г, 0,20 ммоль, 1 экв.) в сухом CH2Cl2 (3 мл) при 0°С прибавляют сухой пиридин (0,026 мл, 0,325 ммоль, 1,6 экв.), а затем ацетилхлорид (0,023 мл, 0,325 ммоль, 1,6 экв.). Реакционную смесь нагревают до 23°С и перемешивают в течение 18 ч. Затем смесь концентрируют и очищают с помощью хроматографии на колонке Biotage (4:1, гексан : EtOAc) и получают неочищенный продукт - NCbz-ацетат (108 мг), который гидрируют в неочищенный искомый продукт с помощью методики, аналогичной использованной для получения соединения Примера 23а и соединения Примера 23b из Соединения 48. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (5:95, МеОН : EtOAc) и получают соединение Примера 49 (0,079 г, 79% за две стадии), МС с электрораспылением [M+1]+ 490,1.
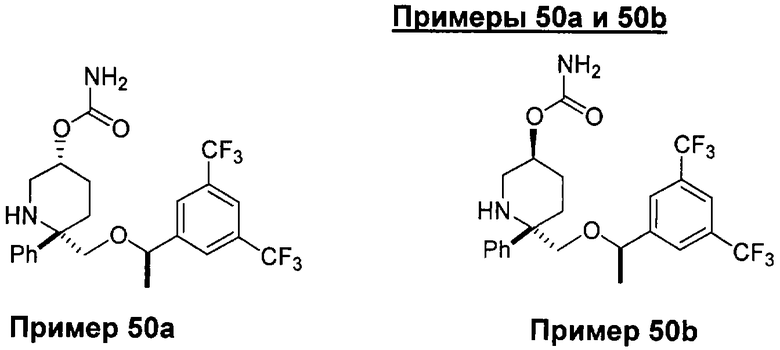
К раствору NCbz-спирта - соединения Примера 46 (0,223 г, 0,385 ммоль, 1 экв.) в CH2Cl2 (8 мл) при 0°С прибавляют трихлорацетилизоцианат (0,055 мл, 0,46 ммоль, 1,2 экв.). Реакционную смесь перемешивают при 0°С в течение 15 мин и затем концентрируют в вакууме. Остаток растворяют в СН3ОН (7 мл) и прибавляют воду (5 мл). Смесь охлаждают до 0°С и прибавляют К2СО3 (0,16 г, 1,16 ммоль, 3 экв.). Реакционную смесь перемешивают при 0°С в течение 1 ч и затем нагревают до 23°С и перемешивают в течение 18 ч. Затем реакционную смесь концентрируют в вакууме и к остатку прибавляют воду (100 мл) прибавляют и смесь экстрагируют с помощью CH2Cl2 (100 мл×2). Объединенные органические слои сушат (Na2SO4), фильтруют, концентрируют и получают неочищенный продукт NCbz-карбамат (232 мг), который гидрируют с получением смеси искомых продуктов - соединений Примеров 50а и 50b с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48. Смесь двух продуктов очищают с помощью хроматографии на колонке Biotage (1:4, гексан : EtOAc) и получают чистое соединение Примера 50а и чистое соединение Примера 50b.
МС с электрораспылением [M+1]+ 491,1 для соединения Примера 50а (менее полярный изомер);
МС с электрораспылением [M+1]+ 491,1 для соединения Примера 50b (более полярный изомер).
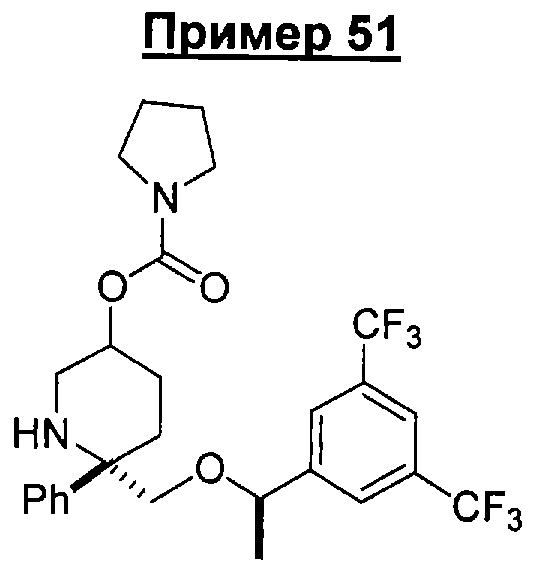
Смесь NCbz-спирта - Соединения 46 (0,2 г, 0,344 ммоль, 1 экв.), 1,4-диоксана (3 мл), 1-пирролидинкарбонилхлорида (0,076 мл, 0,69 ммоль, 2 экв.) и сухого пиридина (0,084 мл, 1,03 ммоль, 3 экв.) нагревают в запаянной трубке при 100°С в течение 18 ч. Реакционную смесь охлаждают до 23°С и разбавляют с помощью EtOAc (150 мл). Смесь промывают водой (100 мл) и органический слой сушат (Na2SO4), фильтруют и концентрируют и получают неочищенный продукт - NCbz-карбамат (232 мг), который гидрируют с получением смеси искомых продуктов - соединений Примеров 51а и 51b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48. Смесь двух продуктов очищают с помощью хроматографии на колонке Biotage (2:3, гексан : EtOAc) и получают менее полярный изомер, соединение Примера 51а и более полярный изомер, соединение Примера 51b.
МС с электрораспылением [M+1]+ 545,1 для соединения Примера 51а;
МС с электрораспылением [М+1]+ 545,1 для соединения Примера 51b.
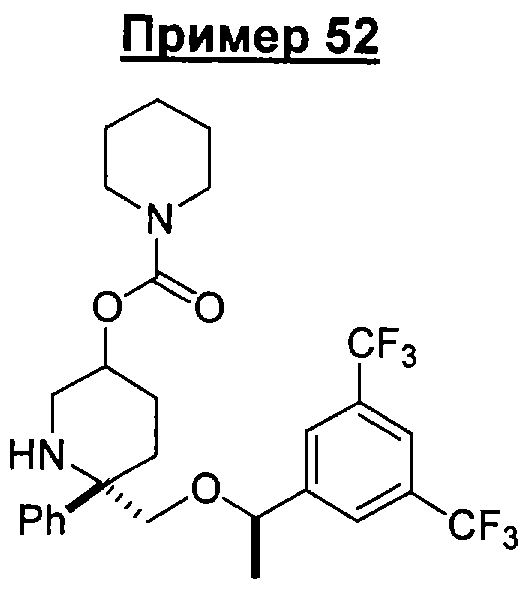
Соединение Примера 52а и соединение Примера 52b получают из Соединения 46 с помощью методики, аналогичной использованной для получения соединения Примера 51а и соединения Примера 51b, и с использованием 1-пиперидинкарбонилхлорида вместо 1-пирролидинкарбонилхлорида.
МС с электрораспылением [M+1]+ 559,1 для соединения Примера 52а (менее полярный изомер);
МС с электрораспылением [M+1]+ 559,1 для соединения Примера 52b (более полярный изомер).
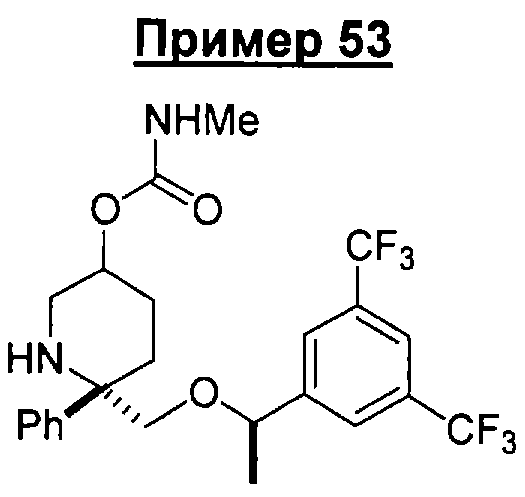
Соединение Примера 53а и соединение Примера 53b получают из Соединения 46 с помощью методики, аналогичной использованной для получения соединения Примера 51а и соединения Примера 51b, и с использованием метилизоцианата вместо 1-пирролидинкарбонилхлорида.
МС с электрораспылением [М+1]+ 505,1 для соединения Примера 53а (менее полярный изомер);
МС с электрораспылением [М+1]+ 505,1 для соединения Примера 53b (более полярный изомер).
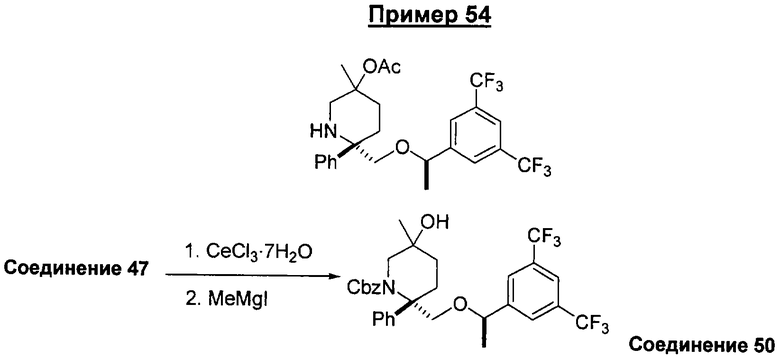
CeCl3 (0,186 г, 0,5 ммоль, 2,1 экв.) помещают в КДК объемом 25 мл и нагревают в вакууме при 140°С в течение 2 ч. Колбу охлаждают до 23°С в атмосфере N2, прибавляют сухой THF (2 мл) и полученную суспензию перемешивают при 23°С в течение 18 ч. Смесь затем охлаждают до 140°С и прибавляют СН3MgI (0,159 мл, 0,476 ммоль, 2 экв.) и перемешивают при 0°С в течение 1 ч. По каплям прибавляют раствор Соединения 47 (0,138 г, 0,238 ммоль, 1 экв.) в сухом THF (2,5 мл) и реакционную смесь перемешивают в атмосфере N2 при 0°С в течение 0,5 ч. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl (50 мл) и реакционную смесь экстрагируют с помощью EtOAc (100 мл×2). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Смесь очищают с помощью хроматографии на колонке Biotage (4:1, гексан : Et2O) и получают NCbz-спирт - Соединение 50 (0,115 г, 80%).
NCbz-спирт - Соединение 50 превращают в искомый продукт - соединение Примера 54 (выход 63% за две стадии) с помощью методики, аналогичной использованной для получения соединения Примера 49 из Соединения 46. МС с электрораспылением [М+1]+ 504,1 для соединения Примера 54.
 К раствору Соединения 47 (0,1 г, 0,173 ммоль, 1 экв.) в сухом пиридине (1 мл) прибавляют метоксиламингидрохлорид (0,058 г, 0,69 ммоль, 4 экв.) и реакционную смесь перемешивают при 23°С в течение 18 ч. Реакцию останавливают водой (50 мл) и реакционную смесь экстрагируют с помощью CH2Cl2 (100 мл×2). Объединенные органические слои сушат (Na2SO4), фильтруют, концентрируют и получают NCbz-оксим (0,102 г, 97%), который гидрируют и получают неочищенный продукт - соединение Примера 55 с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48, за тем исключением, что реакцию проводят в атмосфере подаваемого из баллона Н2 при комнатной температуре, а не в приборе для гидрирования Парра при 40 фунт-сила/дюйм2. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (4:1, EtOAc : гексан) и получают соединение Примера 55 (0,063 г, 79%), МС с электрораспылением [М+1]+ 475,1.
К раствору Соединения 47 (0,1 г, 0,173 ммоль, 1 экв.) в сухом пиридине (1 мл) прибавляют метоксиламингидрохлорид (0,058 г, 0,69 ммоль, 4 экв.) и реакционную смесь перемешивают при 23°С в течение 18 ч. Реакцию останавливают водой (50 мл) и реакционную смесь экстрагируют с помощью CH2Cl2 (100 мл×2). Объединенные органические слои сушат (Na2SO4), фильтруют, концентрируют и получают NCbz-оксим (0,102 г, 97%), который гидрируют и получают неочищенный продукт - соединение Примера 55 с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48, за тем исключением, что реакцию проводят в атмосфере подаваемого из баллона Н2 при комнатной температуре, а не в приборе для гидрирования Парра при 40 фунт-сила/дюйм2. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (4:1, EtOAc : гексан) и получают соединение Примера 55 (0,063 г, 79%), МС с электрораспылением [М+1]+ 475,1.
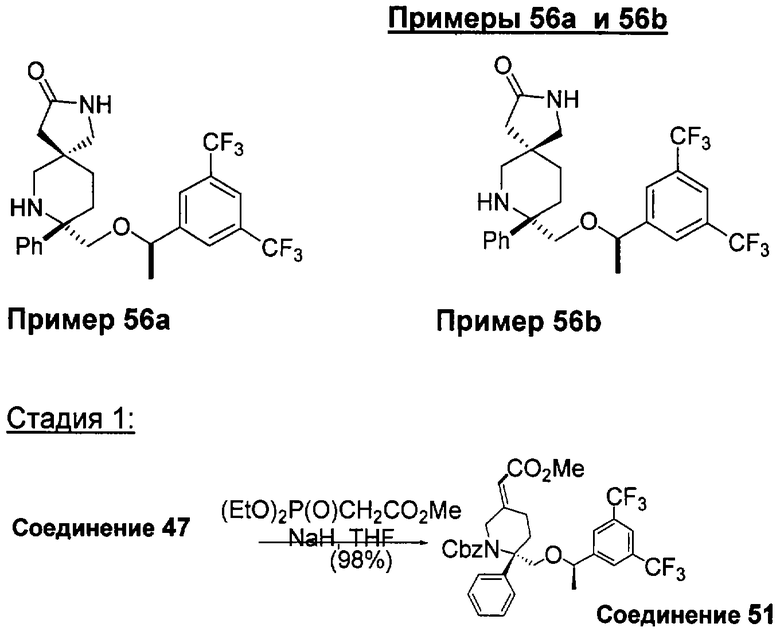
К суспензии NaH (1,8 г, 44,5 ммоль, 60% в масле) в THF (200 мл) при 0°С в атмосфере N2 прибавляют метилдиэтилфосфоноацетат (8,2 мл, 44,5 ммоль). Смесь перемешивают при 0°С в течение 15 мин и прибавляют раствор кетона - Соединения 47 (8,6 г, 14,8 моль) в THF (50 мл). Смеси дают нагреться до комнатной температуры и перемешивают в течение 1 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-EtOAc, 4:1 (об./об.)] дает ненасыщенный сложный эфир - Соединение 51 (9,2 г, 98%) в виде бесцветного масла. МС с электрораспылением [М+1]+=636,1.
Стадия 2:
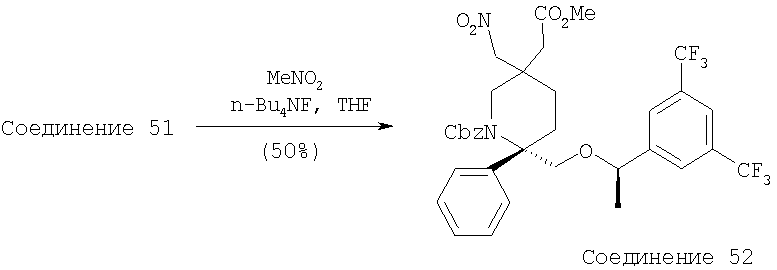
Смесь ненасыщенного сложного эфира - Соединения 51 (9,2 г, 14,5 ммоль) и тетрабутиламмонийфторида (145 мл, 1,0 М в THF) в СН3NO2 кипятят с обратным холодильником в течение 2 ч. Смесь охлаждают до комнатной температуры и реакцию останавливают с помощью насыщенного раствора NH4Cl. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-ацетон, 9:1 (об./об.)] дает менее полярный алкен (4,1 г, 45%) в виде бесцветного масла. Продолжение элюирования с помощью той же системы растворителей дает более полярный сложный нитроэфир - Соединение 52 (5,1 г, 50%) в виде бесцветного масла. МС с электрораспылением [М+1]+ 670,1.
Стадия 3:
Смесь Соединения 52 (5,1 г, 7,32 ммоль), каталитического количества Pd(OH)2 (20% на угле) и каталитического количества Ni Ренея (50% взвесь в воде) встряхивают в приборе для гидрирования Парра при 50 фунт-сила/дюйм2 в течение ночи. Затем смесь фильтруют через слой целита и ВЭЖХ с использованием Chiralcel OD [гексан-изопропанол, 9:1 (об./об.)] дает менее полярный изомер - соединение Примера 56а в виде белого вспененного вещества. МС с электрораспылением [М+1]+=501,1. Продолжение элюирования с помощью той же системы растворителей дает более полярный изомер - соединение Примера 56b в виде бесцветного масла. МС с электрораспылением [М+1]+=501,1.
Пример 57
К раствору этилпропиолата (0,27 мл, 2,69 ммоль) в THF (10 мл) при -78°С в атмосфере N2, прибавляют трет-бутиллитий (1,6 мл, 2,69 ммоль, 1,7 М в пентане). Смесь перемешивают при -78°С в течение 10 мин и прибавляют раствор Соединения 47 (519 мг, 0,90 ммоль) в THF (5 мл). Смесь перемешивают при -78°С в течение 1 ч, затем реакцию останавливают с помощью НОАс при -78°С. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-EtOAc, 4:1 (об./об.)] дает бесцветное масло. Масло растворяют в EtOH и прибавляют каталитическое количество палладия (10% на угле). Смесь встряхивают в приборе для гидрирования Парра при 45 фунт-сила/дюйм2 в течение ночи. Затем смесь фильтруют через слой целита, растворители удаляют в вакууме и получают бесцветное масло. Масло растворяют в толуоле и прибавляют каталитическое количество p-TsOH. Смесь кипятят с обратным холодильником в течение ночи. После охлаждения до комнатной температуры реакцию останавливают с помощью насыщенного раствора NaHCO3. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (250 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и получают смесь соединений Примеров 57а и 57b в виде бесцветного масла. Разделение с помощью хроматографии на колонке [гексан-эфир, 1:2 (об./об.)] дает менее полярный содержащийся в меньшем количестве изомер - соединение Примера 57а (67 мг, 15%) в виде белого вспененного вещества. МС с электрораспылением [М+1]+=502,1. Продолжение элюирования с помощью той же системы растворителей дает более полярный содержащийся в большем количестве изомер - соединение Примера 57b (134 мг, 30%) в виде белого твердого вещества. МС с электрораспылением [М+1]+=502,1.
К раствору соединения Примера 57а (112 мг, 0,22 ммоль) в THF (5 мл) при -78°С в атмосфере N2 прибавляют бис(триметилсилил)амид лития (1,1 мл, 1,12 ммоль, 1,0 М в THF). Смесь перемешивают при -78°С в течение 1 ч и прибавляют СН3I (70 мкл, 1,12 ммоль). Смесь перемешивают при -78°С в течение 1 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (100 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-эфир, 3:1 (об./об.)] дает соединение Примера 58 (92 мг, 78%) в виде бесцветного масла. МС с электрораспылением [M+1]+=530,1.
Соединение Примера 59 (75%) получают из соединения Примера 57b способом, аналогичным использованному для получения соединения Примера 58 из соединения Примера 57а. МС с электрораспылением [М+1]+=530,1.
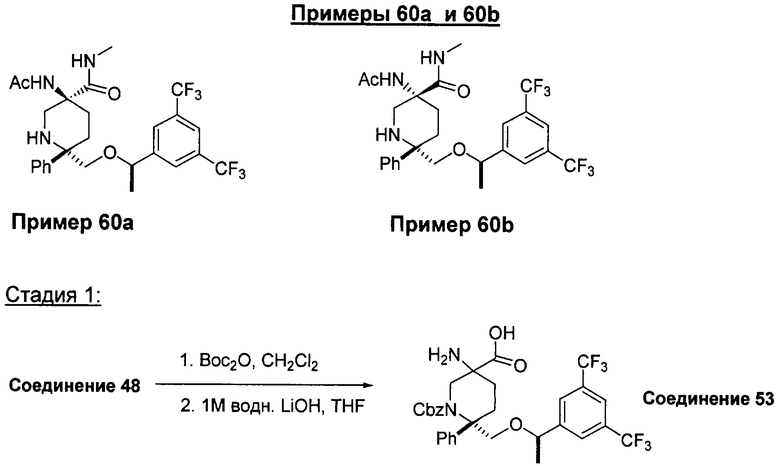
К раствору Соединения 48 (0,5 г, 0,77 ммоль, 1 экв.) в CH2Cl2 (30 мл) прибавляют ди-трет-бутилдикарбонат (0,37 г, 1,69 ммоль, 2,2 экв.), а затем DMAP (0,035 г, 0,286 ммоль, 0,37 экв.) и реакционную смесь перемешивают при 23°С в течение 18 ч. Затем реакционную смесь фильтруют через тонкий слой диоксида кремния с использованием (1:1, гексан : EtOAc) и концентрируют в вакууме и получают diBoc-гидантоин (0,59 г, 90%). diBoc-гидантоин (0,59 г, 0,7 ммоль, 1 экв.) растворяют в THF (30 мл) и прибавляют 1 М водный раствор LiOH (5,56 мл, 5,56 ммоль, 8 экв.). Реакционную смесь перемешивают при 23°С в течение 18 ч. К реакционной смеси прибавляют насыщенный водный раствор NaHCO3 и экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (Na2SO4), фильтруют и концентрируют и получают неочищенное Соединение 53 (0,52 г), которое используют в следующей реакции без дополнительной очистки.
Стадия 2:
К смеси неочищенного Соединения 53 (0,52 г) с пиридином (3 мл) и THF (2 мл) при 0°С прибавляют ацетилхлорид (0,072 мл, 1 ммоль, 1,2 экв.), реакционную смесь нагревают до 23°С и перемешивают в течение 18 ч. Затем реакционную смесь концентрируют и очищают с помощью хроматографии на колонке Biotage (5:95, МеОН : EtOAc) и получают желтое масло - N-ацетилированный продукт (0,31 г, 0,456 ммоль, 1 экв.), который растворяют в THF (10 мл). Прибавляют 2 М раствор СН3NH2 в THF (2,3 мл, 4,6 ммоль, 10 экв.) и реакционную смесь перемешивают при 23°С в течение 18 ч. Смесь разбавляют с помощью EtOAc (100 мл) и промывают с помощью насыщенного водного раствора NaHCO3 (100 мл). Органический слой сушат (Na2SO4), фильтруют, концентрируют и получают неочищенный NCbz-амид, который гидрируют и получают смесь двух изомеров соединений Примеров 60а и 60b с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 43b из Соединения 48. Смесь двух продуктов разделяют на колонке для ВЭЖХ ChiralPak AD с использованием (1:9, IPA : гексан) и получают более полярный изомер - чистое соединение Примера 60а. МС с электрораспылением [М+1]+=546,1, и менее полярный изомер - чистое соединение Примера 60b, МС с электрораспылением [М+1]+=546,1.
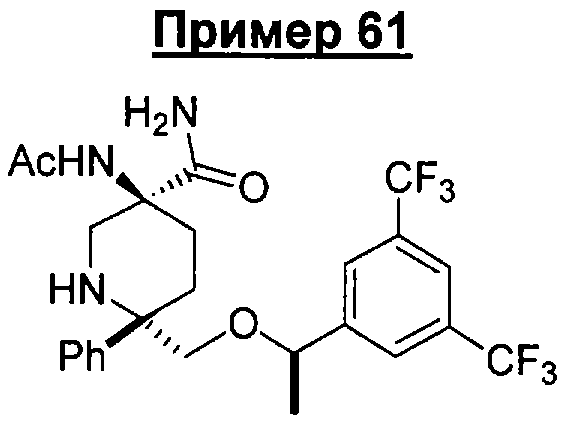
Соединение Примера 61 получают из соединения Примера 43а с помощью методики, аналогичной использованной для получения соединений Примеров 60а и 60b из Соединения 48, но с использованием раствора аммиака (0,5 М в 1,4-диоксане) вместо раствора CH3NH2 (2 М в THF).
МС с электрораспылением [М+1]+=532,1.
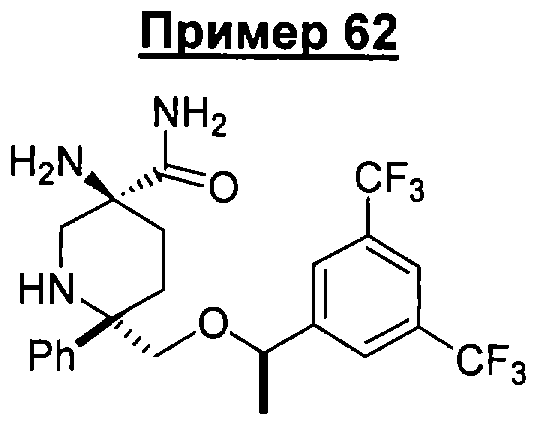
Стадия 1:
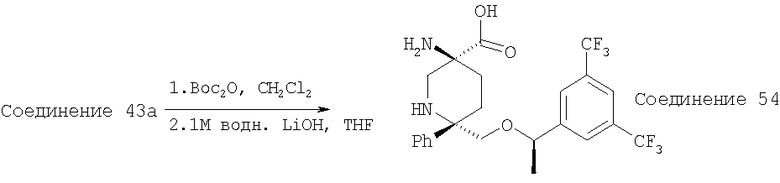
Соединение 54 получают из соединения Примера 43а с помощью методики, аналогичной использованной для получения Соединения 53 из Соединения 48. Соединение 54 используют в следующей реакции без дополнительной очистки.
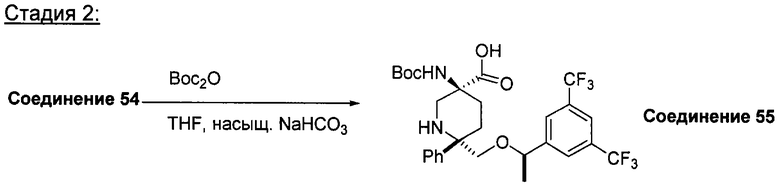
К смеси Соединения 54 (0,5 г, 1,02 ммоль, 1 экв.) в THF (30 мл) прибавляют насыщенный водный раствор NaHCO3, а затем ди-трет-бутилдикарбонат (0,58 г, 2,65 ммоль, 2,6 экв.). Реакционную смесь перемешивают при 23°С в течение 18 ч. Смесь охлаждают до 0°С и прибавляют 10% водный раствор лимонной кислоты (20 мл) и полученную смесь экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (Na2SO4), фильтруют, концентрируют и получают неочищенное Соединение 55 (0,93 г), которое используют в следующей реакции без дополнительной очистки.
Стадия 3:
К раствору Соединения 55 (0,93 г, 1,57 ммоль, 1 экв.) в CH2Cl2 (15 мл) прибавляют DIEA (0,83 мл, 4,72 ммоль, 3 экв.), а затем РуВОР (1,23 г, 2,4 ммоль, 1,3 экв.). Через 15 мин к реакционной смеси прибавляют 0,5 М раствор аммиака в 1,4-диоксане (31,5 мл, 15,75 ммоль, 10 экв.) и перемешивают при 23°С в течение 18 ч. Реакцию останавливают водой (100 мл) и экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (Na2SO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (1:10:89, Et3N : МеОН : EtOAc) и получают NBoc-амид, который растворяют в CH2Cl2 (10 мл) и охлаждают до 0°С. Прибавляют TFA (6 мл) и реакционную смесь нагревают до 23°С и перемешивают в течение 2 ч. Реакцию осторожно останавливают с помощью насыщенного водного раствора NaHCO3 (100 мл) и разбавляют с помощью CH2Cl2 (100 мл). Органический слой отделяют, сушат (Na2SO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (10:90, МеОН : EtOAc) и получают искомый продукт - соединение Примера 62 (0,18 г, 35% за три стадии), МС с электрораспылением [М+1]+=490,1.

Соединение Примера 63 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13 и с использованием хлорангидрида циклопропанкарбоновой кислоты вместо пропионилхлорида, а также с использованием DIEA (1,3 экв.). МС с электрораспылением [М+1]+=558,1.
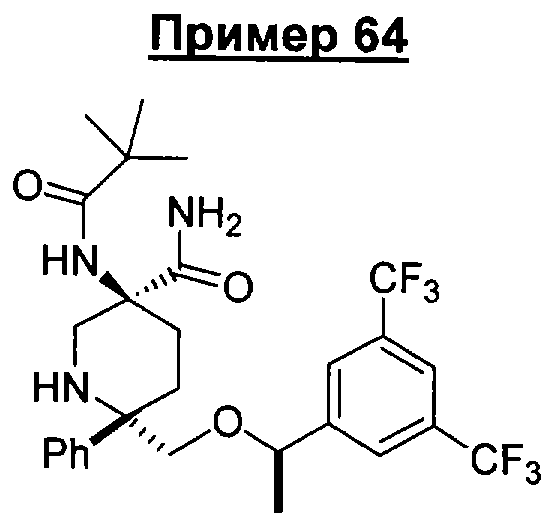
Соединение Примера 64 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13 и с использованием трет-бутилхлорида вместо пропионилхлорида. МС с электрораспылением [М+1]+=574,1.
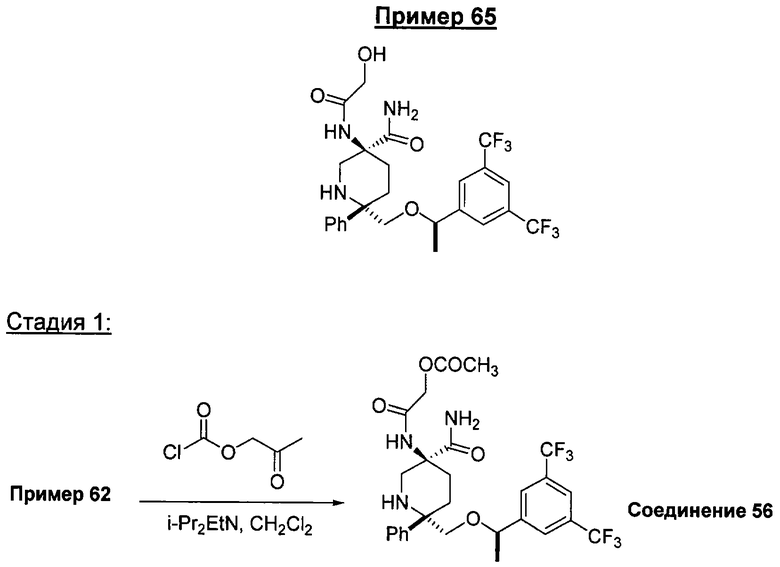
Соединение 56 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13, но с использованием ацетоксиацетилхлорида вместо пропионилхлорида. Неочищенное Соединение 56 используют в следующей реакции без дополнительной очистки.
Стадия 2:
Неочищенное Соединение 56 растворяют в МеОН (5 мл), КНСО3 (3 экв.) прибавляют и реакционную смесь перемешивают при 23°С в течение 18 ч. Реакционную смесь концентрируют и очищают с помощью хроматографии на колонке Biotage (10:90, МеОН : EtOAc) и получают искомый продукт - соединение Примера 65, МС с электрораспылением [М+1]+=548,1.
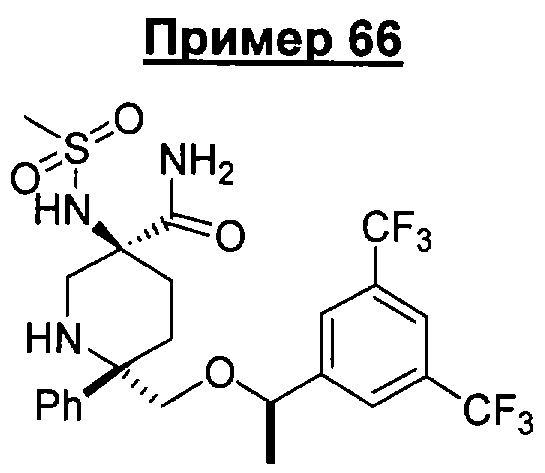
Соединение Примера 66 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13, и с использованием СН3SO2Cl вместо пропионилхлорида.
МС с электрораспылением [М+1]+=568,1 для соединения Примера 66.
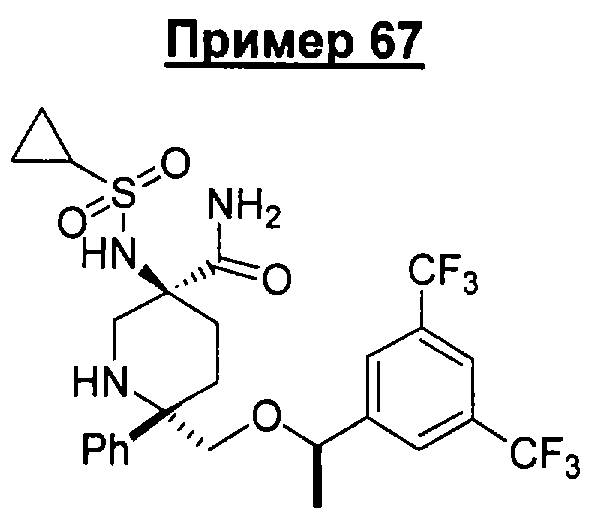
Соединение Примера 67 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13, но с использованием циклопропилсульфонилхлорида вместо пропионилхлорида. МС с электрораспылением [М+1]+=594,1.
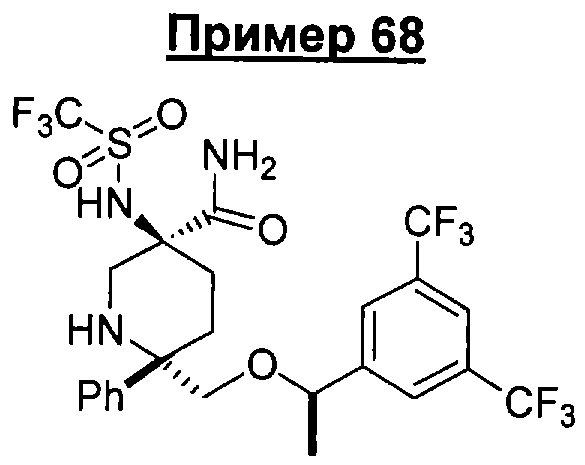
Соединение Примера 68 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13, но с использованием трифторметансульфонового ангидрида вместо пропионилхлорида. МС с электрораспылением [М+1]+=622,1 для соединения Примера 68.
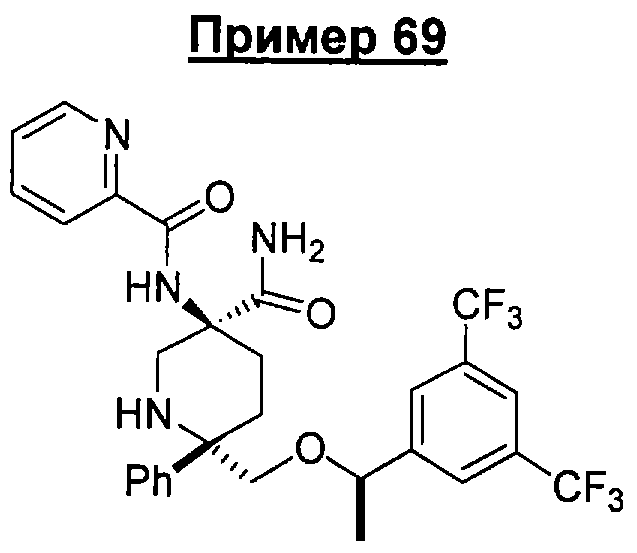
Соединение Примера 69 получают из соединения Примера 62 с помощью методики, аналогичной использованной для получения соединения Примера 14 из соединения Примера 13, и с использованием никотиноилхлорида вместо пропионилхлорида. МС с электрораспылением [М+1]+=595,1 для соединения Примера 69.
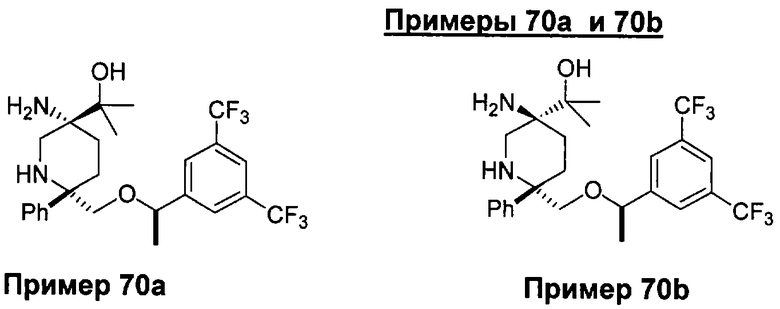
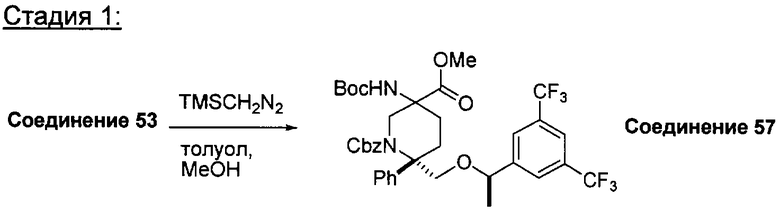
К смеси Соединения 53 (4 г, 5,52 ммоль, 1 экв.), толуола (46 мл) и МеОН (18 мл) при 0°С прибавляют TMSCH2N2 (2 М раствор в гексане, 13,8 мл, 27,6 ммоль, 5 экв.) и полученный раствор перемешивают при 0°С в течение 30 мин. Затем реакционную смесь концентрируют и очищают с помощью хроматографии на колонке Biotage (2:1, гексан : EtOAc) и получают Соединение 57 (1,8 г, 44%).
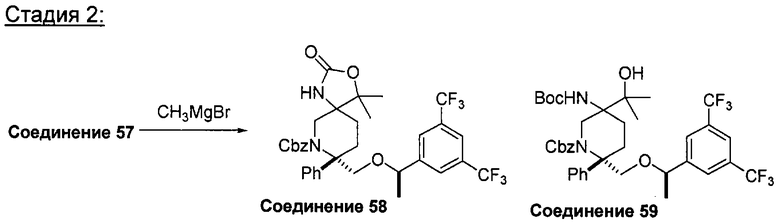
К смеси Соединения 57 (1 г, 1,35 ммоль, 1 экв.) в сухом THF (18 мл) при 0°С прибавляют СН3MgBr (1 М раствор в н-бутиловом эфире, 3,24 мл, 3,24 ммоль, 2,4 экв.) и полученный раствор перемешивают при 0°С в течение 30 мин. Затем реакционную смесь нагревают до 23°С и перемешивают в течение 18 ч. Реакцию останавливают насыщенным водным раствором NaHCO3 (100 мл) и экстрагируют с помощью EtOAc (200 мл). Органический слой отделяют, сушат (Na2SO4), фильтруют и концентрируют. Смесь очищают с помощью хроматографии на колонке Biotage (2:1, гексан EtOAc) и получают более полярное Соединение 58 (0,52 г, 56%) и менее полярное Соединение 59 (0,31 г, 34%).
Стадия 3:
У Соединения 59 с помощью TFA удаляют защитную группу с использованием методики, описанной при получении соединения Примера 62. Полученный NCbz-аминоспирт гидрируют и получают смесь двух изомеров соединений Примеров 70а и 70b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48. Смесь двух продуктов разделяют на колонке для ВЭЖХ ChiralCel OD с использованием (1:9, IPA : гексан) и получают менее полярный изомер - соединение Примера 70а, МС с электрораспылением [М+1]+ 505,1, и более полярный изомер - соединение Примера 70b, МС с электрораспылением [М+1]+ 505,1.
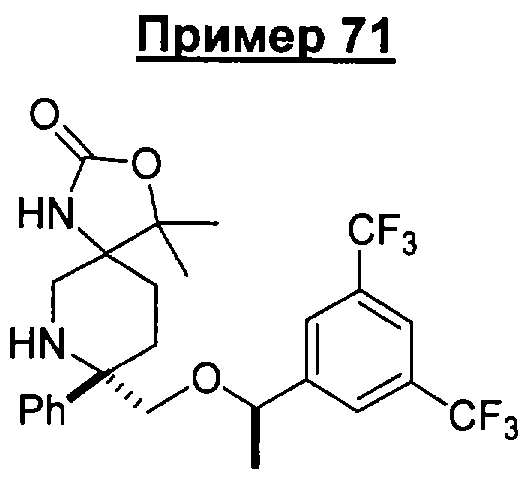
Соединение 58 гидрируют с получением смеси искомых продуктов - соединений Примеров 71а и 71b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48. Смесь двух продуктов очищают с помощью хроматографии на колонке Biotage (1:1, гексан : EtOAc) и получают чистый менее полярный изомер - соединение Примера 71а, МС с электрораспылением [М+1]+ 531,1, и чистый более полярный изомер - соединение Примера 71b, МС с электрораспылением [M+1]+ 531,1.
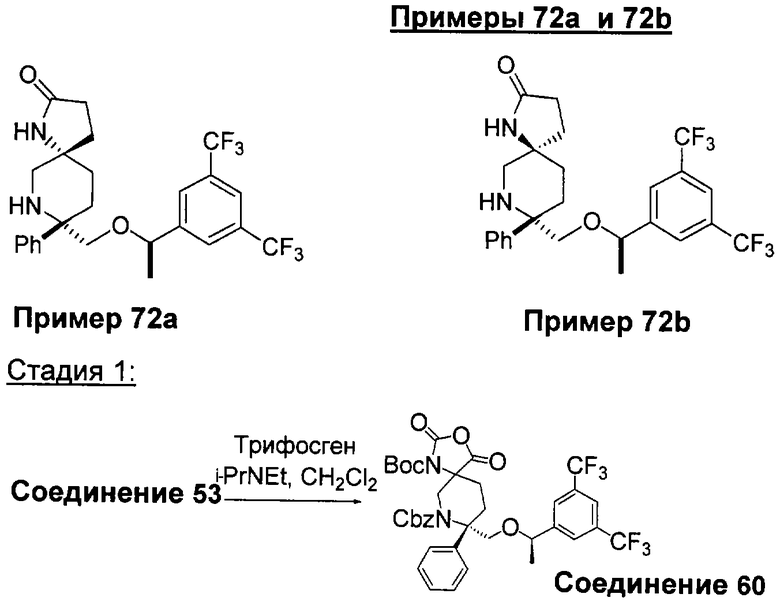 К раствору неочищенного Соединения 53 (19 г) в CH2Cl2 (300 мл) при комнатной температуре прибавляют DIEA (15 мл, 0,087 моль), а затем трифосген (4,34 г, 0,015 моль). Смесь перемешивают при комнатной температуре в течение 18 ч и фильтруют через слой диоксида кремния. Растворители удаляют в вакууме и получают неочищенное Соединение 60 в виде желтого масла, которое используют в следующей реакции без дополнительной очистки.
К раствору неочищенного Соединения 53 (19 г) в CH2Cl2 (300 мл) при комнатной температуре прибавляют DIEA (15 мл, 0,087 моль), а затем трифосген (4,34 г, 0,015 моль). Смесь перемешивают при комнатной температуре в течение 18 ч и фильтруют через слой диоксида кремния. Растворители удаляют в вакууме и получают неочищенное Соединение 60 в виде желтого масла, которое используют в следующей реакции без дополнительной очистки.
Стадия 2:
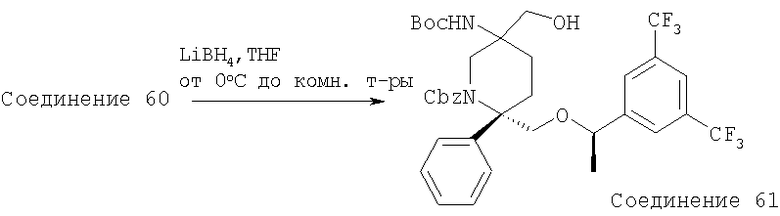
К неочищенному Соединению 60 в THF (200 мл) при 0°С небольшими порциями прибавляют LiBH4 (1,26 г, 0,058 моль). Смесь перемешивают при комнатной температуре в течение 18 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (100×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-EtOAc, 4:1 (об./об.)] дает Соединение 61 (12,9 г, суммарный выход 62%) в виде белого вспененного вещества.
Стадия 3:
Оксалилхлорид (4,2 мл, 0,048 моль) прибавляют к раствору DMSO (6,8 мл, 0,096 моль) в СН2Cl2 (300 мл) при -78°С в атмосфере N2. Смесь перемешивают при -78°С в течение 15 мин, а затем прибавляют раствор Соединения 61 (8,5 г, 0,012 моль) в CH2Cl2 (100 мл). Смесь перемешивают при -78°С в течение еще 1 ч и прибавляют Et3N (23,5 мл). Охлаждающую баню удаляют и смесь нагревают до комнатной температуры, а затем реакцию останавливают с помощью насыщенного раствора NaHCO3. Слои разделяют и водный слой экстрагируют с помощью СН2Cl2 (150 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Удаление растворителей в вакууме дает альдегид в виде желтого масла. К смеси NaH (1,44 г, 0,036 моль) в THF при 0°С прибавляют метилдиэтилфисфоноацетат (6,6 мл, 0,036 моль). Смесь перемешивают при 0°С в течение 15 мин и прибавляют раствор альдегида в THF (100 мл). Охлаждающую баню удаляют и смесь перемешивают при комнатной температуре в течение 1 ч. Реакцию останавливают с помощью насыщенного раствора NH4Cl. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-EtOAc, 4:1 (об./об.)] дает сложный эфир в виде белого вспененного вещества. Сложный эфир растворяют в EtOH (100 мл) и прибавляют каталитическое количество палладия (1,28 г, 10% на угле). Смесь встряхивают в атмосфере H2 (50 фунт-сила/дюйм2) в течение 2 суток. Затем к смеси прибавляют каталитическое количество Pd(OH)2 (20% на угле) и смесь повторно встряхивают в атмосфере H2 (50 фунт-сила/дюйм2) в течение 5 ч. Затем смесь фильтруют через слой целита, растворители удаляют в вакууме и получают белое вспененное вещество. Затем вспененное вещество растворяют в CH2Cl2 (200 мл) и прибавляют TFA (8,9 мл, 0,12 моль). Смесь перемешивают при комнатной температуре в течение 18 ч и охлаждают при 0°С, а затем нейтрализуют с помощью насыщенного раствора NaHCO3. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и получают желтое масло. Масло растворяют в СН3ОН (50 мл) и прибавляют каталитическое количество К2СО3 (166 мг, 0,0012 моль). Смесь нагревают при 60°С в течение 2 ч. После охлаждения до комнатной температуры смесь фильтруют через слой диоксида кремния и растворители удаляют в вакууме. Очистка с помощью хроматографии на колонке (EtOAc) дает смесь двух изомеров - соединений Примеров 72а и 72b (2,3 г, суммарный выход 38%) в виде белого вспененного вещества. Разделение с помощью ВЭЖХ с использованием Chiralcel OD [гексан-изопропанол, 95:5 (об./об.)] дает менее полярный, содержащийся в большем количестве изомер - соединение Примера 72а в виде белого вспененного вещества. МС с электрораспылением [М+1]+=501,1. Продолжение элюирования с помощью той же системы растворителей дает более полярный, содержащийся в меньшем количестве изомер - соединение Примера 72b в виде бесцветного масла. МС с электрораспылением [М+1]+=501,1.
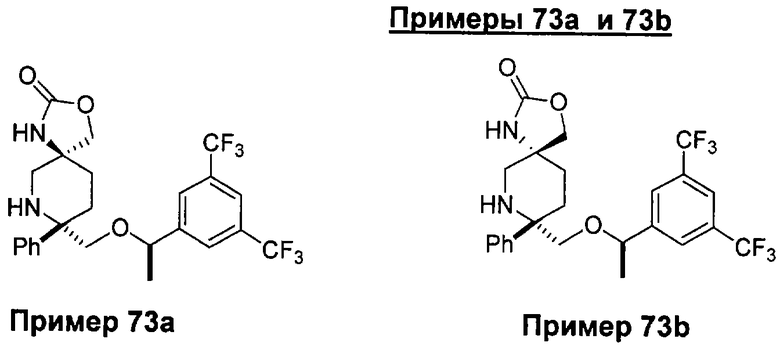
К раствору Соединения 61 (3 г, 4,22 ммоль, 1 экв.) в DMF (60 мл) при 0°С прибавляют NaH (60% в минеральном масле, 0,122 г, 5,07 ммоль, 1,2 экв.), смеси дают нагреться до 23°С и перемешивают в течение 45 мин. Реакцию останавливают водой (100 мл) и экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (2:1, гексан : EtOAc) и получают искомый продукт, который гидрируют и получают смесь двух изомеров - соединений Примеров 73а и 73b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48. Смесь двух продуктов разделяют на колонке для ВЭЖХ ChiralPak AD с использованием (5:95, IPA : гексан) и получают чистый менее полярный изомер - соединение Примера 73а и более полярный изомер - соединение Примера 73b.
МС с электрораспылением [М+1]+=503,1 для соединения Примера 73а;
МС с электрораспылением [М+1]+=503,1 для соединения Примера 73b.
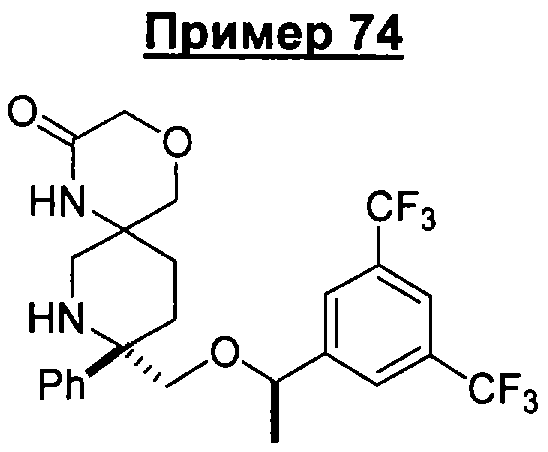
Соединение 61 (1,68 г, 2,36 ммоль, 1 экв.) растворяют в CH2Cl2 (50 мл), прибавляют TFA (5,46 мл, 70,9 ммоль, 30 экв.) и реакционную смесь перемешивают при 23°С в течение 2,5 ч. Реакцию осторожно останавливают с помощью насыщенного водного раствора NaHCO3 (150 мл) и разбавляют с помощью CH2Cl2 (100 мл). Органический слой отделяют, сушат (Na2SO4), фильтруют, концентрируют и получают неочищенный аминоспирт (1,4 г, 97%). Продукт (0,32 г, 0,524 ммоль, 1 экв.) растворяют в сухом THF (10 мл) и прибавляют NaH (60% в минеральном масле, 0,025 г, 0,63 ммоль, 1,2 экв.). Реакционную смесь перемешивают при 23°С в течение 5 мин и затем прибавляют этилхлорацетат (0,062 мл, 0,576 ммоль, 1,1 экв.) и реакционную смесь перемешивают в течение 2,5 ч. Реакцию осторожно останавливают с помощью насыщенного водного раствора NaHCO3 (100 мл) и разбавляют с помощью EtOAc (200 мл). Органический слой отделяют, сушат (Na2SO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (2:3, гексан : EtOAc) и получают продукт (0,1 г, 32%), который гидрируют и получают смесь двух изомеров - соединений Примеров 74а и 74b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48. Смесь двух продуктов разделяют на колонке для ВЭЖХ ChiralCel OD с использованием (1:9, IPA : гексан) и получают чистое соединение Примера 74а, МС с электрораспылением [M+1]+ 517,1, и чистое соединение Примера 74b, MC с электрораспылением [М+1]+ 517,1.
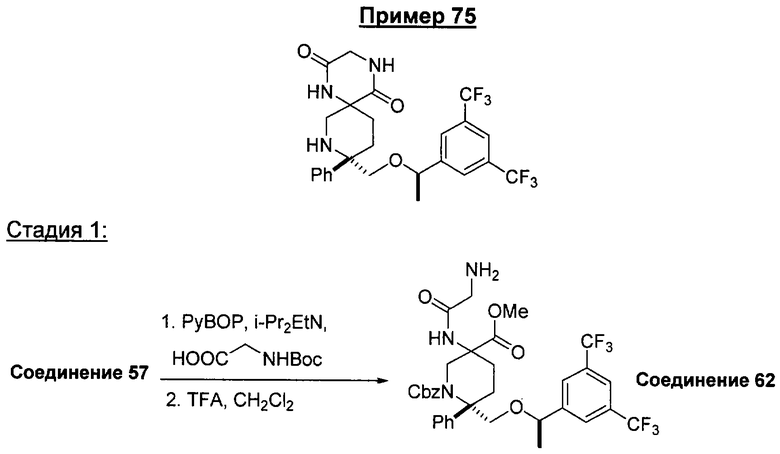
Соединение 57 превращают в Соединение 62 (выход за две стадии 72%) с использованием сочетания с помощью PyBOP и последующего удаления защитной группы с помощью TFA, как это описано при получении соединения Примера 62 из Соединения 55, но с использованием Соединения 57 (1 экв.) вместо аммиака и NH-Boc-глицина (2 экв.) вместо Соединения 55.
Стадия 2:
Соединение 62 (0,5 г, 0,72 ммоль, 1 экв.) растворяют в МеОН (10 мл) и прибавляют Et3N (1 мл, 7,2 ммоль, 10 экв.). Полученную смесь нагревают при 23°С в течение 18 ч. Затем реакционную смесь концентрируют и очищают с помощью хроматографии на колонке Biotage (EtOAc) и получают NCbz-дикетопиперазин (0,33 г), который гидрируют и получают смесь двух изомеров - соединений Примеров 75а и 75b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48. Смесь двух продуктов разделяют на колонке для ВЭЖХ ChiralPak AD с использованием (5:95, IPA : гексан) и получают чистый менее полярный изомер - соединение Примера 75а (0,03 г, выход за две стадии 8%), МС с электрораспылением [М+1]+ 530,1, и более полярный изомер - соединение Примера 75b (0,04 г, выход за две стадии 11%), МС с электрораспылением [М+1]+ 530,1.
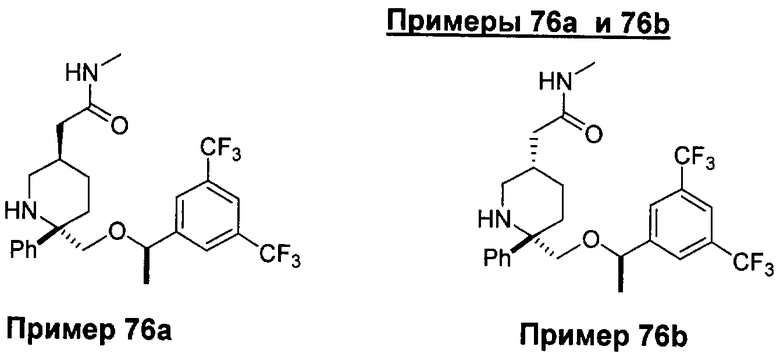
Соединение 51 (3,66 г, 5,76 ммоль, 1 экв.) гидрируют с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 43b из Соединения 48, гидрированный продукт (2,85 г) обрабатывают с помощью СН3NH2 (2 М раствор в СН3ОН, 200 мл) и перемешивают при 23°С в течение 18 ч. Затем реакционную смесь концентрируют, очищают с помощью хроматографии на колонке Biotage (1:9, МеОН : EtOAc) и получают смесь двух изомеров соединений Примеров 76а и 76b. Смесь двух изомеров разделяют на колонке для ВЭЖХ ChiralPak AD с использованием (5:95, IPA : гексан) и получают менее полярный изомер - соединение Примера 76b, МС с электрораспылением [М+1]+ 503,1, более полярный изомер - соединение Примера 76а, МС с электрораспылением [М+1]+ 503,1.
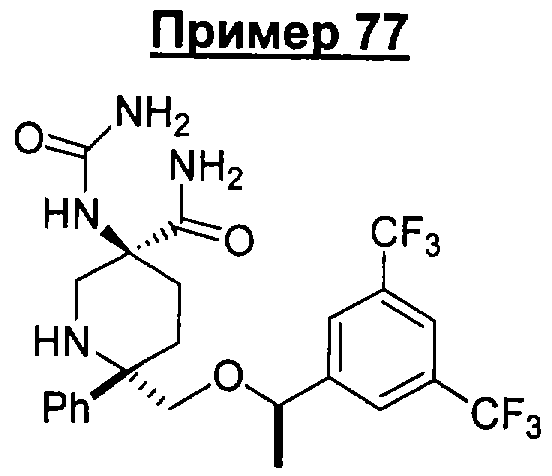
Соединение Примера 62 (0,07 г, 0,133 ммоль, 1 экв.) растворяют в CH2Cl2 (3 мл) и прибавляют DIEA (0,03 мл, 0,147 ммоль, 1,1 экв.), а затем 4-метоксифенил-(pmb)-изоцианат (0,021 мл, 0,147 ммоль, 1,1 экв.) и реакционную смесь перемешивают при 23°С в течение 18 ч. Затем реакционную смесь концентрируют, обрабатывают с помощью СН2CN (3 мл) и воды (1 мл) и смесь охлаждают до 0°С. Прибавляют нитрат аммония-церия (0,24 г, 0,44 ммоль, 4 экв.) и реакционную смесь перемешивают при 0°С в течение 45 мин. Реакцию останавливают насыщенным водным раствором NaHCO3 (100 мл) и экстрагируют с помощью EtOAc (200 мл). Органический слой отделяют, сушат (Na2SO4), фильтруют и концентрируют. Смесь очищают с помощью хроматографии на колонке Biotage (15:85, МеОН : EtOAc) и получают соединение Примера 77 (0,03 г, 42%), МС с электрораспылением [М+1]+ 533,1.
Примеры 78а и 78b
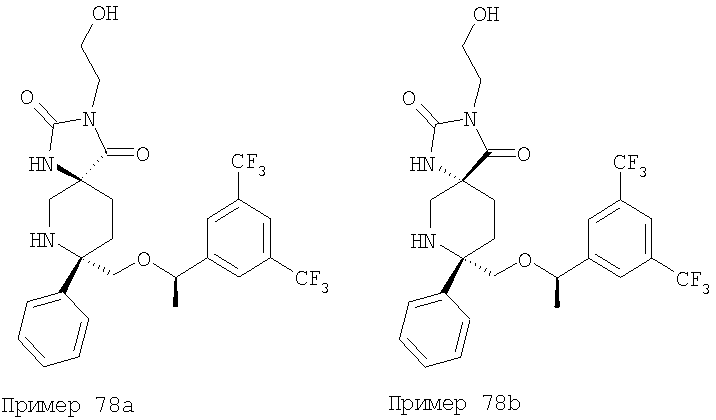
Стадия 1:
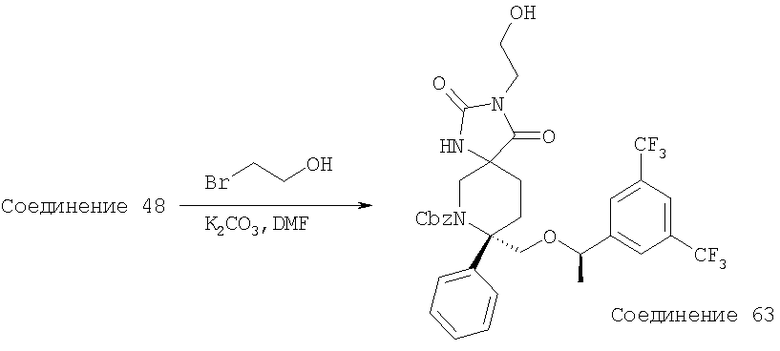
В высушенной на огне КДК объемом 15 мл Соединение 48 (0,25 г, 0,385 ммоль, 1 экв.) в DMF (1 мл) прибавляют к К2СО3 (0,106 г, 0,77 ммоль, 2 экв.), а затем 2-бромэтанол (0,033 мл, 0,46 ммоль, 1,2 экв.) и смесь перемешивают в течение 2 ч при комнатной температуре, затем нагревают при 50°С в течение 6 ч. За протеканием реакции следят с помощью ТСХ (60/40, EtOAc/гексан). Реакционную смесь охлаждают до 0°С, реакцию останавливают с помощью Н2O, разбавляют с помощью EtOAc и промывают рассолом. Органические слои объединяют и сушат над Na2SO4, фильтруют и концентрируют. Реакционную смесь очищают с помощью колонки Biotage с использованием смеси от 2:3 EtOAc/гексан до 3:2 EtOAc/гексан для элюирования Соединения 63 в виде смеси двух изомеров (0,258 г, 97%), МС с электрораспылением [М+1]+ 694,1.
Стадия 2:
К раствору Соединения 63 (0,25 г, 0,36 ммоль, 1 экв.) в безводном МеОН прибавляют (5,5 мл) 20% Pd(OH)2/C (0,08 г). Реакционную смесь продувают током N2, а затем током Н2 и перемешивают в течение 18 ч в атмосфере Н2. За протеканием реакции следят с помощью ТСХ (60/40, EtOAc/гексан). Катализатор отфильтровывают через слой целита, раствор концентрируют и получают неочищенный продукт. Вещество обрабатывают с помощью флэш-хроматографии с использованием колонки Biotage (80:20, EtOAc/гексан). Изомеры разделяют и получают соединение Примера 78а и соединение Примера 78b (0,13 г, 63%).
МС с электрораспылением [M+1]+ 560,1 для соединения Примера 78а (менее полярный изомер);
МС с электрораспылением [M+1]+ 560,1 для соединения Примера 78b (более полярный изомер).
Примеры 79а и 79b
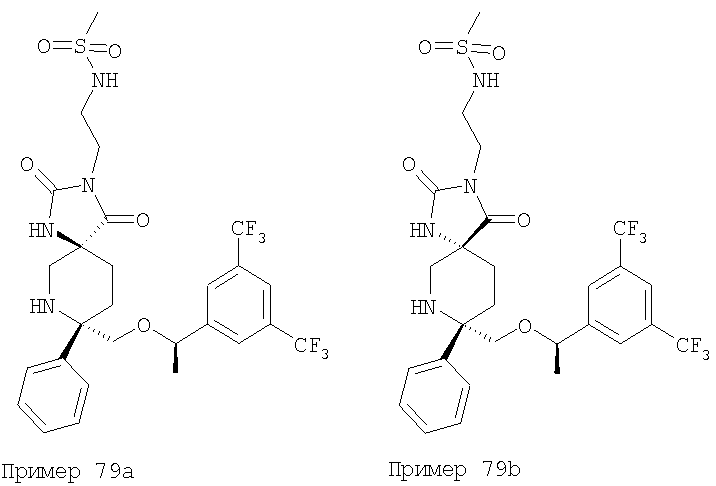
Стадия 1:
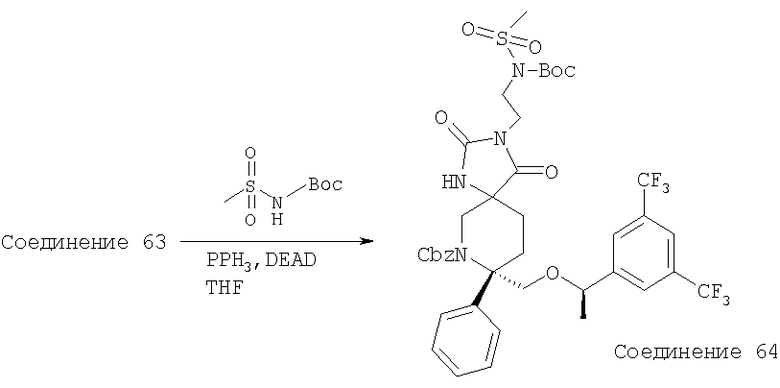
N-(Boc)-метансульфонамид (0,041 г, 0,43 ммоль, 1,5 экв.) растворяют в сухом THF (1 мл) и прибавляют трифенилфосфин (0,228 г, 0,43 ммоль, 3 экв.). Полученный раствор перемешивают в атмосфере N2 и прибавляют раствор Соединения 63 (0,2 г, 0,29 ммоль, 1 экв.) в THF, а затем диэтилазодикарбоксилат (DEAD) (0,12 мл, 0,26 ммоль, 2,5 экв.). За протеканием реакции следят с помощью ТСХ (60/40, EtOAc/гексан). После завершения реакции реакционную смесь концентрируют и получают желтое масло, которое обрабатывают с помощью флэш-хроматографии с использованием колонки Biotage (1:1, EtOAc/гексан) для элюирования конечного продукта, Соединения 64, в виде смеси двух изомеров (0,24 г, 95%), МС с электрораспылением [М+1]+ 871,1.
Стадия 2:
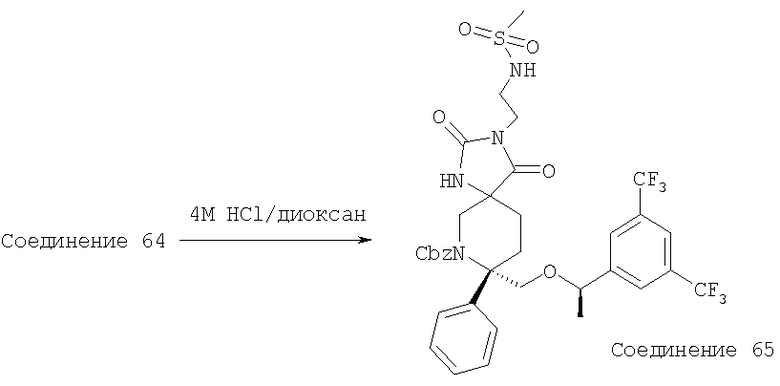
К раствору Соединения 64 (0,25 г, 0,29 ммоль, 1 экв.) в сухом CH2Cl2 (10 мл) при 0°С прибавляют 4 М HCl в диоксане (0,755 мл, 2,9 ммоль, 10 экв.). Реакционную смесь нагревают до комнатной температуры и за протеканием реакции следят с помощью ТСХ (60/40, EtOAc/гексан). После завершения реакции ее останавливают водой, реакционную смесь разбавляют с помощью CH2Cl2, промывают насыщенным раствором NaHCO3, сушат над Na2SO4 и получают неочищенное Соединение 65 в виде смеси двух изомеров (0,2 г, 89%). Неочищенное вещество далее используют без какой-либо очистки. МС с электрораспылением [М+1]+ 771,1 для соединения 65.
Стадия 3:
К раствору Соединения 65 (0,2 г, 0,26 ммоль, 1 экв.) в МеОН (5 мл) прибавляют 10% Pd/C, а затем формиат аммония (0,082 г, 1,3 ммоль, 5 экв.). Реакционную смесь кипятят с обратным холодильником в атмосфере N2 в течение 3 ч, затем охлаждают до комнатной температуры, фильтруют через целит и концентрируют. Остаток растворяют в EtOAc, промывают с помощью NaHCO3 и сушат над Na2SO4. Неочищенное вещество обрабатывают с помощью препаративной тонкослойной хроматографии. Выделяют оба изомера и получают чистое соединение Примера 79а и соединение Примера 79b (0,06 г, суммарный выход для обоих изомеров 36%).
МС с электрораспылением [М+1]+ 637,1 для соединения Примера 79а (менее полярный изомер);
МС с электрораспылением [М+1]+ 637,1 для соединения Примера 79b (более полярный изомер).
Пример 80
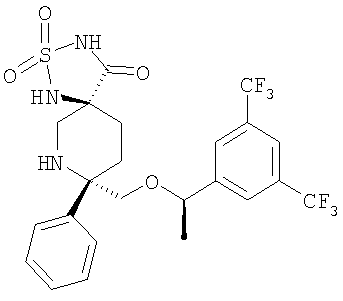
К раствору соединения Примера 62 (0,079 г, 0,127 ммоль, 1 экв.) в сухом CH2Cl2 (1 мл) прибавляют Et3N (0,108 мл, 0,76 ммоль, 6 экв.). Реакционную смесь охлаждают до 0°С и перемешивают в течение 15 мин. К реакционной смеси в течение 5 мин очень медленно прибавляют SO2Cl2 (0,011 мл, 0,133 ммоль, 1,05 экв.). Реакционную смесь перемешивают в течение 10 ч и за протеканием реакции следят с помощью ТСХ (9:1, EtOAc/СН3ОН). Реакционную смесь разбавляют с помощью EtOAc, промывают с помощью NaHCO3 и сушат над Na2SO4. Неочищенный продукт обрабатывают с помощью препаративной тонкослойной хроматографии и выделяют соединение Примера 80 (0,015 г, 20%), МС с электрораспылением [M+1]+ 552,1.
Пример 81
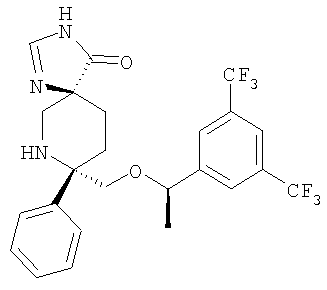
Стадия 1:
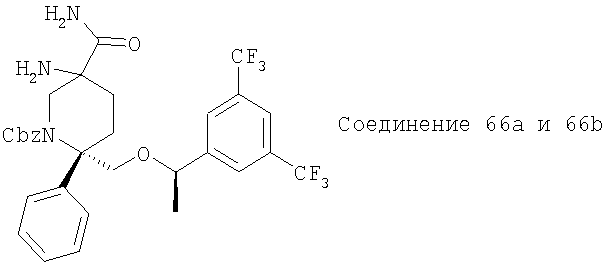
Соединения 66а и 66b получают из Соединения 48 с помощью методики, аналогичной использованной для получения соединения Примера 62 из соединения Примера 43а.
Стадия 2:
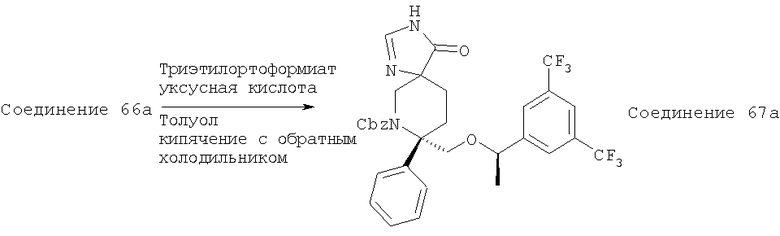
К раствору Соединения 66а (0,34 г, 0,545 ммоль, 1 экв.) в сухом толуоле (14 мл) прибавляют НОАс (0,17 мл), а затем триэтилортоформиат (0,363 мл, 2,18 ммоль, 4 экв.). Раствор кипятят с обратным холодильником в течение 12 ч и за протеканием реакции следят с помощью ТСХ (9:1, EtOAc/СН3ОН). Реакционную смесь охлаждают до 0°С, реакцию останавливают с помощью Н2О, разбавляют с помощью EtOAc, промывают с помощью NaHCO3 и сушат над Na2SO4. Неочищенное вещество обрабатывают с помощью флэш-хроматографии с использованием колонки Biotage (60:40, EtOAc/гексан) для элюирования Соединения 67а (0,272 г, 79%), МС с электрораспылением [М+1]+ 634,1.
Стадия 3:
Соединение Примера 81 получают из Соединения 67а с помощью методики, аналогичной использованной для получения соединений Примеров 79а и 79b из Соединения 65. МС с электрораспылением [M+1]+ 500,1 для соединения Примера 81.
Пример 82
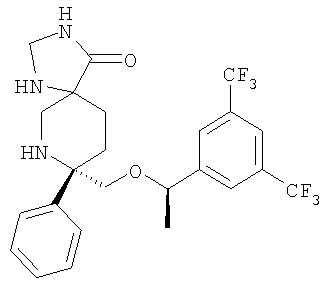
Стадия 1:
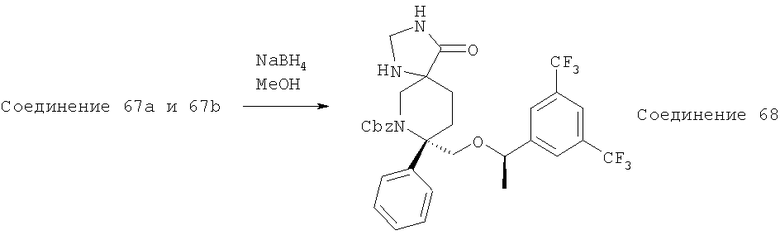
К раствору смеси двух изомеров Соединения 67 (0,27 г, 0,426 ммоль, 1 экв.) в сухом СН3ОН (3 мл) прибавляют NaBH4 (0,048 г, 1,28 ммоль, 3 экв.). Реакционную смесь продувают при добавлении реагента и перемешивают в течение 5 ч в атмосфере N2. За протеканием реакции следят с помощью ТСХ (60/40, EtOAc/гексан) и после завершения реакции ее останавливают с помощью НОАс, реакционную смесь концентрируют, разбавляют с помощью EtOAc, промывают с помощью NaHCO3 и сушат над Na2SO4. Неочищенный продукт является смесью двух изомеров Соединения 68 (0,25 г, 92%) и затем его используют без дополнительной очистки. МС с электрораспылением [M+1]+ 636,1 для Соединения 68.
Стадия 2:
Соединение Примера 82а (менее полярный изомер) и соединение Примера 82b (более полярный изомер) (0,12 г, 61%) получают из Соединения 68 с помощью методики, аналогичной использованной для получения соединений Примеров 79а и 79b из Соединения 65.
МС с электрораспылением [M+1]+ 502,1 для соединения Примера 82а,
МС с электрораспылением [М+1]+ 502,1 для соединения Примера 82b.
Пример 83
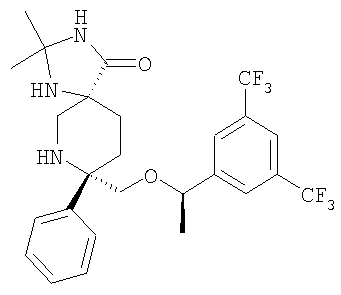
Стадия 1:
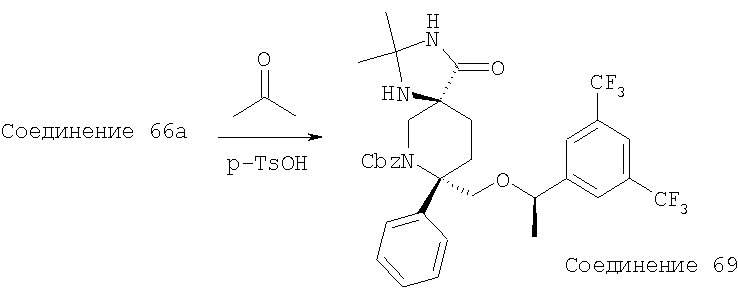
К раствору Соединения 66а (0,1 г, 0,16 ммоль, 1 экв.) в МеОН (0,5 мл) в КДК объемом 25 мл прибавляют ацетон (0,352 мл, 0,48 ммоль, 3 экв.) и p-TsOH (0,06 г, 0,32 ммоль, 2 экв.). Реакционную смесь кипятят с обратным холодильником в течение 12 ч и за протеканием реакции следят с помощью масс-спектрометрического анализа. После завершения реакции реакционную смесь концентрируют, разбавляют с помощью EtOAc, промывают с помощью NaHCO3, сушат над Na2SO4 и получают Соединение 69 (0,1 г, 94%). Неочищенный продукт далее используют без какой-либо очистки.
Стадия 2:
Соединение Примера 83 (0,026 г, 33%) получают из Соединения 69 с помощью методики, аналогичной использованной для получения соединений Примеров 79а и 79b из Соединения 65. МС с электрораспылением [M+1]+ 530,1 для соединения Примера 83.
Пример 84а и Пример 84b
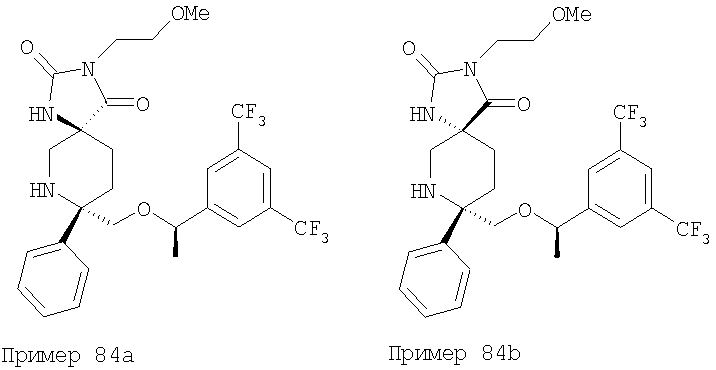
Соединение Примера 84а и соединение Примера 84b получают с помощью методики, аналогичной использованной для получения соединений Примеров 78а и 78b, но с использованием 2-бромэтилметилового эфира вместо 2-бромэтанола.
МС с электрораспылением [М+1]+ 574,1 для соединения Примера 84а,
МС с электрораспылением [M+1]+ 574,1 для соединения Примера 84b.
Пример 85
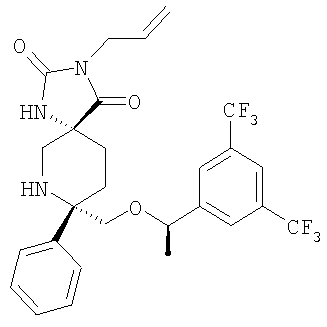
Соединение Примера 85 (46 мг, 88%) получают из соединения Примера 43b с помощью методики, аналогичной использованной для получения Соединения 63, но с использованием аллилбромида вместо 2-бромэтанола. МС с электрораспылением [М+1]+ 556,1.
Пример 86а и Пример 86b
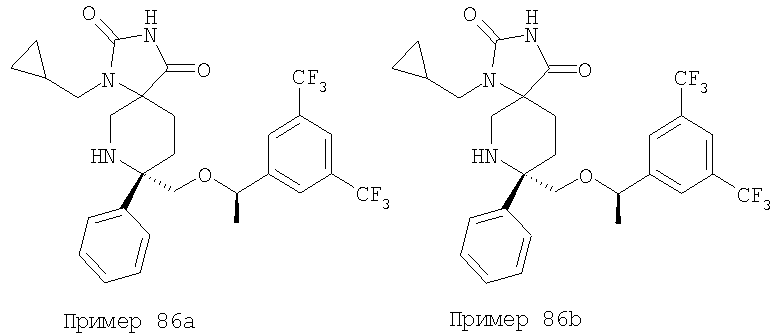
Стадия 1:
Соединение 70 (1,14, 99%) получают из Соединения 48 с помощью методики, аналогичной использованной для получения Соединения 63, но с использованием п-метоксибензилхлорида вместо 2-бромэтанола. МС с электрораспылением [М+1]+ 770,2.
Стадия 2:
К раствору Соединения 70 (0,19 г, 0,25 ммоль, 1 экв.) в 1,0 мл безводного DMF при 0°С прибавляют NaH (60% дисперсия в минеральном масле, 0,012 г, 0,30 ммоль, 1,2 экв.). Через 5 мин баню со льдом удаляют и реакционную смесь перемешивают в течение 30 мин, а затем прибавляют бромметилциклопропан (0,029 мл, 0,30 ммоль, 1,2 экв.). Через 20 ч реакцию останавливают с помощью насыщенного раствора NH4Cl и реакционную смесь разбавляют с помощью EtOAc. Слои разделяют и органический слой один раз промывают рассолом, сушат над Na2SO4, фильтруют, концентрируют и получают Соединение 71 (0,11 г, 99%), МС с электрораспылением [М+1]+ 824,2.
Стадия 3:
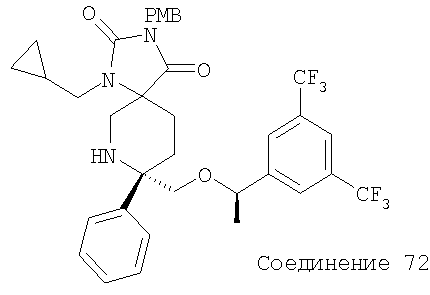
Соединение 72 (0,24 г, 90%) получают из Соединения 71 с помощью методики, аналогичной использованной для получения соединений Примеров 78а и 78b из Соединений 63. МС с электрораспылением [М+1]+ 690,1.
Стадия 4:
К раствору Соединения 72 (0,24 г, 0,34 ммоль, 1 экв.) в 5,0 мл СН3CN и 1,7 мл воды при 0°С прибавляют нитрат аммония-церия (0,79 г, 1,4 ммоль, 4 экв.). Через 5 мин баню со льдом удаляют и реакционную смесь перемешивают при комнатной температуре в течение 17 ч. Реакцию останавливают водой и разбавляют с помощью EtOAc. Слои разделяют и органический слой промывают водой (100 мл×2), сушат над Na2SO4, фильтруют, концентрируют и получают желтое масло. Очистка с помощью хроматографии на колонке Biotage при элюировании в градиентном режиме с растворителями от 20% EtOAc/гексан до 30% EtOAc/гексан и до 50% EtOAc/гексан дает диастереоизомерную смесь соединений Примеров 86а и 86b (15 мг, 8%), чистый изомер менее полярного соединения Примера 86а (14 мг, 7%), МС с электрораспылением [M+1]+ 570,1, и чистый изомер более полярного соединения Примера 86b (16 мг, 9%), МС с электрораспылением [M+1]+ 570,1.
Пример 87
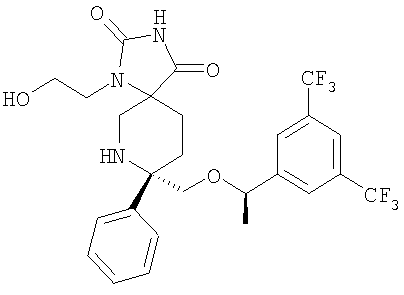
Стадия 1:
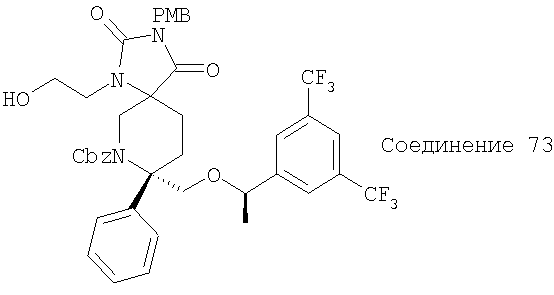
Соединение 73 (0,20 г, 64%) получают из Соединения 48 с помощью методики, аналогичной использованной для получения Соединения 63. МС с электрораспылением [М+1]+ 814,19.
Стадия 2:
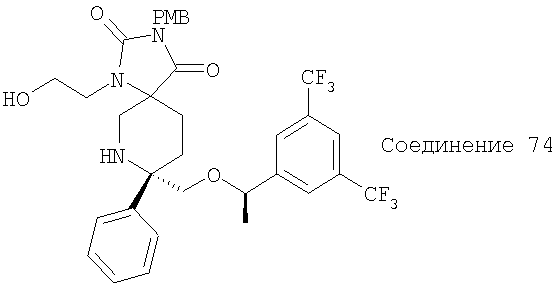
Соединение 74 (0,16 г, 96%) получают из Соединения 73 с помощью методики, аналогичной использованной для получения соединений Примеров 78а и 78b из Соединения 63. МС с электрораспылением [М+1]+ 680,1.
Стадия 3:
Соединения Примеров 87а и 87b получают из Соединения 74 с помощью методики, аналогичной использованной для получения соединений Примеров 86а и 86b из Соединения 72, но используют очистку с помощью колонки Gilson с элюированием смесью вода/CH3CN вместо хроматографии на колонке Biotage и выделяют диастереоизомерную смесь соединений Примеров 87а и 87b (92 мг, 71%). Разделение с помощью ВЭЖХ 50 мг смеси на колонке ChiralCel OD с использованием смеси (90/10) гексан/IPA в качестве элюента дает диастереоизомерную смесь соединений Примеров 87а и 87b (11 мг), чистый изомер первого элюирующегося продукта - соединение Примера 87а (10 мг), МС с электрораспылением [М+1]+ 560,1, чистый изомер второго элюирующегося продукта - соединение Примера 87b (11 мг), МС с электрораспылением [М+1]+ 570,1.
Пример 88
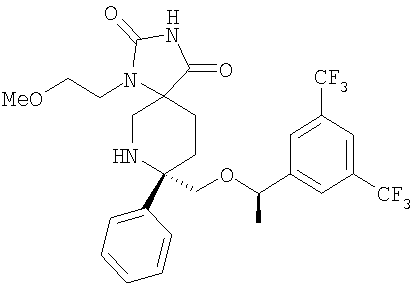
Диастереоизомерную смесь соединений Примеров 88а и 88b (22% суммарный выход за три стадии из Соединения 70) получают с помощью методики, аналогичной использованной для получения соединений Примеров 86а и 86b, но с использованием 2-бромэтилметилового эфира вместо бромметилциклопропана. Разделение с помощью ВЭЖХ 50 мг смеси диастереоизомеров на колонке ChiralCel OD с использованием (90/10) гексан/IPA в качестве элюента дает чистый изомер первого элюирующегося продукта - соединение Примера 88а (16 мг), МС с электрораспылением [M+1]+ 574,3, и чистый изомер второго элюирующегося продукта - соединение Примера 88b (29 мг), МС с электрораспылением [M+1]+ 574,3.
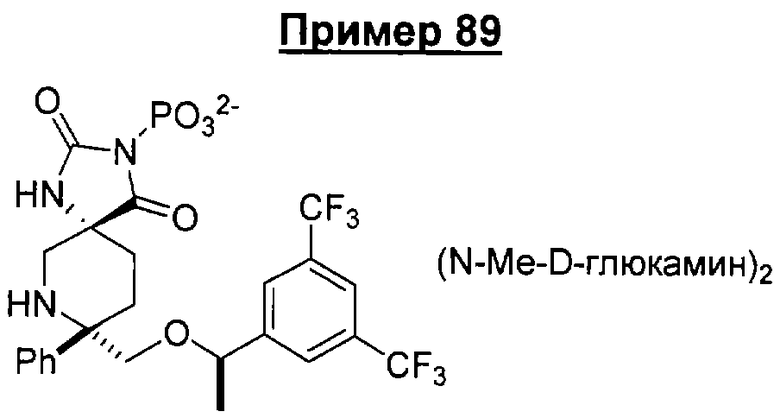
К раствору соединения Примера 43b (0,68 г, 1,32 ммоль, 1 экв.) в DMF (7 мл) при 0°С прибавляют NaH (60% в минеральном масле, 0,105 г, 2,64 ммоль, 2 экв.) и смесь перемешивают при 0°С в течение 15 мин. Прибавляют тетрабензилпирофосфат (1,42 г, 2,64 ммоль, 2 экв.) и реакционную смесь перемешивают при 0°С в течение 15 мин, затем нагревают до 23°С и перемешивают в течение 1 ч. Реакцию останавливают насыщенным водным раствором NaHCO3 (100 мл) и экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (Na2SO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (2:1, гексан : EtOAc) и получают N-фосфорированный гидантоин (0,24 г), который растворяют в МеОН (10 мл); прибавляют N-Me-D-глюкамин (0,119, 0,619 ммоль, 2 экв.), а затем 10% Pd-C (0,021 г). Полученную смесь встряхивают в приборе для гидрирования Парра в атмосфере Н2 при 40 фунт-сила/дюйм2 в течение 18 ч. Реакционную смесь фильтруют через слой целита и целит промывают с помощью МеОН. Полученный раствор концентрируют в вакууме. Остаток растворяют в EtOAc (100 мл) и экстрагируют водой (100 мл), водный слой подвергают лиофильной сушке и получают искомый продукт - соединение Примера 89 в виде соли N-Me-D-глюкамина (0,22 г, 21% за две стадии).
Пример 90
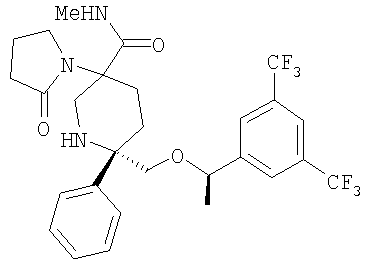
Стадия 1:
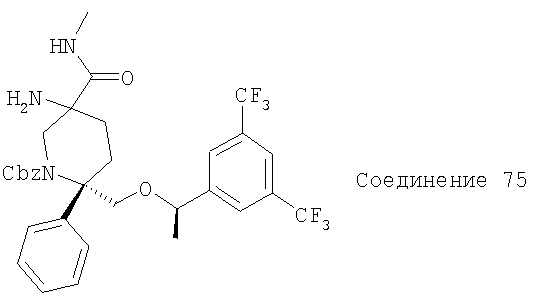
Соединение 75 получают из Соединения 48 с помощью методики, аналогичной использованной для получения Соединения 66 из Соединения 48.
Стадия 2:
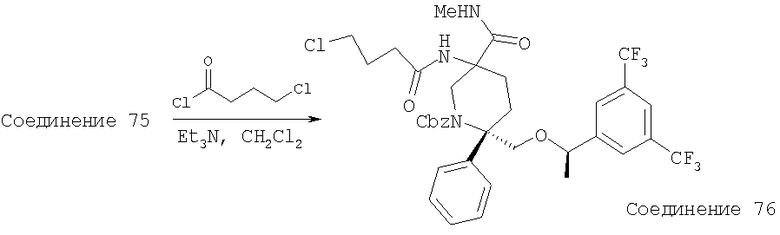
К раствору диастереоизомерного Соединения 75 (0,10 г, 0,16 ммоль, 1 экв.) в 2,0 мл безводного CH2Cl2 при 0°С прибавляют Et3N (0,033 мл, 0,24 ммоль, 1,5 экв.) и 4-хлорбутирилхлорид (0,017 мл, 0,17 ммоль, 1,1 экв.). Через 6 ч реакцию останавливают с помощью насыщенного раствора NH4Cl и разбавляют с помощью EtOAc. Слои разделяют и органический слой один раз промывают рассолом, сушат над Na2SO4, фильтруют, концентрируют и получают Соединение 76 в виде смеси диастереоизомеров (0,12 г, 100%). МС с электрораспылением [М+1]+ 742,2.
Стадия 3:
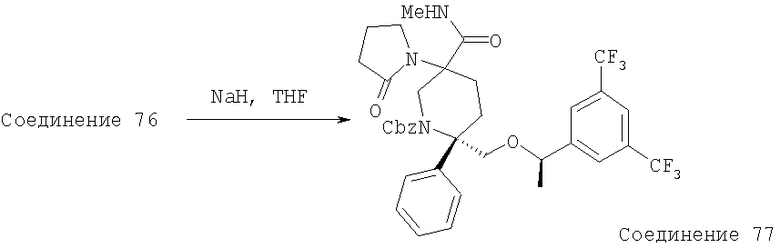
К раствору Соединения 76 (0,12 г, 0,16 ммоль, 1 экв.) в 1,0 мл безводного THF при комнатной температуре прибавляют NaH (60% дисперсия в минеральном масле, 0,010 г, 0,24 ммоль, 1,5 экв.). Через 3 ч реакцию останавливают с помощью насыщенного раствора NH4Cl и разбавляют с помощью EtOAc. Слои разделяют и органический слой один раз промывают рассолом, сушат над Na2SO4, фильтруют, концентрируют и получают Соединение 77 (0,10 г, 88%). МС с электрораспылением [M+1]+ 706,2.
Стадия 4:
Соединения Примеров 90а и 90b получают из Соединения 77 с помощью методики, аналогичной использованной для получения соединения Примера 83 из Соединения 69, но вместо препаративной тонкослойной хроматографии очистку проводят с помощью хроматографии на колонке Biotage. Получают диастереоизомерную смесь соединений Примеров 90а и 90b (26 мг, 32%): менее полярный продукт - соединение Примера 90а (20 мг, 25%), МС с электрораспылением [М+1]+ 572,1, более полярный продукт - соединение Примера 90b (14 мг, 17%), МС с электрораспылением [М+1]+ 572,1.
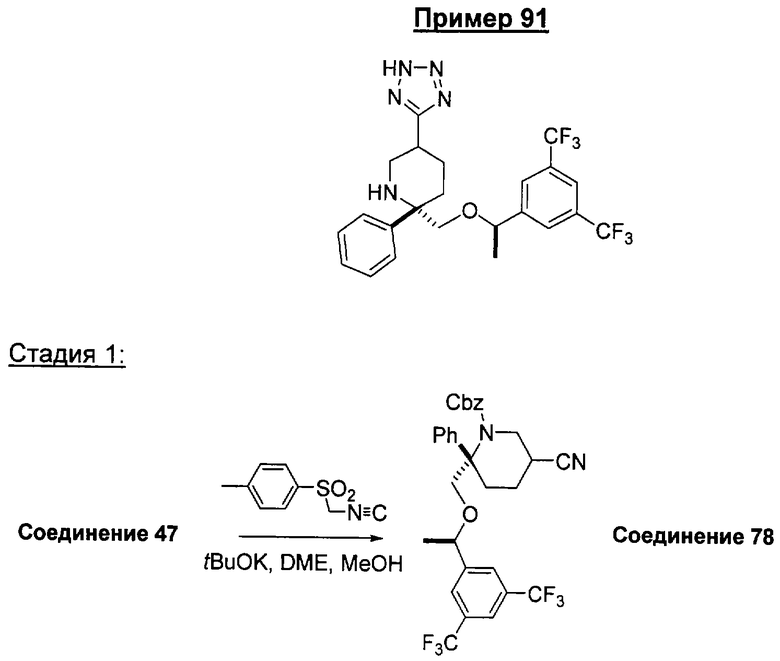
К раствору Соединения 47 (1 г, 1,73 ммоль, 1 экв.) и тозилметилизоцианида (374 мг, 1,9 ммоль, 1,1 экв.) в безводном диметиловом эфире этиленгликоля (11 мл) при -30°С прибавляют безводный MeOH (0,15 мл), а затем прибавляют трет-бутоксид калия (426 мг, 3,8 ммоль, 2,2 экв.). После перемешивания при температуре от -30 до 10°С в течение 7 ч реакционную смесь пропускают через целит. Слой целита тщательно промывают с помощью Et2O. Фильтрат концентрируют и остаток очищают на колонке с силикагелем и получают искомое Соединение 78 (470 мг, 46%).
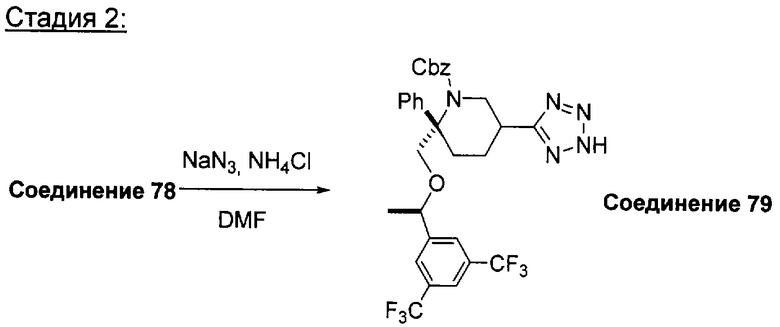
Раствор Соединения 78 (125,7 мг, 0,21 ммоль, 1 экв.) и NH4Cl (68,3 мг, 1,28 ммоль, 6 экв.) и NaN3 (69,2 мг, 1,06 ммоль, 5 экв.) в безводном DMF (1,2 мл) в атмосфере N2 нагревают при 115°С в течение ночи. Смесь концентрируют, а затем подкисляют с помощью HCl (6 н., 10 мл) и экстрагируют с помощью EtOAc (15 мл×3). Объединенные органические растворы сушат над Na2SO4, фильтруют и выпаривают в вакууме. Остаток очищают на колонке с силикагелем и получают Соединение 79 (67 мг, 50%).
Стадия 3:
Раствор Соединения 79 (65 мг, 0,103 ммоль, 1 экв.) в EtOH (1,5 мл) обрабатывают с помощью 10% Pd-C (107 мг, 0,1 ммоль, 1, экв.) и 1,4-циклогексадиена (0,5 мл, 5,29 ммоль, 50 экв.). Смесь нагревают при 85°С в течение 10 мин, затем пропускают через целит. Слой целита промывают с помощью МеОН. Фильтрат концентрируют в вакууме, остаток очищают на колонке с силикагелем и получают соединение Примера 91 (11 мг, 21%). МС с электрораспылением [М+1]+ 500,1.
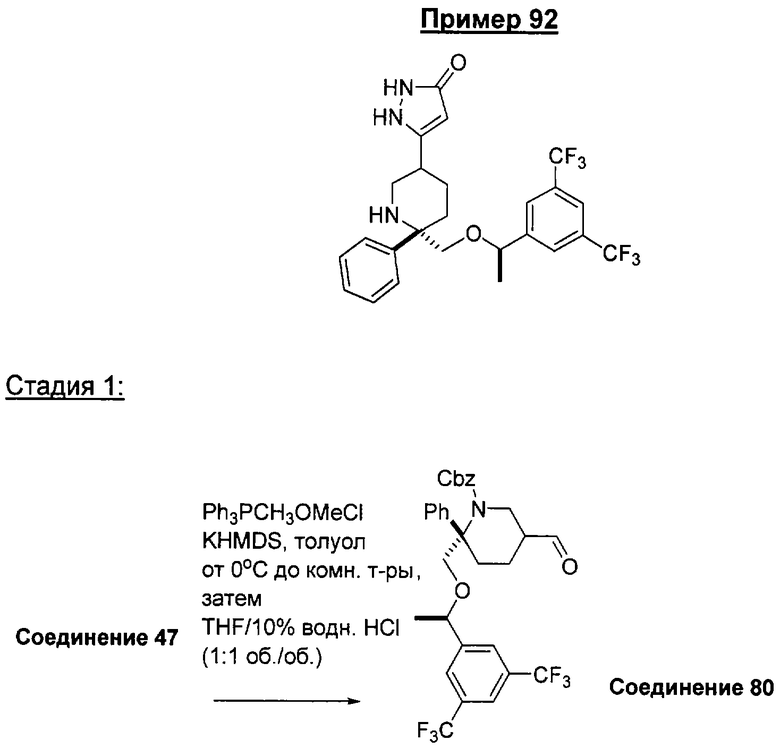
Соединение 80 (выход 72%) получают с помощью методики, аналогичной использованной для получения Соединения 23, с использованием Соединения 47 вместо Соединения 3.
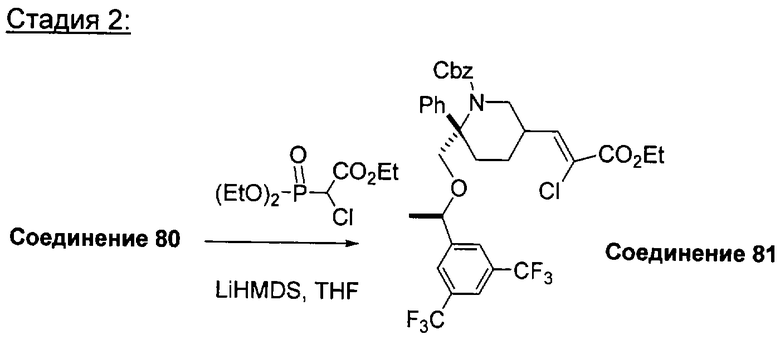
К раствору триэтил-2-хлор-2-фосфоноацетата (73 мкл, 0,34 ммоль, 1,05 экв.) в безводном THF (1,5 мл) при -78°С по каплям прибавляют LiHMDS (0,35 мл, 0,35 ммоль, 1,1 экв., 1 н. раствор в THF). Раствор перемешивают в течение 20 мин, а затем через канюлю прибавляют раствор Соединения 80 (192 мг, 0,32 ммоль, 1 экв.) в сухом THF (1 мл). Его перемешивают при -78°С в течение 2 ч затем реакцию останавливают с помощью насыщенного водного раствора NH4Cl и экстрагируют с помощью Et2O. Органический слой сушат над MgSO4, фильтруют, концентрируют и получают неочищенный продукт, который очищают на колонке с силикагелем и получают Соединение 81 (127 мг, 57%).
Стадия 3:
К раствору Соединения 81 (127 мг, 0,18 ммоль, 1,0 экв.) в EtOH (1 мл) прибавляют H2NNH2 (35 мкл, 1,1 ммоль, 6 экв.). Его перемешивают в течение 3 ч и затем концентрируют в вакууме. Неочищенный продукт повторно растворяют в EtOH (3 мл) и обрабатывают с помощью 10% Pd/C (40 мг, 0,036 ммоль, 0,2 экв.) и гидрируют в течение ночи. Катализатор отфильтровывают и промывают с помощью МеОН. Фильтрат концентрируют и получают неочищенный остаток, который очищают на колонке с силикагелем и получают менее полярный изомер - соединение Примера 92а (14,2 мг, 15%), МС с электрораспылением [М+1]+ 514,1, и более полярный изомер - соединение Примера 92b (28,1 мг, 30%), МС с электрораспылением [М+1]+ 514,1.
Пример 93
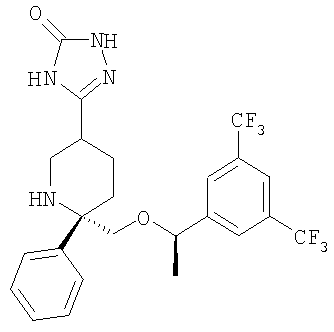
Стадия 1:
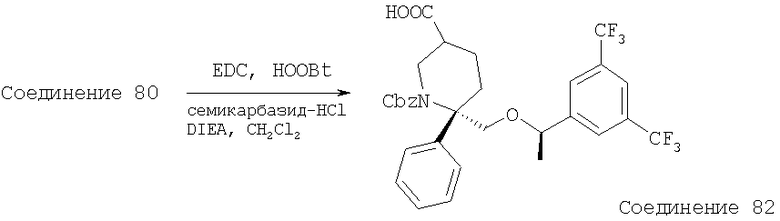
Соединение 80 (0,74 г, 1,25 ммоль, 1 экв.), растворяют в t-BuOH (20 мл) и 2-метил-2-бутадиене (7 мл). К этому раствору прибавляют свежеприготовленный раствор NaClO2 (1,13 г, 12,5 ммоль, 10 экв.) в 20% (об./мас.) водном растворе NaH2PO4. Реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Затем ее разбавляют с помощью EtOAc (200 мл) и органический слой отделяют, сушат (Na2SO4), фильтруют, концентрируют и получают неочищенное Соединение 82, которое используют в следующей реакции без дополнительной очистки.
Стадия 2:
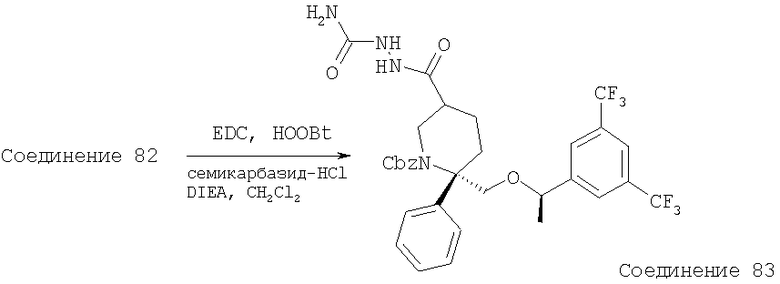
К раствору диастереоизомерного Соединения 82 (0,33 г, 0,54 ммоль, 1 экв.) в 2 мл безводного CH2Cl2 при комнатной температуре последовательно прибавляют DIEA (0,11 мл, 0,65 ммоль, 1,2 экв.), DEC (0,21 г, 1,1 ммоль, 2 экв.), 3-гидрокси-1,2,3-бензотриазин-4(ЗН)-он (0,18 г, 1,1 ммоль, 2 экв.) и семикарбазидгидрохлорид (0,072 г, 0,65 ммоль, 1,2 экв.). Через 3 ч по данным ТСХ [гексан-EtOAc, 1:1 (об./об.)] присутствует исходная карбоновая кислота и прибавляют дополнительное количество DIEA (0,11 мл, 0,65 ммоль, 1,2 экв.). Через 2 дня реакцию останавливают с помощью насыщенного раствора NaHCO3 и разбавляют с помощью EtOAc. Слои разделяют и органический слой промывают один раз водой и рассолом, сушат над Na2SO4, фильтруют, концентрируют и получают оранжевое масло. Очистка с помощью хроматографии на колонке Biotage при элюировании в градиентном режиме с растворителями от 50% EtOAc/гексан до 80% EtOAc/гексан, до EtOAc, до 5% МеОН/EtOAc дает Соединение 83 в виде смеси диастереоизомеров (0,19 г, 54%) МС с электрораспылением [М+1]+ 667,07.
Стадия 3:
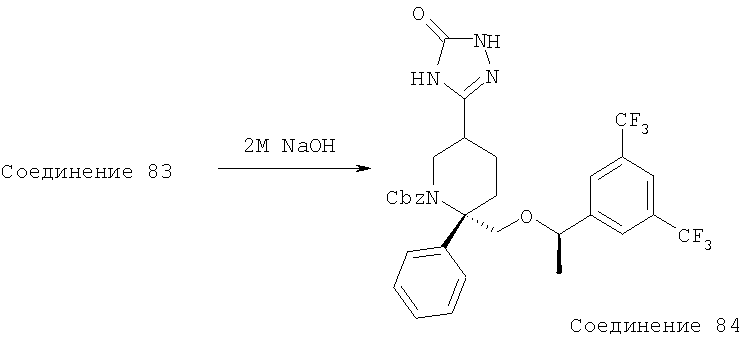
Раствор Соединения 83 (0,17 г, 0,26 ммоль, 1 экв.) в 8 мл 2,0 М раствора NaOH кипятят с обратным холодильником. Через 15 ч смеси дают нагреться до комнатной температуры и ее нейтрализуют с помощью 1,0 М HCl до рН 6. Водный раствор разбавляют с помощью EtOAc и слои разделяют. Органический слой один раз промывают рассолом, сушат над Na2SO4, фильтруют, концентрируют и получают желтое масло (0,16 г). Очистка с помощью хроматографии на колонке Biotage при элюировании с помощью 3% МеОН/EtOAc дает Соединение 84 в виде смеси диастереоизомеров (0,12 г, 71%). МС с электрораспылением [М+1]+ 649,2.
Стадия 4:
Менее полярный продукт - соединение Примера 93а и более полярный продукт - соединение Примера 93b (32 мг и 44 мг, суммарный выход для обоих изомеров 88%) получают из Соединения 84 с помощью методики, аналогичной использованной для получения соединений Примеров 79а и 79b из Соединения 65, но вместо препаративной тонкослойной хроматографии очистку проводят с помощью хроматографии на колонке Biotage.
МС с электрораспылением [M+1]+ 515,3 для соединения Примера 93а.
МС с электрораспылением [М+1]+ 515,3 для соединения Примера 93b.
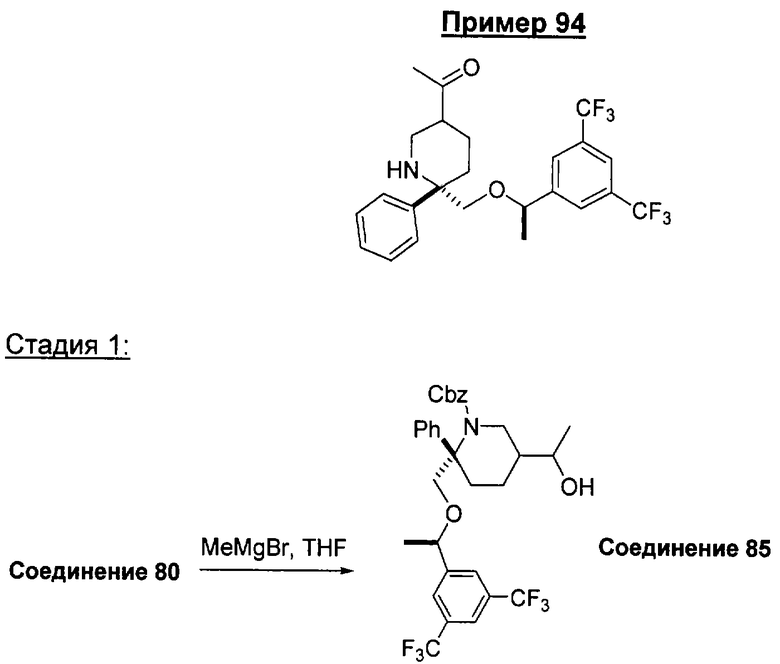
К раствору Соединения 80 (550 мг, 0,98 ммоль, 1 экв.) в безводном THF (6 мл) при -10°С, по каплям прибавляют СН3MgBr (1,24 мл, 3,7 ммоль, 4 экв., 3,0 М раствор в Et2O). Раствор перемешивают при температуре от -10 до 10°С в течение 30 мин. Растворение в воде дает неочищенный продукт, который очищают на колонке с силикагелем и получают Соединение 85 (236 мг, 42%).
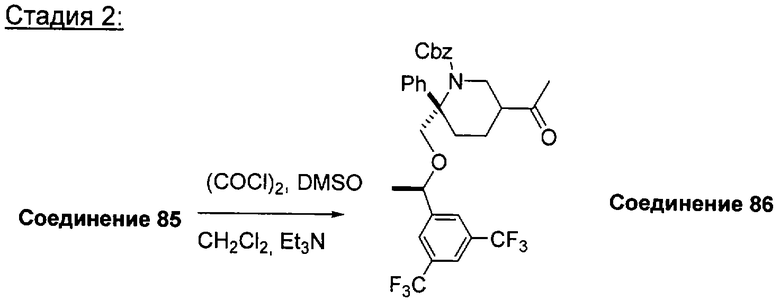
К раствору DMSO (0,11 мл, 1,55 ммоль, 4 экв.) в безводном CH2Cl2 (5 мл) при -78°С по каплям прибавляют оксалилхлорид (0,067 мл, 0,78 ммоль, 2 экв.). Раствор перемешивают в течение 15 мин, а затем через канюлю прибавляют раствор Соединения 85 (236 мг, 0,387 ммоль, 1 экв.) в сухом CH2Cl2 (1 мл). Его перемешивают при -78°С в течение 1 ч, затем по каплям прибавляют Et3N (0,37 мл, 2,71 ммоль, 7 экв.). После перемешивания при -78°С в течение 30 мин охлаждающую баню удаляют и реакционную смесь нагревают до комнатной температуры. Реакцию останавливают с помощью насыщенного водного раствора NH4Cl и реакционную смесь экстрагируют с помощью СН2Cl2. Органический слой сушат над MgSO4, фильтруют, концентрируют и получают неочищенный продукт, который очищают на колонке с силикагелем и получают Соединение 86 (140 мг, 60%).
Стадия 3:
К раствору Соединения 86 (1,0 экв.) в EtOH (3 мл) прибавляют 10% Pd/C (0,4 экв.) и смесь гидрируют в течение ночи в атмосфере подаваемого из баллона Н2. Катализатор отфильтровывают и промывают с помощью МеОН. Фильтрат концентрируют и получают неочищенный остаток, который очищают на колонке с силикагелем и получают менее полярный изомер - соединение Примера 94а (24 мг, 22%), МС с электрораспылением [М+1]+ 474,1, и более полярный изомер - соединение Примера 94b (32 мг, 29%), МС с электрораспылением [M+1]+ 474,1.
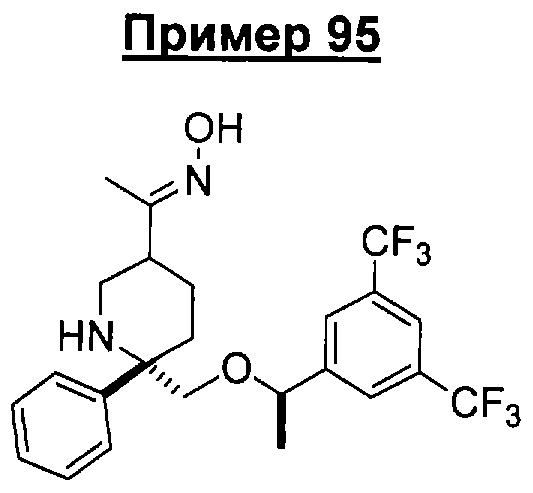
К раствору соединения Примера 94а (16 мг, 0,033 ммоль, 1 экв.) в безводном EtOH (1 мл) прибавляют гидроксиламингидрохлорид (18 мг, 0,26 ммоль, 7,7 экв.) и NaOAc (5 мг, 0,061 ммоль, 1,8 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем концентрируют досуха. Остаток повторно растворяют в Et2O и промывают с помощью насыщенного водного раствора NaHCO3. Органические слои сушат над MgSO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают на колонке с силикагелем и получают соединение Примера 95 (12 мг, 74%), МС с электрораспылением [M+1]+ 489,1.
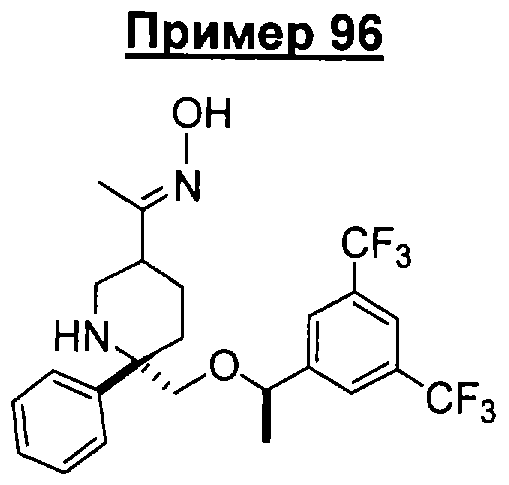
Соединение Примера 96 (13 мг, 50%) получают с помощью методики, аналогичной использованной для получения соединения Примера 95, но с использованием соединения Примера 94b вместо соединения Примера 94а. МС с электрораспылением [М+1]+ 489,1.
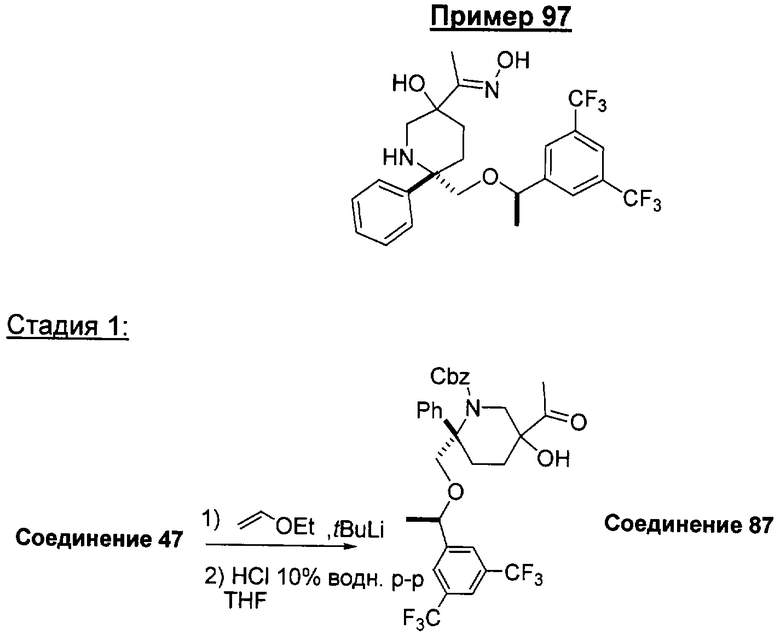
К раствору этилвинилового эфира (0,5 мл, 4,83 ммоль, 12 экв.) в безводном THF (6 мл) при -78°С по каплям прибавляют tBuLi (0,73 мл, 1,24 ммоль, 3 экв., 1,7 н. раствор в пентане). Раствор перемешивают при охлаждении на бане при -10°С до исчезновения оранжевой окраски. Его повторно охлаждают до -78°С и через канюлю прибавляют раствор Соединения 47 (240 мг, 0,41 ммоль, 1 экв.) в сухом THF (1 мл). Его перемешивают при -78°С в течение 1,5 ч, затем реакцию останавливают с помощью насыщенного водного раствора NH4Cl и экстрагируют с помощью Et2O. Органический слой сушат над MgSO4, фильтруют, концентрируют и получают неочищенный продукт, который повторно растворяют в THF (6 мл) и обрабатывают с помощью 10% водного раствора HCl (0,8 мл). Его перемешивают при комнатной температуре в течение ночи. Обработка водным раствором щелочи дает неочищенный продукт, который очищают на колонке с силикагелем и получают Соединение 87 (100 мг, 39%).
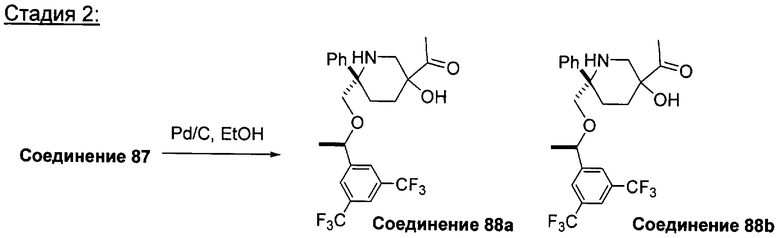
Соединение 88а и Соединение 88b получают с помощью методики, аналогичной использованной для получения соединений Примеров 94а и 94b с использованием Соединения 87 вместо Соединения 86. Разделение на хиральной колонке ВЭЖХ дает Соединение 88а (13 мг, 18%) и Соединение 88b (10 мг, 14%).
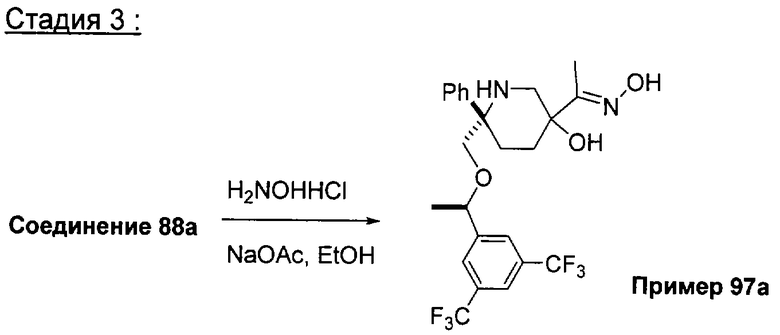
Соединение Примера 97а (9,7 мг, 71%) получают с помощью методики, аналогичной использованной для получения соединения Примера 95, с использованием Соединения 88а вместо соединения Примера 94а. МС с электрораспылением [М+1]+ 505,1.
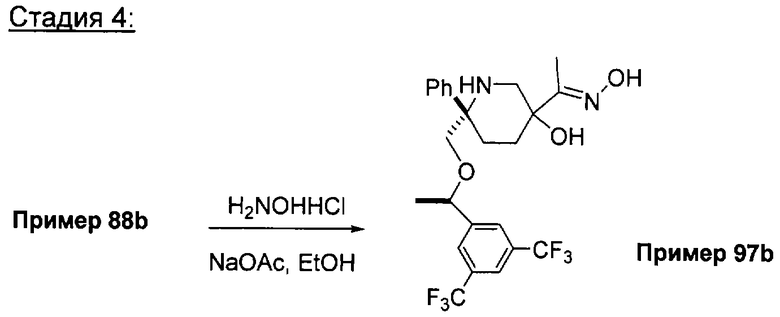
Соединение Примера 97b (10,7 мг, 100%) получают с помощью методики, аналогичной использованной для получения соединения Примера 95, с использованием Соединения 88b вместо соединения Примера 94а. МС с электрораспылением [M+1]+ 505,1.
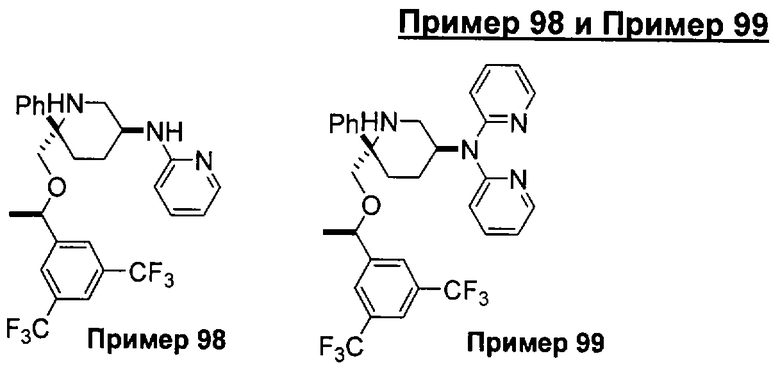
К соединению Примера 13 (340 мг, 0,76 ммоль) в 1 мл толуола прибавляют Pd2(dba)3 (27,8 мг, 0,03 ммоль), BINAP (37,8 мг, 0,06 ммоль), 2-бромпиридин (73 мкл, 0,76 ммоль) и NaOtBu (102 мг, 1,065 ммоль). Смесь концентрируют в вакууме и в колбу подают N2. Эту процедуру повторяют еще один раз. Темно-коричневый раствор нагревают при 90°С в течение 16 ч. Его охлаждают до 23°С и реакцию останавливают с помощью 2 мл буферного раствора с рН 7. Раствор экстрагируют с помощью EtOAc (10 мл×2). Органические слои сушат над Na2SO4 и концентрируют. Разделение с помощью ВЭЖХ дает соединение Примера 98, МС с электрораспылением [М+1]+ 524,1, и соединение Примера 99, МС с электрораспылением [М+1]+ 601,1.
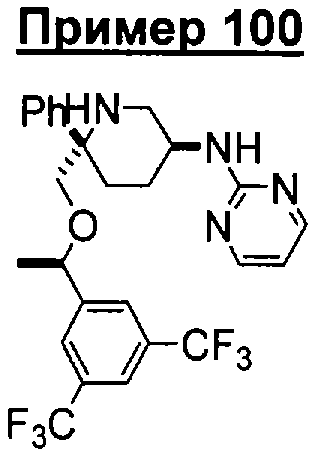
Соединение Примера 100 получают с использованием методики, аналогичной использованной для получения соединения Примера 98, с использованием 2-бромпиримидина вместо 2-бромпиридина. МС с электрораспылением [М+1]+ 525,1.
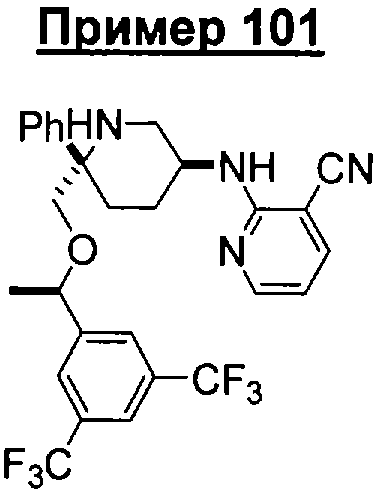
Соединение Примера 101 получают с использованием методики, аналогичной использованной для получения соединения Примера 98, с использованием 2-хлор-3-цианпиридина вместо 2-бромпиридина. МС с электрораспылением [М+1]+ 549,1.
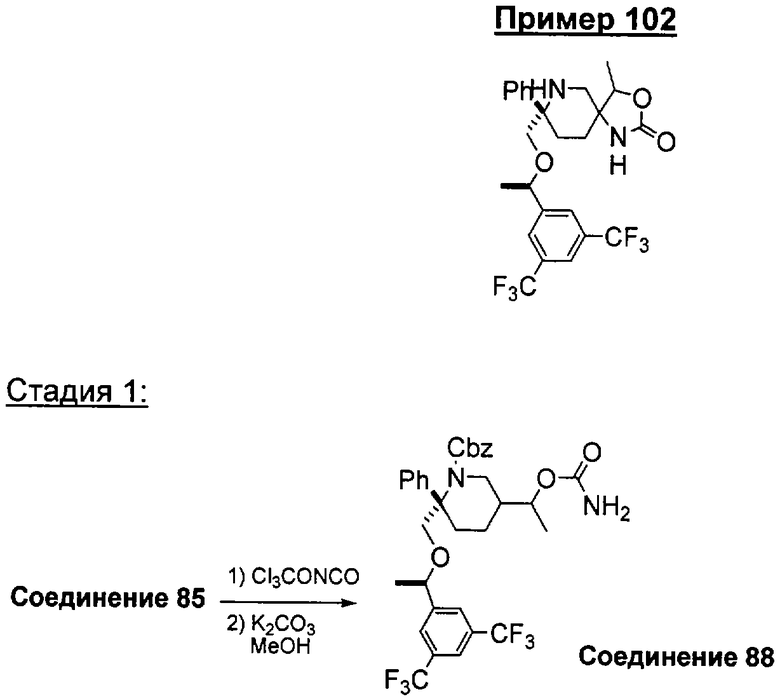
К Соединению 85 (429 мг, 0,704 ммоль) в CH2Cl2 (3,5 мл) при 0°С по каплям прибавляют Cl3CONCO (100 мл, 0,844 ммоль). Раствор перемешивают при 0°С в течение 2 ч. Затем растворитель удаляют и остаток растворяют в МеОН (4 мл) и H2O (1 мл). Прибавляют К2СО3 (1,0 г) и суспензию перемешивают в течение 14 ч. Затем смесь разбавляют с помощью 3 мл воды, концентрируют для удаления МеОН. Остаток экстрагируют с помощью EtOAc (10 мл×3). Объединенные органические слои концентрируют, пропускают через короткую колонку с силикагелем и получают продукт - Соединение 88 (420 мг, 91%).
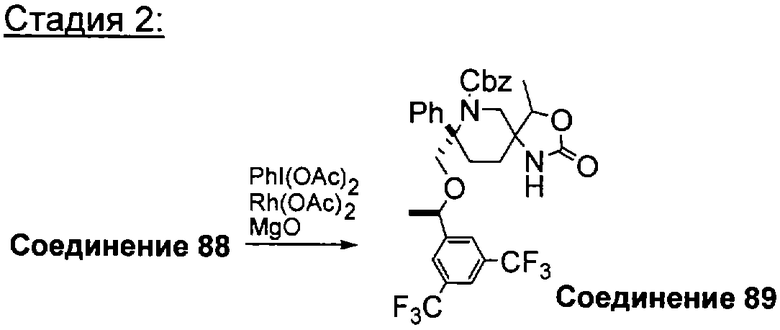
К соединению Примера 88 (290 мг, 0,446 ммоль) в CH2Cl2 (3 мл) прибавляют PhI(ОАс)2 (131 мг, 0,625 ммоль), Rh2(OAc)4 (12,9 мг, 0,022 ммоль) и MgO (26,4 мг, 1,0 ммоль). Суспензию нагревают при 40°С в течение 16 ч, затем охлаждают до 23°С. Прибавляют целит (0,5 г) и суспензию перемешивают в течение 5 мин. Смесь фильтруют и промывают с помощью EtOAc. Объединенные фильтраты концентрируют и очищают с помощью хроматографии на силикагеле, получают Соединение 89 (60 мг, 31%).
Стадия 3:
Соединение 89 переносят в прибор для гидрирования Парра с использованием 5 мл EtOH. Прибавляют 10% Pd-C (10%, 60 мг) и суспензию гидрируют при 40 фунт-сила/дюйм2 в течение ночи. Реакционную смесь фильтруют и концентрируют. Остаток разделяют с использованием ВЭЖХ на колонке OD, элюируя с помощью (1:9) IPA/гексан, и получают два изомера, менее полярный изомер - соединение Примера 102а, МС с электрораспылением [М+1]+ 517,1, и более полярный изомер - соединение Примера 102b, МС с электрораспылением [M+1]+ 517,1.
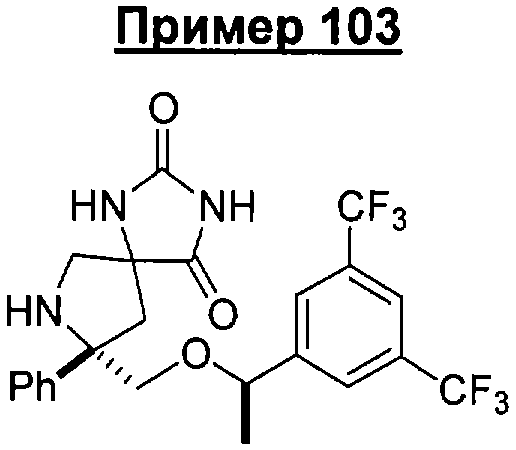
Стадия 1:
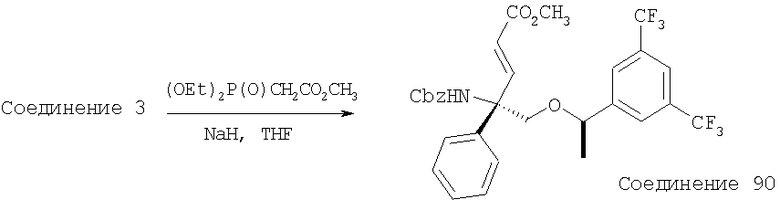
К смеси метилдиэтилфосфоноацетата (9,5 мл, 51,77 ммоль, 3 экв.) в сухом THF (100 мл) при 0°С в атмосфере N2 прибавляют NaH (60% в минеральном масле, 1,24 г, 51,77 мл, 3 экв.). После перемешивания при 0°С в течение 15 мин прибавляют раствор Соединения 3 (10 г, 17,26 ммоль, 1 экв.) в THF (250 мл). Смесь нагревают до 23°С, перемешивают в течение 1 ч и затем реакцию останавливают с помощью насыщенного водного раствора NaHCO3 (100 мл). Смесь экстрагируют с помощью EtOAc (100 мл×3). Объединенные органические слои сушат (MgSO4) и фильтруют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (4:1, гексан : EtOAc, затем 1:1, гексан : EtOAc) и получают Соединение 90 (8,88 г, 86%), МС с электрораспылением [М+1]+ 596,1.
Стадия 2:
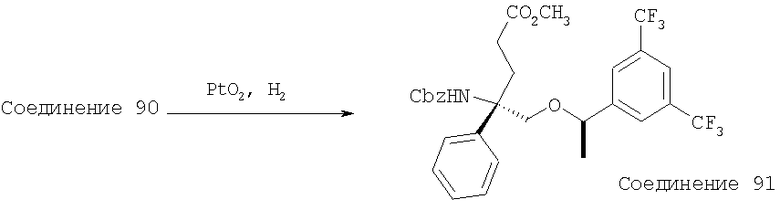
Соединение 91 получают из Соединения 90 с помощью методики, аналогичной использованной для получения Соединения 44 из Соединения 42. Неочищенное Соединение 91 используют в следующей реакции без дополнительной очистки.
Стадия 3:
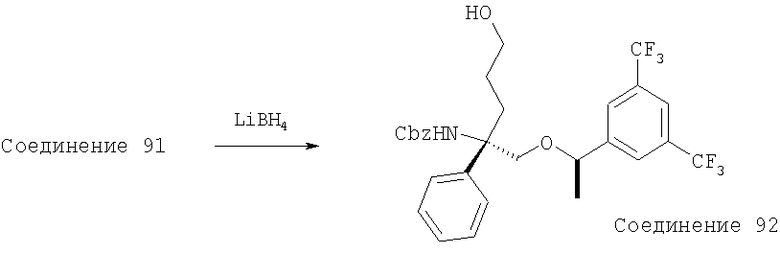
К раствору Соединения 91 (8,8 г, 14,73 ммоль, 1 экв.) в сухом THF (150 мл) прибавляют LiBH4 (0,58 г, 26,51 ммоль, 1,8 экв.) и реакционную смесь перемешивают при 0°С в течение 2 ч. Реакционную смесь охлаждают до 0°С на бане со льдом и реакцию останавливают с помощью насыщенного раствора NaHCO3 (50 мл). Реакционную смесь экстрагируют с помощью EtOAc (3×100 мл). Объединенные органические слои сушат (Na2SO4), фильтруют, концентрируют и получают неочищенное Соединение 92 (8,2 г), МС с электрораспылением [М+1]+ 570,1, которое используют в следующей реакции без дополнительной очистки.
Стадия 4:
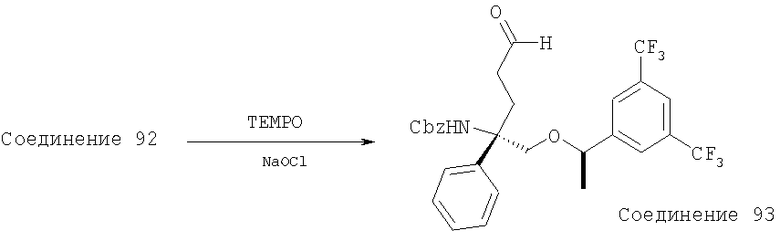
К раствору Соединения 92 (8,2 г, 14,4 ммоль, 1,0 экв.) в EtOAc (150 мл) при 0°С прибавляют насыщенный водный раствор NaHCO3 (150 мл) и реакционную смесь перемешивают в течение 10 мин при 0°С. К реакционной смеси прибавляют NaBr (1,5 г, 14,4 ммоль, 0,01 экв.), а затем TEMPO (0,0225 г, 0,144 ммоль, 0,1 экв.) и хлорную известь (5,25% в H2O, 20,4 мл, 14,4 ммоль, 1,0 экв.). Реакционную смесь перемешивают в течение 15 мин при 0°С. За протеканием реакции следят с помощью ТСХ с использованием (1:2) EtOAc/гексан, которая указывает на присутствие исходного вещества. К реакционной смеси прибавляют дополнительное количество NaOCl (2 мл) и перемешивают в течение 15 мин при 0°С, затем реакцию останавливают с помощью насыщенного раствора Na2S2O3 (20 мл). Реакционную смесь экстрагируют с помощью EtOAc (150 мл×3). Объединенные органические слои сушат над (MgSO4), фильтруют, концентрируют и получают неочищенное Соединение 93 (8 г), которое используют в следующей реакции без дополнительной очистки.
Стадия 4:
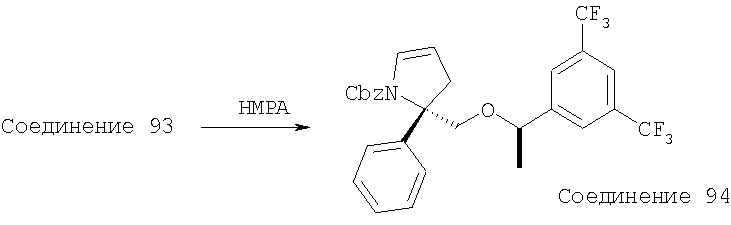
Смесь Соединения 93 (8 г, 14,1 ммоль, 1,0 экв.) и НМРА (50 мл) нагревают при 170°С в течение 2 ч. Реакционную смесь охлаждают до 23°С и реакцию останавливают водой (50 мл). Реакционную смесь экстрагируют с помощью Et2O (150 мл×3). Объединенные органические слои сушат над (MgSO4), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке Biotage (7:3, гексан : EtOAc) и получают Соединение 94 (3,8 г, 40% за три стадии), МС с электрораспылением [М+1]+ 550,1.
Стадия 5:
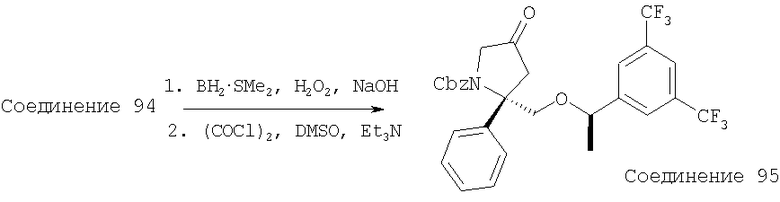
Соединение 95 получают из Соединения 94 с помощью методики, аналогичной использованной для получения Соединения 47 из Соединения 45. МС с электрораспылением [М+1]+ 566,1.
Стадия 6:
Соединение 95 превращают в менее полярный изомер - соединение Примера 103а, МС с электрораспылением [M+1]+ 502,1, и более полярный изомер - соединение Примера 103b, МС с электрораспылением [М+1]+ 502,1, с помощью методики, аналогичной использованной для получения соединения Примера 43а и соединения Примера 44b из Соединения 47.
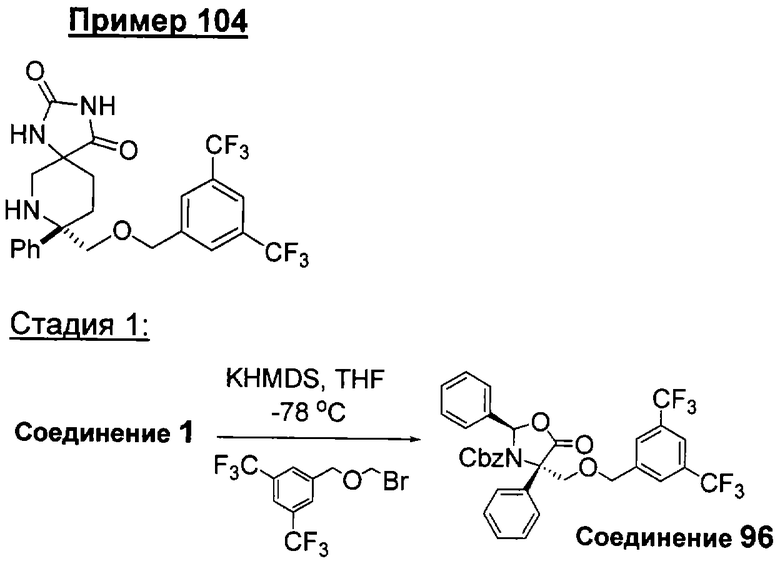
Соединение 96 получают из Соединения 1 с помощью методики, аналогичной использованной для получения Соединения 3 из Соединения 1. МС с электрораспылением [М+1]+ 566,1 для Соединения 106.
Стадия 2:
Соединение 96 превращают в смесь соединения Примера 104а и соединения Примера 104b с помощью методики, аналогичной использованной для получения соединений Примеров 43а и 44b из Соединения 2. Смесь двух изомеров разделяют на колонке ВЭЖХ ChiralPak AD с использованием (5:95, IPA : гексан) и получают чистый менее полярный изомер - соединение Примера 104а, МС с электрораспылением [М+1]+ 502,1, и более полярный изомер - соединение Примера 104b, МС с электрораспылением [М+1]+ 502,1.
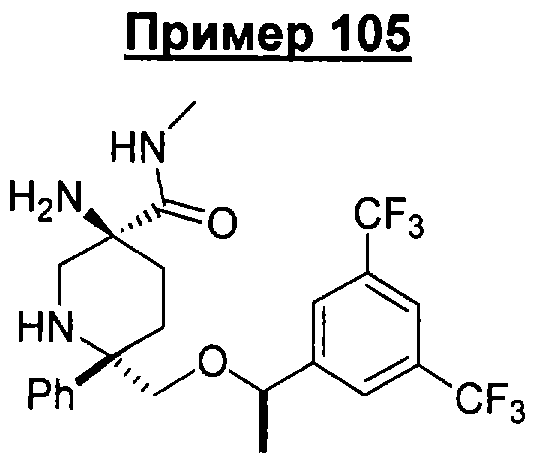
Соединение Примера 105 получают из Соединения 54 с помощью методики, аналогичной использованной для получения Соединения 62 из Соединения 54, но с использованием CH3NH2 (2 М в THF) вместо аммиака (0,5 М в 1,4-диоксане). МС с электрораспылением [М+1]+ 504,1.
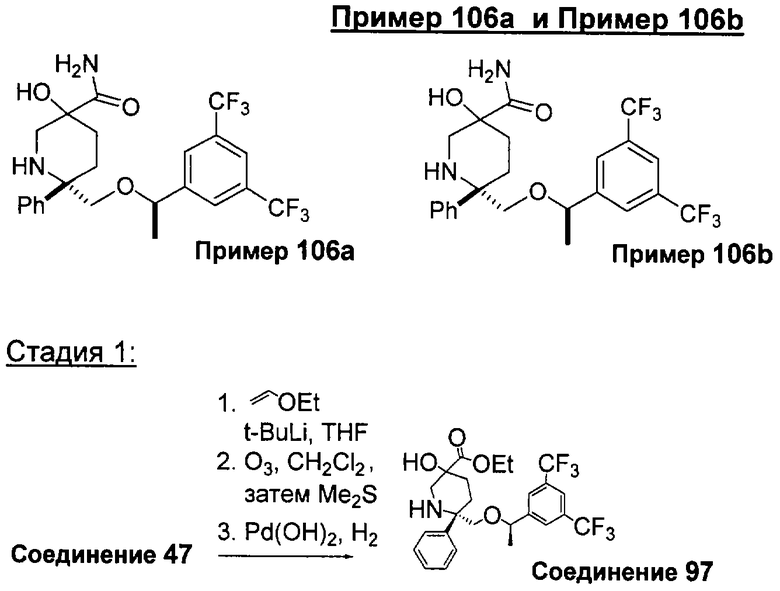
К раствору этилвинилового эфира (2,51 мл, 26,1 ммоль) в THF (50 мл) при -78°С в атмосфере N2 прибавляют t-BuLi (6,6 мл, 11,2 ммоль, 1,7 М в пентане). Смесь нагревают до 0°С и перемешивают, пока окраска раствора не перейдет в бледно-желтую. Затем смесь повторно охлаждают до -78°С и прибавляют раствор Соединения 47 (2,16 г, 3,73 ммоль) в THF (20 мл). Смесь перемешивают при -78°С в течение 1 ч, а затем реакцию останавливают с помощью насыщенного раствора NaHCO3. К смеси прибавляют воду и Et2O. Слои разделяют и водный слой экстрагируют с помощью Et2O (200 мл×2). Объединенные органические слои сушат (К2СО3, Na2SO4) и фильтруют. Растворители удаляют в вакууме и получают спирт в виде желтого масла. Спирт растворяют в CH2Cl2 (20 мл) и через раствор при -78°С пропускают озон, пока не исчезнет бледно-синяя окраска. Прибавляют (СН3)2S (2,7 мл, 37,3 ммоль) и смесь нагревают до комнатной температуры. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [CH2Cl2] дает сложный эфир в виде бесцветного масла. Сложный эфир растворяют в EtOH (20 мл) и прибавляют каталитическое количество Pd(OH)2 (20% на угле). Смесь встряхивают в приборе для гидрирования Парра при 45 фунт-сила/дюйм2 в течение ночи. Затем смесь фильтруют через слой целита, растворители удаляют в вакууме и получают бесцветное масло. Разделение с помощью хроматографии на колонке [гексан-EtOAc, 4:1 (об./об.)] дает Соединение 97 в виде бесцветного масла.
Стадия 2:
Соединение 97 растворяют в СН3ОН (10 мл) и через раствор в течение 30 мин пропускают аммиак. Смесь перемешивают при комнатной температуре в течение ночи, растворители удаляют в вакууме и получают желтое масло. Разделение с помощью ВЭЖХ с использованием Chiralcel OD [гексан-изопропанол, 9:1 (об./об.)] дает менее полярный содержащийся в большем количестве изомер - соединение Примера 106а (суммарный выход 15%) в виде белого вспененного вещества. МС с электрораспылением [М+1]+=491,1. Продолжение элюирования с помощью той же системы растворителей дает более полярный, содержащийся в меньшем количестве изомер - соединение Примера 106b (суммарный выход 10%) в виде белого вспененного вещества. МС с электрораспылением [М+1]+=491,1.
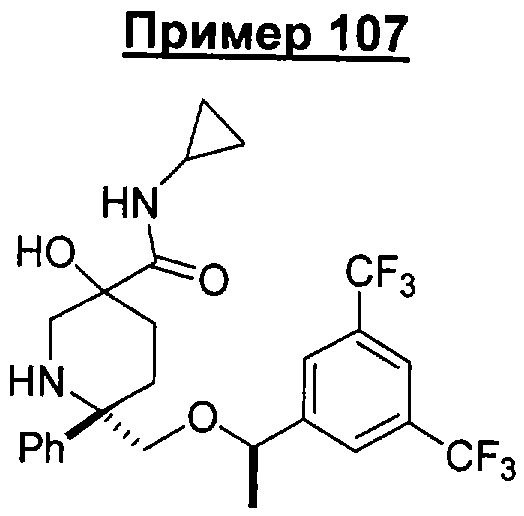
К раствору циклопропиламина (17 мкл, 0,20 ммоль) в толуоле (1 мл) при комнатной температуре в атмосфере N2 прибавляют Al(СН3)3 (0,1 мл, 0,20 ммоль, 2,0 М в толуоле). Смесь перемешивают при комнатной температуре в течение 20 мин и прибавляют раствор Соединения 97 (20 мг, 0,040 ммоль) в толуоле (1 мл). Смесь нагревают при 60°С в течение ночи и охлаждают до комнатной температуры. Прибавляют EtOAc и реакцию останавливают с помощью насыщенного раствора тартрата натрия-калия. Слои разделяют и водный слой экстрагируют с помощью EtOAc (100 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексан-EtOAc, 2:1 (об./об.)] дает соединение Примера 107 (11 мг, 56%) в виде бесцветного масла. МС с электрораспылением [М+1]+ 531.
Соединение Примера 108а и соединение Примера 108b получают из Соединения 97 с помощью методики, аналогичной использованной для получения соединения Примера 107 из Соединения 97, но с использованием 2,2,2-трифторэтиламина вместо циклопропиламина. МС с электрораспылением [M+1]+ 573,1 для менее полярного изомера - соединения Примера 108а и МС с электрораспылением [M+1]+ 573,1 для более полярного изомера - соединения Примера 108b.
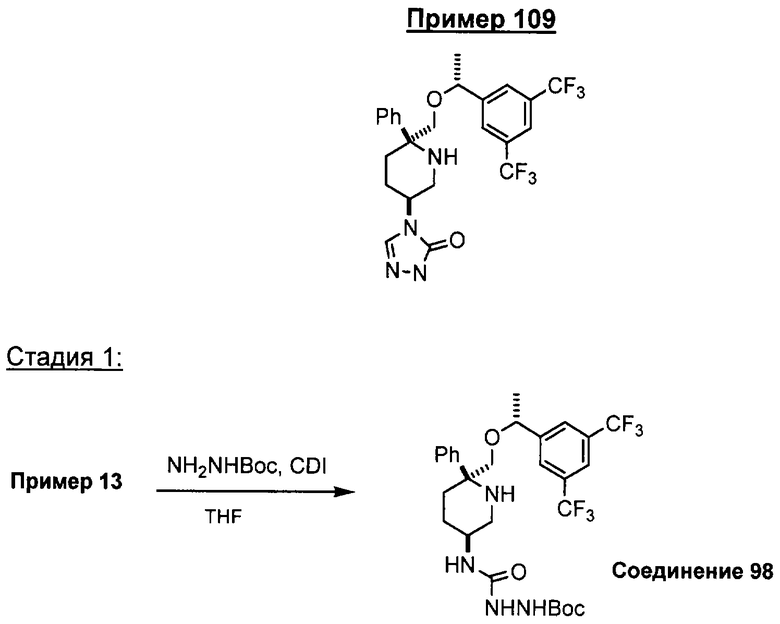
К раствору диамина - соединения Примера 13 (150 мг, 0,336 ммоль, 1 экв.) в безводном THF (5 мл) при 0°С прибавляют трет-бутилкарбазин (44,4 мг, 0,336 ммоль, 1 экв.), а затем CDI (65,4 мг, 0,404 ммоль, 1,2 экв.). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 ч. Затем реакционную смесь концентрируют и очищают на колонке Biotage (5:95, МеОН/EtOAc) и получают Соединение 98 (170 мг, 84%), МС с электрораспылением [M+1]+ 605,3.
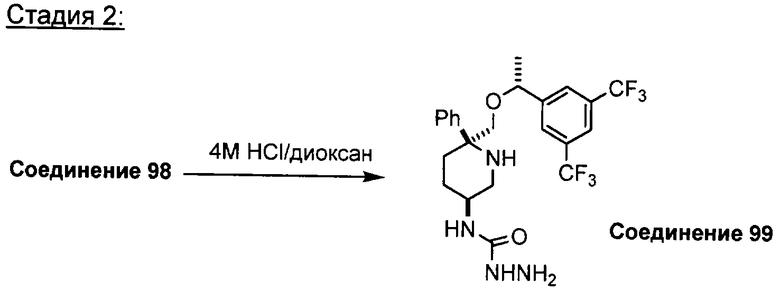
К раствору Соединения 98 (170 мг, 0,281 ммоль, 1 экв.) в безводном CH2Cl2 (15 мл) при 0°С прибавляют 4 М раствор HCl в 1,4-диоксане (0,7 мл, 2,81 ммоль, 10 экв.). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 18 ч. Реакцию останавливают с помощью насыщенного водного раствора NaHCO3 (100 мл) и экстрагируют с помощью EtOAc (2×150 мл). Органический слой сушат (Na2SO4), фильтруют и концентрируют. Неочищенный продукт Соединение 99 используют в следующей реакции без дополнительной очистки.
Стадия 3:
К раствору Соединения 99 (160 мг, 0,317 ммоль, 1 экв.) в безводном DMF (5 мл) прибавляют формамидинацетат (165 мг, 1,6 ммоль, 5 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Прибавляют НОАс (0,091 мл, 1,6 ммоль, 5 экв.) и реакционную смесь нагревают при 80°С в течение 6 ч. Затем реакционную смесь охлаждают до комнатной температуры, выливают в EtOAc (200 мл) и промывают водой (3×100 мл). Органический слой сушат (Na2SO4), фильтруют и концентрируют. Неочищенную смесь очищают на колонке Gilson (1:9, Н2O/СН3CN) и получают соединение Примера 109 (50 мг, 35%), МС с электрораспылением [М+1]+ 515,3.
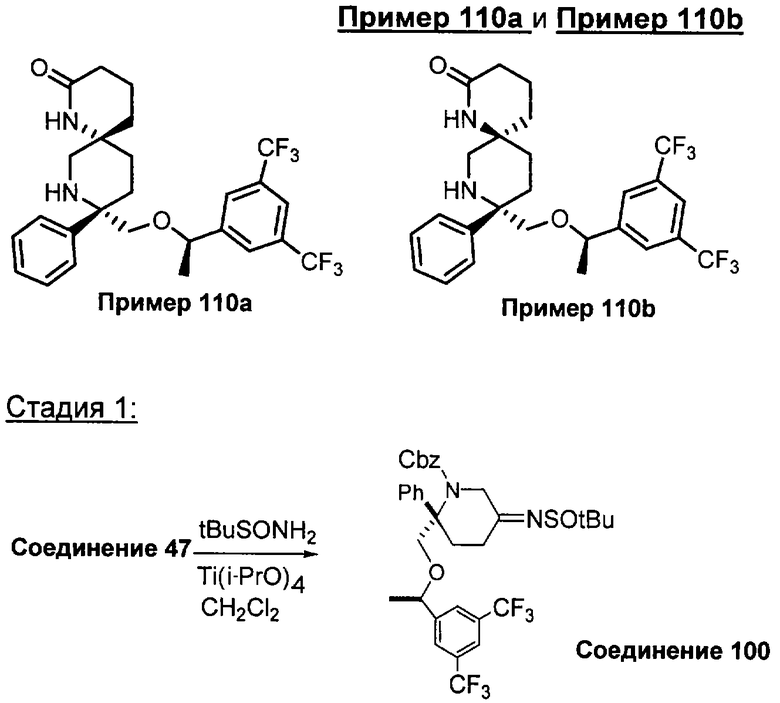
По методике, аналогичной использованной в Примере 11, стадия 2, Соединение 47 превращают в соответствующий сульфинимин - Соединение 100.
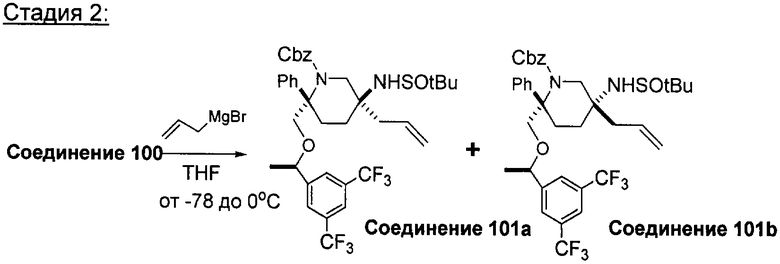
По методике, аналогичной использованной в Примере 11, стадия 3, Соединение 100 превращают в сульфинамид - Соединения 101 а и 101b.
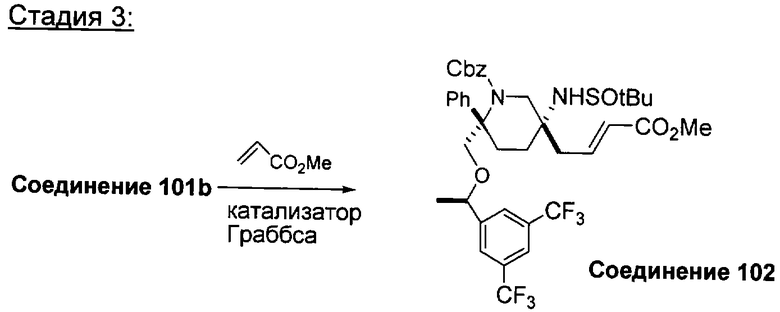
В грушевидную колбу объемом 15 мл помещают Соединение 101b (140 мг, 0,193 ммоль, 1 экв.) и СН2Cl2 (1мл). К этому бледно-желтому раствору прибавляют катализатор Граббса (13,7 мг, 0,016 ммоль, 0,084 экв.) и метилакрилат (21 мкл, 0,232 ммоль, 1,2 экв.). Полученный красноватый раствор нагревают при 40°С в течение ночи и реакцию останавливают с помощью метилсульфоксида (0,2 мл). После перемешивания при комнатной температуре в течение 20 ч его разбавляют с помощью Et2O и промывают водой. Органический слой сушат над MgSO4, фильтруют и концентрируют. Остаток очищают на колонке с силикагелем и получают Соединение 102 (100 мг, 66%).
Стадия 4:
В КДК помещают Соединение 102 (100 мг, 0,128 ммоль, 1 экв.) в EtOH (3 мл) и Pd(OH)2 на угле (90 мг, 0,128 ммоль, 1 экв., 20 мас.%). К верхней части колбы присоединяют баллон с водородом и смесь гидрируют в течение ночи. Реакционную смесь осторожно пропускают через воронку с целитом и слой целита осторожно промывают с помощью МеОН. Фильтрат концентрируют, затем повторно растворяют в МеОН (2 мл), обрабатывают с помощью HCl (2 мл, 4,0 М в 1,4-диоксане), перемешивают при комнатной температуре в течение 2 ч, затем повторно концентрируют, повторно растворяют в МеОН (5 мл), обрабатывают с помощью избытка К2СО3 и нагревают при 50°С в течение 3 ч, фильтруют, концентрируют и полученный остаток очищают на колонке с диоксидом кремния и получают соединение Примера 110а (42 мг, 64%), МС с электрораспылением [М+1]+ 515,1. Соединение Примера 110b (49%) получают по аналогичной методике, но с использованием Соединения 101а. МС с электрораспылением [М+1]+ 515,1.
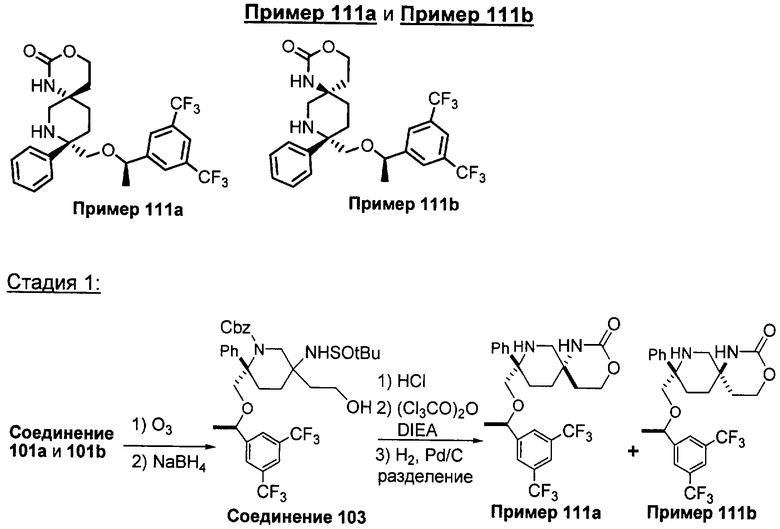
В КДК помещают смесь Соединений 101а и 101b (180 мг, 0,248 ммоль, 1,0 экв.) и CH2Cl2 (2 мл). Этот бледно-оранжевый раствор охлаждают до -78°С и затем через него пропускают О3. После того как раствор станет бледно-синим, раствор реакционной смеси перемешивают при -78°С в течение 10 мин, затем его продувают током N2 для удаления О3. Затем растворитель осторожно удаляют. Остаток растворяют в EtOH, а затем прибавляют NaBH4 (120 мг). Раствор перемешивают при комнатной температуре в течение 12 ч. Реакцию останавливают с помощью раствора NH4Cl. Реакционную смесь экстрагируют с помощью EtOAc (3×10 мл). Органический раствор промывают рассолом, сушат, концентрируют и получают Соединение 103, которое используют в следующей реакции без дополнительной очистки.
Неочищенное Соединение 103 растворяют в МеОН (2 мл) и охлаждают до 0°С, а затем прибавляют HCl (6 мл, 4 н. в диоксане). После перемешивания в течение 3 ч растворители удаляют и остаток повторно растворяют в 3 мл CH2Cl2, а затем прибавляют DIEA (178 мкл). Раствор охлаждают до 0°С, прибавляют трифосген (36 мг), реакционной смеси дают нагреться до комнатной температуры и ее перемешивают в течение 3 ч. Затем ее разбавляют с помощью EtOAc, промывают с помощью 5% HCl, NaHCO3 (водный раствор) и рассола. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт гидрируют и получают смесь соединений Примеров 111а и 111b. Смесь разделяют с помощью препаративной ТСХ (5% МеОН в СН2Cl2) и получают соединение Примера 111а (менее полярное) и соединение Примера 111b (более полярное). МС с электрораспылением - соединение Примера 111а [М+1]+ 517,1; соединение Примера 111а [M+1]+ 517,1.
Пример 112
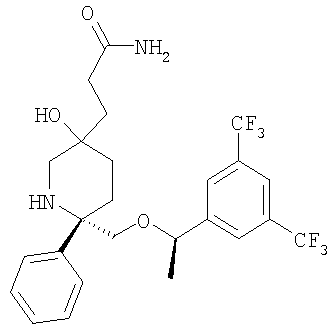
К раствору этилпропиолата (83 мкл, 0,82 ммоль) в THF (2 мл) при -78°С в атмосфере N2 прибавляют трет-бутиллитий (0,48 мл, 0,82 ммоль, 1,7 М в пентане). Смесь перемешивают при -78°С в течение 10 мин и прибавляют раствор Соединения 47 (158 мг, 0,27 ммоль) в THF (1 мл). Смесь перемешивают при -78°С в течение 1 ч, а затем реакцию останавливают с помощью НОАс при -78°С. К смеси прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и очистка с помощью хроматографии на колонке [гексаны-EtOAc, 4:1 (об./об.)] дает бесцветное масло (112 мг, 61%). Масло растворяют в EtOH и прибавляют каталитическое количество палладия (10% на угле). Затем смесь встряхивают в приборе для гидрирования Парра при 45 фунт-сила/дюйм2 в течение ночи. Затем смесь фильтруют через слой целита и растворители удаляют в вакууме, получают сложный эфир в виде бесцветного масла. Масло растворяют в СН3ОН (10 мл) и через раствор в течение 30 мин пропускают аммиак. Смесь перемешивают при комнатной температуре в течение ночи и растворители удаляют в вакууме. Очистка с помощью хроматографии на колонке [СН3ОН-EtOAc, 1:9 (об./об.)] дает соединение Примера 112 в виде бесцветного масла (54 мг, 61%). МС с электрораспылением [M+1]+=519,1.
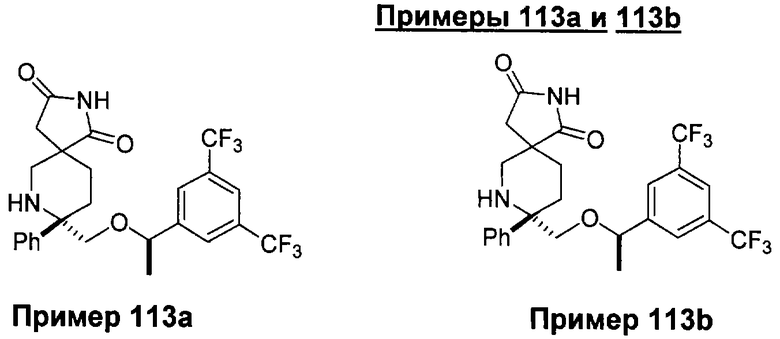
К раствору Соединения 51 (1,97 г, 3,10 ммоль) в CH2Cl2 (50 мл) при -78°С прибавляют DIBAL-H (9,3 мл, 9,3 ммоль, 1,0 М в толуоле). Смесь перемешивают при -78°С в течение 1 ч, а затем реакцию останавливают с помощью насыщенного раствора тартрата натрия-калия. Смесь нагревают до комнатной температуры и прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (200 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и хроматография на колонке [гексан-EtOAc, 3:1 (об./об.)] дает аллиловый спирт (1,6 г, 85%) в виде бесцветного масла.
Аллиловый спирт (1,6 г, 2,63 ммоль) растворяют в триэтилортоацетате (30 мл) и прибавляют каталитическое количество пропионовой кислоты. Смесь нагревают в запаянной трубке при 130°С в течение ночи. Растворители удаляют в вакууме и хроматография на колонке [гексан-Et2O, 5:1 (об./об.)] дает алкен (891 мг, 50%) в виде бесцветного масла.
Алкен (891 мг, 1,31 ммоль) растворяют в CH2Cl2 (20 мл) и охлаждают при -78°С. Через раствор пропускают О3, пока не исчезнет бледно-синяя окраска раствора. Реакционную смесь продувают током N2, пока не образуется бесцветный раствор. Прибавляют метилсульфид (1 мл) и смесь нагревают до комнатной температуры. Растворители удаляют в вакууме, и хроматография на колонке [гексаны-EtOAc, 5:1 (об./об.)] дает альдегид (800 мг, 90%) в виде бесцветного масла.
Альдегид (280 мг, 0,41 ммоль) растворяют в изопрене (2,4 мл) и трет-бутиловом спирте (7 мл) при комнатной температуре. Прибавляют раствор хлорита натрия (414 мг, 4,12 ммоль) в дигидрофосфате натрия (4 мл, 20 мас.% в воде). Смесь энергично перемешивают при комнатной температуре в течение 2 ч. Прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (250 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и получают неочищенную кислоту в виде желтого масла.
Неочищенную кислоту растворяют в CH2Cl2 (10 мл) при комнатной температуре и прибавляют диизопропиламин (0,22 мл, 1,24 ммоль), а затем РуВОР (322 мг, 0,62 ммоль). Смесь перемешивают при комнатной температуре в течение 20 мин, а затем прибавляют раствор аммиака в диоксане (8 мл, 4,12 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, а затем реакцию останавливают с помощью насыщенного раствора NaHCO3. Прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (250 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и получают неочищенный амид в виде желтого масла.
Неочищенный амид растворяют в СН3ОН (10 мл) и прибавляют Pd(OH)2 (20% на угле). Смесь перемешивают в атмосфере Н2 (из баллона) в течение 4 ч. Твердое вещество отфильтровывают через слой целита, растворители удаляют в вакууме и получают неочищенный аминоамид в виде желтого масла.
Неочищенный аминоамид растворяют в СН3ОН и прибавляют избыток NaOCH3. Смесь нагревают при 60°С в течение 1 ч, а затем реакцию останавливают с помощью насыщенного раствора NH4Cl. Прибавляют воду и EtOAc. Слои разделяют и водный слой экстрагируют с помощью EtOAc (250 мл×2). Объединенные органические слои сушат (MgSO4) и фильтруют. Растворители удаляют в вакууме и хроматография на колонке [гексан-EtOAc, 2:1 (об./об.)] дает менее полярный изомер - соединение Примера 113а (20 мг, суммарный выход за 4 стадии 9%) в виде белого вспененного вещества. МС с электрораспылением [М+1]+=515,1. Продолжение элюирования с помощью той же системы растворителей дает более полярный изомер - соединение Примера 113b (25 мг, суммарный выход за 4 стадии 12%) в виде бесцветного масла. МС с электрораспылением [M+1]+=515,1.
Приведенное выше описание не имеет целью подробное изложение всех модификаций и вариантов настоящего изобретения. Специалисты в данной области техники должны понять, что без отклонения от сущности изобретения в описанные выше варианты осуществления могут быть внесены изменения. Поэтому следует понимать, что настоящее изобретение не ограничивается описанными выше конкретными вариантами осуществления, а подразумевается, что оно охватывает модификации, которые входят в объем и сущность настоящего изобретения, как это определено в приведенной ниже формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2408591C9 |
| ТРИЦИКЛИЧЕСКИЕ ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2293734C9 |
| ПОЛИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ АНТАГОНИСТА РЕЦЕПТОРА H, И СПОСОБ ЛЕЧЕНИЯ С ЕГО ПРИМЕНЕНИЕМ | 2001 |
|
RU2301231C2 |
| ПРОИЗВОДНЫЕ ИНДЕНА В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2004 |
|
RU2381209C2 |
| ОКСА- И ТИАЗОЛПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИДИАБЕТИЧЕСКИХ АГЕНТОВ И АГЕНТОВ ПРОТИВ ОЖИРЕНИЯ | 2000 |
|
RU2279427C2 |
| ЗАМЕЩЕННЫЕ ПИРИДИНЫ И ПИРИДАЗИНЫ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ ИНГИБИРОВАТЬ АНГИОГЕНЕЗ | 2000 |
|
RU2260008C2 |
| ТРИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ ТРОМБИНОВОГО РЕЦЕПТОРА | 2003 |
|
RU2329264C9 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ СОДЕРЖАЩИЕ, И ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ CCR5 | 2000 |
|
RU2299206C9 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| АНАЛОГИ ЦИКЛОПАМИНА | 2007 |
|
RU2486194C2 |
Изобретение относится к новым соединениям - производным пирролидина или пиперидина общей формулы (I)
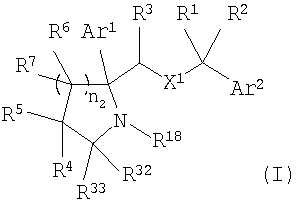
или их фармацевтически приемлемым солям,
где Ar1, Ar2, X1, R1, R2, R3, R4, R5, R6, R7, R18, R32, R33, n2 являются такими, как определено в формуле изобретения. Соединения являются антагонистами NK1. 23 з.п. ф-лы.
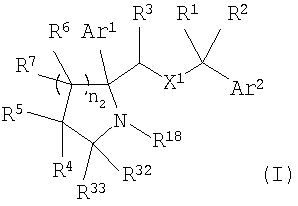
или их фармацевтически приемлемая соль, где
Ar1 и Ar2 означают группу
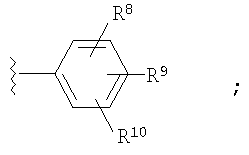
X1 означает -О-,
R1 и R2 независимо выбраны из группы, включающей Н, C1-C6 алкил;
R3 выбран из Н;
каждый R6 выбран из Н;
каждый R7 выбран из Н;
n2 равно от 1 до 4;
R4 и R5 независимо выбраны из группы, включающей 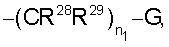
где n1 равно от 0 до 5; и
G означает Н, -ОН, -O-(C1-C6алкил), -NR13R14, -NR12SO2R13, -NR12C(O)R14, -NR12(C(O)NR13R14), -C(O)NR13R14, C(O)OR13, -OC(O)R14, -OC(O)NR13R14, -C(=NOR14)(R13), 5-членный гетероциклоалкенил, содержащий N в качестве гетероатомов и необязательно содержащий от 1 до 4 заместителей, независимо выбранных из группы, включающей R30 и R31,
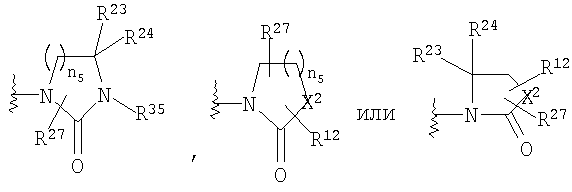
или R4 и R5 совместно означают =NOR12; или
R4 и R5 совместно с атомом углерода, к которому они оба присоединены, образуют 4- - 6-членный гетероциклоалкильный или гетероциклоалкенильный цикл, содержащий от 1 до 3 групп, независимо выбранных из группы, включающей X2, при условии, что хотя бы один X2 означает -NR35-, -О-, или -SO2-, цикл необязательно содержит от 1 до 6 заместителей, независимо выбранных из группы, включающей R30 и R31;
при условии, что R4 и R5 оба не выбраны из группы, включающей Н, алкил и циклоалкил;
также при условии, что, когда один из R4 и R5 означает -ОН, то другой из R4
и R5 не означает алкил;
R8, R9 и R10 независимо выбраны из группы, включающей Н, -CF3;
R12 означает Н, C1-C6 алкил;
R13 и R14 независимо выбраны из группы, включающей Н, C1-C6 алкил,
С3-С8 циклоалкил, -СН2CF3 и 5- или 6-членный гетероарил, содержащий атомы N и/или О в качестве гетероатомов, необязательно замещенный CN-группой; или
R13 и R14 совместно с атомом азота, к которому оба они присоединены, образуют 4- - 6-членный незамещенный насыщенный цикл, где один из атомов углерода цикла необязательно заменен на 0 в качестве гетероатома;
R15 означает C1-C6 алкил;
R18 означает Н;
R23 и R24 означают Н;
R27 означает Н или C1-C6 алкил;
R28 и R29 независимо выбраны из группы, включающей Н и C1-C2 алкил;
R30 и R31 независимо выбраны из группы, включающей Н, C1-C6 алкил;
или
R30 и R31 совместно с атомом углерода, к которому оба они присоединены, образуют =O;
R32 и R33 означают Н;
R35 означает Н, (С3-С8)циклоалкил(С1-С6)алкил, -Р(O)(ОН)2, аллил, гидрокси(С2-С6)алкил, (С1-С6)алкокси(С1-С6)алкил, или
-(CH2)2-N(R12)-SO2-R15, где R12 и R15 имеют вышеуказанное значение;
X2 означает -NR35-, -О-, -СН2, -SO2-;
n5 равно от 1 до 3;
или их диастереоизомер, энантиомер, стереоизомер, региостереоизомер, поворотный изомер или таутомер.
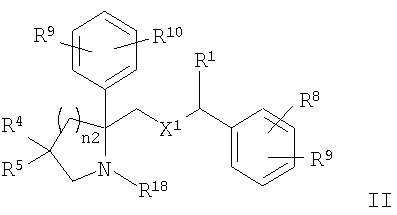
в которой X1 означает -О-, R8 и R9 означают -CF3, R9 и R10 означают Н, n2 равно 1 или 2 и R1, R4, R5 и R18 имеют указанное в п.1 значение.
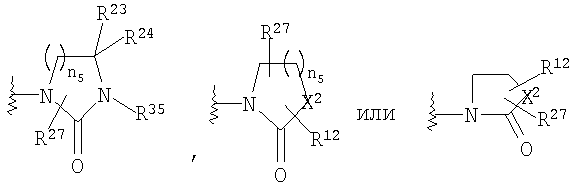
где R12-R14, R23, R24, R27, R35, X2 и n имеют указанное в п.1 значение.
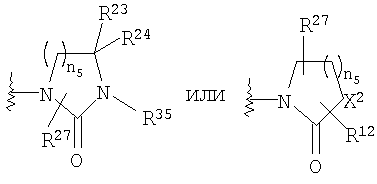
где R12-R14, R23, R24, R27, R35 и n5 имеют указанное в п.1 значение, Х2 означает -О-, -CH2- или -NR35-, где R35 имеет указанное в п.1 значение, R5 означает группу где n1 означает О и G означает -C(O)OR13 или -C(O)NR13R14.
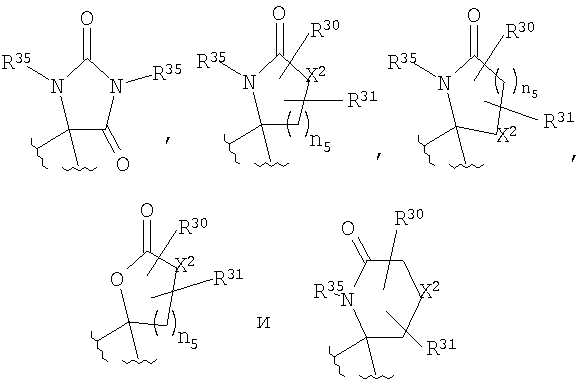 ,
,
где R35 означает Н, (С3-С8)циклоалкил(С1-С6)алкил или гидрокси(С1-С6)алкил; n5 равно 1, 2 или 3; X2 означает -NR35-, -CH2- или -О-, R30 означает Н или C1-C6 алкил; и R31 означает Н или C1-C6 алкил.
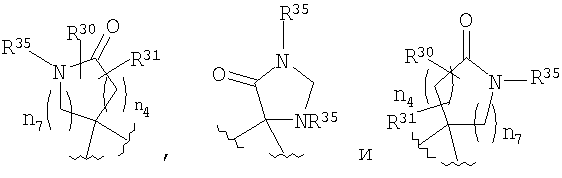
где R30 означает Н или C1-C6 алкил; R31 означает Н или C1-C6 алкил; каждый R35 независимо выбран из группы, включающей Н, (С3-С8)циклоалкил(С1-С6)алкил и гидрокси(С1-С6)алкил; n4 и n7 независимо равны 0-3, при условии, что сумма n4 и n7 равна 1-3.
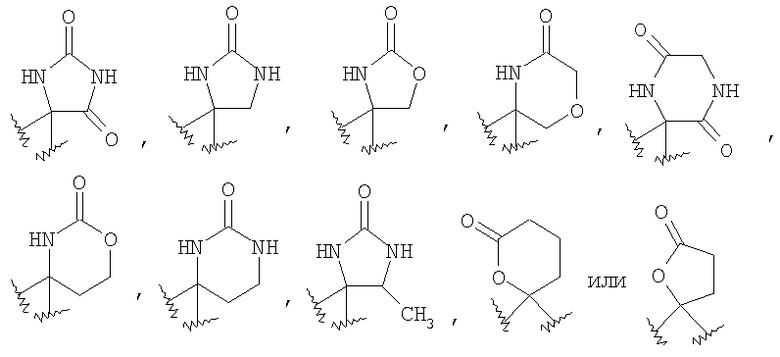 .
.
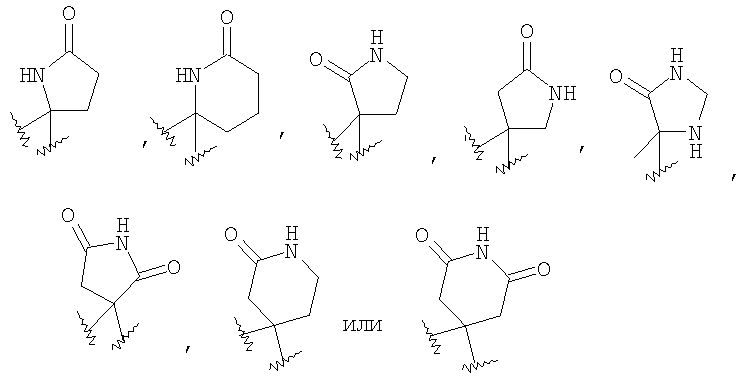 .
.
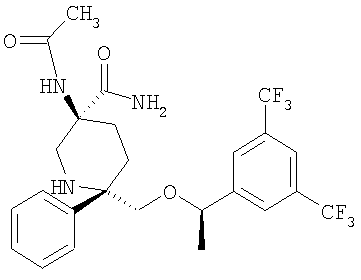 .
.
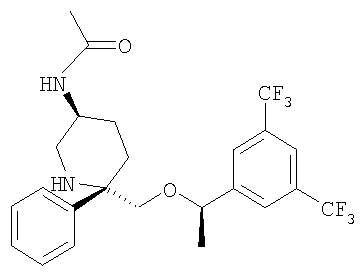 .
.
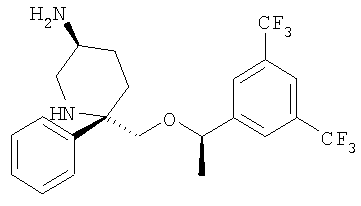 .
.
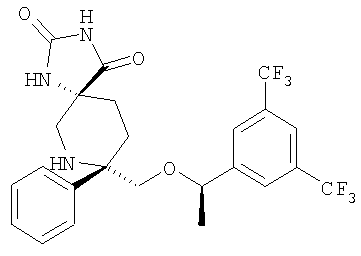 .
.
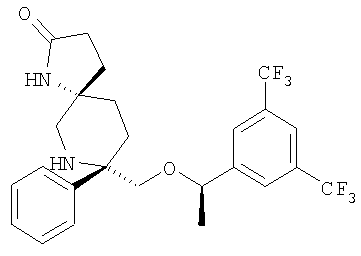 .
.
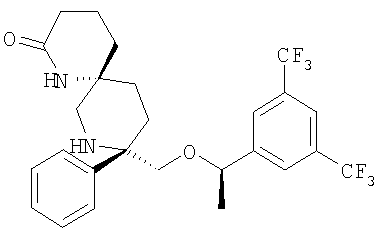 .
.
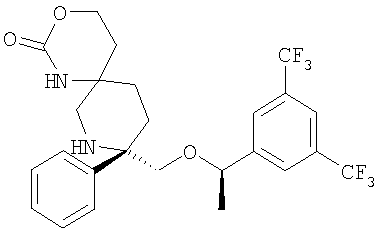 .
.
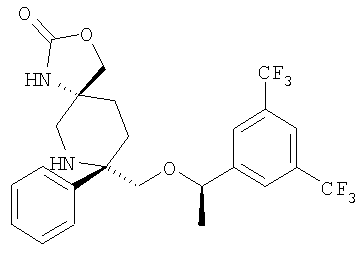 .
.
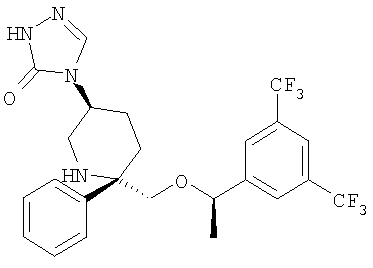 .
.
| Wu et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

