Область применения изобретения
Изобретение относится к новым производным 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты, их стереоизомерам и их фармакологически приемлемым солям, обладающим прекрасной антибактериальной активностью, а также к способам их получения. Изобретение, кроме того, относится к фармацевтическому препарату, содержащему эффективное количество производного и применимому для лечения инфекционных заболеваний, к способу лечения и к промежуточным соединениям для синтеза производных.
Описание прототипов
Ципрофлоксацин относится к хорошо документированному антибактериальному средству с хинолиновым ядром, замещенным в положении 1 циклопропильной группой (Каталог фирмы Мерк, II-ое издание, 2315).
Попытки улучшить ципрофлоксацин были направлены на введение разнообразных заместителей в положения 5, 7 и 8, однако настоящее изобретение относится к впервые полученным производным хинолона с аминогруппой в 5 положении и метильной группой в 8 положении в сочетании с пирролидинильной группой в 7 положении.
В настоящее время антибактериальная активность производных хинолона либо недостаточна, а если и достаточна, то сопровождается тяжелыми нежелательными эффектами, такими как фототоксичность, хромосомная аберрация, судороги и т. д., в силу чего последние средства создают проблему безопасности.
Следующие ссылки документально подтверждают вышеуказанные проблемы, связанные с хинолоновыми антибактериальными средствами.
1) "Хинолоновые противомикробные средства", 2-ое издание, глава 26, ред. D.C. Hooper, J.S. Wolfson,
Американское общество микробиологии, Вашингтон Д. К., 1993, стр. 489 (речь идет о фототоксичности, хромосомной аберрации, судорогах и т.д.).
2) Анализ на мутагенность, 2 (3), стр. 154 (1993) (хромосомная аберрация и т.д.).
3) Environ. Mol. Mutagen, 13, стр. 238 (1989) (хромосомная аберрация и т.д.).
Нижеследующим подчеркивается связь конкретных показателей применяемых заместителей в отдельных положениях с указанными затруднениями. К примеру, известно, что введение сравнительно объемистого заместителя, такого как атом хлора или метильная группа в 8 положение хинолинового ядра желательны для противомикробной активности, однако многие соединения, имеющие в 8 положении в качестве заместителя атом хлора, создают тяжелые нежелательные эффекты, такие как фототоксичность, хромосомная аберрация и т.д., а соединения с метильным заместителем также создают тяжелые нежелательные эффекты, такие как хромосомная аберрация и т.д. Применение подобных соединений связано с большими затруднениями с точки зрения их безопасности.
К широко применяемым в 5 положении заместителям относятся аминогруппа, атом галогена или метильная группа и т.д., но указанные заместители обладают недостатком, заключающимся в понижении противомикробной активности, а кроме того создают тяжелые нежелательные эффекты, такие как фототоксичность, хромосомная аберрация и т.д., вследствие чего возникает проблема безопасности.
Кроме того, введение в 7 положение пиперазинильной группы не создает достаточной противомикробной активности, а введение 3-аминопирролидинильной группы, обладающей достаточной противомикробной активностью, приводит к возникновению тяжелых нежелательных эффектов, таких как хромосомная аберрация и т.д., т.е. снова возникает проблема безопасности.
EP 0248361 раскрывает фармацевтическую композицию, проявляющую актибактериальную синергическую активность. Указанная композиция включает производное цефалоспорина (I) и производное антибиотика пенема (II). Композиция проявляет синергически увеличенную активность против широкого спектра грам-положительных и отрицательных бактерий, включая (1)-устойчивые штаммы и анаэробы.
Краткое изложение сущности изобретения.
Исследования, направленные на решение вышеуказанной проблемы, привели к настоящему изобретению, а именно, созданию производных 1-циклопропил-6-фтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты с 5-аминогруппой, 8-метилом и 7-пирролидинилом в хинолиновом ядре.
Соединения настоящего изобретения обладают очень эффективной антибактериальной активностью, более того, их применение не приводит к тяжелым нежелательным эффектам фитотоксичности, хромосомной аберрации, судорогам и т. д. вопреки тому, что можно было ожидать на основе ранее предпринятых попыток. Кроме того, соединения настоящего изобретения характеризуются прекрасным тканевым распределением, что позволяет быстро достигать высокой концентрации соединения в целевых тканях легких, почек и т.д., являющихся объектом лечения.
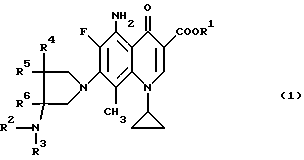
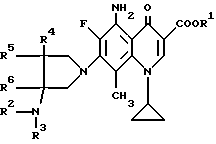
Согласно настоящему изобретению даются новые производные 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты общей формулы (I):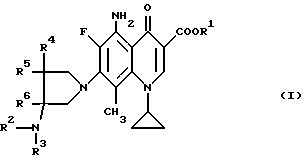
где R1 представляет водород или низший алкил;
R2 представляет водород, низший алкил, низший алканоил, галогенированный низший алканоил или остаток эфира карбоновой кислоты; R3 представляет водород или низший алкил;
R4, R5 и R6 каждый независимо представляет водород или низший алкил, или двое из R4, R5 и R6 могут совместно образовать -(CH2)n-группу, где n = 1 или 2, их стереиозомеры или их фармакологически приемлемые соли.
Даются также способ получения таких соединений, фармацевтические препараты, содержащие эффективные количества этих соединений и способы лечения инфекционных заболеваний путем введения больным эффективного количества указанных соединений.
Согласно еще одному воплощению настоящего изобретения даются новые производные 8-метилхинолин-3-карбоновой кислоты формулы (II):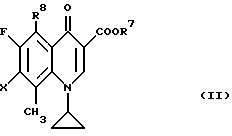
где R7 представляет низший алкил;
R8 представляет нитрогруппу или аминогруппу;
X представляет атом галогена,
являющихся эффективными промежуточными соединениями для получения вышеупмомянутых соединений формулы (I).
Подробное описание изобретения
Рекомендуемыми воплощениями настоящего изобретения даются следующие соединения общей формулы (I);
1) где R1, R2 и R3 каждый представляет атом водорода,
2) где R4 и R5 совместно образуют -(CH2)2-группу,
3) где R4, R5 и R6 каждый представляет атом водорода,
4) где R4-метил; R5 и R6 каждый представляет атом водорода. Кроме того, рекомендуемые воплощения настоящего изобретения включают способ получения указанных соединений, фармацевтические препараты, содержащие эффективное количество таких соединений, и способы лечения инфекционных заболеваний путем введения больным эффективного количества этих соединений, а также промежуточные соединения для их получения.
В вышеприведенных формулах (I) и (II) низший алкил, представленный R1, R2, R3R4, R5, R6 и R7, содержит 1-4 атома углерода, например; метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, или трет-бутил и т.д.; R2 представляет также низший алканоил, галогенированный низший алканоил или остаток эфира карбоновой кислоты. Если R2 представляет низший алканоил, то такая группа содержит 1-6 атомов углерода, например: формил, ацетил, пропаноил, бутироил или триметилацетил и т.д.; если R2 представляет галогенированный низший алканоил, то такая группа состоит из 1-4 атомов углерода и 1-5 атомов галогена, причем отдельные атомы галогена включают атомы фтора, хлора, брома и т.д., примеры таких групп включают: фторацетил, дифторацетил, трифторацетил, хлорацетил, дихлорацетил, трихлорацетил и т.д.; если R2 представляет остаток эфира карбоновой кислоты, такой остаток является алкоксикарбонилом или арилоксикарбонилом, например: бензилоксикарбонилом, этоксикарбонилом, метоксикарбонилом, трет-бутоксикарбонилом и т.д.; атом галогена, представленный X, может быть, например: атомом фтора, атомом хлора, атомом брома и т.д.
Кроме того, двое из R4, R5 и R6 совместно образуют -(CH2)n-группу (где n = 1 или 2), например, R4 и R5 объединяются с образованием -CH2-группы или -(CH2)2-группы, точно также R5 и R6 соединяются вместе с образованием -CH2-группы или -(CH2)2-группы.
Соединения настоящего изобретения общей формулы (I) при желании могут быть превращены в фармакологически приемлемые соли, и полученные соли могут быть затем вновь переведены в свободные соединения формулы (I).
Фармакологически приемлемые соли соединений настоящего изобретения общей формулы (I) могут быть солями, образованными добавлением кислот, и солями, образованными добавлением щелочей. Примеры солей с кислотами включают соли минеральных кислот, например: гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, фосфат и т.д., и соли органических кислот, например: ацетат, малеат, фумарат, цитрат, оксалат, малат, метансульфонат, п-толуолсульфонат, манделат, 10-камфорсульфонат, тартрат, лактат и т.д. Примеры солей с щелочами включают соли неорганических щелочей, например: соли натрия, калия, кальция, магния, аммония и т.д., и соли органических оснований, например: соли этаноламина, N,N-диалкиламина и т.д.
Соединения вышеприведенной общей формулы (I) имеют один или несколько асимметрических атомов углерода, такая молекула соединения, любые стереоизомеры или смеси стереоизомеров охватываются объемом изобретения.
Следующие соединения могут быть приведены в качестве реальных примеров производных 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты, но ими не исчерпываются примеры возможных соединений настоящего изобретения.
(1) 5-Амино-7-(7-амино-5-азаспиро/2.4/гепт-5-ил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота.
(2) 5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-7-(7-метиламино-5-азаспиро/ 2,4/гепт-5-ил/-4-оксохинолин-3-карбоновая кислота.
(3) 5-Амино-1-циклопропил-7-(7-диметиламино-5-азаспиро/2,4/-гепт-5-ил)-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота.
(4) 5-Амино-7-(3-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4- оксохинолин-3-карбоновая кислота,
(5) 5-Амино-7-(3-амино-4-метил-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота.
(6) 5-Амино-7-(3-амино-4,4-диметил-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота.
(7) 5-Амино-7-(3-амино-3-метил-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота,
(8) 5-Амино-7-(3-амино-4-метилен-1-пирролидинил)-1-циклопропил-6-фтор-1,4 дигидро-8-метил-4-оксохинолин-3-карбоновая кислота,
(9) 5-Амино-7-(1-амино-3-азабицикло/3.1.0/гекс-3-ил)-1-цикло-пропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота,
(10) 5-Амино-1-циклопропил-7-(3-диметиламино-1-пирролидинил)-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота,
(11) 5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-7-(3-метиламино-1- пирролидинил)-4-оксохинолин-3-карбоновая кислота,
их стереоизомеры или их фармакологически приемлемые соли.
Из перечисленных соединений особенно рекомендуются соединения (1), (4) и (5), их стереоизомеры и их фармацевтически приемлемые соли.
Согласно настоящему изобретению даются разнообразные способы получения новые производные 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты вышеприведенной общей формулы (I), в том числе и разъясняемые ниже способы. Нижеследующие способы получения не следует рассматривать, как исчерпывающие.
Согласно одному из примеров способа получения настоящего изобретения соединения общей формулы (I) могут быть получены реакцией в растворителе производного 7-галогенированной хинолин-3-карбоновой кислоты общей формулы (III)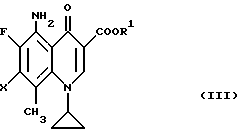
где R1 и X принимают вышеуказанные значения,
с производным пирролидина общей формулы (IV):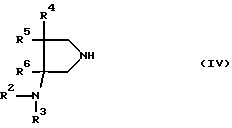
где R2, R3, R4, R5 и R6 принимают вышеуказанные значения,
в присутствии или отсутствие основания с последующим, если необходимо, гидролизом.
В способе настоящего изобретения может быть использован любой приемлемый растворитель. Примеры инертных растворителей включают: спирты, например: метанол, этанол, н-пропанол, изопропанол, трет-бутанол и т.д; апротонные полярные растворители, например: ацетонитрил, N,N-диметилформамид, N-метил-2-пирролидон, диметилсульфоксид, гексаметилфосфоростриамид и т.д.; ароматические углеводороды, например: бензол, толуол и т.д.; органические основания, например: пиридин, пиколин, лутидин, коллидин и т.д; или смеси указанных растворителей. Могут быть использованы следующие основания: триэтиламин, N,N-диизопропилэтиламин, 1,8-диазабицикло/5.4.0/-7-ундецен, 1,2,2,6,6-пентаметилпиперидин, 1,4-диазабицикло/2.2.2/-октан, карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия и т.д. Кроме того, если органическое основание применяют в качестве растворителя, такое основание может быть использовано вместо вышеперечисленных.
Реакция может быть проведена в температурном интервале от температуры бани со льдом до температуры кипения, применяемого в реакции растворителя.
Гидролиз может быть осуществлен известными способами в присутствии кислоты или щелочи. В реакции кислотного гидролиза могут применяться такие кислоты, как соляная кислота, серная кислота и т.д., а в реакции щелочного гидролиза могут применяться такие основания, как гидроксид натрия, гидроксид калия и т.д. Указанные кислоты или основания могут применяться в виде водных растворов или же в виде раствора в органическом растворителе, например: метаноле, этаноле, н-бутаноле, втор-бутаноле, трет-бутаноле и т.д., возможно с добавлением воды.
Реакция гидролиза может быть осуществлена в температурном интервале от комнатной температуры до температуры кипения применяемого в реакции растворителя.
Согласно второму примеру препаративного способа настоящего изобретения соединения вышеприведенной формулы (I) могут быть получены реакцией в растворителе производного борной кислоты общей формулы (V):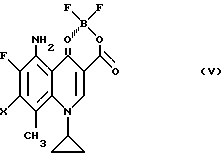
где X принимает вышеуказанные значения,
с производным пирролидина вышеприведенной формулы (IV) в присутствии или отсутствие основания с последующим, если необходимо, разрушением хелата в протонном полярном растворителе в присутствии или отсутствие основания.
В реакции соединений общей формулы (V) с соединениями общей формулы (IV) может быть использован любой приемлемый растворитель. Примеры инертных растворителей включают: спирты, например: метанол, этанол, н-пропанол, изопропанол, н-бутанол и т.д.; апротонные полярные растворители, например: ацетонитрил, N, N-диметиформамид, N-метил-2-пирролидон, диметилсульфоксид, гексаметилфосфоротриамид и т. д. ; ароматические углеводороды, например, бензол, толуол и т.д; органические основания, например: пиридин, пиколин, лутидин, коллидин и т.д.; галогенированные углеводороды, например: дихлорметан, 1,2-дихлорэтан, хлороформ и т.д. или смеси любых из перичисленных растворителей. Могут применяться следующие основания: триэтиламин, N,N-диизопропилэтиламин, 1,8-диазабицикло/5.4.0/-7-ундецен, 1,2,2,6,6-пентаметилпиперидин, 1,4-диазабицикло-/2.2.2/октан, карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия и т.д. Кроме того, при использовании органического основания в качестве растворителя такое основание может применяться вместо вышеупомянутых. Реакция может быть осуществлена в температурном интервале от температуры бани со льдом до температуры кипения применяемого в реакции растворителя.
Применяемые в разрушении хелата протонные полярные растворители включают: спирты, например: метанол, этанол, н-пропанол, изопропанол, н-бутанол и т. д. , воду или смеси указанных растворителей, или смеси апротонных растворителей, например: ацетонитрила, N,N-диметилформамида, N-метил-2-пирролидона, диметилсульфоксида, гексаметилфосфортриамида, бензола, толуола, пиридина, пиколина, лутидина, коллидина, дихлорметана, 1,2-дихлорэтана, хлороформа и т.д. с протонным полярным растворителем, например: спиртом, водой и т. д. Реакция может быть проведена в температурном интервале от температуры бани со льдом до температуры кипения применяемого в реакции растворителя.
Согласно третьему примеру препаративного способа настоящего изобретения соединения вышеприведенной формулы (I), где R2 - атом водорода, могут быть получены гидролизом соединения формулы (I), где R2 представляет низший алканоил или галогенированный низший алканоил, или обработкой соединения формулы (I), где R2 представляет остаток эфира карбоновой кислоты, в растворителе или без растворителя кислотой в присутствии или отсутствие отщепляющего катион агента.
Гидролиз может быть проведен известными методами в присутствии кислоты или щелочи. В реакции кислотного гидролиза могут быть использованы такие кислоты, как соляная кислота, серная кислота и т.д., а в реакции щелочного гидролиза могут применяться такие основания, как гидроксид натрия, гидроксид калия и т.д. Указанные кислоты или основания могут применяться в виде водного раствора или же в виде раствора в органическом растворителе, например: метаноле, этаноле, н-бутаноле, втор-бутаноле, трет-бутаноле и т.д., возможно с добавлением воды. Реакция гидролиза может быть проведена в температурном интервале от комнатной температуры до температуры кипения применяемого в реакции растворителя.
Удаление остатка эфира карбоновой кислоты может быть осуществлено в таком растворителе, как, например, уксусная кислота, этилацетат, диоксин, вода, метанол, этанол или их смеси; в качестве отщепляющего катион агента возможно применение, например: анизола, тиоанизола и т.д.; в качестве кислоты возможно применение хлористоводородной кислоты, бромистоводородной кислоты, трифторуксусной кислоты и т. д. Удаление остатка эфира карбоновой кислоты может быть проведено в температурном интервале от температуры бани со льдом до температуры кипения применяемого растворителя.
Согласно четвертому примеру препаративного способа настоящего изобретения соединения настоящего изобретения общей формулы (I), где R2 и/или R3 каждый представляет низший алкил, могут быть получены реакцией соединения формулы (I), где R2 и/или R3 каждый представляет атом водорода, с галогенированным низшим алкилом в растворителе в присутствии или отсутствие основания или с альдигедом нижеследующей общей формулы (VI):
R9-CHO (VI)
где R9 представляет атом водорода или низший алкил,
в присутствии муравьиной кислоты.
В данном препаративном способе, в случае применения галогенированного низшего алкила могут применяться такие растворители, как, например: N,N-диметилформамид, ацетон, этанол, тетрагидрофуран, бензол, хлороформ и т.д. и такие основания, как, например: триэтиламин, карбонат калия и т.д. В случае применения альдегида общей формулы (VI) примеры таких альдегидов включают: формальдегид, ацетальдегид, пропиональдегид и т.д., и желательно применение формальдегида в виде водного раствора (формалина), а в случае ацетальдегида или пропиональдегида желательно их применение в нитробензоле в качестве растворителя. Все указанные реакции могут быть проведены в температурном интервале от комнатной температуры до температуры кипения применяемого в реакции растворителя.
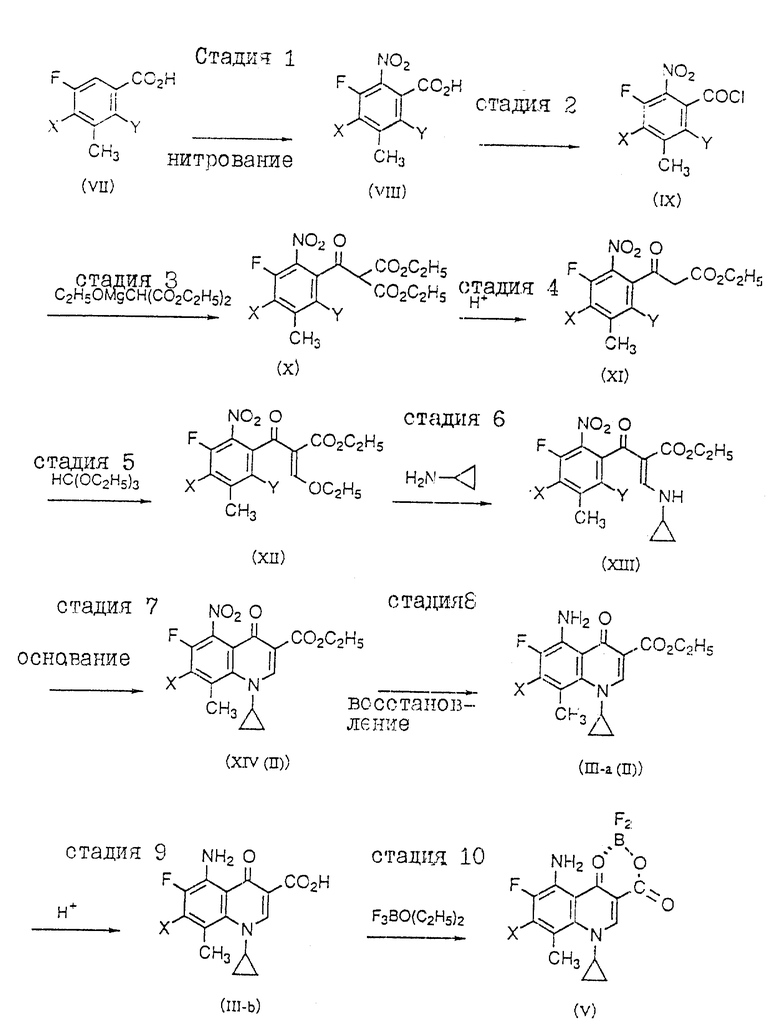
В препаративном способе настоящего изобретения исходные соединения вышеприведенных формул (III) и (V) могут быть синтезированы нижеследующим способом, подробности которого приведены в последующих примерах.
Кроме того, соединения общей формулы (VII), приведенной ниже, являются известными соединениями (см. нерассмотренную патентную публикацию Японии N 62-215572).
где X принимает вышеуказанные значения и Y представляет атом галогена.
Стадия I).
3-Метил-2,4,5-тригалогенбензойную кислоту (VII) нитруют с образованием соединения (VIII). На данной стадии в качестве нитрующего средства возможно применение азотной кислоты, нитрата калия, нитрата аммония и т.д., а в качестве растворителя можно применять серную кислоту, уксусную кислоту, уксусный ангидрид, трифторуксусную кислоту и т.д.
Стадия 2).
Соединение (VIII) обрабатывают хлорирующим средством, например: тионилхлоридом, оксалилхлоридом и т. д. в растворителе или без оного, например: хлороформе, хлористом метилене, 1,2-дихлорэтане и т.д., в присутствии или отсутствие N,N-диметилформамида с образованием хлорангидрида (IX).
Стадия 3). Соединение (IX) и диэтиловый эфир этоксимагниймалоновой кислоты (получен отдельно из этанола, диэтилмалоната и магния) конденсируют в растворителе, например: бензоле, толуоле и т.д. с получением соответственно соединения (X).
Стадия 4).
Соединение (X) гидролизуют и декарбоксилируют нагреванием с водой в присутствии кислоты, например: соляной кислоты, серной кислоты, п-толуолсульфокислоты и т.д. с получением соответственно соединения (XI).
Стадия 5).
Проводят реакцию соединения (XI) с этилортоформатом в присутствии или отсутствие кислоты Льюиса, например: хлорида цинка и т.д. в уксусном ангидриде с получением соответственно соединения (XII).
Стадия 6).
Реакцией соединения (XII) с циклопропиламином в растворителе получают соединение (XIII). На данной стадии может применяться любой приемлемый инертный растворитель, в том числе: спирты, например: метанол, этанол, и т. д. ; галогенированные углеводороды, например: хлороформ, 1,2-дихлорэтан и т. д. ; ароматические углеводороды, например: бензол, толуол и т.д. или апротонные полярные растворители, например: ацетонитрил, N,N-диметилформамид и т.д.
Стадия 7).
Соединение (XIII) циклизуют действием основания в растворителе в присутствии или отсутствие катализатора с получением соответственно соединения (XIV) (II). На данной стадии в качестве основания возможно применение карбоната калия, гидрида натрия, трет-бутоксида калия и т.д.; в качестве растворителя могут применяться простые эфиры, например: диоксан, тетрагидрофуран и т.д. или апротонные полярные растворители, например: ацетонитрил, N, N-диметилформамид и т.д.; в качестве катализатора можно применять краун-эфиры, тетрабутиламмонийбромид, бензилтриэтиламмонийбромид и т.д.
Стадия 8).
Соединение (XIV) (II)) восстанавливают в присутствии катализатора, например, никеля Ренея, палладия на угле, оксида платины и т.д. или восстанавливают в кислотных условиях металлами, например: железа, олова, цинка и т. д. с получением соответственно соединения (III-a(II)). На данной стадии в качестве растворителя могут применяться уксусная кислота, вода, метанол, этанол, N,N-диметилформамид и т.д., а в реакции с металлами могут применяться такие кислоты, как хлористоводородная кислота, уксусная кислота, бромистоводородная кислота и т.д.
Стадия 9).
Соединение (III-a(II) гидролизуют в растворителе, например: воде, уксусной кислоте, спирта, водном спирте и т.д. в кислотных условиях, в присутствии, например: хлористоводородной кислоты, уксусной кислоты, бромистоводородной кислоты и т.д. с получением соответственно соединения (III-b).
Стадия 10).
Реакцией соединения (III-b) с эфиратом трехфтористого бора в растворителе, например: эфире, ацетоне, метилизобутилкетоне и т.д. получают соответственно соединение (V).
Фармацевтический препарат, содержащий эффективное количество нового производного 5-амино-8-метил-7-пирролидинлхинолин-3-карбоновой (одного или нескольких) вышеприведенной формулы (I), его стереоизомера или его фармакологически приемлемой соли, полученных вышеприведенным способом, может иметь вид капсул, таблеток, мелких гранул, просто гранул, порошка, сиропа и т.д. для перорального введения или иметь вид инъекций, суппозитория, глазных капель, глазной мази, ушного раствора или дерматологической дозированной формы. Фармацевтический препарат настоящего изобретения может быть приготовлен смешиванием фармацевтически приемлемой добавки с производным 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты, его стереоизомером или его фармакологически приемлемой солью с последующей обычной обработкой смеси. При получении фармацевтического препарата, пригодного для перорального введения, или суппозитория добавка может представлять собой разбавитель, например: лактозу, D-маннит, кукурузный крахмал, кристалличекую целлюлозу и т.д.; структуратор, например: карбоксиметилцеллюлозу, кальций карбоксиметилцеллюлозу и т.д.; связующее средство, например: гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон и т. д.; смазку, например: стеарат магния, тальк и т.д.; кроющее средство, например: гидроцилцеллюлозу, сахарозу, оксид титана и т.д.; пластификатор, например: полиэтиленгликоль и т. д. или основу, например: полиэтиленгликоль или твердый жир и т.д. Фармацевтический препарат, пригодный для инъекций или применяемый в виде глазных капель или ушных капель, может содержать носители, например: солюбилизирующее средство или растворитель, например: дистиллированную воду для инъекций, солевой раствор, пропиленгликоль и т.д., применяемых для приготовления водных препаратов или препаратов для приготовления водного раствора перед употреблением; регулирующее pH средство, например: неорганические и органические кислоты и основания; придающие изотоничность средства, например: хлорид натрия, глюкозу, глицерин и т.д.; стабилизаторы и т.д. При изготовлении фармацевтического препарата, применяемого в виде глазной мази или дерматологического средства, могут применяться добавки, например, приемлемые фармацевтические ингредиенты, такие как белый петролатум, макроголь, глицерин, жидкий парафин, ткань и т.д., применяемые при получении мазей, кремов или припарок.
Применение фармацевтического препарата настоящего изобретения заключается во введении больному вышеописанного препарата перорально или парентерально. Доза препарата для взрослого больного, как правило, составляет 10-1000 мг в день при пероральном введении или 1-500 мг при парентеральном введении; доза может быть увеличена или уменьшена в зависимости от состояния подлежащего лечению больного.
Фармакологическое действие.
Соединения настоящего изобретения считаются 5-аминированными, 8-метилированными или 7-пирролидинилированными аналогами следующих ссылочных соединений. Превосходство соединений настоящего изобретения над ссылочными соединениями превосходит ожидания, основанные на известном уровне техники.
Создатели настоящего изобретения обнаружили, что одновременное введение аминогруппы, метильной или пирролидинильной групп соответственно в 5-, 8- и 7-положение хинолонового ядра снижает хромосомную аберрационную активность таких соединений. Подобное влияние заместителей до сего времени не было известно, и его невозможно было ожидать на основе известного уровня техники, а именно, на основе структурного превращения ссылочных соединений. Это показано следующими результатами анализа хромосомной аберрации (методика анализа приводится ниже).
Полученные результаты приведены в таблицах 1-3 (см. в конце описания).
Следующие соединения применялись в качестве ссылочных.
Ссылочное соединение A: 7-((S)-7-амино-5-азаспиро/2,4/-гепт-5-ил)-1- циклопропил-6-фтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты гидрохлорид (известное соединение, нерассмотренная патентная публикация Японии N 95176 (1991).
Ссылочное соединение B: 5-амино-7-((S)-7-амино-5-азаспиро-/2,4/гепт-5-ил)-1-циклопропил-6-фтор-1,4- дигидро-4-оксохинолин-3-карбоновой кислоты гидрохлорид (новое соединение).
Ссылочное соединение C: 7-((S)-7-амино-5-азаспиро/2,4/гепт-5-ил)-циклопропил-6-фтор-1,4-дигидро- 8-метил-4-оксохинолин-3-карбоновая кислота (известное соединение, нерассмотренная патентная публикация Японии N 95176 /1991).
Ссылочное соединение D: 7-((S)-3-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-4-оксохинолин- 3-карбоновая кислота (известное соединение, нерассмотренная патентная публикация Японии N 258855/1988).
Ссылочное соединение E: 5-амино-7-((S)-3-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-4-оксохинолин-3-карбоновая кислота (новое соединение).
Ссылочное соединение F: 7-((S)-3-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-8-метил- 4-оксохинолин-3-карбоновая кислота (новое соединение).
Ссылочное соединение G: 5-амино-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-пиперазинилхинолин-3- карбоновая кислота (известное соединение, нерассмотренная патентная публикация Японии N 28157/1999).
Ссылочное соединение H: 1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-пиперазинилхинолин-3- карбоновой кислоты гидрохлорид (известное соединение, нерассмотренная патентная публикация Японии N 215572/1987).
Ссылочное соединение I: 5-амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-пиперазинил-3- карбоновой кислоты гидрохлорида (известное соединение, нерассмотренная патентная публикация Японии N 215572/1987).
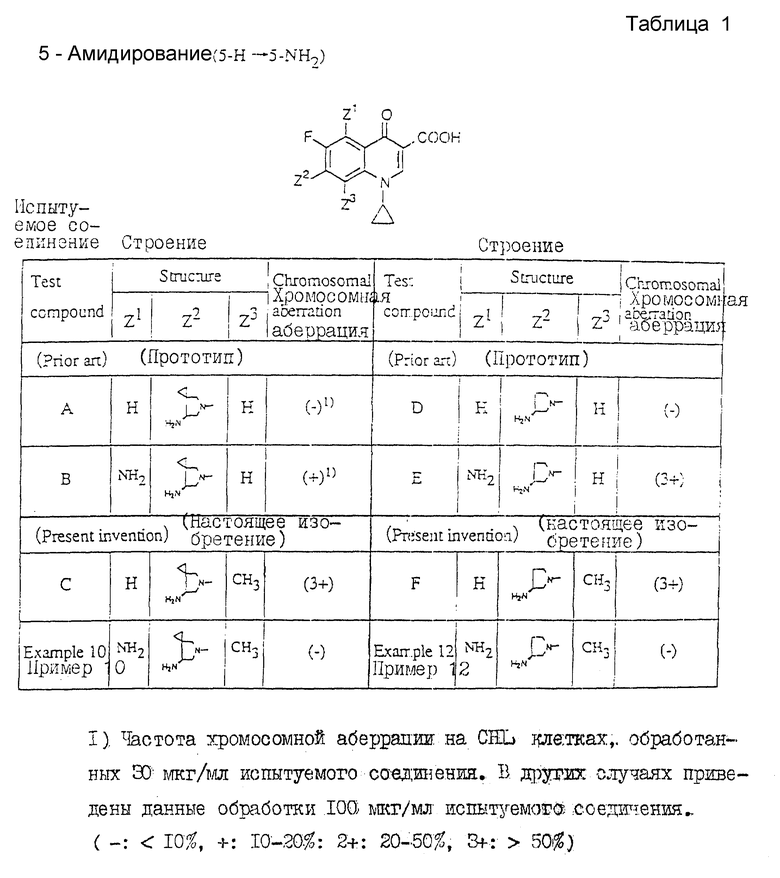
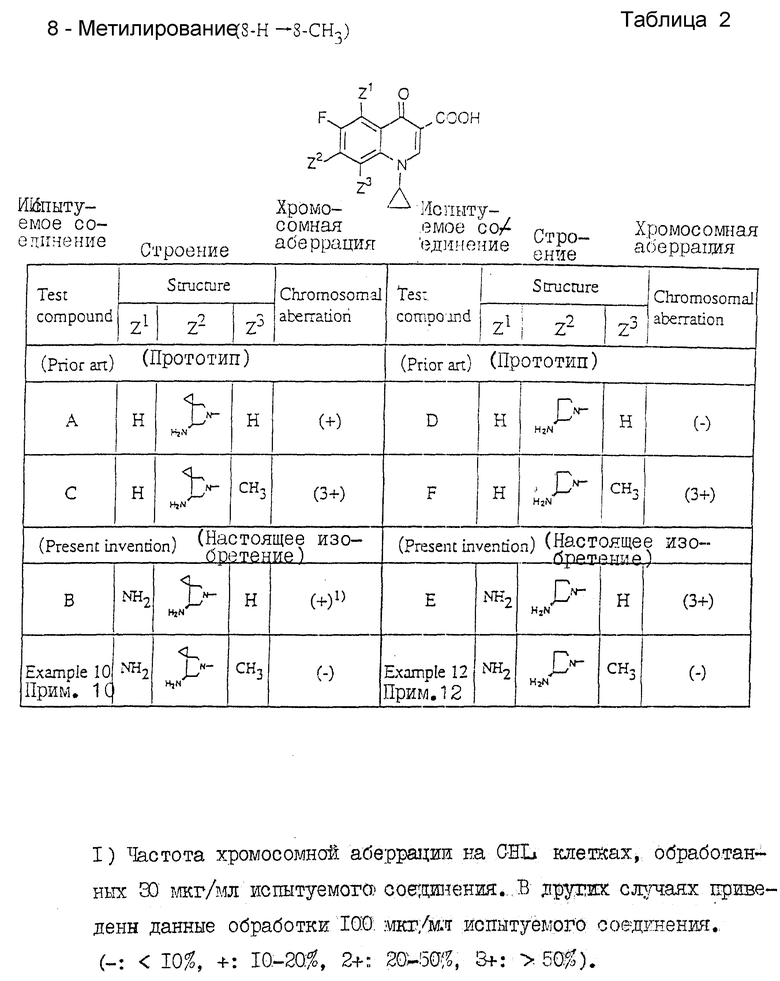
1) 5-Амидирование (5-H _____→ 5NH2)
Таблица 1 показывает следующее.
Наблюдение относительной активности ссылочных соединений A и B ((-) ___→ (+)), D и E ((-) ___→ (3+)) должно привести к заключению о том, что введение аминогруппы в 5-положение хинолонового ядра должно увеличивать хромосомную аберрационную активность.
Таким образом, следует ожидать, что соединение настоящего изобретения (примеры 10 и 12), рассматриваемые, как 5-аминированные аналоги ссылочных соединений C и F, должны проявлять более высокую активность, чем ссылочные соединения C и F соответственно, характеризующиеся высокой хромосомной аберрационной активностью. Вопреки таким ожиданиям соединения настоящего изобретения характеризуются (-). Подобные результаты не могли быть предсказаны на основе прототипа.
2) Метилирование (8-H ___→ 8-Me)
Таблица 2 показывает следующее.
На основе рассмотрения относительной активности ссылочных соединений A и C ((+) ___→ (3+)), D и F ((-) ___→ (3+)) можно ожидать, что введение метильной группы в 8-положение хинолонового ядра должно увеличить хромосомную аберрационную активность.
Таким образом, можно ожидать, что соединения настоящего изобретения (примеры 10 и 12), рассматриваемые, как 8-метилированные аналоги ссылочных примеров B и E, будут обладать более высокой активностью по сравнению с ссылочными примерами B и E соответственно, проявляющими хромосомную аберрационную активность. Вопреки таким ожиданиям соединения настоящего изобретения характеризуются (-). Подобные результаты не могли быть предвидены на основе прототипа.
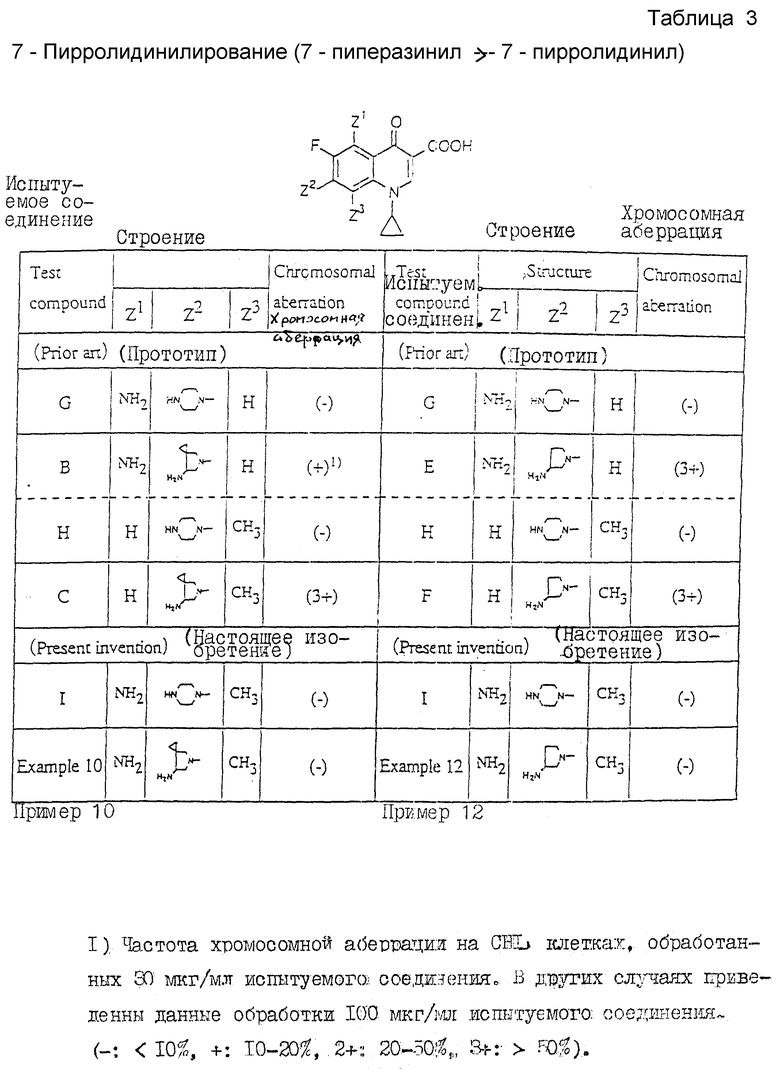
3) 7-Пирролидинилирование (7-пиперазинил ___→ 7-пирролидинил)
Из таблицы 3 следует.
На основе рассмотрения относительной активности ссылочных соединений 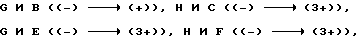 можно ожидать, что замена пиперазинильной группы в 7-положиении хинолонового ядра пирролидинильной группой должно повысить хромосомную аберрационную активность.
можно ожидать, что замена пиперазинильной группы в 7-положиении хинолонового ядра пирролидинильной группой должно повысить хромосомную аберрационную активность.
Таким образом, ожидается, что соединения настоящего изобретения (примеры 10 и 12), рассматриваемые, как 7-пирролидинилированные аналоги ссылочного соединения 1, будут давать положительные результаты при более высокой активности, чем у ссылочного соединения 1. Вопреки таким ожиданиям соединения настоящего соединения характеризуются (-). Подобные результаты невозможно предвидеть на основе прототипа. (Особая характеристика соединений настоящего изобретения).
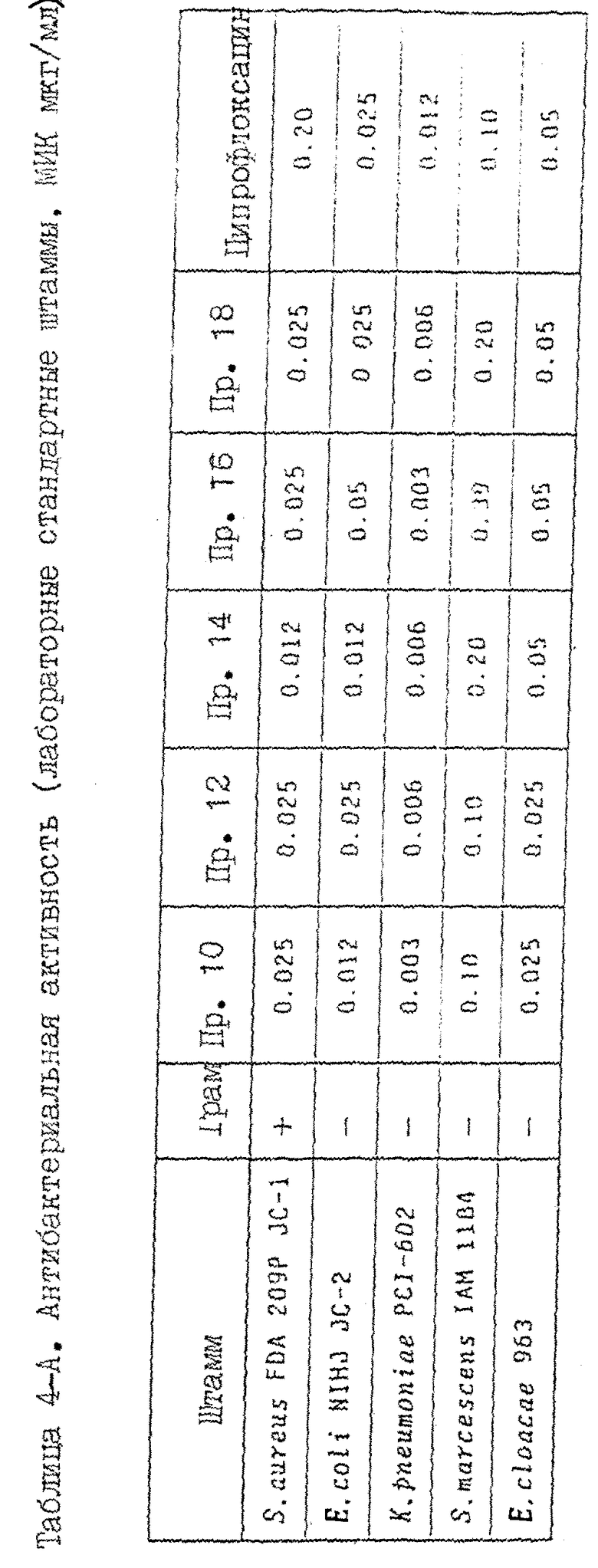
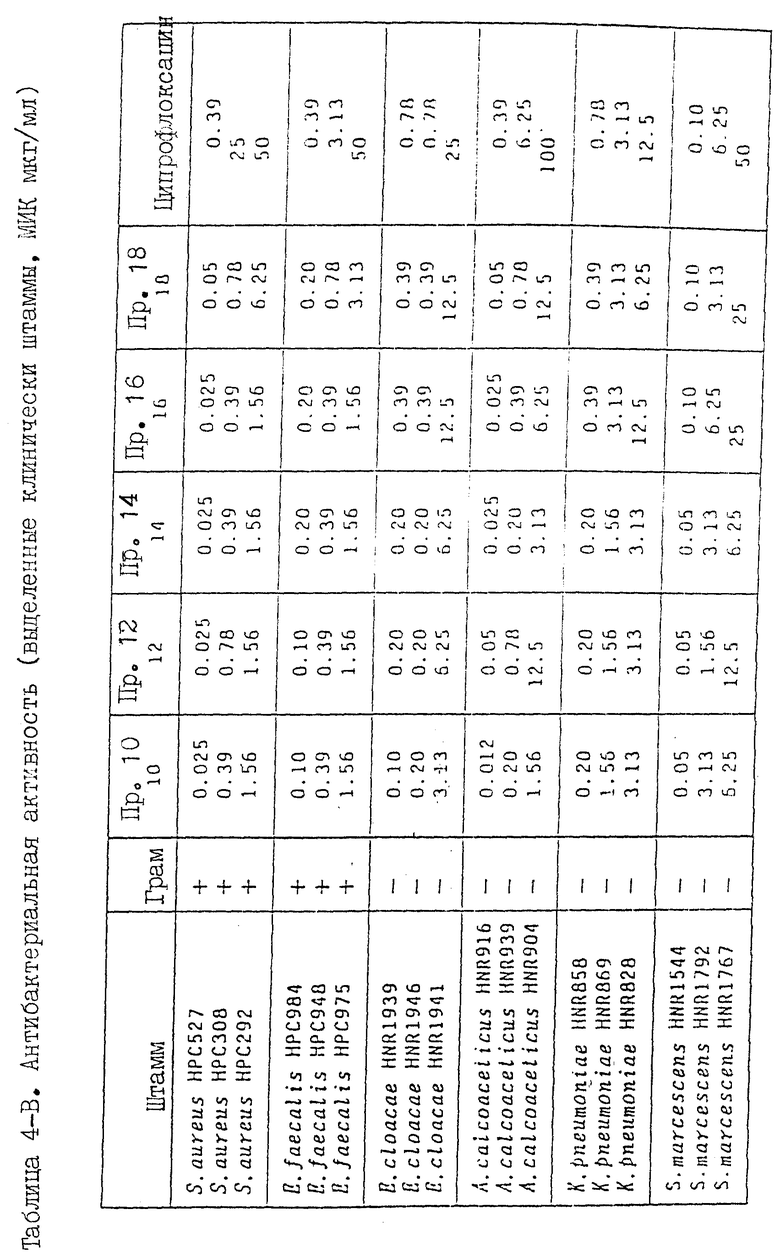
Прекрасное действие соединений настоящего изобретения суммировано в таблицах 4-6, (см. в конце описания) в которых приведены результаты анализа следующих показателей: антибактериальной активности пролабораторных стандартных штаммов и клинически выделенных штаммов, реально вызывающих инфекционные заболевания, хромосомной аберрации, индуцирование микронуклеуса, фототоксичность, индуцирование судорог и тканевое распределение. Ссылочным соединением служил ципрофлоксацин (Каталог фирмы Мерк, II-ое издание, N 2315).
1. Антибактериальная активность
Минимальную ингибирующую концентрацию (МИК) испытуемых соединений определяют по методике с разбавлением агара, приведенной в стандартном методе Японского общества химиотерапии (Chemotherapy (Токио), 29, I, 76 (1981)). Применялись следующие штаммы:
Staphylococcus aureus (S. aureus)
Enterococcus faecalis (E. faecalis)
Escherichia coli (E. coli)
Klebsiella pheumoniae (K. pneumoniae)
Serratia marcescens (S. marcescens)
Enterobacfer cloacae (E. cloacae)
Acinetobacter calcoaceticus (A. calcoaceticus)
Полученные результаты приведены в таблицах 4-A и 4-B.
Соединения настоящего изобретения проявляют прекрасную антибактериальную активность по отношению к выделенным клинически штаммам, и полученные результаты превосходит результаты для ссылочного соединения (ципрофлоксацина). В частности, отмечается заметная разница в активности относительно грамм-положительных бактерий.
2. Анализ хромосомной аберрации
Анализ хромосомной аберрации проводят на клеточной линии легких китайского хомячка (CHL). В качестве положительного контроля используют 2-(2-фурил)-3-(5-нитро-2-фурил)акриламид. Клетки, обработанные испытуемыми соединениями, культивируют 6 ч при 37oC в увлажненном воздухе с 5% CO2. После обработки в течение 6 ч клетки промывают. К промытым клеткам добавляют свежую среду и культивирование продолжают еще 18 ч. Для сбора метафазных клеток за 2 ч до фиксации хромосомного препарата к культуре добавляют кольцемид. Частота хромосомной аберрации на CHL клетках, обработанных 100 мкг/мл испытуемых соединений, приведена в таблице 5-A.
Полученные для всех соединений настоящего изобретения данные во всех случаях характеризуются (-).
3. Анализ микронуклеуса
В данном анализе используют BDFI мышей в возрасте 9 недель. Испытуемые соединения инъектируют внутрибрюшинно в дозе 250 мг/кг. Спустя 24 ч мышей умерщвляют и собирают костный мозг бедренной кости. Мазки костного мозга фиксируют на слайдах метанолом и по обычной методике окрашивают Гиемза. Для каждого зверька под микроскопом подсчитывают число микроядерных полихроматичных эритроцитов (МНПХЭ) на 1000 полихроматичных эритроцитов (ПХЭ). В качестве положительного контроля применяют циклофосфамид. Вероятность возникновения МНПХЭ (% числа МНПХЭ на 1000 ПХЭ) показана в таблице 5-B.
Соединения настоящего изобретения не создают статистического значимого повышения вероятности возникновения МНПХЭ в сравнении с контролем (солевой раствор).
4. Фототоксичность
Самцам морских свинок линии Хартли вводят внутривенно испытуемые соединения в дозе 10 мг/кг и сразу же облучают со спины УФ-излучением в течение 90 минут. Через 24 ч после УФ облучения исследует эритемы на спинках. Число морских свинок с эритемами указано в таблице 5-C.
Ни одно соединение настоящего изобретения не отличается фототоксичностью.
5. Судороги
1) Внутрибрюшинное (в.б.) введение
Закрепленным самцам мышей ICR в возрасте пяти недель перорально вводят фенбуфен в дозе 100 мг/кг. Тридцатью минутами позже мышам инъектируют внутрибрюшинно испытуемые соединения в дозе 100 мг/кг. После этого регистрируют наступление судорог. Число мышей с судорогами указано в таблице 5-C.
Ни одно соединение настоящего изобретения не вызывает каких-либо судорог.
2) Интрацеребральновентрикуляторное (и.ц.в.) введение
Самцов крыс линии Вистар весом 180-220 г анестезируют пентобарбиталом натрия (45 мг/кг, в.б.) и фиксируют в стереотаксичном аппарате. Для интрацеребральновентрикулярной инъекции каждой крысе имплантируют направляющую канюлю из нержавеющей стали диаметром 0,6 мм, канюля расположена на 1,5 мм выше левого бокового желудочка головного мозга (A: 6,2, R:1, H: +1) по атласу De Groot (1959). Направляющую канюлю закрепляют на черепе зубным цементом и замыкают тонким зондом из нержавеющей стали диаметром 0,3 мм. Для предотвращения инфекции внутримышечно инъектируют 10000 единиц калийпенициллина G. Крыс оставляют на несколько дней в покое, чтобы оправиться от хирургической операции.
Для определения судорог 30 минут спустя после внутрибрюшинной инъекции 50 мг/кг фенбуфена, вводят 20 мкг испытуемого соединения. Соединение вводят через канюлю из нержавеющей стали диаметром 0,3 мм с полиэтиленовым катетером, который на 1,5 мм длиннее направляющей канюли, с тем, чтобы катетер попадал в правый желудочек головного мозга (H: +1). В каждом исследовании испытаниям подвергают трех крыс, и отсутствие или появление признаков судорог наблюдают по меньшей мере четыре часа. Положение интрасеребровентрикулярной канюли подтверждают инъектированием 10 мкл 1% голубого Эванса с последующим рассечением головного мозга каждой участвующей в опытах крысы.
Число подверженных судорогам крыс указано в таблице 5-C.
Ни одно соединение настоящего изобретение не вызывает каких-либо судорог.
(Ссылки)
De Groot, J (1959). Передний мозг крыс с стереотоксичных координатах. Ver. Kon. Ned. Acad. Wet., Natuurkunde 52, 1-40.
6. Тканевое распределение
В экспериментах используются крысы фермы Спраг-Доули в возрасте семи недель. Зафиксированным на ночь крысам вводят перорально испытуемые соединения в дозе 5 мг/кг. С интервалом 0,083, 0,25, 0,5, 1, 2, 4, 6, 8, 12 и 24 ч после введения крыс анестезируют эфиром и из брюшной аорты отбирают образцы крови. Из образцов крови обычным путем получают образцы плазмы. После сбора крови удаляют легкие и почки и гомогенируют с 4 мл и 7 мл соответственно 1 М HCl-цитратного буфера (pH 4).
Концентрацию испытуемых соединений в биологических жидкостях (плазма и каждая анализируемая ткань) определяют методом ЖХВД. К 0,5 мл плазмы или 0,5 г каждого тканевого гомогената добавляют соляную кислоту и эфир. Смесь встряхивают и центрифугируют. После удаления органической фазы к водной фазе добавляют водн. раствор NaOH, фосфатный буфер (pH 7) и хлороформ. Смесь встряхивают и центрифугируют. Органическую фазу затем концентрируют. Остаток растворяют и подвергают ЖХВД. Концентрации испытуемых соединений в каждой биологической жидкости при Tмакс (время максимальной концентрации в плазме) приведены в таблице 6.
(Условия ЖХВД)
Колонка - TSK-гель ODS 80TM
Подвижная фаза - 0,03 М фосфатный буфер-CH3CN (3:1, pH 2,5)
Скорость потока - 1,2 мл/мин
Инъектируемый объем - 100 мкл
Детектирование - УФ при 308 нм
Концентрация соединения настоящего изобретения в легких и почках (подлежащие лечению органы) была соответственно в 16 и 7,4 раза выше концентрации ципрофлоксацина. Кроме того, отношения концентраций (ткань/плазма) для соединения настоящего изобретения также были выше соответственно в 8,6 и 4,2 раза отношений для ципрофлоксацина. Полученные данные указывают на то, что соединения настоящего изобретения отличаются хорошим тканевым распределением.
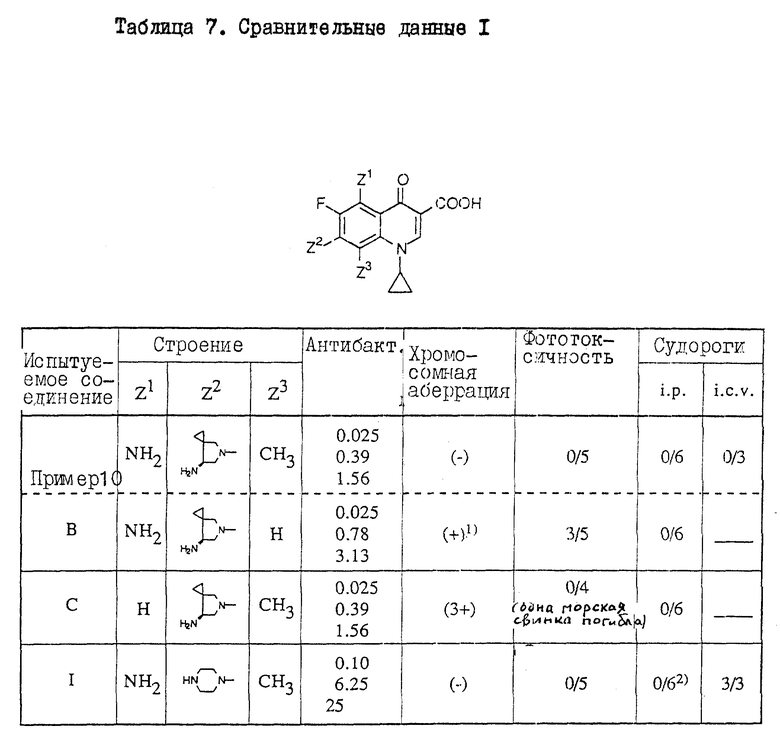
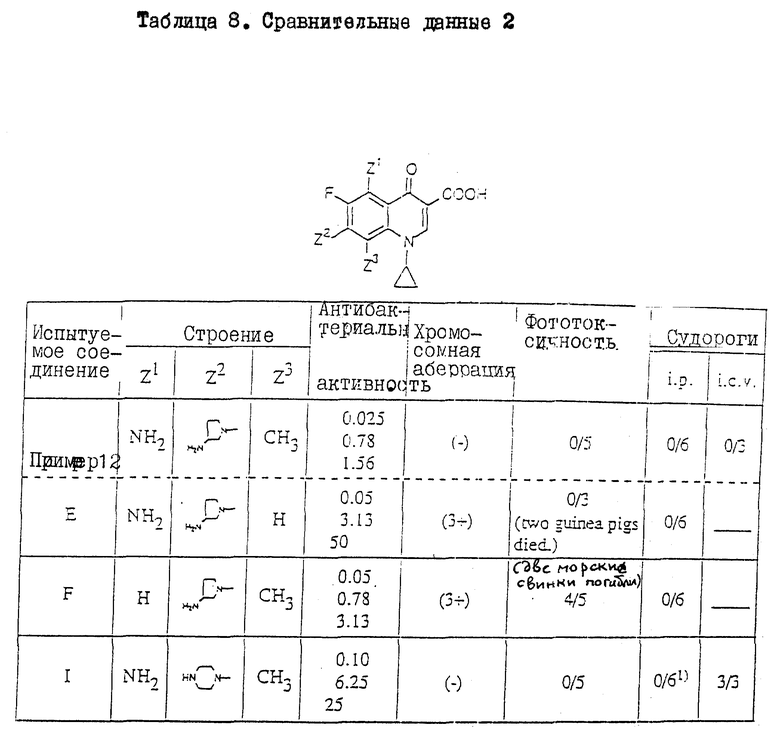
Прекрасное действие соединений настоящего изобретения сравнивалось с действием аналогичных соединений, имеющих только один отличающийся заместитель из 5-, 7- и 8-заместителей в соединениях настоящего изобретения. Результаты сравнения приведены в таблицах 7 и 8 (см. в конце описания).
Все данные по антибактериальной активности, хромосомной аберрационной активности, фототоксичности и индуцирование судорог получены теми же методами анализа, что и приведенные выше. Из этих результатов данные по антибактериальной активности, приведенные в таблицах 7 и 8, представлены величинам МИК (минимальный ингибирующей концентрации) относительно выделенных клинически штаммов (HPC527, HPC308 и HPC292) S. aureus, являющейся характерной грам-положительной бактерией.
Из таблицы 7 следует:
Соединение настоящего изобретения (пример 10) проявляет более высокую антибактериальную активность, чем аналогичное ссылочное соединение B (содержит только один отличающийся заместитель в 8-положении, чем и отличается от соединения настоящего изобретения). У соединений настоящего изобретения отсутствует также токсичность (хромосомная аберрация и фототоксичность), наблюдаемая у соединения B.
Соединение настоящего изобретения (пример 10) проявляет прекрасную антибактериальную активность, столь же высокую, что и у аналогичного ссылочного соединения C (имеет только один отличающийся заместитель в 5-положении, чем и отличается от соединения настоящего изобретения), и кроме того не показывает токсичности (хромосомная аберрация), наблюдаемой у ссылочного соединения C. Хотя ссылочное соединение C, как и соединение настоящего изобретения, не проявляет фототоксичности, введение ссылочного соединения С вызвало смерть одной из пяти морских свинок. Полученные результаты показывают, что ссылочное соединение C более токсично, чем соединение настоящего изобретения.
Соединение настоящего изобретения (пример 10) проявляет гораздо более высокую антибактериальную активность, чем аналогичное ссылочное соединение 1 (имеет только один отличающийся заместитель в 7-положении, чем и отличается от соединения настоящего изобретения), и кроме того не показывает токсичности (судороги), наблюдаемой у ссылочного соединения 1.
Уже состоялась презентация на ICAAC (31-ая межнаучная конференция по антибактериальным средствам и химиотерапии. Чикаго, Иллинойс, реферат N 1507 (1991)), касающаяся высокой хромосомной аберрационной активности ссылочного соединения C.
Таблица 8 иллюстрирует следующее.
Соединение настоящего изобретения (пример 12) проявляет гораздо более высокую антибактериальную активность, чем ссылочное соединение E (имеет только один отличающийся заместитель в 8-положении, чем и отличается от соединения настоящего изобретения), и кроме того не показывает токсичности (хромосомная аберрация), наблюдаемой у ссылочного соединения E. Хотя ссылочное соединение E, как и соединение настоящего изобретения, не проявляет фототоксичности, введение ссылочного соединения E привело к гибели двух из пяти морских свинок. Полученные результаты показывают, что ссылочное соединение E гораздо более токсично, чем соединение настоящего изобретения.
Соединение настоящего изобретения (пример 12) проявляет более высокую антибактериальную активность, чем ссылочное соединение F (имеет только один отличающийся заместитель в 5-положении, чем и состоит его отличие от соединения настоящего изобретения), и кроме того не показывает токсичности (хромосомная аберрация и фототоксичность), наблюдаемой у ссылочного соединения F.
Соединение настоящего изобретения (пример 12) проявляет гораздо более высокую антибактериальную активность, чем аналогичное ссылочное соединение 1 (имеет только один отличающийся заместитель в 7-положении, чем и отличается от соединения настоящего изобретения), и кроме того не показывает токсичности (судороги), наблюдаемой у ссылочного соединения 1.
Токсичность (например: хромосомная аберрация, фототоксичность и судороги), характерная для некоторых антибактериальных средств с хинолиновым ядром, создает серьезные затруднения клиническому применению таких средств. Соединения настоящего изобретения решают эту проблему, вследствие чего обладают большим потенциалом в качестве следующего поколения антибактериальных средств.
Антибактериальная активность: МИК (мкг/мл) испытуемого соединения по отношению к 3 штаммам S. aureus: HPC527, HPC308 и HPC292 (верхние величины: HPC527, средние величины: HPC308, нижние величины: HPC292).
Хромосомная аберрация: частота хромосомной аберрации на CHL клетках, обработанных 100 мкг/мл испытуемого соединения. (-: < 10%, +: 10-20%, 2+: 20-50%, 3+: > 50%).
Фототоксичность: Морские свинки, 10 мг/кг, в.б.
Судороги: мыши, 100 мг/кг в.б. и крысы, 20 мкг, и.ц.в.
1) Все участвующие в экспериментах мыши имели симптомы седативного эффекта, считающегося предшественником судорог. Антибактериальная активность: МИК (мкг/мл) испытуемого соединения по отношению к 3 штаммам S. aureus: HPC527, HPC308 и HPC292 (верхние величины: HPC527, средние величины HPC308, нижние величины: HPC292).
Хромосомная аберрация: частота хромосомной аберрации на CHL клетках, обработанных 100 мкг/мл испытуемого соединения. (-: < 10%, +: 10-20%, 2+: 20-50%, 3+: > 50%).
Фототоксичность: морские свинки, 10 мг/кг, в.в.
Судороги: мыши. 100 мг/кг, в.б. и крысы, 20 мкг, и.ц.в.
Примеры
Настоящее изобретение далее иллюстрируется следующими примерами. Примеры приведены лишь с целью иллюстрации, и их не следует рассматривать, как исчерпывающие.
Пример 1
2,4,5-Трифтор-3-метил-6-нитробензойная кислота
К перемешиваемой смеси 370 мл конц. серной кислоты и 61,2 мл 70%-ной азотной кислоты добавляют при перемешивании порциями при 55-70oC 36,6 гр 2,4,5-трифтор-3-метибензойной кислоты. После выдерживания 2 часа при комнатной температуре реакционную смесь вливают в лед и экстрагируют изопропиловым эфиром. Объединенные экстракты промывают рассолом, сушат, выпаривают и получают в виде желтых кристаллов 30,6 г целевого соединения.
ЯМР-спектр δ (CD3OC) ч/млн.: 2,29 (3H, т., J = 2 Гц).
Пример 2
Диэтиловый эфир (2,4,5-трифтор-3-метил-6-нитробензоил)малоновой кислоты
Суспензию 27 г 2,4,5-трифтор-3-метил-6-нитробензойной кислоты, 19,5 мл оксалилхлорида и нескольких капель N,N-диметилформамида в 270 мл хлористого метилена перемешивают 2 часа при комнатной температуре. Испарением реакционной смеси получают 2,4,5-трифтор-3-метил-6-нитробензоилхлорид. Отдельно к суспензии 3,08 г магния и нескольких капель четыреххлористого углерода в 6,4 мл абсолютного этанола при 50oC по каплям прибавляют раствор 19,2 мл диэтилмалоната в 12 мл абсолютного этанола и затем перемешивают 1,5 часа при той же температуре. Реакционную смесь испаряют, растворяют в толуоле и вновь испаряют. К раствору полученного остатка в 30 мл толуола при охлаждении льдом прибавляют по каплям раствор 2,4,5-трифтор-3-метил-6-нитробензоилхлорида, полученного ранее, в 30 мл толуола. После выдерживания 2 часа при комнатной температуре к реакционной смеси добавляют 100 мл 5%-ной серной кислоты и полученный раствор экстрагируют диэтиловым эфиром. Объединенные органические экстракты промывают рассолом, сушат и после испарения получают в виде коричневого масла 4, 7,3 целевого соединения.
ЯМР-спектр δ (CDCl3) ч/млн: 1,1 (3H, т, J = 7,5 Гц), 1,38 (3H, т., J = 7,5 Гц), 2,33 (3H, т, J = 2 Гц), 3,36, 14,18 (в целом 1H, каждый с), 4,07 (2H, к, J = 7,5 Гц), 4,38 (2H, к, J = 7,5 Гц).
Пример 3
Этиловый эфир (2,4,5-трифтор-3-метил-6-нитробензол)уксусной кислоты.
Суспензию 45,3 г диэтилового эфира (2,4,5-трифтор-3-метил-6-нитробензоил)малоновой кислоты и 30 мг п-толуолсульфокислоты в 120 мл воды кипятят 50 минут. После охлаждения реакционную смесь экстрагируют диэтиловым эфиром. Объединенные органические экстракты сушат после промывания рассолом и испарением получают в виде коричневого масла 34,2 г целевого соединения.
ЯМР-спектр δ/ (CDCl3) ч/млн: 1,26, 1,34 (всего 3H, каждый т, J = 7 Гц), 2,33, 2,35 (всего 3H, каждый т, J = 2,5 Гц), 3,91, 5,48, 12,34 (всего 2H, каждый с), 4,2, 4,28 (всего 2H, каждый к, J = 7 Гц).
Пример 4
Этиловый эфир 3-циклопропиламино-2-/2,4,5-трифтор-3-метил-6-нитробензоил)акриловой кислоты
Смесь 31,9 г этилового эфира (2,4,5-трифтор-3-метил-6-нитробензоил)уксусной кислоты, 26,2 мл этилорфторомата и 23,8 мл уксусного ангидрида кипятят 1 час. Испарением реакционно смеси получают 46,2 г этилового эфира 3-этокси-2-(2,4,5-трифтор-3-метил-6-нитробензоил)акриловой кислоты в виде коричневого масла. К раствору 45,4 полученного соединения в 328 мл этанола при охлаждении льдом и перемешивании по каплям прибавляют 9,6 мл циклопропиламина. После выдерживания 30 минут при комнатной температуре реакционную смесь испаряют и очисткой остатка колоночной хроматографией (силикагель, н-гексан-хлористый метилен (1: 1) получают в виде желтых кристаллов 28,8 г целевого соединения. Перекристаллизацией из изопропилового эфира получают желтые иглы с т.пл. 115-115,5oC.
Анализ для C16H15F3N2O5:
Вычислено %: C 51,62, H 4,06, N 7,52
Найдено %: C 51,57, H 3,92m, N 7,53
Пример 5
Этиловый эфир 1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-5-нитро-4-оксохинолин- 3-карбоновой кислоты
К раствору 27,1 г этилового эфира 3-циклопропиламино-2-(2,4,5-трифтор-3-метил-6-нитробензоил)акриловой кислоты в 270 мл 1,4-диоксана порциями прибавляют 3,2 г гидрида натрия (60%-ная дисперсия в минеральном масле) и перемешивают один час при комнатной температуре. К реакционной смеси добавляют 300 мл воды, выпавшие в осадок кристаллы отделяют фильтрованием и получают в виде бесцветных кристаллов 19,5 г целевого соединения, перекристаллизацией которого из N,N-диметилформамида получают бесцветные иглы с т.пл. 260-263oC.
Анализ для C16H14F2N2O5:
Вычислено %: C 54,55, H 4,01, N 7,95.
Найдено %: C 54,51, H 4,00, N 7,90
Пример 6
Этиловый эфир 5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин- 3-карбоновой кислоты
Суспензию 18,5 г этилового эфира 1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-5-нитро-4-оксохинолин- 3-карбоновой кислоты, 10 мл никеля Ренея в 300 мл уксусной кислоты гидрируют 1,5 часа при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают и полученный фильтрат испаряют. К остатку добавляют 150 мл 10%-го водного раствора карбоната калия и смесь экстрагируют хлористым метиленом. Объединенные органические экстракты сушат и испарением получают 14,8 г целевого соединения в виде слегка желтых кристаллов, перекристаллизацией которых из ацетонитрила получают слегка желтые иглы с т.пл. 182,5-185,5oC.
Анализ для C16H16F2N2O3:
Вычислено %: C 59,62, H 5,00, N 8,69.
Найдено %: C 59,74, H 5,08, N 8,60
Пример 7
5-Амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин- 3-карбоновая кислота
Смесь 14,8 г этилового эфира 5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты, 37,2 мл соляной кислоты и 150 мл 90%-ной уксусной кислоты кипятят 2 часа. После охлаждения осадившиеся кристаллы отфильтровывают и промывают водой с получением 11,8 г целевого соединения в виде желтых кристаллов. Последующей перекристаллизацией из N, N-диметилформамида получают желтые кристаллы с т.пл. 290,5oC (разл.).
Анализ для C14H12F2N2O3:
Вычислено %: C 57,15, H 4,11, N 9,52.
Найдено %: C 57,10, H 4,03, N 9,53
Пример 8
/5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоксилато-O3, O4/дифторборон (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоновой кислоты BF2 хелат).
Смесь 5 г 5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин- 3-карбоновой кислоты, 3,13 мл эфирата трехфтористого бора и 75 мл метилизобутилкетона кипятят 1 час. После охлаждения осадившиеся кристаллы отфильтровывают и промыванием диэтиловым эфиром получают 5,38 г целевого соединения в виде желтых кристаллов.
ЯМР-спектр δ (ДМСО-d6) ч/млн: 1,08-1,15 (2H, м), 1,21-1,3 (2H, м), 2,67 (3H, д, J= 2,5 Гц), 4,52-4,59 (1H, м), 7,28 (2H, ш.с), 9,1 (1H, с).
Пример 9
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-((S)-7- трифторацетиламино-5-азаспиро/2,4/гепт-5-ил)хинолин-3-карбоновая кислота
Смесь 2,13 г /5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоксилато-O3,O4/дифторборона, 2,28 г гидрохлорида (S)-7-трифторацетиламино-6-азаспиро/2,4/-гептана ([α]
Анализ для C22H22F4N4O4:
Вычислено: C 54,77, H 4,6, N 11,61%
Найдено: C 54,57, H 4,7, N 11,56%
Удельное вращение /α/
Пример 10
5-Амино-7-((S)-7-амино-5-азаспиро/2,4, гепт-5-ил)-1-циклопропил- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
Смесь 0,26 г 5-амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-((S)-7- трифторацетиламино-5-азаспиро/2,4/-гепт-5-ил)хинолин-3-карбоновой кислоты, 0,18 г гидроксида калия и 1,8 мл воды перемешивают 0,5 часа при комнатной температуре. Реакционную смесь нейтрализуют 10%-ной соляной кислотой до pH 8, выпавшие в осадок кристаллы отфильтровывают и после промывания водой получают 0,21 г целевого соединения, перекристаллизацией которого из ацетонитрила получают 0,16 г желтых призм с т.пл. 216,5-218oC
Анализ для C20H22FN4O3:
Вычислено %: C 62,16, H 6, N 14,50
Найдено %: C 62,13, H 6, N 14,64
Удельное вращение /α/
Пример 11
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-((S)- 3-трифтороацетиламино-1-пирролидинил)хинолин-3-карбоновая кислота
Смесь 2,5 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4- оксохинолин-3-карбоксилато-O3, O4)дифторборона, 3,2 г гидрохлорида (S)-3-трифторацетиламинопирролидина /α/
Анализ C20H20F4N4O4:
Вычислено %: C 52,63, H 4,42, N 12,28
Найдено %: C 52,64, H 4,37, N 12,35.
Удельное вращение /α/
Пример 12
5-Амино-7-((S)-3-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро- 4-оксохинолин-3-карбоновой кислоты гидрохлорид
Смесь 0,62 г 5-амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-((S)- трифторацетиламино-1-пирролидинил)хинолин-3-карбоновой кислоты, 0,57 г гидроксида калия и 10 мл воды перемешивают 1 час при комнатной температуре. Реакционную смесь нейтрализуют 10%-ной соляной кислотой и испаряют. Остаток разбавляют этанолом, нерастворимые вещества отфильтровывают и полученный фильтрат испаряют. К раствору остатка в ацетоне добавляют этанольный раствор хлористого водорода и фильтрованием образовавшихся кристаллов получают 0,53 г желтого кристаллического продукта. Последующей перекристаллизацией продукта из метанола получают 40 мг целевого соединения в виде желтых кристаллов с т.пл. 263,5oC (разл.).
Анализ для C18H21FN4O3 • HCl:
Вычислено %: C 54,48, H 5,59, N 14,12
Найдено %: C 54,22, H 5,61, N 13,88
Удельное вращение /α/
Пример 13
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-7-(цис-4-метил-3- трифторацетиламино-1-пиролидинил)-4-оксохинолин-3-карбоновая кислота
Смесь 4 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолни-3- карбоксилато-O3, O4)дифторборона, 4,08 г гидрохлорида цис-4-метил-3-трифторацетиламинопироолидина, 5,09 мл N,N-диизопропилэтиламина и 16 мл диметилсульфоксида перемешивают 3 дня при 30oC. К реакционной смеси при охлаждении льдом добавляют воду и хлористый метилен и полученную смесь перемешивают при комнатной температуре. Выпавшие в осадок кристаллы отфильтровывают и промыванием хлористым метиленом получают 0,99 г желтовато-коричневых кристаллов (A). Слой хлористого метилена в фильтрате промывают водой и рассолом, сушат над сульфатом натрия и испаряют. Обработкой остатка хлористым метиленом получают 1,15 г желтовато-коричневых кристаллов (B). Фильтрат испаряют и остаток очищают колоночной хроматографией (силикагель, хлористый метилен-метанол (100:1) с получением 0,25 г желтовато-коричневых кристаллов (C). Смесь 2,39 г полученных кристаллов (A, B и C), 2,42 мл триэтиламина, 48 мл метанола и 24 мл 1,2-дихлорэтана кипятят 9 часов и затем испаряют. К остатку добавляют воду и смесь подкисляют 10%-ной соляной кислотой до pH 4. Выпавшие в осадок кристаллы отфильтровывают и после промывания водой, изопропанолом и диэтиловым эфиром получают 2,24 г целевого соединения в виде желтых кристаллов. Последующей перекристаллизацией кристаллов из смеси N,N-диметилформамида с этанолом получают желтые иглы с т.пл. 253-254,5oC.
Анализ для C21H22F4N4O4:
Вычислено: C 53,62, H 4,71, N 11,91%
Найдено: C 53,41, H 4,92, N 11,70%
Пример 14
5-Амино-7-(цис-3-амино-4-метил-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксоинолин-3-карбоновая кислота.
Смесь 2 г 5-амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-7-(цис-4-метил-3- трифторацетиламино-1-пирролидинил)-4-оксохинолин-3-карбоновой кислоты, 1,4 г гидроксида калия и 14 мл воды перемешивают 1 час при комнатной температуре и затем нейтрализуют 10%-ной водной соляной кислотой до pH 8. Выпавшие в осадок кристаллы отфильтровывают и после промывания водой, изопропанолом и диэтиловым эфиром получают 1,65 г целевого соединения в виде желтых кристаллов. Последующей перекристаллизацией кристаллов из смеси хлористого метилена с метанолом получают 1,32 г желтых призм с т.пл. 213,5-215oC.
Анализ для C19H23FN4O3:
Вычислено %: C 60,95, H 6,19, N 14,96
Найдено %: C 60,83, H 6,35, N 14,83.
Пример 15
5-Амино-1-циклопропил-7-((S)-4,4-диметил-3-трифторацетил-амино-1-пирролидинил)- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота.
Смесь 4 г (5-амино-1-цикопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоксилато-O3,O4)дифторборона, 4,32 г гидрохлорида (S)-4,4-диметил-3-трифторацетиламинопирролидина (/α/
Анализ для C22H24F4N4O4:
Вычислено %: C 54,54, H 4,99, N 11,57
Найдено %: C 54,33, H 4,88, N 11,63.
Удельное вращение /α/
Пример 16
5-Амино-7-((S)-3-амино-4,4-диметил-1-пирролидинил)-1-циклопропил-6-фтор- 1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
Смесь 0,47 г 5-амино-1-циклопропил-7-((S)-4,4-диметил-3-трифторацетиламино-1-пирролидинил)- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты, 0,32 г гидроксида калия и 3,2 мл воды перемешивают 1 час при комнатной температуре. Реакционную смесь нейтрализуют 10%-ной соляной кислотой до pH 8 и экстрагируют хлористым метиленом. Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия и испаряют. Обработкой остатка смесью ацетона с диэтиловым эфиром получают 0,3 г целевого соединения в виде желтых кристаллов. Последующей перекристаллизацией кристаллов из ацетонитрила получают 0,18 г желтых игл с т.пл. 191,5-193oC
Анализ для C20H25FN4O3:
Вычислено: C 61,84, H 6,49, N 14,42%
Найдено: C 61,70, H 6,51, N 14,32%
Удельное вращение /α/
Пример 17
5-Амино-7-(3-трет-бутоксикарбониламино-3-метил-1-пироолидинил)-1-циклопропил- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота.
Смесь 3 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин- 3-карбоксилато-O3, O4)-дифторборона, 2,11 г 3-трет-бутоксикарбониламино-3-метилпирролидина, 1,53 мл N,N-диизопропилэтиламина и 12 мл диметилсульфоксида перемешивают 2,5 дня при 30oC. Реакционную смесь разбавляют водой и экстрагируют хлористым метиленом. Объединенные органические экстракты промывают рассолом, сушат над сульфатом натрия и испаряют. Остаток очищают колоночной хроматографией (силикагель, хлористый метилен-метанол (99:1)) и получают 0,97 г желтых кристаллов. Смесь 0,97 полученных кристаллов, 1 мл триэтиламина и 40 мл метанола кипятят 2,5 часа и затем испаряют. К остатку добавляют воду, выпавшие в осадок кристаллы отфильтровывают и после промывания водой получают 0,84 г целевого соединения в виде желтых кристаллов, перекристаллизацией которых из ацетонитрила получают 0,76 г желтых игл с т. пл. 198-201oC.
Анализ для C24H21FN4O5:
Вычислено %: C 60,75, H 6,58, N 11,81
Найдено %: C 60,43, H 6,66, N 11,56.
Пример 18
5-Амино-7-(3-амино-3-метил-1-пирролидинил)-1-циклопропил-6-фтор- 1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
К 0,76 г 5-амино-7-(3-трет-бутоксикарбониламино-3-метил-1-пирролидинил)-1-циклопропил- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты добавляют 1,1 мл соляной кислоты и затем перемешивают 2 часа при комнатной температуре. К реакционной смеси при охлаждении льдом добавляют 0,89 г гидроксида калия в 1,8 мл воды и образовавшуюся смесь нейтрализуют 10%-ной соляной кислотой до pH 8. Выпавшие в осадок кристаллы отфильтровывают и промыванием водой получают 0,33 г целевого соединения в виде желтых кристаллов. Последующей перекристаллизацией продукта из смеси хлористого метилена с метанолом получают 0,3 г желтых кристаллов с т.пл. 217-221oC.
Анализ для C19H23FN4O3 • 1/4 H2O:
Вычислено %: C 60,23, H 5,26, N 14,79.
Найдено %: C 59,98, H 6,25, N 14,53.
Пример 19
5-Амино-7-((S)-7-трет-бутоксикарбониламино-5-азаспиро/2,4/- гепт-5-ил)-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоновая кислота
Смесь 6 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил- 4-оксохинолин-3-карбоксилато-O3, O4)дифторборона, 5,59 г (S)-7-трет-бутоксикарбониламино-5-азаспиро/2,4/гептана (/α/
Анализ для C25H31FN4O5:
Вычислено %: C 61,72, H 6,42, N 11,52
Найдено %: C 61,71, H 6,48, N 11,39
Удельное вращение /α/
Пример 20
5-Амино-7-((S)-7-амино-5-азаспиро/2,4/гепт-5-ил)-1-циклопропил-6- фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
К 8,8 г 5-амино-7((S)-7-трет-бутоксикарбониламино-5-азаспиро/2,4/гепт-5-ил)- 1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты добавляют 11 мл соляной кислоты и затем перемешивают 1,5 часа при комнатной температуре. К реакционной смеси при охлаждении льдом добавляют раствор 10,5 г гидроксида калия в 32 мл воды и полученную смесь нейтрализуют 10%-ной соляной кислотой до pH 8. Образовавшиеся кристаллы отфильтровывают и промывают водой. Кристаллы разбавляют хлористым метиленом, неорганические вещества отфильтровывают и полученный фильтрат испаряют. Ополаскиванием диэтиловым эфиром получают 5,53 г целевого соединения в виде желтых кристаллов. Перекристаллизацией из ацетонитрила получают желтые призмы, идентифицированные, как соединение примера 10.
По обычной методике получены следующие соли.
Мутансульфонат
Внешний вид: желтые или (EtOH-H2O)
т.пл. 263-264oC (разл.)
Анализ для C20H23FN4O3 • CH4O3S:
Вычислено: C 52,27, H 5,64, N 11,61%
Найдено: C 52,02, H 5,54, N 11,53%
Удельное вращение /α/
п-Толуолсульфонат
Внешний вид: желтые кристаллы (EtOH) т.пл. 188-189,5oC.
Анализ для C20H23FN4O3 • C7H8O3S • 1/2 H2O:
Вычислено %: C 57,13, H 5,68, N 9,87
Найдено %: C 56,95, H 5,85, N 9,77
Удельное вращение /α/
Гидрохлорид
Внешний вид: желтые кристаллы (EtOH-H2O)
т.пл. 276-280oC (разл.)
Анализ для C20H23FN4O3 • HCl:
Вычислено %: C 56,80, H 5,72, N 13,25
Найдено %: C 56,72, H 5,79, N 13,04.
Пример 21
5-амино-7-(3-трет-бутоксикарбониламино-3-метил-1-пирролидинил)-1- циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота (изомер A).
Смесь 3 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин- 3-карбоксилато-O3, O4)дифторборона, 2,11 г 3-трет-бутоксикарбониламино-3-метилпирролидина (изомер A, /α/
Анализ для C24H31FN4O5:
Вычислено %: C 60,75, H 6,58, N 11,81
Найдено %: C 60,63, H 6,55, N 11,80
Удельное вращение /α/
Пример 22
5-Амино-7-(3-амино-3-метил-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты (изомер A) метансульфонат
К 0,8 г 5-амино-7-(3-трет-бутоксикарбониламино-3-метил-1-пирролидинил)-1- циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты (изомер A, получение см. пример 21) при охлаждении льдом добавляют 0,98 мл соляной кислоты, после чего перемешивают 2 часа при комнатной температуре. К реакционной смеси добавляют 0,93 г гидроксида калия в 3,1 мл воды и перемешивают 1 час при комнатной температуре. Образовавшуюся смесь нейтрализуют 10%-ной соляной кислотой до pH 8. Выпавшие в осадок кристаллы отфильтровывают и после промывания водой получают 0,55 г желтых кристаллов, которые по обычной методике превращают в метансульфонат. Перекристаллизацией из смеси этанола с водой (9:1) получают 0,43 г целевого соединения в виде желтых игл с т.пл. 261-262,5oC.
Анализ для C19H23FN4O3 • CH4O3S:
Вычислено %: C 51,05, H 5,78, N 11,91
Найдено %: C 50,89, H 5,93, N 11,78.
Удельное вращение /α/
Пример 23
5-Амино-7-(3-трет-бутоксикарбониламино-3-метил-1-пирролидинил)-1- циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота (изомер B)
Смесь 3 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоксилато-O3,O4)дифторборона, 3-трет-бутоксикарбониламино-3-метилпирролидина (2,11 г, изомер B, [α]
Анализ для С24H31FN4O5:
Вычислено %: C 60,75, H 6,58, N 11,81
Найдено %: C 60,85, H 6,57, N 11,76.
Удельное вращение /α/
Пример 24
5-Амино-7-(3-амино-3-метил-1-пирролидинил)-1-циклопропил-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты (изомер B) метансульфонат
К 0,7 г 5-амино-7-(3-трет-бутоксикарбониламино-3-метил-1-пирролидинил)-1-циклопропил- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты (изомер B, получение см. пример 23) при охлаждении льдом добавляют 0,86 мл соляной кислоты, после чего перемешивают 2 часа при комнатной температуре. К реакционной смеси добавляют раствор 0,82 г гидроксида калия в 2,7 мл воды и затем перемешивают 1 час при комнатной температуре. Образовавшуюся смесь нейтрализуют 10%-ной соляной кислотой до pH 8. Выпавшие в осадок кристаллы отфильтровывают и после промывания водой получают 0,48 г желтых кристаллов, которые по обычной методике превращают в метансульфонат. Перекристаллизацией из смеси этанола с водой (9:1) получают 0,31 г целевого соединения в виде желтых игл с т.пл. 260,5-262oC.
Анализ для C19H23FN4O3 • CH4O3S:
Вычислено %: C 51,05, H 5,78, N 11,91
Найдено %: C 50,75, H 5,88, N 11,69
Удельное вращение /α/
Пример 25
5-Амино-7-((S)-3-трет-бутоксикарбониламино-1-пирролидинил)-1-циклопропил- 6-фтор-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
Смесь 2 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин- 3-карбоксилато-O3, O4)дифторборона, 1,63 г (S)-3-трет-бутоксикарбониламинопирролидина (/α/
Анализ для C23H29FN4O5:
Вычислено %: C 59,99, H 6,35, N 12,17.
Найдено %: C 59,98, H 6,45, N 11,99
Удельное вращение /α/
Пример 26
5-Амино-7-((S)-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-8- метил-4-оксохинолин-3-карбоновая кислота
К 4,07 г 5-амино-7-((S)-3-трет-бутоксикарбониламино-1-пирролидинил)-1-циклопропил- 6-фтор-1,4-дигидро-8-метил-4-оксо-хинолин-3-карбоновой кислоты при комнатной температуре добавляют 5,2 мл соляной кислоты, после чего перемешивают 30 минут при комнатной температуре. К реакционной смеси при охлаждении льдом добавляют раствор 4,9 г гидроксида натрия в 16 мл воды с установлением pH 11, после чего получают смесь нейтрализуют 10%-ной соляной кислотой до pH 8. Водный слой декантируют, а масло ополаскивают небольшим количеством метанола. Кристаллы отфильтровывают и после промывания изопропанолом получают 3,05 г желтых кристаллов. Кристаллы разбавляют этанолом, нерастворимые вещества отфильтровывают и полученный фильтрат испаряют. Остаток разбавляют смесью хлористого метилена с метанолом (19:1), нерастворимые вещества отфильтровывают и после испарения фильтрата получают 2,58 г целевого соединения в виде желтого кристаллического продукта, перекристаллизацией которого из смеси хлористого метилена с метанолом получают бледно-желтые кристаллы с т.пл. 202-204oC (разл.).
Анализ для C18H21FN4O3 • H2O:
Вычислено %: C 57,13, H 6,13, N 14,81
Найдено %: C 57,36, H 5,91, N 14,70
Удельное вращение /α/
По обычной методике получены следующие соли.
Метансульфонат
Внешний вид: желтые иглы (MeOH)
т.пл. 280-281,5oC (разл.)
Анализ для C18H21FN4O3 • CH4O3S • 1/4 H2O:
Вычислено %: C 49,5, H 5,58, N 12,15
Найдено %: C 49,5, H 5,58, N 12,03
Удельное вращение /α/
п-Толуолсульфонат
Внешний вид: желтые иглы (изо-PrOH-H2O)
т.пл. 238-241oC (разл.)
Анализ C18H21FN4O3 • C7H8O3S • 1/2H2O:
Вычислено :% C 55,44, H 5,58, N 10,34
Найдено :% C 55,47, H 5,56 N 10,22
Удельное вращение /α/
Пример 27
5-Амино-1-циклопропил-7-((S)-3-диметиламино-1-пирролидинил)-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
Смесь 3 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил- 4-оксохинолин-3-карбоксилато-O3, O4)дифторборона, 1,2 г ((S)-3-диметилламинопирролидина (/α/
Анализ для C20H25FN4O3:
Вычислено :% C 61,84, H 6,49, N 14,42
Найдено %: C 61,72, H 6,46, N 14,44.
Удельное вращение /α/
Пример 28
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-/3-(трифторацетил) (метил)амино-1-пирролидинил/хинолин-3-карбоновая кислота
Смесь 3 г (5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолином-3- карбоксилато-O3, O4/дифторборона, 2,44 г гидрохлорида 3-(трифторацетил)(метил)аминопирролидина, 3,36 мл N,N-диизопропилэтиламина и 12 мл диметилсульфоксида перемешивают 4 дня при 30oC. Осадок отфильтровывают, полученный фильтрат разбавляют водой и образовавшиеся кристаллы отфильтровывают. Промыванием кристаллов водой и этилацетатом получают желтовато-коричневый кристаллический продукт, очисткой которого колоночной хроматографией (силикагель, хлористый метилен и хлористый метилен-метанол (50:1)) получают 0,92 г желтовато-оранжевых кристаллов. Смесь 0,9 г полученных кристаллов, 0,91 мл триэтиламина, 18 мл метанола и 9 мл 1,2-дихлорэтана кипятят 5 часов и затем испаряют. К остатку добавляют воду и фильтрованием выпавших в осадок кристаллов получают 0,77 г желтого кристаллического продукта, перекристаллизацией которого из метанола получают желтые кристаллы с т.пл. 189-190oC.
Анализ для C21H22F4N4O4:
Вычислено %: C 53,62, H 4,71, N 11,91
Найдено %: C 53,50, H 4,42, N 11,84
Пример 29
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-7-(3-метил-амино-1- пирролидинил)-4-оксохинолин-3-карбоновая кислота
Смесь 0,6 г 5-амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-/3-(трифторацетил) метил)амино-1-пирролидинил/хинолин-3-карбоновой кислоты, 0,38 г гидроксида калия и 3,8 мл воды перемешивают 1 час при комнатной температуре. Реакционную смесь нейтрализуют 10%-ной соляной кислотой до pH 8-9, выпавшие в осадок кристаллы отфильтровывают и после промывания водой получают 0,47 г целевого соединения в виде желтых кристаллов, перекристаллизацией которых из метанола получают желтые столбики с т.пл. 200,5-202oC.
Анализ для C19H23FN4O3:
Вычислено %: C 60,95, H 6,19, N 14,96
Найдено %: C 60,78, H 6,17, N 15,01
Пример 30
5-Амино-7-((S)-7-трет-бутоксикарбониламино-7-азаспиро/2,4/-гепт-5-ил)- 1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-хинолин-3-карбоновая кислота
Смесь 20 г 5-амино-1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-4-оксохинолин-3- карбоновой кислоты, 28,9 г (S)-7-третбутоксикарбониламино-5-азаспиро/2,4/гептана (/α/
Пример 31
Этиловый эфир 1-циклопропил-6,7-дифтор-1,4-дигидро-8-метил-5-нитро-4-оксохинолин-3- карбоновой кислоты
К раствору 10 г этилового эфира 3-циклопропиламино-2-(2,4,5-трифтор-3-метил-6-нитробензоил)акриловой кислоты и 0,1 г 18-краун-6-эфира в 100 мл тетрагидрофурана добавляют 8,04 г карбоната калия и полученную смесь перемешивают 23 часа при комнатной температуре. Образовавшиеся кристаллы отфильтровывают и после промывания тетрагидрофураном, водой и ацетоном получают 8,56 г целевого соединения. Перекристаллизацией из N,N-диметилформамида получают бесцветные иглы, идентифицированные как соединения примера 5.
Пример 32
Фармацевтический препарат настоящего изобретения в виде таблеток получают по обычной методике использованием следующих компонентов:
Соединение примера 10 - 110 мг
Лактоза - сколько необходимо
Кукурузный крахмал - 34 мг
Стеарат магния - 2 мг
Гидроксипропилметилцеллюлоза - 8 мг
Полиэтиленгликоль 6000 - 0,5 мг
Оксид титана - 0,5 мг - 210 мг
Пример 33
Фармацевтический препарат настоящего изобретения получают обычным образом в виде капсул использованием следующих компонентов:
Соединение примера 10 - 110 мг
Лактоза - сколько необходимо
Карбоксиметилцеллюлоза - 15 мг
Гидроксипропилцеллюлоза - 2 мг
Стеарат магния - 2 мг - 160 мг
Пример 34
Фармацевтический препарат настоящего изобретения в виде порошка получают обычным способом использованием следующих компонентов:
Соединение примера 10 - 110 мг
Лактоза - сколько необходимо
D-Маннит - 500 мг
Гидроксипропилцеллюлоза - 5 мг
Тальк - 5 мг - 1000 мг
Пример 35
Фармацевтический препарат настоящего изобретения получают в виде инъекций обычным способом использованием следующих компонентов:
Соединение примера 10 - 50 мг
Глюкоза - 1000 мг
Соляная кислота - сколько необходимо
Дистиллированная вода для инъекций - сколько необходимо - 20 мл
Пример 36
Фармацевтический препарат настоящего изобретения получают обычным способом в виде супозитория использованием следующих компонентов:
Соединение примера 10 - 100 мг
Твердый жир - 1300 мг - 1400 мг
Пример 37
Фармацевтический препарат настоящего изобретения получают обычным способом в виде мази использованием следующих компонентов:
Соединение примера 10 - 5 мг
Белый петролатум - Сколько необходимо
Жидкий парафин - 70 мг - 1000 мг
Ссылочные соединения (A-1) синтезированы по методикам, приведенным в примерах 9 и 10.
Ссылочное соединение A
7-((S)-7-Амино-5-азаспиро/2,4/-гепт-5-ил)-1-циклопропил-6-фтор-1,4- дигидро-4-оксохинолин-3-карбоновой кислоты гидрохлорид
Внешний вид: бледно-желтые иглы
т.пл. 284-288oC (разл.)
Ссылочное соединение B
5-Амино-7-((S)-7-амино-5-азоспиро/2,4/-гепт-5-ил)-1-цикло-пропил-6-фтор- 1,4-дигидро-4-оксохинолин-3-карбоновой кислоты гидрохлорид
Внешний вид: бледно-желтые кристаллы
т.пл. 276-279oC (разл.)
Ссылочное соединение C
7-((S)-7-Амино-5-азаспиро/2,4/гепт-5-ил)-1-циклопропили-6-фтор-1,4- дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты
Внешний вид: бесцветные кристаллы
т.пл. 176,5-178oC.
Ссылочное соединение D
7-((S)-3-Амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-4- оксохинолин-3-карбоновая кислота
Внешний вид: бесцветные кристаллы
т.пл. 253-254oC (разл.)
Ссылочное соединение E
5-Амино-7-((S)-3-амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-4- оксохинолин-3-карбоновая кислота
Внешний вид: бледно-желтовато-коричневые кристаллы
т.пл. 226-228,5oC (разл.).
Ссылочное соединение F
7-((S)-3-Амино-1-пирролидинил)-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4- оксохинолин-3-карбоновая кислота
Внешний вид: бледно-коричневые кристаллы
т.пл. 192-193,5oC (разл.).
Ссылочное соединение G
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-4-оксо-7-пиперазинилхинолин- 3-карбоновая кислота
Внешний вид: бледно-желтые иглы
т.пл. 213-214,5oC.
Ссылочное соединение H
1-Циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-пиперазинилхинолин- 3-карбоновой кислоты гидрохлорид
Внешний вид: бледно-коричневые иглы
Т.пл. 279-281oC (разл.)
Ссылочное соединение I
5-Амино-1-циклопропил-6-фтор-1,4-дигидро-8-метил-4-оксо-7-пиперазинилхинолин- 3-карбоновой кислоты гихрохлорид
Внешний вид: желтые иглы
т.пл. > 300oC.




Производные 5-амино-8-метил-7-пирролидинилхинолин-3-карбоновой кислоты формулы I, где R1 - H; R2 - H, низший алкил, галогенированный низший алканоил или остаток эфира карбоновой кислоты; R3 - водород или низший алкил или два из R4, R5, R6 могут совместно образовывать -(CH2)n - группу, где n = 2, их стереоизомеры или их фармакологически приемлемые соли, обладают прекрасной антибактериальной активностью. 6 с. и 11 з.п. ф-лы, 11 табл.
где R1 представляет атом водорода;
R2 выбирают из группы, включающей атом водорода, низший алкил, галогенированный низший алканоил и остаток эфира карбоновой кислоты;
R3 выбирают из группы, включающей атом водорода и низший алкил;
R4, R5 и R6 каждый независимо представляет атом водорода или низший алкил или два из R4, R5 и R6 могут совместно образовать - (CH2)n-группу, где n = 2,
их стереоизомеры или их фармакологически приемлемые соли.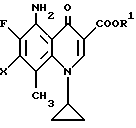
где Х представляет атом галогена и R1 принимает вышеуказанные значения,
с производным пирролидина общей формулы IV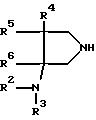
где R2, R3, R4, R5 и R6 принимают указанные значения,
с последующим, если необходимо, гидролизом.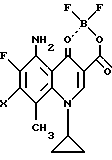
где Х представляет атом галогена,
с производным пирролидина общей формулы IV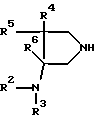
где R2, R3, R4, R5 и R6 принимают указанные значения,
с последующим, если необходимо, дехелатированием.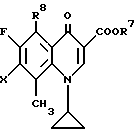
где R7 - низший алкил;
R8 - нитрогруппа или аминогруппа;
Х - атом галогена.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ получения 1-этилимидазолов | 1973 |
|
SU501671A3 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ПОЛУАВТОМАТ ДЛЯ ПРОДАЖИ БИЛЕТОВ | 0 |
|
SU248361A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, ч | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Ротационный фильтр-пресс для отжатия торфяной массы, подвергшейся коагулированию, и т.п. работ | 1924 |
|
SU204A1 |
| ПЕЧНОЙ ЖЕЛЕЗНЫЙ РУКАВ (ТРУБА) | 1920 |
|
SU199A1 |

