Область изобретения
Настоящее изобретение относится к ингибиторам ферментативной активности дипептидилпептидазы IV и ферментов, подобных дипептидилпептидазе IV, и, более конкретно, к фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений для лечения рака и опухолей. Настоящее изобретение относится особенно к способу ингибирования метастазирования и опухолевой колонизации.
Предпосылки к созданию изобретения
Дипептидилпептидаза IV (DPIV) представляет собой сериновую протеиназу, которая отщепляет N-концевые дипептиды от пептидной цепи, содержащей, предпочтительно, остаток пролина в предпоследнем положении. Хотя биологическая роль DPIV у млекопитающих еще не до конца установлена, полагают, что она играет важную роль в метаболизме нейропептидов, активации Т-кеток и проникновении ВИЧ в лимфоидные клетки.
Кроме того, было установлено, что DPIV отвечает за инактивацию глюкагон-подобного пептида-1 (GLP-1) и глюкозозависимого инсулинотропного пептида, известного также как желудочно-ингибирующий пептид (GIP). Поскольку GLP-1 является главным стимулятором секреции инсулина поджелудочной железой и оказывает прямое благоприятное действие на утилизацию глюкозы, в WO 97/40832 и US 6 303 661 ингибирование DPIV и DPIV-подобной ферментативной активности, как было показано, представляет привлекательный подход к лечению инсулиннезависимого сахарного диабета (NIDDM).
Настоящее изобретение относится к новому применению ингибиторов DPIV для профилактики и лечения состояний, опосредованных ингибированием DPIV и DPIV-подобных ферментов, в частности для профилактики и лечения рака и опухолей и для профилактики и лечения метастазирования и опухолевой колонизации, и фармацевтическим композициям, например, пригодным для ингибирования DPIV и DPIV-подобных ферментов, и к способу ингибирования указанной ферментативной активности.
Настоящее изобретение относится к способу лечения, в частности к способу профилактики и лечения рака, опухолей, метастазов и опухолевой колонизации, а также к соединениям и композициям для применения в указанном способе. Дипептидилпептидаза IV (DPIV) представляет собой пост-пролиновую (в меньшей степени, пост-аланиновую, пост-сериновую или пост-глициновую) отщепляющую сериновую протеиназу, которая обнаруживается в различных тканях организма, включая почки, печень и кишечник.
Известно, что ингибиторы DPIV могут быть пригодными для лечения нарушенной толерантности к глюкозе и сахарного диабета (международная патентная заявка, номер публикации WO 99/61431, Pederson R.A. et al., Diabetes, 1998, Aug; 47(8):1253-8 и Pauly R.P. et al., Metabolism 1999 Mar; 48(3):385-9). В частности, WO 99/61431 раскрывает ингибиторы DPIV, включающие аминокислотный остаток и тиазолидиновую или пирролидиновую группу, и их соли, особенно, L-трео-изолейцилтиазолидин, L-алло-изолейцилтиазолидин, L-трео-изолейцилпирролидин, L-алло-изолейцилтиазолидин, L-алло-изолейцилпирролидин и их соли.
Дополнительными примерами низкомолекулярных ингибиторов дипептидилпептидазы IV являются такие агенты, как производные тетрагидроизохинолин-3-карбоксамида, N-замещенные 2-цианопиролы и -пирролидины, N-(N'-замещенный глицил)-4-цианотиазолидины, аминоацил-бороно-пролил-ингибиторы, циклопропил-конденсированные пирролидины и гетероциклические соединения. Ингибиторы дипептидилпептидазы IV описаны в US 6380398; US 6011155; US 6107317; US 6110949; US 6124305; US 6172081; WO 95/15309; WO 99/61431; WO 99/67278; WO 99/67279; DE 19834591; WO 97/40832; DE 196 16 486 C 2; WO 98/19998; WO 00/07617; WO 99/38501; WO 99/46272; WO 99/38501; WO 01/68603; WO 01/40180; WO 01/81337; WO 01/81304; WO 01/55105; WO 02/02560 и WO 02/14271, целиком включенных в настоящий документ в качестве ссылок, особенно в том, что касается указанных ингибиторов, их определения, применения и производства.
Термин "DPIV-подобные ферменты" относится к ферментным белкам, структурно и/или функционально родственным DPIV/CD26 (Sedo & Malik, Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochimica et Biophysica Acta 2001, 36506: 1-10). По существу, указанная небольшая группа ферментов была выделена во время процесса высвобождения дипептидов H-Xaa-Pro и дипептидов H-Xaa-Ala из N-конца олиго- или полипептидов. Они имеют общую черту в том, что они несут позиции Pro, также Ala, Ser, Thr и другие аминокислоты с малыми гидрофобными боковыми цепями, такие как Gly или Val. Гидролитическая эффективность выстраивается следующим образом: Pro>Ala " Ser, Thr " Gly, Val. Данные белки доступны только в таких малых количествах, что можно установить только пост-Pro или пост-Ala расщепление. В то время как белки DPIV, DP II, FAPα (Seprase), DP 6, DP 8 и DP 9 являются структурно родственными и демонстрируют высокую гомологичность последовательности, аттрактин представляет собой чрезвычайно функциональный DPIV-подобный фермент, отличающийся сходной активностью и характером ингибирующего действия.
DPIV-подобные ферменты описаны также в WO 01/19866, WO 02/04610, WO 02/34900 и WO 02/31134. WO 01/19866 раскрывает новую человеческую дипептидиламинопептидазу (DPP8), обладающую структурным и функциональным сходством с DPIV и белком, активирующим фибробласты (FAP). WO 02/34900 раскрывает новую дипептидилпептидазу 9 (DPP9), обладающую значительной гомологичностью с аминокислотными последовательностями DPIV и DPP8. WO 02/31134 раскрывает три DPIV-подобных фермента, DPRP1, DPRP2 и DPRP3. Секвенирование выявило, что DPRP1 является идентичной DPP8, как описано в WO 01/19866, что DPRP2 является идентичной DPP9 и что DPRP3 является идентичной KIAA1492, как описано в WO 02/04610.
DPIV и DPIV-подобные ферменты в иммунофизиологии и при раке
Дипептидилпептидаза IV (DPIV; ЕС 3.4.14.5; CD26) CD26 представляет собой Mr 110 000 поверхностный гликопротеин, обладающий рядом различных функциональных свойств, которые проявляются в различных тканях, включая эпителиальные клетки и субпопуляции лейкоцитов (Mentlein, 1999). Кроме того, она представляет собой связанную с мембраной эктопептидазу, которая проявляет свою активность во внеклеточном домене и которая способна отщеплять N-концевые дипептиды от полипептидов с L-полином или L-аланином на предпоследней позиции.
Патомеханизмы злокачественных опухолей
Злокачественные опухоли представляют собой группу из более 150 заболеваний, которые характеризуются неконтролируемым ростом патологических клеток в организме. Нормальные клетки становятся патологическими, когда они подвергаются воздействию канцерогенов, таких как радиация или определенные лекарственные средства или химические вещества. Они также могут стать злокачественными (раковыми), если они были поражены определенными вирусами или когда они получили еще не до конца понятный внутренний сигнал. После того, как клетки стали злокачественными, они делятся более быстро, чем обычные. Затем они часто образуют массы, называемые опухолями, которые инвазируют близлежащие ткани и мешают нормальному функционированию организма. Злокачественные клетки имеют также тенденцию распространяться к другим частям организма, где они могут образовывать вторичную опухоль.
Механизмы метастазирования
Результат метастазирования зависит от многочисленных взаимодействий в ткани-мишени и зависит от микроокружения, включая молекулы клеточной адгезии, хемокины или гидродинамические эффекты, а также многие другие факторы. Помимо этого, очень быстрое привлечение лейкоцитов и специфичные клеточные ответы в области опухоли могут играть важную роль в ранней защите хозяина от злокачественной опухоли. Данные ранние изменения могут играть главную роль в исходе метастатической болезни и могут расширить имеющиеся в настоящее время представления о резистентности хозяина против метастазирования.
WO 99/47152 раскрывает способ подавления злокачественного фенотипа или индуцирования апоптоза злокачественных клеток у субъекта, включающий внедрение в злокачественную клетку нуклеиновой кислоты, кодирующей белок - дипептидилпептидазу IV или белок-α, активирующий фибробласты, что подавляет злокачественный фенотип рака. WO 99/47152 также раскрывает способ индуцирования экспрессии дипептидилпептидазы IV или белка-α, активирующего фибробласты, включающий введение субъекту фармацевтической композиции, включающей терапевтически эффективное количество агента, способного активировать транскрипцию гена дипептидилпептидазы IV или гена белка-α, активирующего фибробласты, и фармацевтически приемлемый носитель или разбавитель.
Современные способы лечения злокачественных опухолей и адгезии опухолевых клеток
Современные способы лечения злокачественных опухолей включают хирургическую операцию, химиотерапию, облучение и другие способы лечения, включая иммунотерапию. Иммунотерапия состоит из использования или модификации естественных механизмов организма - в большинстве случаев иммунных механизмов - для борьбы со злокачественной опухолью. Химиотерапия убивает злокачественные клетки с помощью лекарственных средств или гормонов. После перорального приема или введения с помощью инъекции химиотерапевтические агенты используют при целом ряде злокачественных опухолей. Их можно использовать в отдельности или в комбинации с оперативным вмешательством или облучением или и с тем и с другим. Химиотерапия представляет собой общепринятый способ разрушения трудных для выявления злокачественных клеток, которые распространились по организму и циркулируют в нем. Анемия (низкое содержание эритроцитов) представляет собой частый побочный эффект химиотерапии и может вызывать такие симптомы, как чрезвычайная утомляемость, головокружение или одышка. Эпоэтин альфа (Procrit®, Epogen®) - рекомбинантный эритропоэтин, который стимулирует образование эритроцитов, является официальным лекарственным средством, имеющимся в распоряжении для лечения анемии, вызванной химиотерапией.
Иммунотерапия использует собственную иммунную систему организма или другие части организма для разрушения злокачественных клеток. Данная форма лечения все еще интенсивно изучается в ходе клинических испытаний; до настоящего времени она не является широко доступной для большинства пациентов, страдающих злокачественными опухолями. Различные используемые иммунологические агенты включают вещества, вырабатываемые организмом (такие как интерфероны, интерлейкины и фактор некроза опухолей), и вещества, получаемые в лабораторных условиях (такие как моноклональные антитела и вакцины). Иммунологические агенты работают различным образом и могут использоваться независимо от других форм лечения или в комбинации с ними.
Ингибиторы ангиогенеза как антиметастатические лекарственные средства в иммунотерапии
Ингибиторы ангиогенеза представляют собой лекарственные средства, которые блокируют развитие новых кровеносных сосудов. Солидные опухоли не могут расти без образования новых кровеносных сосудов. Блокирование развития новых кровеносных сосудов пресекает снабжение опухоли кислородом и питательными веществами.
В настоящее время несколько ингибиторов ангиогенеза проходят клинические испытания на людях. Ткань злокачественной опухоли не может расти или распространяться (давать метастазы) без образования новых кровеносных сосудов. Кровеносные сосуды снабжают ткани кислородом и питательными веществами, необходимыми для выживания и роста.
Краткое описание изобретения
Настоящее изобретение относится к новому применению ингибиторов DPIV формул 1-12 и их соответствующих фармацевтически приемлемых кислотно-аддитивных солевых форм для профилактики и лечения рака и опухолей. В более предпочтительном варианте осуществления настоящего изобретения соединения по настоящему изобретению являются пригодными для предотвращения и ингибирования метастазирования и опухолевой колонизации.
Сокращение экспрессии эктопептидазы DPIV и отсутствие DPIV-подобной активности в легких мутантных крыс F344, не имеющих ферментативной активности и экспрессии DPIV, приводит к снижению адгезии раковых клеток и метастазирования в легкие. Клеточную адгезию и рост сингенной аденокарциномы молочной железы MADB106 у крыс F344 in vivo изучали на крысах F344 после острого и хронического лечения DPIV-лигандами in vivo. Изучали мутантные сублинии F344, не обладающие ферментативной активностью DPIV, и F344 дикого типа. Постоянная внутрижелудочная инфузия изолейцилцианопирролидина TFA и фумарата изолейцилтиазолидина через осмотические мининасосы в течение двух недель дозозависимым образом уменьшала индуцированную злокачественной опухолью потерю массы тела и количество колоний опухоли на поверхности легких. Таким образом, метастазирование MADB106 уменьшалось с помощью постоянного лечения с использованием различных ингибиторов DPIV (фумарата изолейцилтиазолидина; изолейцилцианопиррилидина TFA), что предполагает наличие эффектов защитного класса у двух различных ингибиторов/лигандов DPIV. Возможно, фумарат изолейцилтиазолидина и изолейцилцианопиррилидин TFA защищают от метастазирования посредством взаимодействия с процессами клеточной адгезии, посредством модификации механизмов клеточной защиты хозяина, посредством модулирования ангиогенеза, посредством прямых воздействий на злокачественные клетки или посредством повышенных уровней субстратов DPIV, которые непрямым образом опосредуют защитные эффекты.
Краткое описание чертежей
Фиг. 1: Влияние однократной инъекции фумарата изолейцилтиазолидина на легочные метастазы у крыс F344. Меченные витальным красителем (карбоксифлуоресцеином; CFSE) опухолевые клетки MADB106 инъецировали в латеральную хвостовую вену и извлекали легкие через 30 мин после инокуляции. Положительные по CFSE опухолевые клетки в легких подсчитывали с помощью иммуногистологии и визуализирующего анализа. Данные представляют средние величины ± СКО среднего; достоверной разницы по сравнению с контролями, получавшими физиологический раствор, обнаружено не было.
Фиг. 2: Влияние однократной инъекции изолейцилцианопиррилидина TFA на адгезию опухолевых клеток через 30 мин после инъекции крысам F344USA. Меченные CFSE злокачественные клетки MADB106 инъецировали в латеральную хвостовую вену и извлекали легкие через 30 мин после инокуляции. Положительные по CFSE опухолевые клетки в легких подсчитывали с помощью иммуногистологии и визуализирующего анализа. Данные представляют средние величины ± СКО среднего; достоверной разницы по сравнению с контролями, получавшими физиологический раствор, обнаружено не было.
Фиг. 3: Влияние однократной инъекции фумарата валилпирролидина на адгезию опухолевых клеток через 30 мин после инъекции крысам F344USA. Меченные CFSE клетки аденокарциномы MADB106 инъецировали в латеральную хвостовую вену и извлекали легкие через 30 мин после инокуляции. Положительные по CFSE опухолевые клетки в легких подсчитывали с помощью иммуногистологии и визуализирующего анализа. Данные представляют средние величины ± СКО среднего; достоверной разницы по сравнению с контролями, получавшими физиологический раствор, обнаружено не было.
Фиг. 4: Влияние постоянной внутрижелудочной инфузии фумарата изолейцилтиазолидина на изменение массы тела в граммах у крыс F344, имеющих метастазы в легких. Показано дозозависимое уменьшение потери массы тела после постоянной инфузии различных доз фумарата изолейцилтиазолидина у крыс F344 через 2 недели после инъекции опухолевых клеток MADB106. Однофакторный ANOVA показал достоверное влияние на массу тела, которое стало достоверным при анализе post-hoc при дозах 0,4 мг и 4 мг. Данные представляют средние величины ± СКО среднего; p < 0,05 отражает достоверную разницу по сравнению с контролями SHAM, получавшими физиологический раствор, по определению Fisher PLSD.
Фиг. 5: Влияние постоянной внутрижелудочной инфузии фумарата изолейцилтиазолидина на количество опухолевых колоний в легких у крыс F344. Показано дозозависимое уменьшение количества опухолевых колоний в легких после постоянной инфузии различных доз фумарата изолейцилтиазолидина у крыс F344 через 2 недели после инъекции опухолевых клеток MADB106. Однофакторный ANOVA показал достоверное влияние, которое стало достоверным при анализе post-hoc при дозе 4 мг. Данные представляют средние величины ± СКО среднего; p < 0,05 отражает достоверную разницу по сравнению с контролями SHAM, получавшими физиологический раствор, по определению Fisher PLSD.
Фиг. 6: Влияние постоянной внутрижелудочной инфузии фумарата изолейцилтиазолидина, изолейцилцианопирролидина TFA и фумарата валилпирролидина на количество опухолевых колоний в легких у крыс F344. Показано достоверное уменьшение количества опухолевых колоний в легких после постоянной инфузии фумарата изолейцилтиазолидина и изолейцилцианопирролидина TFA у крыс F344 через 2 недели после инъекции опухолевых клеток MADB106. Данные представляют средние величины ± СКО среднего; p < 0,05 отражает достоверную разницу по сравнению с контролями SHAM, получавшими физиологический раствор, по определению ANOVA и Fisher PLSD.
Подробное описание изобретения
Настоящее изобретение относится к области ингибирования дипептидилпептидазы IV (DPIV) и, более конкретно, к новому применению ингибиторов DPIV и DPIV-подобной ферментативной активности для профилактики и лечения рака и опухолей, в частности для профилактики и ингибирования метастазирования и опухолевой колонизации, и к фармацевтическим композициям, содержащим указанные соединения.
В противоположность другим предложенным ранее способам, настоящее изобретение особо относится к перорально доступной терапии низкомолекулярными ингибиторами дипептидилпептидазы IV. Настоящее изобретение представляет новый подход к профилактике и лечению рака и метастатической болезни. Он является щадящим по отношению к пациенту, коммерчески доступным и подходящим для применения в терапевтическом режиме, особенно при заболеваниях человека.
Спонтанные мутации гена DPIV, наблюдающиеся в сублиниях крыс F344, обеспечивают модель для изучения роли DPIV в адгезии и колонизации опухолей. Мутации у крыс F344 приводят к отсутствию ферментативной активности DPIV и обнаруживаются в сублиниях из Германии (GER) и Японии (JAP) (Thompson et al., 1991; Tsuji et al., 1992), в то время как крысы из питомников США демонстрируют значительную ферментативную активность. У крыс F344JAP замена G633R в белке DPIV приводит к выраженному уменьшению экспрессии мутантного неактивного фермента (Cheng et al., 1999; Tsuji et al., 1992), в то время как другая DPIV-орицательная сублиния F344GER экспрессирует неактивный мутантный фермент (Thompson et al., 1991).
На основе данных открытий, изучение роли экспрессии и ферментативной активности DPIV при раке, согласно настоящему изобретению установило, что пероральное введение ингибиторов DPIV приводит к уменьшению метастазов в легких и колонизации.
Целью настоящего изобретения является разработка ингибиторов и/или лигандов дипептидилпептидазы IV, которые обладают высокой биодоступностью. В другом предпочтительном варианте осуществления настоящее изобретение относится к ингибиторам DPIV, которые демонстрируют точно предсказуемое время активности в ткани-мишени.
Примерами специфичных в отношении мишени, перорально доступных низкомолекулярных агентов являются пролекарства стабильных и нестабильных ингибиторов дипептидилпептидазы IV общей формулы А-В-С, где А представляет аминокислоту, В представляет химическую связь между А и С или аминокислоту, а С представляет нестабильный или стабильный ингибитор дипептидилпептидазы IV, соответственно. Они описаны в WO 99/67278 и WO 99/67279, идеи которых, касающиеся, получения, определения, применения и производства пролекарств, целиком включены в настоящий документ в качестве ссылок. Особо включены в настоящий документ в качестве ссылок подробные определения А, В и С.
Настоящее изобретение относится к новому способу, при котором снижение активности фермента дипептидилпептидазы (DPIV или CD26) или DPIV-подобной ферментативной активности, или связывание DPIV-специфичного лиганда вызывает подавляющее опухоль или иммуностимулирующее действие в организмах млекопитающих, индуцированное эффекторами фермента, и приводит, как следствие, к уменьшению роста или адгезии злокачественных клеток. Подобное лечение будет приводить к уменьшению или задержке адгезии злокачественных клеток (метастазирования) или роста опухоли. Как следствие, млекопитающим, имеющим злокачественную опухоль, лечение ингибиторами активности DPIV или DPIV-подобных ферментов приносит пользу.
Способ и применение по настоящему изобретению для профилактики и лечения злокачественной опухоли и связанных с ней нарушений у животного, включая человека, который в этом нуждается, включают противораковые эффекты посредством связывания или посредством ингибирования DPIV или родственной ферментативной активности, с использованием ингибитора или лиганда указанных ферментов. Пероральное введение ингибитора DPIV может быть предпочтительным при большинстве обстоятельств.
Настоящее изобретение далее иллюстрируется с помощью следующих примеров, касающихся противоракового и антиметастатического действия пониженной DPIV-подобной активности и/или связывания в исследовании in vivo адгезии злокачественных клеток (пример 13), а также в исследованиях колонизации злокачественных опухолей (пример 14).
В одном показательном примере настоящее изобретение относится к применению дипептидных соединений и соединений, аналогичных дипептидным соединениям, которые образуются из аминокислоты и тиазолидиновой или пирролидиновой группы, и их солей, далее в настоящем документе называемых дипептидными соединениями. Предпочтительно, аминокислота и тиазолидиновая или пирролидиновая группа связаны друг с другом посредством амидной связи.
Особенно подходящими для целей по настоящему изобретению являются дипептидные соединения, в которых аминокислота, предпочтительно, выбрана из натуральной аминокислоты, такой как, например, лейцин, валин, глутамин, глутаминовая кислота, пролин, изолейцин, аспарагины и аспарагиновая кислота.
Дипептидные соединения, используемые по настоящему изобретению, в концентрации (дипептидных соединений) 10 мкМ демонстрируют снижение активности дипептидилпептидазы IV или DPIV-аналогичных ферментов по меньшей мере на 10%, особенно, по меньшей мере на 40%. Часто требуется также снижение активности по меньшей мере на 60% или по меньшей мере на 70%. Предпочтительные эффекторы могут также демонстрировать снижение активности максимум на 20% или 30%.
Предпочтительными соединениями являются L-алло-изолейцилтиазолидин, L-трео-изолейцилпирролидин и их соли, особенно, соли фумаровой кислоты, а также L-алло-изолейцилпирролидин и его соли. Особенно предпочтительными соединениями являются глутаминилпирролидин и глутаминилтиазолидин формул 1 и 2:

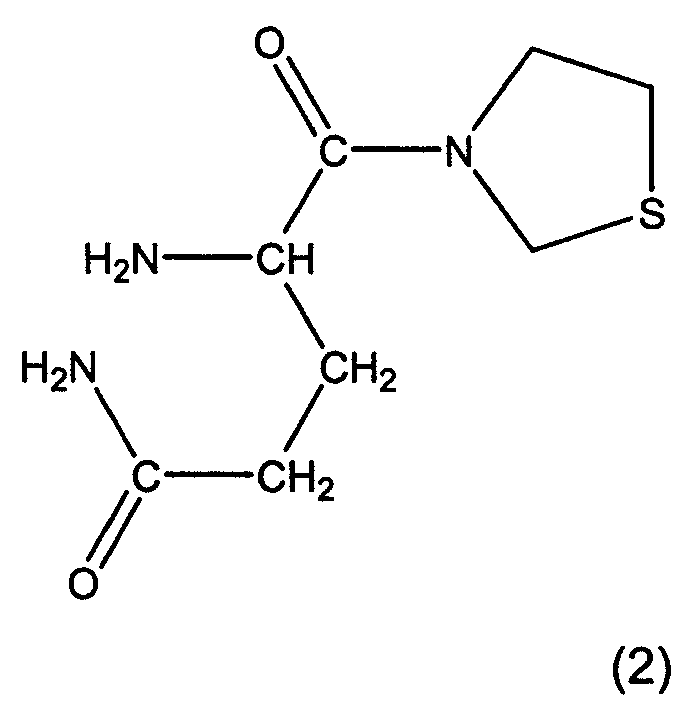
Другие предпочтительные соединения представлены в таблице 1.
Соли дипептидных соединений могут быть представлены в молярном соотношении дипептидного (-аналогичного) компонента и солевого компонента 1:1 или 2:1. Такой солью, например, является (Ile-Thia)2 фумаровая кислота.
Структуры других предпочтительных дипептидных соединений
В другом предпочтительном варианте осуществления настоящее изобретение относится к применению пептидных соединений формулы 3, пригодных для конкурентного модулирования катализа дипептидилпептидазой IV:

где A, B, C, D и Е независимо представляют собой любые аминокислотные части, включая протеиногенные аминокислоты, непротеиногенные аминокислоты, L-аминокислоты и D-аминокислоты, и где Е и/или D могут отсутствовать.
Другие условия, касающиеся формулы (3):
А представляет собой аминокислоту, за исключением D-аминокислоты,
В представляет собой аминокислоту, выбранную из Pro, Ala, Ser, Gly, Hyp, ацетидин-(2)-карбоновой кислоты и пипеколиновой кислоты,
С представляет собой любую аминокислоту, за исключением Pro, Hyp, ацетидин-(2)-карбоновой кислоты, пипеколиновой кислоты, и за исключением N-алкилированных аминокислот, например N-метилвалина и саркозина,
D представляет собой любую аминокислоту или отсутствует,
Е представляет собой любую аминокислоту или отсутствует,
либо:
С представляет собой любую аминокислоту, за исключением Pro, Hyp, ацетидин-(2)-карбоновой кислоты, пипеколиновой кислоты, за исключением N-алкилированных аминокислот, например N-метилвалина и саркозина, и за исключением D-аминокислоты;
D представляет собой любую аминокислоту, выбранную из Pro, Ala, Ser, Gly, Hyp, ацетидин-(2)-карбоновой кислоты и пипеколиновой кислоты,
Е представляет собой любую аминокислоту, за исключением Pro, Hyp, ацетидин-(2)-карбоновой кислоты, пипеколиновой кислоты и за исключением N-алкилированных аминокислот, например N-метилвалина и саркозина.
Примерами аминокислот, которые можно использовать в настоящем изобретении, являются L- и D-аминокислоты, N-метиламинокислоты; алло- и трео-формы Ile и Thr, которые могут, например, представлять собой α-, β- или ω-аминокислоты, из которых предпочтительными являются α-аминокислоты.
Примерами аминокислот в формуле изобретения и описании являются:
аспарагиновая кислота (Asp), глутаминовая кислота (Glu), аргинин (Arg), лизин (Lys), гистидин (His), глицин (Gly), серин (Ser) и цистеин (Cys), треонин (Thr), аспарагин (Asn), глутамин (Gln), тирозин (Tyr), аланин (Ala), пролин (Pro), валин (Val), изолейцин (Ile), лейцин (Leu), метионин (Met), фенилаланин (Phe), триптофан (Trp), гидроксипролин (Hyp), бета-аланин (бета-Ala), 2-аминооктановая кислота (Aoa), азетидин-(2)-карбоновая кислота (Ace), пипеколиновая кислота (Pip), 3-аминопропионовая, 4-аминомасляная и т.п., альфа-аминоизомасляная кислота (Aib), саркозин (Sar), орнитин (Orn), цитрулин (Cit), гомоаргинин (Har), трет-бутилаланин (t-butyl-Ala), трет-бутилглицин (t-butyl-Gly), N-метилизолейцин (N-Melle), фенилглицин (Phg), циклогексилаланин (Cha), норлейцин (Nle), цистеиновая кислота (Cya) и метионинсульфоксид (MSO), ацетил-Lys, модифицированные аминокислоты, такие как фосфорилсерин (Ser(P)), бензилсерин (Ser(Bzl)) и фосфорилтирозин (Tyr(P)), 2-аминомасляная кислота (Abu), аминоэтилцистеин (AECys), карбоксиметилцистеин (Cmc), дегидроаланин (Dha), дегидроамино-2-масляная кислота (Dhb), карбоксиглутаминовая кислота (Gla), гомосерин (Hse), гидроксилизин (Hyl), цис-гидроксипролин (cisHyp), транс-гидроксипролин (transHyp), изовалин (Iva), пироглутаминовая кислота (Pyr), норвалин (Nva), 2-аминобензойная кислота (2-Abz), 3-аминобензойная кислота (3-Abz), 4-аминобензойная кислота (4-Abz), 4-(аминометил)бензойная кислота (Amb), 4-(аминометил)циклогексанкарбоновая кислота (4-Amc), пеницилламин (Pen), 2-амино-4-цианомасляная кислота (Cba), циклоалканкарбоновые кислоты.
Примерами ω-аминокислот являются, например: 5-Ara (аминоралеровая кислота), 6-Ahx (аминогексановая кислота), 8-Aoc (аминооктановая кислота), 9-Anc (аминованоевая кислота), 10-Adc (аминодекановая кислота), 11-Aun (аминоундекановая кислота), 12-Ado (аминододекановая кислота).
Дополнительные аминокислоты следующие: инданилглицин (Igl), индолин-2-карбоновая кислота (Idc), октагидроиндол-2-карбоновая кислота (Oic), диаминопропионовая кислота (Dpr), диаминомасляная кислота (Dbu), нафтилаланин (1-Nal), (2-Nal), 4-аминофенилаланин (Phe(4-NH2)), 4-бензоилфенилаланин (Bpa), дифенилаланин (Dip), 4-бромфенилаланин (Phe(4-Br)), 2-хлорфенилаланин (Phe(2-Cl)), 3-хлорфенилаланин (Phe(3-Cl)), 4-хлорфенилаланин (Phe(4-Cl)), 3,4-хлорфенилаланин (Phe(3,4-Cl2)), 3-фторфенилаланин (Phe(3-F)), 4-фторфенилаланин (Phe(4-F)), 3,4-фторфенилаланин (Phe(3,4-F2)), пентафторфенилаланин (Phe(F5)), 4-гуанидинфенилаланин (Phe(4-guanidino)), гомофенилаланин (hPhe), 3-йодфенилаланин (Phe(3-J)), 4-йодфенилаланин (Phe(4-J)), 4-метилфенилаланин (Phe(4-Ме)), 4-нитрофенилаланин (Phe-4-NO2)), бифенилаланин (Bip), 4-фосфонометилфенилаланин (Pmp), циклогексилглицин (Ghg), 3-пиридинилаланин (3-Pal), 4-пиридинилаланин (4-Pal), 3,4-дигидропролин (A-Pro), 4-кетопролин (Pro(4-keto)), тиопролин (Thz), изонипекотиновая кислота (Inp), 1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота (Tic), пропаргилглицин (Pra), 6-гидроксинорлейцин (NU(6-OH), гомотирозин (hTyr), 3-йодтирозин (Tyr(3-J)), 3,5-дийодтирозин (Tyr(3,5-J2)), d-метилтирозин (Tyr(Me)), 3-NO2-тирозин (Tyr(3-NO2)), фосфотирозин (Tyr(PO3H2)), алкилглицин, 1-аминоиндан-1-карбоновая кислота, 2-аминоиндан-2-карбоновая кислота (Aic), 4-аминометилпиррол-2-карбоновая кислота (Py), 4-аминопирролидин-2-карбоновая кислота (Abpc), 2-аминотетралин-2-карбоновая кислота (Atc), диаминоуксусная кислота (Gly(NH2)), диаминомасляная кислота (Dab), 1,3-дигидро-2Н-изоинолкарбоновая кислота (Disc), гомоциклогексилаланин (hCha), гомофенилаланин (hPhe или Hof), транс-3-фенилазетидин-2-карбоновая кислота, 4-фенилпирролидин-2-карбоновая кислота, 5-фенилпирролидин-2-карбоновая кислота, 3-пиридилаланин (3-Pya), 4-пиридилаланин (4-Pya), стирилаланин, тетрагидроизохинолин-1-карбоновая кислота (Tiq), 1,2,3,4-тетрагидроноргарман-3-карбоновая кислота (Tpi), β-(2-тиенил)аланин (Tha).
Другие аминокислотные замены аминокислот, закодированных в генетическом коде, также могут быть включены в пептидные соединения в рамках настоящего изобретения и могут быть классифицированы с помощью указанной общей схемы.
Протеиногенные аминокислоты определяют как натуральные, полученные из белков α-аминокислоты. Непротеиногенные аминокислоты определяют как все остальные аминокислоты, которые не составляют блоки распространенных натуральных белков.
Полученные пептиды можно синтезировать в форме свободной С-концевой кислоты или в форме С-концевого амида. Свободные кислотные пептиды или амиды могут изменяться посредством модификаций боковых цепей. Указанные модификации боковых цепей включают, но не ограничиваются, например, образование гомосерина, образование пироглутаминовой кислоты, образование дисульфидной связи, дезамидирование остатков аспарагина или глутамина, метилирование, трет-бутилирование, трет-бутоксикарбонилирование, 4-метилбензилирование, тиоанизилирование, тиокрезилирование, бенцилоксиметилирование, 4-нитрофенилирование, бенцилоксикарбонилирование, 2-нитробенкоилирование, 2-нитросульфенилирование, 4-толуолсульфонилирование, пентафторфенилирование, дифенилметилирование, 2-хлорбензилоксикарбонилирование, 2,4,5-тихлорфенилирование, 2-бромбензилоксикарбонилирование, 9-фуоренилметилоксикарбонилирование, трифенилметилирование, 2,2,5,7,8-пентаметилхроман-6-сульфонилирование, гидроксилирование, окисление метионина, формилирование, ацетилирование, анизилирование, бенцилирование, бенкоилирование, трифторацетилирование, карбоксилирование аспарагиновой кислоты или глутаминовой кислоты, фосфорилирование, сульфатирование, цистеинилирование, гликозилирование пентозами, дезоксигексозами, гексозаминами, гексозами или N-ацетилгексозаминами, фарнезилирование, миристоолизирование, биотинилирование, пальмитоилирование, стеароилирование, геранилгеранилирование, глутатионилирование, 5'-аденозилирование, АДФ-рибозилирование, модификация с N-гликолилнейраминовой кислотой, N-ацетилнейраминовой кислотой, пиридоксальфосфатом, липоевой кислотой, 4'-фосфопантетеином или N-гидроксисукцинимидом.
В соединениях формулы (3) аминокислотные части A, B, C, D и Е присоединены, соответственно, к соседнему фрагменту амидными связями обычным образом, согласно стандартной номенклатуре, так, что амино-конец (N-конец) аминокислот (пептида) рисуют слева, а карбокси-конец аминокислот (пептида) рисуют справа (С-конец).
Известные пептидные субстраты пролин-специфичной сериновой протеиназы дипептидилпептидазы IV in vitro представляли собой трипептиды Diprotin A (Ile-Pro-Ile), Diprotin B (Val-Pro-Leu), и Diprotin C (Val-Pro-Ile). Установлено, что соединения, описанные в настоящем документе, действуют как субстраты дипептидилпептидазы IV in vivo у млекопитающих, и, в фармакологических дозах, ингибируют физиологический круговорот эндогенных субстратов посредством конкурентного катализа.
Особенно предпочтительные соединения по настоящему изобретению, которые пригодны в качестве модуляторов дипептидилпептидазы IV и DPIV-подобных ферментов, включают такие соединения, которые имеют величины Кi для связывания DPIV, эффективные для ингибирования DPIV in vivo после в/в и/или п/о введения крысам Wistar.
Другие предпочтительные соединения представляют собой пептидилкетоны формулы 4:
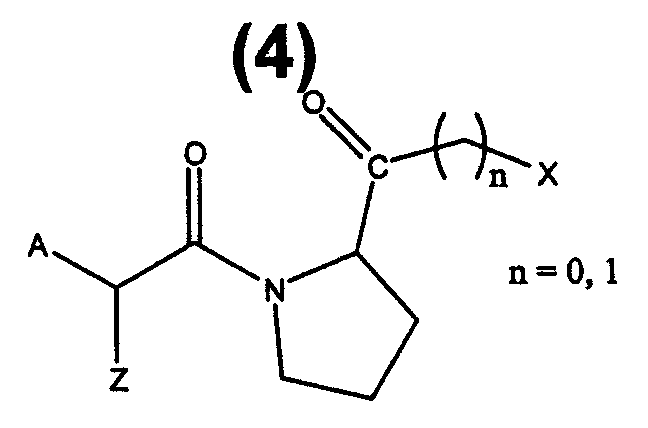
где A выбран из:
Х1 представляет собой Н или ацильную или оксикарбонильную группу, включая все аминокислоты и пептидные остатки,
Х2 представляет собой Н, -(CH)n-NH-C5H3N-Y с n = 2-4 или C5H3N-Y (двухвалентный пиридильный остаток), а Y выбран из H, Br, Cl, I, NO2 или CN,
Х3 представляет собой Н или фенильный или пиридильный остаток, незамещенный или замещенный одним, двумя или более остатками алкила, алкоксила, галогена, нитро, циано или карбоксила,
Х4 представляет собой Н или фенильный или пиридильный остаток, незамещенный или замещенный одним, двумя или более остатками алкила, алкоксила, галогена, нитро, циано или карбоксила,
Х5 представляет собой Н или алкильный, алкоксильный или фенильный остаток,
Х6 представляет собой Н или алкильный остаток,
при n = 1
Х выбран из H, OR2, SR2, NR2R3, N+R2R3R4,
где R2 выбран из ацильных остатков, которые являются незамещенными или замещенными одним, двумя или более алкильным, циклоалкильным, арильным или гетероарильным остатками, или выбран из аминокислот и пептидных остатков, или алкильных остатков, которые являются незамещенными или замещены одним, двумя или более алкильным, циклоалкильным, арильным и гетероарильным остатками,
R3 выбран из алкильных и ацильных функциональных групп, где R2 и R3 могут являться частью одной или более кольцевых структур насыщенных и ненасыщенных карбоциклических или гетероциклических структур,
R4 выбран из алкильных остатков, где R2 и R4 или R3 и R4 могут являться частью одной или более кольцевых структур насыщенных и ненасыщенных карбоциклических или гетероциклических структур,
при n = 0
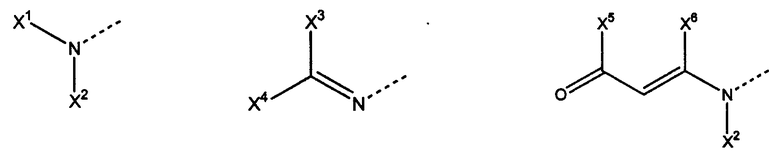
Х выбран из
где В представляет собой: O, S, NR5, где R5 представляет собой Н, алкилиден или ацил,
C, D, E, F, G, H независимо выбраны из ненасыщенных и насыщенных алкильных, оксиалкильных, тиоалкильных, аминоалкильных, карбонилалкильных, ацильных, карбамоильных, арильных и гетероарильных остатков;
при n = 0 и n = 1
Z выбран из Н, или алкильного остатка с разветвленной или неразветвленной цепью из С1-С9, или алкенильного остатка с разветвленной или одинарной цепью из С2-С9,циклоалкильного остатка из С3-С8, циклоалкенильного остатка из С5-С7, арильного или гетероарильного остатка, или боковой цепи, выбранной из всех боковых цепей всех натуральных аминокислот или их производных.
Кроме того, согласно настоящему изобретению, соединения формул 5, 6, 7, 8, 9, 10 и 11, включая все их стереоизомеры и фармацевтически приемлемые соли, описаны и могут применяться:

где R1 представляет собой Н, разветвленный или линейный С1-С9 алкильный остаток, разветвленный или линейный С2-С9 алкенильный остаток, С3-С8 циклоалкильный, С5-С7 циклоалкенильный, арильный или гетероарильный остаток или боковую цепь натуральной аминокислоты или ее производного,
R3 и R4 выбраны из Н, гидроксила, алкила, алкокси, нитро, циано или галогена,
А представляет собой Н или изостеру карбоновой кислоты, такой как функциональная группа, выбранная из CN, SO3H, CONHOH, PO3R5R6, тетразола, амида, сложного эфира, ангидрида, тиазола и имидазола,
В выбран из
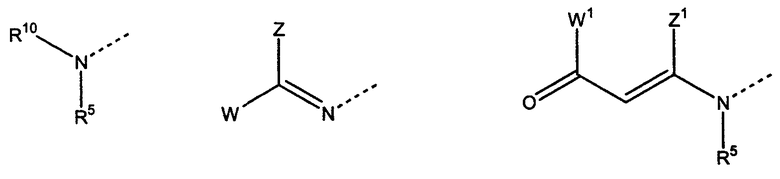
где R5 представляет собой Н, -(CH)n-NH-C5H3N-Y c n = 2-4 и C5H3N-Y (двухвалентный пиридильный остаток) с Y = H, Br, Cl, I, NO2 или CN,
R10 представляет собой Н, ацильный, оксикарбонильный или аминокислотный остаток,
W представляет собой Н или фенильный или пиридильный остаток, незамещенный или замещенный одним, двумя или более остатками алкила, алкоксила, галогена, нитро, циано или карбоксила,
W1 представляет собой Н, алкильный, алкоксильный или фенильный остаток,
Z представляет собой Н или фенильный или пиридильный остаток, незамещенный или замещенный одним, двумя или более остатками алкила, алкоксила, галогена, нитро, циано или карбоксила,
Z1 представляет собой Н или алкильный остаток,
D представляет собой циклический С4-С7 алкильный остаток, С4-С7 алкенильный остаток, который может быть замещен одной, двумя или более алкильными группами, или циклический 4-7-членный гетероалкильный или циклический 4-7-членный гетероалкенильный остаток,
Х2 представляет собой O, NR6, N+(R7)2 или S,
c Х3 по Х12 независимо выбраны из CH2, CR8R9, NR6, N+(R7)2, O, S, SO и SO2, включая все насыщенные и ненасыщенные структуры,
R6, R7, R8, R9 независимо выбраны из Н, разветвленного или линейного С1-С9 алкильного остатка, разветвленного или линейного С2-С9 алкенильного остатка, С3-С8 циклоалкильного остатка, С5-С7 циклоалкенильного остатка, арильного или гетероарильного остатка,
при следующих условиях:
в формуле 6: Х6 представляет собой CH, если А не является Н,
в формуле 7: Х10 представляет собой C, если А не является Н,
в формуле 8: Х7 представляет собой CH, если А не является Н,
в формуле 9: Х12 представляет собой C, если А не является Н.
В описании и формуле изобретения выражение "ацил" может обозначать С1-20 ацильный остаток, предпочтительно С1-8 ацильный остаток, и, особенно предпочтительно, С1-4 ацильный остаток, "циклоалкил" может обозначать С3-12 циклоалкильный остаток, предпочтительно С4, С5 или С6 циклоалкильный остаток, "карбоциклический" может обозначать С3-12 карбоциклический остаток, предпочтительно С4, С5 или С6 карбоциклический остаток. "Гетероарильный" определяется как арильный остаток, в котором от 1 до 4, предпочтительно 1, 2 или 3, атома кольца заменены такими гетероатомами, как N, S или О. "Гетероциклический" определяется как циклоалкильный остаток, в котором 1, 2 или 3 атома кольца заменены такими гетероатомами как N, S или О. "Пептиды" выбраны из пептидов размером от дипептидов до декапептидов, предпочтительно, представляют собой дипептиды, трипептиды, тетрапептиды и пентапептиды. Аминокислоты для образования "пептидов" могут быть выбраны из аминокислот, перечисленных выше.
В силу широкого распространения в организме белка и обширного ряда механизмов, в которых участвуют DPIV, активность DPIV и белки, родственные DPIV, системное лечение (энтеральное или парентеральное введение) ингибиторами DPIV может приводить к ряду нежелательных побочных эффектов.
Проблема, которую предстояло решить, состояла в создании соединений, которые можно использовать для направленного влияния на локально ограниченные патофизиологические и физиологические процессы. Проблема настоящего изобретения особо заключается в получении локально ограниченного ингибирования DPIV и DPIV-аналогичной активности для целей направленного вмешательства в регулирование активности локально активных субстратов.
Указанная проблема решена, согласно настоящему изобретению, с помощью соединений общей формулы (12)

где A представляет собой аминокислоту, имеющую по меньшей мере одну функциональную группу в боковой цепи,
В представляет собой химическое соединение, ковалентно связанное с по меньшей мере одной функциональной группой в боковой цепи А,
С представляет собой тиазолидиновую, пирролидиновую, цианопирролидиновую, гидроксипролиновую, дегидропролиновую или пиперидиновую группу, связанную с А амидной связью.
Данные соединения можно использовать для уменьшения иммунных, аутоиммунных нарушений или нарушений, связанных с центральной нервной системой.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения используются фармацевтические композиции, включающие по меньшей мере одно соединение общей формулы (12) и по меньшей мере одно обычное вспомогательное вещество, подходящее для места действия.
Предпочтительно, А представляет собой α-аминокислоту, особенно натуральную α-аминокислоту, имеющую одну, две или более функциональных групп в боковой цепи, предпочтительно треонин, тирозин, серин, аргинин, лизин, аспарагиновую кислоту, глутаминовую кислоту или цистеин.
Предпочтительно, В представляет собой олигопептид, имеющий цепь длиной до 20 аминокислот, полиэтиленгликоль, имеющий молярную массу до 20 000 г/моль, необязательно замещенный органический амин, амид, спирт, кислоту или ароматическое соединение, имеющее от 8 до 50 атомов С.
В описании и формуле изобретения выражение "алкил" может обозначать С1-50 алкильную группу, предпочтительно С6-30 алкильную группу, особенно С8-12 алкильную группу; например, алкильная группа может представлять собой метильную, этильную, пропильную, изопропильную или бутильную группу. Выражение "алк", например, в выражении "алкокси", и выражение "алкан", например, в выражении "алканоил" определяются так же как и "алкил"; ароматические соединения предпочтительно представляют замещенную или, необязательно, незамещенную фенильную, бензильную, нафтильную, бифенильную или антраценовую группу, которые предпочтительно имеют по меньшей мере 8 атомов С; выражение "алкенил" может обозначать С2-10 алкенильную группу, предпочтительно С2-6 алкенильную группу, которая имеет двойную связь (связи) в любом желательном месте и может быть замещенной или незамещенной; выражение "алкинил" может обозначать С2-10 алкинильную группу, предпочтительно, С2-6 алкинильную группу, которая имеет тройную связь (связи) в любом желательном месте и может быть замещенной или незамещенной; выражение "замещенный" или "заместитель" может обозначать любое желательное замещение одной или более, предпочтительно одной или двумя, алкильной, алкенильной, алкинильной, одно- и многовалентной ацильной, алканоильной, алкоксиалканоильной или алкоксиалкильной группами; упомянутые выше заместители могут, в свою очередь, иметь одну или более (но предпочтительно, не иметь) алкильную, алкинильную, одно- и многовалентную ацильную, алканоильную, алкоксиалканоильную или алкоксиалкильную группы в качестве боковых групп; органические амины, амиды, спирты или кислоты, имеющие от 8 до 50 атомов С, предпочтительно от 10 до 20 атомов С, могут иметь формулы (алкил)2N- или (алкил)NH-, -CO-N(алкил)2 или -CO-NH(алкил), -алкил-ОН или алкил-СООН.
Несмотря на протяженную боковую цепь, соединения формулы (12) все еще могут связываться с активным центром фермента дипептидилпептидазы IV и аналогичных ферментов, но уже не транспортируются активно транспортером пептидов PepT1. Сниженная или сильно ограниченная таким образом транспортабельность соединений по настоящему изобретению приводит, в идеале, к локальному или направленному на определенный участок ингибированию DPIV и DPIV-подобной ферментативной активности.
Соединения формулы (12) или другие соединения и пролекарства, используемые в соответствии с настоящим изобретением, могут быть представлены или использоваться, соответственно, в форме рацематов или в форме энантиомерно чистых соединений, предпочтительно в форме L-трео или L-алло, что касается части А формулы (12).
Путем удлинения/расширения модификаций боковой цепи, например, более семи атомов углерода, становится, соответственно, возможным получение резкого снижения транспортабельности (см. пример 12). Примеры в таблице 12.1 ясно показывают, что с увеличением пространственного размера боковых цепей происходит снижение транспортабельности веществ. Путем пространственного расширения боковых цепей, например, свыше размера группы атомов однозамещенного фенильного радикала, гидроксиламинового радикала или аминокислотного остатка, становится возможным, согласно настоящему изобретению, модифицировать или подавлять транспортабельность веществ-мишеней.
Согласно настоящему изобретению, соединения формулы (12) ингибируют сайт-специфичным образом DPIV или DPIV-подобную ферментативную активность в организме млекопитающего. Соответственно, возможным является эффективное влияние на локальные физиологические и патофизиологические условия (воспаление, псориаз, артрит, аутоиммунные заболевания, аллергии, злокачественную опухоль, метастазы) при резком уменьшении побочных эффектов.
Предпочтительными соединениями формулы (12) являются соединения, в которых олигопептиды имеют длину цепи от 3 до 15, особенно от 4 до 10, аминокислот, и/или полиэтиленгликоли имеют молярную массу по меньшей мере 250 г/моль, предпочтительно по меньшей мере 1500 г/моль, и до 15 000 г/моль, и/или необязательно замещенные органические амины, амиды, спирты, кислоты или ароматические соединения имеют по меньшей мере 12 атомов С и, предпочтительно, до 30 атомов С.
Соединения по настоящему изобретению можно преобразовывать и использовать как кислотно-аддитивные соли, особенно фармацевтически приемлемые кислотно-аддитивные соли. Фармацевтически приемлемая соль обычно принимают форму, в которой основная боковая цепь аминокислоты протонирована неорганической или органической кислотой. Примеры органических или неорганических кислот включают хлористоводородную, бромистоводородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, щавелевую, памовую, 2-нафталинсульфоновую, п-толуолсульфоновую, циклогексансульфаминовую, салициловую, сахариновую или трифторуксусную кислоту. Все фармацевтически приемлемые кислотно-аддитивные солевые формы соединений формул от (1) до (12) входят в объем настоящего изобретения.
С точки зрения тесной взаимосвязи между свободными соединениями и соединениями в форме их солей, какое бы соединение ни упоминалось в данном контексте, имеется в виду также соответствующая соль, при условии, что она существует или является пригодной при указанных обстоятельствах.
Настоящее изобретение также включает в свой объем пролекарства соединений по настоящему изобретению. В общем, указанные пролекарства будут представлять собой функциональные производные соединений, которые легко превращаются in vivo в желаемое терапевтически активное соединение. Таким образом, в указанных случаях применение настоящего изобретения будет включать в себя лечение различных описанных заболеваний с помощью пролекарственных вариантов одного или более заявленных соединений, которые превращаются в описанное выше соединение in vivo после введения субъекту. Обычные процедуры отбора и получения подходящих пролекарственных производных описаны, например, в "Design of Prodrugs", изд. H. Bundgaard, Elsevier, 1985, и в патентных заявках DE 198 28 113 и DE 198 28 114, которые целиком включены в настоящий документ в качестве ссылок.
В том случае, когда соединения или пролекарства по настоящему изобретению имеют по меньшей мере один хиральный центр, они могут, соответственно, существовать как энантиомеры. В том случае, когда соединения или пролекарства имеют два или более хиральных центра, они могут существовать также как диастереоизомеры. Следует понимать, что все указанные изомеры и их смеси входят в объем настоящего изобретения. Кроме того, некоторые из кристаллических форм соединений или пролекарств могут существовать как полиморфы и в качестве таковых также включены в настоящее изобретение. Помимо этого, некоторые из соединений могут образовывать сольваты с водой (т.е. гидраты) или обычными органическими растворителями, и указанные сольваты также входят в объем настоящего изобретения.
Соединения, включая их соли, могут также быть получены в форме их гидратов или могут включать другие растворители, которые использовались для их кристаллизации.
Как указано выше, соединения и пролекарства по настоящему изобретению и соответствующие им фармацевтически приемлемые кислотно-аддитивные солевые формы являются пригодными для ингибирования DPIV и DPIV-подобной ферментативной активности. Способность соединений и пролекарств по настоящему изобретению и соответствующих им фармацевтически приемлемых кислотно-аддитивных солевых форм ингибировать DPIV и DPIV-подобную ферментативную активность можно продемонстрировать с помощью анализа активности DPIV для определения величин Ki и величин IC50in vitro, как описано в примерах 7 и 8.
Способность соединений по настоящему изобретению и соответствующих им фармацевтически приемлемых кислотно-аддитивных солевых форм ингибировать DPIV in vivo можно продемонстрировать с помощью перорального или внутрисосудистого введения крысам Wistar, как описано в примере 11. Соединения по настоящему изобретению ингибируют активность DPIV in vivo и после перорального и после внутрисосудистого введения крысам Wistar.
DPIV присутствует во многих органах и тканях млекопитающих, например в щеточной каемке кишечника (Gutschmidt S. et al., "In situ" - measurements of protein contents in the brush border region along rat jejunal villi and their correlations with four enzyme activities. Histochemistry 1981, 72 (3), 467-79), экзокринном эпителии, гепатоцитах, почечных канальцах, эндотелии, миофибробластах (Feller A.C. et al., A monoclonal antibody detecting dipeptidylpeptidase IV in human tissue. Virchows Arch. A. Pathol. Anat. Histopathol. 1986; 409 (2):263-73), нервных клетках, латеральных мембранах некоторых видов поверхностного эпителия, например в фаллопиевых трубах, матке и пузырьковой железе, в цитоплазме просвета, например, эпителия пузырьковой железы и в мукоцитах бруннеровых желез (Hartel S. et al., Dipeptidyl peptidase (DPP) IV in rat organs. Comparison of immunohistochemistry and activity histochemistry. Histochemistry 1988; 89 (2): 151-61), репродуктивных органах, например придатке семенника и ампуле, семенных пузырьках и их секретах (Agrawal & Vanha-Perttula, Dipeptidyl peptidases in bovine reproductive organs and secretions. Int. J. Androl. 1986, 9 (6): 435-52). В сыворотке крови человека присутствуют две молекулярные формы дипептидилпептидазы (Krepela E. et al. Demonstration of two molecular forms of dipeptidyl peptidase IV in normal human serum. Physiol. Bohemoslov. 1983, 32 (6): 486-96). Сывороточная высокомолекулярная форма DPIV экспрессируется на поверхности активированных Т-клеток (Duke-Cohan J.S. et al. Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L released from activated T cells. J. Immunol. 1996, 156(5): 1714-21).
Соединения и пролекарства по настоящему изобретению и соответствующие им фармацевтически приемлемые кислотно-аддитивные солевые формы способны ингибировать DPIV in vivo. В одном варианте осуществления настоящего изобретения все молекулярные формы, гомологи и эпитопы DPIV из всех тканей и органов млекопитающих, а также те, которые еще не открыты, входят в объем настоящего изобретения.
Из редкой группы пролин-специфичных протеиназ DPIV, как сначала предполагали, являлась единственным связанным с мембраной ферментом, специфичным в отношении пролина как предпоследнего остатка на амино-конце полипептидной цепи. Однако недавно были идентифицированы другие молекулы, даже структурно не гомологичные DPIV, но несущие соответствующую ферментативную активность. DPIV-подобные ферменты, которые идентифицированы к настоящему времени, представляют собой, например, белок-α, активирующий фибробласты, дипептидилпептидазу IV β, белок, подобный дипептидиламинопептидазе, N-ацетилированную α-связанную кислую дипептидазу, неактивную клеточную пролиновую дипептидазу, дипептидилпептидазу II, аттрактин и белок, родственный дипептидилпептидазе IV (DPP 8), и описаны в обзорной статье Sedo & Malik (Sedo & Malik, Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochimica et Biophysica Acta 2001, 36506: 1-10). Другие DPIV-пдобные ферменты описаны в WO 01/19866, WO 02/04610 и WO 02/34900. WO 01/19866 описывает новую человеческую дипептидиламинопептидазу (DPP8), имеющую структурное и функциональное сходство с DPIV и белком, активирующим фибробласты (FAP). Фермент, подобный дипептидилпептидазе IV, описанный в WO 02/04610, хорошо известен специалистам. В базе данных генного банка данный фермент зарегистрирован под номером KIAA1492. В другом предпочтительном варианте осуществления настоящего изобретения все молекулярные формы, гомологи и эпитопы белков, обладающих DPIV-подобной ферментативной активностью, из всех тканей и органов млекопитающих, а также те, которые еще не открыты, входят в объем настоящего изобретения.
Способность соединений и пролекарств по настоящему изобретению и соответствующих им фармацевтически приемлемых кислотно-аддитивных солевых форм ингибировать DPIV-подобные ферменты можно продемонстрировать с помощью анализа ферментативной активности для определения величин Kiin vitro, как описано в примере 9. Величины Ki соединений по настоящему изобретению по сравнению со свиной дипептидилпептидазой II были определены как Ki = 8,52·10-5 М ± 6,33·10-6 М для глутаминилпирролидина и Ki = 1,07·10-5 М ± 3,81·10-7 М для глутаминилтиазолидина.
В другом варианте осуществления соединения и пролекарства по настоящему изобретению и соответствующие им фармацевтически приемлемые кислотно-аддитивные солевые формы обладают только низкой или вовсе не обладают ингибирующей активностью в отношении не-DPIV и не-DPIV-подобных пролин-специфичных ферментов. Как описано в примере 10 на примере глутаминилтиазолидина и глутаминилпирролидина, ингибирования дипептидилпептидазы I и пролилолигопептидазы выявлено не было. Против пролидазы оба соединения показали значительно более низкую эффективность по сравнению с DPIV. Величины IC50 против пролидазы были определены как IC50 > 3 мМ для глутаминилтиазолидина и как IC50 = 3,4·10-4 ± 5,63·10-5 для глутаминилпирролидина.
Настоящее изобретение относится к способу профилактики или лечения состояния, опосредованного модуляцией DPIV или DPIV-пдобной ферментативной активности, у субъекта, который в этом нуждается, который включает введение любого из соединений по настоящему изобретению или его фармацевтических композиций в количестве и при режиме дозирования, терапевтически эффективных для лечения указанного состояния. Кроме того, настоящее изобретение включает применение соединений и пролекарств по настоящему изобретению и соответствующих им фармацевтически приемлемых кислотно-аддитивных солевых форм для изготовления лекарственного средства для профилактики или лечения состояния, опосредованного модуляцией активности DPIV, у субъекта. Соединение можно вводить пациенту любым общепринятым путем, включая, без ограничения, внутривенный, пероральный, подкожный, внутримышечный, внутрикожный, парентеральный пути и их комбинации.
В еще одном показательном варианте осуществления настоящее изобретение относится к составам для соединений формул от 1 до 12 и соответствующих им фармацевтически приемлемых пролекарств и кислотно-аддитивных солевых форм, в фармацевтических композициях.
Термин "субъект", используемый в настоящем документе, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно к человеку, которое является объектом лечения, наблюдения или эксперимента.
Термин "терапевтически эффективное количество", используемый в настоящем документе, означает такое количество активного соединения или фармацевтического агента, которое вызывает биологический или медицинский ответ в системе ткани, у животного или человека, которого добивается исследователь, ветеринар, врач или другой клиницист, и который включает облегчение симптомов заболевания или расстройства, по поводу которого осуществляется лечение.
Используемый в настоящем документе термин "композиция" включает продукт, включающий заявленные соединения в терапевтически эффективных количествах, а также любой другой продукт, который получается, прямо или опосредованно, из комбинаций заявленных соединений.
Для изготовления фармацевтических композиций, используемых в настоящем изобретении, одно или более соединений формул 1 - 12 или соответствующих им фармацевтически приемлемых пролекарств или кислотно-аддитивных солевых форм в качестве активного ингредиента гомогенно смешивают с фармацевтическим носителем, в соответствии с обычными методиками изготовления лекарственных средств; указанный носитель может иметь самые разнообразные формы, в зависимости от формы препарата, желательного для введения, например, пероральным или парентеральным путем, таким как внутримышечный. При изготовлении композиций в виде лекарственной формы для перорального введения можно использовать любые обычные фармацевтические среды. Так жидкие препараты для перорального введения, такие как, например, суспензии, эликсиры и растворы, подходящие носители и добавки, могут включать воду, гликоли, масла, спирты, корригенты, консерванты, красители и т.п.; твердые препараты для перорального введения, такие как, например, порошки, капсулы, мягкие желатиновые капсулы и таблетки, подходящие носители и добавки, включают крахмалы, сахара, разбавители, гранулирующие агенты, смазывающие агенты, связывающие агенты, разрыхлители и т.п. В силу простоты введения таблетки и капсулы представляют собой наиболее выгодные дозированные лекарственные формы для перорального введения; в данном случае используются твердые фармацевтические носители. Если желательно, таблетки можно покрывать сахаром или энтеросолюбильной оболочкой с помощью стандартных технологий. Для парентеральных препаратов носитель обычно будет включать стерильную воду, хотя можно включать и прочие ингредиенты, необходимые, например, для таких целей, как улучшение растворимости или для консервации.
Также можно изготавливать суспензии для инъекций; в данном случае можно использовать подходящие жидкие носители, суспендирующие агенты и т.п. Фармацевтические композиции в данном случае будут содержать, на дозированную единицу, например таблетку, капсулу, порошок, инъекцию, чайную ложку и т.п., количество активного ингредиента, необходимое для доставки эффективной дозы, как описано выше. Фармацевтические композиции в данном случае будут содержать, на дозированную единицу, например таблетку, капсулу, порошок, инъекцию, суппозиторий, чайную ложку и т.п., приблизительно от 0,01 мг до 1000 мг (предпочтительно, приблизительно от 5 до 500 мг) и могут вводиться в дозе приблизительно от 0,1 до 300 мг/кг массы тела в день (предпочтительно, от 1 до 50 мг/кг в день). Дозировки, однако, можно изменять в зависимости от того, что требуется пациенту, тяжести состояния, по поводу которого осуществляется лечение, и используемого соединения. Можно использовать как ежедневное введение, так и периодическое введение. Обычно дозу будет регулировать лечащий врач на основе характеристик пациента, его/ее состояния и желательного терапевтического эффекта.
Предпочтительно, указанные композиции имеют вид дозированных лекарственных форм, таких как таблетки, пилюли, капсулы, порошки, гранулы, стерильные растворы или суспензии для парентерального введения, аэрозоли или жидкие спреи с отмеренной дозой, капли, ампулы, устройства для самостоятельных инъекций или суппозитории; для перорального, парентерального, интравазального, сублингвального или ректального введения или для введения посредством ингаляции или инсуффляции. Альтернативно, композиция может быть представлена в форме, подходящей для введения один раз в неделю или один раз в месяц; например, нерастворимую соль активного соединения, такую как соль деканоат, можно адаптировать для создания депонированного препарата для внутримышечной инъекции. Для изготовления твердых композиций, таких как таблетки, главный активный ингредиент идеально смешивают с фармацевтическим носителем, например обычными ингредиентами для таблетирования, такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, двухкальциевый фосфат или смолы, и другими фармацевтическими разбавителями, например с водой, для получения предварительной твердой композиции, содержащей гомогенную смесь соединения по настоящему изобретению или его фармацевтически приемлемой соли. При упоминании указанных предварительных композиций как гомогенных, имеют в виду, что активный ингредиент идеально равномерно диспергирован в композиции, таким образом, что композицию можно легко разделить на равные по эффективности лекарственные формы, такие как таблетки, пилюли и капсулы. Указанную предварительную твердую композицию можно затем разделить на дозированные лекарственные формы таких типов, которые описаны выше, содержащие приблизительно от 0,01 до 1000 мг, предпочтительно приблизительно от 5 до 500 мг активного ингредиента по настоящему изобретению.
Таблетки или пилюли новой композиции можно снабжать оболочкой или изготавливать каким-либо другим образом, чтобы получать лекарственную форму, обладающую преимуществами пролонгированного действия. Например, таблетка или пилюля может иметь внутренний и внешний дозировочный компонент, причем последний в форме оболочки, покрывающей первый компонент. Указанные два компонента могут быть отделены друг от друга энтеросолюбильным слоем, который препятствует разрушению в желудке и позволяет внутреннему компоненту пройти интактным в двенадцатиперстную кишку или отсрочить его высвобождение. Для указанных энтеросолюбильных слоев или покрытий можно использовать разнообразные материалы, включая ряд полимерных кислот с такими материалами как шеллак, цетиловый спирт и ацетат целлюлозы.
Указанные жидкие формы, в которые можно выгодно включать новые композиции по настоящему изобретению для перорального введения или введения путем инъекции, включают водные растворы, сиропы с подходящими корригентами, водные или масляные суспензии и содержащие корригенты эмульсии на основе съедобных масел, таких как масло семян хлопчатника, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические средства. Подходящие диспергирующие или суспендирующие агенты для водных суспензий включают синтетические и натуральные смолы, такие как трагакант, аравийская камедь, альгинат, декстран, натрий-карбоксиметилцеллюлозу, метилцеллюлозу, поливинилпирролидон или желатин.
В том случае, если в процессе получения соединений по настоящему изобретению получаются смеси стереоизомеров, указанные изомеры можно разделить с помощью обычных методик, таких как препаративная хроматография. Соединения могут быть получены в рацемической форме, или можно получать отдельные энантиомеры с помощью как энантиоспецифичного синтеза, так и посредством разделения. Соединения можно, например, разделять на энантиомеры посредством стандартных методик, таких как образование диастереоизомерных пар путем образования соли с оптически активной кислотой, такой как (-)-ди-п-толуол-d-винная кислота или (+)-ди-п-толуол-l-винная кислота, с последующей фракционной кристаллизацией и регенерацией свободного основания. Соединения можно также разделять посредством образования диастереоизомерных сложных эфиров или амидов, с последующим хроматографическим разделением и удалением хирального вспомогательного агента. Альтернативно, соединения можно разделять с использованием хиральной колонки ВЭЖХ.
Во время любых процессов получения соединений по настоящему изобретению может быть необходимым и/или желательным защищать чувствительные или реакционные группы любых участвующих в процессах молекул. Этого можно достичь с помощью обычных защитных групп, таких как те, которые описаны в Protective Groups in Organic Chemistry, изд. J.F.W. McOmie, Plenum Press, 1973; и T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991, целиком включенных в настоящий документ в качестве ссылок. Защитные группы могут быть удалены на последующих подходящих стадиях, с использованием способов, известных специалистам.
Способ лечения состояний, модулированных дипептидилпептидазой IV и DPIV-подобными ферментами, описанный в настоящем изобретении, можно также осуществлять с использованием фармацевтической композиции, включающей одно или более соединений, как описано в настоящем документе, и фармацевтически приемлемый носитель. Фармацевтическая композиция может содержать приблизительно от 0,01 мг до 1000 мг, предпочтительно приблизительно от 5 до 500 мг, соединения (соединений), и может изготавливаться в любой форме, удобной для избранного способа введения. Носители включают необходимые и инертные фармацевтические наполнители, включая, без ограничения, связывающие агенты, суспендирующие агенты, смазывающие агенты, корригенты, подсластители, консерванты, красители и покрытия. Композиции, удобные для перорального введения, включают твердые формы, такие как пилюли, таблетки, мягкие желатиновые капсулы, капсулы (каждые из которых включают композиции с немедленным высвобождением, высвобождением через определенный период времени и с замедленным высвобождением), гранулы и порошки, а также жидкие формы, такие как растворы, сиропы, эликсиры, эмульсии и суспензии. Формы, пригодные для парентерального введения, включают стерильные растворы, эмульсии и суспензии.
Предпочтительно введение соединений по настоящему изобретению в виде однократной суточной дозы или суммарную суточную дозу можно вводить разделенными дозами два, три или четыре раза в день. Кроме того, соединения по настоящему изобретению можно вводить в интраназальной форме посредством местного применения подходящих интраназальных носителей или посредством накожных пластырей для чрескожного введения, хорошо известных специалистам. Для введения в форме системы чрескожной доставки введение дозы будет, разумеется, скорее непрерывным, чем прерывистым, а величину дозы будет необходимо соответственно модифицировать, чтобы получить желательные терапевтические эффекты.
Более предпочтительно, для перорального введения в форме таблетки или капсулы активный лекарственный компонент можно комбинировать с пероральным, нетоксичным, фармацевтически приемлемым носителем, таким как этанол, глицерин, вода и т.п. Помимо этого, если это желательно или необходимо, в смесь можно также включать подходящие связывающие агенты, смазывающие агенты, разрыхляющие агенты и окрашивающие агенты. Подходящие связывающие агенты включают, без ограничения, крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, натуральные и синтетические смолы, такие как аравийская камедь, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и другие соединения, известные специалистам.
Жидкие формы удобны в содержащих корригенты суспендирующих или диспергирующих агентах, таких как синтетические или натуральные смолы, например трагакант, аравийская камедь, метилцеллюлоза и т.п. Для парентерального введения желательны стерильные суспензии и растворы. В случае, когда желательно внутривенное введение, применяют изотоничные препараты, которые обычно содержат подходящие консерванты.
Соединения по настоящему изобретению можно также вводить в форме систем липосомной доставки, таких как малые однослойные пузырьки, большие однослойные пузырьки и многослойные пузырьки. Липосомы могут быть сформированы из ряда фосфолипидов, таких как холестерин, стериламин или фосфатидилхолины, с использованием способов, хорошо известных специалистам.
Соединения по настоящему изобретению можно также связывать с растворимыми полимерами в качестве нацеливаемых носителей лекарственных средств. Указанные полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатком пальмитоила. Кроме того, соединения по настоящему изобретению можно сопрягать с классом биоразлагаемых полимеров, пригодных для достижения контролируемого высвобождения лекарственного средства, например, с полимолочной кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианакрилатами и поперечно-связанными или амфипатическими блок-сополимерами гидрогелей.
Соединения по настоящему изобретению можно вводить в виде любой из указанных композиций и согласно схемам введения, принятым в данной области, когда бы ни потребовалось лечение упомянутых расстройств.
Суточная доза продуктов может варьироваться в широких пределах от 0,01 до 1000 мг на взрослого человека в день. Для перорального введения композиции предпочтительно изготавливают в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100, 150, 200, 250, 500 и 1000 миллиграмм активного ингредиента для симптоматического подбора дозы для пациента, лечение которого осуществляется. Эффективное количество лекарственного средства обычно поставляется при уровне доз приблизительно от 0,1 мг/кг до 300 мг/кг массы тела в день. Предпочтительно, указанные пределы составляют приблизительно от 1 до 50 мг/кг массы тела в день. Соединения можно вводить от 1 до 4 раз в день.
Оптимальные дозы, которые следует вводить, может легко определить специалист, и они будут варьироваться в зависимости от конкретного используемого соединения, способа введения, количества активного ингредиента, содержащегося в препарате, биодоступности в силу способа введения и степени развития болезненного состояния. Кроме того, при подборе дозировок обычно принимают во внимание факторы, связанные с пациентом, подвергаемым лечению, включая возраст, массу тела, характер питания пациента, а также время введения.
Соединения или композиции по настоящему изобретению можно принимать до приема пищи, во время приема пищи и после приема пищи.
При приеме во время еды соединения или композиции по настоящему изобретению можно смешивать с пищей или принимать в виде отдельной лекарственной формы, как описано выше.
Примеры
Пример 1: Синтез дипептидных соединений
1.1 Общий синтез соли изолейцилтиазолидина
Вос-защищенную аминокислоту BOC-Ile-OH помещают в этилацетат и порцию охлаждают приблизительно до -5°C. По каплям добавляют N-метилморфолин, по каплям добавляют хлорид пивалиновой кислоты (в лабораторных условиях) или неогексаноилхлорид (в масштабах пробного или промышленного производства) при постоянной температуре. Реакционную смесь перемешивают в течение нескольких минут для активации. По каплям один за другим добавляют N-метилморфолин (лабораторные условия) и гидрохлорид тиазолидина (лабораторные условия), добавляют тиазолидин (масштаб пробный или промышленный). Обработку в лаборатории производят обычным путем с использованием солевых растворов, в промышленных условиях серию очищают растворами NaOH и CH3COOH.
Удаление ВОС-защитной группы осуществляют с использованием HCl/диоксана (лабораторные условия) или H2SO4 (пробный и промышленный масштаб). В условиях лаборатории гидрохлорид кристаллизуют из EtOH/эфира.
В пробных и промышленных условиях свободный амин получают добавлением NaOH/NH3. Фумаровую кислоту растворяют в горячем этаноле, по каплям добавляют свободный амин и в осадок выпадает фумарат (Ile-Thia)2 (М = 520,71 гмоль-1). Анализ изомеров и энантиомеров осуществляют посредством электрофореза.
1.2 Синтез свободного основания глутаминилпирролидина
Ацилирование:
N-бензилоксикарбонилглутамин (2,02 г, 7,21 ммоль) растворяют в 35 мл ТГФ и доводят до температуры -15°C. В полученную смесь добавляют CAIBE (изобутилхлорформиат) (0,937 мл, 7,21 ммоль) и 4-метилморфолин (0,795 мл, 7,21 ммоль) и раствор перемешивают в течение 15 мин. Образование смешанного ангидрида контролируют посредством ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10°C добавляют пирролидин (0,596 мл, 7,21 ммоль). Смесь доводят до комнатной температуры и перемешивают в течение ночи.
Обработка:
Полученный осадок отфильтровывают и растворитель выпаривают. Полученное масло помещают в этилацетат (20 мл) и промывают насыщенным раствором гидросульфата натрия, а затем насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой отделяют, высушивают и выпаривают. Чистоту полученного продукта проверяют посредством ТСХ (элюент: CHCl3/MeOH: 9/1).
Выход: 1,18 г, воскоподобное твердое вещество.
Расщепление:
1,18 г полученного твердого Z-защищенного соединения растворяют в 40 мл абсолютного этанола. В раствор добавляют приблизительно 20 мг Pd на угле (10%, FLUKA) и суспензию встряхивают в атмосфере водорода в течение 3 ч. За ходом реакции наблюдают с помощью ТСХ (элюент: CHCl3/MeOH: 9/1). По окончании реакции защитную группу удаляют, чтобы получить свободное основание.
Выход: 99%.
Чистоту проверяют с помощью ТСХ: н-бутанол/АсОН/вода/этилацетат: 1/1/1/1, Rf = 0,4. Идентичность продукта реакции проверяют с помощью анализа ЯМР.
1.3 Синтез гидрохлорида глутаминилтиазолидина
Ацилирование:
N-трет-бутилоксикарбонилглутамин (2,0 г, 8,12 ммоль) растворяют в 5 мл ТГФ и доводят до температуры -15°C. В полученную смесь добавляют CAIBE (изобутилхлорформиат) (1,06 мл, 8,12 ммоль) и 4-метилморфолин (0,895 мл, 8,12 ммоль) и раствор перемешивают в течение 15 мин. Образование смешанного ангидрида контролируют посредством ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10°C добавляют еще один эквивалент 4-метилморфолина (0,895 мл, 8,12 ммоль) и гидрохлорида тиазолидина (1,02 мл, 8,12 ммоль). Смесь доводят до комнатной температуры и перемешивают в течение ночи.
Обработка:
Полученный осадок отфильтровывают и растворитель выпаривают. Полученное масло помещают в хлороформ (20 мл) и промывают насыщенным раствором гидросульфата натрия, а затем насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой отделяют, высушивают и выпаривают. Чистоту полученного продукта проверяют посредством ТСХ (элюент: CHCl3/MeOH: 9/1).
Выход: 1,64 г, твердое вещество.
Расщепление:
640 мг полученного твердого Вос-защищенного соединения растворяют в 3,1 мл ледяной HCl в диоксане (12,98 М, 20 эквивалентов) и оставляют на льду. За ходом реакции наблюдают с помощью ТСХ (элюент: CHCl3/MeOH: 9/1). По окончании реакции растворитель удаляют, а полученное масло помещают в метанол и снова выпаривают. Затем полученное масло высушивают над оксидом фосфора V и дважды растирают с диэтиловым эфиром. Чистоту проверяют посредством ВЭЖХ.
Выход: 0,265 г.
Чистоту проверяют с помощью ВЭЖХ. Идентичность продукта реакции проверяют с помощью анализа ЯМР.
1.4 Синтез гидрохлорида глутаминилпирролидина
Ацилирование:
N-трет-бутилоксикарбонилглутамин (3,0 г, 12,18 ммоль) растворяют в 7 мл ТГФ и доводят до температуры -15°C. В полученную смесь добавляют CAIBE (изобутилхлорформиат) (1,6 мл, 12,18 ммоль) и 4-метилморфолин (1,3 мл, 12,18 ммоль) и раствор перемешивают в течение 15 мин. Образование смешанного ангидрида контролируют посредством ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10°C добавляют 1 эквивалент пирролидина (1,0 мл, 12,18 мл). Смесь доводят до комнатной температуры и перемешивают в течение ночи.
Обработка:
Полученный осадок отфильтровывают и растворитель выпаривают. Полученное масло помещают в хлороформ (20 мл) и промывают насыщенным раствором гидросульфата натрия, а затем насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой отделяют, высушивают и выпаривают. Чистоту полученного продукта проверяют посредством ТСХ (элюент: CHCl3/MeOH: 9/1).
Выход: 2,7 г твердого вещества.
Расщепление:
2,7 г полученного твердого вещества растворяют в 13,0 мл ледяной HCl в диоксане (12,98 М, 20 эквивалентов) и оставляют на льду. За ходом реакции наблюдают с помощью ТСХ (элюент: CHCl3/MeOH: 9/1). По окончании реакции растворитель удаляют, а полученное масло помещают в метанол и снова выпаривают. Затем полученное масло высушивают над оксидом фосфора V и дважды растирают с диэтиловым эфиром.
Выход: 980 мг.
Чистоту проверяют с помощью ВЭЖХ. Идентичность продукта реакции проверяют с помощью анализа ЯМР.
Пример 2: Изучение химических свойств отобранных дипептидных соединений
2.1 Определение температуры плавления
Температуру плавления определяли с помощью микроскопа Kofler с нагревающейся платформой от Leica Aktiengesellschaft, величины не корректировали, или на аппарате DSC (Heumann-Pharma).
2.2 Оптическое вращение
Величины вращения фиксировали на различных длинах волн на "Polarimeter 341" или выше, от компании Perkin-Elmer.
2.3 Условия измерения для масс-спектроскопии
Масс-спектры фиксировали посредством электрораспыляющей ионизации (ESI) на "API 165" или "API 365" от компании PE Sciex. Измерения осуществляли с приблизительной концентрацией с=10 мкг/мл, вещество помещали в МеОН/Н2О 50:50, 0,1% НСО2Н, проводили с помощью распылительного насоса (20 мкл/мин). Измерения производили в положительном режиме [M+H]+, вольтаж ESI составлял U = 5600 V.
2.4 Результаты
2.4.1 Изучение фумарата изолейцилтиазолидина (изомеров)
Данные ЯМР и ВЭЖХ подтверждают идентичность изучавшегося вещества.
2.4.2. Изучение других солей изолейцилтиазолидина
Пример 3: Синтез трипептидов Xaa-Pro-Yaa
Все синтезы осуществляют на синтезаторе пептидов SP 650 (Labortec AG) с использованием стратегии Fmoc/tBu. Защищенные аминокислоты приобретают у компании Novabiochem или Bachem. Трифторуксусную кислоту (TFA) приобретают у компании Merck, триизопропилсилан (TIS) приобретают у компании Fluka.
С предварительно нагруженной Fmoc-Yaa-Wang смолы (2,8 г/уровень замещения 0,57 ммоль/г) снимают защиту с использованием 20% пиперидина/N,N-диметилформамида (DMF). После промывания DMF раствор 2 экв. (1,1 г) Fmoc-Pro-ОН растворяют в DMF (12 мл растворителя на грамм смолы). Добавляют 1,04 г тетрафторбората 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU) и 4 экв. 1,11 мл N,N-дизопропилэтиламина (DIEA) и помещают в реакционный сосуд. Смесь встряхивают при комнатной температуре в течение 20 минут. Затем цикл связывания повторяют. Затем промывают DMF, дихлорметаном, изопропанолом и диэтиловым эфиром и полученную Fmoc-Pro-Ile-Wang смолу высушивают, а затем делят на 6 частей перед присоединением последнего аминокислотного производного.
Защитную группу Fmoc удаляют, как описано выше. После этого 0,54 ммоль Вос-аминокислоты, 0,54 ммоль TBTU и 0,108 ммоль DIEA в DMF встряхивают в течение 20 мин. Цикл связывания повторяют. В конце пептидную смолу промывают и высушивают, как описано выше.
Пептид отщепляют от смолы в течение 2,5 ч с использованием смеси трифторуксусной кислоты (TFA), содержащей следующие скавенджеры: TFA/H2O/триизопропилсилан (TIS) = 9,5/0,25/0,25.
Выход сырых пептидов составляет в среднем 80-90%. Сырой пептид очищают посредством ВЭЖХ на колонке Nucleosil C18 (7 мкм, 250×21,20 мм, 100 А), с использованием линейного градиента 0,1% TFA/H2O с возрастающей концентрацией 0,1% TFA/ацетонитрила (от 5% до 65% за 40 мин) при скорости 6 мл/мин.
Чистый пептид получают лиофильной сушкой, идентифицируют с помощью электрораспыляющей масс-спектрометрии и анализа ВЭЖХ.
3.1 Результаты - Идентификация трипептидов Xaa-Pro-Yaa после химического синтеза
2 Условия ВЭЖХ с ОФ:
колонка: LiChrospher 100 RP 18 (5 мкм), 125 х 4 мм
прием (У/Ф): 14 нм
градиентная система: цетонитрил (ACN)/H2O (0,1% TFA), от 5% до 50% ACN в течение 15 мин
поток: мл/мин
k'= (tr-t0)/t0
t0 = 1,16 мин
трет-бутил-Gly определяется как:
Ser(Bzl) и Ser(Р) определяются как бензилсерин и фосфорилсерин, соответственно.
Tyr(Р) определяется как фосфорилтирозин.
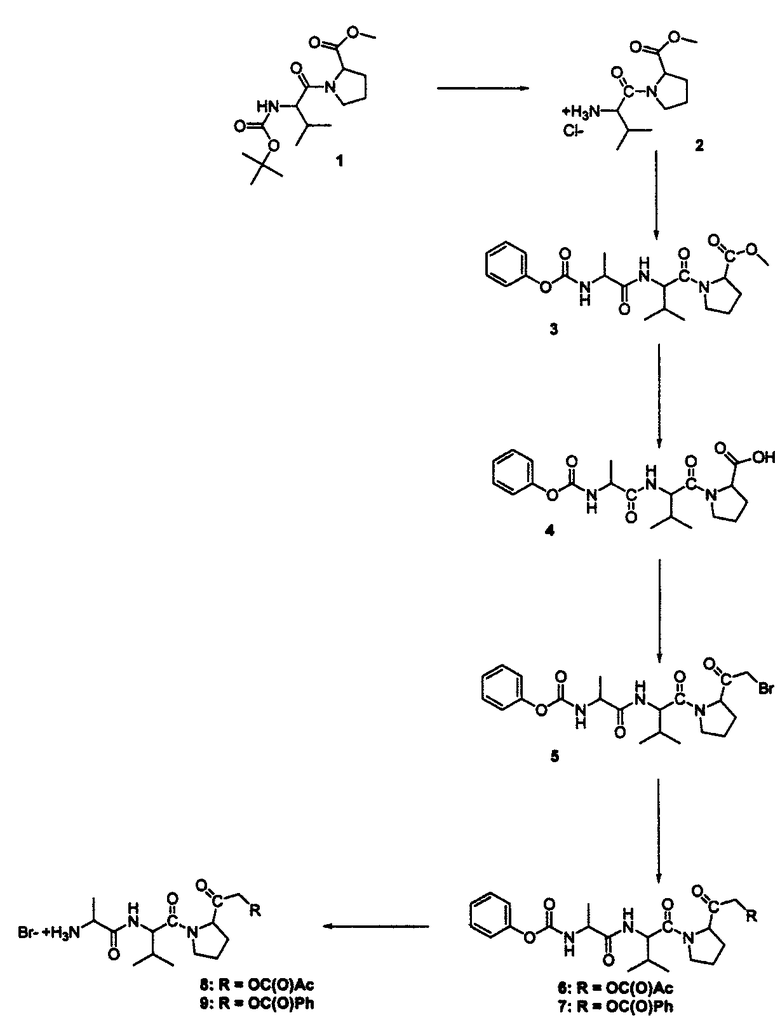
Пример 4: Синтез пептидилкетонов
H-Val-Pro-OMe·HCl 2
Boc-Val-OH (3,00 г, 13,8 ммоль) растворяют в 10 мл сухого ТГФ и охлаждают до -15°C. К смеси добавляют CAIBE (1,80 мл, 13,8 ммоль) и NMM (1,52 мл, 13,8 ммоль) и раствор перемешивают до окончания образования смешанного ангидрида. Затем температуру смеси доводят до -10°C и добавляют NMM (1,52 мл, 13,8 ммоль), а затем H-Pro-OMe·HCl (2,29 г, 13,8 ммоль). Смесь достигает комнатной температуры, после чего ее оставляют на ночь. После удаления растворителя и обычной обработки полученный сложный эфир 1берут в работу без дополнительного изучения его свойств. Сложный эфир 1растворяют в HCl/HOAc ( 5 мл, 6 н) и оставляют при 0°C до окончания удаления группы Вос. Затем растворитель удаляли и на полученное масло действовали диэтиловым эфиром с получением твердого вещества белого цвета 2.
Выход: 2,5 г, 80%.
Z-Ala-Val-Pro-OMe (3)
Z-Ala-ОН (3,5 г, 15,7 ммоль) и 2 (4,18 г, 15,7 ммоль) обрабатывают как описано выше для 1, с получением твердого вещества белого цвета 3.
Выход: 4,2 г, 64%.
Z-Ala-Val-Pro-OH (4)
Вещество 3(4,2 г, 9,6 ммоль) растворяют в 30 мл смеси вода/ацетон (1/5 об./об.) и добавляют 11,6 мл NaOH (1 н.). По окончании реакции органический растворитель удаляют выпариванием и полученный раствор разбавляют 15 мл раствора NaHCO3 (насыщенного). Затем смесь трижды экстрагируют 10 мл этилового эфира уксусной кислоты. Затем рН раствора доводят до 2 добавлением HCl (15% в воде). Полученную смесь трижды экстрагируют 30 мл этилового эфира уксусной кислоты. Органический слой отделяют и трижды промывают насыщенным солевым раствором, высушивают (Na2SO4) и выпаривают.
Выход: 3,5 г, 87%.
Z-Ala-Val-Pro-CH2-Br (5)
Соединение 4(2,00 г, 4,76 ммоль) растворяют в 15 мл сухого ТГФ и превращают в смешанный ангидрид (см. соединение 1) с использованием CAIBE (0,623 мл, 4,76 ммоль) и NMM (0,525 мл, 4,76 ммоль). Образовавшийся осадок отфильтровывают и охлаждают до -15°C. Затем в раствор по каплям добавляют диазометан (23,8 ммоль в 30 мл эфира) в атмосфере аргона. Смесь оставляют на 1 ч при 0°C, после чего добавляют 1,27 мл HBr (33% в АсОН) и раствор перемешивают в течение 30 мин при комнатной температуре. Затем добавляют 70 мл эфира и смесь промывают 20 мл воды. Органический слой отделяют, высушивают (Na2SO4) и выпаривают.
Выход (сырой): 1,8 г, 80%.
Z-защищенные ацилоксиметиленкетоны
Кислоту (2 экв.) растворяют в DMF и добавляют эквимолярное количество KF. Суспензию оставляют перемешиваться при комнатной температуре в течение 1 часа. Затем добавляют бромметиленовый компонент (1 экв.) и раствор оставляют перемешиваться в течение ночи. Затем растворитель удаляют в условиях вакуума и полученное масло растворяют в хлороформе и промывают насыщенным солевым раствором. Затем органический слой отделяют, высушивают (Na2SO4) и растворитель удаляют. Продукт очищают колоночной хроматографией с использованием силикагеля и смеси гептан/хлороформ.
Z-Ala-Val-Pro-CH2О-С(О)-СН3 (6)
Уксусная кислота (230 мкл, 4,02 ммоль), KF (0,234 г, 4,02 ммоль), 5 (1,00 г, 2,01 ммоль).
Выход: 0,351 г, 36%.
Z-Ala-Val-Pro-CH2О-С(О)-Ph (7)
Бензойная кислота (0,275 г, 2,25 ммоль), KF (0,131 мг, 2,25 ммоль), 5 (0,56 г, 1,13 ммоль).
Выход: 0,34 г, 56%.
Снятие защиты
Z-защищенное соединение растворяют в HBr/AcOH и перемешивают. По окончании реакции добавляют эфир, образовавшийся белый осадок отфильтровывают и высушивают.
Н-Ala-Val-Pro-CH2О-С(О)-СН3·HBr (8)
6 (0,351 г, 0,73 ммоль)
Выход: 0,252 г, 98%.
Н-Ala-Val-Pro-CH2О-С(О)Ph·HBr (9)
7 (0,34 г, 0,63 ммоль)
Выход: 0,251 г, 99%.
Пример 5: Синтез циклоалкилкетонов

Вос-изолейциналь (2)
Оксалилхлорид (714 мкл, 8,28 ммоль) растворяют в 10 мл сухого дихлорметана и доводят температуру раствора до -78°C. Затем по каплям добавляют DMSO (817 мкл, 8,28 ммоль). Раствор перемешивают в течение 20 мин при -78°C. Затем добавляют соединение 1 (1,00 г, 4,6 ммоль) и смесь перемешивают в течение 20 мин. Затем добавляют TEA (2,58 мл, 18,4 ммоль) и смесь оставляют стоять до достижения ею комнатной температуры. Смесь разбавляют смесью гексан/этилацетат (2/1 об./об.) и добавляют 10 мл HCl (10% в воде). Органический слой отделяют и водную фазу экстрагируют 20 мл метиленхлорида. Все органические слои собирают и промывают последовательно насыщенным солевым раствором и водой, затем высушивают. Продукт очищают колоночной хроматографией с использованием силикагеля и смеси гептан/хлороформ.
Выход: 0,52 г, 52%.
трет-бутил-N-1-[циклопентил(гидрокси)метил]-2-метилбутилкарбамат 3
Соединение 2 (0,52 г, 2,42 ммоль) растворяют в 10 мл сухого ТГФ и охлаждают до 0°C. Затем добавляют циклопентилмагнийбромид (1,45 мл 2 М раствора). По окончании реакции добавляют воду (2 мл) и раствор нейтрализовывают добавлением водного раствора HCl. Затем добавляют метиленхлорид и органический слой отделяют и высушивают (Na2SO4). После выпаривания полученное масло используют без дополнительного изучения его свойств.
трет-бутил-N-[1-(циклопентилкарбонил)-2-метилбутил]карбамат (4)
Соединение 3 (0,61 г, 2,15 ммоль) обрабатывают, как соединение 1. Оксалилхлорид (333 мкл, 3,87 ммоль), DMSO (382 мкл, 5,37 ммоль), ТЕА (1,2 мл, 8,59 ммоль).
Выход: 0,180 г, 30%
Хлорид 1-циклопентил-3-метил-1-оксо-2-пентанаминия (5)
Соединение 4 (0,18 г, 0,63 ммоль) растворяют в 2 мл HCl (7 н. в диоксане). По окончании реакции растворитель удаляют, и полученное масло очищают колоночной хроматографией на силикагеле с использованием градиента хлороформ/метанол/вода. Полученное масло растирают с эфиром.
Выход: 0,060 г, 54%
Пример 6: Синтез ингибиторов DPIV с модифицированной боковой цепью
6.1 Синтез Вос-глутамилтиазолидина (Boc-Glu-Thia)
Взаимодействие Boc-Glu(OMe)-OH с Thia·HCl согласно способу В (способы см. в разделе 6.4), гидролиз Boc-Glu(OMe)-Thia согласно способу G.
6.1.1 Аналитические данные для Boc-Glu-Thia
Выход
т. пл.
Конц. Растворитель
B+G
62%
115-118°C
c = 1 метанол
H:6,96/6,82
N:8,80/8,59
Система А: хлороформ/метанол 90:10
Система В: бензол/ацетон/уксусная кислота 25:10:0,5
Система С: н-бутанол/ЕА/ уксусная кислота/Н2О 1:1:1:1
2 Условия разделения ВЭЖХ
Колонка: Nucleosil С-18, 7 мкм, 250 мм х 21 мм
Элюент: изократный, 40% ACN/вода/0,1% TFA
Скорость потока: 6 мл/мин
λ = 220 нм
6.2 Вос-глутамилтиазолидины с модифицированной боковой цепью
Boc-Glu-Thia модифицируют по γ-карбоновой кислоте путем введения радикалов различного размера. Радикалы присоединяют через их аминогруппу путем образования амидной связи с γ-карбоновой кислотой с помощью ряда способов связывания, в зависимости от используемого радикала. Следующие аминокомпоненты присоединяют к Boc-Glu-Thia с использованием заявленных способов:
В двух случаях очистка продуктов реакции отличается от общего описания синтеза.
Boc-Glu(Gly5)-Thia
Продукт выпадает в осадок из смеси уже при перемешивании в течение ночи; впоследствии его отфильтровывают и промывают 0,1 н. HCl и обильными количествами воды, а затем высушивают над Р4О10 в условиях вакуума.
Boc-Glu(PEG)-Thia
В противоположность общей процедуре, исходные материалы для синтеза растворяют в 500-кратном избытке DMF. По окончании реакции DMF полностью удаляют в условиях вакуума, а остаток растворяют в большом количестве метанола. После того как наливают эфир для образования верхнего слоя, продукт выпадает в осадок вместе с непрореагировавшим PEG. Тонкую очистку осуществляют разделением с помощью препаративной ВЭЖХ на колонке для гель-фильтрации (Pharmazia, Sephadex G-25, 90 мкм, 260 мм - 100 мм).
Условия разделения: элюент: вода; скорость потока: 5 мл/мин; λ = 220 нм.
6.2.2 Данные синтеза для Boc-глутамилтиазолидинов с модифицированной боковой цепью
Mr
Выход
ТСХ/Rf/
система
т. пл.
Конц. Растворитель
Rt [мин]/ система
49%
H:6,38
N:14,31
86%
0,09/C разлож.
с 202°C
H:6,18/6,11
N:16,24/16,56
52-53°C
Колонка: Nucleosil С-18, 7 мкм, 250 мм х 21 мм
Элюент: ACN/вода/0,1% TFA
Скорость потока: 6 мл/мин
λ = 220 нм
6.3 Глутамилтиазолидины с модифицированной боковой цепью
N-концевые Вос-защитные группы отщепляют от соединений, описанных в таблице 6.2.2, с использованием способа F. Вещества, модифицированные производными Gly, очищенные препаративной ВЭЖХ, присутствуют в виде трифторацетатов. H-Glu(PEG)-Thia очищают на гель-фильтрационной колонке так же, как его Вос-защищенный предшественник.
6.3.1 Данные синтеза для глутамилтиазолидинов с модифицированной боковой цепью
Mr
Выход
ТСХ/Rf/
система
т. пл.
Конц. Растворитель
Rt [мин]/ система
94%
0,32/C
91-94°C
с = 1
метанол
H:4,80/4,78
N:13,91/13,43
98%
0,25/C
105-107°C
H:4,90/4,79
N:15,88/15,39
Колонка: Nucleosil С-18, 7 мкм, 250 мм х 21 мм
Элюент: ACN/вода/0,1% TFA
Градиент: 20% ACN → 90% ACN в течение 30 мин.
Скорость потока: 6 мл/мин
λ = 220 нм
не опр. - не определялось или не поддается определению
6.4 Общие процедуры синтеза
Способ А: Присоединение через пептидную связь способом смешанного ангидрида, с использованием CFIBE в качестве активирующего реагента
10 ммоль N-терминально защищенной аминокислоты или пептида растворяют в 20 мл абсолютного ТГФ. Раствор охлаждают до -15°C ± 2°C. В каждом случае при перемешивании добавляют последовательно 10 ммоль N-MM и 10 ммоль изобутилового эфира хлормуравьиной кислоты, при строгом соблюдении указанного температурного режима. Приблизительно через 6 мин добавляют 10 ммоль аминокомпонента. В случае, когда аминокомпонент представляет собой соль, к реакционной смеси добавляют еще 10 ммоль N-MM. Реакционную смесь затем перемешивают в холодном состоянии в течение 2 ч и в течение ночи при комнатной температуре.
Реакционную смесь концентрируют на роторном испарителе, помещают в ЕА, промывают 5% раствором KH2SO4, насыщенным раствором NaHCO3 и насыщенным раствором NaCl и высушивают над Na2SO4. После удаления растворителя в условиях вакуума соединение перекристаллизовывают из ЕА/пентана.
Способ В: Присоединение через пептидную связь способом смешанного ангидрида, с использованием хлорангидрида пивалиновой кислоты в качестве активирующего реагента
10 ммоль N-терминально защищенной аминокислоты или пептида растворяют в 20 мл абсолютного ТГФ. Раствор охлаждают до 0°C. В каждом случае при перемешивании добавляют последовательно 10 ммоль N-MM и 10 ммоль хлорида пивалиновой кислоты, при строгом соблюдении указанного температурного режима. Приблизительно через 6 мин смесь охлаждают до -15°C и по достижении более низкой температуры добавляют 10 ммоль аминокомпонента. В случае, когда аминокомпонент представляет собой соль, к реакционной смеси добавляют еще 10 ммоль N-MM. Реакционную смесь затем перемешивают в холодном состоянии в течение 2 ч и в течение ночи при комнатной температуре.
Дальнейшую обработку осуществляют, как в способе А.
Способ С: Присоединение через пептидную связь, с использованием TBTU в качестве активирующего реагента
10 ммоль N-терминально защищенной аминокислоты или пептида и 10 ммоль С-терминально защищенного аминокомпонента растворяют в 20 мл абсолютного DMF. Раствор охлаждают до 0°C. В каждом случае при перемешивании добавляют последовательно 10 ммоль DIPEA и 10 ммоль TBTU. Реакционную смесь перемешивают в течение одного часа при 0°C, и в течение ночи при комнатной температуре. DMF полностью удаляют в условиях вакуума и дальнейшую обработку продукта осуществляют, как в способе А.
Способ D: Синтез активного сложного эфира (N-гидроксисукцинимидного эфира)
10 ммоль N-терминально защищенной аминокислоты или пептида и 10 ммоль гидроксисукцинимида растворяют в 20 мл абсолютного THF. Раствор охлаждают до 0°C и при перемешивании добавляют 10 ммоль дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение еще 2 ч при 0°C, а затем в течение ночи - при комнатной температуре. Полученную N,N'-дициклогексилмочевину отфильтровывают и растворитель удаляют в условиях вакуума, а оставшийся продукт перекристаллизовывают из ЕА/пентана.
Способ Е: Присоединение через амидную связь с использованием N-гидроксисукцинимидных эфиров
10 ммоль С-терминально незащищенного аминокомпонента вводят в раствор NaHCO3 (20 ммоль в 20 мл воды). При комнатной температуре и при перемешивании, медленно, по каплям добавляют 10 ммоль N-терминально защищенного N-гидроксисукцинимидного эфира, растворенного в 10 мл диоксана. Перемешивание реакционной смеси продолжают в течение ночи и растворитель затем удаляют в условиях вакуума.
Дальнейшую обработку осуществляют как в способе А.
Способ F: Отщепление Вос-защитной группы
3 мл 1,1 н. HCl/ледяной уксусной кислоты (способ F1) или 3 мл 1,1 н. HCl/диоксана (способ F2) или 3 мл 50% TFA в DCM (способ F3) добавляют к 1 ммоль пирролидина, тиазолидина или пептида с Boc-защищенной аминокислотой. Отщепление при КТ отслеживают с помощью ТСХ. По окончании реакции (приблизительно 2 ч) соединение осаждают в форме гидрохлорида с использованием абсолютного диэтилового эфира и выделяют отсасыванием и высушивают над Р4О10 в условиях вакуума. С использованием метанола/эфира продукт перекристаллизовывают или повторно осаждают.
Способ G: Гидролиз
1 ммоль метилового эфира пептида растворяют в 10 мл ацетона и 11 мл 0,1 М раствора NaOH и перемешивают при комнатной температуре. За ходом гидролиза следят с помощью ТСХ. По окончании реакции ацетон удаляют в условиях вакуума. Оставшийся водный раствор подкисляют концентрированным раствором KH2SO4 до рН 2-3. Продукт затем несколько раз экстрагируют ЕА; объединенные фракции этилацетата промывают насыщенным раствором NaCl и высушивают над NaSO4, а растворитель удаляют в условиях вакуума. Осуществляют перекристаллизацию из ЕА/пентана.
Пример 7: Определение Ki
Для определения Ki используют дипептидилпептидазу IV из почки свиньи, обладающую специфичной активностью против глицилпролил-4-нитроанилина 37,5 ЕД/мг и концентрацию фермента 1,41 мг/мл в маточном растворе.
Смесь для анализа:
100 мкл испытуемого соединения с концентрацией в пределах 1·10-5 М - 1·10-8 М, соответственно, смешивают с 50 мкл глицилпролил-4-нитроанилина с различными концентрациями (0,4 мМ, 0,2 мМ, 0,1 мМ, 0,05 мМ) и 100 мкл HEPES (40 мМ, рН 7,6; ионная концентрация = 0,125). Смесь для анализа предварительно инкубируют при 30°C в течение 30 мин. После предварительного инкубирования добавляют 20 мкл DPIV (в разбавлении 1:600) и осуществляют измерение появления желтого окрашивания вследствие высвобождения 4-нитроанилина при 30°C и λ = 405 нм в течение 10 мин, с использованием планшет-ридера (HTS7000 plus, Applied Biosystems, Weiterstadt, Германия).
Величины Ki рассчитывали с помощью программы Graphit, версия 4.0.13, 4.0.13 и 4.0.15 (Erithacus Software, Ltd, UK).
7.1 Результаты - величины Ki ингибирования DPIV
трет-бутил-Gly определяется как:
Ser(Bzl) и Ser(Р) определяются как бензилсерин и фосфорилсерин, соответственно.
Tyr(Р) определяется как фосфорилтирозин.
Пример 8: Определение величин IC50
10 мкл маточного раствора ингибитора смешивали с 100 мкл буфера (HEPES рН 7,6) и 50 мкл субстрата (Gly-Pro-pNA, конечная концентрация 0,4 мМ) и предварительно инкубировали при 30°C. Реакцию начинали добавлением 20 мкл очищенной свиной DPIV. Образование продукта pNA измеряли на 405 нм в течение 10 мин с использованием планшет-ридера HTS 7000Plus (Perkin Elmer) и рассчитывали углы наклона. Конечные концентрации ингибитора составляли от 1 мМ до 30 нМ. Для расчета IC50 использовали программу GraFit 4.0.13 (Erithacus Software).
8.1 Результаты - определение величин IC50
трет-бутил-Gly определяется как:
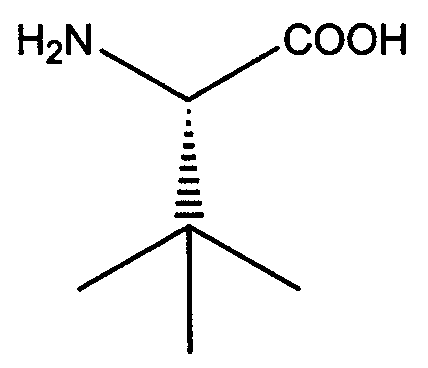
Ser(Bzl) и Ser(Р) определяются как бензилсерин и фосфорилсерин, соответственно.
Tyr(Р) определяется как фосфорилтирозин.
Пример 9: Ингибирование DPIV-подобных ферментов - дипептидилпептидазы II
DP II (3.4.14.2) высвобождает N-концевые дипептиды из олигопептидов, если N-конец не протонирован (McDonald, J.K., Ellis, S. & Reilly, T.J., 1966, J. Biol. Chem., 241, 1494-1501). Pro и Ala в положении Р1 являются предпочтительными остатками. Ферментативная активность описана как DPIV-подобная активность, но DP II имела оптимум при кислых значениях рН. Использованный фермент был выделен из почки свиньи.
Анализ:
100 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1·10-4 М - 5·10-8 М смешивали со 100 мкл буферного раствора (40 мМ HEPES, рН 7,6, 0,015 Brij, 1 мМ DTT), 50 мкл раствора лизилаланиламинометилкумарина (5 мМ) и 20 мкл свиной DP II (разведенной в 250 раз буферным раствором). Флуоресценцию измеряли при 30°C и λexciatation = 380 нм, λemission = 465 нм в течение 25 мин, с использованием планшет-ридера (HTS7000 plus, Applied Biosystems, Weiterstadt, Германия). Величины Ki рассчитывали с помощью программы Graphit 4.0.15 (Erithacus Software, Ltd, UK) и определили как Ki = 8,52·10-5 М ± 6,33·10-6 М для глутаминилпирролидина и Ki = 1,07·10-5 М ± 3,81·10-7 М для глутаминилтиазолидина.
Пример 10: Перекрестно-реагирующие ферменты
Глутаминилпирролидин и глутаминилтиазолидин изучали на предмет перекрестно-реагирующей активности против дипептидилпептидазы I, пролилолигопептидазы и пролидазы.
Дипептидилпептидаза I (DP I, катепсин С):
DP I или катепсин С представляет собой лизосомальную цистеиновую протеиназу, которая отщепляет дипептиды от N-конца своих субстратов (Gutman, H.R. & Fruton, J.S., 1948, J. Biol. Chem., 174, 851-858). Ее классифицируют как цистеиновую протеиназу. Использовавшийся фермент приобрели у компании Qiagen (Qiagen GmbH, Hilden, Германия). Для того чтобы получить полностью активный фермент, фермент разбавляли в 1000 раз буфером MES с рН 5,6 (40 мМ MES, 4 мМ DTT, 4 мМ KCl, 2 мМ EDTA, 0,015% Brij) и предварительно инкубировали в течение 30 мин при 30°C.
Анализ:
50 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1·10-5 М - 1·10-7 М смешивали с 110 мкл буферно-ферментной смеси. Смесь для анализа предварительно инкубировали при 30°С в течение 15 мин. После предварительной инкубации добавляли 100 мкл гистидилсерилнитроанилина (2·10-5 М) и осуществляли измерение появления желтого окрашивания вследствие высвобождения β-нитроанилина при 30°С и λexciatation = 380 нм, λemission = 465 нм в течение 10 мин, с использованием планшет-ридера (HTS7000 plus, Applied Biosystems, Weiterstadt, Германия).
Для расчета величин IC50 использовали программу Graphit 4.0.15 (Erithacus Software, Ltd., UK). Ингибирования ферментативной активности DP I глутаминилпирролидином или глутаминилтиазолидином выявлено не было.
Пролилолигопептидаза (РОР)
Пролилолигопептидаза (ЕС 3.4.21.26) представляет собой эндопротеазу серинового типа, которая расщепляет пептиды на N-концевой части связи Xaa-Pro (Walter, R., Shlank, H., Glass, J.D., Schwartz, I.L. & Kerenyl, T.D., 1971, Science, 173, 827-829). Субстратами являются пептиды с молекулярной массой до 3000 Да. Использованный фермент представлял собой рекомбинантную человеческую пролилолигопептидазу. Рекомбинантную экспрессию осуществляли на E. coli в обычных условиях, как описано повсеместно в научной литературе.
Анализ:
100 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 1·10-4 М - 5·10-8 М смешивали с 100 мкл буферного раствора (40 мМ HEPES, рН 7,6, 0,015 Brij, 1 мМ DTT) и 20 мкл раствора РОР. Смесь для анализа предварительно инкубировали при 30°C в течение 15 мин. После предварительной инкубации добавляли 50 мкл раствора глицилпролилпролил-4-нитроанилина (0,29 мМ) и осуществляли измерение появления желтого окрашивания вследствие высвобождения 4-нитроанилина при 30°C и λ = 405 нм в течение 10 мин, с использованием планшет-ридера (sunrise, Tecan, Crailsheim, Германия). Для расчета величин IC50 использовали программу Graphit 4.0.15 (Erithacus Software, Ltd., UK). Ингибирования активности РОР глутаминилпирролидином или глутаминилтиазолидином выявлено не было.
Пролидаза (X-Pro-дипептидаза)
Пролидаза (ЕС 3.4.13.9) была впервые описана (Bergmann, M. & Fruton, J.S., 1937, J. Biol. Chem. 189-202). Пролидаза высвобождает N-концевую аминокислоту из дипептидов Xaa-Pro и имеет оптимум действия при рН от 6 до 9.
Пролидазу из почки свиньи (ICN Biomedicals, Eschwege, Германия) растворяли (1 мг/мл) в буфере для анализа (20 мМ NH4(CH3COO)2, 3 мМ MnCl2, рН 7,6). Для получения полностью активного фермента раствор инкубировали при комнатной температуре в течение 60 мин.
Анализ:
450 мкл глутаминилпирролидина или глутаминилтиазолидина с концентрацией в пределах 5·10-3 М - 5·10-7 М смешивали с 500 мкл буферного раствора (20 мМ NH4(CH3COO)2, рН 7,6) и 250 мкл Ile-Pro-OH (0,5 мМ в смеси для анализа). Смесь для анализа предварительно инкубировали при 30°C в течение 5 мин. После предварительной инкубации добавляли 75 мкл раствора пролидазы (разведенной в буфере для анализа 1:10) и осуществляли измерение появления при 30°C и λ = 220 нм в течение 20 мин, с использованием фотометра UV/Vis, UV1 (Thermo Spectronic, Cambridge, UK).
Для расчета величин IC50 использовали программу Graphit 4.0.15 (Erithacus Software, Ltd., UK). Указанные величины были определены как IC50 > 3 мМ для глутаминилтиазолидина и как IC50 = 3,4·10-4 М ± 5,63·10-5 М для глутаминилпирролидина.
Пример 11: Определение DPIV-ингибирующей активности после внутрисосудистого и перорального введения крысам Wistar
Животные
Самцов крысы Wistar (Shoe: Wist(Sho)) с массой тела от 250 до 350 г приобретали у компании Tierzucht Schonwalde, Германия).
Условия содержания
Животных содержали в отдельных клетках в обычных условиях при контролируемой температуре (22 ± 2°C) и световом режиме 12 часов света/12 часов темноты (свет с 6 часов утра). Животные получали стандартный сухой корм (ssniff® Soest, Германия) и водопроводную воду, подкисленную HCl, ad libitum.
Катетеризация сонной артерии
После периода адаптации к условиям содержания в течение одной недели или более, в сонную артерию крыс Wistar помещали катетер под общим наркозом (и/п инъекция 0,25 мл/кг массы тела Rompun® [2%], BayerVital, Германия, и 0,5 мл/кг массы тела Ketamin 10, Atarost GmbH & Co., Twistringen, Германия). Животных оставляли для выздоровления на одну неделю. Катетеры промывали гепаринизированным физиологическим раствором (100 МЕ/мл) три раза в неделю. В случае плохой работы катетера у данного животного, катетеризировали сонную артерию на противоположной стороне. Через неделю восстановительного периода данное животное вновь включали в эксперимент. В случае плохой работы второго катетера данное животное исключали из эксперимента. Отбирали новое животное, и эксперименты продолжались по плану, с началом по меньшей мере через 7 дней после катетеризации.
Схема эксперимента
Крысам с нормальной работой катетера вводили плацебо (1 мл физиологического раствора, 0,154 моль/л) или испытуемое соединение пероральным или внутрисосудистым (интраартериальным) путем. После ночного воздержания от пищи отбирали 100 мкл образцы гепаринизированной артериальной крови в точках -30, -5 и 0 мин. Испытуемое вещество перед введением растворяли в 1,0 мл физиологического раствора (0,154 моль/л) и вводили в 0 мин перорально, через желудочный зонд (75 мм; Fine Science Tools, Heidelberg, Германия) или внутрисосудистым путем. В случае перорального введения через артериальный катетер вводили дополнительно 1 мл физиологического раствора. В случае интраартериального введения катетер немедленно промывали 30 мкл физиологического раствора и дополнительно вводили 1 мл физиологического раствора перорально через желудочный зонд.
После введения плацебо или испытуемых веществ отбирали образцы артериальной крови через 2,5, 5, 7,5, 10, 15, 20, 40, 60 и 120 мин из катетера, помещенного в сонную артерию, у нефиксированных и находящихся в сознании крыс. Все образцы крови собирали в охлажденные на льду пробирки Eppendorf (Eppendorf-Netheler-Hinz, Hamburg, Германия), содержавшие 10 мкл 1 М натрий-цитратного буфера (рН 3,0), для определения активности DPIV в плазме крови. Пробирки Eppendorf немедленно центрифугировали (12000 об./мин в течение 2 мин, на центрифуге Hettich Zentrifuge EBA 12, Tuttlingen, Германия). Фракции плазмы хранили на льду до проведения анализа или замораживали при -20°С до проведения анализа. Все образцы плазмы снабжали этикетками, содержавшими следующие сведения:
- Кодовый номер
- Номер животного
- Дата забора крови
- Время забора крови
Аналитические методы
Смесь для анализа для определения активности DPIV в плазме крови состояла из 80 мкл реагента и 20 мкл образца плазмы. Кинетическое измерение образования желтого продукта 4-нитроанилина из субстрата глицилпролил-4-нитроанилина осуществляли на 390 нм в течение 1 мин при 30°C после 2 мин предварительной инкубации при указанной температуре. Активность DPIV выражали в мЕД/мл.
Статистические методы
Статистические оценки и графики получали с помощью программы PRISM® 3.02 (GraphPad Software, Inc.). Все параметры анализировали в описательной манере, включая среднее и СО.
11.1 Результаты - ингибирование DPIV in vivo в tmax
Пример 12: Действие глутамилтиазолидинов с модифицированной боковой цепью как плохо транспортирующихся ингибиторов DPIV
Были синтезированы глутамилтиазолидины с модифицированной боковой цепью, имеющие структуру H-Glu(X)-Thia, в которых в качестве Х использовали полиэтиленгликоль или глициновые олигомеры с различной длиной цепи (см. способ А примера описания синтеза). Изучались характеристики связывания указанных производных и их транспортабельность транспортером пептидов РерТ1.
Неожиданно было установлено, что модификации боковой цепи изменяют характеристики связывания соединений с DPIV только в незначительной степени. Напротив, способность ингибиторов транспортироваться транспортером пептидов вследствие модификации боковой цепи резко понизилась.
Ингибиторы DPIV и DPIV-подобных ферментов с модифицированной боковой цепью, таким образом, хорошо подходят для достижения сайт-направленного ингибирования DPIV в организме.
12.1 Результаты: Транспортабельность отобранных ингибиторов DPIV
аминокислотные тиазолидины
H-Glu-Thia
1,1
35 ± 13
H-Glu(Gly3)-Thia
H-Glu(Gly5)-Thia
H-Glu(PEG)-Thia
8,54
> 10
> 10
не опр.3
не опр.3
не опр.3
2 Транспортные характеристики на РерТ1-экспрессирующих ооцитах X. leavis - посредством методики двухэлектродного зажима, I = внутренние токи, генерируемые транспортом
Пример 13: Изучение адгезии злокачественных клеток in vivo
С помощью нового анализа адгезии in vivo, в котором используются преимущества меченных прижизненным красителем опухолевых клеток и их выявления в ткани-мишени in situ (von Horsten et al., 2000), в данном примере изучают, отличается ли адгезия in vivo опухолевых клеток MADB106 у леченных крыс дикого типа F344 и сублиний F344 c мутациями гена DPIV.
Животные, инъекция опухолевых клеток и обработка легких
Сублинии F344USA, F344JAP и F344GER получали из питомника центрального вивария ганноверской медицинской школы, Германия. Все сублинии представляли собой одно поколение, содержавшееся в особых, лишенных патогенных микроорганизмов, помещениях при 25°C с режимом освещения 12 ч света - 12 часов темноты (свет с 7 ч утра), с неограниченным доступом к пище и воде. Точное количество животных, взятых в эксперимент, указывается величиной F и составляет по меньшей мере четыре животных на каждое условие и точку времени.
Культура клеток, инъекция опухолевых клеток, вскрытие животных и иммуногистохимическое исследование осуществляли, как описано ранее (von Horsten et al., 2000). Кратко, опухолевые клетки MADB106, полученные во время лог-фазы опухолевого роста, инъецировали в латеральную вену хвоста и легкие удаляли после этого через различные промежутки времени. Для количественного определения опухолевых клеток in situ на ранних стадиях после инъекции (30 мин), клетки окрашивали прижизненным красителем с использованием производного флуоресцеина сукцинимидилового эфира диацетата 5-(и -6)-карбоксифлуоресцеина (CFSE) перед инъекцией. Для количественного определения колоний на поверхности легких на более поздних стадиях (2 недели после инокуляции опухолевых клеток), в вырезанные единым блоком легкие и сердце инъецировали 8 мл раствора Bouin (72% насыщенный раствор пикриновой кислоты, 23: формальдегид и 5% ледяная уксусная кислота) и фиксировали тем же раствором до того, как подсчитывать узелки на поверхности легких (см. ниже).
Эксперименты
Были проведены три эксперимента:
Влияние однократной инъекции фумарата изолейцилтиазолидина (2 мг в/в + фумарат изолейцилтиазолидина 2 мг и/п) на колонизацию опухоли в легких у крыс F344USA дикого типа;
Влияние однократной инъекции изолейцилцианопирролидина (0,1 мг в/в + 0,1 мг и/п) на адгезию опухолевых клеток в легких у крыс F344USA дикого типа;
Влияние однократной инъекции фумарата валилпирролидина (0,1 мг в/в + 0,1 мг и/п) на адгезию опухолевых клеток в легких у крыс F344USA дикого типа.
Иммуногистохимическое исследование меченных CFSE опухолевых клеток в легких
Окраска с использованием иммунной метки меченных CFSE опухолевых клеток MADB106 производилась посредством характеризации c помощью mAb внутриклеточного антигена CFSE (анти-CFSE; mAb DE1, Boehringer, Mannheim, Германия; мышь, 1:100). Для иммуногистохимического исследования выполняли одно или два последовательных окрашивания АРААР, как было описано ранее (von Horsten et al., 2000). В исследование включали контрольные срезы, в которых одно или оба первичных антитела были опущены.
Количественное определение опухолевых мишеней: in vivo/in situ анализ клеточной адгезии
Прижизненный краситель (карбоксифлуоресцеин, CFSE), которым метили опухолевые клетки MADB, позволяет определить количество опухолевых клеток и клеток NK в толстых срезах легочной ткани посредством стереологии in situ (von Horsten et al., 2000). В настоящем исследовании заявители делали тонкие срезы (8 мкм) тех же легких (n = 10) и выполняли дополнительный подсчет с помощью микроскопа посредством визуального анализа DE1-положительных клеток. Это было сделано для того, чтобы еще более упростить ранее утвержденную технику стереологического количественного определения. Таким образом, в настоящем исследовании оценку DE1-пложительных опухолевых клеток в легочной ткани от различных сублиний через 30 мин после инокуляции опухоли выполняли, используя подход визуального анализа. Подсчитывались все меченные CFSE опухолевые клетки MADB106 и субпопуляции лейкоцитов в сетке линзы окуляра (Zeiss Kpl-W 12,5x; сетка 0,75 х 0,75 мм = 0,5625 мм2/на сетку, с использованием объектива Zeiss Neofluar, x10, NA = 0,3). Каждую правую верхнюю долю легких рассекали на 6 случайно выбранных неприлегающих друг к другу уровнях. С каждого уровня оценивали три среза. В среднем исследовали 30 квадратов сетки на срез (т.е. 0,5625 мм2/на сетку х 30 квадратов х 3 среза х 6 уровней), что дает площадь 3,04 см2 на каждое животное.
Количественное определение макрометастазов в легких
Для количественного определения колоний на поверхности легких на более поздних стадиях (2 недели после инокуляции опухолевых клеток) в вырезанные единым блоком легкие и сердце инъецировали 8 мл раствора Bouin (72% насыщенный раствор пикриновой кислоты, 23: формальдегид и 5% ледяная уксусная кислота) и фиксировали тем же раствором до того, как подсчитывать узелки на поверхности легких. Исследовали три площади на легкое с использованием стандарта (1 см2) и количество колоний на поверхности легких выражали как среднее/см2, согласно методике Wexler (Wexler, 1966).
Статистический анализ
Данные исследования адгезии in vivo анализировали посредством односторонних ANOVA и post hoc тестов Фишера PLSD, если это было необходимо. Звездочкой помечены достоверные эффекты post hoc по сравнению с контролями, получавшими физиологический раствор (имитацию воздействия), полученные с помощью теста Фишера PLSD. Все данные представлены как среднее ± СО среднего.
Результаты
Влияние однократной инъекции фумарата изолейцилтиазолидина на колонизацию опухоли в легких у крыс F344USA
Количество опухолевых колоний на поверхности легких послеоднократного введения фумарата изолейцилтиазолидина крысам F344 через 2 недели после инъекции опухолевых клеток MADB показано на фиг. 1. Однофакторный ANOVA показал отсутствие достоверного влияния (F(1,12)=3,2; p = 0,1 n.s.) на количество колоний. Тенденция к уменьшению количества колоний у экспериментальных крыс была очевидной.
Влияние однократной инъекции изолейцилцианопирролидина TFA на адгезию опухолевых клеток в легких у крыс F344USA
Среднее количество CFSE-положительных клеток в легочной ткани через 30 мин после инокуляции опухолевых клеток MADB после леченияизолейцилцианопирролидином TFA показано на фиг. 2. ANOVA показал отсутствие достоверного эффекта "лечения" (F(1,18) = 0,1; p = 0,8 n.s.).
Влияние однократной инъекции фумарата валилпирролидина на адгезию опухолевых клеток в легких у крыс F344USA
Среднее количество CFSE-положительных клеток в легочной ткани через 30 мин после инокуляции опухолевых клеток MADB106 после лечениявалилпирролидином показано на фиг. 3. ANOVA показал отсутствие достоверного эффекта "лечения" (F(1,18) = 0,6; p = 0,5 n.s.).
Обсуждение
Адгезия и колонизация опухолевых клеток достоверно изменяется после однократной инъекции фумарата изолейцилтиазолидина только у мутантных сублиний F344, что наводит на мысль о взаимодействии лиганда с мутантной DPIV и опухолевыми клетками. Поскольку лиганд не влияет достоверным образом на адгезию опухоли у крыс F344USA дикого типа, это может означать, что соединение не взаимодействует с сайтом связывания клеток MADB106.
Пример 14: Исследования колонизации злокачественных клеток
В предыдущем примере было показано, что адгезия опухолевых клеток MADB106 достоверно изменяется после однократной инъекции фумарата изолейцилтиазолидина только у мутантных сублиний F344, но не у крыс F344USA, экспрессирующих DPIV, дикого типа. Ингибиторы/лиганды DPIV могут воздействовать на рост метастазов опухоли, демонстрируя свойства, сходные со свойствами химиотерапевтических соединений и/или иммунотерапевтических соединений. В противоположность этому, данный пример изучает, отличается ли колонизация опухолевых клеток MADB106 у крыс F344 дикого типа, получающих постоянное лечение ингибитором DPIV.
Животные, инъекция опухолевых клеток и обработка легких
Крыс F344USA получали из питомника центрального вивария ганноверской медицинской школы, Германия. Все крысы принадлежали к одному поколению, содержавшееся в особых, лишенных патогенных микроорганизмов, помещениях при 25°C с режимом освещения 12 ч света - 12 часов темноты (свет с 7 ч утра), с неограниченным доступом к пище и воде. Точное количество животных, взятых в эксперимент, указывается величиной F и составляет по меньшей мере четыре животных на каждое условие и точку времени.
Культура клеток, инъекция опухолевых клеток, вскрытие животных и иммуногистохимическое исследование осуществляли, как описано ранее (von Horsten et al., 2000). Кратко, 1 х 106 опухолевых клеток MADB106, полученных во время лог-фазы опухолевого роста, инъецировали в латеральную вену хвоста и легкие удаляли после этого через различные промежутки времени. Для количественного определения опухолевых клеток in situ на ранних стадиях после инъекции (30 мин), клетки окрашивали прижизненным красителем с использованием производного флуоресцеина сукцинимидилового эфира диацетата 5-(и -6)-карбоксифлуоресцеина (CFSE) перед инъекцией. Для количественного определения колоний на поверхности легких на более поздних стадиях (2 недели после инокуляции опухолевых клеток) в вырезанные единым блоком легкие и сердце инъецировали 8 мл раствора Bouin (72% насыщенный раствор пикриновой кислоты, 23: формальдегид и 5% ледяная уксусная кислота) и фиксировали тем же раствором до того, как подсчитывать узелки на поверхности легких (см. ниже).
Эксперименты
Были проведены два эксперимента:
Влияние хронической инфузии различных доз фумарата изолейцилтиазолидина (0 мг, 0,04 мг, 0,4 мг, 4 мг/24 ч интрагастрально через имплантированные осмотические мининасосы) на изменение массы тела и колонизацию опухоли в легких у крыс F344USA дикого типа.
Влияние хронической инфузии фумарата изолейцилтиазолидина (4 мг/24 ч интрагастрально через имплантированные осмотические мининасосы), изолейцилцианопирролидина TFA (0,1 мг/24 ч интрагастрально через имплантированные осмотические мининасосы) и фумарата валилпирролидина (0,1 мг/24 ч интрагастрально через имплантированные осмотические мининасосы) на колонизацию опухоли в легких у крыс F344USA дикого типа.
Имплантация осмотических мининасосов для хронической интрагастральной инфузии соединений
Осмотические мининасосы (модель Alzet 2ML4; скорость потока 2,5 мкл/ч; Alza Corporation), через которые вводили постоянные количества различных соединений, предварительно асептически заполненные физиологическим раствором или ингибитором DPIV, помещали подкожно в абдоминальную область. Мининасосы были соединены с канюлей посредством полиэтиленовой трубки. Канюлю имплантировали интрагастрально с увеличившимся от нагревания концом канюли в просвете желудка.
Количественное определение макрометастазов в легких
Для количественного определения колоний на поверхности легких на более поздних стадиях (2 недели после инокуляции опухолевых клеток) в вырезанные единым блоком легкие и сердце инъецировали 8 мл раствора Bouin (72% насыщенный раствор пикриновой кислоты, 23: формальдегид и 5% ледяная уксусная кислота) и фиксировали тем же раствором до того, как подсчитывать узелки на поверхности легких. Исследовали три площади на легкое с использованием стандарта (1 см2) и количество колоний на поверхности легких выражали как среднее/см2, согласно методике Wexler (Wexler, 1966).
Статистический анализ
Данные исследования прибавки массы тела и количества опухолевых колоний на легочной поверхности in vivo анализировали посредством односторонних ANOVA и post hoc тестов Фишера PLSD, если это было необходимо. Звездочкой помечены достоверные эффекты post hoc по сравнению с контролями, получавшими физиологический раствор (имитацию воздействия), полученные с помощью теста Фишера PLSD. Все данные представлены как среднее ± СО среднего.
Результаты
Влияние хронической инфузии различных доз фумарата изолейцилтиазолидина (0 мг, 0,04 мг, 0,4 мг, 4 мг/24 ч) на колонизацию опухоли в легких
Изменение массы тела после хронической инфузии различных доз фумаратаизолейцилтиазолидина у крыс F344 через 2 недели после инъекции опухолевых клеток MADB106 представлено на фиг. 4. Однофакторный ANOVA показал достоверное влияние (F(3,22)=3,5; p = 0,03) на массу тела, которое стало достоверным при анализе post hoc при дозах 0,4 мг и 4 мг.
Количество опухолевых колоний на поверхности легких после хронической инфузии различных доз фумаратаизолейцилтиазолидина у крыс F344 через 2 недели после инъекции опухолевых клеток MADB106 представлено на фиг. 5. Однофакторный ANOVA показал достоверное влияние (F(3,22)=3,8; p = 0,03) на количество колоний, которое стало достоверным при анализе post hoc при дозе 4 мг.
Влияние хронической инфузии фумарата изолейцилтиазолидина, изолейцилцианопирролидина TFA и фумарата валилпирролидина на колонизацию опухоли в легких
Количество опухолевых колоний на поверхности легких после хронической инфузии фумаратаизолейцилтиазолидина, изолейцилцианопирролидина TFA и фумарата валилпирролидина у крыс F344 через 2 недели после инъекции опухолевых клеток MADB106 представлено на фиг. 6. Однофакторный ANOVA показал достоверное влияние (F(3,20)=3,8; p = 0,03) на количество колоний, которое стало достоверным при анализе post hoc для соединений изолейцилцианопирролидина TFA и фумаратаизолейцилтиазолидина.
Обсуждение
Метастазы MADB106 уменьшаются при хроническом лечении различными ингибиторами DPIV (фумаратомизолейцилтиазолидина, изолейцилцианопирролидином TFA), предполагая эффекты защитного класса. Возможно, фумаратизолейцилтиазолидина и изолейцилцианопирролидин TFA защищают от метастазов как посредством взаимодействия с процессами адгезии клеток, так и посредством модификации защитных клеточных механизмов хозяина. Возможно также, что лечение ингибитором DPIV оказывает цитостатическое действие. Указанные антиметастатические эффекты подтверждают биологические свойства ингибиторов DPIV для лечения рака и метастатической болезни.
СПИСОК ЛИТЕРАТУРЫ

| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОНИЖЕНИЯ КРОВЯНОГО ДАВЛЕНИЯ | 2002 |
|
RU2305553C2 |
| СПОСОБ ДЛЯ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ И НЕЙРОПСИХОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2001 |
|
RU2286149C2 |
| СПОСОБ ДЛЯ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ И НЕЙРОПСИХОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2006 |
|
RU2416406C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2827936C2 |
| НОВЫЙ ПОДХОД К ЛЕЧЕНИЮ РАКА С ПРИМЕНЕНИЕМ ИММУНОМОДУЛЯЦИИ | 2016 |
|
RU2817047C2 |
| АНАЛОГ ЭКСЕНАТИДА | 2020 |
|
RU2823623C1 |
| КОНЪЮГАТЫ АНТИТЕЛО-STING АГОНИСТ И ИХ ПРИМЕНЕНИЕ В ИММУНОТЕРАПИИ | 2020 |
|
RU2826228C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА, ЭКСПРЕССИРУЮЩЕГО IGF-1R | 2016 |
|
RU2728568C2 |
| КОНЪЮГАТ АНТИТЕЛО К B7H3-АНАЛОГ ЭКЗАТЕКАНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2019 |
|
RU2785664C2 |
| ЭФФЕКТОРЫ ДИПЕПТИДИЛПЕПТИДАЗУ IV | 1999 |
|
RU2309161C2 |
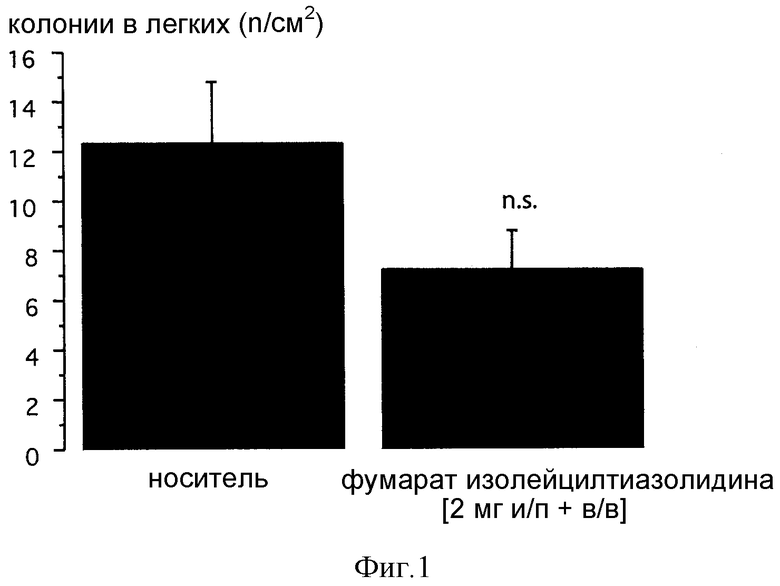

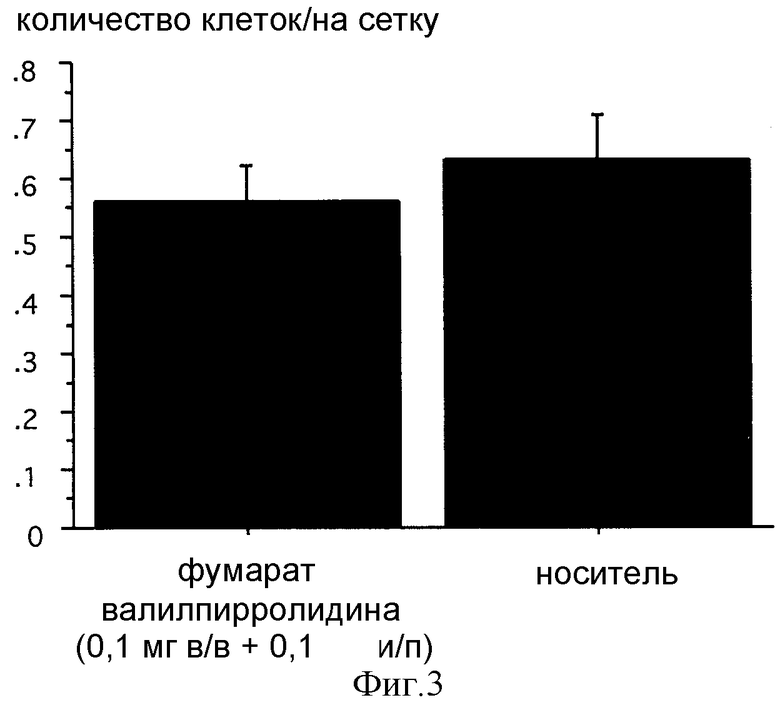
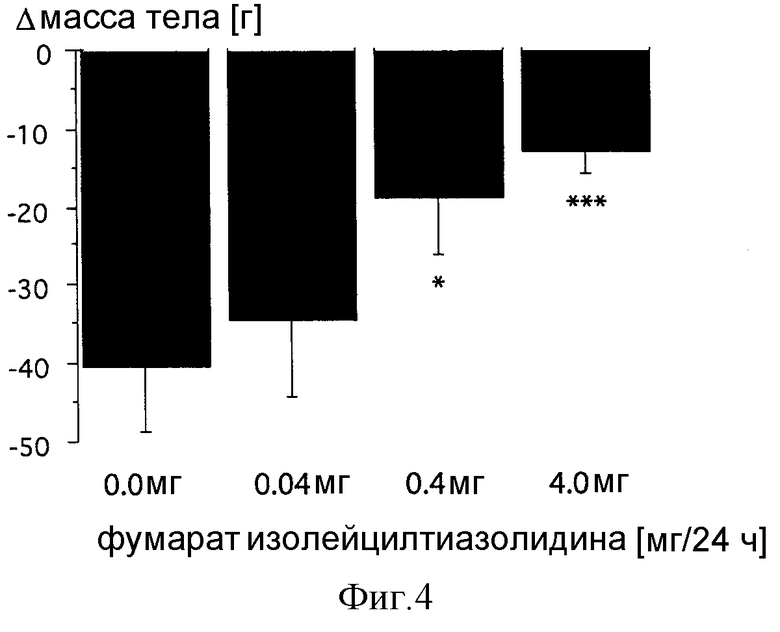

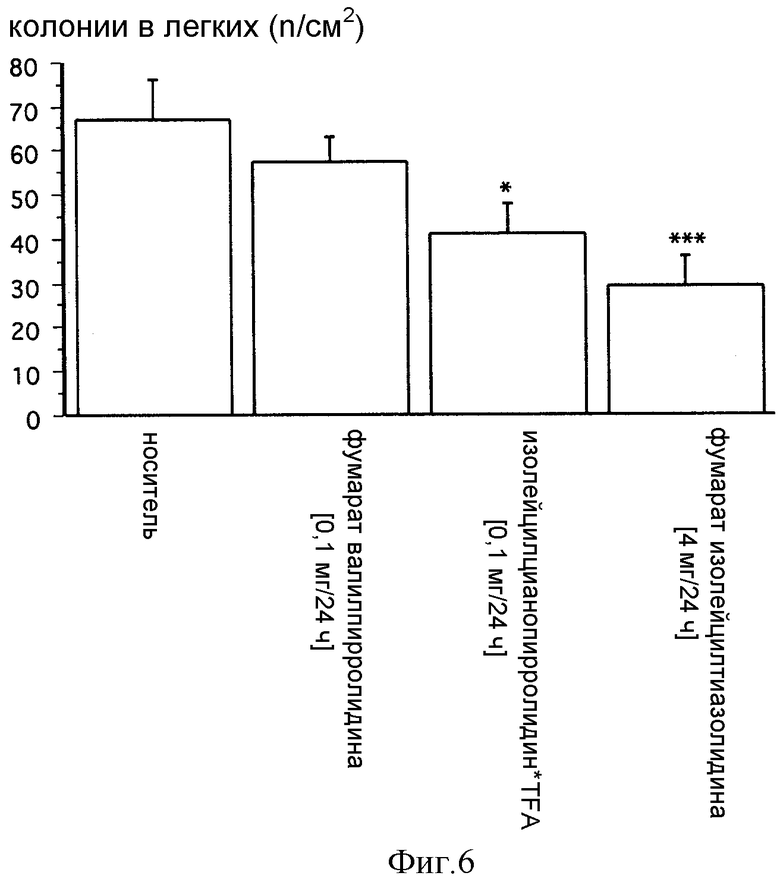
Настоящее изобретение относится к новому применению ингибиторов DPIV (изолейцилтиазолидина, изолейцилцианопирролидина, валилпирролидина) и их соответствующих фармацевтически приемлемых кислотно-аддитивных солевых форм для лечения и профилактики колонизации опухолевых клеток и/или метастазов. Перечисленные соединения уменьшали адгезию опухолевых клеток в легких крыс и приводили к уменьшению у них количества колоний опухолевых клеток, уменьшали метастазы. 2 з.п. ф-лы, 6 ил.
| DE19826972 А1, 23.12.1999 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| реферат Entrez PubMed: Reinhold D.et al | |||
| Inhibitors of dipeptidyl peptidase IV (DP IV, CD26) specifically suppress proliferation and modulate cytokine production of strongly CD26 expressing U937 cells | |||
| Immunobiology | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| [найдено on line] | |||

