ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка претендует на приоритет китайской патентной заявки № 201910908468.3, поданной в Национальное управление интеллектуальной собственности Китая 25 сентября 2019 г. и озаглавленной «АНАЛОГ ЭКСЕНАТИДА», которая полностью включена в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
[0002] Настоящее изобретение относится к аналогу эксенатида и его применению, и аналог представляет собой аналог глюкагоноподобного пептида-1 (GLP-1).
УРОВЕНЬ ТЕХНИКИ
[0003] Среди неинфекционных заболеваний диабет занимает третье место по распространенности после сердечно-сосудистых и цереброваскулярных заболеваний и опухолей. Всемирная организация здравоохранения (ВОЗ) прогнозирует, что к 2030 году в мире количество людей, страдающих от диабета, превысит 360 миллионов, из которых более 90% будут составлять пациенты с диабетом II типа. GLP-1, секретин, секретируемый кишечными L-клетками, имеет такие функции, как стимуляция секреции инсулина, ингибирование высвобождения глюкагона, стимуляция пролиферации островковых β-клеток, индукция регенерации островковых β-клеток, предотвращение апоптоза островковых β-клеток, улучшение чувствительности к инсулину и увеличение утилизации глюкозы, что играет важную роль в возникновении и развитии сахарного диабета II типа. У больных сахарным диабетом II типа нарушен «инкретиновый эффект», что в основном проявляется в том, что повышение концентрации GLP-1 после еды меньше, чем у нормальных людей, но стимулирование секреции инсулина и гипогликемические эффекты существенно не нарушены. Следовательно, GLP-1 можно использовать в качестве важной мишени для лечения диабета II типа. Кроме того, функция GLP-1 зависит от концентрации глюкозы, а его гипогликемические свойства являются основой и гарантией клинической безопасности, что устраняет опасения, что существующие препараты и схемы лечения диабета могут вызывать тяжелую гипогликемию у пациентов. Таким образом, GLP-1 имеет широкие перспективы применения в области лечения диабета.
[0004] Однако клиническое применение GLP-1 также сталкивается с огромными проблемами. GLP-1, продуцируемый человеческим организмом, очень нестабилен и легко расщепляется в организме дипептидилпептидазой IV (DPP-IV). GLP-1 имеет короткое время жизни в плазме, что ограничивает его клиническое применение. Кроме того, многие пациенты с диабетом II типа плохо воспринимают ежедневные инъекции, поэтому разработка безопасных и эффективных аналогов GLP-1, которые можно вводить один раз в неделю, имеет большие перспективы.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0005] В настоящем изобретении предложен аналог эксенатида и его применение, и аналог представляет собой аналог глюкагоноподобного пептида-1 (GLP-1).
[0006] Для достижения вышеуказанной цели, в настоящем изобретении, во-первых, предложено соединение, имеющее структуру I, его фармацевтически приемлемую соль, его сольват, его хелат или его нековалентный комплекс, его пролекарство или любую их смесь.
AA1-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-
Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-
Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-AA2(R)-AA3
Структура I
[0007] AA1 в структуре I представляет собой:
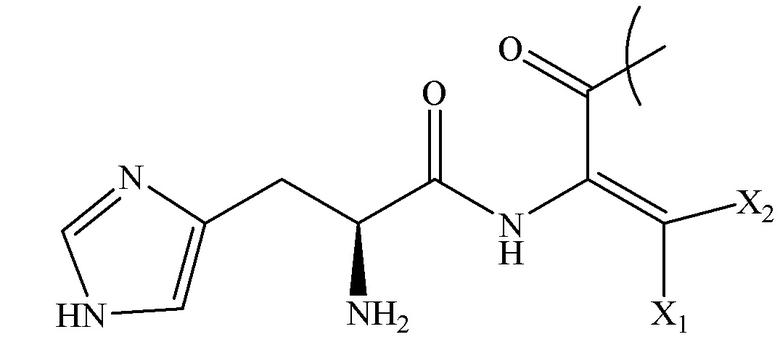 ,
,
X1 и X2, каждый независимо, выбирают из группы, состоящей из H,
CH3, CH(CH3)2, C(CH3)3, CH(CH2CH3)2, C(CH2CH3)3, CH(CH2CH2CH3)2, C(CH2CH2CH3)3, CH(CH(CH3))2, C(CH(CH3))3,
CH2CH3, CH2CH(CH3)2, CH2C(CH3)3, CH2CH(CH2CH3)2, CH2C(CH2CH3)3, CH2CH(CH2CH2CH3)2, CH2C(CH2CH2CH3)3, CH2CH(CH(CH3))2, and CH2C(CH(CH3))3;
[0008] AA2 из структуры I выбирают из группы, состоящей из Lys, Dah, Orn, Dab и Dap;
[0009] AA3 из структуры I представляет собой NH2 или OH; и
[0010] R из структуры I представляет собой HO2C(CH2)n1CO-(γGlu)n2-(PEGn3(CH2)n4CO)n5-,
n1 представляет собой целое число от 10 до 20,
n2 представляет собой целое число от 1 до 5,
n3 представляет собой целое число от 1 до 30,
n4 представляет собой целое число от 1 до 5, и
n5 представляет собой целое число от 1 до 5.
[0011] Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение по настоящему изобретению, а также к применение фармацевтической композиции по настоящему изобретению в производстве лекарственного средства для лечения заболевания.
[0012] Предпочтительно, фармацевтическая композиция используется в производстве лекарственного средства для лечения заболевания, при этом заболевание выбирают из группы, состоящей из диабета II типа, нарушения толерантности к глюкозе, диабета I типа, ожирения, гипертонии, метаболического синдрома, дислипидемии, когнитивного расстройства, атеросклероза, инфаркта миокарда, ишемической болезни сердца, сердечно-сосудистого заболевания, инсульта, синдрома раздраженного кишечника, диспепсии, язвы желудка, фиброзного заболевания печени, фиброзного заболевания легких и их комбинации.
[0013] Предпочтительно, фармацевтическую композицию используют в производстве лекарственного средства для лечения диабета II типа с отсроченной эффективностью и/или для предотвращения обострения диабета II типа.
[0014] Предпочтительно, фармацевтическую композицию используют в производстве лекарственного средства для снижения потребления пищи, уменьшения апоптоза β-клеток, усиления функции островковых β-клеток, увеличения количества β-клеток и/или восстановления чувствительности β-клеток к глюкозе.
[0015] Настоящее изобретение также относится к способу регулирования уровня глюкозы в крови в организме путем введения соединения нуждающемуся в этом субъекту.
[0016] Больше аспектов настоящего изобретения более подробно описано ниже, или некоторые из них могут быть проиллюстрированы в вариантах осуществления настоящего изобретения.
[0017] Если не указано иное, количество различных компонентов и условия реакции, используемые в настоящем документе, могут быть интерпретированы как «примерные» или «приблизительные» в любом случае. Соответственно, если не указано иное, числовые параметры, приведенные ниже и в формуле изобретения, являются приблизительными параметрами, и могут быть получены другие числовые параметры из-за различий в стандартных ошибках в соответствующих экспериментальных условиях.
[0018] Когда существуют разногласия или сомнения между химической структурной формулой и химическим названием соединения в данном документе, для точного определения соединения используется химическая структурная формула. Описанное в настоящем документе соединение может содержать один или несколько хиральных центров и/или двойные связи и т.п., а также может существовать в виде стереоизомеров, включая изомеры по двойной связи (такие как геометрические изомеры), оптически активные энантиомеры или диастереомеры. Соответственно, любая химическая структура в рамках данного описания, будь то часть или вся структура, содержащая подобные структуры, указанные выше, включает все возможные энантиомеры и диастереомеры соединения, включая любые чистые стереоизомеры (такие как чистые геометрические изомеры, чистые энантиомеры или чистые диастереомеры) и любую смесь этих изомеров. Эти рацемические и стереоизомерные смеси также могут быть дополнительно разделены на составляющие их энантиомеры или стереоизомеры специалистами в данной области с использованием различных методов разделения или способов хирального молекулярного синтеза.
[0019] Соединения, имеющие структуру I, включают, но не ограничиваются ими, оптические изомеры, рацематы и/или смеси этих соединений. В приведенном выше случае чистый энантиомер или диастереомер, такой как оптический изомер, может быть получен путем асимметричного синтеза или разделения рацематов. Разделение рацематов можно осуществить различными способами, такими как обычная перекристаллизация с агентом, способствующим разделению, или хроматография. Кроме того, соединения, имеющие структуру I, также включают цис- и/или транс-изомеры с двойными связями.
[0020] Соединение по настоящему изобретению включает, но не ограничивается ими, соединения со структурой I и все различные фармацевтически приемлемые формы. Различные фармацевтически приемлемые формы включают различные фармацевтически приемлемые соли, сольваты, комплексы, хелаты, нековалентные комплексы, пролекарства на основе вышеуказанных веществ и любую смесь вышеуказанных форм.
[0021] Соединение, имеющее структуру I по настоящему изобретению, обладает стабильными свойствами, плохо расщепляется дипептидилпептидазой IV (DPP-IV) в организме, является аналогом GLP-I длительного действия и оказывает значительное гипогликемическое действие.
ПОДРОБНОЕ ОПИСАНИЕ
[0022] В настоящем изобретении раскрыт аналог глюкагоноподобного пептида-1 (GLP-1) и его применение, и специалисты в данной области техники могут изучить содержание настоящего изобретения и надлежащим образом улучшить соответствующие параметры для достижения целей изобретения. Следует особо отметить, что все подобные замены и модификации очевидны для специалистов в данной области техники, и считается, что они включены в настоящее изобретение. Способ по настоящему изобретению был описан посредством предпочтительных вариантов осуществления, и очевидно, что соответствующие лица могут достигать целей изобретения и применять способы по настоящему изобретению посредством изменений или соответствующих модификаций и комбинаций соединения и способа получения, описанных в настоящем документе, не отклоняясь от содержания, сути и объема настоящего изобретения.
[0023] Названия, соответствующие аббревиатурам, используемым в настоящем изобретении, приведены в нижеследующей таблице:
Пример 1. Получение соединения 1
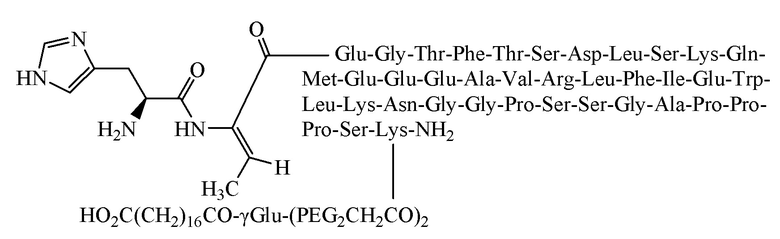
[0024] Способ получения включает в себя: получение пептида на смоле способом твердофазного полипептидного синтеза, затем отщепление пептида от смолы с получением неочищенного продукта и, наконец, очистку неочищенного продукта с получением чистого продукта; где стадия получения пептида на смоле способом твердофазного полипептидного синтеза заключается в последовательном присоединении защищенной аминокислоты или фрагмента в соответствии с заданной последовательностью к смоле-носителем с помощью реакции сочетания на твердой фазе для получения пептида на смоле.
В указанном выше способе получения количество Fmoc-защищенной аминокислоты или защищенного аминокислотного фрагмента в 1,2-6 раз, предпочтительно в 2,5-3,5 раза превышает общее количество молей загружаемой смолы.
[0026] В указанном выше способе получения степень замещения смолы-носителя составляет 0,2-1,0 ммоль/г смолы, предпочтительно, 0,3-0,5 ммоль/г смолы.
[0027] В качестве предпочтительного варианта осуществления настоящего изобретения способ синтеза сочетанием на твердой фазе включает в себя: снятие защитной группы Fmoc с защищенной аминокислоте на смоле, полученной на предыдущей стадии, которую затем подвергают реакции сочетания со следующей защищенной аминокислотой. Время снятия защиты Fmoc составляет 10-60 мин, предпочтительно, 15-25 мин. Время реакции сочетания составляет 60-300 мин, предпочтительно, 100-140 мин.
[0028] Реакция сочетания требует добавления конденсирующего реагента, и конденсирующий реагент выбирают из группы, состоящей из DIC (N,N-диизопропилкарбодиимида), N,N-дициклогексилкарбодиимида, бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфата, 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата, бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата и О-бензотриазол-N,N,N',N'-тетраметилурония тетрафторбората; предпочтительно, N,N-диизопропилкарбодиимида. Молярное количество конденсирующего реагента в 1,2-6 раз, предпочтительно, в 2,5-3,5 раза превышает общее количество молей аминогрупп в аминосмоле.
[0029] Реакция сочетания требует добавления активирующего агента, и активирующий агент выбирают из группы, состоящей из 1-гидроксибензотриазола и N-гидрокси-7-азабензотриазола, предпочтительно, 1-гидроксибензотриазола. Количество активирующего агента в 1,2-6 раз, предпочтительно, в 2,5-3,5 раза превышает общее количество молей аминогрупп в аминосмоле.
[0030] В качестве предпочтительного варианта осуществления настоящего изобретения реагент для снятия защиты Fmoc представляет собой смешанный раствор PIP/DMF (пиперидин/N,N-диметилформамид), причем содержание пиперидина в смешанном растворе составляет 10-30% (V). Количество агента для удаления Fmoc составляет 5-15 мл на грамм аминосмолы, предпочтительно, 8-12 мл на грамм аминосмолы.
[0031] Предпочтительно, содержащую пептиды смолу подвергают кислотному гидролизу при отщеплении пептида от смолы и удалении защитной группы боковой цепи, получая неочищенный продукт.
[0032] Кроме того, предпочтительно, агент, используемый для кислотного гидролиза пептида на смоле, представляет собой смесь трифторуксусной кислоты (TFA), 1,2-этандитиола (EDT) и воды, и объемный процент каждого компонента в смеси составляет: TFA 80-95%, EDT 1-10%, и остальное составляет вода.
[0033] Более предпочтительно, объемный процент каждого компонента в смешанном растворителе составляет: TFA 89-91%, EDT 4-6%, а остальное составляет вода. Оптимальное объемное процентное содержание каждого компонента в смешанном растворителе составляет: TFA 90%, EDT 5%, и остальное составляет вода.
[0034] Количество кислотного гидролизующего агента составляет 4-15 мл кислотного гидролизующего агента на грамм пептида на смоле; предпочтительно, требуется 7-10 мл кислотного гидролизующего агента на грамм пептида на смоле.
Время расщепления с использованием кислотного гидролизующего агента составляет 1-6 часов при комнатной температуре, предпочтительно, 3-4 часа.
Кроме того, неочищенный продукт очищают с помощью высокоэффективной жидкостной хроматографии и сушат вымораживанием, получая чистый продукт.
1. Синтез пептида на смоле
[0037] Защищенные аминокислоты, показанные в таблице ниже, последовательно связывали с амидной BHHA-смолой Ринка в качестве смолы-носителя путем снятия защиты Fmoc и реакции сочетания с получением пептида на смоле. Защищенные аминокислоты, используемые в этом примере, являются следующими:
[0038] (1) Присоединение первой защищенной аминокислоты в основной цепи
[0039] 0,03 моль первой защищенной аминокислоты и 0,03 моль HOBt растворяли в соответствующем количестве DMF. Затем к раствору защищенной аминокислоты в DMF при перемешивании медленно добавляли 0,03 моль DIC, перемешивали и проводили реакцию при комнатной температуре в течение 30 мин, получая активированный раствор защищенной аминокислоты для последующего использования.
[0040] С 0,01 моль амидной MBHA-смолы Ринка (степень замещения составляла около 0,4 ммоль/г) удаляли защиту с помощью 20%-го раствора PIP/DMF в течение 25 минут, промывали и фильтровали, получая смолу со снятой защитной группой Fmoc.
[0041] Активированный раствор первой защищенной аминокислоты добавляли к смоле со снятой защитной группой Fmoc для реакции сочетания в течение 60-300 мин, промывали и фильтровали для получения смолы, содержащей первую защищенную аминокислоту.
[0042] (2) Связывание защищенных аминокислот со 2-й по 40-ю в основной цепи
[0043] Указанные выше соответствующие защищенные аминокислоты со 2-й по 40-ю последовательно присоединяли тем же способом, что и описанный выше для связывания первой защищенной аминокислоты в основной цепи, получая смолу, содержащую 40 аминокислот в основной цепи.
[0044] (3) Связывание первой защищенной аминокислоты в боковой цепи
[0045] 0,03 моля защищенной 1-й аминокислоты из боковой цепи и 0,03 моля HOBt растворяли в соответствующем количестве DMF. 0,03 моль DIC медленно добавляли к раствору защищенной аминокислоты в DMF при перемешивании, перемешивали и оставляли реакцию при комнатной температуре на 30 мин, получая активированный раствор защищенной аминокислоты.
[0046] 2,5 ммоль тетракистрифенилфосфин-палладия и 25 ммоль фенилсилана растворяли в соответствующем количестве дихлорметана, проводили реакцию снятия защиты в течение 4 часов, промывали и фильтровали, получая смолу со снятой защитной группой Alloc для последующего использования.
[0047] Активированный раствор защищенной 1-й аминокислоты из боковой цепи добавляли к смоле со снятой защитной группой Alloc для реакции сочетания в течение 60-300 мин, промывали и фильтровали, получая смолу, содержащей защищенную 1-ю аминокислоту из боковой цепи.
[0048] (4) Связывание со 2-й по 4-ю защищенных аминокислот в боковой цепи
[0049] Защищенные аминокислоты со 2-й по 4-ю, соответствующие боковой цепи, и монозащищенную жирную кислоту последовательно присоединяли тем же способом, что описан выше для связывания первой защищенной аминокислоты в основной цепи, получая пептид на смоле.
2. Получение неочищенного продукта
[0050] К вышеуказанному пептиду на смоле добавляли отщепляющий агент с объемным соотношением TFA:вода:EDT=95:5:5 (10 мл отщепляющего агента/г смолы), равномерно перемешивали и проводили реакцию при перемешивании при комнатной температуре. температура в течение 3 ч. Реакционную смесь фильтровали через воронку с песчаным сердечником, собирали фильтрат и трижды промывали смолу небольшим количеством TFA. Фильтраты объединяли, концентрировали при пониженном давлении, осаждали добавлением безводного эфира, трижды промывали безводным эфиром, удаляли жидкость и сушили осадок, получая неочищенный продукт в виде беловатого порошка.
3. Получение чистого продукта
[0051] К указанному выше неочищенному продукту добавляли воду и перемешивали. pH раствора доводили до 8,0 водным раствором аммиака до тех пор, пока неочищенный продукт полностью не растворялся. Раствор фильтровали через смешанную микропористую фильтрующую мембрану 0,45 мкм для последующей очистки.
[0052] Очистку проводили с помощью высокоэффективной жидкостной хроматографии. Хроматографический материал набивки колонки для очистки представлял собой силикагель С18 10 мкм для обращеннофазовой хроматографии. Система подвижной фазы представляла собой 0,1%TFA/вода-0,1%TFA/ацетонитрил. Скорость потока в хроматографической колонке 30мм*250мм составляла 20 мл/мин. Для элюции применяли градиентную систему, а нанесение осуществляли циклом для очистки. Раствор неочищенного продукта наносили на хроматографическую колонку и затем элюировали подвижной фазой. Элюат, содержащий основной пик, собирали и выпаривали для удаления ацетонитрила, получая очищенный промежуточный концентрат.
[0053] Очищенный промежуточный концентрат фильтровали через мембрану 0,45 мкм для последующего использования. Для солевого обмена использовали высокоэффективную жидкостную хроматографию. Система подвижной фазы представляла собой 1%-ю уксусную кислоту в воде/ацетонитрил. Хроматографический материал набивки колонки для очистки представлял собой силикагель С18 10 мкм для обращеннофазовой хроматографии. Скорость потока в хроматографической колонке 30мм*250мм составляла 20 мл/мин (соответствующая скорость потока может быть отрегулирована в зависимости от параметров хроматографической колонки). Использовали способ градиентного элюирования и циклического наноса образца. Образец наносили на хроматографическую колонку и затем элюировали подвижной фазой. Элюаты собирали и анализировали спектр, наблюдая изменение поглощения. Элюат, содержащий основной пик при солевом обмене, собирали и определяли чистоту с использованием аналитической жидкостной хроматографии. Элюат, содержащий основной пик при солевом обмене, объединяли и концентрировали при пониженном давлении, получая водно-уксусный раствор чистого продукта, который затем лиофилизировали, получая 7,7 г очищенного продукта с чистотой 95,8%, общим выходом 15,2% и молекулярной массой 5056,2 (100% М+Н).
Пример 2. Получение соединения 2
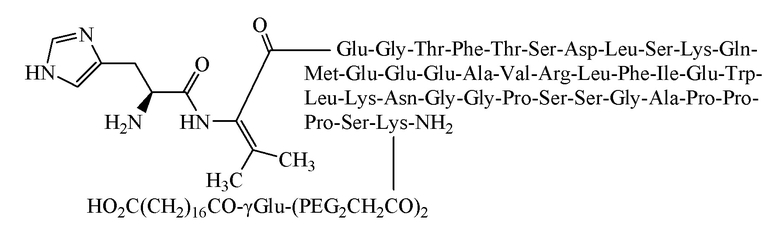
[0054] Способ получения был таким же, как в примере 1, и использовали следующие защищенные аминокислоты:
[0055] Было получено 6,2 г очищенного продукта с чистотой 95,8%, общим выходом 12,2% и молекулярной массой 5070,6 (100% М+Н).
Пример 3. Получение соединения 3
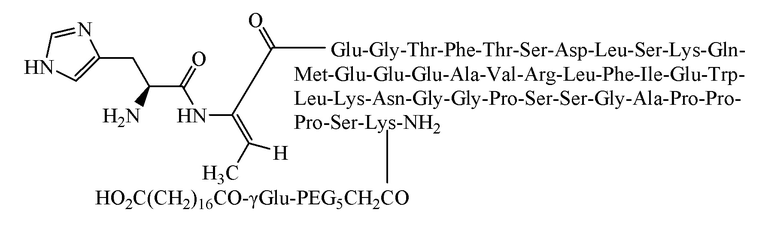
[0056] Способ получения был таким же, как в примере 1, и использовали следующие защищенные аминокислоты:
[0057] Было получено 8,9 г очищенного продукта с чистотой 98,5%, общим выходом 17,6% и молекулярной массой 5043,2 (100% М+Н).
Пример 4. Получение соединения 4
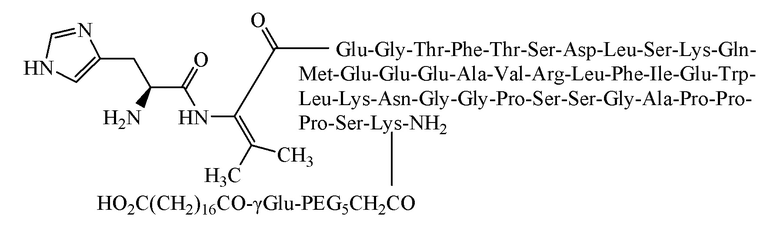
[0058] Способ получения был таким же, как в примере 1, и использовали следующие защищенные аминокислоты:
[0059] Было получено 5,2 г очищенного продукта с чистотой 96,3%, общим выходом 10,3% и молекулярной массой 5057,6 (100% М+Н).
Пример 5. Получение соединения 5
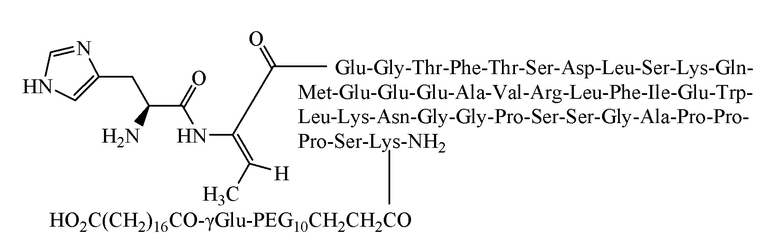
[0060] Способ получения был таким же, как в примере 1, и использовали следующие защищенные аминокислоты:
[0061] Было получено 5,6 г очищенного продукта с чистотой 97,6%, общим выходом 10,6% и молекулярной массой 5263,8 (100% М+Н).
Пример 6. Получение соединения 6
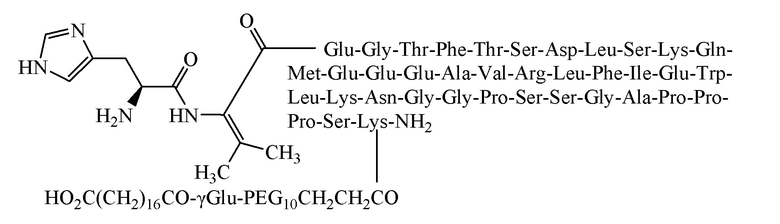
[0062] Способ получения был таким же, как в примере 1, и использовали следующие защищенные аминокислоты:
[0063] Было получено 4,5 г очищенного продукта с чистотой 97,1%, общим выходом 8,5% и молекулярной массой 5277,6 (100% М+Н).
Пример 7. Обнаружение активности
1. Способ
[0064] GLP-1R, в основном присутствующий на поверхности островковых β-клеток, представляет собой рецептор, связанный с G-белком (GPCR). При стимуляции специфическим агонистом GLP-1R может активировать внутриклеточный аденилатциклазный путь, повышать уровень cAMP и, в итоге, приводить к продукции и высвобождению инсулина. При стимуляции стабильной клеточной линии, экспрессирующей GLP-1R, тестируемым агонистом уровень внутриклеточного cAMP будет быстро увеличиваться, и относительные световые единицы (RLU) после стимуляции клеток при каждой дозе определяются методом хемилюминесценции для расчета EC50 агониста. Этот способ определения активности обычно используется для анализа активности агонистов рецептора GLP-1 в Китаре и в мире.
[0065] В эксперименте использовали стабильную клеточную линию CHO-K1, экспрессирующую GLP-1R. Клетки стимулировали различными концентрациями агониста, измеряли относительные световые единицы после стимуляции клеток каждой дозой и получали биологическую активность агониста.
2. Результаты
[0066] Результаты приведены в таблице ниже.
Пример 8. Предварительное определение фармакокинетических свойств
[0067] Каждое соединение испытывали в двух группах введения. Использовали крыс SD, по 4 самца и 4 самки в каждой группе, всего 8 крыс.
[0068] Группа внутривенных инъекций в хвостовую вену: доза составляла 1 мг/кг. Образцы крови собирали из глазничной вены крыс перед введением (0 ч) и через 30 мин, 1 ч, 2 ч, 4 ч, 8 ч, 24 ч, 48 ч, 96 ч и 144 ч после введения. Образцы плазмы собирали после центрифугирования.
[0069] Группа подкожного введения: доза составляла 1 мг/кг. Образцы крови собирали из глазничной вены крыс перед введением (0 ч) и через 1 ч, 2 ч, 3 ч, 4 ч, 8 ч, 24 ч, 48 ч, 96 ч и 144 ч после введения. Образцы плазмы собирали после центрифугирования.
[0070] Концентрации соответствующих соединений в образцах плазмы крыс SD определяли с помощью ЖХ/МС. После внутривенного и подкожного введения период полувыведения соединений у крыс SD, которым соединение вводили подкожно (п/к), приведен в следующей таблице:
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| ПЕПТИД GLP-1 С ПРИСОЕДИНЕННОЙ ОЛИГОСАХАРИДНОЙ ЦЕПЬЮ | 2008 |
|
RU2539829C2 |
| ПРОИЗВОДНОЕ АНАЛОГА GLP-1 ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2565536C2 |
| Сайт-специфически монопегилированные аналоги эксендина и способ их получения | 2012 |
|
RU2625015C2 |
| МУЛЬТИАГОНИСТЫ И ИХ ПРИМЕНЕНИЕ | 2022 |
|
RU2833609C2 |
| ПЕПТИД ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА И ЕГО ОСЛОЖНЕНИЙ | 2014 |
|
RU2573933C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОПЕПТИДА, ИМЕЮЩЕГО СИАЛИРОВАННУЮ САХАРНУЮ ЦЕПЬ, И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО СИАЛИЛГЛИКОАСПАРАГИНА | 2012 |
|
RU2586524C2 |
| МУЛЬТИРЕЦЕПТОРНЫЙ АГОНИСТ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2816492C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
Настоящее изобретение относится к области биотехнологии и может быть использовано в медицине. Изобретение раскрывает новый аналог пептида эксенатида. В сравнении с природным гормоном ГПП1 и эксенатидом аналог согласно настоящему изобретению обладает увеличенным временем полувыведения из организма субъекта. Изобретение может быть использовано при лечении заболеваний, ассоциированных с нарушениями биологической функции гормона ГПП-1, в частности при лечении метаболических заболеваний, например диабета (I или II тип), дислипидемии, нарушения толерантности к глюкозе и др. 4 н. и 2 з.п. ф-лы, 4 табл., 8 пр.
1. Аналог эксенатида для агонистической стимуляции рецептора глюкагоноподобного пептида-1 (GLP-1), имеющий структуру I:
где AA1 в структуре I представляет собой:
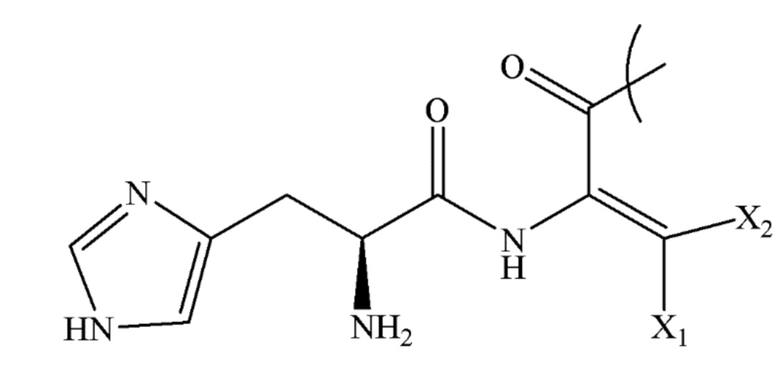 ,
,
X1 и X2, каждый независимо, выбирают из группы, состоящей из H, CH3, CH(CH3)2, C(CH3)3, CH(CH2CH3)2, C(CH2CH3)3, CH(CH2CH2CH3)2, C(CH2CH2CH3)3, CH(CH(CH3))2, C(CH(CH3))3, CH2CH3, CH2CH(CH3)2, CH2C(CH3)3, CH2CH(CH2CH3)2, CH2C(CH2CH3)3, CH2CH(CH2CH2CH3)2, CH2C(CH2CH2CH3)3, CH2CH(CH(CH3))2 и CH2C(CH(CH3))3;
AA2 из структуры I выбирают из группы, состоящей из Lys, Dah, Orn, Dab и Dap;
AA3 из структуры I представляет собой NH2 или OH; и
R из структуры I представляет собой HO2C(CH2)n1CO-(γGlu)n2-(PEGn3(CH2)n4CO)n5-,
n1 представляет собой целое число от 10 до 20,
n2 представляет собой целое число от 1 до 5,
n3 представляет собой целое число от 1 до 30,
n4 представляет собой целое число от 1 до 5, и
n5 представляет собой целое число от 1 до 5.
2. Аналог эксенатида по п.1, который представляет собой его фармацевтически приемлемую соль, его сольват или любую их смесь.
3. Применение аналога эксенатида по п.1 или 2 для лечения заболевания, где заболевание выбрано из группы, состоящей из диабета II типа, нарушения толерантности к глюкозе, диабета I типа, ожирения, дислипидемии и их комбинации.
4. Применение аналога эксенатида по п.3 для лечения диабета II типа с отсроченной эффективностью и/или предотвращения обострения диабета II типа.
5. Применение аналога эксенатида по п.1 или 2 для снижения потребления пищи.
6. Применение аналога эксенатида по п.1 или 2 для снижения апоптоза β-клеток, усиления функции островковых β-клеток, увеличения количества β-клеток и/или восстановления чувствительности β-клеток к глюкозе.
| CN 106554404 A1, 05.04.2017 | |||
| WO 2017178829 A1, 19.10.2017 | |||
| WO 2018004283 A3, 04.01.2018 | |||
| НОВЫЕ АНАЛОГИ ГЛЮКАГОНА | 2011 |
|
RU2559320C2 |

