Область техники
Настоящее изобретение относится к ингибиторам активности дипептидилпептидазы IV и дипептидилпептидаза IV-подобных ферментов и, в частности, к фармацевтическим композициям, содержащим вышеуказанные соединения, и применению указанных соединений для понижения уровней кровяного давления у млекопитающих и (лечения) связанных с этим расстройств.
Предшествующий уровень техники
Дипептидилпептидаза IV (DPIV) - сериновая протеаза, которая отщепляет N-концевые дипептиды от пептидной цепи, содержащей предпочтительно остаток пролина в предпоследнем положении. Хотя биологическая роль DPIV в системах млекопитающих полностью не установлена, полагают, что она играет важную роль в метаболизме нейропептидов, активации Т-клеток и проникновении ВИЧ (HIV) в лимфоидные клетки.
Настоящее изобретение обеспечивает новое использование DPIV-ингибиторов для профилактики и лечения состояний, опосредованных ингибированием DPIV и DPIV-подобных ферментов, в частности для понижения уровней кровяного давления и лечения родственных нарушений, и фармацевтических композиций, например, полезных для ингибирования DPIV и DPIV-подобных ферментов, и способ ингибирования активности вышеуказанных ферментов.
Данное изобретение относится к способу лечения, в частности к способу снижения уровней кровяного давления у млекопитающих и к соединениям и композициям для использования в вышеупомянутом способе. Дипептидилпептидаза IV (DPIV; EC 3.4.14.5; CD26) представляет сериновую протеиназу, расщепляющую пептидные связи, следующие после пролина (в меньшей степени после аланина, после серина или после глицина), которая экспрессируется в ряде тканей, включая эпителиальные клетки и подгруппу лейкоцитов. Кроме того, она представляет ассоциированную с мембраной эктопептидазу, которая демонстрирует свою активность во внеклеточном домене.
Примерами низкомолекулярных ингибиторов дипептидилпептидазы IV являются такие средства, как: производные тетрагидроизохинолин-3-карбоксамида, N-замещенные 2-цианопиролы и -пирролидины, N-(N'-замещенный глицил)-2-цианопирролидины, N-(замещенный глицил)тиазолидины, N-(замещенный глицил)-4-цианотиазолидины, амино-ацил-бороно-пролилингибиторы, циклопропил-конденсированные пирролидины и гетероциклические соединения. Ингибиторы дипептидилпептидазы IV описаны в патенте США 6380398, патенте США 6011155; патенте США 6107317; патенте США 6110949; патенте США 6124305; патенте США 6172081; международной публикации WO 95/15309, международной публикации WO 99/61431, WO 99/67278, международной публикации WO 99/67279, патенте DE 19834591, международной публикации WO 97/40832, патенте DE 196 16 486 C 2, международных публикациях WO 98/19998, WO 00/07617, WO 99/38501, WO 99/46272, WO 99/38501, WO 01/68603, WO 01/40180, WO 01/81337, WO 01/81304, WO 01/55105, WO 02/02560 и WO 02/14271, на описания которых здесь ссылаются во всей их полноте, особенно относительно вышеуказанных ингибиторов, их определения, применений и их получения.
Термин DPIV-подобные ферменты относится к структурно и/или функционально DPIV/СD26-родственным ферментным белкам (Sedo & Malik, Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochimica et Biophysica Acta 2001 36506: 1-10). По существу, эта небольшая группа, как было выявлено, высвобождает H-Xaa-Pro-Дипептиды и H-Xaa-Ala-Дипептиды от N-конца олиго- или полипептидов. Они демонстрируют общую особенность, заключающуюся в том, что они действуют в Pro-положении, а также Ala, Ser, Thr и других аминокислот с небольшими гидрофобными боковыми цепями, таким как Gly или Val. Гидролитическая активность располагается в следующий ряд Pro > Ala " Ser, Thr " Gly, Val. Те же самые белки были доступны в таких небольших количествах, что можно было установить только пост-Pro и пост-Ala расщепление. В то время как белки DPIV, DP II, FAPa (Seprase), DP 6, DP 8 и DP 9 структурно родственны и демонстрируют высокую гомологию последовательностей, аттрактин является экстраординарно функциональным DPIV-подобным ферментом, характеризующимся аналогичной активностью и схожей картиной ингибирования.
Другие DPIV-подобные ферменты раскрываются в международных патентных публикациях WO 01/19866, WO 02/04610, WO 02/34900 и W0 02/31134. WO 01/19866 раскрывает новую человеческую дипептидиламинопептидазу (DPP8) со структурным и функциональным сходствами с DPIV и фибробласт-активирующий белок (FAP). В международной публикации WO 02/34900 раскрывается новая дипептидилпептидаза 9 (DPP9), структура аминокислотной последовательности которой в значительной степени схожа с аминопоследовательностями DPIV и DPP8. В международной публикации WO 02/31134 раскрываются три DPIV-подобных фермента, DPRP1, DPRP2 и DPRP3. Секвенирование показало, что DPRPI идентичен DPP8, как раскрывается в международной публикации WO 01/19866, что DPRP2 идентичен DPP9, а DPRP3 идентичен K1AA1492, как раскрывается в международной публикации WO 02/04610.
Высокое кровяное давление (гипертензия) является бессимптомным состоянием, и это аномально высокое давление в артериях увеличивает риск возникновения таких проблем, как: удар, аневризма, сердечная недостаточность, сердечный приступ и поражение почек. Для многих людей слово "гипертензия" означает чрезмерное напряжение, нервозность или стресс. В медицинских терминах, однако, гипертензия относится к состоянию повышенного кровяного давления независимо от причины его проявления. Ее называют "бесшумный киллер", так как гипертензия обычно не вызывает симптомов, непосредственно связанных с заболеванием, на протяжении многих лет, до тех пор, пока не повреждается жизненно важный орган. Высокое кровяное давление определяют как систолическое давление в состоянии покоя, которое в среднем составляет 140 мм Hg или больше, диастолическое давление в состоянии покоя, которое в среднем равняется 90 мм Hg или больше, или и то и другое. При высоком кровяном давлении обычно оба и систолическое и диастолическое давления являются повышенными.
В качестве вторичного действия сахарного диабета происходит поражение нервов, которые регулируют кровяное давление и процессы пищеварения. Это приводит к колебаниям кровяного давления; к осложнениям, связанным с глотанием, и изменению функции желудочно-кишечного тракта с приступами диареи. Кроме того, как вторичное действие сахарного диабета в отдельных участках тканей артериальной стенки происходит образование атеросклеротических бляшек, которые могут блокировать артерии больших и средних размеров в сердце, головном мозге, ногах и пенисе. Стенки небольших кровеносных сосудов повреждаются до такой степени, что сосуды не транспортируют кислород нормальным образом и могут допускать утечку.
Другие определения и классификация высокого кровяного давления представлены в Merck Manual of Medical Information-Home Edition, Merck & Co., 2000. В тех случаях, когда систолическое и диастолическое давления человека соответствуют различным категориям, для классифицирования кровяного давления используют более высокую категорию. Например, 160/92 классифицируют как гипертензию 2 стадии, и 180/120 классифицируют как гипертензию 4 стадии. Оптимальное кровяное давление, которое сводит к минимуму риск возникновения проблем, связанных с сердечно-сосудистыми заболеваниями, ниже 120/80 мм Hg. Однако необычно низкие значения давлениядолжны учитываться.
Если человек имеет высокое кровяное давление, соответствующее 3 стадии (тяжелая), или оно носит устойчивый характер и не лечится, то возникают такие симптомы, как: головная боль, утомление, тошнота, рвота, одышка, возбужденное состояние и размытое зрение из-за поражения головного мозга, глаз, сердца и почек. Иногда у людей с высоким кровяным давлением наступает состояние дремоты и даже развивается кома, вызванные отеком мозга. Это состояние, называемое гипертонической энцефалопатией, требует экстренного лечения.
Отсутствие систематического лечения высокого кровяного давления у людей увеличивает риск развития болезни сердца (таких как сердечная недостаточность или сердечный приступ), почечной недостаточности и удара в раннем возрасте. Высокое кровяное давление - наиболее важный фактор риска удара. Кроме того, оно является одним из трех основных факторов риска для развития сердечного приступа (инфаркта миокарда), которые человек может регулировать; другими двумя факторами являются курение и высокий уровень холестерина.
Раскрытие изобретения
Настоящее изобретение обеспечивает новые применения DPIV-ингибиторов формул 1-12 и их соответствующих фармацевтически приемлемых аддитивных солей кислот для снижения кровяного давления или лечения связанных с этим расстройств у млекопитающих.
Пониженная экспрессия эктопептидазы DPIV и недостаток DPIV-подобной активности у мутантных крыс F344, испытывающих недостаток в ферментативной активности и экспрессии DPIV, приводит к снижению кровяного давления. Были испытаны мутантные сублинии F344, испытывающие недостаток в ферментативной активности DPIV, и дикие линии типаF344. Хроническая внутрижелудочная инфузия изолейцил-цианопирролидин ТФК и изолейцил-тиазолидин фумарата при помощи осмотических мининасосов на протяжении двух недель снижала кровяное давление у крыс доза-зависимым образом. Таким образом, кровяное давление снижается при хроническом лечении с использованием различных ингибиторов DPIV (изолейцил-тиазолидин фумарат; изолейцил-цианопирролидин ТФК), что дает основание предположить проявление сходных защитных действий со стороны двух различных DPIV-ингибиторов/лигандов. Возможно, изолейцил-тиазолидин фумарат и изолейцил-цианопирролидин ТФК защищают от высокого кровяного давления посредством увеличения уровней субстратов DPIV, которые косвенно опосредуют соответствующие действия.
Настоящее изобретение относится к новому способу, в котором уменьшение активности фермента дипептидилпептидазы (DPIV или СD26) или активности DPIV-подобных ферментов в крови млекопитающих специфическими эффекторами фермента приводит к снижению деградации эндогенных или экзогенно вводимых инсулинотропных пептидов (инкретинов), пептидного гормона, секретируемого желудком/глюкоза-зависимого инсулинотропного полипептида 1-42 (GIP1-42) и глюкагон-подобного Пептида-1 7-36 амида(GLP-17-36)(или аналогов этих пептидов). Уменьшение концентрации этих пептидов или их аналогов, являющееся результатом их деградации DPIV и DPIV-подобными ферментами, будет в соответствии с этим снижено или замедлено.
В результате повышения стабильности эндогенных или экзогенно вводимых инкретинов или их аналогов, вызванной уменьшением DPIV-активности, их инсулинотропные действия повышаются, что приводит к ярко выраженному стимулированию секреции инсулина из панкреатических островков Лангерганса и более быстрому удалению глюкозы из крови. В результате толерантность к глюкозе повышается.
Как следствие, метаболические аномалии, ассоциированные с сахарным диабетом, включая аномалии углеводного и липидного обмена, глюкозурию и диабетический кетоацидоз, и хронические изменения, такие как: микроваскулярная и макроваскулярная болезнь, полиневропатия и диабетическая ретинопатия, которые являются следствием длительных повышенных концентраций циркулирующей глюкозы, предотвращаются или облегчаются и, в частности, снижаются высокие уровни кровяного давления.
Настоящее изобретение представляет новый подход к снижению повышенных концентраций глюкозы в крови и повышенных уровней кровяного давления. Это простой, коммерчески выгодный и пригодный к использованию для лечения, особенно болезней человека, которые вызваны повышенными или экстраординарными уровнями глюкозы в крови и/или кровяного давления.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Более глубокое понимание настоящего изобретения можно получить, ссылаясь на сопроводительные чертежи, где:
Фиг.1 демонстрирует MALDI-TOF-анализ DPIV-катализируемого гидролиза GIP1-42 (a) и GLP7˜36 (b) и ингибирование их гидролиза изолейцил-тиазолидином.
Фиг.2демонстрирует ВЭЖХ-анализ присутствия в сыворотке метаболитов GLP-1 в присутствии ингибитора DPIV, изолейцил-тиазолидина, in vivo.
Фиг.3демонстрируетвлияниеDPIV-ингибитора, изолейцил-тиазолидина на различные параметры в крови вн.дуод. (i.d.)-глюкозостимулированной крысы.
Фиг.4демонстрирует влияние хронического перорального лечения тучных (fa/fa) VDF крыс линии Zucker DPIV-ингибитором, изолейцил-тиазолидином на уровни глюкозы в крови натощак в течение 12-недельного приема лекарственного средства.
Фиг.5 демонстрирует влияние хронического лечения тучных (fa/fa) VDF крыс линии Zucker DPIV-ингибитором, изолейцил-тиазолидином на систолическое кровяное давление в пределах 8-недельного применения лекарственного средства (систолическое кровяное давление измеряют, используя способ наложения манжеты на хвост).
Фиг.6демонстрирует дозозависимое снижение уровней глюкозы в крови у диабетических крыс линии Zucker после перорального введения 5 мг/кг, 15 мг/кг, 50 мг/кг м.т. глутаминилпирролидина и плацебо соответственно;
Фиг.7 демонстрирует дозозависимое снижение уровней глюкозы в крови у диабетических крыс линии Zucker после перорального введения 5 мг/кг, 15 мг/кг, 50 мг/кг м.т. глутаминилтиазолидина и плацебо соответственно;
Фиг.8 демонстрирует химическую структуру пироглутаминил-тиазолидина, продукта деградации, обнаруженного после перорального введения глутаминилтиазолидина крысам линии Wistar; и
Фиг.9демонстрирует хроматограмму экстракта плазмы крысы, полученного после перорального введения глутаминилтиазолидина тучным крысам линии Zucker. Пик при 2,95 мин представляет глутаминилтиазолидин и пик при 6,57 мин представляет пироглутаминилтиазолидин.
Детальное описание изобретение
Целью настоящего изобретения является разработка простого и нового способа снижения уровня глюкозы в крови и/или кровяного давления, в котором уменьшение активности фермента дипептидилпептидазы (DPIV или СD26) или активности DPIV-подобных ферментов в крови млекопитающих, индуцируемое эффекторами фермента, приводит к снижению деградации эндогенных (или экзогенно вводимых) инсулинотропных пептидов (инкретинов), пептидного гормона, секретируемого желудком/глюкозозависимого инсулинотропного полипептида 1-42 (GIP1-42) и глюкагон-подобного пептида-1 амида (7-36) (GLP-17-36) (или аналогов этих пептидов). В соответствии с этим уменьшение концентрации этих пептидов или их аналогов, являющееся результатом их деградации DPIV и DPIV-подобными ферментами, будет снижаться или замедляться.
Настоящее изобретение основано на неожиданном установлении того факта, что уменьшение ферментативной активности дипептидилпептидазы IV (DPIV или СD26) или активности DPIV-подобных ферментов в теле млекопитающих in vivo приводит к повышению толерантности к глюкозе и к снижению высокого кровяного давления.
Авторами было обнаружено, что:
1. Снижение активности дипептидилпептидазы IV (DPIV или СD26) или DPIV-подобных ферментов ведет к повышению стабильности глюкозостимулированных эндогенно выделяемых или экзогенно вводимых инкретинов (или их аналогов) с вытекающим отсюда следствием, что введение эффекторов DPIV или DPIV-подобных белков может быть использовано для регулирования деградации инкретинов, участвующих в циркуляции.
2. Повышенная биологическая стабильность инкретинов (или их аналогов) приводит к изменению реакции инсулина.
3. Повышенная стабильность циркулирующих инкретинов, вызванная уменьшением дипептидилпептидазы IV (DPIV или СD26) или DPIV-подобных ферментов, приводит к последующему изменению инсулинзависимой утилизации (удалению) глюкозы, указывая на то, что толерантность к глюкозе может быть повышена применением DPIV-эффекторов.
4. Высокое кровяное давление снижается.
Соответственно данное изобретение относится к использованию эффекторов активности дипептидилпептидазы IV (DPIV) или DPIV-подобных ферментов для снижения повышенных уровней глюкозы в крови и/или кровяного давления, таких как: уровни, обнаруживаемые у млекопитающих, демонстрирующих клинически несоответствующие базальную и пост-обеденную гипергликемию. Применение в соответствии с изобретением, в частности, характеризуется введением эффекторов активности DPIV или DPIV-подобных ферментов для предотвращения или облегчения патологических аномалий метаболизма млекопитающих, таких как: глюкозурия, гиперлипидемия, диабетический кетоацидоз, диабетическая ретинопатия и сахарный диабет. В другом предпочтительном варианте осуществления изобретение относится к способу снижения повышенных уровней глюкозы в крови у млекопитающих, таких как уровни, обнаруживаемые у млекопитающего, демонстрирующего клинически несоответствующие базальную и пост-обеденную гипергликемию, включающему введение млекопитающему, при необходимости такого лечения, терапевтически эффективного количества эффектора активности дипептидилпептидазы IV (DPIV) или DPIV-подобных ферментов.
В другом предпочтительном варианте осуществления изобретение касается эффекторов активности дипептидилпептидазы IV (DPIV) или DPIV-подобных ферментов, применяемых в способе снижения повышенных уровней глюкозы в крови и/или кровяного давления у млекопитающих, таких как уровни, обнаруживаемые у млекопитающих, демонстрирующих клинически несоответствующие базальную и пост-обеденную гипергликемию.
Предлагаемые эффекторы DPIV и DPIV-подобных ферментов согласно настоящему изобретению могут быть использованы в фармацевтических составах как ингибиторы ферментов, субстраты, псевдосубстраты, ингибиторы экспрессии гена DPIV, связывающие белки или антитела белков ферментов-мишеней или в виде комбинации таких различных соединений, которые снижают концентрацию DPIV белка и DPIV-подобного белка или активность ферментов у млекопитающих. Эффекторами согласно изобретению являются, например, ингибиторы DPIV, такие как производные дипептидов или дипептидные миметики, как например аланилпиролидид, изолейцилтиазолидин, а также псевдосубстрат N-валил-пролил, O-бензоил-гидроксиламин. Такие соединения известны из литературы [DEMUTH, H-U., Recent developments in the irreversible inhibition of serine and cysteine proteases. J. Enzyme Inhibition 3, 249 (1990)] или они могут быть синтезированы согласно способам, описанным в литературе.
Способ согласно изобретению представляет новый подход к снижению повышенной концентрации глюкозы, циркулирующей в крови млекопитающих, и к снижению высоких уровней кровяного давления.
Настоящее изобретение относится к области ингибирования дипептидилпептидазы IV (DPIV) и, в частности к новому использованию ингибиторов активности DPIV и DPIV-подобных ферментов для снижения высоких уровней кровяного давления или (лечения) связанных с этим расстройств у млекопитающих, и фармацевтических композиций, содержащих вышеуказанные соединения.
В отличие от других предложенных способов в данной области настоящее изобретение обеспечивает перорально доступную терапию низкомолекулярными ингибиторами дипептидилпептидазы IV. Настоящее изобретение представляет новый подход к снижению уровней кровяного давления и лечению связанных с этим нарушений у млекопитающих. Этот подход удобен для пользователя, коммерчески выгоден и пригоден для использования в лечебной схеме, особенно касающейся болезней человека.
Исходя из полученных данных, исследование роли экспрессии DPIV и ферментативной активности DPIV в регулировании кровяного давления согласно изобретению позволило установить, что пероральное введение ингибиторов DPIV приводит к уменьшению уровней кровяного давления.
Цель настоящего изобретения состоит в разработке ингибиторов дипептидилпептидазы IV и/или лигандов, которые обладали бы высокой биодоступностью. В другом предпочтительном варианте осуществления настоящее изобретение обеспечивает ингибиторы DPIV, которые обладают точно предсказуемым временем активности в ткани-мишени.
Примерами перорально доступных средств с низкой молекулярной массой являются пролекарства стабильных и нестабильных ингибиторов дипептидилпептидазы IV общей формулы A-B-C, где A представляет аминокислоту, B представляет химическую связь между A и C или аминокислоту и С представляет нестабильный или стабильный ингибитор дипептидилпептидазы IV соответственно. Такие соединения описаны в международных патентных публикациях WO 99/67278 и WO 99/67279, на раскрытия которых относительно обеспечения, определения, использования и получения пролекарств здесь ссылаются во всей их полноте. Особенно это касается детального описания определений А, В и С.
Настоящее изобретение относится к новому способу, в котором уменьшение активности фермента, дипептидилпептидазы (DPIV или CD26) или активности DPIV-подобных ферментов, или где связывание специфического лиганда DPIV оказывают благотворные действия в организмах млекопитающих, индуцированные эффекторами фермента, и ведет как причинное следствие к снижению кровяного давления у млекопитающего. В результате млекопитающие, имеющие повышенное кровяное давление, будут получать пользу от лечения ингибиторами активности DPIV и DPIV-подобных ферментов.
Способ и применение согласно настоящему изобретению включает профилактику повышения кровяного давления или снижение кровяного давления и родственных расстройств у животного, включая человека, путем ингибирования DPIV, или активностей родственных ферментов, используя ингибитор или лиганд вышеуказанных ферментов. В большинстве обстоятельств пероральное введение ингибитора DPIV может быть предпочтительным.
В дальнейшем настоящее изобретение иллюстрируется, ссылаясь на нижеследующие примеры, сфокусированные на действии в направлении снижения кровяного давления и уровней глюкозы в крови, вызванном уменьшением DPIV-подобной активности и/или связыванием.
В одном иллюстративном варианте воплощения настоящее изобретение относится к применению дипептид-подобных соединений и соединений, подобных дипептидным соединениям, которые образованы из аминокислоты и тиазолидиновой или пирролидиновой группы, и их солей, упоминаемых в дальнейшем как дипептид-подобные соединения. Предпочтительно аминокислота и группа тиазолидина или пирролидина связаны амидной связью.
Особенно подходящими для этой цели согласно изобретению являются дипептидные соединения, в которых аминокислота предпочтительно выбрана из природной аминокислоты, такой как, например, лейцин, валин, глутамин, глутаминовая кислота, пролин, изолейцин, аспарагины и аспарагиновая кислота.
Дипептид-подобные соединения, используемые согласно изобретению, демонстрируют при концентрации (дипептидных соединений) 10 мкМ уменьшение активности дипептидилпептидазы IV или активностей DPIV-подобных ферментов, по крайней мере, на 10%, особенно, по крайней мере, на 40%. Зачастую требуется уменьшение активности, по крайней мере, на 60% или, по крайней мере, на 70%. Предпочтительные эффекторы могут также демонстрировать уменьшение активности максимально на 20% или 30%.
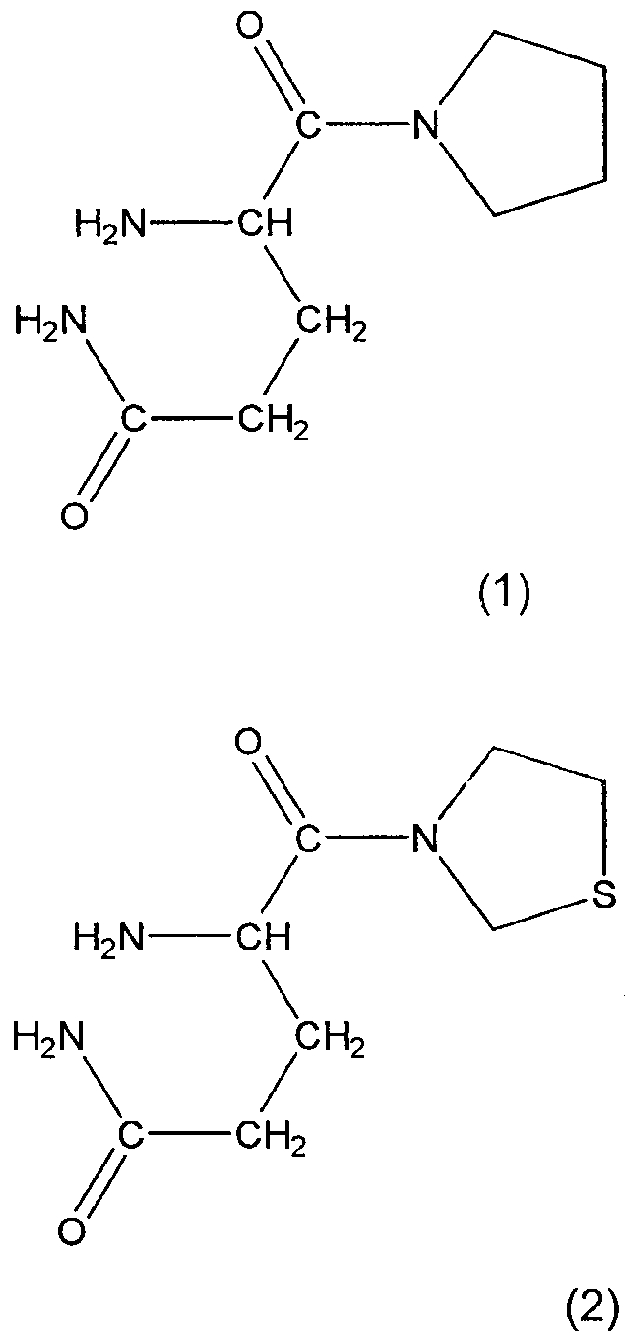
Предпочтительными соединениями являются N-валил-пролил, O-бензоил-гидроксиламин, аланил-пирролидин, изолейцил-тиазолидин так же как и их оптические изомеры L-алло-изолейцил-тиазолидин, L-трео-изолейцил-пирролидин и их соли, особенно соли фумаровой кислоты, и L-алло-изолейцил-пирролидин и его соли. Особенно предпочтительными соединениями являются глутаминилпирролидин и глутаминилтиазолидин формул 1 и 2:
Другие предпочтительные соединения представлены в Таблице 1.
Соли дипептид-подобных соединений могут быть в молярном отношении дипептид(-аналог) компонент к компоненту соли 1:1 или 2:1.
Такой солью является, например, (Ile-Thia)2 фумаровая кислота.
Таблица 1: Структуры дополнительных дипептидных соединений
В другом предпочтительном варианте воплощения настоящее изобретение представляет применение пептидных соединений формулы 3, полезных для конкурентной модуляции катализа дипептидилпептидазой IV:
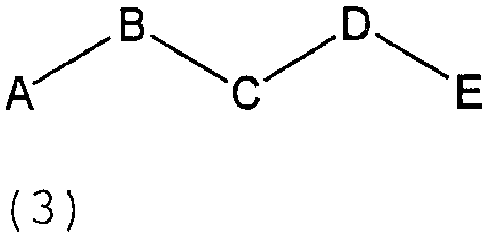
где A, B, C, Dи E представляют независимо любые аминокислотные части, включая протеиногенные аминокислоты, не-протеиногенные аминокислоты, L-аминокислоты и D-аминокислоты, и где E и/или D могут отсутствовать.
Дополнительные условия в отношении формулы (3):
A представляет аминокислоту, исключая D-аминокислоту,
B представляет аминокислоту, выбранную из Pro, Ala, Ser, Gly, Hyp, азетидин-(2)-карбоновой кислоты и пипеколиновой кислоты,
C представляет любую аминокислоту, исключая Pro, Hyp, азетидин-(2)-карбоновую кислоту, пипеколиновую кислоту и исключая N-алкилированные аминокислоты, например N-метилвалин и саркозин,
D представляет любую аминокислоту или отсутствует, и
E представляет любую аминокислоту или отсутствует,
или:
C представляет любую аминокислоту, кроме Pro, Hyp, азетидин-(2)-карбоновой кислоты, пипеколиновой кислоты, кроме N-алкилированных аминокислот, например, N-метилвалина и саркозина, и кроме D-аминокислоты;
D представляетлюбую аминокислоту, выбранную изPro, Ala, Ser, Gly, Hyp, азетидин-(2)-карбоновой кислоты и пипеколиновой кислоты, и
E представляет любую аминокислоту, кроме Pro, Hyp, азетидин-(2)-карбоновой кислоты, пипеколиновой кислоты и, исключая N-алкилированные аминокислоты, например, N-метилвалин и саркозин.
Примерами аминокислот, которые могут быть использованы в настоящем изобретении, являются L и D-аминокислоты, N-метил-аминокислоты; алло- и трео-формы Ile и Thr, которые могут, например, быть a-, Я- и ω-аминокислотами, среди которых a-аминокислоты являются предпочтительными.
Примерами аминокислот для формулы изобретения и описания являются:
аспарагиновая кислота (Asp), глутаминовая кислота (Glu), аргинин (Arg), лизин (Lys), гистидин (His), глицин (Gly), серин (Ser) и цистеин (Cys), треонин (Thr), аспарагин (Asn), глутамин (Gln), тирозин (Tyr), аланин (Ala), пролин (Pro), валин (Val), изолейцин (Ile), лейцин (Leu), метионин (Met), фенилаланин (Phe), триптофан (Trp), гидроксипролин (Hyp), бета-аланин (бета-Ala), 2-аминооктановая кислота (Aoa), азетидин-(2)-карбоновая кислота (Ace), пипеколиновая кислота (Pip), 3-аминопропионовая кислота, 4-аминомасляная кислота и т.д., альфа-аминоизомасляная кислота (Aib), саркозин (Sar), орнитин (Orn), цитруллин (Cit), гомоаргинин (Har), т-бутилаланин (т-бутил-Ala), т-бутилглицин (т-бутил-Gly), N-метилизолейцин (N-MeIle), фенилглицин (Phg), циклогексилаланин (Cha), норлейцин (Nle), цистеиновая кислота (Cya) и метионин сульфоксид (MSO), Ацетил-Lys, модифицированные аминокислоты, такие как: фосфорил-серин (Ser(P)), бензил-серин (Ser(Bzl)) и фосфорил-тирозин (Tyr(P)), 2-аминомасляная кислота (Abu), аминоэтилцистеин (AECys), карбоксиметилцистеин (Cmc), дегидроаланин (Dha), дегидроамино-2-масляная кислота (Dhb), карбоксиглутаминовая кислота (Gla), гомосерин (Hse), гидроксилизин (Hyl), цис-гидроксипролин (цисHyp), транс-гидроксипролин (трансHyp), изовалин (Iva), пироглутаминовая кислота (Pyr), норвалин (Nva), 2-аминобензойная кислота (2-Abz), 3-аминобензойная кислота (3-Abz), 4-аминобензойная кислота (4-Abz), 4-(аминометил)бензойная кислота (Amb), 4-(аминометил)циклогексанкарбоновая кислота (4-Amc), Пеницилламин (Pen), 2-амино-4-цианомасляная кислота (Cba), циклоалкан-карбоновые кислоты.
Примерами ω-аминокислот являются, например: 5-Ara (аминовалериановая кислота), 6-Ahx (аминогексановая кислота), 8-Aoc (аминооктановая кислота), 9-Anc (аминованоевая кислота), 10-Adc (аминодекановая кислота), 11-Aun (аминоундекановая кислота), 12-Ado (аминододекановая кислота).
Дополнительными аминокислотами являются: инданилглицин (Igl), индолин-2-карбоновая кислота (Idc), октагидроиндол-2-карбоновая кислота (Oic), диаминопропионовая кислота (Dpr), диаминомасляная кислота (Dbu), нафтилаланин (1-Nal), (2-Nal), 4-аминофенилаланин (Phe(4-NH2)), 4-бензоилфенилаланин (Bpa), дифенилаланин (Dip), 4-бромфенилаланин (Phe(4-Br)), 2-хлорфенилаланин (Phe(2-Cl)), 3-хлорфенилаланин (Phe(3-Cl)), 4-хлорфенилаланин (Phe(4-Cl)), 3,4-хлорфенилаланин (Phe(3,4-Cl2)), 3-фторфенилаланин (Phe(3-F)), 4-фторфенилаланин (Phe(4-F)), 3,4-фторфенилаланин (Phe(3,4-F2)), пентафторфенилаланин (Phe(F5)), 4-гуанидинофенилаланин (Phe(4-гуанидино)), гомофенилаланин (hPhe), 3-иодфенилаланин (Phe(3-J)), 4-иодфенилаланин (Phe(4-J)), 4-метилфенилаланин (Phe(4-Me)), 4-нитрофенилаланин (Phe-4-N02)), бифенилаланин (Bip), 4-фосфонометилфенилаланин (Pmp), циклогексилглицин (Ghg), 3-пиридинилаланин (3-Pal), 4-пиридинилаланин (4-Pal), 3,4-дегидропролин (A-Pro), 4-кетопролин (Pro(4-кето)), тиопролин (Thz), изонипекотиновая кислота (Inp), 1,2,3,4,-тетрагидроизохинолин-3-карбоновая кислота (Tic), пропаргилглицин (Pra), 6-гидроксинорлейцин (NU(6-OH)), гомотирозин (hTyr), 3-иодотирозин (Tyr(3-J)), 3,5-дииодотирозин (Tyr(3,5-J2)), d-метил-тирозин (Tyr(Me)), 3-N02-тирозин (Tyr(3-N02)), фосфотирозин (Tyr(PO3H2)), алкилглицин, 1-аминоиндан-1-карбокси кислота, 2-аминоиндан-2-карбокси кислота (Aic), 4-амино-метилпиррол-2-карбоновая кислота (Py), 4-амино-пирролидин-2-карбоновая кислота (Abpc), 2-аминотетралин-2-карбоновая кислота (Atc), диаминоуксусная кислота (Gly(NH2)), диаминомасляная кислота (Dab), 1,3-дигидро-2H-изоинол-карбоновая кислота (Disc), гомоциклогексилаланин (hCha), гомофенилаланин (hPhe oder Hof), транс-3-фенил-азетидин-2-карбоновая кислота, 4-фенил-пирролидин-2-карбоновая кислота, 5-фенил-пирролидин-2-карбоновая кислота, 3-пиридилаланин (3-Pya), 4-пиридилаланин (4-Pya), стирилаланин, тетрагидроизохинолин-1-карбоновая кислота (Tiq), 1,2,3,4-тетрагидроноргарман-3-карбоновая кислота (Tpi), Я-(2-тиенил)аланин (Tha).
Другие замены аминокислот для аминокислот, кодированных в генетическом коде, могут быть также включены в пептидные соединения в пределах объема изобретения и могут быть классифицированы внутри этой общей схемы.
Термин протеиногенные аминокислоты означает a-аминокислоты, являющиеся производными природных белков. Термин не-протеиногенные аминокислоты означает все другие аминокислоты, которые не являются строительными блоками обычных природных белков.
Результирующие пептиды могут быть синтезированы как в форме со свободной С-концевой кислотой, так и форме С-концевого амида. Пептиды со свободной кислотой или амиды могут варьироваться путем модификаций их боковых цепей. Такие модификации боковой цепи включают, например, но ими не ограничиваются, образование гомосерина, образование пироглутаминовой кислоты, образование дисульфидной связи, деамидирование остатков аспарагина или глутамина, метилирование, т-бутилирование, т-бутилоксикарбонилирование, 4-метилбензилирование, тиоанизилирование, тиокрезилирование, бензилоксиметилирование, 4-нитрофенилирование, бензилоксикарбонилирование, 2-нитробензоилирование, 2-нитросульфенилирование, 4-толуолсульфонилирование, пентафторфенилирование, дифенилметилирование, 2-хлорбензилоксикарбонилирование, 2,4,5-трихлорфенилирование, 2-бромбензилоксикарбонилирование, 9-флуоренилметилоксикарбонилирование, трифенилметилирование, 2,2,5,7,8-пентаметилхроман-6-сульфонилирование, гидроксилирование, окисление метионина, формилирование, ацетилирование, анизилирование, бензилирование, бензоилирование, трифтороацетилирование, карбоксилирование аспарагиновой кислоты или глутаминовой кислоты, фосфорилирование, сульфатирование, цистеинилирование, гликолизация пентозами, дезоксигексозами, гексозаминами, гексозами или N-ацетилгексозаминами, фарнезилирование, муристолизирование, биотинилирование, пальмитоилирование, стеароилирование, геранилгеранилирование, глутатионилирование, 5'-аденозилирование, ADP-рибозилирование, модификация N-гликолилнейраминовой кислотой, N-ацетилнейраминовой кислотой, пиридоксаль-фосфатом, липоевой кислотой, 4'-фосфопантетином, или N-гидроксисукцинимидом.
В соединениях формулы (3) аминокислотные части A, B, C, D, и E соответственно присоединены к соседней части амидными связями обычным способом согласно стандартной номенклатуре так, что амино-конец (N-конец) аминокислот (пептида) изображается слева (N-конец) и карбоксил-конец аминокислот изображается справа (С-конец).
До настоящего изобретения как пептидные субстраты пролин-специфичной сериновой протеазы дипептидилпептидазы IV in vitro были известнытрипептиды Diprotin A (Ile-Pro-Ile), Diprotin B (Val-Pro-Leu) и Diprotin C (Val-Pro-Ile). Авторами неожиданно было обнаружено, что соединения, раскрываемые здесь выше и ниже, действуют как субстраты дипептидилпептидазы IV in vivo у млекопитающего и в фармакологических дозах снижают кровяное давление и облегчают патологические аномалии метаболизма у млекопитающих, такие, как: глюкозурия, гиперлипидемия, метаболический ацидоз и сахарный диабет, посредством конкурентного катализа.
Особенно предпочтительные соединения настоящего изобретения, которые используют в качестве модуляторов дипептидилпептидазы IV и DPIV-подобных ферментов, включают такие соединения, которые демонстрируют Ki-значения для связывания DPIV, эффективны в ингибировании DPIV in vivo после внутривенного (i.v.) и/или перорального (p.o.) введения крысам линии Wistar.
Другими предпочтительными соединениями являются пептидилкетоны формулы 4:
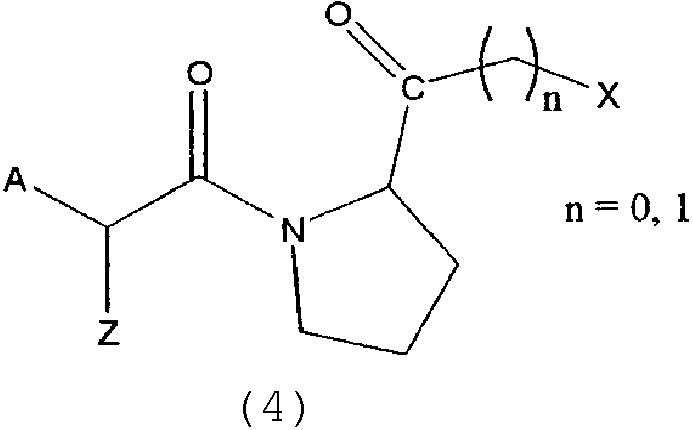
где A выбран из

X1 представляет H или ацильную или оксикарбонильную группу, включая все аминокислотны или пептидные остатки,
X2 представляет H, -(CH)n-NH-C5H3N-Y с n=2-4 или C5H3N-Y (дивалентный остаток пиридила) и Y выбран из H, Br, Cl, I, NO2 или CN,
Х3 представляет H или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген, нитро, циано или карбокси группами,
X4 представляет H или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген, нитро, циано или карбокси группами,
X5 представляет H или алкил, алкокси или фенил,
X6 представляет H или алкил;
для n=1
X выбран из H, OR2, SR2, NR2R3, N+R2R3R4, где
R2 означает ацильные остатки, которые являются незамещенными или замещенными одним, двумя или несколькими алкилом, циклоалкилом, арилом или гетероарилом, или обозначает все аминокислоты и пептидные остатки, или алкильные остатки, которые незамещены или замещены одним, двумя или несколькими алкилами, циклоалкилами, арилами и гетероарильными остатками,
R3 обозначает алкильные и ацильные функции, где R2 и R3 могут быть частью одной или нескольких кольцевых структур ненасыщенных и насыщенных карбоциклических или гетероциклических структур,
R4 означает алкильные остатки, где R2 и R4 или R3 и R4 могут быть частью одной или нескольких кольцевых структур насыщенных и ненасыщенных карбоциклических или гетероциклических структур;
для n=0
X выбран из:
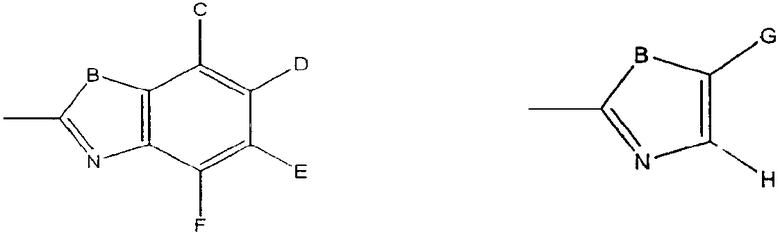
где B означает О, S, NR5, где R5 представляет H, алкилиден или ацил,
C, D, E, F, G, H независимо выбраны из незамещенных и замещенных алкильных, оксиалкильных, тиоалкильных, аминоалкильных, карбонилалкильных, ацильных, карбамоильных, арильных и гетероарильных остатков; и
для n=0 и n=1
Z выбран из H, или С1-С9 алкила с разветвленной или одинарной цепью или С2-С9 алкенила с разветвленной или одинарной цепью, С3-С8 циклоалкила, С5-С7 циклоалкенила, арил- или гетероарильного остатка, или боковой цепи, выбранной из всех боковых цепей всех природных аминокислот или их производных.
Кроме того, согласно настоящему изобретению раскрываются соединения формул 5, 6, 7, 8, 9, 10 и 11, включая все стереомеры и их фармацевтически приемлемые соли, и все они могут быть использованы:
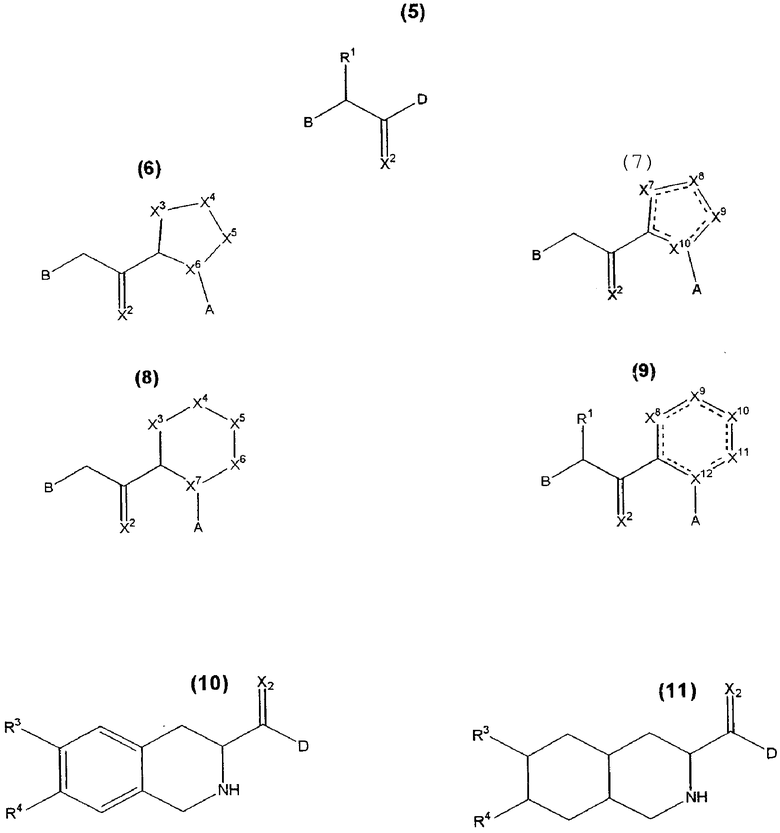
где R1 представляет H, разветвленный или прямой С1-С9 алкил, разветвленный или прямой С2-С9 алкенил, С3-С8 циклоалкил, С5-С7 циклоалкенил, арил или гетероарил или боковую цепь природной аминокислоты или ее производного,
R3 и R4 выбраны из H, гидрокси, алкила, алкокси, арилокси, нитро, циано или галогена,
A представляет H или изостер карбоновой кислоты, функциональную группу, выбранную из CN, SO3H, CONHOH, P03R5R6, тетразола, амида, сложного эфира, ангидрида, тиазола и имидазола,
B выбран из
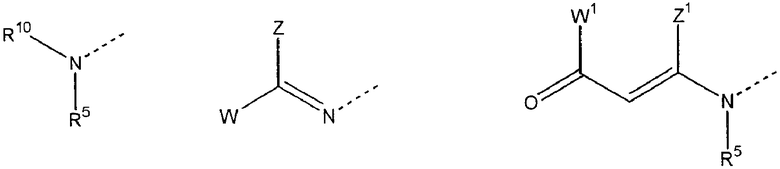
где R5 представляет H, -(CH)n-NH-C5H3N-Y c n=2-4 и C5H3N-Y (дивалентный остаток пиридила) с Y=H, Br, CL, I, NO2 или CN,
R10 представляет H, ацил, оксикарбонил или остаток аминокислоты,
W представляет H или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген, нитро, циано или карбокси группами,
W1 представляет H, алкил, алкокси или фенил,
Z представляет H или фенил или пиридил, незамещенный или замещенный одним, двумя или несколькими алкилом, алкокси, галогеном, нитро, циано или карбокси остатками,
Z' представляет H или алкил,
D представляет циклический C4-C7 алкил, C4-C7 алкенил, который может быть незамещенным или замещенным одной, двумя или несколькими группами, или циклический (4-7)-членный гетероалкил или циклический (4-7)-членный гетероалкенил,
X2 представляет О, NR6, N+(R7)2, или S,
X3 до X12 независимо выбраны из CH2, CR8R9, NR6, N+(R7)2, О, S, SO и SO2, включая все насыщенные или ненасыщенные структуры,
R6, R7, R8, R9 независимо выбраны из H, разветвленного или прямого C1-C9 алкила, разветвленного или прямого C2-C9 алкенила, C3-C8 циклоалкила, C5-C7 циклоалкенила, арила или гетероарила,
при следующих условиях:
Формула 6: X6 представляет CH, если A не является H,
Формула 7: X10 представляет C, если А не является H,
Формула 8: X7 представляет CH, если A не является H,
Формула 9: X12 представляет C, если A не является H,
На протяжении всего описания и формулы изобретения выражение "ацил" может означать С1-20 ацильный остаток, предпочтительно С1-8 ацильный остаток и особенно предпочтителен С1-4 ацильный остаток, "циклоалкил" может означать C3-12 циклоалкильный остаток, предпочтительно С4, С5 или С6 циклоалкильный остаток, "карбоциклический" может означать С3-12 карбоциклический остаток, предпочтительно С4, С5 или С6 карбоциклический остаток. "Гетероарил" обозначает арильный остаток, где 1 до 4, предпочтительно 1, 2 или 3 атомов кольца заменены гетероатомами, такими как N, S или О. "Гетероцикл" означает циклоалкильный остаток, где 1, 2 или 3 атомов кольца заменены гетероатомами, такими как N, S или О. "Пептиды" выбирают от дипептидов до декапептидов, предпочтительно дипептиды, трипептиды, тетрапептиды и пентапептиды. Аминокислоты для образования "пептидов" могут быть выбраны из аминокислот, перечисленных выше.
Из-за широкого распределения белка в организме и разнообразия механизмов, в которых задействованы DPIV, DPIV-активность и DPIV-родственные белки, системная терапия (интеральное или парентеральное введение) DPIV-ингибиторами может привести к ряду нежелательных побочных действий.
Кроме того, задачей, которую предстоит решить, является разработка соединений, которые можно было бы использовать для целенаправленного воздействия на локально проявляющиеся патофизиологические и физиологические процессы. Задача изобретения, в частности, заключается в обеспечении локально ограниченного ингибирования DPIV или DPIV-аналогичной активности с целью целенаправленного вмешательства в регулирование активности локально активных субстратов.
Данная задача решается в соответствии с изобретением при помощи соединений общей формулы (12)

где A представляет аминокислоту, имеющую, по крайней мере, одну функциональную группу в боковой цепи,
B представляет химическое соединение, ковалентно связанное с, по крайней мере, одной функциональной группой боковой цепи А,
C представляет тиазолидин, пирролидин, цианопирролидин, гидроксипролин, дегидропролин или пиперидиновую группу, связанную с А амидной связью.
Соединения можно, например, использовать для снижения кровяного давления, воздействуя на DPIV или DPIV-подобные ферменты эндотелия кровеносных сосудов.
В соответствии с предпочтительным вариантом осуществления изобретения, фармацевтические композиции используют, включающие, по крайней мере, одно соединение общей формулы (12) и, по крайней мере, один обычный адъювант, приемлемый для места действия.
Предпочтительно A представляет a-аминокислоту, особенно природную a-аминокислоту, имеющую одну, две или больше функциональных групп в боковой цепи, предпочтительно треонин, тирозин, серин, аргинин, лизин, аспарагиновую кислоту, глутаминовую кислоту или цистеин.
Предпочтительно В представляет олигопептид с длиной цепи вплоть до 20 аминокислот, полиэтиленгликоль, имеющий молярную массу вплоть до 20000 г/моль, необязательно замещенный органический амин, амид, спирт, кислоту или ароматическое соединение, имеющее от 8 до 50 атомов С.
На протяжении всего текста описания и формулы изобретения выражение "алкил" означает С1-50 алкильную группу, предпочтительно С6-30 алкильную группу, особенно С8-12 алкильную группу; например, алкильной группой может быть метильная, этильная, пропильная, изопропильная или бутильная группа. Выражение "алк", например, в выражении "алкокси" и выражение "алкан", например, в выражении "алканоил" означает "алкил"; ароматические соединения представляют предпочтительно замещенные или необязательно незамещенные фенил, бензил, нафтил, бифенил или антраценовые группы, которые предпочтительно имеют, по крайней мере, 8 атомов С; выражение "алкенил" может означать С2-10 алкенильную группу, предпочтительно С2-6 алкенильную группу, которая имеет двойную связь(и) в любом желаемом местоположении и может быть замещенной или незамещенной; выражение "алкинил" может означать C2-10 алкинильную группу, предпочтительноС2-6 алкинильную группу, которая имеет тройную связь(и) в любом желаемом местоположении и может быть замещенной или незамещенной; выражение "замещенный" или заместитель может означать любое требуемое замещение одним или несколькими, предпочтительно одной или двумя, алкил, алкенил, алкинил, моно- или мультивалентными ацил-, алканоил-, алкоксиалканоил- или алкоксиалкильными группами; вышеупомянутые заместители могут, в свою очередь, иметь одну или несколько (но предпочтительно не иметь) алкил, алкенил, алкинил, моно- или мультивалентных ацил, алканоил, алкоксиалканоил или алкоксиалкильных групп в качестве боковых групп; органические амины, амиды, спирты или кислоты, каждый из которых имеет от 8 до 50 атомов С, предпочтительно от 10 до 20 атомов С, могут иметь формулы (алкил)2N- или алкил-NH-, -CO-N(алкил)2 или -CO-NH(алкил), -алкил-OH или -алкил-COOH.
Несмотря на расширенную функцию боковой цепи, соединения формулы (12) могут все же связываться с активным центром фермента дипептидилпептидазы IV и аналогичных ферментов, однако, они не могут транспортироваться далее активно пептидным транспортером PepT1. Полученная пониженная или весьма ограниченная транспортируемость согласно изобретению ведет к локальному или целенаправленному ингибированию активности DPIV и DPIV-подобного фермента.
Соединения формулы (12) или другие соединения и пролекарства, используемые в соответствии с изобретением, могут присутствовать или использоваться соответственно в форме рацематов или в форме энантиомерно чистых соединений, предпочтительно в L-трео- или L-алло-форме в отношении части А формулы (12).
Путем введения ответвлений в боковую цепь, например, включающих свыше семи атомов углерода, можно соответствено достигнуть резкого снижения транспортируемости (смотри Пример 12). Примеры в Таблице 12.1 показывают, что при увеличении пространственного размера боковых цепей имеет место снижение транспортируемости веществ. Путем пространственного и стерического увеличения размера боковых цепей, например, свыше размера атомной группы монозамещенного фенильного радикала, радикала гидроксиламина или аминокислотного остатка можно в соответствии с изобретением изменять или подавлять транспортируемость целевых веществ.
Согласно изобретению, соединения формулы (12) ингибируют активность DPIV или DPIV-подобного фермента в теле млекопитающего сайт-специфичным образом. В соответствии с этим возможно эффективное воздействие на локальные физиологические и патофизиологические состояния (воспаление, псориаз, артрит, аутоиммунные заболевания, аллергии, рак, метастазы, кровяное давление в эндотелии кровеносных сосудов), при этом существенно снижая побочные действия.
Предпочтительными соединениями формулы (12) являются соединения, где олигопептиды имеют длины цепей от 3 до 15 аминокислот, в особенности от 4 до 10, и/или полиэтиленгликоли имеют молярные массы, по крайней мере, 250 г/моль, предпочтительно, по крайней мере, 1500 г/моль и вплоть до 15000 г/моль, и/или необязательно замещенные органические амины, амиды, спирты, кислоты или ароматические соединения имеют, по крайней мере, 12 атомов С и предпочтительно вплоть до 30 атомов С.
Соединения настоящего изобретения могут быть превращены в аддитивные соли кислоты и использоваться в виде указанных солей, особенно в виде фармацевтически приемлемых аддитивных солей кислоты. Фармацевтически приемлемая соль обычно принимает форму, в которой основная боковая цепь аминокислот протонирована неорганической или органической кислотой. Репрезентативные органические или неорганические кислоты включают хлористоводородную, бромистоводородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, малеиновую, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, щавелевую, памоевую, 2-нафталинсульфоновую, п-толуолсульфоновую, циклогексансульфаминовую, салициловую, сахариновую или трифторуксусную кислоту. Все формы фармацевтически приемлемых аддитивных солей кислот входят в объем настоящего изобретения.
Ввиду тесной связи между свободными соединениями и соединениями в форме их солей всякий раз, когда в этом контексте упоминается соединение, предполагается и соответствующая соль, при условии, что при данных обстоятельствах это возможно или целесообразно.
Кроме того, настоящее изобретение включает в свой объем пролекарства соединений данного изобретения. В общем такие пролекарства представляют собой функциональные производные соединений настоящего изобретения, которые легко могут превращаться in vivo в желаемое терапевтически активное соединение. В соответствии с этим, в этих случаях использование настоящего изобретение будет охватывать и лечение различных расстройств, описанных здесь, вариантами (разновидностями) пролекарств из одного или нескольких заявляемых соединений, которые превращаются in vivo в вышеупомянутое конкретное соединение после их введения субъекту. Обычные процедуры для отбора и получения подходящих производных пролекарств описаны, например, в "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985 и патентных заявках DE 19828 113 и DE 19828 114, на которые здесь ссылаются.
В том случае, когда заявляемые соединения или пролекарства имеют, по крайней мере, один хиральный центр, они могут соответственно существовать в виде энантиомеров. В том случае, когда соединения или пролекарства обладают двумя или большим числом хиральных центров, они могут, кроме того, существовать в виде диастереомеров. Следует иметь в виду, что все вышеуказанные изомеры или их смеси входят в объем настоящего изобретения. Кроме того, некоторые из кристаллических форм соединений или пролекарств могут существовать в виде полиморфных модификаций, и, предполагается, что такие модификации также охватываются настоящим изобретением. В дополнение к этому некоторые из соединений могут образовывать сольваты с водой (т.е. гидраты) или с обычными органическими растворителями, и такие сольваты, как предполагается, также не выходят за рамки объема данного изобретения.
Соединения, включая их соли, могут быть также получены в форме их гидратов, или они включают другие растворители, используемые для их кристаллизации.
Как указывалось выше, соединения или пролекарства настоящего изобретения и их соответствующие формы фармацевтически приемлемых аддитивных солей кислоты являются полезными для ингибирования активности DPIV и DPIV-подобных ферментов. Способность соединений и пролекарств настоящего изобретения и их соответствующих форм фармацевтически приемлемых аддитивных солей кислот ингибировать активность DPIV и DPIV-подобных ферментов может быть продемонстрирована, используя испытание активности DPIV для определения значений Ki и значений IC50in vitro, как описано в примерах 7 и 8.
Способность соединений настоящего изобретения и их соответствующих фармацевтически приемлемых форм аддитивных солей кислоты ингибировать DPIV in vivo может быть продемонстрирована введением их пероральным или внутрисосудистым путем крысам линии Wistar, как описано в примере 11. Соединения настоящего изобретения ингибируют активность DPIV in vivo после их введения крысам линии Wistar как перорально, так и внутрисосудисто.
DPIV присутствует в широком ряде органов и тканей млекопитающих, например щеточная кайма эпителиальной ткани кишечника (Gutschmidt S et al., "In situ" - измерения содержания белка в области щеточной каймы вдоль ворсинок тощей кишки у крыс и их корреляции с активностями четырех ферментов. Histochemistry 1981, 72 (3), 467-79), эндокринный эпителий, гепатоциты, почечные канальцы, эндотелий, миофибробласты (Feller A.C. et al., A monoclonal antibody detecting dipeptidylpeptidase IV in human tissue. Virchows Arch. A. Pathol. Anat. Histopathol. 1986; 409 (2):263-73), нервные клетки, латеральные мембраны некоторых поверхностных эпителиев, например фаллопиевы трубы, матка и везикулярная железа, в люминальной цитоплазме, например эпителий везикулярной железы, и в клетках слизистой дуоденальных желез (Hartel S. et al., Dipeptidyl peptidase (DPP) IV in rat organs. Comparison of immunohistochemistry and activity histochemistry. Histochemistry 1988; 89 (2): 151-61), репродуктивные органы, например, хвостообразный отдел эпидидимиса и ампулы (cauda epididymis and ampulla), семенные везикулы и их секреты (Agrawal & Vanha-Perttula, Dipeptidyl peptidases in bovine reproductive organs and secretions. Int. J. Androl. 1986, 9 (6): 435-52). В человеческой сыворотке присутствуют две молекулярные формы дипептидилпептидазы (Krepela E. et al., Demonstration of two molecular forms of dipeptidyl peptidase IV in normal human serum. Physiol. Bohemoslov. 1983, 32 (6): 486-96). Сывороточная высокомолекулярная форма DPIV экспрессируется на поверхности активированных Т-клеток (Duke-Cohan J.S. et al., Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L released from activated T cells. J. Immunol. 1996, 156 (5): 1714-21).
Соединения и пролекарства настоящего изобретения и их соответствующие формы фармацевтически приемлемых солей кислоты способны ингибировать DPIV in vivo. В одном варианте осуществления настоящего изобретения все молекулярные формы, гомологи и эпитопы из всех тканей и органов млекопитающих, также и те, которые еще не раскрыты, как предполагается, входят в объем данного изобретения.
Среди редкой группы пролин-специфичных протеаз, как первоначально считали, DPIV является единственным мембрано-связанным ферментом, специфичным в отношении пролина как предпоследнего остатка на амино-конце полипептидной цепи. Однако недавно были идентифицированы другие молекулы, в структурном отношении не-гомологичные DPIV, но несущие соответствующую ферментативную активность. К DPIV-подобным ферментам, которые идентифицированы в настоящее время, относятся, например, фибробласт-активирующий белок a, дипептидилпептидаза IV Я, дипептидиламинопептидаза-подобный белок, N-ацетилированная a-связанная кислая дипептидаза, латентная клеточная пролиндипептидаза, дипептидилпептидаза II, аттрактин и дипептидилпептидаза IV-родственный белок (DPP 8), и они описаны в обзорной статье Sedo & Malik (Sedo & Malik, Dipeptidyl peptidase V-like molecules: homologous proteins or homologous activities? Biochimica et Biophysica Acta 2001 36506: 1-10). Другие DPIV-подобные ферменты раскрываются в международных публикациях WO 01/19866, WO 02/04610 и WO 02/34900. В WO 01/19866 раскрывается новая человеческая дипептидиламинопептидаза (DPP8), имеющая структурные и функциональные сходства с DPIV, и фибробласт-активирующий белок (FAP). Дипептидилпептидаза IV-подобный фермент, приведенный в WO 02/04610, хорошо известен в данной области. В базе данных Банка генов этот фермент зарегистрирован как KIAA1492. В другом предпочтительном варианте осуществления настоящего изобретения все молекулярные формы, гомологи и эпитопы белков, включая активность DPIV-подобного фермента, из всех тканей и органов млекопитающих, также и тех структур, которые еще не раскрыты, как предполагается, входят в объем данного изобретения.
Способность соединений и пролекарств настоящего изобретения и их соответствующих фармацевтически приемлемых форм аддитивных солей кислоты ингибировать DPIV-подобные ферменты может быть продемонстрирована, используя испытание активности фермента для определения значений Kiin vitro, как описано в примере 9. Были определены значения Ki соединений настоящего изобретения в отношении свиной дипептидилпептидазы II для глутаминилпирролидина как Ki = 8,52·10-5 M ± 6,33·10-6 M и для глутаминилтиазолидина как Ki = 1,07·10-5 M ± 3,81·10-7 M.
В другом варианте воплощения соединения и пролекарства настоящего изобретения и их соответствующие формы фармацевтически приемлемых аддитивных солей кислоты имеют лишь низкую (если она вообще присутствует) ингибирующую активность в отношении не-DPIV и не-DPIV-подобных пролин-специфичных ферментов. Как описано в примере 10, в случае глутаминилтиазолидина и глутаминилпирролидина, никакого ингибирования дипептидилпептидазы I и пролилолигопептидазы не было обнаружено. В отношении пролидазы оба соединения продемонстрировали заметную более низкую эффективность по сравнению с DPIV. Были определены значения IC50 в отношении пролидазы для глутаминилтиазолидина как IC50 > 3 мМ и для глутаминилпирролидина как IC50 = 3,4·10-4 M ± 5,63·10-5.
Настоящее изобретение обеспечивает способ профилактики или лечения состояния, опосредованного модуляциейактивностиDPIV или DPIV-подобного фермента, у субъекта, при необходимости такого лечения, который включает введение любого из соединений настоящего изобретения или фармацевтических композиций на их основе в количестве и по схеме приема, терапевтически эффективных для лечения вышеупомянутого состояния. Кроме того, настоящее изобретение включает применение соединений и пролекарств данного изобретения и их соответствующих фармацевтически приемлемых аддитивных солей кислот для получения лекарственного средства для профилактики или лечения у субъекта состояния, опосредованного модуляцией активности DPIV. Соединение можно вводить пациенту любым обычным путем введения, включая, но не ограничиваясь ими, внутривенный, пероральный, внутримышечный, внутрикожный, парентеральный пути введения и их комбинации.
В другом иллюстративном варианте воплощения настоящее изобретение обеспечивает составы соединений формул 1-12 и их соответствующих фармацевтически приемлемых аддитивных солей кислот для фармацевтических композиций.
Термин "субъект", используемый здесь, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который представлял объект лечения, наблюдения или эксперимента.
Используемый здесь термин "терапевтически эффективное количество" означает такое количество активного соединения или фармацевтического средства, которое вызывает биологическую или лечебную реакцию в системе тканей, животного или человека, которой добивается исследователь, ветеринар, лечащий врач или другой клиницист, и эта реакция включает облечение симптомов заболевания или состояния, подлежащего лечению.
Используемый здесь термин "композиция", как предполагают, охватывает продукт, включающий заявляемые соединения в терапевтически эффективных количествах, а также любой продукт, который является непосредственно или косвенно результатом комбинаций из заявляемых соединений.
Для получения фармацевтических композиций, используемых в настоящем изобретении, одно или несколько соединений формул 1-12 или их соответствующих фармацевтически приемлемых пролекарств или аддитивных солей кислот в качестве активного ингредиента тщательно смешивают с фармацевтическим носителем с получением однородной смеси в соответствии с обычными способами приготовления фармацевтических композиций,и этот носитель может принимать целый ряд форм в зависимости от того, какая лекарственная форма препарата требуется для выбранного пути введения, например для перорально или парентерального пути, такого как внутримышечное. Для получения композиций для пероральной лекарственной формы можно использовать любую из обычных фармацевтических сред. Так, для жидких пероральных препаратов, таких как, например, суспензии, эликсиры и растворы, подходящие носители и добавки могут преимущественно включать воду, гликоли, масла, спирты, ароматизирующие средства, консерванты, красители и т.п.; для твердых пероральных препаратов, таких как, например, порошки, капсулы, гель-капсулы и таблетки, подходящие носители и добавки включают крахмалы, сахара, разбавители, гранулирующие средства, лубриканты, связующие, дезинтегрирующие средства и т.п. Из-за легкости при введении таблетки и капсулы являются наиболее выгодной пероральной дозированной единичной формой, и в этом случае используют твердые фармацевтические носители. При необходимости таблетки могут быть покрыты сахарным покрытием или энтеросолюбильным покрытием обычными способами. В случае парентеральных препаратов носитель обычно включает стерильную воду для других целей, например, чтобы способствовать растворению или для консервации, могут быть включены и другие компоненты.
Кроме того, можно получить инъецируемые суспензии, и в этом случае могут быть использованы соответствующие жидкие носители, суспендирующие средства и т.п. Фармацевтические композиции, о которых здесь идет речь, обычно содержат в расчете на дозированную единицу, например, таблетку, капсулу, порошок, инъекцию, полную чайную ложку и т.п., такое количество активного компонента, которое необходимо для доставки эффективной дозы, как описано выше. Фармацевтические композиции, о которых здесь идет речь, обычно содержат в расчете на дозированную единицу, например, таблетку, капсулу, порошок, инъекцию, полную чайную ложку и т.п., от около 0,01 мг до около 1000 мг (предпочтительно от около 5 до около 500 мг) и их могут назначать при дозировке от около 0,1 до около 300 мг/кг массы тела в день (предпочтительно от 1 до 50 мг/кг в день). Однако дозировки могут варьироваться в зависимости от потребности пациентов, тяжести состояния, подлежащего лечению, и конкретного соединения, подлежащего применению. Может быть использовано либо ежедневное введение либо пост-периодическое дозирование. Обычно дозировка регулируется лечащим врачом, исходя из особенностей личности пациента, его/ее состояния и ожидаемого терапевтического действия.
Предпочтительно эти композиции представлены в дозированных лекарственных формах, таких как: таблетки, пилюли, капсулы, порошки, гранулы, стерильные парентеральные растворы или суспензии, дозируемый аэрозоль или жидкий спрей, капли, ампулы, автоматические устройства для инъецирования или суппозитории; для перорального, парентерального, интраназального, сублингвального или ректального введения, или для введения путем ингаляции или инсуффляции. Альтернативно композиция может быть представлена в форме, подходящей для введения один раз в неделю или один раз в месяц; например, нерастворимая соль активного соединения, такая как деканоатная соль, может быть использована с получением депо-препарата для внутримышечной инъекции. Для получения твердых композиций, таких как таблетки, основной активный компонент тщательно смешивают с фармацевтическим носителем, например обычными компонентами для таблетирования, такими как: кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, дикальций фосфат или камеди, и другими фармацевтическими разбавителями, например, водой, с получением твердой пре-композиции, содержащей гомогенную смесь соединения настоящего изобретения или его фармацевтически приемлемую соль. При упоминании этих вышеуказанных пре-композиций как гомогенных, подразумевается, что активный компонент идеально диспергирован равномерно по всей композиции так, что композиция может быть легко подразделена на в равной степени эффективные лекарственные формы, такие как таблетки, пилюли и капсулы. Указанная твердая пре-композиция затем может быть подразделена на единичные дозированные формы описанного выше типа, содержащие от около 0,01 до около 1000 мг, предпочтительно от около 5 до около 500 мг активного компонента настоящего изобретения.
Таблетки или пилюли новой композиции могут быть с определенной целью покрыты или иным образом приготовлены, чтобы обеспечить лекарственную форму, преимущество которой реализовалось пролонгированным действием. Например, таблетка или пилюля может включать внутренний дозированный и наружный дозированный компонент, при этом последний служит оболочкой, покрывающей первый. Два компонента могут быть разделены энтеросолюбильным слоем, который служит для того, чтобы воспрепятствовать распаду в желудке, и позволяет проникнуть неповрежденным внутреннему компоненту в двенадцатиперстную кишку или способствует его замедленному высвобождению. Различные вещества могут быть использованы для вышеуказанных энтеросолюбильных слоев или покрытий, такие вещества включают ряд полимерных кислот с такими веществами как шеллак, цетиловый спирт и ацетат целлюлозы.
Указанные жидкие формы, в которые новые композиции настоящего изобретения могут быть преимущественно включены для введения пероральным путем или при помощи инъекции, включают водные растворы, соответсвующим образом ароматизированные сиропы, водные или масляные суспензии и ароматизированные эмульсии со съедобными маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и аналогичные фармацевтические наполнители. Подходящие диспергирующие или суспендирующие средства для водных суспензий включают синтетические или природные смолы, такие как: трагакант, гуммиарабик, альгинат, декстран, натрий карбоксиметилцеллюлоза, метилцеллюлоза, поливинилпирролидон или желатин.
В тех случаях, когда способы получения соединений согласно изобретению приводят к смеси стереоизомеров, указанные изомеры могут быть разделены обычными методами, такими как препаративная хроматография. Соединения могут быть получены в рацемической форме, или индивидуальные энантиомеры можно получить либо энантиоспецифическим синтезом либо путем разделения. Соединения могут, например, быть разделены на их энантиомеры-компоненты обычными способами, такими как образование диастереомерных пар при солеобразовании с оптически активной кислотой, такой как (-)-ди-п-толуоил-d-винная кислота и/или (+)-ди-п-толуоил-l-винная кислота, с последующей фракционированной кристаллизацией и регенерацией свободного основания. Кроме того, соединения могут быть разделены путем образования диастереомерных сложных эфиров или амидов, с последующим хроматографическим разделением и удалением хирального вспомогательного вещества. Альтернативно соединения можно разделить, используя ВЭЖХ колонку, наполненную хиральным сорбентом.
Во время любого из способов получения соединений настоящего изобретения может потребоваться или может быть желательно использование защиты чувствительных или реакционноспособных групп на любой из рассматриваемых молекул. Это может быть достигнуто при помощи обычных защитных групп, таких как группы, описанные в Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; and T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991, включенные здесь как ссылки. Защитные группы могут быть удалены на удобной последующей стадии, используя известные в данной области способы.
Способ лечения состояний, модулируемых дипептидилпептидазой IV и DPIV-подобными ферментами, описанными в настоящем изобретении, может быть также осуществлен, используяфармацевтическую композицию, включающую одно или несколько соединений, определенных здесь, и фармацевтически приемлемый носитель. Фармацевтическая композиция может содержать от около 0,01 мг до 1000 мг, предпочтительно от около 5 до около 500 мг, соединения (соединений)и может быть приготовлена в любой форме, подходящей для выбранного способа введения. Носители включают необходимые и инертные фармацевтические эксципиенты, включая, но не ограничиваясь ими, связующие, суспендирующие средства, лубриканты, ароматизаторы, подслащивающие вещества, консерванты, красители и покрытия. Композиции, подходящие для перорального введения, включают твердые формы, такие как пилюли, таблетки, карлеты, капсулы (при этом каждая включает составы незамедлительного высвобождения, нормированного по времени высвобождения и пролонгированного высвобождения), гранулы и порошки; и жидкие формы, такие как растворы, сиропы, эликсиры, эмульсии и суспензии. Формы, используемые для парентерального введения, включают стерильные растворы, эмульсии и суспензии.
Преимущественно соединения настоящего изобретения могут вводиться в виде однократной суточной дозы или суммарная суточная дозировка может вводиться частями, в виде небольших доз, два, три или четыре раза в день. Кроме того, соединения настоящего изобретения можно вводить в интраназальной форме путем местного использования подходящих интраназальных наполнителей или при помощи трансдермальных накладок (пластырей), хорошо известных специалистам в данной области. При введении с использованием трансдермальной системы доставки дозирование должно быть, конечно, непрерывным, а не прерывистым на протяжении всей схемы приема лекарственного средства, и в соответствии с этим содержание активного вещества потребует внесения изменений для того, чтобы достичь желаемого терапевтического действия.
В случае перорального введения в форме таблетки или капсулы более предпочтительно, когда активный компонент лекарственного средства может быть смешан с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Кроме того, если требуется или необходимо, в смесь могут быть также включены подходящие связующие; лубриканты, дезинтегрирующие средства и красители. Подходящие связующие включают без ограничения крахмал, желатин, природные сахара, такие как глюкоза или беталактоза, подсластители из кукурузы, натуральные и синтетические камеди, такие как гуммиарабик, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Дезинтеграторы включают без ограничения крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и другие соединения, известные в данной области.
Подходящие жидкие формы включают в свой состав ароматизированные суспендирующие или диспергирующие средства, такие как синтетические и природные камеди, например трагакант, акация, метилцеллюлоза и т.п. Для парентерального введения требуются стерильные суспензии и растворы. В случае внутривенного введения используют изотонические препараты, которые обычно содержат подходящие консерванты.
Кроме того, соединения настоящего изобретения могут быть введены в форме липосомных систем доставки, таких как небольшие одноламеллярные везикулы, большие одноламеллярные и многоламеллярные везикулы. Липосомы можно получить из целого ряда фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины, используя способы, описанные в данной области.
Соединения настоящего изобретения могут быть также связаны с растворимыми полимерами, выступающими в качестве носителей, способных осуществлять направленный транспорт лекарственного средства. Такие полимеры могут включать поливинилпирролидон, сополипер пирана, полигидроксиметакрилатамидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоил-остатком. Кроме того, соединения настоящего изобретения могут быть связаны с классом биодеградируемых полимеров, используемых для создания систем контролируемого высвобождения лекарственного средства, например полиактиновая кислота, поли-эксилон-капролактон, полигидроксибутириновая кислота, полиортоэстеры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блоксополимеры гидрогелей.
Соединения данного изобретения можно вводить в составе любой из вышеупомянутых композиций и в соответствии со схемами приема лекарственного средства, установленными в данной области, всякий раз, когда требуется лечение адресных нарушений.
Суточная доза продуктов может варьироваться в широком диапазоне от 0,01 до 1,000 мг в день для взрослого человека. В случае перорального введения композиции предпочтительно обеспечиваются в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100, 150, 200, 250, 500 и 1000 мг активного компонента, для того, чтобы можно было симптоматически корректировать дозу пациенту, который подвергается лечению. Эффективное количество лекарственного средства обычно обеспечивается при уровне дозировки от около 0,1 мг/кг до около 300 мг/кг веса тела в день. Предпочтительно уровень дозировки лежит в диапазоне от около 1 до около 50 мг/кг веса тела в день. Соединения можно принимать по схеме от 1 до 4 раз в день.
Оптимальные дозы, подлежащие введению, могут быть легко определены специалистами в данной области и они обычно варьируются в зависимости от конкретного используемого соединения, способа введения, содержания активного компонента в препарате, биодоступности, обусловленной способом введения, и прогрессирования состояния заболевания. Кроме того, при корректировке доз следует учитывать факторы, имеющие отношение к конкретному пациенту, подлежащему лечению, включая возраст пациента, вес, диету и время введения.
Соединения или композиции настоящего изобретения можно принимать до еды, например за 1 час, 30, 15 или 5 мин до еды или питья, при приеме пищи или после еды.
При приеме во время еды соединения или композиции настоящего изобретения могут быть добавлены в пищу или их можно принимать в виде отдельной лекарственной формы, как описано выше.
ПРИМЕРЫ
Пример 1: Синтез дипептид-подобных соединений
1.1. Общий синтез соли изолейцил-тиазолидина
Вос-защищенную аминокислоту Вос-Ile-OH помещают в этилацетат и полученную смесь охлаждают до около -5°C. По каплям при постоянной температуре добавляют N-метиоморфолин, хлорангидрид пивалиновой кислоты (установка лабораторного масштаба) или неогексаноилхлорид (установка полузаводского масштаба) добавляют по каплям при постоянной температуре. Реакционную смесь перемешивают в течение нескольких минут для активации. Последовательно по каплям добавляют N-метилморфолин (лабораторный масштаб) и тиазолидин гидрохлорид (лабораторный масштаб) добавляют тиазолидин (полузаводской масштаб). Обработку в лаборатории осуществляют обычным способом, используя солевые растворы, в случае установки полузаводского масштаба смесь очищают растворами NaOH и CH3COOH.
Удаление защитной группы Boc проводят, используя смесь HCl/диоксан (лабораторный масштаб) или H2S04 (полузаводской масштаб). В лаборатории гидрохлорид кристаллизуют из смеси EtOH/простой эфир.
В случае установки полузаводского масштабасвободный амин получают, добавляя смесь NaOH/NH3. Фумаровую кислоту растворяют в горячем этаноле, добавляют по каплям свободный амин и (Ile-Thia)2 фумарат (M = 520,71 гмоль-1) осаждается. Анализ изомеров и энантиомеров осуществляют электрофорезом.
1.2. Синтез свободного основания глутаминилпирролидина
Ацилирование
N-Бензил-оксикарбонилглутамин (2,02 г, 7,21 ммоль) растворяют в 35 мл ТГФ и охлаждают до -15°C. В указанную смесь добавляют CAIBE (изобутилхлорформиат) (0,937 мл, 7,21 ммоль) и 4-метилморфолин (0,795 мл, 7,21 ммоль) и раствор перемешивают в течение 15 мин. Образование смешанного ангидрида контролируют при помощи ТСХ (TLC) (элюент: CHCl3/MeOH: 9/1). После нагревания до -10°C добавляют пирролидин (0,596 мл, 7,21 ммоль). Смеси дают возможность нагреться до комнатной температуры и перемешивают на протяжении ночи.
Обработка
Образовавшийся осадок отфильтровывают и растворитель выпаривают. Полученное масло растворяют в этилацетате (20 мл) и промывают насыщенным раствором гидросульфата натрия, затем насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой отделяют, сушат и упаривают. Полученный продукт контролируют на чистоту ТСХ (элюент: CHCl3/MeOH: 9/1).
Выход: 1,18 г, воскообразное твердое вещество
Расщепление
1,18 г полученного твердого Z-защищенного соединения растворяют в 40 мл абсолютного этанола. В раствор добавляют прибл. 20 мг Pd на угле (10%, FLUKA) и суспензию встряхивают в атмосфере водорода в течение 3 ч. Течение реакции контролируют при помощи ТСХ (элюент: CHCl3/MeOH: 9/1). После завершения реакции растворитель удаляют, получая свободное основание.
Выход: 99%
Чистоту контролируют при помощи ТСХ: смесь н-бутанол/AcOH/вода/этилацетат. 1/1/1/1, Rf = 0,4. Идентичность продукта реакции контролируют методом ЯМР.
1.3. Синтез глутаминилтиазолидин гидрохлорида
1.4. Ацилирование
N-т-Бутил-оксикарбонилглутамин (2,0 г, 8,12 ммоль) растворяют в 5 мл ТГФ и охлаждают до -15°C. В эту смесь добавляют CAIBE (изобутилхлорформиат) (1,06 мл, 8,12 ммоль) и 4-метилморфолин (0,895 мл, 8,12 ммоль) и раствор перемешивают в течение 15 мин. Образование смешанного ангидрида контролируют ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10°C добавляют еще эквивалент 4-метилморфолина (0,895 мл, 8,12 ммоль) и тиазолидин гидрохлорид (1,02 г, 8,12 ммоль). Смеси дают возможность нагреться до комнатной температуры и перемешивают на протяжении ночи.
Обработка
Образовавшийся осадок отфильтровывают и растворитель выпаривают. Полученное масло поглощают хлороформом (20 мл) и промывают насыщенным раствором гидросульфата натрия, а затем насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой отделяют, сушат и упаривают. Полученный продукт анализируют на чистоту при помощи ТСХ (элюент: CHCl3/MeOH: 9/1).
Выход: 1,64 г, твердое вещество
Расщепление
640 мг полученного твердого Вос-защищенного соединения растворяют в 3,1 мл охлажденной льдом смеси HCl в диоксане (12,98М, 20 эквивалентов) и оставляют на льду. Течение реакции контролируют при помощи ТСХ (элюент: CHCl3/MeOH: 9/1). После завершения реакции растворитель удаляют и затем полученное масло растворяют в метаноле и снова выпаривают. После этого полученное масло сушат над оксидом фосфора (V) и растирают два раза с диэтиловым эфиром. Чистоту контролируют при помощи ВЭЖХ (HPLC).
Выход: 0,265 г
Чистоту контролируют при помощи ВЭЖХ. Идентичность продукта реакции контролируют методом ЯМР.
1.4. Синтез глутаминилпирролидин гидрохлорида
Ацилирование
N-т-Бутил-оксикарбонилглутамин (3,0 г, 12,18 ммоль) растворяют в 7 мл ТГФ и охлаждают до -15°C. В эту смесь добавляют CAIBE (изобутилхлорформиат)) (1,6 мл, 12,18 ммоль) и 4-метилморфолин (1,3 мл, 12,18 ммоль) и раствор перемешивают в течение 15 мин. Образование смешанного ангидрида контролируют ТСХ (элюент: CHCl3/MeOH: 9/1). После нагревания до -10°C добавляют 1 эквивалент пирролидина (10 мл, 12,18 ммоль). Смеси дают возможность нагреться до комнатной температуры и перемешивают на протяжении ночи.
Обработка
Образовавшийся осадок отфильтровывают и растворитель выпаривают. Полученное масло поглощают хлороформом (20 мл) и промывают насыщенным раствором гидросульфата натрия, затем насыщенным раствором бикарбоната натрия, водой и насыщенным раствором соли. Органический слой отделяют, сушат и упаривают. Полученный продукт контролируют при помощи ТСХ (элюент: CHCl3/MeOH: 9/1).
Выход: 2,7 г, твердое вещество
Расщепление
2,7 г полученного твердого вещества растворяют в 13,0 мл охлажденной льдом смеси HCl в диоксане (12,98M, 20 эквивалентов) и оставляют на льду. Течение реакции контролируют при помощи ТСХ (элюент: CHCl3/MeOH: 9/1). После завершения реакции растворитель удаляют и полученное масло поглощают метанолом и снова выпаривают. После этого полученное масло сушат над оксидом фосфора (V) и растирают два раза с диэтиловым эфиром.
Выход: 980 мг
Чистоту контролируют при помощи ВЭЖХ. Иденичность продукта реакции контролируют анализом методом ЯМР.
Пример 2: Химическое исследование выбранных дипептидных соединений
2.1. Определение точки плавления
Точки плавления определяют на микроскопе, снабженном нагревательной платформой от Leica Aktiengesellschaft, значения приводятся без коррекции, или на установке DSC (HeumannPharma).
2.2. Оптическое вращение
Значения величин вращения плоскости поляризации света регистрируют при различных длинах волн на "Polarimeter 341" иливыше от компании Perkin-Elmer.
2.3. Условия измерения для масс-спектроскопии
Масс-спектры регистрируют методом электрораспылительной ионизации (ESI) на приборах "API 165" или "API 365" компании PE Sciex. Операцию осуществляют, используя соответствующую концентрацию c=10 мкг/мл, вещество растворяют в смеси MeOH/H20 50:50, 0,1% HCO2H, введение образцов осуществляют при помощи распылительного насоса (20 мкл/мин). Измерения проводят с регистрацией положительных ионов [M+H]+, напряжение ESI U=5600В.
2.4. Результаты
2.4.1. Исследование изолейцил-тиазолидин фумарата (изомер)
(405 нм)
(380 нм)
(380 нм)
IT*F = изолейцил-тиазолидин фумарат
Данные ЯМР и ВЭЖХ подтвердают индентичность рассматриваемого вещества
2.4.2. Исследование других солей изолейцил-тиазолидина
Пример 3: Синтез Xaa-Pro-Yaa трипептидов
Все синтезы осуществляют на пептидном синтезаторе SP 650 (Labortec AG), применяя Fmoc/тBu-стратегию. Защищенные аминокислоты поставлялись от Novabiochem или Bachem, трифторуксусная кислоту (TFA) закупали от Merck, триизопропилсилан (TIS) закупали от Fluka.
С предварительно загруженной Fmoc-Yaa-Wang смолы (2,8 г, степень замещения 0,57 ммоль/г) снимают защиту, используя смесь 20% пиперидин/N,N-диметилформамид (ДМФ, DMF). После промывания ДМФ раствор 2 экв. (1,1 г) Fmoc-Pro-OH растворяют в ДМФ (12 мл растворителя на грамм смолы). Добавляют 2 экв. (1,04 г) 2-(1H- бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторбората (TBTU) и 4 экв. (1,11 мл) N,N-диизопропилэтиламина (ДИЭА, DIEA) и помещают в реакционный сосуд. Смесь встряхивают при комнатной температуре в течение 20 мин. Затем цикл присоединения повторяют. После последующего промывания ДМФ дихлорметаном, изопропанолом и диэтиловым эфиром полученную Fmoc-Pro-Ile-Wang смолу сушат и затем делят на шесть частей до присоединения последнего производного аминокислоты.
Fmoc-защитную группу удаляют, как описано выше. После этого 0,54 ммоль Вос-аминокислоты, 0,54 ммоль TBTU и 0,108 ммоль ДИЭА (DIEA) в ДМФ встряхивают в течение 20 мин. Цикл присоединения повторяют. Наконец, смолу с пептидом промывают и сушат, как описано выше.
Пептид отщепляют от смолы, используя смесь трифторуксусной кислоты (ТФК, TFA) в течение 2,5 ч, содержащую следующие поглотители: ТФК/H2O/триизопропилсилан (TIS)=9,5/0,25/0,25.
Выходы неочищенных пептидов составляли 80-90%, в среднем. Неочищенный пептид очищают при помощи ВЭЖХ на колонке Nucleosil С18 (7 мкм, 250*21,20 мм, 100 А), используя линейный градиент 0,1% ТФК/H2O при увеличении концентрации 0,1% ТФК/ацетонитрил (от 5% до 65% в течение 40 мин) при скорости потока 6 мл/мин.
Чистый пептид получают лиофилизацией, идентифицируют методами электрораспылительной масс-спектрометрии и ВЭЖХ.
3.1. Результаты - Идентификация трипептидов Xaa-Pro-Yaa после химического синтеза
[M+H+]
1 [M+H+] определяли методом электрораспылительной масс-спектрометрии в режиме регистрации положительных ионов.
2 ОФ-ВЭЖХ, условия:
колонка: LiChrospher 100 RP 18 (5 мкм), 125 x 4 мм
детектирование (УФ), 214 нмm
градиентное элюирование, система: ацетонитрил (АЦН, ACN)/H20 (0,1% ТФК) от 5% АЦН до 50% за 15 мин,
поток: 1 мл/мин
k'=(tr-to)/to
to=1,16 мин
т-бутил-Gly означает:
Ser(Bzl) и Ser(P) означают бензилсерин и фосфорилсерин, соответственно. Tyr(P) означает фосфорилтирозин.
Пример 4: Синтез пептидилкетонов
H-Val-Pro-OMe*HCl 2
Вос-Val-OH (3,00 г, 13,8 ммоль) растворяют в 10 мл ТГФ и охлаждают вплоть до -15°С. К смеси добавляют CAIBE (изобутилхлорформиат, 1,80 мл, 13,8 ммоль) и NMM (1,52 мл, 13,8 ммоль) и раствор перемешивают до тех пор, пока не завершится образование смешанного ангидрида. Затем смесь доводят до -10°С и добавляют NMM (1,52 мл, 13,8 ммоль), а затем H-Pro-OMe*HCl (2,29 г, 13,8 ммоль). Смеси дают возможность достичь комнатной температуры и оставляют на протяжении ночи. После удаления растворителя и обычной обработки имеют полученный сложный эфир 1 без дополнительного охарактеризовывания. Сложный эфир 1 растворяют в смеси HCl/HOAc (5 мл, 6N) и оставляют при 0°С до тех пор, пока удаление Бок-группы не завершится. Затем растворитель удаляют и полученное масло обрабатывают диэтиловым эфиром, получая белое твердое вещество 2.
Выход: 2,5 г, 80%
Z-Ala-Val-Pro-OMe 3
Z-Ala-OH (3,5 г, 15,7 ммоль) и 2 (4,18 г,15,7 ммоль) подвергают обработке таким же способом, как упомянуто выше для 1, получая 3 в виде белого твердого вещества.
Выход: 4,2 г, 64%
Z-Ala-Val-Pro-OH 4
3 (4,2 г 9,6 ммоль) растворяют в 30 мл смеси вода/ацетон (1/5 об/об) и добавляют 11,6 мл NaOH (1N). После завершения реакции органический растворитель удаляют выпариванием и полученный раствор разбавляют 15 мл насыщенного раствора NaHCO3. Затем смесь экстрагируют три раза 10 мл этилового эфира уксусной кислоты. После этого рН раствора доводят до 2, добавляя HCl (15% в воде). Полученную смесь экстрагируют три раза 30 мл этилового эфира уксусной кислоты. Органический слой отделяют и промывают три раза насыщенным раствором соли, сушат (Na2SO4) и упаривают.
Выход: 3,5 г, 87%
Z-Ala-Val-Pro-CH2-Br 5
4 (2,00 г, 4,76 ммоль) растворяют в 15 мл ТГФ и превращают в смешанный ангидрид (смотри соединение 1), используя CAIBE (0,623 мл, 4,76 ммоль) и NMM (0,525 мл, 4,76 ммоль). Образовавшийся осадок отфильтровывают и охлаждают вплоть до -15°С. Затем в раствор по каплям в атмосфере аргона добавляют диазометан (23,8 ммоль в 30 мл простого эфира). После выдерживания смеси в течение 1 ч при 0°С добавляют 1,27 мл HBr (33% в AcOH) и раствор перемешивают в течение 30 мин при комнатной температуре. После этого добавляют 70 мл простого эфира и смесь промывают 20 мл воды. Органический слой отделяют и сушат (Na2SO4) и упаривают.
Выход (неочищенный): 1,8 г, 80%
Z-защищенные ацилоксиметиленкетоны
Кислоту (2 экв.) растворяют в ДМФА (DMF) и добавляют эквимолярное количество KF. Суспензии дают возможность перемешиваться при комнатной температуре в течение 1 ч. Затем добавляют бромметилен (1 экв.) и раствору дают возможность перемешиваться на протяжении ночи. После этого растворитель удаляют в вакууме и полученное масло растворяют в хлороформе и промывают насыщенным раствором соли. Затем органический слой отделяют, сушат (Na2SO4) и растворитель удаляют. Продукт очищают колоночной хроматографией, используя силикагель и смесь гептан/хлороформ в качестве элюента.
Z-Ala-Val-Pro-CH2О-C(O)-CH36
Уксусная кислота (230 мкл, 4,02 ммоль), KF (0,234 г, 4,02 ммоль), 5 (1,00 г, 2,01 ммоль).
Выход: 0,351 г, 36%
Z-Ala-Val-Pro-СН2О-C(O)-Ph 7
Бензойная кислота (0,275 г, 2,25 ммоль), KF (0,131 мг, 2,25 ммоль), 5 (0,56 г, 1,13 ммоль).
Выход: 0,34 г, 56%
Снятие защиты
Z-защищенное соединение растворяют в смеси HBr/AcOH и перемешивают. После того как реакция завершится, добавляют простой эфир, образовавшийся белый осадок отфильтровывают и сушат.
H-Ala-Val-Pro-CH2O-C(O)CH3* HBr 8
6 (0,351 г, 0,73 ммоль)
Выход: 0,252 г, 98%
H-Ala-Val-Pro-СН2О-С(О)Ph*HBr 9
7 (0,34 г, 0,63 ммоль)
Выход: 0,251 г, 99%
Пример 5: Синтез циклоалкилкетонов
Вос-изолейциналь 2
Оксалилхлорид (714 мкл, 8,28 ммоль) растворяют в 10 мл сухого дихлорметана и охлаждают до -78°С. Затем по каплям добавляют ДМСО (DMSO) (817 мкл, 8,28 ммоль). Раствор перемешивают в течение 20 мин при -78°С. Затем добавляют 1 (1,00 г, 4,6 ммоль) и смесь перемешивают в течение 20 мин. После этого добавляют ТФК (2,58 мл, 18,4 ммоль) и смеси дают возможность достичь комнатной температуры. Смесь разбавляют смесью гексан/этилацетат (2/1 об/об)и добавляют 10 мл HCl (10% в воде). Органический слой отделяют и водную фазу экстрагируют 20 мл метиленхлорида. Органические слои объединяют и промывают насыщенным раствором соли, затем водой и затем сушат. Продукт очищают колоночной хроматографией, используя силикагель и смесь гептан/хлороформ в качестве элюента.
Выход: 0,52 г, 52%
трет-Бутил N-1-[циклопентил(гидрокси)метил]-2-метил-бутилкарбамат 3
2 (0,52 г, 2,42 ммоль) растворяют в 10 мл сухого ТГФ и охлаждают вплоть до 0°С. Затем добавляют циклопентилмагнийбромид (1,45 мл 2M раствора). После завершения реакции добавляют 2 мл воды и раствор нейтрализуют, добавляя водную HCl. Затем добавляют метиленхлорид и органический слой отделяют и сушат (Na2SO4). После упаривания полученное масло используют без дополнительного охарактеризовывания.
трет-Бутил N-[1-(циклопентилкарбонил)-2-метилбутил]карбамат 4
3 (0,61 г, 2,15 ммоль) обрабатывают подобно 1. Оксалилхлорид (333 мкл, 3,87 ммоль), ДМСО (382 мкл, 5,37 ммоль), ТФК (1,2 мл, 8,59 ммоль).
Выход: 0,180 г, 30%
1-циклопентил-3-метил-1-оксо-2-пентанамин хлорид 5
4 (0,18 г, 0,63 ммоль) растворяют в 2 мл HCl (7N в диоксане). После завершения реакции, растворитель удаляют и полученное масло очищают колоночной хроматографией на силикагеле, используя градиентное элюирование смесью хлороформ/метанол/вода. Полученное масло растирают с простым эфиром.
Выход: 0,060 г, 54%
Пример 6: Синтез ингибитора DPIV с модифицированной боковой цепью
6.1. Синтез Вос-глутамилтиазолидина (Вос-Glu-Thia)
Реакция (взаимодействие) Вос-Glu(OMe)-OH с Thia*HCl согласно способу В (смотри раздел 6.4 для способов), гидролиз Вос-Glu(OMe)-Thia согласно способу G.
6.1.1 Аналитические данные для Boc-Glu-Thia
Mr;
Способ синтеза;
Выход
т.пл.
найдено) %
Rt [мин]/
система
B+G;
62%
0,52/A1 0,42/B1
115-1180С
c=1; метанол
H:6,96/6,82
N:8,80/8,59
1 Тонкослойная хроматография
Система A: хлороформ/метанол 90:10
Система B: бензол/ацетон/уксусная кислота 25:10:0,5
Система C: н-бутанол/ЭА/уксусная кислота/H2O 1:1:1:1
2 ВЭЖХ, условия разделения:
Колонка: Nucleosil C-18, 7 мк, 250 мм х 21 мм
Элюент: изократическое элюирование, 40 % АЦН/вода/0,1% ТФК
Скорость потока: 6 мл/мин
λ=220 нм
6.2. Вос-глутамилтиазолидины с модифицированной боковой цепью
Вос-Glu-Thia модифицируют в положении функции γ-карбоновой кислоты введением радикалов с различной длиной цепи. Радикалы присоединяют при помощи их аминогруппы с образованием амидной связи с функцией γ-карбоновой кислоты, при этом используют различные способы присоединения в зависимости от (природы) радикала. Ниже представлены аминокомпоненты, которые присоединяют к Вос-Glu-Thia с указанием используемого для этого способа:
(смотри раздел 3.4)
В 2 случаях очистка продуктов реакции отличается от общего описания синтеза.
Вос-Glu(Gly5)-Thia
Продукт осаждается из смеси уже при перемешивании на протяжении ночи; в дальнейшем его отфильтровывают и промывают 0,1N HCl и обильными количествами воды и затем сушат над P4010 в вакууме.
Вос-Glu(ПЭГ)-Thia
В отличие от общей процедуры исходные вещества для синтеза растворяют в 500-кратном избытке ДМФА. После завершения реакции, ДМФА полностью удаляют в вакууме, и остаток растворяют в большом количестве метанола. После добавления простого эфира с образованием верхнего слоя продукт осаждается вместе с непрореагировавшим ПЭГ. Тонкую очистку осуществляют путем разделения с помощью препаративной ВЭЖХ на гель-фильтрационной колонке (Pharmazia, Sephadex G-25, 90 мкм, 260 мм - 100 мм). Условия разделения: элюент: вода; скорость потока: 5 мл/мин; λ=220 нм.
6.2.2. Данные синтеза для модифицированных по боковой цепи Boc-глутамилтиазолидинов
Mr;
Выход
Т.пл.
Концентрация
Растворитель
(Рассчит./
найдено)%
Rt [мин]/ система
49%
H:6,38
N:14,31
603,64
86%
0,09/С;
разл.
от 202°С
H:6,18/6,11
N:16,24/16,5
(акцент на массу)
52-53°С
2 Условия разделения ВЭЖХ:
Колонка: Nucleosil C-18, 7 мк, 250 мм x 21 мм
Элюент: изократическое элюирование, 40% АЦН/вода/0,1% ТФК
Скорость потока: 6 мл/мин
λ=220 нм
6.3. Глутамилтиазолидины с модифицированной боковой цепью
N-концевые Вос защитные группы отщепляют от соединений, описанных в Таблице 6.2.2, используя способ F. Вещества, модифицированные Gly-производными, очищают путем разделения препаративной ВЭЖХ и они имеются в виде трифторацетатов. H-Glu(PEG)-Thia очищают на гель-фильтрационной колонке таким же способом, как Boc-защищенный предшественник.
6.3.1. Данные синтеза для модифицированных в боковой цепи глутамилтиазолидинов
Mr
Выход
ТСХ/Rf/ система
Т.пл.
Концентрация
Растворитель
Rt[мин]/ система
503,45
94%
0,32/C
91-940С
c=1
метанол
H:4,80/4,78
N:13,91/13,43
617,55
98%
0,25/C
105-107°С
H:4,90/4,79
N:15,88/15,39
(акцент на массу)
3 Условия разделения ВЭЖХ:
Колонка: Nucleosil C-18, 7 мк, 250 мм x 21 мм
Элюент: АЦН/вода/0,1% ТФК
Градиент: 20%АЦН 90% АЦН на протяжении 30 мин
Скорость потока: 6 мл/мин
λ=220 нм
не опред. - не определяли или не определяемо
6.4. Общие способы синтеза
Способ A: Присоединение с образованием пептидной связи методом смешанных ангидридов, используя CFIBE в качестве активирующего реагента
10 ммоль аминокислоты или пептида с N-защищенным концом растворяют в 20 мл абсолютного ТГФ. Раствор охлаждают до -15°С ± 2°С. При перемешивании в каждом случае последовательно добавляют 10 ммоль NMM и 10 ммоль изобутилового эфира хлормуравьиной кислоты, строго поддерживая установленный интервал температур. Спустя приблизительно 6 мин добавляют 10 ммоль аминокомпонента. В тех случаях, когда аминокомпонентом является соль, затем к реакционной смеси добавляют дополнительно 10 ммоль NMM. Затем реакционную смесь перемешивают в течение 2 ч в холодном состоянии и на протяжении ночи при комнатной температуре.
Реакционную смесь концентрируют, используя роторный испаритель, поглощают в ЭА (этилацетат, EA), промывают 5% раствором KH2SO4, насыщенным раствором NaHCO3 и насыщенным раствором NaCl и сушат над NaSO4. После удаления растворителя в вакууме соединение перекристаллизовывают из смеси ЭА/пентан.
Способ B: Присоединение с образованием пептидной связи методом смешанных ангидридов, используя хлорангидрид пивалиновой кислоты в качестве активирующего реагента
10 ммоль аминокислоты или пептида с N-защищенным концом растворяют в 20 мл абсолютного ТГФ. Раствор охлаждают до 0°С. При перемешивании в каждом случае последовательно добавляют 10 ммоль NMM и 10 ммоль хлорангидрида пивалиновой кислоты, строго поддерживая установленный интервал температуры. Спустя приблизительно 6 мин смесь охлаждают до -15°С, и сразу после того как была достигнута эта низкая температура, добавляют 10 ммоль аминокомпонента. В тех случаях, когда аминокомпонентом является соль, затем к реакционной смеси добавляют дополнительно 10 ммоль NMM. Затем реакционную смесь перемешивают в течение 2 ч в холодном состоянии и на протяжении ночи при комнатной температуре.
Дальнейшую обработку осуществляют так же как в Способе А.
Способ C: Присоединение с образованием пептидной связи, используя TBTU в качестве активирующего реагента
10 ммоль аминокислоты или пептида с N-защищенным концом и 10 ммоль аминокомпонента с С-защищенным концом растворяют в 20 мл абсолютного ДМФА (DMF). Раствор охлаждают до 0°С. При перемешивании в каждом случае последовательно добавляют 10 ммоль DIPEA and 10 ммоль TBTU. Реакционную смесь перемешивают в течение 1 ч при 0°С и затем на протяжении ночи при комнатной температуре. ДМФА полностью удаляют в вакууме, и продукт обрабатывают, как описано в Способе А.
Способ D: Синтез активированного сложного эфира (сложный эфир N-гидроксисукцинимида)
10 ммоль аминокислоты или пептида с N-защищенным концом и 10 ммоль N-гидроксисукцинимида растворяют в 20 мл абсолютного ТГФ. Раствор охлаждают до 0°С и при перемешивании добавляют 10 ммоль дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение еще 2 ч при 0°С и затем на протяжении ночи при комнатной температуре. Полученную N,N'-дициклогексилмочевину отфильтровывают и растворитель удаляют в вакууме и оставшийся после этого продукт перекристаллизовывают из смеси ЭА/пентан.
Способ E: Присоединение (с образованием) амидной связи, используя сложные эфиры N-гидроксисукцинимида
10 ммоль аминокомпонента с незащищенным С-концом вводят в раствор NaHCO3 (20 ммоль в 20 мл воды). При комнатной температуре и при перемешивании, медленно по каплям добавляют 10 ммоль сложного эфира N-гидроксисукцинимида с N-защищенным концом, растворенного в 10 мл диоксана. Перемешивание реакционной смеси продолжают на протяжении ночи и затем растворитель удаляют в вакууме.
Дальнейшую обработку осуществляют так же как в Способе А.
Способ F: Отщепление Вос-защитной группы
3 мл смеси 1,1N HCl/ледяная уксусная кислота (Метод F1) или 3 мл смеси 1,1N HCI/диоксан (Метод F2) или 3 мл 50% ТФК в ДХМ (DCM) (Метод F3) добавляют к1 ммолю Вос-защищенная аминокислота - пирролидин, тиазолидин или пептид. Расщепление при КТ контролируют посредством ТСХ. После завершения реакции (приблизительно 2 ч) соединение осаждают в форме гидрохлорида, используя абсолютный диэтиловый эфир, и выделяют отсасыванием и сушат над P4О10 в вакууме. Используя смесь метанол/простой эфир, продукт перекристаллизовывают или переосаждают.
Способ G: Гидролиз
1 ммоль метилового (сложного) эфира пептида растворяют в 10 мл ацетона и 11 мл 0,1M раствора NaOH и перемешивают при комнатной температуре. Ход гидролиза контролируют при помощи ТСХ. После окончания реакции ацетон удаляют в вакууме. Оставшийся водный раствор подкисляют, используя концентрированный раствор KH2SO4, до тех пор, пока рН не достигнет 2-3. Затем продукт экстрагируют несколько раз, используя ЭА (EA); объединенные этилацетатные фракции промывают насыщенным раствором NaCI и сушат над Na2SO4, и растворитель удаляют в вакууме. Осуществляют кристаллизацию из смеси ЭА/пентан.
Пример 7: определение Ki
Для определения Ki используют дипептидилпептидазу IV, выделенную из свиной почки, с удельной активностью в отношении глицилпролил-4-нитроанилина 37,5 Е/мг и концентрацией фермента 1,41 мг/мл в исходном растворе.
Смесь для анализа:
100 мкл испытываемого соединения в диапазоне концентраций 1·10-5M -1·10-8M соответственно смешивают с 50 мкл глицилпролил-4-нитроанилина при различных концентрациях (0,4 мМ, 0,2 мМ, 0,1 мМ, 0,05 мМ) и 100 мкл HEPES (40 мМ, pH 7,6; ионная сила =0,125). Аналитическую смесь предварительно инкубируют при 30°С в течение 30 мин. После предварительной инкубации добавляют 20 мкл DPIV (1:600 разбавленный) и осуществляют контроль за развитием желтой окраски, обусловленной выделением 4-нитроанилина, при 30°С и при λ=405 нм в течение 10 мин, используя спектрофотометр для прочтения планшетов (HTS7000 plus, Applied Biosystems, Weiterstadt, Germany).
Значения Ki рассчитывают, используя Graphit версию 4.0.13, 4.0.13 и 4.0.15 (Erithacus Software, Ltd, UK),
7.1. Результаты - значенияKi ингибирования DPIV
т-бутил-Gly означает:

Ser(Bzl) и Ser(P) означают бензил-серин и фосфорил-серин, соответственно, Tyr(P) означает фосфорил-тирозин.
Пример 8: Определение IC50-Значений
100 мкл исходного раствора ингибитора смешивают с 100 мкл буфера (HEPES pH 7,6) и 50 мкл субстрата (Gly-Pro-pNA, конечная концентрация 0,4 мМ) и предварительно инкубируют при 30°C. Реакцию инициируют добавлением 20 мкл очищенной свиной DPIV. Образование pNA продукта определяют при 405 нм на протяжении 10 мин, используя HTS 7000Plus спектрофотометр для чтения планшетов (Perkin Elmer), и рассчитывают наклоны. Конечные концентрации ингибитора колеблятся между 1 мМ и 30 нМ. Для расчета IC50 используют GraFit версию 4.0.13 (Erithacus Software).
8.1. Результаты - Определение значений IC50
t-butyl-Gly означает:
Ser(Bzl) и Ser(P) означают бензил-серин и фосфорил-серин соответственно, Tyr(P) означает фосфорил-тирозин
Пример 9: Ингибирование DPIV-подобных ферментов - дипептидилпептидазы II
DP II (3.4.14.2) высвобождает N-концевые дипептиды из олигопептидов, если N-конец не протонирован (McDonald, J,K., Ellis, S, & Reilly, T,J., 1966, J, Biol, Chem,, 241, 1494-1501). Pro и Ala в P1-положении являются предпочтительными остатками. Ферментативная активность описана как DPIV-подобная активность, но DP II имеет оптимум рН в кислой области. Используемый фермент выделяют из свиной почки.
Анализ:
100 мкл глутаминилпирролидина или глутаминилтиазолидина в диапазоне концентраций 1·10-4M - 5·10-8M смешивают с 100 мкл буферного раствора (40 мМ HEPES, pH 7,6, 0,015% Brij, 1 мМ DTT), 50 мкл раствора лизилаланиламинометилкумарина (5 мМ) и 20 мкл свиной DP II (250-кратное разбавление в буферном растворе). Измерение флуоресценции осуществляют при 30°С и λвозбуждения=380 нм, λизлучения=465 нм в течение 25 мин, используя ридер-планшет (HTS7000plus, Applied Biosystems, Weiterstadt, Germany). Значения Ki рассчитывают, используя Graphit версию 4.0.15 (Erithacus Software, Ltd., UK) и определяют для глутаминилпирролидина Ki=8,52·10-5M ± 6,33·10-6M и для глутаминилтиазолидина Ki=1,07·10-5M ± 3,81·10-7M.
Пример 10: Ферменты перекрестных взаимодействий
Глутаминилпирролидин и глутаминилтиазолидин были испытаны на их эффективность перекрестных взаимодействий в отношении дипептидилпептидазы I, пролил олигопептидазы и пролидазы.
Дипептидилпептизаза I (DP I, катепсин C),
DP I или катепсин C представляет лизосомальную цистеиновую протеиназу, которая отщепляет дипептиды от N-конца их субстратов (Gutman, H. R. & Fruton, J.S., 1948, J. Biol: Chem., 174, 851-858).Ее классифицируют как цистеиновую протеиназу. Используемый фермент закупают от Qiagen (Qiagen GmbH, Hilden, Germany). Чтобы получить полностью активный фермент, фермент разбавляют в 1000 раз в MES буфере рН 5,6 (40 мМ MES, 4 мМ DTT, 4 мМ KCI, 2мМ EDTA, 0,015% Brij) и предварительно инкубируют в течение 30 мин при 30°С.
Анализ:
50 мкл глутаминилпирролидина или глутаминилтиазолидина в диапазоне концентраций 1·10-5M - 1·10-7M смешивают с 110 мкл смеси буфер-фермент. Смесь для анализа предварительно инкубируют при 300С в течение 15 мин. После предварительной инкубации добавляют 100 мкл гистидилсерил-β-нитроанилина (2·10-5M) и осуществляют контроль за развитием желтой окраски, обусловленной высвобождением β-нитроанилина при 30°С и λвозбуждения=380 нм, λизлучения=465 нм в течение 10 мин, используя ридер-планшет (HTS7000plus, Applied Biosystems, Weiterstadt, Germany).
Значения IC50 рассчитывают, используя Graphit версию 4.0.15 (Erithacus Software, Ltd,, UK). Никакого ингибирования активности фермента DP I глутаминилпирролидином или глутаминилтиазолидином обнаружено не было.
Пролилолигопептидаза (POP)
Пролилолигопептидаза представляет эндопротеиназу серинового типа, которая отщепляет пептиды при N-концевой части Xaa-Pro связи (Walter, R., Shiank, H., Glass, J. D., Schwartz, I.L. & Kerenyi, T.D., 1971, Science, 173, 827-829). Субстратами являются пептиды с молекулярной массой вплоть до 3000 Да. Используемым ферментом была человеческая пролилолигопептидаза. Рекомбинантную экспрессию осуществляли в E. coli в стандартных условиях, как это описано в другом месте, имеющем отношение к данной области.
Анализ:
100 мкл глутаминилпирролидина или глутаминилтиазолидина в диапазоне концентраций 1·10-4M - 5·10-8M смешивают с 100 мкл буферного раствора (40мМ HEPES, pH 7,6, 0,015% Brij, 1 мМ DTT) и 20 мкл раствора POP. Смесь для анализа предварительно инкубируют при 30°С в течение 15 мин. После предварительного инкубирования добавляют 50 мкл раствора глицилпролилпролил-4-нитроанилина (0,29 мМ) и осуществляют контроль за развитием желтой окраски, обусловленной высвобождением 4-нитроанилина при 30°С и λ=405 нм в течение 10 мин, используя ридер-планшет (Sunrise, Tecan Crailsheim, Germany). Значения IC50 рассчитывают, используя Graphit версию 4.0.15 (Erithacus Software, Ltd., UK). Никакого ингибирования активности POP глутаминилпирролидином или глутаминилтиазолидином обнаружено не было.
Пролидаза (X-Pro дипептидаза)
Пролидаза (EC 3.4.13.9) впервые была описана Bergmann & Fruton (Bergmann, M. & Fruton, JS, 1937, J. Biol. Chem. 189-202). Пролидаза высвобождает N-концевую аминокислоту из Xaa-Pro дипептидов и имеет оптимум рН между 6 и 9.
Пролидазу из свиной почки (ICN Biomedicals, Eschwege, Germany) растворяют (1 мг/мл) в аналитическом буфере (20 мМ NH4(CH3С00)2, 3 мМ MnCl2, pH 7,6). Чтобы получить полностью активный фермент, раствор инкубируют в течение 60 мин при комнатной температуре.
Анализ:
450 мкл глутаминилпирролидина или глутаминилтиазолидина в диапазоне концентраций 5·10-3M - 5·10-7M смешивают с 500 мкл буферного раствора (20 мМ NH4(CH3С00)2, pH 7,6) и 250 мкл Ile-Pro-OH (0,5 мМ в смеси для анализа). Смесь для анализа предварительно инкубируют при 30°С в течение 5 мин. После предварительного инкубирования добавляют 75 мкл пролидазы (1:10 разбавленной в буфере для анализа) и осуществляют измерение при 30°С и λ=220 нм в течение 20 мин, используя UV/Vis фотометр, UV1 (Thermo Spectronic, Cambridge, UK).
Значения IC50 рассчитывают, используя версию Graphit 4.0.15 (Erithacus Software, Ltd., UK). Они составляли IC50>3 мМ для глутаминилтиазолидина и для глутаминилпирролидина IC50 = 3,4·10-4 М± 5,63·10-5М.
Пример 11: Определение ингибирующей активности соединений изобретения в отношении DPIV после их внутрисосудистого и перорального введения крысам линии Wistar
Животные
Крыс-самцов линии Wistar (ShOe: Wist(Sho)) с массой тела в диапазоне между 250 и 350 г закупают от Tierzucht Schönwalde (Schönwalde, Germany).
Условия содержания
Животных содержали в клетках, по одной особи в клетку, в обычных условиях с контролируемой температурой (22±2°С) при цикле свет/темнота 12/12 ч (освещение с 06:00 до полудня). С доступом к стандартному гранулированному корму (ssniff® Soest, Germany) и водопроводной воде, подкисленной HCl, по желанию.
Введение катетера в сонную артерию
Спустя ≥ одной недели адаптации в указанных условиях содержания крысам линии Wistar в сонную артерию имплантируют катетеры под общей анестезией (вн.бр. инъекция 0,25 мл/кг массы тела Rompun® [2 %], BayerVital, Germany и 0,5 мл/кг массы тела Ketamin 10, Atarost GmbH & Co., Twistringen, Germany). Животным дают возможность прийти в себя в течение одной недели. Катетеры промывают смесью гепарин-физиологический раствор (100 МЕ/мл) три раза в неделю. В случае дисфункции катетера, вводят второй катетер в контралатеральную сонную артерию соответствующей крысы. Через одну неделю после второго хирургического вмешательства указанное животное вводят в исследование. В случае дисфункции второго катетера животное выводят из исследования. Подбирают новое животное, а эксперименты продолжают в запланированной последовательности, начиная, по крайней мере, 7 дней спустя после имплантации катетера.
Схема эксперимента
Крысам с неповрежденной функцией катера вводят плацебо (1 мл физиологического раствора, 0,154 моль/л) или испытываемое соединение пероральным путем и внутрисосудистым (внутриартериальным) путем.
После голодания на протяжении ночи 100 мкл пробы гепаринизированной артериальной крови отбирают на -30, -5, и 0 мин. Приготавливают свежий раствор испытываемого соединения в 1,0 мл физиологического раствора (0,154 моль/л) и вводят в момент времени 0 либо перорально через зонд для искусственного питания (75 мм; Fine Science Tools, Heidelberg, Germany) либо внутрисосудистым путем. В случае перорального введения в артериальный катетер инъецируют дополнительный объем 1 мл физиологического раствора. В случае внутриартериального введения катетер немедленно промывают 30 мкл физиологического раствора и перорально дают через зонд для искусственного кормления дополнительный 1 мл физиологического раствора.
После применения плацебо или испытываемых веществ отбирают образцы артериальной крови при 2,5; 5; 7,5; 10, 15, 20, 40, 60 и 120 мин из катетера в сонной артерии у крыс, находящихся в сознании и естественном состоянии. Все образцы крови собирали в охлаждаемые льдом пробирки Eppendorf (Eppendorf-Netheler-Hinz, Hamburg, Germany), заполненные 10 мкл буфера - 1М цитрата натрия (pH 3,0), для определения активности DPIV в плазме. Пробирки Eppendorf сразу центрифугируют (12000 об/мин в течение 2 мин, Hettich Zentrifuge EBA 12, Tuttlingen; Germany). Фракции плазмы хранят на льду до анализа или замораживают при -20°С до анализа. Все образцы плазмы метили с указанием следующих данных:
- кодовый номер
- номер животного
- дата отбора проб
- время отбора проб
Аналитические методы
Смесь для анализа на определение активности DPIV в плазме содержала 80 мкл реагента и 20 мкл образца плазмы. Кинетическое измерение образования желтого продукта - 4-нитроанилина - из субстрата глицилпролил-4-нитроанилина осуществляли при 390 нм в течение 1 мин при 30°С после предварительной инкубации в течение 2 мин при той же температуре. Активность DPIV выражали в мЕ/мин.
Статистические методы
Статистические оценки и графики были выполнены с помощью PRISM® 3.02 (GraphPad Software, Inc.). Все параметры представлены в виде общепринятых в описательной статистике определений, включая среднее (значение) и СО (стандартное отклонение).
11.1. Результаты - in vivo ингибирование DPIV при tмакс
(мг/кг)
Пример 12: Действие модифицированных в боковой цепи глутамилтиазолидинов в качестве затрудненно-транспортируемых DPIV-ингибиторов
Были синтезированы модифицированные в боковой цепи глутамилтиазолидины, имеющие структуру H-Glu(X)-Thia, при этом в качестве Х используют полиэтиленгликоль и олигомеры глицина с различными длинами цепей (см. Способ А примера описания синтеза). Были исследованы характеристики связывания этих производных и их транспортируемость пептидным транспортером PepT1.
Неожиданно было обнаружено, что модификации боковой цепи изменяют характеристики связывания соединений с DPIV только в незначительной степени. В противоположность этому способность ингибиторов транспортироваться пептидным транспортером значительно уменьшается при модификации их боковой цепи.
Поэтому модифицированные в боковой цепи ингибиторы DPIV или DPIV-подобных ферментов хорошо подходят для достижения целенаправленного ингибирования DPIV в определенном месте (органе-мишени) живого организма.
12.1. Результаты: Транспортируемость выбранных ингибиторов DPIV
аминокислота- тиазолидины
H-Glu-Thia
1,1
35 ± 13
H-Glu(Gly3)-Thia
H-Glu(Gly5)-Thia
H-Glu(ПЭГ)-Thia
8,54
> 10
> 10
не опред.3
не опред.3
не опред.3
1Эффективные концентрации соединений, ингибирующие связывание3H-D-Phe-Ala (80 мМ) с клетками P. pastoris, экспрессирующими PepT1, на 50% (значения EC50).
2Транспортные характеристики в PepT1-экспрессирующих ооцитах X. leavis - методом двухточечного измерения разности потенциалов, I=внутренние токи, генерированные транспортом
Пример 13: Ингибирование катализируемого DPIV гидролиза инкретинов GIP1-42 и GLP-17-36in vitro
Можно подавить in vitro гидролиз инкретинов, вызываемый DPIV и DPIV-подобной ферментативной активностью, используя очищенный фермент или объединенную человеческую сыворотку (фиг.1).
Согласно настоящему изобретению полное подавление катализируемого ферментом гидролиза обоих пептидных гормонов достигается in vitro инкубированием 30 мМ GIP1-42 или 30 мМ GLP-17-36 и 20 мМ изолейцилтиазолидина (1а), обратимого DPIV-ингибитора, в 20% смешанной сыворотки при рН 7,6 и 30°С на протяжении 24 ч (1b и 1с, оба верхних спектра). Синтетический GIP1-42 (5 мМ) и синтетический GLP-17-36 (15 мкМ) инкубировали с человеческой сывороткой (20 %) в 0,1 мМ TRICINE Буфере при pH 7,6 и 30°С в течение 24 ч. Пробы инкубационных анализируемых смесей (в случае GIP1-42 2,5 пкмоль и в случае GLP-17-36 7,5 пкмоль) отбирали в различные интервалы времени. Пробы подвергали сокристаллизации, используя в качестве матрицы 2',6'-дигидроксиацетофенон, и анализировали MALDI-TOF-масс-спектрометрией. Спектры (Fig. 1) отображают накопления 250 отдельных лазерных импульсов на образец.
(1b) Сигнал m/z4980,1±5,3 соответствует DPIV-субстрат GIP1-42(M 4975,6) и сигнал массы m/z4745,2±5,5 соответствует продукту GIP3-42(M 4740,4), высвобожденному под воздействием DPIV.
(1c) Сигнал m/z3325,0±1,2 соответствует DPIV-субстрат GLP-17-36(M 3297,7) и сигнал массы m/z 3116,7±1,3 соответствует продукту GLP-19-36(M 3089,6), высвобожденному под воздействием DPIV.
В контрольных испытаниях, не содержащих никакого ингибитора, инкретины оказались практически полностью деградированными (фиг.1b и 1c, оба нижних спектра).
Пример 14:Ингибирование деградации GLP17-36 DPIV-ингибитором, изолейцил-тиазолидином, in vivo.
Анализ метаболизма природных инкретинов (в данном случае GLP-17-36) в циркуляции (кровообращении) крысы в присутствии или отсутствии DPIV-ингибитора изолейцил-тиазолидина (вн. сосуд. инъекция 1,5М ингибитора в 0,9% физиологическом растворе) и в контроле. Никакой деградации инсулинотропного пептидного гормона GLP-17-36 не происходит при концентрации 0,1 мг/кг ингибитора изолейцил-тиазолидина у обработанных животных (n=5) во время течения эксперимента (фиг.2).
Чтобы провести анализ метаболитов инкретинов в присутствии и отсутствии DPIV-ингибитора, испытываемые и контрольные животные получали дополнительную вн.сосуд. инъекцию 50-100 пМ(pM) 125I-GLP-17-36 (удельная активность около 1 мкКи/пМ) спустя 20 мин после первоначального вн.сосуд. введения ингибитора и/или введения физиологического раствора. Образцы крови отбирали после 2-5 мин времени инкубации и плазму экстрагировали, используя 20% ацетонитрил. В дальнейшем пептидный экстракт разделяли на ОФ-ВЭЖХ. Множественные фракции элюата собирали между 12-18 мин и считывали на γ-счетчике. Данные представлены в виде числа импульсов в минуту (cpm) относительно максимума.
Пример 15:Модуляция инсулиновых ответов и уменьшения уровней глюкозы в крови после вн. сосуд. введения DPIV-ингибитора, изолейцил-тиазолидина in vivo.
На фигуре представлены реакции циркулирующих (в крови) глюкозы и инсулина на интрадуоденальное (и.д.) введение глюкозы крысам в присутствии или отсутствии изолейцил-тиазолидина (0,1 мг на кг). Наблюдается более быстрое снижение концентрации циркулирующей глюкозы у животных, которые получили DPIV-эффекторы, по сравнению с необработанными животными. Наблюдаемый эффект носит дозозависимый характер и является обратимым после прекращения вливания 0,05 мг/мин DPIV-ингибитора изолейцил-тиазолидина на 1 кг массы крысы. В отличие от и.д. глюкозостимулированных животных после вн. сосуд. введения такого же количества глюкозы контрольным животным, подвергнутым обработке ингибитором, никакого сравнимого с вышеуказанным эффектом действия не наблюдалось. На фиг.3 представлены вышеуказанные зависимости, отображающие ингибитор-зависимые изменения выбранных плазменных параметров: A - DPIV-активность, B - уровень инсулина в плазме, C - уровень глюкозы в плазме.
Пример 16: Влияние хронического лечения тучных крыс линии Zucker на уровень глюкозы в крови натощак во время 12-недельного перорального применения лекарственного средства.
Хроническое применение DPIV-ингибитора изолейцил-тиазолидина приводит к драматическому снижению и практически к нормализации уровня глюкозы в крови натощак у выбранной животной модели - диабетических крыс (фиг.4).
Животные.
Шесть пар однопометных тучных крыс-самцов(fa/fa)VDF Zucker были беспорядочно распределены либо в контрольную группу либо в группу, подлежащую обработке (изолейцил-тиазолидин фумарат), при массе тела 440 г (возраста 11±0,5 недель). Животных содержали по одному, при цикле 12 ч.свет/темнота (освещение с 6.00 до полудня), со свободным доступом к стандартному корму для крыс и воде.
Протокол суточного контроля и введения лекарственного средства
Группа, подлежащая обработке, получала 10 мг/кг изолейцил-тиазолидин фумарата при помощи перорального зонда дважды в день (8:00 до полудня и 5:00 после полудня) в течение 100 дней, в то время как контрольные животные получали параллельные дозы наполнителя, состоящие из 1% раствора целлюлозы. Каждые два дня оценивали массу тела, уровень глюкозы утром и вечером и потребление пищи и воды. Пробы крови для определения глюкозы отбирали из хвоста и концентрацию определяли, используя анализатор глюкозы SureStep (Lifescan Canada Ltd., Burnaby).
Протокол для ежемесячной оценки глюкозотолерантности
Каждые четыре недели от начала эксперимента проводили пероральный глюкозотолерантный тест (ПГТТ, OGTT): животных подвергали голоданию после 17 ч дозирования и получали 1 г/кг глюкозы перорально. Этот период времени эквивалентен ˜12 периодам полужизни циркулирующего изолейцил-тиазолидин фумарата.
Пример 17: Влияние хронической пероральной обработки тучных крыс линии Zucker DPIV-ингибитором, изолейцил-тиазолидином на систолическое кровяное давление
Хроническое применение DPIV-ингибитора, изолейцил-тиазолидин фумарата приводит к стабилизации систолическго кровяного давления в выбранной животной модели - диабетических крысах (фиг.5).
Животные
Шесть пар (одного помета) тучных крыс-самцов (fa/fa)VDF Zucker были беспорядочно распределены либо в контрольную группу либо в группу, подлежащую обработке (изолейцил-тиазолидин фумарат), при массе тела 440 г (возраста 11±0,5 недель). Животных содержали по одному, при цикле 12 ч.свет/темнота (освещение с 6.00 до полудня), при свободном доступе к стандартному корму для крыс и воде.
Протокол суточного контроля и введения лекарственного средства
Группа, подлежащая обработке, получала 10 мг/кг изолейцил-тиазолидин фумарата при помощи перорального зонда дважды в день (8:00 до полудня и 5:00 после полудня) в течение 100 дней, в то время как контрольные животные получали параллельные дозы наполнителя, состоящие из 1% раствора целлюлозы. Систолическое кровяное давление измеряют еженедельно, используя способ наложения манжеты на хвост).
Испытываемые животные (n=5, крысы-самцы линии Wistar, 200-225 г) сначала получали 1,5M изолейцил-тиазолидина в 0,9% физиологическом растворе () или такой же объем чистого 0,9% физиологического раствора (¦) (контрольная группа n=5). Испытываемую группу дополнительно подвергали инфузии ингибитора 0,75 М/мин на протяжении 30 мин экспериментального времени (*). Контрольную группу подвергали на протяжении такого же интервала времени инфузии 0,9% физиологического раствора, не содержащего ингибитор. В начальный момент времени t=0 всем животным вводили вн.д.(i.d.) дозу глюкозы 1 г/кг (40% раствор декстрозы, мас./об.). Пробы крови отбирали у всех испытываемых животных через 10-минутные интервалы времени. Глюкозу анализировали, используя цельную кровь (анализатор Lifescan One Touch II), в то время как активность DPIV и концентрацию инсулина анализировали в плазме. Радиоиммунный анализ инсулина чувствителен в диапазоне 10 и 160 мЕ/мл [PEDERSON, R.A., BUCHAN, A.M.J., ZAHEDI-ASH, S., CHEN, C.B. & BROWN, J.C Reg. Peptides, 3, 53-63 (1982)]. Активность DPIV оценивали спектрофотометрически[DEMUTH, H.-U. and HEINS, J., On the catalytic Mechanism of Dipeptidyl Peptidase IV. in Dipeptidyl Peptidase IV (CD26) in Metabolism and the Immune Response (B. Fleischer, Ed.) R.G. Landes, Biomedical Publishers, Georgetown, 1-35 (1995)]. Все данные представлены как среднее ± с.с.о. (среднее стандартное отклонение).
Пример 18: Исследование воздействия перорального введения возрастающих доз глутаминилпирролидина на тучных крысах линии Zucker
Животные
N=30 крыс-самцов линии Zucker (fa/fa), средний возраст 11 недель (5-12 недель), средняя масса тела 350 г (150-400 г) были закуплены от Charles River (Sulzfeld, Germany).
После доставки крыс выдерживали в течение >12 недель до тех пор, пока почти все тучные крысы линии Zucker не обнаруживали черты явно выраженного сахарного диабета. Группа из N=8 животных была подвергнута испытанию трех возрастающих доз глутаминилпирролидина в сравнении с плацебо (физиологический раствор).
Условия содержания
Животных содержали в клетках, по одной особи в клетку, в стандартизованных условиях с контролируемой температурой (22±2°С) при цикле свет/темнота 12/12 ч (освещение с 06:00 до полудня). Доступ к стандартному стерильному гранулированному корму (ssniff® Soest, Germany) и водопроводной воде, подкисленной HCl, был свободным.
Введение катетера в сонную артерию
Тучные крысы линии Zucker возраста 24-31 (средний: 25 недель) недель, адаптированные к условиям содержания, были соответствующим образом подготовлены для исследования.
Тучным крысам линии Zucker в сонную артерию имплантируют катетеры под общей анестезией (вн.бр. инъекция 0,25 мл/кг массы тела Rompun® [2 %], BayerVital, Germany и 0,5 мл/кг массы тела Ketamin 10, Atarost GmbH & Co., Twistringen, Germany). Животным дают возможность прийти в себя в течение одной недели. Катетеры промывают смесью гепарин-физиологический раствор (100 МЕ/мл) три раза в неделю.
Схема эксперимента
Группам из N=8 тучных крыс линии Zucker вводят плацебо (1 мл физиологического раствора, 0,154 моль/л) или возрастающие дозы глутаминилпирролидина (5, 15 и 50 мг/кг массы тела). 375 мг глутаминилпирролидина растворяют в 1000 мкл ДМСО (E. Merck, Darmstadt; Germany [Dimethyl sulfoxide p.a.]). Добавляют 10 мл физиологического раствора и 1 мл аликвоты, каждая содержащая 34,09 мг глутаминилпирролидина, хранят при -20°С. Для препарата из испытываемого вещества дозозависимые аликвоты разбавляют в физиологическом растворе.
После голодания на протяжении ночи тучным крысам линии Zucker вводили плацебо или испытываемое вещество перорально при помощи зонда для искусственного кормления (15 G, 75 мм; Fine Science Tools, Heidelberg, Germany) на -10 мин. Пероральный глюкозотолерантный тест (ПГТТ, OGTT) с 2 г глюкозы/кг м.т. (40% раствор, B. Braun Melsungen, Melsungen, Germany) применяли на ±0 мин при помощи второго питательного зонда. Образцы венозной крови из хвостовой вены отбирали при -30 мин, -15 мин, ±0 мин и при 5, 10, 15, 20, 30, 40, 60, 90 и 120 мин в 20-мкл стеклянные капилляры, которые помещали в стандартные пробирки, наполненные 1 мл раствора для определения уровня глюкозы в крови.
Все образцы крови метили с указанием следующих данных:
- кодовый номер
- номер животного
- дата отбора пробы
- время отбора пробы
Аналитические методы
Уровни глюкозы определяли, используя глюкозооксидазный метод (Super G Glucose analyzer; Dr. Müller Gerätebau, Freital, Germany)
Статистические методы
Статистические оценки и графики были выполнены с помощью PRISM® 3.02 (GraphPad Software, Inc.). Все параметры представлены в виде общепринятых в описательной статистике определений, включая среднее (значение) и СО.
Влияние лечения на глюкозотолерантность
Диабетические крысы линии Zucker, подвергнутые обработке плацебо, демонстрировали сильное отклонение в сторону повышенных уровней глюкозы в крови, что указывает на интолерантность к глюкозе, свойственную больным с явно выраженным сахарным диабетом. Введение 5 мг глутаминилпирролидина/кг м.т. приводило к ограниченному повышению глюкозотолерантности у диабетических крыс Zucker. Значительное снижение повышенных уровней глюкозы в крови и повышение глюкозотолерантности достигалось после введения 15 мг/кг и 50 мг глутаминилпирролидина/кг м.т. (см. фиг.6).
Пример 19:Исследование воздействия перорального введения возрастающих доз глутаминилтиазолидина на тучных крысах линии Zucker
Животные:
N=30 крыс-самцов линии Zucker (fa/fa), средний возраст 11 недель (5-12 недель), средняя масса тела 350 г (150-400 г) были закуплены от Charles River (Sulzfeld, Germany).
После доставки крыс выдерживали в течение >12 недель до тех пор, пока почти все тучные крысы линии Zucker не обнаруживали черты явно выраженного сахарного диабета. Группа из N=8 животных была подвергнута испытанию трех возрастающих доз глутаминилтиазолидина в сравнении с плацебо (физиологический раствор).
Условия содержания
Животных содержали в клетках, по одной особи в клетку, в стандартизированных условиях с контролируемой температурой (22±2°С) при цикле свет/темнота 12/12 ч (освещение с06:00 до полудня). Доступ к стандартному стерильному гранулированнному корму (ssniff® Soest, Germany) и водопроводной воде, подкисленной HCl, был свободным.
Введение катетера в сонную артерию
Тучные крысы линии Zucker возраста 24-31 (среднее значение: 25 недель) недель, адаптированные к условиям содержания, были соответствующим образом подготовлены для исследования.
Тучным крысам линии Zucker в сонную артерию имплантировали катетеры под общей анестезией (вн.бр. инъекция 0,25 мл/кг массы тела Rompun® [2 %], BayerVital, Germany и 0,5 мл/кг массы тела Ketamin 10, Atarost GmbH & Co., Twistringen, Germany). Животным предоставили возможность прийти в себя в течение одной недели. Катетеры промывали смесью гепарин-физиологический раствор (100 МЕ/мл) три раза в неделю.
Схема эксперимента
Группам из N=8 тучных крыс линии Zucker вводили плацебо (1 мл физиологического раствора, 0,154 моль/л) или возрастающие дозы глутаминилтиазолидина (5, 15 и 50 мг/кг массы тела). Соответствующие количества глутаминилтиазолидина растворяли в 1000 мкл физиологического раствора.
После голодания на протяжении ночи тучным крысам линии Zucker вводили плацебо или испытываемое вещество перорально при помощи питательного зонда (15 G, 75 мм; Fine Science Tools, Heidelberg, Germany) на -10 мин. Пероральный глюкозотолерантный тест (ПГТТ, OGTT) с 2 г глюкозы/кг м.т. (40% раствор, B. Braun Melsungen, Melsungen, Germany) применяли на ±0 мин при помощи второго питательного зонда. Образцы венозной крови из хвостовой вены отбирали на -30 мин, -15 мин, ±0 мин и на 5, 10, 15, 20, 30, 40, 60, 90 и 120 мин в 20-мкл стеклянные капилляры, которые помещали в стандартные пробирки, наполненные 1 мл раствора, для определения уровня глюкозы в крови.
Все образцы крови метили с указанием следующих данных:
- кодовый номер
- номер животного
- дата отбора пробы
- время отбора пробы
Аналитические методы
Уровни глюкозы определяли, используя глюкозооксидазный метод (Super G Glucose analyzer; Dr. Müller Gerätebau, Freital, Germany)
Статистические методы
Статистические оценки и графики были выполнены с помощью PRISM® 3.02 (GraphPad Software, Inc.). Все параметры представлены в виде общепринятых в описательной статистике определений, включая среднее (значение) и СО.
Влияние лечения на глюкозотолерантность:
Диабетические крысы Zucker, подвергнутые обработке плацебо, демонстрировали сильное отклонение в сторону повышенных уровней глюкозы в крови, что указывает на интолерантность к глюкозе, свойственную субъектам с явно выраженным сахарным диабетом. Введение 5 мг/кг, 15 мг/кг и 50 мг глутаминилтиазолидина/кг м.т. приводило к дозозависимому снижению повышенных уровней глюкозы в крови и повышению глюкозотолерантности у диабетических крыс линии Zucker (см. фиг.7).
Пример 20: In vivo инактивация глутаминилтиазолидина после перорального введения крысам линии Wistar
Животные/Схема эксперимента
Глутаминилтиазолидин вводят крысам линии Wistar перорально, как описано в примере 9.
Аналитические методы
После применения плацебо или глутаминилтиазолидина у крыс, находящихся в сознании и в естественном состоянии, отбирают пробы артериальной крови из катетера, имплантированного в сонную актерию, при 2,5;5; 7,5; 10, 15, 20, 40, 60 и 120 мин с целью обнаружения образования продуктов деградации глутаминилтиазолидина.
Для анализа, для выделения из плазмы представляющих интерес соединений используют простой твердофазный способ экстракции на C18 картриджах. Экстракты анализируют, используя комбинированный метод анализа - жидкостную хроматографию с обращенной фазой на колонке Lichrospher 60 RP Select B в сочетании с тамдемной масс-спектрометрией, функционирующей в режиме APCI (химическая ионизация при атмосферном давлении), с регистрацией положительных ионов. Для количественной оценки используют метод внутреннего стандарта.
Результаты
После перорального введения глутаминилтиазолидина крысам линии Wistar была обнаружена деградация соединения. Используя ЖХ/МС, продукт деградации смогли определить как пироглутаминилтиазолидин. См. фиг.8 и 9.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2002 |
|
RU2299066C2 |
| ЭФФЕКТОРЫ ДИПЕПТИДИЛПЕПТИДАЗУ IV | 1999 |
|
RU2309161C2 |
| СПОСОБ УЛУЧШЕНИЯ ПЕРЕДАЧИ СИГНАЛА В ОСТРОВКАХ ЛАНГЕРГАНСА ПРИ САХАРНОМ ДИАБЕТЕ И ЕГО ПРОФИЛАКТИКЕ | 2001 |
|
RU2261096C2 |
| НОВЫЕ АНТИДИАБЕТИЧЕСКИЕ АГЕНТЫ | 2000 |
|
RU2265012C2 |
| СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ-4, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2015 |
|
RU2589258C1 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ MASP, И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2840441C2 |
| СТАБИЛИЗИРОВАННЫЕ ПО ОТНОШЕНИЮ К ПРОТЕАЗАМ АЦИЛИРОВАННЫЕ АНАЛОГИ ИНСУЛИНА | 2009 |
|
RU2571857C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2004 |
|
RU2385723C2 |
| ПРОЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ С ВЫСОКОЙ СТЕПЕНЬЮ ПРОНИКНОВЕНИЯ НА ОСНОВЕ ПЕПТИДОВ И РОДСТВЕННЫХ ПЕПТИДАМ СОЕДИНЕНИЙ | 2010 |
|
RU2627065C2 |
| АНАЛОГ ЭКСЕНАТИДА | 2020 |
|
RU2823623C1 |
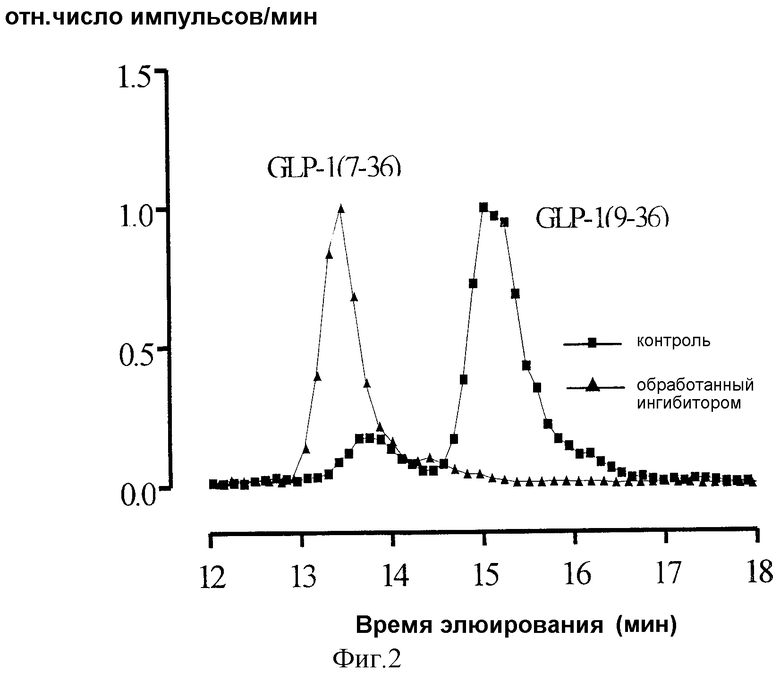
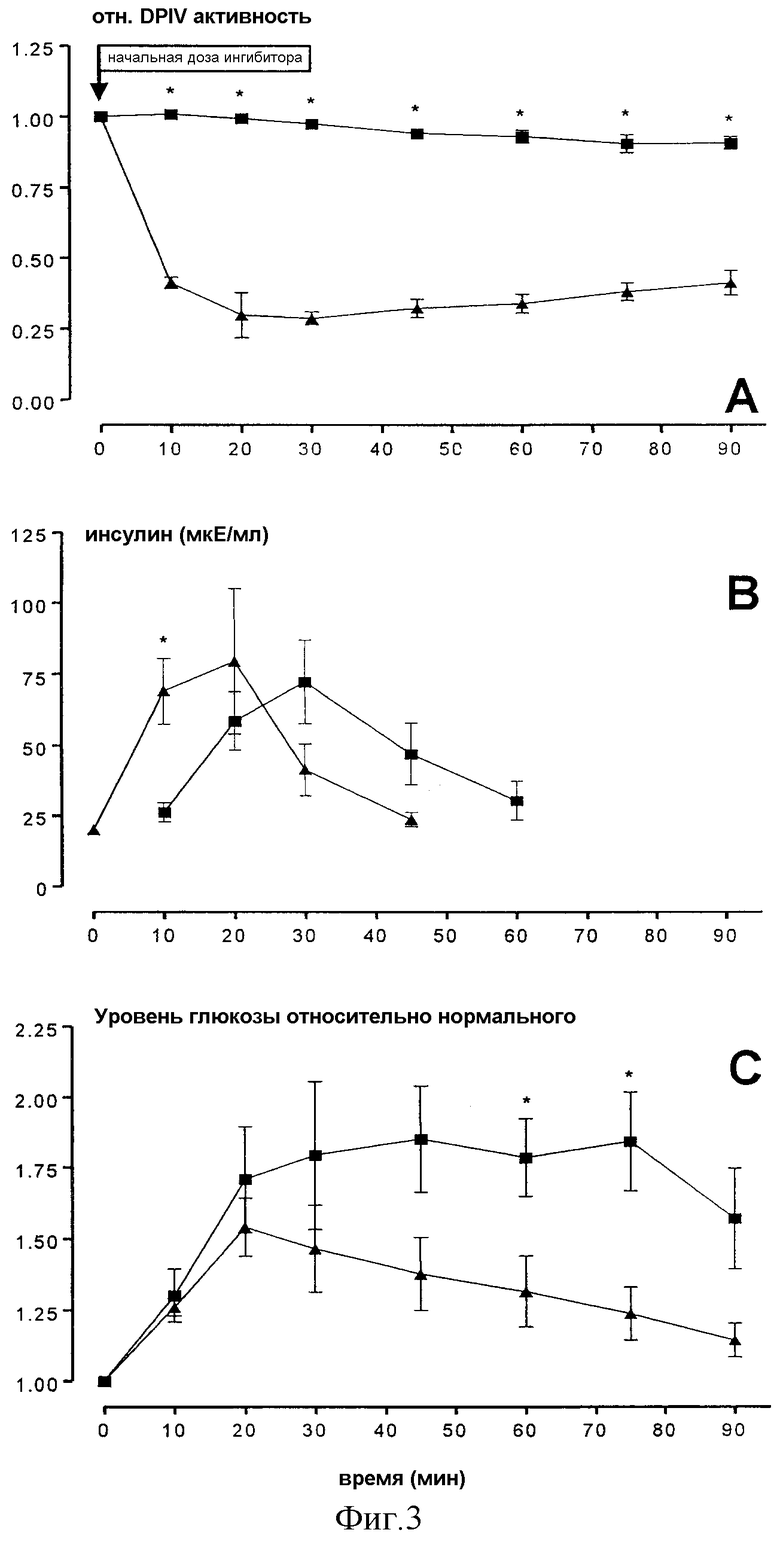
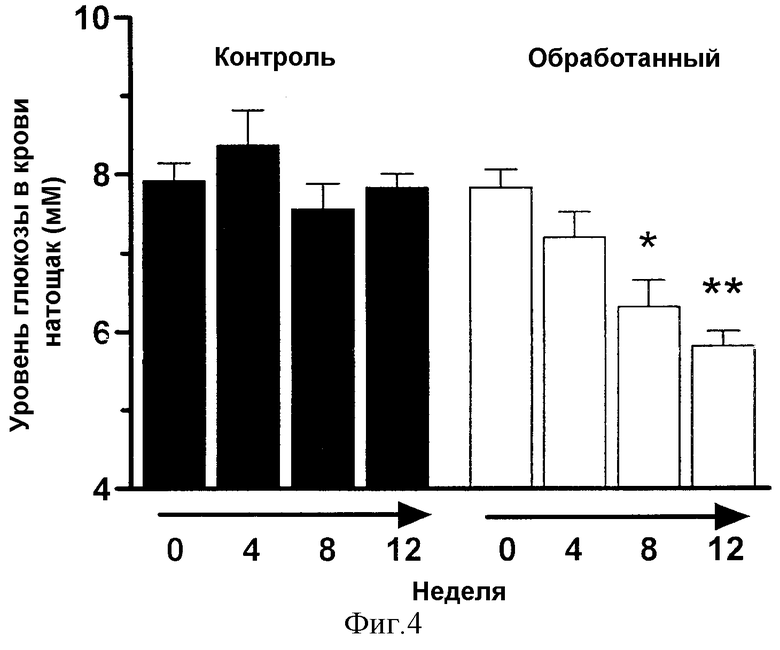
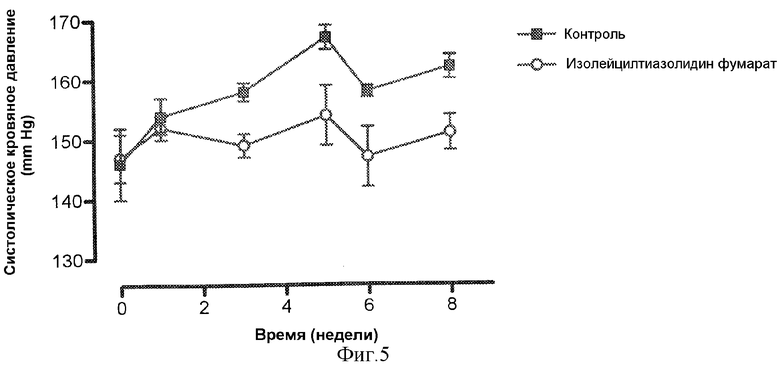
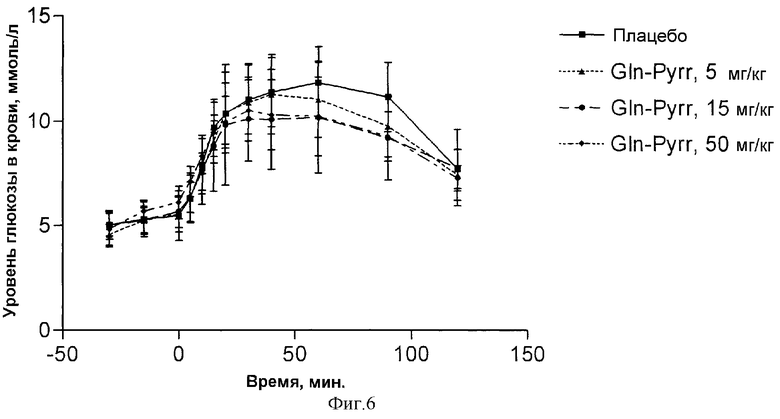

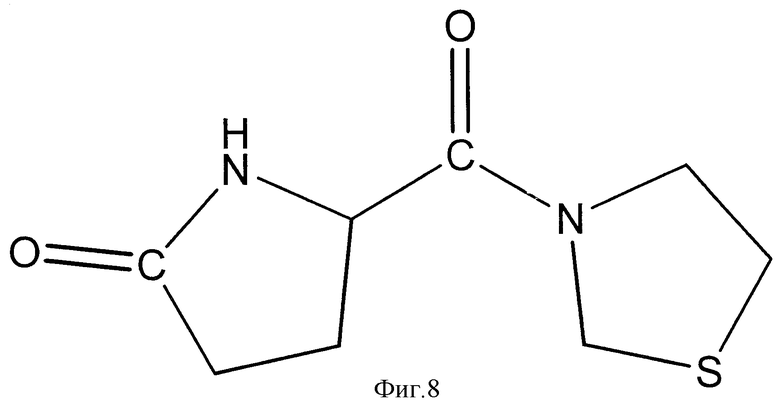
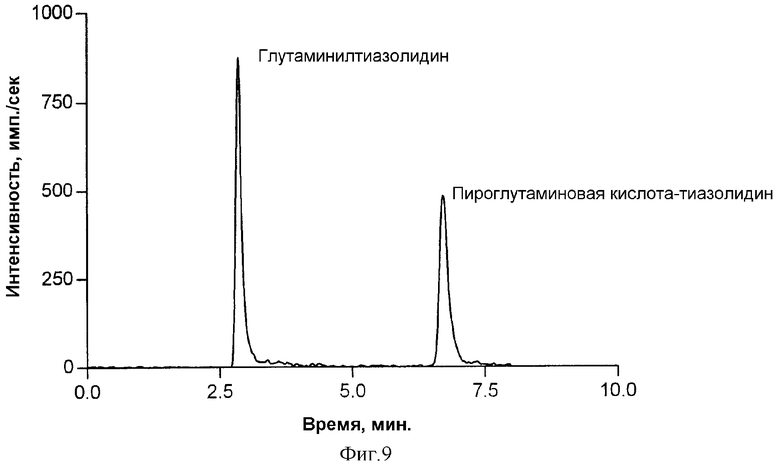
Предложено применение ингибиторов дипептидилпептидазы (DPIV-ингибиторов) и, в частности, изолейцил-тиазолидина в качестве активного компонента фармацевтической композиции для понижения уровней кровяного давления у млекопитающих с диабетом. Показана стабилизация систолического давления у диабетических крыс и снижение его уровней со 170 (контрольные животные без изолейцил-тиазолидина) до 150 мм рт. ст. 8 з.п. ф-лы, 9 ил., 11 табл.
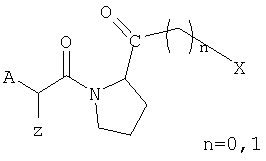
включая все стереомеры, и его фармацевтически приемлемые соли,
где А выбран из
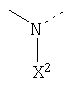
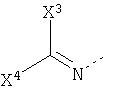

и X1 представляет собой Н или ацильную или оксикарбонильную группу или остаток аминокислоты или пептида,
X2 представляет собой Н, -(CH)n-NH-C5H3N-Y с n=2-4 или C5H3N-Y (дивалентный остаток пиридила) и Y выбран из Н, Br, Cl, I, NO2 или CN,
X3 представляет собой Н или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген, нитро, циано или карбокси группами,
X4 представляет собой Н или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген нитро, циано или карбокси группами,
X5 представляет собой Н или алкил, алкокси или фенил,
X6 представляет собой Н или алкил;
для n=1
Х выбран из Н, OR2, SR2, NR2R3, N+R2R3R4, где
R2 означает ацильные остатки, которые являются незамещенными или замещенными одним, двумя или несколькими алкилом, циклоалкилом, арилом или гетероарильными остатками, или обозначает все аминокислоты и пептидные остатки, или алкильные остатки, которые незамещены или замещены одним, двумя или несколькими алкилами, циклоалкилами, арилами и гетероарильными остатками,
R3 обозначает алкильные и ацильные функции, где R2 и R3 могут быть частью одной или нескольких кольцевых структур насыщенных и ненасыщенных карбоциклических или гетероциклических структур,
R4 означает алкильные остатки, где R2 и R4 или R3 и R4 могут быть частью одной или нескольких кольцевых структур насыщенных и ненасыщенных карбоциклических или гетероциклических структур;
для n=0
Х выбран из
где В означает О, S, NR5, где R5 представляет собой Н, алкилиден или ацил,
С, D, Е, F, G, Н независимо выбраны из незамещенных и замещенных алкилов, оксиалкилов, тиоалкилов, аминоалкилов, карбонилалкилов, ацилов, карбамоил, арил и гетероарильных остатков; и
для n=0 и n=1
Z выбран из Н, C1-C9 алкила с разветвленной или одинарной цепью или С2-С9 алкенила с разветвленной или одинарной цепью, С3-C8 циклоалкила, С5-С7 циклоалкенила, арил- или гетероарильного остатка, или боковой цепи, выбранной из боковых цепей природных аминокислот.

где R1 представляет собой Н, разветвленный или прямой C1-C9 алкильный остаток, разветвленный или прямой C2-C9 алкенильный остаток, С3-С8 циклоалкил-, С5-С7 циклоалкенил-, арил- или гетероарильный остаток или боковую цепь природной аминокислоты,
R3 и R4 независимо выбраны из Н, гидрокси, алкила, алкокси, арилокси, нитро, циано или галогена,
А представляет собой Н или изостер карбоновой кислоты, функциональную группу, выбранную из CN, SO3Н, CONHOH, РО3R5R6, тетразола, амида, сложного эфира, ангидрида, тиазола и имидазола;
В выбран из
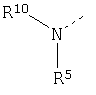
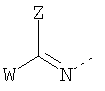
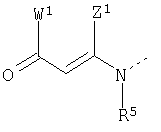
где R5 представляет собой Н, -(CH)n-NH-C5H3N-Y с n=2-4 и С5Н3N-Y (дивалентный остаток пиридила) с Y=H, Br, Cl, I, NO2 или CN,
R10 представляет собой Н, ацил, оксикарбонил или остаток аминокислоты
W представляет собой Н или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген, нитро, циано или карбокси группами,
W1 представляет собой Н, алкил, алкокси или фенил,
Z представляет собой Н или фенил или пиридил, незамещенный или замещенный одной, двумя или несколькими алкил, алкокси, галоген, нитро, циано или карбокси группами,
Z' представляет собой Н или алкил,
D представляет собой циклический С4-С7 алкил, С4-С7 алкенильный остаток, который может быть незамещенным или замещенным одним, двумя или несколькими алкильными группами, или циклический (4-7)-членный гетероалкил или циклический (4-7)-членный гетероалкенильный остаток,
X2 представляет собой О, NR6, N+(R7)2 или S,
X3 до X12 независимо выбраны из СН2, CR8R9, NR6, N+(R7)2, О, S, SO и SO2, включая все насыщенные и ненасыщенные структуры,
R6, R7, R8, R9 независимо выбраны из Н, разветвленного или прямого C1-C9 алкильного остатка, разветвленного или прямого С2-С9 алкенильного остатка, С3-C8 циклоалкильного остатка, С5-С7 циклоалкенильного остатка, арил или гетероарильного остатка,
при следующих условиях:
формула 6: X6 представляет СН, если А не является Н,
формула 7: Х10 представляет С, если А не является Н,
формула 8: Х7 представляет СН, если А не является Н,
формула 9: Х12 представляет С, если А не является Н.
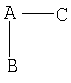
включая все стереомеры и их фармацевтически приемлемые соли, где
А представляет α-аминокислоту, в частности природную α-аминокислоту, имеющую, по крайней мере, одну функциональную группу в боковой цепи, выбранную из группы, состоящей из треонина, тирозина, серина, аргинина, лизина, аспарагиновой кислоты, глутаминовой кислоты или цистеина,
В представляет химическое соединение, ковалентно связанное, по крайней мере, с одной функциональной группой боковой цепи А, в частности
олигопептид, имеющий длину цепи вплоть до 20 аминокислот, или
полиэтиленгликоль, имеющий молярную массу вплоть до 20000 г/моль,
необязательно замещенный органический амин, амид, спирт, кислоту или ароматическое соединение, имеющее от 8 до 50 атомов С, и
С представляет тиазолидин, пирролидин, цианопирролидин, гидроксипролин, дегидропролин или пиперидиновую группу, связанную с А через амидную связь.
| реферат Entrez PubMed: Papies В | |||
| et al | |||
| Isoenzyme (lactate dehydrogenase, aspartate aminotransferase) and dipeptidyl peptidase IV activity changes in blood plasma likely indicative of organ involvement due to arterial hypertension | |||
| Cor Vasa | |||
| Циркуль-угломер | 1920 |
|
SU1991A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

