Область техники
Настоящее изобретение относится к способу получения феноксизамещенных 2-пиридоновых соединений, которые применимы в качестве промежуточных продуктов для получения медицинского и сельскохозяйственного препарата, в частности гербицидного соединения.
Уровень техники
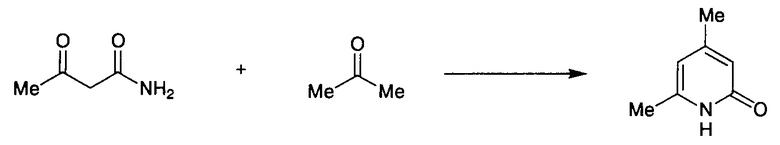
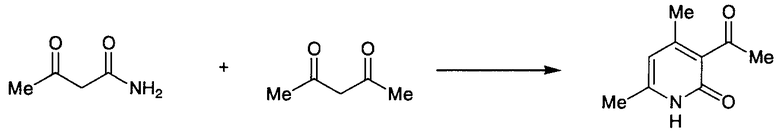
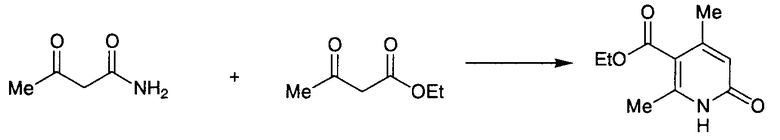
Известно, что 2-пиридоновые соединения могут быть получены взаимодействием 3-оксобутанамида с различными кетонами. В частности, 4,6-диметил-2-пиридон получен взаимодействием 3-оксобутанамида с ацетоном в присутствии полифосфорной кислоты; 3-ацетил-4,6-диметил-2-пиридон получен взаимодействием 3-оксобутанамида с пентан-2,4-дионом в присутствии хлористого водорода или полифосфорной кислоты; и 5-этоксикарбонил-4,6-диметил-2-пиридон получен взаимодействием 3-оксобутанамида с этилацетатом в присутствии полифосфорной кислоты (Chem. Pharm. Bull. 28(7) 2244-2247 (1989), J. Chem. Soc. (C), 1967, 1836-1839).
Описание изобретения
Авторы настоящего изобретения интенсивно изучили способ получения 2-пиридонового соединения, замещенного в положении 3 необязательно замещенным фенокси, которое применимо в качестве промежуточного продукта для получения медицинского и сельскохозяйственного препарата, в частности гербицидного соединения. В результате авторы установили, что 3-фенокси-2-пиридон, имеющий один заместитель в положении 3, может быть получен взаимодействием 2-фенокси-3-оксобутанамида, в котором фенокси может быть замещенным, с определенным производным малонового альдегида или с малоновым альдегидом, что составляет настоящее изобретение.
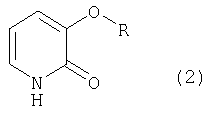
В настоящем изобретении предлагается способ (в дальнейшем называемый как данный способ) получения пиридонового соединения формулы (2) (в дальнейшем называемого как данное пиридоновое соединение):
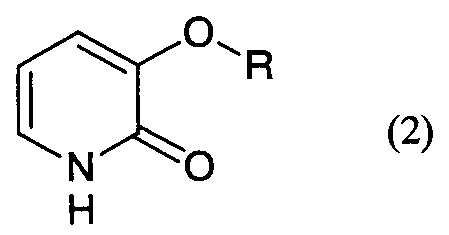
где R определен ниже;
который включает взаимодействие амидного соединения формулы (1) (в дальнейшем называемого как данное амидное соединение):
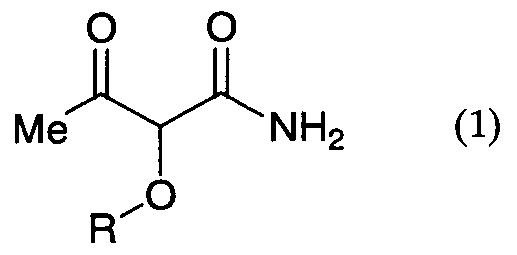
где R представляет необязательно замещенный фенил;
по меньшей мере, с одним соединением (в дальнейшем именуемым как данное производное малонового альдегида), выбранным из группы, включающей 3-алкоксипропеналь формулы (3), 3,3-диалкоксипропаналь формулы (4), 1,3,3-триалкокси-1-пропен формулы (5), 1,1,3,3-тетраалкоксипропан формулы (6) и малоновый альдегид:


где R9 представляет алкил (например, С1-С3алкил, такой как метокси, этокси и т.п.);
в присутствии протонной кислоты.
Кроме того, в настоящем изобретении предлагается также способ получения пиридонового соединения формулы (В):
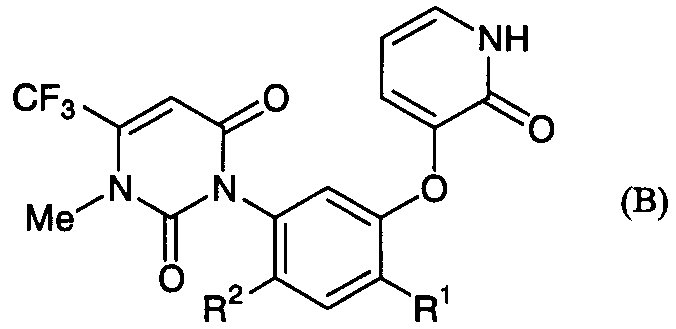
где R1 и R2 определены ниже;
с использованием амидного соединения формулы (А):
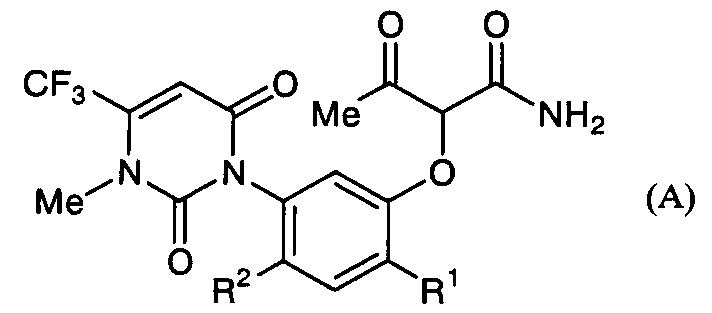
где R1 представляет атом галогена или нитрогруппу и R2 представляет атом водорода или атом галогена;
в качестве данного амидного соединения.
Заместитель в необязательно замещенном фенокси данного амидного соединения, которое является исходным соединением для данного способа, и данное пиридоновое соединение включают, например, атом галогена (т.е. атом фтора, атом хлора, атом брома и т.п.); алкил (например, С1-С4алкил, такой как метил, этил, пропил, изопропил т.п.); алкокси (например, С1-С4алкокси, такой как метокси, этокси, пропокси, изопропокси и т.п.); галогеналкил (например, трифторметил, пентафторэтил); нитро; циано; и 5-6-членный гетероциклический радикал (например, 1,2,3,6-тетрагидро-2,6-диоксопиримидин-1-ил, 1,6-дигидро-6-оксопиридазин-1-ил и т.п.). Необязательно замещенный фенокси включает, например, фенокси и фенокси, показанные на схеме:
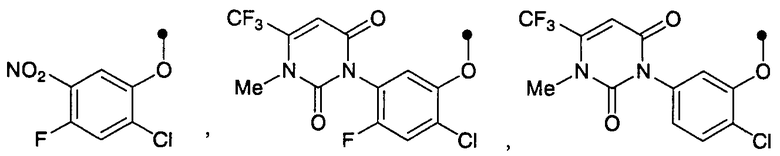
где R1 и R2 определены выше.
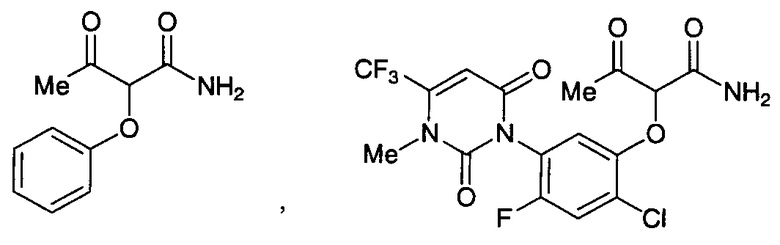
Данное амидное соединение включает, например, соединение, показанное на схеме:
Данное производное малонового альдегида представляет собой малоновый альдегид или обычные продукты конденсации малонового альдегида и спирта, среди которых, в частности, по меньшей мере, одно производное малонового альдегида выбрано из группы, включающей 3-алкоксипропеналь формулы (3), 3,3-диалкоксипропаналь формулы (4), 1,3,3-триалкокси-1-пропен формулы (5), 1,1,3,3-тетраалкоксипропан формулы (6). 3-Алкоксипропеналь формулы (3) включает, например, 3-метоксипропеналь и 3-этоксипропеналь. 3,3-Диалкоксипропаналь формулы (4) включает, например, 3,3-диметоксипропаналь и 3,3-диэтоксипропаналь. 1,3,3-Триалкокси-1-пропен формулы (5) включает, например, 1,3,3-триметокси-1-пропен и 1,3,3-триэтокси-1-пропен. 1,1,3,3-Тетраалкоксипропан формулы (6) включает, например, 1,1,3,3-тетраметоксипропан и 1,1,3,3-тетраэтоксипропан.
Для данного способа 1,1,3,3-тетраалкоксипропан формулы (6) является предпочтительным соединением как производное малонового альдегида по причине его доступности и прочего.
Протонная кислота, предназначенная для применения по данному способу, представляет собой соединение, обладающее сильной тенденцией отдавать протон, в соответствии с теорией кислот и оснований Бренстеда, основанной на предоставлении и акцептировании протона. В частности, протонная кислота включает галогеноводороды (например, хлористый водород, бромистый водород и т.п.), фосфорную кислоту, полифосфорную кислоту, серную кислоту, тригалогенуксусную кислоту (например, трихлоруксусную кислоту, трифторуксусную кислоту и т.п.), сульфоновую кислоту (например, хлорсульфоновую кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, трифторметансульфоновую кислоту и т.п.) и их смеси, причем предпочтительно протонные кислоты имеют значения рКа (константа диссоциации кислоты в воде), меньшие или равные 2,5.
Реакция по данному способу может быть проведена в растворителе. Растворитель, предназначенный для применения, включает, например, ароматические углеводороды, такие как толуол, ксилол и т.п.; галогенированные ароматические углеводороды, такие как хлорбензол, дихлорбензол, бензотрифторид и т.п.; галогенированные алифатические углеводороды, такие как хлороформ, 1,2-дихлорэтан и т.п.; спирты, такие как гексафторизопропанол и т.п.; и их смеси.
В реакции обычно используют от 1 до 10 моль данного производного малонового альдегида и количество кислоты от каталитического до избыточного (например, от 0,1 до 1000 моль, предпочтительно, от 1 до 10 моль), по отношению к 1 моль данного амидного соединения.
Температура проведения реакции обычно находится в интервале от 0 до 150°C, предпочтительно от 20 до 100°C, время реакции обычно составляет от 0,5 до 72 часов, причем указанные условия реакции зависят от количества и типов применяемых протонных кислот.
Реакцию проводят при добавлении данного производного амидного соединения и данного производного малонового альдегида к протонной кислоте или к протонной кислоте, разбавленной вышеупомянутым растворителем. В данном случае допустимо, что вся протонная кислота применяется сразу в начале реакции; и часть протонной кислоты применяется в начале реакции, а остаток добавляют по ходу течения реакции. Данное амидное соединение и данный малоновый альдегид могут быть добавлены сразу в протонную кислоту или в протонную кислоту, разбавленную вышеупомянутым растворителем; но, предпочтительно, данное амидное соединение и данный малоновый альдегид следует добавлять постепенно в соответствии с течением реакции.
Ход реакции можно контролировать, например, отбирая пробу из реакционной смеси и исследуя ее хроматографически (например, с помощью тонкослойной хроматографии, высокоэффективной жидкостной хроматографии и т.п.) для анализа остаточных количеств амидного соединения формулы (1) в реакционной смеси.
Данное пиридоновое соединение может быть выделено из реакционной смеси после завершения реакции с помощью следующих процедур, которые включают:
1. Разбавление реакционной смеси после завершения реакции гидрофобным органическим растворителем; промывание смеси насыщенным водным раствором хлорида натрия, насыщенным водным раствором бикарбоната натрия или т.п.; сушка полученного органического слоя; и концентрирование органического слоя для полного удаления растворителя.
2. Разбавление реакционной смеси после завершения реакции гидрофобным органическим растворителем; промывание смеси насыщенным водным раствором хлорида натрия, насыщенным водным раствором бикарбоната натрия или т.п.; частичное концентрирование органического слоя при температуре от 80 до 120°C и охлаждение; фильтрование и сушка полученного твердого вещества.
3. Частичное концентрирование реакционной смеси после завершения реакции; выливание реакционной смеси в смесь воды и гидрофильного органического растворителя, взятых в любом соотношении; фильтрование и сушка полученного твердого вещества.
4. Выливание реакционной смеси после завершения реакции в воду; доведение рН водного слоя приблизительно до нейтральных значений; удаление органического растворителя азеотропной перегонкой; и сушка полученного твердого вещества.
Гидрофобный органический растворитель, предназначенный для применения в процедуре последующей обработки, включает, например, эфиры, такие как этилацетат и т.п.; галогенированные алифатические углеводороды, такие как хлороформ и т.п.; галогенированные ароматические углеводороды, такие как хлорбензол, дихлорбензол, бензотрифторид и т.п.; кетоны, такие как метилизобутилкетон и т.п.; и их смеси. Гидрофильный органический растворитель включает спирты, такие как метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и т.п.
Выделенное данное пиридоновое соединение может быть очищено хроматографически, перекристаллизацией, промыванием небольшим количеством растворителя и т.п.
Данное амидное соединение, предназначенное для применения в данном способе, может быть получено, например, взаимодействием фенольного соединения формулы (7) с амидным соединением формулы (8) (например, с 2-хлор-3-оксобутанамидом):
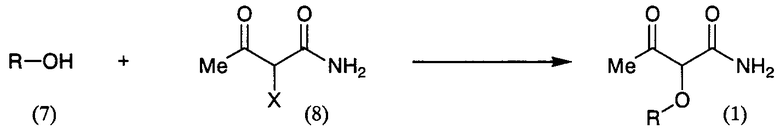
где Х представляет атом галогена, а R определен выше.
Данную реакцию обычно проводят в растворителе в присутствии основания. Растворитель, предназначенный для применения, включает, например, ароматические углеводороды, такие как толуол, ксилол и т.п.; амиды кислот, такие как N,N-диметилформамид и т.п. Основание, предназначенное для применения, включает, например, неорганические основания, такие как карбонат натрия, карбонат калия и т.п.; третичные амины, такие как триэтиламин, трибутиламин и т.п.
В реакции обычно используют от 1 до 1,5 моль амидного соединения формулы (8) и от 1 до 3 моль основания по отношению к 1 моль фенольного соединения формулы (7).
Температура проведения реакции обычно находится в интервале от 20 до 150°C, время реакции обычно составляет от 0,5 до 24 часов.
Данное амидное соединение может быть выделено из реакционной смеси после завершения реакции, например, разбавлением реакционной смеси органическим растворителем, промыванием смеси насыщенным водным раствором хлорида натрия, насыщенным водным раствором бикарбоната натрия или т.п., сушкой и концентрированием полученного органического слоя. Выделенное амидное соединение может быть очищено хроматографически, перекристаллизацией и т.п.
Данное производное малонового альдегида, предназначенное для применения в данном способе, представляет собой известное соединение или может быть получено способом, описанным в литературе.
В качестве публикаций указаны нижеследующие материалы.
Japanese Publication of unexamined application S52-97905, для 3-алкоксипропеналя формулы (3);
J. Chem. Soc., Chem. Commun., (20) 1421-1422 (1991), для 3,3-диалкоксипропаналя формулы (4);
Tetrahedron Lett., 29 (29) 3597-3598 (1988), для 1,3,3-триалкокси-1-пропена формулы (5);
J. Org. Chem., 53 (13) 2920-2925 (1988), для 1,1,3,3-тетраалкоксипропана формулы (6);
J. Org. Chem., 50 3585-3592 (1985), для малонового альдегида.
Данное пиридоновое соединение, полученное данным способом, применимо в качестве промежуточного продукта для медицинского и сельскохозяйственного продукта. Например, пиридиновое соединение формулы (D) может быть получено взаимодействием соединения формулы (В) с эфирным соединением диазоуксусной кислоты формулы (С) в присутствии соли родия (II), трифторида бора, п-толуолсульфоновой кислоты или т.п. Полученное пиридиновое соединение формулы (D) применяется в качестве активного ингредиента гербицидной композиции (Публикация европейской патентной заявки ЕР1122244А1).
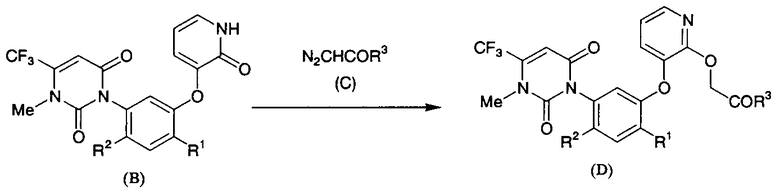
где R3 представляет С1-С6алкокси.
Реакцию в присутствии соли родия (II) обычно проводят в растворителе при температуре от 60 до 120°C и длительности реакции от минуты до 72 часов. Растворитель, предназначенный для применения, включает, например, галогенированные углеводороды, такие как 1,2-дихлорэтан и т.п. Обычно количество диазоацетатного соединения формулы (С) составляет от 0,5 до 2 моль и количество соли родия (II) составляет от 0,01 до 0,05 по отношению к 1 моль соединения формулы (В). Эти количества могут быть изменены в соответствии с условиями реакции. Соль родия (II), предназначенная для применения, включает, например, димер трифторацетата родия (II).
После завершения реакции пиридиновое соединение формулы (D) может быть выделено из реакционной смеси последующей обработкой, такой как фильтрование реакционной смеси и концентрирование фильтрата; разбавление реакционной смеси органическим растворителем и отделение от водного раствора бикарбоната натрия; сушка и концентрирование полученного органического слоя и т.п. Выделенное пиридиновое соединение может быть очищено хроматографически и т.п.
Настоящее изобретение далее будет проиллюстрировано следующими примерами получения и т.п.; однако настоящее изобретение не ограничивается данными примерами. Кроме того, в следующем описании слово «часть» означает массовую часть.
Пример получения 1
В атмосфере азота 57 мг 3-оксо-2-феноксибутанамида и 49 мг 1,1,3,3-тетраметоксипропана добавляли к 2 мл 25% раствора бромистого водорода в уксусной кислоте, смесь перемешивали при 50°C в течение 2,5 часов и при 100°C в течение 2 часов. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 36 мг 3-фенокси-2-пиридона. 1Н-ЯМР (395,75МГц, CDCl3) δ (ч/млн.): 6,18 (т, J=6,9Гц, 1H), 6,92 (дд, J=7,5, 1,7Гц, 1H), 7,08 (шир.д, J=7,8Гц, 2H), 7,15 (шир.т, J=7,5Гц, 1H), 7,19 (дд, J=6,7, 1,7Гц, 1H), 7,36 (шир.т, J=7,5Гц, 2H), 13,7 (шир.с, 1H).
Пример получения 2
В атмосфере азота 108 мг амидного соединения формулы (F):
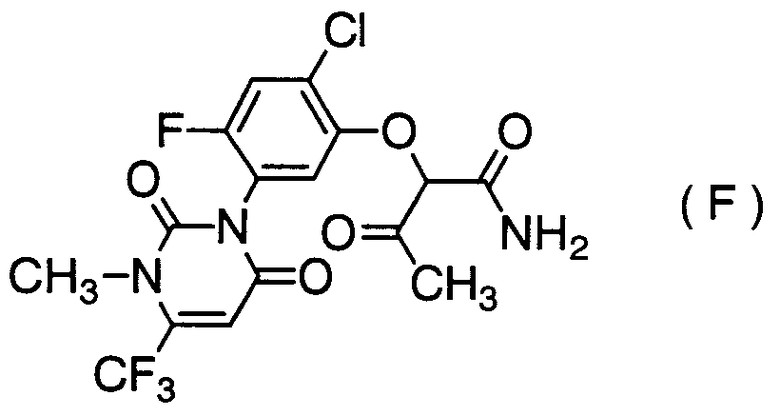
и 53 мг 1,1,3,3-тетраметоксипропана добавляли к 2 мл 85% водного раствора фосфорной кислоты и смесь перемешивали при 50°C в течение 9 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 30 мг пиридонового соединения формулы (G):
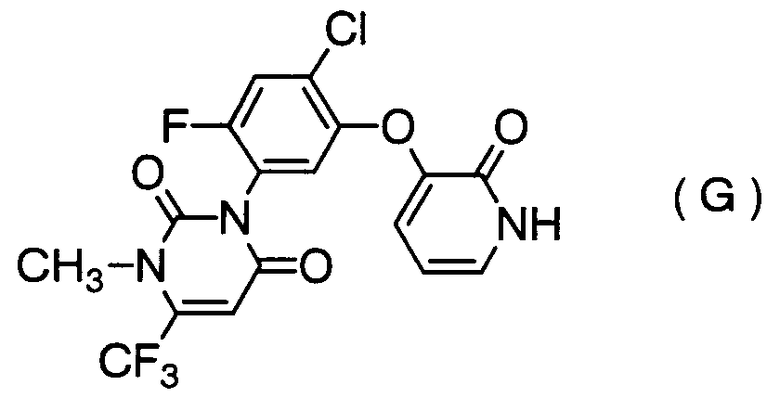
1Н-ЯМР (395,75МГц, CDCl3) δ (ч/млн.): 3,52 (с, 3H), 6,21 (т, J=6,9Гц, 1H), 6,31 (с, 1H), 6,95 (с, 1H), 7,00 (дд, J=7,3, 1,5Гц, 1H), 7,26 (дд, J=6,5, 1,7Гц, 1H), 7,38 (д, J=9,1Гц, 1H), 13,4 (шир.с, 1H).
Пример получения 3
В атмосфере азота 100 мг амидного соединения формулы (F) и 49 мг 1,1,3,3-тетраметоксипропана добавляли к 1,4 г полифосфорной кислоты и смесь перемешивали при 100°C в течение 3 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры. В реакционную смесь добавляли 150 мл этилацетата и 10 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 50 мл насыщенного водного раствора хлорида натрия, дважды 50 мл насыщенного водного раствора бикарбоната натрия и один раз 35 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 38 мг пиридонового соединения формулы (G).
Пример получения 4
В атмосфере азота 101 мг амидного соединения формулы (F) и 38 мг 1,1,3,3-тетраметоксипропана добавляли к 2 мл трифторуксусной кислоты, смесь перемешивали при 70°C в течение 9 часов. В указанный период времени в смесь добавляли 38 мг 1,1,3,3-тетраметоксипропана каждые 1,5 часа (общее количество 1,1,3,3- тетраметоксипропана составляло 228 мг). Реакционную смесь оставляли охлаждаться до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 44 мг пиридонового соединения формулы (G).
Пример получения 5
В атмосфере азота 102 мг амидного соединения формулы (F) и 50 мг 1,1,3,3-тетраметоксипропана добавляли к 2 мл 12,5% раствора бромистого водорода в уксусной кислоте, смесь перемешивали при 50°C в течение 2,5 часов и при 80°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 96 мг пиридонового соединения формулы (G).
Пример получения 6
В атмосфере азота 103 мг амидного соединения формулы (F) и 50 мг 1,1,3,3-тетраметоксипропана добавляли к 1 мл 48% бромистоводородной кислоты, смесь перемешивали при 80°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 31 мг пиридонового соединения формулы (G).
Пример получения 7
В атмосфере азота 104 мг амидного соединения формулы (F) и 51 мг 1,1,3,3-тетраметоксипропана добавляли к 2 мл 1 моль/л хлористого водорода в уксусной кислоте, смесь перемешивали при 80°C в течение 1,5 часов и при 100°C в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 34 мг пиридонового соединения формулы (G).
Пример получения 8
В атмосфере азота 109 мг амидного соединения формулы (F) и 53 мг 1,1,3,3-тетраметоксипропана добавляли к 0,89 г трихлоруксусной кислоты, смесь перемешивали при 100°C в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 24 мг пиридонового соединения формулы (G).
Пример получения 9
В атмосфере азота 119 мг амидного соединения формулы (F) и 34 мг 3-метоксипропеналя добавляли к 2 мл 12,5% раствора бромистого водорода в уксусной кислоте, смесь перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 80 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 20 мл насыщенного водного раствора бикарбоната натрия и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 100 мг пиридонового соединения формулы (G).
Пример получения 10
В атмосфере азота смесь 117 частей амидного соединения формулы (F), 57 частей 1,1,3,3-тетраметоксипропана и 211 частей уксусной кислоты добавляли к 628 частям 30% раствора бромистого водорода в уксусной кислоте, который предварительно охлаждали до 10-15°C, и смесь перемешивали при 50°C в течение 8 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении до 240 массовых частей. Смесь остатка и 69 частей метанола добавляли по каплям к смеси 1160 частей ледяной воды и 316 частей метанола при 0-3°C. Доводили рН смеси до 7,3 с помощью 40% водного раствора гидроксида натрия и насыщенного водного раствора бикарбоната натрия. Смесь перемешивали при комнатной температуре полдня и затем фильтровали. Отфильтрованный осадок промывали три раза 240 частями воды и сушили при пониженном давлении. К сухому осадку добавляли 343 части метанола, содержимое перемешивали в течение 1 часа при нагревании с обратным холодильником, охлаждали до комнатной температуры и фильтровали. Отфильтрованный осадок промывали 114 частями метанола, сушили при пониженном давлении и получали 96 частей пиридонового соединения формулы (G) (содержание: 94%).
Пример получения 11
В атмосфере азота смесь 48 частей амидного соединения формулы (F), 21 части 1,1,3,3-тетраметоксипропана, 36 частей серной кислоты и 1037 частей хлорбензола перемешивали при 80°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2250 частей этилацетата и 2500 частей ледяной воды и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 32 части пиридонового соединения формулы (G).
Пример получения 12
В атмосфере азота смесь 96 частей амидного соединения формулы (F), 42 частей 1,1,3,3-тетраметоксипропана, 49 частей серной кислоты и 1618 частей толуола перемешивали при 60°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2250 частей этилацетата и 2500 частей ледяной воды и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 65 частей пиридонового соединения формулы (G).
Пример получения 13
В атмосфере азота смесь 48 частей амидного соединения формулы (F), 21 части 1,1,3,3-тетраметоксипропана, 33 частей метансульфоновой кислоты и 1037 частей хлорбензола перемешивали при 80°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2250 частей этилацетата и 2500 частей ледяной воды и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 34 части пиридонового соединения формулы (G).
Пример получения 14
В атмосфере азота смесь 112 частей амидного соединения формулы (F), 49 частей 1,1,3,3-тетраметоксипропана, 78 частей метансульфоновой кислоты и 1894 частей толуола перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2250 частей этилацетата и 2500 частей ледяной воды и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 85 частей пиридонового соединения формулы (G).
Пример получения 15
В атмосфере азота смесь 96 частей амидного соединения формулы (F), 42 частей 1,1,3,3-тетраметоксипропана, 55 частей хлорсульфоновой кислоты и 746 частей хлороформа перемешивали при 60°C в течение 2,5 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2255 частей этилацетата, 500 частей ледяной воды и 1200 частей насыщенного водного раствора бикарбоната натрия и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 73 части пиридонового соединения формулы (G).
Пример получения 16
В атмосфере азота смесь 96 частей амидного соединения формулы (F), 42 частей 1,1,3,3-тетраметоксипропана, 81 части хлорсульфоновой кислоты и 2070 частей хлорбензола перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2250 частей этилацетата и 2500 частей ледяной воды и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 73 части пиридонового соединения формулы (G).
Пример получения 17
В атмосфере азота смесь 48 частей амидного соединения формулы (F), 21 части 1,1,3,3-тетраметоксипропана, 41 части моногидрата п-толуолсульфоновой кислоты и 1037 частей хлорбензола перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры. В реакционную смесь добавляли 2250 частей этилацетата и 2500 частей ледяной воды и смесь разделяли. Органический слой последовательно промывали дважды 1200 частями воды, один раз 1200 частями насыщенного водного раствора бикарбоната натрия и один раз 1200 частями насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: метанол/этилацетат = 5/95) и получали 32 части пиридонового соединения формулы (G).
Следующий способ получения исходного соединения, используемого в Примере получения, представленном выше, будет проиллюстрирован справочным примером.
Справочный пример 1
В атмосфере азота 1,57 г 2-хлор-3-оксобутиламида, 1,08 г фенола и 1,7 мл триэтиламина добавляли к 20 мл N,N-диметилформамида, смесь перемешивали при 80°C в течение 6 часов и при 100°C в течение 4 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры. В реакционную смесь добавляли 100 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 20 мл насыщенного водного раствора хлорида натрия, дважды 20 мл хлористоводородной кислоты (1 моль/л) и один раз 20 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/этилацетат = 6/4) и получали 0,44 г 3-оксо-2-феноксибутиламида.
3-Оксо-2-феноксибутиламид
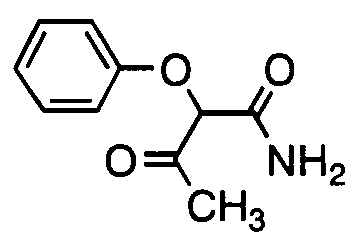
т.пл.: 107,1°C
Справочный пример 2
В атмосфере азота 8,92 г 2-хлор-3-оксобутанамида, 20,3 г соединения формулы (Н):
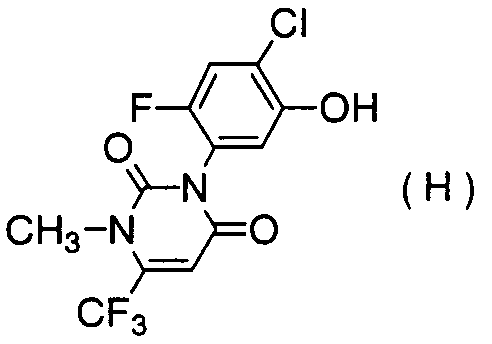
и 16,7 мл триэтиламина добавляли к 120 мл N,N-диметилформамида, смесь перемешивали при 70°C в течение 1 часа и при 100°C в течение 4,5 часов. Реакционную смесь оставляли охлаждаться до комнатной температуры. В реакционную смесь добавляли 200 мл этилацетата и 30 мл насыщенного водного раствора хлорида натрия и смесь разделяли. Органический слой промывали последовательно один раз 30 мл насыщенного водного раствора хлорида натрия, дважды 30 мл хлористоводородной кислоты (1 моль/л) и один раз 30 мл насыщенного водного раствора хлорида натрия; сушили над безводным сульфатом магния и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/этилацетат = 6/4) и получали 17,9 г соединения формулы (F).
Соединение формулы (F)
т.пл.: 192,3°C
Следующий способ получения гербицидного соединения с применением соединения формулы (G) в качестве исходного соединения, полученного в справочном примере, представленном выше, будет проиллюстрирован справочным примером.
Справочный пример 3
В 15 мл дихлорэтана добавляли 0,5 г соединения формулы (G) и 8 мг димера трифторацетата родия (II), затем к смеси добавляли по каплям 0,15 г метилдиазоацетата при 80°C в течение 3 часов, после чего смесь перемешивали при 80°C в течение 1 часа и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/этилацетат = 3/1-0/1) и получали 0,18 г непрореагировавшего исходного соединения формулы (G) и 0,34 г 3-(2-хлор-4-фтор-5-[3-метил-2,6-диоксо-4-(трифторметил)-1,2,3,6-тетрагидропиримидин-1-ил]фенокси)-2-(метоксикарбонилметокси)пиридина:
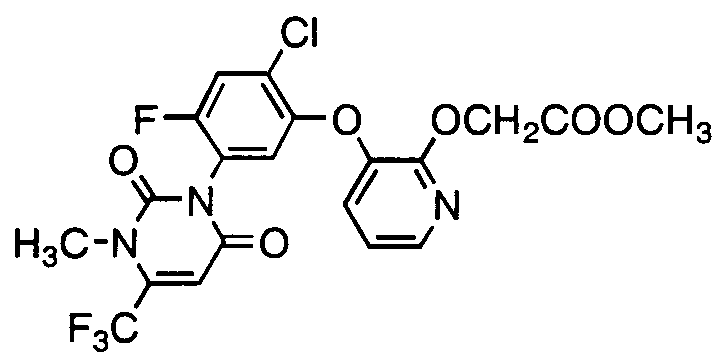
т.пл.: 52,2°C
1Н-ЯМР (300МГц, CDCl3, TMS δ (ч/млн.): 3,50 (3H, кв, J=1,0Гц), 3,70 (3H, с), 4,90 (1H, д, J=15,8Гц), 4,97 (1H, д, J=15,8Гц), 6,29 (1H, с), 6,90-6,95 (2H, м), 7,32 (1H, дд, J=1,9Гц, 7,7Гц), 7,37 (1H, д, J=8,7Гц), 7,92 (1H, дд, J=1,9Гц, 4,9Гц).
Промышленное применение
Представленное пиридоновое соединение может быть получено из данного амидного соединения по данному способу.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ N-АЦИЛАНТРАНИЛОВОЙ КИСЛОТЫ ИЛИ ЕГО СОЛЬ | 2010 |
|
RU2529860C2 |
| ИНГИБИТОРЫ FXIA, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2020 |
|
RU2802878C1 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2016 |
|
RU2687245C1 |
| НОВОЕ ДИЗАМЕЩЕННОЕ 1,2,4-ТРИАЗИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2726410C2 |
| НОВОЕ ДИЗАМЕЩЕННОЕ 1,2,4-ТРИАЗИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2693624C2 |
| ПРОИЗВОДНОЕ АМИДА ИНДАЗОЛАКРИЛОВОЙ КИСЛОТЫ | 2008 |
|
RU2444515C2 |
| НОВЫЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ (1) | 2005 |
|
RU2330021C2 |
| ПРОИЗВОДНОЕ ТИЕНОПИРАЗОЛА, ИМЕЮЩЕЕ ИНГИБИРУЮЩУЮ ФОСФОДИЭСТЕРАЗУ 7 (PDE 7) АКТИВНОСТЬ | 2005 |
|
RU2376309C2 |
| ДИТИОЛАНОВЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА НА ИХ ОСНОВЕ | 1998 |
|
RU2165932C2 |
Изобретение относится к усовершенствованному способу получения феноксизамещенных 2-пиридоновых соединений формулы (2), которые могут быть использованы в качестве промежуточного продукта для получения медицинского или сельскохозяйственного препарата, в частности гербицидного соединения.
Предлагаемый способ получения феноксизамещенных 2-пиридоновых соединений формулы (2)
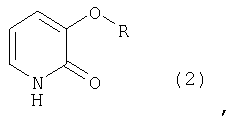
где R представляет фенил или радикал следующей формулы
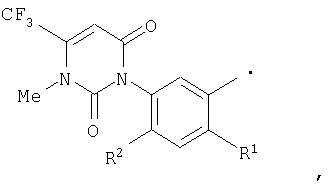
где R1 представляет атом галогена или нитро и R2 представляет водород или атом галогена, включает взаимодействие амидного соединения формулы (1)
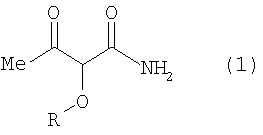
где R определен выше,
по меньшей мере, с одним соединением, выбранным из группы, включающей соединения формул (3)-(6) и малоновый альдегид:
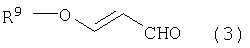
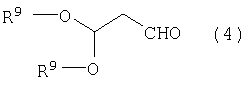
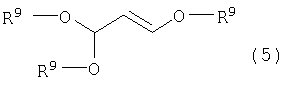
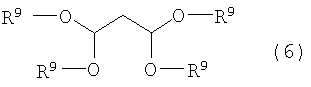
где R9 представляет алкил,
в присутствии протонной кислоты.
Технический результат изобретения - упрощение процесса за счет использования более доступного малонового альдегида или его производного формулы (3)-(6) вместо кетона. 5 з.п. ф-лы.
где R представляет фенил или радикал следующей формулы:
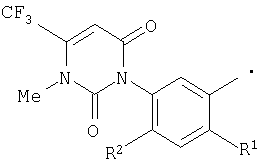
где R1 представляет атом галогена или нитро, и R2 представляет водород или атом галогена, который включает взаимодействие амидного соединения формулы (1)
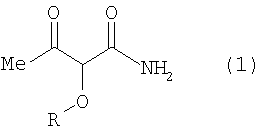
где R определен выше, по меньшей мере, с одним соединением, выбранным из группы, включающей 3-алкоксипропеналь формулы (3), 3,3-диалкоксипропаналь формулы (4), 1,3,3-триалкокси-1-пропен формулы (5), 1,1,3,3-тетраалкоксипропан формулы (6) и малоновый альдегид
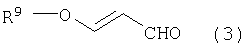
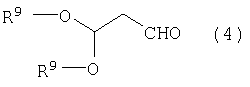
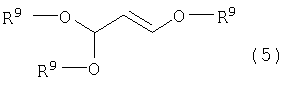
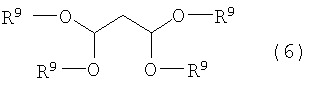
где R9 представляет алкил, в присутствии протонной кислоты.
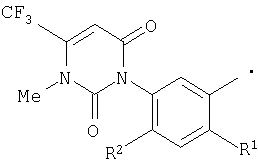
где R1 представляет атом галогена или нитро, и R2 представляет водород или атом галогена.
| ПРОИЗВОДНЫЕ ПИРИДИН-2-ОНА ИЛИ ПИРИДИН-2-ТИОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, АНТИМИКРОБНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОДАВЛЕНИЯ РОСТА МИКРООРГАНИЗМОВ | 1994 |
|
RU2131415C1 |
| Устройство для контроля переговоров абонентов в системе с временным уплотнением | 1975 |
|
SU1122244A3 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| ПРИВОДНОЙ МЕХАНИЗМ ШТОРНОГО ОБТЮРАТОРА КИНОАППАРАТА | 0 |
|
SU255047A1 |

