Настоящее изобретение относится к объектам, охарактеризованным в формуле изобретения, а именно к перфторалкилсодержащим металлическим комплексам с полярными остатками общей формулы I, к способу их получения и к их применению в ЯМР- и рентгенодиагностике, радионуклидной диагностике и лучевой терапии, в МРТ-лимфографии (МРТ=магнитно-резонансная томография), а также в качестве контрастных веществ для визуализации пулов крови. Предлагаемые в изобретении соединения наиболее пригодны для применения при внутривенной лимфографии, для диагностики опухолей и для визуализации инфарктов и некрозов.
В методах, основанных на ядерном магнитном резонансе, вторым по значению элементом после водорода является фтор, что обусловлено следующими факторами:
1) фтор обладает высокой восприимчивостью, составляющей 83% от восприимчивости водорода,
2) фтор имеет лишь один ЯМР-активный изотоп,
3) фтор характеризуется аналогичной водороду резонансной частотой, что позволяет анализировать оба элемента с помощью одной и той же аппаратуры,
4) фтор биологически инертен,
5) фтор не содержится в биологическом материале (за исключением зубов) и поэтому может применяться в качестве зонда или контрастного вещества на не создающем паразитных сигналов фоне.
Благодаря этим свойствам фтор находит самое широкое применение в диагностике, основанной на методе ядерного магнитного резонанса, например в 19F-томографии, функциональной диагностике и спектроскопии, что отражено в соответствующей патентной литературе.
Так, например, в патенте US 4639364 (на имя Mallinckrodt) были предложены трифторметансульфонамиды для применения в качестве контрастных веществ в 19F-томографии:
CF3SO2NH2 и
CF3SO2NH-СН2-(СНОН)4-СН2OH.
Равным образом к 19F-томографии относится и патент DE 4203254 (на имя Max-Planck-Gesellschaft), в котором предложено производное анилина приведенной ниже формулы:

19F-томография является объектом заявки WO 93/07907 (на имя Mallinckrodt), в которой также предложены производные фенила для применения в качестве контрастных веществ:
Для использования в 19F-томографии были предложены также соединения со значительно более простой структурой. Так, например, в патенте US 4586511 (на имя Children's Hospital Medical Center) описан перфтороктилбромид формулы
CF3(CF2)7-Br,
в патенте ЕР 307863 (на имя Air Products) заявлен перфтор-15-краун-5-эфир формулы

а в патенте US 4588279 (на имя University of Cincinnati, Children's Hospital Research Foundation) описаны перфторуглеродные соединения, такие как перфторциклононан или -октан, простые перфорированные эфиры, такие как тетрагидрофуран формулы
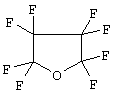
или простые диэфиры, такие как диэфир перфторпропиленгликоля формулы

Аналогичным образом для применения в 19F-томографии предназначены описанные в заявке WO 94/22368 (на имя Molecular Biosystems) соединения, например

которые в качестве фторсодержащего остатка содержат перфтор-1Н,1Н-неопентильную группу.
Структуры другого типа с более широким спектром применения в диагностике представлены в патенте US 5362478 (на имя VIVORX), в котором заявлена предназначенная для использования в томографии комбинация фторуглерод/полимерная оболочка. Согласно этому патенту предлагается применять перфторнонан и сывороточный альбумин человека. Подобная комбинация, как было установлено, позволяет, кроме того, использовать атом фтора в качестве зонда для локального измерения температуры и для определения парциального давления кислорода.
Перфторуглероды, предназначенные для определения содержания кислорода, описаны также в патенте US 4586511.
В патенте DE 4008179 (на имя Schering) предлагается использовать фторсодержащие бензолсульфонамиды в качестве рН-зондов:

Для применения в ЯМР-диагностике в качестве повышающих контрастность веществ предназначены также соединения, содержащие атомы йода и фтора и описанные, например, в заявках WO 94/05335 и WO 94/22368 (обе на имя Molecular Biosystems):
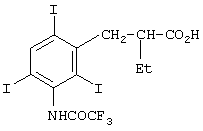

Для применения в 19F-томографии предназначена также комбинация фтор-парамагнитный ион металла, при этом речь идет о комплексах с открытой цепью, в качестве примера которых в заявке WO 94/22368 (на имя Molecular Biosystems) названы соединения формулы
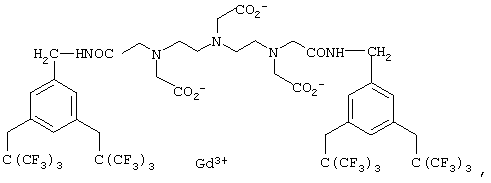
а в ЕР 292306 (на имя TERUMO Kabushiki Kaisha) указаны соединения формулы

а также о циклических соединениях, описанных в ЕР 628316 (на имя TERUMO Kabushiki Kaisha):
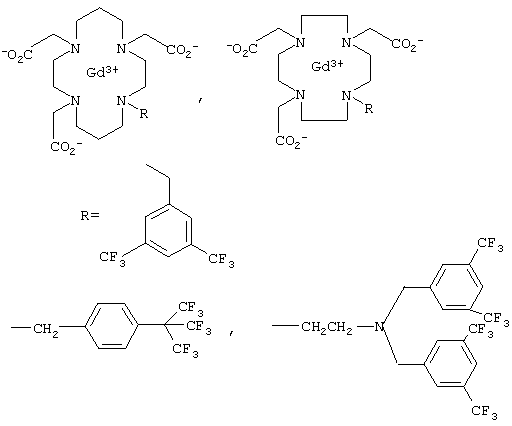
Для ЯМР-спектроскопических измерений температуры в DE 4317588 (на имя Schering) было предложено также использовать следующую комбинацию из атома фтора и редкоземельного элемента:
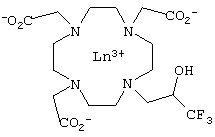
где Ln обозначает редкоземельный элемент La, Pr, Dy или Eu.
Если в соединениях, содержащих фтор и йод, между обоими ядрами взаимодействие не происходит, то в соединениях, содержащих фтор и парамагнитные центры (радикалы, ионы металлов), происходит достаточно интенсивное взаимодействие, которое проявляется в сокращении времени релаксации ядра фтора. Степень проявления такого эффекта зависит от числа неспаренных электронов иона металла (Gd3+>Mn2+>Fe3+>Cu2+) и от расстояния между парамагнитным ионом и 19F-атомом.
Чем больше число неспаренных электронов иона металла и чем ближе они расположены к фтору, тем существеннее сокращается время релаксации ядра фтора.
Сокращение времени релаксации в функции удаленности от парамагнитного иона наблюдается у всех ядер с нечетным спиновым числом, в том числе и у протона, и поэтому гадолиниевые соединения находят широкое применение в качестве контрастных веществ в ЯМР-томографии (Magnevist®, Prohance®, Omniscan®, Dotarem®).
При 1Н-МР-томографии, однако, определяют время релаксации T1 или Т2 протонов, т.е. прежде всего протонов воды, а не время релаксации ядер фтора, и используют полученные данные для визуализации. Количественной мерой, характеризующей сокращение времени релаксации T1, является релаксационность, выражаемая в л/ммоль-с. Для сокращения времени релаксации с успехом применяют комплексы парамагнитных ионов. В следующей таблице приводятся данные о релаксационности некоторых коммерчески доступных препаратов:
В этих соединениях происходит только взаимодействие между протонами и ионом гадолиния. Выявленная для указанных контрастных веществ релаксационность в воде составляет примерно 4 л/ммоль-с.
Таким образом, при МР-томографии с успехом могут применяться и фторсодержащие соединения, предназначенные для 19F-томографии, где используется сокращенное время релаксации ядра фтора, и не содержащие фтор соединения, у которых определяют время релаксации протонов воды.
Неожиданный эффект, связанный с введением перфторуглеродсодержащего остатка в парамагнитное контрастное вещество, т.е. эффект, связанный с приданием соединениям, которые использовали в методах протонной визуализации, свойств тех соединений, которые до настоящего времени рассматривались как пригодные только для применения в методах визуализации, основанных на использовании фтора, проявляется также в быстром возрастании релаксационности протонов воды. В результате этот показатель достигает значений, составляющих 10-50 л/ммоль-с, тогда как аналогичные значения у некоторых коммерчески доступных продуктов составляют, как следует из вышеприведенной таблицы, от 3,5 до 3,8 л/ммоль-с.
Из заявки DE 19603033.1 уже известны перфторалкилсодержащие комплексы металлов. Однако возможности применения этих соединений ограничены, поскольку во многих случаях они не позволяют достичь удовлетворительных результатов. С учетом этого в настоящее время, как и прежде, сохраняется необходимость в контрастных веществах, предназначенных для визуализации злокачественных опухолей, лимфатических узлов и некротических тканей.
Злокачественные опухоли часто метастазируют в регионарные лимфатические узлы, причем этот процесс может также охватывать несколько уровней лимфоузлов. Так, в частности, метастазы в лимфатические узлы были обнаружены примерно у 50-69% всех пациентов со злокачественными опухолями (см. Elke, Lymphographie, Radiologische Diagnostik in Klinik und Praxis, под ред. Frommhold, Stender, Thurn, том IV, изд-во Thieme Verlag, Stuttgart, 7-е изд., 1984, cc.434-496). Возможность диагностики метастазирования в лимфатические узлы имеет важное значение для терапии онкологических заболеваний и прогнозирования их развития. Современные методы визуализации (компьютерная томография, ультразвуковое исследование и магнитно-резонансная томография) не позволяют с достаточной высокой точностью и надежностью распознавать лимфогенное метастазирование злокачественных опухолей, поскольку в большинстве подобных случаев в качестве критериев соответствующего диагноза могут использоваться только размеры лимфатического узла. В результате такие методы просто не позволяют отличить небольшие метастазы в неувеличенных лимфатических узлах (<2 см) от гиперплазии лимфоузлов, не пораженных злокачественной опухолью (см. Steinkamp и др., Sonographie und Kernspintomographie: Differentialdiagnostik von reaktiver Lymphknotenvergröberung und Lymphknotenmetastasen am Hals, Radiol. diagn. 33 (1992), с.158).
С учетом этого представляется целесообразным обеспечить при применении специфических контрастных веществ возможность дифференцировать лимфатические узлы, пораженные метастазами, и гиперпластические лимфатические узлы.
В качестве примера известного инвазивного метода визуализации можно назвать прямую лимфорентгенографию (инъекция масляной суспензии контрастного вещества в подготовленный соответствующим образом лимфатический сосуд), которая, однако, в настоящее время используется лишь в редких случаях и которая позволяет визуализировать только некоторые пути оттока лимфы.
В экспериментах, проводимых на животных, используют также декстраны с флуоресцентной меткой с целью обеспечить после их интерстициального введения возможность наблюдения оттока лимфы. Все метки, используемые после их интерстициального/внутрикожного введения для визуализации лимфатических протоков и лимфатических узлов, представляют собой вещества в виде твердых частиц ("макрочастицы", например эмульсии и суспензии нанокристаллов) либо крупные полимеры (см. также WO 90/14846). Однако все известные в настоящее время из литературы композиции вследствие их недостаточной местной и системной переносимости, равно как и их малой подвижности в лимфе, обусловливающей неудовлетворительную эффективность диагностики, все еще остаются не оптимальными для применения в непрямой лимфографии.
Поскольку визуализация лимфатических узлов имеет важное значение для раннего обнаружения метастазов у онкологических больных, существует необходимость в лимфоспецифичных композициях контрастных веществ, которые позволяли бы своевременно и надежно диагностировать соответствующие изменения в лимфатической системе.
Для достижения требуемого эффекта при использовании контрастных веществ целесообразно обеспечить не только их предельно высокую концентрацию в лимфе и высокую стабильность, но и максимально равномерное накапливание в лимфе в нескольких уровнях лимфатической системы, что имеет важное для постановки точного диагноза значение. Вместе с тем контрастное вещество должно быстро и полностью выводиться из организма с целью минимизировать его отрицательное воздействие на весь организм в целом. Действие контрастного вещества должно начинать проявляться по возможности уже через несколько часов после его введения, что является важным условием в радиологической практике. Столь же важным требованием, предъявляемым к контрастным веществам, является их хорошая переносимость.
Не менее актуальна и потребность в лимфоспецифичных контрастных веществах, которые позволяли бы за один сеанс диагностического исследования визуализировать и первичную опухоль, и возможное ее метастазирование в лимфатические узлы.
Еще важными задачами медицины являются обнаружение, локализация и наблюдение за некрозами и инфарктами. Так, в частности, инфаркт миокарда является не стационарным, а динамическим процессом, длящимся в течение продолжительного промежутка времени (от нескольких недель до нескольких месяцев). Это сердечно-сосудистое заболевание протекает примерно в три стадии, которые невозможно четко разграничить, поскольку они накладываются одна на другую, соответственно плавно переходят одна в другую. Длительность первой стадии, на которой происходит развитие инфаркта миокарда, составляет примерно первые 24 часа после его начала, в течение которых разрушение ткани распространяется подобно ударной волне (аналогично явлению волнового фронта) от субэндокарда к миокарду. Вторая стадия, на которой развитие инфаркта, как такового, уже закончилось, включает стабилизацию области, в которой в процессе заживления пораженной инфарктом ткани происходит образование волокон (фиброз). Третья стадия, которая соответствует полному заживлению затронутой инфарктом ткани, начинается после замены всей разрушенной ткани фиброзной рубцовой тканью. В этот период происходит активная реструктуризация.
На сегодняшний день не существует ни одного метода, который позволял бы с высокой точностью и надежностью диагностировать текущую фазу инфаркта миокарда у живого пациента. Для оценки же инфаркта миокарда решающее значение имеет информация о том, насколько велика доля утраченной при инфаркте ткани и в каком именно месте, поскольку от этой информации зависит тип терапии.
Инфаркты, как известно, поражают не только миокард, но и другие ткани, прежде всего головной мозг.
Если затронутая инфарктом ткань в некоторой степени поддается заживлению, то при некрозе, т.е. при локально ограниченном омертвении ткани, можно лишь предотвратить или по меньшей мере смягчить его вредные последствия для остального организма. Возникновение некрозов может быть обусловлено самыми различными причинами и, в частности, травмами, воздействием химикалиев, дефицитом кислорода или же облучением. Аналогично инфаркту наличие информации о степени и типе некроза имеет важное значение для выбора последующих врачебных мер.
С учетом этого уже достаточно давно предпринимались попытки повысить эффективность локализации, т.е. определения местонахождения, инфарктов и некрозов за счет применения контрастных веществ при неинвазивных методах, таких как сцинтиграфия или ЯМР-томография. При этом большое число опубликованных работ посвящено экспериментальным исследованиям по использованию порфиринов для визуализации некрозов. Однако полученные в ходе подобных исследований результаты носят противоречивый характер. Так, в частности, в работе Winkelman и Hoyes, опубликованной в Nature, 200 (1967), с.903, говорится о селективном накоплении марганец-5,10,15,20-тетракис(4-сульфонатофенил)порфирина (ТФПС) в некротической области опухоли.
В отличие от этого в работе Lyon и др. (Magn. Res. Med. 4 (1987), с.24) говорится о том наблюдавшемся этими авторами эффекте, что марганец-ТФПС распределяется по существу по всему организму, а именно накапливается в почках, печени, опухоли и лишь незначительно в мышечных тканях. Особый интерес при этом представляет тот факт, что концентрация указанного вещества в опухоли достигает максимума только на 4-й день после введения и то лишь после повышения дозы с 0,12 ммолей/кг до 0,2 ммолей/кг. Поэтому авторами делается также вывод о неспецифическом накоплении ТФПС в опухолевой ткани. В работе Bockhurst и др., опубликованной в Acta Neurochir. 60 (1994, дополн.), с.347, вновь говорится о селективном связывании Mn-ТФПС с опухолевыми клетками.
В свою очередь по результатам исследований, проводившихся Foster и др. (J. Nucl. Med. 26 (1985), с.756), было установлено, что 111In-5,10,15,20-тетракис(4-N-метилпиридиний)порфирин (ТМПиП) накапливается не в некротической области, а в окружающих ее живых краевых слоях. На основании этого можно было бы сделать очевидный вывод о наличии взаимодействия между порфирином и тканью, что, однако, не обязательно соответствует действительности.
В работе Ni и др., опубликованной в Circulation, т.90, №4, часть 2, с.1468, Реферат №2512 (1994), сообщалось о возможности визуализации затронутых инфарктом областей с помощью марганец-тетрафенилпорфирина (Mn-ТФП) и гадолиний-мезопорфирина (Gd-МП). Согласно заявке WO 95/31219 оба этих вещества использовались для визуализации инфарктов и некрозов. В этой заявке, авторами которой являются Marchal и Ni, говорится (см. пример 3), что при использовании соединения Gd-МП содержание металла в пораженной инфарктом почке находилось на том же уровне, что и в неинфарцированном органе, тогда как в миокарде содержание металла в инфарцированной ткани (см. пример 1) в девять раз превышало его содержание в неинфарцированной ткани. Неожиданным является при этом тот факт, что при МРТ соотношение между интенсивностью сигнала, формируемого инфарцированной тканью, и интенсивностью сигнала, формируемого здоровой тканью, в обоих случаях находилось на сравнительно высоком уровне и составляло 2,10 и 2,19 соответственно. Другие металлопорфирины описаны в заявке DE 19835082 (на имя Sobering AG).
Порфирины обладают тенденцией накапливаться в коже, что приводит к ее фотосенсибилизации. Подобная сенсибилизация может сохраняться в течение нескольких дней, а иногда и в течение нескольких недель. В этом заключается нежелательный побочный эффект, проявляющийся при применение порфиринов в качестве диагностикумов. Помимо этого, порфирины имеют лишь исключительно низкий терапевтический индекс, поскольку, например, в случае Mn-ТФПС его действие проявляется только при его использовании в дозе 0,2 ммоля/кг, тогда как его летальная доза ЛД50 составляет уже 0,5 ммоля/кг.
Другие контрастные вещества, не являющиеся производными порфиринового каркаса и предназначенные для визуализации некрозов и инфарктов, описаны в заявках DE 19744003 (на имя Schering AG), DE 19744004 (на имя Schering AG) и WO 99/17809 (на имя EPIX). Однако до настоящего времени все еще нет соединений, которые можно было бы достаточно эффективно применять в качестве контрастных веществ для визуализации инфарктов и некрозов.
Исходя из вышеизложенного, в основу настоящего изобретения была положена задача предложить контрастные вещества, которые были бы пригодны для применения прежде всего в МРТ-лимфографии, а также для диагностики опухолей и визуализации некрозов и инфарктов.
Указанная задача решается согласно изобретению с помощью перфторалкилсодержащих комплексов с полярными остатками общей формулы I
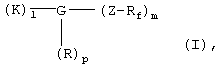
в которой
Rf обозначает перфторированную, прямую или разветвленную углеродную цепь формулы -CnF2nE,
где Е представляет собой концевой атом фтора, хлора, брома, йода или водорода,
n обозначает числа 4-30,
К обозначает металлический комплекс общей формулы II

в которой
R1 представляет собой атом водорода или эквивалент иона металла с порядковым номером 21-29 и 58-71,
при условии, что по меньшей мере два радикала R1 обозначают эквиваленты ионов металлов,
R2 и R3 независимо друг от друга обозначают водород, С1-С7алкил, бензил, фенил, -СН2ОН или -СН2ОСН3 и
U представляет собой -С6Н4-O-СН2-ω-, -(CH2)1-5-ω-, фениленовую группу, -СН2-NHCO-СН2-СН(СН2СООН)-С6Н4-ω-, -C6H4-(OCH2CH2)0-1-N(CH2COOH)-CH2-ω- или необязательно прерванную одним или несколькими атомами кислорода, 1-3 -NHCO-группами, 1-3 -CONH-группами и/или замещенную 1-3 -(СН2)0-5СООН-группами С1-С12алкиленовую или С7-С12-С6Н4-O-группу, при этом ω обозначает место присоединения к -СО-,
или общей формулы III

в которой
R1 имеет вышеуказанные значения,
R4 обозначает водород или указанный для R1 эквивалент иона металла и
U1 обозначает -С6Н4-O-СН2-ω-, где ω обозначает место присоединения к -СО-,
или общей формулы IV

в которой R1 и R2 имеют указанные выше значения,
или общей формулы VA или VB
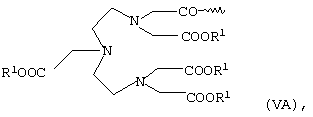

в которых R1 имеет указанные выше значения,
или общей формулы VI
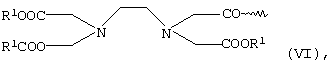
в которой R1 имеет указанные выше значения,
или общей формулы VII

в которой
R1 имеет указанные выше значения, а
U1 обозначает -С6H4-О-СН2-ω-, где ω обозначает место присоединения к -СО-,
при этом необязательно присутствующие в остатке К свободные кислотные группы необязательно могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот либо в виде амидов аминокислот,
G обозначает по меньшей мере трехкратно функционализованный остаток, выбранный из следующих остатков a)-i):
(а)
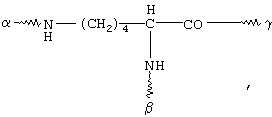
(b)
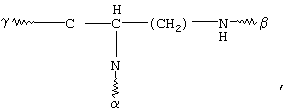
(с)
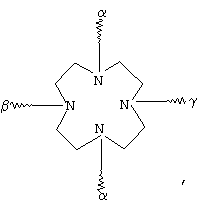
(d)

(е)

(О

(g)

где α обозначает место присоединения G к комплексу К, β обозначает место присоединения G к остатку R, а γ обозначает место присоединения G к остатку Z,
Z обозначает группу

γ-С(O)СН2O(СН2)2-ε,
где γ обозначает место присоединения Z к остатку G, а ε обозначает место присоединения Z к перфторированному остатку Rf,
R представляет собой полярный остаток, выбранный из комплексов К общих формул II-VII, причем в этом случае R1 обозначает атом водорода или эквивалент иона металла с порядковым номером 20-29, 31-33, 37-39, 42-44, 49 или 57-83, а остатки R2, R3, R4, U и U1 имеют указанные выше значения, причем в том случае, когда G представляет собой остаток формулы (с) или (d), а R представляет собой комплекс, выбранный из комплексов общих формул II и V, R не может быть идентичен остатку К общей формулы I, если Z представляет собой δ-С(O)СН2O(СН2)2-ε,
или представляет собой остаток фолиевой кислоты или присоединенную через -СО-, SO2- или прямую связь к остатку G углеродную цепь с 2-30 С-атомами, которая является прямой или разветвленной, насыщенной или ненасыщенной и которая необязательно прервана 1-10 атомами кислорода, 1-5 -NHCO-группами, 1-5 -CONH-группами, 1-2 атомами серы, 1-5 -NH-группами или 1-2 фениленовыми группами, которые необязательно могут быть замещены 1-2 ОН-группами, 1-2 NH2-группами, 1-2 -СООН-группами или 1-2 -SO3Н-группами, или необязательно замещена 1-8 ОН-группами, 1-5 -СООН-группами, 1-2 SO3Н-группами, 1-5 NH2-группами, 1-5 С1-С4алкоксигруппами, и
l, m, p независимо друг от друга обозначают целые числа 1 или 2.
Если предлагаемое в изобретении соединение предназначено для его применения при ЯМР-диагностике, то ион металла формирующей сигнал группы должен быть парамагнитным. К подобным ионам относятся прежде всего двух- и трехвалентные ионы элементов с порядковыми номерами 21-29, 42, 44 и 58-70. В качестве примера пригодных для применения в указанных целях ионов можно назвать ионы хрома(III), железа(II), кобальта(II), никеля(II), меди(II), празеодима(III), неодима(III), самария(III) и иттербия(III). Наиболее предпочтительны при этом с учетом их высокого магнитного момента ионы гадолиния(III), тербия(III), диспрозия(III), гольмия(III), эрбия(III), железа(III) и марганца(II).
Для применения предлагаемых в изобретении соединений в медицинской радиологии (радиоизотопная диагностика и лучевая терапия) ион металла должен быть радиоактивным. Для применения в этих целях пригодны, например, радиоизотопы элементов с порядковыми номерами 27, 29, 31-33, 37-39, 43, 49, 62, 64, 70, 75 и 77. Предпочтительны при этом технеций, галлий, индий, рений и иттрий.
Если предлагаемое в изобретении соединение предназначено для его применения при рентгенодиагностике, то в качестве иона металла предпочтительно использовать элемент с более высоким порядковым номером с целью обеспечить достаточно высокую степень поглощения рентгеновских лучей. Было установлено, что для этой цели пригодны диагностические средства, содержащие физиологически совместимую комплексную соль с ионами металлов порядковых номеров 25, 26 и 39, а также 57-83. Предпочтительны при этом ионы марганца(II), железа(II), железа(III), празеодима(III), неодима(III), самария(III), гадолиния(III), иттербия(III) или висмута(III), прежде всего ионы диспрозия(III) и иттрия(III).
Присутствующие при определенных условиях в R кислотные атомы водорода, т.е. атомы, которые не замещены центральным ионом, необязательно могут быть полностью или частично заменены на катионы неорганических и/или органических оснований или аминокислот либо амидов аминокислот. В качестве примера приемлемых неорганических катионов можно назвать ион лития, ион калия, ион кальция и прежде всего ион натрия. Приемлемыми катионами органических оснований являются, в частности, таковые первичных, вторичных или третичных аминов, таких, например, как этаноламин, диэтаноламин, морфолин, глюкамин, N,N-диметилглюкамин и прежде всего N-метилглюкамин. В качестве примера приемлемых катионов аминокислот можно назвать катионы лизина, аргинина и орнитина, а также амиды в основном кислых или нейтральных аминокислот.
К наиболее предпочтительным соединениям общей формулы I относятся соединения, содержащие макроцикл К общей формулы II, III, VB или VII.
Остаток U в металлическом комплексе К предпочтительно обозначает -СН2- или -С6Н4-O-СН2-ω-, где ω представляет собой место присоединения к -СО-.
Указанные в качестве значений R2 и R3 алкильные группы в макроцикле общей формулы II могут иметь прямую или разветвленную цепь. При этом в качестве примеров можно назвать метил, этил, пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил. Предпочтительно R2 и R3 независимо друг от друга обозначают водород или С1-С4алкил. В одном из особо предпочтительных вариантов R2 обозначает метил, а R3 обозначает водород.
Указанная в качестве значения R2 или R3 бензильная группа или фенильная группа в макроцикле К общей формулы II может быть также замещена в кольце.
Полярный остаток R в общей формуле I в одном из предпочтительных вариантов представляет собой комплекс К, которым предпочтительно помимо Gd3+- или Mn2+-комплекса может также являться Са2+-комплекс. Наиболее предпочтительны в качестве полярных остатков R комплексы К общих формул II, III, VA и VII. В качестве R1 они в особо предпочтительном варианте содержат эквивалент иона металла с порядковым номером 20, 25 или 64.
Согласно еще одному предпочтительному варианту полярный остаток R имеет следующие значения: -С(O)СН2СН2SO3Н, -С(O)СН2OCH2СН2OCH2СН2OH, -С(O)СН2OCH2СН2OH, -С(O)СН2OCH2СН(ОН)СН2OH, -С(O)СН2NH-С(O)СН2СООН, -С(O)СН2СН(ОН)СН2OH, -С(O)СН2OCH2СООН, -SO2CH2CH2COOH, -С(O)-С6Н3-(м-СООН)2, -С(O)СН2O(СН2)2-С6Н3-(м-СООН)2, -С(O)СН2O-С6Н4-м-SO3Н, -С(O)СН2NHC(O)СН2NHC(O)СН2OCH2СООН, -С(O)СН2OCH2СН2OCH2СООН, -С(O)СН2OCH2СН(ОН)СН2O-СН2СН2OH, -С(O)СН2OCH2СН(ОН)СН2OCH2-СН(ОН)-СН2OH, -С(O)СН2SO3Н, -С(O)СН2СН2СООН, -С(O)СН(ОН)СН(ОН)СН2OH, -С(O)СН2O[(СН2)2O]1-9-СН3, -С(O)СН2O[(СН2)2O]1-9-Н, -С(O)СН2OCH(СН2OH)2, -С(O)СН2OCH(СН2OCH2СООН)2, -С(O)-С6Н3-(м-ОСН2СООН)2, -СО-СН2O-(СН2)2O(СН2)2O-(СН2)2O(СН2)2OCH3, предпочтительно -С(O)СН2O[(СН2)2O]4-СН3.
В другом предпочтительном варианте полярный остаток R представляет собой остаток фолиевой кислоты.
Среди предлагаемых в изобретении соединений общей формулы I предпочтительны далее соединения, в которых Rf обозначает -CnF2n+1, n предпочтительно обозначает числа от 4 до 15. Наиболее предпочтительны остатки -C4F9, -С6F13, -C8F17, -C12F29 и -C14F29, а также остатки рассмотренных в примерах соединений.
По меньшей мере трехкратно функционализованный остаток G в общей формуле I, который является "скелетом", в одном из предпочтительных вариантов осуществления изобретения обозначает остаток лизина (а) или (b).
Z обозначает указанный для общей формулы I линкер, при этом предпочтительна группа

Способ получения перфторалкилсодержащих металлических комплексов с полярными остатками общей формулы I
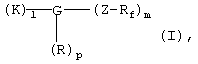
в которой К, G, R, Z, Rf, I, m и р имеют указанные выше значения, заключается в том, что карбоновую кислоту общей формулы IIa
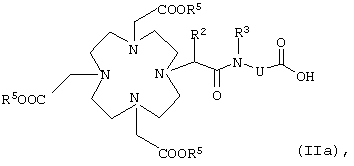
в которой R5 обозначает эквивалент иона металла с порядковым номером 21-29, 31-33, 37-39, 42-44, 49 или 57-83 или карбоксизащитную группу, а R2, R3 и U имеют указанные выше значения,
или карбоновую кислоту общей формулы IIIa

в которой R4, R5 и U1 имеют указанные выше значения,
или карбоновую кислоту общей формулы IVa
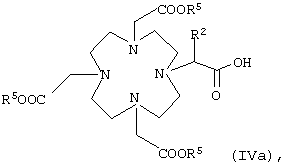
в которой R5 и R2 имеют указанные выше значения,
или карбоновую кислоту общей формулы Va либо Vb


где R5 имеет указанные выше значения,
или карбоновую кислоту общей формулы VIa

в которой R5 имеет указанные выше значения,
или карбоновую кислоту общей формулы VIIa

в которой R5 и U1 имеют указанные выше значения,
необязательно в активированной форме, подвергают по известной методике взаимодействию в условиях реакции сочетания с амином общей формулы VIII
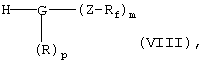
в которой G, R, Z, Rf, m и р имеют указанные выше значения,
и затем при необходимости отщепляют присутствующие при определенных условиях защитные группы с получением в результате комплекса металла общей формулы I либо, если R5 представляет собой защитную группу, после отщепления таких защитных групп на следующей стадии подвергают взаимодействию по известной методике по меньшей мере с одним оксидом металла или солью металла с порядковым номером 21-29, 31-33, 37-39, 42-44, 49 или 57-83, после чего при необходимости присутствующие при определенных условиях кислотные атомы водорода замещают катионами неорганических и/или органических оснований, аминокислот или амидов аминокислот.
Используемые в описанных выше реакциях карбоновые кислоты общих формул IIa-VIIa либо являются известными соединениями, либо их получают по описанным в примерах методам. Так, в частности, способ получения карбоновых кислот общей формулы IIа известен из DE 19652386. Карбоновые кислоты общей формулы IIIa можно получать аналогично примеру 4, приведенному ниже в настоящем описании. Способ получения карбоновых кислот общей формулы IVa описан в DE 19728954.
Предшественником соединений общей формулы VA является N3-(2,6-диоксоморфолиноэтил)-N6-(этоксикарбонилметил)-3,6-диазапробковая кислота, которая описана в ЕР 263059.
Соединения общей формулы VB являются производными изомерной диэтилентриаминпентауксусной кислоты (ДТПК), которая присоединена через находящуюся у центрального N-атома уксусную кислоту. Эта ДТПК описана в DE 19507819 и DE 19508058.
Соединения общей формулы VI являются производными N-(карбоксиметил)-N-[2-(2,6-диоксо-4-морфолинил)этил]глицина, способ получения которого описан в J. Am. Oil. Chem. Soc., 59 (2) (1982), c.104-107.
Соединения общей формулы VII являются производными 1-(4-карбоксиметоксибензил)этилендиаминтетрауксусной кислоты, способ получения которой описан в US 4622420.
Получение аминов общей формулы VIII подробно описано в примерах, приведенных ниже в настоящем описании, и поэтому указанные соединения можно получать аналогично рассмотренным в этих примерах методам.
Предлагаемые в изобретении металлические комплексы пригодны, как было установлено, для применения при ЯМР- и рентгенодиагностике, а также при радионуклидной диагностике и лучевой терапии. Объектом изобретения в соответствии с этим является также применение предлагаемых перфторалкилсодержащих металлических комплексов с полярными остатками для получения контрастных веществ, предназначенных для использования при ЯМР- и рентгенодиагностике, прежде всего при лимфографии, для диагностики опухолей и визуализации инфарктов и некрозов, а также при радионуклидной диагностике и лучевой терапии. Предлагаемые в изобретении соединения наиболее пригодны для применения при интерстициальной и прежде всего при внутривенной лимфографии. Наряду с этим они могут использоваться также для визуализации внутрисосудистой полости (в качестве контрастных веществ для визуализации пулов крови).
Объектом изобретения являются также фармацевтические средства, содержащие по меньшей мере одно физиологически совместимое соединение по изобретению, необязательно в сочетании с обычно используемыми в галеновых препаратах добавками.
Предлагаемые в настоящем изобретении соединения отличаются исключительно хорошей системной совместимостью и высокой степенью накопления (концентрации) в лимфатических узлах трех последовательно расположенных уровней (что особенно важно при внутривенной лимфографии). Благодаря этому они особенно пригодны для использования в МРТ-лимфографии.
Соединения по изобретению пригодны также для выявления и локализации заболеваний сосудов, поскольку они при их введении во внутрисосудистую полость распределяются только в ней. Предлагаемые в изобретении соединения позволяют с помощью ЯМР-томографии четко дифференцировать ткань с хорошим и плохим кровотоком и тем самым диагностировать ишемию. В равной степени при применении контрастных веществ по изобретению удается также четко разграничить инфарцированную ткань вследствие ее анемии и окружающую ее здоровую или ишемическую ткань. Подобная возможность имеет особое значение в тех случаях, когда, например, необходимо точно установить, имеет ли место инфаркт миокарда или речь идет об ишемии.
По сравнению с использовавшимися до настоящего времени в качестве контрастных веществ для визуализации пулов крови макромолекулярными соединениями, такими, например, как Gd-ДТПК-полилизин, соединения по изобретению обладают также более высокой T1-релаксационностью и в соответствии с этим для них характерен более высокий уровень интенсивности сигнала при ЯМР-визуализации. Поскольку, кроме того, такие соединения удерживаются в крови в течение более длительного промежутка времени, их можно вводить также в относительно малых дозах (например ≤50 мкмолей Gd на 1 л циркулирующей в организме крови). Однако не менее важное преимущество предлагаемых в изобретении соединений состоит в том, что они быстро и практически полностью выводятся из организма.
Помимо этого, было установлено, что соединения по изобретению накапливаются в областях с повышенной проницаемостью сосудов, в частности в опухолях, и в соответствии с этим они позволяют получать информацию о перфузии тканей, дают возможность определять объем крови в тканях, селективно сокращать время релаксации, соответственно период загустения крови, и визуализировать проницаемость кровеносных сосудов. Получать такого рода физиологическую информацию при применении внеклеточных контрастных веществ, таких, например, как Gd-ДТПК (Magnevist®), невозможно. С учетом этого появляется также возможность использовать соединения по изобретению в таких современных методах визуализации, как ЯМР-томография и компьютерная томография, в частности для специфического диагностирования злокачественных опухолей, для раннего контроля за ходом лечения при цитостатической, противовоспалительной или вазодилатационной терапии, для раннего выявления областей со сниженным кровоснабжением (например, в миокарде), для ангиографии при заболеваниях сосудов, а также для выявления и диагностики асептических или инфекционных воспалений.
Предлагаемые в изобретении фармацевтические средства получают по известной технологии суспендированием или растворением комплексных соединений по изобретению, при необходимости в сочетании с обычно используемыми в галеновых препаратах добавками, в водной среде, после чего полученную суспензию или полученный раствор необязательно подвергают стерилизации. В качестве указанных добавок могут использоваться, например, физиологически совместимые буферы (такие, как трометамин), комплексообразователи (такие, как диэтилентриаминпентауксусная кислота) или слабые комплексы либо соответствующие предлагаемым в изобретении металлическим комплексам Са-комплексы или при необходимости электролиты, такие как хлорид натрия, или при необходимости антиоксиданты, такие как аскорбиновая кислота.
Если для энтерального, соответственно парентерального, введения либо для иных целей предусматривается применять суспензии или растворы предлагаемых в изобретении средств в воде или физиологическом солевом растворе, то их смешивают с одним либо несколькими обычно используемыми в галеновых препаратах вспомогательными веществами (например, с метилцеллюлозой, лактозой, маннитом) и/или с одним или несколькими поверхностно-активными веществами (например, с лецитином, Tween®, Myrj®) и/или с одним или несколькими корригентами (например, с эфирными маслами).
В принципе фармацевтические средства по изобретению можно также получать без выделения комплексов. Однако в любом случае в процессе образования хелатных соединений следует особенно тщательно соблюдать условия, которые практически полностью исключают возможность присутствия в предлагаемых в изобретении комплексах ионов металла, не образовавших комплекс и обладающих токсическим действием. В этих целях процесс получения комплексных соединений можно контролировать путем титрования с помощью, например, цветных индикаторов, таких как ксиленоловый оранжевый. В соответствии с этим настоящее изобретение относится также к способу получения комплексных соединений и их солей. В крайнем случае выделенный комплекс можно подвергать очистке.
При применении предлагаемых в изобретении средств in vivo их можно вводить совместно с пригодным для этой цели носителем, таким, например, как сыворотка или физиологический раствор поваренной соли, и совместно с другим белком, таким, например, как сывороточный альбумин человека (САЧ).
Предлагаемые в изобретении средства предназначены преимущественно для парентерального, предпочтительно внутривенного, введения. Вместе с тем в зависимости от исследуемого объекта - сосудов или тканей - они могут также предназначаться для внутрисосудистого или интерстициального/внутрикожного введения.
Фармацевтические средства по изобретению содержат предпочтительно от 0,1 мкмоля до 2 молей комплекса на 1 л и предназначены для введения в дозах, составляющих, как правило, от 0,0001 до 5 ммолей/кг.
Предлагаемые в изобретении средства отвечают самым различным требованиям, которыми определяется их пригодность для применения в качестве контрастных веществ в ЯМР-томографии. Так, в частности, такие средства позволяют за счет увеличения интенсивности сигнала после их перорального или парентерального введения повысить информативность изображения, полученного с помощью ЯМР-томографа. Помимо этого, такие средства обладают высокой эффективностью, которая необходима для снижения концентрации вводимых в организм чужеродных веществ до минимально возможного уровня, и вместе с тем обладают хорошей переносимостью, которая необходима для сохранения неинвазивного характера исследований.
Благодаря высокой степени растворимости предлагаемых в изобретении средств в воде и их малой осмолярности появляется возможность получать на их основе высококонцентрированные растворы, что позволяет поддерживать объемную перегрузку системы кровообращения в допустимых пределах и компенсировать разбавление таких растворов жидкостями организма. Предлагаемые в изобретении средства обладают далее не только высокой стабильностью in vitro, но и проявляют неожиданно высокую стабильность in vivo, благодаря чему высвобождение или обмен связанных в комплексе ионов, которые в принципе являются токсичными, происходит лишь крайне медленно в течение того промежутка времени, за который новые контрастные вещества полностью выводятся из организма.
Обычно предлагаемые в изобретении средства при их использовании в качестве ЯМР-диагностикумов применяют в дозах от 0,0001 до 5 ммолей/кг, предпочтительно от 0,005 до 0,5 ммоля/кг.
Предлагаемые в изобретении комплексные соединения могут, кроме того, эффективно применяться в качестве реагентов с магнитной восприимчивостью и реагентов сдвига в ЯМР-спектроскопии in vivo.
Средства по изобретению благодаря их оптимальным радиоактивным свойствам и высокой стабильности содержащихся в них комплексных соединений пригодны также для применения в качестве диагностикумов при радионуклидной диагностике. Более подробно применение таких средств и их дозировка описаны, например, в публикации "Radiotracers for Medical Applications", изд-во CRC-Press, Boca Raton, Florida.
Предлагаемые в изобретении соединения и средства могут применяться также в позитронно-эмиссионной томографии, где используют эмиттирующие протоны изотопы, такие, например, как 43Sc, 44Sc, 52Fe, 55Co, 68Ga и 86Y (см. W.D.Heiss и М.Е.Phelps, Positron Emission Tomography of Brain, изд-во Springer Verlag, Berlin, Heidelberg, New York (1983)).
Соединения по изобретению могут, как неожиданно было установлено, применяться также для дифференциации злокачественных и доброкачественных опухолей без гематоэнцефалического барьера.
Соединения по изобретению отличаются также тем, что они полностью выводятся из организма, проявляя тем самым хорошую переносимость.
Поскольку предлагаемые в изобретении соединения накапливаются в злокачественных опухолях (отсутствие диффузии в здоровые ткани, но высокая проницаемость опухолевых сосудов), их можно также применять в дополнение к лучевой терапии злокачественных опухолей. Отличие лучевой терапии от соответствующей диагностики состоит лишь в количестве и типе используемого изотопа. Целью при этом является разрушение опухолевых клеток под воздействием мощного коротковолнового излучения с предельно малым радиусом действия. При этом используется взаимодействие содержащихся в комплексных соединениях металлов (таких, например, как железо или гадолиний) с ионизирующим излучением (например с рентгеновскими лучами) или с нейтронным излучением. Благодаря этому эффекту удается значительно повысить локальную дозу облучения в том месте, где находится металлический комплекс (например, в опухолях). Для обеспечения такой же дозы облучения в злокачественной ткани применение подобных металлических комплексов позволяет существенно снизить дозу облучения здоровых тканей и предотвратить тем самым нежелательные для пациентов побочные действия. Поэтому предлагаемые в изобретении конъюгаты металлических комплексов пригодны также для применения в качестве радиосенсибилизирующих веществ при лучевой терапии злокачественных опухолей (например, за счет использования эффектов Мессбауэра или при нейтронозахватной терапии). В качестве примера приемлемых испускающих β-излучение ионов можно назвать 46Sc, 47Sc, 48Sc, 72Ga, 73Ga и 90Y. В качестве приемлемых испускающих α-излучение ионов с малым периодом полураспада можно назвать, например, 211Bi, 212Bi, 213Bi и 214Bi, предпочтителен из которых 212Bi. Испускающим протоны и электроны ионом является 158Gd, который можно получать из 157Gd путем захвата нейтронов.
Если предлагаемое в изобретении средство предназначено для применения при лучевой терапии в соответствии с методикой, предложенной R.L.Mills и др. (см. Nature, том 336 (1988), с.787), то центральный ион должен быть производным мессбауэровского изотопа, такого, например, как 57Fe или 151Eu.
При применении предлагаемых в изобретении средств in vivo их можно вводить совместно с пригодным для этой цели носителем, таким, например, как сыворотка или физиологический раствор поваренной соли, и совместно с другим белком, таким, например, как сывороточный альбумин человека. Дозировка при этом зависит от типа целлюлярного нарушения, используемого иона металла и типа метода визуализации.
Предлагаемые в изобретении средства предназначены преимущественно для парентерального, предпочтительно внутривенного, введения. Вместе с тем, как указывалось выше, в зависимости от исследуемого объекта - сосудов или тканей - они могут также предназначаться для внутрисосудистого или интерстициального/внутрикожного введения.
Предлагаемые в изобретении средства пригодны также для применения в качестве рентгеноконтрастных веществ, при этом особо следует отметить, что при их применении в биохимически-фармакологических исследованиях не наблюдается никаких признаков анафилактических реакций, известных для йодсодержащих контрастных веществ. Эти средства благодаря их оптимальным свойствам поглощать излучение в диапазоне более высоких напряжений на трубке наиболее пригодны для применения в цифровой субтракционной технике.
Как правило, предлагаемые в изобретении средства при их использовании в качестве рентгеноконтрастных веществ применяют аналогично, например, диатризоату меглумина в дозах от 0,1 до 5 ммолей/кг, предпочтительно от 0,25 до 1 ммоля/кг.
При применении предлагаемых в изобретении соединений удается прежде всего достичь их более высокой концентрации в крови по сравнению с внеклеточными контрастными веществами. После внутривенного введения они распределяются только во внутрисосудистой полости, в чем состоит их основное преимущество перед внеклеточными контрастными веществами.
Примеры осуществления изобретения
Пример 1а
2-N-трифторацетил-6-N-бензилоксикарбониллизин
100 г (356,7 ммоля) 6-N-бензилоксикарбониллизина растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и в течение 24 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 128,9 г (96% от теории) бесцветного кристаллического порошка.
Элементный анализ:
Пример 1б
[1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-6-N-бензилоксикарбониллизина
К 125 г (332 ммоля) соединения, указанного в заголовке примера 1а, и 188,7 г (332 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 800 мл тетрагидрофурана при 0°С добавляют 164,2 г (0,664 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 286 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
Пример 1в
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбониллизина
Раствор 280 г (302,2 ммоля) соединения, указанного в заголовке примера 1б, в 2000 мл этанола при 0°С барботируют в течение часа газообразным аммиаком. Затем смесь перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 243,5 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
Пример 1г
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина
К 50 г (60,20 ммоля) соединения, указанного в заголовке примера 1 в, и 7,10 г (70 ммолей) триэтиламина в 350 мл дихлорметана при 0°С по каплям добавляют раствор 19,93 г (70 ммолей) хлорангидрида 3,6,9,12,15-пентаоксагексадекановой кислоты в 50 мл дихлорметана и перемешивают в течение 3 ч при 0°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 15:1).
Выход: 53,7 г (93% от теории) бесцветного вязкого масла.
Элементный анализ:
Пример 1д
[1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина
50 г (52,15 ммоля) соединения, указанного в заголовке примера 1г, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 1е
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина, Gd-комплекс
20 г (24,25 ммоля) соединения, указанного в заголовке примера 1д, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)]пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 28,21 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 2а
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)-6-карбонилметил]-2-N-[3,6,9,12,15-пентаоксагексадеканоил)лизина
К раствору 20 г (24,08 ммоля) соединения, указанного в заголовке примера 1д, 14,88 г (24,08 ммоля) бис(трет-бутилового эфира) 3,9-бис(трет-бутилоксикарбонилметил-3,6,9-триазаундекан-1,11-дикарбоновой кислоты и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 31,61 г (91% от теории) вязкого масла.
Элементный анализ:
Пример 2б
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[6-карбонилметил-3,9-бис(карбоксилатометил)-3,6,9-триазаундекандикарбоновая кислота-1-карбокси-11-карбоксилато]-2-N-(3,6,9,12,15-пентаоксагексадеканоил)лизина, Gd-комплекс, натриевая соль
30 г (20,8 ммоля) соединения, указанного в заголовке примера 2а, растворяют в 300 мл трифторуксусной кислоты и в течение 5 ч перемешивают при комнатной температуре. После этого упаривают досуха, остаток растворяют в 300 мл воды и значение рН устанавливают на 2,5 с помощью 10%-ного водного NaOH. Далее добавляют 3,77 г (10,4 ммоля) оксида гадолиния и перемешивают в течение 3 ч при 60°С. После этого смеси дают охладиться до комнатной температуры и значение рН устанавливают на 7,4 с помощью едкого натра. Затем упаривают досуха и остаток очищают на силикагеле RP-18 (элюент: градиент воды/ацетонитрила).
Выход: 19,18 г (67% от теории) бесцветного, аморфного твердого вещества.
Содержание воды 9,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 3а
[1-(4-перфтороктилсульфонилпиперазин]амид лизина
20 г (24,08 ммоля) соединения, указанного в заголовке примера 1в, растворяют в 300 мл этанола и добавляют 4 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 16,77 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 3б
[1-(4-перфтороктилсульфонилпиперазин]амид 2,6-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
10 г (14,36 ммоля) соединения, указанного в заголовке примера 3а, 3,34 г (29 ммолей) N-гидроксисукцинимида, 2,54 г (ммоля) хлорида лития и 18,26 г (29 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 12,38 г (60 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 19,02 г (69% от теории) бесцветного твердого вещества.
Содержание воды: 11,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 4а
Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенилуксусной кислоты
К 200 г (1,204 моля) метилового эфира 4-гидроксифенилуксусной кислоты и 212 г (2 моля) карбоната натрия в 2000 мл ацетона добавляют 233,8 г (1,4 моля) этилового эфира 2-бромуксусной кислоты и в течение 5 ч кипятят с обратным холодильником. Твердое вещество отфильтровывают и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: н-гексан/этиловый эфир уксусной кислоты в соотношении 15:1).
Выход: 288,5 г (95% от теории) бесцветного масла.
Элементный анализ:
Пример 4б
Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенил-2-бромуксусной кислоты
К 285 г (1,13 моля) указанного в заголовке примера 4а соединения, растворенного в 2000 мл четыреххлористого углерода, добавляют 201 г (1,13 моля) N-бромсукцинимида и 100 мг дибензилпероксида и в течение 8 ч кипятят с обратным холодильником. Далее охлаждают на ледяной бане, выпавший в осадок сукцинимид отфильтровывают и фильтрат досуха упаривают в вакууме. Остаток очищают на силикагеле (элюент: н-гексан/ацетон в соотношении 15:1).
Выход: 359,2 г (96% от теории) бесцветного вязкого масла.
Элементный анализ:
Пример 4в
Метиловый эфир 2-[4-(3-оксаэтилпропионат)]фенил-2-[1-(1,4,7,10-тетраазациклододекан-7-ил]уксусной кислоты
К 603 г (3,5 моля) 1,4,7,10-тетраазациклододекана в 6000 мл хлороформа добавляют 350 г (1,057 моля) соединения, указанного в заголовке примера 4б, и перемешивают в течение ночи при комнатной температуре. Далее трижды экстрагируют 3000 мл воды, органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток без дополнительной очистки используют в последующей реакции (пример 4г).
Выход: 448 г (количественный) вязкого масла. Элементный анализ:
Пример 4г
2-[4-(3-оксапропионовая кислота)]фенил-2-[1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота
445 г (1,053 моля) соединения, указанного в заголовке примера 4в, и 496 г (5,27 моля) хлоруксусной кислоты растворяют в 4000 мл воды. Затем значение рН устанавливают на 10 с помощью 30%-ного водного едкого натра. Далее смесь нагревают до 70°С и значение рН добавлением 30%-ного водного едкого натра поддерживают на уровне 10. Смесь перемешивают в течение 8 ч при 70°С. После этого значение рН устанавливают на 13 и в течение 30 мин кипятят с обратным холодильником. Затем раствор охлаждают на ледяной бане и добавлением концентрированной соляной кислоты значение рН устанавливают на 1. После этого досуха упаривают в вакууме. Остаток растворяют в 4000 мл метанола и перемешивают в течение часа при комнатной температуре. Выпавшую в осадок поваренную соль отфильтровывают, фильтрат упаривают досуха и остаток очищают на силикагеле RP-18 (элюент: градиент воды/этанола/ацетонитрила).
Выход: 403 г (69% от теории) бесцветного твердого вещества.
Содержание воды: 10,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 4д
2-[4-(3-оксапропионовая кислота)]фенил-2-[1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота, Gd-комплекс
К 400 г (721,3 ммоля) соединения, указанного в заголовке примера 4г, в 2000 мл воды добавляют 130,73 г (360,65 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Раствор фильтруют и фильтрат лиофилизуют.
Выход: 511 г (количественный) аморфного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 4е
[4-перфтороктилсульфонил)пиперазин]амид 2,6-N,N'-бис{2-[4-(3-оксапропионил)фенил]-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-ил]уксусная кислота]лизина, дигадолиниевый комплекс, динатриевая соль
10 г (14,36 ммоля) соединения, указанного в заголовке примера 3а, 3,45 г (30 ммолей) N-гидроксисукцинимида, 2,54 г (60 ммолей) хлорида лития и 21,26 г (30 ммолей) соединения, указанного в заголовке примера 4д, растворяют при умеренном нагревании в 250 мл диметилсульфоксида. Далее при 10°С добавляют 16,51 г (80 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила). Это вещество растворяют в небольшом количестве воды, значение рН устанавливают на 7,4 с помощью едкого натра и лиофилизуют.
Выход: 21,02 г (69% от теории) бесцветного твердого вещества.
Содержание воды: 11,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 5а
[1-(4-перфтороктилсульфонил)пиперазин]амид 2,6-N,N'-бис[6-карбонилметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина
К раствору 10 г (14,36 ммоля) соединения, указанного в заголовке примера 3а, 18,53 г (30 ммолей) бис(трет-бутилового эфира) 3,9-бис(трет-бутилоксикарбонилметил)-6-карбоксиметил-3,6,9-триазаундекан-1,11-дикарбоновой кислоты и 3,45 г (30 молей) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 10,32 г (50 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 19,60 г (72% от теории) вязкого масла.
Элементный анализ:
Пример 5б
2,6-N,N-бис[6-карбонилметил-3,9-бис(карбоксилатометил)-3,6,9-триазаундекандикарбоновая кислота-1-карбокси-11-карбоксилатолизин-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс, натриевая соль
15 г (7,91 моля) соединения, указанного в заголовке примера 5а, растворяют в 50 мл хлороформа и добавляют 200 мл трифторуксусной кислоты. Смесь перемешивают в течение 10 мин при комнатной температуре. После этого досуха упаривают в вакууме и остаток растворяют в 150 мл воды. Далее добавляют 2,87 г (7,91 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Затем смеси дают охладиться до комнатной температуры и значение рН устанавливают на 7,4 с помощью 2н. едкого натра. Раствор досуха упаривают в вакууме и очищают на RP-18 (элюент: градиент воды/этанола/ацетонитрила).
Выход: 8,11 г (57% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 9,6%.
Элементный анализ (в пересчете на безводное вещество):
Пример 6а
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[6-карбокси]метил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина
К раствору 20 г (24,08 ммоля) соединения, указанного в заголовке примера 1в, 14,88 г (24,08 ммоля) бис(трет-бутилового эфира) 3,9-бис(трет-бутилоксикарбонилметил)-6-карбоксиметил-3,6,9-триазаундекан-1,11-дикарбоновой кислоты и 2,88 г (25 ммолей) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 8,25 г (40 молей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 27,21 г (79% от теории) вязкого масла.
Элементный анализ:
Пример 6б
[1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[карбонилметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина
25 г (17,48 ммоля) соединения, указанного в заголовке примера 6а, растворяют в 350 мл этанола и добавляют 5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 22,66 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 6в
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[6-карбонилметил-3,9-бис(трет-бутилоксикарбонилметил)-3,6,9-триазаундекан-1,11-дикарбоновая кислота-бис(трет-бутиловый эфир)]лизина, Gd-комплекс
20 г (15,43 ммоля) соединения, указанного в заголовке примера 6б, 1,78 г (15,43 ммоля) N-гидроксисукцинимида, 1,48 г (35 ммолей) хлорида лития и 9,72 г (15,43 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановая кислота-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 150 мл диметилсульфоксида. Далее при 10°С добавляют 5,16 г (25 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2500 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 22,94 г (78% от теории) бесцветного твердого вещества.
Содержание воды: 7,9%.
Элементный анализ (в пересчете на безводное вещество):
Пример 6г
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-2-N-[6-карбонилметил-3,9-бис(карбоксилатометил)-3,6,9-триазаундекан-дикарбоновая кислота-карбокси-11-карбоксилато-z]лизина, дигадолиниевый комплекс, натриевая соль
20 г (10,49 ммоля) соединения, указанного в заголовке примера 6в, растворяют в 200 мл трифторуксусной кислоты. Смесь перемешивают в течение 60 мин при комнатной температуре. Далее досуха упаривают в вакууме и остаток растворяют в 150 мл воды. Затем добавляют 1,90 г (5,25 ммоля) оксида гадолиния и перемешивают в течение 5 ч при 80°С. Смеси дают охладиться до комнатной температуры и значение рН устанавливают на 7,4 с помощью едкого натра. Раствор досуха упаривают в вакууме и очищают на силикагеле RP-18 (элюент: градиент воды/этанола/ацетонитрила).
Выход: 11,89 г (61% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 10,2%.
Элементный анализ (в пересчете на безводное вещество):
Пример 7а
а) трет-бутиловый эфир 5,6-бис(бензилокси)-3-оксагексановой кислоты
100 г (376,2 ммоля) 1,2-ди-О-бензилглицерина [полученного согласно Chem. Phys. Lipids, 43(2) (1987), c.113-127 ] и 5 г гидросульфата тетрабутиламмония растворяют в смеси из 400 мл толуола и 200 мл 50%-ного водного едкого натра. Далее при 0°С в течение 30 мин по каплям добавляют 78 г (400 ммолей) трет-бутилового эфира 2-бромуксусной кислоты и затем перемешивают в течение 3 ч при 0°С. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: N-гексан/ацетон в соотношении 20:1).
Выход: 133,4 г (94% от теории) бесцветного масла.
Элементный анализ:
Пример 7б
5,6-бис(бензилокси)-3-оксагексановая кислота
130 г (336,4 ммоля) соединения, указанного в заголовке примера 7а, растворяют в 200 мл дихлорметана и при 0°С добавляют 100 мл трифторуксусной кислоты. Смесь перемешивают в течение 4 ч при комнатной температуре и затем упаривают досуха. Остаток кристаллизуют из пентана/диэтилового эфира.
Выход: 102,2 г (92% от теории) воскоподобного твердого вещества.
Элементный анализ:
Пример 7в
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
50 г (60,20 ммоля) соединения, указанного в заголовке примера 1в, 6,93 г (60,20 ммоля) N-гидроксисукцинимида, 5,09 г (120 ммолей) хлорида лития и 37,91 г (60,20 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан- 10-пентаноил-3-аза-4-оксо-5-метил-5-ила растворяют при умеренном нагревании в 400 мл диметилсульфоксида. Далее при 10°С добавляют 20,63 г (100 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 75,53 г (87% от теории) бесцветного твердого вещества.
Содержание воды: 10,1%.
Элементный анализ (в пересчете на безводное вещество):
Пример 7г
[1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил]лизина, Gd-комплекс
70 г (48,53 ммоля) соединения, указанного в заголовке примера 7в, растворяют в смеси из 500 мл воды и 100 мл этанола и добавляют 5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 63,5 г (количественный) бесцветного твердого вещества.
Содержание воды: 9,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 7д
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[5,6-бис(бензилокси)-3-оксагексаноил]-2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
10 г (7,64 ммоля) соединения, указанного в заголовке примера 7г, 3,30 г (10 ммолей) соединения, указанного в заголовке примера 7б, 0,85 г (20 ммолей) хлорида лития и 1,15 г (10 ммолей) N-гидроксисукцинимида растворяют при умеренном нагревании в 150 мл диметилсульфоксида. Далее при 10°С добавляют 3,10 г (15 ммолей) N,N'-дициклогексилкарбодиимида и перемешивают в течение 8 ч при комнатной температуре. Реакционный раствор сливают в 2000 мл ацетона и выделяют выпавший осадок. Указанное в заголовке соединение очищают на силикагеле RP-18 (элюент: градиент воды/этанола/ацетонитрила).
Выход: 11,14 г (90% от теории) бесцветного аморфного твердого вещества.
Содержание воды: 4,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 7е
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-(5,6,-дигидрокси-3-оксагексаноил)-2-Н-[1,4,7-трискарбоксилатометил)-1,4,7,10-тетраазациклододекан-10-пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
10 г (6,17 ммоля) соединения, указанного в заголовке примера 7д, растворяют в 200 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 8,89 г (количественный) бесцветного твердого вещества.
Содержание воды: 3,1%.
Элементный анализ (в пересчете на безводное вещество):
Пример 8а
1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[5,6-бис(бензилокси)-3-оксагексаноил]лизина
К раствору из 20 г (24,08 ммоля) соединения, указанного в заголовке примера 1в, 9,91 г (30 ммолей) соединения, указанного в заголовке примера 7б, и 3,45 г (30 ммолей) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 9,28 г (45 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 24,50 г (89% от теории) вязкого масла.
Элементный анализ:
Пример 8б
[1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(5,6-дигидрокси-3-оксагексаноил)лизина
20 г (17,5 ммоля) соединения, указанного в заголовке примера 8а, растворяют в 300 мл этанола и добавляют 5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 17,65 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 8в
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизина, Gd-комплекс
15 г (14,87 ммоля) соединения, указанного в заголовке примера 8б, 1,73 г (15 ммолей) N-гидроксисукцинимида, 1,27 г (30 ммолей) хлорида лития и 9,48 г (15 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановая кислота-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 100 мл диметилсульфоксида. Далее при 10°С добавляют 5,16 г (25 моля) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 1500 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 19,28 г (80% от теории) бесцветного твердого вещества.
Содержание воды: 10,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 9а
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-[2,6-N,N'-бис(бензилоксикарбонил)лизил]лизина
20 г (24,08 ммоля) соединения, указанного в заголовке примера 1в, и 2,53 г (25 ммолей) триэтиламина растворяют в 200 мл тетрагидрофурана (ТГФ) и добавляют 14,46 г (27 ммолей) паранитрофенолового эфира ди-N,N'-Z-лизина. Смесь перемешивают в течение 5 ч при 50°С. После этого досуха упаривают в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 28,07 г (95% от теории) бесцветного твердого вещества.
Элементный анализ:
Пример 9б
[1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(лизил)лизина, тригидробромид
К 25 г (20,37 ммоля) соединения, указанного в заголовке примера 9а, добавляют 100 мл бромистоводородной кислоты в ледяной уксусной кислоте (48%-ной) и перемешивают в течение 2 ч при 40°С. Далее охлаждают до 0°С, по каплям добавляют 1500 мл диэтилового эфира и выпавшее в осадок твердое вещество отфильтровывают. После сушки в вакууме (60°С) получают 21,52 г (99% от теории) кристаллического твердого вещества светло-желтого цвета.
Элементный анализ:
Пример 9в
[1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-[2,6-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]лизил]лизина, тригадолиниевый комплекс
31,49 г (50 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил)-5-ил)пентановой кислоты, 6,91 г (60 ммолей) N-гидроксисукцинимида и 4,24 г (100 ммолей) хлорида лития растворяют при умеренном нагревании в 350 мл диметилсульфоксида. Далее при 10°С добавляют 16,51 г (80 ммолей) N,N-дициклогексилкарбодиимида и перемешивают в течение 5 ч при 10°С. К этой смеси добавляют 10 г (9,37 ммоля) соединения, указанного в заголовке примера 9б, и 3,03 г (30 ммолей) триэтиламина и перемешивают в течение 12 ч при 60°С. Далее смеси дают охладиться до комнатной температуры и после этого ее сливают в 3000 мл ацетона. Выпавший осадок отфильтровывают и очищают на силикагеле RP-18 (элюент: градиент воды/этанола/ацетонитрила).
Выход: 16,7 г (67% от теории) бесцветного твердого вещества.
Содержание воды: 7,9%.
Элементный анализ (в пересчете на безводное вещество):
Пример 10а
а) 1,7-бис(бензилоксикарбонил)-4-(3,6,9,12,15-пентаоксагексадеканоил)-1,4,7,10-тетраазациклододекан
К 18,13 г (68,1 ммоля) 3,6,9,12,15-пентаоксагексадекановой кислоты и 30 г (68,1 ммоля) 1,7-ди-Z-циклена, полученного согласно Z. Kovacs и А.D. Sherry, J. Chem. Soc. chem. Commun., 2 (1995), с.185, в 300 мл тетрагидрофурана при 0°С добавляют 24,73 г (100 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 19,13 г (42% от теории) бесцветного твердого вещества.
Элементный анализ:
Пример 10б
1,7-бис(бензилоксикарбонил)-4-(3,6,9,12,15-пентаоксагексадеканоил)-10-(2Н,2Н,4Н,5Н,5Н-3-оксаперфтортридеканоил)-1,4,7,10-тетраазациклододекан
К 18 г (26,91 ммоля) соединения, указанного в заголовке примера 10а, и 14,05 г (26,91 ммоля) 2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридекановой кислоты, полученной согласно DE 19603033, в 300 мл тетрагидрофурана при 0°С добавляют 12,36 г (50 ммолей) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 21,51 г (67% от теории) бесцветного твердого вещества.
Элементный анализ:
Пример10в
1-(3,6,9,12,15-пентаоксагексадеканоил)-7-(2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридеканоил)-1,4,7,10-тетраазациклододекан
20 г (16,77 ммоля) соединения, указанного в заголовке примера 1г, растворяют в 200 мл этанола и добавляют 2,5 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 15,5 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 10г
1,7-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-4-(3,6,9,12,15-пентаоксагексадеканоил)-10-(2Н,2Н,4Н,4Н,5Н,5Н-3-оксаперфтортридеканоил)-1,4,7,10-тетраазациклододекан, Gd-комплекс
15 г (16,22 ммоля) соединения, указанного в заголовке примера 10в, 4,60 г (40 ммолей) N-гидроксисукцинимида, 3,39 г (80 ммолей) хлорида лития и 25,19 г (40 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановой кислоты растворяют при умеренном нагревании в 300 мл диметилсульфоксида. Далее при 10°С добавляют 24,73 г (100 ммолей) ЭЭДХ и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 19,86 г (57% от теории) бесцветного твердого вещества.
Содержание воды: 11,3%.
Элементный анализ (в пересчете на безводное вещество):
Пример 11а
1-[(4-перфтороктилсульфонил)пиперазин]амид 3,5-динитробензойной кислоты
К 20 г (35,2 ммоля) перфтороктилсульфонилпиперазина и 8,1 г (80 ммолей) триэтиламина, растворенным в 200 мл дихлорметана, при 0°С по каплям добавляют раствор 8,76 г (38 ммолей) 3,5-динитробензоилхлорида в 55 мл дихлорметана и перемешивают в течение 3 ч при 0°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 15:1).
Выход: 24,96 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
Пример 11б
[1-(4-перфтороктилсульфонил)пиперазин]амид 3,5-диаминобензойной кислоты
20 г (26,23 ммоля) соединения, указанного в заголовке примера 11а, растворяют в 400 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 18,43 г (количественный) твердого вещества кремового цвета.
Элементный анализ:
Пример 11в
[1-(4-перфтороктилсульфонил)пиперазин]амид 3,5-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]бензойной кислоты, Gd-комплекс
10 г (14,24 ммоля) соединения, указанного в заголовке примера 11б, 3,45 г (30 ммолей) N-гидроксисукцинимида, 2,54 г (60 молей) хлорида лития и 18,89 г (30 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)пентановой кислоты растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 10,32 г (50 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 2000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 19,74 г (72% от теории) бесцветного твердого вещества.
Содержание воды: 11,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 12
а) трет-Бутиловый эфир 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканкарбоновой кислоты
25,0 г (53,8 ммоля) 1Н,1Н,2Н,2Н-перфтор-1-деканола [поставляется фирмой Lancaster] растворяют в 250 мл абсолютного толуола и при комнатной температуре смешивают с каталитическим количеством (примерно 0,75 г) гидросульфата тетра-н-бутиламмония. Далее при 0°С добавляют в целом 7,55 г (134,6 ммоля, 2,5 эквивалента в пересчете на количество используемого спиртового компонента) высокодисперсного порошкового гидроксида калия, а затем 15,73 г (80,7 ммоля, 1,5 эквивалента в пересчете на количество используемого спиртового компонента) трет-бутилового эфира бромуксусной кислоты и оставляют на 2 ч для перемешивания при 0°С. Полученный таким путем реакционный раствор затем перемешивают в течение 12 ч при комнатной температуре и для переработки смешивают с этиловым эфиром уксусной кислоты общим количеством 500 мл и 250 мл воды. Органическую фазу отделяют и дважды промывают водой. После сушки органической фазы над сульфатом натрия соль отделяют вакуум-фильтрацией и растворитель отгоняют в вакууме. Полученный маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/гексана (в соотношении 1:10) в качестве элюента.
Выход: 26,3 г (84,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
б) 3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканкарбоновая кислота
20,0 г (34,58 ммоля) соединения, указанного в заголовке примера 12а, при перемешивании суспендируют при комнатной температуре в 200 мл смеси, состоящей из метанола и 0,5-молярного едкого натра в соотношении 2:1, и затем нагревают до 60°С. После проведения реакции в течение 12 ч при 60°С прозрачную реакционную смесь для переработки нейтрализуют смешением с катионообменной смолой Amberlite® IR 120 (Н+-форма), ионит удаляют вакуум-фильтрацией и полученный таким путем метанольно-водный фильтрат перегоняют в вакууме до получения сухого остатка. Полученный аморфно-маслянистый остаток очищают на силикагеле с использованием этилового эфира уксусной кислоты/н-гексана (в соотношении 1:3) в качестве элюента.
Выход: 16,0 г (88,6% от теории) указанного выше в заголовке соединения в виде бесцветного и высоковязкого масла.
Элементный анализ:
в) 1,7-бис{[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан}диэтилентриамин, дигадолиниевый комплекс
2,48 г (3,94 ммоля, 2,05 молярных эквивалента в пересчете на количество используемого диэтилентриамина) описанного в заявке DE 19728954 С1 в примере 31h Gd-комплекса 10-(4-карбокси-1-метил-2-оксо-3-азабутил)-1,4,7,10-тетраазациклододекан-1,4,7-триуксусной кислоты и 167 мг безводного хлорида лития (3,94 ммоля) при 40°С растворяют при перемешивании в 40 мл абсолютного диметилсульфоксида и при этой температуре смешивают с N-гидроксисукцинимидом общим количеством 453 мг (3,94 ммоля). После охлаждения до комнатной температуры полученный таким путем реакционный раствор смешивают с 814 мг (3,946 ммоля) N,N'-дициклогексилкарбодиимида и в течение 2 ч перемешивают при комнатной температуре. Полученную суспензию активированного эфира затем смешивают с 198,3 мг (1,92 ммоля) диэтилентриамина, растворенного в 5 мл абсолютного диметилсульфоксида, и перемешивают в течение 12 ч при комнатной температуре. Для переработки реакционную смесь смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившуюся дициклогексилмочевину и фильтрат для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость 3000 Да). Ретентат (вещество, не прошедшее через фильтрационную мембрану) в завершение лиофилизуют.
Выход: 1,85 г (72,7% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 3,89%.
Элементный анализ (в пересчете на безводное вещество):
г) 1,7-бис{[1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-илпентаноил)]-1,4,7,10-тетраазациклододекан}-4-(3-окса-2Н,2Н,4Н,4Н,5Н,5Н-перфтортридеканоил)диэтилентриамин, дигадолиниевый комплекс
К раствору 3,23 г (2,44 ммоля) соединения, указанного в заголовке примера 12в, в смеси из 30 мл диметилсульфоксида и 3 мл тетрагидрофурана при 50°С в атмосфере азота по каплям добавляют 1,27 г (2,44 ммоля) указанного в заголовке примера 12б соединения, растворенного в смеси из 15 мл тетрагидрофурана и 15 мл диметилсульфоксида. После этого при 0°С порциями добавляют в целом 1,80 г (3,66 ммоля) ЭЭДХ [2-этокси-1-этоксикарбонил-1,2-дигидрохинолин] и оставляют на ночь для перемешивания при комнатной температуре. Полученный реакционный раствор затем смешивают с достаточным количеством ацетона до полного осаждения указанного выше в заголовке соединения, осадок отделяют вакуум-фильтрацией, сушат, растворяют в воде, отфильтровывают нерастворившиеся компоненты и фильтрат подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да), что обеспечивает полное обессоливание указанного в заголовке соединения и одновременно его очистку от низкомолекулярных компонентов. Ретентат в завершение лиофилизуют.
Выход: 3,54 г (79,4% от теории) в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 5,87%.
Элементный анализ (в пересчете на безводное вещество):
Пример 13
а) 2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизин
100,0 г (356,7 ммоля) 6-N-бензилоксикарбонил-L-лизина растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и в течение 24 ч перемешивают при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 128,9 г (96% от теории) бесцветного кристаллического порошка.
Температура плавления: 98,5°С.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-трифторацетил-6-N-бензилоксикарбонил-L-лизина
К 125,0 г (332,0 ммоля) соединения, указанного в заголовке примера 1а, и 188,7 г (332,0 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 750 мл тетрагидрофурана при 0°С добавляют 164,2 г (0,664 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 286,0 г (93% от теории) бесцветного твердого вещества.
Температура плавления: 92°С.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-L-лизина
Раствор 280,0 г (302,2 моля) соединения, указанного в заголовке примера 1б, в 2000 мл этанола при 0°С барботируют в течение часа газообразным аммиаком. После этого смесь перемешивают в течение 4 ч при 0°С. Далее упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 243,5 г (97,0% от теории) аморфного твердого вещества.
Элементный анализ:
г) [1-(4-перфтороктилсульфонил)пиперазин]амид L-лизина
200,0 г (240,8 ммоля) полученного в примере 13в соединения растворяют в 1000 мл этанола, смешивают с 5,0 г катализатора Перлмана (20%-ный Pd/C) и гидрируют при комнатной температуре в атмосфере водорода (1 атм) до тех пор, пока не прекратится поглощение водорода. После этого катализатор удаляют вакуум-фильтрацией, тщательно промывают этанолом (трижды порциями примерно по 100 мл) и досуха концентрируют в вакууме. Таким путем получают указанное в заголовке соединение в виде высоковязкого масла желтого цвета.
Выход: 162,5 г (96,9% от теории).
Элементный анализ:
д) [1-(4-перфтороктилсульфонил)пиперазин]амид 6N-2N-бис{4-[2,3-бис(N,N-бис(трет-бутилоксикарбонилметил)амино)пропил]фенил}-3-оксапропионил-L-лизина
5,25 г (7,72 ммоля) 4-[2,3-бис-(N,N-бис(трет-бутилоксикарбонилметил)амино)пропил]фенил}-3-оксапропионовой кислоты и 781,0 мг (7,72 ммоля) триэтиламина растворяют в 50 мл метиленхлорида. Далее при -15°С по каплям в течение 5 мин добавляют раствор 1,16 г (8,5 ммоля) изобутилового эфира хлормуравьиной кислоты в 10 мл метилхлорида и перемешивают еще в течение 20 мин при -15°С. Далее раствор охлаждают до -25°С, в течение 30 мин по каплям добавляют раствор из 2,68 г (3,86 ммоля) указанного в заголовке примера 13г соединения и 2,12 г (21,0 ммоль) триэтиламина в 70 мл тетрагидрофурана, после чего перемешивают еще в течение 30 мин при -15°С, а затем в течение ночи при комнатной температуре. Для переработки растворитель отгоняют в вакууме и полученный маслянистый остаток растворяют в 250 мл хлороформа. Хлороформную фазу дважды экстрагируют 10%-ным водным раствором хлорида аммония порциями по 100 мл, органическую фазу сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: метиленхлорид/этанол в соотношении 20:1).
Выход: 5,37 г (68,8% от теории) бесцветного и высоковязкого масла.
Элементный анализ:
е) [1-(4-перфтороктилсульфонил)пиперазин]амид 6N-2N-бис{4-[2,3-бис(N,N-бис(карбоксилатометил)амино)пропил]фенил}-3-оксапропионил-L-лизина, октанатриевая соль
5,0 г (2,47 ммоля) соединения, указанного в заголовке примера 13д, растворяют в 60 мл абсолютного дихлорметана. После этого полученный раствор при 0°С по каплям смешивают с трифторуксусной кислотой общим количеством 75 мл. После проведения реакции в течение 12 ч при комнатной температуре смесь досуха концентрируют в вакууме. Полученный остаток смешивают с 100 мл воды и вновь перегоняют в вакууме до получения сухого остатка. Полученный таким путем остаток растворяют в 200 мл дистиллированной воды и водный, содержащий продукт раствор указанного выше в заголовке соединения дважды экстрагируют диэтиловым эфиром порциями по 60 мл. Общий объем полученного водного содержащего продукт раствора доводят смешением с водой до 300 мл, отфильтровывают нерастворившиеся компоненты и фильтрат подвергают ультрафильтрации через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да), что обеспечивает полное обессоливание указанного в заголовке соединения и одновременно его очистку от низкомолекулярных компонентов. Общий объем ретентата смешением с водой доводят до 200 мл и затем значение рН этого раствора устанавливают на 10,0 с помощью 15%-ного едкого натра. Щелочной водный содержащий продукт раствор в завершение лиофилизуют.
Таким путем получают 4,0 г (92,8% от теории) указанного в заголовке соединения в виде октанатриевой соли в форме аморфного лиофилизата.
Содержание воды: 5,37%.
Элементный анализ (в пересчете на безводное вещество):
ж) [1-(4-перфтороктилсульфонил)пиперазин]амид 6N-2N-бис{4-[2,3-бис(N,N-бис(карбоксиметил)амино)пропил]фенил}-3-оксапропионил-L-лизина, димарганцевый комплекс, тетранатриевая соль
1,94 г (1,11 ммоля) соединения, указанного в заголовке примера 13е, растворяют в 100 мл дистиллированной воды и значение рН полученного раствора устанавливают на 4,0 смешением с 1-молярной водной соляной кислотой. Далее при 80°С порциями смешивают с 0,25 г (2,22 ммоля) карбоната марганца(II). Полученный таким путем реакционный раствор затем в течение 5 ч кипятят с обратным холодильником. После охлаждения до комнатной температуры значение рН водного содержащего продукт раствора устанавливают при интенсивном перемешивании на 7,2 смешением с 1н. едким натром и для обессоливания и для очистки от низкомолекулярных компонентов пропускают через ультрафильтрационную мембрану AMICON® YM-3 (предельная проницаемость: 3000 Да). Ретентат в завершение лиофилизуют.
Выход: 1,80 г (92,0% от теории) указанного в заголовке соединения в виде бесцветного лиофилизата.
Н2О-содержание (при использовании реактива Карла Фишера): 7,28%.
Элементный анализ (в пересчете на безводное вещество):
Пример 14
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-(бензилоксикарбонил)-2-N-[(N-птероил)-L-глутаминил]лизина
20 г (45,31 ммоля) фолиевой кислоты растворяют в 300 мл диметилсульфоксида и при 10°С добавляют 9,49 г (46 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение ночи при комнатной температуре. Далее к этой смеси добавляют 29,1 г (35 ммолей) соединения, указанного в заголовке примера 1в, и 20 мл пиридина и перемешивают в течение 3 ч при 50°С. Затем охлаждают до комнатной температуры и добавляют смесь из 1500 мл диэтилового эфира и 1500 мл ацетона. Выпавший осадок отфильтровывают и очищают на силикагеле RP-18 (элюент: градиент воды/этанола/тетрагидрофурана).
Выход: 21,59 г (38% от теории) желтого твердого вещества.
Содержание воды: 2,1%.
Элементный анализ (в пересчете на безводное вещество):
6) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-[(N-птероил)-L-глутаминил]лизина
К 20 г (15,95 ммоля) соединения, указанного в заголовке примера 14а, добавляют 200 мл бромистоводородной кислоты в ледяной уксусной кислоте (48%-ной) и перемешивают в течение 2 ч при 40°С. Далее охлаждают до 0°С, по каплям добавляют 2000 мл диэтилового эфира и выпавшее в осадок твердое вещество отфильтровывают. После сушки в вакууме (60°С) получают 18,96 г (99% от теории) кристаллического твердого вещества желтого цвета.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил]-2-N-[(N-птероил)-L-глутаминил]лизина, Gd-комплекс
0,92 г (8 ммолей) N-гидроксисукцинимида, 0,68 г (16 молей) хлорида лития и 5,04 г (8 ммолей) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-(3-аза-4-оксо-5-метил-5-ил)-1,4,7-10-тетраазациклододекана растворяют при умеренном нагревании в 80 мл диметилсульфоксида. Далее при 10°С добавляют 2,06 г (10 молей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение 3 ч при комнатной температуре. К этому реакционному раствору добавляют 5 г (4,16 ммоля) соединения, указанного в заголовке примера 14б, и 10 мл пиридина. Смесь перемешивают в течение ночи при комнатной температуре. Затем раствор сливают в 1000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и после этого очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила). Продукт растворяют в небольшом количестве воды, значение рН устанавливают на 7,4 с помощью едкого натра и лиофилизуют.
Выход: 3,87 г (53% от теории) желтого твердого вещества.
Содержание воды: 5,8%.
Элементный анализ (в пересчете на безводное вещество):
Пример 15
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(3,6,9,12-тетраоксатридеканоил)лизина
К 50 г (60,20 ммоля) указанного в заголовке примера 1в соединения и 7,10 г (70 ммолей) триэтиламина, растворенным в 350 мл дихлорметана, при 0°С по каплям добавляют раствор 16,85 г (70 ммолей) хлорангидрида 3,6,9,12-тетраоксатридекановой кислоты в 50 мл дихлорметана и перемешивают в течение 3 ч при 0°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 15:1).
Выход: 30,94 г (92% от теории) бесцветного, вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(3,6,9,12-тетраоксатридеканоил)лизина
53,96 г (52,15 ммоля) соединения, указанного в заголовке примера 15а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(3,6,9,12-тетраоксатридеканоил)лизина, Gd-комплекс
21,84 (24,25 ммоля) соединения, указанного в заголовке примера 15б, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 28,21 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 16
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N-(пропил-3-сульфоновая кислота)лизина
К 50 г (60,20 ммоля) соединения, указанного в заголовке примера 1в, и 7,10 г (70 ммолей) триэтиламина, растворенным в 250 мл сухого тетрагидрофурана, при 50°С по каплям добавляют раствор 7,33 г (60 молей) пропансультона в 50 мл тетрагидрофурана и перемешивают в течение 3 ч при 60°С. Далее добавляют 200 мл 5%-ной водной соляной кислоты и перемешивают в течение 5 мин при комнатной температуре. Органическую фазу отделяют, сушат над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 15:1).
Выход: 45,16 г (79% от теории) бесцветного вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N-(пропил-3-сульфоновая кислота)лизина
49,68 г (52,15 ммоля) соединения, указанного в заголовке примера 16а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 42,69 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(пропил-3-сульфоновая кислота)лизина, Gd-комплекс
19,85 г (24,25 ммоля) соединения, указанного в заголовке примера 16б, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 28,13 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество)
Пример 17
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-бензилоксикарбонил-2-N,N-бис(пропил-3-сульфоновая кислота)лизина
К 50 г (60,20 ммоля) соединения, указанного в заголовке примера 1в, и 12,14 г (120 ммолей) триэтиламина, растворенным в 250 мл сухого тетрагидрофурана, при 50°С по каплям добавляют раствор 14,65 г (120 ммолей) 1,3-пропансультона в 100 мл тетрагидрофурана и перемешивают в течение 3 ч при 60°С. Далее добавляют 400 мл 5%-ной водной соляной кислоты, перемешивают в течение 5 мин при комнатной температуре, смешивают с хлоридом натрия, органическую фазу отделяют, сушат ее над сульфатом магния и досуха упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 15:1).
Выход: 52,24 г (81% от теории) бесцветного вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 2-N,N-бис(пропил-3-сульфоновая кислота)лизина
53,74 г (52,15 ммоля) соединения, указанного в заголовке примера 17а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 49,06 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) [1-(4-перфтороктилсульфонил)пиперазин]амид 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N,N-бис(пропил-3-сульфоновая кислота)лизина, Gd-комплекс, динатриевая соль
38,76 г (24,25 ммоля) соединения, указанного в заголовке примера 17б, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 31,63 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 18
а) 5-бензиловый эфир N-трифторацетил-L-глутаминовой кислоты
100 г (421,5 ммоля) 5-бензилового эфира L-глутаминовой кислоты растворяют в смеси из 1000 мл этилового эфира трифторуксусной кислоты и 500 мл этанола и перемешивают в течение 24 ч при комнатной температуре. После этого упаривают досуха и остаток кристаллизуют из диизопропилового эфира.
Выход: 140,47 г (96% от теории) бесцветного кристаллического порошка.
Элементный анализ:
б) N-бис(2-гидроксиэтил)амид 5-бензилового эфира 2-N-трифторацетил-L-глутаминовой кислоты
К раствору 24,9 г (24,08 ммоля) соединения, указанного в заголовке примера 18а, 2,53 г (24,08 ммоля) диэтаноламина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 9,11 г (90% от теории) вязкого масла.
Элементный анализ:
в) N-бис(2-гидроксиэтил)моноамид N-трифторацетил-L-глутаминовой кислоты
21,92 г (52,15 ммоля) соединения, указанного в заголовке примера 18б, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 43,0 г (количественный) бесцветного твердого вещества.
Элементный анализ:
г) Трифторацетил-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
К 10,96 г (33,2 ммоля) соединения, указанного в заголовке примера 18а, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 30,93 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
д) L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 30,24 г (30,22 ммоля) указанного в заголовке примера 18б в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 26,55 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
e) N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс
211,96 г (24,25 ммоля) соединения, указанного в заголовке примера 18д, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 27,43 г (81% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 19
а) N-диметилбис(1,1-дигидроксиметил)амид 5-бензилового эфира N-трифторацетил-L-глутаминовой кислоты
К раствору 8,03 г (24,08 ммоля) соединения, указанного в заголовке примера 18а, 3,98 г (24,08 ммоля) диметилбис(1,1-дигидроксиметил)амина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 110,53 г (91% от теории) вязкого масла.
Элементный анализ:
б) [1-(4-перфтороктилсульфонил)пиперазин]амид 5-бензилового эфира N-трифторацетил-L-глутаминовой кислоты
25,05 г (52,15 ммоля) соединения, указанного в заголовке примера 19а, растворяют в 500 мл этанола и добавляют 6 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 20,36 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) N-трифторацетил-L-глутаминовая кислота-N-диметилбис(1,1-дигидроксиметил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
К 12,96 г (33,2 ммоля) соединения, указанного в заголовке примера 19б, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 800 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 28,42 г (91% от теории) бесцветного твердого вещества.
Элементный анализ:
г) L-глутаминовая кислота-N-[диметилбис(1,1-дигидроксиметил)]амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 28,41 г (30,2 ммоля) соединения, указанного в заголовке примера 19в, в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 24,74 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
д) 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-N-[диметилбис(1,1-дигидроксиметил)амид]-5-[1-(4-перфтороктилсульфонил)пиперазин] амид, Gd-комплекс
20,48 г (24,25 ммоля) соединения, указанного в заголовке примера 19г, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанолы/ацетонитрила).
Выход: 29,05 г (83% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество):
Пример 20
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 5-бензилового эфира N-трифторметилацетил-L-глутаминовой кислоты
К 11,06 г (33,2 ммоля) соединения, указанного в заголовке примера 18а, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 27,28 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 5-[1-[4-перфтороктилсульфонил)пиперазин]амид N-трифторацетил-L-глутаминовой кислоты
21,92 г (52,15 ммоля) соединения, указанного в заголовке примера 18а, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 41,37 г (количественный) бесцветного твердого вещества.
Элементный анализ:
в) N-трифторацетил-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
К раствору 24,9 г (24,08 ммоля) соединения, указанного в заголовке примера 18а, 2,53 г (24,08 ммоля) диэтаноламина и 2,77 г (24,08 ммоля) N-гидроксисукцинимида в 150 мл диметилформамида при 0°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида. Смесь перемешивают в течение 3 ч при 0°С, а затем в течение ночи при комнатной температуре. Выпавшую в осадок мочевину отфильтровывают, фильтрат досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/этанол в соотношении 20:1).
Выход: 9,11 г (90% от теории) вязкого масла.
Элементный анализ:
г) L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид
Раствор 26,61 г (30,22 ммоля) соединения, указанного в заголовке примера 18в, в 200 мл этанола при 0°С в течение часа барботируют газообразным аммиаком. Смесь затем перемешивают в течение 4 ч при 0°С. После этого упаривают досуха и остаток выделяют из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме (50°С).
Выход: 23,93 г (97% от теории) аморфного твердого вещества.
Элементный анализ:
д) N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовая кислота-N-бис(2-гидроксиэтил)амид-5-[1-(4-перфтороктилсульфонил)пиперазин]амид, Gd-комплекс
16,43 г (24,25 ммоля) соединения, указанного в заголовке примера 20г, 2,79 г (24,25 ммоля) N-гидроксисукцинимида, 2,12 г (50 ммолей) хлорида лития и 15,27 г (24,25 ммоля) Gd-комплекса 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)пентановая кислота]-1,4,7,10-тетраазациклододекана растворяют при умеренном нагревании в 200 мл диметилсульфоксида. Далее при 10°С добавляют 8,25 г (40 ммолей) N,N-дициклогексилкарбодиимида и затем перемешивают в течение ночи при комнатной температуре. Раствор сливают в 3000 мл ацетона и перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают хроматографией (силикагель RP-18, элюент: градиент воды/этанола/ацетонитрила).
Выход: 28,10 г (83% от теории) бесцветного твердого вещества.
Содержание воды: 11,0%.
Элементный анализ (в пересчете на безводное вещество)
Пример 21
а) [1-(4-перфтороктилсульфонил)пиперазин]амид 5-бензилового эфира N-трифторацетилглутаминовой кислоты
К 11,06 г (33,2 ммоля) соединения, указанного в заголовке примера 18а, и 18,87 г (33,2 ммоля) 1-перфтороктилсульфонилпиперазина (полученного согласно DE 19603033) в 80 мл тетрагидрофурана при 0°С добавляют 16,42 г (66,4 ммоля) ЭЭДХ (этиловый эфир 2-этокси-1,2-дигидрохинолин-1-карбоновой кислоты) и перемешивают в течение ночи при комнатной температуре. После этого досуха упаривают в вакууме и хроматографируют на силикагеле (элюент: дихлорметан/метанол в соотношении 20:1).
Выход: 27,28 г (93% от теории) бесцветного твердого вещества.
Элементный анализ:
б) 5-[1-[4-перфтороктилсульфонил)пиперазин]амид N-трифторацетил-L-глутаминовой кислоты
21,92 г (52,15 ммоля) соединения, указанного в заголовке примера 21а, растворяют в 500 мл этанола и добавляют 3 г палладиевого катализатора (10%-ный Pd/C). После этого гидрируют при комнатной температуре. Затем катализатор отфильтровывают и фильтрат досуха упаривают в вакууме.
Выход: 41,37 г (количественный) бесцветного твердого вещества.
Элементный анализ:
Пример 22. Распределение предлагаемого в изобретении контрастного вещества из примера 3 в органах (включая накопление в опухоли и лимфатических узлах) после его внутривенного введения крысам с раком предстательной железы
После внутривенного введения крысам (копулятивный инбридинг, за 12 дней до проведения опытов животным внутримышечно имплантировали клетки рака предстательной железы линии Dunning R3327 MAT-Lu) соединения из примера 3 из расчета 100 мкмолей общего гадолиния на 1 кг веса тела через 10 мин, 1 ч и 24 ч после введения (п.в.) определяли содержание металла в различных органах, в опухоли, а также в лимфатических узлах (данные, по которым сгруппированы в виде данных для брызжеечных и периферических лимфатических узлов) (в таблице указаны средние значения ± среднеквадратичное отклонение, n=3).
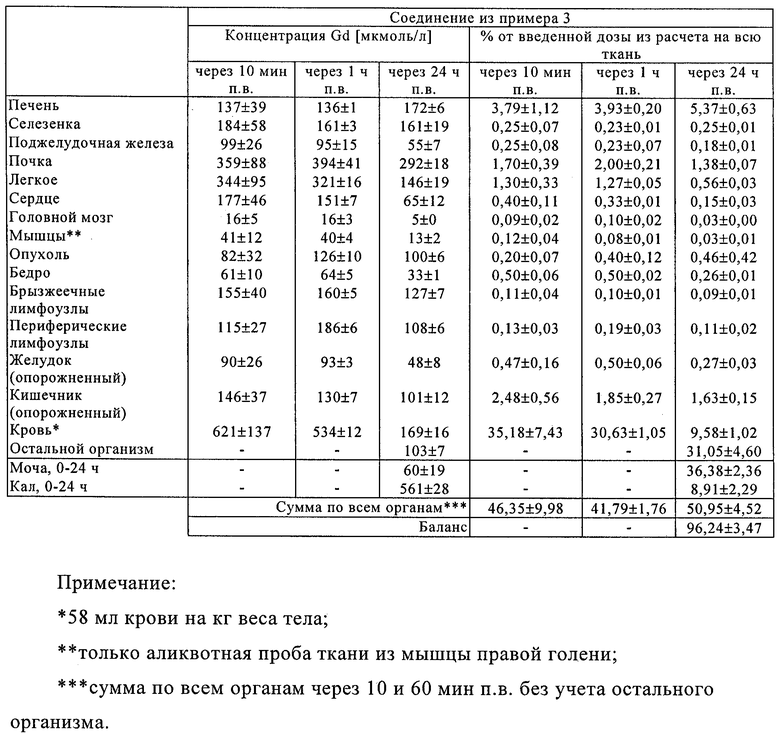
Пример 23. Картина инфаркта (MRT) после внутривенного введения контрастного вещества крысам
На снимках приводятся MR-фотографии сердца (in vivo и post morten) через 24 ч после внутривенного введения 100 мкмолей Gd/кг KGW соединения, указанного в заголовке примера 6г, крысам с острым индуцированным инфарктом сердца. Контрастное вещество вводили через 24 ч после окклюзии (закупорки) коронарной артерии. Примененная в этом примере модель инфаркта миокарда непосредственно сопоставима с моделью экспериментально индуцированного инфаркта почек, при котором происходит окклюзия ветви почечной артерии. T1-нагруженные, EKG-развернутые спин-эхо-снимки (1,5 Т, TR (эффективное): 400 мс, ТЕ: 12 мс, NA: 4, матрица: 128''128, толщина слоя: 2,5 мм) наглядно показывают сильное усиление сигнала в области инфаркта. Последующая индукция острого инфаркта миокарда подтверждается NBT-окрашиванием.
Этим экспериментом можно продемонстрировать пригодность соединений по изобретению в качестве маркеров некроза.
1. 24 ч после инъекции, in vivo, осевой
2. 25 ч после инъекции, post morten, осевой
3. 25 ч после инъекции, post morten, коронарный
4. 25 ч после инъекции, срез, ех vivo
5. 25 ч после инъекции, срез, NBT-окрашивание
6. Соединение, указанное в заголовке примера 6г: 100 мкмолей/кг, внутривенно, SE, TR/TE 400/12 мс, стрелка: инфаркт миокарда
Пример 24 Снимки лимфатических узлов (MRT) после внутривенного введения контрастного вещества морским свинкам
На снимках приводятся MR-фотографии подколенных лимфатических узлов до введения контрастного вещества и через 24 ч после внутривенного введения 100 мкмолей Gd/кг KGW соединения, указанного в заголовке примера 6г, морским свинкам. Иммунную систему животных стимулировали внутримышечным введением эмульсии желтка яйца (по 0,1 мл в бедро и голень через 6 суток) с целью увеличения размеров и массы лимфатических узлов. T1-нагруженные, турбо-спин-эхо-снимки (1,5 Т, последовательность: T1-TSE, TR 666 мс, ТЕ: 12 мс) наглядно показывают сильное усиление сигнала в здоровой ткани лимфатических узлов уже через короткое время после инъекции. Так увеличение подколенных и паховых лимфатических узлов составляет 201±9% через 15 мин после инъекции, 209±23% через 60 мин и 82±24% через 24 ч после инъекции. Фактор интенсивности сигнала между лимфатическими узлами и мышцей составляет 1,5±0,1 через 15 после инъекции и через 24 ч после инъекции достигает значения 1,6±0,1.
1. Базовая линия,
2. 15 мин после инъекции,
3. 24 ч после инъекции,
4. Соединение, указанное в заголовке примера 6г: 0,2 ммоля/кг, внутривенно, T1-TSE, TR/TE 666/12 мс, ТА 3:49,
морские свинки со стимулированными лимфатическими узлами, стрелка: подколенный лимфатический узел.
Пример 25. Картина инфаркта (MRT) после внутривенного введения контрастного вещества крысам
На снимках приводятся MR-фотографии сердца (in vivo и post morten) через 25 ч после внутривенного введения 100 мкмолей Gd/кг KGW соединения, указанного в заголовке примера 9в, крысам с острым индуцированным инфарктом сердца. Контрастное вещество вводили через 24 ч после окклюзии (закупорки) коронарной артерии. Примененная в этом примере модель инфаркта миокарда непосредственно сопоставима с моделью экспериментально индуцированного инфаркта почек, при котором происходит окклюзия ветви почечной артерии. T1-нагруженные, EKG-развернутые спин-эхо-снимки (1,5 Т, TR (эффективное): 400 мс, ТЕ: 12 мс, NA: 4, матрица: 128''128, толщина слоя: 2,5 мм) наглядно показывают сильное усиление сигнала в области инфаркта. Последующая индукция острого инфаркта миокарда подтверждается NBT-окрашиванием.
Этим экспериментом можно продемонстрировать пригодность соединений по изобретению в качестве маркеров некроза.
1. 25 ч после инъекции, in vivo, осевой
2. 25 ч после инъекции, post morten, осевой
3. 25 ч после инъекции, post morten, коронарный
4. 25 ч после инъекции, срез, ех vivo
5. 25 ч после инъекции, срез, NBT-окрашивание
6. Соединение, указанное в заголовке примера 9в: 100 мкмолей/кг, внутривенно, SE, TR/TE 400/12 мс, стрелка: инфаркт миокарда
Пример 26. Снимки лимфатических узлов (MRT) после внутривенного введения контрастного вещества морским свинкам
На снимках приводятся MR-фотографии подколенных лимфатических узлов до введения контрастного вещества и через 24 ч после внутривенного введения 50 мкмолей Gd/кг KGW соединения, указанного в заголовке примера 9 г, морским свинкам. Иммунную систему животных стимулировали внутримышечным введением эмульсии желтка яйца (по 0,1 мл в бедро и голень через 6 суток) с целью увеличения размеров и массы лимфатических узлов. Т1-нагруженные, турбо-спин-эхо-снимки (1,5 Т, последовательность: T1-TSE, TR 666 мс, ТЕ: 12 мс) наглядно показывают сильное усиление сигнала в здоровой ткани лимфатических узлов уже через короткое время после инъекции. Так увеличение подколенных и паховых лимфатических узлов составляет 84-87% через 15 мин после инъекции, 80-98% через 60 мин и 48-69% через 24 ч после инъекции. Фактор интенсивности сигнала между лимфатическими узлами и мышцей составляет 1,5 через 15 после инъекции и через 24 ч после инъекции достигает значения 1,8.
1. Базовая линия,
2. 15 мин после инъекции,
3. 24 ч после инъекции,
4. Соединение, указанное в заголовке примера 9в: 50 мкмолей/кг, внутривенно, T1-TSE, TR/TE 666/12 мс, ТА 3:49, морские свинки со стимулированными лимфатическими узлами, стрелка: подколенный лимфатический узел.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОМПЛЕКСЫ С ОСТАТКАМИ САХАРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2280644C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ В МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ БЛЯШЕК, ОПУХОЛЕЙ И НЕКРОЗОВ | 2001 |
|
RU2290206C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2242477C2 |
| ХЕЛАТЫ МЕТАЛЛОВ, ИМЕЮЩИЕ ПЕРФТОРИРОВАННЫЙ ПЭГ РАДИКАЛ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2470014C2 |
| СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2002 |
|
RU2305096C2 |
| КАСКАДНЫЕ ПОЛИМЕРНЫЕ КОМПЛЕКСЫ, ИСХОДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2166501C2 |
| КОМПЛЕКСЫ КАСКАДНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩЕЕ ИХ ДИАГНОСТИЧЕСКОЕ КОНТРАСТНОЕ СРЕДСТВО И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2197495C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ ПРИ МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ ВНУТРИСОСУДИСТЫХ ТРОМБОВ | 2003 |
|
RU2328310C2 |
| НОВЫЕ ХЕЛАТНЫЕ СОЕДИНЕНИЯ ГАДОЛИНИЯ С ВЫСОКОЙ РЕЛАКСИВНОСТЬЮ ДЛЯ ПРИМЕНЕНИЯ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ | 2017 |
|
RU2755181C2 |
| КОНЪЮГАТЫ, ПОЛУЧЕННЫЕ ИЗ КОМПЛЕКСОВ МЕТАЛЛОВ И ОЛИГОНУКЛЕОТИДОВ, СРЕДСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТЫ, ИХ ИСПОЛЬЗОВАНИЕ В РАДИОДИАГНОСТИКЕ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2165771C2 |


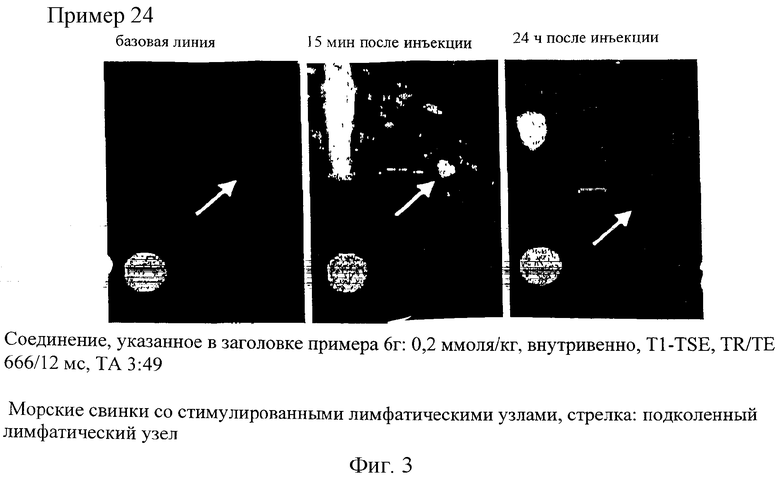
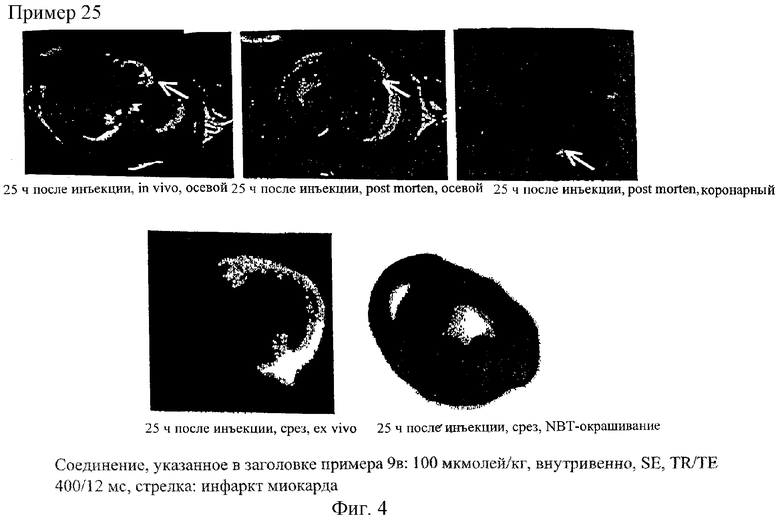
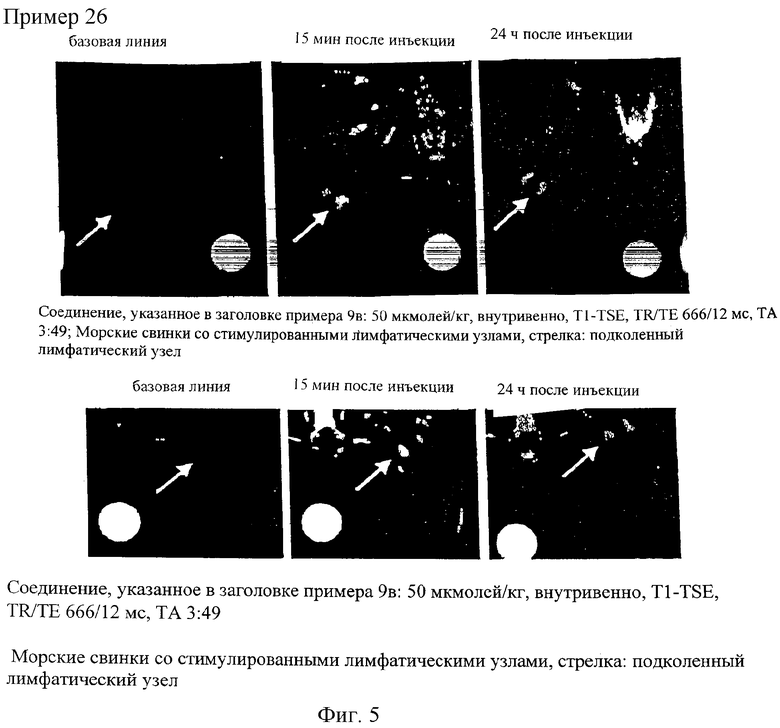
Описываются перфторалкилсодержащие комплексы с полярными остатками общей формулы (I)

где R - полярный остаток, G - трехфункциональный остаток, перфторированная углеродная цепь, К - комплекс металла, a Z - линкерная группа. Комплексы пригодны для применения при внутривенной лимфографии, для диагностики опухолей, а также для визуализации инфарктов и некрозов. Описан способ их получения. 3 н. и 13 з.п. ф-лы, 1 табл., 5 ил.

в которой Rf обозначает перфторированную, прямую или разветвленную углеродную цепь формулы -CnF2n, в которой n означает целое число от 4 до 30;
К обозначает металлический комплекс общей формулы II
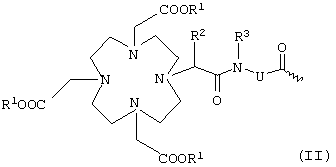
в которой R1 представляет собой атом водорода или эквивалент иона металла с порядковым номером 21-29 и 58-71 при условии, что по меньшей мере два радикала R1 обозначают эквиваленты ионов металлов;
R2 и R3 независимо друг от друга обозначают водород, С1-С4алкил, бензил, фенил, -CH2OH или -СН2ОСН3 и
U представляет собой -С6Н4О-СН2-ω-, -(CH2)1-5-ω-, -C6H4-(OCH2CH2)0-1-N(CH2COOH)-CH2-ω- или необязательно прерванную одним или несколькими атомами кислорода, 1-3 -NHCO-группами, 1-3 -CONH-группами и/или замещенную 1-3 -(СН2)0-5СООН-группами С1-С12алкиленовую или С7-С12-С6Н4-O-группу, при этом ω обозначает место присоединения к -СО-,
или общей формулы III
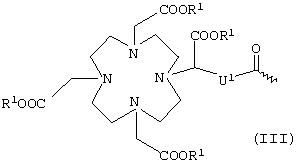
в которой R1 имеет вышеуказанные значения;
R4 обозначает водород или указанный для R1 эквивалент иона металла и
U1 обозначает -С6Н4-O-СН2-ω-, где ω обозначает место присоединения к -СО-,
или общей формулы IV
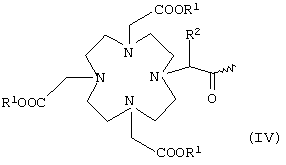
в которой R1 и R2 имеют указанные выше значения,
или общей формулы VB

в которой R1 имеет указанные выше значения,
или общей формулы VII

в которой R1 имеет указанные выше значения, а
U1 обозначает -С6Н4О-CH2-ω-, где ω обозначает место присоединения к -СО-,
при этом необязательно присутствующие в остатке К свободные кислотные группы необязательно могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот либо в виде амидов аминокислот,
G обозначает по меньшей мере трехкратно функционализованный остаток, выбранный из следующих остатков a)-i):
(a)

(b)

(c)
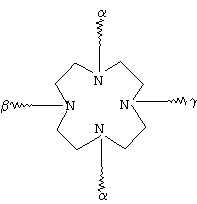
(d)
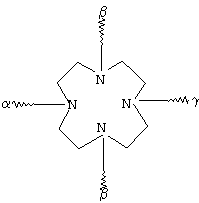
(e)

(f)

(g)
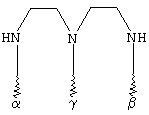
(h)
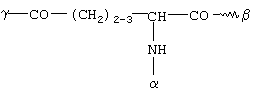

где а обозначает место присоединения G к комплексу К;
β обозначает место присоединения G к остатку R;
γ обозначает место присоединения G к остатку Z,
Z обозначает группу

γ-C(O)CH2O(CH2)2-ε,
где γ обозначает место присоединения Z к остатку G;
ε обозначает место присоединения Z к перфторированному остатку Rf,
R представляет собой полярный остаток, выбранный из комплексов К общих формул II-VII, причем в этом случае R1 обозначает атом водорода или эквивалент иона металла с порядковым номером 21-29 и 58-71, а остатки R2, R3, R4, U и U1 имеют указанные выше значения, причем в том случае, когда G представляет собой остаток формулы (с) или (d), a R представляет собой комплекс, выбранный из комплексов общих формул II и V, R не может быть идентичен остатку К общей формулы I, если Z представляет собой δ-С(O)СН2O(СН2)2-ε,
или представляет собой остаток фолиевой кислоты или присоединенную через -СО-, SO2- или прямую связь к остатку G углеродную цепь с 2-30 С-атомами, которая является прямой или разветвленной, насыщенной или ненасыщенной и которая необязательно прервана 1-10 атомами кислорода, 1-5 -NHCO-группами, 1-5 -CONH-группами, 1-2 атомами серы, 1-5 -NH-группами или 1-2 фениленовыми группами, которые необязательно могут быть замещены 1-2 ОН-группами, 1-2 NH2-группами, 1-2 -СООН-группами или 1-2 -SO3Н-группами, или необязательно замещена 1-8 ОН-группами, 1-5 -СООН-группами, 1-2 SO3Н-группами, 1-5 NH2-группами, 1-5 С1-С4алкоксигруппами, и
l, m, p независимо друг от друга обозначают целые числа 1 или 2.

в которой К, G, R, Z, Rf, I, m и р имеют указанные в п.1 значения,
отличающийся тем, что карбоновую кислоту общей формулы IIa
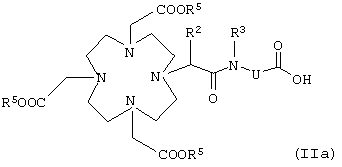
в которой R5 обозначает эквивалент иона металла с порядковым номером 21-29 и 58-71 либо карбоксизащитную группу, а R2, R3 и U имеют указанные выше значения,
или карбоновую кислоту общей формулы IIIa
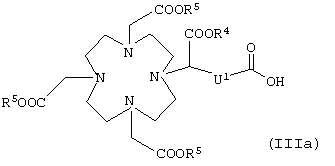
в которой R4, R5 и U1 имеют указанные выше значения,
или карбоновую кислоту общей формулы IVa

где R5 и R2 имеют указанные выше значения,
или карбоновую кислоту общей формулы Va либо Vb


где R5 имеет указанные выше значения,
или карбоновую кислоту общей формулы VIa
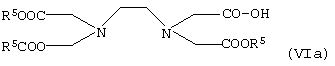
в которой R5 имеет указанные выше значения,
или карбоновую кислоту общей формулы VIIa
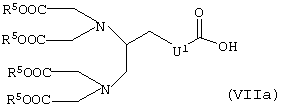
в которой R5 и U1 имеют указанные выше значения,
необязательно в активированной форме, подвергают по известной методике взаимодействию в условиях реакции сочетания с амином общей формулы VIII
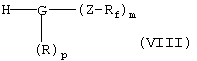
в которой G, R, Z, Rf, m и р имеют указанные выше значения,
и затем при необходимости отщепляют присутствующие при определенных условиях защитные группы с получением в результате комплекса металла общей формулы I либо, если R5 представляет собой защитную группу, после отщепления таких защитных групп на следующей стадии подвергают взаимодействию по известной методике по меньшей мере с одним оксидом металла или солью металла с порядковым номером 21-29 и 58-71, после чего при необходимости присутствующие при определенных условиях кислотные атомы водорода замещают катионами неорганических оснований.
| DE 19603033 А, 24.07.1997 | |||
| Рудоскат | 1977 |
|
SU628316A1 |
| DE 4317588 A1, 01.12.1994 | |||
| DE 19608278 А, 28.08.1997 | |||
| DE 19729013 А, 04.02.1999 | |||
| WO 9916474 А, 08.04.1999. | |||

