Данное изобретение относится к объектам, охарактеризованным в формуле изобретения, а именно к хелатам металлов, имеющим перфторированный ПЭГ радикал, к способам их получения и к их применению в ЯМР- и рентгенодиагностике, радионуклидной диагностике и лучевой терапии, и в МРТ лимфографии. Хелаты металлов, имеющие перфторированный ПЭГ радикал, применяются в магнитно-резонансной томографии (МРТ) для распознавания различных физиологических и патофизиологических структур и, таким образом, для улучшения диагностической информации, например местоположения и тяжести заболевания, для выбора и оценки результата направленной терапии и для профилактики.
Соединения в соответствии с изобретением наиболее пригодны для лимфографии, для диагностики опухолей и для визуализации инфарктов и некрозов и отличаются исключительно хорошей переносимостью.
В области ядерного магнитного резонанса известна возможность применения некоторых фторсодержащих соединений для визуализации. Однако такие соединения предлагаются исключительно для применения в фтор-19 томографии и пригодны только для этого применения. Такие соединения описаны, например, в патенте US 4,639,364 (Mallinckrodt), DE 4203254 (Max-Planck-Gesellschaft), WO 93/07907 (Mallinckrodt), US 4,586,511 (Children's Hospital Medical Center), EP 307863 (Air Products), US 4,588,279 (University of Cincinnati, Children's Hospital Research Foundation) и WO 94/22368 (Molecular Biosystems).
Другие фторсодержащие соединения, которые можно применять для визуализации, описаны в US 5,362,478 (VIVORX), патенте US 4,586,511, DE 4008179 (Schering), WO 94/05335 и WO 94/22368 (обе - Molecular Biosystems), EP 292306 (TERUMO Kabushiki Kaisha), EP 628316 (TERUMO Kabushiki Kaisha) и DE 4317588 (Schering).
Если в соединениях, содержащих фтор и йод, между обоими ядрами взаимодействие не происходит, то в соединениях, содержащих фтор и парамагнитные центры (радикалы, ионы металлов), происходит достаточно интенсивное взаимодействие, которое проявляется в сокращении времени релаксации ядра фтора. Степень проявления такого эффекта зависит от числа неспаренных электронов иона металла (Gd3+>Mn2+>Fe3+>Cu2+) и от расстояния между парамагнитным ионом и 19F-атомом.
Чем больше число неспаренных электронов иона металла и чем ближе они расположены к фтору, тем существеннее сокращается время релаксации ядра фтора.
Сокращение времени релаксации в функции удаленности от парамагнитного иона наблюдается у всех ядер с нечетным спиновым числом, в том числе и у протона, и поэтому гадолиниевые соединения находят широкое применение в качестве контрастных веществ в магнитно-резонансной томографии (Magnevist®, Prohance®, Omniscan®, Dotarem®).
При 1H-МР-томографии (1Н МРТ), однако, определяют время релаксации Т1 или Т2 протонов, т.е. прежде всего протонов воды, а не время релаксации ядер фтора, и используют полученные данные для визуализации. Количественной мерой, характеризующей сокращение времени релаксации, является релаксационность [л/ммоль·с]. Для сокращения времени релаксации с успехом применяют комплексы парамагнитных ионов. В нижеследующей таблице приводятся данные о релаксационности некоторых коммерчески доступных препаратов:
В этих соединениях происходит только взаимодействие между протонами и ионом гадолиния. Выявленная для указанных контрастных веществ релаксационность в воде составляет примерно 4 [л/ммоль·с].
Таким образом, при МР-томографии с успехом могут применяться и фторсодержащие соединения, предназначенные для 19F-томографии, где используется сокращенное время релаксации ядра фтора, и не содержащие фтор соединения, у которых определяют время релаксации протонов воды.
Неожиданный эффект, связанный с введением перфторуглеродсодержащего остатка в парамагнитное контрастное вещество, т.е. эффект, связанный с приданием соединениям, которые использовали в методах протонной визуализации, свойств тех соединений, которые до настоящего времени рассматривались как пригодные только для применения в методах визуализации, основанных на использовании фтора, проявляется также в быстром возрастании релаксационности протонов воды. В результате этот показатель достигает значений, составляющих 10-50 л/ммоль·с, тогда как аналогичные значения у некоторых коммерчески доступных продуктов составляют, как следует из вышеприведенной таблицы, от 3,5 до 3,8 л/ммоль·с.
Перфторалкилсодержащие комплексы металлов уже известны из заявок DE 19603033.1, WO 99/01161, DE 19914101, DE 10040381, DE 10040858. Однако возможности применения этих соединений ограничены, поскольку во многих случаях переносимость указанных соединений недостаточна. С учетом этого сохраняется необходимость в контрастных веществах для МРТ, которые обладают исключительно хорошими визуализирующими свойствами и в то же время имеют хорошую переносимость, что необходимо для достижения неинвазивного характера исследований. Это важно, например, при диагностике опухолей, в том числе отдаленных метастазов, когда достигается распределение контрастного вещества по всему телу.
Злокачественные опухоли часто метастазируют в регионарные лимфатические узлы, причем этот процесс может также охватывать несколько уровней лимфоузлов. Так, в частности, метастазы в лимфатические узлы были обнаружены примерно у 50-69% всех пациентов со злокачественными опухолями (Elke, Lymphographie [Лимфография], в: Frommhold, Stender, Thurn (Eds.), Radiologische Diagnostik in Klinik und Praxis [Радионуклидная диагностика в клинических исследованиях и в практике], том IV, Thieme Verlag, Stuttgart, 7-е изд., 434-496, 1984). Возможность диагностики метастазирования в лимфатические узлы имеет важное значение для терапии онкологических заболевания и прогнозирования их развития. Современные методы визуализации (компьютерная томография (КТ), ультразвуковое исследование (УЗИ) и магнитно-резонансная томография (МРТ)) не позволяют с достаточной высокой точностью и надежностью распознавать лимфогенное метастазирование злокачественных опухолей, поскольку в большинстве подобных случаев в качестве критериев соответствующего диагноза могут использоваться только размеры лимфатического узла. В результате такие методы просто не позволяют отличить небольшие метастазы в не увеличенных лимфатических узлах (<2 см) от гиперплазии лимфоузлов, не пораженных злокачественной опухолью (Steinkamp и др., Sonographie und Kernspintomographie: Differentialdiagnostik von reaktiver Lymphknoten-vergröRerung und Lymphknotenmetastasen am Hals [Сонография и магнитно-резонансная томография: дифференциальная диагностика реактивного увеличения лимфатических узлов и метастазов в лимфатические узлы шеи], Radiol. Diagn. 33: 158, 1992).
С учетом этого представляется целесообразным обеспечить при применении специфических контрастных веществ возможность дифференцировать лимфатические узлы, пораженные метастазами, и гиперпластические лимфатические узлы.
Прямая лимфорентгенография (инъекция масляной суспензии контрастного вещества в подготовленный лимфатический сосуд) известна в качестве инвазивного метода, который используется лишь в редких случаях и позволяет визуализировать только некоторые пути оттока лимфы.
В экспериментах, проводимых на животных, используют также декстраны с флуоресцентной меткой с целью обеспечить после их интерстициального введения возможность наблюдения оттока лимфы. Таким образом, характерно, что все обычные метки, используемые после их интерстициального/внутрикожного введения для распознавания лимфатических протоков и лимфатических узлов, представляют собой вещества в виде твердых частиц ("макрочастицы", например эмульсии и суспензии нанокристаллов) либо крупные полимеры (см. также WO 90/14846). Однако все известные в настоящее время из литературы композиции вследствие их недостаточной местной и системной переносимости, равно как и их малой подвижности в лимфе, обусловливающей неудовлетворительную эффективность диагностики, все еще остаются не оптимальными для применения в непрямой лимфографии.
Поскольку визуализация лимфатических узлов имеет первоочередную важность для раннего обнаружения метастазов у онкологических больных, существует необходимость в лимфоспецифичных препаратах контрастных веществ для диагностики соответствующих изменений в лимфатической системе, которые характеризовались бы очень хорошей переносимостью. В контексте настоящего изобретения, понятие лимфатическая система включает как лимфатические узлы, так и лимфатические сосуды. Вещества согласно настоящему изобретению являются вследствие этого пригодными для диагностики изменений лимфатической системы, предпочтительно для диагностики изменений лимфатических узлов и/или лимфатических сосудов, в частности, диагностики метастазов в лимфатических узлах.
Для достижения требуемого эффекта при использовании контрастных веществ целесообразно обеспечить не только их предельно высокую концентрацию в лимфе и высокую стабильность, но и максимально равномерное накопление в лимфе в нескольких уровнях лимфатической системы, что имеет важное для постановки точного диагноза значение. Вместе с тем контрастное вещество должно быстро и полностью выводиться из организма с целью минимизировать нагрузку на весь организм в целом. Действие контрастного вещества должно начинать проявляться по возможности уже через несколько часов после его введения, что является важным условием в радиологической практике. Также необходима их хорошая переносимость.
Не менее актуальна и потребность в лимфоспецифичных контрастных веществах, которые позволяли бы за один сеанс диагностического исследования визуализировать и первичную опухоль, и возможное ее метастазирование в лимфатические узлы.
Еще одной из важных задач медицины является обнаружение, определение расположения и наблюдение за некрозами и инфарктами. Так, в частности, инфаркт миокарда является не стационарным, а динамическим процессом, длящимся в течение продолжительного промежутка времени (от нескольких недель до нескольких месяцев). Это сердечно-сосудистое заболевание протекает примерно в три стадии, которые невозможно четко разграничить, поскольку они накладываются одна на другую, соответственно плавно переходят одна в другую. Длительность первой стадии, на которой происходит развитие инфаркта миокарда, составляет примерно первые 24 часа после его начала, в течение которых разрушение ткани распространяется подобно ударной волне (аналогично явлению волнового фронта) от субэндокарда к миокарду. На второй стадии, на которой развитие инфаркта как такового уже закончилось, включает стабилизацию области, в которой в процессе заживления пораженной инфарктом ткани происходит образование волокон (фиброз). Третья стадия, которая соответствует полному заживлению затронутой инфарктом ткани, начинается после замены всей разрушенной ткани фиброзной рубцовой тканью. В этот период протекает активная реструктуризация.
На сегодняшний день не существует ни одного метода, который позволял бы с высокой точностью и надежностью диагностировать текущую фазу инфаркта миокарда у живого пациента. Для оценки же инфаркта миокарда решающее значение имеет информация о том, насколько велика доля утраченной при инфаркте ткани и в каком именно месте, поскольку от этой информации зависит тип терапии.
Инфаркты, как известно, поражают не только миокард, но и другие ткани, прежде всего головной мозг.
Если затронутая инфарктом ткань в некоторой степени поддается заживлению, то при некрозе, т.е. при локально ограниченном омертвении ткани, можно лишь предотвратить или по меньшей мере смягчить его вредные последствия для остального организма. Возникновение некрозов может быть обусловлено самыми различными причинами, в частности травмами, воздействием химикалиев, дефицитом кислорода или же облучением. Аналогично инфаркту наличие информации о степени и типе некроза имеет важное значение для выбора последующих врачебных мер.
Попытки повысить эффективность определения местонахождения инфарктов и некрозов за счет применения контрастных веществ в неинвазивных методах, таких как сцинтиграфия или магнитно-резонансная томография, предпринимались уже достаточно давно. При этом большое число опубликованных работ посвящено экспериментальным исследованиям по использованию порфиринов для визуализации некрозов. Полученные в ходе подобных исследований результаты, однако, носят противоречивый характер. Кроме того, порфирины склонны откладываться в коже, что вызывает фотосенсибилизацию.
Подобная сенсибилизация может сохраняться в течение нескольких дней, а иногда и в течение нескольких недель. В этом заключается нежелательный побочный эффект, проявляющийся при применении порфиринов в качестве диагностикумов. Помимо этого порфирины имеют лишь исключительно низкий терапевтический индекс, поскольку, например, в случае Mn-ТФПС его действие проявляется только при его использовании в дозе 0,2 ммоль/кг, тогда как его летальная доза ЛД50 составляет уже 0,5 ммоль/кг.
Контрастные вещества, не являющиеся производными порфиринового каркаса и предназначенные для визуализации некрозов и инфарктов, описаны в заявках DE 19744003 (Schering AG), DE 19744004 (Schering AG) и WO 99/17809 (EPIX). Однако до настоящего времени все еще нет соединений, которые можно было бы достаточно эффективно применять в качестве контрастных веществ для визуализации инфарктов и некрозов.
Аналогичные трудности существуют и с соединениями, применяемыми для диагностики тромбов или артериосклеротических бляшек: нет соединений, которые можно было бы достаточно эффективно применять в качестве контрастных веществ для визуализации тромбов или артериосклеротических бляшек и которые, в то же время, характеризовались бы исключительно хорошей переносимостью.
Исходя из вышеизложенного, цель изобретения заключалась в том, чтобы предложить контрастные вещества, которые с одной стороны имели бы исключительно хорошие визуализирующие свойства в качестве контрастных веществ для МРТ, и являлись бы пригодными, в частности, для визуализации опухолей и некрозов, и/или лимфографии, и/или визуализации пулов крови, и/или для распознавания тромбов или артериосклеротических бляшек, и, в то же время, характеризовались бы исключительно хорошей переносимостью.
Цель изобретения достигается с помощью хелатов металлов, включающих
a) по меньшей мере один перфторированный ПЭГ радикал, и
b) по меньшей мере один хелатирующий радикал, и
c) по меньшей мере один эквивалент иона металла порядкового номера 21-29, 31-33, 37-39, 42-44, 49 или 57-83 и их соли.
В предпочтительном варианте осуществления изобретения, хелаты металлов содержат перфторированный ПЭГ радикал и хелатирующий радикал.
В другом предпочтительном варианте осуществления изобретения, хелаты металлов содержат перфторированный ПЭГ радикал и 2 хелатирующих радикала.
В особенно предпочтительном варианте осуществления изобретения, настоящее изобретение относится к хелатам металлов согласно формулы I:
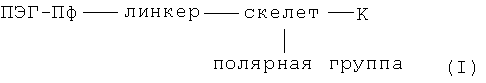
где
Таким же образом, изобретение дополнительно охватывает промежуточные соединения для получения вышеупомянутых хелатов металлов, где промежуточные соединения содержат
a) по меньшей мере один перфторированный ПЭГ радикал, и
b) по меньшей мере один хелатирующий радикал, где перфторированный ПЭГ радикал и хелатирующий радикал имеют указанные выше значения и при условии, что промежуточные соединения не содержат эквивалент иона металла порядкового номера 21-29, 31-33, 37-39, 42-44, 49 или 57-83.
Предпочтительными промежуточными соединениями для получения вышеупомянутых хелатов металлов согласно формулы I являются соединения, изображаемые формулой Ia:
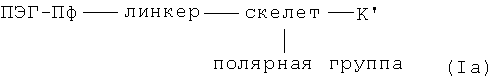
где
Особенно предпочтительные варианты промежуточных соединений соответствуют предпочтительным вариантам хелатов металлов при условии, что промежуточные соединения не заняты эквивалентом иона металла порядкового номера 21-29, 31-33, 37-39, 42-44, 49 или 57-83.
Предпочтительные варианты перфторированного ПЭГ радикала хелатов металлов и промежуточных соединений в соответствии с изобретением
В предпочтительном варианте осуществления изобретения, хелаты металлов и промежуточные соединения содержат перфторированный ПЭГ радикал, имеющий 4-30 С атомов, в частности 4-20 С-атомов.
В особенно предпочтительном варианте осуществления изобретения, перфторированный ПЭГ радикал является линейным. В частности, линейные перфторированные ПЭГ радикалы, имеющие 6-12 С атомов, являются предпочтительными, а наиболее предпочтительны имеющие 7, 8, 9, 10, или 11 С атомов.
В другом особенно предпочтительном варианте осуществления изобретения, перфторированный ПЭГ радикал является разветвленным. В частности, разветвленные перфторированные ПЭГ радикалы, имеющие 8-16 С атомов, являются предпочтительными, а наиболее предпочтительны имеющие 9, 10, 11, 12, 13, или 14 С атомов.
В наиболее предпочтительном варианте осуществления изобретения, ПЭГ радикал имеет следующую формулу XXI:
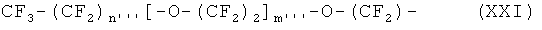
где
n''' означает целое число между 0 и 6, предпочтительно 0, 1, 2 или 3,
и m''' означает целое число между 1 и 14, предпочтительно 2 и 9, в частности предпочтительно 2, 3, 4 или 5.
Предпочтительные варианты хелатирующего радикала хелатов металлов и промежуточных соединений в соответствии с изобретением
В предпочтительном варианте осуществления изобретения, хелаты металлов и промежуточные соединения отличаются тем, что хелатирующий радикал является циклическим или имеет открытую цепь.
В особенно предпочтительном варианте осуществления изобретения, хелатирующий радикал является циклическим, в частности хелатирующий радикал представляет собой DOTA-радикал или его производное.
Наиболее предпочтительно, циклический хелатирующий радикал, имеющий комплексированный ион металла, выбирают из следующих радикалов:
- хелатирующий радикал общей формулы II:
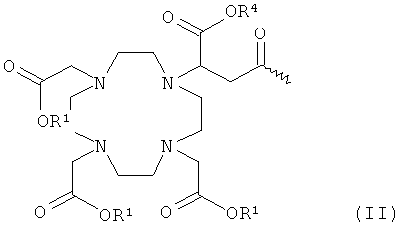
в которой
R1 означает атом водорода или эквивалент иона металла порядкового номера 21-29, 31-33, 37-39, 42-44, 49 или 57-83 при условии, что по меньшей мере два радикала R1 обозначают эквиваленты ионов металлов,
R4 означает водород или эквивалент иона металла, указанный для R1, и
U1 означает -С6Н4-O-СН2-ω- или группу -(СН2)р'-, где ω обозначает место присоединения к -СО- и р' означает целое число между 1 и 4;
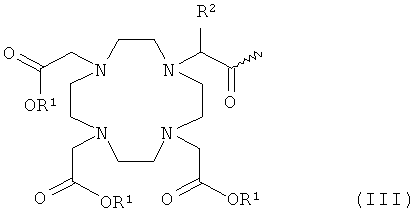
- хелатирующие радикалы общей формулы III:
где
R1 имеет указанное выше значение, и
R2 означает водород, С1-С7-алкил, бензил, фенил, -CH2OH или -СН2ОСН3;
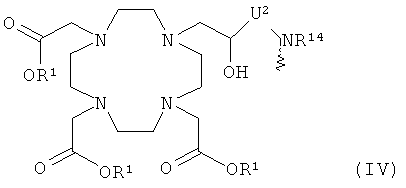
- хелатирующий радикал общей формулы IV:
в которой
R1 имеет указанное выше значение,
R14 означает Н или C1-C4алкил, и
U2 означает неразветвленную или разветвленную, насыщенную или ненасыщенную C1-С20 алкиленовую группу, которая необязательно включает имино, фениленовые, фениленокси, фениленимино, амидные, гидразидные, карбонильные или сложноэфирные группы, атом(-ы) кислорода, серы и/или азота, и является необязательно замещенной гидроксильными, меркапто, оксо, тиоксо, карбоксильными, карбоксиалкильными, сложноэфирными и/или аминогруппой(-ами);
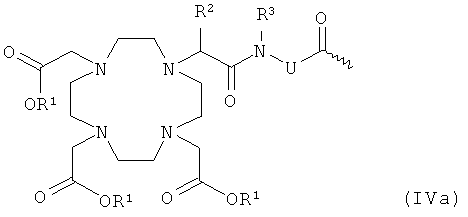
- хелатирующий радикал общей формулы IVa:
в которой
R1 имеет указанное выше значение,
R2 и R3 независимо друг от друга означают водород, C1-С7-алкил, бензил, фенил, -CH2OH или -CH2OCH3, и
U означает -С6Н4-O-СН2-ω-, -(CH2)1-5-ω, фениленовую группу, -СН2-NHCO-СН2-СН(СН2СООН)-С6Н4-ω-, -С6Н4-(ОСН2СН2)0-1-N(CH2COOH)-CH2-ω или С1-С12-алкиленовую или -(СН2)7-12-С6Н4-O-группу, необязательно прерванную одним или несколькими атомами кислорода, 1-3 -NHCO-, 1-3 -CONH группами и/или замещенную 1-3 -(СН2)0-5СООН группами, где ω обозначает место присоединения к -СО-;
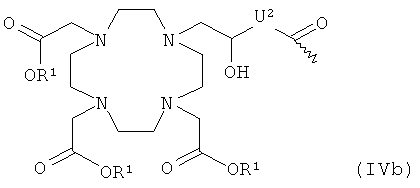
- хелатирующий радикал общей формулы IVb:
в которой
R1 и U2 имеют указанные выше значения;
где свободные кислотные группы, необязательно присутствующие в хелатирующем радикале, могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот, или амидов аминокислот.
Радикал U в хелате К общей формулы IVa предпочтительно означает -СН2- или С6Н4-O-СН2-ω, где ω обозначает место присоединения к -СО-.
В дальнейшем предпочтительном варианте осуществления изобретения, хелатирующий радикал имеет открытую цепь, в частности представляет собой DTPA радикал или его производное, или хелатор на основе катехоламида (САМ), терефталамида (ТАМ), гидроксипиридона (НОРО) и/или гидроксипиримидона (HOPY) или их производных.
В частности, хелатирующий радикал с открытой цепью, имеющий комплексированный ион металла, выбирают из следующих радикалов:
- хелатирующие радикалы общей формулы Va или Vb:
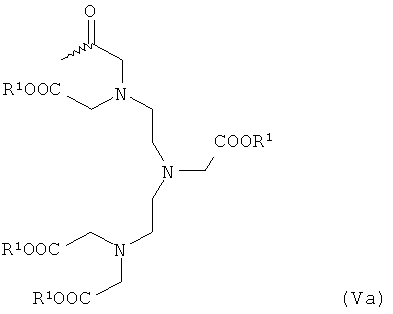
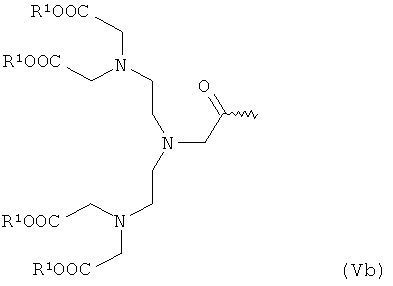
в которой R1 имеет указанное выше значение,
- хелатирующие радикалы общей формулы VI:
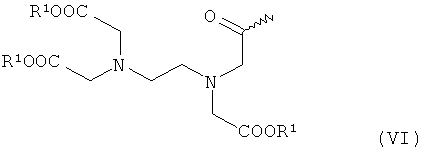
в которой R1 имеет указанное выше значение,
- хелатирующие радикалы общей формулы VII:
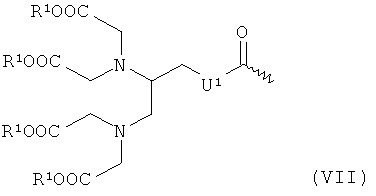
в которой R1 и U1 имеют указанные выше значения, где ω обозначает место присоединения к -СО-;
- хелатирующие радикалы общей формулы VIII:
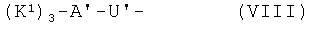 ,
,
в которой К1 независимо друг от друга означают радикал
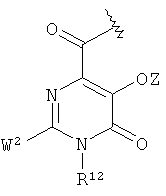 ,
, 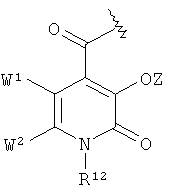 или
или 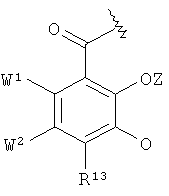
и в которой
Z имеет значение R1,
R12 означает атом водорода или неразветвленный или
разветвленный, насыщенный или ненасыщенный C1-10-алкильный радикал, который является необязательно прерванным 1-3 атомами кислорода, 1-3 атомами азота, 1-2 -CONH- и/или 1-3 -NR5-радикалами, является необязательно замещенным 1-4 гидроксильными группами, 1-2 карбоксильными группами (которые необязательно присутствуют в защищенной форме), 1-2 -SO3H группами (которые необязательно присутствуют в защищенной форме), 1-2 -РО3Н2 группами и/или 1-2 атомами галогена и/или в котором необязательно 1-2 атомов углерода присутствуют в виде карбонильных групп, где алкильный радикал или часть алкильного радикала может находиться в циклической форме,
R13 означает атом водорода, неразветвленный или разветвленный, насыщенный или ненасыщенный С1-10-алкильный радикал, который является необязательно прерванным 1-3 атомами кислорода, 1-3 атомами азота и/или 1-3 -NR5- радикалами, является необязательно замещенным 1-2 гидроксильными группами, 1-2 карбоксильными группами, 1-2 -SO3H группами, 1-2 -РО3Н2 группами и/или 1-2 атомами галогена и/или в котором необязательно 1-2 атомов углерода присутствуют в виде карбонильной группы, где алкильный радикал или часть алкильного радикала может находиться в циклической форме, -СООН, галоген, -CONR5R6, -SO3H или -РО3Н2,
R5 и R6 независимо друг от друга означают атом водорода или неразветвленный, разветвленный или циклический, насыщенный или ненасыщенный C1-10-алкильный радикал, который является необязательно замещенным 1-4 гидроксильными группами или прерван 1-2 атомами кислорода,
W1 и W2 независимо друг от друга означают радикал R1 или -CONR5R6,
A' означает радикал
 или
или 
где положения α изображают места связи к радикалу К1 и положения β изображают места связи к радикалу U', и
U' означает прямую связь или неразветвленный, разветвленный или циклический, насыщенный или ненасыщенный C1-20-алкиленовый радикал, который необязательно прерван 1-3 атомами кислорода, 1-3 атомами серы, 1-3 атомами азота, 1-3 -NR5- радикалами, 1-3 -NHCO-радикалами, 1-3-CONH-радикалами, 1-2-СО-радикалами, 1-3-O-Р-(=O)(-ОН)-O- радикалами и/или 1-2 ариленовыми радикалами, необязательно замещенными 1-3 неразветвленными, разветвленными или циклическими, насыщенными или ненасыщенными C1-6-алкильными радикалами, 1-3 гидроксильными группами, 1-3 карбоксильными группами, 1-3 арильными группами, 1-3 атомами галогена и/или 1-3 -O-C1-6-алкильными группами, где алкильный радикал является неразветвленным, разветвленным или циклическим, насыщенным или ненасыщенным, и/или в котором необязательно 1-3 атомов углерода могут присутствовать в виде карбонильных групп, где алкиленовый радикал или часть алкиленового радикала может находиться в циклической форме, а 1-4 атом(-ов) углерода - в виде карбонильных групп,
- хелатирующий радикал, включающий каркасный радикал, присоединенный к 3 радикалам общей формулы IX:
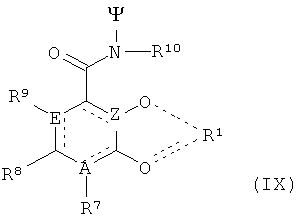
где
R7, R8 и R9 независимо друг от друга выбирают из Н, линейной или разветвленной C1-С6-алкильной группы, которая необязательно может быть прерванной 1-4 атомами кислорода, 1-4 атомами серы, 1-4 атомами азота, 1-4 -NR3-радикалами, 1-4 -NHCO- радикалами, 1-4 -CONH-радикалами, 1-2 -СО-радикалами, 1-4 -O-Р-(=O)(-ОН)-O-радикалами и/или 1-2 ариленовыми радикалами, является необязательно замещенной 1-3 неразветвленными, разветвленными или циклическими, насыщенными или ненасыщенными С1-10-алкильными радикалами, 1-3 гидроксильными группами, 1-3 карбоксильными группами, 1-3 арильными группами, 1-3 атомами галогена и/или 1-3 -O-C1-6-алкильными группами, где алкильный радикал является неразветвленным, разветвленным или циклическим, насыщенным или ненасыщенным, и/или в котором необязательно 1-3 атомов углерода могут присутствовать в виде карбонильных групп, где алкиленовый радикал или часть алкиленового радикал может находиться в циклической форме, замещенной или незамещенной арильной группы или аралкильной группы, замещенной или незамещенной C1-C6-гетероалкильной группы, или гидроксильной, карбоксильной, амидной, сложноэфирной и аминогруппы, где если А означает азот, то R7 может быть отличный от амино, и если Е означает азот, то R9 отсутствует, и
где один из 3 радикалов согласно формуле (IX) - R7, или R8, или R9 является двухвалентной группой, которая присоединяет хелатирующий радикал, имеющий комплексированный ион металла к скелету,
R10 означает группу, выбранную из Н, замещенной или незамещенной C1-С6-алкильной группы, замещенной или незамещенной арильной группы, замещенной или незамещенной С1-С6-гетероалкильной группы, или гидроксильной группы, карбоксильной группы, амидной группы и сложноэфирной группы, и
А, Е и Z независимо друг от друга выбирают из углерода и азота
Ψ означает связь к каркасу, и
для того чтобы образовывать хелатор в соответствии со смыслом настоящего изобретения, должны присутствовать по меньшей мере 3 радикала формулы (IX), причем эти 3 радикала могут быть одинаковыми или разными.
Предпочтительным каркасом является триэтиленаминовый радикал следующей формулы:
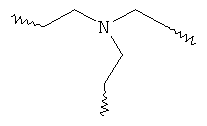
Хелатирующими радикалами, образующимися посредством его, являются TREN-производные.
Особенно предпочтительными хелатирующими радикалами являются TREN-бис-НОРО-ТАМ радикалы и их производные, TREN-трис-НОРО радикалы, ТРЕМ-бис-НОРО-НОРУ радикалы, TREN-трис-HOPY, TREN-бис-HOPY-TAM радикалы.
В предпочтительном варианте осуществления изобретения, в одном из 3 радикалов согласно формуле (IX) R7 означает двухвалентную группу, которая соединяет хелатирующий радикал с комплексированным ионом металла к скелету.
В особенно предпочтительном варианте осуществления изобретения, 3 радикала согласно формуле (IX) выбирают из следующих радикалов:
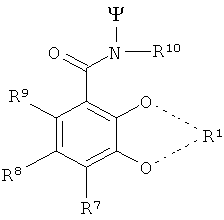
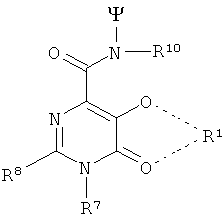
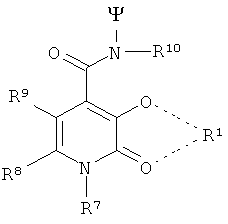
Особенно предпочтительными TREN-бис-НОРО-ТАМ радикалами являются радикалы следующей формулы:
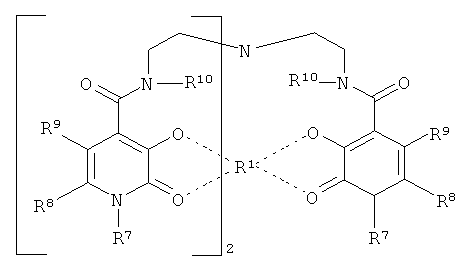
Особенно предпочтительными являются те TREN-бис-НОРО-ТАМ радикалы, в которых R7 в ТАМ-радикале означает двухвалентную группу, которая соединяет хелатирующий радикал с комплексированным ионом металла к скелету.
В особенно предпочтительном варианте осуществления изобретения, двухвалентной группой, которая соединяет хелатирующий радикал с комплексированным ионом металла к скелету, является группа -С(O)-.
В другом предпочтительном варианте осуществления изобретения, R8 и R9 независимо друг от друга означают Н или С1-С4-алкильные группы или C1-С6-гидроксиалкильные группы.
Особенно предпочтительными соединениями являются соединения, включающие хелатный радикал К общей формулы IVa.
В предпочтительном варианте осуществления изобретения, U2 означает С1-С6 алкиленовую цепь, которая необязательно прервана 1-2 -NHCO- группами и/или 1-1 O-атомами и которая может быть замещена 1-3 -ОН группами.
Радикал U2 в комплексе металла К особенно предпочтительно представляет собой
- линейную алкиленовую группу, имеющую 1-6 С атомов, в частности 2, 3 или 4 С атомов, или
- линейную алкиленовую группу, имеющую 1-6 С атомов, в частности 2, 3 или 4 С атомов, которая является прерванной 1 О атомом, или
- линейную алкиленовую группу, имеющую 1-6 С атомов, в частности 2, 3 или 4 С атомов, которая содержит -NHCO- группу.
В особенно предпочтительном варианте осуществления изобретения, U2 означает этиленовую группу.
Алкильные группы R2 и R3 в макроцикле общей формулы IVa могут быть прямыми или разветвленными. В качестве примера можно упомянуть метил, этил, пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил. Предпочтительно, R2 и R3 независимо друг от друга означают водород или C1-C4-алкил.
В наиболее предпочтительном варианте осуществления изобретения, R2 означает метил и R3 означает водород.
Бензильная группа или фенильная группа R2 или R3 в хелате К общей формулы IVa также может быть замещена в кольце.
Соединения формул VIII и IX в соответствии с изобретением включают радикалы катехола. Эти радикалы способствуют координированию иона металла или выравниванию зарядов. Вследствие этого Z означает или атом водорода, или эквивалент иона металла.
Радикал гидроксипиридинона или гидроксипиримидона, который может представлять собой К' в общей формуле VIII, в предпочтительном варианте осуществления изобретения несет заместитель R12, который означает атом водорода или неразветвленный или разветвленный, насыщенный или ненасыщенный C1-10-алкильный радикал, который необязательно прерван 1-3 атомами кислорода, 1-3 атомами азота и/или 1-3-NR5- радикалами, необязательно замещен 1-4 гидроксильными группами, 1-2 карбоксильными группами (которые необязательно присутствуют в защищенной форме), 1-2 -SO3H группами (которые необязательно присутствуют в защищенной форме), 1-2 -РО3Н2- группами и/или 1-2 атомами галогена и/или в котором необязательно 1-2 атомов углерода присутствуют в виде карбонильной группы, где алкильный радикал или часть алкильного радикала может находиться в циклической форме.
Предпочтительно, R12 означает атом водорода или неразветвленный или разветвленный, предпочтительно неразветвленный C1-5-алкильный радикал, который может быть прерван 1-2 атомами кислорода или 1-2 -CONH- группами и/или может быть необязательно замещен 1-4 гидроксильными группами, карбоксильной группой и/или группой -SO3H. Предпочтительными примерами R12 являются -Н, -CH2-CO-NH2, -СН3, -СН2-СН3, -СН2-СН2-СН3, -СН(СН3)-СН3, -С(СН3)(СН3)-СН3, -СН2-ОН, -СН2-СН2-ОН, -СН2-СН2-O-СН3, -СН2-СООН, -CH2-COOt-But, -СН2-СООСН2-С6Н5, -СН2-СН2-SO3H, -СН2-СН2-СН2-SO3H, -СН2-СН2-СН2-СН2-SO3H, -СН2-СН(ОН)-СН2-ОН, -СН2-СН2-O-СН2-СН2-O-СН3, -СН2-СН2-O-СН2-СН2-ОН, -СН2-СН2-O-СН2-СООН и -СН[СН2-O-СН-(СН2-ОН)2]2.
-Н, метоксиэтил, метил, -CH2-CO-NH2 и -СН2-СООН, в частности -СН2-СО-NH2, метоксиэтил и метил, являются особенно предпочтительными.
W1 и W2 независимо друг от друга означают радикал R12, где R12 принимает вышеприведенные значения и также включает ранее указанные предпочтительные радикалы. Особенно предпочтительно, W1 и W2 независимо означают атом водорода или неразветвленный или разветвленный, предпочтительно неразветвленный C1-5-алкильный радикал, в частности атом водорода или метильный радикал. Например, один из W1 и W2 может быть атомом водорода, а другой из W1 и W2 может быть метильным радикалом, или W1 и W2 оба могут быть атомом водорода.
Радикал катехола, которым альтернативно может быть К' в формуле VIII, несет заместитель R13. Последним может быть атом водорода, неразветвленный или разветвленный, насыщенный или ненасыщенный С1-10-алкильный радикал, который необязательно прерван 1-3 атомами кислорода, 1-3 атомами азота и/или 1-3 -NR5- радикалами, необязательно замещен 1-2 гидроксильными группами, 1-2 карбоксильными группами, 1-2 -SO3H группами, 1-2 -РО3Н2 группами и/или 1-2 атомами галогена и/или в котором необязательно 1-2 атомов углерода присутствуют в виде карбонильной группы, где алкильный радикал или часть алкильного радикала может находиться в циклической форме, -СООН, галоген, -CONR5R6, -SO3H или -PO3H2. Предпочтительными алкильными радикалами и алкильными радикалами, которые замещены и прерваны гетероатомами, в случае R13 являются те, что описаны выше для радикала R3. Фтор, хлор, бром и йод являются пригодными в качестве галогена.
Упомянутые ранее радикалы R5 и R6 независимо друг от друга означают атом водорода или неразветвленный, разветвленный или циклический, насыщенный или ненасыщенный C1-6-алкильный радикал, который является необязательно замещенным 1-2 гидроксильными группами. Пригодными C1-6-алкильными радикалами в случае R5 и R6 являются, в частности, метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, гексил, циклогексил, 2-гидроксиэтил и -СН[СН2-O-СН-(СН2-ОН)2]2.
В одном варианте осуществления настоящего изобретения, U' в формуле (VIII) означает фениленовый или циклогексиленовый радикал или неразветвленный, или разветвленный, насыщенный С1-10-алкиленовый радикал, который может быть прерван атомом кислорода,-NR5-радикалом, одним или двумя амидными радикалами и/или фениленовым радикалом и в котором один или два атома углерода могут присутствовать в виде карбонильных групп. Неразветвленный или разветвленный, насыщенный C1-4-алкиленовый радикал, в котором один или два атома углерода присутствуют в виде карбонильных групп, является наиболее предпочтительным.
Например, U' может быть выбран из группы, состоящей из -СН2-СН2-СО-, -CH2-CH2-CO-NH-CH2-CH2-CO-, -CH2-CO-NH-CH2-CO-, -СН(СН3)-СО-NH-CH2-CO-NH-CH2-CH2-CO-, -(СН2)4-СО-, -(СН2)4-NH-СО-СН2-СН2-СО- и -(CH2)4-NH-CO-CH2-O-CH2-CO-, где эти радикалы, в направлении чтения, слева связывают остаток А', а справа - скелет.
Радикалы формулы (VIII) и их получение известно из DE 102004062258.2.
Радикалы формулы (IX) и их получение известно из WO 03/016923.
Предпочтительные варианты линкера хелатов металлов в соответствии с изобретением (согласно формулы I) и промежуточных соединений (согласно формулы Ia)
В предпочтительном варианте осуществления изобретения, линкер представляет собой углеродную цепь, имеющую 1-15 С атомов, которая может быть линейной или разветвленном, насыщемной или ненасыщенной и которая необязательно прерывается 1-5 атомами кислорода, 1-3-NHCO-группами, 1-3-CONH-группами, 1-2 атомами серы, 1-4 -NH- группами и/или 1-2 фениленовыми группами, которые необязательно могут быть замещены 1-2 ОН группами, 1-2 NH2 группами, 1-2 -СООН группами или 1-2 -SO3H группами, и которая необязательно замещена 1-6 ОН группами, 1-5 -СООН группами (которые необязательно присутствуют в защищенной форме), 1-2 SO3H группами (которые необязательно присутствуют в защищенной форме), 1-3 NH2 группами и/или 1-3 С1-С4-алкокси группами.
В особенно предпочтительном варианте осуществления изобретения, линкер представляет собой группу формулы X:
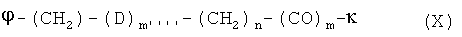
где
D означает О или S,
n означает целое число между 1 и 15,
m и m'''' независимо друг от друга означают или 0, или 1,
φ означает место присоединения линкера к ПЭГ-Пф, и
κ означает место присоединения линкера к скелету.
В предпочтительном варианте линкера согласно формуле X, m=0 и n=2-4, особенно предпочтительно n=2.
В дальнейшем предпочтительном варианте осуществления изобретения D означает кислород.
В другом предпочтительном варианте осуществления изобретения, m=1 и n=1-3.
Предпочтительно, m'''' означает 1.
Предпочтительные варианты скелета хелатов металлов в соответствии с изобретением (согласно формуле I) и промежуточных соединений (согласно формуле Ia)
В предпочтительном варианте осуществления изобретения, скелет представляет собой фосфор- и/или азотсодержащий радикал, особенно предпочтительно азотсодержащий радикал, наиболее предпочтительно азотсодержащий радикал, выбранный из: аминокислот, имеющих боковую функциональную цепь, таких как аспарагиновая кислота, глутаминовая кислота, серин, цистеин, орнитин, лизин и 2,4-диаминомасляная кислота, и алкилен-диаминовых радикалов и их производные, азота и 3,5-диаминобензойной кислоты.
В особенно предпочтительном варианте осуществления изобретения, скелет выбирают из следующих групп XIa-XIm:
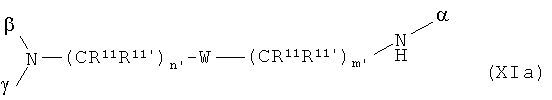
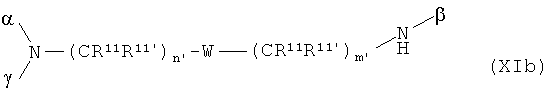
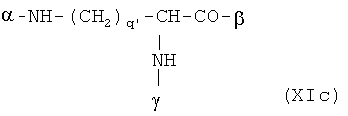
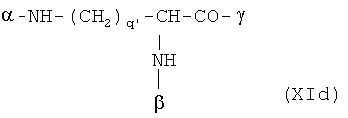
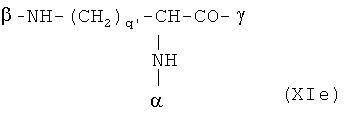
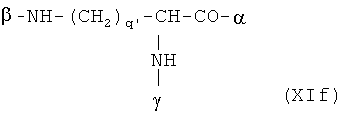
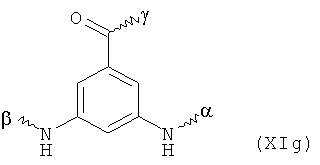
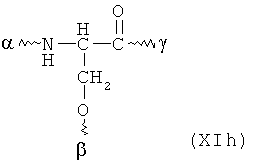
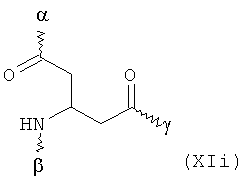
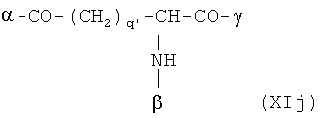
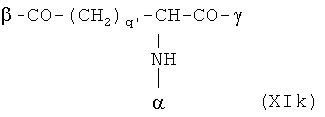
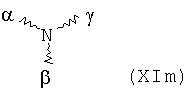
где
n' и m' независимо друг от друга означают целое число между 0 и 4, и m'+n' принимает значение ≥1, и
R11 и R11' независимо друг от друга означают или -H или -ОН, где когда m'+n'>1, каждая из групп -(CR11R11')- может быть одинаковой или разной, и
W означает или прямую связь, -O-, или фениленовую группу, которая необязательно может быть замещена 1-4 гидроксильными группами, и
q' принимает значение или 1, 2, 3, или 4,
где α означает место присоединения скелета к хелату К, β означает место присоединения скелета к полярной группе и γ означает место присоединения скелета к линкеру.
Предпочтительными хелатами металлов являются те, что имеют скелет (XIb), (XIc), (XIe) и (XIm).
Предпочтительные варианты полярной группы хелатов металлов в соответствии с изобретением (согласно формуле I) и промежуточных соединений (согласно формуле Ia)
В другом предпочтительном варианте осуществления изобретения, полярная группа представляет собой моносахаридный радикал, имеющий 5 или 6 С атомов или олигосахаридный радикал, предпочтительно глюкозу, маннозу, галактозу, рибозу, арабинозу, или ксилозу, или их дезоксисахара, такие как, например, 6-дезоксигалактоза (фукоза), или 6-дезоксиманноза (рамноза), или их пералкилированные производные. Глюкоза, манноза и галактоза, в частности манноза, являются особенно предпочтительными, причем моно- или олигосахаридный радикал присоединен к скелету через группу Q, где Q имеет значение группы, выбранной из:
δ-СО-(СН2)n''-ε,
δ-NH-(СН2)n''-ε, или
δ-(СН2)m''-ε
где
n'' означает целое число от 1 до 5, и
m'' означает целое число от 1 до 6, и
δ показывает место присоединения к скелету, и
ε обозначает место присоединения к моно- или олигосахаридному радикалу.
В другом предпочтительном варианте осуществления изобретения,
полярная группа означает радикал, выбранный из хелатных радикалов общих формул II-IX,
где
R1 здесь означает атом водорода или эквивалент иона металла порядкового номера 20-29, 31-33, 37-39, 42-44, 49 или 57-83, и
R2, R3, R4, R5, R6, R7, R8, R9, R10, R12, R13, K', A', U, U', U2, U1 и p'
имеют указанные выше значения,
или углеродную цепь, имеющую 1-30 С атомов, присоединенную к скелету через -СО-, -NR14- или прямую связь, которая может быть линейной или разветвленной, насыщенной или ненасыщенной, и которая необязательно прерывается 1-10 атомами кислорода, 1-5 -NHCO- группами, 1-5 -CONH- группами, 1-2 атомами серы, 1-5 -NH- группами или 1-2 фениленовыми группами, которые необязательно могут быть замещены 1-2 ОН группами, 1-2 NH2 группами, 1-2 -СООН группами или 1-2 -SO3H группами, и которая необязательно замещена 1-10 ОН группами, 1-5 -СООН группами, 1-2 SO3H группами, 1-5 NH2 группами, 1-5 С1-С4-алкокси группами,
R14 означает водород или C1-C4 алкил.
В особенно предпочтительном варианте осуществления настоящего изобретения, полярную группу выбирают из следующих радикалов:
-C(O)CH2O[(CH2)2O]pR'
-C(O)CH2OCH[CH2OCH(CH2OR')2]2
-C(O)CH2OCH2CH[CH2OCH(CH2OR')2]2
-R''N[(CH2)2O]pR'
-N{[(CH2)2O]pR'}2
-R''NCH2CH(OH)CH2OH
-N[CH2CH(OH)CH2OH]2
-R''NCH(CH2OH)CH(OH)CH2OH
-N[CH(CH2OH)CH(OH)CH2OH]2
-R''NCH[CH2OCH(CH2OR')2]2
-R''NCH2CH[CH2OCH(CH2OR')2]2
-R''NCH2CH2OCH[CH2OCH(CH2OR')2]2
-R''NCH2CH2OCH2CH[CH2OCH(CH2OR')2]2
-N{CH[CH2OCH(CH2OR')2]2}2
-N{CH2CH[CH2OCH(CH2OR')2]2}2
-R''NCH2CH(OH)CH(OH)CH(OH)CH(OH)CH2OH
-N[CH2CH(OH)CH(OH)CH(OH)CH(OH)CH2OH]2
или комплекса формулы (IVa),
где
R1, R2, R3 и U принимают значения, указанные выше для формулы (IVa),
р принимает значение или 1, 2, 3, 4, 5, 6, 7, 8, или 9, и
R' означает или -Н, или -СН3, и
R'' означает или Н, или С1-С4-алкильный радикал.
Предпочтительно, р принимает значение 1, 2, 3 или 4.
Упомянутые здесь полярные радикалы являются коммерческими продуктами или их получают в соответствии со способами, описанными в литературе: Cassel и др., Eur. J. Org. Chem., 2001, 5, 875-896; Whitessides и др., JACS, 1994, 5057-5062; Voegtle и др., Liebigs Ann. Chem., 1980, 858-862; Liu и др., Chem. Commun., 2002, 594; Mitchell и др., Heterocyclic Chem., 1984, 697-699; Bartsch и др., J. Org. Chem., 1984, 4076-4078; Keana и др., J. Org. Chem., 1983, 2647-2654.
В наиболее предпочтительном варианте осуществления изобретения, полярная группа означает присоединенный к скелету радикал формулы:
-C(O)CH2O[(CH2)2O]pR'
в которой
р и R' имеют указанные выше значения, где особенно предпочтительно R' означает группу -СН3.
Предпочтительные варианты ионов металлов хелатов металлов в соответствии с изобретением
Если соединение в соответствии с изобретением предназначено для применения при ЯМР-диагностике, то ион металла формирующей сигнал группы должен быть парамагнитным. К подобным ионам относятся прежде всего двух- и трехвалентные ионы элементов с порядковыми номерами 21-29, 42, 44 и 58-70. Пригодными ионами являются, например, ионы хрома(III), железа(II), кобальта(II), никеля(II), меди(II), празеодима(III), неодима(III), самария(III) и иттербия(III). Наиболее предпочтительны при этом с учетом их высокого магнитного момента ионы гадолиния(III), тербия(III), диспрозия(III), гольмия(III), эрбия(III), железа(III) и марганца(II).
Для применения соединений в соответствии с изобретением в медицинской радиологии (радиоизотопная диагностика и лучевая терапия) ион металла должен быть радиоактивным. Пригодными радиоизотопами являются, например, радиоизотопы элементов с порядковыми номерами 27, 29, 31-33, 37-39, 43, 49, 62, 64, 70, 75 и 77. Предпочтительными при этом являются технеций, галлий, индий, рений и иттрий.
Если соединение в соответствии с изобретением предназначено для применения при рентгенодиагностике, то в качестве иона металла предпочтительно использовать ион элемента с более высоким порядковым номером с целью обеспечить достаточно высокую степень поглощения рентгеновских лучей. Было установлено, что для этой цели пригодны диагностические средства, содержащие физиологически переносимую комплексную соль с ионами металлов порядковых номеров 25, 26 и 39, а также 57-83.
Предпочтительными при этом являются ионы марганца(II), железа(II), железа(III), празеодима(III), неодима(III), самария(III), гадолиния(III), иттербия(III) или висмута(III), прежде всего ионы диспрозия(III) и иттрия(III).
Необязательно присутствующие в R1 кислотные атомы водорода, т.е. атомы, которые не замещены центральным ионом, необязательно могут быть полностью или частично заменены на катионы неорганических и/или органических оснований или аминокислот либо амидов аминокислот. Пригодными неорганическими катионами являются, например, ион лития, ион калия, ион кальция и, в частности, ион натрия. Пригодными катионами органических оснований являются, среди прочего, таковые первичных, вторичных или третичных аминов, таких, например, как этаноламин, диэтаноламин, морфолин, глюкамин, N,N-диметилглюкамин и, в частности, N-метилглюкамин. Пригодными катионами аминокислот являются, например, катионы лизина, аргинина и орнитина, а также амиды в остальном кислых или нейтральных аминокислот.
Соединения в соответствии с изобретением являются особенно пригодными для применения при ЯМР- и рентгенодиагностике, радионуклидной диагностике и лучевой терапии и при МРТ лимфографии. Хелаты металлов, имеющие перфторированный ПЭГ радикал, являются, в частности, пригодными для применения в магнитно-резонансной томографии (МРТ) для распознавания различных физиологических и патофизиологических структур и, таким образом, для улучшения диагностической информации, например местоположения и тяжести заболевания, для выбора и оценки результата направленной терапии и для профилактики заболеваний и расстройств.
Подходящие заболевания и нарушения, с учетом возможного применения соединений согласно изобретению, включают опухолевые заболевания, в частности, касательно обнаружения и определения параметров первичных опухолей, отдаленных метастазов, метастазов в лимфоузлы и некрозов, сердечно-сосудистые заболевания, в частности, касательно определения изменения диаметра сосуда, такие как стенозы и аневризмы, артериосклероз, касательно обнаружения артериосклеротических бляшек, тромбоэмболические болезни, инфаркты, воспаления, в частности, артрит, остеомиелит, язвенный колит, а также повреждения нервов.
В особенно предпочтительном варианте осуществления изобретения, вещества в соответствии с изобретением применяют при МРТ лимфографии.
В другом особенно предпочтительном варианте, вещества в соответствии с изобретением применяют для визуализации пулов крови.
В особенно предпочтительном варианте осуществления изобретения, вещества в соответствии с изобретением применяют для визуализации некрозов или опухолей.
Изобретение также относится к фармацевтическим композициям, которые содержат по меньшей мере одно физиологически переносимое соединение в соответствии с изобретением, необязательно в сочетании с обычно используемыми в галеновых препаратах добавками.
Соединения настоящего изобретения отличаются исключительно хорошей переносимостью и, в то же время, исключительно хорошими визуализирующими свойствами. Таким образом они особенно пригодны для системного применения при МРТ, в частности при МРТ лимфографии и при визуализации опухолей. Соединения имеют исключительно хорошую системную переносимость.
Получение фармацевтических композиций в соответствии с изобретением ведут по известной технологии путем суспендирования или растворения комплексных соединений в соответствии с изобретением - при необходимости при добавлении обычно используемых в галеновых препаратах добавок - в водной среде, после чего полученную суспензию или полученный раствор необязательно подвергают стерилизации. Пригодными добавками являются, например, физиологически приемлемые буферы (такие как, например, трометамин), комплексообразователи либо слабые комплексы (такие как, например, диэтилентриаминпентауксусная кислота или Са-комплексы, соответствующие предлагаемым в изобретении комплексам металлов) или - при необходимости - электролиты, такие как, например, хлорид натрия, или - при необходимости - антиоксиданты, такие как, например, аскорбиновая кислота.
Если для энтерального, соответственно парентерального введения либо для иных целей необходимы суспензии или растворы композиций в соответствии с изобретением в воде или физиологическом солевом растворе, то их смешивают с одним либо несколькими обычно используемыми в галеновых препаратах вспомогательными веществами [например, с метилцеллюлозой, лактозой, маннитом] и/или с одним или несколькими поверхностно-активными веществами [например, с лецитином, Tween®, Myrj®] и/или с вкусовой(-ыми) добавкой(-ами) для коррекции вкуса [например, с эфирными маслами].
В принципе фармацевтические композиции в соответствии с изобретением можно также получать без выделения комплексов. Однако в любом случае в процессе образования хелатных соединений следует особенно тщательно соблюдать условия, которые практически полностью исключают возможность присутствия в предлагаемых в изобретении комплексах ионов металла, не образовавших комплекс и обладающих токсическим действием.
Это можно гарантировать, например, путем проведения контрольных титрований во время процесса получения, например, с помощью цветных индикаторов, таких как ксиленоловый оранжевый. В соответствии с этим изобретение относится также к способу получения комплексных соединений и их солей. В крайнем случае выделенный комплекс можно подвергать очистке.
В случае введения композиций в соответствии с изобретением in vivo, их можно вводить совместно с пригодным носителем, таким, например, как сыворотка или физиологический раствор поваренной соли, и совместно с другим белком, таким, например, как сывороточный альбумин человека (САЧ).
Композиции в соответствии с изобретением обычно вводятся парентерально, предпочтительно внутривенно. Вместе с тем, они могут также вводится внутрисосудисто или интерстициально/внутрикожно, в зависимости от исследуемого объекта - сосудов или тканей.
Фармацевтические композиции в соответствии с изобретением предпочтительно содержат от 0,1 мкмоль до 2 моль комплекса на л и предназначены для введения в дозах, составляющих, как правило, от 0,0001 до 5 ммоль/кг.
Композиции в соответствии с изобретением отвечают самым различным требованиям, которыми определяется их пригодность для применения в качестве контрастных веществ в магнитно-резонансной томографии. Такие средства позволяют за счет увеличения интенсивности сигнала после их перорального или парентерального введения повысить информативность изображения, полученного с помощью магнитно-резонансного томографа. Помимо этого они обладают высокой эффективностью, которая необходима для снижения концентрации вводимых в организм чужеродных веществ до минимально возможного уровня, и вместе с тем обладают исключительно хорошей переносимостью, которая необходима для сохранения неинвазивного характера диагностики.
Благодаря высокой степени растворимости композиций в соответствии с изобретением в воде и их малой осмоляльности появляется возможность получать на их основе высококонцентрированные растворы, что позволяет поддерживать объемную перегрузку системы кровообращения в допустимых пределах и компенсировать разбавление жидкостями организма. Более того, композиции в соответствии с изобретением обладают далее не только высокой стабильностью in vitro, но и проявляют неожиданно высокую стабильность in vivo, благодаря чему высвобождение или обмен связанных в комплексах ионов, которые в принципе являются токсичными, происходит лишь крайне медленно в течение того промежутка времени, за который новые контрастные вещества полностью выводятся из организма.
Обычно композиции в соответствии с изобретением при их использовании в качестве ЯМР-диагностикумов применяют в дозах от 0,0001 до 5 ммоль/кг, предпочтительно от 0,005 до 0,5 ммоль/кг.
Комплексные соединения в соответствии с изобретением могут, кроме того, эффективно применяться в качестве реагентов с магнитной восприимчивостью и реагентов сдвига в ЯМР-спектроскопии in vivo.
Композиции в соответствии с изобретением, благодаря оптимальным радиоактивным свойствам и высокой стабильности содержащихся в них комплексных соединений, пригодны также для применения в качестве диагностикумов при радионуклидной диагностике. Детали такого применения и дозировка описаны, например, в публикации "Radiotracers for Medical Applications", изд-во CRC-Press, Boca Raton, Florida.
Соединения и композиции в соответствии с изобретением могут применяться также в позитронно-эмиссионной томографии, где используют испускающие протоны изотопы, такие, например, как 43Sc, 44Sc, 52Fe, 55Со, 68Ga и 86Y (см. W.D.Heiss и М.Е.Phelps, Positron Emission Tomography of Brain, изд-во Springer Verlag, Berlin, Heidelberg, New York (1983)).
Соединения в соответствии с изобретением отличаются в особенности тем, что они полностью выводятся из организма и таким образом исключительно хорошо переносятся. Таким образом, могут задействоваться отличные визуализирующие свойства и неинвазивный характер диагностики может быть сохранен.
Поскольку соединения в соответствии с изобретением накапливаются в злокачественных опухолях (отсутствие диффузии в здоровые ткани, но высокая проницаемость опухолевых сосудов), их можно также применять в дополнение к лучевой терапии злокачественных опухолей. Отличие лучевой терапии от соответствующей диагностики состоит лишь в количестве и типе используемого изотопа. Целью при этом является разрушение опухолевых клеток под воздействием мощного коротковолнового излучения с предельно малым радиусом действия. При этом задействуется взаимодействие содержащихся в комплексных соединениях металлов (таких, например, как железо или гадолиний) с ионизирующим излучением (например, с рентгеновскими лучами) или с нейтронным излучением. Благодаря этому эффекту удается значительно повысить локальную дозу облучения в том месте, где находится комплекс металла (например, в опухолях). Для обеспечения такой же дозы облучения в злокачественной ткани применение подобных комплексов металлов позволяет существенно снизить дозу облучения здоровых тканей и предотвратить тем самым нежелательные для пациентов побочные действия. Поэтому конъюгаты комплексов металлов в соответствии с изобретением пригодны также для применения в качестве радиосенсибилизирующих веществ при лучевой терапии злокачественных опухолей (например, за счет использования эффектов Мессбауэра или при нейтронозахватной терапии). Пригодными испускающими β-излучение ионами являются, например, 46Sc, 47Sc, 48Sc, 72Ga, 73Ga и 90Y. Пригодными испускающими α-излучение ионами с малым периодом полураспада являются, например, 211Bi, 212Bi, 213Bi и 214Bi, предпочтителен из которых 212Bi. Пригодным испускающим протоны и электроны ионом является 158Gd, который можно получать из 157Gd путем захвата нейтронов.
Если композиция в соответствии с изобретением предназначена для применения при лучевой терапии в соответствии с методикой, предложенной R.L.Mills и др. (см. Nature, том 336 (1988), с.787), то центральный ион должен быть производным мессбауэровского изотопа, такого, например, как 57Fe или 151Eu.
В случае применения композиций в соответствии с изобретением in vivo их можно вводить совместно с пригодным для этой цели носителем, таким, например, как сыворотка или физиологический раствор поваренной соли, и совместно с другим белком, таким, например, как сывороточный альбумин человека. Дозировка при этом зависит от типа целлюлярного нарушения, используемого иона металла и типа метода визуализации.
Композиции в соответствии с изобретением обычно вводятся парентерально, предпочтительно внутривенно. Вместе с тем - как уже указывалось выше - они могут также вводиться внутрисосудисто или интерстициально/внутрикожно, в зависимости от исследуемого объекта - сосудов или тканей.
Композиции в соответствии с изобретением также пригодны для применения в качестве рентгеноконтрастных веществ, при этом особо следует отметить, что при их применении в биохимических/фармакологических исследованиях не наблюдается никаких признаков анафилактических реакций, известных для йодсодержащих контрастных веществ. Эти средства благодаря их оптимальным свойствам поглощать излучение в диапазоне более высоких напряжений на трубке особенно пригодны для применения в цифровой субтракционной технике.
В общем, композиции в соответствии с изобретением при их использовании в качестве рентгеноконтрастных веществ применяют аналогично, например, диатризоату меглумина в дозах от 0,1 до 5 ммоль/кг, предпочтительно от 0,25 до 1 ммоль/кг.
Выражение "эквивалент иона металла", используемый в данной заявке, является обычным и известным специалисту в области комплексной химии термином. Эквивалент иона металла означает эквивалент ионов металла, которые могут присоединяться, например, к карбоксилатным группам вместо водорода. Например, Gd3+ может присоединяться к 3-м карбоксилатным группам, то есть 1/3 Gd3+ соответствует эквиваленту иона металла R1 в формуле (II), (III), (IV), (IVa), (IVb), (Va), (Vb), (VI) или (VII), если металлом является гадолиний.
"ПЭГ радикал" в соответствии со смыслом настоящего изобретения означает одновалентный линейный или разветвленный алкильный радикал, имеющий вплоть до 30 С атомов и включающий по меньшей мере один этиленоксидный радикал. Предпочтительно, радикал является линейным. Предпочтительно, радикал содержит 1-14 этиленоксидных радикалов. Особенно предпочтительными ПЭГ радикалами являются те, в которых все присутствующие этиленоксидные радикалы имеют следующую формулу:
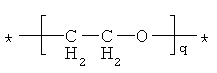
где q означает число этиленоксидных радикалов.
"Перфторированный ПЭГ радикал" в соответствии со смыслом настоящего изобретения означает одновалентный радикал, производный от ПЭГ радикала, причем радикал является перфторированным.
"Полярная группа" в соответствии со смыслом настоящего изобретения означает радикал, включающий функциональные группы, которые путем специфического распределения электронов вещества согласно изобретению дают значительный электрический дипольный момент. Такие группы являются причиной сродства к другим полярным химическим соединениям (см. также межмолекулярные силы) и они вследствие этого также являются ответственными за гидрофильные свойства веществ в соответствии с изобретением. Полярные радикалы имеют электрический дипольный момент и поляризованную ковалентную связь.
"TREN" в соответствии со смыслом настоящего изобретения является сокращением от трис(аминоэтил)амина.
"НОРО" в соответствии со смыслом настоящего изобретения является сокращением от гидроксипиридинона.
"HOPY" в соответствии со смыслом настоящего изобретения является сокращением от гидроксипиримидинона.
"ТАМ" в соответствии со смыслом настоящего изобретения является сокращением от терефталамида.
"Хелатор" в соответствии со смыслом настоящего изобретения означает вещество, образующее комплекс с по меньшей мере одним ионом металла с порядковым номером 21-29, 31-33, 37-39, 42-44, 49 или 57-83, имеющий константу стабильности по меньшей мере 1015, предпочтительно по меньшей мере 1018. Константу стабильности определяют, как описано в публикации (Martell, А.Е.; Motekaitis, R.J. The Determination and Use of Stability Constants, 2-e изд.; VCH: New York, 1992).
Примерное описание путей синтеза
Кроме того, изобритение относится к способу получения перфтор-ПЭГ-содержащих комплексов металлов общей формулы I
с К в значении комплекса металла одного из общих формул II, III, IVa, IVb, Va, Va, VI-VIII, и линкером, скелетом, полярной группой и ПЭГ-Пф, в указанном выше значении, отличающемуся тем, что известным по существу способом карбоновую кислоту общей формулы II
в которой R1 означает эквивалент иона металла порядкового номера 21-29, 31-33, 37-39, 42-44, 49 или 57-83 или защитную для карбоксила группу, и R4 и U1 имеют упомянутые выше значения,
или карбоновую кислоту общей формулы III
в которой R1 и R2 имеют упомянутые значения,
или карбоновую кислоту общей формулы IVa или IVb
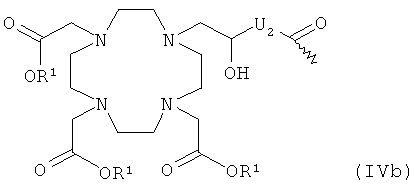
где R1, R2, R3 и U, U2 имеют упомянутые значения,
или карбоновую кислоту общей формулы Va или Vb
где R1 имеет указанное выше значение,
или карбоновую кислоту общей формулы VI
в которой R1 имеет упомянутое значение
или карбоновую кислоту общей формулы VII
в которой R1 и U1 имеют упомянутые значения,
или карбоновую кислоту общей формулы VIII
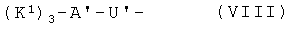
в которой К1 и А' имеют упомянутые значения,
и U' содержит концевой радикал - остаток карбоновой кислоты,
в необязательно активированной форме, вводят в реакцию сочетания с амином общей формулы XIIa
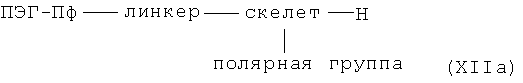
в которой линкер, скелет, полярная группа и ПЭГ-Пф имеют указанные выше значения, и, необязательно, затем удаляют необязательно присутствующие защитные группы с получением комплекса металла общей формулы I
или
если R1 означает защитную группу, после удаления этой защитной группы на последующей стадии вводят в реакцию известным по существу способом с по меньшей мере одним оксидом металла или солью металла элемента с порядковым номером 21-29, 31-33, 37-39, 42-44, 49 или 57-83, и впоследствии, при необходимости, необязательно присутствующие кислые атомы водорода замещают катионами неорганических и/или органических оснований, аминокислот или амидов аминокислот.
Этот способ получения комплексов металлов/амидов карбоновых кислот известен из DE 19652386.
Используемую в реакции сочетания смесь комплекса металла/карбоновой кислоты, которая содержит необязательно присутствующие карбоксильные и/или гидроксильные группы в защищенной форме, и по меньшей мере одного способствующего растворению вещества в количестве вплоть до 5, предпочтительно 0.5-2 молярных эквивалентов в пересчете на комплекс металла/карбоновой кислоты, можно получить или на дополнительной стадии реакции и выделять (например, путем упаривания, сублимационной сушки или распылительной сушки водного или смешивающегося с водой раствора компонентов или же путем осаждения из подобного раствора органическим растворителем) и затем подвергать взаимодействию в ДМСО с дегидратирующим реагентом и необязательно со вспомогательным агентом сочетания, или можно получать in situ, необязательно путем добавления способствующего(-их) растворению вещества(-в), дегидратирующего реагента и, необязательно, вспомогательного агента сочетания.
С целью предварительной обработки (активирования кислоты), реакционный раствор, приготовленный одним из этих способов, выдерживают в течение 1-24, предпочтительно 3-12 часов при температурах от 0 до 50°С, предпочтительно при комнатной температуре.
Далее прибавляют амин общей формулы XIIa
в которой линкер, скелет, полярная группа и ПЭГ-Пф имеют указанные выше значения, без растворителя или в растворенном виде, например, в диметилсульфоксиде, спиртах, таких как, например, метанол, этанол, изопропанол или их смеси, формамиде, диметилформамиде, воде или смесях указанных растворителей, предпочтительно в диметилсульфоксиде, в воде или в смешанных с водой растворителях. Для осуществления амидного сочетания, полученную таким путем реакционную смесь выдерживают при температурах от 0 до 70°С, предпочтительно от 30 до 60°С, в течение от 1 до 48, предпочтительно от 8 до 24 ч.
В некоторых случаях, как было установлено, амин в реакции предпочтительно использовать в виде его соли, например в виде гидробромида или гидрохлорида. Для высвобождения амина добавляют основание, такое как, например, триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, трипропиламин, трибутиламин, гидроксид лития, карбонат лития, гидроксид натрия или карбонат натрия.
После этого необязательные все еще присутствующие защитные группы отщепляют.
Выделение продукта реакции ведут в соответствии с методиками, известными специалисту в данной области техники, предпочтительно путем осаждения органическими растворителями, предпочтительно ацетоном, 2-бутаноном, диэтиловым эфиром, этиловым эфиром уксусной кислоты, метил-трет-бутиловым эфиром, изопропанолом или их смесями. Последующую очистку можно проводить, например, путем хроматографии, кристаллизации или ультрафильтрации.
Пригодными способствующими растворению веществами являются соли щелочных металлов, соли щелочноземельных металлов, соли триалкиламмония, соли тетраалкиламмония, мочевины, N-гидроксиимиды, гидроксиарилтриазолы, замещенные фенолы и соли гетероциклических аминов. В качестве примера можно назвать следующие вещества: хлорид лития, бромид лития, йодид лития, бромид натрия, йодид натрия, метансульфонат лития, метансульфонат натрия, п-толуолсульфонат лития, п-толуолсульфонат натрия, бромид калия, йодид калия, хлорид натрия, бромид магния, хлорид магния, йодид магния, п-толуолсульфонат тетраэтиламмония, п-толуолсульфонат тетраметиламмония, п-толуолсульфонат пиридиния, п-толуолсульфонат триэтиламмония, 2-морфолиноэтилсульфоновую кислоту, 4-нитрофенол, 3,5-динитрофенол, 2,4-дихлорфенол, N-гидроксисукцинимид, N-гидроксифталимид, мочевину, тетраметилмочевину, N-метилпирролидон, формамид, а также циклические мочевины, при этом пять указанных первыми соединений являются предпочтительными.
В качестве дегидратирующих реагентов можно использовать все известные специалистам средства. При этом в качестве примера можно назвать карбодиимиды и ониевые реагенты, такие как дициклогексилкарбодиимид (ДЦКД), 1-этил-3-(3-диметиламинопропил)карбодиимидгидроксихлорид (ЭДК), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (БОФ) и гексафторфосфат O-(бензотриазол-1-ил)-1,1,3,3-тетраметилурония (ГБТУ), предпочтительно ДЦКД.
Ниже в качестве примера представлены литературные источники, в которых описаны пригодные методы.
- Активирование карбоновых кислот. Обзор в Houben-Weyl, Methoden der Organischen Chemie [Методы органической химии], том XV/2, Georg Thieme Verlag Stuttgart, 1974 (и J.Chem. Research (S) 1996, 302).
- Активирование карбодиимидами. R.Schwyzer and H.Kappeler, Helv. 46: 1550 (1963).
- Е.Wünsch и др., В.100: 173 (1967).
- Активирование карбодиимидами/гидроксисукцинимидом: J. Am. Chem. Soc. 86: 1839 (1964) и J. Org. Chem. 53: 3583 (1988). Synthesis 453 (1972).
- Ангидридный метод, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин: В.Belleau и др., J. Am. Chem. Soc., 90: 1651 (1986), H.Kunz и др., Int. J.Pept. Prot. Res., 26: 493 (1985) и J.R.Voughn, Am. Soc. 73: 3547 (1951).
- Имидазолидный метод: B.F.Gisin, R.B.Menifield, D.C.Tosteon, Am. Soc. 91: 2691 (1969).
- Методы с хлорангидридами кислот, тионилхлорид: Helv., 42: 1653 (1959).
- Оксалилхлорид: J. Org. Chem., 29: 843 (1964).
Необязательно используемыми вспомогательными агентами сочетания являются все пригодные, известные специалисту в данной области агенты (Houben-Weyl, Methoden der organischen Chemie, том XV/2, Georg Thieme-Verlag, Stuttgart, 1974). В качестве примера при этом можно назвать 4-нитрофенол, N-гидроксисукцинимид, 1-гидроксибензотриазол, 1-гидрокси-7-азабензотриазол, 3,5-динитрофенол и пентафторфенол. Среди этих соединений предпочтительны 4-нитрофенол и N-гидроксисукцинимид; наиболее предпочтительным является первый из указанных реагентов.
Отщепление защитных групп осуществляют с помощью методов, известных специалисту в данной области, например путем гидролиза, гидрогенолиза, щелочного омыления сложных эфиров щелочью в водно-спиртовом растворе при температуре от 0 до 50°С, кислотного омыления минеральными кислотами или, в случае, например, сложных трет-бутиловых эфиров, с помощью трифторуксусной кислоты [Protective Groups in Organic Synthesis, 2-е изд., T.W.Greene и P.G.M. Wuts, John Wiley and Sons, Inc. New York, 1991], а в случае простых бензиловых эфиров - посредством водорода в присутствии палладия на угле.
Соединения в соответствии с изобретением общей формулы I
с К в значении комплекса металла общей формулы (IV) и линкером, скелетом, полярной группой и ПЭГ-Пф, в указанном выше значении, получают путем реакции сочетания амина общей формулы IV
в которой R1 означает эквивалент иона металла порядкового номера 21-29, 31-33, 37-39, 42-44, 49 или 57-83 или защитную для карбоксила группу, и U2 и R14 в указанном выше значении,
с необязательно активированной карбоновой кислотой общей формулы XIIb
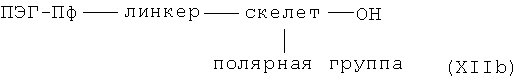
где линкер, скелет, полярная группа и ПЭГ-Пф имеют указанные выше значения,
и, необязательно, последующего удаления необязательно присутствующей защитной группы с получением комплекса металла общей формулы I
или
если R1 означает защитную группу, после удаления этой защитной группы, путем введения на последующей стадии в реакцию известным по существу способом с по меньшей мере одним оксидом металла или солью металла элемента с порядковым номером 21-29, 31-33, 37-39, 42-44, 49 или 57-83, и впоследствии, при необходимости, замещения необязательно присутствующих кислых атомов водорода катионами неорганических и/или органических оснований, аминокислот или амидов аминокислот.
Используемые карбоновые кислоты общих формул IIa-VIIa либо являются известными соединениями, либо их получают по описанным в примерах методам, см. DE 10040381 и DE 10040858. Так, в частности, получение карбоновых кислот общей формулы IIa известно из DE 19652386. Амины общей формулы IV можно получать, как описано в WO 95/17451.
Соединения общей формулы XIIa+b
со скелетом в значении
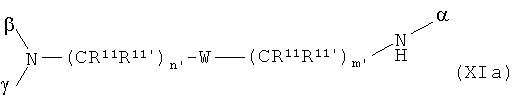
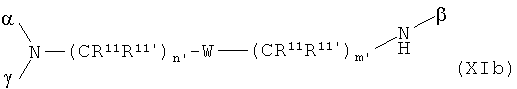
где α означает место присоединения скелета к хелату К, β означает место присоединения скелета к полярной группе и γ означает место присоединения скелета к линкеру, получают с помощью реакции гидрофильных карбоновых кислот R, описанных выше, согласно методам синтеза амидов, известных специалисту в данной области, с аминами общей формулы XIIIa
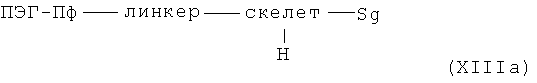
или в случае гидрофильных аминов R, описанных выше, согласно методам синтеза амидов, известных специалисту в данной области, с карбоновыми кислотами общей формулы XIIIb
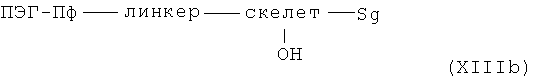
с Sg в значении защитной группы и линкером, скелетом и ПЭГ-Пф в указанном выше значении.
Отщепление защитных групп осуществляют с помощью методов, известных специалисту в данной области, например путем гидролиза, гидрогенолиза, щелочного омыления сложных эфиров щелочью в водно-спиртовом растворе при температурах от 0 до 50°С, кислотного омыления минеральными кислотами или, в случае, например, сложных трет-бутиловых эфиров, с помощью трифторуксусной кислоты [Protective Groups in Organic Synthesis, 2-е изд., T.W.Greene и P.G.M.Wuts, John Wiley and Sons, Inc. New York, 1991], а в случае простых бензиловых эфиров - посредством водорода в присутствии палладия на угле.
которые являются производными от соединений общей формулы XIIa+b,
со скелетом в значении
получают с помощью реакции дважды защищенных аминокислот общей формулы XIV
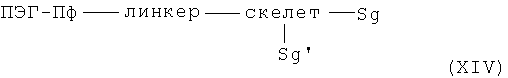
с Sg и Sg' в значении защитных групп, где Sg и Sg' могут быть отщеплены в разных условиях, и реакционным линкером, скелетом и ПЭГ-Пф в указанном выше значении.
Отщепление защитных групп осуществляют в соответствии с методами, известными специалисту в данной области и описанными выше.
Соединения общей формулы (XIV) получают с помощью реакции дважды защищенных аминокислот общей формулы XVa+b
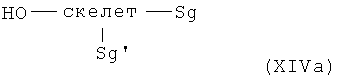
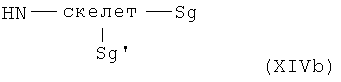
согласно методам синтеза амидов, известных специалисту в данной области, в случае (XVa) с аминами общей формулы XVIa,
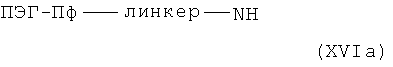
или, в случае (XVb) с кислотами общей формулы XVIb,
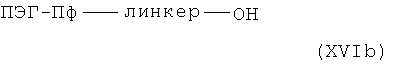
Такие дважды защищенные аминокислоты общей формулы (XVa+b) являются коммерческими продуктами (Bachem).
Соединения общей формулы XIIa
со скелетом в значении
получают с помощью реакции кислоты общей формулы XVIb
с гидрофильными первичными аминами R, описанными выше, согласно методам синтеза амидов, известных специалисту в данной области.
Соединения общей формулы XIIIa
которые являются производными от соединений общей формулы XIIa,
со скелетом в значении
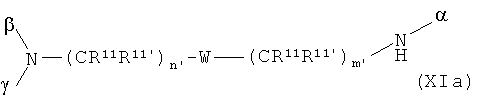
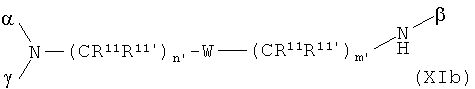
получают с помощью реакции монозащищенных диаминов общей формулы XVII
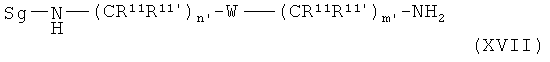
с R11, R11', n', W и m' в указанном выше значении и с Sg в значении защитной группы, с нуклеофилами общей формулы XVIc,
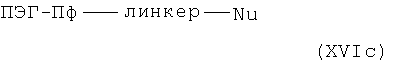
в которой Nu означает нуклеофуг, в присутствии основания и необязательно катализаторов межфазного переноса. В качестве нуклеофуга, радикалы -Cl, -Br, -I,
-OTs, -OMs, -OSO2CF3, -OSO2C4F9 или -OSO2C8F17, например, могут присутствовать в алкилирующем реагенте общей формулы XVIc.
Монозащищенные диамины общей формулы (XVII) известны из литературы и описаны в следующих публикациях
- Atwell и др., Synthesis, 1984, 1032-1033.
- Koenig и др., Eur. J. Org. Chem., 2002, 3004-3014.
- Boeijen и др., J. Org. Chem., 2001, 8454-8462.
- Spivak и др., J. Org. Chem., 1999, 4627-4634.
- Pittelkov и др., Synthesis, 2002, 2195-2202.
- Katchalski и др., J. Am. Chem. Soc., 1951, 1829.
- патент BASF AG, DE 1130803.
Кислоты общей формулы (XVIb) можно получить путем растворения спиртов общей формулы XIX
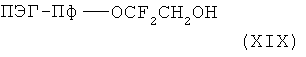
в несмешивающемся с водой органическом растворителе и взаимодействия с алкилирующим реагентом общей формулы (XX)
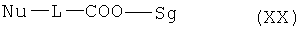 ,
,
в которой Nu означает нуклеофуг, L означает -(СН2)-z (где z=1-5), -CH2-CHOH-, или
-СН(СНОН-CH2OH)-СНОН-СНОН-, и Sg означает защитную группу,
в присутствии основания и необязательно катализатора межфазного переноса. В качестве нуклеофуга, радикалы -Cl, -Br, -I, -OTs, -OMs, -OSO2CF3, -OSO2C4F9 или
-OSO2C8F17, например, могут присутствовать в алкилирующем реагенте общей формулы XVIII.
Защитная группа представляет собой обычную защитную группу для кислоты. Эти защитные группы хорошо знакомы специалисту в данной области техники (Protective Groups in Organic Syntheses, второе изд., T.W.Greene and P.G.M. Wuts, John Wiley & Sons Inc., New York 1991).
Реакцию в соответствии с изобретением можно проводить при температурах 0-50°С, предпочтительно от 0°С до комнатной температуры. Время реакции составляет от 10 минут до 24 часов, предпочтительно от 20 минут до 12 часов.
Основание добавляют либо в твердом виде, предпочтительно в виде тонкоизмельченного порошка, либо в виде 10-70%-ного, предпочтительно 30-50%-ного водного раствора. Предпочтительными основаниями при этом являются NaOH и КОН.
В качестве органического не смешивающегося с водой растворителя в предлагаемом в изобретении методе алкилирования можно использовать, например, толуол, бензол, CF3-бензол, гексан, циклогексан, диэтиловый эфир, тетрагидрофуран, дихлорметан, МТБЭ или их смеси.
Межфазные катализаторы, используемые в способе в соответствии с изобретением, представляют собой известные по их применению в этих целях соли четвертичного аммониевого основания либо фосфония или, альтернативно, краун-эфиры, такие, например, как[15]-краун-5 или [18]-краун-6. Предпочтительны при этом соли четвертичного аммониевого основания с четырьмя идентичными или различными углеводородными группами у катиона, выбранными из метила, этила, пропила, изопропила, бутила и изобутила. Углеводородные группы у катиона должны быть достаточно большими для того, чтобы обеспечить хорошую растворимость алкилирующего агента в органическом растворителе. В соответствии с изобретением, особенно предпочтительно при этом использовать N(бутил)4 +-Cl-, N(бутил)4 +-HSO4 -, а также N(метил)4 +-Cl-.
Многочисленные примеры спиртов общей формулы (XIX) описаны в US 3293306.
Амины общей формулы (XVIa) могут быть получены согласно следующему способу: из соответствующей кислоты общей формулы (XVIb) путем реакции с первичными аминами или аммиаком согласно методам синтеза амидов, известных специалисту в данной области, и последующего восстановления, известным по существу способом, используя диборан или алюмогидрид лития.
Нуклеофилы общей формулы (XVIc) могут быть получены согласно следующему способу: из соответствующей кислоты общей формулы (XVIb) путем восстановления, известным по существу способом, используя DIBAL или алюмогидрид лития с получением соответствующих вторичных спиртов. Указанные спирты затем превращают в соответствующие нуклеофилы по реакции Мицунобу [О.Mitsunobu, Synthesisis, 1981, 1-28].
Благодаря их исключительно хорошей переносимости и их фармакокинетическим свойствам, таким как очень высокое содержание контрастного вещества в ранние моменты времени после введения и быстрое выведение с мочой, соединения в соответствии с изобретением являются особенно пригодными для распознавания полостей, содержащих кровь, например в качестве средства для распознавания пулов крови.
Примеры
Пример 1
а) 2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадекановая кислота
К 100 г (182.45 ммоль) 1Н,1Н,-3,6,9-триоксаперфтор-1-тридеканола (Apollo) и 20.5 г (365 ммоль) тонкоизмельченного гидроксида калия и каталитическому количеству (2 г) бисульфата тетра-н-бутиламмония в 800 мл толуола при 0°С добавляют 53.4 г (275 ммоль) трет-бутилбромацетата и смесь перемешивают при этой температуре в течение 2 ч и при комнатной температуре в течение 12 ч. Реакционный раствор обрабатывают 1500 мл этилацетата и 800 мл воды. Органическую фазу отделяют и дважды промывают водой порциями по 500 мл, затем сушат над сульфатом магния и упаривают досуха в вакууме. Остаток суспендируют в смеси, состоящей из 1200 мл метанола и 0.5 М раствора гидроксида натрия в соотношении 2:1, и затем нагревают при 60°С в течение 12 ч. Для выделения продукта реакции, реакционную смесь нейтрализуют с помощью обработки катионообменной смолой Amberlite IR 120 (H+ форма), отфильтровывают от ионита, упаривают досуха и хроматографируют на силикагеле (элюент: этилацетат/гексан 1:3).
Выход: 57.6 г (52% от теории) бесцветного воскообразного вещества.
b) N-метиламид(2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадекановой кислоты)
К 50 г (82.49 ммоль) соединения, указанного в заголовке примера 1а, в 500 мл дихлорметана добавляют 15.3 г (120 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 400 мл дихлорметана, через раствор при 0°С в течение приблизительно 2 ч пропускают газообразный метиламин и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 400 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 47.9 г (94% от теории) бесцветного воскообразного вещества.
с) N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)амин
45 г (72.68 ммоль) соединения, указанного в заголовке примера 1b, в 150 мл ТГФ обрабатывают 50 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 100 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 300 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 300 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 300 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 33.9 г (77% от теории) бесцветного масла.
d) [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амид 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина
К 18.82 г (50 ммоль) 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина (полученного в соответствии с ЕР 01/08498) и 30.31 г (50 ммоль) соединения, указанного в заголовке примера 1с, в 200 мл ТГФ при 0°С добавляют 24.7 г (100 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 38.6 г (80% от теории) бесцветного вязкого масла.
е) [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амид 6-N-бензилоксикарбонил-L-лизина
Через раствор 38 г (39.44 ммоль) соединения, указанного в заголовке примера 1d, в 250 мл этанола при 0°С в течение 1 ч пропускают газообразный аммиак, и смесь затем перемешивают при 0°С в течение 4 ч. Смесь упаривают досуха в вакууме и при перемешивании из воды осаждают остаток. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 34.3 г (98% от теории) аморфного твердого вещества.
f) [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)-маннопираноза]-L-лизина
К раствору 32.0 г (36.89 ммоль) соединения, указанного в заголовке примера 1е, и 22.09 г (36.89 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии с WO 99/01160 А1) и 4.25 г (36.89 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 9.51 г (46.11 ммоль) дициклогексилкарбодиимида и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 39.6 г (74% от теории) бесцветного вязкого масла.
g) [N-метил-(1H,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амид 2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
К раствору 38.0 г (26.24 ммоль) соединения, указанного в заголовке примера 1f, в 600 мл этанола добавляют 5.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 25.2 г (количественный) бесцветного твердого вещества.
h) Гадолиниевый комплекс 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизин-[N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амида
20 г (20.76 ммоль) соединения, указанного в заголовке примера 1g, 2.39 г (20.76 ммоль) N-гидроксисукцинимида, 1.76 г (41.52 ммоль) хлорида лития и 13.07 г (20.76 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) при слабом нагревании растворяют в 200 мл диметилсульфоксида. При 10°С добавляют 5.35 г (25.95 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 21.5 г (62% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.5%
Пример 2
а) N-2-(метоксиэтил)-амид(2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадекановой кислоты)
К 10 г (16.5 ммоль) соединения, указанного в заголовке примера 1а, в 100 мл дихлорметана добавляют 2.55 г (20 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 2.48 г (33 ммоль) 2-метоксиэтиламина (Aldrich) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 9.9 г (90% от теории) бесцветного воскообразного вещества.
b) N-2-метоксиэтил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-амин
9.5 г (14.32 ммоль) соединения, указанного в заголовке примера 2а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.5 г (91% от теории) бесцветного масла.
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
8 г (12.32 ммоль) соединения, указанного в заголовке примера 2b, 1.42 г (12.32 ммоль) N-гидроксисукцинимида, 1.04 г (24.64 ммоль) хлорида лития и 7.76 г (12.32 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 3.18 г (15.4 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 9.2 г (56% от теории) бесцветного твердого вещества. Содержание воды (по Карлу Фишеру): 5.8%
Пример 3
а) 2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридекановая кислота
К 100 г (251.21 ммоль) 1Н,1Н,-3,6,9-триоксаперфтор-1-деканола (Apollo) и 28.1 г (500 ммоль) тонкоизмельченного гидроксида калия и каталитическому количеству (2 г) бисульфата тетра-н-бутиламмония в 800 мл толуола при 0°С добавляют 72.8 г (375 ммоль) трет-бутилбромацетата и смесь перемешивают при этой температуре в течение 2 ч и при комнатной температуре в течение 12 ч. Реакционный раствор обрабатывают 1500 мл этилацетата и 800 мл воды. Органическую фазу отделяют и дважды промывают водой порциями по 500 мл, затем сушат над сульфатом магния и упаривают досуха в вакууме. Остаток суспендируют в смеси, состоящей из 1200 мл метанола и 0.5 М раствора гидроксида натрия в соотношении 2:1 и затем нагревают при 60°С в течение 12 ч. Для выделения продукта реакции, реакционную смесь нейтрализуют с помощью обработки катионообменной смолой Amberlite IR 120 (Н+ форма), отфильтровывают от ионита, упаривают досуха и хроматографируют на силикагеле (элюент: этилацетат/гексан 1:3).
Выход: 67.5 г (59% от теории) бесцветного масла.
b) N-метиламид(2H,2H,4H,4H,-3,6,9,12-тетраоксаперфтортридекановой кислоты)
К 40 г (87.70 ммоль) соединения, указанного в заголовке примера 3а, в 500 мл дихлорметана добавляют 15.3 г (120 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 400 мл дихлорметана, через раствор при 0°С в течение приблизительно 2 ч пропускают газообразный метиламин и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 400 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 35.4 г (86% от теории) бесцветного масла.
с) N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)амин
34 г (72.47 ммоль) соединения, указанного в заголовке примера 3b, в 150 мл ТГФ обрабатывают 50 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 100 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 300 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 300 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 300 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 27.9 г (85% от теории) бесцветного масла.
d) [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)]амид 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина
К 18.82 г (50 ммоль) 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина (полученного в соответствии с ЕР 01/08498) и 22.76 г (50 ммоль) соединения, указанного в заголовке примера 3с, в 200 мл ТГФ при 0°С добавляют 24.7 г (100 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 31.7 г (78% от теории) бесцветного вязкого масла.
e) [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)]амид 6-N-бензилоксикарбонил-L-лизина
Через раствор 30 г (36.88 ммоль) соединения, указанного в заголовке примера 3d, в 250 мл этанола при 0°С в течение 1 ч пропускают газообразный аммиак, и смесь затем перемешивают при 0°С в течение 4 ч. Смесь упаривают досуха в вакууме и остаток осаждают из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 25.2 г (95% от теории) аморфного твердого вещества.
f) [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)-маннопираноза]-L-лизина
К раствору 24.0 г (33.45 ммоль) соединения, указанного в заголовке примера 3е, и 20.03 г (33.45 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии с WO 99/01160 А1) и 3.85 г (33.45 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 8.63 г (41.81 ммоль) дициклогексилкарбодиимида и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 34.2 г (79% от теории) бесцветного вязкого масла.
g) [N-метил-(1Н,1Н,2Н,2Н,-4Н,4Н,3,6,9,12-тетраоксаперфтортридецил)]амид2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
К раствору 33.0 г (25.42 ммоль) соединения, указанного в заголовке примера 3f, в 600 мл этанола добавляют 5.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 20.6 г (количественный) бесцветного твердого вещества.
h) Гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)]амида 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
15 г (18.67 ммоль) соединения, указанного в заголовке примера 3g, 2.15 г (18.67 ммоль) N-гидроксисукцинимида, 1.58 г (37.34 ммоль) хлорида лития и 11.76 г (18.67 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 4.82 г (23.34 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 15.6 г (55% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.0%
Пример 4
а) N-2-(метоксиэтил)-амид(2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридекановой кислоты)
К 10 г (21.92 ммоль) соединения, указанного в заголовке примера 3а, в 100 мл дихлорметана добавляют 3.19 г (25 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 3.29 г (43.84 ммоль) 2-метоксиэтиламина (Aldrich) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 9.5 г (84% от теории) бесцветного воскообразного вещества.
b) N-2-метоксиэтил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)амин
9.0 г (17.54 ммоль) соединения, указанного в заголовке примера 4а, в 50 мл 15 ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают 20 при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 25 10:1).
Выход: 7.5 г (86% от теории) бесцветного масла.
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
7.0 г (14.02 ммоль) соединения, указанного в заголовке примера 4b, 1.61 г (14.02 ммоль) N-гидроксисукцинимида, 1.19 г (28.04 ммоль) хлорида лития и 8.83 г (14.02 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 3.62 г (17.53 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 8.5 г (51% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.7%
Пример 5
а) 1Н,1Н,-3,6,9-триокса-2,5,8-триметилперфтордодекан-1-ол
К 100 г (150.58 ммоль) 3,6,9-триокса-2,5,8-триметилперфтортридеканоилфторида (Oakwood) в 500 мл диоксана добавляют 3.76 г (99.4 ммоль) борогидрида натрия и смесь перемешивают при 60°С в течение 2 ч. Реакционный раствор выливают в 500 мл воды со льдом, и трижды экстрагируют диэтиловым эфиром порциями по 300 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и хроматографируют на силикагеле (элюент: этилацетат/гексан 1:2).
Выход: 83.1 г (85% от теории) бесцветного масла.
b) 2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадекановая кислота
К 50 г (77.15 ммоль) соединения, указанного в заголовке примера 5а, и 8.5 г (152 ммоль) тонкоизмельченного гидроксида калия и каталитическому количеству (1 г) бисульфата тетра-н-бутиламмония в 400 мл толуола при 0°С добавляют 22.3 г (115 ммоль) трет-бутилбромацетата и смесь перемешивают при этой температуре в течение 2 ч и при комнатной температуре в течение 12 ч. Реакционный раствор обрабатывают 1000 мл этилацетата и 500 мл воды. Органическую фазу отделяют и дважды промывают водой порциями по 300 мл, затем сушат над сульфатом магния и упаривают досуха в вакууме. Остаток суспендируют в смеси, состоящей из 800 мл метанола и 0.5 М раствора гидроксида натрия в соотношении 2:1 и затем нагревают при 60°С в течение 12 ч. Для выделения продукта реакции, реакционную смесь нейтрализуют с помощью обработки катионообменной смолой Amberlite IR 120 (Н+ форма), отфильтровывают от ионита, упаривают досуха и хроматографируют на силикагеле (элюент: этилацетат/гексан 1:3).
Выход: 24.0 г (44% от теории) бесцветного воскообразного вещества.
с) N-метиламид(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфтор-пентадекановой кислоты)
К 21 г (29.74 ммоль) соединения, указанного в заголовке примера 5b, в 200 мл дихлорметана добавляют 5.1 г (40 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 200 мл дихлорметана, через раствор при 0°С в течение приблизительно 2 ч пропускают газообразный метиламин и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 200 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 17.3 г (81% от теории) бесцветного воскообразного вещества.
d) N-метил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)-амин
16.5 г (22.94 ммоль) соединения, указанного в заголовке примера 5с, в 50 мл ТГФ обрабатывают 20 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 30 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/15 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 13.6 г (84% от теории) бесцветного воскообразного вещества.
e) [N-метил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)]амид 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина
К 7.53 г (20 ммоль) 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина (полученного в соответствии с ЕР 01/08498) и 14.10 г (20 ммоль) соединения, указанного в заголовке примера 5d, в 200 мл ТГФ при 0°С добавляют 9.88 г (40 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 17.5 г (82% от теории) аморфного твердого вещества.
f) [N-метил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)]амид 6-N-бензилоксикарбонил-L-лизина
Через раствор 17 г (15.98 ммоль) соединения, указанного в заголовке примера 5е, в 100 мл этанола при 0°С в течение 1 ч пропускают газообразный аммиак, и смесь затем перемешивают при 0°С в течение 4 ч. Смесь упаривают досуха в вакууме и остаток осаждают из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 14.9 г (97% от теории) аморфного твердого вещества.
g) [N-метил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)-маннопираноза]-L-лизина
К раствору 14.3 г (14.78 ммоль) соединения, указанного в заголовке примера 5f, и 8.85 г (14.78 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии c WO 99/01160 А1) и 1.70 г (14.78 ммоль) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 3.81 г (18.48 ммоль) дициклогексилкарбодиимида, смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 18.8 г (82% от теории) бесцветного вязкого масла.
h) [N-метил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)]амид 2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
К раствору 18.0 г (11.63 ммоль) соединения, указанного в заголовке примера 5g, в 300 мл этанола добавляют 3.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 12.4 г (количественный) бесцветного твердого вещества.
i) Гадолиниевый комплекс [N-метил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)]амида 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
11.8 г (11.20 ммоль) соединения, указанного в заголовке примера 5h, 1.29 г (11.20 ммоль) N-гидроксисукцинимида, 0.95 г (22.40 ммоль) хлорида лития и 7.05 г (11.20 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Sobering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.89 г (14.00 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 11.7 г (58% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.4%
Пример 6
а) N-2-(метоксиэтил)амид(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфтор-пентадекановой кислоты)
К 10 г (14.16 ммоль) соединения, указанного в заголовке примера 5b, в 100 мл дихлорметана добавляют 2.55 г (20 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 2.13 г (28.32 ммоль) 2-метоксиэтиламина (Aldrich) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюентэтилацетат/гексан 1:1).
Выход: 10.1 г (93% от теории) бесцветного воскообразного вещества.
b) N-2-метоксиэтил-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)амин
9.5 г (12.45 ммоль) соединения, указанного в заголовке примера 6а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.2 г (88% от теории) бесцветного масла.
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(2Н,2Н,4Н,4Н,-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
7.6 г (10.14 ммоль) соединения, указанного в заголовке примера 6b, 1.17 г (10.14 ммоль) N-гидроксисукцинимида, 0.86 г (20.28 ммоль) хлорида лития и 6.39 г (10.14 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.62 г (12.68 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 7.7 г (52% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.0%
Пример 7
а) 1Н,1Н,-3,6,9,12-тетраокса-2,5,8,11-тетраметилперфторпентадекан-1-ол
К 100 г (120.46 ммоль) 3,6,9,12-тетраокса-2,5,8,11-тетраметилперфтортридеканоилфторида (Oakwood) в 500 мл диоксана добавляют 3.01 г (79.5 ммоль) борогидрида натрия и смесь перемешивают при 60°С в течение 2 ч. Реакционный раствор выливают в 500 мл воды со льдом, и трижды экстрагируют диэтиловым эфиром порциями по 300 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и хроматографируют на силикагеле (элюент: этилацетат/гексан 1:2).
Выход: 87.3 г (89% от теории) бесцветного масла.
b) 3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадекановая кислота
К 50 г (61.41 ммоль) соединения, указанного в заголовке примера 7а, и 6.88 г (123 ммоль) тонкоизмельченного гидроксида калия и каталитическому количеству (1 г) бисульфат тетра-н-бутиламмония в 400 мл толуола при 0°С добавляют 17.8 г (92 ммоль) трет-бутилбромацетата и смесь перемешивают при этой температуре в течение 2 ч и при комнатной температуре в течение 12 ч. Реакционный раствор обрабатывают 1000 мл этилацетата и 500 мл воды. Органическую фазу отделяют и дважды промывают водой порциями по 300 мл, затем сушат над сульфатом магния и упаривают досуха в вакууме. Остаток суспендируют в смеси, состоящей из 800 мл метанола и 0.5 М раствора гидроксида натрия в соотношении 2:1 и затем нагревают при 60°С в течение 12 ч. Для выделения продукта реакции, реакционную смесь нейтрализуют с помощью обработки катионообменной смолой Amberlite IR 120 (H+ форма), отфильтровывают от ионита, упаривают досуха и хроматографируют на силикагеле (элюент: этилацетат/гексан 1:3).
Выход: 20.9 г (39% от теории) бесцветного воскообразного вещества.
с) N-метиламид (3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадекановой кислоты)
К 20 г (22.93 ммоль) соединения, указанного в заголовке примера 7b, в 200 мл дихлорметана добавляют 5.1 г (40 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 200 мл дихлорметана, через раствор при 0°С в течение приблизительно 2 ч пропускают газообразный метиламин и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 200 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 15.0 г (74% от теории) бесцветного воскообразного вещества.
d) N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)амин
14.5 г (16.38 ммоль) соединения, указанного в заголовке примера 7с, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и смесь нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 30 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/15 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 12.6 г (88% от теории) бесцветного воскообразного вещества.
е) [N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)]амид 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина
К 3.76 г (10 ммоль) 6-N-бензилоксикарбонил-2-N-трифторацетил-L-лизина (полученного в соответствии с ЕР 01/08498) и 8.71 г (10 ммоль) соединения, указанного в заголовке примера 7d, в 200 мл ТГФ при 0°С добавляют 4.94 г (20 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 10.5 г (85% от теории) аморфного твердого вещества.
f) [N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)]амид 6-N-бензилоксикарбонил-L-лизина
Через раствор 10 г (8.13 ммоль) соединения, указанного в заголовке примера 7е, в 100 мл этанола при 0°С в течение 1 ч пропускают газообразный аммиак, и смесь затем перемешивают при 0°С в течение 4 ч. Смесь упаривают досуха в вакууме и остаток осаждают из воды путем перемешивания. Твердое вещество отфильтровывают и сушат в вакууме при 50°С.
Выход: 9.1 г (99% от теории) аморфного твердого вещества.
g) [N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)]амид 6-N-бензилоксикарбонил-2-N-[1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)-маннопираноза]-L-лизина
К раствору 8.5 г (7.50 ммоль) соединения, указанного в заголовке примера 7f, и 4.49 г (7.50 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии c WO 99/01160 А1) и 863 мг (7.50 ммоль) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 1.93 г (9.38 ммоль) дициклогексилкарбодиимида и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 10.8 г (84% от теории) бесцветного вязкого масла.
h) [N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)]амид 2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
К раствору 10.2 г (5.95 ммоль) соединения, указанного в заголовке примера 7g, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 7.3 г (количественный) бесцветного твердого вещества.
i) Гадолиниевый комплекс [N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)]амида 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
6.8 г (5.58 ммоль) соединения, указанного в заголовке примера 7h, 642 мг (5.58 ммоль) N-гидроксисукцинимида, 473 мг (11.16 ммоль) хлорида лития и 3.51 г (5.58 ммоль) 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана, гадолиниевый комплекс (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 100 мл диметилсульфоксида. При 10°С добавляют 1.44 г (6.98 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 7.1 г (64% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.5%
Пример 8
а) N-2-(метоксиэтил)амид (3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтор-октадекановой кислоты)
К 10 г (11.47 ммоль) соединения, указанного в заголовке примера 7b, в 100 мл дихлорметана добавляют 2.55 г (20 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 1.72 г (22.94 ммоль) 2-метоксиэтиламина (Aldrich) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 9.5 г (89% от теории) бесцветного воскообразного вещества.
b) N-2-метоксиэтил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)-амин
9.0 г (9.68 ммоль) соединения, указанного в заголовке примера 8а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и смесь нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола, и смесь перемешивают при комнатной температуре в течение 1 ч и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.1 г (91% от теории) бесцветного масла.
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
7.5 г (8.19 ммоль) соединения, указанного в заголовке примера 8b, 943 мг (8.19 ммоль) N-гидроксисукцинимида, 694 мг (16.38 ммоль) хлорида лития и 5.16 г (8.19 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.11 г (10.24 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 7.9 г (58% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.3%
Пример 9
а) 1-N-(бензилоксикарбонил)-1Н,1Н,2Н,2Н,5Н,5Н,7Н,7Н-3-аза-4-оксо-6,9,12,15-тетраоксаперфторгексадециламин
К 10 г (21.92 ммоль) соединения, указанного в заголовке примера 3а, в 100 мл дихлорметана добавляют 3.19 г (25 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 8.52 г (43.84 ммоль) N-бензилоксикарбонилэтилендиамина (Atwell и др., Synthesis, 1984, 1032-1033) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 11.0 г (79% от теории) бесцветного воскообразного вещества.
b) 1-N-(бензилоксикарбонил)-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н,7Н,7Н-3-аза-6,9,12,15-тетраоксаперфторгексадециламин
10.6 г (16.76 ммоль) соединения, указанного в заголовке примера 9а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола, перемешивают при комнатной температуре в течение 1 ч и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.4 г (81% от теории) бесцветного воскообразного вещества.
с) N-[2-(бензилоксикарбонил)аминоэтил-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)-2-[2-(2-метоксиэтокси)этокси]ацетамид
К раствору 8 г (12.94 ммоль) соединения, указанного в заголовке примера 9b, и 2.31 г (12.94 ммоль) [2-(2-метоксиэтокси)этокси]уксусной кислоты (Aldrich) и 1.49 г (12.94 ммоль) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 3.34 г (16.18 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 8.5 г (84% от теории) бесцветного вязкого масла.
d) N-2-(аминоэтил)-N-(1H,1H,2H,2H,4H,4H-3,6,9,12-тетраоксаперфтортридецил)-2-[2-(2-метоксиэтокси)этокси]ацетамид
К раствору 8.2 г (10.53 ммоль) соединения, указанного в заголовке примера 9с, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 6.8 г (количественный) бесцветного твердого вещества.
e) Гадолиниевый комплекс N-{[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-аминоэтил}-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)-2-[2-(2-метоксиэтокси)этокси]ацетамида
6.0 г (9.31 ммоль) соединения, указанного в заголовке примера 9d, 1.07 г (9.31 ммоль) N-гидроксисукцинимида, 789 мг (18.62 ммоль) хлорида лития и 5.86 г (9.31 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 100 мл диметилсульфоксида. При 10°С добавляют 2.4 г (11.64 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 8.2 г (66% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.1%
Пример 10
а) 1-N-(бензилоксикарбонил)-1Н,1Н,2Н,2Н,5Н,5Н,7Н,7Н-3-аза-4-оксо-6,9,12,15-тетраокса-8,11,14-триметилперфтороктадециламин
К 15 г (21.24 ммоль) соединения, указанного в заголовке примера 5b, в 100 мл дихлорметана добавляют 3.19 г (25 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 8.26 г (42.48 ммоль) N-бензилоксикарбонилэтилендиамина (Atwell и др., Synthesis, 1984, 1032-1033) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 13.9 г (74% от теории) бесцветного воскообразного вещества.
b) 1-N-(бензилоксикарбонил)-1Н,1Н,2Н,2Н,4Н,4Н,5Н,5Н,7Н,7Н-3-аза-6,9,12,15-тетраокса-8,11,14-триметилперфтороктадециламин
13.5 г (15.30 ммоль) соединения, указанного в заголовке примера 10а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола, и смесь перемешивают при комнатной температуре в течение 1 ч и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 9.0 г (68% от теории) бесцветного воскообразного вещества.
с) N-[2-(бензилоксикарбонил)аминоэтил-N-(1Н,1Н,2Н,2Н,4Н,4Н-6,9,12,15-тетраокса-8,11,14-триметилперфторпентадецил)-2-[2-(2-метоксиэтокси)этокси]ацетамид
К раствору 8.5 г (9.79 ммоль) соединения, указанного в заголовке примера 10b, и 1.74 г (9.79 ммоль) [2-(2-метоксиэтокси)этокси]уксусной кислоты (Aldrich) и 1.13 г (9.79 ммоль) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 2.52 г (12.24 ммоль) дициклогексилкарбодиимида и смесь перемешивают при 0°С в течение 3 ч и затем перемешивают при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 8.1 г (80% от теории) бесцветного вязкого масла.
d) N-2-(аминоэтил)-N-(1Н,1Н,2Н,2Н,4Н,4Н-6,9,12,15-тетраокса-8,11,14-триметилперфторпентадецил)-2-[2-(2-метоксиэтокси)этокси]ацетамид
К раствору 8.0 г (7.78 ммоль) соединения, указанного в заголовке примера 10с, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 7.0 г (количественный) бесцветного твердого вещества.
e) Гадолиниевый комплекс N-{[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-аминоэтил}-N-(1Н,1Н,2Н,2Н,4Н,4Н-6,9,12,15-тетраокса-8,11,14-триметилперфторпентадецил)-2-[2-(2-метоксиэтокси)этокси]-ацетамида
6.5 г (7.27 ммоль) соединения, указанного в заголовке примера 10d, 837 мг (7.27 ммоль) N-гидроксисукцинимида, 616 мг (14.54 ммоль) хлорида лития и 4.58 г (7.27 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 100 мл диметилсульфоксида. При 10°С добавляют 1.87 г (9.09 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 8.1 г (69% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.7%
Пример 11
а) сложный метиловый эфир 6-N-(бензилоксикарбонил)-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
К 10 г (21.92 ммоль) соединения, указанного в заголовке примера 3а, в 100 мл дихлорметана добавляют 3.19 г (25 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, обрабатывают 8.07 г (27.4 ммоль) сложным метиловым эфиром 6-N-бензилоксикарбонил-L-лизина (Bachem) и 2.75 г (27.4 ммоль) триэтиламина и затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 13.5 г (84% от теории) бесцветного воскообразного вещества.
b) сложный метиловый эфир 2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
К раствору 13.0 г (17.75 ммоль) соединения, указанного в заголовке примера 11а, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме. Выход: 10.7 г (количественный) бесцветного твердого вещества.
с) сложный метиловый эфир 6-N-[1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопираноза]-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
К раствору 10.3 г (17.21 ммоль) соединения, указанного в заголовке примера 11b, 10.30 г (17.21 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии c WO 99/01160 А1), 1.98 г (17.21 ммоль) N-гидроксисукцинимида и 3.47 г (34.42 ммоль) триэтиламина в 200 мл диметилформамида при 0°С добавляют 4.44 г (21.51 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 16.6 г (82% от теории) бесцветного вязкого масла.
d) 6-N-[1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопираноза]-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизин
16.0 г (13.57 ммоль) соединения, указанного в заголовке примера 11с растворяют в 100 мл метанола и 25 мл 2 н. раствора гидроксида калия и перемешивают при комнатной температуре в течение 16 ч. Смесь подкисляют 2 н. соляной кислотой, концентрируют в вакууме и трижды экстрагируют этилацетатом порциями по 50 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 12.4 г (78% от теории) бесцветного твердого вещества.
е) Гадолиниевый комплекс [1,4,7-трис-(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(2-гидрокси-3-ил)]амида 6-N-(1-O-α-d-карбонилметилманнопираноза)-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
12.0 г (10.30 ммоль) соединения, указанного в заголовке примера 11d, 1.18 г (10.30 ммоль) N-гидроксисукцинимида, 873 мг (20.60 ммоль) хлорида лития и 5.91 г (10.30 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[3-амино-2-гидроксипропил]-1,4,7,10-тетраазациклододекана (WO 95/17451, Schering AG) растворяют при слабом нагревании в 200 мл диметилформамида. При 10°С добавляют 2.66 г (12.88 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 48 ч. Выпавшую в осадок мочевину отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток растворяют в 100 мл метанола, обрабатывают 2.0 г палладиевого катализатора (10% Pd/C) и гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток переносят в небольшое количество воды, отфильтровывают нерастворимые компоненты, и фильтрат затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила). Выход: 7.3 г (49% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.5%
Элементный анализ (в пересчете на безводное вещество):
Пример 12
а) сложный метиловый эфир 6-N-(бензилоксикарбонил)-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраокса-5,8,11-триметилперфторпентадеканоил)-L-лизина
К 15 г (21.24 ммоль) соединения, указанного в заголовке примера 5а, в 100 мл дихлорметана добавляют 3.19 г (25 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, обрабатывают 7.82 г (26.55 ммоль) сложного метилового эфира 6-N-бензилоксикарбонил-L-лизина (Bachem) и 2.66 г (26.55 ммоль) триэтиламина и затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 16.7 г (80% от теории) бесцветного воскообразного вещества.
b) сложный метиловый эфир 2-N-(2H,2H,4H,4H-3,6,9,12,-тетраокса-5,8,11-триметилперфторпентадеканоил)-L-лизина
К раствору 16.0 г (16.29 ммоль) соединения, указанного в заголовке примера 12а, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 13.9 г (количественный) бесцветного твердого вещества.
с) сложный метиловый эфир 6-N-{2-[2-(2-метоксиэтокси)этокси]ацетил}-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраокса-6,8,11-триметилперфторпентадеканоил)-L-лизина
К раствору 13.5 г (15.91 ммоль) соединения, указанного в заголовке примера 12b, 2.83 г (15.91 ммоль) [2-(2-метоксиэтокси)этокси]уксусной кислоты (Aldrich) и 1.83 г (15.91 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 4.10 г (19.89 ммоль) дициклогексилкарбодиимида, смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 12.4 г (77% от теории) бесцветного вязкого масла.
d) 6-N-{2-[2-(2-метоксиэтокси)этокси]ацетил}-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраокса-5,8,11-триметилперфторпентадеканоил)-L-лизин
12.0 г (11.89 ммоль) соединения, указанного в заголовке примера 12с, растворяют в 100 мл метанола и 25 мл 2 н. раствора гидроксида калия и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь подкисляют 2 н. соляной кислотой, концентрируют в вакууме и трижды экстрагируют этилацетатом порциями по 50 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 10.8 г (91% от теории) бесцветного твердого вещества.
е) Гадолиниевый комплекс [1,4,7-трис-(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(2-гидрокси-3-ил)]амид 6-N-{2-[2-(2-метоксиэтокси)этокси]ацетил}-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраокса-5,8,11-триметилперфторпентадеканоил)-L-лизина
10.0 г (10.06 ммоль) соединения, указанного в заголовке примера 12d, 1.16 г (10.06 ммоль) N-гидроксисукцинимида, 861 мг (20.12 ммоль) хлорида лития и 5.86 г (10.06 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[3-амино-2-гидроксипропил]-1,4,7,10-тетраазациклододекана (WO 95/17451, Schering AG) растворяют при слабом нагревании в 200 мл диметилформамида. При 10°С добавляют 2.62 г (12.57 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 48 ч. Выпавшую в осадок мочевину отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 9.1 г (54% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.2%
Пример 13
а) 6-N-бензилоксикарбонил-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизин
10 г (13.65 ммоль) соединения, указанного в заголовке примера 11а, растворяют в 100 мл метанола и 25 мл 2 н. раствора гидроксида калия и смесь перемешивают при комнатной температуре в течение 18 ч. Смесь подкисляют 2 н. соляной кислотой, упаривают досуха и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 9.4 г (96% от теории) бесцветного твердого вещества.
b) (2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амид 6-N-бензилоксикарбонил-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
К раствору 9.0 г (12.53 ммоль) соединения, указанного в заголовке примера 13а, и 2.60 г (19.12 ммоль) (2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амина (Whitessides и др., JACS, 1994, 5057-5062) и 1.44 г (12.53 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 3.23 г (15.66 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.99 г (79% от теории) бесцветного вязкого масла.
с) (2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амид 2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
К раствору 8.7 г (9.58 ммоль) соединения, указанного в заголовке примера 13b, в 100 мл этанола добавляют 1.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 7.43 г (количественный) бесцветного твердого вещества.
d)Гадолиниевый комплекс (2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амида 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(2Н,2Н,4Н,4Н-3,6,9,12,-тетраоксаперфтортридеканоил)-L-лизина
7.0 г (9.05 ммоль) соединения, указанного в заголовке примера 13с, 1.04 г (9.05 ммоль) N-гидроксисукцинимида, 767 мг (18.10 ммоль) хлорида лития и 5.70 г (9.05 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 100 мл диметилсульфоксида. При 10°С добавляют 2.33 г (11.31 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 7.7 г (57% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.7%
Пример 14
а) [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)-метил]амид 3,5-динитробензойной кислоты
К 10 г (21.97 ммоль) соединения, указанного в заголовке примера 3с, и 4.5 г (44 ммоль) триэтиламина, растворенных в 200 мл дихлорметана, при 0°С по каплям добавляют раствор 5.76 г (25 ммоль) динитробензоилхлорида в 100 мл дихлорметана и смесь перемешивают при 0°С в течение 3 ч. Смесь обрабатывают 250 мл 0.5 М соляной кислоты, и затем перемешивают при комнатной температуре в течение 10 мин. Органическую фазу отделяют, сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: гексан/этилацетат 3:1).
Выход: 12.1 г (85% от теории) бесцветного твердого вещества.
b) [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)-метил]амид 3,5-диаминобензойной кислоты
К раствору 11.7 г (18.02 ммоль) соединения, указанного в заголовке примера 14а, в 300 мл этанола добавляют 3.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме. Выход: 10.6 г (количественный) желтоватого твердого вещества.
с) Гадолиниевый комплекс [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амида 3,5-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]бензойной кислоты
10.0 г (16.97 ммоль) соединения, указанного в заголовке примера 14b, 3.91 г (33.94 ммоль) N-гидроксисукцинимида, 2.88 г (67.88 ммоль) хлорида лития и 21.37 г (33.94 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 8.75 г (42.43 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 48 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 17.7 г (53% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.8%
Пример 15
а) сложный метиловый эфир N-бензилоксикарбонил-3-{2-[2-(2-метоксиэтокси)этокси]этил}-L-серина
К раствору 11.76 г (50 ммоль) метил-N-бензилоксикарбонил-L-азиридинкарбоксилата (Aldrich) и 4.85 г (23.36 ммоль) 2-[2-(2-метоксиэтокси)этокси]-этанола (Aldrich) в 100 мл дихлорметана при 0°С по каплям добавляют 10 мл 10% раствора эфирата трифторида бора в хлороформе и смесь перемешивают при комнатной температуре в течение 6 ч. Реакционный раствор упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 15.4 г (77% от теории) бесцветного масла.
b) N-бензилоксикарбонил-3-{2-[2-(2-метоксиэтокси)этокси]этил}-L-серин
15.0 г (37.55 ммоль) соединения, указанного в заголовке примера 15а, растворяют в 100 мл метанола и 50 мл 2 н. раствора гидроксида калия и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь подкисляют 2 н. соляной кислотой, концентрируют в вакууме и трижды экстрагируют этилацетатом порциями по 50 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 12.9 г (89% от теории) бесцветного твердого вещества.
с) 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид N-бензилоксикарбонил-3-{2-[2-(2-метоксиэтокси)этокси]этил}-L-серина
К 10 г (25.95 ммоль) соединения, указанного в заголовке примера 15b, и 11.82 г (25.95 ммоль) соединения, указанного в заголовке примера 3с, в 100 мл ТГФ при 0°С добавляют 12.35 г (50 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 17.3 г (81% от теории) бесцветного вязкого масла.
d) 1-[(1Н,11-1,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид 3-{2-[2-(2-метоксиэтокси)этокси]этил}-L-серина
К раствору 15.0 г (18.16 ммоль) соединения, указанного в заголовке примера 15 с, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 12.5 г (количественный) бесцветного твердого вещества.
е) Гадолиниевый комплекс 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амида N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-3-{2-[2-(2-метоксиэтокси)этокси]этил}-L-серина
10.0 г (14.53 ммоль) соединения, указанного в заголовке примера 15d, 1.67 г (14.53 ммоль) N-гидроксисукцинимида, 1.22 г (29.06 ммоль) хлорида лития и 9.15 г (14.53 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 3.75 г (18.16 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 11.9 г (58% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 8.0%
Пример 16
а) 5-бензиловый сложный эфир, 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид N-трет-бутилоксикарбонил-L-глутаминовой кислоты
К 6.75 г (20 ммоль) 5-бензилового сложного эфира N-mpem-бутилоксикарбонил-L-глутаминовой кислоты (Bachem) и 9.10 г (20 ммоль) соединения, указанного в заголовке примера 3с, в 200 мл ТГФ при 0°С добавляют 9.88 г (40 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 13.2 г (85% от теории) бесцветного вязкого масла.
b) 5-бензиловый сложный эфир, 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид L-глутаминовой кислоты
К раствору 12.0 г (15.49 ммоль) соединения, указанного в заголовке примера 16а в 50 мл дихлорметана при 0°С добавляют 25 мл трифторуксусной кислоты и смесь затем перемешивают при комнатной температуре в течение 4 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 9.3 г (89% от теории) аморфного твердого вещества.
с) 5-бензиловый сложный эфир, N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}ацетил)-1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид L-глутаминовой кислоты
К раствору 8.5 г (12.60 ммоль) соединения, указанного в заголовке примера 16b, и 3.07 г (13.86 ммоль) {2-[2-(2-метоксиэтокси)этокси]этокси}уксусной кислоты (Voegtie и др., Liebigs Ann. Chem., 1980, 858-862) и 1.60 г (13.86 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 3.58 г (17.33 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 9.2 г (83% от теории) бесцветного вязкого масла.
Элементный анализ:
d) N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}ацетил)-1-[(1H,1H,2H,2H,4H,4H,-3,6,9,12-тетраоксаперфтортридецил)метил]амид L-глутаминовой кислоты
К раствору 9.0 г (10.24 ммоль) соединения, указанного в заголовке примера 16 с, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 8.1 г (количественный) бесцветного твердого вещества.
е) Гадолиниевый комплекс 5-{[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(2-гидрокси-3-ил)]амидо}-N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}ацетил)-1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амида L-глутаминовой кислоты
7.5 г (9.51 ммоль) соединения, указанного в заголовке примера 16d, 1.09 г (9.51 ммоль) N-гидроксисукцинимида, 799 мг (19.02 ммоль) хлорида лития и 5.46 г (9.51 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[3-амино-2-гидроксипропил]-1,4,7,10-тетраазациклододекана (WO 95/17451, Schering AG) растворяют при слабом нагревании в 200 мл диметилформамида. При 10°С добавляют 2.42 г (11.89 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 48 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 8.5 г (61% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 8.1%
Элементный анализ (в пересчете на безводное вещество):
Пример 17
а) 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид N-трет-бутилоксикарбонил-L-глутаминовой кислоты
К раствору 12.0 г (15.49 ммоль) соединения, указанного в заголовке примера 16а, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) v смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 10.6 г (количественный) бесцветного твердого вещества.
b) 5-(2-{2-[2-(2-метоксиэтокси)этокси]-этокси}этил)амид, 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)-метил]амид N-трет-бутилоксикарбонил-L-глутаминовой кислоты
К раствору 10.0 г (14.61 ммоль) соединения, указанного в заголовке примера 17а, и 3.03 г (14.61 ммоль) (2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амина (Whitessides и др., JACS, 1994, 5057-5062) и 2.27 г (14.61 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 3.77 г (18.26 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 10.7 г (84% от теории) бесцветного вязкого масла.
с) 5-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амид, 1-[(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид L-глутаминовой кислоты
К раствору 10.0 г (11.45 ммоль) соединения, указанного в заголовке примера 17b, в 50 мл дихлорметана при 0°С добавляют 25 мл трифторуксусной кислоты и смесь затем перемешивают при комнатной температуре в течение 4 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.5 г (96% от теории) аморфного твердого вещества.
d) Гадолиниевый комплекс 5-(2-{2-[2-(2-метоксиэтокси)этокси]-этокси}этил)амида, 1-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)-метил]амида N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовой кислоты
8.0 г (10.34 ммоль) соединения, указанного в заголовке примера 17с, 1.19 г (10.34 ммоль) N-гидроксисукцинимида, 869 мг (20.68 ммоль) хлорида лития и 6.51 г (10.34 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.67 г (12.93 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 9.0 г (59% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.5%
Пример 18
а) [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид L-2-бензилоксикарбониламино-4-аминомасляной кислоты
К 17.62 г (50 ммоль) L-2-бензилоксикарбониламино-4-трет-бутилоксикарбониламиномасляной кислоты (Bachem) и 22.76 г (50 ммоль) соединения, указанного в заголовке примера 3с, в 200 мл ТГФ при 0°С добавляют 24.7 г (100 ммоль) EEDQ (этил-2-этокси-1,2-дигидрохинолин-1-карбоксилат) и смесь перемешивают при комнатной температуре в течение 16 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 80 мл дихлорметана, обрабатывают при 0°С 40 мл трифторуксусной кислоты, и затем перемешивают при комнатной температуре в течение 4 ч. Смесь упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 25.2 г (73% от теории) бесцветного вязкого масла.
b) [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]-амид L-2-бензилоксикарбониламино-4-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}ацетил)-аминомасляной кислоты
К раствору 20 г (29.01 ммоль) соединения, указанного в заголовке примера 18а, и 6.45 г (29.01 ммоль) {2-[2-(2-метоксиэтокси)этокси]этокси}уксусной кислоты (Voegtie и др., LiebigsAnn. Chem., 1980, 858-862) и 3.34 г (29.01 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 7.48 г (36.26 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 20.7 г (80% от теории) бесцветного вязкого масла.
с) [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амид L-2-амино-4-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}ацетил)аминомасляной кислоты
К раствору 20.0 г (22.38 ммоль) соединения, указанного в заголовке примера 18b в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 17.1 г (количественный) бесцветного твердого вещества.
d) Гадолиниевый комплекс [(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфтортридецил)метил]амида L-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-амино-4-(2-{2-[2-(2-метоксиэтокси)этокси]-этокси}ацетил)аминомасляной кислоты
15.0 г (19.75 ммоль) соединения, указанного в заголовке примера 18с, 2.27 г (19.75 ммоль) N-гидроксисукцинимида, 1.68 г (39.50 ммоль) хлорида лития и 12.43 г (19.75 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 5.09 г (24.69 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 23.4 г (59% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.5%
Пример 19
а) Гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амида 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-6-N-бензилоксикарбонил-L-лизина
50.0 г (57.64 ммоль) соединения, указанного в заголовке примера 1е, 6.63 г (57.64 ммоль) N-гидроксисукцинимида, 4.88 г (115.28 ммоль) хлорида лития и 36.30 г (57.64 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, ScheringAG, (пример 1)) растворяют при слабом нагревании в 400 мл диметилсульфоксида. При 10°С добавляют 14.87 г (72.05 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 5000 мл диэтилового эфира и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол/водн. аммиак 10:5:1).
Выход: 57.4 г (64% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 4.8%
b) Гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амида 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-лизина
К раствору 55 г (35.4 ммоль) соединения, указанного в заголовке примера 19а, в 600 мл метанола и 100 мл воды добавляют 5.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 50.7 г (количественный) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.0%
с) Гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амида 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-6-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
К раствору 10.0 г (6.99 ммоль) соединения, указанного в заголовке примера 19b, и 4.19 г (6.99 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии с WO 99/01160 А1) и 806 мг (6.99 ммоль) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 1.80 г (8.74 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток растворяют в 100 мл метанола, обрабатывают 2.0 г палладиевого катализатора (10% Pd/C) и гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток переносят в небольшое количество воды, отфильтровывают нерастворимые компоненты, и фильтрат затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 7.5 г (64% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.2%
Пример 20
а) Гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]-амида 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-6-N-{2-[2-(2-метоксиэтокси)этокси]ацетил}-L-лизина
К раствору 14.31 г (10.0 ммоль) соединения, указанного в заголовке примера 19b, и 1.96 г (11.0 ммоль) [2-(2-метоксиэтокси)этокси]уксусной кислоты (Aldrich) и 1.27 г (11.0 ммоль) N-гидроксисукцинимида в 100 мл диметилформамида при 0°С добавляют 2.84 г (13.75 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток переносят в небольшое количество воды, отфильтровывают нерастиворимые компоненты, и фильтрат затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 12.4 г (77% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.7%
Пример 21
а) N-[2-(2-метоксиэтокси)этил]амид (2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадекановой кислоты)
К 10 г (16.5 ммоль) соединения, указанного в заголовке примера 1а, в 100 мл дихлорметана добавляют 2.55 г (20 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 3.93 г (33 ммоль) 2-(метоксиэтокси)этиламина (Whitesides и др., JACS, 1994, 5057-5062) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 11.2 г (96% от теории) бесцветного воскообразного вещества.
b) N-[2-(2-метоксиэтокси)этил]-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)амин
10.5 г (14.85 ммоль) соединения, указанного в заголовке примера 21а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, и остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.4 г (82% от теории) бесцветного масла.
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-[2-(2-метоксиэтокси)этил]амид}-1,4,7,10-тетраазациклододекана
8 г (11.54 ммоль) соединения, указанного в заголовке примера 21b, 1.33 г (11.54 ммоль) N-гидроксисукцинимида, 974 мг (23.08 ммоль) хлорида лития и 7.26 г (11.54 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.98 г (14.43 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 8.4 г (52% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.8%
Пример 22
а) N-{2-[2-(2-метоксиэтокси)этокси]этил}амид(2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадекановой кислоты)
К 10 г (16.5 ммоль) соединения, указанного в заголовке примера 1а, в 100 мл дихлорметана добавляют 2.55 г (20 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 100 мл дихлорметана, добавляют 5.39 г (33 ммоль) 2-[2-(метоксиэтокси)этокси]этиламина (Whitesides и др., JACS, 1994, 5057-5062) и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 100 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:1).
Выход: 11.4 г (92% от теории) бесцветного воскообразного вещества.
b) N-{2-[2-(2-метоксиэтокси)этокси]этил}-(1H,1H,2H,2H,4H,4H,-3,6,9,12-тетраоксаперфторгексадецил)амин
10.0 г (13.31 ммоль) соединения, указанного в заголовке примера 22а, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток 5 хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 8.6 г (88% от теории) бесцветного масла.
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-{2-[2-(2-метоксиэтокси)этокси]-этил}амид]-1,4,7,10-тетраазациклододекана
8 г (10.43 ммоль) соединения, указанного в заголовке примера 22b, 1.20 г (10.43 ммоль) М-гидроксисукцинимида, 880 мг (20.86 ммоль) хлорида лития и 6.56 г (10.43 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.69 г (13.04 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 8.4 г (56% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.5%
Пример 23
а) [1-O-α-d-(2,3,4,6-тетра-O-бензил)маннопиранозил]ацетамид
К 40 г (66.81 ммоль) 1-O-α-d-карбонилметил-(2,3,4,6-тетра-O-бензил)маннопиранозы (полученной в соответствии с WO 99/01160 А1) в 300 мл дихлорметана добавляют 11.45 г (90 ммоль) оксалилхлорида и смесь перемешивают при комнатной температуре в течение 14 ч. Смесь упаривают досуха в вакууме, остаток растворяют в 400 мл дихлорметана, газообразный аммиак пропускают через раствор при 0°С в течение приблизительно 2 ч и смесь затем перемешивают при комнатной температуре в течение 4 ч. Реакционный раствор обрабатывают 400 мл 1 н. соляной кислоты и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют, сушат над сульфатом магния и упаривают досуха в вакууме. Остаток хроматографируют на силикагеле (элюент: этилацетат/гексан 1:2).
Выход: 34.1 г (85% от теории) бесцветного масла.
b) 2-[1-O-α-d-(2,3,4,6-тетра-O-бензил)маннопиранозил]этиламин
33 г (55.21 ммоль) соединения, указанного в заголовке примера 23а, в 100 мл ТГФ обрабатывают 30 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 100 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 200 мл этанола / 100 мл этаноламина и перемешивают при 60°С в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 300 мл раствора гидроксида натрия крепостью 5% и трижды экстрагируют дихлорметаном порциями по 300 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 26.2 г (81% от теории) бесцветного твердого вещества.
с) N-{2-[1-O-α-d-(2,3,4,6-тетра-O-бензил)маннопиранозил]этил}амид (2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадекановой кислоты)
К раствору 11.16 г (19.12 ммоль) соединения, указанного в заголовке примера 23b, и 11.59 г (19.12 ммоль) соединения, указанного в заголовке примера 1а, и 2.2 г (19.12 ммоль) N-гидроксисукцинимида в 200 мл диметилформамида при 0°С добавляют 4.93 г (23.90 ммоль) дициклогексилкарбодиимида, и смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 15.7 г (71% от теории) бесцветного вязкого масла.
d) N-{2-[1-O-α-d-(2,3,4,6-тетра-O-бензил)маннопиранозил]этил}-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)амин
15.0 г (12.80 ммоль) соединения, указанного в заголовке примера 23с, в 50 мл ТГФ обрабатывают 15 мл 10 М борандиметилсульфида (в ТГФ) и нагревают в емкости с обратным холодильником в течение 5 ч. Смесь охлаждают до 0°С, по каплям добавляют 20 мл метанола и перемешивают при комнатной температуре в течение 1 ч, и затем упаривают досуха в вакууме. Остаток переносят в смесь 100 мл этанола/50 мл 1 М соляной кислоты и перемешивают при 40°С в течение 14 ч. Смесь упаривают досуха в вакууме, остаток переносят в 100 мл раствора гидроксида натрия крепостью 5% и смесь трижды экстрагируют дихлорметаном порциями по 100 мл. Объединенные органические фазы сушат над сульфатом магния, упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 10:1).
Выход: 12.2 г (82% от теории) бесцветного масла.
е) N-[2-(1-O-α-d-маннопиранозил)этил]-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)амин
К раствору 11.5 г (9.93 ммоль) соединения, указанного в заголовке примера 23d, в 200 мл этанола добавляют 2.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 24 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 7.90 г (количественный) бесцветного твердого вещества.
f) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-{[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-[2-(1-O-α-d-маннопиранозил)этил]амид}-1,4,7,10-тетраазациклододекана
7 г (8.78 ммоль) соединения, указанного в заголовке примера 23е, 1.01 г (8.78 ммоль) N-гидроксисукцинимида, 741 мг (17.56 ммоль) хлорида лития и 5.52 г (8.78 ммоль) гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана (WO 98/24775, Schering AG, (пример 1)) растворяют при слабом нагревании в 200 мл диметилсульфоксида. При 10°С добавляют 2.26 г (10.98 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 7.7 г (58% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.9%
Пример 24
а) 10-(5-оксотетрагидрофуран-2-илметил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекан
К 12.0 г (34.6 ммоль) 1,4,7-трис-(карбоксиметил)-1,4,7,10-тетраазациклододекана (DОЗА) в 50 мл воды добавляют 8.3 г (207.6 ммоль) гидроксида натрия. Туда же по каплям добавляют раствор 5.02 г (43.25 ммоль) 3-оксиранилпропионовой кислоты (Dakoji и др., J. Am. Chem. Soc., 1996, 10971-10979) в смеси 50 мл н-бутанол/50 мл 2-пропанол и раствор нагревают при 80°С в течение 24 ч. Реакционный раствор упаривают досуха в вакууме, и остаток обрабатывают 300 мл воды и значение рН устанавливают на 3 с помощью 3 н. соляной кислоты. Затем, смесь трижды экстрагируют н-бутанолом порциями по 200 мл, объединенные бутанольные фазы упаривают досуха в вакууме и остаток очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 13.6 г (79% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 10.4%
b) Гадолиниевый комплекс 10-(5-оксотетрагидрофуран-2-илметил)-1,4,7-трис(карбоксиметил)-1,4,7,10-тетраазациклододекана
12.0 г (24.2 ммоль) соединения, указанного в заголовке примера 23а, растворяют в 100 мл воды и 1 мл уксусной кислоты, обрабатывают 4.39 г (12.1 ммоль) оксида гадолиния и перемешивают при 80°С в течение 6 ч. Раствор фильтруют, упаривают досуха и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 13.8 г (89% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.5%
с) Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(4-гидрокси-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
2.5 г (3.85 ммоль) соединения, указанного в заголовке примера 2b, и 3.70 г (5.78 ммоль) соединения, указанного в заголовке примера 23b, растворяют в 50 мл метанола и смесь перемешивают при температуре 50°С в течение 48 ч. Смесь упаривают досуха и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 3.89 г (75% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 7.2%
Пример 25
а) мононатриевая соль гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[(4-(R)-карбоксилато-4-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана и мононатриевая соль гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-{[(R)-(2-карбоксилатоэтил)ил]-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид}-1,4,7,10-тетраазациклододекана
2.5 г (3.85 ммоль) соединения, указанного в заголовке примера 2b, 493 мг (4.82 ммоль) триэтиламина и 3.84 г (4.82 ммоль) гадолиниевого комплекса монопентафторфенил-2-(R)-2-[4,7,10-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-1-ил]пентандикарбоксилата, (WO 2005/0014154, EPIX PHARMACEUTICALS, INC., (пример 9: EP-2104-15-Pfp)) растворяют в 50 мл диметилсульфоксида и перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 1000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила). Содержащие продукт фракции упаривают, растворяют в воде, нейтрализуют 0.1 н. раствором гидроксида натрия и затем лиофилизуют.
Выход: 2.01 г (37% от теории) бесцветного твердого вещества в виде смеси региоизомеров в соотношении 3:2.
Содержание воды (по Карлу Фишеру): 8.5%
Пример 26
а) комплекс 1,4,7-трис(трет-бутоксикарбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана с бромидом натрия
20 г (30.80 ммоль) соединения, указанного в заголовке примера 2b, 1.42 г (12.32 ммоль) N-гидроксисукцинимида и 23.0 г (30.80 ммоль) комплекса 1,4,7-трис(трет-бутоксикарбоксилатометил)-10-[1-карбокси-3-аза-4-оксо-5-метилпентан-5-ил]-1,4,7,10-тетраазациклододекана с бромидом натрия (WO 98/24775, Schering AG, (пример 1d)) растворяют при слабом нагревании в 400 мл диметилформамида. При 10°С добавляют 3.18 г (15.4 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Выпавшую в осадок мочевину отфильтровывают, фильтрат упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/метанол 20:1).
Выход: 27.7 г (65% от теории) бесцветного твердого вещества.
b) 1,4,7-трис(карбоксиметил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-М-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекан
25 г (18.14 ммоль) соединения, указанного в заголовке примера 25а, растворяют в 150 мл трифторуксусной кислоты и перемешивают при комнатной температуре в течение 5 ч. Смесь упаривают досуха, переносят в воду и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 17.1 г (81% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 4.7%
Пример 27
a) Y комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
2.0 г (1.81 ммоль) соединения, указанного в заголовке примера 25а, растворяют в 50 мл воды и 1 мл уксусной кислоты, обрабатывают 387 мг (1.99 ммоль) хлорида иттрия и перемешивают при 80°С в течение 6 ч. Смесь нейтрализуют аммиаком, упаривают досуха и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 1.92 г (84% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 5.5%
Пример 28
a) Dy-комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
2.0 г (1.81 ммоль) соединения, указанного в заголовке примера 25а, растворяют в 50 мл воды и 1 мл уксусной кислоты, обрабатывают 534 мг (1.99 ммоль) хлорида диспрозия и перемешивают при 80°С в течение 6 ч. Смесь нейтрализуют аммиаком, упаривают досуха и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила). Выход: 2.14 г (87% от теории) бесцветного твердого вещества. Содержание воды (по Карлу Фишеру): 6.1%
Пример 29
а) Yb-комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана
2.0 г (1.81 ммоль) соединения, указанного в заголовке примера 25а, растворяют в 50 мл воды и 1 мл уксусной кислоты, обрабатывают 555 мг (1.99 ммоль) хлорида иттербия и перемешивают при 80°С в течение 6 ч. Смесь нейтрализуют аммиаком, упаривают досуха и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 2.10 г (84% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 6.7%
Пример 30
а) Бензил-4-бензилоксикарбониламино-5-[бис(2-бензилоксикарбониламиноэтил)амино]-пентанкарбоксилат
17.87 г (50 ммоль) Z-Glu-(OBn)-OH (Bachem) растворяют в 200 мл метиленхлорида и при -78°С по каплям в течение 30 мин добавляют раствор 15.5 г (55 ммоль) ангидрида трифторметансульфоновой кислоты (Aldrich) и 6.97 г (65 ммоль) 2,6-диметилпиридина (Aldrich) в 100 мл метиленхлорида и смесь перемешивают при 0°С в течение 3 ч. Реакционную смесь дважды экстрагируют водой со льдом порциями по 100 мл и органическую фазу сушат, используя сульфат натрия. Сырой продукт затем при -20°С по каплям добавляют к раствору 18.57 г (50 ммоль) N,N''-ди-Z-диэтилентриамина (Пика) и 12.9 г (100 ммоль) этилдиизопропиламина в 200 мл метиленхлорида и смесь перемешивают при -20°С в течение 6 ч. Далее, смесь перемешивают при комнатной температуре в течение дополнительных 24 ч. Реакционную смесь дважды экстрагируют 150 мл воды, и органическую фазу сушат, используя сульфат натрия, упаривают досуха и хроматографируют на силикагеле (гексан/этилацетат 5:1). Содержащие продукт фракции объединяют и упаривают.
b) 4-амино-5-[бис(2-аминоэтил)амино]пентанкарбоновая кислота
14.2 г (20 ммоль) бензил-4-бензилоксикарбониламино-5-[бис(2-бензилоксикарбонил-аминоэтил)амино]пентанкарбоксилата растворяют в 300 мл изопропанола, обрабатывают 30 мл воды и добавляют 3 г палладиевого катализатора (10% Pd/C). Смесь гидрируют при 50°С в течение 8 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме.
Выход: 4.35 г (количественный) бесцветного порошка.
с) 1-(натрий сульфонатобутил)-4-карбокси-3-бензилокси-6-метил-1[H]-пиридин-2-он
4.31 г (15 ммоль) 4-этоксикарбонил-3-бензилокси-6-метил-1[Н]-пиридин-2-она (международная заявка на патент WO 03/016923, пример 2) в 15 мл ДМФА обрабатывают 0.41 г (17 ммоль) гидроксида лития и, после добавления 2.04 г (15 ммоль) 1,4-бутансультона, перемешивают в течение ночи при комнатной температуре. Далее, растворитель отгоняют, остаток обрабатывают 50 мл 2 н. раствора гидроксида натрия и смесь перемешивают при комнатной температуре в течение 6 ч. Значение рН раствора устанавливают на 3 путем добавления ионообменной смолы Amberlite® IR-120 (H+) и лиофилизируют. Лиофилизат хроматографируют на колонке Lichroprep RP-18 (элюент: вода).
Содержащие продукт фракции объединяют и упаривают досуха.
d) 1-(натрий сульфонатобутил)-4-(4-нитрофенилоксикарбонил)-3-бензилокси-6-метил-1[Н]-пиридин-2-он
2.09 г (5 ммоль) соединения, указанного в заголовке примера 30 с, и 765 мг (5.5 ммоль) нитрофенола растворяют в 30 мл ДМФА, обрабатывают 1 мл этилдиизопропиламина и 1.77 г (5.5 ммоль) тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и перемешивают в течение ночи при комнатной температуре. Реакционную смесь упаривают досуха и хроматографируют на силикагеле (изопропанол). Содержащие продукт фракции объединяют и упаривают.
е) 5-[бис-(2-{[1-(натрий сульфонатобутил)-3-бензилокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]-4-{[1-(натрий сульфонатобутил)-3-бензилокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}пентанкарбоновая кислота
2.15 г (4 ммоль) соединения, указанного в заголовке примера 30d, и 262 мг (1.2 ммоль) 4-амино-5-[бис(2-аминоэтил)амино]пентанкарбоновой кислоты (пример 30b) растворяют в 50 мл ДМФА, обрабатывают 870 мкл (5 ммоль) этилдиизопропиламина и перемешивают при комнатной температуре в течение трех дней. Реакционную смесь упаривают досуха и хроматографируют на колонке Lichroprep RP-18 (градиент воды/ацетонитрила). Содержащие продукт фракции объединяют и упаривают.
f) 5-[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]-4-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}пентанкарбоновая кислота
К раствору 1.42 г (1 ммоль) соединения, указанного в заголовке примера 30е, в 100 мл этанола добавляют 1.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 48 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток вводили в реакцию комплексообразования без дополнительного определения характеристик.
g) Гадолиниевый комплекс 5-[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]-4-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}пентанкарбоновой кислоты
К 1.15 г (1 ммоль) соединения, указанного в заголовке примера 30f, в 50 мл воды при рН 8.5 (рН-стат) добавляют 371 мг (1 ммоль) гексагидрата хлорида гадолиния и смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь упаривают досуха и хроматографируют на колонке Lichroprep RP-18 (градиент воды/ацетонитрила). Содержащие продукт фракции объединяют и упаривают.
Содержание воды (по Карлу Фишеру): 8.1%
h) Гадолиниевый комплекс [N-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)]амида 5-[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]-4-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}пентанкарбоновой кислоты
1.48 г (2.28 ммоль) соединения, указанного в заголовке примера 2b, 263 мг (2.28 ммоль) N-гидроксисукцинимида и 3.0 г (2.28 ммоль) соединения, указанного в заголовке примера 30g, растворяют при слабом нагревании в 50 мл диметилсульфоксида. При 10°С добавляют 588 мг (2.85 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 2.15 г (46% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 4.8%
Пример 31
а) [бис(2-{[1-(натрий сульфонатобутил)-3-бензилокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]этиламин
6.45 г (12 ммоль) соединения, указанного в заголовке примера 30d, и 876 мг (6 ммоль) трис(2-аминоэтил)амина растворяют в 50 мл ДМФА, обрабатывают 2.6 мл (15 ммоль) этилдиизопропиламина и перемешивают при комнатной температуре в течение трех дней. Реакционную смесь упаривают досуха и хроматографируют на колонке Lichroprep RP-18 (градиент воды/ацетонитрила).
Содержащие продукт фракции объединяют и упаривают.
b) моноамид 2,3-бисбензилокси-N-{[бис(2-{[1-(натрий сульфонатобутил)-3-бензилокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]этил}-терефталевой кислоты
4.4 г (5.65 ммоль) соединения, указанного в заголовке примера 31а, и 4.91 г (8.47 ммоль) (2,3-бисбензилокси)-1,4-(бис-2-тиоксотиазолидин-3-карбонил)бензола (Raymond и др., Inorg. Chem. (2003), (42), 4930) растворяют в 100 мл метиленхлорида и перемешивают при комнатной температуре в течение трех дней. Реакционную смесь экстрагируют 100 мл 1 н. раствора гидроксида натрия и 100 мл насыщенного раствора хлорида натрия, и. органическую фазу сушат, используя сульфат натрия, упаривают досуха и хроматографируют на колонке Lichroprep RP-18 (градиент воды/ацетонитрила). Содержащие продукт фракции объединяют и упаривают.
с) моноамид 2,3-дигидрокси-М-{[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]этил}терефталевой кислоты
К раствору 5.2 г (3.94 ммоль) соединения, указанного в заголовке примера 31b, в 100 мл этанола добавляют 1.0 г палладиевого катализатора (10% Pd/C) и смесь гидрируют при комнатной температуре в течение 48 ч. Катализатор отфильтровывают и фильтрат упаривают досуха в вакууме. Остаток вводили в реакцию комплексообразования без дополнительного определения характеристик.
d) Гадолиниевый комплекс моноамида 2,3-дигидрокси-N-{[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-ди гидропиридин-4-карбонил]амино}этил)амино]этил}-терефталевой кислоты
К 3.75 г (3.94 ммоль) соединения, указанного в заголовке примера 31с, в 100 мл воды при рН 8.5 (рН-стат) добавляют 1.46 г (3.94 ммоль) гексагидрата хлорида гадолиния и смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь упаривают досуха и хроматографируют на колонке Lichroprep RP-18 (градиент воды/ацетонитрила). Содержащие продукт фракции объединяют и упаривают.
Содержание воды (по Карлу Фишеру): 8.0%
е) Гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н,-3,6,9,12-тетраоксаперфторгексадецил)]амида 6-N-(2,3-дигидрокси-N-{[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]этил}-терефталил)-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина
2.10 г (2.20 ммоль) соединения, указанного в заголовке примера 1g, 254 мг (2.28 ммоль) N-гидроксисукцинимида и 2.5 г (2.20 ммоль) соединения, указанного в заголовке примера 30g, растворяют при слабом нагревании в 50 мл диметилсульфоксида. При 10°С добавляют 580 мг (2.79 ммоль) дициклогексилкарбодиимида и смесь перемешивают при комнатной температуре в течение 16 ч. Раствор выливают в 2000 мл ацетона и затем перемешивают в течение 10 мин. Выпавшее в осадок твердое вещество отфильтровывают и затем очищают с помощью хроматографии (RP-18; элюент: градиент воды/ацетонитрила).
Выход: 3.15 г (65% от теории) бесцветного твердого вещества.
Содержание воды (по Карлу Фишеру): 5.3%
Пример 32
Релаксационность
Время релаксации Т1 и Т2 протонов воды и плазмы (бычьей) с возрастающими концентрациями вещества из примера 2 с) определяли при 40°С, используя импульсный спектрометр ЯМР (прибор Minispec PC 20) при напряженности поля 0.47 тесла (табл.1).
Исходя из R1 релаксационностей воды при высоких и низких концентрациях может быть определена критическая концентрация мицеллообразования (ККМ), составляющая 0.02 ммоль Gd/л. Релаксационность плазмы является большей, чем воды, и указывает на связывание белков.
Пример 33
Острая токсичность после одноразового внутривенного введения препарата мышам (эксплоративная)
После внутривенного введения веществ примеров 2с), 21с) и 22с) мышам (n=3; скорость введения; 2 мл/мин) эксплоративным способом определяли острую системную переносимость (LD50). В каждом случае, изучали влияние нескольких доз, причем период наблюдений составлял 7 дней. Ожидаемая средняя острая токсичность составляла 2.5 ммоль Gd/кг массы тела для вещества из примера 2с) и >10.0 ммоль Gd/кг массы тела для вещества из примера 21с) и 22с).
Пример 34
Высвобождение гистамина после одноразового внутривенного введения крысам
После внутривенного введения веществ примеров 2с) и 22с) крысам (n=3) в разные моменты времени определяли высвобождение гистамина. Для этого, из сонной артерии до и через 10, 30 и 60 минут после введения контрастного вещества отбирали кровь, и содержащийся в плазме гистамин определяли с помощью ELISA-системы. Определенные значения концентрации гистамина можно видеть в таблице 2.
Значения концентрации гистамина у контрольных, здоровых крыс были в пределах нормы, известных из литературы. Соединения в соответствии с изобретением не вызывали значительного высвобождения гистамина.
Пример 35
Выведение препарата после внутривенного введения крысам
После внутривенного введения соединений примеров 2с), 21с) и 22с) крысам (n=3) из расчета 50 мкмоль общего гадолиния/кг массы тела, содержание металла в выделениях - моче и фекалиях, и в теле (остальное тело) фракционно определяли с помощью атомной эмиссионной спектрометрии (ICP-AES) вплоть до 14 дней после введения (Таблица 3).
Пример 36
Кинетика плазмы после внутривенного введения препарата крысам
После внутривенного введения крысам (n=3) веществ примеров 2с), 21с) и 22с), из расчета 50 мкмоль общего гадолиния/кг массы тела, были взяты пробы крови с помощью катетера в общей сонной артерии в различные моменты времени (вплоть до 24 ч после введения) и определено содержание металла с помощью атомной эмиссионной спектрометрии (ICP-AES), которое пересчитывали в соответствующие значения для плазмы, используя коэффициент пересчета (0.625). Фармакокинетические параметры (таблица 4) вычисляли путем специального программного обеспечения (WinNonlin) исходя из концентраций в плазме.
Пример 37
Распознавание лимфатических узлов с помощью МРТ после внутривенного введения крысам
На фигурах показаны примерные, полученные при МР-томографии изображения подколенных лимфатических узлов, зарегистрированные в различные моменты времени после внутривенного введения крысам вещества из примера 2с) - фигура 1, вещества из примера 21с) - фигура 2, и вещества из примера 22с) - фигура 3, из расчета 50 мкмоль Gd/кг массы тела. T1 - взвешенные изображения, полученные так называемым методом быстрого спин-эха (1.5 Т; последовательность: T1-TSE; TR 451 мс, ТЕ 8.7 мс) иллюстрируют значительное увеличение интенсивности сигнала в функционирующей ткани лимфатического узла, наблюдаемое уже через короткий интервал времени после инъекции (вплоть до 60 минут после инъекции).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ В МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ БЛЯШЕК, ОПУХОЛЕЙ И НЕКРОЗОВ | 2001 |
|
RU2290206C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОПЛЕКСЫ С ПОЛЯРНЫМИ ОСТАТКАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО | 2001 |
|
RU2289579C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОМПЛЕКСЫ С ОСТАТКАМИ САХАРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2280644C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2242477C2 |
| СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2002 |
|
RU2305096C2 |
| НОВЫЕ ХЕЛАТНЫЕ СОЕДИНЕНИЯ ГАДОЛИНИЯ С ВЫСОКОЙ РЕЛАКСИВНОСТЬЮ ДЛЯ ПРИМЕНЕНИЯ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ | 2017 |
|
RU2755181C2 |
| ПРИМЕНЕНИЕ ПЕРФТОРАЛКИЛСОДЕРЖАЩИХ КОМПЛЕКСОВ МЕТАЛЛОВ В КАЧЕСТВЕ КОНТРАСТНЫХ ВЕЩЕСТВ ПРИ МАГНИТНО-РЕЗОНАНСНОЙ ТОМОГРАФИИ ДЛЯ ВИЗУАЛИЗАЦИИ ВНУТРИСОСУДИСТЫХ ТРОМБОВ | 2003 |
|
RU2328310C2 |
| КАСКАДНЫЕ ПОЛИМЕРНЫЕ КОМПЛЕКСЫ, ИСХОДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2166501C2 |
| КОМПЛЕКСЫ КАСКАДНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩЕЕ ИХ ДИАГНОСТИЧЕСКОЕ КОНТРАСТНОЕ СРЕДСТВО И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2197495C2 |
| КОНЪЮГАТЫ, ПОЛУЧЕННЫЕ ИЗ КОМПЛЕКСОВ МЕТАЛЛОВ И ОЛИГОНУКЛЕОТИДОВ, СРЕДСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТЫ, ИХ ИСПОЛЬЗОВАНИЕ В РАДИОДИАГНОСТИКЕ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2165771C2 |


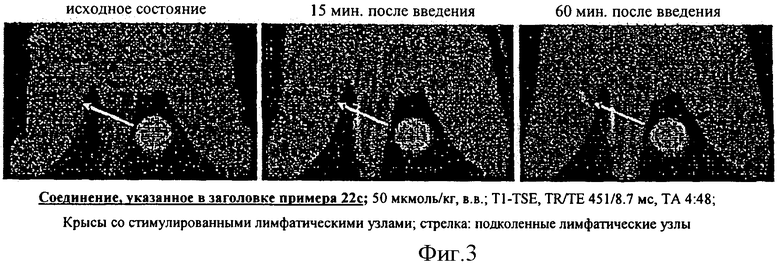
Изобретение относится к хелатам металлов формулы I:
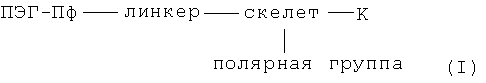
где ПЭГ-Пф означает перфторированный ПЭГ радикал, имеющий 4-30 атомов углерода, включающий а) по меньшей мере один перфторированный ПЭГ радикал формулы XXI
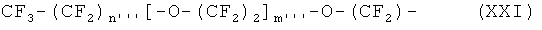
где n''' означает целое число между 0 и 6, m''' означает целое число между 1 и 14; линкер означает линкерную группу, которая соединяет ПЭГ-Пф радикал со скелетом; скелет означает трехвалентный радикал, который представляет собой азотсодержащий радикал, выбранный из аминокислот, имеющих боковую функциональную цепь, алкилендиаминового радикала и его производных, азота и 3,5-диаминобензойной кислоты, К означает хелатный радикал, состоящий из хелатирующего радикала, и по меньшей мере одного эквивалента иона металла порядкового номера 57-83. Также предложено применение хелатов металлов для получения контрастных веществ. Изобретение позволяет получить контрастные вещества для МРТ, обладающие хорошими визуализирующими свойствами и хорошей переносимостью. 6 н. и 3 з.п. ф-лы, 3 ил., 4 табл., 37 пр.
1. Хелаты металлов формулы I:
где ПЭГ-Пф означает перфторированный ПЭГ радикал, имеющий 4-30 атомов углерода, включающий
а) по меньшей мере один перфторированный ПЭГ радикал формулы XXI
где n''' означает целое число между 0 и 6, и
m''' означает целое число между 1 и 14, и
линкер означает линкерную группу, которая соединяет ПЭГ-Пф радикал со скелетом, и представляет собой углеродную цепь, имеющую 1-15 С атомов, которая может быть линейной или разветвленной, насыщенной или ненасыщенной, и которая необязательно прерывается 1-5 атомами кислорода, 1-3 -NHCO- группами, 1-3 -CONH- группами, 1-2 атомами серы, 1-4 -NH-группами и/или 1-2 фениленовыми группами, которые необязательно могут быть замещены 1-2 ОН группами, 1-2 NH2 группами, 1-2 -СООН группами или 1-2 -SO3Н группами, и которая необязательно замещена 1-6 ОН группами, 1-5 -СООН группами (которые необязательно присутствуют в защищенной форме), 1-2 SO3H группами (которые необязательно присутствуют в защищенной форме), 1-3 NH2 группами и/или 1-3 С1-С4-алкоксигруппами, скелет означает трехвалентный радикал, который представляет собой азотсодержащий радикал, выбранный из аминокислот, имеющих боковую функциональную цепь, алкилендиаминового радикала и его производных, азота и 3,5-диаминобензойной кислоты,
К означает хелатный радикал, состоящий из хелатирующего радикала, и по меньшей мере одного эквивалента иона металла порядкового номера 57-83, причем хелатирующий радикал представляет собой DOTA-радикал или его производное, где свободные кислотные группы, необязательно присутствующие в хелатирующем радикале, необязательно могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот, или амидов аминокислот, или хелатирующий радикал представляет собой DTPA радикал или его производное, или хелатирующий радикал на основе катехоламида (САМ), терефталамида (ТАМ), гидроксипиридона (НОРО) и/или гидроксипиримидона (HOPY) или их производных, и
полярная группа означает полярную группу.
2. Хелаты металлов по п.1, отличающиеся тем, что перфторированный ПЭГ радикал содержит 4-30 С атомов.
3. Хелаты металлов по одному из пп.1 и 2, отличающиеся тем, что хелатирующий радикал является циклическим, где свободные кислотные группы, необязательно присутствующие в хелатирующем радикале, необязательно могут быть представлены в виде солей органических и/или неорганических оснований или аминокислот, или амидов аминокислот.
4. Гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-{(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-[2-(2-метоксиэтокси)этил]амид} -1,4,7,10-тетраазациклододекана.
5. Гадолиниевый комплекс 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-(α-d-карбонилметилманнопираноза)-L-лизин-[N-метил-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)]амида,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизин-[N-метил-(2Н,2Н,4Н,4Н-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)]амида,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(2Н,2Н,4Н,4Н-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс [N-метил-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)]амида 6-N[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)кислота-N-(3,6,9,12,15-пентаокса-5,8,11,14-тетраметилперфтороктадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс N-{[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-аминоэтил}-N-(1H,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)-2-[2-(2-метоксиэтокси)этокси]ацетамида,
гадолиниевый комплекс N-{[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-аминоэтил}-N-(1H,1H,2H,2H,4H,4H-6,9,12,15-тетраокса-8,11,14-триметилперфторпентадецил)-2-[2-(2-метоксиэтокси)этокси]-ацетамида,
гадолиниевый комплекс [1,4,7-трис-(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(2-гидрокси-3-ил)]амида 6-N-(1-O-α-d-карбонилметилманнопираноза)-2-N-(2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридеканоил)-L-лизина,
гадолиниевый комплекс [1,4,7-трис-(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(2-гидрокси-3-ил)]амид 6-N-{2-[2-(2-метоксиэтокси)этокси]ацетил}-2-N-(2Н,2Н,4Н,4Н-3,6,9,12-тетраокса-5,8,11-триметилперфторпентадеканоил)-L-лизина,
гадолиниевый комплекс (2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)амида 6-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-2-N-(2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридеканоил)-L-лизина,
гадолиниевый комплекс [(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)метил]амида 3,5-N,N'-бис[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]бензойной кислоты,
гадолиниевый комплекс 1-[(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)метил]амида N-[1,4,7-трис(карбоксилатометил)-
1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-3-{2-[2-(2-метоксиэтокси)этокси]этил}-L-серина,
гадолиниевый комплекс 5-{[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(2-гидрокси-3-ил)]амидо}-N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}ацетил)-1-[(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)метил]амида L-глутаминовой кислоты,
гадолиниевый комплекс 5-(2-{2-[2-(2-метоксиэтокси)этокси]-этокси}этил)амида, 1-[(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)-метил]амида N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-L-глутаминовой кислоты,
гадолиниевый комплекс [(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфтортридецил)метил]амида L-2-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-амино-4-(2-{2-[2-(2-метоксиэтокси)этокси]-этокси}ацетил)аминомасляной кислоты,
гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)]амида 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-6-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина,
гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)]-амида 2-N-[1,4,7-трис(карбоксилатометил)-1,4,7,10-тетраазациклододекан-10-N-(пентаноил-3-аза-4-оксо-5-метил-5-ил)]-6-N-{2-[2-(2-метоксиэтокси)этокси]ацетил}-L-лизина,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-{2-[2-(2-метоксиэтокси)этокси]-этил}амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-{[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-[2-(1-O-α-d-маннопиранозил)этил]амид}-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс 1,4,7-трис(карбоксилатометил)-10-[(4-гидрокси-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
мононатриевая соль гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-[(4-(R)-карбоксилато-4-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана и мононатриевая соль гадолиниевого комплекса 1,4,7-трис(карбоксилатометил)-10-{[(R)-(2-карбоксилатоэтил)ил]-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид}-1,4,7,10-тетраазациклододекана,
Y комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
Dy-комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
Yb-комплекс 1,4,7-трис(карбоксилатометил)-10-[(3-аза-4-оксо-5-метил-5-ил)-кислота-N-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)амид]-1,4,7,10-тетраазациклододекана,
гадолиниевый комплекс [N-(1H,1H,2H,2H,4H,4H-3,6,9,12-тетраоксаперфторгексадецил)-N-(2-метоксиэтил)]амида 5-[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]-4-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}пентанкарбоновой кислоты,
гадолиниевый комплекс [N-метил-(1Н,1Н,2Н,2Н,4Н,4Н-3,6,9,12-тетраоксаперфторгексадецил)]амида 6-N-(2,3-дигидрокси-N-{[бис(2-{[1-(натрий сульфонатобутил)-3-гидрокси-6-метил-2-оксо-1,2-дигидропиридин-4-карбонил]амино}этил)амино]этил}-терефталил)-2-N-(1-O-α-d-карбонилметилманнопираноза)-L-лизина.
6. Хелаты металлов по пп.1, 4 и 5 для применения при ЯМР- и рентгенодиагностике.
7. Применение хелатов металлов по пп.1, 4 и 5 для получения контрастных веществ для визуализации инфарктов и некрозов.
8. Применение хелатов металлов по пп.1, 4 и 5 для получения контрастных веществ для применения при лимфографии для диагностики изменений в лимфатической системе.
9. Применение хелатов металлов по пп.1, 4 и 5 для получения контрастных веществ для диагностики воспалительных заболеваний.
| ПРОТИВОИЗНОСНАЯ ПРИСАДКА К СМАЗОЧНЫМ СРЕДАМ И ТОПЛИВУ | 2002 |
|
RU2206605C1 |
| JP 2004101881 A, 02.04.2004 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ КОМПЛЕКСЫ С ОСТАТКАМИ САХАРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2280644C2 |

