Данное изобретение относится к новому антагонисту рецептора гистамина и применению антагониста рецептора гистамина для снижения внутричерепного давления (ICP), в частности для предотвращения и лечения повышенного внутричерепного давления и/или вторичной ишемии, вызываемых, например, черепно-мозговой травмой, более конкретно травматическим (TBI) и нетравматическим повреждением головного мозга.
TBI представляет серьезную проблему в развитых странах. В США каждый год около 500000 человек получают черепно-мозговые травмы, которые являются достаточно тяжелыми и требуют госпитализации. Указанные травмы сопровождаются высокой смертностью, при этом примерно у 80000 человек с травматическим повреждением головного мозга жизненно важные функции не восстанавливаются в течение всей последующей жизни, у 5000 человек возникает эпилепсия и 2000 человек постоянно находятся в вегетативном состоянии. В настоящее время TBI является главной причиной смерти и инвалидности у молодежи, и затраты на лечение таких травм составили в 1989 г. более 25 миллиардов долларов.
Первичное необратимое повреждение после черепно-мозговой травмы включает кровоизлияние, контузию, некроз нервных клеток и диффузное повреждение аксонов. Подобное повреждение в сочетании с возможным угнетением сердечно-сосудистой деятельности и дыхания может вызвать острые вторичные поражения, включающие отек (поражающий кровеносные сосуды и/или клетки), вторичное кровотечение, изменение объема церебральной крови (CBV), нарушение ауторегуляции церебрального кровотока (CBF) и ишемию. Отек, кровотечение и повышение CBV увеличивают общий объем головного мозга и вследствие этого внутричерепное давление (ICP). Это в свою очередь может привести к дальнейшему развитию ишемии, инфаркта и в тяжелых случаях к образованию грыжи ствола мозга с возможным острым угнетением дыхания и летальным исходом. Поэтому лечение в случае TBI должно быть направлено на прерывание последовательности патологических явлений, уменьшение объема головного мозга и снижение внутричерепного давления. Целью лекарственного лечения является также предотвращение опасного для жизни вторичного повышения внутричерепного давления, что часто происходит, например, после острой фазы травмы или восстановления сердечной деятельности.
В настоящее время клинические средства снижения внутричерепного давления весьма ограничены. Стандартные схемы лечения включают хирургическое дренирование желудочков головного мозга, поддержание нормального кровяного давления, вливание маннита, гипервентиляцию легких и введение высоких доз барбитурата. Побочные эффекты нехирургических методов лечения включают ишемию головного мозга, вредное воздействие на внутричерепное давление и повышенный риск бактериальных инфекций и сепсиса. Кроме того, различные соединения, обладающие отличительными механизмами воздействия (например, антагонистическое воздействие на брадикинин и кальций, ингибирование окислительного стресса, блокада рецепторов глутамата и противоэпилептическое воздействие), прошли II и III стадии клинических испытаний или все еще находятся на стадии исследования (причем основное внимание уделяется общему результату, а не ICP). До настоящего времени не было получено разрешение на применение соединения для экстренного лечения внутричерепного давления (K.K. Jain, Chapter 4: Neuroprotection in Acute Trauma, 'Neuroprotection in CNS Disorders: Commercial Opportunities'. A Jain PharmaBiotech Report: 65-73, 2000). Совершенно очевидно, что существует потребность в фармацевтических препаратах и/или методах лечения повышенного внутричерепного давления (ICP) и/или вторичной ишемии, вызываемых, в частности, черепно-мозговой травмой, более конкретно травматическим повреждением головного мозга (TBI).
Авторы настоящего изобретения обнаружили, что производные замещенного тетрациклического имидазола общей формулы (I) обладают антагонистической активностью в отношении Н1- и/или Н2-рецепторов гистамина. Кроме того, установлено, что указанные соединения особенно пригодны для снижения внутричерепного давления (ICP), в частности, для профилактики и лечения повышенного внутричерепного давления и/или вторичной ишемии, вызываемых, в частности, черепно-мозговой травмой, более конкретно травматическим (TBI) и нетравматическим повреждением головного мозга.
Кроме того, авторы настоящего изобретения обнаружили, что соединения, являющиеся антагонистами Н1- и/или Н2-рецепторов гистамина (обычно называемые антигистаминными средствами), пригодны также для снижения внутричерепного давления (ICP), в частности, для предотвращения и лечения повышенного внутричерепного давления и/или вторичной ишемии, вызываемых, в частности, травмой головного мозга, более конкретно травматическим (TBI) и нетравматическим повреждением головного мозга.
Отличительной особенностью всех соединений является то, что они способны быстро снижать внутричерепное давление при введение в кровоток млекопитающего, в частности, при внутривенном введении. Весьма полезным и очень важным фактором является то, что, снижая ICP, указанные соединения оказывают незначительное воздействие или вообще не воздействуют на кровяное давление, в частности, такие соединения не снижают кровяное давление, что является наиболее желательным свойством потенциального лекарственного средства.
До настоящего времени не были получены антагонисты Н1- и/или Н2-рецепторов гистамина, предназначенные для снижения ICP, в частности, для снижения ICP после травмы. Mohanty et al., in Journal of the Neurological Sciences, 1989, 90:87-97 установили, что гистамин играет определенную роль в образовании вызванного травмой отека головного мозга. Повышение содержания воды в головном мозге и уровней гистамина в плазме и головном мозге предотвращали путем предварительного введения циметидина, являющегося антагонистом Н2-рецептора гистамина. Однако мейпирамин (антагонист Н1-рецептора гистамина) не вызывал снижения повышенного содержания воды в головном мозге, при этом уровни гистамина в плазме и головном мозге оставались высокими. До сих пор не было исследовано воздействие антагонистов гистамина на ICP, в частности, на экстренное снижение повышенного внутричерепного давления после травмы, а также на кровяное давление.
Не ограничивая себя вышеизложенными теоретическими фактами, авторы настоящего изобретения считают, что с учетом того, что антагонисты рецепторов гистамина способны снижать нормальное внутричерепное давление при отсутствии отека мозга, а также того, что антагонисты рецепторов гистамина не влияют или лишь незначительно влияют на кровяное давление, то есть не вызывают эффект, приписываемый периферической вазодилатации, следует признать, что механизм действия указанных антагонистов не сводится к простому уменьшению отека головного мозга и вазодилатации, в частности, к оказанию таких воздействий, которые, как было известно раньше, присущи антигистаминным средствам.
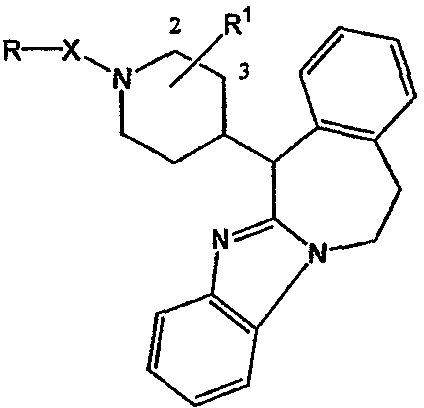
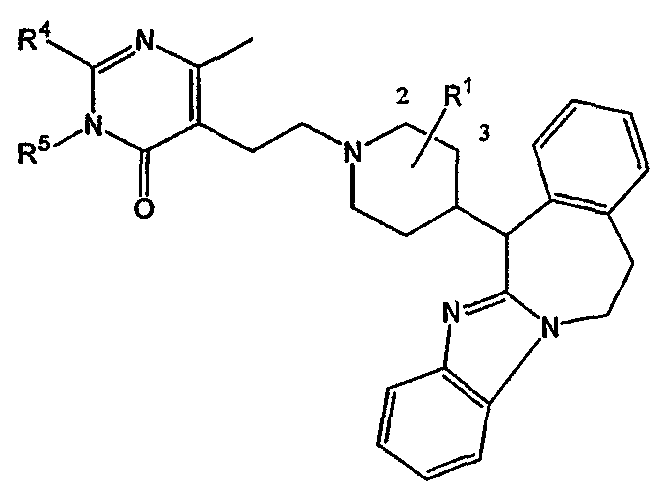
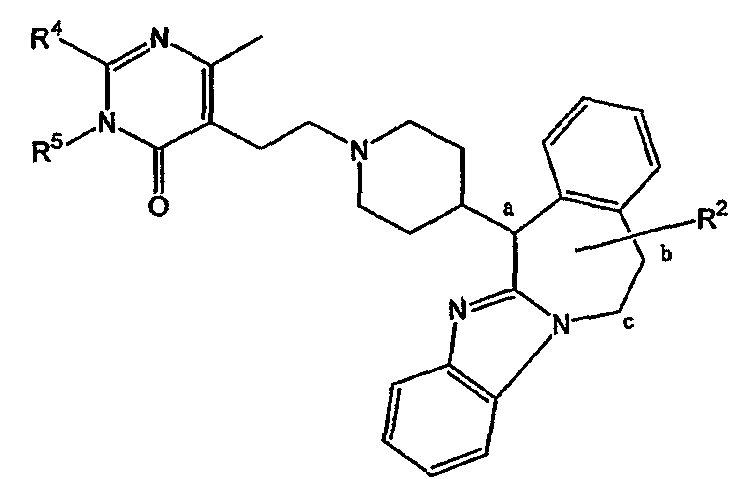
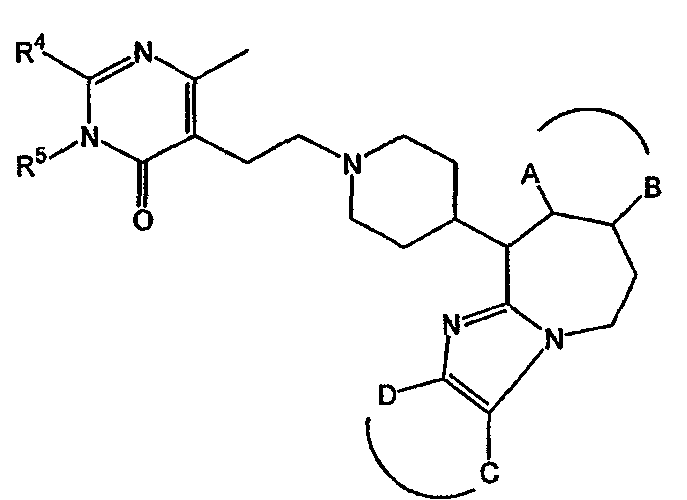
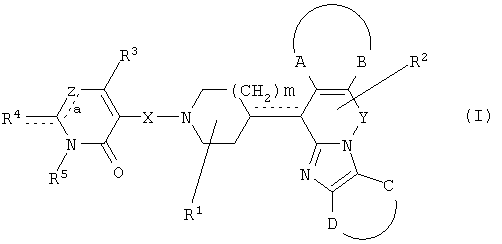
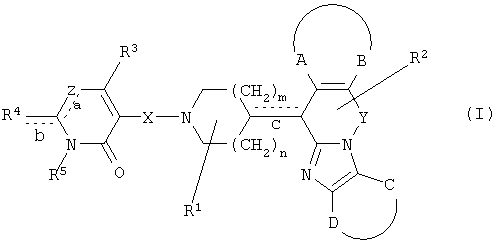
Таким образом, целью настоящего изобретения является создание производных замещенного тетрациклического имидазола общей формулы (I), предназначенных для использования в качестве антагониста гистамина, в частности, в качестве антагониста Н1-рецептора гистамина, более конкретно антагониста Н1- и Н2-рецепторов гистамина,
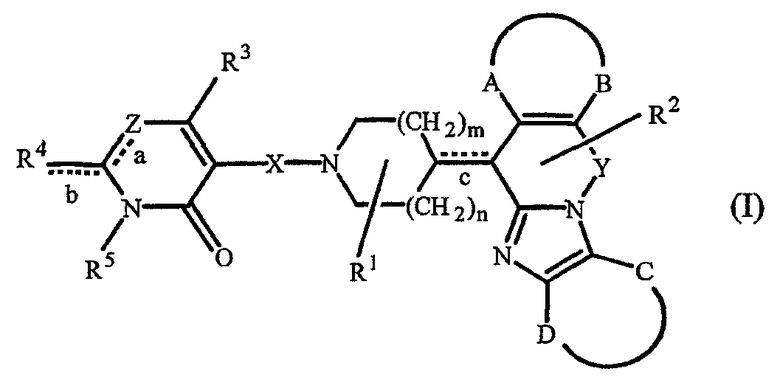
их фармацевтически приемлемых солей присоединения кислоты или основания, стереохимически изомерных форм и N-оксидов, где
m равно 1 или 2;
n равно 0, 1 или 2;
a, b, c независимо означают простую или двойную связь;
Х означает ковалентную связь или двухвалентный С1-6 алкандиильный радикал, в котором одна или несколько групп -СН2- могут быть необязательно заменены -О-, -S-, -CO- или -NR7-,
где R7 означает водород, алкил, Ar, Ar-алкил, Het, Het-алкил, гидроксиалкил, алкилокси, алкилоксиалкил, алкилоксиалкилоксиалкил, аминоалкил, моно- или диалкиламиноалкил, формил, алкилкарбониламиноалкил, алкилкарбонилоксиалкил, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонил, алкиламинокарбонилалкил, гидроксиалкилоксиалкил, аминокарбонил, аминокарбонилалкил, алкилоксикарбонил или алкилкарбонилоксиалкилоксиалкил;
Y означает двухвалентный С1-4 алкандиильный или С2-4 алкендиильный радикал;
Z означает N, когда а означает двойную связь и b означает простую связь, или N-R7, когда а означает простую связь, b означает двойную связь и R7 имеет указанные выше значения;
R1, R2 независимо означают водород, гидрокси, алкил, алкилокси, Ar, Ar-алкил, ди(Ar-)алкил, Het или Het-алкил;
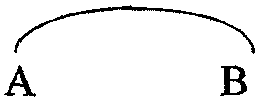
-А-В- независимо означает двухвалентный радикал формулы
где R8 независимо означает водород, галоген, гидрокси, алкил или алкилокси;
Е означает двухвалентный радикал формулы -О-, -S- или -NR7-, где R7 имеет указанные выше значения;
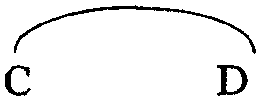
-C-D- независимо означает двухвалентный радикал формулы
где R8 имеет указанные выше значения;
R3 означает водород, галоген, гидрокси, алкил, алкилокси, Ar, Ar-алкил, ди(Ar-)алкил, Het или Het-алкил;
R4 означает водород, алкил, амино, алкиламино, Ar-амино, Het-амино, алкилкарбониламино, Ar-карбониламино, Het-карбониламино, алкиламинокарбониламино, Ar-аминокарбониламино, Het-аминокарбониламино, алкилоксиалкиламино, Ar-оксиалкиламино или Het-оксиалкиламино;
R5 означает водород или алкил;
или R4 и R5 вместе могут образовывать двухвалентный радикал формулы
где R7 и R8 имеют указанные выше значения;
R9, R10 независимо означают водород, алкил, галоген, галогеналкил;
или R9 и R10 вместе могут образовывать двухвалентный радикал формулы -CR8=CR8-CR8=CR8-, и
М означает двухвалентный радикал формулы -СН2-, -О-, -S- или -NR7-, где R7 имеет указанные выше значения.
В данной заявке Ar означает гомоцикл, выбранный из группы нафтила и фенила, которые, каждый, необязательно замещены 1, 2 или 3 заместителями, независимо выбранными из группы, включающей гидрокси, галоген, циано, нитро, амино, моно- или диалкиламино, алкил, галогеналкил, алкилокси, галогеналкилокси, карбоксил, алкилоксикарбонил, аминокарбонил и моно- или диалкиламинокарбонил. Ar предпочтительно означает нафтил или фенил, необязательно замещенный 1 заместителем, который независимо выбран из группы, включающей галоген или алкил.
В данной заявке Het означает моноциклический гетероцикл, выбранный из группы, включающей пирролил, пиразолил, имидазолил, фуранил, тиенил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридинил, пиримидинил, пиразинил и пиридазинил, или бициклический гетероцикл, выбранный из группы, включающей хинолинил, хиноксалинил, индолил, бензимидазолил, бензоксазолил, бензизоксазолил, бензотиазолил, бензизотиазолил, бензофуранил и бензотиенил; причем каждый моноциклический и бициклический гетероцикл может быть необязательно замещен у атома углерода галогеном, гидрокси, алкилом или алкилокси. Het предпочтительно означает пиридинил, пиразинил или индолил.
В данной заявке алкил означает насыщенный углеводородный радикал с прямой или разветвленной цепью, имеющий 1-6 атомов углерода; циклический насыщенный углеводородный радикал, имеющий 3-6 атомов углерода; циклический насыщенный углеводородный радикал, имеющий 3-6 атомов углерода, присоединенных к насыщенному углеводородному радикалу с прямой или разветвленной цепью, имеющему 1-6 атомов углерода, где каждый атом углерода может быть необязательно замещен галогеном, гидрокси, алкилокси или оксо. Алкил предпочтительно означает метил, этил или циклогексилметил.
В данной заявке галоген является заместителем, выбранным из группы, включающей фтор, хлор, бром и йод; галогеналкил означает насыщенный углеводородный радикал с прямой или разветвленной цепью, имеющий 1-6 атомов углерода, или циклический насыщенный углеводородный радикал, имеющий 3-6 атомов углерода, в котором один или несколько атомов углерода замещены одним или несколькими атомами галогенов. Галоген предпочтительно является фтором или хлором, и галогеналкил предпочтительно является трифторметилом.
Предпочтительную группу соединений составляют соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, в которых -А-В- означает двухвалентный радикал формулы (а-1) или (а-3), где Е означает двухвалентный радикал формулы -О-, -S- или -NR7-, где R7 означает водород, R8 означает водород, -C-D- означает двухвалентный радикал формулы (b-1) или (b-2), где R8 означает водород и Y означает двухвалентный радикал формулы -СН2-, -СН2-СН2- или -СН=СН-.
Другую группу предпочтительных соединений формулы (I) составляют соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, в которых m и n, оба равны 1.
Другую группу предпочтительных соединений формулы (I) составляют соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, в которых R1 и R2 независимо означают водород, алкил, Ar-алкил, Het или Het-алкил.
Еще одну группу предпочтительных соединений формулы (I) составляют соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, в которых Х означает двухвалентный радикал формулы -СН2-СН2- или -СН2-СН2-СН2-.
Еще одну группу предпочтительных соединений формулы (I) составляют соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, в которых R3 означает водород или алкил, Z означает N-R7, где R7 означает водород или алкил, а означает простую связь и b означает двойную связь, и R4 и R5 вместе образуют двухвалентный радикал формулы (с-1), (с-3), (с-5), (с-7), (с-8) или (с-10), где R7 и R8 означают водород.
Еще одну группу предпочтительных соединений формулы (I) составляют соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, в которых R3 означает водород или алкил, Z означает N-R7, где R7 означает водород или алкил, а означает простую связь и b означает двойную связь, R4 и R5 вместе образуют двухвалентный радикал формулы (с-1), (с-3), (с-5), (с-7), (с-8) или (с-10), где R7 и R8 означают водород и R9 и R10 вместе образуют двухвалентный радикал формулы -CR8=CR8-CR8=CR8-, где R8 означает водород.
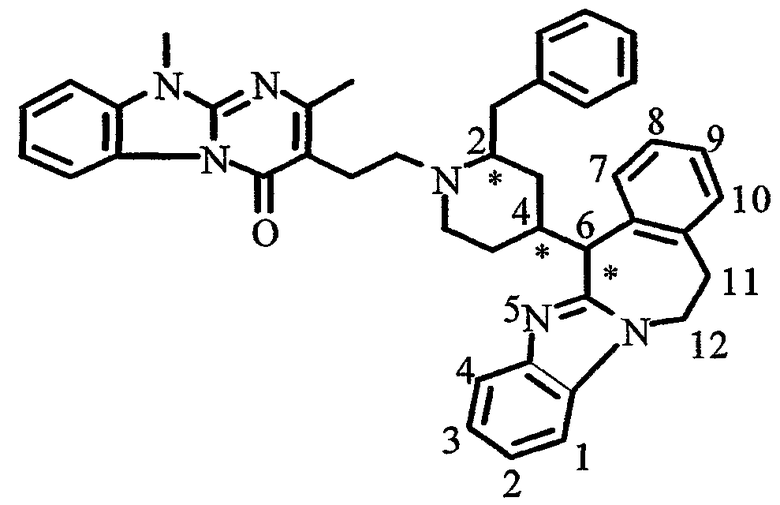
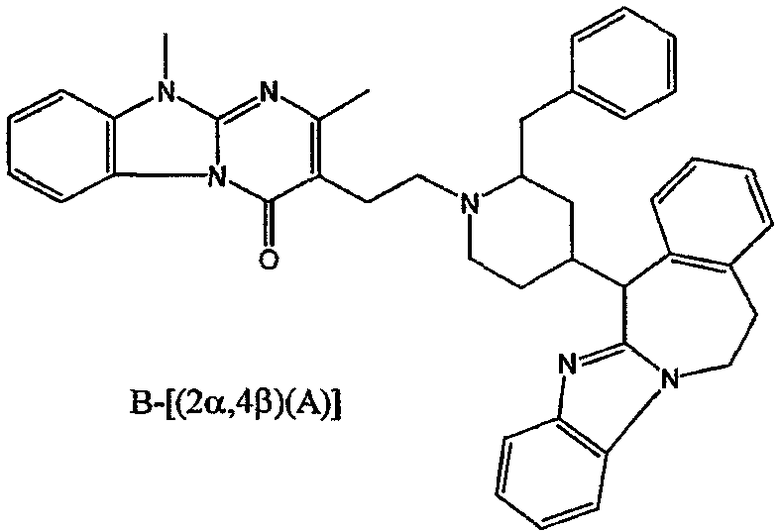
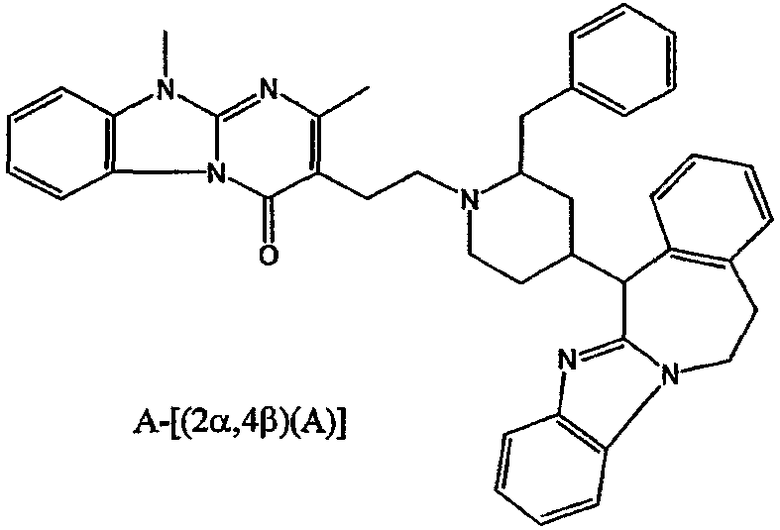
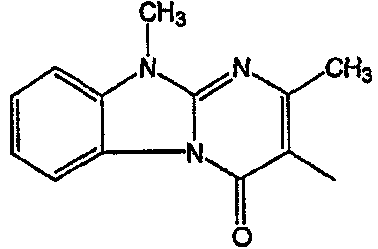
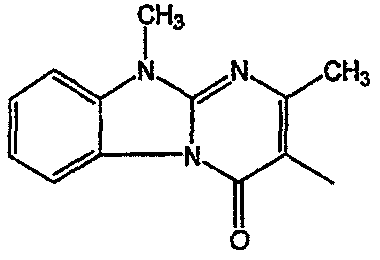
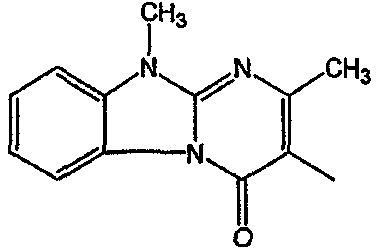
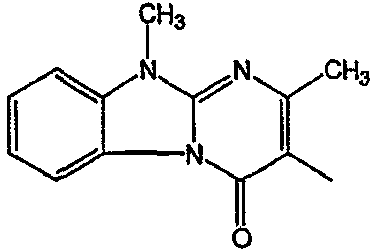
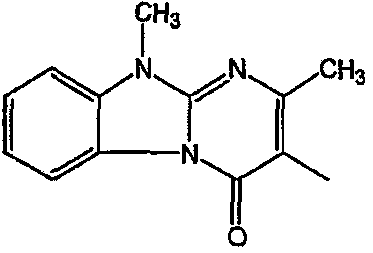
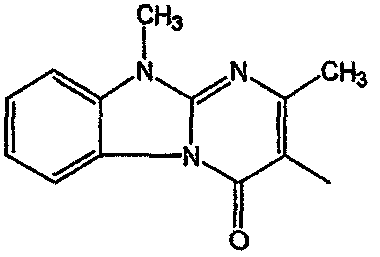
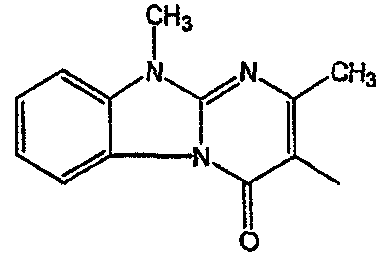
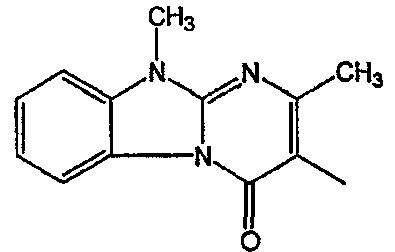
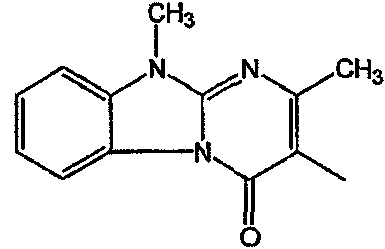
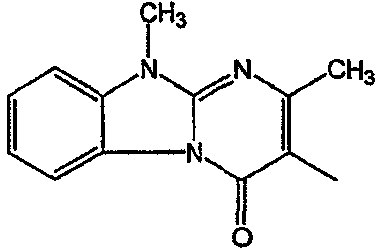
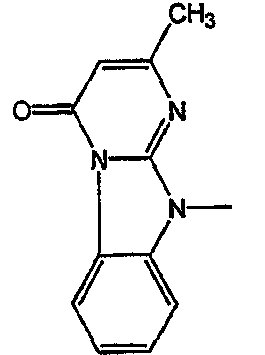
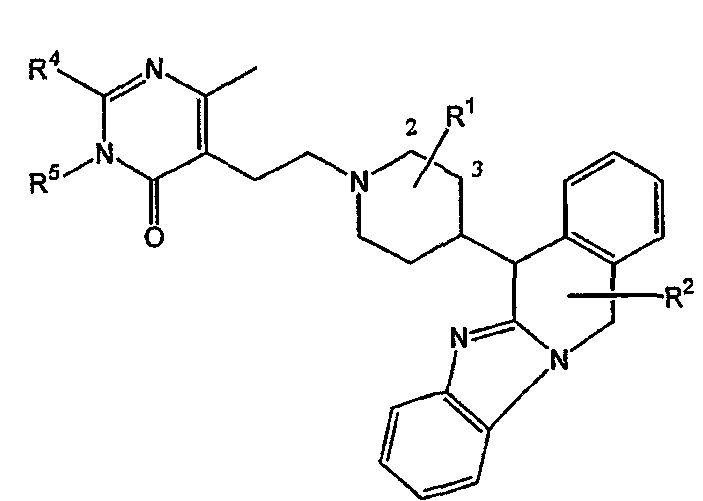
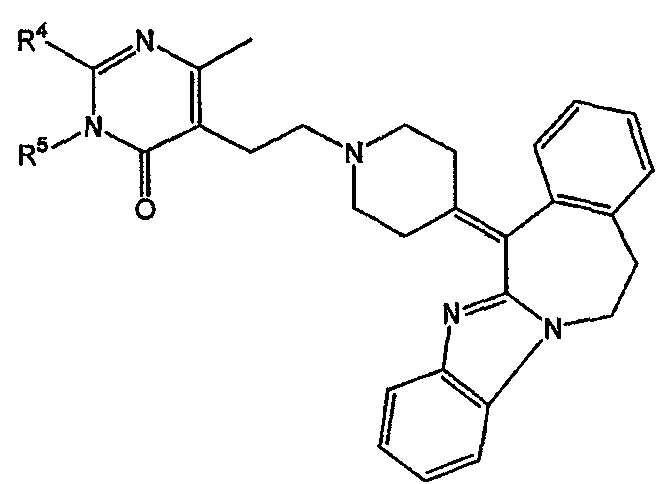
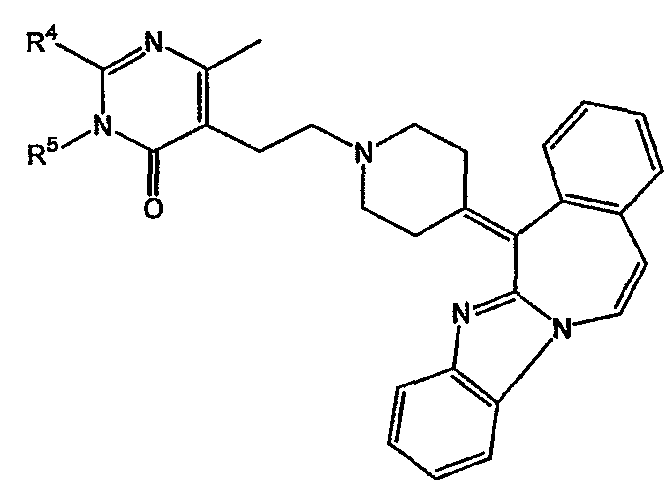
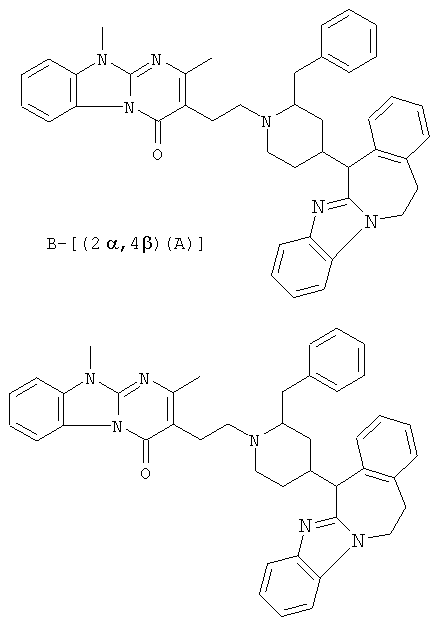
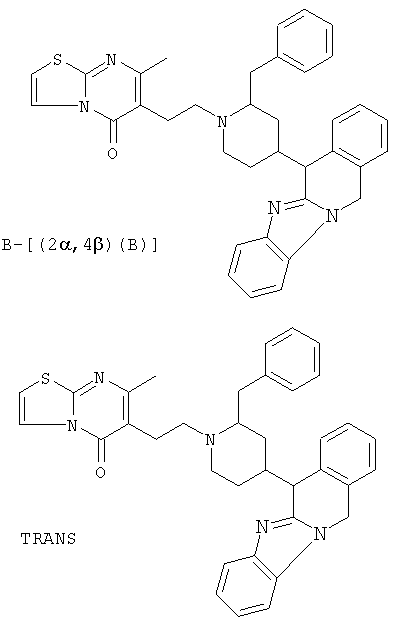
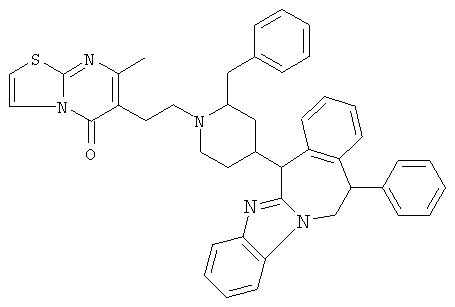
В частности, наиболее предпочтительным является соединение 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-он, его фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды.
Фармацевтически приемлемые кислотно-аддитивные соли включают терапевтически активные нетоксичные кислотно-аддитивные соли, которые могут образовывать соединения формулы (I). Указанные кислотно-аддитивные соли можно получить, обрабатывая основную форму соединений формулы (I) соответствующими кислотами, например, неорганическими кислотами, такими как галогенводородная кислота, в частности, хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; органическими кислотами, такими как уксусная кислота, гидроксиуксусная кислота, пропановая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, цикламиновая кислота, салициловая кислота, п-аминосалициловая кислота и памовая кислота.
Соединения формулы (I), содержащие кислотные протоны, могут быть также превращены в терапевтически активные нетоксичные основно-аддитивные соли в результате обработки соответствующими органическими и неорганическими основаниями. Соответствующие основные соли включают, например, соли аммония, соли щелочных и щелочноземельных металлов, в частности, соли лития, натрия, калия, магния и кальция, соли с органическими основаниями, такие как соли бензатина, N-метил-D-глюкамина, гибрамина, и соли с аминокислотами, такими как аргинин и лизин.
И наоборот, указанные кислотно- или основно-аддитивные соли могут быть превращены в свободные формы в результате обработки соответствующим основанием или кислотой.
Термин "аддитивная соль" в значении, используемом в данной заявке, означает также сольваты, которые могут образовывать соединения формулы (I), и их соли. Такими сольватами являются, например, гидраты и алкоголяты.
Среди кислотно-аддитивных солей наиболее предпочтительным соединением является гидрат (1:1) (Е)-2-бутендиоата 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-она (2:3), включая все стереоизомерные формы указанного соединения.
Особенно предпочтительными соединениями являются (А)[(2α,4β)(А)]-энантиомер, (В)[(2α,4β)(А)]-энантиомер и смесь указанных энантиомеров соединений 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-β][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-она и гидрата (1:1) (Е)-2-бутендиоата 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-β][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-она (2:3).
N-оксиды соединений формулы (I) являются соединениями формулы (I), в которых один или несколько атомов азота окислены в так называемые N-оксиды, в частности, такие N-оксиды, в которых один или несколько атомов азота пиперидинильного радикала в формуле (I) представляют собой N-окислен.
Термин "стереохимические изомерные формы" в используемом значении означает все возможные изомерные формы, которые могут иметь соединения формулы (I). За исключением особо оговоренных случаев, данное химическое обозначение соединений включает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. В частности, стереогенные центры могут иметь R- или S-конфигурацию; заместители двухвалентных циклических (частично) насыщенных радикалов могут иметь цис- или транс-конфигурацию. Соединения, включающие двойные связи, могут иметь Е- или Z-стереохимическую структуру в положении указанной двойной связи. Совершенно очевидно, что стереохимически изомерные формы соединений формулы (I) входят в объем данного изобретения.
В соответствии с условными обозначениями номенклатуры CAS при наличии в молекуле двух стереогенных центров известной абсолютной конфигурации идентификатор R или S присваивается (на основе правила последовательности Кана-Инголда-Прелога) хиральному центру с наименьшим номером, который является центром отсчета. Для обозначения конфигурации второго стереогенного центра используют относительные идентификаторы [R*,R*] или [R*,S*], где R* всегда определяется как центр отсчета, [R*,R*] обозначает центры с одинаковой хиральностью и [R*,S*] обозначает центры с разной хиральностью. Например, если хиральный центр с наименьшим номером в молекуле имеет S-конфигурацию и второй центр имеет R-конфигурацию, идентификатор стереоизомера должен определяться как S-[R*,S*]. При использовании символов "α" и "β": положение заместителя с наивысшим приоритетом в положении асимметричного атома углерода в кольцевой системе, имеющей наименьший номер кольца, всегда условно находится в положении "α" средней плоскости, определяемой кольцевой системой. Положение заместителя с наивысшим приоритетом в положении другого асимметричного атома углерода в кольцевой системе относительно положения заместителя с наивысшим приоритетом в положении атома отсчета обозначается символом "α", если указанный заместитель находится с той же стороны средней плоскости, определяемой кольцевой системой, или символом "β", если указанный заместитель находится с другой стороны средней плоскости, определяемой кольцевой системой.
Когда связь в положении "с" является простой связью, соединения формулы (I) и некоторые промежуточные соединения имеют по крайней мере два стереогенных центра в своей структуре. Когда R1 не является водородом, моноциклическое N-кольцо в формуле (I) имеет еще один стереогенный центр. Таким образом можно получить 8 стереохимически разных структур.
Соединения формулы (I), полученные описанными ниже способами, можно синтезировать в форме рацемических смесей энантиомеров, которые могут быть отделены друг от друга при помощи методов разделения, известных в данной области. Рацемические соединения формулы (I) можно превратить в соответствующие диастереомерные соли, осуществляя взаимодействие с приемлемой хиральной кислотой. Указанные диастереомерные соли затем разделяют, например, селективной или фракционной кристаллизацией и выделяют из них энантиомеры путем щелочной обработки. Альтернативный метод разделения энантиомерных форм соединений формулы (I) включает применение жидкостной хроматографии с использованием хиральной стационарной фазы. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ при условии выполнения стереоспецифического взаимодействия. Если необходимо получить специфический стереоизомер, указанное соединение предпочтительно синтезируют стереоспецифическими методами получения. При осуществлении указанных методов преимущественно используют энантиомерно чистые исходные вещества.
Соединения формулы (I) могут также существовать в таутомерной форме. Несмотря на то, что такие формы не отражены в полной мере в приведенной выше формуле, они входят в объем настоящего изобретения. Например, соединения формулы (I), в которой R5 означает Н, могут существовать в соответствующей таутомерной форме.
В объем настоящего изобретения входят также производные соединения (обычно именуемые "пролекарствами") фармакологически активных соединений по данному изобретению, которые расщепляются in vivo с образованием соединений по данному изобретению. Пролекарства обычно (но не всегда) оказывают менее сильное воздействие на рецептор-мишень по сравнению с соединениями, в которые они превращаются в результате расщепления. Пролекарства особенно пригодны в тех случаях, когда химические или физические свойства желаемого соединения затрудняют или делают неэффективным процесс его введения. Например, требуемое соединение может плохо растворяться, плохо переноситься через эпителий слизистой оболочки или иметь нежелательно короткий период полувыведения из плазмы. Пролекарства более подробно рассмотрены в статьях Stella, V.J. et al., "Prodrugs", Drug Delivery Systems, 1985, pp. 112-176, and Drugs, 1985, 29, pp. 455-473.
Пролекарственные формы фармакологически активных соединений по данному изобретению обычно представляют собой соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, имеющие кислотную группу, которая этерифицирована или амидирована. К таким этерифицированным кислотным группам относятся группы формулы -COORх, где Rх означает С1-6 алкил, фенил, бензил или одну из нижеследующих групп:
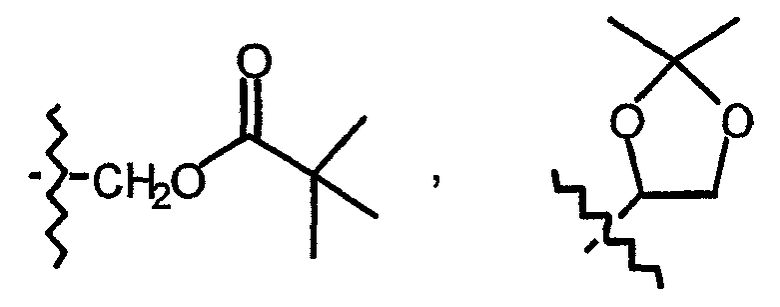
Амидированные группы включают группы формулы -CONRyRz, где Ry означает Н, С1-6 алкил, фенил или бензил и Rz означает -ОН, Н, С1-6 алкил, фенил или бензил.
Соединения по данному изобретению, содержащие аминогруппу, могут быть получены с использованием кетона или альдегида, такого как формальдегид, с образованием основания Манниха. Данное основание гидролизуют при помощи реакционной кинетики первого порядка в водном растворе.
Соединения формулы (I) можно получить, выполняя последовательность стадий, каждая из которых известна специалисту в данной области. Получение указанных соединений описано в одновременно рассматриваемой заявке, которая включена в данное описание изобретения в качестве ссылки.
Помимо снижения внутричерепного давления (ICP) соединения формулы (I) и их производные пригодны также для лечения других заболеваний, обусловленных Н1- и Н2-рецепторами гистамина, в частности, для иммуномодуляции у млекопитающих, подавления повышенной чувствительности и/или воспалительных реакций, лечения и предотвращения аллергических заболеваний, таких как ринит, крапивница, астма, анафилаксия и тому подобные, и лечения заболеваний желудочно-кишечного тракта, таких как язвы, диспепсия, разные рефлюксы и тому подобные. Таким образом, данное изобретение относится также к применению антагониста рецептора гистамина формулы (I) и его производных для получения лекарственных средств для иммуномодуляции у млекопитающих, подавления повышенной чувствительности и/или воспалительных реакций, лечения и профилактики аллергических заболеваний и заболеваний желудочно-кишечного тракта.
Другим объектом данного изобретения является новое применение антагонистов Н1- и/или Н2-рецепторов гистамина для экстренного снижения внутричерепного давления (ICP), в частности, повышенного ICP, более конкретно, критически повышенного ICP и/или предотвращения повышения ICP и вторичной ишемии вследствие травмы головного мозга. При этом особенно важно, что антагонисты Н1- и/или Н2-рецепторов гистамина не влияют на кровяное давление или вызывают незначительное понижение или повышение кровяного давления.
В соответствии с данным изобретением антагонисты Н1- и/или Н2-рецепторов гистамина являются соединениями и их производными формулы (I) или известными антагонистами Н1- и/или Н2-рецепторов гистамина, которые образуют обособленную и ограниченную группу лекарственных средств, уже известных в данной области.
До настоящего времени антагонисты Н1-рецептора гистамина обычно использовались для иммуномодуляции у млекопитающих и подавления повышенной чувствительности и/или воспалительных реакций. В частности, антагонист Н1-рецептора гистамина выбран из группы, включающей акривастин, алимемазин, антазолин, астемизол, азатадин, азеластин, бромфенирамин, буклизин, карбиноксамин, каребастин, цетиризин, хлорциклизин, хлорфенирамин, циннаризин, клемастин, клемизол, клоцинизин, клонидин, циклизин, ципрогептадин, дескарбоэтоксилоратидин, дексхлорфенирамин, дименгидринат, диметинден, диметотиазин, дифенгидрамин, дифенилпиралин, доксиламин, эбастин, эфлетиризин, эпинастин, фексофенадин, гидроксизин, кетотифен, левокабастин, лоратидин, меклизин, меквитазин, метдилазин, миансерин, мизоластин, ниапразин, ноберастин, норастемизол, оксатомид, оксомемазин, фенбензамин, фенирамин, пикумаст, прометазин, пириламин, темеластин, терфенадин, тримепразин, трипеленнамин и трипролидин, их производные и смеси двух или более любых вышеуказанных средств.
До настоящего времени антагонисты Н2-рецептора гистамина обычно использовали для лечения млекопитающих, страдающих некоторыми заболеваниями желудочно-кишечного тракта, такими как язвы, диспепсия, разные рефлюксы и тому подобные. В частности, антагонист Н2-рецептора гистамина выбран из группы, включающей ранитидин, циметидин, фамотидин, низатидин, тиотидин, золантидин, их производные и смеси двух или более любых вышеуказанных средств.
Кроме того, антагонисты рецепторов гистамина могут оказывать антагонистическое действие на Н1- и/или Н2-рецепторы гистамина; к таким антагонистам, в частности, относятся ритансерин или соединения формулы (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды.
Несмотря на то, что все соединения вызывают значительное снижение ICP, установлено, что нижеследующие соединения не влияют на кровяное давление или вызывают незначительное понижение кровяного давления: кетотифен, хлорциклизин, прометазин, пириламин, дифенилгидрамин, хлорфенирамин и золантадин.
Для оценки антагонистического воздействия на гистамин соединений по настоящему изобретению могут быть выполнены исследования in vitro с моделированием соответствующих рецепторов.
Для оценки биологической активности соединений по настоящему изобретению могут быть выполнены исследования in vivo. Для этой цели была создана клинически обоснованная модель травматического повреждения головного мозга у крыс (модель закрытой черепно-мозговой травмы) и использована для испытания соединений по данному изобретению (K. Engelborghs et al., Temporal changes in intracranial pressure in a modified experimental model of closed head injury, J. Neurosurg., 89: 796-806, 1998; K. van Rossem et al., Brain oxygenation after experimental closed head injury, Adv. Exp. Med. Biol., 471: 209-215, 1999; K. Engelborghs et al., Impaired autoregulation of cerebral blood flow in an experimental model of traumatic brain injury, J. Neurotrauma, 17(8):667-677, 2000). В одном исследовании повышенное внутричерепное давление вызывали у кроликов, повреждая холодом кору головного мозга.
Антагонисты рецепторов гистамина по данному изобретению, включающие соединения формулы (I) и известные в настоящее время антагонисты Н1-, Н2- и Н1/Н2-рецепторов гистамина, могут быть использованы для получения разных фармацевтических препаратов. В качестве примеров приемлемых композиций можно привести все композиции, обычно используемые для получения лекарственных средств, предназначенных для системного введения. Для получения фармацевтических композиций по данному изобретению эффективное количество определенного соединения, необязательно в форме аддитивной соли, используемого в качестве активного ингредиента, однородно смешивают с фармацевтически приемлемым носителем, который может иметь разные формы в зависимости от формы требуемого препарата. Указанные фармацевтические композиции предпочтительно получают в виде стандартной дозированной лекарственной формы, пригодной, в частности, для перорального или парентерального введения в виде инъекций. Например, при получении композиции в виде дозированной лекарственной формы для перорального введения можно использовать любые фармацевтические среды, такие как, например, вода, гликоли, масла, спирты и тому подобные, в случае жидких пероральных препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, разбавители, смазывающие вещества, связывающие вещества, вещества, улучшающие распадаемость, и тому подобные, в случае порошков, пилюль, капсул и таблеток. Благодаря легкости введения таблетки и капсулы являются наиболее предпочтительными дозированными лекарственными формами для перорального введения, в которых используют твердые фармацевтические носители. При получении композиций для парентерального введения в качестве носителя обычно используют стерильную воду, по крайней мере в значительной степени, хотя для улучшения растворимости в композицию могут быть включены другие ингредиенты. Например, можно получить инъекционные растворы, в которых носителем является физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Кроме того, можно получить инъекционные суспензии, в которых могут быть использованы соответствующие жидкие носители, суспендирующие агенты и тому подобные. В объем настоящего изобретения входят также твердые препараты, которые непосредственно перед применением должны быть превращены в жидкие препараты.
Вышеуказанные фармацевтические композиции предпочтительно получают в виде стандартной дозированной лекарственной формы, которая характеризуется легкостью введения и однородностью доз. Стандартная дозированная лекарственная форма в используемом здесь значении представляет физически раздельные формы, используемые в качестве унифицированных доз, причем каждая форма содержит заранее определенное количество активного ингредиента, необходимое для достижения желаемого терапевтического эффекта, в сочетании с требуемым фармацевтическим носителем. Примерами таких дозированных лекарственных форм являются таблетки (включая таблетки с насечкой или таблетки с покрытием), капсулы, пилюли, пакетики с порошком, облатки, суппозитории, инъекционные растворы или суспензии и тому подобные, а также несколько вышеуказанных форм в одной упаковке, отделенных друг от друга. Для простого и быстрого введения вышеуказанные фармацевтические композиции наиболее предпочтительно получают в виде раствора или суспензии для инъекций или вливаний.
Приведенные ниже примеры иллюстрируют настоящее изобретение, не ограничивая его объем.
Экспериментальная часть
Для некоторых соединений не была экспериментально определена абсолютная стереохимическая конфигурация одного или нескольких стереогенных атомов углерода. В подобных случаях стереохимически изомерная форма, выделенная первой, обозначена как "А", и выделенная второй, обозначена как "В" без дальнейшей ссылки на действительную стереохимическую конфигурацию. Однако указанные изомерные формы "А" и "В" могут быть точно охарактеризованы специалистом в данной области методами, хорошо известными в данной области, такими как, например, дифракция рентгеновских лучей.
Например, для 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-она можно определить 8 возможных стереохимически изомерных форм, которые представлены ниже:
В данном описании изобретения "DMF" (ДМФА) означает N,N-диметилформамид, "DIPE" означает диизопропиловый эфир, "THF" (ТГФ) означает тетрагидрофуран, "MIBK" означает метилизобутилкетон, "DIPA" означает диизопропиламин.
А. Получение промежуточных соединений
Пример А1
а) Получение промежуточного соединения 1
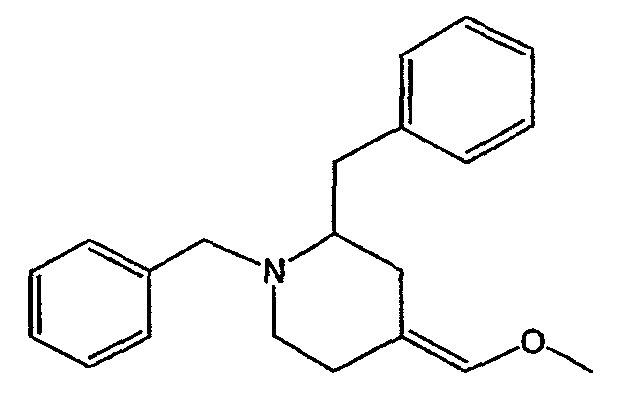
Использовали сухую стеклянную посуду. Смесь хлорида (метоксиметил)трифенилфосфония (0,35 моль) в ТГФ (чистота для анализа) (молекулярные сита) (2 л) перемешивали при -50°С в потоке N2. Затем по каплям добавляют 2,5 M BuLi/гексан (0,35 моль) и смесь перемешивают при -25°С в течение 30 минут. Раствор 1,2-бис(фенилметил)-4-пиперидинона (0,35 моль) в ТГФ добавляли по каплям при -25°С. Смесь оставляли нагреваться до комнатной температуры, перемешивали при комнатной температуре в течение ночи и разлагали водой. Органический растворитель выпаривали. Водный концентрат экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/СН3ОН, 97,5/2,5). Чистые фракции собирали и растворитель выпаривали. Выход: 121 г энантиомерной смеси 4-(метоксиметилен)-1,2-бис(фенилметил)пиперидина (промежуточное соединение 1) (100%).
b) Получение промежуточного соединения 2
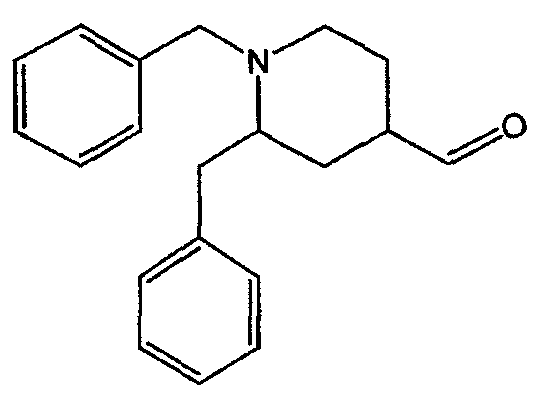
Смесь промежуточного соединения 1 (0,35 моль) в ТГФ (500 мл) перемешивали до полного растворения. Добавляли Н2О (900 мл) и 38% HCl (чистота для анализа) (100 мл). Смесь перемешивали и нагревали с обратным холодильником в течение 3 часов. Органический растворитель выпаривали. Водный концентрат подщелачивали К2СО3 и экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/СН3ОН, 97/3). Чистые фракции собирали и растворитель выпаривали. Выход: 81 г энантиомерной смеси 1,2-бис(фенилметил)-4-пиперидинкарбоксальдегида (промежуточное соединение 2) (79%).
с) Получение промежуточного соединения 3
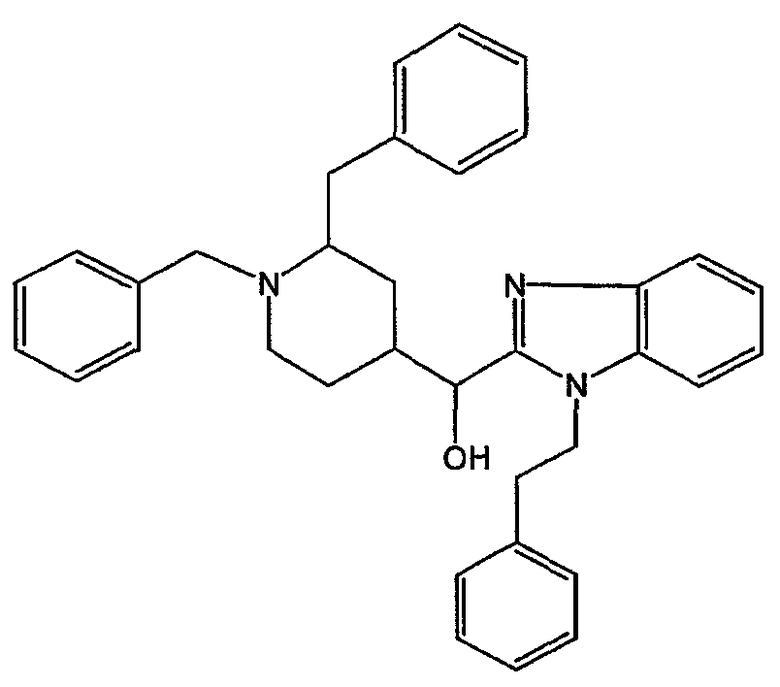
Смесь DIPA (0,33 моль) в ТГФ (чистота для анализа) (предварительно высушенный на молекулярных ситах) (2 л) перемешивали при -78°С в потоке N2. Затем по каплям добавляли 2,5 М BuLi/гексан (0,276 моль). Смесь перемешивали при -78°С в течение 15 минут. По каплям добавляли раствор 1-(2-фенилэтил)-1Н-бензимидазола (0,276 моль) в ТГФ. Смесь перемешивали при -78°С в течение 1 часа. По каплям добавляли раствор промежуточного соединения 2 (0,276 моль) в ТГФ. Смесь перемешивали при -78°С в течение 1 часа, оставляли нагреваться до комнатной температуры, перемешивали при комнатной температуре в течение ночи и разлагали водой. Органический растворитель выпаривали. Водный концентрат экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/СН3ОН, от 95/5 до 90/10). Чистые фракции собирали и растворитель выпаривали. Выход: 113 г α-[1,2-бис(фенилметил)-4-пиперидинил]-1-(2-фенилэтил)-1Н-бензимидазол-2-метанола (промежуточное соединение 3) (79%).
d) Получение промежуточного соединения 4
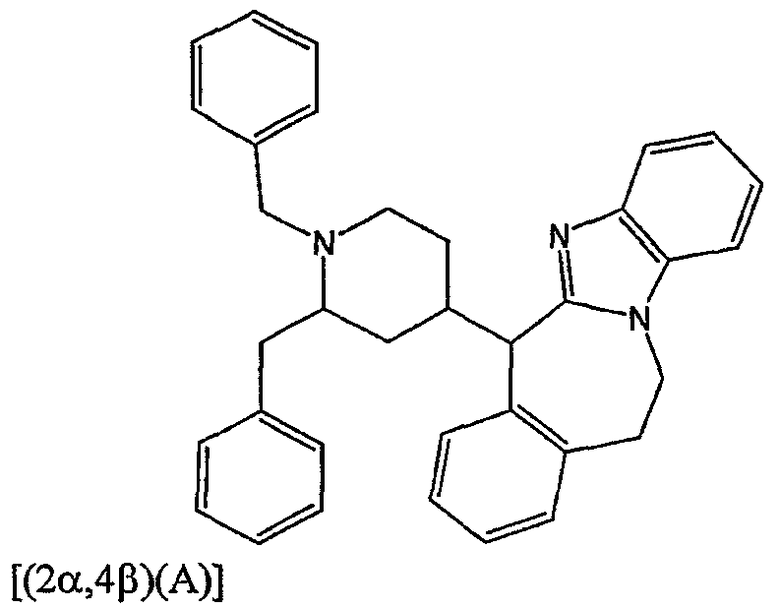
Смесь промежуточного соединения 3 (0,22 моль) в трифторметансульфоновой кислоте (750 мл) перемешивали при 110°С в течение 7 часов. Затем смесь охлаждали, выливали на лед, подщелачивали 50% NaOH и экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток кристаллизовали из CH3CN. Смесь фильтровали. Осадок и фильтрат очищали отдельно хроматографией на колонке с силикагелем (элюент: CH2Cl2/СН3ОН, от 98,5/1,5 до 95/5). Собирали четыре чистые фракции и их растворители выпаривали. Остатки кристаллизовали из CH3CN. Осадки отфильтровывали и сушили. Выход: 16 г фракции 1 [(2α,4β(А)]-6-[1,2-бис(фенилметил)-4-пиперидинил]-11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепина (промежуточное соединение 4) (14,6%), 19,5 г фракции 2 [(2α,4β)(В)]-6-[1,2-бис(фенилметил)-4-пиперидинил]-11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепина (17,8%), 8,66 г фракции 3 [(2α,4α)(А)]-6-[1,2-бис(фенилметил)-4-пиперидинил]-11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепина (7,9%) и 7,74 г фракции 4 [(2α,4α)(В)]-6-[1,2-бис(фенилметил)-4-пиперидинил]-11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепина (8,9%).
е) Получение промежуточного соединения 5
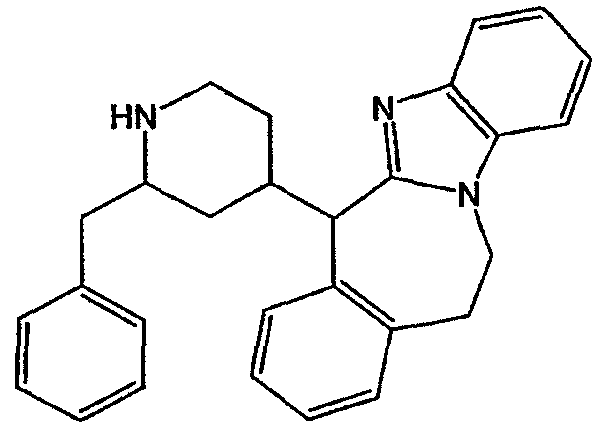
Смесь промежуточного соединения 4 (0,0305 моль) в метаноле (150 мл) гидрировали при 50°С в течение ночи, используя 10% Pd/C (1 г) в качестве катализатора. После поглощения Н2 (1 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток кристаллизовали из СН3CN. Осадок отфильтровывали и сушили. Выход: 11,66 г [(2α,4β)(А)]-11,12-дигидро-6-[2-(фенилметил)-4-пиперидинил]-6Н-бензимидазо[2,1-b][3]бензазепина (промежуточное соединение 5) (94%).
Пример А2
а) Получение промежуточного соединения 6
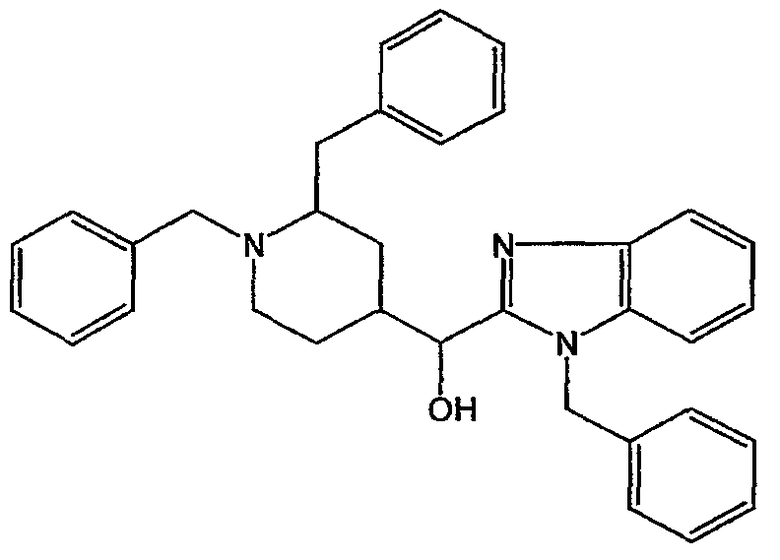
Использовали сухую стеклянную посуду. Смесь DIPA (0,22 моль) в ТГФ (чистота для анализа) (предварительно высушенный на молекулярных ситах) (1400 мл) перемешивали при -70°С в потоке N2. Затем по каплям добавляют 2,5 М BuLi (0,185 моль) и смесь перемешивали при -70°С в течение 15 минут. 1-(фенилметил)-1Н-бензимидазол (0,185 моль), растворенный в ТГФ, добавляли по каплям при -70°С и смесь перемешивали при -70°С в течение 1 часа. Промежуточное соединение 2 (0,185 моль), растворенное в ТГФ, добавляли по каплям при -70°С. Смесь перемешивали при -70°С в течение 1 часа, затем медленно доводили до комнатной температуры, перемешивали при комнатной температуре в течение ночи и разлагали Н2О. Органический растворитель выпаривали. Водный концентрат экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/СН3ОН, 95/5). Чистые фракции собирали и растворитель выпаривали. Выход: 91 г промежуточного соединения 6 (98%).
b) Получение промежуточного соединения 7
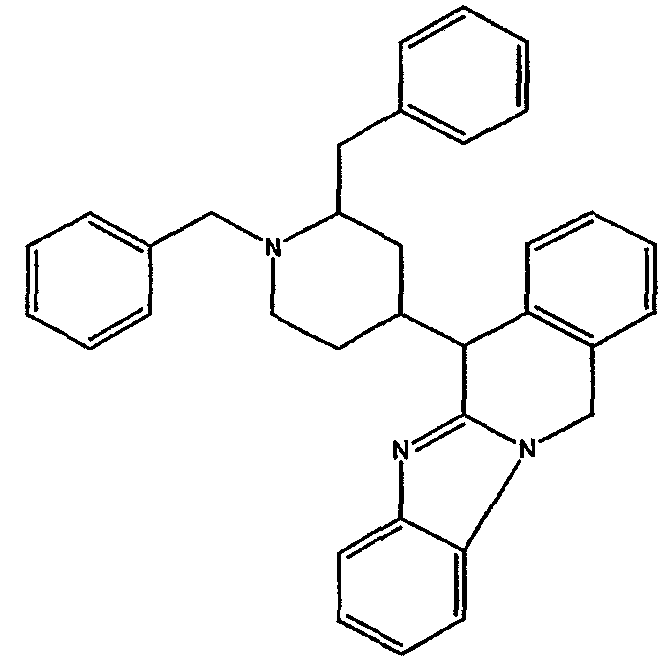
Смесь промежуточного соединения 6 (0,18 моль) в трифторметансульфоновой кислоте (700 мл) перемешивали при 120°С в потоке N2 в течение 18 часов. Смесь охлаждали, выливали на лед, подщелачивали 50% NaOH и экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/(СН3ОН/NH3), 99/1). Чистые фракции собирали и растворитель выпаривали. Выход: 40 г промежуточного соединения 7 (46%).
с) Получение промежуточного соединения 8
и получение промежуточного соединения 9
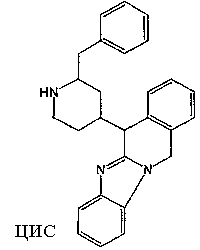
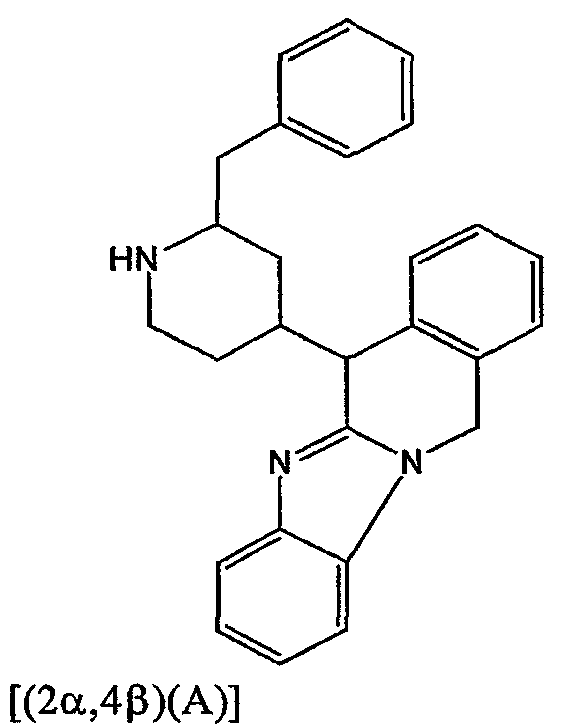
Смесь промежуточного соединения 7 (0,081 моль) в метаноле (200 мл) гидрировали при 50°С, используя 10% Pd/C (2 г) в качестве катализатора. После поглощения Н2 (1 экв.) катализатор отфильтровывали и фильтрат упаривали. Полученную фракцию очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/(СН3ОН/NH3), 97/3). Собирали две чистые фракции и их растворители выпаривали. Выход: фракция 1 и 12,5 г промежуточного соединения 9 (цис-изомеры) (36%). Фракцию 1 кристаллизовали из CH3CN. Осадок отфильтровывали и сушили. Выход: 4,44 г промежуточного соединения 8 (14%) в виде [(2α,4β)(А)]-рацемата.
Пример А3
а) Получение промежуточного соединения 10
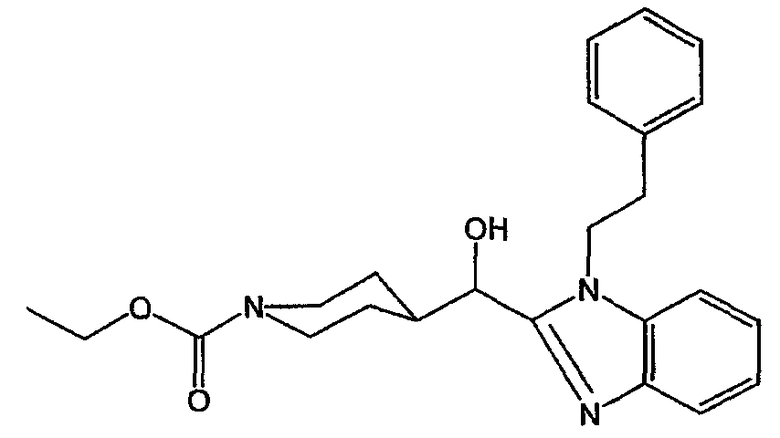
Смесь DIPA (0,1 моль) в ТГФ (100 мл) перемешивали в потоке N2. Смесь охлаждали до -70°С и порциями добавляли 2,5 М BuLi/гексан (40 мл). Температуру доводили до -30°С, перемешивая смесь в течение 10 минут. Затем смесь охлаждали до -70°С. При указанной температуре по каплям добавляли раствор 1-(фенилэтил)-1Н-бензимидазола (0,1 моль) в ТГФ (50 мл) и смесь перемешивали в течение 2 часов при -70°С. Затем по каплям добавляли этил-4-формил-1-пиперидинкарбоксилат (0,1 моль) и смесь перемешивали в течение 30 минут при -70°С. Смесь оставляли нагреваться до комнатной температуры и продолжали перемешивать еще 30 минут. Реакционную смесь разлагали водой и упаривали. Остаток перемешивали в воде и полученную смесь экстрагировали CH2Cl2. Органический слой отделяли, сушили, фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/СН3ОН, 98/2). Чистые фракции собирали и растворитель выпаривали. Выход: 38 г этил-4-[гидрокси[1-(2-фенилэтил)-1Н-бензимидазол-2-ил]метил]-1-пиперидинкарбоксилата (промежуточное соединение 10).
b) Получение промежуточного соединения 11
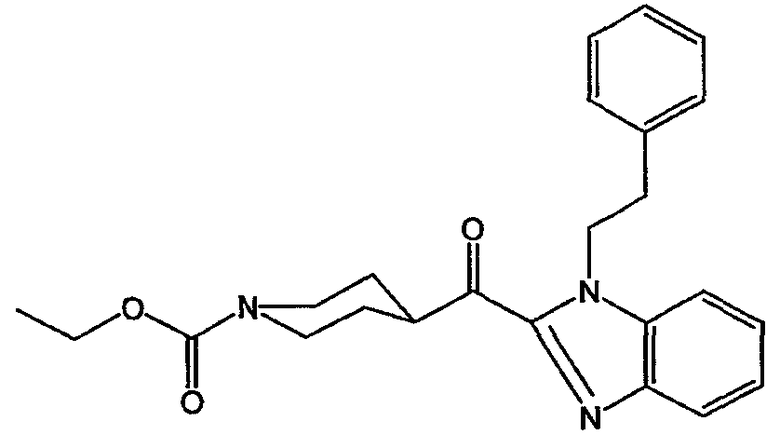
Смесь промежуточного соединения 10 (0,011 моль) и MnO2 (15 г) в CH2Cl2 (150 мл) перемешивали в течение ночи при комнатной температуре. MnO2 отфильтровывали над дикалитом. Указанную реакцию осуществляли во второй раз, используя такие же количества реагентов. Смесь перемешивали в течение ночи. MnO2 отфильтровывали над дикалитом. Фильтрат упаривали. Выход: 4,5 г этил-4-[[1-(2-фенилэтил)-1Н-бензимидазол-2-ил]карбонил]-1-пиперидинкарбоксилата (промежуточное соединение 11).
с) Получение промежуточного соединения 12
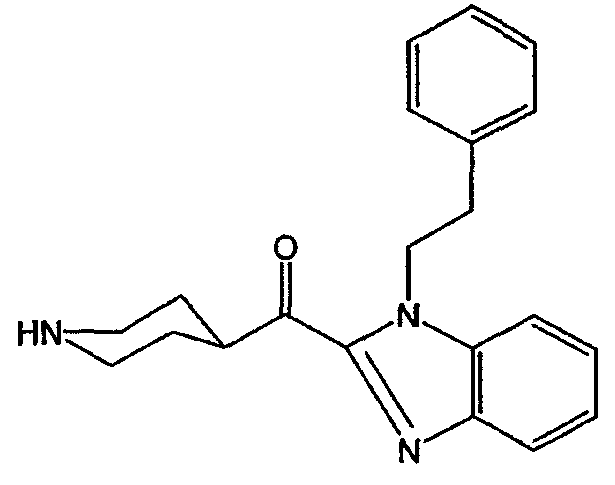
Смесь промежуточного соединения 11 (0,011 моль) и 48% водного раствора HBr (25 мл) перемешивали в течение 10 часов при 80°С. Растворитель выпаривали. Остаток перемешивали в кипящем 2-пропаноле, охлаждали и образовавшийся осадок отфильтровывали и сушили. Образец (1 г) перекристаллизовывали из этанола. Кристаллы отфильтровывали и сушили. Выход: 0,5 г [1-(2-фенилэтил)-1Н-бензимидазол-2-ил](4-пиперидинил)метанон-дигидробромида (промежуточное соединение 12) (т.п. 261,9°С).
d) Получение промежуточного соединения 13
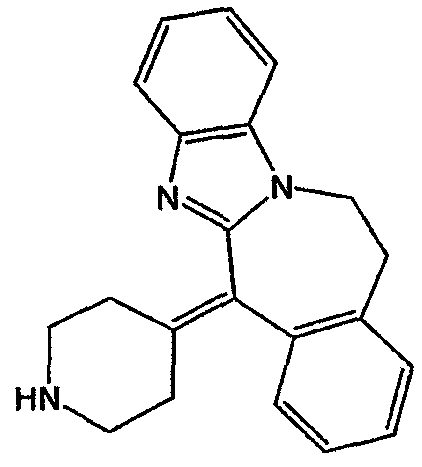
Трифторметансульфоновую кислоту (150 мл) перемешивали в потоке N2. Порциями добавляли промежуточное соединение 12 (0,1 моль) и полученную реакционную смесь перемешивали в течение 20 часов при 100°С (в потоке N2). Реакционную смесь охлаждали, выливали на лед (1 кг) и полученную смесь нейтрализовали 50% NaOH при перемешивании и охлаждении. Реакционную смесь экстрагировали CH2Cl2. Образовывался осадок. Органический слой отделяли. Осадок отфильтровывали и перекристаллизовывали из CH3CN. Кристаллы отфильтровывали и снова перекристаллизовывали из CH3CN. Кристаллы отфильтровывали и сушили. Выход: 3,0 г трифторметансульфоната 11,12-дигидро-6-(4-пиперидинилиден)-6Н-бензимидазо[2,1-b][3]бензазепина (2:3). Отделенную органическую жидкость объединяли с маточными слоями, сушили, фильтровали и растворитель выпаривали. Остаток (37 г) растворяли в воде/этаноле, подщелачивали 50% NaOH и экстрагировали CH2Cl2. Отделенный органический слой сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток перемешивали в 2-пропаноне/DIPE, отфильтровывали и сушили. Выход: 16,2 г 11,12-дигидро-6-(4-пиперидинилиден)-6Н-бензимидазо[2,1-b][3]бензазепина (промежуточное соединение 13) (т.п. 180,3°С).
Пример А4
а) Получение промежуточного соединения 14
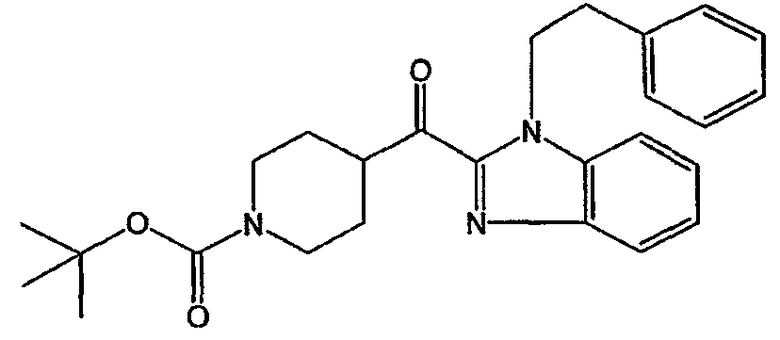
Использовали сухую стеклянную посуду. Смесь DIPA (1,1 моль) в ТГФ (чистота для анализа) (предварительно высушенный на молекулярных ситах) (3000 мл) перемешивали при -78°С в потоке N2. По каплям добавляли 1,5 М BuLi в гексане (1,05 моль) при -70°С и смесь перемешивали при -70°С в течение 20 минут. 1-(фенилэтил)-1Н-бензимидазол (1 моль), растворенный в ТГФ, добавляли по каплям при -78°С и смесь перемешивали при -78°С в течение 1 часа. 4-этил-1-(1,1-диметил)-1,4-пиперидиндикарбоксилат (1,1 моль), растворенный в ТГФ, добавляли по каплям при -70°С. Смесь перемешивали при -78°С в течение 1 часа, доводили до комнатной температуры, перемешивали при комнатной температуре в течение ночи и разлагали Н2О. Органический растворитель выпаривали. Водный концентрат экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток кристаллизовали из CH3CN. Осадок отфильтровывали и сушили. Выход: 350 г промежуточного соединения 14 (81%).
b) Получение промежуточного соединения 15
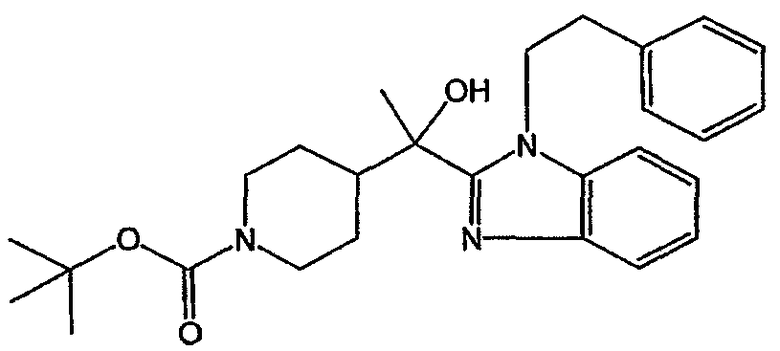
Реакцию выполняли в атмосфере N2. Метилмагнийхлорид (0,0165 моль, 8,2 мл, 2,0 М/ТГФ) добавляли по каплям к раствору промежуточного соединения 14 (0,0150 моль) в ТГФ (90 мл) и перемешивали при комнатной температуре. Полученную реакционную смесь перемешивали в течение 2 часов. Добавляли воду. Органический растворитель выпаривали и водный концентрат экстрагировали CH2Cl2. Отделенный органический слой сушили, фильтровали и растворитель выпаривали. Остаток (6 г) кристаллизовали из СН3CN. Осадок отфильтровывали и сушили. Выход: 4,3 г промежуточного соединения 15 (64%).
с) Получение промежуточного соединения 16
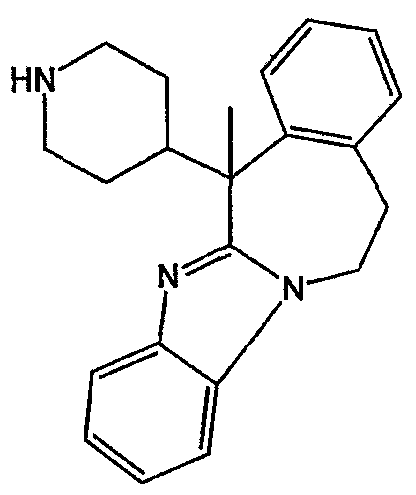
Смесь промежуточного соединения 15 (0,0076 моль) в трифторметансульфоновой кислоте (29 мл) перемешивали в течение 48 часов при комнатной температуре. Реакционную смесь выливали в воду. Полученную смесь подщелачивали К2СО3. Водный слой экстрагировали CH2Cl2. Отделенный органический слой сушили, фильтровали и растворитель выпаривали. Остаток очищали хроматографией на короткой открытой колонке с силикагелем (элюент: CH2Cl2/(CH3OH/NH3), 90/10). Чистые фракции собирали и растворитель выпаривали. Выход: 2 г промежуточного соединения 16 (79%).
Пример А5
а) Получение промежуточного соединения 17
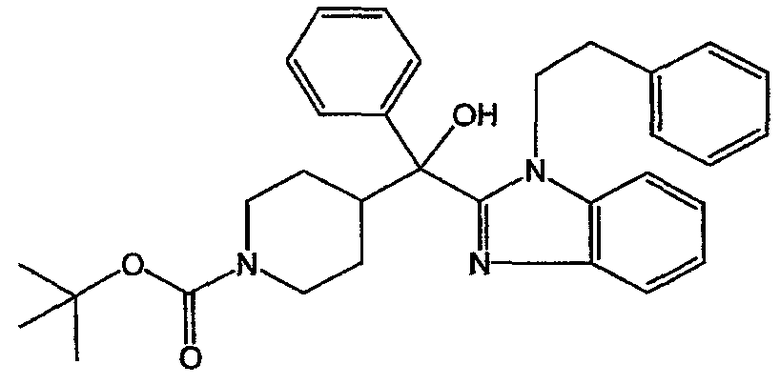
Реакцию выполняли в атмосфере N2. Фенилмагнийхлорид (0,0440 моль) добавляли к раствору промежуточного соединения 14 (0,0400 моль) в ТГФ (200 мл) и перемешивали при комнатной температуре. Полученную реакционную смесь перемешивали в течение одного часа. Добавляли воду. Органический растворитель выпаривали и водный концентрат экстрагировали CH2Cl2. Отделенный органический слой сушили, фильтровали и растворитель выпаривали. Остаток объединяли с аналогично полученным веществом и все вещество (20 г) кристаллизовали из CH3CN. Осадок отфильтровывали и сушили. Выход: 20 г промежуточного соединения 17 (98%).
b) Получение промежуточного соединения 18
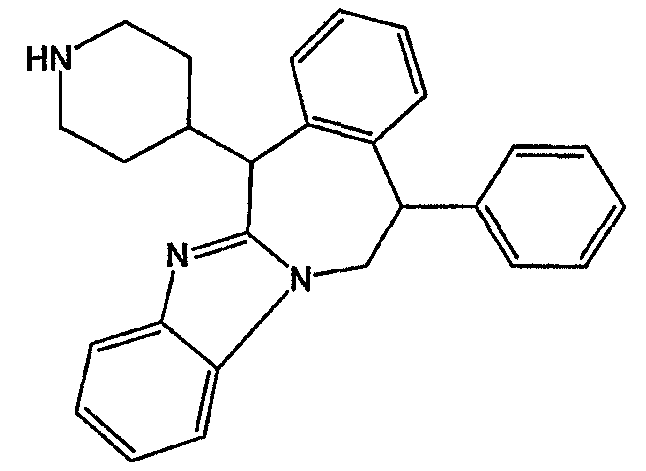
Смесь промежуточного соединения 17 (0,0360 моль) в трифторметансульфоновой кислоте (120 мл) перемешивали в течение 24 часов, повышая температуру от 0°С до комнатной температуры. Реакционную смесь выливали в воду. Полученную смесь подщелачивали 50% NaOH и экстрагировали CH2Cl2. Отделенный органический слой сушили, фильтровали и растворитель выпаривали. Остаток кристаллизовали из CH3CN, отфильтровывали и очищали хроматографией на короткой открытой колонке с силикагелем (элюент: CH2Cl2/(CH3OH/NH3), 90/10). Чистые фракции собирали и растворитель выпаривали. Выход: 11 г промежуточного соединения 18 (78%) (т.п. 270,7°С).
Пример А6
а) Получение промежуточного соединения 19
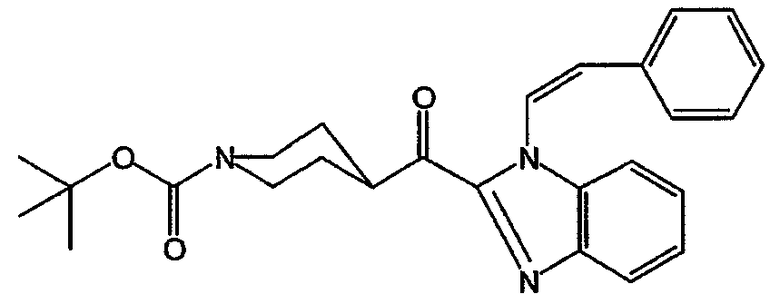
Смесь 1-(2-фенилэтенил)-1Н-бензимидазола (0,04 моль) в ТГФ (100 мл) перемешивали в потоке N2 и охлаждали до -70°С. По каплям добавляли 2,5 М BuLi/гексан (0,04 моль) при -70°С и продолжали перемешивать смесь при -70°С еще 30 минут. По каплям добавляли раствор 4-этил-1-(1,1-диметилэтил)-1,4-пиперидиндикарбоксилата (0,04 моль) в ТГФ и смесь перемешивали в течение 1 часа при -70°С. Полученную смесь оставляли нагреваться до комнатной температуры, разлагали водой и экстрагировали CH2Cl2. Отделенный органический слой сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали хроматографией на колонке с силикагелем (элюент: CH2Cl2/CH3CN, от 97/3 до 94/6). Собирали две фракции и растворитель выпаривали. Остаток второй фракции кристаллизовали из DIPE/CH3CN. Кристаллы отфильтровывали и сушили. Выход: 7,0 г (1,1-диметилэтил) (Z)-4-[[1-(2-фенилэтенил)-1Н-бензимидазол-2-ил]карбонил]-1-пиперидинкарбоксилата (41%) (промежуточное соединение 19) (т.п. 155,8°С).
b) Получение промежуточного соединения 20
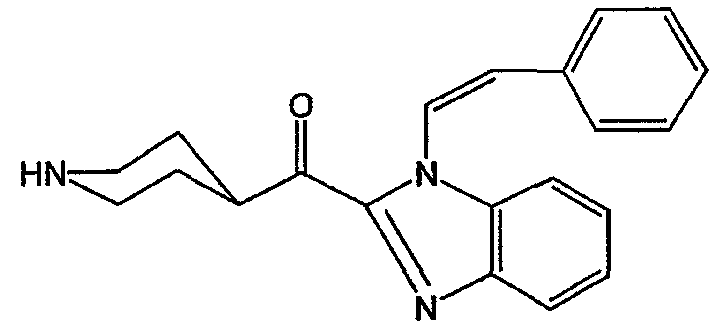
Смесь промежуточного соединения 19 (0,043 моль) в трифторуксусной кислоте (130 мл) перемешивали в течение 1/2 часа при комнатной температуре. Реакционную смесь выливали в диэтиловый эфир. Осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Выход: 18 г трифторацетата (Z)-[1-(2-фенилэтенил)-1Н-бензимидазол-2-ил](4-пиперидинил)метанона (1:1) (промежуточное соединение 20) (94,0%) (т.п. 202,2°С).
с) Получение промежуточного соединения 21
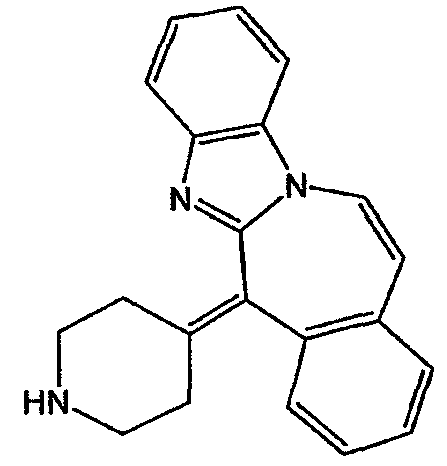
Смесь промежуточного соединения 20 (0,0276 моль), AlCl3 (0,187 моль) и NaCl (0,187 моль) перемешивали в течение 1 часа при 150°С (расплав). Реакционную смесь разлагали в смеси льда, воды и 50% NaOH. Полученную смесь экстрагировали дихлорметаном, органический слой отделяли, сушили, фильтровали и упаривали. Остаток (4,3 г) очищали на стеклянном фильтре с силикагелем (элюент: СН2Cl2/(CH3OH/NH3), 90/10). Чистые фракции собирали и растворитель выпаривали. Остаток превращали в соль (Е)-2-бутендиоевой кислоты (2:3) в этаноле. Полученную соль отфильтровывали и сушили. Выход: 1,8 г (Е)-2-бутендиоата 6-(4-пиперидинилиден)-6Н-бензимидазо[2,1-b][3]бензазепина (2:3) (13,4%) (промежуточное соединение 21) (т.п. 229,4°С).
Пример А7
а) Получение промежуточного соединения 22
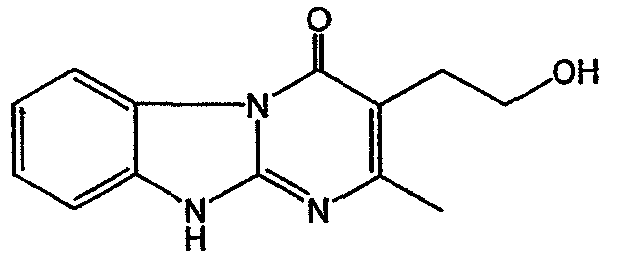
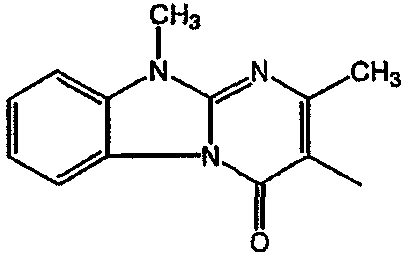
Смесь 2-амин-1Н-бензимидазола (0,04 моль), 3-ацетилдигидро-2(3Н)-фуранона (0,53 моль) и 4-метилбензолсульфоновой кислоты (4 г) в ксилоле (930 мл) перемешивали, нагревали с обратным холодильником в течение ночи и охлаждали. Осадок отфильтровывали и перемешивали в Н2О (200 мл), Na2CO3 (5 г) и CH2Cl2 (500 мл). Осадок отфильтровывали, кипятили в СН3ОН, отфильтровывали и сушили. Выход: 47,4 г 3-(2-гидроксиэтил)-2-метилпиримидо[1,2-α]бензимидазол-4(10Н)-она (промежуточное соединение 22).
b) Получение промежуточного соединения 23
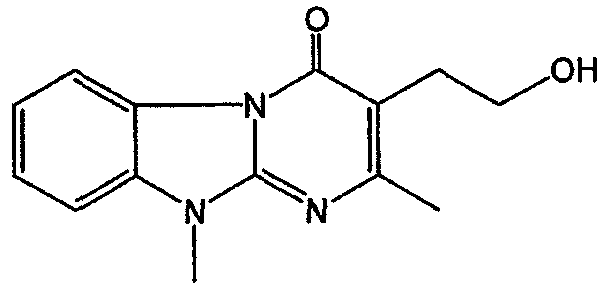
Смесь промежуточного соединения 22 (0,025 моль) и К2СО3 (чистота для анализа) (0,03 моль) в ДМФА (70 мл) перемешивали при 50°С. По каплям добавляли метилиодид (0,03 моль). Смесь перемешивали при 50°С в течение 4 часов и охлаждали. Растворитель выпаривали. Остаток кипятили в СН3ОН. Осадок отфильтровывали и сушили. Остаток очищали ВЭЖХ на силикагеле (элюент: CH2Cl2/(CH3OH/NH3), 97/3). Собирали две чистые фракции и их растворители выпаривали. Выход: 2,08 г 3-(2-гидроксиэтил)-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-она (промежуточное соединение 23).
с) Получение промежуточного соединения 24
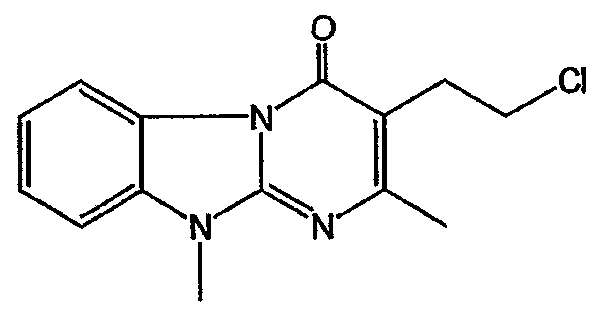
Смесь промежуточного соединения 23 (0,02 моль) и SOCl2 (0,06 моль) в CHCl3 (50 мл) перемешивали, нагревали с обратным холодильником в течение 4 часов и охлаждали. Добавляли Н2О. Смесь подщелачивали К2СО3 и разделяли на слои. Водный слой экстрагировали CH2Cl2. Объединенный органический слой сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток кристаллизовали из CH3CN. Осадок отфильтровывали и сушили. Выход: 3,44 г промежуточного соединения 24.
В. Получение конечных соединений
Пример В1
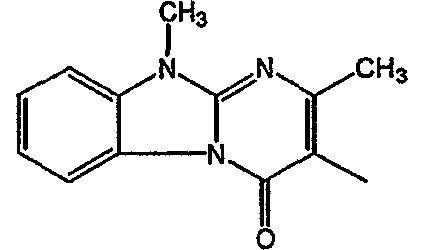
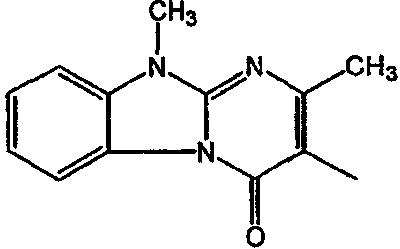
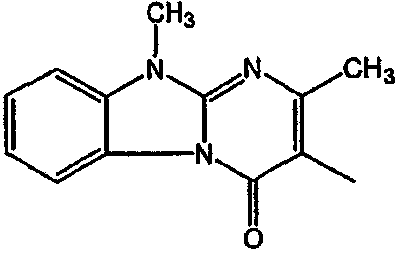
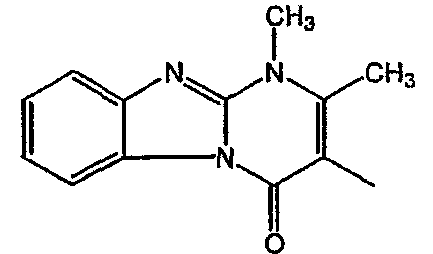
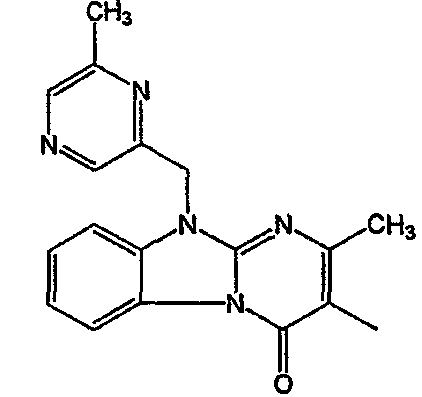
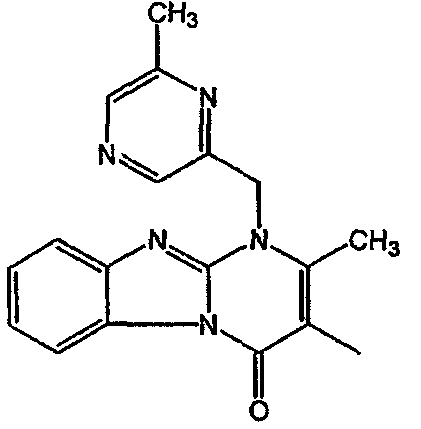
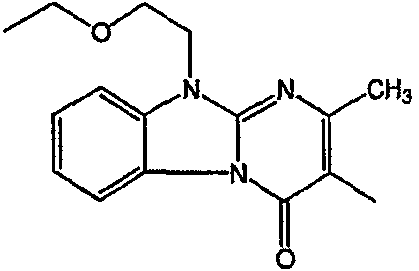
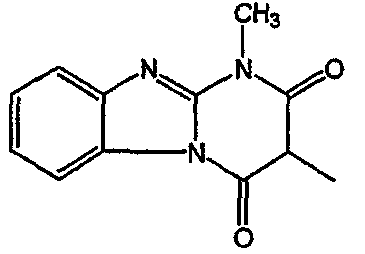
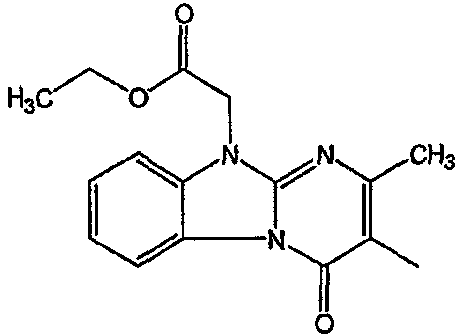
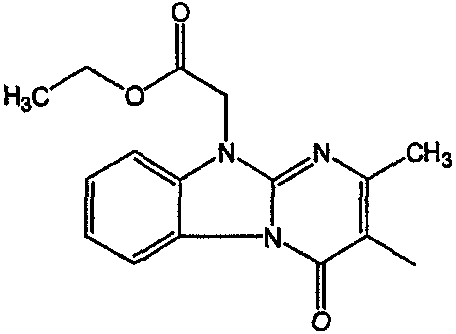
Получение соединения 1
и получение соединения 2
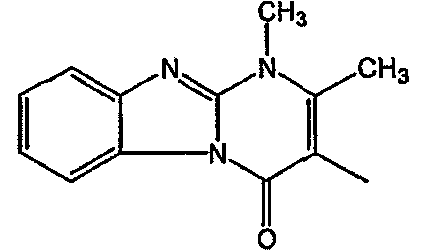
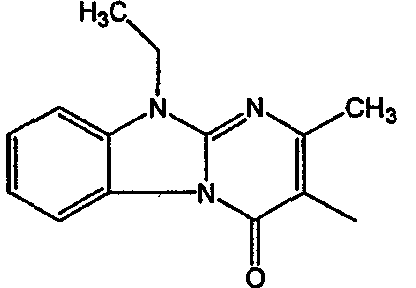
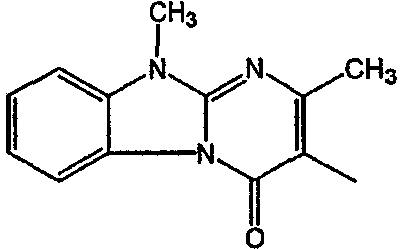
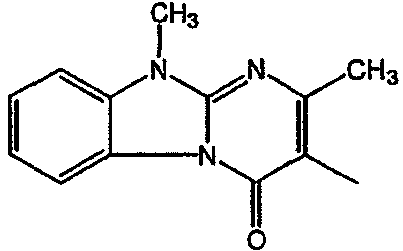
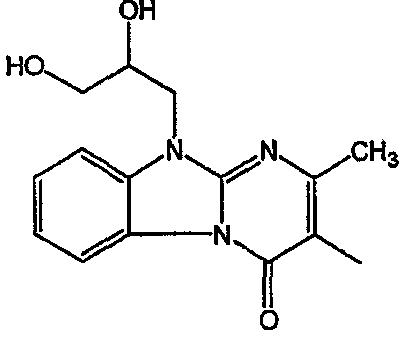
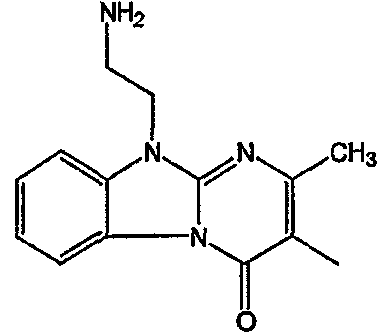
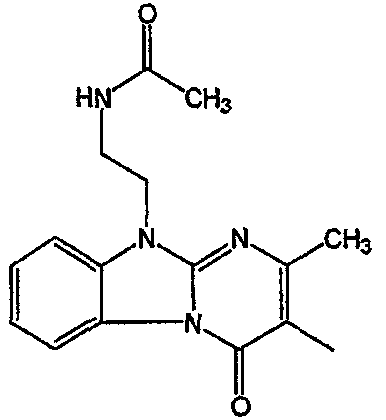
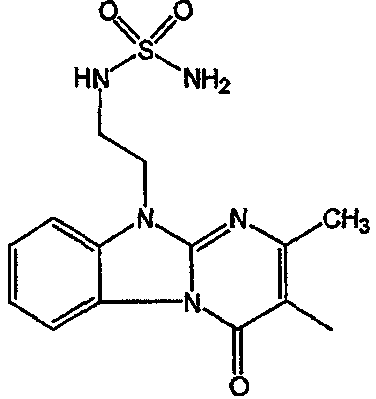





С. Фармакологические примеры
С1. Определение in vitro антагонистической активности в отношении Н1- и Н2-рецепторов гистамина
Исследования связывания рецептора с меченым лигандом выполняли in vitro для определения связывания выбранных соединений с меченым лигандом, используя препарат ткани, обогащенный конкретным рецептором, то есть Н1- или Н2-рецептором гистамина. Ткань, используемая для Н1-рецептора гистамина, представляла собой клетки яичника китайского хомячка (СНО), постоянно трансфецированные Н1-рецептором гистамина человека. Только дифенгидрамин испытывали с использованием клеток коры головного мозга морской свинки. Конкурентное ингибирование [3H]-пириламина испытуемыми соединениями исследовали, инкубируя меченый лиганд в низкой (нМ) концентрации с небольшим образцом препарата ткани (0,2-5 мл; 1-5 мг ткани) в забуференной среде, содержащей испытуемые соединения, растворенные в ДМСО, в диапазоне концентраций, отличающихся по крайней мере на 4 порядка величины от значения pIC50, установленного на основании кривой ингибирования. Антагонистическую активность в отношении Н2-рецептора гистамина испытывали по существу так же, как антагонистическую активность в отношении Н1-рецептора гистамина, используя клетки полосатого тела морской свинки и [125I]АРТ в качестве меченого лиганда в концентрации 0,1 нМ. Инкубацию производили в течение 150 минут при 22°С.
Все соединения по настоящему изобретению обладали антагонистической активностью в отношении Н1-рецептора гистамина, соответствующей значению pIC50, равному 5 или большей величине. Несколько соединений обладали антагонистической активностью в отношении Н1-рецептора гистамина, соответствующей значению pIC50, равному 6 или большей величине. Указанные соединения приведены в таблице 9. Кроме того, установлено, что коммерчески производимый типичный антагонист Н1-рецептора гистамина (дифенгидрамин) обладает лишь незначительно более высокой антагонистической активностью в отношении Н1-рецептора гистамина по сравнению с большинством соединений по данному изобретению. Далее установлено, что коммерчески производимые антагонисты Н2-рецептора гистамина (ранитидин и циметидин) характеризуются антагонистической активностью в отношении Н2-рецептора гистамина в диапазоне (умеренно высокой) активности соединений по данному изобретению в отношении Н2-рецептора гистамина. Некоторые соединения, приведенные в таблице 9, в том числе коммерчески производимые соединения, были также испытаны in vivo в отношении их способности снижать внутричерепное давление (ICP).
Результаты исследования модели антагонистической активности в отношении Н1- и Н2-рецепторов гистамина
С.2. Фармакология in vivo
Модель закрытой черепно-мозговой травмы (CHI)
Для испытания соединений по данному изобретению и коммерчески производимых соединений была использована клинически обоснованная модель травматического повреждения головного мозга у крыс. Вышеуказанная модель, имитирующая несколько клинических признаков травматического повреждения головного мозга, таких как повышенное внутричерепное давление, пониженное давление мозгового кровоснабжения, морфологические изменения, включающие диффузное поражение аксонов, некроз нервных клеток и контузию, нарушение ауторегуляции мозгового кровотока и уменьшение оксигенации головного мозга, была использована для исследования лекарственных средств, снижающих внутричерепное давление. Травму вызывали у интубированных, анестезированных изофлураном (1,5% изофлурана в смеси 30% О2 и 70% N2O) крыс Sprague-Dawley (380-400 г), которых помещали в стереотаксическом положении на стол, смонтированных на 4 пружинах. 400-граммовый стальной цилиндр, защищенный кремниевым диском с диаметром 9 мм, опускали в падении на незащищенный череп с высоты 70 см или 50 см (соответственно "тяжелая" и "средней тяжести" черепно-мозговая травма). Область нанесения удара была сцентрирована между брегмой и ламбдой. Внутричерепное давление регистрировали при помощи зонда с микродатчиком Кодмана, вводимого в теменную область коры головного мозга. В случае как тяжелой черепно-мозговой травмы, так и черепно-мозговой травмы средней тяжести внутричерепное давление повышалось сразу же после травмы и оставалось повышенным в течение нескольких дней. Модель тяжелой черепно-мозговой травмы была использована для оценки фармакологического действия лекарственных средств сразу же после травмы (метод отсеивания). Выживших и пришедших в себя после анестезии крыс исследовали по типу модели черепно-мозговой травмы средней тяжести. В фармакологических исследованиях были использованы животные с патологическим внутричерепным давлением от 12,5 до 35 мм Hg. Изменения внутричерепного давления, среднего артериального кровяного давления (МАВР) и давления мозгового кровоснабжения СРР (=МАВР-СРР) выражали в виде процентного значения от исходной величины в момент начала лечения.
Метод отсеивания, используемый для оценки соединений по данному изобретению
Четыре подвергаемых лечению группы по 3 крысы в каждой группе каждую неделю сравнивали с 3 животными, которым вводили физиологический раствор. Так как обычные статистические методы требуют большего числа животных, был использован последовательный метод. Последовательные методы включают несколько ступеней отбора. На каждой ступени выбирали как можно более однородную группу животных. Животных произвольно распределяли для введения лекарственных средств или физиологического раствора. Такой метод позволял принять решение о прекращении введения лекарственного средства, признании лекарственного средства активным или использовании новой группы животных на следующей ступени. Принимая во внимание биологически обоснованный уровень активности, который должен быть обнаружен, была определена и зафиксирована ожидаемая часть ложных положительных и отрицательных результатов. Был использован критерий последовательной оценки групп животных, основанный на двойной выборке. Трехступенчатый последовательный метод с использованием относительно небольшого числа животных на каждой ступени оказался оптимальным. Несмотря на изменчивость отдельных данных, указанный метод позволил состоятельно оценить стандартное лечение маннитом как активное, в то время как контрольные животные были отсеяны. Клинически обоснованные внутривенные дозы маннита (3 г в течение 45 минут) значительно снижали внутричерепное давление (среднее снижение составило примерно 20%).
Результаты метода отсеивания
(1) Испытуемые соединения вводили в виде болюса в дозе 1 мг/кг в течение 1 минуты с последующим вливанием 0,5 мг/кг/мин в течение 44 минут; растворители вводили в виде болюса в дозе 0,4 мл в течение 1 минуты с последующим вливанием 0,2 мл/мин в течение 44 минут; маннит вводили в виде вливания 67 мг/кг/мин в течение 45 минут.
(2) Дельта-изменение в % означает среднее изменение относительного внутричерепного давления от исходного уровня за период лечения.
(3) Решение основано на последовательной статистической оценке.
CD=гидроксипропил-β-циклодекстрин, используемый в качестве растворителя.
Н2Т = винная кислота, используемая в качестве растворителя.
Маннит1-5: маннит оценивали 5 раз в отдельных испытаниях (положительные контрольные образцы). В таблице приведены результаты всех испытаний.
Дальнейшие исследования
В таблице 11 показаны изменения некоторых важных физиологических параметров у крыс, зарегистрированные во время лечения после тяжелой закрытой черепно-мозговой травмы. Лечение, которое начинали через 20 минут после тяжелой черепно-мозговой травмы, включало введение дозы, равной 0,5 мг/кг/мин, в течение 10 минут с последующим введением 0,1 мг/кг/мин в течение 50 минут.
Изменение важных физиологических параметров у крыс во время лечения после тяжелой закрытой черепно-мозговой травмы
* = Значения, существенно отличающиеся от группы, получавшей растворитель (р<0,05, критерий Даннета)
Растворитель: 10% гидроксипропил-бета-циклодекстрин, винная кислота, NaOH и маннит в пирогенной воде; рН 4; осмотическое давление 312-314 mOsm/кг; концентрация соединения 2 мг/мл
Соединение: гидрат (1:1) (Е)-2-бутендиоата 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-она (2:3)
Соединение 1: (В)[(2α,4β)(А)]
Соединение 2: (А)[(2α,4β)(А)]
Рацемат (соединение 1 и соединение 2): (2α,4β)(А), то есть рацемическая смесь соединения 1 и соединения 2
ICP: внутричерепное давление
МАВР: среднее артериальное кровяное давление
СРР: давление мозгового кровоснабжения
ETCO2: конечное количество СО2, обмениваемого за одно дыхание
Воздействие соединения 2 на МАВР гораздо менее выражено при введении данного соединения в виде непрерывного вливания в количестве 0,1 мг/кг/мин. В данном случае отсутствует пик кровяного давления и не наблюдаются увеличения МАВР более чем на 20% (среднее увеличение МАВР в конце вливания составляет 9%, n=6). Максимальное снижение внутричерепного давления при указанной дозе сравнимо с величиной, наблюдаемой в том случае, когда вливанию предшествует введение "ударной дозы", равной 5 мг/кг, в течение 10 минут, но время, необходимое для достижения такого эффекта, является более продолжительным (в среднем 30 минут).
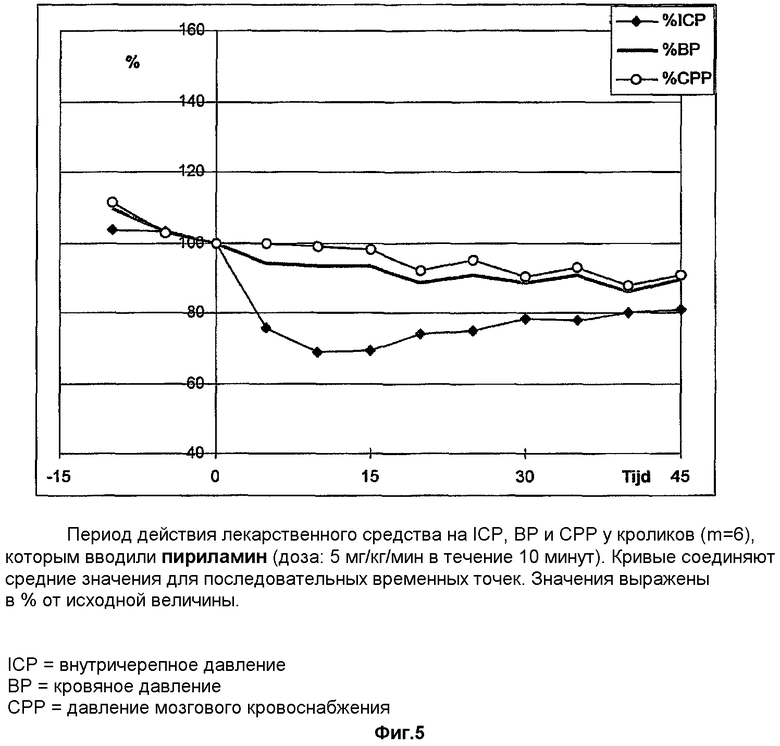
Воздействие ранитидина и дифенгидрамина на внутричерепное давление
Ранитидин вливали крысам, используемым в качестве модели закрытой черепно-мозговой травмы, в дозе 2 мг/кг/мин в течение 6 минут после нанесения тяжелой черепно-мозговой травмы. Растворитель (NaCl+H2T) вводили в том же количестве. В каждой группе подвергали лечению 6 крыс. Ранитидин вызывал статистически значимое более выраженное снижение внутричерепного давления по сравнению с группой животных, которым вводили растворитель (снижение на 7,7% по сравнению с 0,5%, что является статистически значимым показателем при р=0,013). Процентное значение снижения внутричерепного давления высчитывали в виде % изменения ICP, зарегистрированного в начале лечения и в конце вливания. Существенного изменения кровяного давления не наблюдалось.
Дифенгидрамин вливали крысам, используемым в качестве модели закрытой черепно-мозговой травмы, в дозе 1 мг/кг/мин в течение 10 минут после нанесения тяжелой черепно-мозговой травмы. Лечению подвергали трех крыс. Дифенгидрамин вызывал 34% снижение внутричерепного давления без какого-либо значительного влияния на кровяное давление.
Сравнительные исследования с использованием агонистов
Для сравнения испытывали два коммерчески производимых агониста Н2-рецептора гистамина (димаприт и импромидин), которые вливали нетравмированным крысам в дозе 0,5 мг/кг/мин в течение 10 минут для димаприта и в дозах, увеличенных до 3,75 мг/кг/час, для импромидина. Не наблюдалось никакого эффекта. При введении димаприта в высокой дозе, равной 2 мг/кг/мин, в течение 10 минут и импромидина в виде болюса в дозе 0,5 мг/кг наблюдалось снижение кровяного давления и ICP, которые восстанавливались после лечения.
На основании полученных данных был сделан вывод о том, что антагонисты Н1- и/или Н2-рецепторов гистамина вызывают снижение ICP и при этом не оказывают существенного влияния на кровяное давление.
Эксперименты с использованием коммерчески производимых антагонистов Н1- и Н2-рецепторов гистамина
Несколько коммерчески производимых антагонистов Н1- и Н2-рецепторов гистамина вливали крысам, используемым в качестве модели закрытой черепно-мозговой травмы, в дозе 0,5 мг/кг/мин в течение 10 минут после нанесения тяжелой черепно-мозговой травмы. Растворитель (NaCl+H2T) вводили в том же количестве. В каждой группе подвергали лечению 6 крыс. Результаты изменения ICP и кровяного давления в течение первых 15 минут суммированы в таблице 12.
Действие коммерчески производимых антагонистов Н1- и Н2-рецепторов гистамина
Действие соединения 1 в зависимости от дозы
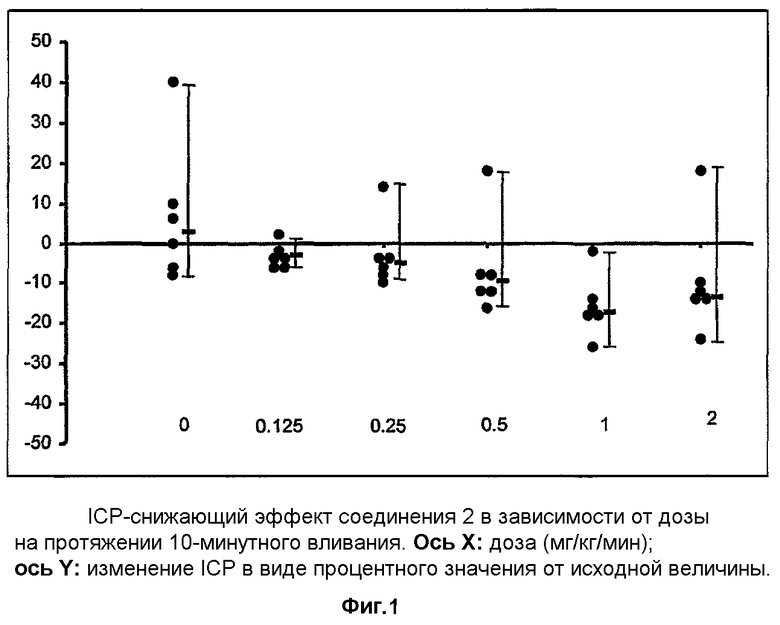
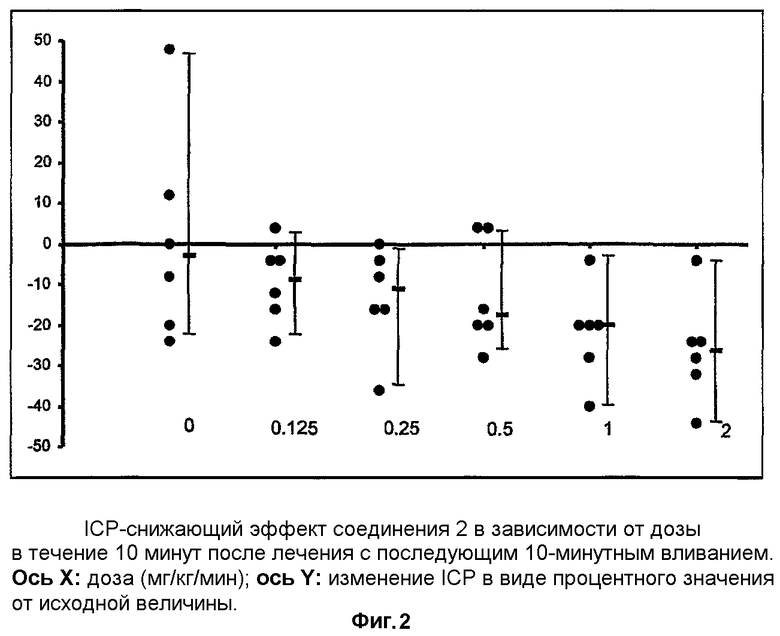
Результаты слепого, полностью рандомизированного исследования соединения 1, вводимого в разных дозах (0,125, 0,25, 0,5, 1 и 2 мг/кг/мин) в виде 10 минутного вливания крысам, используемым в качестве модели закрытой черепно-мозговой травмы, показывают, что в процессе лечения соединение 1 вызывает значительное снижение ICP в зависимости от дозы (фиг.1). Начиная с дозы 1 мг/кг/мин, соединение 1 вызывает статистически значимое более выраженное снижение ICP по сравнению с группой животных, которым вводили растворитель. Значительное воздействие на ICP в зависимости от дозы сохранялось в течение 10 минут после вливания (фиг.2).
Действие соединения 2, соединения 1 и рацемата (соединение 1 и соединение 2) на концентрацию гемоглобина и оксигенацию головного мозга
Спектроскопия в ближней инфракрасной области (NIRS) головного мозга крыс "in vivo" позволяет произвести неинвазивное количественное определение насыщения гемоглобина головного мозга кислородом (HbSat) и общей концентрации гемоглобина в головном мозге ([HbTot]). Указанный последним параметр является мерой объема мозговой крови (CBV). Кроме того, можно определить изменение окислительно-восстановительного состояния митохондриального фермента цитохром-оксидазы (CytOx), являющегося показателем оксигенации тканей.
Все соединения 2, 1 и рацемат (соединение 1 и соединение 2) не оказывают значительного воздействия на [HbTot] при введении через 24 часа после черепно-мозговой травмы средней тяжести в виде внутривенного вливания в дозе 0,5 мг/кг/мин в течение 10 минут с последующим вливанием 0,1 мг/кг/мин в течение 45 минут. Только соединение 2 вызывает небольшое, но статистически значимое снижение HbSat. Соединение 1 и рацемат (соединение 1 и соединение 2) не воздействуют на HbSat. При введении указанной дозы все соединения не оказывают воздействия на окислительно-восстановительное состояние CytOx. Полученные результаты показывают, что в созданных экспериментальных условиях сосудосуживающее действие на кровеносные сосуды головного мозга, если и имеет место, является весьма ограниченным, и оксигенация тканей не подвергается опасному воздействию.
Влияние анестезии на действие соединения 2
Эффект лечения соединением 2 (внутривенное вливание в дозе 0,1 мг/кг/мин в течение 30 минут) через 24 часа после травмы средней тяжести исследовали с использованием разных анестезирующих средств (изофлуран, хлоральгидрат, пентобарбитал). При использовании в качестве анестезирующего средства хлоральгидрата (внутрибрюшинное вливание 400 мг/кг) ICP снижается до 75% от исходного значения и МАВР (среднее артериальное кровяное давление) постепенно увеличивается до 110% от исходного значения (средние величины, n=6). Указанные воздействия сравнимы с результатами, наблюдаемыми при анестезии изофлураном. При использовании пентобарбитала (внутрибрюшинное вливание 60 мг/кг) соединение 2 вызывает значительное постепенное повышение МАВР до 141% от исходного значения в конце вливания, при этом ICP снижается до 64% от исходного значения (средние величины, n=6). Полученные результаты показывают, что аналогичные воздействия на ICP и МАВР наблюдаются при разных типах анестезии. То, что указанное соединение значительно снижает ICP при анестезии пентобарбиталом, имеет важное значение, так как барбитураты часто применяются для лечения субъектов с травматическим повреждением головного мозга. Благодаря тому, что барбитураты также снижают ICP, при использовании данного соединения можно получить важный дополнительный эффект.
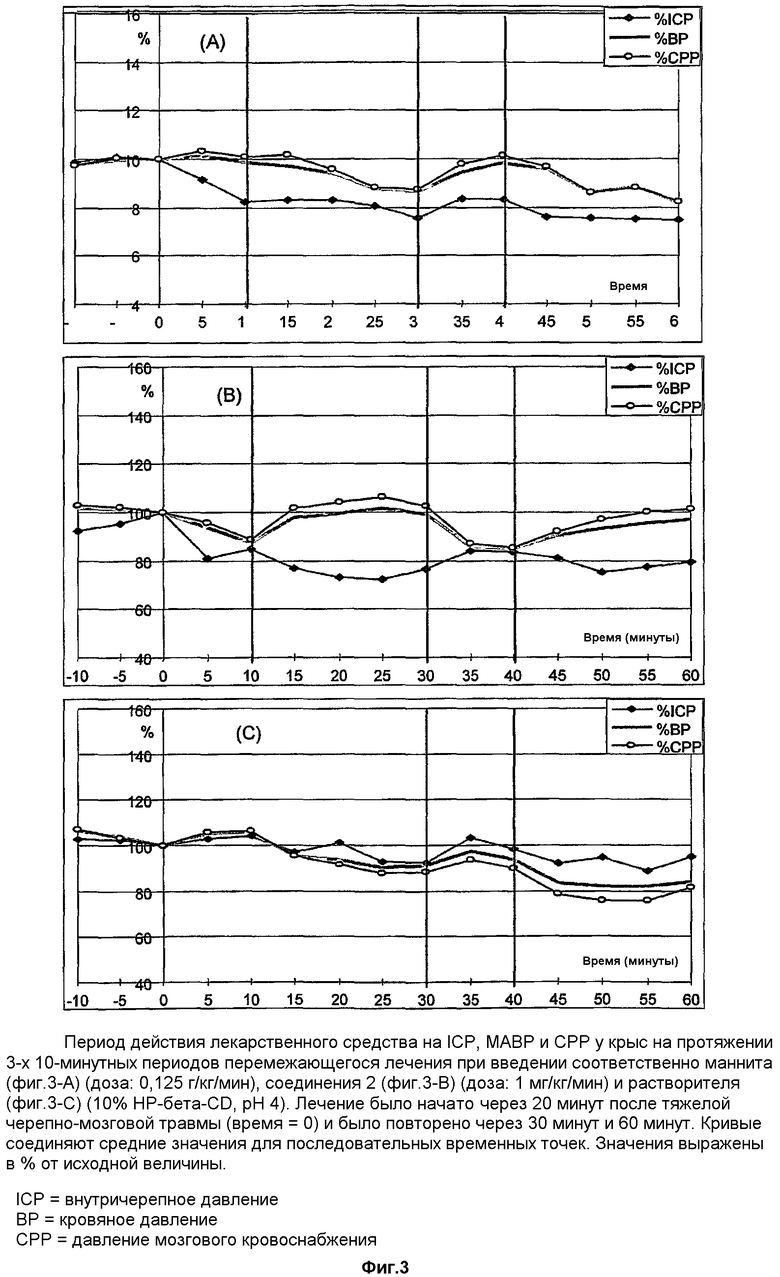
Влияние повторного введения соединения 1 и маннита на повышенное внутричерепное давление у травмированных крыс
Соединение 1 вводили 2 раза с 20-минутными промежутками в виде внутривенного вливания в дозе 1 мг/кг/мин в течение 10 минут, начиная через 20 минут после нанесения тяжелой черепно-мозговой травмы.
Маннит вводили внутривенно с такими же временными параметрами, что и соединение 1, в дозе 0,125 г/кг/мин. Контрольные животные получали только растворитель (содержащий 10% НР-β-CD, рН 4).
Вливание соединения 1 вызывало быстрое снижение внутричерепного давления (фиг.3). Данный эффект усиливался после окончания каждого периода вливания. Кровяное давление снижается во время введения соединения 1, но восстанавливается после окончания лечения. Такой результат отличается от воздействия, оказываемого маннитом, который вызывает снижение ICP и увеличение кровяного давления во время каждого вливания с последующим снижением кровяного давления после окончания каждой процедуры. Только у животных, которым вводили соединение 1, наблюдается четко выраженное различие между изменением кровяного давления и ICP. В отличие от этого, у животных, получавших маннит, происходят более или менее параллельные изменения кровяного давления и внутричерепного давления. Такой результат свидетельствует о том, что фармакологическое действие соединения 1 отличается от действия маннита.
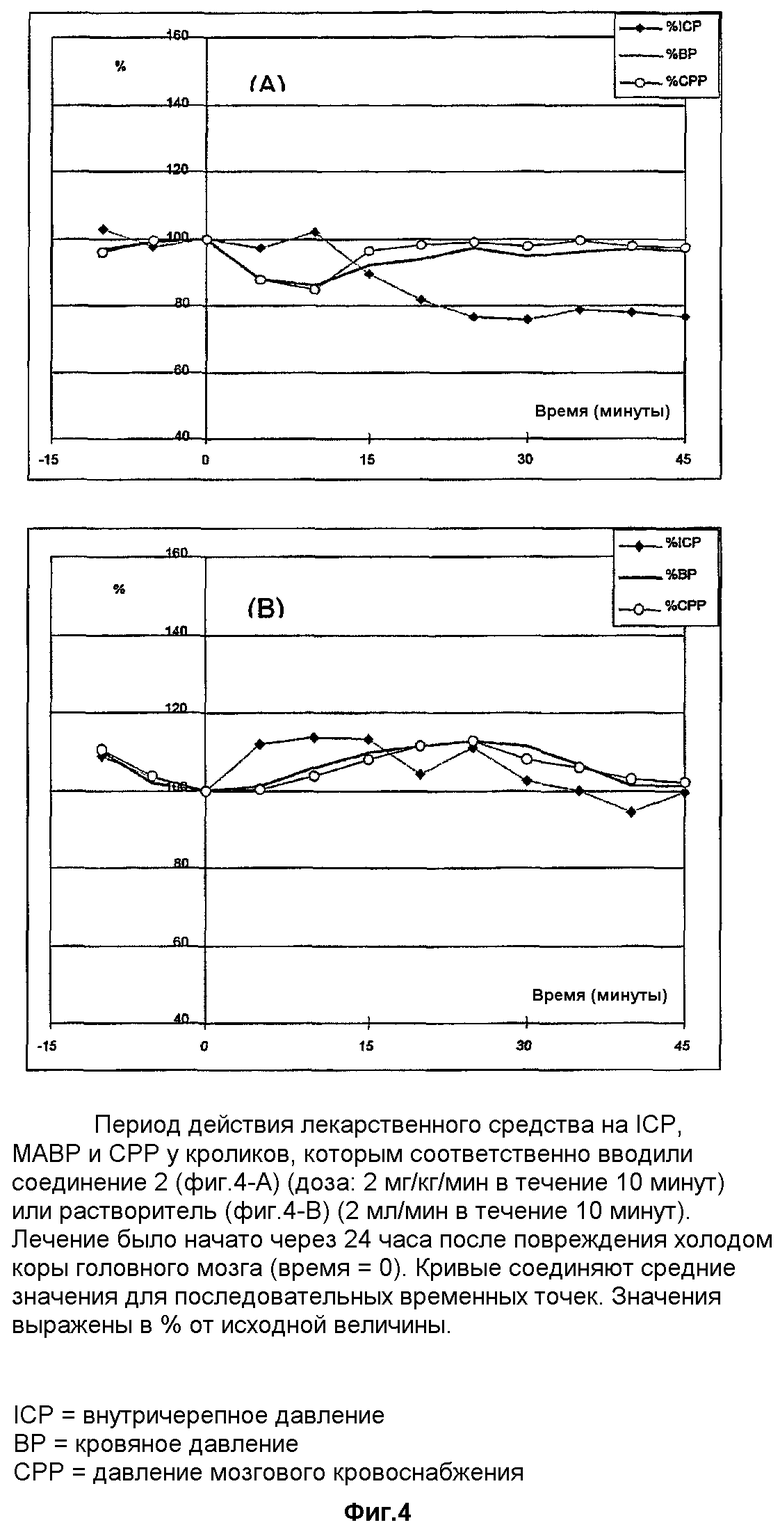
Действие соединения 1 на повышение внутричерепного давления у кроликов в результате повреждения холодом коры головного мозга
У взрослых кроликов вызывали криогенные поражения с целью патологического изменения внутричерепного давления вследствие отека тканей. Прут из нержавеющей стали с диаметром 8 мм прикладывали к заранее определенным участкам обнаженного черепа кроликов, находящихся под глубоким наркозом, и охлаждали жидким азотом в течение 10 минут. Через один день животных снова анестезировали и постоянно регистрировали ICP и кровяное давление аналогично измерению подобных показателей у крыс. После стабилизации показателей в течение 15 минут соединение 1 вливали в дозе 2 мг/кг/мин в течение 10 минут. Растворитель (преклинический препарат, содержащий 10% НР-β-CD, рН 4) вводили в дозе 2 мл/мин в течение 10 минут.
Во время вливания соединения 1 кровяное давление снижалось и, хотя не происходило экстренного снижения ICP, наблюдалось противодействие повышению ICP в отличие от животных, которым вводили растворитель (фиг.4). После окончания вливания лекарственного средства кровяное давление возвращалось к исходному значению и наблюдалось значительное снижение ICP, которое оставалось стабильным на протяжении всего периода регистрации.
Полученные результаты показывают, что данное соединение снижает ICP не только у грызунов, но и у других видов животных, а также при патологических повреждениях, отличающихся от закрытой черепно-мозговой травмы.
Действие соединения 1 на внутричерепное давление у нетравмированных животных
Крысы
Испытывали действие соединения 2, соединения 1 и рацемата (соединение 1 и соединение 2) на ICP, МАВР и СРР у анестезированных нетравмированных крыс. Указанные соединения вводили внутривенно в такой же дозе, что и травмированным крысам (0,5 мг/кг/мин в течение 10 минут с последующим введением 0,1 мг/кг/мин в течение 50 минут). Результаты сравнимы с величинами, полученными у травмированных животных.
Выводы
Результаты, полученные у травмированных животных, животных с повреждением головного мозга холодом и нетравмированных животных, показывают, что испытанные соединения являются активными в случае разных поражений и даже при отсутствии поражений. Область применения указанных соединений, по-видимому, включает разные патологические состояния, для которых характерно повышение внутричерепного давления.
Приложение 1
Приложение 2
Получение конечных соединений WO 02/100862)
Пример В1
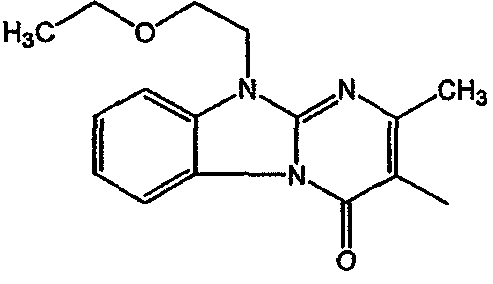
Получение соединения 1 и соединения 2
А[(2α,4β)(А)]
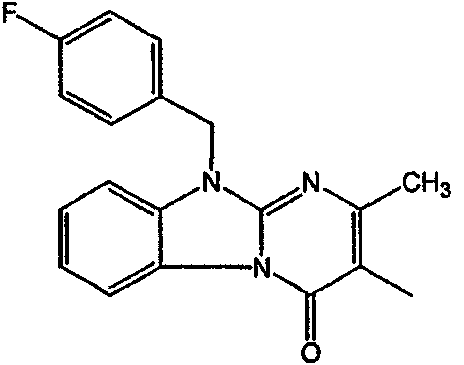
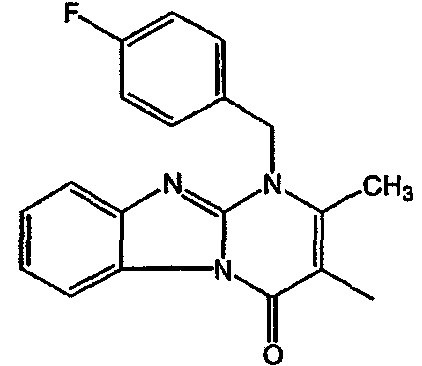
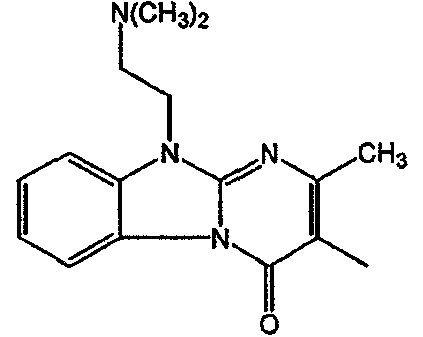
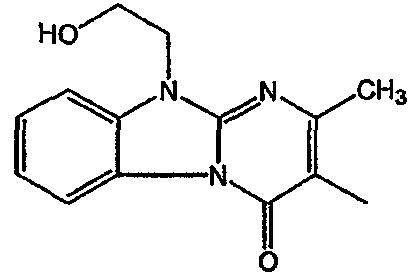
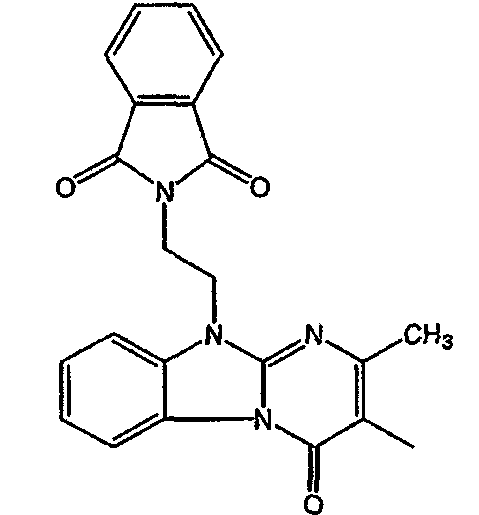
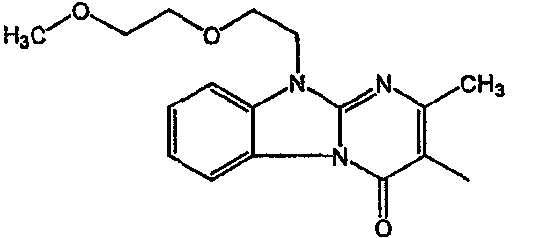
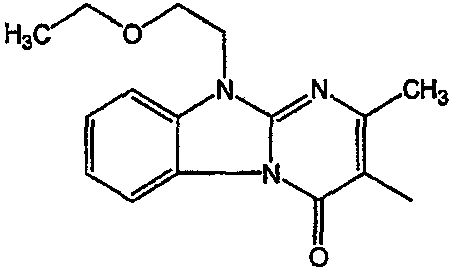
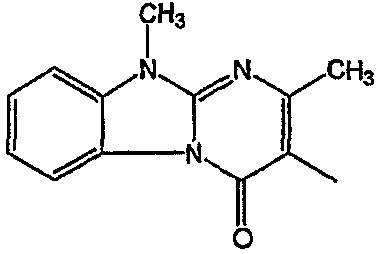
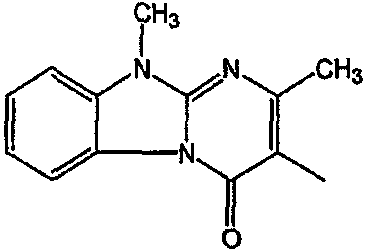
Смесь промежуточного соединения 24 (0,021 моль), промежуточного соединения 5 (0,015 моль), Na2CO (0,021 моль) и KI (1 г) в MIBK (500 мл) перемешивали и кипятили с обратным холодильником в течение 72 часов. Растворитель выпаривали. Осадок распределяли в воде и CH2Cl2. Слои разделяли. Водный слой повторно экстрагировали CH2Cl2. Отделенный органический слой сушили (MgSO4), фильтровали и раствор выпаривали. Осадок очищали в стеклянной воронке с силикагелем (элюент: CH2CI2/(СН3ОН/NH3, 97/3 к 94/6). Фракции собирали и растворитель выпаривали. Осадок перекристаллизовывали из СН3CN, отфильтровывали и сушили. Эту фракцию (6,95 г) разделяли на энантиомеры над Chiralcel OD (элюент: гексан/(C2H5OH + 0,04% Et3N) 58/42). Чистые фракции собирали и растворитель выпаривали. Фракцию 1 растворяли в in 2-пропанол/этанол (95/5) и превращали в соль (Е)-2-бутендикарбоновой кислоты (2:3). Осадок отфильтровывали и сушили. Эту фракцию сушили. Выход: 2,2 г (A)[(2α,4β)(А)] 3-[2-(4-(11,12-дигидро-6H-бензимидазо[2,1-b] [3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-a] бензимидазол-4 (10H)-он (соединение 2) (45%). Фракцию 2 растворяли в 2-пропанол/этаноле (95/5) и превращали в соль (Е)-2-бутендикарбоновой кислоты (2:3).Осадок отфильтровывали и сушили. Эту фракцию сушили. Выход: 2,1 г (B) [(2α,4β)(А)] 3-[2-(4-(11,12-дигидро-6H-бензимидазо[2,1-b] [3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-а] бензимидазол-4 (10H)-он(Е)-2-бутендиоата (2:3) гидрат (1:1) (соединение 1) (43%).
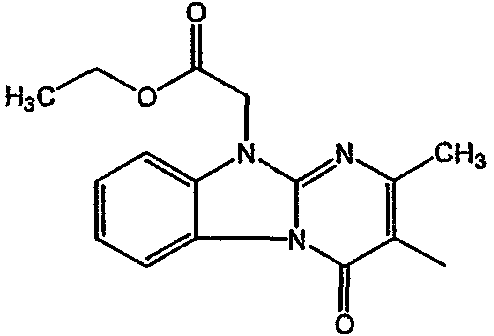
Пример В2
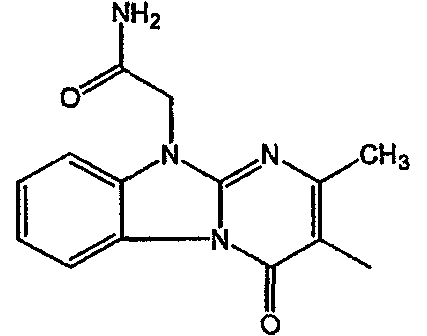
Получение соединения 3 и соединения
Реакцию проводят в токе N2.
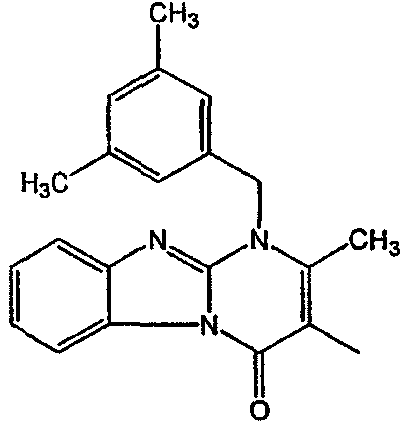
Смесь 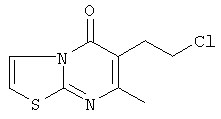 моль), производного 8 (0,0092 моль), NaHCO3 (0,0185 моль) и KI (1 г) в MIK (200 мл) перемешивали и кипятили с обратным кипятильником в течение нескольких часов. Растворитель выпаривали. Осадок помещали в H2O и CH2Cl2 и смесь разделяли на слои. Водный слой экстрагировали СН2Cl2. Объединенный органический слой сушили (MgSO4), фильтровали и выпаривали. Осадок очищали в стеклянном фильтре с силикагелем (элюент: CH2CI2/(CH3OH/NH3) 97/3). Чистые фракции собирали и раствор выпаривали. Осадок снова очищали ВЭЖХ с помощью RP 18 (элюент: [NH4OAc (0,5% в H2O)/CH3CN 90/10]/CH3CN 60/40). Две органические фракции собирали и их органические растворители выпаривали. Водные концентрации экстрагировали H2Cl2. Органические слои разделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Выход: Фракция 1 и 0,98 г соединения 4 (транс-изомер) (Фракция 2, 18%). Фракцию 1 перекристаллизовывали из СН3CN. Осадок отфильтровывали и сушили. Выход: 0,8 г соединение 3 ([(2α,4β)(В)]-энантиомер)(15%).
моль), производного 8 (0,0092 моль), NaHCO3 (0,0185 моль) и KI (1 г) в MIK (200 мл) перемешивали и кипятили с обратным кипятильником в течение нескольких часов. Растворитель выпаривали. Осадок помещали в H2O и CH2Cl2 и смесь разделяли на слои. Водный слой экстрагировали СН2Cl2. Объединенный органический слой сушили (MgSO4), фильтровали и выпаривали. Осадок очищали в стеклянном фильтре с силикагелем (элюент: CH2CI2/(CH3OH/NH3) 97/3). Чистые фракции собирали и раствор выпаривали. Осадок снова очищали ВЭЖХ с помощью RP 18 (элюент: [NH4OAc (0,5% в H2O)/CH3CN 90/10]/CH3CN 60/40). Две органические фракции собирали и их органические растворители выпаривали. Водные концентрации экстрагировали H2Cl2. Органические слои разделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Выход: Фракция 1 и 0,98 г соединения 4 (транс-изомер) (Фракция 2, 18%). Фракцию 1 перекристаллизовывали из СН3CN. Осадок отфильтровывали и сушили. Выход: 0,8 г соединение 3 ([(2α,4β)(В)]-энантиомер)(15%).
Пример В3
Получение соединения 5

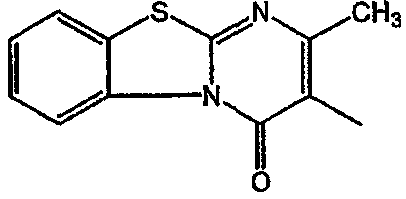
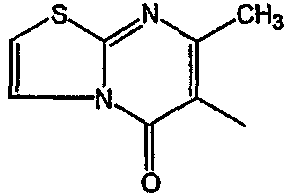
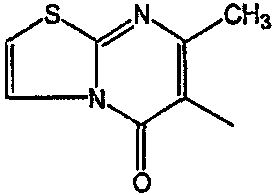
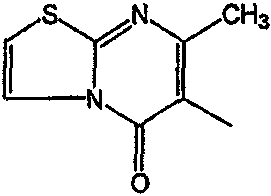
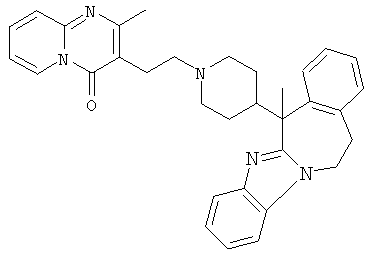
Смесь (0,01 мол), промежуточное соединение 13 (0,01 моль), Na2СО3 (0,025 моль)и KI (каталитическое количество) в MIBK (200 мл) перемешивали при 120°С. Реакционную смесь охлаждали и фильтровали с помощью дикалита. Фильтрат выпаривали. Осадок очищали на стеклянном фильтре с силикагелем (элюент: CH2CI2/СН3ОН 96/4). Чистые фракции собирали и растворитель выпаривали. Осадок (3,9 г) перекристаллизовывали из СН3CN. Продукт отфильтровывали и сушили. Выход: 2,3 г 6-[2-[4-(11,12-дигидро-6H-бензимидазо[2,1-b][3]бензазепин-6-илиден)-1-пиперидинил]этил]-7-метил-5H-тиазоло[3,2-а]пиримидин-5-она (соединения 5) (45,3%) (т.пл. 224,9°С).
Пример В4
Получение соединения 6
Смесь промежуточного соединения 16 (0,0023 моль),  (0,0046 моль), Na2CO3 (0,0046 моль) и KI (0,0046 моль) в MIBK перемешивали в течение 24 часов. Реакционную смесь подвергали гидролизу водой и экстрагировали CH2Cl2. Осадок очищали колоночной хроматографией с силикагелем (элюент: CH2CI2/(CH3OH/NH3) 95/5), затем ВЭЖХ с сликагелем (элюент: СН2CI2/CH3OH/NH3) 97/3). Чистые фракции собирали и растворитель выпаривали. Выход: 0,250 г соединения 6 (23%) (т.пл: 133,9°С).
(0,0046 моль), Na2CO3 (0,0046 моль) и KI (0,0046 моль) в MIBK перемешивали в течение 24 часов. Реакционную смесь подвергали гидролизу водой и экстрагировали CH2Cl2. Осадок очищали колоночной хроматографией с силикагелем (элюент: CH2CI2/(CH3OH/NH3) 95/5), затем ВЭЖХ с сликагелем (элюент: СН2CI2/CH3OH/NH3) 97/3). Чистые фракции собирали и растворитель выпаривали. Выход: 0,250 г соединения 6 (23%) (т.пл: 133,9°С).
Пример В5
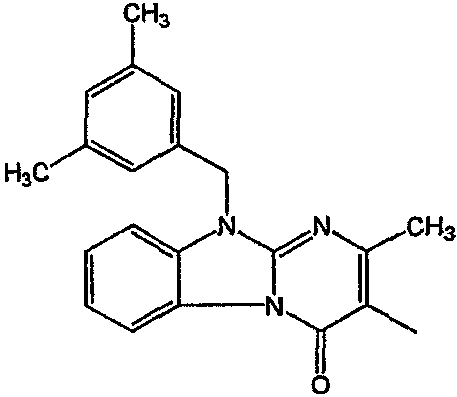
Смесь промежуточного соединения 18 (0,0063 моль), (0,0127 моль), Na2CO3 (0,0127 моль) и KI (0,0127 моль) в MIBK (200 мл) перемешивали и нагревали с обратным холодильником в течение ночи. Добавляли H2O и смесь экстрагировали CH2Cl2. Осадок очищали ВЭЖХ с помощью силикагеля (элюент: CH2CI2/(СН3ОН/NH3) 95/5). Чистые фракции собирали и растворитель выпаривали. Осадок промывали CH3CN и сушили. Выход: 1 г соединения 7 (28%) (т.пл: 213,2°С).
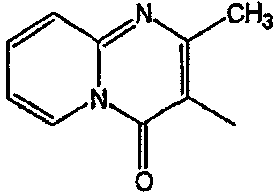
Пример В6
Получение соединения 8
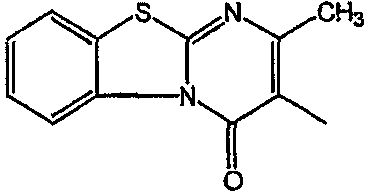
Смесь 6-(2-хлорэтил)-7-метил-5H-тиазоло [3,2-а]пиримидин-5-она (0,012 моль), промежуточное соединение 13 (0,01 моль), Na2CO3 (0,01 моль) и KI (0,01 г) в MIBK (200 мл) перемешивали и кипятили с обратным холодильником в течение ночи. Реакционную смесь выливали в воду. Слои разделяли и водный слой повторно экстрагировали 4-метил-2-пентаноном. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали. Осадок очищали на стеклянном фильтре с силикагелем (элюент: СН2CI2/СН3ОН/(СН3ОН/NH3) 90/10/1). Очищенные фракции собирали и растворитель выпаривали. Осадок перекристаллизовывали из ацетонитрила. Продукт отфильтровывали и сушили. Выход: 2,2 г 6-[2-[4-(6H-бензимидазо[2,1-b][3]бензазепин-6-илиден)-1-пиперидинил]этил]-7-метил-5H-тиазоло [3,2-а]пиримидин-5-она (соединение 8) (43,5%) (т.пл.: 178,8°С).
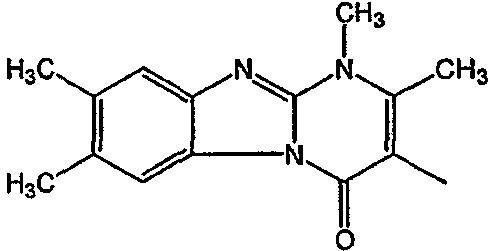
Соединения, представленные в таблицах, были получены способами, аналогичными одному из способов согласно примерам В1-В6.
Изобретение описывает соединения общей формулы I /значения радикалов см. в формуле изобретения/, являющиеся новыми антагонистами рецептора гистамина. Предпочтительным соединением является 3-[2-[4-(11,12-дигидро-6Н-бензимидазо[2,1-b][3]бензазепин-6-ил)-2-(фенилметил)-1-пиперидинил]этил]-2,10-диметилпиримидо[1,2-α]бензимидазол-4(10Н)-он, или его соли, изомеры и N-оксиды. Соединения полезны для предотвращения и лечения повышенного черепного давления (ICP) и/или вторичной ишемии, вызываемых черепно-мозговой травмой. 9 н. и 4 з.п. ф-лы, 5 ил., 13 табл.
его фармацевтически приемлемые кислотно- или основно-аддитивные соли, стереохимически изомерные формы и N-оксиды, где
m равно 1;
n равно 1;
a, b, с независимо означают простую или двойную связь;
Х означает двухвалентный С1-5 алкандиильный радикал, в котором одна или несколько групп -CH2- могут быть необязательно заменены -СО- или -NR7-, где
R7 означает алкилкарбонил;
Y означает -CH2-, -CH2-CH2-; или -СН=СН-;
Z означает N, когда а означает двойную связь и b означает
простую связь, или N-R7, когда а означает простую связь и R7 выбран из группы алкил, пиридинилалкил, фенилалкил и пиразинилалкил;
R1, R2 независимо означают водород, алкил, нафтилметил, изоиндолил и фенил;
-А-В- независимо означает двухвалентный радикал формулы
где R8 означает водород;
Е означает двухвалентный радикал формулы -S- или -NR7-, где R7 означает алкил;
-C-D- независимо означает двухвалентный радикал формулы
где R8 означает водород;
R3 означает алкил;
R4 означает амино, алкиламино, пиридинилалкиламино, фенилкарбониламино, алкиламинокарбониламино или алкилоксиалкиламино;
R5 означает алкил;
или R4 и R5 вместе могут образовывать радикал формулы
где R7 означает алкил, бензил, пиридинилалкил, алкилоксиалкил, пиразинилалкил, алкилоксиалкилоксиалкил, моно- или диалкиламиноалкил, и алкилоксикарбонилалкил, гидроксиалкил, изоиндол-1,3-дионил, аминокарбонилалкил, гидроксиалкилоксиалкил, алкилкарбонилоксиалкилоксиалкил, аминоалкил, алкил-карбониламиноалкил или алкилоксиалкил;
R8 означает водород, алкил, галоген, или галогеналкил;
R9, R10 независимо означают водород, алкил, галоген, галогеналкил;
или R9 и R10 вместе могут образовывать радикал формулы -CR8=CR8-CR8=CR8-, где R8 означает водород; и
М означает двухвалентный радикал формулы -O-, -S- или -NR7-, где R7 означает алкил.
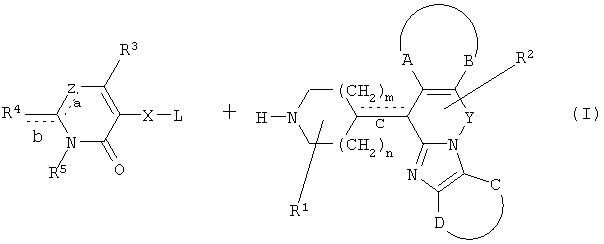
где все возможные заместители являются такими как они определены для формулы (I) и L представляет любую пригодную рекционноспособную уходящую группу, в частности галоген или сульфонилокси.
| US 6251894 B1, 26.06.2001 | |||
| WO 9913871 A, 25.03.1999 | |||
| Способ получения олигофениленов | 1977 |
|
SU675889A1 |

