Изобретение относится к области генной и белковой инженерии и может быть использовано в анализе формиата в сложных образцах и в процессах ферментативного синтеза оптически активных соединений или физиологически активных веществ с использованием дегидрогеназ.
Получены новые мутантные формы формиатдегидрогеназы (ФДГ) бактерий, характеризующиеся повышенной по сравнению с соответствующими ферментами дикого типа термостабильностью. Все предложенные мутеины характеризуются тем, что природный аминокислотный остаток глутаминовой кислоты в положении, соответствующем положению 106 в последовательности формиатдегидрогеназы из Mycobacterium vaccae N10, заменен на остаток аргинина. Данная замена может быть использована в комбинации с другими мутациями, обеспечивающими приобретение формиатдегидрогеназами новых или улучшенных свойств
Настоящее изобретение относится к новому белку, в частности мутантным формиатдегидрогеназам из бактерий, которые обладают повышенной термостабильностью по сравнению с соответствующим ферментом дикого типа, к ДНК, кодирующей мутантные формиатдегидрогеназы, к применению данных ферментов в аналитических тестах на наличие формиата, к наборам, включающим их и в процессах ферментативного синтеза оптически активных соединений или физиологически активных веществ с помощью дегидрогеназ.
НАД+-зависимая формиатдегидрогеназа катализирует окисление формиата до углекислого газа при сопряженном восстановлении НАД+ до НАДН.
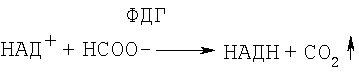
Исключительно высокую специфичность формиатдегидрогеназы к формиату используют для определения формиата в сложных смесях. Необратимость реакции, катализируемой этим ферментом, и широкий рН-оптимум активности формиатдегидрогеназы позволяют очень эффективно его использовать для регенерации НАДН в процессах синтеза оптически активных соединений с использованием дегидрогеназ. В качестве примера можно привести крупномасштабный процесс получения терт-L-лейцина из триметилпирувата с использованием лейциндегидрогеназы с системой регенерации НАДН на основе формиатдегидрогеназы из дрожжей Candida boidinii (Bommarius et al., Tetrahedron Asymmetry 1995, 6, 2851-2852).
Формиатдегидрогеназы широко распространены в природе и могут быть получены из бактерий, дрожжей, низших грибов и высших растений. Подробный список известных источников формиатдегидрогеназ можно найти в обзоре (Tishkov V., Popov V. Protein engineering of formate dehydrogenase. Biomolecular Engineering, 2006, v.23, N2-3, p.89-110). Однако поскольку многие из генов, кодирующих данные ферменты, были клонированы и секвенированы, их можно также получить с использованием технологии рекомбинантных ДНК. Последовательности рекомбинантной ДНК, кодирующие ферменты, используют для трансформации микроорганизмов, таких как E.coli, которые затем экспрессируют желаемый ферментный продукт.
Недостатком природных формиатдегидрогеназ и рекомбинантных формиатдегидрогеназ дикого типа можно признать их низкую каталитическую активность и относительно невысокую химическую и температурную стабильность. Особенно это актуально для ферментов из дрожжей, низших грибов и высших растений. Наиболее высокой химической и температурной стабильностью обладает формиатдегидрогеназа из бактерий Pseudomonas sp.101. Для этого фермента описаны мутантные формы, в которых остатки серина в положениях 131, 160, 184 и 228 (все они относятся к альфа-спиральным участкам структуры фермента) заменены на остатки аланина (Rojkova et. al. Bacterial formate dehydrogenase. Increasing the enzyme thermal stability by hydrophobization of alpha helices. FEBS Letters - 1999, v.445, N1, p.183-188). Полученные мутантные ферменты с одной, двумя и суммарно с четырьмя аминокислотным заменами превосходили по термостабильности ФДГ дикого типа из Pseudomonas sp.101 на 10-60%. Для формиатдегидрогеназы из C.boidinii известны мутантные формы, которые превосходят соответствующий фермент дикого типа по активности, химической и температурной стабильности (Slusarczyk H., Felber S., Kula M. - R., Pohl M., 2003. Novel mutants of formate dehydrogenase from Candida boidinii. US Patent Application Publication US 2003/0157664, 21.09.2003), которые однако по всем этим параметрам уступают мутантной PseФДГ с заменой Cys255Ala (Одинцева Е.Р., Попова А.С., Рожкова A.M., Тишков В.И. Роль остатков цистеина в стабильности бактериальной формиатдегидрогеназы. Вестн. Моск. Ун-та. Сер.2, Химия, 2002, т.43, №6, с.356-359).
Техническая задача, решаемая посредством предлагаемого технического решения, состоит в получении мутантной формы формиатдегидрогеназы с улучшенными характеристиками.
Технический результат, получаемый при реализации предлагаемого технического решения, состоит в повышении термостабильности полученной формы формиатдегидрогеназы.
Для достижения указанного технического результата предложено использовать рекомбинантную термостабильную формиатдегидрогеназу, характеризующуюся аминокислотной последовательностью, которая соответствует аминокислотной последовательности формиатдегидрогеназы дикого типа с заменой природного остатка глутаминовой кислоты в положении, соответствующем положению 106 в последовательности формиатдегидрогеназы Mycobacterium vaccae N10, остатком аргинина, введение которого обеспечивает повышение термостабильности модифицированного фермента. Последовательность указанной формиатдегидрогеназы дикого типа предпочтительно соответствует формиатдегидрогеназе Mycobacterium vaccae N10, Pseudomonas sp.101, Thiobacillus sp.KNK65MA, Ancylobacter aquaticus sp.KNK607M, Moraxella sp.C-1, Paracoccus denitrificans PD1222, Paracoccus sp.12-A, Hyphomicrobium sp.JC17, Sinorhizobium meliloti 1021. Наиболее предпочтительно последовательность формиатдегидрогеназы дикого типа является последовательностью фермента, получаемого из Mycobacterium vaccae N10, Pseudomonas sp.101 или Moraxella sp.C-1. Преимущественно белок обладает, по меньшей мере, 80%-ной гомологией по отношению к формиатдегидрогеназе из Mycobacterium vaccae N10, Pseudomonas sp.101 или Moraxella sp.C-1 и содержит остаток глутаминовой кислоты в положении, соответствующем положению 106 в аминокислотной последовательности формиатдегидрогеназы из Mycobacterium vaccae N10. Кроме того, помимо замены природного остатка глутаминовой кислоты на остаток аргинина в положении, соответствующем положению 106 в аминокислотной последовательности формиатдегидрогеназы из Mycobacterium vaccae N10, она может содержать, по меньшей мере, одну дополнительную замену, обеспечивающую улучшение термостабильности, химической стабильности, кинетических свойств, попарное или одновременное улучшение всех вышеуказанных свойств по сравнению с соответствующей формиатдегидрогеназой дикого типа.
Белки по изобретению включают как формиатдегидрогеназы дикого типа, так и рекомбинантные. Они обычно обладают 70% гомологией по отношению к последовательностям дикого типа, как таковые у фермента Mycobacterium vaccae N10, Pseudomonas sp.101 или Moraxella sp.C-1, в том смысле, что, по меньшей мере, 70% аминокислот, находящихся в ферментах дикого типа, находятся в белках по изобретению. Такие белки могут обладать более высокой степенью гомологии, в частности, по меньшей мере, 80%, и наиболее предпочтительно, по меньшей мере, 90% по отношению к ферментам дикого типа, перечисленным выше. Гомологию определенной последовательности по отношению к последовательностям по изобретению можно оценить с использованием метода множественного сравнительного анализа первичной структуры, описанного Lipman and Pearson (Lipman D.J. & Pearson W.R. Rapid and Sensitive Protein Similarity Searches, Science, 1985, vol.227, pp.1435-1441). Следует высчитать «оптимизированную» процентную схему по следующим параметрам для алгоритма Липмана-Пирсона: ktup =1, пробел =4 и длина пробела =12. Последовательности, для которых требуется оценить гомологию, следует использовать в качестве «испытуемой последовательности», что означает, что основную последовательность для сравнения, такую как последовательность из Mycobacterium vaccae N10, Pseudomonas sp.101 или Moraxella sp.C-1 следует вводить в алгоритм первой. Мутантная форма формиатдегидрогеназы по изобретению может быть получена из Mycobacterium vaccae N10, Pseudomonas sp.101, Thiobacillus sp.KNK65MA, Ancylobacter aquaticus sp.KNK607M, Moraxella sp.C-1, Paracoccus denitrificans PD1222, Paracoccus sp.12-A, Hyphomicrobium sp.JC17, Sinorhizobium meliloti 1021, некультивируемых морских гамма-протеобактерий ЕВАС20Е09.
Экспериментально установлено, что рекомбинантные бактериальные формиатдегидрогеназы, полученные из Mycobacterium vaccae N10, Pseudomonas sp.101 и Moraxella sp.C-1 и содержащие замену Glu106Arg, обладают повышенной термостабильностью по сравнению с ферментами дикого типа. Также экспериментально установлено, что введение замены Glu106Arg в мутантную форму формиатдегидрогеназы из Pseudomonas sp.101 с другими аминокислотными заменами, обеспечивающими повышение термостабильности фермента, приводит к получению нового мутантного фермента с еще более высокой термостабильностью.
В дальнейшем способы получения и преимущества полученной модифицированной формиатдегидрогеназы будут рассмотрены с использованием примеров ее реализации.
Пример 1.
Получение мутантной формиатдегидрогеназы Mycobacterium vaccae N10 с повышенной термостабильностью с использованием неупорядоченного мутагенеза.
Все генно-инженерные манипуляции, если не оговорено особо, осуществлены в соответствии с Манниатис Е., Фрич. Э., Сэмбрук Дж. Молекулярное клонирование, М. - Мир, 1984 или в соответствии с инструкциями фирм-производителей приборов, наборов и других реагентов. Все эти методы хорошо известны специалистам в данной области.
Неупорядоченный мутагенез полноразмерного гена ФДГ из Mycobacterium vaccae N10 проводили с использованием полимеразной цепной реакции (ПНР) в условиях, обеспечивающих возникновение ошибок в последовательности ДНК (Caldwell R.С., Joyce G.F. Randomization of Genes Mutagenesis. PCR Methods Appl.2, 1992, 28-33) (error-prone PCR). В качестве матрицы для проведения неупорядоченного мутагенеза использовали плазмиду pFDH6, в которой ген формиатдегидрогеназы находится под контролем тандема двух промоторов - lac- и tac. Плазмида pFDH6 получена из коммерчески доступной плазмиды pUC119, в которую по сайтам рестрикции HindIII и BamHI встроен фрагмент ДНК размером 81 основание, вырезанный по тем же сайтам рестрикции из другой коммерчески доступной плазмиды pDR540 и содержащий tac-промотор (подробно получение этой плазмиды см. в Tishkov V.I., Galkin A.G., Fedorchuk V.V., Savitsky P.A., Rojkova A.M., Gieren H., Kula M. - R. Pilot scale production and isolation of recombinant NAD+- and NADP+-specific formate dehydrogenase. Biotechnology & Bioengineering - 1999, v.64, N2, p.187-193). Ген ФДГ из Mycobacterium vaccae N10 встраивали в плазмиду pFDH6 по сайтам рестрикции BamHI и EcoRI, которые вводили с использованием ПЦР по хорошо известной методике соответственно перед рибосомсвязывающим участком и сразу за стоп-кодном. В результате получили плазмиду pMycFDH6. Ее выделили из клеток Е.coli с использованием набора для выделения плазмид NucleoSpin® Plus согласно инструкции фирмы-производителя (Macherey-Nagel GmbH & Со. KG).
Для проведения ПЦР с ошибками использовали пару следующих праймеров:
pFDH6For 5′-CAT GAT TAC GCC AAG CTT ACT C-3′
pFDH6Rev 5′-AAC GAC GGC CAG TGAATTCAC-3′
Последовательности этих праймеров являются частью последовательности плазмиды pFDH6 и могут быть использованы для введения неупорядоченных мутаций с использованием ПЦР в любой ген, клонированный в эту плазмиду. Реакционная смесь для проведения ПЦР с ошибками имела следующий состав (таблица 1).
Реакцию проводили в тонкостенной пластиковой пробирке объемом 0,5 мл (Eppendorf, Германия). Для предотвращения испарения реакционной смеси и ее конденсации на крышке в пробирку добавляли 50 мкл минерального масла. Пробирку прогревали 5 мин при 95°С и затем проводили реакцию ПЦР с ошибками по следующей программе: 1-я стадия - 95°С, 60 с; 2-я стадия - 50°С, 45 с и 3-я стадия - 72°С, 2 мин, всего 25-35 циклов. После этого реакционную смесь выдерживают еще 5 мин при 72°С. Температуру на второй стадии выбирают на 3-5 градусов ниже Tm температуры плавления дуплексов, образуемых праймерами.
Продукты ПЦР очищают или с использованием предназначенных для этого наборов (например, NucleoSpin Extract (Macherey-Nagel GmbH & Co. KG)) или с использованием препаративного электрофореза в агарозном геле. Затем их обрабатывают рестриктазами BamHI и EcoRI в течение 6 часов при 37°С, очищают от коротких фрагментов препаративным электрофорезом в агарозном геле с последующей экстракцией нужной полосы из агарозы с использованием набора NucleoSpin Extract (Macherey-Nagel GmbH & Co. KG) и клонируют в исходную плазмиду, из которой предварительно по тем же сайтам рестрикции BamHI и EcoRI удален фрагмент с геном ФДГ из Mycobacterium vaccae N10 дикого типа. Полученную библиотеку мутантных клонов экспрессировали в штамме Е.coli TG1 (генотип supE hsdΔ5 thi Δ(lac-proAB) F′[traD36 proAB+ lacIq lacZΔM15])). Плазмида pMycFDH6 имеет ген бета-лактамазы и придает клеткам E.coli, содержащим эту плазмиду, резистентность к ампициллину.
Штамм Е.coli TG1 трансформировали полученной библиотекой электропорацией с использованием генератора импульсов BIORAD для Е.coli и выращивали в течение ночи при 37°С на агаре LB, содержащем ампициллин в концентрации 150 мкг/мл и 0,1 мМ изопропил-β-D-тиогалактозида (ИПТГ). Диаметр колоний в конце культивирования составлял около 1 мм.
По окончании культивирования клетки заливали 1,5% раствором агарозы с буфером для лизиса №1 (0,1 М фосфатный буфер; рН 7,0, 0,2% Тритон Х-100, 10 мМ ЭДТА) в количестве, достаточном для образования слоя толщиной около 2 мм. Перед заливкой температура агарозы не должна превышать 55-60°С. После затвердевания агарозы чашки 5 раз промывали буфером для лизиса №1 и 5 раз 0,1 М фосфатным буфером, рН 7,0 (толщина слоя раствора на чашке 5 мм).
Далее колонии окрашивали по активности. Для этого готовили проявляющий раствор №1 (0,1 М фосфатный буфер, содержащий 1,5 М формиат натрия, 0,2 г/л феназинметасульфата, 2 г/л нитротетразолия синего, рН 7,0. Раствор покрывает чашки слоем толщиной 1-2 мм. После инкубации чашек с проявляющим раствором №1 в течение 10 мин в темноте при постоянном перемешивании к ним добавляли проявляющий раствор №2 (60 мг/мл НАД+ в 0,1 М фосфатном буфере, рН 7,0) из расчета 1 часть проявляющего раствора №2 на 50 частей проявляющего раствора №1. Далее чашки инкубируют в темноте при 30°С при перемешивании (примерно 15-30 мин) пока не станет заметно образование черно-коричневого гало вокруг колоний. Затем чашки промывают дважды раствором 0,1 М фосфатного буфера, рН 7,0 и подсушивают, чтобы можно было отобрать колонии для дальнейшего культивирования.
Колонии, содержащие активную формиатдегидрогеназу, отбирают и культивируют в течение ночи при 37°С в 96-луночных микропланшетах в 0,2 мл среды 2YT, содержащей ампициллин в концентрации 150 мкг/мл. Полученные растворы колоний в микропланшетах использовали как запасные для дальнейшей работы.
Для поиска клонов, содержащих формиатдегидрогеназу с повышенной термостабильностью, проводили повторное культивирование. Для этого из лунок запасных микропланшетов отбирали по 50 мкл суспензии клеток и переносили в новый микропланшет, содержащий в каждой лунке по 0,2 мл среды 2YT, ампициллин в концентрации 150 мкг/мл и 0,1 мМ ИПТГ. Микропланшеты с клетками инкубировали в течение ночи при 37°С при постоянном перемешивании. После окончания культивирования клетки в микропланшетах осаждали центрифугированием (1600 g, 15 мин, 20°С), удаляли из лунок культуральную жидкость и ресуспендировали в 200 мкл буфера для лизиса №2 (0,1 М фосфатный буфер, рН 8,0, яичный лизоцим 0,1 мг/мл и Тритон Х-100, 0,1 объемных %). Лизис клеток проводили в течение 2 часов при 35°С при постоянном перемешивании. Клеточный дебрис осаждали с использованием центрифугирования (5000 g, 15 мин, 20°С). 25 мкл супернатанта переносили в новый микропланшет для измерения исходной активности (АO). Для этого в каждую лунку нового микропланшета добавляли еще по 175 мкл фосфатного буфера, рН 7,0, содержащего 0,35 М формиата натрия и 1,2 мг/мл НАД+ Реакцию регистрировали при 30°С по увеличению поглощения на 340 нм на анализаторе микропланшетов (Spectramax® Plus, MWG Biotech). Еще 50 мкл супернатанта переносили в другой микропланшет для проведения ПЦР, содержащих в каждой лунке по 50 мкл 0,1 М фосфатного буфера, рН 7,0. Микропланшет помещали в термоблок прибора для проведения ПЦР в микропланшетах (Techne 96, Techne) и инкубировали в течение 20 мин при определенной температуре (обычно 60-63°С). Затем микропланшет охлаждали в смеси воды и льда в течение 5 мин, при необходимости удаляли осадок центрифугированием и отбирали 50 мкл для определения остаточной активности (А20).
Из клонов E.coli, экспрессирующих МусФДГ с повышенной термостабильностью, выделяли плазмидную ДНК, как описано выше, и затем ее секвенировали для того, чтобы определить мутации, ответственные за термостабильность фермента. Секвенирование ДНК проводили на автоматическом секвенаторе ДНК ABI PRISMTM модели 377 и реакционного набора для циклического секвенирования с терминаторами ABI PRISMTM BigDyeTM (Perkin Elmer Applied Biosystems), который основан на дидезокситерминационном методе [F.Sanger et al., Proc. Natl. Acad. Sci. USA. 1977, 74, 5463-5467].
В результате данной работы идентифицировали новый мутант МусФДГ Glu106Arg. Количественную оценку эффекта такой замены на термостабильность фермента проводили согласно процедуре, описанной в примере 5. Полученная мутантная формиатдегидрогеназа была на 18% более стабильна, чем фермент дикого типа (табл.2).
Пример 2.
Получение мутантной MorФДГ с заменой Glu106Arg с использованием направленного мутагенеза.
Направленный мутагенез проводили по методу Кункеля (Kunkel Т.А. Rapid and efficient site-derected mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA - 1985, v.82, p.488-492) с использованием уридиновой матрицы pMorFDH6 в одноцепочечной форме с использованием праймера MORE106R, несущего требуемую замену
5′-CGC-CTT-GGC-GAT GCG NCG GGC GGT CAG ATA GGC-3′
Жирным шрифтом выделен триплет, обеспечивающий мутацию Glu106Arg. Буква N означает, что в этом положении может быть любое из 4 оснований (А, Т, G или С).
Для получения плазмиды pMorFDH6 в одноцепочечной форме с заменой основания Т на U использовали специальную линию клеток E.coli RZ1023. Протрансформированные плазмидой pMorFDH6 клетки Е.coli RZ1023, инокулировали в 4 мл среды 2YT, содержащей 100 мкг/мл ампициллина и 5 мкг/мл уридина, и растили при 37°С в течение 8-10 часов при аэрировании. Далее клетки инфицировали фагом-помощником М13К07 (количество бляшкообразующих частиц КБЕ 109-1010) при множественности инфекции 10 и инкубировали при 37°С в течение 30 минут. Затем всю инкубированную смесь помещали в 20 мл среды 2YT (содержащей 100 мкг/мл ампициллина, 100 мкг/мл канамицина, 5 мкг/мл уридина и 10 мкг/мл тиамина) и растили при 37°С в течение 12 часов при сильном аэрировании. Клетки центрифугировали на центрифуге Eppendorff 5403 при 10000 об/мин в течение 15 минут. Надосадочную жидкость переносили в чистую центрифужную пробирку, добавляли 0,2 объема 20% раствора ПЭГ-6000 и 2,5 М NaCI, инкубировали при 0°С в течение 1 часа и центрифугировали при 10000 об/мин в течение 30 минут. Осадок фаговых частиц ресуспендировали в 400 мкл деионизованной воды и переносили раствор в чистую пробирку на 1,5 мл. Далее к нему добавляли 400 мкл смеси фенола с хлороформом (в соотношении 1:1), перемешивали и центрифугировали при 14000 об/мин в течение 15 минут. Отбирали верхнюю фазу и повторяли экстракцию смесью фенола с хлороформом еще раз. Затем к верхней фазе добавляли 400 мкл хлороформа, перемешивали и центрифугировали при 14000 об/мин в течение 15 минут. ДНК осаждали добавлением двойного объема изопропанола и центрифугировали при 14000 об/мин в течение 30 минут. Осадок промывали 1 мл охлажденного 70% этанола и центрифугировали в тех же условиях. ДНК высушивали на воздухе в течение 10-15 минут и растворяли в 30 мкл деионизованной воды.
Для проведения реакции фосфорилирования к 25 пикомоль сухого праймера добавляли 2 мкл 10-кратного фосфорилирующего буфера (700 мМ Tris-HCl, 100 мМ MgCl2, pH 7,6), 2 мкл раствора АТФ (10 мМ), 2 мкл ДТТ (50 мМ), 2 мкл полинуклеотидкиназы фага Т4 (10 ед/мкл), 12 мкл деионизованной воды и инкубировали в течение 2 часов при температуре 37°С. Затем к 10 мкл раствора фосфорилированного праймера добавляли 2 мкл (0,025 пикомоль) уридиновой матрицы, 2 мкл 10-кратного буфера для отжига (200 мМ Tris-HCl, 100 мМ MgCl2, 500 мМ NaCl, pH 7,5) и 8 мкл деионизованной воды. Полученный раствор помещали в стакан с кипящей водой и оставляли на столе медленно охлаждаться до комнатной температуры. Для проведения реакции синтеза второй цепи ДНК в реакционную смесь добавляли 3 мкл 10-кратного буфера для синтеза (100 мМ Tris-HCl, 20 мМ ДТТ, по 5 каждого dNTP, 10 мМ АТФ), 1,5 мкл ДНК-полимеразы фага Т4 (10 ед/мкл), 1,5 мкл ДНК-лигазы фага Т4 (400 ед/мкл), 4 мкл деионизованной воды и инкубировали последовательно 5 минут при 0°С, 5 минут при комнатной температуре и 2-3 часа при 37°С.
Полученной в результате мутагенеза реакционной смесью трансформировали клетки Е.coli TG1, как это указано в примере 1. Выделение плазмид и их секвенирование также проводили аналогично примеру 1. Из 6 выделенных плазмид все 6 содержали только требуемую замену Glu106Arg. Количественное определение изменения термостабильности мутантной MorФДГ Glu106Arg проводили, как описано в примере 5. Мутантная MorФДГ Glu106Arg была на 11% более стабильна, чем фермент дикого типа (табл.2).
Пример 3.
Получение мутантной PseФДГ с заменой Glu106Arg с использованием направленного мутагенеза.
Данный пример идентичен примеру 2 за исключением того, что в качестве матрицы для мутагенеза использовали плазмиду pPseFDH6 и праймер
5′-GGC CTT GGC GAT GCG NCG GGG CGT CAG АТА G-3′
Этот же праймер может быть использован для введения замены Glu106Arg в гене МусФДГ.
Количественное определение изменения термостабильности мутантной PSeФДГ Glu106Arg проводили, как описано в примере 5. Полученная мутантная PSeФДГ с заменой Glu106Arg была на 15% более стабильна, чем PseФДГ дикого типа (табл.2).
Пример 4.
Получение многоточечной мутантной PSeФДГ с заменами Ser(131, 160, 184, 228)Ala и Glu106Arg.
Данный пример идентичен примеру 3 за исключением того, что замену Glu106Arg вводили в ген мутантной PSeФДГ, в котором по сравнению с геном PseФДГ дикого типа остатки Ser в положениях 131, 160, 184 и 228 заменены на остатки Ala. В качестве исходной матрицы использовали плазмиду pPseFDH6T4, получение которой описано в Rojkova, A.M., et al. Bacterial formate dehydrogenase. Increasing the enzyme thermal stability by hydrophobization of alpha helices. FEBS Letters - 1999, v.445, N1, p.183-188.
Полученная мутантная PseФДГ с заменами Ser(131, 160, 184, 228)Ala и Glu106Arg была на 85% более стабильна, чем PseФДГ дикого типа (табл.2).
Пример 5.
Точное определение эффекта стабилизации формиатдегидрогеназы за счет замены Glu106Arg.
Для точной оценки эффекта стабилизации формиатдегидрогеназы за счет замены Glu106Arg мутантный фермент и формиатдегидрогеназу дикого типа выделяли в высокоочищенном состоянии и определяли константы скорости термоинактивации при определенной температуре (выше 54°С).
Методика очистки абсолютно одинакова для всех приведенных выше рекомбинантных формиатдегидрогеназ и не зависит как от источника гена (Mycobacterium vaccae N10, Pseudomonas sp.101 или Moraxella sp.C-1), так и от типа фермента (мутантный или дикого типа). Биомассу клеток E.coli TG1, содержащую целевой фермент, получали культивированием единичной колонии в колбах объемом 1 л, содержащих 250 мл среды 2YT и 150 мкг/мл ампициллина при 37°С при непрерывном перемешивании (110 об/мин). По достижении поглощения среды на 600 нм величины А600 0,6-0,9 в колбы добавляли ИПТГ до концентрации 0,5 мМ и культивировали еще 12 часов. Клетки собирали центрифугированием (2500g, 15 мин, 4°С). Затем их ресуспендировали в 0,1 М фосфатном буфере, рН 7,0 для получения 20% суспензии (общий объем 10 мл). Для предварительного разрушения клетки подвергали замораживанию-размораживанию и далее обрабатывали ультразвуком в течение 5 мин при охлаждении льдом. Осадок удаляли центрифугированием (10000g, 30 мин, 4°С), а к супернатанту (объем 8 мл) добавляли при интенсивном перемешивании 6,4 мл 4М раствора сульфата аммония в 0,1 М фосфатном буфере, рН 7,0. Полученный раствор оставляли на ночь при 4°С, образующийся осадок удаляли центрифугированием (10000g, 30 мин, 4°С). Супернатант наносили на колонку 1×10 см с Фенилсефарозой Fast Flow (Pharmacia Biotech, Германия), уравновешенной 1,5 М раствором сульфата аммония в 0,1 М фосфатном буфере, рН 7,0. После нанесения фермента на колонку ее промывали 1,2 М раствором сульфата аммония в 0,1 М фосфатном буфере, рН 7,0 (раствор А) до нулевого поглощения на 280 нм выходящего из колонки раствора (70-100 мл). Фермент элюировали с колонки линейным нисходящим градиентом (50 мл раствора А - 50 мл 0,1 М фосфатного буфера, рН 7,0). Собирали фракции, содержащие мутантный фермент или формиатдегидрогеназу дикого типа, и концентрировали на мембране РМ10 в ячейке для концентрирования объемом 10 мл (мембрана и ячейка производства Amicon, США) до объема 1 мл. Полученный фермент наносили на колонку 1×100 см с Sephacryl S-200 (Pharmacia Biotech, Германия), уравновешенную 0,1 М фосфатным буфером, рН 7,0. Собирали фракции по 2 мл, определяли их поглощение на 280 нм A280 на спектрофотометре Shimadzu 1601 PC (Shimadzu GmBH, Германия) и измеряли величину ферментативной активности на 340 нм при 30°С на том же спектрофотометре (0,1 М фосфатный буфер, рН 7,0, 03 М формиат натрия, 1 мг/мл НАД+). Отбирали фракции, имеющие постоянное отношение (активность/А280). Чистота полученных препаратов составляет не менее 95% согласно данным аналитического электрофореза в 12% полиакриламидном геле в присутствии додецилсульфата натрия. Электрофорез проводили на приборе фирмы BioRad согласно инструкции фирмы-производителя.
Для определения константы скорости термоинактивации формиатдегидрогеназы дикого типа и мутантного фермента с заменой Glu106Arg для каждого типа фермента готовили серию из 12-15 пластиковых пробирок объемом 1,5 мл (Eppendorf, Германия), содержащих по 60 мкл раствора фермента в концентрации 0,2-1 мг/мл в 0,1 М фосфатном буфере, рН 7,0. Пробирки помещали в водяной термостат при определенной температуре (54-64°С, точность термостатирования ±0,1 град) и через заданные промежутки времени отбирали по 1 пробирке, охлаждали в холодной воде (0-10°С) в течение 5 мин, центрифугировали 3 мин при 10000g и определяли величину остаточной ферментативной активности А, как указано выше. Затем строили график зависимости натурального логарифма остаточной активности ln(А/А0) от времени и из тангенса угла наклона рассчитывают величину константы скорости инактивации kin. В качестве примера приведены зависимости для PSeФДГ дикого типа и его мутантной формы с заменой Glu106Arg. В таблице 2 приведены значения констант скоростей инактивации для всех изученных ФДГ.
Полученная мутантная форма формиатдегидрогеназы обладает повышенной относительно исходной формы термостабильностью.
| название | год | авторы | номер документа |
|---|---|---|---|
| МУТАНТНАЯ РАСТИТЕЛЬНАЯ ФОРМИАТДЕГИДРОГЕНАЗА (ВАРИАНТЫ) | 2014 |
|
RU2557299C1 |
| МУТАНТНАЯ ФОРМИАТДЕГИДРОГЕНАЗА (ВАРИАНТЫ) | 2013 |
|
RU2545966C1 |
| МУТАНТНАЯ ФОРМИАТДЕГИДРОГЕНАЗА (ВАРИАНТЫ) | 2012 |
|
RU2522819C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДА pHpFDH_HisA, ОБЕСПЕЧИВАЮЩАЯ СИНТЕЗ В КЛЕТКАХ Escherichia coli ФОРМИАТДЕГИДРОГЕНАЗЫ МЕТИЛОТРОФНЫХ ДРОЖЖЕЙ Hansenula polymorpha (HpFDH_HisA) И РЕКОМБИНАНТНЫЙ ШТАММ E.сoli BL21(DE3)/pHpFDH_HisA ВКПМ В-14559 - ПРОДУЦЕНТ HpFDH_HisA | 2023 |
|
RU2829762C1 |
| МУТАНТНАЯ ОКСИДАЗА D-АМИНОКИСЛОТ | 2009 |
|
RU2412998C1 |
| МУТАНТНАЯ ФОРМА КСИЛОГЛЮКАНАЗЫ ИЗ ASPERGILLUS CERVINUS С ПОВЫШЕННОЙ ПО СРАВНЕНИЮ С ФЕРМЕНТОМ ДИКОГО ТИПА ТЕРМОСТАБИЛЬНОСТЬЮ, РЕКОМБИНАНТНЫЙ ШТАММ ESCHERICHIA COLI, ПРОДУЦИРУЮЩИЙ ЭТУ КСИЛОГЛЮКАНАЗУ, И СПОСОБ МИКРОБИОЛОГИЧЕСКОГО СИНТЕЗА МУТАНТНОЙ КСИЛОГЛЮКАНАЗЫ ИЗ ASPERGILLUS CERVINUS НА ОСНОВЕ ЭТОГО ШТАММА | 2022 |
|
RU2815837C1 |
| РЕКОМБИНАНТНЫЙ ШТАММ ДРОЖЖЕЙ PICHIA PASTORIS - ПРОДУЦЕНТ СЕКРЕТИРУЕМОЙ КСИЛОГЛЮКАНАЗЫ СЕМЕЙСТВА GH12, КОДИРУЕМОЙ МУТИРОВАННЫМ ГЕНОМ, И СПОСОБ МИКРОБИОЛОГИЧЕСКОГО СИНТЕЗА СЕКРЕТИРУЕМОЙ КСИЛОГЛЮКАНАЗЫ НА ОСНОВЕ ЭТОГО ШТАММА | 2023 |
|
RU2833908C1 |
| МУТАНТНАЯ ОКСИДАЗА D-АМИНОКИСЛОТ (ВАРИАНТЫ) | 2012 |
|
RU2507262C1 |
| МУТАНТНЫЕ ОКСИДАЗЫ D-АМИНОКИСЛОТ | 2007 |
|
RU2362806C2 |
| ЛЮЦИФЕРАЗЫ | 1995 |
|
RU2192467C2 |
Изобретение относится к биотехнологии и представляет собой рекомбинантную формиатдегидрогеназу, которая имеет аминокислотную последовательность, которая соответствует аминокислотной последовательности формиатдегидрогеназы дикого типа, содержащей остаток глутаминовой кислоты в положении, соответствующем положению 106 в последовательности формиатдегидрогеназы Mycobacterium vaccae N10, в котором остаток глутаминовой кислоты заменен на остаток аргинина. Данная замена может быть использована в комбинации с другими мутациями, обеспечивающими приобретение формиатдегидрогеназами новых и/или улучшенных свойств. Изобретение позволяет получить новую формиатдегидрогеназу, которая обладает повышенной по сравнению с ферментами термостабильностью. 3 з.п. ф-лы, 2 табл.
| GALKIN A | |||
| et al., Cloning of formate dehydrogenase gene from a methanol-utilizing bacterium Mycobacterium vaccae №10 Appl | |||
| Microbiol | |||
| Biotechnol | |||
| Приспособление для плетения проволочного каркаса для железобетонных пустотелых камней | 1920 |
|
SU44A1 |
| US 20030157664, | |||

