Настоящее изобретение относится к способу получения кристаллов, средняя крупность которых находится в заданных пределах и максимальный размер которых не превышает заданного значения, к получаемым этим способом кристаллам и к их применению в фармацевтических препаратах, прежде всего в препаратах с низкой дозировкой действующего вещества.
Из ЕР 0648778 А2 известны производные 11β-бензальдоксимэстра-4,9-диена. В этой заявке описаны синтез и очистка этих соединений. Однако в указанной заявке ничего не говорится ни о кристаллизации, ни о формообразования кристаллов. Указанные соединения, как и большинство стероидов, кристаллизуют из соответствующего растворителя. Известно, однако, что при традиционной кристаллизации, проводимой при охлаждении или методом вытеснения, образуется крупнозернистый кристаллизат с не установленным детально гранулометрическим составом.
К препаратам с низкой дозировкой действующего вещества, т.е. содержащим его лишь в минимальных количествах, например от 0,1 до 2 мас.%, предъявляются особые требования касательно однородности распределения в них действующего вещества (CUT от англ. "content uniformity test", анализ однородности содержания) и кинетики его растворения. Для этих препаратов с низким содержанием действующего вещества характерно то, что входящее в их состав в крайне малых количествах действующее вещество в значительной мере разбавлено другими ингредиентами лекарственного средства. Для сохранения однородности распределения действующего вещества на практически постоянном уровне средний размер его частиц не должен превышать некоторого максимально допустимого предела, а статистический разброс размеров частиц не должен быть слишком широким. Эти максимально допустимые размеры частиц действующего вещества зависят от его дозировки и методики его введения в организм, и их можно определить статистическим путем. Кроме того, при получении препаратов с низкой дозировкой действующего вещества следует учитывать и тот факт, что мелкие частицы растворяются в желудке быстрее по сравнению с крупными частицами. Поэтому размеры частиц действующего вещества не должны превышать некоторого определенного предела, что является необходимым условием, обеспечивающим соблюдение требований касательно кинетики растворения действующего вещества (растворение более 70% действующего вещества по истечении 45 мин).
Для соответствия указанным требованиям, предъявляемым к препаратам с низкой дозировкой действующего вещества в отношении однородности его распределения и кинетики растворения, кристаллизаты до настоящего времени микронизируют по традиционной технологии в струйной мельнице. При этом образуются кристаллы со средним размером от 1,5 до 3 мкм. Однако при подобном подходе наблюдается чрезмерное увеличение поверхности кристаллов и ее термодинамическое активирование, обусловленные частичной аморфизацией (переходом в аморфное состояние), соответственно существенными нарушениями в структуре решетки. Эти физические изменения инициируют химическую дестабилизацию действующего вещества не только как такового (т.е. в чистом виде), но и прежде всего в составе фармацевтических препаратов.
Карбаматная функция вышеназванных производных 11β-бенальдоксимэстра-4,9-диена при отщеплении этиламина и СО2 трансформируется в нитрил. Применение микронизатов тем самым приводит к получению лекарственных форм, в которых действующее вещество оказывается недостаточно стабильным в условиях, указанных в рекомендациях ICH (при 40°С и 70%-ной относительной влажности воздуха).
Хотя снижение давления при микронизации и позволяет достичь некоторого увеличения средних размеров частиц действующего вещества, тем не менее оно приводит и к нежелательному расширению диапазона их разброса. При этом следует учитывать и тот факт, что для нормальной работы мельницы обязательно требуется определенное минимальное давление. Таким образом, воздействовать на форму твердого вещества с целью повышения его химической стабильности за счет выбора соответствующих параметров микронизации, если таковое вообще возможно, удается лишь в самой минимальной степени.
Исходя из вышеизложенного в основу настоящего изобретения была положена задача разработать способ получения кристаллов, который не имел бы известных из уровня техники недостатков и позволял получать прежде всего кристаллы, удовлетворяющие требованиям, предъявляемым к препаратам с низкой дозировкой действующего вещества.
Указанная задача решается согласно изобретению благодаря предлагаемому в нем способу получения кристаллов, средняя крупность которых находится в заданных пределах и максимальный размер которых не превышает заданных значений, заключающемуся в том, что пересыщенный раствор 11β-бензальдоксимэстра-4,9-диена формулы (1)
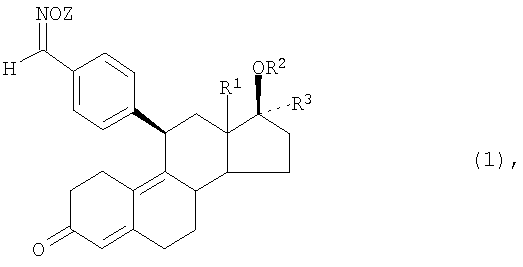
в которой R1 представляет собой атом водорода или алкильный остаток с 1-6 атомами углерода,
R2 представляет собой атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода, ацильный остаток с 1-10 атомами углерода или остаток -CONHR4 либо -COOR4, где R4 обозначает атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода,
R3 представляет собой атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода, остаток -(CH2)n-СН2Х, где n обозначает 0, 1 или 2, Х обозначает атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода, атом фтора, хлора, брома или иода, циано-, азидо- или родановую группу, остаток OR5 или SR5, где R5 представляет собой атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода или ацильный остаток с 1-10 атомами углерода, остаток OR5, где R5 имеет указанные выше значения,
остаток -(CH2)o-CH=CH(CH2)p-R6, где о обозначает 0, 1, 2 или 3, а р обозначает 0, 1 или 2 и R6 представляет собой атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода, гидроксильную группу, алкоксигруппу или ацилоксигруппу с 1-10 атомами углерода,
остаток -(CH2)qC=CR7, где q обозначает 0, 1 или 2, а R7 представляет собой атом водорода, атом фтора, хлора, брома или иода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода или ацильный остаток с 1-10 атомами углерода,
Z обозначает атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода, ацильный остаток с 1-10 атомами углерода, остаток -CONHR4 либо -COOR4, где R4 представляет собой атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода, или атом щелочного металла либо щелочноземельного металла, а также их фармацевтически приемлемые соли, подвергают в процессе кристаллизации мокрому измельчению с помощью предназначенного для подобного измельчения устройства с получением в результате суспензии первичных зерен.
Предлагаемый в изобретении способ неожиданно позволяет получать кристаллы, которые обладают достаточно высокой стабильностью и параметры гранулометрического состава которых можно регулировать с учетом требований фармацевтики, предъявляемых к препаратам с низкой дозировкой действующего вещества касательно однородности распределения в них действующих веществ (CUT-показатель) и кинетики их растворения, и которые тем самым удовлетворяют всем указанным условиям. Кроме того, предлагаемое в изобретении решение позволяет с высокой точностью и воспроизводимостью получать частицы (кристаллы) действующего вещества с определенным, пригодным для его применения в соответствующей дозировке гранулометрическим составом. Еще одно преимущество предлагаемого в изобретении способа состоит в простоте, быстроте и экономичности его проведения.
В одном из предпочтительных вариантов осуществления предлагаемого в изобретении способа используют соединение 11β-{4-[(этиламинокарбонил)оксиминометил]фенил}-17β-метокси-17α-метоксиметилэстра-4,9-диен-3-он (обозначаемый далее как соединение J956). При использовании этого соединения при осуществлении предлагаемого в изобретении способа вышеописанные преимущества проявляются особенно эффективно.
Ниже изобретение более подробно рассмотрено со ссылкой на прилагаемые к описанию чертежи, на которых показано:
на фиг.1 - ИК-спектр 11β-{4-[(этиламинокарбонил)оксиминометил]фенил}-17β-метокси-17α-метоксиметилэстра-4,9-диен-3-она (соединения J956),
на фиг.2 - образование нитрила в соединении J956 в функции крупности кристаллов и
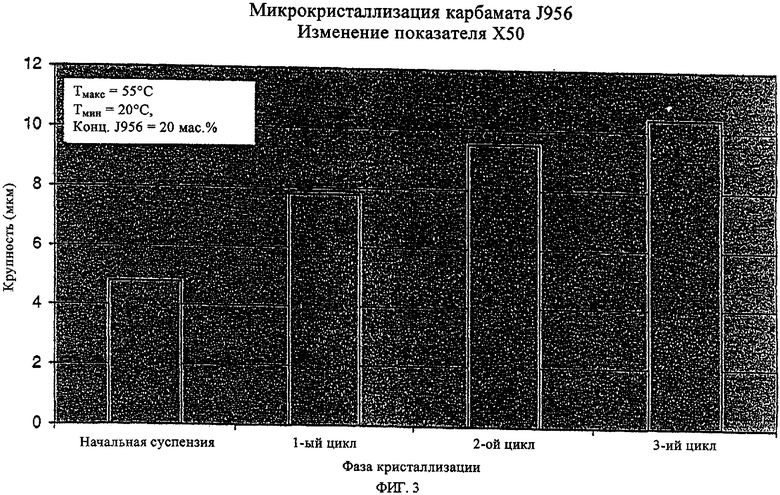
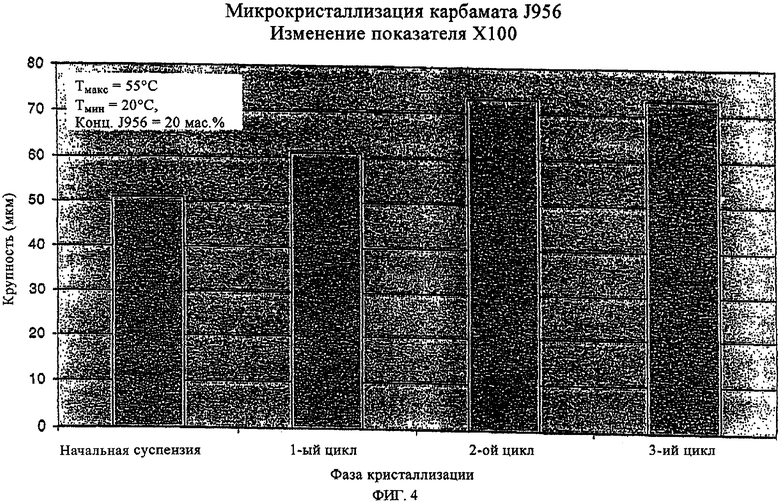
на фиг.3 и 4 - увеличение крупности кристаллов при кристаллизации предлагаемым в изобретении способом.
Средний размер или средняя крупность кристаллов предпочтительно составляет от 3 до 25 мкм, прежде всего от 7 до 15 мкм. Максимальный размер кристаллов предпочтительно не превышает 100 мкм, прежде всего не превышает 80 мкм. Понятие "максимальный размер кристаллов" означает при этом, что размер ни одного из кристаллов не превышает указанного значения. Преимущество, связанное с ограничением средней крупности кристаллов и их максимального размера указанными пределами, состоит в возможности целенаправленно подбирать гранулометрический состав кристаллов таким образом, чтобы он отвечал предъявляемым к препаратам с низкой дозировкой действующего вещества фармацевтическим требованиям касательно CUT-показателя и кинетики растворения.
Предлагаемый в изобретении способ предусматривает применение пересыщенного раствора соединения формулы (1). Иными словами, этот раствор содержит соединение формулы (1) в растворенном в соответствующем растворителе виде. В качестве растворителя можно использовать и смеси различных растворителей. Применяемый при осуществлении предлагаемого в изобретении способа пересыщенный раствор, который можно получать, например, путем переохлаждения, содержит растворенное вещество в концентрации, превышающей максимально возможную для такого раствора в условиях термического равновесия. При осуществлении предлагаемого в изобретении способа могут применяться пересыщенные растворы, в которых образование зародышей (центров кристаллизации) происходит спонтанно.
В одном из предпочтительных вариантов осуществления предлагаемого в изобретении способа пересыщенный раствор содержит соединение формулы (1) в количестве от 10 до 30 мас.%, прежде всего в количестве примерно 20 мас.%, в пересчете на массу пересыщенного раствора. При использовании подобных пересыщенных растворов вышеописанные преимущества предлагаемого в изобретении способа проявляются наиболее ярко.
В качестве растворителя для получения пересыщенного раствора предпочтительно использовать этилацетат, который зарекомендовал себя как наиболее пригодный для получения пересыщенных растворов соединений формулы (1).
Пересыщенные растворы можно получать по обычной методике. В предпочтительном варианте пересыщенный раствор получают растворением соединения формулы (1) в соответствующем растворителе при температуре ниже температуры его кипения с последующим охлаждением полученного раствора до температуры выше точки его замерзания. При применении при осуществлении предлагаемого в изобретении способа этилацетата, являющегося предпочтительным растворителем для получения пересыщенного раствора, нагревать раствор можно, например, до температуры порядка 70°С до полного растворения соединения формулы (1) в этилацетате и образования прозрачного раствора. Период времени, затрачиваемого на последующее охлаждение раствора до температуры в интервале от примерно 50 до 10°С, прежде всего от 30 до 35°С, может составлять от 10 мин до 1 ч, прежде всего от 15 до 30 мин. Параметры, необходимые для получения соответствующего пересыщенного раствора с использованием отличного от этилацетата растворителя, можно определить опытным путем исходя из приведенных выше данных проведением несложных экспериментов.
Кристаллизацию целесообразно проводить в закрытой емкости, снабженной мешалкой. В качестве примера при этом можно назвать известные, широко используемые в технике кристаллизаторы.
При осуществлении предлагаемого в изобретении способа в процессе кристаллизации образующиеся кристаллы одновременно подвергают мокрому измельчению (мокрому размолу) с помощью соответствующего устройства. Кристаллизация из пересыщенного раствора может начинаться сразу же после начала мокрого размола. Пригодными для подобного мокрого размола устройствами являются среди прочих диспергаторы с мелющими телами и гомогенизаторы, такие как гомогенизаторы роторно-статорного типа (центробежные гомогенизаторы), мельницы с мешалкой, валковые дробилки и коллоидные мельницы.
Согласно изобретению кристаллы, как указывалось выше, получают кристаллизацией из растворителя либо смеси растворителей, предпочтительно из образованного путем охлаждения пересыщенного этилацетатного раствора, при этом в начальной фазе кристаллизации либо вскоре после ее начала, либо до ее начала в дополнение к перемешиванию обычной мешалкой осуществляют мокрый размол с помощью предназначенного для этой цели устройства, например с помощью гомогенизатора роторно-статорного типа или коллоидной мельницы. Подобное устройство для мокрого размола может размещаться непосредственно в кристаллизационной емкости в качестве дополнительного смесителя либо в циркуляционном контуре кристаллизатора. При использовании гомогенизатора роторно-статорного типа окружная скорость ротора может составлять от 10 до 50 м/с, предпочтительно от 20 до 40 м/с. За счет воздействия на пересыщенный раствор дополнительной энергией, создаваемой при мокром размоле, прежде всего при использовании гомогенизатора роторно-статорного типа, существенно повышается скорость образования вторичных зародышей и тем самым значительно ограничивается рост кристаллов. Помимо этого в узком зазоре между ротором и статором, в котором (зазоре) действуют высокие усилия сдвига, происходит дробление возможно образующихся агломератов. В результате создаются условия для образования мелких первичных зерен, размер которых в зависимости от установленной степени пересыщения раствора и окружной скорости ротора составляет от 3 до 5 мкм и максимальный размер которых не превышает 25-60 мкм. Получения кристаллов с подобными параметрами их крупности уже вполне может оказаться достаточным для их применения для получения препаратов с низкой дозировкой действующего вещества.
Для возможности получения согласно требованиям фармацевтики с соответствующей целевой точностью и высокой воспроизводимостью и кристаллов большей крупности с определенным гранулометрическим составом суспензию первичных зерен предпочтительно подвергать обработке в температурном режиме с циклически изменяемой (периодически увеличиваемой и уменьшаемой) температурой. С этой целью полученную суспензию мелкокристаллических первичных зерен нагревают до температуры Тмакс, лежащей ниже температуры, при которой достигается предел растворимости присутствующих в суспензии первичных зерен, а затем медленно охлаждают до температуры Тмин, лежащей выше точки замерзания суспензии. В процессе нагревания присутствующая в суспензии первичных зерен мелкозернистая или -кристаллическая фракция растворяется, а в процессе последующего охлаждения выкристаллизовывается, наслаиваясь или нарастая на присутствующую крупнозернистую или -кристаллическую фракцию. В результате происходит определенный сдвиг гранулометрического состава в сторону бóльших значений крупности зерен, соответственно кристаллов. Температуру Тмакс предпочтительно выбирать с таким расчетом, чтобы обеспечить растворение в используемом растворителе от 10 до 90 мас.%, прежде всего от 20 до 50 мас.%, наиболее предпочтительно порядка 30 мас.%, первичных зерен. Относительное количество растворяемых первичных зерен выбирают в зависимости от заданного гранулометрического состава получаемых кристаллов, который в свою очередь определяется типом препарата с низкой дозировкой действующего вещества. При растворении большой доли первичных зерен получают соответственно кристаллы большей крупности.
В одном из предпочтительных вариантов осуществления предлагаемого в изобретении способа температуру Тмин выбирают с таким расчетом, чтобы обеспечить возможность преимущественно полной повторной кристаллизации растворенных первичных зерен. В оптимальном варианте для сведения потерь соединений формулы (1) к минимуму практически все растворенное количество первичных зерен должно выкристаллизовваться на еще остающихся в суспензии не растворившимися первичных зернах.
Суспензию первичных зерен предпочтительно охлаждать от Тмакс до Тмин в течение промежутка времени, составляющего от 1 мин до 10 ч, прежде всего от 0,5 до 2 ч.
Скорость изменения температуры при охлаждении суспензии первичных зерен следует при этом регулировать таким образом, чтобы свести к минимально возможному повторное образование зародышей кристаллизации. Величина подобного укрупнения кристаллов (зерен) за один цикл изменения температуры зависит от количества растворившегося за один цикл нагревания кристаллизата, которое в свою очередь определяется уровнем обеих температур Тмакс и Тмин относительно температуры, при которой достигается предел растворимости, и от концентрации твердого вещества в суспензии. Подобный цикл нагрева-охлаждения суспензии первичных зерен можно повторять необходимое число раз, предпочтительно от 1 до 10 раз, до достижения требуемого гранулометрического состава. Регулируемыми параметрами при этом являются температура Тмакс, температура Тмин и число циклов. Чем ниже должна быть величина требуемого укрупнения кристаллов, тем ниже следует выбирать температуру Тмакс. Таким путем можно постепенно, малыми приращениями приближать размер кристаллов к требуемому конечному значению. Параметры растворения соответствующей доли кристаллизата в фазах нагрева суспензии первичных зерен подбирают при этом с таким расчетом, чтобы при последующем охлаждении суспензии происходило лишь самое минимальное укрупнение кристаллов максимального диаметра и преимущественно происходило укрупнение кристаллов меньшего размера. Так, например, при растворении и повторной кристаллизации 40% от всего количества соединения J956, выкристаллизовавшегося из 20%-ного по массе раствора этилацетата, средний диаметр кристаллов (Х50) возрастает с 4,9 до 7,8 мкм, тогда как укрупнения кристаллов максимального размера (X100) практически не наблюдается. Сказанное означает, что распределение кристаллов по крупности при возрастании среднего значения их размеров (Х50) существенно сужается. Наличие подобного эффекта особенно предпочтительно с точки зрения фармацевтического применения получаемых предлагаемым в изобретении кристаллов и прежде всего с точки зрения достижения соответствующих CUT-показателей и кинетики растворения.
После обработки суспензии первичных зерен в температурном режиме с циклически изменяемой температурой полученную суспензию кристаллов можно подвергнуть фильтрации, а затем промыть кристаллы соответствующим растворителем, который растворяет кристаллы соединения формулы (1) лишь в незначительных количествах, составляющих, например, менее 1 мас.%. В качестве примера пригодных для применения в этих целях растворителей можно назвать метил-трет-бутиловый эфир, гексан, гептан, воду и смеси двух или более таких растворителей. Благодаря этому при последующей сушке, которую предпочтительно проводить непосредственно в фильтровальной установке с использованием сушильного газа или в вакууме, удается избежать образования "мостиков" и агломерации частиц.
Сушку можно проводить методом конвекционной или вакуумной сушки в неподвижном либо подвижном слое сыпучего высушиваемого материала.
Если традиционные фильтрация и сушка по каким-либо причинам оказываются затруднительны и отрицательно влияют на полученный при кристаллизации гранулометрический состав кристаллов, как, например, в случае очень мелкой зернистости, полученный при фильтрации и промытый осадок на фильтре в другом варианте можно взмучивать в суспендирующей жидкости, которая обладает предельно малой способностью растворять соединение формулы (1), например способна растворять его в количестве не более 1 мас.%, и в качестве которой предпочтительно использовать воду. Из полученной суспензии соединение формулы (1) можно переводить в высушенную твердую форму путем распылительной сушки.
Объектом настоящего изобретения являются далее кристаллы соединения формулы (1), получаемые предлагаемым в изобретении способом. Касательно получения предлагаемых в изобретении кристаллов этим способом справедливы приведенные выше детальные пояснения, относящиеся к рассмотренному выше предлагаемому в изобретении способу.
Полученные предлагаемым в изобретении способом кристаллы стероида 11β-{4-[(этиламинокарбонил)оксиминометил]фенил}-17β-метокси-17α-метоксиметилэстра-4,9-диен-3-она (ниже обозначаемого также как соединение J956), являющегося одним из конкретных соединений формулы (1), анализировали рентгеновской порошковой дифрактометрией, данные которой приведены в таблице 1, и ИК-спектрометрией, полученный при которой ИК-спектр показан на фиг.1.
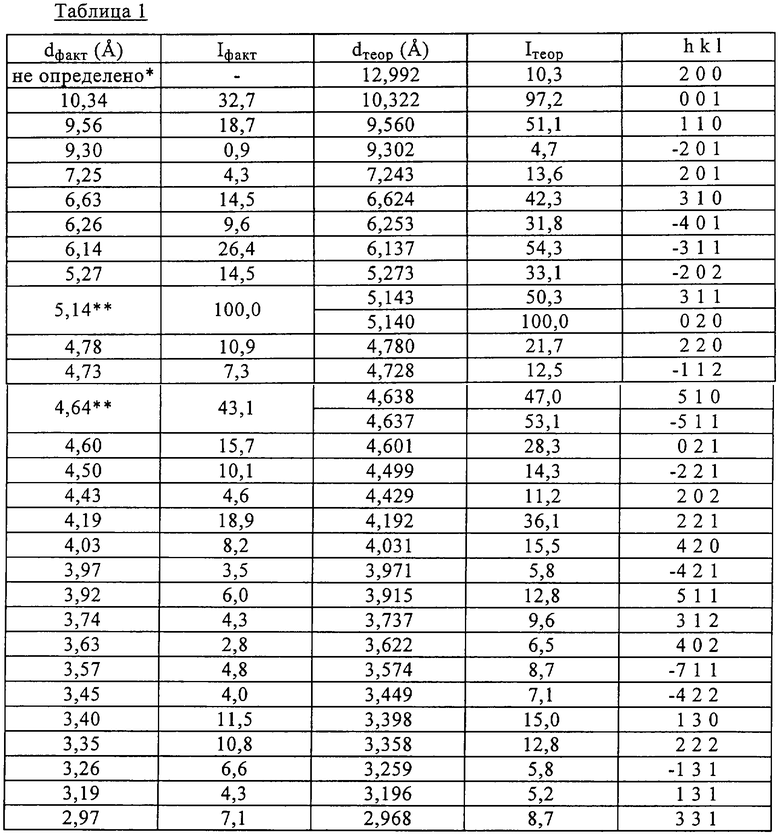
Соединение J956, как известно, может существовать в двух кристаллических формах, только одна из которых, однако, является фармацевтически приемлемой. Эту фармацевтически приемлемую кристаллическую форму соединения J956 получают предлагаемым в изобретении способом, и именно для нее в таблице 1 представлены соответствующие данные ее анализа рентгеновской порошковой дифрактометрией. Согласно приведенным в этой таблице данным расхождение между теоретическими и фактическими значения межплоскостного расстояния d составляет менее 1%.
Настоящее изобретение относится далее к фармацевтическим препаратам, содержащим получаемые предлагаемым в нем способом кристаллы соединений формулы (1). В качестве фармацевтически эффективных лекарственных форм, прежде всего для перорального применения, могут использоваться, например, твердожелатиновые капсулы или таблетки с покрытием либо без него. Лекарственные формы, получаемые с использованием микрокристаллического стероида формулы (1), не должны оказывать отрицательного воздействия на химическую и кристаллическую стабильность микрокристаллов. Обеспечить соблюдение этого условия можно следующим образом:
- предусмотреть для лекарственных форм защиту от света, например, за счет использования окрашенных оболочек для капсул или за счет нанесения окрашенного покрытия,
- не использовать способствующих увеличению поверхности вспомогательных веществ, таких как высокодисперсный диоксид кремния,
- по возможности не использовать никаких растворителей или вспомогательных веществ либо использовать в качестве таковых только воду и/или
- поддерживать содержание влаги в лекарственной форме на минимальном уровне за счет тщательной сушки.
В таблице 2 в качестве примера представлена возможная рецептура капсулы.
В таблице 3 в качестве примера представлена возможная рецептура таблетки.
Один из существенных результатов, которого позволяет достичь настоящее изобретение, состоит в возможности получения микрокристаллов стероидов формулы (1), которые обладают гораздо более высокой химической стабильностью по сравнению с известными на сегодняшний день микронизатами, поскольку, во-первых, они имеют меньшую удельную поверхность и, во-вторых, имеют не нарушенную предлагаемым в изобретении способом кристаллизации и высококристаллическую поверхность.
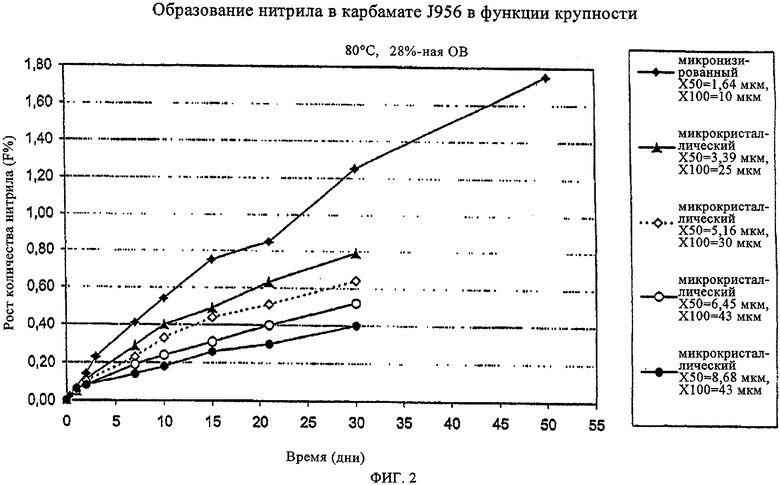
На фиг.2 в графическом виде представлена стабильность полученных предлагаемым в изобретении способом микрокристаллов в сравнении с известными микронизатами касательно образования нитрила при высокой термической нагрузке (80°С, 28%-ная относительная влажность (OB)). Согласно приведенному на указанном чертеже графику полученные предлагаемым в изобретении способом микрокристаллы с увеличением их размеров обладают по сравнению с полученным известным способом микронизатом гораздо более высокой стабильностью, что проявляется в сниженной степени образования нитрила.
Еще один результат, которого позволяет достичь настоящее изобретение, состоит в том, что получаемые предлагаемым в нем способом микрокристаллы стероидов формулы (1) по своему гранулометрическому составу и растворимости удовлетворяют фармацевтическим требованиям, предъявляемым к готовым лекарственным формам в отношении CUT-показателя и кинетики растворения.
Данные, полученные на примере капсулы и таблетки, содержащих по 1 мг действующего вещества (составы которых приведены выше), свидетельствуют о том, что полученные показатели нисколько не уступают аналогичным показателям, получаемым при использовании микронизированного твердого вещества (см. таблицы 4 и 5).
Еще один из важных результатов, которого позволяет достичь настоящее изобретение, состоит в возможности целенаправленного получения с высокой воспроизводимостью предлагаемым в нем способом стероидов формулы (1) с отвечающим требованиям фармацевтики гранулометрическим составом. На фиг.3 и 4 в графическом виде представлено изменение размеров кристаллов в сторону увеличения при их кристаллизации предлагаемым в изобретении способом. При этом основное преимущество состоит в значительном уменьшении разброса крупности кристаллов и в существенно меньшей степени увеличения максимальной крупности кристаллов, несмотря на многократное увеличение их средних размеров. Подобный эффект способствует достижению хороших CUT-показателей даже для препаратов с низкой дозировкой действующего вещества.
Еще одно преимущество изобретения состоит в сохранении полученного в суспензии гранулометрического состава кристаллов и в их высушенном состоянии.
**Суспензия соединения J956 в смеси вода/этанол (в массовом соотношении 90:10) с 10 мас.% микрокристаллов соединения J956.
Еще одним объектом изобретения является, таким образом, фармацевтический препарат, который представляет собой изготовленную с использованием полученных предлагаемым в изобретении способом микрокристаллов химически стабильную и фармацевтически эффективную лекарственную форму.
Лекарственные формы с предлагаемыми в изобретении микрокристаллами стероидов формулы (1) могут успешно применяться в следующих случаях. Стероиды формулы (1), прежде всего соединение J956, представляют собой обладающие антигестагенным действием вещества, которые при той же активности, которая присуща соединению RU 486 (мифепристону) по отношению к прогестероновому рецептору (ПР), проявляют в сравнении с RU 8486 заметно более слабую антиглюкокортикоидную активность. Соединение J956, обозначаемое как мезопрогестин, является соединением, которое обладает in vivo как агонистической, так и антагонистической активностью по отношению к прогестероновому рецептору. Добиться соответствующих функциональных состояний с помощью гестагенов и антигестагенов не удается. Соединение J956 пригодно для применения прежде всего при следующих показаниях. Это соединение, которое при определенных условиях можно использовать совместно с эстрогеном, пригодно для получения лекарственного средства, предназначенного для женской контрацепции, а также оно может использоваться для лечения и предупреждения доброкачественных, гормонзависимых гинекологических расстройств, например для лечения таких гинекологических расстройств, как эндометриоз, фиброиды матки, послеоперационные перитонеальные спайки, дисфункциональные маточные кровотечения (метроррагия, меноррагия) и дисменорея. Суточная доза мезопрогестина может составлять от 0,5 до 100 мг, предпочтительно от 5,0 до 50 мг, наиболее предпочтительно от 10 до 25 мг. Соединение J956 может применяться также, при определенных условиях совместно с эстрогеном, в качестве фармацевтического компонента для получения соответствующего лекарственного средства, предназначенного для гормонзаместительной терапии (ГЗТ), а также для восполнения дефицита гормонов и для лечения симптоматических проявлений нарушения гормональной регуляции.
Для получения экспериментальных данных использовали следующие аналитические методы и аппаратуру.
Рентгеновская порошковая дифрактометрия
Для получения соответствующих данных использовали порошковый дифрактометр STADIP фирмы STOE с монохроматическим Кα1-излучением меди (λ=1,540598 Å) и германиевым монохроматором при 3°≤2θ<35°.
ИК-спектроскопия
Для данного анализа использовали прибор NICOLET 20 SXB с фотоакустическим детектором типа МТЕС (KBr, 8t, 90 секунд).
Гранулометрический состав
Sympatec HELOS (H0445), система сухого диспергирования (RODOS), давление 2 бара.
ЖХВР (жидкостная хроматография высокого разрешения)
Степень чистоты определяли следующим образом:
Определение остаточного содержания растворителя газовой хроматографией в паровом объеме
Автоматическая система для газовой хроматографии с HS40 Perkin Elmer, колонка DB-Wax, 30 м на 0,23 мм, пламенно-ионизационный детектор.
Влагосодержание определяли по методу Карла Фишера.
Анализ однородности содержания действующего вещества
Содержимое отдельных капсул определяли согласно Фармакопее США и европейской фармакопее после вымывания с помощью ЖХВР с внешней калибровкой:
Высвобождение действующего вещества
Высвобождение действующего вещества исследовали в 1000 мл воды с 0,3% додецилсульфата натрия при скорости вращения мешалки 100 об/мин. Содержимое капсул определяли с помощью ЖХВР с внешней калибровкой:
Ниже изобретение более подробно рассмотрено на примерах, не ограничивающих его объем.
Пример 1
В стеклянном реакторе с анкерной мешалкой и двойной нагревательной/охлаждающей рубашкой 250 г карбамата J956 при 70°С растворяют в 1100 мл этилацетата с образованием прозрачного раствора. Затем раствор в течение 30 мин охлаждают до 35°С. Далее задействуют центробежный диспергатор (тип Ultra Turrax), работающий при 8000-13000 об/мин в циркуляционном режиме. Через 2-5 мин начинается кристаллизация. Диспергатор продолжает работать еще в течение 10 мин, после чего его выключают. Полученную первичную суспензию сначала нагревают до 55°С, а затем в течение 1 ч 20 мин охлаждают до 20°С. Данную операцию повторяют еще два раза. Далее суспензию фильтруют через фритту и промывают 500 мл холодного метил-трет-бутилового эфира. В завершение фильтровальный осадок подвергают вакуум-фильтрации с использованием воздуха для его сушки.
Таким путем получили микрокристаллы следующего гранулометрического состава:
Остаточное содержание растворителей: 0,016% метил-трет-бутилового эфира, 0,24% этилацетата.
Пример 2
В колбе для сульфирования с лопастной мешалкой и термостатированной нагревательной/охлаждающей баней при 70°С растворяют 50 г соединения J956 в 200 г этилацетата с образованием прозрачного раствора. Затем раствор в течение 15 мин охлаждают до 35°С. Затем в колбу помещают центробежный диспергатор (тип Ultra Turrax) и приводят его во вращение с частотой 12000-16000 об/мин. Через 2 мин начинается кристаллизация. Диспергатор работает еще в течение 10 мин, после чего его выключают. Эту полученную начальную суспензию нагревают до 50°С и затем в течение 1 ч охлаждают до 20°С. Данный процесс повторяют еще дважды. После этого суспензию фильтруют через фритту и промывают 100 мл метил-трет-бутилового эфира. Осадок на фильтре тщательно промывают 1000 мл воды и затем взмучивают в 300 г воды. В завершение суспензию подвергают распылительной сушке в лабораторной распылительной сушилке с двухкомпонентной форсункой (QVF/Yamato) при следующих условиях:
В осадительной камере распылительной сушилки получили микрокристаллы следующего гранулометрического состава:
Остаточное содержание воды равно 0,13%, а этилацетата 0,12%.
Пример 3
Получение капсул с микрокристаллическим карбаматом J956
Микрокристаллический карбамат J956 смешивают в соответствующем смесителе (например, в контейнерной мешалке) с микрокристаллической целлюлозой. Затем добавляют стеарат магния и перемешивание повторяют. Необходимо проконтролировать отсутствие воды в используемых аппаратах. В завершение смесь с помощью машины для заполнения капсул (например, KFMIIIC фирмы Harro Höflinger) расфасовывают в твердожелатиновые капсулы размера 3.
Пример 4
Получение таблеток в оболочке (филм-таблеток) с микрокристаллическим карбаматом J956
Микрокристаллический карбамат J956 смешивают в грануляторе (например, в грануляторе с псевдоожиженным слоем типа GPCG 3.1 фирмы Glatt) с лактозой и кукурузным крахмалом. На эту смесь напыляют раствор мальтодекстрина в воде и образующийся гранулят сушат (температура приточного воздуха 70°С). Затем гранулят смешивают с Na-карбоксиметилкрахмалом и монобегенатом глицерина и прессуют из него сердцевину таблеток массой по 150 мг. На эту сердцевину с помощью машины для нанесения покрытий (например, Driacoater 500 фирмы Driam) напыляют суспензию соответствующих покровных веществ в воде и образующуюся пленку сушат (масса пленки 4 мг, температура приточного воздуха 70°С, потери филм-таблетки при сушке составляют 3%).
Описывается способ получения кристаллов, средняя крупность которых находится в заданных пределах от 3 до 25 мкм и максимальный размер которых не превышает 100 мкм, заключающийся в том, что в процессе кристаллизации пересыщенный раствор соединения, представляющего собой 11β-бензальдоксимэстра-4,9-диен, подвергают мокрому измельчению с помощью соответствующего, предназначенного для подобного мокрого измельчения устройства с получением в результате суспензии первичных зерен. Описываются также кристаллы, получаемые описываемым в изобретении способом, и содержащее эти кристаллы фармацевтическое средство. 3 н. и 11 з.п.ф-лы, 4 ил., 8 табл.
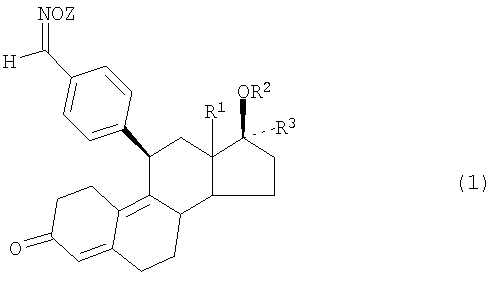
в которой R1 представляет собой атом водорода или алкильный остаток с 1-6 атомами углерода,
R2 представляет собой атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода, ацильный остаток с 1-10 атомами углерода или остаток -CONHR4 либо -COOR4, где R4 обозначает атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода,
R3 представляет собой атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода, остаток -(СН2)n-СН2Х, где n обозначает 0, 1 или 2, Х обозначает атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода, атом фтора, хлора, брома или иода, циано-, азидо- или родановую группу, остаток OR5 или SR5, где R5 представляет собой атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода или ацильный остаток с 1-10 атомами углерода, остаток OR5, где R5 имеет указанные выше значения,
остаток -(CH2)o-CH=CH(CH2)p-R6, где о обозначает 0, 1, 2 или 3, а р обозначает 0, 1 или 2 и R6 представляет собой атом водорода, алкильную, арильную, аралкильную либо алкиларильную группу с 1-10 атомами углерода, гидроксильную группу, алкоксигруппу или ацилоксигруппу с 1-10 атомами углерода,
остаток -(CH2)qC=CR7, где q обозначает 0, 1 или 2, а R7 представляет собой атом водорода, атом фтора, хлора, брома или иода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода или ацильный остаток с 1-10 атомами углерода,
Z обозначает атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода, ацильный остаток с 1-10 атомами углерода, остаток -CONHR4 либо -COOR4, где R4 представляет собой атом водорода, алкильный, арильный, аралкильный либо алкиларильный остаток с 1-10 атомами углерода, или атом щелочного металла либо щелочноземельного металла, а также их фармацевтически приемлемые соли,
подвергают в процессе кристаллизации мокрому измельчению с помощью предназначенного для подобного мокрого измельчения устройства с получением в результате суспензии первичных зерен.
| Инерционный аккумулятор | 1977 |
|
SU648778A1 |
| ЕР 1157996 А, 28.11.2001 | |||
| US 5534270 А, 09.07.1996. | |||

