Область техники, к которой относится изобретение
Данное изобретение относится к соединениям 7-тио-замещенного-3-нитро-1,2,4-триазоло-[5,1-c]-1,2,4-триазин-4(1H)-она для применения в лечении вирусных инфекций, которые вызываются РНК-содержащими вирусами с одноцепочечной РНК отрицательной полярности [в дальнейшем - оцРНК], оцРНК-вирусами, отличными от вирусов гриппа А и гриппа В, и методам лечения таких вирусных инфекций. Кроме того, данное изобретение относится к способам изготовления упомянутых соединений 7-тио-замещенного 3-нитро-1,2,4-триазоло-[5,1-c]-1,2,4-триазин-4(1H)-она.
Уровень техники
РНК-содержащие вирусы с одноцепочечной РКН отрицательной полярности (оцРНК), такие как оцРНК-содержащие вирусы, относящиеся к порядку Mononegavirales, например, вирусы семейства Рабдовирусов (лат. Rhabdoviridae), в частности, вирус бешенства (лат. Rabies), семейства Филовирусов (лат. Filoviridae), в частности, вирус Эбола, и семейства Парамиксовирусов (лат. Paramyxoviridae), в частности, вирус кори, и другие оцРНК-содержащие вирусы, принадлежащие к неопределенным семействам, особенно, к семействам Аренавирусов (лат. Arenaviridae), Буниавирусов (лат. Bunyaviridae) и Ортомиковирусов (лат. Orthomyxoviridae), а также другие неопределенные оцРНК-содержащие вирусы, такие как Дельтавирус, вызывают множество заболеваний в дикой природе, у домашних животных и человека. Эти оцРНК-содержащие вирусы, принадлежащие к различным семействам, они разнообразны в генном и антигенном отношении, демонстрируют совершенно различные тропизмы к тканям и обладают огромным патогенным потенциалом.
Например, филовирусы, относящиеся к порядку Mononegavirales, в частности, вирусы Эбола и Марбург (лат. Marburgviruses), являются одними из самых смертельно опасных и деструктивных вирусов в мире. Отдельный повод для беспокойства представляют филовирусы, которые в потенциале могут быть рассеяны в виде аэрозоли и, таким образом, использованы в качестве биологического оружия.
Род вирусов Эбола включает пять видов: эболавирусы Заир (лат. Zaire), Судан (лат. Sudan), Рестон (лат. Reston), леса Тай (лат. Taï Forest) и Бундибуго (лат. Bundibugyo). В частности, эболавирусы Заир, Судан и Бундибуго вызывают сильную, часто смертельную вирусную геморрагическую лихорадку у людей и нечеловекообразных приматов.
В течение более чем 30 лет с эболавирусом связывают периодические вспышки геморрагической лихорадки в Центральной Африке, которая приводит к серьезным заболеваниям у зараженных пациентов. При подобных вспышках летальность составляла от 50% в случае эболавируса Судан и до 90% в случае эболавируса Заир ((Sanchez et al., Filoviridae: Marburg and Ebola Viruses, in Fields Virology, стр. 1409-1448 (Lippincott Williams & Wilkins, Philadelphia)). В ноябре 2007 г. во время вспышки лихорадки Эбола в районе Бундибуго Уганды, по поблизости от границы с Демократической Республикой Конго, был обнаружен пятый вид эболавируса - Бундибуго. Летальность при этом составила ок. 25% (Towner et al., PLoS Pathog., 4(11) :e1000212 (2008)). Эболавирус вида Заир также привел к уничтожению популяций диких обезьян в том же регионе Африки (Walsh et al., Nature, 422:611-614 (2003)).
При заражении эболавирусом заболевание начинается внезапно и характеризуется высокой температурой, головными болями, болями в суставах, мышцах, горле, общей слабостью, диареей, рвотой, а также болями в животе. У некоторых пациентов могут наблюдаться сыпь на коже, покраснение глаз, икота, внутренние и внешние кровотечения. В течение одной недели после заражения вирусом большинство пациентов испытывают боль в груди, полиорганную недостаточность, впадают в шок и умирают. Перед смертью некоторые больные слепнут и страдают от обширных кровотечений.
Еще одним примером одноцепочечных РНК-содержащих вирусов являются вирус кори, относящийся к роду Morbilllivirus, и вирус бешенства из рода Lyssavirus.
Род Lyssavirus, принадлежащий к семейству Рабдовирусов, включает одиннадцать признанных видов вирусов, один из них - вирус бешенства. Бешенство известно с древних времен, самое раннее упоминание об этом вирусном заболевании в Старом Свете относится к 2300 г. до н.э. Бешенство представляет угрозу здоровью и в настоящее время, причина - в недостатке эффективных мер контроля популяций животных-носителей инфекции, а также в недоступности вакцинации во многих странах. Вирус бешенства распространен во всем мире среди млекопитающих-носителей инфекции, включая хищников и летучих мышей. Ежегодно сообщается о множестве случаев передачи вируса бешенства от животных людям (например, через укус животного). От бешенства умирает более 50000 человек в год, особенно в Азии и Африке.
Таким образом, сохраняется необходимость в противовирусных соединениях, которые можно эффективно использовать для лечения вирусных инфекций, вызванных оцРНК-вирусами и отличных от инфекций вирусов гриппа А и гриппа В.
Раскрытие изобретения.
Таким образом, основной предмет изобретения - противовирусное соединение для применения в лечении вирусных инфекций, вызванных РНК-содержащими вирусами с одноцепочечной РНК отрицательной полярности [в дальнейшем - оцРНК] и отличных от инфекций вирусов гриппа А и гриппа В, в котором указанное противовирусное соединение выражается общей формулой (I) [в дальнейшем - противовирусное соединение (A),], либо его фармацевтически приемлемая соль
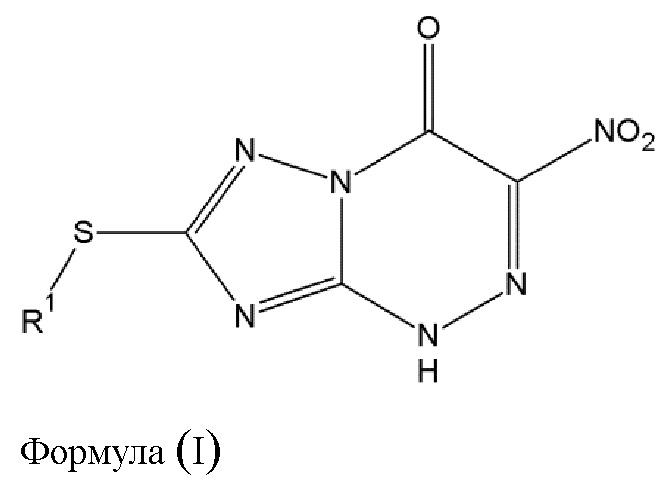
где
- R1 выбирается из алкильной группы с числом атомов углерода от 1 до 12, которая в некоторых случаях замещается атомом галогена или гидроксильной группой, циклоалкильной группы, включающей от 3 до 12 атомов углерода, которая в некоторых случаях замещается атомом галогена или гидроксильной группой, арильной группой, алкиларильной группой, или циклогетероалкильной группы с числом атомов углерода от 3 до 12.
Дополнительная цель изобретения - предоставить способы изготовления указанного противовирусного соединения или его фармацевтически приемлемой соли.
Еще одна цель данного изобретения - предоставить способы лечения вирусных инфекций, вызванных оцРНК-содержащими вирусами и отличных от инфекций вируса гриппа А и гриппа В, как упоминалось выше, за счет применения противовирусного соединения (A) или его фармацевтически приемлемой соли.
Кроме того, еще одна цель данного изобретения - предоставить фармацевтические композиции, включающие противовирусное соединение (А) или его фармацевтически приемлемую соль.
Антивирусное соединение (A) и его фармацевтически приемлемая соль
В контексте настоящего документа термин "алкил" имеет самое широкое значение, общепринятое в данной области знаний, и может включать фрагменты, которые являются линейными, разветвленными, либо комбинацией указанных видов.
В контексте настоящего документа термин "циклоалкильная группа, включающая от 3 до 12 атомов углерода", означает неароматическое кольцо на основе углерода, состоящее из не менее трех и не более двенадцати атомов углерода. Примеры циклоалкильной группы на основе атомов углерода в количестве от 3 до 12, включают, помимо прочего, циклопропил, циклобутил, циклопентил, циклогексил и т.д.
В контексте настоящего документа термин "арильная группа" означает ароматическую группу на основе углерода, включающую, помимо прочего, фенил, толил, ксилил, куменил, нафтил и т.д. Термин "ароматический" также включает "гетероарильную группу" - ароматическую группу, имеющую не менее одного гетероатома, входящего в кольцо ароматической группы. Примеры гетероатома - это, помимо прочего, атомы азота, кислорода, серы и фосфора. К гетероарильным группам, помимо прочего, относятся фурил, тиенил, пирролил, имидазолил, пиридил, пиразил, пиримидинил, индолил, карбазолил, изоксазолил, изотиазолил и т.д. Арильная группа может быть замещенной или незамещенной. Арильная группа может быть замещена одной или более группами, включая, помимо прочего, алкил, алкинил, алкенил, арил, галид, нитро-, амино-, эфир, кетон, альдегид, гидрокси-, карбоновую кислоту или алкокси-.
В контексте настоящего документа термин "алкиларильная группа" означает, предпочтительно, алкиларильную группу с числом атомов углерода от 6 до 20, включая, помимо прочего, бензил, фенэтил, нафтилметил, в некоторых случаях замещенную одной или более группами, включая, помимо прочего, алкил, алкил, алкинил, алкенил, арил, галид, нитро-, амино-, эфир, кетон, альдегид, гидрокси-, карбоновую кислоту или алкокси-.
В контексте настоящего документа термин "гетероцилоалкильная группа с числом атомов углерода от 3 до 12" означает циклоалкильную группу в определении, данном выше, в которой, как минимум, один из атомов углерода в кольце замещен гетероатомом, например, помимо прочего, атомом азота, кислорода, серы или фосфора. Примеры гетероциклоалкильных групп на основе атомов углерода в количестве от 3 до 12, включают, помимо прочего, морфолин, пиперидин, n-метил-пиперазин, и т.д.
Предпочтительно, R1 в противовирусном соединении (A) или его фармацевтически приемлемой соли, выбирается из алкильной группы с числом атомов углерода от 1 до 8, в некоторых случаях замещенной атомом галогена или гидроксильной группой, циклоалкильной группы с числом атомов углерода от 3 до 6, в некоторых случаях замещенной атомом галогена или гидроксильной группой, фенилом, бензилом, морфолином или имидазолилом. Более предпочтительно, R1 выбирается из алкильной группы с числом атомов углерода от 1 до 4, циклогексила, фенила или бензила. Еще более предпочтительно, R1 выбирается из метила, этила, пропила или изопропила. Наиболее предпочтительный вариант, R1 - это метильная группа.
Противовирусное соединение (A), подробно описанное выше, и его фармацевтически приемлемая соль в состоянии подавлять распространение оцРНК-содержащих вирусов, отличных от вирусов гриппа А и гриппа В и, таким образом, лечить или предупреждать развитие инфекций, вызываемых оцРНК-содержащими вирусами и отличных от инфекций вирусов гриппа А и гриппа В.
В некоторых предлагаемых вариантах данного изобретения, оцРНК-содержащие вирусы, отличные от вирусов гриппа А и гриппа В, предпочтительно, выбираются среди оцРНК-содержащих вирусов, принадлежащих к порядку Mononegavirales, среди оцРНК-содержащих вирусов, принадлежащих к неопределенному семейству, выбранному из группы семейств Ареновирусов, Буниавирусов и Ортомиксовирусов, либо среди неопределенных оцРНК-содержащих вирусов, таких как Дельтавирусы
В одном из предлагаемых вариантов данного изобретения, оцРНК-содержащие вирусы, отличные от вирусов гриппа А и гриппа В, принадлежащие к порядку Mononegavirales, выбираются из группы, состоящей из семейств Борнавирусов, Рабдовирусов, Филовирусов и Паромиксовирусов.
В более предпочтительном предлагаемом варианте изобретения, оцРНК-содержащий вирус выбирается из группы, состоящей из борнавируса, Lyssavirus, вируса везикулярного стоматита, эболавируса, вируса Марбург, вируса Нипах, вируса бешенства, вирусов рода Respirovirus, рода Rubulavirus, Metapneumovirus и Pneumovirus.
Эболавирус, предпочтительно, выбирается из группы, включающей эболавирусы родов Заир, Судан и Бундибуго.
Вирус рода Lyssavirus, предпочтительно, выбирается из группы, состоящей из вируса бешенства, Mokola, Duvenhage и Australian bat lyssavirus.
Вирус рода Pneumovirus, предпочтительно, выбирается из группы, включающей респираторно-синцитиальный вирус человека и респираторно-синцитиальный вирус крупного рогатого скота.
В еще одном предлагаемом варианте данного изобретения, оцРНК-содержащие вирусы, отличные от вирусов гриппа А и гриппа В, предпочтительно, выбираются среди оцРНК-содержащих вирусов, принадлежащих к неопределенному семейству, выбранному из группы семейств Ареновирусов, Буниавирусов и Ортомиксовирусов.
В более предпочтительном варианте настоящего изобретения, оцРНК-содержащий вирус выбирается из группы, состоящей из вирусов рода Mammarenavirus, Hantavirus, Nairovirus, Orthobunyavirus, Phlebovirus, вируса гриппа С, вируса гриппа D, вирусов рода Quaranjavirus и Thogotovirus.
В некоторых предлагаемых вариантах данного изобретения, оцРНК-содержащий вирус выбирается из группы, состоящей из вирусов рода Mammarenavirus, Hantavirus, Nairovirus, Orthobunyavirus, Phlebovirus, Quaranjavirus и Thogotovirus.
В настоящем изобретении, фармацевтически приемлемые соли антивирусного соединения (A), подробно описанного выше, не ограничены при условии их способности лечить или предупреждать инфекции, вызванные вышеупомянутыми специфическими оцРНК-содержащими вирусами.
В предпочтительном варианте данного изобретения противовирусное соединение (A), подробно описанное выше, имеет форму фармацевтически приемлемой соли.
Подразумевается, что противовирусное соединение (А), подробно описанное выше и обладающее достаточной кислотной функцией, включает соответствующие фармацевтически приемлемые соли неорганических и органических оснований.
Таким образом, в данном изобретении термин "фармацевтически приемлемые соли" означает соли, приготовленные из фармацевтически приемлемых нетоксичных оснований, включая неорганические и органические основания. Соли, полученные из органических оснований, в частности, включают соли натрия, калия, лития, аммиака, кальция, магния, железа, цинка, марганца, алюминия, трехвалентного железа, трехвалентного марганца и т.п. Особо предпочтительны соли натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных, третичных и четверичных аммониевых соединений, замещенных аминов, включая встречающиеся в природе замещенные амины, циклических аминов и катионообменных смол, например, триэтиламин, трипропиламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, лизин, аргинин, гистидин, кофеин, прокаин, N-этилпиперидин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилгликамин, теобромин, пурины, пиперазин, пиперидин, полиаминная смола, и т.п. Особо предпочтительны аргинин и лизин.
Кроме того, подразумевается, что названные фармацевтически приемлемые соли противовирусного соединения (А) могут быть в форме сольвата. Среди сольватов следует, в частности, упомянуть сольваты, полученные из гидратов.
Термин "сольват" обозначает кристаллическую структуру, включающую стехиометрические или нестехиометрические количества растворителя. Если сольват - это гидрат, указанным растворителем является вода.
Например, если фармацевтически приемлемые соли противовирусного соединения (А) получают в смеси воды и органического растворителя, который способен смешиваться с водой, названные фармацевтически приемлемые соли противовирусного соединения (А) имеют форму гидрата.
Способы изготовления противовирусного соединения (а) или его фармацевтически приемлемой соли
Еще одной целью данного изобретения являются усовершенствованные способы изготовления противовирусного соединения (а) или его фармацевтически приемлемой соли.
Антивирусное соединение (А) или его фармацевтически приемлемую соль по данному изобретению можно синтезировать рядом способов, известных в данной области знаний. Известные способы, в частности, описаны В.Л. Русиновым и др. в "Химико-фармацевтическом журнале", сентябрь 1990 г., том 24, Вып. 9, стр. 646-650, Роспатенты №№ 2294936 и 2536874, полное содержание указанных документов включено в данный документу путем отсылки.
В данных способах, противовирусное соединение (A), либо его фармацевтически приемлемую соль получают из 5-амино-3-метилтио-1,2,4-триазола, который подвергают диазотированию, в результате которого образуется соль диазония, после чего осуществляют азосочетание указанной соли диазония с α-нитроэфирами, в частности, с этилнитроацетатом. Известно, что выход последнего соединения низкий. Его обычно синтезируют путем нитрования ацетоуксусного эфира азотоуксусным ангидридом, при этом выход составляет от 27 до 47% (см. Journal of the Chemical Society, 1958, p.2276-82 и Bulletin de la Societe Chimique de France, 31, 847-854; 1904). Таким образом, упомянутый способ характеризуется низким выходом и взрывоопасностью азотоуксусного ангидрида.
Известные способы получения 5-амино-3-метилтио-1,2,4-триазола, в частности, описаны исследователями А.В. Долженко и др. в "Гетероциклах", 2007, том 71, № 2, стр. 429-436, Компаунд, и СМ Десенко и др., Фолио:Харьков, 1998, стр. 122-123.
Одним из недостатков этих известных методов является необходимость синтеза 5-амино-3-метилтио-1,2,4-триазола, ключевого прекурсора 3-метилтио-1,2,4-триазол-5-ил-диазония. Способ, описанный А.В. Долженко и др., предусматривает синтез 5-амино-3-метилтио-1,2,4-триазола с использованием токсичного сернистого углерода (CS2) и образованием нестабильного промежуточного соединения цианамида. В способе, описанном С.М. Десенко, синтез 5-амино-3-метилтио-1,2,4-триазола включает алкилирование 5-амино-3-меркапто1,2,4-триазола йодистым метилом. Недостаток последнего способа - низкий выход ключевого промежуточного соединения на этапе алкилирования.
Учитывая вышеизложенное, необходимы усовершенствованные способы изготовления антивирусного соединения (А) или его фармацевтически приемлемой соли, подробно описанных выше.
Найдены новые способы изготовления антивирусного соединения (А) или его фармацевтически приемлемой соли, которые позволяют увеличить выход и эффективность при снижении затрат на изготовление, в частности, по сравнению с известными способами, в которых в качестве сырья используется 5-амино-3-метилтио-1,2,4-триазол.
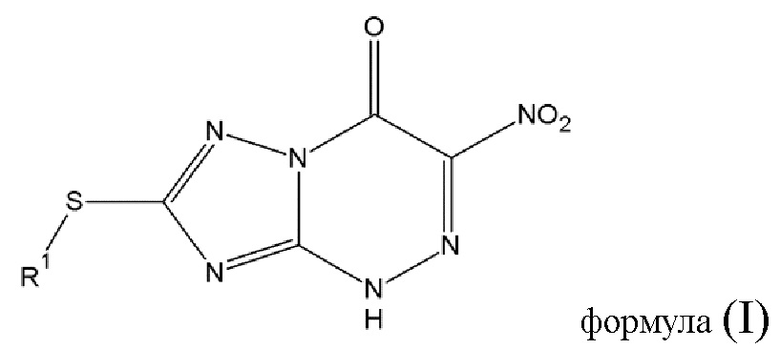
В связи с этим, изобретение относится к способу изготовления противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли
где
- R1 выбирается из алкильной группы с числом атомов углерода от 1 до 12, которая в некоторых случаях замещена галогеном или гидроксильной группой, циклоалкильной группы, включающей от 3 до 12 атомов углерода, которая в некоторых случаях замещена галогеном или гидроксильной группой, арильной группой, алкиларильной группой, или циклогетероалкильной группы с числом атомов углерода от 3 до 12.
Изготовление включает следующие этапы:
Этап 1. Диазотирование соединения (B) общей формулы (II):
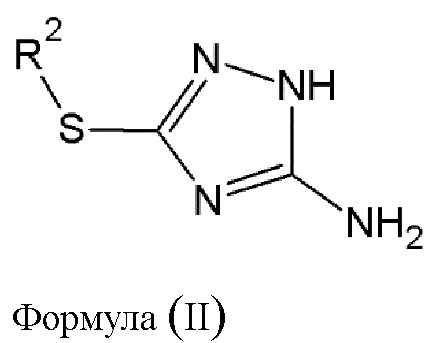
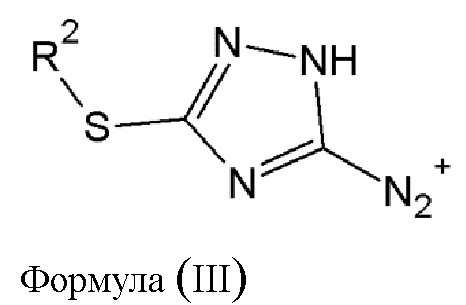
где R2 выбирается из водорода, либо R2 имеет то же значение, что и R1, в определении, данном выше, в результате чего образуется соединение диазония общей формулы (III) или его фармацевтически приемлемая соль:
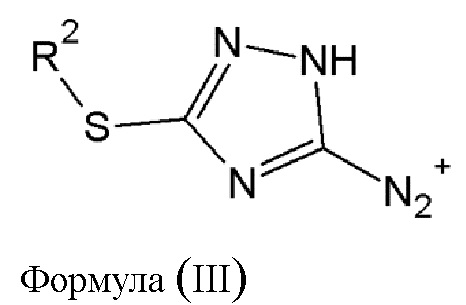
где R2 имеет то же значение, что и в определении, данном выше.
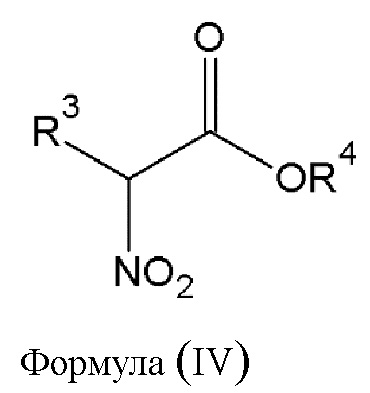
Этап 2. Реакция конденсации соединения диазония общей формулы (III) или его фармацевтически приемлемой соли, полученных на этапе 1, с не менее чем одним α-нитроэфиром общей формулы (IV), приведенной ниже,
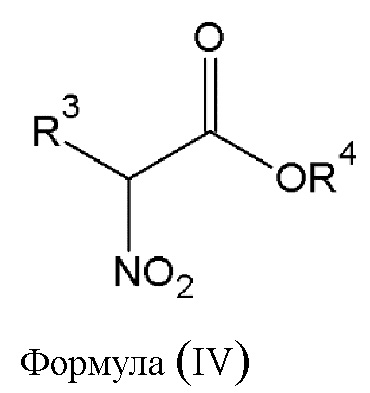
где
- R3 выбирается независимым образом из группы, состоящей из следующих элементов:
• -(CO)-OR5, где R5 - водород или алкильная группа с числом атомов углерода от 1 до 24, которая в некоторых случаях замещена гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой;
• -(CO)-R6, где R6 - алкильная группа с числом атомов углерода от 1 до 12, которая в некоторых случаях замещена гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой;
• -O-(CO)-Y, где Y - алкильная группа с числом атомов углерода от 1 до 12, которая в некоторых случаях замещена гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой;
- R4 - алкильная группа с числом атомов углерода от 1 до 24, которая в некоторых случаях замещена атомом галогена, арильной группой или аралкильной группой, алкенильной группой.
Кроме того, подразумевается, что все представленные выше определения и предпочтения в равной степени применимы к способу изготовления противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли, подробно описанным выше, а также в равной степени относятся ко всем описанным ниже вариантам.
Предпочтительно, R4 - это алкильная группа с числом атомов углерода от 1 до 12, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, R4 это алкильная группа с числом атомов углерода от 1 до 8, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно - алкильная группа с числом атомов углерода от 1 до 6, еще более предпочтительно - алкильная группа с числом атомов углерода от 1 до 4. В наиболее предпочтительном варианте, R4 выбирается из метила, этила, пропила или изопропила.
Предпочтительно, R5 - это алкильная группа с числом атомов углерода от 1 до 12, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, R5 это алкильная группа с числом атомов углерода от 1 до 8, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно - алкильная группа с числом атомов углерода от 1 до 6, еще более предпочтительно - алкильная группа с числом атомов углерода от 1 до 4. В наиболее предпочтительном варианте, R5 выбирается из метила, этила, пропила или изопропила.
Предпочтительно, R6 - это алкильная группа с числом атомов углерода от 1 до 8, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, R6 это алкильная группа с числом атомов углерода от 1 до 6, еще более предпочтительно - алкильная группа с числом атомов углерода от 1 до 4. В наиболее предпочтительном варианте, R6 выбирается из метила, этила, пропила или изопропила.
Предпочтительно, Y - это алкильная группа с числом атомов углерода от 1 до 8, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, это алкильная группа с числом атомов углерода от 1 до 6, еще более предпочтительно - алкильная группа с числом атомов углерода от 1 до 4. В наиболее предпочтительном варианте, Y выбирается из метила, этила, пропила или изопропила.
Предпочтительный α-нитроэфир общей формулы (IV), подробно описанный выше и пригодный для использования в реакции конденсации соединения диазония общей формулы (III), либо его фармацевтически приемлемая соль, - это диметил нитромалонат, диэтил нитромалонат, дипропил нитромалонат, диизопропил нитромалонат, дибутил нитромалонат, диизобутил нитромалонат, ди-втор-бутил нитромалонат, ди-трет-бутил нитромалонат, 2-нитро-метилацетоацетат, 2-нитро-этилацетоацетат, 2-нитро-пропилацетоацетат, 2-нитро-изопропилацетоацетат, 2-нитро-бутилацетоацетат, 2-нитро-изобутилацетоацетат, 2-нитро-втор-бутилацетоацетат, 2-нитро-трет-бутилацетоацетат, более предпочтительно - диметил нитромалонат, диэтил нитромалонат, 2-нитро-метилацетоацетат, 2-нитро-этилацетоацетат.
В некоторых вариантах предлагаемого способа, R2 - это водород, а α-нитроэфир имеет общую формулу (IV), представленную ниже,
где R3 - это -(CO)-OR5, где R5 это алкильная группа с числом атомов углерода от 1 до 24, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, предпочтительно, R5 это алкильная группа с числом атомов углерода от 1 до 12, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, R5 это алкильная группа с числом атомов углерода от 1 до 8, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, алкильная группа с числом атомов углерода от 1 до 6, и еще более предпочтительно, алкильная группа с числом атомов углерода от 1 до 4. Еще более предпочтительно, R5 - это метил, этил, пропил или изопропил. Наиболее предпочтительный вариант, R5 это этил.
В некоторых вариантах предлагаемого способа, R2 равен R1, по данному выше определению, и α-нитроэфир имеет общую формулу (IV), представленную ниже,
где R3 - это -(CO)- R6 где R6 - это алкильная группа с числом атомов углерода от 1 до 12, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, предпочтительно, R6 это алкильная группа с числом атомов углерода от 1 до 8, которая в некоторых случаях замещается гидроксильной группой, аминогруппой, атомом галогена, арильной группой или аралкильной группой, более предпочтительно, R6 - алкильная группа с числом атомов углерода от 1 до 6, еще более предпочтительно, алкильная группа с числом атомов углерода от 1 до 4, еще более предпочтительно, R6 - это метил, этил, пропил или изопропил. Наиболее предпочтителен метил.
Диазотирование соединения (B) общей формулы (II), подробно описанного выше, можно осуществлять с помощью принятых в отрасли способов диазотирования, предпочтительно с использованием азотистокислого натрия (NaNO2) или нитрозилсерной кислоты (HNO5S) в кислой среде с участием неорганических кислот, например, соляной, серной или фосфорной кислоты или их смеси, либо органических кислот, например, уксусной, трифторуксусной или пропионовой кислоты или их смеси. Кроме того, можно успешно использовать смеси неорганических кислот с органическими.
Как правило, диазотирование соединения (B) общей формулы (II), подробно описанного выше, проводится при температуре не ниже -25°C, предпочтительно, не ниже -20°C, более предпочтительно, не ниже -15°C. Оно обычно проводится при температуре не выше 15°C, более предпочтительно. не выше 10°C, более предпочтительно, при температуре не выше 5°C. Наиболее предпочтительной является температура от -10°C до 5°C.
По желанию, можно использовать водные растворы азотистокислого натрия (NaNO2) или нитрозилсерной кислоты (HNO5S), в частности, водный раствор азотистокислого натрия (NaNO2) в форме дробленого льда.
Следует отметить, что использование так называемого "холодного" способа диазотирования, описанного выше, позволяет обеспечить безопасность способа по настоящему изобретению, исключив образование пены с возможной последующей детонацией из-за повышения давления в системе, например, образование пены - потеря субстанции - распад азотсодержащего вещества.
В реакции конденсации соединения диазония общей формулы (III) или его фармацевтически приемлемой соли с не менее, чем одним α-нитроэфиром общей формулы (IV), подробно описанным выше, на этапе 2 способа по настоящему изобретению, молярное соотношение соединения диазония общей формулы (III), подробно описанного выше, и α-нитроэфира общей формулы (IV), подробно описанного выше, составляет, преимущественно, от 0,5:2 до 2:0,5, предпочтительно, от 0,7:1,5 до 1,5:0,7, и, более предпочтительно, от 0,8:1,2 до 1,2:0,8, еще более предпочтительно, от 0,9:1 до 1:0,9. Наиболее предпочтительное значение молярного соотношения - ок. 1:1.
В одном из предлагаемых вариантов, на этапе 2 реакция конденсации соединения диазония общей формулы (III), либо его фармацевтически приемлемой соли, с не менее чем одним α-нитроэфиром общей формулы (IV), подробно описанным выше, осуществляется в присутствии, как минимум, одного фармацевтически приемлемого нетоксичного неорганического или органического основания.
Типичные примеры таких фармацевтически приемлемых нетоксичных неорганических оснований могут включать гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия, гидроксид лития; гидроксиды щелочноземельных металлов, например, гидроксид кальция, гидроксид магния; основные соли щелочных металлов, например, карбонат натрия, гидрокарбонат натрия, карбонат калия, гидрокарбонат калия; аммиачное соединение. Предпочтительные фармацевтически приемлемые нетоксичные неорганические основания - гидроксид натрия и гидроксид калия. Наиболее предпочтительное фармацевтически приемлемое неорганическое основание - это гидроксид натрия.
Типичные примеры таких фармацевтически приемлемых нетоксичных органических оснований могут включать первичные, вторичные, третичные и четверичные амины, замещенные амины, включая встречающиеся в природе замещенные амины, циклических амины и катионообменные смолы, например, триэтиламин, трипропиламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, лизин, аргинин, гистидин, кофеин, прокаин, N-этилпиперидин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилгликамин, теобромин, пурины, пиперазин, пиперидин, полиаминная смола, и т.п. Предпочтительные фармацевтически приемлемые органические основания - аргинин и лизин. Наиболее предпочтительное фармацевтически приемлемое органическое основание - это аргинин.
Количество упомянутых фармацевтически приемлемых нетоксичных неорганических или органических оснований выбирают таким образом, чтобы величина pH реакционной смеси составляла между 6,0 и 12,0, предпочтительно, между 7,0 и 11,0, наиболее предпочтительно - между 7,5 и 10,5.
Способ введения таких фармацевтически приемлемых нетоксичных неорганических или органических оснований на этапе 2 выбирается таким образом, чтобы температура реакционной смеси оставалась ниже 10°C, предпочтительно, ниже 5°C.
Как правило, конденсация соединения диазония общей формулы (III), подробно описанного выше, с не менее, чем одним α-нитроэфиром общей формулы (IV), подробно описанным выше, проводится при температуре не выше 15°C, предпочтительно, не выше 10°C, более предпочтительно, не выше 5°C. Оно обычно проводится при температуре не ниже -25°C, более предпочтительно, не ниже -15°C, более предпочтительно, при температуре не ниже -10°C. Наиболее предпочтительной является температура от -10°C до 5°C.
По желанию, можно использовать, как минимум один α-нитроэфир общей формулы (IV), подробно описанный выше, в сочетании с растворителем, выбранным из спирта, например, метанола, этанола, н-пропилового спирта, изопропилового спирта, трет-бутилового спирта, н-бутилового спирта, изобутилового спирта и т.п.; эфира, например, диметилового эфира, диэтилового эфира, дипропилового эфира, диизопропиловым эфира, ди-трет-бутилового эфира, диизобутилоого эфира, дибутилового эфира, ди-втор-бутилового эфира, диоксана, тетрагидрофурана, диметоксиметана, диметоксиэтана, диметоксипропана и т.п.; кетона, например, диметилкетона, диэтилкетона, дипропилкетона, метилэтилкетона и т.п.; серосодержащего растворителя, например, диметилсульфоксида, диметилсульфона, дифенилсульфона, диэтилсульфоксида, диэтилсульфона, диизопропилсульфона, тетрагидротиофена-1, 1-диоксида (который часто называют тетраметилсульфоном или сульфоланом), тетрагидротиофен-1-моноксила и т.п.; азотсодержащего полярного апротонного растворителя, например, диметилацетомида, диметилформамида и N-метил пирролидинона (т.е. NMP) и т.п.; и их смесей.
При необходимости, растворитель обычно используют в избытке. По преимуществу, растворитель используют в количестве, в десять раз, а предпочтительно - в пятнадцать раз превышающем количество, как минимум, одного α-нитроэфира общей формулы (IV), подробно описанного выше.
По желанию, можно использовать спирт в виде водного спиртового раствора, органический растворитель, либо в сочетании с не менее, чем одним алкоксидом щелочного металла.
Типичные примеры алкоксидов щелочных металлов включают, в частности, помимо прочего, метилат натрия, изопропилат натрия, этилат калия и т.п.
В рамках данного изобретения, термин "спиртовой раствор" означает растворы спирта в воде, в соленой воде или в любом другом водном растворе минеральной соли.
В случае, если R2 - это водород, то на этапе 2 способа изготовления противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли образуется 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-он или его фармацевтически приемлемая соль.
В данном варианте предлагаемого способа, 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-он или его фармацевтически приемлемая соль, получаемые на этапе 2, далее подвергаются реакции алкилирования с алкилирующим агентом формулы (V): R1-X или формулы (VI) R1-Z, где R1 имеет значение, определение которого дано выше, X - атом галогена или кислород, содержащий функциональные группы, такие как OTos и OTMS, а Z -сульфатная группа (т.е. O-S(=O)2-O) или карбонатная группа (т.е. O-C(=O)-O), предпочтительно, сульфатная группа, в результате чего образуется противовирусное соединение (A) с формулой (I) или его фармацевтически приемлемая соль.
X, предпочтительно, выбирается из следующей группы: фтор, бром, хлор, йод, OTos или OTMS; более предпочтительный вариант X - это фтор, бром, хлор, или йод, наиболее предпочтительно, X - это йод.
Предпочтительно, R1 выбирается из алкильной группы с числом атомов углерода от 1 до 4, циклогексила, фенила или бензила, Х - это фтор, бром, хлор, или йод. Более предпочтительно, R1 выбирается из метила, этила, пропила или изопропила, Х - это йод.
В другом предпочтительном варианте, R1 выбирается из алкильной группы с числом атомов углерода от 1 до 4, циклогексила, фенила или бензила, а Y - это сульфатная группа (т.е. O-S(=O)2-O) или карбонатная группа (т.е. O-C(=O)-O).
В другом предпочтительном варианте, R1 выбирается из метила, этила, пропила или изопропила, Y - это сульфат.
В данном изобретении фармацевтически приемлемой солью 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-она может быть фармацевтически приемлемая моносоль, фармацевтически приемлемая дисоль или их смесь.
В контексте настоящего документа дисоль 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-она означает дианионную форму соединения 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-он в комбинации с двумя фармацевтически приемлемыми катионами, каждый из которых получают независимо друг от друга, из фармацевтически приемлемых нетоксичных неорганических или органических оснований, подробно описанных выше.
В контексте настоящего документа моносоль 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-она означает моноанионную форму 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-она в комбинации с одним катионом, полученным из фармацевтически приемлемых нетоксичных неорганических или органических оснований, подробно описанных выше.
Фармацевтически приемлемые нетоксичные неорганические катионы, в частности, включают катионы натрия, калия, лития, аммиака, кальция, магния, железа, цинка, марганца, алюминия, трехвалентного железа, трехвалентного марганца и т.п. Особо предпочтительным является катион натрия. Фармацевтически приемлемые нетоксичные органические катионы, в частности, включают катионы первичных, вторичных, третичных и четверичных аммониевых соединений, замещенных аминов, включая встречающиеся в природе замещенные амины, циклических аминов и катионообменных смол, например, триэтиламин, трипропиламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, лизин, аргинин, гистидин, кофеин, прокаин, N-этилпиперидин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилгликамин, теобромин, пурины, пиперазин, пиперидин, полиаминная смола, и т.п. Особо предпочтительны катионы аргинина и лизина.
В предпочтительном варианте данного изобретения вещество 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-он имеет вид дисоли, описанной выше.
Как правило, алкилирование 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-она, либо его моносоли или дисоли проводится при комнатной температуре.
Однако, авторы обнаружили, что способ изготовления антивирусного соединения (A) общей формулы (I) или его фармацевтически приемлемой соли, состоящий из трех этапов, а именно, диазотирования 5-амино-3-метилтио-1,2,4-триазола (т.е. соединения (B) общей формулы (II), имеющего R2 - галоген), в результате чего образуется 3-меркапто-1,2,4-триазол-5-ил-диазоний или его моносоль, как описано выше, (т.е. этап 1), затем реакции конденсации 3-меркапто-1,2,4-триазол-5-ил-диазония или его моносоли с не менее, чем одним α-нитроэфиром общей формулы II, как подробно описано выше (т.е. этап 2), а после чего, алкилирования 7-меркапто-3-нитро-1,2,4-триазоло[5,1-c]-1,2,4-триазин-4(1H)-она или его моносоли, либо дисоли, как описано выше, может быть реализован без выделения промежуточных продуктов, образующихся в предлагаемом способе. При этом, такой предлагаемый одноступенчатый способ позволяет увеличить выход и эффективность, а также уменьшить затраты на изготовление, в частности, по сравнению с известными многоступенчатыми способами, в частности, описанными в работах исследователей В.Л. Русинова и др. в "Химико-фармацевтическом журнале", сентябрь 1990г., том 24, Вып. 9, стр. 646-650, и RU №№ 2294936 и RU 2536874, упоминавшихся выше.
В случае, если R2 равен R1, в определении, данном выше в данном документе, то противовирусное соединение (A) общей формулы (I) или его фармацевтически приемлемая соль образуются на этапе 2 способа, в результате чего можно исключить последний этап - алкилирование.
Имеется также дополнительная необходимость в том, чтобы иметь возможность легко, с высоким выходом и эффективно, синтезировать соединение (B) общей формулы (IIR1), где R2 равен R1, как показано ниже.
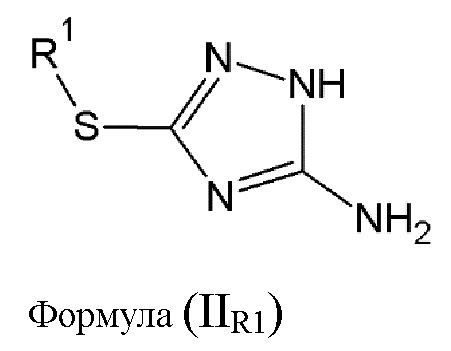
Однако, авторы данного изобретения обнаружили, что выполнив все вышеупомянутые условия, можно получить усовершенствованный способ изготовления соединения (B) общей формулы (IIR1).
Еще одной целью настоящего изобретения является способ изготовления соединения (B) общей формулы (IIR1), состоящий из реакции конденсации аминогуанидина с тио-соединением общей формулы (VII):
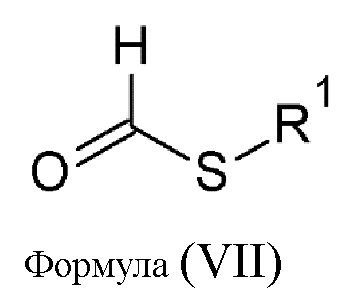
где R1 выбирается из алкильной группы с числом атомов углерода от 1 до 12, которая в некоторых случаях замещается атомом галогена или гидроксильной группой, циклоалкильной группы, включающей от 3 до 12 атомов углерода, которая в некоторых случаях замещается атомом галогена или гидроксильной группой, арильной группой, алкиларильной группой, или циклогетероалкильной группы с числом атомов углерода от 3 до 12.
Определения и предпочтения, представленные выше для предлагаемого способа изготовления противовирусного соединения (A) общей формулы (I) или его фармацевтически приемлемой соли, в равной степени применимы к способу изготовления исходного материала, а именно, соединения (B) общей формулы (IIR1).
Предпочтительные тио-соединения общей формулы (VII) выбираются из следующей группы: S-метил тиоформиат, S-этил тиоформиат, S-пропил тиоформиат, S-изопропил тиоформиат, S-бутил тиоформиат, S-изобутил тиоформиат, S-втор-бутил тиоформиат, S-трет-бутил тиоформиат, S-бензил тиоформиат, S-фенил тиоформиат, S-циклопропил тиоформиат, S-циклобутил тиоформиат, S-циклопентил тиоформиат, S-циклогексил тиоформиат. Предпочтительно использовать S-метил тиоформиат.
Реакцию конденсации аминогуанидина с тио-соединением общей формулы (VII), в частности, метилтиоформиатом, можно с успехом провести в присутствии основания. При этом можно использовать неорганическое, либо органическое основание. Предпочтительно использовать органическое основание. Органическое основание можно выбрать из группы, включающей азотсодержащие гетероциклические соединения, например, пиридин, хинолин или пиколин; и третичные основания, например, триэтиламин, диметиланилин, диэтиланилин и 4-диметиламинопиридин. Среди них наиболее предпочтительными являются пиридин, триэтиламин, диметиланилин, диэтиланилин и 4-диметиламинопиридин. Особенно предпочтителен пиридин. Эти основания можно использовать по отдельности или в сочетании, в виде смеси.
Неорганические основания можно выбрать из группы, включающей гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид цезия, гидроксиды щелочноземельных металлов, например, гидроксид кальция, гидроксид бария, гидроксид магния, гидроксид стронция, и основные соли щелочных металлов, например, карбонат натрия, гидрокарбонат натрия, карбонат калия, гидрокарбонат калия. Предпочтительное основание - гидроксид натрия.
Фармацевтическая композиция (P)
Данное изобретение также относится к фармацевтической композиции [далее - фармацевтическая композиция (P)] для лечения или профилактики инфекций, вызванных оцРНК-содержащими вирусами и отличных от инфекций вирусов гриппа А и гриппа В, как описано выше, которая включает в качестве действующего вещества соединение (A) общей формулы (I), или его фармацевтически приемлемую соль, описанные выше.
Кроме того, подразумевается, что все представленные выше определения и предпочтения в равной степени применимы к предлагаемой фармацевтической композиции (P), описанной выше
Предлагаемая фармацевтическая композиция (P) может также содержать один или несколько фармацевтически приемлемых носителей.
Фармацевтически приемлемые носители, пригодные для использования в фармацевтической композиции (P), в частности, включают, помимо прочего, наполнители, расширители, связывающие и увлажняющие компоненты, разрыхлители, поверхностно-активные и смазывающие вещества, и т.п.
Предлагаемая фармацевтическая композиция (P) может также включать один или несколько разбавителей, одно или несколько вспомогательных веществ, которые обычно используются в зависимости от лекарственной формы препарата.
Фармацевтическая композиция (P) может быть обработана до получения различных форм в соответствии с целью лечения. Типичные примеры - таблетки, пилюли, порошки, жидкости, суспензии, капсулы, суппозитории, инъекции (жидкости, суспензии), спреи, аэрозоли, пары, микрокапсулы с замедленным высвобождением и т.д.
Предлагаемая фармацевтическая композиция (P) может, соответственно, включать действующее вещество, соединение (A) общей формулы (I), или его фармацевтически приемлемую соль, подробно описанные выше, в пропорции, выбранной из широкого диапазона, обычно от 1,0×10-5 до ок. 100,0% масс., предпочтительно, от ок. 1,0×10-4 до ок 99,0% масс., относительно общей массы фармацевтической композиции (P). Кроме того, подразумевается, что пропорцию соединения (A) общей формулы (I), ил его фармацевтически приемлемой соли, как правило, определяют исходя из способа введения фармацевтической композиции (P) (например, в твердой лекарственной форме, в жидкой лекарственной форме).
Предлагаемая фармацевтическая композиция (P) может также содержать один или несколько дополнительных ингредиентов.
Неограничивающие примеры подходящих дополнительных ингредиентов, в частности, включают, буферы, изотонирующие средства, хелатообразующие вещества, красители, консерванты, отдушки, ароматизаторы, подсластители, другие препараты, и т.п.
Среди буферов можно, в частности, упомянуть буферы, приготовленные из фосфорных кислот, уксусных кислот, лимонных кислот, ε-аминокапроновых кислот, глютаминовой кислоты и их солей (например, солей щелочных металлов и солей щелочноземельных металлов, таких как соли натрия, калия, кальция, магния и т.д.).
Среди изотонирующих средств можно, в частности, упомянуть средства на основе хлорида натрия, хлорида калия, сахаридов, глицерина и т.д.
Среди хелатообразующих веществ можно, в частности, упомянуть вещества на основе эдетата натрия, лимонной кислоты, Trilon® (соль ЭДТК), унитиола ((RS)-2,3-бис(сульфанил)пропан-1-сульфонат) и т.п.
Кроме того, при использовании в качестве жидкого лекарственного препарата предлагаемую фармацевтическую композицию (P) можно для целей консервации подвергать сублимационной сушке, а перед использование растворять в водном водосодержащем буфере, физиологическом растворе и т.п. с целью получения необходимой концентрации.
Предлагаемую фармацевтическую композицию (P) можно также выпускать в твердых лекарственных формах, например, в таблетках, пилюлях, порошках, гранулах, капсулах и т.п., и в жидких формах, например, в растворах, суспензиях, эмульсиях, сиропах и т.п. Кроме того, такие композиции можно выпускать в формах для орального, парентерального, назального, вагинального применения, в форме суппозиториев, таблеток для рассасывания и т.п. с использованием традиционных методов смешивания, составления рецептуры и приготовления.
В частности, в рецептуре таблеток можно использовать следующие носители вышеупомянутого лекарственного вещества: вспомогательные вещества, например, лактозу, мягкий белый сахар, хлорид натрия, глюкозу, мочевину, крахмал, карбонат кальция, каолин, кристаллическую целлюлозу, кремниевую кислоту, силикат кальция, фосфат калия и т.д.; связующие вещества, например, воду, этанол, пропанол, сахарный сироп, жидкую глюкозу, жидкий крахмал, растворы желатина, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, метилцеллюлозу, поливинилпирролидон, и т.д..; разрыхлители, например, натрий-карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлозу, гидроксипропилцеллюлозу с низкой степенью замещения, обезвоженный крахмал, алгинат натрия, агар в порошке, ламинаран в порошке, гидрокарбонат натрия, карбонат кальция, кросповидон и т.д.; поверхностно-активные вещества, например, полиоксиэтиленовые эфиры сорбита и жирной кислоты, лаурилсульфат натрия, моноглицериды стеариновой кислоты и т.д.; ингибиторы расслоения, например, мягкий белый сахар, стеарин, масло какао, гидрогенизированные жиры и т.д.; усилители поглощения, например, четвертичные аммониевые основания, лаурилсульфат натрия и т.д.; увлажняющие компоненты, такие как глицерин, крахмал и т.д.; адсорбенты, например, крахмал, лактозу, каолин, бентонит, коллоидную кремниевую кислоту и т.д.; смазывающие вещества, например, тальк очищенный, стеараты, например, стеарат магния, борную кислоту в порошке, полиэтиленгликоль и т.д.; и т.п.
Таблетки можно также выпускать в форме таблеток с оболочкой, покрытых традиционной или пленочной оболочкой, например, таблеток в сахарной облатке, таблеток с желатиновой пленочной оболочкой, таблеток, покрытых кишечнорастворимой оболочкой, таблеток с пленочной оболочкой и двухслойных или многослойных таблеток.
В рецептуре пилюль можно использовать такие носители лекарственного средства, как глюкозу, лактозу, масло-какао, отвержденное растительное масло, каолин, тальк и т.д.; связующие вещества, такие как аравийскую камедь в порошке, трагакант в порошке, желатин, этанол и т.д..; и разрыхлители, такие как ламинаран, агар и т.д.
Капсулы можно получать традиционным способом - путем смешивания вышеупомянутых различных носителей с предлагаемым действующим веществом и последующего заполнения полученной смесью твердых или мягких желатиновых капсул и т.п.
Жидкие лекарственные формы для орального применения могут содержать обычно используемые в таких случаях инертные разбавители, например, водосодержащие фармацевтически приемлемые растворы, эмульсии, суспензии, сиропы, эликсиры и т.д.; а также могут дополнительно содержать вспомогательные вещества, например, смачивающие агенты, эмульсии, суспендирующие агенты и т.д. Для приготовления таких лекарственных форм можно использовать традиционные методики.
Жидкие лекарственные формы для парентерального применения, например, водные или неводные растворы, эмульсии, суспензии и т.д., можно получать с использованием разбавителей, таких как вода, этиловый спирт, пропиленгликоль, полиэтиленгликоль, этоксилированный изостеариловый спирт, поликсилированный изостеариловый спирт, полиоксиэтиленовые эфиры сорбита и жирной кислоты растительные масла, например, оливковое масло и т.д. Можно также добавлять инъекционные органические эфиры, например, этилолеат. Кроме того, можно добавлять обычно используемые в таких случаях солюбилизирующие средства, буферы, смачивающие агенты, эмульгаторы, суспендирующие агенты, консерванты, диспергирующие агенты. Стерилизацию можно проводить, например, путем фильтрации, когда препарат пропускают через бактериальный фильтр; смешивают со стерилизующим агентом; подвергают облучению; термообработке и (или) т.п. Препарат может также выпускаться в форме стерилизуемой твердой композиции, которую перед непосредственным использованием можно растворять в стерильной воде или другой аналогичной среде, подходящей для стерилизации.
В рецептуре суппозиториев и форм для вагинального применения можно использовать такие носители лекарственного вещества, как полиэтиленгликоль, масло какао, высшие спирты, эфиры высших спиртов, желатин, полусинтетический глицерид и т.д.
Фармацевтические композиции (P) для выпуска в виде спреев, аэрозолей, паров, назальных и сублингвальных форм можно приготовлять с использованием стандартных вспомогательных веществ традиционными методами.
Способы введения предлагаемых фармацевтических композиций (P) не ограничены и, соответственно, устанавливаются в зависимости от формы выпуска; возраста, пола, других параметров состояния пациента; выраженности симптомов и т.д. Например, таблетки, пилюли, жидкости, суспензии, эмульсии, гранулы и капсулы вводятся орально, а инъекционные формы - внутривенно по отдельности или в сочетании с жидкостями, обычно используемыми при инфузионной терапии, например, содержащими глюкозу, аминокислоты и т.д.; при необходимости, их вводят внутримышечно, внутрикожно, подкожно ил внутрибрюшинно. Суппозитории вводятся ректально, формы для вагинального применения - вагинально, назальные формы вводятся интраназально, а сублингвальные формы - интраорально.
Дозировка предлагаемых фармацевтических композиций (P) не ограничена и выбирается из широкого диапазона в зависимости от желаемой эффективности лечения, способа введения, периода лечения, возраста, поля и других параметров состояния пациента. Фармацевтические композиции (P) обычно вводятся в дозе от ок. 1,0×10-4 мг до ок 9500,0 мг, предпочтительно, от ок. 1,0×10-3 мг до ок. 1000,0 мг, более предпочтительно от ок. 0,01 мг до ок. 500,0 мг, и наиболее предпочтительно - от ок. 0,1 мг до ок. 100,0 мг действующего вещества на 1 кг массы тела в день для взрослых, одной или несколькими порциями в день.
Примеры
Ниже приводится более подробное описание изобретения на следующий примерах, цель которых - лишь проиллюстрировать, а не ограничить объем изобретения.
Пример 1. Однореакторный синтез натриевой соли 7-метилтио-3-нитро [1, 2, 4] триазоло [5,1-c] [1, 2, 4] триазин -4 (1H)-она
Этап 1: Диазотирование соединения (В): Для получения раствора (в дальнейшем - раствор [1]) растворили 5,8 г (0,05 M) 5-амино-3-меркапто-1,2,4-триазола в 6,7 мл азотной кислоты (15 M) и 12 мл воды. Указанный раствор [1] охладили до -7°C. Затем к раствору [1] частями по 0,5 мл добавили 40%-ный раствор азотистокислого натрия, таким образом, общее количество азотистокислого натрия в смеси составило 3,8 г.
Этап 2: Конденсация соединения диазония с α-нитроэфиром: К полученной на этапе 1 соли диазония добавили 8,54 мл диэтил нитромалоната. После выдержки в течение пяти минут к реакционной смеси медленно, по каплям добавляли охлажденный раствор гидроксида натрия до тех пор, пока значение pH не достигло 9-10 (далее - раствор [2]). Полученный в результате раствор [2] перемешивали в течение 1 часа при 0°C, а затем в течение 2 часов - при комнатной температуре.
Этап 3: алкилирование. К раствору [2], полученному на этапе 2, добавили 6,23 мл (0,1 моль) йодистого метила. Полученную смесь перемешивали в течение 1 часа при комнатной температуре и отфильтровали. Полученный осадок с успехом кристаллизовали из воды и сушили на воздухе. Схема реакции представлена на схеме 1.
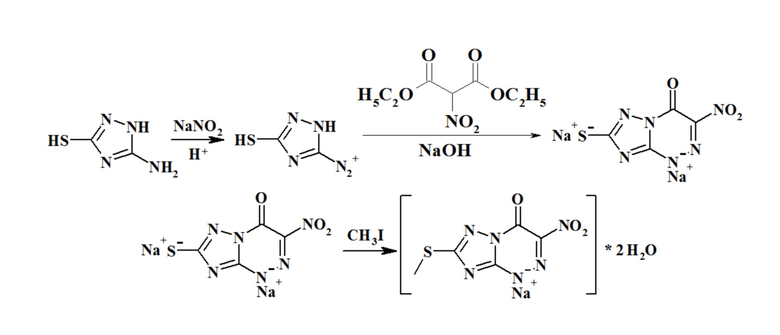
Вход составил 9,87 г (69%).
Физико-химические характеристики натриевой соли 7-метилтио-3-нитро [1, 2, 4] триазоло [5,1-c] [1, 2, 4] триазин -4 (1H)-она: желтый кристаллический порошок, растворимый в воде, ацетоне, диметилсульфоксиде, диметилформамиде, нерастворим в хлороформе; Tплавления = 300°C, 1H ЯМР спектр, δ, мд, растворитель ДМСО-d6: 2.62 (3H, s, SCH3); ИК-спектр, n, см-1: 3535 (OH), 1649 (C=O), 1505 (NO2), 1367 (NO2); обнаружено, %: C - 20.86, H 2.51, N 29.28; C5H7N6NaO5S; Вычислено, %: C - 20.98, H 2.47, N 29.36.
Пример 2. Синтез натриевой соли 7-метилтио-3-нитро [1, 2, 4] триазоло [5,1-c] [1, 2, 4] триазин -4 (1H)-она
В данном примере синтез включает 3 этапа: на первом этапе получили 5-амино-3-меркапто-1,2,4-триазол (т.е. соединение (B)) путем конденсации аминогуанидина с тио-соединением, являющимся производным муравьиной кислоты, HC(=O)S-R, где -R - метил. На втором этапе из 5-амино-3-меркапто-1,2,4-триазола получили соответствующую соль диазония. На третьем этапе проведена реакция соли диазония с α-нитроэфиром, 2-нитроацетоуксусным эфиром, в результате чего был получен 7-метилтио-3-нитро [1, 2, 4] триазоло [5,1-c] [1, 2, 4] триазин -4 (1H)-он. Ниже приведено более подробное описание этих этапов.
Этап 1: Синтез соединения (В): В реакционной колбе, снабженной мешалкой и противоточным конденсатором, в атмосфере инертного газа (азот, аргон), к 400 мл абсолютного пиридина добавили 20 г (0,1 моль) аминогуанидина и 7,6 г (0,1 моль) метилтиоформиата. Реакционную смесь выдержали в течение 4 часов при температуре 115°C, после чего перенесли в дистиллированную воду и несколько раз промыли водой. Промытую смесь высушили над нутч-фильтром в вакууме. Провели рекристаллизацию из этанола. Схема реакции представлена на схеме 2.
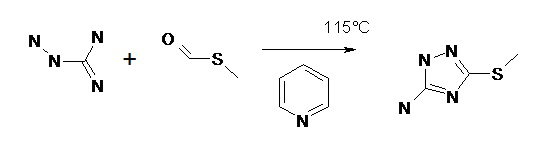
Вход составил 19,3 г (70%).
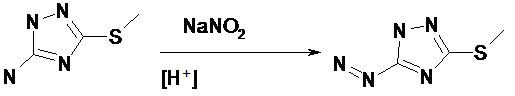
Этап 2: Диазотирование соединения (B): Для получения раствора (в дальнейшем - раствор [3]) 26 г (0,1 моль) 5-амино-3-метилтио-1,2,4-триазола (полученного на этапе 1) растворили в 32 мл азотной кислоты (0,1 моль) и 200 мл воды. Раствор перемешали и охладили до -5°C. В отдельной емкости приготовили 0,1 M раствор азотистокислого натрия, растворив 16 г азотистокислого натрия в 100 мл воды. Раствор азотистокислого натрия поместили в морозильную камеру до образования льда, после чего лед раздробили. Далее, раствор [3] и азотистокислый натрий в виде дробленого льда переместили в реактор объемом 1 л. Смесь перемешивали в течение 1 часа при температуре 0°C. Низкая температура и разное фазовое состояние двух компонентов реакции (жидкой и твердой) обеспечивали низкую скорость и постепенность реакции диазотирования на границе раздела фаз. Окончание процесса диазотирования определили с помощью йодо-крахмального теста (доказательство отсутствия азотистокислого натрия в свободном состоянии).
Схема реакции представлена на схеме 3.
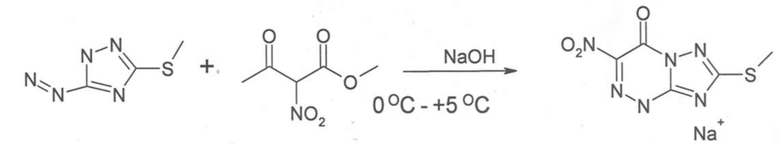
Этап 3: Конденсация соединения диазония с α-нитроэфиром: Для получения раствора (раствор [4]) 17,5 г метил 2- нитроацетоацетата смешали с 300 мл изопропанола. Раствор [4] смешали с солью диазония, полученной на этапе 2. Смесь охладили до температуры 0°C. При 0°C, к реакционной смеси добавляли 10%-ный раствор гидроксида натрия (чтобы нейтрализовать остатки нитрита и ацетата) до получения выраженной щелочной реакции (значение pH - между 8 и 9). Температуру регулировали и поддерживали на уровне ниже +5°C. Полученную в результате смесь перемешивали в течение 1 часа. Осадок отфильтровали и высушили на воздухе. Вход составил 78%.
Схема реакции представлена на схеме 4.
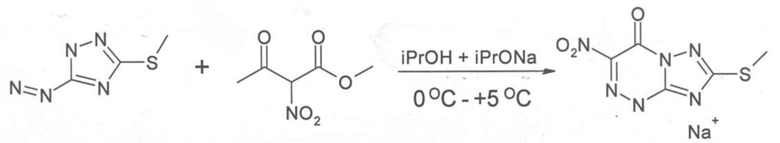
Пример 3: Синтез натриевой соли 7-метилтио-3-нитро [1, 2, 4] триазоло [5,1-c] [1, 2, 4] триазин -4 (1H)-она
Синтез натриевой соли 7-метилтио-3-нитро-1,2,4-триазоло [5,1-c]-1,2,4-триазин-7-она может осуществлять, как показано в Примере 2, только на Этапе 2 вместо водно-спиртового раствора использовать спирт с щелочью (например, гидроксид натрия). Выход противовирусного соединения (А) (натриевая соль 7-метилтио-3-нитро [1, 2, 4] триазоло [5,1-c] [1, 2, 4] триазин -4 (1H)-она) можно увеличить до 83%.
Схема реакции представлена на схеме 5.
Изобретение относится к способу изготовления противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли, в которой R1 выбран из алкильной группы с числом атомов углерода от 1 до 4, и изготовление включает следующие этапы: этап 1 - диазотирование соединения (В) общей формулы (II), в которой R2 выбирается из водорода, при котором соединение (В) растворяют в азотной кислоте, охлаждают до -7°С и добавляют азотнокислый натрий, в результате чего образуется соединение диазония общей формулы (III) или его фармацевтически приемлемая соль; этап 2 - реакция конденсации соединения диазония общей формулы (III) или его фармацевтически приемлемой соли, полученных на этапе 1, с α-нитроэфиром общей формулы (IV), в которой R3 выбирается из -C(O)-OR5, где R5 - алкильная группа с числом атома азота от 1 до 4, R4 - алкильная группа с числом атомов углерода от 1 до 4, в среде охлажденного раствора гидроксида натрия при рН реакционной смеси 9-10, при перемешивании и температуре 0°С в течение 1 часа, а потом в течение 2 часов при комнатной температуре; этап 3 - алкилирование полученного на этапе 2 соединения или его фармацевтически приемлемой соли алкилирующим агентом формулы (V): R1-X, где R1 представляет алкильную группу с числом атомов углерода от 1 до 4, X представляет атом галогена, при комнатной температуре, при перемешивании с получением противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли. Изобретение также относится к иному способу изготовления противовирусного соединения (А) общей формулы (I) и к способу изготовления соединения (В) общей формулы (IIR1). Технический результат: разработан одноступенчатый способ изготовления противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли без выделения промежуточных продуктов, позволяющий увеличить выход целевого продукта. 3 н. и 4 з.п. ф-лы, 3 пр.
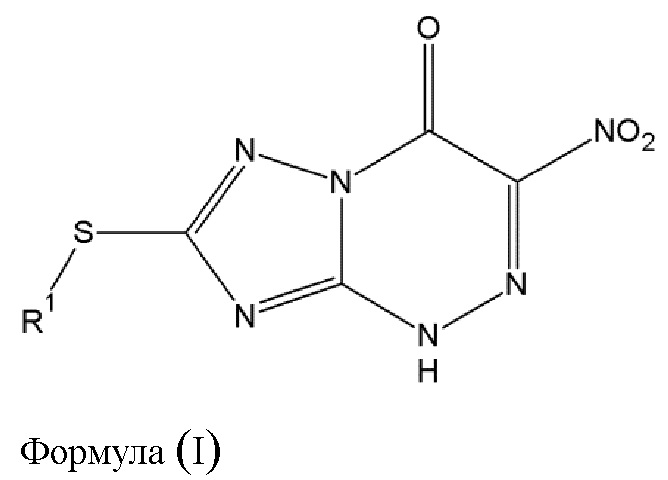

1. Способ изготовления противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли
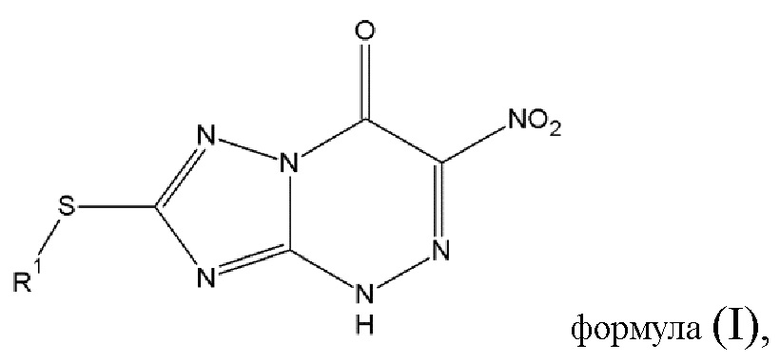
где R1 выбран из алкильной группы с числом атомов углерода от 1 до 4,
и изготовление включает следующие этапы:
этап 1 - диазотирование соединения (В) общей формулы (II)
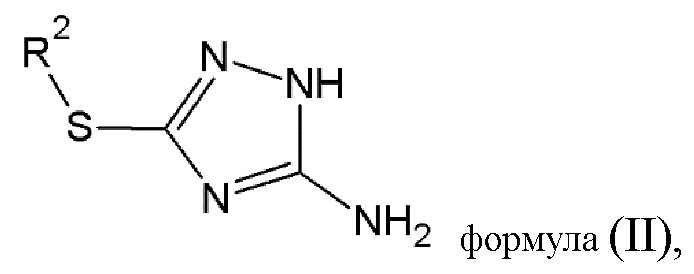
где R2 выбирается из водорода,
при котором соединение (В) растворяют в азотной кислоте, охлаждают до -7°С и добавляют азотнокислый натрий,
в результате чего образуется соединение диазония общей формулы (III) или его фармацевтически приемлемая соль:
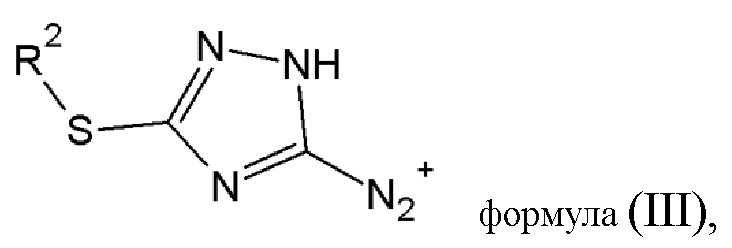
где R2 имеет то же значение, что и в определении, данном выше;
этап 2 - реакция конденсации соединения диазония общей формулы (III) или его фармацевтически приемлемой соли, полученных на этапе 1, с α-нитроэфиром общей формулы (IV)
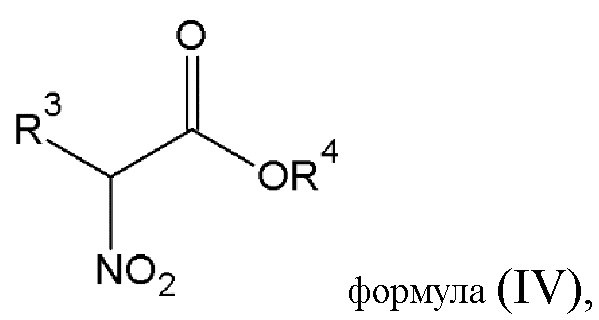
где R3 выбирается из
-(CO)-OR5, где R5 - алкильная группа с числом атомов углерода от 1 до 4;
R4 - алкильная группа с числом атомов углерода от 1 до 4;
в среде охлажденного раствора гидроксида натрия при рН реакционной смеси 9-10, при перемешивании и температуре 0°С в течение 1 часа, а потом в течение 2 часов при комнатной температуре;
этап 3 - алкилирование полученного на этапе 2 соединения или его фармацевтически приемлемой соли алкилирующим агентом формулы (V): R1-X, где R1 представляет алкильную группу с числом атомов углерода от 1 до 4, X представляет атом галогена, при комнатной температуре, при перемешивании с получением противовирусного соединения (А) общей формулы (I) или его фармацевтически приемлемой соли.
2. Способ по п. 1, отличающийся тем, что R1 выбирают из метила, или этила, или пропила, или изопропила.
3. Способ по п. 1 или 2, отличающийся тем, что R1 - метильная группа.
4. Способ по любому пп. 1-3, отличающийся тем, что R3 - (CO)-OR5, где R5 - алкильная группа с числом атомов углерода от 1 до 4.
5. Способ изготовления противовирусного соединения (А) общей формулы (I)
или его фармацевтически приемлемой соли осуществляется при R1, выбранном из алкильной группы с числом атомов углерода от 1 до 4, и включает следующие этапы:
этап 1 – диазотирование соединения (В) общей формулы (II)
где R2 равно R1, в определении, данном выше,
при котором соединение (B) растворяют в азотной кислоте, охлаждают до -5°C и добавляют азотнокислый натрий в форме дробленого льда, смесь перемешивают при температуре 0°С, в результате чего образуется соединение общей формулы (III) или его фармацевтически приемлемая соль:
где R2 имеет то же значение, что и в определении, данном выше;
этап 2 - реакция конденсации соединения диазония общей формулы (III) или его фармацевтически приемлемой соли, полученных на этапе 1, с α-нитроэфиром общей формулы (IV)
где R3 выбирается из
-(CO)-R6, где R6 - алкильная группа с числом атомов углерода от 1 до 4;
R4 - алкильная группа с числом атомов углерода от 1 до 4;
в среде изопропанола при охлаждении реакционной смеси до 0°С, затем к реакционной смеси добавляют раствор гидроксида натрия до рН реакционной смеси 8-9, смесь перемешивают при температуре ниже +5°С в течение 1 часа.
6. Способ по любому из пп. 1-5, отличающийся тем, что молярное соотношение соединения диазония общей формулы (III) и α-нитроэфира общей формулы (IV) составляет от 0,5:2 до 2:0,5, предпочтительно от 0,9:1 до 1:0,9.
7. Способ изготовления соединения (B) общей формулы (IIR1)
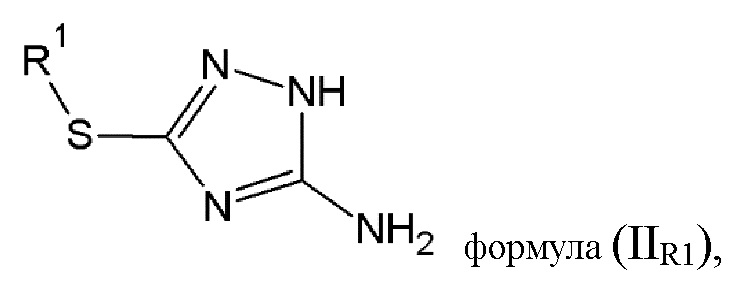
включающий реакцию конденсации аминогуанидина с тио-соединением общей формулы (VII)
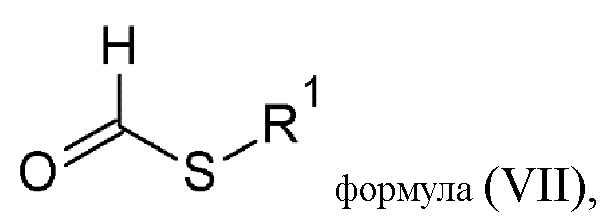
где R1 выбран из алкильной группы с числом атомов углерода от 1 до 4,
в атмосфере инертного газа в среде пиридина при перемешивании в течение 4 часов при температуре 115°С.
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ 2-МЕТИЛТИО-6-НИТРО-1,2,4-ТРИАЗОЛО[5,1-c]-1,2,4-ТРИАЗИН-7-ОНА, ДИГИДРАТА | 2007 |
|
RU2343154C2 |
| Русинов В.Л | |||
| и др | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| НАТРИЕВАЯ СОЛЬ 2-МЕТИЛТИО-6-НИТРО-1,2-4-ТРИАЗОЛО[5,1-C]-1,2,4-ТРИАЗИН-7(4H)-ОНА, ДИГИДРАТ, ОБЛАДАЮЩАЯ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2294936C1 |
| НАТРИЕВАЯ СОЛЬ 2-н-ПРОПИЛТИО-6-НИТРО-1,2,4-ТРИАЗОЛО[5,1-с]-1,2,4-ТРИАЗИН-7-ОНА ДИГИДРАТ И НАТРИЕВАЯ СОЛЬ 2-н-БУТИЛТИО-6-НИТРО-1,2,4-ТРИАЗОЛО[5,1-с]-1,2,4-ТРИАЗИН-7-ОНА ДИГИДРАТ | 2008 |
|
RU2402552C2 |
| НАТРИЕВАЯ СОЛЬ 2-ЭТИЛТИО-6-НИТРО-1,2,4-ТРИАЗОЛО[5,1-c]-1,2,4-ТРИАЗИН-7-ОНА ДИГИДРАТ | 2008 |
|
RU2404182C2 |
| Устройство для срезки голов железобетонных свай | 1977 |
|
SU737568A1 |
| Способ получения 3-амино-5-метилмеркапто-1,2,4-триазола | 1981 |
|
SU1002291A1 |

