Область применения относится к медицине, а именно к молекулярно-генетическим методам диагностики наследственных заболеваний.
Наиболее близким аналогом метода диагностики 3-М синдрома в якутской популяции является молекулярно-генетический способ идентификации мутации в гене Куллин 7 (CUL7) при 3-М синдроме, включающий определение методом ПЦР в гене CUL7, картированием в хромосоме 6р21.1., 25 различных мутаций у обследуемых из 29 семей с 3-М синдромом. В результате найдены нонсенс и миссенс - мутации R1445X и Н1464Р, играющие важную роль в задержке внутриутробного роста у человека (Huber С. et al. Identification of mutations in 3-M syndrome / Nat. Genet. - 2005. - Oct.37 (10). - P.1119-24).
Метод диагностики 3-М синдрома, описанный Huber и др., был применен для диагностики 3-М синдрома в якутской популяции на 39 больных с 3-М синдромом. Ни одна из описанных в данном источнике мутаций не характерна для якутской популяции. В гене CUL7 выявлена другая мутация 4582 insT (Arg 1528 Ser), характерная для якутской популяции. Данный метод не позволяет выявить мутацию 4582 insT (Arg 1528 Ser), характерную для якутской популяции, тем самым не позволяет выставить точный и окончательный диагноз заболевания, дать точный генетический риск заболевания для потомства в семье.
С целью устранения недостатков метода, описанного Huber и др., предложен способ диагностики 3-М синдрома путем молекулярно-генетического анализа посредством ПЦР реакции с применением оригинальных праймеров F-GTATGCACTTAGGGCCAGAG; R-CACACTCCTACAGGCTACAC для выявления мутации 4582 insT (Arg 1528Ser) в гене Куллин 7 (CUL7), характерной для якутской популяции, с последующим проведением анализа полиморфизма длин рестрикционных фрагментов с использованием эндонуклеазы HinfI и при наличии фрагментов длиной 218 п.н. и 175, 26 п.н. диагностируют носительство мутантного гена в гомозиготном состоянии (больной), при наличии фрагментов - 218, 175, 114, 104 и 26 п.н. диагностируют носительство мутантного гена в гетерозиготном состоянии (здоровый носитель мутации), а при длине фрагментов 175, 114, 104, 26 п.н. - норма (здоровый).
Описание метода
Предложенный способ диагностики 3-М синдрома в якутской популяции осуществляется следующим образом.
У больного производится забор крови из локтевой вены в пробирку, содержащую ЭДТА в количестве 7-10 мл.
Выделение ДНК из лейкоцитов периферической крови выполняют стандартным методом с помощью фенол - хлороформного метода.
Для диагностики 3-М синдрома в якутской популяции (быстрой идентификации мутации 4582 insT (Arg 1528 Ser) в гене CUL7, характерной для якутской популяции) амплификация необходимых фрагментов ДНК проводится методом ПЦР на программируемом термоциклере MJ Research РТС-200 в 25 мкл объеме реакционной смеси следующего состава: 0,1-1 мкг геномной ДНК; по 25 пкмоль оригинального олигопраймера (F-GTATGCACTTAGGGCCAGAG; R-CACACTCCTACAGGCTACAC), по 200 мкМ каждого нуклеозидтрифосфата; 0,5 единиц активности ДНК-полимеразы (Applied Biosystems), буфер для ПНР-реакции фирмы «Applied Biosystems» (500 мМ KCI, 100 мМ Tris-HCI, pH 8,3; 15 мМ МСI2; 0,01% gelatin), бетаин 1:2, 20-30 мкл минерального масла. ПНР проводится при следующих условиях: первоначальная денатурация - 95° - 15 минут, 35 циклов амплификации-: 94° - 1 минута, 50-55° - 1 минута, 72° - 1 минута. Заключительная элонгация - 72° - 10 минут. Результаты ПЦР оцениваются в 1% агарозном геле.
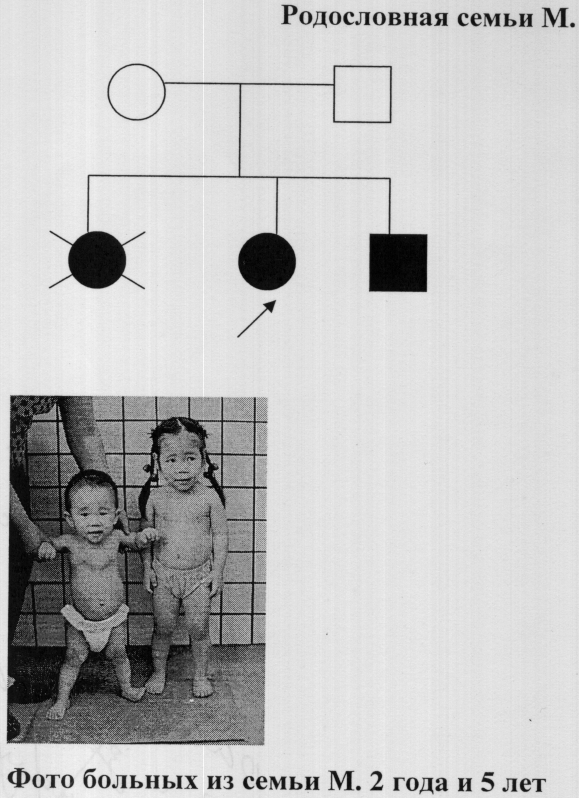
Установление гетерозиготного носительства мутантного гена у родственников пробанда проводится методом анализа полиморфизма длин рестрикционных фрагментов с использованием эндонуклеазы Hinf I (Toyobo). Реакция проводится согласно протоколу фирмы-производителя. Результаты амплификации после ПДРФ - анализа оцениваются методом электрофореза в 2% агарозном Nusieve геле (чертеж).
В норме должны образовываться фрагменты 175, 114, 104, 26 п.н., в случае носительства в гомозиготном состоянии (больной) - вместо фрагментов 114 и 104 образуются фрагменты длиной 218 п.н. и 175, 26 п.н. В случае гетерозиготного носительства мутации (здоровый носитель) длины фрагментов будут 218, 175, 114, 104 и 26 п.н.
Быстрый способ диагностики 3-М синдрома испытан на 39 больных с 3-М синдромом, 35 их родственниках и 20 здоровых индивидах якутской национальности. Все больные были гомозиготами по данной мутации и имели аллели длиной 218 п.н. и 175, 26 п.н., 30 родственников из 35 были гетерозиготными носителями мутации и имели длины фрагментов 218, 175, 114, 104 и 26 п.н., остальные 5 из группы родственников и 20 здоровых имели фрагменты длиной 175, 114, 104, 26 п.н. Чертеж.
Данный способ диагностики 3-М синдрома в якутской популяции позволяет выставить точный диагноз, проводить пренатальную ДНК - диагностику и проспективное медико-генетическое консультирование для вступающих в брак. Предлагаемый нами способ диагностики 3-М синдрома прост в выполнении, точен и доступен для выполнения в молекулярно-генетической лаборатории.
Клинические примеры заболевания:
Пример 1
Пациент К.Р., 14 - 09 - 2001 г.р.
Адрес: Республика Саха, г.Якутск, ул. Крупская 3, кв.3
Национальность: саха
Жалобы на низкий рост, отставание в физическом развитии с рождения.
Анамнез болезни: Впервые был осмотрен в отделении патологии новорожденных врачами-генетиками медико-генетической консультации и выставлен диагноз: Метафизарная хондродистрофия, тип Маккьюсика. До 2006 года ребенок наблюдался в медико-генетической консультации Республиканской больницы №1 - НЦМ Минздрава Республики Саха(Якутия) с клиническим диагнозом: Метафизарная хондродистрофия, тип Маккьюсика. После проведения молекулярно-генетического анализа был выставлен окончательный диагноз: 3-М синдром с аутосомно-рецессивным типом наследования.
Анамнез жизни: Ребенок от 3 беременности, протекавшей без патологии, 2 физиологических родов в сроке 38 недель беременности с ростом - 44 см, весом - 2,980 кг. Закричал после реанимационных мероприятий, к груди приложен на 5 сутки, переведен на 5 сутки в отделение патологии новорожденных Республиканской больницы №1 - НЦМ.
Перенесенные заболевания: ОРВИ, ожоговая болезнь в 2, 5 года.
Аллергологический анамнез: спокоен.
Психомоторное развитие: головку удерживает с 5 месяцев, ходит с 12 месяцев. Говорит с 2,5 лет.
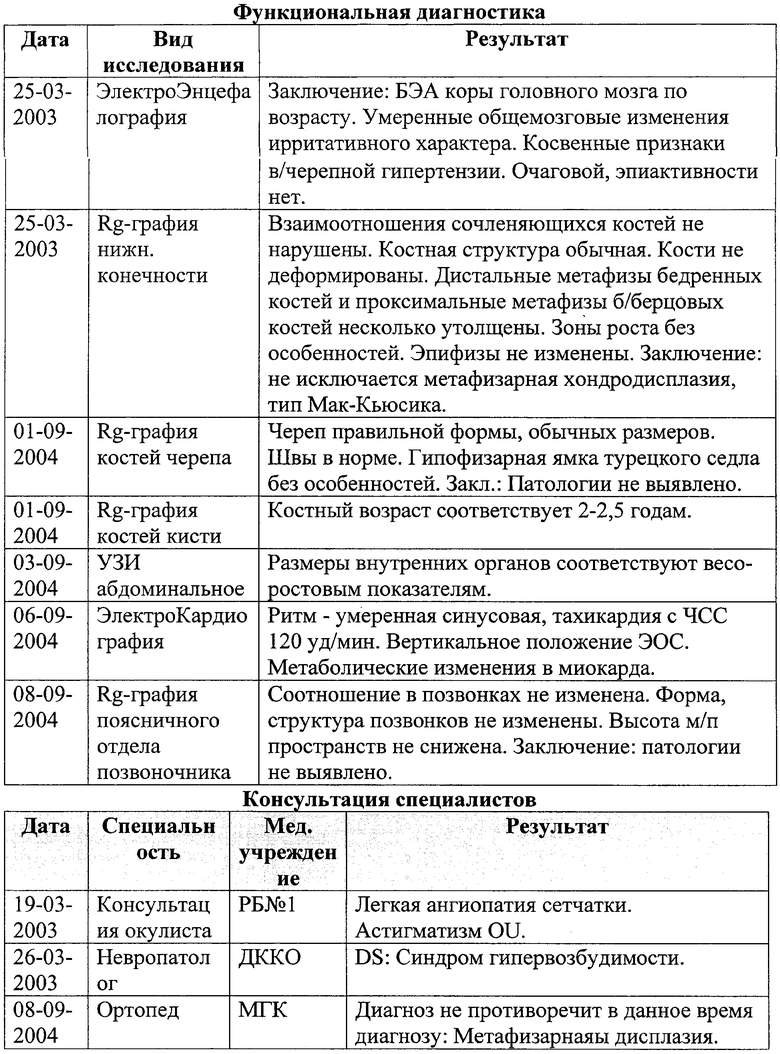
По родословной: наследственность не отягощена, родители не являются родственниками, в семье еще 1 ребенок - здоровый, от первого брака. Родители по национальности оба саха. Рост родителей- средний.
Фенотип
Телосложение непропорциональное, голова большая по отношению к туловищу. Рост низкий, нанизм. Голова гидроцефальной формы, лицо треугольное, большие миндалевидные глаза, гипоплазия средней трети лица, толстые губы, гипотелоризм, длинный фильтр. Короткий нос, гипоплазия скуловых костей. Деформация грудной клетки по типу «груди сапожника», широкая и укороченная грудина. Конечности пропорциональные, микромелия верхняя и нижняя. Поясничный гиперлордоз. Выступающие пятки на стопах. Мышечная гипотония. Сердечные тоны ритмичные, ясные. Живот большой, мягкий, безболезненный. Печень и селезенка не увеличены. НПО по мужскому типу. Стул и диурез в норме.
Результаты лабораторных исследований:
Заключение № 21
по молекулярно - генетическому исследованию геномной ДНК
На базе отделения неврологии Института мозга Университета г.Ниигаты (Япония) проведена косвенная ДНК-диагностика следующих заболеваний:
1. Метафизарной хондродистрофии Маккьюсика (в гене RMPR, с использованием полиморфных динуклеотидных маркеров D9S163, 99АС, СА1, D9S1804).
2. Нанизма Мюллибрея (в локусе гена MUL с использованием полиморфных динуклеотидных маркеров D17S1606, 95-СА, 272а-СА, 52-CA, D17S924).
3. Болезни Ларона (в гене GH с использованием полиморфных динуклеотидных маркеров D5S628, D5S634).
Пациент в локусах генов этих заболеваний по данным полиморфным маркерам не является гомозиготой, следовательно, все эти заболевания исключены молекулярно-генетическим методом.
Проведено полное секвенирование гена Куллин 7 (Cullin7, CUL7). В 25 экзоне гена Куллин 7 (Cullin7, CUL7), вызывающего наследственное заболевание 3-М Синдром выявлена нонсенс мутация - инсерция Т (тимина) в положении 4582. Проведена ДНК-диагностика методом ПНР с последующим проведением ПДРФ - анализа и использованием рестриктазы HinfI и выявлено наличие фрагментов длиной 218 п.н. и 175, 26 п.н., что является показателем носительства мутантного гена в гомозиготном состоянии (больной).
Заключение: Таким образом, по результатам молекулярно-генетического анализа диагноз метафизарной хондродисплазии Маккьюсика снят. В результате молекулярно-генетического анализа (найдена мутации в гене CULT) выставлен окончательный диагноз: 3 - М синдром (OMIM-273750).
Медицинское заключение
Диагноз метафизарной хондродисплазии, тип Маккьюсика на основании результатов ДНК-диагностики снят. Учитывая клинические данные(пренатальную и постнатальную гипоплазию, пропорционально низкий рост, лицевые дизморфии, нормальный интеллект и отсутствие эндокринных нарушений), лабораторно- функциональные исследования, молекулярно-генетический анализ (идентифицирована точковая мутацию - инсерцию тимина в 25 экзоне гена CUL7, ответственного за развитие 3-М синдрома) выставлен
ЗАКЛЮЧИТЕЛЬНЫЙ ДИАГНОЗ: 3-М синдром с аутосомно-рецессивным типом наследования (наследственный нанизм (OMIM - 73750). Генетический риск для сибсов 25%.

Пример 2.
Пациентка М.Т.
Дата рождения: 27-07-1999
Адрес: Республики Саха(Якутия), Верхневилюйский улус, село Хоро.
Жалобы на низкий рост, резкое отставание в росте с рождения.
Анамнез заболевания: Ребенок был осмотрен в возрасте 2 недель в медико-генетической консультации и выставлен диагноз: Метафизарная хондродистрофия, тип Маккьюсика. До 2006 года ребенок наблюдался в медико-генетической консультации Республиканской больницы - НЦМ Минздрава Республики Саха(Якутия) с клиническим диагнозом: Метафизарная хондродистрофия, тип Маккьюсика. После проведения молекулярно-генетического анализа был выставлен окончательный диагноз: 3-М синдром.
Анамнез жизни: Ребенок от 2 беременности, протекавшей во 2 половине с гестозом. Была направлена на стационарное лечение в сроке 37 недель, по УЗИ плода - задержка внутриутробного развития. От 2 оперативных родов на 38 неделе с весом - 1860, ростом - 35 см, Апгар - 2/6 баллов. Состояние при рождении тяжелое, переведен в отделение патологии новорожденных РБ №1-НЦМ.
Перенесенные заболевания: ОРВИ.
Аллергологический анамнез: спокоен.
Психомоторное развитие: Головку держит с 5 месяцев, сидит - с 8 месяцев, самостоятельно ходит - с 14 месяцев, первые зубы - с 6 месяцев.
Из родословной: Беременность-1 завершилась мертворожденным плодом женского пола, с весом - 1700 и гидроцефальной формой головы. Сибс с таким же фенотипом.
Фенотип
Телосложение непропорциональное, голова большая относительно туловища, макроцефалия, туловище укорочено. Низкий рост, нанизм. Кожные покровы смуглые, чистые. Голова ближе к гидроцефальной форме. Лоб выступает. Волосы темные. Лицо треугольное, переносье упрощено. Нос короткий с выступающими носовыми ходами. Фильтр длинный. Шея короткая. Грудная клетка широкая, укорочена. Микромелия верхняя и нижняя. ЧПС на обеих кистях. Сердечные тоны ритмичные, ясные, шумов нет. Живот увеличен, печень и селезенка не увеличены. Поясничный гиперлордоз. Выступающие пятки на стопах. НПО по женскому типу. Стул и диурез в норме
Результаты лабораторных исследований:
Заключение N 23
по молекулярно-генетическому исследованию геномной ДНК
На базе отделения неврологии Института мозга Университета г.Ниигаты (Япония) проведена косвенная ДНК-диагностика следующих заболеваний:
1. Метафизарной хондродистрофии Маккьюсика (в гене RMPR, с использованием полиморфных динуклеотидных маркеров D9S163, 99АС, СА1, D9S1804).
2. Нанизма Мюллибрея (в локусе гена MUL с использованием полиморфных динуклеотидных маркеров D17S1606, 95-СА, 272а-СА, 52-CA, D17S924).
3. Болезни Ларона (в гене GH с использованием полиморфных динуклеотидных маркеров D5S628, D5S634).
Пациент в локусах генов этих заболеваний по данным полиморфным маркерам не является гомозиготой, следовательно, все эти заболевания исключены молекулярно-генетическим методом.
Проведено полное секвенирование гена Куллин 7 (Cullin7, CUL7). В 25 экзоне гена Куллин 7 (Cullin7, CUL7), вызывающего наследственное заболевание 3-М Синдром, выявлена нонсенс мутация - инсерция Т (тимина) в положении 4582. Проведена ДНК-диагностика методом ПЦР с последующим проведением ПДРФ - анализа и использованием рестриктазы Hinfl и выявлено наличие фрагментов длиной 218 п.н. и 175, 26 п.н., что является показателем носительства мутантного гена в гомозиготном состоянии (больной).
Заключение: Таким образом, по результатам молекулярно-генетического анализа диагноз метафизарной хондродисплазии Маккьюсика снят. В результате молекулярно-генетического анализа (найдена мутации в гене CUL7) выставлен окончательный диагноз: 3-М синдром (OMIM-273750).
Медицинское заключение
Диагноз метафизарной хондродисплазии, тип Маккьюсика на основании ДНК-диагностики снят. Учитывая клинические данные исследований (пренатальную и постнатальную гипоплазию, пропорционально низкий рост, лицевые дизморфии, нормальный интеллект и отсутствие эндокринных нарушений), лабораторно-функциональные и молекулярно-генетический анализ (идентифицированную точковую мутацию - инсерцию тимина в 25 экзоне гена CUL 7, ответственного за развитие 3-М синдрома) выставлен
ЗАКЛЮЧИТЕЛЬНЫЙ ДИАГНОЗ: 3-М синдром с аутосомно-рецессивным рецессивным типом наследования (наследственный нанизм (OMIM - 273750). Генетический риск для сибсов 25%.
Пациент М. Д, сибс М.Т.
Дата рождения: 27-07-1999
Адрес: Республики Саха(Якутия), Верхневилюйский улус, село Хоро,
Жалобы на низкий рост, резкое отставание в росте с рождения.
Анамнез заболевания: Ребенок был осмотрен в возрасте 4 месяцев в медико-генетической консультации и выставлен диагноз: Метафизарная хондродистрофия, тип Маккьюсика. До 2006 года ребенок наблюдался в медико-генетической консультации Республиканской больницы - НЦМ Минздрава Республики Саха(Якутия) с клиническим диагнозом: Метафизарная хондродистрофия, тип Маккьюсика. После проведения молекулярно-генетического анализа был выставлен окончательный диагноз: 3-М синдром.
Анамнез жизни: Ребенок от 3 беременности, протекавшей во 1 половине - с токсикозом, во 2 половине с гестозом. От 3 оперативных родов на 38 неделе с весом - 1860, ростом - 35 см, Апгар - 2/6 баллов. Состояние при рождении тяжелое, переведен в отделение патологии новорожденных РБ №1 - НЦМ.
Перенесенные заболевания: ОРВИ
Аллергологический анамнез: спокоен.
Психомоторное развитие: Головку держит с 3 месяцев, сидит - с 7 месяцев, самостоятельно ходит - с 14 месяцев, первые зубы - с 6 месяцев.
Из родословной: Беременность - 1 завершилась мертворожденным плодом женского пола, с весом - 1700 и гидроцефальной формой головы. Сибс с таким же фенотипом.
Фенотип
Телосложение непропорциональное, голова большая относительно туловища, макроцефалия, туловище укорочено. Низкий рост, нанизм. Кожные покровы смуглые, чистые. Голова ближе к гидроцефальной форме. Лоб выступает. Волосы темные. Лицо треугольное, переносье упрощено, гипоплазия средней части лица. Нос короткий с выступающими носовыми ходами. Фильтр длинный. Шея короткая. Грудная клетка широкая, укорочена. Микромелия верхняя и нижняя. ЧПС на обеих кистях. Выступающие пятки на стопах. Сердечные тоны ритмичные, ясные, шумов нет. Живот увеличен, печень и селезенка не увеличены. Поясничный гиперлордоз. НПО по мужскому типу. Стул и диурез в норме.
Результаты лабораторных исследований:
Заключение N 24
по молекулярно-генетическому исследованию геномной ДНК
На базе отделения неврологии Института мозга Университета г.Ниигаты (Япония) проведена косвенная ДНК-диагностика следующих заболеваний:
1. Метафизарной хондродистрофии Маккьюсика (в гене RMPR, с использованием полиморфных динуклеотидных маркеров D9S163, 99АС, СА1, D9S1804).
2. Нанизма Мюллибрея (в локусе гена MUL с использованием полиморфных динуклеотидных маркеров D17S1606, 95-СА, 272а-СА, 52-CA, D17S924).
3. Болезни Ларона (в гене GH с использованием полиморфных динуклеотидных маркеров D5S628, D5S634).
Пациент в локусах генов этих заболеваний по данным полиморфным маркерам не является гомозиготой, следовательно, все эти заболевания исключены молекулярно-генетическим методом.
Проведено полное секвенирование гена Куллин 7 (Cullin7, CUL7). В 25 экзоне гена Куллин 7 (Cullin7, CUL7), вызывающего наследственное заболевание 3-М Синдром выявлена нонсенс мутация - инсерция Т (тимина) в положении 4582 / Проведена ДНК-диагностика методом ПЦР с последующим проведением ПДРФ - анализа и использованием рестриктазы Hinfl и выявлено наличие фрагментов длиной 218 п.н. и 175, 26 п.н., что является показателем носительства мутантного гена в гомозиготном состоянии (больной).
Заключение: Таким образом, по результатам молекулярно-генетического анализа диагноз метафизарной хондродисплазии Маккьюсика снят. В результате молекулярно-генетического анализа (найдена мутации в гене CUL7) выставлен окончательный диагноз: 3 М синдром (OMIM-273750).
Медицинское заключение
Диагноз метафизарной хондродисплазии, тип Маккьюсика на основании ДНК-диагностики снят.Учитывая клинические данные (пренатальную и постнатальную гипоплазию, пропорционально низкий рост, лицевые дизморфии, нормальный интеллект и отсутствие эндокринных нарушений), лабораторно- функциональные исследований и молекулярно-генетический анализ (идентифицированную точковую мутацию - инсерцию тимина в 25 экзоне гена CUL7, ответственного за развитие 3-М синдрома) выставлен ЗАКЛЮЧИТЕЛЬНЫЙ ДИАГНОЗ: 3-М синдром с аутосомно-рецессивным типом наследования, (наследственный нанизм (OMIM - 273750). Генетический риск для сибсов 25%.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ одновременной диагностики наследственных заболеваний | 2015 |
|
RU2627115C2 |
| Способ диагностики наследственной моторно-сенсорной нейропатии 6С типа | 2023 |
|
RU2826719C1 |
| Способ диагностики варианта SOPH c.5741G>A в гене NBAS | 2024 |
|
RU2819985C1 |
| Способ диагностики нейронального цероидного липофусциноза 6 типа | 2021 |
|
RU2784293C1 |
| Способ диагностики мукополисахаридоз-плюс синдрома | 2022 |
|
RU2798695C1 |
| Способ диагностики точечных мутаций в нативной ДНК с применением оксида графена | 2015 |
|
RU2614111C1 |
| СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ | 2010 |
|
RU2448163C2 |
| Способ ДНК-диагностики аутосомно-рецессивной глухоты-103 | 2019 |
|
RU2727684C1 |
| Способ ДНК-диагностики врожденной формы катаракты (CTRCT18) | 2017 |
|
RU2648464C1 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ РИСКА СУБАРАХНОИДАЛЬНОГО КРОВОИЗЛИЯНИЯ ВСЛЕДСТВИЕ РАЗРЫВА АНЕВРИЗМЫ СОСУДОВ ГОЛОВНОГО МОЗГА У ЛИЦ АЗИАТСКОЙ РАСЫ | 2015 |
|
RU2627643C2 |
Изобретение относится к области медицины, а именно к молекулярно-генетическим методам диагностики наследственных заболеваний, а именно способу диагностики 3-М синдрома в якутской популяции. Сущность способа заключается в том, что при проведении ПЦР реакции использованы оригинальные праймеры для выявления мутации 4582 insT (Arg 1528 Ser) в гене Куллин 7 (CUL7), характерной для якутской популяции, с последующим проведением ПДРФ - анализа и использованием рестриктазы HinfI и наличие фрагментов длиной 218 п.н. и 175, 26 п.н. указывает на носительство мутантного гена в гомозиготном состоянии (больной), наличие фрагментов - 218, 175, 114, 104 и 26 п.н. является показателем носительства мутантного гена в гетерозиготном состоянии (здоровый носитель мутации), а наличие фрагментов длиной 175, 114, 104, 26 п.н. - является нормой (здоровый). Преимущество изобретения заключается в разработке быстрого и точного способа диагностики 3-М синдрома у больных и их родственников в отягощенных семьях, пренатальной ДНК-диагностики, и проспективного медико-генетического консультирования для вступающих в брак. 4 ил.
Способ диагностики 3-М синдрома путем молекулярно-генетического анализа посредством ПЦР, отличающийся тем, что при диагностике 3-М синдрома в якутской популяции с аутосомно-рецессивным типом наследования с применением праймеров F-GTATGCACTTAGGGCCAGAG; R-CACACTCCTACAGGCTACAC проводят амплификацию геномной ДНК, выделенную из лейкоцитов периферической крови и анализ полиморфизма длин рестрикционных фрагментов с использованием эндонуклеазы Hinfl, и при наличии фрагментов длиной 218 п.н. и 175, 26 п.н. диагностируют носительство мутантного гена Куллин 7 (CUL7) с мутацией 4582 insT (Arg 1528 Ser) в гомозиготном состоянии (больной), при наличии фрагментов -218, 175, 114, 104 и 26 п.н. диагностируют носительство мутантного гена в гетерозиготном состоянии и при длине фрагментов 175, 114, 104, 26 п.н. диагностируют нормальный ген (здоровый).
| HUBER С | |||
| et al., Identification of mutations in CUL7 in 3-M syndrome, Nat | |||
| Genet, 2005, oct., 37 (10), 1119-1124 | |||
| БОЧКОВ Н.П., Клинико-генеалогический метод диагностики - сочетание общего (стандартного) клинического и генеалогических методов, Клиническая генетика | |||
| Учебник для студентов медицинских Вузов, 2001, с.59-60, 84-85 | |||
| ELLIOT AM | |||
| et al., |

