Изобретение относится к области медицины, в частности, к молекулярно-генетическим методам диагностики наследственного заболевания - нейронального цероидного липофусциноза 6 типа (НЦЛ 6).
Нейрональный цероидный липофусциноз (НЦЛ) представляет собой наиболее распространенную группу нейродегенеративных заболеваний детского возраста, характеризующихся накоплением аутофлуоресцентных липопигментов в различных тканях (см. CLN6 disease caused by the same mutation originating in Pakistan has varying pathology / R. Guerreiro, J.T. Bras, M. Vieira et al. // European Journal Paediatric Neurololgy. - 2013. - Vol. 17, N 6. - P.657-660). Частота встречаемости НЦЛ в мире составляет от 1 до 8 на 100 000 новорожденных (см. Batten disease: biochemical and molecular characterization revealing novel PPT1 and TPP 1 gene mutations in Indian patients / J. Sheth, M. Mistri, R. Bhavsar et al. // BMC Neurology. – 2018. – Vol. 18, N 203. - P.1-11). В настоящее время идентифицировано 14 типов НЦЛ (НЦЛ1-НЦЛ14), среди них в мире наиболее часто встречаются типы НЦЛ 1, НЦЛ 2 и НЦЛ 3 (см. Next-Generation Analysis Reveals Novel Pathogenic Variants in Four Chinese Siblings with Late-Infantile Neuronal Ceroid Lipofuscinosis / X-T. Ren, X-H. Wang, Ch-H. Ding et al. // Frontiers in Genetics. – 2019. – Vol. 10. - P.1-10). В России наиболее распространенными являются НЦЛ 2 и НЦЛ 7 типов (см. Neuronal ceroid lipofuscinosis in the Russian population: Two novel mutations and the prevalence of heterozygous carriers / A.A. Kozina, E.G. Okuneva, N.V. Baryshnikova et al. // Mol Genet and Genomic Med. - 2020. – Vol. 8, N 7. - P.1-11). Всего описано 13 генов, которые вызывают НЦЛ (CLN1/PPT1, CLN2/TPP1, CLN3, CLN4/DNAJC5, CLN5, CLN6, CLN7/MFSD8, CLN8, CLN10/CTSD, CLN11/GRN, CLN12/ATP13A2, CLN13/CTSF и CLN14/KCTD7) (см. Warrier V Genetic basis and phenotypic correlations of the neuronal ceroid lipofuscinoses / V. Warrier, M. Vieir, S Mole // Biochim. Biophys. Acta. - 2013. - Vol. 1832. - P.1827-1830; Cell biology and function of neuronal ceroid lipofuscinosis-related proteins / K. Kollman, K. Uusi-Rauva, E. Scifo et al. // Biochim. Biophys. Acta. – 2013. – Vol. 1832. - P.1866-1881). Большинство типов НЦЛ - это аутосомно-рецессивные заболевания, за исключением типа, вызванного мутацией в гене CLN4/DNAJC5, при котором заболевание имеет аутосомно-доминантный характер наследования (см. Haltia M, Goebel H.H. The neuronal ceroid-lipofuscinoses: A historical introduction // Biochimica et Biophysica Acta. - 2013. - Vol. 1832, N 11. - P.1795-1800). В этих идентифицированных генах выявлены более 500 патогенных вариантов, которые являются причиной заболевания (см. Batten disease: biochemical and molecular characterization revealing novel PPT1 and TPP 1 gene mutations in Indian patients / J. Sheth, M. Mistri, R. Bhavsar et al. // BMC Neurology. – 2018. – Vol. 18, N 203. - P.1-11; Next-Generation Analysis Reveals Novel Pathogenic Variants in Four Chinese Siblings with Late-Infantile Neuronal Ceroid Lipofuscinosis / X-T. Ren, X-H. Wang, Ch-H. Ding et al. // Frontiers in Genetics. – 2019. – Vol. 10. - P.1-10).
Нейрональный цероидный липофусциноз 6 типа (OMIM 601780) относится к поздним инфантильным формам НЦЛ и вызывается мутациями в гене CLN6. Ген CLN6 расположен на хромосоме 15q22-23 и кодирует белок, состоящий из 311 аминокислотных белков с 7 трансмембранными доменами (см. Variant late infantile ceroid lipofuscinoses associated with novel mutations in CLN6 / N. Canelli, B. Garavaglia B, A. Simonati et al. // Biochemical and Biophysical Research Communications. – 2009. – Vol. 379. - P.892-897). В гене CLN6 согласно базе данных «NCL Mutation and Patient Database» (см. NCL Mutation and Patient Database. – URL: https://www.ucl.ac.uk/ncl-disease/mutation-and-patient-database (дата обращения 15.11.2020)) было установлено более 130 патогенных мутаций в разных популяциях, ответственных за причину широкой генетической гетерогенности заболевания. Белок CLN6 участвует в эндоцитозе лизосомальных белков, селективном транспорте липидов и белков, связанных с функцией и подкислением лизосом, и аутофагии (см. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins / K. Kollman, K. Uusi-Rauva, E. Scifo et al. // Biochim. Biophys. Acta. – 2013. – Vol. 1832. - P.1866-1881; Cárcel-Trullols J. Cell biology of the NCL proteins: What they do and don’t do / J. Cárcel-Trullols, A.D. Kovács, D.A. Pearce // Biochimica et Biophysica Acta. - 2015. - Vol.1852, N 10. - P.2242-2255). Возраст начала заболевания при НЦЛ 6 типа варьирует от 18 месяцев до 8 лет. Тип наследования заболевания аутосомно-рецессивный. Клинически дебютирует в виде прогрессирующей неврологической симптоматики – атаксии, эпилептических припадков, регресса психомоторного развития, нарушения зрения (см. Mole S, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease) // Biochim Biophys Acta Mol Basis Dis. - 2015. - Vol. 1852, N 10. - P.2237-2241). Пациенты с НЦЛ 6 типа были зарегистрированы в нескольких странах мира, в таких как Чешская Республика, Хорватия, Португалия, Америка, Европа, Индия, Пакистан, Турция, Китай и Япония (см. Spectrum of CLN6 mutations in variant late infantile neuronal ceroid lipofuscinosis / J.D. Sharp, R.B. Wheeler, K.A. Parker et al. // Human Mutation. – 2003. – Vol. 22, N 1. - P.35-42; The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland / S.J. Moore, D.J. Buckley D.J, A. MacMillan et al. // Clin. Genet. - 2008. - Vol. 74, N 3. - P.213-222; Two novel CLN6 mutations in variant late-infantile neuronal ceroid lipofuscinosis patients of Turkish origin / E. Siintola, M. Topcu, A. Kohlschütter et al. // Clin. Genet. – 2005. – Vol. 68, N 2. - P.167-173; First Japanese variant of late infantile neuronal ceroid lipofuscinosis caused by novel CLN6 mutations / R. Sato, T. Inui, W. Endo et al. // Brain Development. - 2016. -Vol. 38, N 9. - P.852–856; A first CLN6 variant case of late infantile neuronal ceroid lipofuscinosis caused by a homozygous mutation in a boy from China: a case report / G. Sun, F. Yao, Z. Tian et al. // BMC Medical Genetics. - 2018. - Vol. 19, N 177. - P.1-5). В отечественной литературе исследований, посвященных нейрональному цероидному липофусцинозу, немного, например, источники: Neuronal ceroid lipofuscinosis in the Russian population: Two novel mutations and the prevalence of heterozygous carriers / A.A. Kozina, E.G. Okuneva, N.V. Baryshnikova et al. // Mol Genet and Genomic Med. - 2020. – Vol. 8, N 7. - P.1-11; The first three Russian cases of classical, late-infantile, neuronal ceroid lipofuscinosis / A. Lavrov, E. Ilyna, E. Zakharova et al. // European Journal of Pediatric Neurology. - 2002. - Vol. 6, N 3. - P.161-164.
При анализе уровня техники были выявлены изобретения с применением методики ПЦР-ПДРФ для диагностики других наследственных и мультифакториальных заболеваний, например, см. № RU №2315310 (кл. G01N 33/50, С12Q 1/68, опубл. 20.01.2008), RU №2742956 (кл. C12Q 1/68, G01N 33/48, C12N 15/00, опубл. 12.02.2021), RU №2727684 (кл. C12Q 1/68, опубл. 22.07.2020), RU №2688180 (кл. G01N 33/50, C12Q 1/68, опубл. 21.05.2019), RU №2648464 (кл. G01N 33/50, C12Q 1/68, опубл. 26.03.2018), RU №2671156 (кл. C12N 15/00, G01N 33/00, опубл. 29.10.2018), RU №2697398 (кл. G01N 33/50, C12Q 1/6876, опубл. 14.08.2019), RU №2723090 (кл. C12Q 1/68, опубл. 08.06.2020), RU №2723585 (кл. C12Q 1/68, опубл. 16.06.2020).
Кроме того, известен ДНК-микрочип для молекулярно-генетического исследования человека (см. RU № 137553, кл. C12Q 1/68, опубл. 20.02.2014), представляющий собой пластину с нанесенными на нее олигонуклеотидными зондами, содержащими участки ДНК, позволяющими диагностировать не менее 10 генетических заболеваний, при этом в панель включен нейрональный цероидный липофусциноз 1 типа.
При этом прямых аналогов к способу диагностики НЦЛ 6 типа методом ПЦР-ПДРФ из уровня техники не выявлено.
Задача, на решение которой направлено заявленное изобретение, является создание способа диагностики нейронального цероидного липофусциноза 6 типа.
Технический результат, получаемый при решении поставленной задачи, выражается в повышении достоверности диагностики на наличие или отсутствие гетерозиготной/гомозиготной мутации c.396dupT (p.Val133CysfsTer18) в 4 экзоне гена CLN6, определении генотипа человека в биологическом материале (цельная кровь), а также повышении доступности подобных исследований, поскольку заявленное решение может быть осуществлено на стандартном известном оборудовании.
Для решения поставленной задачи предложен способ диагностики носительства мутантного гена CLN6 с мутацией c.396dupT (p.Val133CysfsTer18), приводящего к нейрональному цероидному липофусцинозу 6 типа с аутосомно-рецессивным типом наследования в якутской популяции, путем молекулярно-генетического анализа посредством полимеразной цепной реакции, включающий амплификацию геномной ДНК с применением праймеров F- GACAGTGCCCTCACCTAGC; R- CTGGCCTGCTAAAGGGACC, выделенной из лейкоцитов периферической крови, и анализ полиморфизма длин рестрикционных фрагментов, где при длине фрагментов 469 п.н. диагностируют носительство мутантного гена в гомозиготном состоянии (больной), при длине фрагментов 469, 224 и 245 п.н. - носительство мутантного гена в гетерозиготном состоянии (здоровый носитель мутации) и при длине фрагментов 224 и 245 п.н. диагностируют нормальный ген (здоровый).
Сопоставительный анализ признаков заявленного решения с признаками аналогов свидетельствует о соответствии заявленного решения критерию «новизна».
Совокупность признаков изобретения обеспечивает решение заявленной технической задачи, а именно, получение молекулярно-генетического способа диагностики наследственного заболевания нейронального цероидного липофусциноза 6 типа, наиболее часто встречаемого у детей.
Заявленный способ диагностики нейронального цероидного липофусциноза 6 типа в якутской популяции позволяет выставить точный диагноз, проводить пренатальную ДНК-диагностику и проспективное медико-генетическое консультирование для вступающих в брак. Техническое решение простое в выполнении, точное и доступное для выполнения в условиях молекулярно-генетической лаборатории.
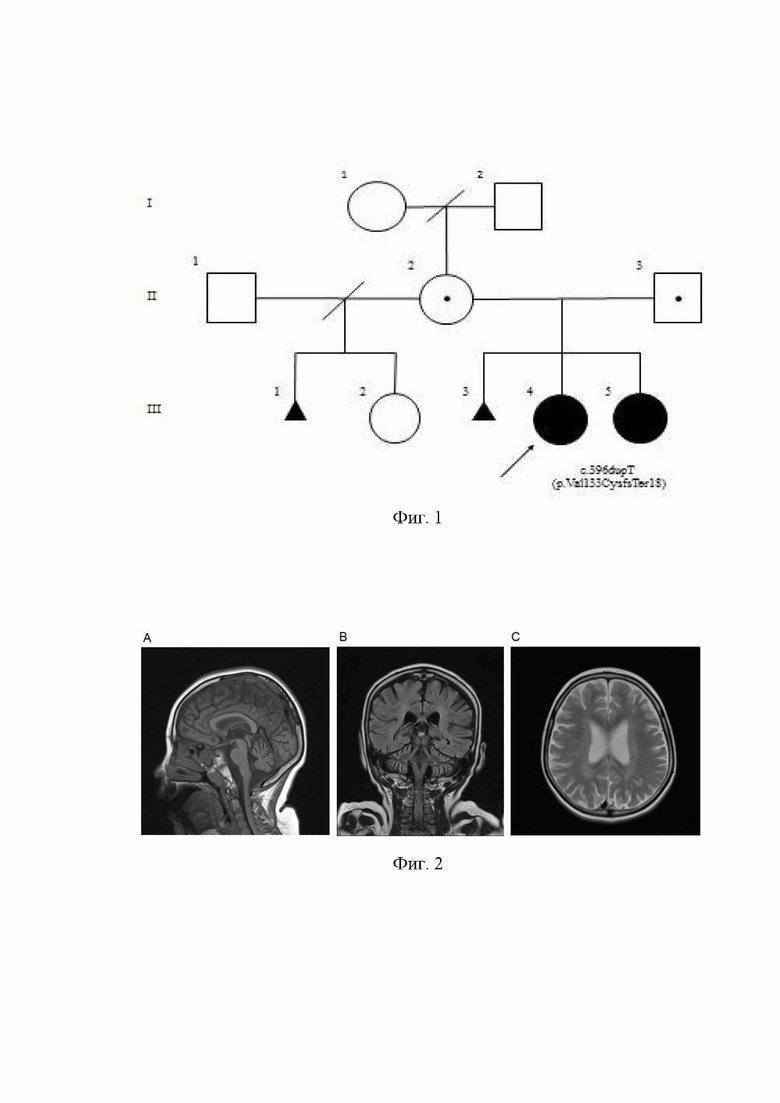
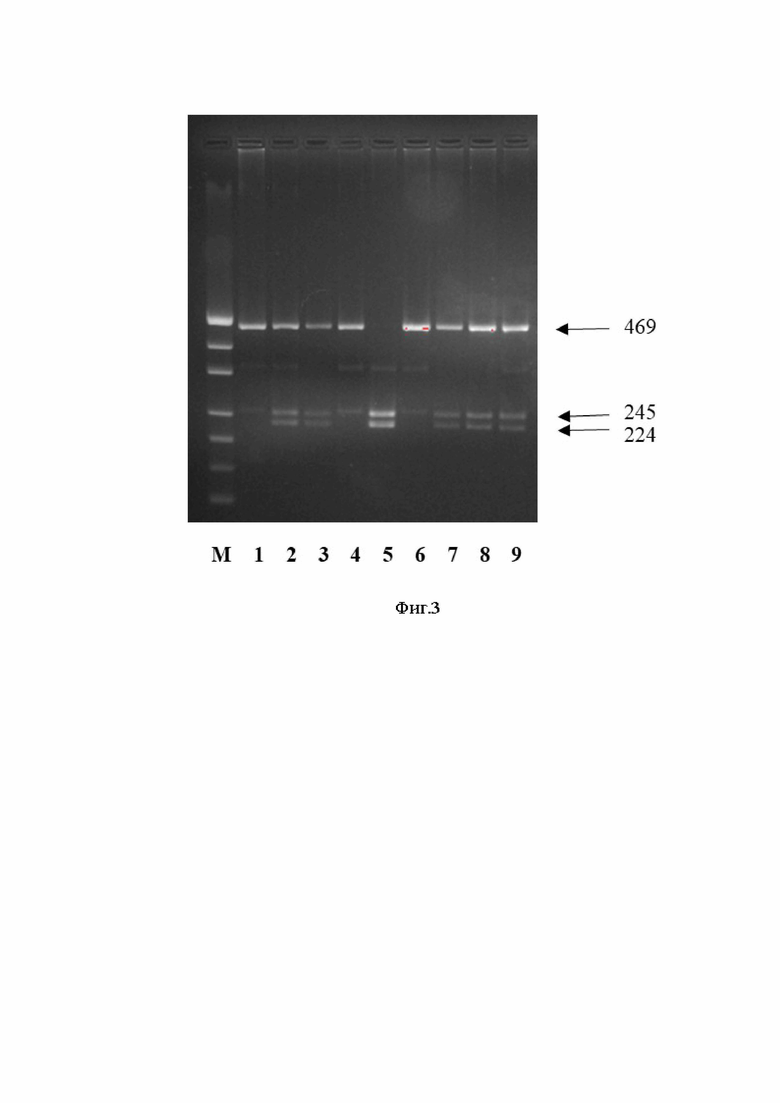
Заявленное техническое решение иллюстрируется чертежом, где на фигуре 1 показана родословная пациента с нейрональным цероидным липофусцинозом (НЦЛ) 6 типа (для примера); на фигуре 2 - МРТ головного мозга пробанда в возрасте 6 лет (для примера), где A - сагиттальное T1-взвешенное изображение, B - коронарное Т2-взвешенное изображение, C - осевое T2-взвешенное изображение FLAIR; на фигуре 3 - диагностика нейронального цероидного липофусциноза 6 типа в якутской популяции (c.396dupT (p.Val133CysfsTer18) после ПДРФ – анализа в 3% агарозном геле (для примера), где М – маркер pUC19/Msp I; 1- больной, гомозигота по мутантному аллелю, имеет аллель 469 п.н.; 2- мать, гетерозигота по мутантному аллелю, имеет аллель 469, 224 и 245 п.н.; 3- отец, гетерозигота по мутантному аллелю, имеет аллель 469, 224 и 245 п.н.; 4- больной сибс, гомозигота по мутантному аллелю, имеет аллель 469 п.н.; 5- здоровый, имеет аллели 224 и 245 п.н.; 6- больной, гомозигота по мутантному аллелю, имеет аллель 469 п.н.; 7-9- родственники больного, гетерозиготы по мутантному аллелю, имеют аллели 469, 224 и 245 п.н.
Заявленный способ диагностики НЦЛ 6 типа включает следующие этапы:
- у больного производят забор крови из локтевой вены в пробирку, содержащую ЭДТА в количестве 7-10 мл;
- выделение ДНК из лейкоцитов периферической крови выполняют стандартным методом с помощью фенол-хлороформного метода;
- для диагностики НЦЛ 6 типа (быстрой идентификации мутации c.396dupT (p.Val133CysfsTer18) в 4 экзоне гена CLN6, характерной для якутской популяции), амплификацию необходимых фрагментов ДНК проводят методом ПЦР на программируемом термоциклере S1000 Touch (BioRad) в объеме 20 мкл реакционной смеси следующего состава: 1 мкл геномной ДНК, по 1,0 мкл праймеров F и R, 2 мкл 10х ПЦР буфер + MgCl2 , 2 мкл dNTP (1.5 мM), 2,0 мкл MgCl2 (25 mM), 10,8 мкл бетаина, 0,2 мкл Taq-полимеразы (5 ед/мкл). ПЦР проводится при следующих условиях: первоначальная денатурация - 95° - 3 минуты, 34 цикла амплификации: 95° - 30 секунд, 66° - 30 секунд, 72° - 1 минуту. Заключительная элонгация - 72° - 5 минут.
- результаты ПЦР оценивают в 1% агарозном геле;
- установление гетерозиготного носительства мутантного гена у родственников пробанда проводят методом анализа полиморфизма длин рестрикционных фрагментов с использованием эндонуклеазы PflFI (New England Biolabs Inc.). Реакция проводят в 20,0 мкл объеме реакционной смеси следующего состава: рестриктаза PflFI 1,0 мкл, Буфер CutSmart или Tango 10X 2,0 мкл, деионизированная вода 7,0 мкл, амплификат 10 мкл. Далее устанавливают в термостат при температуре 37° на ночь;
- результаты амплификации после ПДРФ-анализа оценивают методом электрофореза в 3% агарозном геле. В норме должны образовываться фрагменты длиной 224 и 245 п.н., в случае носительства в гомозиготном состоянии (больной) – вместо фрагментов 224 и 245 образуются фрагменты длиной 469 п.н. В случае гетерозиготного носительства мутации (здоровый носитель) длины фрагментов составят 469, 224 и 245 п.н.
Разработанный способ диагностики нейронального цероидного липофусциноза 6 типа апробирован на 19 больных, их 42 родственниках и 30 здоровых индивидах якутской национальности. Все больные были гомозиготами по данной мутации и имели аллели длиной 469 п.н., их родственник были гетерозиготными носителями мутации и имели длины фрагментов 469, 224 и 245 п.н., остальные из группы родственников и здоровых имели фрагменты длиной 224 и 245 п.н.
Клинический пример заболевания. Пробанд (III-4): 8-летняя девочка, рожденная от неродственного брака. Родители пробанда якутской этнической группы, здоровые. Родословная семьи пробанда (III-4) показана на фигуре 1. Стрелка указывает на пробанда (III-4); родители здоровые, являются гетерозиготными носителями мутации (II-2, II-3); сибс (III-5) тоже больна НЦЛ 6 типа.
Пробанд родилась от четвертой беременности, от вторых естественных родов, с весом при рождении 4120 г., ростом 56 см. Оценка по шкале Апгар 8/9 баллов. До трех лет ребенок развивался без особых отклонений, но отмечалась некоторая задержка речевого развития. В возрасте 3 лет 9 месяцев ребенок стал часто падать, особенно на ягодичную область, появилась слабость в ногах. В 4 года больная не могла приседать из-за слабости в ногах, перестала ходить, начала передвигаться на четвереньках, возникли нарушения мелкой моторики, трудности с засыпанием, появились частые миоклонические приступы. Магнитно-резонансная томография (МРТ) головного мозга выявила атрофию полушарий и червя мозжечка. Видео ЭЭГ-мониторинг регистрировал эпиактивность в виде комплексов пик-волна по морфологии доброкачественные эпилептиформные паттерны детства по правому центрально-височному отделу. Была назначена вальпроевая кислота. Молекулярно-генетические исследования на частые атаксии не выявили патологии. На основании неврологических симптомов и данных исследований головного мозга был установлен клинический диагноз лейкодистрофия. В возрасте 6 лет пациентка госпитализирована в отделение психоневрологии с жалобами на нарушение походки, частые падения, задержку психоречевого развития. На фигуре 2 представлена МРТ головного мозга, которая выявила атрофию коры, перивентрикулярную лейкопатию обоих полушарий головного мозга и атрофию мозжечка: (A) Сагиттальное T1-взвешенное изображение: видны атрофия мозжечка, атрофия коры головного мозга; (B) Коронарное Т2-взвешенное изображение: наблюдается гиперинтенсивность перивентрикулярного белого вещества, атрофия мозжечка; (C) Осевое T2-взвешенное изображение FLAIR: корковая атрофия, наблюдается повышенная интенсивность МР-сигнала в перивентрикулярном белом веществе.
По ЭЭГ на фоне замедленной корковой активности с частотной характеристикой 6 Гц регистрировалась пароксизмальная медленно волновая активность с включением множественных нечетких эпилептиформных комплексов по типу острая-медленная волна и дифазных острых волн без четкой локализации. Исследование с помощью тандемной масс-спектрометрии не выявило данных за наследственные аминоацидопатии, органические ацидурии и дефекты митохондриального бетта-окисления. Больная была выписана с диагнозом нейродегенеративное заболевание неясного генеза. В возрасте 8 лет окулистом была выявлена субатрофия зрительного нерва обоих глаз, в неврологическом статусе при осмотре продуктивному контакту была недоступна из-за выраженных когнитивных нарушений, больная самостоятельно не могла сидеть и ходить, наблюдались миоклонические приступы.
У младшей сестры пробанда (III-5), девочки 5 лет, наблюдались сходные симптомы и прогрессирующее течение, которые дебютировали в возрасте 4 лет. МРТ головного мозга выявила изменение интенсивности МР-сигнала в перивентрикулярной области белого вещества головного мозга с обеих сторон. Кроме того, у пациентки на первом году жизни была диагностирована гепатобластома в правой доле печени с метастазами в легкие, по поводу которой она получала комплексное лечение в Российском онкологическом научном центре имени Н.Н. Блохина (Москва). На момент данного исследования у нее наблюдается ремиссия по раку печени; однако неврологические симптомы продолжают прогрессировать, больная не может самостоятельно сидеть и ходить. Офтальмоскопия выявила признаки ангиопатии сетчатки.
Таким образом, с помощью массового параллельного секвенирования (МПС) была выявлена молекулярно-генетическая причина нейронального цероидного липофусциноза 6 типа в якутской семье - гомозиготная мутация c.396dupT (p.Val133CysfsTer18) в гене CLN6, приводящая к сдвигу рамки считывания и явившаяся причиной заболевания.
Подобная мутация также была обнаружена в другой семье, вероятно якутской национальности (см. Neuronal ceroid lipofuscinosis in the Russian population: Two novel mutations and the prevalence of heterozygous carriers / A.A. Kozina, E.G. Okuneva, N.V. Baryshnikova et al. // Mol Genet and Genomic Med. - 2020. – Vol. 8, N 7. - P.1-11).
Преимущество изобретения заключается в разработке быстрого и точного способа диагностики нейронального цероидного липофусциноза 6 типа у больных и их родственников в отягощенных семьях, пренатальной ДНК-диагностики, и проспективного медико-генетического консультирования для вступающих в брак.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ диагностики наследственной моторно-сенсорной нейропатии 6С типа | 2023 |
|
RU2826719C1 |
| Способ диагностики мукополисахаридоз-плюс синдрома | 2022 |
|
RU2798695C1 |
| СПОСОБ ДИАГНОСТИКИ 3-М СИНДРОМА В ЯКУТСКОЙ ПОПУЛЯЦИИ | 2006 |
|
RU2315310C1 |
| Способ диагностики варианта SOPH c.5741G>A в гене NBAS | 2024 |
|
RU2819985C1 |
| СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ | 2010 |
|
RU2448163C2 |
| Способ диагностики мутации 167delT (rs80338942) гена GJB2 | 2020 |
|
RU2739943C1 |
| Способ диагностики мутации c.-23+1G>A (rs80338940) гена GJB2 | 2020 |
|
RU2746055C1 |
| Способ идентификации полиморфизмов Cys1079Gly и Cys1079Phe медь-транспортной АТФ-азы Вильсона | 2020 |
|
RU2756112C1 |
| Способ ДНК-диагностики аутосомно-рецессивной глухоты-103 | 2019 |
|
RU2727684C1 |
| СПОСОБ АНАЛИЗА СОМАТИЧЕСКИХ МУТАЦИЙ В ГЕНЕ PI3K С ИСПОЛЬЗОВАНИЕМ LNA-БЛОКИРУЮЩЕЙ МУЛЬТИПЛЕКСНОЙ ПЦР И ПОСЛЕДУЮЩЕЙ ГИБРИДИЗАЦИЕЙ С ОЛИГОНУКЛЕОТИДНЫМ БИОЛОГИЧЕСКИМ МИКРОЧИПОМ (БИОЧИПОМ) | 2013 |
|
RU2549682C1 |
Изобретение относится к медицине, а именно к лабораторной диагностике, и может быть использовано для диагностики носительства мутантного гена CLN6 с мутацией c.396dupT (p.Val133CysfsTer18), приводящего к нейрональному цероидному липофусцинозу 6 типа с аутосомно-рецессивным типом наследования в якутской популяции. Из лейкоцитов периферической крови выделяют ДНК. Проводят молекулярно-генетический анализ посредством полимеразной цепной реакции с применением праймеров F- GACAGTGCCCTCACCTAGC; R- CTGGCCTGCTAAAGGGACC и анализ полиморфизма длин рестрикционных фрагментов. При длине фрагментов 469 п.н. диагностируют носительство мутантного гена в гомозиготном состоянии. При длине фрагментов 469, 224 и 245 п.н. диагностируют носительство мутантного гена в гетерозиготном состоянии. При длине фрагментов 224 и 245 п.н. диагностируют нормальный ген. Способ обеспечивает быструю и точную диагностику нейронального цероидного липофусциноза 6 типа у больных и их родственников в отягощенных семьях за счет определения наличия или отсутствия гетерозиготной или гомозиготной мутации c.396dupT (p.Val133CysfsTer18) в 4 экзоне гена CLN6. 3 ил., 1 пр.
Способ диагностики носительства мутантного гена CLN6 с мутацией c.396dupT (p.Val133CysfsTer18), приводящего к нейрональному цероидному липофусцинозу 6 типа с аутосомно-рецессивным типом наследования в якутской популяции, путем молекулярно-генетического анализа посредством полимеразной цепной реакции, включающий амплификацию геномной ДНК с применением праймеров F- GACAGTGCCCTCACCTAGC; R- CTGGCCTGCTAAAGGGACC, выделенной из лейкоцитов периферической крови, и анализ полиморфизма длин рестрикционных фрагментов, где при длине фрагментов 469 п.н. диагностируют носительство мутантного гена в гомозиготном состоянии, при длине фрагментов 469, 224 и 245 п.н. - носительство мутантного гена в гетерозиготном состоянии и при длине фрагментов 224 и 245 п.н. диагностируют нормальный ген.
| WO 2012154944 A2, 15.11.2012 | |||
| ГУРЬЕВА П.И | |||
| и др | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| NGS в медицинской генетике | |||
| Вторая международная научно-практическая конференция | |||
| Тезисы конференции, г | |||
| Суздаль | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| Видоизменение прибора с двумя приемами для рассматривания проекционные увеличенных и удаленных от зрителя стереограмм | 1919 |
|
SU28A1 |

