Изобретение относится к области медицины, а именно к молекулярно-генетическим методам диагностики наследственной моторно-сенсорной нейропатии 6С типа (НМСН6С).
Наследственные моторно-сенсорные нейропатии (НМСН), или болезнь Шарко-Мари-Тута (ШМТ) - большая генетически гетерогенная группа заболеваний, характеризующихся симптомами вялого пареза и чувствительных нарушений в дистальных отделах конечностей. Симптомы заболевания, как правило, проявляются на первом или втором десятилетии жизни.
Заболевание может наследоваться по аутосомно-доминантному, аутосомно-рецессивному или рецессивному сцепленному с X-хромосомой типу. Преимущественно встречается аутосомно-доминантный тип наследования, 10-20% всех случаев ШМТ приходится на форму заболевания, сцепленную с X-хромосомой [1].
Распространенность ШМТ в мире составляет в среднем 35 случаев на 100 000 человек. В России, по разным оценкам, частота заболевания в разных регионах варьирует от 7 до 13,3 случаев на 100 000 населения. В целом на долю ШМТ приходится около 80% всех наследственных невропатий в России [1, 2]. Различия данных об эпидемиологии ШМТ обусловлены ее высокой гетерогенностью: клиническая форма болезни может быть обусловлена мутациями в разных локусах или множественными аллелями в одном локусе [2].
Заболевание связано с мутациями, вызывающими дефекты белков в нейронах, что приводит к нарушению передачи нервных импульсов [1]. Они составляют самую многочисленную группу наследственных нервно-мышечных заболеваний, распространенность которых в европейских странах колеблется от 1:10 000 до 1:1215 человек [3].
Давиденков С.Н. в 1932 году определил амиотрофию Шарко-Мари как сборную группу нескольких заведомо неоднородных, но очень сходных наследственно обусловленных форм [4]. В 1968 году на основании клинических, электронейрофизиологических и патоморфологических исследований перонеальные мышечные атрофии были разделены на 2 типа: демиелинизирующий (I) и аксональный (II) [5, 6, 7, 8, 9, 10]. В дальнейшем были описаны сравнительно редкие формы наследственных нейропатий, сочетающихся с поражением других органов и систем. В 1968 г. Dyck и Lambert представили исправленную и дополненную классификацию типов НМСН [5]. Кроме того, предложена классификационная структура ШМТ, основанная на данных электромиографии, типе наследования и пораженного гена (см. Inherited Peripheral Neuropathies [Electronic resource] // Inherited Peripheral Neuropathies Mutation Database. - URL: http://www.molgen.vib-ua.be/CMTMutations/Home/IPN.cfm).
К настоящему времени идентифицировано более 100 генов, обуславливающих развитие наследственных периферических нейропатий. До сих пор описываются новые клинические варианты, а также новые гены, причастные к формированию этой патологии [11]. Некоторые генетические варианты аутосомно-рецессивных НМСН встречаются с наибольшей частотой у пациентов отдельных этнических групп [12].
В Республике Саха (Якутия) болезнь Шарко-Мари-Тута является одной из частых наследственных заболеваний по данным эпидемиологических работ, проведенных разными авторами [13, 14, 15]. Распространенность ШМТ в целом в РС (Я) составляет 11,8 на 100000 населения, а среди якутской этнической группы 18,4 на 100000 населения [15], что выше, чем в среднем по России.
Медико-генетическое консультирование при ШМТ имеет сложности в виду выраженной генетической гетерогенности заболевания при достаточно схожем фенотипе, в связи с чем различные генетические типы ШМТ дифференцируются только на молекулярном уровне.
Клинико-генетическое изучение НМСН активно проводится во всем мире: в Европе [16, 17]; в азиатском регионе [18, 19, 20, 21, 22]; в Северной и Южной Америке [23, 24].
В последнее время такие работы осуществляются и в нашей стране [25, 26, 27, 28].
Для эффективной диагностики в настоящее время применяется генетическое тестирование. В 92% случаев ШМТ у пациентов обнаруживаются мутации четырех генов: PMP22, GJB1, MPZ и MFN2 [29]. Мутации других генов встречаются значительно реже.
Наследственная моторная и сенсорная нейропатия типа 6C с атрофией зрительного нерва (OMIM 618511) вызвана гомозиготной мутацией в гене PDXK (179020) на хромосоме 21q22.
Наследственная моторная и сенсорная нейропатия типа 6C с атрофией зрительного нерва (НМСН6С) - аутосомно-рецессивная аксональная сенсомоторная периферическая нейропатия, характеризующаяся прогрессирующей дистальной мышечной слабостью и атрофией, преимущественно поражающей нижние конечности. Начало нейропатии приходится на первое десятилетие, проявляется затруднениями при ходьбе и беге и сопровождается аналогичным поражением верхних конечностей и рук. Расстройство связано с дистальными сенсорными нарушениями, особенно чувства положения и вибрации, а также с арефлексией. У людей обычно наблюдаются полая стопа, молоткообразные пальцы ног и атрофия внутренних мышц рук. Кроме того, в зрелом возрасте наблюдается прогрессирующая атрофия зрительного нерва и нарушение зрения. Лечение добавками пиридоксаль-5-фосфата (витамина B6) может привести к облегчению симптомов и замедлению прогрессирования заболевания [30].
Челбан и др. в 2019 году сообщили о 4 пациентах из 2 неродственных семей со схожей периферической нейропатией, связанной с атрофией зрительного нерва. Первая семья кипрского происхождения, состоящая из брата 80 лет и сестры 75 лет. Вторая семья шотландско-итальянского происхождения, состоящая из двух сестер в возрасте 31 и 29 лет. У этих 2 неродственных семей с НМСН6С выявили гомозиготные миссенс-мутации в гене PDXK, кодирующий пиридоксаль (PL) киназу. Исследования клеток, полученных от пациентов обеих семей, показали снижение активности пиридоксалькиназы по сравнению с контрольной группой, а у пациентов наблюдалось снижение уровня пиридоксальфосфата в плазме по сравнению с контрольной группой. Результаты соответствовали эффекту потери функции.
Также Натали Келлер и др. в 2020 году сообщили о двух пострадавших людях из кровнородственной семьи (брат 17 лет, сестра 14 лет) с сенсомоторной аксональной невропатией в детстве и первыми признаками атрофии зрительного нерва с новой двуаллельной мутацией PDXK [31].
Таким образом, в мире 6 пациентов из 3 неродственных семей, которые страдают наследственной моторно-сенсорной нейропатией 6С типа.
В отечественной литературе еще не описано пациентов с НМСН 6С типа.
При анализе уровня техники были выявлены изобретения с применением методики ПЦР-ПДРФ для диагностики других наследственных и мультифакториальных заболеваний, например, см. № RU №2315310 (кл. G01N 33/50, C12Q 1/68, опубл. 20.01.2008), RU №2742956 (кл. C12Q 1/68, G01N 33/48, C12N 15/00, опубл. 12.02.2021), RU №2727684 (кл. C12Q 1/68, опубл. 22.07.2020), RU №2688180 (кл. G01N 33/50, C12Q 1/68, опубл. 21.05.2019), RU №2648464 (кл. G01N 33/50, C12Q 1/68, опубл. 26.03.2018), RU №2671156 (кл. C12N 15/00, G01N 33/00, опубл. 29.10.2018), RU №2697398 (кл. G01N 33/50, C12Q 1/6876, опубл. 14.08.2019), RU №2723090 (кл. C12Q 1/68, опубл. 08.06.2020), RU №2723585 (кл. C12Q 1/68, опубл. 16.06.2020).
При этом прямых аналогов к способу диагностики НМСН 6С типа методом ПЦР-ПДРФ из уровня техники не выявлено.
Задачей, на решение которой направлено заявленное изобретение, является создание способа диагностики наследственной моторно-сенсорной нейропатии 6С типа.
Технический результат, получаемый при решении поставленной задачи, выражается в повышении достоверности диагностики на наличие или отсутствие гетерозиготной/гомозиготной мутации c.659G>A (p.Arg220Gln) в 21 хромосоме гена PDXK, определении генотипа человека в биологическом материале (цельная кровь), а также повышении доступности подобных исследований, поскольку заявленное решение может быть осуществлено на стандартном известном оборудовании.
Для решения поставленной задачи предложен способ диагностики носительства мутантного гена PDXK с мутацией c.659G>A (p.Arg220Gln), приводящего к наследственной моторно-сенсорной нейропатии 6С типа с аутосомно-рецессивным типом наследования в якутской популяции, путем молекулярно-генетического анализа посредством полимеразной цепной реакции, включающий амплификацию геномной ДНК с применением праймеров F- CTGAGCAAGGCCACCTTGTG (SEQ ID NO:1); R- CAACTCCTGTCATGGCCTGG (SEQ ID NO:2), выделенной из лейкоцитов периферической крови, и анализ полиморфизма длин рестрикционных фрагментов, где при длине фрагментов 434 п.н. диагностируют носительство мутантного гена в гомозиготном состоянии (больной), при длине фрагментов 434, 265 и 168 п.н. - носительство мутантного гена в гетерозиготном состоянии (здоровый носитель мутации) и при длине фрагментов 265 и 168 п.н. диагностируют нормальный ген (здоровый).
Сопоставительный анализ признаков заявленного решения с признаками аналогов свидетельствует о соответствии заявленного решения критерию «новизна».
Совокупность признаков изобретения обеспечивает решение заявленной технической задачи, а именно, получение молекулярно-генетического способа диагностики наследственной моторно-сенсорной нейропатии 6С типа.
Заявленный способ диагностики наследственной моторно-сенсорной нейропатии 6С типа в якутской популяции позволяет выставить точный диагноз, проводить пренатальную ДНК-диагностику и проспективное медико-генетическое консультирование для вступающих в брак. Техническое решение простое в выполнении, точное и доступное для выполнения в условиях молекулярно-генетической лаборатории.
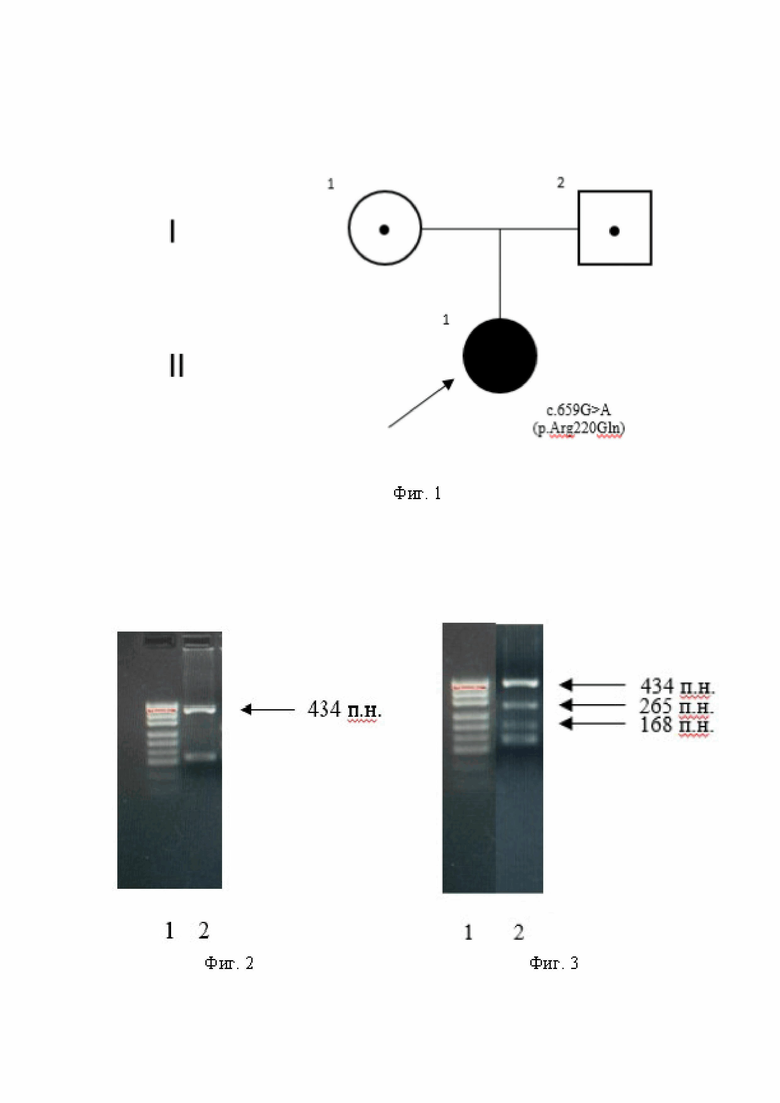
Заявленное техническое решение иллюстрируется чертежом, где на фиг 1 показана родословная пациента с наследственной моторно-сенсорной нейропатией 6С типа (для примера); на фиг. 2-4 - примеры диагностики носительства мутантного гена PDXK с мутацией c.659G>A (p.Arg220Gln).
Заявленный способ диагностики НМСН 6С типа включает следующие этапы:
- у больного производят забор крови из локтевой вены в пробирку, содержащую ЭДТА в количестве 7-10 мл;
- выделение ДНК из лейкоцитов периферической крови выполняют стандартным методом с помощью фенол-хлороформного метода;
- для диагностики НМСН 6С типа (быстрой идентификации мутации c.659G>A (p.Arg220Gln) в гене PDXK, характерной для якутской популяции), амплификацию необходимых фрагментов ДНК проводят методом ПЦР на программируемом термоциклере S1000 Touch (BioRad) в объеме 15 мкл реакционной смеси следующего состава: 1 мкл геномной ДНК, по 0,5 мкл праймеров F и R, 2 мкл 10× ПЦР буфер + MgCl2 , 1,5 мкл dNTP (1.5 мМ), 6,5 мкл бетаина, 0,4 мкл Taq-полимеразы (5 ед/мкл). ПЦР проводится при следующих условиях: первоначальная денатурация - 95° - 3 минуты, 34 цикла амплификации: 95° - 30 секунд, 62,5° - 30 секунд, 72° - 1 минуту. Заключительная элонгация - 72° - 5 минут;
- результаты ПЦР оценивают в 2,5 % агарозном геле;
- установление гетерозиготного носительства мутантного гена у родственников пробанда проводят методом анализа полиморфизма длин рестрикционных фрагментов с использованием эндонуклеазы Bsp13I. Реакцию проводят в 15,0 мкл объеме реакционной смеси следующего состава: рестриктаза Bsp13I 0,5 мкл, Буфер 10X 1,5 мкл, деионизированная вода 3,0 мкл, амплификат 10 мкл. Далее устанавливают в термостат при температуре 37° на ночь;
- результаты амплификации после ПДРФ-анализа оценивают методом электрофореза в 3% агарозном геле. В норме должны образовываться фрагменты длиной 265 и 168 п.н., в случае носительства в гомозиготном состоянии (больной) - вместо фрагментов 265 и 168 образуются фрагменты длиной 434 п.н. В случае гетерозиготного носительства мутации (здоровый носитель) длины фрагментов составят 434, 265 и 168 п.н.
Разработанный способ диагностики наследственной моторно-сенсорной нейропатии 6C типа апробирован на 44 больных и их 80 родственниках. Все больные были гомозиготами по данной мутации и имели аллели длиной 434 п.н., их родственники (родители) были гетерозиготными носителями мутации и имели длины фрагментов 434, 265 и 168 п.н., остальные из группы родственников имели фрагменты длиной 265 и 168 п.н. (норма).
Клинический пример заболевания. Пробанд (II-1): 14-летняя девочка, рожденная от неродственного брака. Родители пробанда якутской этнической группы, здоровые. Родословная семьи пробанда (II-1) показана на фиг. 1. Стрелка указывает на пробанда (II-1); родители здоровые, являются гетерозиготными носителями мутации (I-1, I-2).
Пробанд родилась от второй беременности, от первых оперативных родов, с весом при рождении 3980 г, ростом 53 см. Оценка по шкале Апгар 8/9 баллов. До четырех лет ребенок развивался без особых отклонений. С 4-х лет появились боли в ногах, с 7 лет - нарушение походки, деформация стоп с постепенным прогрессированием. Находилась на амбулаторном обследовании и лечении у невролога и ортопеда с клиническим диагнозом: Моторно-сенсорная нейропатия. Нижний парапарез. В динамике беспокоит слабость в нижних конечностях, нарушение походки, не может бегать. Отмечается мышечная гипотония, голени по типу ног «аиста», стопы деформированы, укорочены, эквино-варусные. Походка по типу степпажа, на носках и пятках не ходит. По данным ЭНМГ: признаки полного поражения сенсорных волокон, умеренно-значительно выраженного поражения моторных волокон периферических нервов верхних и нижних конечностей первично-демиелинизирующего характера (СПИ 34-41 м/с) с вторичными аксональными нарушениями, умеренно-выраженными в верхних конечностях, значительно выраженными в нижних конечностях.
С помощью ПЦР-ПДРФ была выявлена молекулярно-генетическая причина заболевания, обнаружен патогенный вариант c.659G>A в гене PDXK в гомозиготном состоянии, которая вызывает заболевание - наследственная моторно-сенсорная нейропатия 6С типа.
Таким образом, заявленное техническое решение направлено на идентифицирование следующих вариантов состояний:
- носительство мутации c.659G>A (p.Arg220Gln) в гомозиготном состоянии в гене PDXK, при этом будут диагностированы фрагменты длиной 434 п.н.;
- носительство мутации c.659G>A (p.Arg220Gln) в гетерозиготном состоянии в гене PDXK, при этом будут диагностированы фрагменты длиной 434, 265 и 168 п.н.;
- отсутствие носительства мутации c.659G>A (p.Arg220Gln) в гене PDXK, при этом будут диагностированы фрагменты длиной 265 и 168 п.н. (см. фиг. 4).
Примеры диагностики носительства мутантного гена PDXK с мутацией c.659G>A (p.Arg220Gln) (см. фиг. 2-4).
На фиг. 2 приведен пример диагностики носительства мутации c.659G>A (p.Arg220Gln) в гомозиготном состоянии в гене PDXK после ПДРФ-анализа в 3% агарозном геле, при этом диагностируется фрагмент длиной 434 п.н., где 1 - маркер pUC19/Msp I; 2 - носительство мутации c.659G>A (p.Arg220Gln) в гомозиготном состоянии в гене PDXK.
На фиг. 3 приведен пример диагностики носительства мутации c.659G>A (p.Arg220Gln) в гетерозиготном состоянии в гене PDXK после ПДРФ - анализа в 3% агарозном геле, при этом диагностируются фрагменты длиной 434, 265 и 168 п.н., где 1 - маркер pUC19/Msp I; 2 - носительство мутации c.659G>A (p.Arg220Gln) в гетерозиготном состоянии в гене PDXK.
На фиг. 4 приведен пример отсутствия носительства мутации c.659G>A (p.Arg220Gln), при этом диагностируют фрагменты длиной 265 и 168 п.н., где 1 - маркер pUC19/Msp I; 2 - отсутствует носительство мутации c.659G>A (p.Arg220Gln) в гене PDXK (дикий тип, норма). Диагностика выполнена после ПДРФ - анализа в 3% агарозном геле.
Преимущество изобретения заключается в разработке быстрого и точного способа диагностики наследственной моторно-сенсорной нейропатии 6С типа у больных и их родственников в отягощенных семьях, пренатальной ДНК-диагностики, и проспективного медико-генетического консультирования для вступающих в брак.
Список литературы:
1. Галым А. Случай наследственной нейропатии Шарко-Мари-Тута в педиатрии // Журнал «Нейрохирургия и неврология Казахстана». 2017. №2 (47).
2. Гончарова С. И., Шнайдер Наталья Алексеевна Наследственная невропатия Шарко-Мари-Тута: возможности нефармакологического лечения // Физиотерапия, бальнеология и реабилитация. 2013. №6.
3. Barreto L.C., Oliveira F.S., Nunes P.S., de França Costa I.M., Garcez C.A., Goes G.M., Neves E.L., de Souza Siqueira Quintans J., de Souza Araújo A.A. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology. 2016; 46 (3): 157-65. doi: 10.1159/000443706. Epub 2016 Feb 6. PMID: 26849231.
4. Давиденков С.Н. Проблема полиморфизма наследственных болезней нервной системы: клинико-генетическое исследование / С.Н. Давиденков. - Ленинград: Издательство Всесоюзного института экспериментальной медицины, 1934 - 132 с.
5. Dyck P.J., Lambert E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch Neurol. 1968 Jun; 18 (6): 619-25. doi: 10.1001/archneur.1968.00470360041003. PMID: 5652992.
6. Harding A.E., Thomas P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980a; 103: 259-280.
7. Harding A.E., Thomas P.K. Genetic aspects of hereditary motor and sensory neuropathy (types I and II) J Med Genet. 1980b; 17: 329-336.
8. Dyck, P.J., Chance, P., Lebo, R., and Carney, J.A. (1993). Hereditary motor and sensory neuropathies. In Peripheral Neuropathy, P.J. Dyck, P.K. Thomas, et al., eds. (Philadelphia, Pennsylvania: W. B. Saunders Company), pp. 1094-1136.
9. Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК диагностика и медико-генетическое консультирование в неврологии // М. Медицинское информационное агентство. 2002. С.591.
10. Неврология. Национальное руководство. Краткое издание / под ред. Е.И. Гусева, А.Н. Коновалова, А.Б. Гехт. - М.: ГЭОТАР-Медиа, 2014. - 688 с. - ISBN 978-5-9704-2890-0.
11. Щагина О.А., Рыжкова О.П., Чухрова А.Л., Миловидова Т.Б., Гундорова П., Миронович О.Л., Орлова А.А., Орлова М.Д., Поляков А.В. Применение экзомного секвенирования для диагностики наследственных моторно-сенсорных нейропатий. Нервно-мышечные болезни. 2020; 10 (4): 12-26. https://doi.org/10.17650/2222-8721-2020-10-4-12-26
12. Муртазина А.Ф., Щагина О.А., Миловидова Т.Б., Дадали Е.Л., Руденская Г.Е., Курбатов С.А., Федотова Т.В., Никитин С.С., Спарбер П.А., Орлова М.Д., Поляков А.В. Клинико-генетические характеристики болезни Шарко-Мари-Тута типа 4D (типа Lom) в России. Нервно-мышечные болезни. 2020; 10 (2): 39-45. https://doi.org/10.17650/2222-8721-2020-10-2-39-45
13. Ноговицына А.Н. Отягощенность населения Республики Саха (Якутия) наследственной патологией и анализ работы региональной медико-генетической консультации: Автореф. дис. канд. мед. наук. Томск, 2001.- 24 с.
14. Пузырев В.П. Наследственные болезни у якутов / В.П. Пузырев, Н.Р. Максимова // Генетика. - 2008. - Т. 44, № 10. - С. 1317-1324. - EDN JSJRFV.
15. Гурьева П.И. Эпидемиологическая и клинико-генетическая характеристика болезни Шарко-Мари-Тута в Республике Саха (Якутия): дис. канд.мед.наук: 14.01.11. - Красноярск, 2014. - 109 с.
16. Gabrikova D., Mistrik M., Bernasovska J., Bozikova A., Behulova R., Tothova I., Macekova S. Founder mutations in NDRG1 and HK1 genes are common causes of inherited neuropathies among Roma/Gypsies in Slovakia. J Appl Genet. 2013 Nov; 54 (4): 455-60. doi: 10.1007/s13353-013-0168-7. Epub 2013 Aug 31. PMID: 23996628.
17. Šafka Brožková D., Haberlová J., Mazanec R., Laštůvková J. and Seeman P. (2016), HSMNR belongs to the most frequent types of hereditary neuropathy in the Czech Republic and is twice more frequent than HMSNL. Clin Genet, 90: 161-165. https://doi.org/10.1111/cge.12745.
18. Yoshimura A., Yuan J., Hashiguchi A. et al. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. Journal of Neurology, Neurosurgery & Psychiatry Published Online First: 26 September 2018. doi: 10.1136/jnnp-2018-318839.
19. Li L.X., Liu G.L., Liu Z.J., Lu C., Wu Z.Y. Identification and functional characterization of two missense mutations in NDRG1 associated with Charcot-Marie-Tooth disease type 4D. Hum Mutat. 2017 Nov; 38 (11): 1569-1578. doi: 10.1002/humu.23309. Epub 2017 Aug 23. PMID: 28776325.
20. Grewal, Raji P & Peddareddygari, Leema Reddy & Oberoi, Kinsi. (2018). Clinical and Genetic Analysis of an Asian Indian Family with Charcot-Marie-Tooth Disease Type 4C. Case Reports in Neurology. 10. 38-44. 10.1159/000486589.
21. Zhi-Ying Wu, Cong-Xin Chen, Jia-Qi Li et al. Identification and functional characterization of novel GDAP1 variants in Chinese patients with Charcot-Marie-Tooth disease. Authorea. June 02, 2020. DOI: 10.22541/au.159110407.77885838.
22. Dong, Hai-Lin & Li, Jia-Qi & Liu, Gong-Lu & Yu, Hao & Wu, Zhi-Ying. (2021). Biallelic SORD pathogenic variants cause Chinese patients with distal hereditary motor neuropathy. npj Genomic Medicine. 6. 10.1038/s41525-020-00165-6.
23. Pasnoor, Mamatha & Nascimento, Osvaldo & Trivedi, Jaya & Wolfe, Gil & Nations, Sharon & Herbelin, Laura & de Freitas, Marcos & Quintanilha, Giseli & Khan, Saud & Dimachkie, Mazen & Barohn, Rj. (2013). North America and South America (NA-SA) neuropathy project. The International journal of neuroscience. 123. 10.3109/00207454.2013.782026.
24. Brewer M.H., Chaudhry R., Qi J., Kidambi A., Drew A.P., Menezes M.P., Ryan M.M., Farrar M.A., Mowat D., Subramanian G.M., Young H.K., Zuchner S., Reddel S.W., Nicholson G.A., Kennerson M.L. Whole Genome Sequencing Identifies a 78 kb Insertion from Chromosome 8 as the Cause of Charcot-Marie-Tooth Neuropathy CMTX3. PLoS Genet. 2016 Jul 20; 12 (7): e1006177. doi: 10.1371/journal.pgen.1006177. PMID: 27438001; PMCID: PMC4954712.
25. Ильинский В.В., Корнеева В.А., Шаталов П.А. Применение экзомного секвенирования для диагностики наследственных неврологических и психических заболеваний. Журнал неврологии и психиатрии им. С.С. Корсакова. 2015; 115 (1): 45-52. Ilinsky VV, Korneeva VA, Shatalov PA. Application of whole exome sequencing in the diagnosis of hereditary neurological diseases. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2015; 115 (1): 45-52. (In Russ.) https://doi.org/10.17116/jnevro20151151145-52.
26. Дадали Е.Л., Макаов А.Х.-М., Галкина В.А. и др. Наследственная моторно-сенсорная нейропатия, обусловленная мутацией в гене NEFL, в семье из Карачаево-Черкессии. Нервно-мышечные болезни 2016.
27. Сайфуллина Е.В., Магжанов Р.В., Хидиятова И.М., Хуснутдинова Э.К. Анализ первичной диагностики наследственных моторно-сенсорных нейропатий в Республике Башкортостан. Нервно-мышечные болезни. 2017; 7 (1): 37-42. https://doi.org/10.17650/2222-8721-2017-7-1-37-42.
28. Федорова В.С., Смочилин А.Г., Куляхтин А.И., Яковлев А.А., Пушкарев М.С., Гавриченко А.В., Гаврилова Е.А., Гапешин Р.А. Болезнь Шарко-Мари-Тутса: описание двух клинических случаев заболевания у членов одной семьи (отца и дочери) // Ученые записки СПбГМУ им. И.П. Павлова. 2020. №2.
29. Murphy S.M., Laura M., Fawcett K., Pandraud A., Liu Y.T., Davidson G.L., Rossor A.M., Polke J.M., Castleman V., Manji H., Lunn M.P., Bull K., Ramdharry G., Davis M., Blake J.C., Houlden H., Reilly M.M. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. 2012 Jul; 83 (7): 706-10. doi: 10.1136/jnnp-2012-302451. Epub 2012 May 10. PMID: 22577229; PMCID: PMC3736805.
30. Chelban, Viorica & Wilson, Matthew & Chardon, Jodi & Vandrovcova, Jana & Zanetti, Maria Natalia & Zamba-Papanicolaou, Eleni & Efthymiou, Stephanie & Pope, Simon & Conte, Maria & Abis, Giancarlo & Liu, Yo-Tsen & Tribollet, Eloise & Haridy, Nourelhoda & Botía, Juan & Ryten, Mina & Nicolaou, Paschalis & Minaidou, Anna & Christodoulou, Kyproula & Kernohan, Kristin & Shaikh, Mohd. Farooq. (2019). PDXK mutations cause polyneuropathy responsive to pyridoxal 5'-phosphate supplementation. Annals of Neurology. 86.
31. Keller N., Mendoza-Ferreira N., Maroofian R., Chelban V,. Khalil Y., Mills P.B., Boostani R., Torbati P.N., Karimiani E.G., Thiele H., Houlden H., Wirth B., Karakaya M. Hereditary polyneuropathy with optic atrophy due to PDXK variant leading to impaired Vitamin B6 metabolism. Neuromuscul Disord. 2020 Jul; 30 (7): 583-589. doi: 10.1016/j.nmd.2020.04.004. Epub 2020 Apr 29. PMID: 32522499.
--->
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE ST26SequenceListing PUBLIC "-//WIPO//DTD Sequence Listing
1.3//EN" "ST26SequenceListing_V1_3.dtd">
<ST26SequenceListing dtdVersion="V1_3" fileName="Способ диагностики
наследственной моторно-сенсорной нейропатии 6С типа.xml"
softwareName="WIPO Sequence" softwareVersion="2.3.0"
productionDate="2024-01-09">
<ApplicationIdentification>
<IPOfficeCode>RU</IPOfficeCode>
<ApplicationNumberText>2023132791</ApplicationNumberText>
<FilingDate>2023-12-12</FilingDate>
</ApplicationIdentification>
<EarliestPriorityApplicationIdentification>
<IPOfficeCode>RU</IPOfficeCode>
<ApplicationNumberText>2023132791</ApplicationNumberText>
<FilingDate>2023-12-12</FilingDate>
</EarliestPriorityApplicationIdentification>
<ApplicantName languageCode="ru">Федеральное государственное
автономное образовательное учреждение высшего образования
"Северо-Восточный федеральный университет имени М.К.
Аммосова</ApplicantName>
<ApplicantNameLatin>Federalnoe gosudarstvennoe avtonomnoe
obrazovatelnoe uchrezhdenie vysshego obrazovaniia
"Severo-Vostochnyi federalnyi universitet imeni M.K.
Ammosova"</ApplicantNameLatin>
<InventorName languageCode="ru">Анастасия Максимова </InventorName>
<InventorNameLatin>Anastasiia Maksimova</InventorNameLatin>
<InventionTitle languageCode="ru">Способ диагностики наследственной
моторно-сенсорной нейропатии 6С типа</InventionTitle>
<SequenceTotalQuantity>2</SequenceTotalQuantity>
<SequenceData sequenceIDNumber="1">
<INSDSeq>
<INSDSeq_length>20</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..20</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>genomic DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q2">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>Homo sapiens</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>ctgagcaaggccaccttgtg</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
<SequenceData sequenceIDNumber="2">
<INSDSeq>
<INSDSeq_length>20</INSDSeq_length>
<INSDSeq_moltype>DNA</INSDSeq_moltype>
<INSDSeq_division>PAT</INSDSeq_division>
<INSDSeq_feature-table>
<INSDFeature>
<INSDFeature_key>source</INSDFeature_key>
<INSDFeature_location>1..20</INSDFeature_location>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>mol_type</INSDQualifier_name>
<INSDQualifier_value>genomic DNA</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier id="q4">
<INSDQualifier_name>organism</INSDQualifier_name>
<INSDQualifier_value>Homo sapiens</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
</INSDSeq_feature-table>
<INSDSeq_sequence>caactcctgtcatggcctgg</INSDSeq_sequence>
</INSDSeq>
</SequenceData>
</ST26SequenceListing>
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ диагностики нейронального цероидного липофусциноза 6 типа | 2021 |
|
RU2784293C1 |
| СПОСОБ ДИАГНОСТИКИ 3-М СИНДРОМА В ЯКУТСКОЙ ПОПУЛЯЦИИ | 2006 |
|
RU2315310C1 |
| Способ диагностики варианта SOPH c.5741G>A в гене NBAS | 2024 |
|
RU2819985C1 |
| Способ одновременной диагностики наследственных заболеваний | 2015 |
|
RU2627115C2 |
| СПОСОБ ДЕТЕКЦИИ 17 МУТАЦИЙ ГЕНОВ GJB2 И GJB6 ПРИ НАСЛЕДСТВЕННОЙ НЕСИНДРОМАЛЬНОЙ ГЛУХОТЕ | 2010 |
|
RU2448163C2 |
| Способ диагностики мукополисахаридоз-плюс синдрома | 2022 |
|
RU2798695C1 |
| Способ ДНК-диагностики аутосомно-рецессивной глухоты-103 | 2019 |
|
RU2727684C1 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ РИСКА СУБАРАХНОИДАЛЬНОГО КРОВОИЗЛИЯНИЯ ВСЛЕДСТВИЕ РАЗРЫВА АНЕВРИЗМЫ СОСУДОВ ГОЛОВНОГО МОЗГА У ЛИЦ АЗИАТСКОЙ РАСЫ | 2015 |
|
RU2627643C2 |
| Способ дифференциальной и подтверждающей молекулярно-генетической диагностики нейросенсорной тугоухости в популяции чувашей | 2021 |
|
RU2768033C1 |
| Способ ДНК-диагностики врожденной формы катаракты (CTRCT18) | 2017 |
|
RU2648464C1 |

Изобретение относится к области молекулярной биологии. Описан способ диагностики носительства мутантного гена PDXK с мутацией c.659G>A (p.Arg220Gln), приводящего к наследственной моторно-сенсорной нейропатии 6С типа с аутосомно-рецессивным типом наследования в якутской популяции, путем молекулярно-генетического анализа посредством полимеразной цепной реакции. Способ включает амплификацию геномной ДНК с применением праймеров. Технический результат выражается в повышении достоверности диагностики на наличие или отсутствие гетерозиготной/гомозиготной мутации c.659G>A (p.Arg220Gln) в 21 хромосоме гена PDXK. 4 ил., 1 пр.
Способ диагностики носительства мутантного гена PDXK с мутацией c.659G>A (p.Arg220Gln), приводящего к наследственной моторно-сенсорной нейропатии 6С типа с аутосомно-рецессивным типом наследования в якутской популяции, путем молекулярно-генетического анализа посредством полимеразной цепной реакции, включающий амплификацию геномной ДНК с применением праймеров F- CTGAGCAAGGCCACCTTGTG (SEQ ID NO:1); R- CAACTCCTGTCATGGCCTGG (SEQ ID NO:2), выделенной из лейкоцитов периферической крови, и анализ полиморфизма длин рестрикционных фрагментов, где при длине фрагментов 434 п.н. диагностируют носительство мутантного гена в гомозиготном состоянии, при длине фрагментов 434, 265 и 168 п.н. - носительство мутантного гена в гетерозиготном состоянии и при длине фрагментов 265 и 168 п.н. диагностируют нормальный ген.
| KR 102072772 B1, 04.02.2020 | |||
| СПОСОБ ДИАГНОСТИКИ АУТОСОМАЛЬНО-ДОМИНАНТНОЙ ОПТИЧЕСКОЙ НЕЙРОПАТИИ | 2011 |
|
RU2491350C1 |
| Chelban V., Wilson M.P | |||
| et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Annals of Neurology | |||
| Станок для придания концам круглых радиаторных трубок шестигранного сечения | 1924 |
|
SU2019A1 |
| Синхронизирующее устройство для аппарата, служащего для передачи изображений на расстояние | 1920 |
|
SU225A1 |

