Изобретение относится к области технологии получения взрывчатых веществ (ВВ), компонентов порохов и смесевых твердых ракетных топлив (СТРТ).
Нитроизобутилглицеринтринитрат (НИБГТН) относится к классу эфиров нитроспиртов и обладает рядом преимуществ по сравнению с широко применяемым в производстве порохов и ВВ нитроглицерином (НГ). Он обладает более высокой плотностью (1,634 г/см3) и более высокой теплотой образования (ΔН=-54 ккал/моль), что позволяет создавать более мощные энергетические композиции порохов и СТРТ. Очень ценным свойством НИБГТН по сравнению с НГ, является низкая температура кристаллизации (-35°С) по сравнению с температурой -13°С для НГ, что позволяет создавать композиции, пригодные для эксплуатации в широком диапазоне температур. Кроме того, НИБГТН обладает существенно более низкой летучестью, что обеспечивает большую безопасность технологических процессов при изготовлении порохов и СТРТ.
Впервые НИБГТН получен Hofwimmer [Zeitschr. fur Schiess. u. Sprengst. 1912. т.7, 43 Ch. Zbl. 1912. 1265] нитрованием нитроизобутилглицерина (НИБГ) серно-азотной смесью, содержащей 60% серной кислоты и 40% азотной кислоты при массовом соотношении 1:6. Более подробно метод получения НИБГТН был исследован Stettbacher [Nitrocellulose, 1934, т.5, 181-184, 203-206]. Воспроизведение этих методик сотрудниками Пикатинского Арсенала в США [Enciclopedia of Explosives and Related tems, 1978, vol.8, p.112-113; Dover, NewJersy. Ed. B.Fedoroff] показало, что при промывке нитроэфира, отделенного от отработанной кислоты, образуются очень стойкие эмульсии. Использование чистой воды или 0,5÷2,0%-ных водных растворов карбонатов или бикарбонатов натрия или калия, применяемых в традиционных методах получения нитроэфиров, оказалось совершенно неприемлемо для НИБГТИ. Тщательное встряхивание кислого НИБГТН с водными растворами соды дает нерасслаивающиеся в течение 10-12 часов эмульсии. При этом даже после 3-5 - кратной обработки 1-2%-ным водным карбонатом или бикарбонатом натрия удалить кислоту из нитроэфирного слоя не удается. При рН водного слоя 10-12 рН нитроэфирного слоя остается на уровне 1,0÷1,5. Хранение такого кислого нитроэфира сопровождается довольно быстрым выделением окислов азота (1÷2 дня при комнатной температуре). Температура начала интенсивного разложения (ТНИР) такого кислого продукта 60÷80°С делает практически невозможным применение его для производства порохов или СТРТ. Использование растворов сульфита или бисульфита натрия, как рекомендовано в описании к патенту США №2112749 [W. de C.Crater. 1938. Chem. Abst, 1938, т.32, 3964], который выбран нами в качестве наиболее близкого аналога, не позволяет получить нейтральный НИБГТН. Одной из причин такого поведения, возможно, является гидролиз НИБГТН водой, остающейся в нитроэфире после сепарации основного количества промывной жидкости. Хорошо известно [К.К.Андреев, Г.Н.Беспалов «Теория взрывчатых вещества, Оборонгиз, 1963, стр.131-184], что остаточная кислотность резко снижает стабильность нитроэфиров.
Другим принципиальным недостатком известных методов получения НИБГТН является непостоянство такого важного показателя качества НИБГТН, как плотность. При исследовании методов получения НИБГТН было замечено, что при дозировке НИБГ в нитросмесь, содержащую 60% и более серной кислоты, происходит быстрая сепарация образующегося нитроэфира. Дозируемый нитроспирт, попадая в капли нитроэфира, остается длительное время нерастворившимся, что, по-видимому, связано с высокой вязкостью нитроэфира и высокой растворимостью нитроспирта в нитроэфире. Продукт, полученный в таких операциях, как правило, имеет плотность 1,61÷1,62 г/см3, что указывает на присутствие недонитрованного нитроспирта.
Задачей данного изобретения является разработка способа получения НИБГТН, обладающего плотностью не менее 1,63 г/см3 и высокой термической стабильностью с температурой начала интенсивного разложения на уровне не ниже, чем для штатного нитроглицерина (135÷140°С).
Технический результат достигается нитрованием нитроизобутилглицерина смесью азотной и серной кислот, при этом нитроизобутилглицерин растворяют в серно-азотной смеси, обогащенной азотной кислотой, образующийся раствор нитроизобутилглицерин тринитрат обрабатывают серной кислотой или олеумом, и затем стабилизируют в среде инертного растворителя, хлористого метилена или дихлорэтана, безводным нейтрализующим агентом - оксидом кальция или карбонатом кальция.
При содержании азотной кислоты 50% при массовом соотношении нитросмесь/нитроспирт выше 3,0÷1,0 быстро образуется гомогенный раствор нитроэфира в нитросмеси. При этом реакция нитрования практически полностью завершается. Для выделения
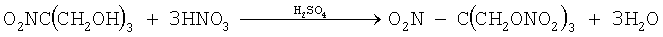
образовавшегося нитроэфира к полученному раствору добавляют серную кислоту или олеум в количестве 1,5÷3,0 частей на 1 часть нитроспирта. При этом содержание серной кислоты в нитромассе повышается до 70÷80%.
В таком растворе НИБГТН растворим очень слабо и быстро сепарируется от отработанной кислоты. Отработанная кислота после отстоя в течение 5÷10 минут сливается на лед. НИБГТН, выделившийся при разбавлении отработанной кислоты в количестве 3÷5% от общего количества, объединяют с основным количеством отсепарированного нитроэфира, промывают 2 раза холодной водой и 1 раз 1%-ным водным раствором соды. При этом слой нитроэфира остается кислым (рН 1,0÷1,5), хотя водный содовый раствор имеет сильную щелочную реакцию (рН 10,0÷11).
Для полного удаления примесей кислот и получения стабильного нейтрального нитроэфира кислый НИБГТН растворяют в 3-5 частях инертного растворителя и обрабатывают полученный раствор безводными, тонко измельченными, сухими нейтрализующими агентами, способными связывать не только кислоты (азотную и серную), но и остаточную воду, содержащуюся в кислом нитроэфире как в растворенном виде, так и в форме мельчайших капель, не сепарирующихся от нитроэфира при водной промывке.
В качестве нейтрализующих агентов можно использовать безводные оксиды кальция или магния, или безводные карбонаты кальция, натрия или калия.
Наилучшие результаты получены при использовании оксида кальция или карбоната кальция при массовом соотношении нейтрализующего агента к нитроэфиру 0,5÷1,0 части на 10 частей нитроэфира. После нейтрализации кислот и удаления примесей воды инертный растворитель удаляют испарением при пониженном давлении и температуре 40÷70°С. Получаемый нитроэфир был совершенно бесцветным, нейтральным (рН 6,5÷7,0) и имел ТНИР 135-140°С, не уступая по этому показателю штатному нитроглицерину.
Кроме оксидов кальция и магния, а также карбонатов кальция, натрия и калия, в качестве нейтрализующих агентов испытывались оксид и карбонат цинка, мочевина и диметилмочевина. Получить нейтральный стабильный НИБГТН в этих случаях не удавалось. Даже после 3-часовой обработки рН получаемого нитроэфира был в пределах 4,0÷5,5. Продукт имел светло-желтую окраску и ТНИР снижалась до 90÷110°С. Результаты приведены в таблице.
В качестве инертного растворителя используют хлористый метилен, дихлорэтан, хлороформ и другие хлорированные углеводороды. Связывание кислот происходит очень быстро, но для полного удаления воды и, соответственно, предотвращения последующего гидролиза требуется перемешивание суспензии в течение 1÷2 часов.
После отфильтровывания твердого нейтрализующего агента и образовавшихся солей растворитель удаляют испарением при пониженном давлении и температуре 40÷70°С, остающийся нитроэфир имеет плотность 1,634÷1,635 г/см3 и представляет собой бесцветную, нейтральную жидкость (рН 6,5÷7,0) и ТНИР 135÷140°С. ТНИР определялась стандартным методом дифференциально-термического анализа.
Следующие примеры иллюстрируют достижение поставленной цели.
Пример 1
Получение НИБГТН проводят по следующей общей методике:
К 30,5 мл нитросмеси, содержащей 60% азотной кислоты и 40% серной кислоты, приготовленной смешением 98,5%-ной азотной кислоты и 20%-ного олеума, дозируют 15,1 г НИБГ при температуре +5÷+10°С за 8-9 минут. К полученному гомогенному раствору добавляют 12 мл 100%-ной серной кислоты или 20%-ного олеума за 1-2 минуты при температуре 5÷10°С. Дают 15-минутную выдержку при этой температуре, затем выливают реакционную массу в делительную воронку и сепарируют отработанную кислоту от НИБГТН. Отработанную кислоту сливают в 200 г измельченного льда, а слой нитроэфира сливают в 50 мл холодной воды. НИБГТН, выделившийся после разбавления отработанной кислоты, объединяют с основной порцией нитроэфира, промывают 2 раза холодной водой по 50 мл и 1 раз 1%-ным раствором соды. рН нитроэфира остается на уровне 1,0÷1,5. К кислому нитроэфиру добавляют 30 мл хлористого метилена, промывают еще 1 раз 20 мл холодной воды. К полученному желтоватому раствору добавляют 3 г оксида кальция и контролируют температуру, не позволяя повышаться выше 25°С. Выдерживают при перемешивании 2 часа. Отфильтровывают твердую фазу, проверяют рН раствора нитроэфира индикаторной бумагой. При значениях рН 6,5÷7,5 растворитель упаривают в вакууме водоструйного насоса при температуре 40÷70°С. Выход НИБГТН составляет 25,4÷26,0 г. (89÷91% от теории). Плотность продукта составляет 1,634 г/см3, показатель преломления - n0 20=1,4930, ТНИР=140°С. Продукт бесцветный, рН после удаления растворителя остается 6,5÷7,0.
Пример 2
К полученному, как в примере 1, НИБГТН после растворения в 30 мл хлористого метилена добавляют 3,0 г карбоната кальция и последующую обработку полученной суспензии продолжают аналогично описанной в примере 1.
Получают 25,4÷25,8 г (89÷90%) бесцветного продукта, рН 6,5÷7,0, плотность - 1,634 г/см3, ТНИР=140°С, n0 20=1,4930.
Пример 3
К полученному, как в примере 1, кислому НИБГТН добавляют 50 мл дихлорэтана и 3,0 г оксида кальция, и дальнейшую обработку проводят, как описано в примере 1. Получают 25,5-26,0 г (89÷91%) бесцветного нитроэфира с рН 6,5÷7,0, плотностью 1,634 г/см3, ТНИР=140°С, n0 20=1,4930.
Результаты, полученные по аналогичной методике с использованием безводных нейтрализующих агентов, приведены в таблице.
Из таблицы видно, что нейтральному НИБГТН (рН 6,5÷7,0) соответствует максимальная ТНИР. Использование оксидов магния, цинка, карбоната магния, мочевины и диметилмочевины также обнаруживает значительный стабилизирующий эффект по сравнению с исходным кислым нитроэфиром, однако ТНИР НИБГТН в этих случаях значительно ниже, чем для нитроглицерина.
Использование твердых карбонатов натрия или калия позволяет получать продукт с ТНИР 135-140°С, однако здесь наблюдается протекание неизвестных побочных реакций на поверхности нейтрализующего агента, что легко обнаруживается по появлению окраски раствора от розовой до ярко красной, особенно заметной на поверхности твердых частиц карбонатов. Как видно из изменения температуры, стабилизация завершается за время около 1 часа.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕЗОПАСНЫЙ СПОСОБ ПОЛУЧЕНИЯ АЛИФАТИЧЕСКИХ ПОЛИНИТРАТОВ СПИРТОВ В ПРОМЫШЛЕННЫХ УСЛОВИЯХ | 2014 |
|
RU2567236C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,4',6',2'',4'',6''-ОКТАНИТРО-МЕТА-ТЕРФЕНИЛА | 2014 |
|
RU2562271C1 |
| Способ получения нитроэфиров | 2019 |
|
RU2689406C1 |
| Безопасный способ получения динитратов вицинальных гликолей | 2017 |
|
RU2688690C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЭТИЛГЕКСИЛНИТРАТА | 2016 |
|
RU2640953C2 |
| Способ получения 2,4-динитробензальдегида | 1962 |
|
SU150832A1 |
| ЦЕТАНПОВЫШАЮЩАЯ ПРИСАДКА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2002 |
|
RU2235118C1 |
| Способ получения органических нитросоединений | 2016 |
|
RU2611009C1 |
| Способ получения 1-β-нитроксиэтил-5-аминотетразола | 1969 |
|
SU1841209A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЭТИЛГЕКСИЛНИТРАТА (ВАРИАНТЫ) | 2011 |
|
RU2472771C1 |
Изобретение относится к области технологии получения компонентов порохов, смесевых твердых ракетных топлив и смесевых взрывчатых веществ. Описывается способ получения нитроизобутилглицеринтринитрата нитрованием нитроизобутилглицерина смесью азотной и серной кислот, при этом нитроизобутилглицерин растворяют в серно-азотной смеси, обогащенной азотной кислотой, образующийся раствор нитроизобутилглицеринтринитрата обрабатывают серной кислотой или олеумом, и затем стабилизируют в среде инертного растворителя, хлористого метилена или дихлорэтана, безводным нейтрализующим агентом - оксидом кальция или карбонатом кальция. Технический результат - разработан способ получения нитроизобутилгрицеринитрата, обладающего плотностью не менее 1,63 г/см3 и высокой термической стабильностью с температурой начала интенсивного разложения на уровне не ниже, чем для штатного нитроглицерина (135-140°С). 2 з.п. ф-лы, 1 табл.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ОРЛОВА Е.Ю | |||
| Химия и технология бризантных взрывчатых веществ | |||
| - М.: Оборонгиз, 1960, с.346 | |||
| НАУМ Ф | |||
| Нитроглицерин | |||
| - М.-Л.: Онти-Госхимтехиздат, 1934, с.208. | |||

