Настоящее изобретение относится к способу получения соединений, прежде всего дисахаридов, используемых для определения активности лактазы в кишечнике без отбора крови.
Область изобретения
Недостаточность или низкая активность лактазы кишечника, которая приводит к недостаточной способности или полной неспособности усваивать лактозу, является редким врожденным метаболическим дефектом, но вместе с тем широко распространенным синдромом у взрослых пациентов. Однако у большинства млекопитающих наблюдается заметное снижение активности лактазы с момента отнятия от груди. У человека, предки которого зависели от значительного потребления молока или молочных продуктов в течение длительного времени, такое снижение является не столь частым. С другой стороны, у младенцев, еще не оторванных от груди, недостаточность или низкая активность лактазы кишечника является довольно редким явлением.
Определение активности лактазы в кишечнике имеет большое значение в педиатрии и гастроэнтерологии. Такой анализ можно проводить непосредственно на образце мембраны слизистой или косвенным образом по уровню сахара в крови или по выдыхаемому водороду после введения субъекту дозы лактазы.
Недостаток прямого определения заключается в том, что анализ является сложным и дорогостоящим методом, поскольку для взятия образцов ткани и их последующего анализа необходимы специальные инструменты и высококвалифицированный персонал, не считая того, что операция является неприятной или отчасти опасной для пациента.
Другие методы определения лактазы основаны на сродстве дисахаридов к лактазе и на том факте, что эти дисахариды являются субстратом лактазы и под действием фермента превращаются в определенные моносахариды, которые легко абсорбируются и выводятся с мочой.
В патенте Испании ES-P-9001680 описано получение дисахарида 4-O-β-галактопиранозил-D-ксилозы формулы (I)
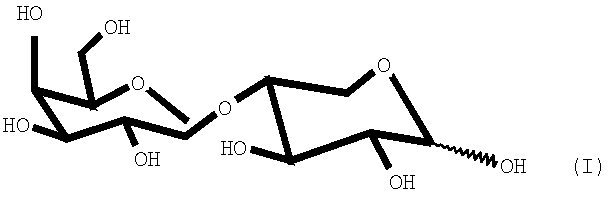
для определения активности лактазы в кишечнике. Указанный дисахарид вводится перорально, действует как субстрат лактазы и, следовательно, гидролизуется в кишечном тракте с образованием ксилозы и галактозы, причем ксилоза абсорбируется и выводится с мочой, где ее определяют простым колориметрическим методом. Количество ксилозы, выводимой с мочой, коррелирует с уровнем лактазы в кишечнике.
Кроме того, в патенте Испании ES-P-9001680 в основном описан метод получения 4-O-β-галактопиранозил-D-ксилозы, который включает синтез бензил-β-D-ксилопиранозида и последующую последовательность реакций, включающую селективную защиту функциональных групп, гликозилирование и удаление защитных групп. Многостадийность процесса, а также применение дорогостоящих реагентов, таких как трифлат серебра на стадии гликозилирования, а также хроматографических колонок на стадии очистки промежуточных соединений и конечного продукта, делают этот процесс слишком дорогостоящим и трудоемким для реализации в препаративном масштабе.
В патентах Испании ES-P-9502185 и ES-P-9701156 описаны ферментативные способы получения смесей дисахаридов галактопиранозилксилозы, которые содержат дисахарид (I) и его региоизомеры 2-O-β-D-галактопиранозил-D-ксилозу и 3-О-β-D-галактопиранозил-D-ксилозу, соответственно, которые имеют следующие формулы:
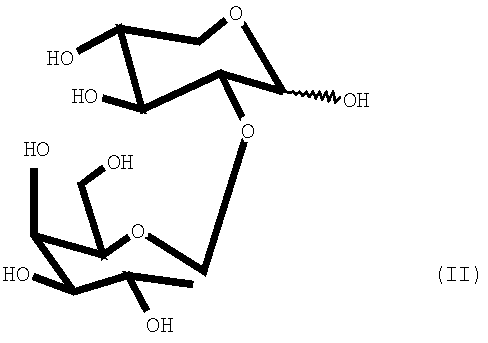
Способы, описанные в патентах Испании ES-P-9502185 и ES-P-9701156, позволяют получить в одну стадию после хроматографической очистки смеси 2-, 3- и 4-O-β-D-галактопиранозил-D-ксилозы, которые используются в качестве субстратов и, следовательно, могут использоваться для определения ферментативной активности лактазы в кишечнике. Несмотря на то что указанные способы выполнимы при наличии соответствующих субстратов и ферментов, их реализация в производственном масштабе связана с рядом проблем, таких как определение наиболее пригодных соотношений региомеров, воспроизводимость получения продукта с указанным соотношением региомеров и определение возможных примесей.
С другой стороны, в статье Gorin и др., The Synthesis of β-Galacto- and β-Glucopyranosyi Disaccharides by Sporobolomyces singularis. Can. J. Chem., 42, 2307-2319 (1964), описан способ микробиологического синтеза ряда дисахаридов, в том числе 2-O-β-D-галактопиранозил-D-ксилозы и 3-O-β-D-галактопиранозил-D-ксилозы. Однако в этой публикации не описано применение различных синтетических дисахаридов.
Объект изобретения
В качестве первого объекта в настоящем изобретении предлагается решение проблем, упомянутых в уровне техники.
В качестве другого объекта в настоящем изобретения предлагается улучшенный способ, который включает любую ферментативную реакцию D-ксилозы с β-D-галактопиранозидным субстратом и последующую стадию выделения и очистки, что позволяет увеличить содержание 4-O-β-D-галактопиранозил-D-ксилозы в конечной смеси, полученной в результате ферментативной реакции, по сравнению с содержанием 2- и 3-O-β-D-галактопиранозил-D-ксилозы, причем 4-D-β-D-галактопиранозил-D-ксилозу можно выделить из конечной смеси простым способом.
Следующим объектом изобретения является 4-O-β-D-галактопиранозил-D-ксилоза, которую можно получить указанным способом, а также композиции, которые включают указанную 4-O-β-D-галактопиранозил-D-ксилозу.
Еще одним объектом изобретения являетсяе применение 4-O-β-D-галактопиранозил-D-ксилозы для получения композиций и растворов, которые применяются при анализе in vivo активности лактазы в кишечнике.
Описание изобретения
Указанные цели достигаются по настоящему изобретению благодаря ферментативному способу получения 4-O-β-D-галактопиранозил-D-ксилозы, который включает:
первую стадию получения первой реакционной смеси, содержащей 2-20 мас.% D-ксилозы, 0,5-5 мас.% β-D-галактопиранозидного субстрата, 75-97,5 мас.% реакционной среды, которая включает буферный раствор с рН от 5,0 до 9,0; добавление к первой реакционной смеси от 10 до 1000 ед. фермента β-D-галактозидазы на 1 г β-D-галактопиранозида и получение второй реакционной смеси;
вторую стадию, на которой вторую реакционную смесь вводят в реакцию при температуре от температуры выше температуры замерзания второй реакционной смеси до 45°С в течение от 2 до 48 ч, с целью получения дисахаридов;
третью стадию, на которой реакцию останавливают при образовании необходимого количества дисахаридов, причем способ обработки выбирают из следующих методов: инактивация β-D-галактозидазы замораживанием второй реакционной смеси при температуре от -20°С до -170°С, инактивация β-D-галактозидазы нагреванием второй реакционной смеси при температуре от 95°С до 110°С; и отделение β-D-галактозидазы от второй реакционной смеси ультрафильтрацией с получением третьей реакционной смеси;
четвертую стадию, на которой агликоновый фрагмент β-D-галактопиранозидного субстрата, использованного на первой стадии, отделяют от третьей реакционной смеси экстракцией или фильтрацией с получением четвертой реакционной смеси;
пятую стадию, включающую выделение фракций, содержащих 4-O-β-D-галактопиранозил-D-ксилозу, причем способ выделения выбирают из следующих двух методов:
добавление к четвертой реакционной смеси целита, экстракцию твердой фазы растворителем и элюирование из колонки первым элюентом, и
добавление к четвертой реакционной смеси активированного угля, фильтрование и элюирование вторым элюентом;
и шестую стадию, на которой фракции, содержащие 4-O-β-D-галактопиранозил-D-ксилозу, кристаллизуют из смеси растворителей, которую выбирают из смесей: ацетон/метанол в соотношении от 5:1 до 20:1 и ацетон/вода в соотношении от 5:1 до 20:1.
Содержание D-ксилозы по изобретению во второй реакционной смеси составляет предпочтительно 7,5 мас.%, содержание β-D-галактопиранозида составляет 1,5 мас.%, а β-D-галактозидазу добавляют в количестве 100 ед. на 1 г β-D-галактопиранозида.
Реакционная среда необязательно может также содержать по меньшей мере один сорастворитель, выбранный из ряда диметилсульфоксид, диметилформамид, диоксан и их смеси, предпочтительно в количестве 20% от реакционной среды. В одном из вариантов изобретения реакционная среда означает буферный раствор с рН 7.
Для обеспечения вопроизводимости реакцию обычно проводят при постоянной температуре. В одном из вариантов способа по изобретению температура реакционной смеси составляет выше температуры замерзания второй реакционной смеси и ниже 40°С. В другом варианте осуществления изобретения реакцию проводят при комнатной температуре, что позволяет получить высокие выходы без охлаждения второй реакционной смеси. Кроме того, реакцию можно проводить при температуре -5°С или 37°С. Реакцию предпочтительно проводят при температуре ниже 0°С, но выше температуры замерзания второй реакционной смеси.
По изобретению β-D-галактопиранозидный субстрат предпочтительно выбирают из орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и лактозы. Фермент β-D-галактозидаза может представлять собой β-D-галактозидазу из E.coli или β-D-галактозидазу из Kluyveramyces lactis (например, такую, как MAXILACT). При использовании в качестве субстрата гал-ONP в качестве продукта реакции образуется орто-нитрофенол, который отделяют экстракцией этилацетатом, если реакцию останавливают нагреванием, или его отделяют обычным фильтрованием, если реакцию останавливают охлаждением.
На третьей стадии способа, если реакцию останавливают замораживанием второй реакционной смеси, температура предпочтительно составляет -75°С. С другой стороны, если на третьей стадии реакцию останавливают нагреванием второй реакционной смеси, температура предпочтительно составляет 100°С.
На пятой стадии 4-O-β-D-галактопиранозил-D-ксилозу выделяют из реакционной смеси с использованием нескольких альтернативных способов.
В первом альтернативном способе из четвертой реакционной смеси удаляют воду, при этом получают остаток, содержащий дисахариды. Остаток ацетилируют, при этом получают перацетилированное производное 4-O-β-D-галактопиранозил-D-ксилозы, которое отделяют хроматографией на колонке с силикагелем. Ацетилирование обычно проводят уксусным ангидридом в пиридине, а деацетилирование перацетилированного производного проводят при катализе метоксидом натрия в метаноле.
Во втором альтернативном способе четвертую реакционную смесь элюируют из колонки первым элюентом, который выбирают из ряда смесей воды с метанолом, этанолом или изопропанолом, предпочтительно смесью вода/изопропанол с содержанием изопропанола от 1% до 10% (об./об.), предпочтительно 2% (об./об.).
Элюирование проводят при фильтровании через колонку с наполнителем, который выбирают из сшитых полимеров декстрана, таких, например, как сефадекс, полимеров акриламида, таких как Биогель, активированного угля или смеси активированный уголь/целит, при этом получают фракции, содержащие 4-О-β-D-галактопиранозил-D-ксилозу.
Перед нанесением на колонку четвертую реакционную смесь предпочтительно концентрируют. В третьем альтернативном способе к четвертой реакционной смеси добавляют целит, полученную смесь концентрируют досуха и остаток подвергают экстракции органическим растворителем в экстракторе Сокслета, а затем элюируют из колонки с наполнителем. В качестве растворителя предпочтительно используют этилацетат. Колонку выбирают из колонок, заполненных сшитыми полимерами декстрана, такими, например, как сефадекс, полимерами акриламида, такими, например, как биогель, активированным углем или смесью активированный уголь/целит. Предпочтительно используют колонку с активированным углем/целитом, причем уголь инактивируют добавлением соляной кислоты.
Преимущество третьего альтернативного способа заключается в том, что почти вся ксилоза в основном, когда она используется в большом избытке, удаляется до нанесения на колонку, что позволяет существенно снизить количество наполнителя и первого элюента. Другим преимуществом третьего способа является абсолютная избирательность экстракции этилацетатом из твердой фазы, в результате чего в жидкой фазе отсутствуют дисахариды и содержатся преимущественно только ксилоза и галактоза.
В четвертом альтернативном способе для выделения продукта на пятой стадии к четвертой реакционной смеси добавляют активированный уголь (вместо использования колонки с наполнителем для отделения агликона от субстрата на четвертой стадии). При этом 4-O-β-D-галактопиранозил-D-ксилоза адсорбируется на активированном угле, после чего ее элюируют вторым элюентом. Указанное элюирование предпочтительно проводят последовательным промыванием сорбента водой и в ступенчатом градиенте концентрации изопропанол/вода: на первой стадии содержание изопропанола составляет от 1% до 3%, на второй стадии от 3% до 5%, а на третьей стадии от 5% до 7%. Предпочтительные концентрации изпропанола составляют 2%, 4% и 6% соответственно. После концентрирования экстракта и кристаллизации из ацетона/воды получают очищенную 4-O-β-D-галактопиранозил-D-ксилозу.
В четвертом альтернативном способе в качестве субстрата предпочтительно используют орто-нитрофенил-β-D-галактопиранозид.
Четвертый альтернативный способ обладает многими преимуществами, например, нет необходимости нагревать вторую реакционную смесь до 100°С с целью остановки реакции, нет необходимости отделять агликон от субстрата методом экстракции на четвертой стадии. Аналогичным образом исключается концентрирование четвертой реакционной смеси и, следовательно, не происходит ее карамелизации. Кроме того, уменьшается количество активированного угля, необходимого для заполнения колонки, а также общее количество элюентов, нет необходимости использовать целит.
На шестой стадии способа по изобретению фракции, содержащие 4-O-β-D-галактопиранозил-D-ксилозу, кристаллизуют из смеси растворителей, которую выбирают из ряда ацетон/метанол в соотношении от 5:1 до 20:1, ацетон /вода в соотношении от 5:1 до 20:1, предпочтительно в соотношении 10:1.
Кроме того, изобретение относится к 4-O-β-D-галактопиранозил-D-ксилозе, полученной вышеуказанным способом, и к композициям и солевым или водным растворам, которые включают 4-O-β-D-галактопиранозил-D-ксилозу, полученную указанным способом, а также к применению 4-O-β-D-галактопиранозил-D-ксилозы для получения композиций и растворов, которые предназначены для определения in vivo содержания лактазы в кишечнике человека.
В таких композициях и растворах 4-O-β-D-галактопиранозил-D-ксилозу используют в смеси с фармацевтически приемлемыми количествами по меньшей мере одного дополнительного компонента, выбранного из обычных фармацевтически приемлемых стабилизаторов, защитных агентов, ароматизаторов, лактозы, желирующих агентов, пластификаторов и консервантов.
4-O-β-D-Галактопиранозил-D-ксилоза или содержащие ее композиции или растворы вводят пероральным способом, при этом ксилозу, которая появляется в моче, определяют спектрофотометрическим способом. Указанный способ используется в качестве специфичного, стандартного и простого диагностического анализа недостаточности активности лактазы без отбора крови.
Варианты осуществления изобретения
Изобретение иллюстрируется следующими примерами, в которых более подробно описаны некоторые вышеуказанные характеритики способа.
Пример 1
Влияние температуры проведения реакции определяли по следующей методике:
к образцам реакционной смеси, содержащей D-ксилозу (125 мг, 500 ммолей) и орто-нитрофенил-β-D-галактопиранозид (25 мг, 50 ммолей) в 1,75 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCl2, 5 мМ меркаптоэтанол), добавляли указанное ниже количество единиц β-галактозидазы из E.coli и инкубировали при указанных температурах.
Увеличение количества фермента необходимо для компенсации снижения скорости реакции при более низкой температуре. Из приведенных данных видно, что реакцию можно проводить при температурах ниже температуры замерзания воды благодаря криоскопическому эффекту, создающемуся в реакционной среде из-за высокой концентрации сахара в образцах.
В каждом образце на каждой стадии процесса определяли соотношение 4-, 2- и 3-О-β-D-галактопиранозил-D-ксилозы методом газовой хроматографии на хроматографе, снабженном пламенно-ионизационным детектором и капиллярной колонкой SE-54 (0,15 мм ×1,5 м, толщина слоя жидкой фазы 0,3 мкм, скорость потока азота 1 мл/мин). Хроматографию проводили в следующем температурном режиме:
Триметилсилильные производные анализируемых образцов получали по следующей методике:
Аликвотную часть (10 мкл) замораживали при -170°С и высушивали лиофильно до получения сухого остатка. Затем к сухому остатку добавляли пиридин (25 мкл), содержащий в качестве внутреннего стандарта бензил-β-D-ксилопиранозид (10 мМ) и N-триметилсилилимидазол (25 мкл), и нагревали при 60°С в течение 30 мин. Различные дисахариды характеризуются следующими временами удерживания:
бензил-β-D-ксилопиранозид 12,04 мин
2-O-β-D-галактопиранозил-D-ксилоза 18,46 и 19,50 мин
3-О-β-D-галактопиранозил-D-ксилоза 18,30 мин
4-O-β-D-галактопиранозил-D-ксилоза 20,35 и 20,50 мин
В табл.1 приведено отношение количества 4-O-β-D-галактопиранозил-D-ксилозы (соединение I) к суммарному количеству ее региоизомеров (2- и 3-О-β-D-галактопиранозил-D-ксилозы) в условиях максимального выхода дисахаридов
Их табл.1 следует, что при снижении температуры реакции наблюдается увеличение содержания 4-O-β-D-галактопиранозил-D-ксилозы.
Пример 2
Влияние величины рН на ход реакции определяли по следующей методике:
получали образцы реакционной смеси, содержащие гал-ONP (25 мг, 50 ммолей), D-ксилозу (125 мг, 500 ммолей), галактозидазу из E.coli (1,6 ед.) в 1,6 мл буферного раствора (50 мМ фосфат калия, рН 8,5, 7 и 5, 1 мМ MgCl2, 5 мМ меркаптоэтанол), и инкубировали при 37°С.
Ход реакции анализировали аналогично тому, как описано в примере 1.
В табл.2 приведено отношение количества 4-O-β-D-галактопиранозил-D-ксилозы (соединение I) к суммарному количеству ее региоизомеров, 2- и 3-О-β-D-галактопиранозил-D-ксилозы
Из табл.2 следует, что при проведении реакции в щелочной среде (рН 8,5) содержание продукта I ниже, чем в нейтральной среде (рН 7), а максимальное содержание достигается при проведении реакции в кислой среде (рН 5).
Пример 3
Для синтеза 4-O-β-D-галактопиранозил-D-ксилозы 6 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 25 г D-ксилозы растворяли в 330 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCl2, 5 мМ меркаптоэтанол), добавляли 2 мг (640 ед.) β-галактозидазы из Е.coli и инкубировали при 30°С при перемешивании на орбитальной мешалке (встряхивателе) до практически полного потребления гал-ONP (приблизительно 4 ч). Контроль за ходом реакции проводили методом ТСХ (элюент: изопропанол/NH3 (30%)/Н2O, 7,5:0,5:2,5) с учетом следующих значений Rf:
Реакцию останавливали нагреванием на водяной бане при 100°С в течение 10 мин, а затем образующийся орто-нитрофенол экстрагировали хлористым метиленом. Водный слой концентрировали досуха и остаток ацетилировали обычным способом (уксусный ангидрид/пиридин, 1:1, при комнатной температуре в течение ночи при перемешивании на магнитной мешалке). Затем реакционную смесь концентрировали, остаток пиридина и уксусного ангидрида удаляли последовательным добавлением и выпариванием толуола. Выпавшие в осадок соли удаляли фильтрованием, фильтрат концентрировали досуха и остаток очищали хроматографией на колонке с силикагелем (элюент:градиент гексан/этилацетат, от 4:1 до 1:1). Сначала из колонки элюировали ацетилированную D-ксилозу, а затем смесь ацетилированных дисахаридов. Фракции, содержащие смесь дисахаридов, концентрировали, остаток растворяли в метаноле, добавляли раствор 1 М MeONa/MeOH и смесь перемешивали до завершения деацетилирования (ход реакции контролировали методом ТСХ, элюент: изопропанол/NH3/Н2О). Смесь нейтрализовали добавлением Амберлита IR-120 (Н+) и концентрировали досуха. Смесь свободных дисахаридов дважды кристаллизовали из МеОН/ацетона, при этом получали 1,07 г очищенной 4-O-β-D-галактопиранозил-D-ксилозы, выход 17% в расчете на исходный гал-ONP. tпл. 171-176°С.
1H ЯМР (D2O): δ 5,17 и 4,58 (2d, 1H, J 3,8 и 7,8 Гц, Н-1α и Н-1β), 4,55 и 4,45 (2d, 1H, J 7,8 Гц, Н-1'), 4,05 (dd, 1H, J 5,3 и 11,6 Гц, Н-5е), 3,38 (dd, 1H, J 10,6 и 11,6 Гц), 3,25 (dd, 1H, J 7,8 и 9,4 Гц, Н-2').
Пример 4
Колонку с активированным углем/целитом получали по следующей методике:
200 г активированного угля (DARCO G-60) и 200 г целита смешивали в сухом состоянии и добавляли воду до получения однородной пасты. Пасту обрабатывали 150 мл HCl (35%) для инактивации активированного угля, а также для удаления следов железа и щелочной золы и промывали водой до нейтральной реакции. Затем пасту помещали в хроматографическую колонку (5×50 см) и уплотняли.
Для синтеза 4-O-β-D-галактопиранозил-D-ксилозы 5 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 25 г D-ксилозы растворяли в 330 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCl2, 5 мМ меркаптоэтанол), добавляли 2 мг (640 ед.) β-галактозидазы из Е.coli и полученный раствор инкубировали при 37°С при перемешивании на орбитальной мешалке (встряхивателе) до практически полного потребления гал-ONP (приблизительно 2 ч). Затем в соответствии с методикой, описанной в примере 3, реакцию останавливали нагреванием при 100°С в течение 10 мин и образующийся орто-нитрофенол экстрагировали этилацетатом. Водный слой концентрировали до приблизительно 50 мл, фильтровали через стекловату и пропускали через колонку с активированным углем/целитом. Сначала из колонки элюировали водой избыток D-ксилозы, а затем в градиенте 2%-10% EtOH/H2O элюировали смесь дисахаридов. Фракции, обогащенные региоизомером 4-O-β-D-галактопиранозил-D-ксилозы, объединяли и концентрировали. Затем добавляли ацетон до появления мутности и полученную смесь выдерживали на холоду. Очищенную 4-O-β-D-галактопиранозил-D-ксилозу получали в виде кристаллического вещества (970 мг, выход 19% в расчете на исходный гал-ONP), спектральные характеристики которого соответствуют данным, полученным для продукта, описанного в примере 3.
Пример 5
Колонку с активированным углем/целитом получали по следующей методике:
200 г активированного угля (DARCO G-60) и 200 г целита смешивали в сухом состоянии и добавляли воду до получения однородной пасты. Пасту обрабатывали 150 мл HCl (35%) для инактивации активированного угля, а также для удаления следов железа и щелочной золы и промывали водой до нейтральной реакции. Затем пасту помещали в хроматографическую колонку (5×50 см) и уплотняли.
Для синтеза 4-O-β-D-галактопиранозил-O-ксилозы 5 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 25 г D-ксилозы растворяли в 330 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCl2, 5 мМ меркаптоэтанол), добавляли 2 мг (640 ед.) β-галактозидазы из Е.coli и полученный раствор инкубировали при 37°С при перемешивании на орбитальной мешалке (встряхивателе) до практически полного потребления гал-ONP (приблизительно 2 ч). Затем в соответствии с методикой, описанной в примере 3, реакцию останавливали нагреванием при 100°С в течение 10 мин и образующийся орто-нитрофенол экстрагировали этилацетатом. Водный слой концентрировали до приблизительно 50 мл, фильтровали через стекловату и пропускали через колонку с активированным углем/целитом.
С целью получения кристаллической 4-O-β-D-галактопиранозил-D-ксилозы сначала элюировали водой избыток D-ксилозы, а затем в градиенте 2%-10% EtOH/Н2О элюировали смесь дисахаридов. Фракции, обогащенные региоизомером 4-O-β-D-галактопиранозил-D-ксилозы, объединяли, концентрировали и растворяли в минимальном количестве воды. Затем по каплям добавляли ацетон до появления мутности и полученную смесь выдерживали при комнатной температуре в течение 2 ч. Через 2 ч при анализе методом ТСХ в супернатанте (прозрачном) было обнаружено наличие некоторого количества 4-O-β-D-галактопиранозил-D-ксилозы. К раствору вновь добавляли ацетон до появления мутности и выдерживали в течение еще 2 ч. Наконец, добавляли следующую порцию ацетона и раствор выдерживали в холодильнике в течение ночи. После этого в полученном супернатанте наблюдалось минимальное количество 4-O-β-D-галактопиранозил-D-ксилозы. Кристаллическую 4-O-β-D-галактопиранозил-D-ксилозу отделяли фильтрованием и промывали ацетоном.
При этом получали очищенную 4-O-β-D-галактопиранозил-D-ксилозу (1,557 мг, выход 30% в расчете на исходный гал-ONP), спектральные характеристики которой соответствуют данным, полученным для продукта, описанного в примере 3.
Пример 6
Колонку с активированным углем/целитом получали по следующей методике:
200 г активированного угля (DARCO G-60) и 200 г целита смешивали в сухом состоянии и добавляли воду до получения однородной пасты. Пасту обрабатывали 150 мл HCl (35%) для инактивации активированного угля, а также для удаления следов железа и щелочной золы и промывали водой до нейтральной реакции. Затем пасту помещали в хроматографическую колонку (5×50 см) и уплотняли.
Для синтеза 4-O-β-D-галактопиранозил-D-ксилозы 5 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 25 г D-ксилозы растворяли в 330 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 6,8, 1 мМ MgCl2, 5 мМ меркаптоэтанол), добавляли 70 ед. β-галактозидазы из Kluyveramyces lactis (MAXILACT®) и полученный раствор инкубировали при 37°С при перемешивании на орбитальной мешалке (встряхивателе) до практически полного потребления гал-ONP (приблизительно 2 ч). Контроль за ходом реакции осуществляли методом ТСХ (элюент:изопропанол/NH3 (30%)/Н2О, 7,5:0,5:2,5) с учетом следующих значений Rf:
В соответствии с методикой, описанной в примере 4, реакцию останавливали нагреванием при 100°С в течение 10 мин, образующийся орто-нитрофенол экстрагировали этилацетатом и реакционную смесь фильтровали для удаления остатков фермента. Водный слой концентрировали в вакууме до объема приблизительно 45 мл и пропускали через колонку с активированным углем/целитом. Сначала из колонки элюировали водой избыток D-ксилозы, а затем в градиенте 2%-10% EtOH/Н2О элюировали смесь дисахаридов. Фракции, обогащенные региоизомером 4-O-β-D-галактопиранозил-D-ксилозы, объединяли и концентрировали. Затем добавляли ацетон до появления мутности и полученную смесь выдерживали при охлаждении. Кристаллическую 4-O-β-D-галактопиранозил-D-ксилозу отделяли фильтрованием на пористом фильтре. При этом получали очищенную 4-O-β-D-галактопиранозил-D-ксилозу (817 мг, выход 16% в расчете на исходный гал-ONP).
Пример 7
Колонку с активированным углем/целитом получали по следующей методике:
200 г активированного угля (DARCO G-60) и 200 г целита смешивали в сухом состоянии и добавляли воду до получения однородной пасты. Пасту обрабатывали 150 мл HCl (35%) для инактивации активированного угля, а также для удаления следов железа и щелочной золы и промывали водой до нейтральной реакции. Затем пасту помещали в хроматографическую колонку (5×50 см) и уплотняли.
Для синтеза 4-O-β-D-галактопиранозил-D-ксилозы 5 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 25 г D-ксилозы растворяли в 330 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCl2 5 мМ меркаптоэтанол), добавляли 80 ед. β-галактозидазы из Е.coli и полученный раствор инкубировали при 37°С при перемешивании на орбитальной мешалке (встряхивателе) в течение 24 ч. Контроль за ходом реакции осуществляли методом ТСХ (элюент:изопропанол/NH3 (30%)/Н2O, 7,5:0,5:2,5) с учетом следующих значений Rf:
В соответствии с методикой, описанной в примере 3, реакцию останавливали нагреванием при 100°С в течение 10 мин, образующийся орто-нитрофенол экстрагировали этилацетатом и реакционную смесь фильтровали для удаления остатков фермента. Водный слой концентрировали в вакууме до объема приблизительно 70 мл и концентрированный раствор пропускали через колонку с активированным углем/целитом. Сначала колонку элюировали 2% изопропанолом/водой (1,3 л), затем 4% изопропанолом/водой (2,6 л), собирая общий объем элюата 3,9 л.
Фракции, обогащенные региоизомером 4-O-β-D-галактопиранозил-D-ксилозы, объединяли, концентрировали до небольшого объема, добавляли ацетон до появления мутности и полученную смесь выдерживали на холоду. Кристаллическую 4-O-β-D-галактопиранозил-D-ксилозу отделяли фильтрованием на пористом фильтре. При этом получали очищенную 4-O-β-D-галактопиранозил-D-ксилозу (1,213 мг, выход 24% в расчете на исходный гал-ONP).
Пример 8
Колонку с активированным углем/целитом получали по следующей методике:
24 г активированного угля (DARCO G-60) и 24 г целита смешивали в сухом состоянии и добавляли воду до получения однородной пасты. Пасту обрабатывали 18 мл HCl (35%) для инактивации активированного угля, а также для удаления следов железа и щелочной золы и промывали водой до нейтральной реакции. Затем пасту помещали в хроматографическую колонку и уплотняли.
Для синтеза 4-O-β-D-галактопиранозил-D-ксилозы 5 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 25 г D-ксилозы растворяли в 330 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCb, 5 мМ меркаптоэтанол), добавляли 80 ед. β-галактозидазы из Е.coli и полученный раствор инкубировали при 37°С при перемешивании на механической мешалке до практически полного потребления гал-ONP (24 ч). Контроль за ходом реакции осуществляли методом ТСХ (элюент: изопропанол/NH3 (30%)/Н2O, 7,5:0,5:2,5) аналогично тому, как описано в примере 7. В соответствии с методикой, описанной в примере 3, реакцию останавливали нагреванием при 100°С в течение 10 мин, охлаждали, образующийся орто-нитрофенол экстрагировали этилацетатом. К водному раствору добавляли целит (40 г) и смесь концентрировали досуха. Твердый остаток экстрагировали этилацетатом (500 мл) в экстракторе Сокслета, снабженном целлюлозным вкладышем. Через 23 ч остаток в виде твердого вещества промывали водой (3×40 мл) и водный экстракт пропускали через колонку с активированным углем/целитом. Сначала колонку элюировали 2% изопропанолом/водой, затем 4% изопропанолом/водой, собирая общий объем элюата 400 мл. Фракции, обогащенные региоизомером 4-O-β-D-галактопиранозил-D-ксилозы, объединяли, концентрировали досуха. Остаток кристаллизовали из ацетона/воды аналогично тому, как описано в примере 7, при этом получали 0,44 г очищенного дисахарида в виде кристаллического вещества.
Пример 9
Для синтеза 4-O-β-D-галактопиранозил-D-ксилозы 4,12 г орто-нитрофенил-β-D-галактопиранозида (гал-ONP) и 20,6 г D-ксилозы растворяли в 272 мл буферного раствора (0,05 М КН2PO4/К2HPO4, рН 7, 1 мМ MgCl2, 5 мМ меркаптоэтанол), добавляли 66 ед. β-галактозидазы из Е.coli и полученный раствор инкубировали при 37°С при перемешивании на механической мешалке до практически полного потребления гал-ONP (21 ч). Реакцию останавливали охлаждением до 0°С и орто-нитрофенол отделяли фильтрованием. Затем к фильтрату добавляли 60 г актививированного угля и полученную смесь перемешивали в течение 30 мин. При анализе супернатанта методом ТСХ было показано, что в растворе отсутствуют дисахариды, поскольку эти соединения адсорбированы на активированном угле. Смесь фильтровали, активированный уголь промывали водой (400 мл), 2% изопропанолом (100 мл), 4% изопропанолом (200 мл) и 6% изопропанолом (200 мл). Фракции, содержащие 4-О-β-D-галактопиранозил-D-ксилозу, концентрировали и остаток (2,38 г) кристаллизовали из ацетона/воды, при этом получали 1,55 г твердого вещества, которое кристаллизовали вторично из той же смеси растворителей аналогично тому, как описано в примере 7, при этом получали 1,32 г (32%) очищенного дисахарида.
Изобретение относится к биотехнологии, в частности к способу получения 4-O-β-D-галактопиранозил-D-ксилозы, используемой для определения in vivo активности лактазы в кишечнике человека. Способ включает взаимодействие D-ксилозы с β-D-галактопиранозидом в течение 2-48 ч в растворе с рН 5-9 при температуре, которая изменяется от температуры замерзания смеси до 45°С, и последующее добавление 10-1000 ед. β-D-галактозидазы на 1 г β-D-галактопиранозида. Реакцию останавливают инактивацией фермента с последующим выделением и кристаллизацией фракций, содержащих 4-O-β-D-галактопиранозил-D-ксилозу, в смеси для кристаллизации, выбранной из ацетона/метанола (5:1-20:1) и ацетона/воды (5:1-20:1). Изобретение позволяет увеличить содержание 4-O-β-D-галактопиранозил-D-ксилозы в конечной смеси. 41 з.п. ф-лы, 2 табл.
первую стадию получения первой реакционной смеси, содержащей 2-20 мас.% D-ксилозы, 0,5-5 мас.% β-D-галактопиранозидного субстрата, 75-97,5 мас.% реакционной среды, которая представляет собой буферный раствор с рН от 5,0 до 9,0;
добавление к первой реакционной смеси от 10 до 1000 ед. фермента β-D-галактозидазы на 1 г β-D-галактопиранозида, и получение второй реакционной смеси;
вторую стадию, на которой вторую реакционную смесь вводят в реакцию при температуре от величины выше температуры замерзания второй реакционной смеси до 45°С в течение от 2 до 48 ч, с получением дисахаридов;
третью стадию, на которой реакцию останавливают при образовании необходимого количества дисахаридов, причем способ обработки выбирают между инактивацией β-D-галактозидазы замораживанием второй реакционной смеси при температуре от -20 до -170°С и инактивацией β-D-галактозидазы нагреванием второй реакционной смеси при температуре от 95 до 110°С; и отделение β-D-галактозидазы от второй реакционной смеси ультрафильтрацией, с получением третьей реакционной смеси;
четвертую стадию, на которой агликоновый фрагмент β-D-галактопиранозидного субстрата, использованного на первой стадии, отделяют от третьей реакционной смеси экстракцией или фильтрацией с получением четвертой реакционной смеси;
пятую стадию, включающую выделение фракций, содержащих 4-O-β-D-галактопиранозил-D-ксилозу, причем пятую стадию выбирают из следующих двух методов:
добавление к четвертой реакционной смеси целита с последующей экстракцией твердой фазы растворителем и элюированием из колонки первым элюентом; и
прямое добавление к четвертой реакционной смеси активированного угля с последующим фильтрованием и элюированием вторым элюентом; а затем
шестую стадию, на которой фракции, содержащие 4-O-β-D-галактопиранозил-D-ксилозу, кристаллизуют из смеси растворителей, которую выбирают из смесей: ацетон/метанол в соотношении от 5:1 до 20:1, и ацетон/вода в соотношении от 5:1 до 20:1.
| СПОСОБ ПОЛУЧЕНИЯ СЫВОРОТКИ РАСТИТЕЛЬНОГО БЕЛКА, ОБОГАЩЕННОЙ ИЗОФЛАВОНАМИ АГЛЮКОНА (ВАРИАНТЫ), СПОСОБ ИЗВЛЕЧЕНИЯ БЕЛКА СЫВОРОТКИ | 1994 |
|
RU2130073C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАНУЛИРОВАННОЙ ШЛАКООБРАЗУЮЩЕЙ СМЕСИ | 1995 |
|
RU2100131C1 |
| Способ получения плевромутилиновых гликозидных производных | 1978 |
|
SU902666A3 |
| US 5994092, 30.11.1999. | |||

