Данное изобретение относится к определенным бициклическим 6-алкилиденпенемам, которые действуют как ингибиторы β-лактамаз широкого спектра. β-Лактамазы гидролизуют β-лактамные антибиотики и как таковые выступают в качестве основной причины бактериальной резистентности. Соединения по данному изобретению, когда их объединяют с β-лактамными антибиотиками, будут обеспечивать эффективное лечение опасных для жизни бактериальных инфекций.
Пенициллины и цефалоспорины являются наиболее часто и широко применяемыми в клинической практике β-лактамными антибиотиками. Однако развитие резистентности различных патогенов к β-лактамным антибиотикам оказывает разрушающее действие на обеспечение эффективного лечения бактериальных инфекций (Coleman, K. Expert Opin. Invest. Drugs 1995, 4, 693; Sutherland, R. Infection 1995, 23 (4) 191; Bush, K, Cur. Pharm. Design 1999, 5, 839-845). Наиболее значительным известным механизмом, имеющим отношение к развитию бактериальной резистентности к β-лактамным антибиотикам, является продуцирование сериновых β-лактамаз класса-А, класса-В и класса-С. Указанные ферменты разрушают β-лактамные антибиотики, что приводит к потере антибактериальной активности. Ферменты класса-А преимущественно гидролизуют пенициллины, тогда как лактамазы класса-С имеют профиль субстрата, благоприятствующий гидролизу цефалоспорина (Bush, K.; Jacoby, G.A.; Medeiros, A.A. Antimicrob. Agents Chemother. 1995, 39, 1211). На данный момент имеются сведения об около 250 различных β-лактамазах (Payne, D.J.: Du, W и Bateson, J.H. Exp. Opin. Invest. Drugs 2000, 247) и существует потребность в новом поколении ингибиторов β-лактамаз широкого спектра. Бактериальная резистентность к указанным антибиотикам могла бы быть значительно уменьшена введением β-лактамного антибиотика в сочетании с соединением, которое ингибирует указанные ферменты.
Коммерчески доступные ингибиторы β-лактамаз, такие как клавулановая кислота, сулбактам и тазобактам, все эффективны против патогенов, продуцирующих лактамазы класса-А. Клавулановая кислота применяется в клинической практике в сочетании с амоксициллином и тикарциллином, так же как сулбактам с ампициллином и тазобактам с пиперациллином. Однако указанные соединения неэффективны против организмов, продуцирующих лактамазы класса-С. Механизм инактивации β-лактамаз класса-А (таких как PCI и ТЕМ-1) объяснен (Bush, K.; Antimicrob. Agents Chemother. 1993, 37, 851; Yang, Y.; Janota, K.; Tabei, K.; Huang, N.; Seigal, M.M.; Lin, Y.I.; Rasmussen, B.A. и Shlaes, D.M. J. Biol. Chem. 2000, 35, 26674-26682).
В 1981 году группа Beecham открыла 6-алкилиденпинемы общей структуры 1 как ингибиторы β-лактамаз. [N.F. Osborne, патент США 4485110 (1984); N.F. Osborne, Европейская патентная заявка 81301683.9, 1981]
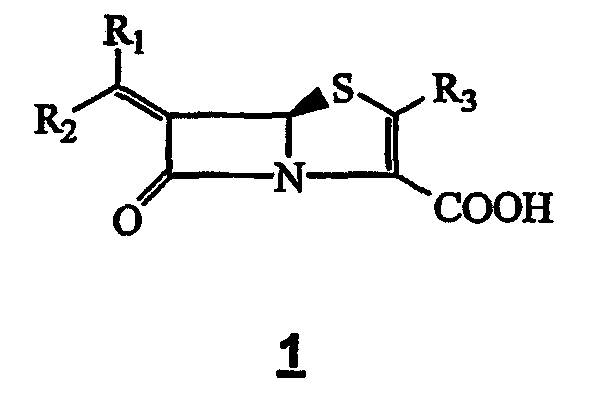
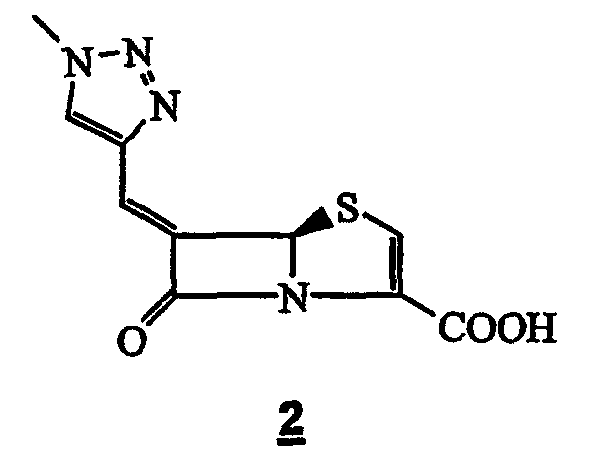
R1 и R2 независимо означают водород, или C1-10-углеводородную группу, или моногетероциклическую группу, и R3 означает водород или органическую группу. После этого та же группа открыла соединения общей формулы 1, где R1 содержит группу 1,2,3-триазола [N.F. Osborne, Европейская патентная заявка 84301255.0]. На следующий год та же группа подала заявки на 3 патента на структуру 1, где R1 означает необязательно замещенную шестичленную или пятичленную систему ароматического монокольца [N.F. Osborne, Европейская патентная заявка 85100520.7; Европейская патентная заявка 85100521.5; Европейская патентная заявка 85300456-2]. Европейская патентная заявка No. 86305585.1 раскрывает синтез и пригодность (Z)-6-(1-метил-1,2,3-триазол-4-илметилен)пенем-3-карбоксилата 2 в качестве ингибитора β-лактамаз класса-А и класса-С.
Европейская патентная заявка 86305584,4 раскрывает получение соединений общей формулы 1, где R1 = неароматическая гетероциклическая группа, и опубликована заявка PCT [N.J. Broom; P.D. Edwards, N.F. Osborne и S. Coulton PCT WO 87/00525], раскрывающая R1 = конденсированную бициклическую гетероароматическую группу. Подобным образом патентные заявки [N.J. Broom; G. Brooks; S. Coulton, Европейская патентная заявка 88311786.3; N.J. Broom; G. Brooks; B.P. Clarke, Европейская патентная заявка 88311787.1) описывают получение и применение соединений общей структуры 1, где R1 означает замещенное пятичленное гетероароматическое кольцо. Способ получения соединений общей формулы 1 описан Coulton и др. [S. Coulton; J.B. Harbridge; N.F. Osborne и G. Walker, Европейская патентная заявка No 87300193.7].
В 1993 году группой Beecham [A.V. Stachulski и R. Walker, PCT WO 93/03042] раскрыто получение и применение соединений общей формулы 1, где R1 = (C1-6)-алкил и R2 = CH2X или COY, где X = галоген или CONR2.
В течение последнего десятилетия группой Beecham получены три патента, описывающие соединения общей формулы 3. [N.J. Broom; F.P. Harrington, PCT WO 94/10178; K. Coleman; J.E. Neale PCT WO 95/28935; K. Coleman; J.E. Neale, РСТ WO 95/17184], где R1 = водород или органическая группа, и R4 и R5 оба могут означать водород или один или несколько заместителей, замещающих атомы водорода в кольцевой системе, показанной ниже.
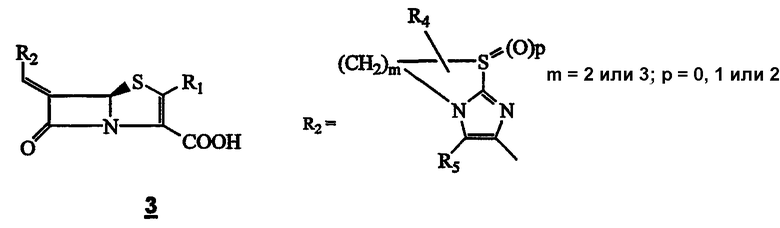
Данное изобретение относится к новым низкомолекулярным соединениям β-лактама широкого спектра и в особенности к классу бициклических гетероарил-замещенных 6-алкилиденпенемов, которые имеют ингибирующие β-лактамазы и антибактериальные свойства. Соединения, следовательно, применимы в лечении бактериальных инфекций у людей и животных либо сами по себе, либо в сочетании с другими антибиотиками.
В соответствии с данным изобретением предложены соединения общей формулы I или их фармацевтически приемлемые соли или гидролизуемые in vivo сложные эфиры R5:
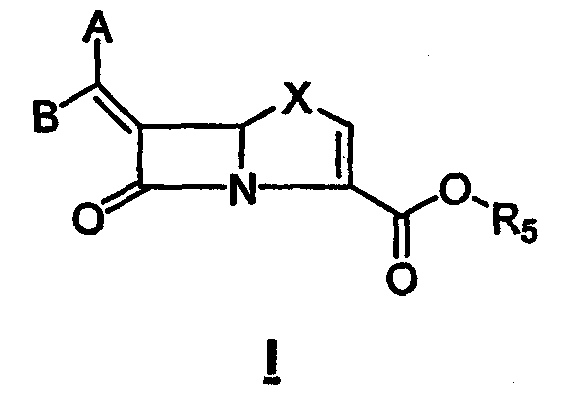
и предпочтительные соединения формулы
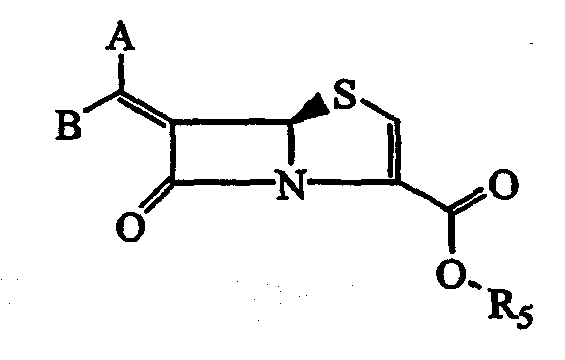
где:
один из A и В означает водород и другой необязательно замещенную конденсированную бициклическую гетероарилгруппу. Выражение "конденсированная бициклическая гетероарилгруппа", используемое в описании и формуле изобретения, подразумевает следующее:
группа, содержащая два конденсированных кольца, одно из которых имеет ароматический характер [т.е. отвечает правилу Huckel (4n+2)], а другое кольцо является неароматическим;
конденсированная бициклическая гетероарилгруппа содержит от одного до шести гетероатомов, выбранных из группы, состоящей из O, S, N и N-R1;
Конденсированная бициклическая гетероарилгруппа связана с остальной частью молекулы через атом углерода в ароматическом кольце, как показано в формуле I;
Ароматическое кольцо конденсированной бициклической гетероарилгруппы содержит от пяти до шести атомов кольца (включая головные атомы мостиковой группы), выбранных из CR2, N, O, S или N-R1. Ароматическое кольцо конденсированной бициклической гетероарилгруппы содержит от 0 до 3 гетероатомов, выбранных из группы, состоящей из O, S, N и N-R1.
Неароматическое кольцо конденсированной бициклической гетероарилгруппы содержит от пяти до восьми атомов кольца (включая головные атомы мостиковой группы), выбранных из CR4R4, N, N-R1, O, S(O)n, где n = 0-2. Неароматическое кольцо конденсированной бициклической гетероарилгруппы содержит от 0 до 4 гетероатомов кольца, выбранных из N, N-R1, O или S(O)n, где n = 0-2.
X означает O или S, предпочтительно S;
R5 означает Н, гидролизуемый in vivo сложный эфир, такой как C1-6-алкил, C5-6-циклоалкил, CHR3OCOC1-C6 или соли, такие как Na, K, Ca; предпочтительно R5 означает H или соль;
R1 означает Н, необязательно замещенный -C1-C6-алкил, необязательно замещенный -арил, необязательно замещенный -гетероарил или моно или бициклические насыщенные гетероциклы, необязательно замещенный -C3-C7-циклоалкил, необязательно замещенный -C3-C6-алкенил, необязательно замещенный -C3-C6-алкинил при условии, что и двойная связь, и тройная связь не должны находиться при атоме углерода, который непосредственно связан с N; необязательно замещенный -C1-C6-перфторалкил, необязательно замещенный группой -S(O)P алкил или арил, где p означает 2, необязательно замещенный -С=O-гетероарил, необязательно замещенный -C=O-арил, необязательно замещенный -C=O(C1-C6)-алкил, необязательно замещенный -C=O(C3-C6)-циклоалкил, необязательно замещенные -C=O моно или бициклические насыщенные гетероциклы, необязательно замещенный C1-C6-алкиларил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный арил-C1-C6-алкил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенные C1-C6-алкил-моно- или бициклические насыщенные гетероциклы, необязательно замещенный арилалкенил из 8-16 атомов углерода, -CONR6R7, -SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный -алкил-O-алкиларил, необязательно замещенный -алкил-O-алкилгетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный C1-C6-алкиларилоксиарил, необязательно замещенный C1-C6-алкиларилоксигетероарил, необязательно замещенные алкиларилоксиалкиламины, необязательно замещенный алкоксикарбонил, необязательно замещенный арилоксикарбонил, необязательно замещенный гетероарилоксикарбонил. Предпочтительными R1 группами являются H, необязательно замещенный алкил, необязательно замещенный арил, -C=O(C1-C6)-алкил, C3-C6-алкенил, C3-C6-алкинил, необязательно замещенный циклоалкил, SO2-алкил, SO2-арил, необязательно замещенные гетероциклы, -CONR6R7 и необязательно замещенный гетероарил.
R2 означает водород, необязательно замещенный C1-C6-алкил, необязательно замещенный C2-C6-алкенил, имеющий 1-2 двойные связи, необязательно замещенный C2-C6-алкинил, имеющий 1-2 тройные связи, галоген, циано, N-R6R7, необязательно замещенный C1-C6-алкокси, гидрокси; необязательно замещенный арил, необязательно замещенный гетероарил, COOR6, необязательно замещенные алкиларилоксиалкиламины, необязательно замещенный арилокси, необязательно замещенный гетероарилокси, необязательно замещенный C3-C6-алкенилокси, необязательно замещенный C3-C6-алкинилокси, C1-C6-алкиламино-C1-C6-алкокси, алкилендиокси, необязательно замещенный арилокси-C1-C6-алкиламин, C1-C6-перфторалкил, необязательно S(O)q-замещенный C1-C6-алкил, необязательно S(O)q-замещенный арил, где q означает 0, 1 или 2, CONR6R7, гуанидино или циклический гуанидино, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный C1-C6-алкилгетероарил, необязательно замещенный гетероарил-C1-C6-алкил, необязательно замещенные C1-C6-алкил-моно- или бициклические насыщенные гетероциклы, необязательно замещенный арилалкенил из 8 до 16 атомов углерода, SO2NR6R7, необязательно замещенный арилалкилоксиалкил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенный арилоксиарил, необязательно замещенный арилоксигетероарил, необязательно замещенный гетероарилоксиарил, необязательно замещенный C1-C6-алкиларилоксиарил, необязательно замещенный C1-C6-алкиларилоксигетероарил, необязательно замещенный арилоксиалкил, необязательно замещенный гетероарилоксиалкил, необязательно замещенные алкиларилоксиалкиламины. Предпочтительными группами R2 являются H, необязательно замещенный алкил, необязательно замещенный алкокси, необязательно замещенный гетероарил, галоген, CN, гидрокси, необязательно замещенный гетероцикл, -CONR6R7, COOR6, необязательно замещенный арил, S(O)q-алкил и S(O)q-арил.
R3 означает водород, C1-C6-алкил, C5-C6-циклоалкил, необязательно замещенный арил, необязательно замещенный гетероарил; предпочтительными группами R3 являются H или C1-C6-алкил;
R4 означает Н, необязательно замещенный C1-C6-алкил, один из R4 означает OH, C1-C6-алкокси, -S-C1-C6-алкил, COOR6, -NR6R7, -CONR6R7; или R4R4 вместе могут быть =O или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать спиро-систему из пяти до восьми членов с наличием или без гетероатомов, выбранных из N, O, S=(O)n (где n = 0 до 2), N-R1; предпочтительными группами R4 являются H, C1-C6-алкил, NR6R7 или R4R4 вместе с атомом углерода, к которому они присоединены, могут образовывать спиро-систему из пяти до восьми членов с наличием или без гетероатомов, например, одного или двух атомов кислорода, азота и серы;
R6 и R7 независимо означают H, необязательно замещенный C1-C6-алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный C1-C6-алкиларил, необязательно замещенный арилалкил, необязательно замещенный гетероарилалкил, необязательно замещенный C1-C6-алкилгетероарил, R6 и R7 вместе могут образовывать 3-7-членную насыщенную кольцевую систему, необязательно имеющую один или два гетероатома, таких как N-R1, O, S=(O)n, n = 0-2. Предпочтительными группами R6 иR7 являются H, C1-C6-алкил, арилалкил, гетероарилалкил или R6 и R7 вместе образуют 3-7-членную насыщенную кольцевую систему, необязательно имеющую один или два гетероатома.
Термин алкил означает алкилгруппы с прямой и с разветвленной цепью, содержащие от 1 до 12 атомов углерода, предпочтительно от 1 до 6 в цепи атомов углерода.
Термин алкенил означает алкенилгруппы с прямой и с разветвленной цепью, содержащие от 2 до 8 атомов углерода в цепи, содержащие по меньшей мере одну двойную связь и не имеющие тройной связи, предпочтительно алкенилгруппа имеет одну или две двойные связи. Такие алкенилгруппы могут существовать в E или Z конформациях; соединения по данному изобретению включают обе конформации. В случае алкенила гетероатомы, такие как O, S или N-R1 не должны присутствовать на атоме углерода, который связан двойной связью;
Термин алкинил включает алкинилгруппы и с прямой, и с разветвленной цепью из 2-6 атомов углерода, содержащие по меньшей мере одну тройную связь, предпочтительно алкинилгруппа имеет одну или две тройные связи. В случае алкинила гетероатомы, такие как O, S или N-R1, не должны присутствовать на атоме углерода, который связан двойной или тройной связью;
Термин циклоалкил относится к алициклической углеводородной группе, имеющей 3-7 атомов углерода. Используемый здесь термин перфторалкил относится к насыщенным алифатическим углеводородным группам и с прямой, и с разветвленной цепью, имеющим по меньшей мере один атом углерода и два или более атомов фтора. Примеры включают CF3, CH2CF3, CF2CF3 и CH(CF3)2. Термин галоген означает Cl, Br, F и I.
Если алкил, алкенил, алкинил или циклоалкил является "необязательно замещенным", возможными являются один или два следующих заместителя: нитро, -арил, -гетероарил, алкоксикарбонил-, -алкокси, -алкоксиалкил, алкил-O-C2-C4-алкил-O-, -циано, -галоген, -гидрокси, -N-R6R7, -COOH, -COO-алкил, -трифторметил, -трифторметокси, арилалкил, алкиларил, R6R7N-алкил-, HO-C1-C6-алкил-, алкоксиалкил-, алкил-S-, -SO2N-R6R7, -SO2NHR6, -CO2H, CONR6R7, арил-O-, гетероарил-O-, -S(O)s-арил (где s = 0-2), -алкил-O-алкил-NR6R7, -алкиларил-O-алкил-N-R6R7, C1-C6-алкил, алкенил, алкинил, циклоалкил, алкоксиалкил-O-, R6R7N-алкил- и -S(O)s-гетероарил (где s = 0-2); Предпочтительные заместители для алкила, алкенила, алкинила и циклоалкила включают галоген, нитро, арил, гетероарил, -COOH, -COO-алкил, алкоксикарбонил-, алкокси, -алкоксиалкил, -циано, гидрокси и -N-R6R7.
Арил означает ароматическую углеводородную часть молекулы, выбранную из группы, состоящей из следующих групп: фенил, α-нафтил, β-нафтил, бифенил, антрил, тетрагидронафтил, флуоренил, инданил, бифениленил, аценафтенил. Предпочтительными арилгруппами являются фенил и бифенил.
Гетероарил означает ароматическую гетероциклическую кольцевую систему (моноциклическую или бициклическую), где гетероарилгруппы выбраны из следующего: (1) фуран, тиофен, индол, азаиндол, оксазол, тиазол, изоксазол, изотиазол, имидазол, N-метилимидазол, пиридин, пиримидин, пиразин, пиррол, N-метилпиррол, пиразол, N-метилпиразол, 1,3,4-оксадиазол, 1,2,4-триазол, 1-метил-1,2,4-триазол, 1H-тетразол, 1-метилтетразол, бензоксазол, бензотиазол, бензофуран, бензизоксазол, бензимидазол, N-метилбензимидазол, азабензимидазол, индазол, хиназолин, хинолин и изохинолин; (2) бициклический ароматический гетероцикл, где кольцо фенила, пиридина, пиримидина или пиридизина является (a) конденсированным с 6-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один атом азота; (b) конденсированным с 5- или 6-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим два атома азота; (c) конденсированным с 5-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один атом азота вместе или с одним атомом кислорода, или с одним атомом серы; или (d) конденсированным с 5-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один гетероатом, выбранный из O, N или S. Предпочтительными гетероарилгруппами являются фуран, оксазол, тиазол, изоксазол, изотиазол, имидазол, N-метилимидазол, пиридин, пиримидин, пиразин, пиррол, N-метилпиррол, пиразол, N-метилпиразол, 1,3,4-оксадиазол, 1,2,4-триазол, 1-метил-1,2,4-триазол, 1H-тетразол, 1-метилтетразол, хинолин, изохинолин и нафтиридин.
Если арил или гетероарил является "необязательно замещенным", возможными заместителями являются один или два из следующих: нитро, -арил, -гетероарил, алкоксикарбонил-, -алкокси, -алкоксиалкил, алкил-O-C2-C4-алкил-O-, -циано, -галоген, -гидрокси, -N-R6R7, -трифторметил, -трифторметокси, арилалкил, алкиларил, R6R7N-алкил-, HO-C1-C6-алкил-, алкоксиалкил-, алкил-S-, -SO2N-R6R7, -SO2NHR6, -CO2H, CONR6R7, арил-O-, гетероарил-O-, -S(O)S-арил (где s = 0-2), -алкил-O-алкил-NR6R7, -алкиларил-O-алкил-N-R6R7, C1-C6-алкил, алкенил, алкинил, циклоалкил, алкоксиалкил-O-, R6R7N-алкил- и -S(O)s-гетероарил (где s = 0-2); Предпочтительные заместители для арила и гетероарила включают алкил, галоген, -N-R6R7, трифторметил, -трифторметокси, арилалкил и алкиларил.
Арилалкил означает арил-C1-C6-алкил-; арилалкилгруппы включают бензил, 1-фенилэтил, 2-фенилэтил, 3-фенилпропил, 2-фенилпропил и тому подобное. Термин "необязательно замещенный" относится к незамещеному или замещенному 1 или 2 заместителями на группе алкил или арил, которые имеют значения, указанные выше.
Алкиларил означает C1-C6-алкиларил. Термин "необязательно замещенный" относится к незамещенным или замещенным 1 или 2 заместителями на арильной или алкильной части молекулы, определения которых даны выше.
Гетероарил-C1-C6-алкил означает замещенную гетероарилом алкилгруппу, где цепь алкила имеет 1-6 атомов углерода (прямая или разветвленная). Алкилгетероарилгруппы включают гетероарил-(CH2)1-6- и тому подобное. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями на алкил- или гетероарилгруппе, определения которых даны выше.
C1-C6-алкилгетероарил означает цепь алкила из 1-6 атомов углерода (прямую или разветвленную), присоединенную к гетероарилгруппе, которая связана с остальной частью молекулы. Например C1-C6-алкилгетероарил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями на алкил- или гетероарилгруппе, определения которых даны выше.
Насыщенные или частично насыщенные гетероциклические группы определены как гетероциклические кольца, выбранные из групп: азиридинил, азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, дигидробензимидазолил, дигидробензофуранил, дигидробензотиенил, дигидробензоксазолил, дигидрофуранил, дигидроимидазолил, дигидроиндолил, дигидроизооксазолил, дигидроизотиазолил, дигидрооксадиазолил, дигидрооксазолил, дигидропиразинил, дигидропиразолил, дигидропиридинил, дигидропиримидинил, дигидропирролил, дигидрохинолинил, дигидротетразолил, дигидротиадиазолил, дигидротиазолил, дигидротиенил, дигидротриазолил, дигидроазетидинил, дигидро-1,4-диоксанил, тетрагидрофуранил, тетрагидротиенил, тетрагидрохинолинил и тетрагидроизохинолинил. Предпочтительными насыщенными или частично насыщенными гетероциклами являются азиридинил, азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, тетрагидрохинолинил, тетрагидроизохинолинил, дигидроимидазолил и дигидроизооксазолил.
C1-C6-алкил-моно- или бициклические насыщенные или частично насыщенные гетероциклы определены как C1-C6-алкилгруппа (с прямой или разветвленной цепью), присоединенная к гетероциклам (определение которых дано ранее) через атом углерода или атом азота, и другой конец алкильной цепи присоединен к остальной части молекулы. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкиле или на гетероциклической части молекулы, определения которых даны выше.
Арилалкилоксиалкил определен как арил-C1-C6-алкил-O-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной и/или арильной частях, определения которых даны выше.
Алкилоксиалкил определен как C1-C6-алкил-O-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной части, определение которой дано выше.
Арилоксиалкил определен как арил-O-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной или арильной части, определение которой дано выше.
Гетероарилалкилоксиалкил определен как гетероарил-C1-C6-алкил-O-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной или гетероарильной части, определения которых даны выше.
Арилоксиарил определен как арил-O-арил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на арильной части, определение которой дано выше.
Арилоксигетероарил определен как арил-O-гетероарил- или -арил-O-гетероарил. При таком определении или арильная часть, или гетероарильная часть может быть присоединена к остальной части молекулы. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на арильной части или на гетероарильной части, определения которых даны выше.
Алкиларилоксиарил определен как арил-O-арил-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на арильной части, определение которой дано выше.
Алкиларилоксигетероарил определен как гетероарил-O-арил-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на арильной части или на гетероарильной части, определения которых даны выше.
Алкиларилоксиалкиламин определен как R6R7N-C1-C6алкил-O-арил-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной или арильной части, определения которых дано выше; R6 и R7 имеют значения, указанные выше.
Алкоксикарбонил определен как C1-C6-алкил-O-C=O-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной части алкоксигруппы, определение которой дано выше.
Арилоксикарбонил определен как арил-O-C=O-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на арильной части, определение которой дано выше.
Гетероарилоксикарбонил определен как гетероарил-O-C=O-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на гетероарильной части, определение которой дано выше.
Алкокси определен как C1-C6-алкил-O-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной части, определение которой дано выше.
Арилокси определен как арил-O-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на арильной части, определение которой дано выше.
Гетероарилокси определен как гетероарил-O-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на гетероарильной части, определение которой дано выше.
Алкенилокси определен как C3-C6-алкен-O-. Пример аллил-O-, группы подобные бут-2-ен-O. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на группе алкена, определение которого дано выше, при условии, что никаких гетероатомов, таких как O, S или N-R1, нет на атоме углерода, который присоединен к двойной связи.
Алкинилокси определен как C3-C6-алкин-O-. Пример CH тройная связь C-CH2-O-, подобные группы. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на группе алкина, определение которого дано выше, при условии, что никаких гетероатомов, таких как O, S или N-R1 нет на атоме углерода, который присоединен к двойной или тройной связи.
Алкиламиноалкокси определен как R6R7N-C1-C6-алкил-O-C1-C6-алкил-, где концевая алкилгруппа, присоединенная к атому кислорода, соединена с остальной частью молекулы. Значения R6 и R7 указаны выше. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной части, определение которой дано выше.
Алкилендиокси определен как -O-CH2-O- или -O-(CH2)2-O-;
Арилоксиалкиламин определен как R6R7N-C1-C6-алкил-O-арил-, где арил присоединен к остальной части молекулы; Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкильной или арильной группе, определения которых даны выше.
Арилалкенил определен как арил-C2-C8-алкен-, при условии, что никакого гетероатома, такого как O, S или N-R1, нет на атоме углерода, который присоединен к двойной связи. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на алкеновой или арильной группе, определения которых даны выше.
Гетероарилоксиалкил определен как гетероарил-O-C1-C6-алкил-. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на гетероарильной части, определение которой дано выше.
Гетероарилоксиарил определен как гетероарил-O-арил-, где арилгруппа присоединена к остальной части молекулы. Термин "необязательно замещенный" относится к незамещенному или замещенному 1 или 2 заместителями, присутствующими на гетероарилгруппе или на арилгруппе, определения которых даны выше.
Алкокси, алкоксиалкил, алкоксиалкилокси и алкилтиоалкилокси являются группами, где алкильная цепь (прямая или разветвленная) состоит из 1-6 атомов углерода. Арилокси, гетероарилокси, арилтио и гетероарилтио являются группами, где арил- и гетероарилгруппы являются такими, как указано выше. Арилалкилокси, гетероарилалкилокси, арилалкилтио и гетероарилалкилтио являются группами, где арил- и гетероарилгруппы являются такими, как указано выше, и где алкильная цепь (прямая или разветвленная) состоит из 1-6 атомов углерода. Арилоксиалкил, гетероарилоксиалкил, арилоксиалкилокси и гетероарилоксиалкилокси являются заместителями, где радикал алкил имеет 1-6 атомов углерода. Термины моноалкиламино и диалкиламино относятся к группам с одной или двумя алкилгруппами, где алкильная цепь имеет 1-6 атомов углерода и группы могут быть одинаковыми или разными. Термины моноалкиламиноалкил и диалкиламиноалкил относятся к моноалкиламино и диалкиламиногруппам с одной или двумя алкилгруппами (одинаковыми или разными), связанными с атомом азота, который присоединен к алкилгруппе из 1-3 атомов углерода.
Примерами конденсированных бициклических гетероарилгрупп являются необязательно замещенные кольцевые системы, такие как одна из следующих:
4,5,6,7-тетрагидротиено[3,2-c]пиридин, необязательно замещенный, например, арилалкилом, таким как бензил; алкоксиарилалкилом, таким как 4-метоксибензил; C1-C6-алкилом, таким как метил; гетероарилалкилом, таким как пиридин-3-илметил; группой арилалкил-CO-, такой как фенилацетил; или группой гетероарил-CO-, такой как пиридин-3-илкарбонил; например, группой алкил-CO-, такой как ацетил;
5,6,7,8-тетрагидроимидазо[1,2-а]пиразин, необязательно замещенный, например, C1-C6-алкилом, таким как метил;
5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин;
6,7-дигидро-5H-пирроло[1,2-a]имидазол;
5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин;
5,6-дигидро-4H-пирроло[1,2-b]пиразол;
4,5,6,7-тетрагидропиразоло[1,5-a]пиридин;
6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин, необязательно замещенный, например, C1-C6-алкилом, таким как метил;
6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин;
4H-5-тиа-1,6a-диазапентален;
7H-имидазо[1,2-c]тиазол;
4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин;
6,7-дигидро-4H-тиено[3,2-c]пиран;
6,7-дигидро-4H-тиено[3,2-c]тиопиран;
6,7-дигидро-4H-тиено[3,2-с]пиридин, необязательно замещенный C2-C7-алкоксикарбонилом;
6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин;
5,6,7,8-тетрагидроимидазо[1,2-a]пиразин, необязательно замещенный арилалкилом, таким как бензил;
5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-с][1,4]тиазин;
4,5,6,7-тетрагидропиразоло[1,5-a]пиразин;
5,6-дигидро-4H-циклопента[b]фуран;
4,5-дигидро-6-тиа-1,7a-диазаинден;
5,6-дигидро-8-H-имидазо[2,1-c][1,4]тиазин;
4H-5-тиа-1,6a-диазапентален;
2,3-дигидропиразоло[5,1-b]тиазол;
2,3-дигидропиразоло[5,1-b]оксазол;
6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин;
6,7-5H-дигидропиразоло[5,1-b]оксазин и
4,5,6,7-тетрагидропиразоло[1,5-a]пиразин, необязательно замещенный, например, группой алкоксиалкил-CO-, такой как 2-метоксиацетил, или группой алкилоксиалкил-CO-, такой как метоксиацетил.
Дополнительными примерами бициклических гетероарилгрупп являются следующие:
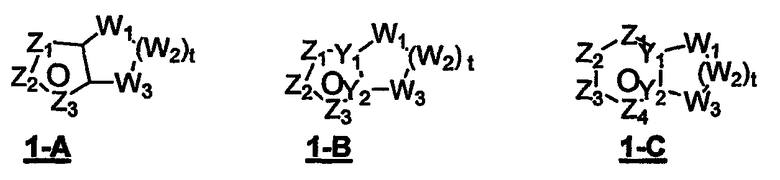
В формуле 1-A Z1, Z2 и Z3 независимо означают CR2, N, O, S или N-R1 и один из Z1-Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле 1. Когда один из Z означает CR2, два других Z могут означать либо два атома N, либо один атом N и O, S, N-R1 в любых сочетаниях без нарушения ароматической структуры; когда два Z = CR2, другой Z может быть необязательно выбран из N, O, S или N-R1 в любых сочетаниях без нарушения ароматической структуры;
W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O, N-R1, C=O при условии, что не может возникнуть ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы; t = 1-4.
В формуле 1-B Z1, Z2 и Z3 независимо означают CR2, N, O, S или N-R1 и один из Z1-Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле 1. Когда один из Z = CR2, тогда два других Z независимо могут быть CR2, N, O, S или N-R1 в любых сочетаниях без нарушения ароматической структуры;
когда два Z = N, тогда другой атом углерода в кольце связан с группой пенема молекулы, как показано в формуле 1.
W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O, N-R1, t = 1-4;
Y1 и Y2 = N или C при условии, что когда ароматическим гетероциклом является имидазол, насыщенное кольцо не может содержать S по соседству с атомом углерода в голове моста.
В формуле 1-C Z1, Z2, Z3 и Z4 независимо означают CR2 или N и один из Z1-Z4 означает углерод и связан с остальной частью молекулы.
W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1 при условии, что не возникает ни S-S, ни O-O, ни S-O связь с образованием насыщенной кольцевой системы; t = 1-4.
Y1 и Y2 независимо означают C или N.
Наиболее предпочтительный вариант формулы 1-A:
1. t = 1-3.
2. В формуле 1-A Z1 означает N, S, N-R1 или O и один из Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле 1.
3. В формуле 1-A Z3 означает N, S, N-R1 или O и один из Z2 или Z1 означает CR2 и другой Z2 или Z1 означает углерод и связан с остальной частью молекулы, как показано в формуле 1.
4. В формуле 1-A Z2 означает N, S, N-R1 или O и один из Z1 или Z3 означает CR2 и другой Z1 или Z3 означает углерод, связанный с остальной частью молекулы, как показано в формуле 1.
5. В формуле 1-A Z1 означает N, N-R1, O или S и Z2 означает N, O или S и Z3 означает углерод, связанный с группой пенема молекулы, как показано в формуле 1.
6. В формуле 1-A Z3 означает N, N-R1, O или S и Z2 означает N, O или S и Z1 означает углерод, связанный с группой пенема молекулы, как показано в формуле I.
7. В формуле 1-A Z1 означает N, N-R1, O или S и Z3 означает N, O или S и Z2 означает углерод, связанный с группой пенема молекулы, как показано в формуле I.
8. В формуле 1-A Z1 означает N, S, N-R1 или O и Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы; W1, W2 и W3 независимо означают CR4R4.
9. В формуле 1-A Z3 означает N, S, N-R1 или O и один из Z2 или Z1 означает CR2 и другой Z2 или Z1 означает углерод и связан с остальной частью молекулы; W1, W2 и W3 независимо означают CR4R4.
10. В формуле 1-A Z2 означает N, S, N-R1 или O и один из Z1 или Z3 означает CR2 и другой Z1 или Z3 означает углерод и связан с остальной частью молекулы; W1, W2 и W3 независимо означают CR4R4.
11. В формуле 1-A Z1 означает N, N-R1, O или S и Z2 означает N, O или S; Z3 означает углерод, связанный с группой пенема молекулы; W1, W2, W3 независимо означают CR4R4.
12. В формуле 1-A Z3 означает N, N-R1, O или S; Z2 означает N, O или S; Z1 означает углерод, связанный с группой пенема молекулы; W1, W2, W3 независимо означают CR4R4.
13. В формуле 1-A Z1 означает N, N-R1, O или S; Z3 означает N, O или S; Z2 означает углерод, связанный с группой пенема молекулы; W1, W2, W3 независимо означают CR4R4.
14. В формуле 1-A Z3 означает N, N-R1, O или S; Z1 означает N, O или S; Z2 означает углерод, связанный с группой пенема молекулы; W1, W2, W3 независимо означают CR4R4.
15. В формуле 1-A Z1 означает N, S, N-R1 или O; один из Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы, t = 1-3; один W2 означает N-R1, O или S(O)n, n = 0-2, и другой W2 означает CR4R4.
16. В формуле 1-A Z3 означает N, S, N-R1 или O; один из Z2 или Z1 означает CR2 и другой Z2 или Z1 означает углерод и связан с остальной частью молекулы, t = 1-3; один W2 означает N-R1, O или S(O)n, n = 0-2, и другой W2 = CR4R4.
17. В формуле 1-A Z2 означает N, S, N-R1 или O; один из Z1 или Z3 означает CR2 и другой Z1 или Z3 означает углерод и связан с остальной частью молекулы; t = 1-3; один W2 означает N-R1, O или S(O)n, n = 0-2 и другой W2 означает CR4R4.
18. В формуле 1-A, когда Z1 = N, N-R1, O или S и Z2 = N, O или S и Z3 = углерод, связанный с группой пенема молекулы, где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2 и другой W2 = CR4R4.
19. В формуле 1-A Z3 = N, N-R1, O или S и Z2 = N, O или S и Z1 = углерод, связанный с группой пенема молекулы, где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2 и другой W2 = CR4R4.
20. В формуле 1-A когда Z1 = N, N-R1, O или S и Z3 = N, O или S и Z2 = углерод, связанный с группой пенема молекулы, где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2 и другой W2 = CR4R4.
21. В формуле 1-A Z1 = N, S, N-R1 или O и Z2 или Z3 = CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы; тогда W1 и W3 = CH2 или оба атома водорода на метиленовой связи могут быть замещены с образованием спиро-системы с наличием или без гетероатомов, выбранных из O, S=(O)n (n = 0-2), N-R1, с образованием от пяти- до восьмичленной циклической системы; t = 1-3; один W2 = N-R1, O или S(O)n, n = 0-2 и другой W2 = CR4R4.
22. В формуле 1-A Z3 = N, S, N-R1 или O и Z2 или Z1 = CR2 и другой Z2 или Z1 означает углерод и связан с остальной частью молекулы; тогда W1 и W3 = CR4R4; где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2 и другой W2 = CR4R4.
23. В формуле 1-A Z2 = N, S, N-R1 или O и Z1 или Z3 = CR2 и другой Z1 или Z3 означает углерод и связан с остальной частью молекулы; тогда W1 и W3 = CR4R4, где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2, и другой W2 = CR4R4.
24. В формуле 1-A, когда Z1 = N, N-R1, O или S и Z2 = N, O или S, тогда Z3 = углерод, связанный с группой пенема молекулы; тогда W1 и W3 = CR4R4, где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2 и другой W2 = CR4R4.
25. В формуле 1-A Z3 = N, N-R1, O или S и Z2 = N, O или S, тогда Z1 = углерод, связанный с группой пенема молекулы; тогда W1 и W3 = CR4R4, где t = 1-3, тогда один W2 = N-R1, O или S(O)n, n = 0-2, и другой W2 = CR4R4.
26. В формуле 1-A когда Z1 = N, N-R1, O или S и Z3 = N, O или S, тогда Z2 = углерод, связанный с группой пенема молекулы; тогда W1 и W3 = CR4R4; t = 1-3; один W2 означает N-R1, O или S(O)n, n = 0-2, и другой W2 означает CR4R4.
27. В формуле 1-A Z3 означает N, N-R1, O или S; Z1 означает N, O или S; Z2 означает углерод, связанный с остальной частью молекулы; W1 и W3 независимо означают CR4R4; t = 1-3; один W2 означает N-R1, O или S(O)n, n = 0-2, и другой W2 означает CR4R4.
Наиболее предпочтительные варианты формулы 1-B:
28. В формуле 1-B t=3.
29. В формуле 1-B Z1 и Z3 означают N; Y1 означает N; Y2 означает C и Z2 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
30. В формуле 1-B Z2 и Z3 означают N; Y1 означает N; Y2 означает C и Z1 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
31. В формуле 1-B Z1 означает N, Y1 означает N, Y2 означает C, один из Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
32. В формуле 1-B Z1 означает N, Y1 означает C, Y2 означает N, один из Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
33. В формуле 1-B Z1 означает N, Y1 означает N, Y2 означает C, один из Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле 1, W1 и W3 независимо означают CR4R4; t = 1-3; один W2 означает N-R1, O, S=(O)n (n = 0-2) и другой W2 означает CR4R4.
34. В формуле 1-B Z1 означает N, Y1 означает C, Y2 означает N, один из Z2 или Z3 означает CR2 и другой Z2 или Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле I; W1 и W3 независимо означают CR4R4; t = 1-3; один W2 означает N-R1, O, S=(O)n (n = 0-2) и другой W2 означает CR4R4.
35. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; один из Z1 или Z2 означает CR2 и другой Z1 или Z2 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
36. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C; один из Z1 или Z3 означает CR2 и другой Z1 или Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
37. В формуле 1-B Z1 и Z2 означают N; Y1 означает N; Y2 означает C и Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
38. В формуле 1-B Z1, Z2 и Z3 независимо означают CR2; Y1 означает C; Y2 означает N; за исключением того, что один из Z1-Z3 означает углерод и связан с остальной частью молекулы, как показано в формуле I.
39. В формуле 1-B Z1 и Z3 означают N; Y1 означает N; Y2 означает C; Z2 означает углерод и связан с остальной частью молекулы, как показано в формуле I; и t = 1-3.
40. В формуле 1-B Z2 и Z3 означают N; Y1 означает N; Y2 означает C; и Z1 означает углерод и связан с остальной частью молекулы, и t = 1-3;
41. В формуле 1-B Z2 и Z3 означают N, Y1 означает C и Y2=N и Z1 означает углерод и связан с остальной частью молекулы и t = 1-3;
42. В формуле 1-B Z2 и Z3 означают N, Y1 означает N; Y2 означает C; Z1 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CH2 или оба атома водорода на метиленовой связи могут быть замещены с образованием спиро-системы с наличием или без гетероатомов, выбранных из O, S(O)n, n = 0-2, N-R1, с образованием от пяти- до восьмичленной циклической системы; t = 1-3 и W2 означает CH2, N-R1, O, S(O)n, где n = 0-2.
43. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2 и Z2 означает атом углерода, связанный с остальной частью молекулы.
44. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z2 означает атом углерода, связанный с остальной частью молекулы; W1, W2 и W3 независимо означают CR4R4; t = 1-3.
45. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z2 означает атом углерода, связанный с остальной частью молекулы; W1 и W3 независимо означают CR4R4; и один из W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4; t = 1-3.
46. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z2 означает атом углерода, связанный с остальной частью молекулы; W1 и W2 независимо означают CR4R4; W3 означает N-R1, O или S(O)n; и t = 2.
47. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z2 означает атом углерода, связанный с остальной частью молекулы; W1 и W3 независимо означают CR4R4; W2 означает N-R1, O или S(O)n; и t = 1.
48. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C; Z3 означает CR2; Z1 означает углерод, связанный с остальной частью молекулы; W1 и W2 означают CH2 или оба атома водорода на метиленовой связи могут быть замещены с образованием спиро-системы с наличием или без гетероатомов, выбранных из O, S(O)n, n = 0-2, N-R1, с образованием от пяти- до восьмичленной циклической системы; W3 означает N-R1, O или S(O)n; и t = 3.
49. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C, Z3 означает CR2; Z1 означает углерод, связанный с остальной частью молекулы; W1 и W3 независимо означают CH2 или оба атома водорода на метиленовой связи могут быть замещены с образованием спиро-системы с наличием или без гетероатомов, выбранных из O, S(O)n, n= 0-2, N-R1, с образованием от пяти- до восьмичленной циклической системы; и один W2 означает N-R1, O или S(O)n, и другой W2 означает CR4R4; и t = 2.
50. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C; Z3 означает CR2; Z1 означает углерод, связанный с остальной частью молекулы; W1 и W3 независимо означают CH2 или оба атома водорода на метиленовой связи могут быть замещены с образованием спиро-системы с наличием или без гетероатомов, выбранных из O, S(O)n, n = 0-2, N-R1, с образованием от пяти- до восьмичленной циклической системы; W2 означает N-R1, O или S(O)n; и t = 1.
51. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z3 означает атом углерода, связанный с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один из W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4, и t = 3.
52. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z3 означает атом углерода, связанный с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4; и t = 2.
53. В формуле 1-B Z2 означает N; Y1 означает N; Y2 означает C; Z1 означает CR2; Z3 означает атом углерода, связанный с остальной частью молекулы; W1 и W3 независимо означают CR4R4; W2 означает N-R1, O или S(O)n; и t = 1.
54. В формуле 1-B Z1 и Z2 означают N; Y1 означает N; Y2 означает C; Z3 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один из W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4; и t = 3.
55. В формуле 1-B Z1 и Z2 означают N; Y1 означает N; Y2 означает C; Z3 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один из W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4; и t = 2.
56. В формуле 1-B Z1 и Z2 означают N; Y1 означает N; Y2 означает C; Z3 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; W2 означает N-R1, O или S(O)n; и t = 1.
57. В формуле 1-B Z1 и Z2 независимо означают CR2; Y1 означает C; Y2 означает N; Z3 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один из W2 означает N-R1, O или S(O)n; другой W2 означает CR4R4 и t = 3.
58. В формуле 1-B Z1 и Z2 независимо означают CR2; Y1 означает C и Y2 означает N и Z3 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; и один W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4 и t = 2.
59. В формуле 1-B Z1 и Z2 независимо означают CR2; Y1 означает C; Y2 означает N; Z3 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; W2 означает N-R1, O или S(O)n; и t = 1.
60. В формуле 1-B Z1 и Z3 независимо означают CR2; Y1 означает C; Y2 означает N; Z2 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один W2 означает N-R1, O или S(O)n; другой W2 означает CR4R4 и t = 3.
61. В формуле 1-B Z1 и Z3 независимо означают CR2; Y1 означает C; Y2 означает N; Z2 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; и один W2 означает N-R1, O или S(O)n и другой W2 означает CR4R4; и t = 2.
62. В формуле 1-B Z1 и Z3 независимо означают CR2; Y1 означает C; Y2 означает N; Z2 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; W2 означает N-R1, O или S(O)n; и t = 1.
63. В формуле 1-B Z3 и Z2 независимо означают CR2; Y1 означает C; Y2 означает N; Z1 означает углерод и связан с остальной частью молекулы; W1 и W2 независимо означают CR4R4; один W2 означает N-R1, O или S(O)n; другой W2 означает CR4R4; и t = 3.
64. В формуле 1-B Z3 и Z2 независимо означают CR2; Y1 означает C; Y2 означает N; Z1 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; один W2 означает N-R1, O или S(O)n; другой W2 означает CR4R4; и t = 2.
65. В формуле 1-B Z3 и Z2 независимо означают CR2; Y1 означает C; Y2 означает N; Z1 означает углерод и связан с остальной частью молекулы; W1 и W3 независимо означают CR4R4; W2 означает N-R1, O или S(O)n; и t = 1.
66. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; один из Z1 и Z2 означает CR2 и другой означает C; W1 означает CR4R4; W2 означает CR4R4; W3 означает CH2, N-R1 или O; и t = 1.
67. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; один из Z1 и Z2 означает CR2 и другой означает C; W1 означает CR4R4; W2 означает C=O; W3 означает N-R1, и t = 1.
68. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; один из Z1 и Z2 означает CR2 и другой означает C; W1 означает N-R1, W2 означает C=O; W3 означает CR4R4; и t = 1.
69. В формуле 1-B Z3 означает N; Y1 означает N; Y2 означает C; один из Z1 и Z2 означает CR2 и другой означает C; W1 означает C=O; W2 означает N-R1; W3 означает CH2; и t = 1.
Наиболее предпочтительные варианты формулы 1-C являются:
70. В формуле 1-C Z1, Z2, Z3 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
71. В формуле 1-C Z1, Z2, Z3 и Z4 независимо означают CR2 и один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 и Y2 = C или N; t = 1-3; W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
72. В формуле 1-C Z1, Z2, Z3 и Z4 независимо означают CR2; Y1 и Y2 означают N; t = 1-3; W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
73. В формуле 1-C Z1 означает N и Z2, Z3 и Z4 независимо означают CR2; Y1 и Y2 означают C; t = 1-3; W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
74. В формуле 1-C Z1 означает N и Z2, Z3 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает C; Y2 означает N; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
75. В формуле 1-C Z2 = N и Z1, Z3 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
76. В формуле 1-C Z2 означает N и Z1, Z3 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает C; Y2 означает N; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
77. В формуле 1-C Z3 означает N; Z1, Z2 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
78. В формуле 1-C Z3 означает N и Z1, Z2 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает C и Y2 означает N; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
79. В формуле 1-C Z4 означает N и Z1, Z2 и Z3 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, О или N-R1.
80. В формуле 1-C Z4 означает N и Z1, Z2 и Z3 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает N; Y2 означает C; t =1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, О или N-R1.
81. В формуле 1-C Z1 означает N и Z2, Z3 и Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, О или N-R1.
82. В формуле 1-C Z1 и Z2 означают N и Z3 или Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает C; Y2 означает N; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
83. В формуле 1-C Z1 и Z3 означают N и Z2 или Z4 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает C; Y2 означает N; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
84. В формуле 1-C Z1 и Z4 означают N и Z2 или Z3 независимо означают CR2; один из Z1-Z4 означает углерод и связан с остальной частью молекулы; Y1 означает N; Y2 означает C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
85. В формуле 1-C Z1, Z2, Z3 означают N и Z4 означает углерод и связан с остальной частью молекулы; Y1 означает C; Y2 означает N; t =1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или H-R1.
86. В формуле 1-C Z1, Z3 и Z4 означают N и Z2 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C; t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
87. В формуле 1-C Z1, Z2 и Z4 означают N и Z3 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C и t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
88. В формуле 1-C Z2, Z3, Z4 означают N и Z1 означает углерод и связан с остальной частью молекулы; Y1 и Y2 означают C и t = 1-3; и W1, W2 и W3 независимо означают CR4R4, S, SO, SO2, O или N-R1.
Наиболее предпочтительными соединениями по данному изобретению являются:
1. (5R,6Z)-6-[(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота;
2. (5R),(6Z)-6-(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
3. (5R),(6Z)-7-оксо-6-(5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
4. (5R,6Z)-6-{[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
5. (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
6. (5R),(6Z)-6-(6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
7. (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
8. (5R),(6Z)-6-(5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
9. (5R)(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
10. (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
11. (5R)(6Z)-6-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
12. (5R)(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
13. (5R)(6Z)-6-(7H-имидазо[1,2-c]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль;
14. (5R,6Z)-7-оксо-6-[(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метилен]-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота;
15. 6-(6,7-дигидро-4H-тиено[3,2-c]пиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота;
16. 6-(6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота;
17. 6-(5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота;
18. 2-(2-карбокси-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-илиденметил)-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновая кислота, этиловый сложный эфир;
19. 7-оксо-6-(6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота;
20. (5R),(6Z)-6-(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
21. (5R,6Z)-7-оксо-6-{[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
22. (5R,6Z)-7-оксо-6-{[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
23. (5R,6Z)-7-оксо-6-{[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
Соединения по данному изобретению обладают ингибирующим свойством в отношении β-лактамазы и антибактериальным свойством и применимы для лечения инфекционных заболеваний людей и животных. Следует отметить, что применение соединений по данному изобретению в сочетании с β-лактамными антибиотиками приводит к повышенной антибактериальной активности (синергический эффект) против организмов, продуцирующих (лактамазы) класса-А и класса-С. β-лактамные антибиотики включают пенициллиновые антибиотики, такие как пиперациллин, амоксициллин, тикарциллин, бензилпенициллины, ампициллин, другие известные пенициллины, и цефалоспорины, такие как цефатризин, цефалоридин, цефалотин, цефазолин, цефалексин, цефрадин, другие известные цефалоспорины, азтреонам и латамоксеф (моксалактам). Наиболее предпочтительно, соединения по данному изобретению использовать с пиперациллином или амоксициллином, который имеет широкий спектр активности против грамположительных и грамотрицательных патогенов.
Соединения по данному изобретению могут использоваться до, одновременно или последовательно с β-лактамным антибиотиком ("совместное введение"). Под использованием подразумевается введение соединения непосредственно или in vivo, например, пролекарства. Когда соединения по данному изобретению вводят совместно с β-лактамным антибиотиком, отношение количества соединения к количеству β-лактамного антибиотика может изменяться в широком диапазоне. Отношение β-лактамного антибиотика к ингибитору β-лактамазы может изменяться от 1:1 до 100:1. Предпочтительно отношение β-лактамного антибиотика к ингибитору β-лактамазы менее чем 10:1. Состав по данному изобретению может быть в форме, подходящей для перорального (РО), внутривенного (IV) или местного введения. Составы по изобретению могут быть в форме таблеток, капсул, кремов, сиропов, суспензии, стерильных растворов, подходящих для инъекции или вливания. Предпочтительно, соединения по данному изобретению вводят совместно с пиперациллином внутривенно или с амоксициллином внутривенно или перорально.
Структурная формула соединений включает любые таутомеры, любые стереоизомеры (кроме случаев, когда стереохимия ясно обозначена) и всякие кристаллические формы.
Определение IC50 для ингибитора на основе пенема
Ингибирующую β-лактамазу активность ингибиторов на основе пенема определяют спектрофотометрически, как описано Bush и др. [Bush, K., Macalintan, C., Rasmussen, B.A., Lee, V. и Yang, Y. Antimicrobial Agents и Chemotherapy 1993, 37, 851]. В анализе использовали гомогенно очищенные β-лактамазы класса А ТЕМ-1 из E. coli и Imi-1 из Enterobacter cloacae, фермент CcrA класса В из Bacteroides fragilis и фермент AmpC класса С из Enterobacter cloacae. Концентрации фермента для ТЕМ-1, Imi-1, CcrA и AmpC были 4,3, 7,1, 1,2 и 2,1 нМ соответственно. Широкий диапазон концентраций ингибитора получали в 50 мМ РО4, рН 7,0, чтобы включить возможные величины IC50. Субстратом, используемым для инициирования реакции фермента, был нитроцефин при 50 мкг/мл в том же буфере, как для ингибитора. Вначале фермент и ингибитор (20 мкл каждого) подвергали предварительной инкубации в течение 10 минут при 25°С перед добавлением объема 160 мкл нитроцефина. Первоначальные степени гидролиза отслеживали в течение 5 минут с помощью молекулярных устройств Spectra Max 250 c кинетическим протоколом программы SoftMax. Показания от Spectra Max 250 выводили и передавали к Microsoft Excel. Процент ингибирования для каждой концентрации ингибитора рассчитывали на основе контрольной активности фермента. Концентрацию ингибитора, которая послужила причиной 50% снижения ферментной активности (IC50), определяли графически.
Испытание антимикробной чувствительности. Активности in vitro антибиотиков определяли методом разбавления микробульона, как рекомендовано Национальным комитетом по клиническим лабораторным стандартам (NCCLS). (NCCLS. 2000. Методы испытаний антимикромной чувствительности разбавлением для бактерий, которые растут аэробно; утвержденные стандарты: М7-А5, том 19, Национальный комитет по клиническим лабораторным стандартам, Vilanova, PA). Для процедуры испытания использовали бульон II Mueller-Hinton (MHBII) (BBL Cockeysville, MD). Микротитрационные планшеты, содержащие 50 мкл на лунку двукратных последовательных разбавлений пиперациллина, объединенных с постоянным количеством (4 мкг/мл) ингибитора β-лактамазы, инокулировали 50 мкл инокулята, чтобы получить соответствующую плотность (105 КОЕ/мл) в конечном объеме 100 мкл. Планшеты инкубировали в течение 18-22 часов при 35°С в присутствии воздуха. Минимальную ингибирующую концентрацию (MIC) для всех изолятов определяли как наименьшую концентрацию антимикробного агента, которая полностью ингибирует рост организма, что обнаруживали невооруженным глазом. Данные MIC, полученные указанной выше процедурой, представлены в таблице 2.
Данные минимальной ингибирующей концентрации (мкг/мл): Inc: 35°С в течение 18 часов
Антибактериальная защита in vivo
Материалы:
Животные:
Самок мышиной расы CD-1, приблизительно 18-22 г, получали от Charles River Laboratories и выдерживали 7 дней до использования. В дополнение у мыши могло быть уменьшенное с помощью цитоксана содержание нейтрофильных гранулоцитов в крови.
Инфекции:
В эксперименте используют клинические изоляты, адаптированные для вызывания инфекции у мышей, включая инфекции штаммами E. coli, K. pneumoniae, M. morganii, E. cloacae, S. marcescens, C. freundii, staphylococci, streptococci, P. aeruginosa и N. gonorrhoeae.
Подготовка:
Животных размещают по пять в клетке со свободным доступом к пище и воде в соответствии с указаниями NIH.
Экспериментальный протокол:
Мышей провоцируют впрыскиванием 0,5 мл интраперитонеально или 0,05 мл интраназально заранее определенного бактериального инокулята, суспендированного в бульоне, физиологическом растворе или свином желудочном муцине (обогащенном сухим бычьим гемоглобином для N. gonorrhoeae). Бактериальный инокулят эквивалентен 10-100 LD50 специфического инфекционного штамма и будет иметь результатом гибель необработанных контрольных животных в пределах 7 дней: "Бактериальная вирулентность на мышах". Антибактериальные дозы (концентрация дозы, полученная двукратными последовательными разбавлениями антибиотика) растворяют или суспендируют в 0,2% водном агаре или метоцеле, забуференный фосфатом физиологический раствор или адъювант вводят перорально, подкожно или внутривенно следующим образом:
а) Перорально или подкожно: Дозу объемом 0,5 мл вводят через 1/2 часа после заражения. Вторая доза может быть введена через 3 ч после заражения для лечения инфекций, вызываемых более вирулентными организмами.
b) Внутривенно: Дозу объемом 0,2 мл вводят через 1/2 часа после заражения. Для лечения инфекций, вызываемых более вирулентными организмами, может быть введено больше доз в течение вплоть до 48 ч. (Внутривенное дозирование не должно превышать 3 дозы/24 ч период.)
с) Пероральная предварительная обработка: В конкретных условиях требуется регулирование рН желудка для того, чтобы повысить устойчивость антибиотика в желудке. Для этой цели 0,5 мл забуференного фосфатом физиологического раствора (рН 7,8, 0,06 М) (или конкретного подходящего адъюванта) вводят перорально через 1/2 часа после заражения, затем, спустя 5 минут, вводят 0,5 мл антибиотика (также перорально), содержащегося в забуференном фосфатом физиологическом растворе (рН 7,8, 0,06 М).
Разновидности животных
Далее следует подробное объяснение в отношении числа животных, необходимых для определения эффективности in vivo:
А) Новые антибиотики испытывают при 5 различных уровнях доз на 5 мышах на уровень дозы при каждом из трех путей введения (пероральном, подкожном и внутривенном). Первоначально три пути введения должны быть исследованы с тем, чтобы определить, абсорбируется ли лекарство перорально и/или который из них является наиболее эффективным путем. Для этого требуется 25 мышей/путь с 3 путями/антибиотик или 75 мышей на новое испытуемое соединение. Один-два новых антибиотика могут быть испытаны за эксперимент (75-150 мышей).
В) Эффективность нового соединения должна быть изучена в сравнении со стандартом или антибиотиком известной эффективности. Известные или предварительно испытанные антибиотики испытывают при 5 уровнях доз с 5 мышами на уровень дозы единственным путем введения в сумме на 25 мышах/антибиотик. Обычно 3-6 антибиотиков может быть испытано за эксперимент (75-150 мышей).
С) Необработанные контроли - в каждом из указанных испытаний необработанных животных заражают 3 различными концентрациями бактериального инокулята с 10 мышами на концентрацию (вцелом 30 мышей на каждое и любое испытание). Эти необработанные контроли используют для определения и поддержания уровня инфекции между 10-100 LD50, как требуется для сравнения испытания с испытанием и для обоснованности.
Определение защитных эффектов антибактериальных агентов
Защитные эффекты антибактериального агента (агентов) измеряют по выжившим инфицированным необработанным животным в сравнении с обработанными животными. Для этого определения животных обследуют через 7 дней после обработки. Перепись выживших осуществляют дважды в день и в это время мертвых, а также умирающих животных удаляют. Отношения выживших за 7 дней из трех отдельных испытаний объединяют для оценки средней эффективной дозы (ED50) с использованием компьютеризованной программы для пробит-анализа (Cleeland, R. and E. Squires. 1991. Evaluation of New Antimicrobials in Vitro and in Experimental Animal Infections. In Antibiotics in Laboratory Medicine", 3-е издание, издано Victor Lorian. Willams and Wilkins Baltimore, Maryland. стр. 752-783). Испытание проводят три раза в отдельные дни, чтобы обеспечить статистически обоснованное число животных и свести к минимуму отклонения в результатах испытаний на основе результатов от одного дня к другому и от испытания к испытанию.
Способ по изобретению
Данное изобретение относится к способу получения соединения формулы I, который содержит подвергание восстановительному элиминированию соединения формулы II:
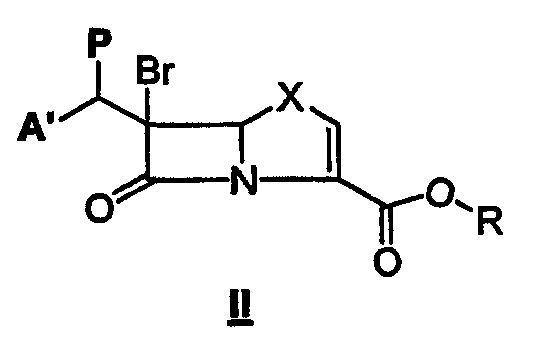
где А' означает А или В, значения которых указаны выше, Х означает О или S, Р означает сложноэфирную уходящую группу, например ацетат, мезилат, трифлат или тозилат, и R означает защитную группу с последующим, если необходимо, удалением защитной группы, чтобы получить соединение формулы I, где R5 означает водород и, если желательно, превращением в фармацевтически приемлемую соль или в сложный эфир, где R5 означает C1-С6-алкил, C5-С6-циклоалкил или CHR3OCOC1-С6-алкил.
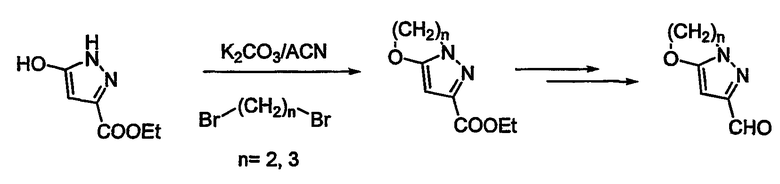
Соединения общей формулы I удобно могут быть получены новым мягким и легким путем, а именно конденсацией подходяще замещенного альдегида 4 с производным 6-бромпенема структуры 1 (схема 1) в присутствии безводного MgBr2 или MgBr2:эфирата и основания, такого как триэтиламин или DBU или DMAP, предпочтительно при температуре от -20°С до -40°С. Промежуточный альдольный продукт 5 может быть функционализован хлорангидридами или ангидридами кислот до ацетата, трифлата или тозилата 6. Соединение 6 может быть спокойно превращено в желательный продукт в процессе восстановительного элиминирования с использованием металла, такого как активированный цинк, и фосфатного буфера при 20°С - 35°С при рН от 6,5 до 8,0. Если защитной группой на кислороде карбоксилата является заместитель п-нитробензил, тогда восстановительное элиминирование и удаление защитной группы могут быть достигнуты за одну стадию. Однако если защитная группа иная, чем заместитель п-нитробензил, может последовать двухстадийная процедура в зависимости от характера защитной группы. Продукт может быть изолирован как свободная кислота или как соль щелочного металла. Вышеупомянутая двухстадийная процедура может быть осуществлена в одну стадию проведением всего процесса в одну стадию без изоляции промежуточного соединения 6. Это наиболее широко распространенная относительно простая и эффективная процедура в смысле выхода и экономической осуществимости. Эта процедура может быть приспособлена для синтеза в промышленном масштабе и является согласуемой с разнообразными альдегидами. В качестве варианта соединение 6 может быть галогенировано при давлении 40 фунт/кв. дюйм в присутствии Pd/C (10%) в THF и 6,5 фосфатного буфера, чтобы получить конечный продукт.
Вышеупомянутая реакция альдольной конденсации является очень гибкой и может быть применена к любому производному бромпенема, где карбоксигруппа защищена иной группой, чем 4-нитробензил. Примерами других защитных групп являются бензил, производное п-метоксибензила, бензгидрол, производные тритила, алкила и аллила. Однако когда защитная группа иная, чем 4-нитробензилгруппа, отдельная стадия удаления защитной группы обязательно должна быть проведена после процедуры восстановительного элиминирования. Химические механизмы, связанные со стадией удаления защитной группы, хорошо известны специалистам в этой области.
Схема 1
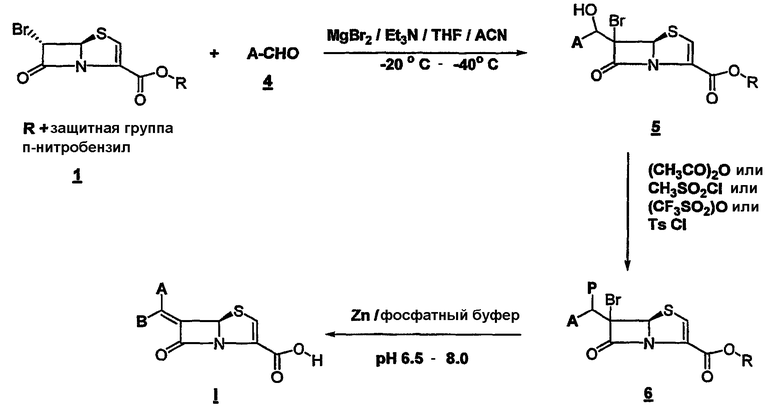
Р = -OMs (мезилат), -OTf (трифлат), -OTs (тозилат)
Необходимые альдегиды 4 для указанных выше превращений могут быть получены из их соответствующих спиртовых производных окислением MnO2 или окислением по Swern. В некоторых случаях необходимая альдегидная функциональность может быть введена непосредственно в гетероциклическую часть молекулы реакцией Vilsmier Haack с использованием DMF/POCl3. Альдегиды, необходимые для данного исследования, могут быть получены, как изображено на схемах 2-5. N-(трет-бутоксикарбонил)- (т.е.) t-Boc-защищенный -4-пиперидон обрабатывают DMF/POCl3, чтобы получить 4-хлор-3-формил-производное (схема 2). Эта реакция может быть проведена на тетрагидро-4Н-пиран-4-оне и соответствующем производном тетрагидро-4Н-пиран-4-она, чтобы получить соответствующие кислородные и серные производные. Эта реакция также может быть проведена на производных пяти-восьми-членных циклических кетонов. Промежуточное хлорформил-соединение может быть подвергнуто взаимодействию с 2-меркапто-этилацетатом, чтобы получить тиено-производное. Сложный эфир может быть превращен в спирт, который может быть превращен в исходную альдегидную функциональность. Схема 3 иллюстрирует получение производного имидазоло-тетрагидропиридина и производного имидазолопиразина. 2-аминопиридин или 2-аминопиразин могут быть подвергнуты взаимодействию с этил-бромпируватом в кипящем этаноле, чтобы получить циклизованное производное (схема 3). Восстановление одного кольца может быть достигнуто.
Схема 2
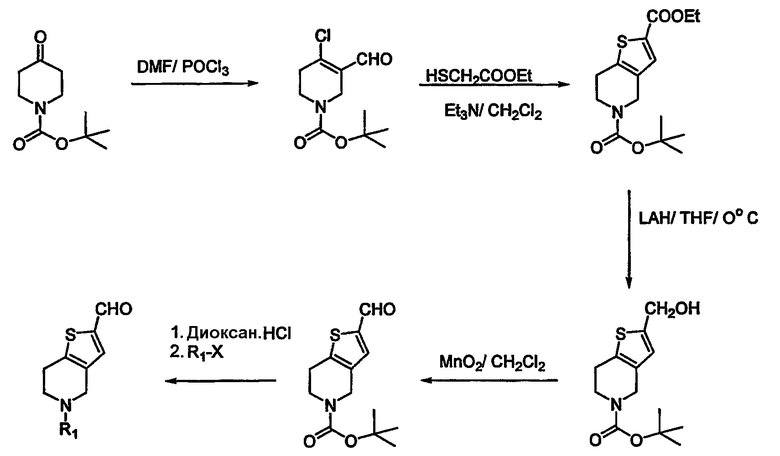
Указанная последовательность может быть проведена исходя из тетрагидро-4Н-пиран-4-она и соответствующего тетрагидро-4Н-тиопиран-4-она. Реакция Vilsmier может быть осуществлена на пяти-восьми-членных циклических кетонах.
Схема 3
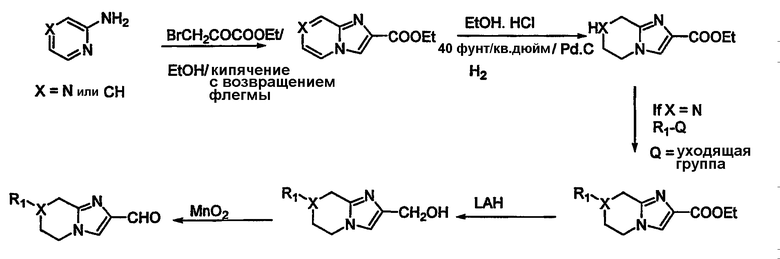
...гидрогенизацией этого над Pd/C при давлении 40 фунт/кв. дюйм в стандартном аппарате. После этого сложноэфирная группа может быть восстановлена до спирта и превращена в альдегид. В случае Х = N промежуточный сложный аминоэфир может быть дериватизирован с использованием подходящего R1Q (где Q означает уходящую группу или конденсирующуюся группу). В случае схемы 3, где R1 = Н, может быть синтез по процедуре, показанной в общих чертах на схеме 4.
Схема 4
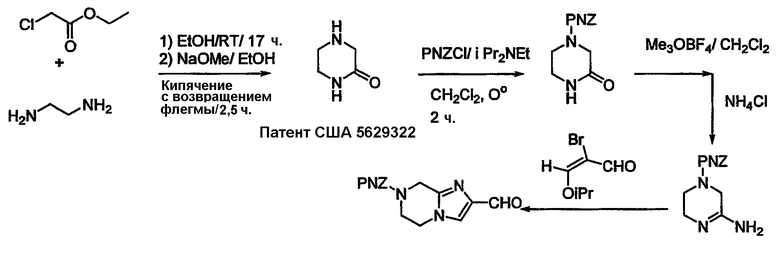
Дополнительные альдегиды могут быть синтезированы, как показано в общих чертах на схемах 5-7.
Схема 5
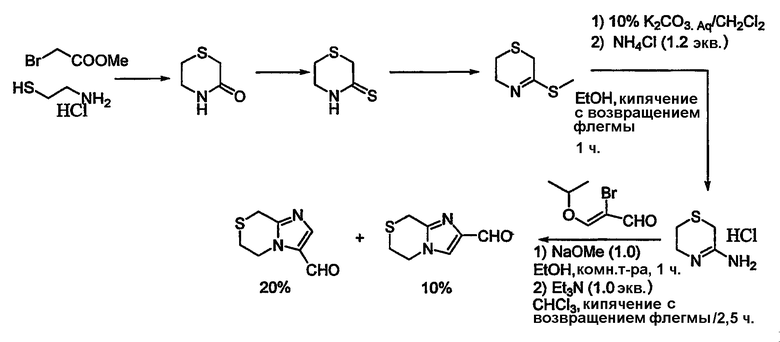
Альдегиды, необходимые для примеров 24-32, 34 и 35 получают путем, показанным на схемах 8-18.
Схема 6
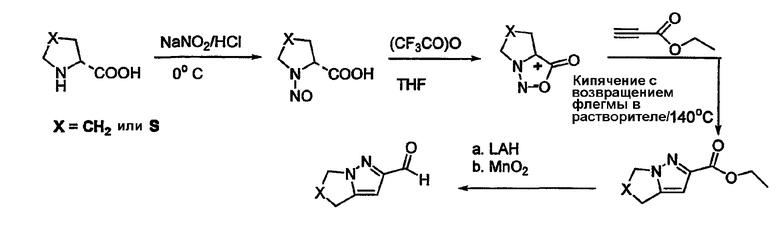
Схема 7
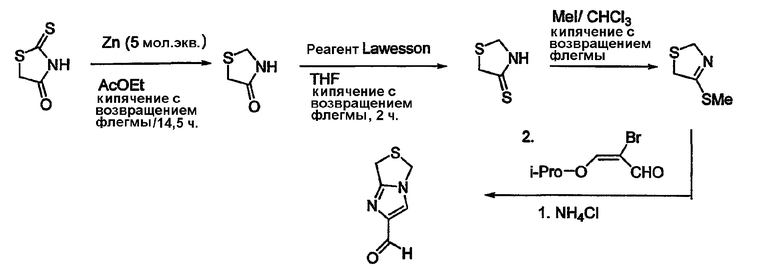
Схема 8
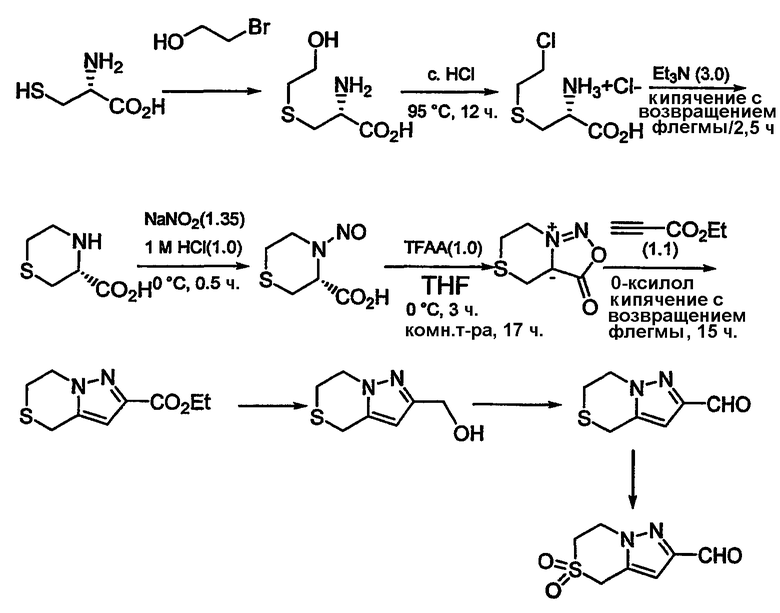
Схема 9
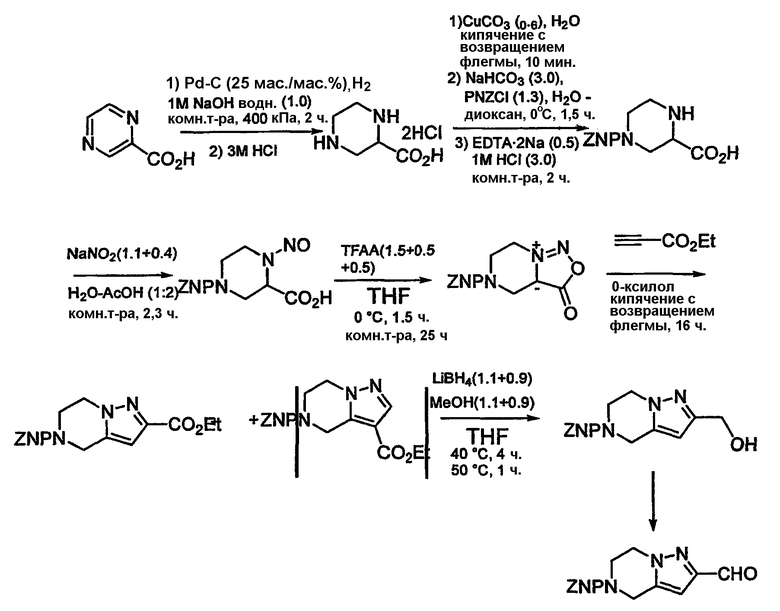
Схема 10
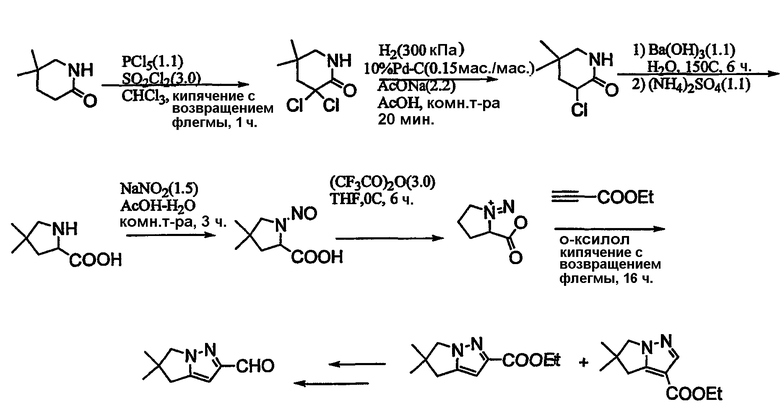
Схема 11
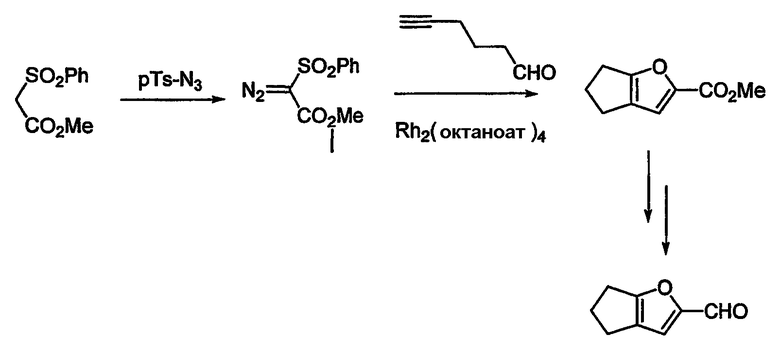
Схема 12
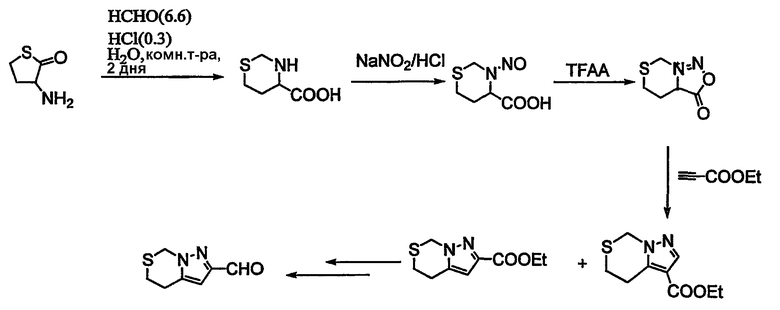
Схема 13
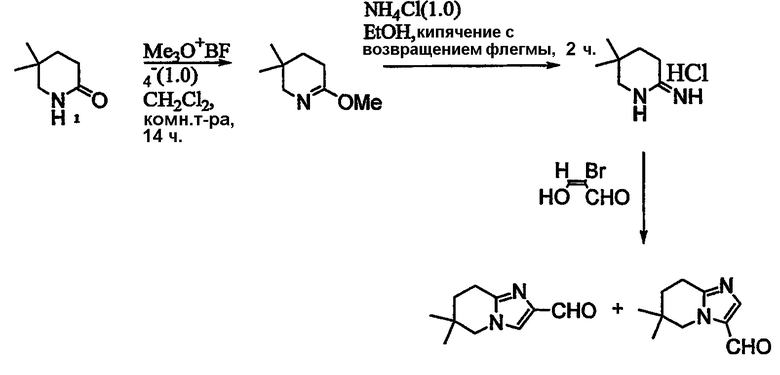
Схема 14
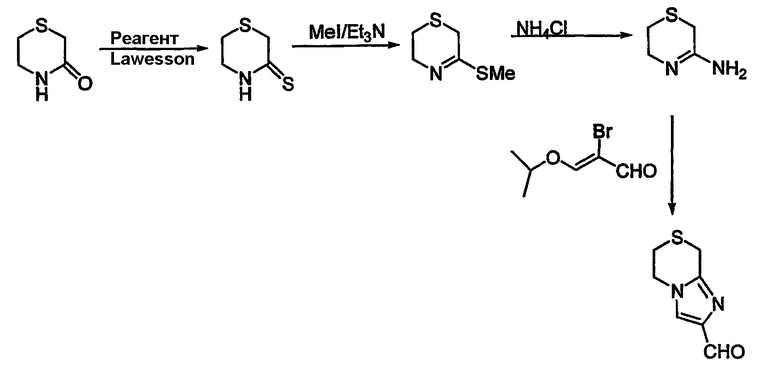
Схема 15
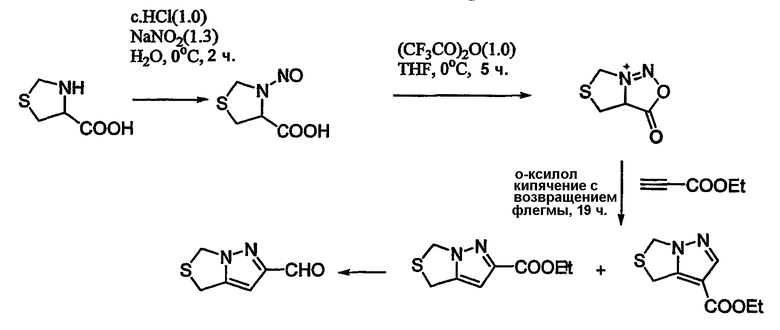
Схема 16
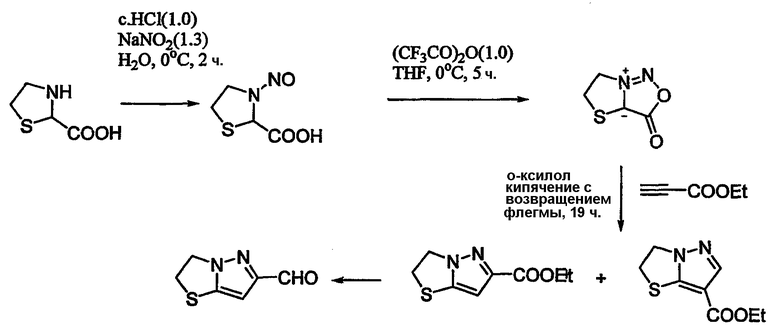
Схема 17
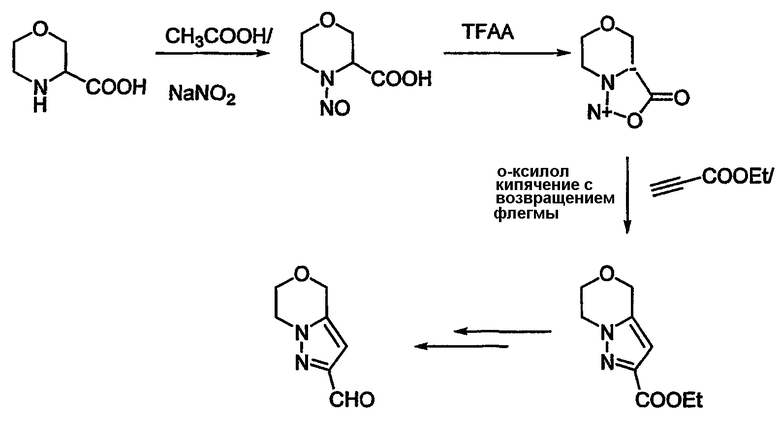
Схема 18
Экспериментальная часть
Пример 1
Получение (5R,6Z)-6-[(5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метилен]-7оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: этил 5-бензоил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбоксилат:
К перемешиваемому сухому DMF (7,3 г, 100 ммоль) медленно добавляют POCl3 (12,25 г, 80 ммоль) при температуре между 0°C и 5°C. После добавления отвержденную массу растворяют в CH2Cl2 (20 мл) и перемешивают при комнатной температуре в течение 2 ч. Снова понижают температуру до 0°C и медленно добавляют 1-бензоил-4-пиперидон в CH2Cl2. После добавления реакционную смесь перемешивают при комнатной температуре в течение 2 ч и выливают на измельченный лед и ацетат натрия. Все это перемешивают в течение 30 минут при комнатной температуре. Экстрагируют CH2Cl2; хорошо промывают водой, сушат над безводным MgSO4 и концентрируют. Сырой продукт растворяют в CH2Cl2 и медленно добавляют смесь этилмеркаптоацетата (9,6 г, 80 ммоль)/Et3N (10,1 г, 100 ммоль) при комнатной температуре. Реакционную смесь кипятят с возвращением флегмы в течение 2 ч и гасят водой. Слой CH2Cl2 хорошо промывают водой, сушат над безводным MgSO4, фильтруют и концентрируют. Продукт очищают колоночной хроматографией на SiO2, элюируя смесью 50% этилацетат-гексан. Желтое масло. Выход: 6,4 г (25%); M+H 316.
Стадия 2: (5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метанол:
К перемешиваемой суспензии LAH (2,0 г) медленно добавляют при 0°C раствор этил 5-бензоил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбоксилата (6,0 г, 19 ммоль) в THF. После добавления реакционную смесь перемешивают в течение 30 минут и гасят насыщенным NH4Cl. Все это разбавляют CHCl3 и фильтруют. Фильтрат промывают насыщенным раствором соли и сушат над безводным MgSO4. Это фильтруют и забирают на следующую стадию без очисток. Выход: 4,5 г, 91%. Желтая жидкость.
Стадия 3: 2-формил (5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин:
К перемешиваемому раствору (5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метанола (4,0 г, 15,4 ммоль) в CH2Cl2 (300 мл) добавляют активированный MnO2 (20 г, избыток) и перемешивают при комнатной температуре в течение 18 ч. В конце реакционную смесь фильтруют через целит и промывают CHCl3. Реакционную смесь хорошо промывают водой, сушат и концентрируют. Находят, что продукт чистый и забирают его на следующую стадию без очисток. Выход: 3,0 г (76%). (M+H: 257).
Стадия 4: 4-нитробензил-6-[(ацетилокси)(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат:
2-Формил (5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин (565 мг, 2,2 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (390 мг, 1,5 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 550 мг, 40%. M+H 687.
Стадия 5: (5R,6Z)-6-[(5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота:
4-Нитробензил-6-[(ацетилокси)(5-бензил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (450 мг, 0,65 ммоль) растворяют в THF (20 мл) и ацетонитриле (10 мл). Свежеактивированную Zn пыль (5,2 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 часов при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C, получая желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой НР21. Вначале колонку элюируют деионизированной водой (2 л) и позднее смесью 10% CAN:вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном и фильтруют. Сушат. Выход: 50 мг, 18% в виде желтых кристаллов, т.пл. 198°C. (M+H) 411.
1H ЯМР (ДМСО-d6) δ д 2,7 (м, 2H), 2,8 (уш.м, 2H), 3,4 (м, 2H), 3,8 (с, 2H), 6,3 (с, 1H), 6,5 (с, 1H), 7,1 (с, 1H), 7,28 (с, 1H), 7,4 (c, 5H).
Пример 2
Получение натриевой соли (5R),(6Z)-6-(7-метил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: этиловый эфир имидазо[1,2-a]пиразин-2-карбоновой кислоты:
Этил-бромпируват (62,9 г) добавляют к раствору в DME (258 мл) 2-аминопиразина (24,8 г) при комнатной температуре и перемешивают в течение 2,5 ч. Реакционную смесь охлаждают до 0°C и перемешивают в течение 30 мин, получая бледно-коричневый осадок. Осадок фильтруют и промывают Et2O, получая бледно-коричневые кристаллы. Суспензию осадка (66,1 г) в EtOH (1,29 л) нагревают при температуре кипения с возвращением флегмы до превращения в прозрачный раствор. После кипячения с возвращением флегмы в течение 2 ч реакционную смесь концентрируют при пониженном давлении, затем смешивают с CHCl3 и насыщенным водным NaHCO3. Смесь фильтруют через прокладку целита и отделенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют CHCl3-MeOH (99/1˜97/3) и собранные фракции концентрируют при пониженном давлении с последующей перекристаллизацией из CHCl3-Et2O. Указанное в заголовке соединение получают в виде бледнорозовых кристаллов. Выход: 10,9 г, 22%).
1H ЯМР (CDCl3) δ д 1,46 (т, 3H, J=7,2 Гц), 4,49 (кв, 2H, J=7,2 Гц), 7,96 (д, 1H, J=4,7 Гц), 8,08 (дд, 1H, J=1,2, 4,7 Гц), 8,26 (с, 1H), 9,21 (д, 1H, J=1,2 Гц).
Стадия 2: гидрохлорид этилового эфира 5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты:
0,46 M HCl - EtOH (169 мл) и 10% Pd-C (50% влажности) (1,37 г) добавляют к раствору в EtOH (546 мл) этилового сложного эфира имидазо[1,2-a]пиразин-2-карбоновой кислоты (13,7 г). Смесь гидрогенизируют под давлением H2 40 фунт/кв. дюйм при комнатной температуре в течение 15 ч. Реакционную смесь фильтруют и Pd-C промывают EtOH. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют CHCl3-MeOH (9/1˜2/1). Указанное в заголовке соединение получают в виде коричневых кристаллов Выход: 10,4 г, 63%.
1H ЯМР (CDCl3) δ д 1,38 (т, 3H, J=7,1 Гц), 3,90 (т, 2H, J=5,7 Гц), 4,40 (кв, 2H, J=7,1 Гц), 4,59 (т, 2H, J=5,7 Гц), 4,80 (с, 2H), 8,20 (с, 1H).
Стадия 3: этиловый сложный эфир 7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты:
Et3N (3,44 мл), 37% HCHO водный (2,02 мл) и NaBH3CN (1,78 г) добавляют последовательно к раствору в MeOH (75 мл) гидрохлорида этилового сложного эфира 5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (5,2 г) при комнатной температуре и перемешивают в течение 3,5 ч в атмосфере азота. Смесь разбавляют CH2Cl2 и промывают 50% водным K2CO3. Органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью CHCl3-ацетон (1/1˜1/2). Указанное в заголовке соединение получают в виде оранжевого масла. Выход: 2,68 г, 57%).
1H ЯМР (CDCl3) δ д 1,37 (т, 3H, J=7,1 Гц), 2,50 (с, 3H), 2,85 (т, 2H, J=5,5 Гц), 3,69 (с, 2H), 4,06 (т, 2H, J=5,5 Гц), 4,36 (т, 2H, J=7,1 Гц), 7,52 (с, 1H).
Стадия 4: 7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид:
1,01 M раствор DIBAL в толуоле (13,6 мл) добавляют к сухому раствору в CH2Cl2 (86 мл) этилового сложного эфира 7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (1,8 г) в атмосфере азота при -78°C и перемешивают в течение 2 ч. Смесь гасят 1 M HCl. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают 50% водным K2CO3 и водный слой экстрагируют CH2Cl2 три раза. Комбинированный органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью CHCl3-MeOH (19/1˜9/1). Указанное в заголовке соединение 5 получают в виде бесцветных кристаллов. Выход: 591 мг, 42%).
1H ЯМР (CDCl3) δ д 2,51 (с, 3H), 2,87 (т, 2H, J=5,5 Гц), 3,70 (с, 2H), 4,10 (т, 2H, J=5,5 Гц), 7,53 (с, 1H), 9,82 (д, 1H, J=1,4 Гц).
Стадия 5: 4-нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (смесь диастереоизомеров):
7-Метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (1,19 г) добавляют к раствору в сухом ацетонитриле (97 мл) безводного MgBr2 (4,05 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (97 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (3,32 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (3,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 4,5 ч при -20°C и обрабатывают уксусным ангидридом (1,36 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 17 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью CHCl3-ацетон (9/1˜2/1). Указанное в заголовке соединение получают как смесь двух диастереоизомеров. Красное масло. Выход: 1,13 г.
1H ЯМР (CDCl3) δ д 1,20 (с, 0,81×3H), 2,24 (с, 0,19×3H), 2,48 (с, 3H), 2,80˜2,84 (м, 2H), 3,57˜3,67 (м, 2H), 3,97˜4,02 (м, 2H), 5,27 (д, 1H, J=13,6 Гц), 5,42 (д, 0,19×1H, J=13,6 Гц), 5,45 (д, 0,81×1H, J=13,6 Гц), 6,07 (с, 0,19×1H), 6,30 (с, 0,81×2H), 6,79 (с, 0,19×1H), 6,80 (с, 0,19×1H), 7,02 (с, 0,81×1H), 7,44 (с, 0,19×1H), 7,47 (с, 0,81×1H), 7,60 (д, 0,19×2H, J=8,2 Гц), 7,62 (д, 0,81×2H, J=8,6 Гц), 8,22˜8,26 (м, 2H).
Стадия 6: натриевая соль (5R),(6Z)-6-(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты:
4-Нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(7-метил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,11 г) растворяют в THF (32 мл) и ацетонитриле (32 мл). Свежеактивированную Zn пыль (4,46 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 48 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита, охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 7,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (20 мл, Mitsubishi Kasei Co. Ltd.) После адсорбирования колонку элюируют H2O-MeCN (1/0˜95/5). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества. Выход: 417 мг, 65%: т.пл. 200°C (с разложением).
1H ЯМР (D2О) δ д 2,32 (с, 3H), 2,79˜2,81 (м, 2H), 3,54 (с, 2H), 3,95 (т, 2H, J=5,6 Гц), 6,39 (с, 1H), 6,85 (с, 1H), 6,87 (с, 1H), 7,26 (с, 1H).
Пример 3
Получение натриевой соли (5R),(6Z)-7-оксо-6-[5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
2-Кетопиперазин
2-Кетопиперазин может быть получен согласно процедурам в патенте США 5629322.
Стадия 1: 4-п-нитробензилоксикарбонил-2-кетопиперазин
48,7% раствор п-нитробензилоксикарбонилхлорида в 1,4-диоксане (10,7 мл) добавляют к раствору в дихлорметане (110 мл) 2-кетопиперазина (2,21 г) и диизопропилэтиламина (4,6 мл) при 0°C и перемешивают в течение 0,5 ч при 0°C. К реакционной смеси добавляют воду (300 мл) и экстрагируют дихлорметаном (3 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируют смесью CHCl3-метанол (30:1) и указанное в заголовке соединение получают в виде белого твердого вещества (7,1 г, количественный выход).
1H ЯМР (д, CDCl3) δ 3,42-3,45 (м, 2H), 3,74 (т, 2H, J=5,4 Гц), 4,19 (с, 2H), 5,26 (с, 2H), 6,39 (уш.с, 1H), 7,52 (д, 2H, J=8,6 Гц), 8,24 (д, 2H, J=8,6 Гц).
Стадия 2: 5-метокси-4-п-нитробензилоксикарбонил-1,2,3,6-тетрагидропиразин:
Тетрафторборат триметилоксония (97%, 3,7 г) добавляют к раствору в сухом дихлорметане (120 мл) 4-п-нитробензилоксикарбонил-2-кетопиперазина (6,7 г) при комнатной температуре и перемешивают в течение 17 часов. Реакционную смесь обрабатывают насыщенным водным раствором гидрокарбоната натрия и органический слой отделяют. Водный слой экстрагируют этилацетатом (3 × 100 мл), затем объединенный органический слой промывают насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и указанное в заголовке соединение получают в виде бледно-коричневого твердого вещества. Выход; 5,7 г, 80,6%.
1H ЯМР (д, CDCl3) δ 3,48 (м, 2H), 3,57 (м, 2H), 3,70 (с, 3H), 3,97 (с, 2H), 5,26 (с, 2H), 7,52 (д, 2H, J=8,7 Гц), 8,23 (д, 2H, J=8,7 Гц).
Стадия 3: 2-имино-4-п-нитробензилоксикарбонил-пиперазин:
Смесь 5-метокси-4-п-нитробензилоксикарбонил-1,2,3,6-тетрагидропиразина (5,7 г) и хлорида аммония (1,6 г) в сухом этаноле (100 мл) нагревают до кипения с возвращением флегмы в течение 4 часов. Реакционную смесь затем концентрируют при пониженном давлении. Дихлорметан (100 мл) добавляют к остатку и экстрагируют водой (3 × 50 мл), затем объединенный органический слой промывают дихлорметаном. Водный слой нейтрализуют 10% водным раствором гидрокарбоната калия и затем экстрагируют дихлорметаном (8 × 50 мл). Объединенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и указанное в заголовке соединение получают в виде белого твердого вещества. Выход: 4,9 г, 91,2%.
1H ЯМР (д, CDCl3) δ 3,49 (уш.с, 4H), 3,98 (уш.с, 2H), 5,26 (с, 2H), 7,52 (д, 2H, J=8,6 Гц), 8,23 (д, 2H, J=8,6 Гц).
Стадия 4: 7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (9) и 7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-3-карбальдегид:
Смесь 2-бром-3-гидроксипропеналя (2,8 г), моногидрата п-толуолсульфоновой кислоты (33 мг) и 2-пропанола (3,5 мл) в циклогексане (28 мл) подвергают азеотропной перегонке, пока температура пара не повысится до 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом ацетонитриле (30 мл). Раствор в сухом ацетонитриле (310 мл) 2-имино-4-п-нитробензилоксикарбонил-пиперазина (4,7 г) добавляют при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч и затем реакционный раствор удаляют в вакууме. Остаток растворяют в этилацетате (170 мл) и добавляют триэтиламин (2,4 мл), затем реакционную смесь нагревают до температуры кипения с возвращением флегмы в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры и затем добавляют воду (170 мл) к реакционной смеси и разделяют смесь. Водный слой экстрагируют дихлорметаном (2 × 100 мл). Объединенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируют смесью CHCl3-метанол (50:1) и указанное в заголовке соединение получают в виде коричневого твердого вещества (выход: 2,9 г, 51,6%) и получают его региоизомер (оранжевое аморфное твердое вещество, выход: 0,8 г, 14,9%.
1H ЯМР (д, CDCl3) δ 3,99 (т, 2H, J=5,4 Гц), 4,14 (т, 2H, J=5,4 Гц), 4,85 (с, 2H), 5,29 (с, 2H), 7,54 (д, 2H, J=8,6 Гц), 7,57 (с, 1H), 8,24 (д, 2H, J=8,6 Гц), 9,85 (с, 1H).
Региоизомер 1H ЯМР (д, CDCl3) δ 3,95 (т, 2H, J=5,4 Гц), 4,44 (т, 2H, J=5,4 Гц), 4,87 (с, 2H), 5,29 (с, 2H), 7,54 (д, 2H, J=8,7 Гц), 7,78 (с, 1H), 8,24 (д, 2H, J=8,7 Гц), 9,71 (с, 1H).
Стадия 5: п-нитробензиловый сложный эфир (5R)-6-[ацетокси(7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты:
Раствор в сухом ацетонитриле (25 мл) 7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегида (1,6 г) добавляют к раствору MgBr2 (2,2 г) в сухом ацетонитриле (55 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Добавляют раствор в сухом THF (80 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,8 г), смесь охлаждают до -20°C, затем добавляют триэтиламин (1,6 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4,4-диметиламинопиридином (58,3 мг) и уксусным ангидридом (0,89 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (320 мл) и водный слой экстрагируют этилацетатом (3 × 160 мл). Органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируют смесью CH2Cl2-ацетон (20:1) и указанное в заголовке соединение получают как смесь двух диастереоизомеров (81:19, коричневое пенистое аморфное твердое вещество. Выход: 2,1 г, 59,9%.
1H ЯМР (д, CDCl3) δ 2,01 (с, 2,43H), 2,24 (с, 0,57H), 3,93-3,96 (м, 2H), 4,02-4,05 (м, 2H), 4,74-4,76 (м, 2H), 5,28 (д, 1H, J=13,5 Гц), 5,28 (с, 2H), 5,45 (д, 1H, J=13,5 Гц), 6,07 (с, 0,19H), 6,29 (с, 0,81H), 6,31 (с, 0,81H), 6,80 (с, 0,19H), 6,83 (с, 0,19H), 7,08 (с, 0,81H), 7,43 (с, 0,19H), 7,46 (с, 0,81H), 7,54 (д, 2H, J=8,6 Гц), 7,61 (д, 2H, J=8,8 Гц), 8,24 (д, 4H, J=8,3 Гц).
Стадия 6: натриевая соль (5R),(6Z)-7-оксо-6-(5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты:
п-Нитробензиловый сложный эфир (5R)-6-[ацетокси(7-п-нитробензилоксикарбонил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (2,0 г) растворяют в THF (63 мл). Свежеактивированную Zn пыль (7,9 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 63 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор фильтруют через прокладку целита и прокладку промывают водой (150 мл) и н-бутанолом (150 мл). Водный слой отделяют и затем органический слой экстрагируют водой (2 × 50 мл). Объединенный органический слой концентрируют до 61 г и подвергают колоночной хроматографии на смоле Diaion HP-21 (80 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 5% водным раствором ацетонитрила. Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества. Выход: 172 мг, 20,1%: т.пл. 150°C (с разложением).
1H ЯМР (д, D2О) δ 3,02 (т, 2H, J=5,6 Гц), 3,82 (с, 2H), 3,89 (д, 2H, J=5,6 Гц), 6,38 (с, 1H), 6,84 (с, 1H), 6,87 (с, 1H), 7,24 (с, 1H); IR (KBr).
Пример 4
Получение (5R,6Z)-6-{[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 5-трет-бутил-2-этил-6,7-дигидротиено[3,2-c]пиридин-2,5(4H)-дикарбоксилат:
5-трет-бутил-2-этил-6,7-дигидротиено[3,2-c]пиридин-2,5(4H)-дикарбоксилат получают согласно процедуре, которая описана в общих чертах в примере 1 (стадия 1) исходя из трет-бутил-1-пиперидинкарбоксилата (9,9 г, 50 ммоль), POCl3 (6,3 г, 40 ммоль) и DMF (3,8 г, 50 ммоль). Промежуточное хлорформил-соединение подвергают взаимодействию с этилмеркаптоацетатом (6,0 г, 50 ммоль) и Et3N. Продукт очищают колоночной хроматографией на SiO2, элюируя смесью 3:1 гексан:этилацетат. Выход: 8,7 г, 56%. Белая жидкость. (M+H) 312.
Стадия 2: трет-бутил-2-(гидроксиметил)-6,7-дигидротиено[3,2-c] пиридин-5(4H)-карбоксилат:
трет-Бутил-2-(гидроксиметил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилат получают согласно процедуре, охарактеризованной в общих чертах в примере 1 (стадия 2). Исходя из 5-трет-бутил-2-этил-6,7-дигидротиено[3,2-c]пиридин-2,5(4H)-дикарбоксилата (1,0 г, 3,21 ммоль) и LiAlH4 (500 мг, избыток) изолируют 807 мг (выход 92%) спиртового производного в виде белой жидкости. (M+H) 270.
Стадия 3: трет-бутил-2-(формил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилат:
трет-Бутил-2-(формил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилат получают согласно процедуре, описанной в общих чертах в примере 1, (стадия 3). Исходя из трет-бутил-2-(гидроксиметил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилата (1,0 г, 3,7 ммоль) в метиленхлориде (100 мл) и активированного MnO2 (5 г, избыток) изолируют 800 г (выход 81%) альдегидного производного в виде коричневого твердого вещества. (M+H) 268.
Стадия 4: 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)-пиридин:
2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)-пиридин получают исходя из трет-бутил-2-(формил)-6,7-дигидротиено[3,2-c]пиридин-5(4H)-карбоксилата (1,0 г, 3,7 ммоль), растворенного в CH2Cl2 (20 мл), MeOH (90% 20 мл) и 1 н. HCl в диоксане (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 48 ч. В конце реакционную смесь концентрируют до сухости и забирают на следующую стадию без очистки. Выход: 750 мг (HCl соль, количественный выход). M+H 168.
Стадия 5: 2-формил-[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин:
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)-пиридина (1,4 г, 5,2 ммоль) в DMF (20 мл) добавляют при комнатной температуре 4-метоксибензилхлорид (0,94 г, 6,2 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток). Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, хорошо промывают водой и сушат над безводным MgSO4. Ее фильтруют и концентрируют. Продукт очищают колоночной хроматографией на SiO2, элюируя этилацетатом. Бледно-желтое масло. Выход: 470 мг, 35%. M+H 288.
Стадия 6: 4-нитробензил-6-[(ацетилокси)[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат:
2-Формил-[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин (574 мг, 2,0 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (390 мг, 1,5 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 550 мг, 40%. M+H 714 и 716.
Стадия 7: (5R,6Z)-6-{[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота:
4-Нитробензил-6-[(ацетилокси)[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (300 мг, 0,42 ммоль) растворяют в THF (20 мл) и ацетонитриле (10 мл). Свежеактивированную Zn пыль (5,2 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C, получая желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой НР21. Вначале колонку элюируют деионизированной водой (2 л) и позднее смесью 10% CAN:вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном и фильтруют. Сушат. Выход: 50 мг, 18%, в виде желтых кристаллов, т.пл. 127°C. (M+H) 441.
1H ЯМР (ДМСО-d6) δ д 2,7 (м, 2H), 2,8 (уш.м, 2H), 3,4 (м, 2H), 3,74 (с, 3H), 3,8 (с, 2H), 6,6 (с, 1H), 6,88 (дд, 2H), 7,14 (с, 1H), 7,24 (дд, 2H), 7,4 (с, 1H), 7,59 (с, 1H).
Пример 5
Получение натриевой соли (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: гидроиодид 5-метилтио-3,6-дигидро-2H-[1,4]тиазина
Гидроиодид 5-метилтио-3,6-дигидро-2H-[1,4]тиазина получают способом, который описан в общих чертах в патенте США 5629322.
Стадия 2: гидрохлорид 3-иминотиоморфолина
Гидроиодид 5-метилтио-3,6-дигидро-2H-[1,4]тиазина (7,1 г) растворяют в 10% водном раствое K2CO3 (150 мл) и водный слой экстрагируют CH2Cl2 (5 × 70 мл). Объединенный органический слой сушат (MgSO4), фильтруют и концентрируют при пониженном давлении. Хлорид аммония (1,7 г) добавляют к полученному остатку в сухом этаноле (128 мл) и нагревают до температуры кипения с возвращением флегмы в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры. Реакционный раствор удаляют в вакууме и получают гидрохлорид иминотиоморфолина в виде коричневого твердого вещества (4,3 г, количественный выход).
1H ЯМР (д, ДМСО-d6) δ 3,15 (т, 2H, J=5,9 Гц), 3,74 (т, 2H, J=5,9 Гц), 3,83 (с, 2H), 8,97 (уш.с, 1H), 9,38 (уш.с, 1H), 9,99 (уш.с, 1H).
Стадия 3: 5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-2-карбальдегид и 5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-3-карбальдегид
Смесь 2-бром-3-гидроксипропеналя (7, 4,3 г), моногидрата п-толуолсульфоновой кислоты (52 мг) и 2-пропанола (5,3 мл) в циклогексане (43 мл) подвергают азеотропной перегонке, пока температура пара не достигнет 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом этаноле (28 мл). Смесь раствора в сухом этаноле (143 мл) гидрохлорида 3-иминотиоморфолина (4,3 г) и 28% раствора в метаноле метилата натрия (5,0 мл) добавляют при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 1 ч и затем реакционный раствор удаляют в вакууме. Остаток растворяют в хлороформе (128 мл) и добавляют триэтиламин (3,6 мл), затем реакционную смесь нагревают до температуры кипения с возвращением флегмы в течение 2,5 ч. Реакционную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении. Остаток растворяют в дихлорметане (300 мл) и промывают 50% водным раствором K2CO3 (2 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируют смесью CHCl3-ацетон (10:1), и получают 5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-карбальдегид (коричневое твердое вещество, 445 мг, 10,3%) и 5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-3-карбальдегид (коричневое твердое вещество, 872 мг, 20,2%).
5,6-Дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-карбальдегид: 1H ЯМР (д, CDCl3) δ 3,07 (т, 2H, J=5,7 Гц), 3,95 (с, 2H), 4,33 (т, 2H, J=5,7 Гц), 7,55 (с, 1H), 9,83 (с, 1H).
5,6-Дигидро-8H-имидазо[2,1-c][1,4]тиазин-3-карбальдегид: 1H ЯМР (д, CDCl3) δ 3,05 (т, 2H, J=5,7 Гц), 3,98 (с, 2H), 4,61 (т, 2H, J=5,7 Гц), 7,73 (с, 1H), 9,69 (с, 1H).
Стадия 4: (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль:
Раствор в сухом ацетонитриле (20 мл) 5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-карбальдегида (392 мг) добавляют к раствору MgBr2 (1,1 г) в сухом ацетонитриле (20 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Раствор в сухом THF (40 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,0 г) добавляют и смесь охлаждают до -20°C, затем добавляют триэтиламин (0,8 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3,5 ч при -20°C и обрабатывают 4-диметиламинопиридином (30 мг) и уксусным ангидридом (0,44 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 14 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (240 мл) и водный слой экстрагируют этилацетатом (3 × 100 мл). Объединенный органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток грубо очищают колоночной хроматографией на силикагеле, элюируют смесью CH2Cl2-ацетон (50:1) и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(5,6-дигидро-8Н-имидазо[2,1-c][1,4]тиазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде твердого вещества.
Полученное указанное твердое вещество очищают колоночной хроматографией на SiO2, элюируя смесью 50% этилацетат:гексан. Полученное бледно-желтое твердое вещество растворяют в THF (17 мл). Свежеактивированную Zn пыль (2,2 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 17 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор фильтруют через прокладку целита и прокладку промывают водой (40 мл) и н-бутанолом (30 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 10 мл). Объединенный органический слой концентрируют до 23 г, добавляют 1 моль/л NaOH, чтобы довести рН до 7,25, и подвергают колоночной хроматографии на смоле Diaion HP-21 (30 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 10% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая натриевую соль (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (168 мг, 20,9%). т.пл. 135°C (с разложением).
1H ЯМР (д, D2О) δ 3,00 (т, 2H, J=5,7 Гц), 3,80 (AB, 2H, J=16,7, 18,1 Гц), 4,19 (т, 2H, J=5,7 Гц), 6,44 (д, 1H, J=0,8 Гц), 6,89 (с, 1H), 6,93 (с, 1H), 7,29 (с, 1H); М+H=322.
Пример 6
Получение натриевой соли (5R),(6Z)-6-(6,7-дигидро-5H-пирроло{1,2-a]имидазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-карбальдегид
28% Метоксид натрия (5,26 г) добавляют к раствору в EtOH (250 мл) гидрохлорида 4,5-дигидро-3H-пиррол-2-иламина (3,27 г) при комнатной температуре. После перемешивания в течение 5 мин при комнатной температуре добавляют к смеси 2-бром-3-пропоксипропеналь (5,79 г) при комнатной температуре, затем реакционную смесь перемешивают в течение 1 ч при комнатной температуре. Реакционную смесь доводят до сухости в вакууме. Остаток растворяют в CHCl3 (300 мл) и добавляют триэтиламин (3,8 мл). Смесь нагревают до кипения с возвращением флегмы в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры, промывают 50% K2CO3, сушат над безводным K2CO3, фильтруют и выпаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируют смесью CHCl3-ацетон (2:1) и получают 6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-карбальдегид (41%, 1,51 г) в виде бледно-желтого твердого вещества.
1H ЯМР (д, CDCl3) δ 2,62-2,7 (м, 2H), 2,90-2,94 (м, 2H), 4,07 (т, 2H, J=7,2 Гц), 7,59 (с,1H), 9,80 (с, 1H).
Стадия 2: (5R),(6Z)-6-(6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль
6,7-Дигидро-5H-пирроло[1,2-a]имидазол-2-карбальдегид (1,36 г) добавляют к раствору в сухом ацетонитриле (155 мл) безводного MgBr2 (5,64 г) в атмосфере аргона при комнатной температуре. Раствор в сухом THF (155 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (3,86 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (4,18 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают уксусным ангидридом (1,89 мл) и DMAP (370 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 14,5 ч при 0°C. Смесь разбавляют этилацетатом и промывают 1 M водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтр промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток растворяют в THF (166 мл) и ацетонитриле (77 мл). Свежеактивированную Zn пыль (23,2 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 243 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8. Фильтрат промывают этилацетатом и водный слой отделяют. Снова добавляют 1 M NaOH к водному слою, чтобы довести рН до 8. Полученную смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (20 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0˜9/1), получая очищенные активные фракции натриевой соли (5R),(6Z)-6-(6,7-дигидро-5H-пирроло[1,2-a]имидазол-2-илметилен)-7-оксо-4-тиа-l-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (681 мг, 24%, pH 7,8). т.пл. 190°C (с разложением).
1H ЯМР (д, D2О): δ 2,48-2,56 (м, 2H), 2,74-2,79 (м, 2H), 3,94-3,99 (м, 2H), 6,47 (д, 1H, J=0,7 Гц), 6,94 (с, 1H), 6,95 (с, 1H), 7,36 (с, 1H); (М+H) 291.
Пример 7
Получение натриевой соли (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: морфолин-3-он
Морфолин-3-он получают способом по патенту США 5349045.
Стадия 2: морфолин-3-тион
Смесь морфолин-3-она (4,7 г) и реагента Lawesson (10,3 г) в сухом THF (94 мл) нагревают до кипения с возвращением флегмы в течение 1,5 ч. Реакционную смесь охлаждают до комнатной температуры и реакционный растворитель удаляют в вакууме. Остаток подвергают колоночной хроматографии на силикагеле и элюируют смесью CHCl3-метанол (50:1), получая желтое твердое вещество. Перекристаллизация сырого продукта из гексана-этилацетата дает указанное в заголовке (4,0 г, 72,2%) в виде желтого порошка.
1H ЯМР (CDCl3) δ 3,45 (т, 2H, J=5,1 Гц), 3,91 (т, 2H, J=5,1 Гц), 4,55 (с, 2H).
Стадия 3: 5-метилтио-3,6-дигидро-2Н-[1,4]оксазин
Смесь морфолин-3-тиона (4,7 г) и метилиодида (13 мл) в сухом CH2Cl2 (140 мл) перемешивают при комнатной температуре в течение 15 ч. Реакционную смесь фильтруют и твердое вещество промывают CH2Cl2. Полученное твердое вещество растворяют в 50% водном растворе K2CO3 (150 мл) и водный слой экстрагируют CH2Cl2 (8 × 100 мл). Объединенный слой CH2Cl2 сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и указанное в заголовке получают в виде бледно-желтого масла (3,6 г, 67,8%).
1H ЯМР (CDCl3) δ 2,32 (с, 3H), 3,71-3,74 (м, 4H), 4,14-4,15 (м, 2H).
Стадия 4: гидрохлорид 3-иминоморфолина
Смесь 5-метилтио-3,6-дигидро-2H-[1,4]оксазина (3,6 г) и хлорида аммония (1,5 г) в сухом этаноле (136 мл) нагревают до кипения с возвращением флегмы в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры. Реакционный растворитель удаляют в вакууме и указанное в заголовке соединение получают в виде бледно-коричневого твердого вещества (3,6 г, 97,7%).
1H ЯМР (ДМСО-d6) 3,34 (м, 2H), 3,86 (т, 2H, J=5,2 Гц), 4,47 (с, 2H).
Стадия 5: 5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-карбальдегид (9) и 5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-3-карбальдегид
Смесь 2-бром-3-гидроксипропеналя (4,1 г), моногидрата п-толуолсульфоновой кислоты (52 мг) и 2-пропанола (5,2 мл) в циклогексане (42 мл) подвергают азеотропной перегонке, пока температура пара не достигнет 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом этаноле (50 мл). Смесь раствора в сухом этаноле (200 мл) гидрохлорида 3-иминоморфолина (3,4 г) и 28% раствора метилата натрия в метаноле (4,8 г) добавляют при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 2 ч и затем реакционный растворитель удаляют в вакууме. Остаток растворяют в хлороформе (125 мл) и добавляют триэтиламин (3,5 мл), затем реакционную смесь нагревают до температуры кипения с возвращением флегмы в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении. Остаток растворяют в дихлорметане (300 мл) и промывают 50% водным раствором K2CO3 (2 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и элюируют смесью CHCl3-ацетон (4:1), получая указанное в заголовке (бледно-оранжевое твердое вещество, 1,4 г, 36,3%) и другой региоизомер (бледно-оранжевое твердое вещество, 609 мг, 16,1%).
Желательный продукт: 1H ЯМР (CDCl3) δ 4,08-4,15 (м, 4H), 4,88 (с, 2H), 7,58 (с, 1H), 9,85 (с, 1H).
Нежелательный региоизомер: 1H ЯМР (CDCl3) δ 4,06 (т, 2H, J=5,2 Гц), 4,40 (т, 2H, J=5,2 Гц), 4,90 (с, 2H), 7,75 (с, 1H), 9,72 (с, 1H).
Стадия 6: (5R),(6Z)-6-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль
Раствор в сухом ацетонитриле (66 мл) 5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-карбальдегида (1,2 г) добавляют к раствору MgBr2 (3,6 г) в сухом ацетонитриле (66 мл) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Раствор в сухом THF (132 мл) п-нитробензил-(5R, 6S)-6-бромпенем-3-карбоксилата (3,4 г) добавляют и смесь охлаждают до -20°C, затем добавляют триэтиламин (2,8 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 4 ч при -20°C и обрабатывают 4-диметиламинопиридином (100 мг) и уксусным ангидридом (1,5 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 18 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (1 л) и водный слой экстрагируют этилацетатом (3 × 500 мл). Объединенный органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного твердого вещества.
Свежеактивированную Zn пыль (14 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 72 мл) к раствору в THF (72 мл) п-нитробензилового сложного эфира (5R)-6-[ацетокси(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2,5 ч при комнатной температуре. Реакционный раствор фильтруют через прокладку целита и прокладку промывают водой (170 мл) и н-бутанолом (170 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 50 мл). Объединенный органический слой концентрируют до 90 г, добавляют 1 моль/л NaOH, чтобы довести рН до 7,5, и подвергают колоночной хроматографии на смоле Diaion HP-21 (120 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 5% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке в виде желтого аморфного твердого вещества (756 мг, 29,1%). Т.пл. 130°C (с разложением).
1H ЯМР (ДМСО-d6) δ 3,98-4,01 (м, 2H), 4,04-4,07 (м, 2H), 4,74 (AB, 2H, J=15,3, 22,9 Гц), 6,40 (д, 1H, J=0,8 Гц), 6,55 (с, 1H), 6,95 (д, 1H, J=0,6 Гц), 7,54 (с, 1H); IR (KBr) 3412, 1741, 1672, 1592, 1549 см-1; λмакс. (H2O) 304 нм.
Пример 8
Получение натриевой соли (5R),(6Z)-6-(5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: этиловый сложный эфир 5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-карбоновой кислоты
Указанное в заголовке соединение получают таким же путем, как Ranganathan и сотрудники (Indian J. Chem. 1991, 30 В, 169-175).
Стадия 2: (5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-илметанол
MeOH (2,73 мл) добавляют к раствору в THF (180 мл) LiBH4 (1,63 г) в атмосфере азота при комнатной температуре и затем к суспензии добавляют этиловый сложный эфир 5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбоновой кислоты (8,11 г) и перемешивают в течение 2 ч при 40°C. Смесь гасят 1 моль/л HCl при pH 1 и перемешивают в течение 1 ч при комнатной температуре. Твердый K2CO3 добавляют к раствору, чтобы довести рН до 8, и смесь экстрагируют AcOEt. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде коричневых кристаллов (4,87 г, 78%).
1H ЯМР (CDCl3) δ 2,44 (т, 1H, J=5,8 Гц), 2,54-2,62 (м, 2H), 2,87 (т, 2H, J=7,4 Гц), 4,10 (т, 2H, J=7,2 Гц), 4,63 (д, 2H, J=5,8 Гц), 5,96 (с, 1H).
Стадия 3: 5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбальдегид
MnO2 (активированный) (24,4 г) добавляют к раствору в CHCl3 (350 мл) (5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-ил)метанола (4,87 г) и кипятят с возвращением флегмы в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. Объем фильтрата уменьшают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (1/1-1/2). Указанное в заголовке соединение получают в виде желтого масла (4,35 г, 91%).
1H ЯМР (CDCl3) δ 2,63-2,71 (м, 2H), 2,95 (т, 2H, J=7,4 Гц), 4,22 (т, 2H, J=7,4 Гц), 6,52 (с, 1H), 9,89 (с, 1H).
Стадия 4: (5R),(6Z)-6-(5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль
5,6-Дигидро-4H-пирроло[1,2-b]пиразол-2-карбальдегид (1,36 г) добавляют к раствору в сухом ацетонитриле (148 мл) безводного MgBr2 (5,52 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (148 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 97%) (3,97 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (4,18 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 4 ч при -20°C и обрабатывают уксусным ангидридом (1,89 мл) и DMAP (123 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 14 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в THF (106 мл) и ацетонитриле (49 мл). Свежеактивированную Zn пыль (22,5 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 155 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (79 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-9/1). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (848 мг, 29%, pH 7,1). Т.пл. 190°C (с разложением).
1H ЯМР (D2О) δ 2,49 (м, 2H), 2,78 (т, 2H, J=7,4 Гц), 4,02 (т, 2H, J=7,4 Гц), 6,01 (с, 1H), 6,29 (с, 1H), 6,90 (с, 2H).
Пример 9
Получение натриевой соли (5R),(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: тетрагидропиридино[1,2-c][1,2,3]оксадиазолон
Концентрированную HCl (1,96 мл) и NaNO2 (2,2 г) добавляют к раствору в H2O (21 мл) DL-пипеколиновой кислоты (3,04 г) в атмосфере азота при 0°C и перемешивают в течение 1 ч. Раствор экстрагируют CH2Cl2 и органический слой промывают насыщенным раствором соли. Смесь сушат над Na2SO4 и концентрируют при пониженном давлении, получая сырую (2RS)-1-нитрозопиперидин-2-карбоновую кислоту в виде бледно-желтых кристаллов.
Трифторуксусный ангидрид (1,93 г) добавляют к раствору в THF (92 мл) сырой (2RS)-1-нитрозопиперидин-2-карбоновой кислоты в атмосфере азота при 0°C и перемешивают в течение 5 ч при 0°C и в течение 2 ч при комнатной температуре. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (1/1-0/1). Указанное в заголовке соединение получают в виде бесцветных кристаллов (1,10 г, 33%).
1H ЯМР (CDCl3) δ 1,93-1,99 (м, 2H), 2,08-2,15 (м, 2H), 2,65 (т, 2H, J=6,5 Гц), 4,26 (т, 2H, J=6,1 Гц).
Стадия 2: этиловый сложный эфир 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-карбоновой кислоты
Этилпропиолат (804 мг) добавляют к раствору в o-ксилоле (15 мл) тетрагидропиридино[1,2-c][1,2,3]оксадиазолона (1,04 г) в атмосфере азота и кипятят с возвращением флегмы в течение 16 ч. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (2/1-1/1). Указанное в заголовке соединение получают в виде желтого масла (871 мг, 65%) и этиловый сложный эфир 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-3-карбоновой кислоты получают в виде желтого масла (345 мг, 26%).
1H ЯМР (CDCl3) δ 1,39 (т, 3H, J=7,1 Гц), 1,84-1,91 (м, 2H), 2,02-2,09 (м, 2H), 2,82 (т, 2H, J=6,4 Гц), 4,22 (т, 2H, J=6,2 Гц), 4,39 (кв, 2H, J=7,1 Гц), 6,53 (с, 1H).
Стадия 3: (4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-ил)метанол
MeOH (0,29 мл) добавляют к раствору в THF (19 мл) LiBH4 (содержание 90%) (174 мг) в атмосфере азота при комнатной температуре, затем к суспензии добавляют этиловый сложный эфир 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-карбоновой кислоты (862 мг) и перемешивают в течение 1 ч при комнатной температуре и 1,5 ч при 40°C. Смесь гасят 1 моль/л HCl при pH 1 и перемешивают в течение 1 ч при комнатной температуре. Твердый K2CO3 добавляют к раствору, чтобы довести рН до 8, и смесь экстрагируют AcOEt. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде бледно-желтого масла (691 мг, 95%).
1H ЯМР (CDCl3) δ 1,80-1,87 (м, 2H), 1,98-2,05 (м, 2H), 2,77 (т, 2H, J=6,4 Гц), 2,81-2,84 (уш., 1H), 4,09 (т, 2H, J=6,1 Гц), 4,62 (д, 2H, J=5,3 Гц), 5,96 (с, 1H).
Стадия 4: 4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-карбальдегид
MnO2 (активированный) (3,36 г) добавляют к раствору в CHCl3 (44 мл) (4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-ил)метанола (673 мг) и кипятят с возвращением флегмы в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. Объем фильтрата уменьшают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (2/1-1/2). Указанное в заголовке соединение получают в виде бледно-желтого масла (510 мг, 77%).
1H ЯМР (CDCl3) δ 1,90 (м, 2H), 2,10 (м, 2H), 2,84 (т, 2H, J=6,4 Гц), 4,23 (т, 2H, J=6,2 Гц), 6,52 (с, 1H), 9,92 (с, 1H).
Стадия 5: (5R)(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиридин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль
4,5,6,7-Тетрагидропиразоло[1,5-a]пиридин-2-карбальдегид (483 мг) добавляют к раствору в сухом ацетонитриле (48 мл) безводного MgBr2 (1,81 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (48 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 97%) (1,28 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (1,35 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (0,61 мл) и DMAP (40 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 16 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в THF (35 мл) и ацетонитриле (16 мл). Свежеактивированную Zn пыль (7,43 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 51 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (105 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-85/15). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (427 мг, 41%, pH 7,7). Т.пл. 190°C (с разложением).
1H ЯМР (D2О) δ 1,67-1,71 (м, 2H), 1,85-1,89 (м, 2H), 2,64 (т, 2H, J=6,3 Гц), 3,97 (т, 2H, J=6,1 Гц), 5,97 (с, 1H), 6,25 (с, 1H), 6,85 (с, 1H), 6,88 (с, 1H).
Пример 10
Получение натриевой соли (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 5-метокси-1-метил-3,6-дигидро-1H-пиразин-2-он
Указанное в заголовке соединение получают таким же путем, как S.Rajappa и B.G.Advani (Tetrahedron. 1973, 29, 1299-1302).
Стадия 2: 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-он
Смесь 5-метокси-1-метил-3,6-дигидро-1H-пиразин-2-она (2,3 г) и хлорида аммония (936 мг) в сухом этаноле (32 мл) перемешивают при комнатной температуре в течение 1 ч и затем кипятят с возвращением флегмы в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и выпаривают при пониженном давлении. Остаток растирают с хлороформом при комнатной температуре в течение 30 мин. Осадок отфильтровывают и сушат в вакууме. Гидрохлорид 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-она получают в виде бледно-коричневого порошка (1,7 г, 66%).
Раствор гидрохлорида 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-она (662 мг) в метаноле (10 мл) дополняют 10% водным раствором гидрокарбоната калия при 0°С и затем перемешивают в течение 40 мин при 0°С. Смесь концентрируют при пониженном давлении. Остаток растирают с хлороформом (18 мл) и метанолом (2 мл) при комнатной температуре в течение 30 мин. Осадок отфильтровывают и сушат в вакууме. Соединение получают в виде бледно-коричневого порошка (515 мг, количественный выход).
1H ЯМР (ДМСО-d6) δ 2,88 (с, 3H), 3,94 (с, 2H), 4,42 (с, 2H).
Стадия 3: 7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид и 7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид
Раствор 2-бром-3-изопропоксипропеналя (1,3 г) в сухом ацетонитриле (60 мл) добавляют к раствору 5-амино-1-метил-3,6-дигидро-1H-пиразин-2-она (782 мг) в сухом ацетонитриле (60 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 20 ч, добавляют триэтиламин (0,95 мл) и затем кипятят с возвращением флегмы в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и затем выпаривают при пониженном давлении. Остаток растворяют в хлороформе (10 мл) и промывают 50% водным раствором K2CO3 (10 мл). Водный слой экстрагируют хлороформом. Органический слой сушат (MgSO4) и фильтруют. Фильтрат выпаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и элюируют смесью CHCl3-MeOH (95:5), получая указанное в заголовке соединение 7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид в виде бледно-желтого твердого вещества (541 мг, 49,1%) и его региоизомер 7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид в виде бледно-желтого твердого вещества (128 мг, 11,6%).
7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид: 1H ЯМР (CDCl3) δ 3,17 (с, 3H), 4,68 (с, 2H), 4,78 (с, 2H), 7,66 (с, 1H), 9,83 (с, 1H).
7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-3-карбальдегид: 1H ЯМР (CDCl3) δ 3,16 (с, 3H), 4,70 (с, 2H), 5,03 (с, 2H), 7,82 (с, 1H), 9,73 (с, 1H).
Стадия 4: (5R,6RS)-6-[ацетокси(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, 4-нитробензиловый сложный эфир
7-Метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (319 мг) добавляют к раствору в сухом ацетонитриле (32 мл) безводного MgBr2 (786 мг) в атмосфере азота при комнатной температуре. Раствор в сухом THF (32 мл) 4-нитро-бензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (687 мг) добавляют к смеси, охлаждают до -20°C и добавляют триэтиламин (0,60 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4-диметиламинопиридином (44 мг) и уксусным ангидридом (0,35 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 20 ч при 0°C. Смесь разбавляют этилацетатом и H2O. После отделения органического слоя водный слой экстрагируют этилацетатом. Органические слои объединяют и промывают 5% водным раствором лимонной кислоты и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем элюируют хлороформом. Указанное в заголовке соединение получают в виде смеси диастереоизомеров (желтого аморфного твердого вещества; 410 мг, 38%).
1H ЯМР (CDCl3) δ 2,03 (с, 0,7×3Н), 2,09 (с, 0,3×3Н), 3,15 (с, 3Н), 4,59-4,62 (м, 2Н), 4,66 (с, 0,3×2Н), 4,67 (с, 0,7×2Н), 5,28 (д, 1Н, J=13,5 Гц), 5,43 (д, 0,3×1Н, J=13,5 Гц), 5,45 (д, 0,7×1Н, J=13,5 Гц), 6,07 (с, 0,3×1Н), 6,28 (с, 0,7×1Н), 6,32 (с, 0,7×1Н), 6,83 (с, 0,3×1Н), 6,86 (с, 0,3×1Н), 7,10 (с, 0,7×1Н), 7,44 (с, 0,3×1Н), 7,47 (с, 0,7×1Н), 7,60 (д, 0,7×2Н, J=8,6 Гц), 7,61 (д, 0,3×2Н, J=8,6 Гц), 8,24 (д, 2Н, J=8,6 Гц).
Стадия 5: натриевая соль (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты и натриевая соль (5R),(6E)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый сложный эфир (5R,6RS)-6-[ацетокси(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (481 мг) растворяют в THF (6,7 мл) и ацетонитриле (3,1 мл). Свежеактивированную Zn пыль (1,92 г) и 0,5 M фосфатный буфер (pH 6,5, 9,9 мл) добавляют к смеси. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор смешивают с этилацетатом и фильтруют через прокладку целита. Прокладку промывают водой и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C и лиофилизуют. Остаток отделяют препаративной ВЭЖХ (Inertsil ODS-2, GL Science Inc., 10 × 250 мм, 0,05 моль/л фосфатный буфер (pH 7,1):CH3CN = 93:7, 4,0 мл/мин). Отделенные фракции натриевой соли (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты и натриевой соли (5R),(6Е)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0 соответственно. Каждый раствор концентрируют в высоком вакууме при 35°C. Каждый концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (60 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем смесью 5% ацетонитрил-вода. Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение - натриевую соль (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (125 мг, 44,4%, т.пл. 115-117°C (с разложением)) и соединение - натриевую соль (5R),(6E)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (19 мг, 6,7%) соответственно.
Соединение (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль 1H ЯМР (δ, D2О) 2,99 (с, 3H), 4,54 (с, 2H), 4,66 (с, 2H), 6,38 (с, 1H), 6,85 (с, 1H), 6,90 (с, 1H), 7,30 (с, 1H).
Соединение (5R),(6Е)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль 1H ЯМР (δ, D2О) 2,94 (с, 3H), 4,45 (с, 2H), 4,56 (с, 2H), 6,22 (с, 1H), 6,48 (с, 1H), 6,94 (с, 1H), 7,69 (с, 1H).
Пример 11
Получение натриевой соли (5R)(6Z)-6-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: (3R)-тиоморфолин-3-карбоновая кислота
Указанное в заголовке соединение получают таким же путем, как Shiraiwa и сотрудники (Biosci. Biotechnol. Biochem. 1998, 62, 2382-2387).
Стадия 2: 3-оксо-3a,4,6,7-тетрагидро-3H-2-окса-5-тиа-1-аза-7a-азониоинденид
NaNO2 (3,14 г) добавляют к раствору в 1 моль/л HCl (33,7 мл) (3R)-тиоморфолин-3-карбоновой кислоты (4,96 г) в атмосфере азота при 0°C и перемешивают в течение 0,5 ч. Раствор экстрагируют CHCl3 (5 раз) и органический слой промывают насыщенным раствором соли. Смесь сушат над MgSO4 и концентрируют при пониженном давлении, получая сырую (3R)-4-нитрозотиоморфолин-3-карбоновую кислоту в виде бледно-желтых кристаллов.
Трифторуксусный ангидрид (7,07 г) добавляют к раствору в THF (169 мл) сырой (3R)-4-нитрозотиоморфолин-3-карбоновой кислоты в атмосфере азота при 0°C и перемешивают в течение 3 ч при 0°C и в течение 17 ч при комнатной температуре. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (1/1-0/1). Указанное в заголовке соединение получают в виде бледно-коричневых кристаллов (3,41 г, 64%).
1H ЯМР (CDCl3) δ 3,15 (т, 2H, J=5,5 Гц), 3,71 (с, 2H), 4,54 (т, 2H, J=5,5 Гц).
Стадия 3: этиловый сложный эфир 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбоновой кислоты
Этилпропиолат (2,33 г) добавляют к раствору в o-ксилоле (72 мл) 3-оксо-3a,4,6,7-тетрагидро-3H-2-окса-5-тиа-1-аза-7a-азониоинденида (3,41 г) в атмосфере азота и кипятят с возвращением флегмы в течение 15 ч. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (2/1-1/1). Указанное в заголовке соединение получают в виде желтого масла (3,13 г, 68%) и другой нежелательный региоизомер этиловый сложный эфир 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-3-карбоновой кислоты получают в виде желтого масла (556 мг, 12%).
1H ЯМР (CDCl3) δ 1,31 (т, 3H, J=7,1 Гц), 3,04 (т, 2H, J=5,7 Гц), 3,81 (с, 2H), 4,32 (кв, 2H, J=7,1 Гц), 4,40 (т, 2H, J=5,7 Гц), 6,54 (с, 1H).
Стадия 4: (6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-ил)метанол
LiBH4 (содержание 90%) (536 мг) и MeOH (0,9 мл) добавляют к раствору в THF (59 мл) этилового сложного эфира 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбоновой кислоты (3,13 г) в атмосфере азота при комнатной температуре и перемешивают в течение 3 ч при 40°C. Смесь гасят 1 моль/л HCl при pH 1 и перемешивают в течение 1 ч при комнатной температуре. Твердый K2CO3 добавляют к раствору, чтобы довести рН до 8, и смесь экстрагируют AcOEt. Органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении, получая указанное в заголовке соединение в виде бледно-желтого масла (2,51 г, количественный выход).
1H ЯМР (CDCl3) δ 2,58 (уш., 1H), 3,07 (т, 2H, J=5,7 Гц), 3,84 (с, 2H), 4,33 (т, 2H, J=5,7 Гц), 4,63 (д, 2H, J=3,9 Гц), 6,05 (с, 1H).
Стадия 5: 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбальдегид
MnO2 (активированный) (11,46 г) добавляют к раствору в CHCl3 (135 мл) (6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-ил)метанола (2,31 г) и кипятят с возвращением флегмы в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (1/1). Указанное в заголовке соединение получают в виде бледно-желтых кристаллов (1,78 г, 78%).
1H ЯМР (CDCl3) δ 3,15 (т, 2H, J=5,8 Гц), 3,90 (с, 2H), 4,48 (т, 2H, J=5,8 Гц), 6,58 (с,1H), 9,92 (с, 1H).
Стадия 6: натриевая соль (5R)(6Z)-6-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбальдегид (841 мг) добавляют к раствору в сухом ацетонитриле (39 мл) безводного MgBr2 (1,88 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (39 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 99,7%) (1,93 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (2,79 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают уксусным ангидридом (0,94 мл) и DMAP (61 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 17 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в THF (83 мл) и ацетонитриле (39 мл). Свежеактивированную Zn пыль (7,72 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 122 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (150 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-85/15). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (371 мг, 22%, pH 8,0). Т.пл. 190°C (с разложением).
1H ЯМР (D2О) δ 3,03 (т, 2H, J=5,7 Гц), 3,75 (с, 2H), 4,22 (т, 2H, J=5,7 Гц), 6,07 (с, 1H), 6,27 (с, 1H), 6,86 (с, 1H), 6,89 (с, 1H).
Пример 12
Получение натриевой соли (5R)(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 3-оксо-3a,4-дигидро-3H,6H-2-окса-5-тиа-1-аза-6a-азонио-3a-пенталенид
Концентрированную HCl (15 мл) и NaNO2 (16,6 г) добавляют к раствору в H2O (166 мл) L-тиопролина (24,3 г) в атмосфере азота при 0°C и перемешивают в течение 2 ч. Раствор экстрагируют CH2Cl2, органический слой сушат над MgSO4 и концентрируют при пониженном давлении, получая сырое N-нитрозо-производное в виде желтого твердого вещества.
Трифторуксусный ангидрид (5,0 мл) добавляют к раствору в THF (350 мл) сырого N-нитрозотиопролина в атмосфере азота при 0°C и перемешивают в течение 5 ч при 0°C. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (1:1). Указанное в заголовке соединение получают в виде бледно-коричневого твердого вещества (4,0 г, 15,1%).
1H ЯМР (CDCl3) δ 4,04 (т, 2H, J=1,7 Гц), 5,40 (т, 2H, J=1,7 Гц).
Стадия 2: этиловый сложный эфир 4H-5-тиа-1,6a-диазапентален-2-карбоновой кислоты
Этилпропиолат (3,1 мл) добавляют к раствору в o-ксилоле (130 мл) 3-оксо-3a,4-дигидро-3H,6H-2-окса-5-тиа-1-аза-6a-азонио-3a-пенталенида (4,0 г) в атмосфере азота и кипятят с возвращением флегмы в течение 19 ч. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (4:1). Указанное в заголовке соединение получают в виде желтого твердого вещества (2,7 г, 49,3%) и этиловый сложный эфир 4H-5-тиа-1,6a-диазапентален-3-карбоновой кислоты получают в виде бледно-желтых кристаллов (1,2 г, 21,7%).
1H ЯМР (CDCl3) δ 1,40 (т, 3H, J=7,1 Гц), 4,11 (д, 2H, J=2,1 Гц), 4,40 (кв, 2H, J=7,1 Гц), 5,24 (т, 2H, J=1,6 Гц), 6,61 (с, 1H)
Стадия 3: (4H-5-тиа-1,6a-диазапентален-2-ил)метанол
LiBH4 (содержание 90%) (459 мг) добавляют к раствору в простом эфире (126 мл) этилового сложного эфира 4H-5-тиа-1,6a-диазапентален-2-карбоновой кислоты (2,5 г) и MeOH (0,77 мл) в атмосфере азота при комнатной температуре, затем кипятят с возвращением флегмы в течение 1,5 ч. Смесь гасят 1 моль/л HCl (25 мл) и перемешивают в течение 1 ч при комнатной температуре. Смесь нейтрализуют насыщенным раствором гидрокарбоната натрия и разделяют. Водный слой экстрагируют дихлорметаном (10 × 25 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют AcOEt. Указанное в заголовке соединение получают в виде бледно-желтого твердого вещества (1,7 г, 87,9%).
1H ЯМР (CDCl3) δ 2,95 (т, 1H, J=5,6 Гц), 4,07 (с, 2H), 4,62 (д, 2H, J=5,1 Гц), 5,13 (т, 1H, J=1,6 Гц), 6,04 (с, 1H).
Стадия 4: 4H-5-тиа-1,6a-диазапентален-2-карбальдегид
Раствор в сухом дихлорметане (8 мл) диметилсульфоксида (2,2 мл) добавляют по каплям к раствору в сухом дихлорметане (110 мл) оксалилхлорида (2,0 мл) при -78°C. Реакционную смесь перемешивают в течение 15 мин при той же температуре. Раствор в сухом дихлорметане (40 мл) (4H-5-тиа-1,6a-диазапентален-2-ил)метанола (1,7 г) добавляют по каплям к реакционной смеси при -78°C и перемешивание продолжают в течение дополнительных 15 мин. Реакционной смеси позволяют нагреться до -45°C и перемешивают в течение 1 ч. Триэтиламин (11,3 мл) добавляют по каплям и реакционной смеси позволяют нагреться до 0°C. Через 20 мин добавляют насыщенный раствор хлорида аммония (50 мл) и воду (100 мл) и проводят разделение. Водный слой экстрагируют AcOEt (3 × 150 мл). Объединенные органические слои промывают водой (200 мл) и насыщенным раствором соли (200 мл), сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью гексан - AcOEt (1:1). Указанное в заголовке соединение получают в виде желтого твердого вещества (1,7 г, количественный выход).
1H ЯМР (CDCl3) δ 4,13 (с, 2H), 5,26 (д, 2H, J=1,4 Гц), 6,59 (с, 1H), 9,90 (с, 1H).
Стадия 5: (5R)(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоноваякислота, натриевая соль
Раствор в сухом ацетонитриле (92 мл) 4H-5-тиа-1,6a-диазапентален-2-карбальдегида (1,7 г) добавляют к раствору в сухом ацетонитриле (92 мл) MgBr2 (5,0 г) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Раствор в сухом THF (184 мл) п-нитробензил-(5R,6S)-6-бромпенем-3-карбоксилата (4,3 г) добавляют и смесь охлаждают до -20°C, затем добавляют триэтиламин (7,4 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4-диметиламинопиридином (138 мг) и уксусным ангидридом (2,1 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Водный раствор 1 моль/л лимонной кислоты (1000 мл) добавляют к реакционной смеси и водный слой экстрагируют этилацетатом (3 × 400 мл). Объединенные органические слои промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(4H-5-тиа-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного твердого вещества.
Свежеактивированную Zn пыль (19,3 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 100 мл) к раствору в THF (100 мл) сырого п-нитробензилового сложного эфира (5R)-6-[ацетокси(4H-5-тиа-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2,5 ч при комнатной температуре. Реакционный раствор фильтруют через прокладку целита и прокладку промывают водой (200 мл) и н-бутанолом (200 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 50 мл). Объединенные органические слои концентрируют до 90 г, добавляют 1 моль/л NaOH, чтобы довести рН до 8,0, и подвергают колоночной хроматографии на смоле Diaion HP-21 resin (180 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 15% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (634 мг, 17,4%, pH 7,25). Т.пл. 150°C (с разложением).
1H ЯМР (D2О) δ 4,00 (с, 2H), 5,09 (с, 2H), 6,14 (с, 1H), 6,36 (с, 1H), 6,91 (с, 1H), 6,92 (с, 1H); IR (KBr) 3381, 1752, 1683, 1600, 1558 см-1; λмакс.(Н2О) 292, 196 нм.
Пример 13
Получение натриевой соли (5R)(6Z)-6-(7H-имидазо{1,2-c]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: тиазолидин-4-он
Указанное в заголовке соединение получают таким же путем, как Marvin M. и Allen R. Harkness. (Tetrahedron Letters. 1994, 35, 6971-6974).
Стадия 2: тиазолидин-4-тион
Реагент Lawesson (33,5 г) добавляют к раствору тиазолидин-4-она (14,2 г) в сухом THF (690 мл) и реакционную смесь кипятят с возвращением флегмы в течение 2 ч. Смесь охлаждают до комнатной температуры и выпаривают при пониженном давлении. Остаток растирают с раствором CHCl3:MeOH = 7:3 (65 мл) при комнатной температуре в течение 30 мин. Осадок отфильтровывают, промывают раствором CHCl3:н-гексан = 7:3 (15 мл) и сушат в вакууме. Тиазолидин-4-тион получают в виде бледно-желтого порошка (10,7 г, 65%).
1H ЯМР (CDCl3) δ 4,08 (с, 2H), 4,70 (с, 2H).
Стадия 3: 4-метилтио-2,5-дигидротиазол
Метилиодид (28,4 г) добавляют к кипящему раствору тиазолидин-4-тиона (9,5 г) в хлороформе (400 мл) и реакционную смесь кипятят с возвращением флегмы в течение 1,5 ч. К реакционной смеси добавляют дополнительный метилиодид (56,8 г) 5-ю порциями с интервалами 30-60 мин. После дополнительного кипячения с возвращением флегмы в течение 1 ч реакционную смесь охлаждают до комнатной температуры. Затем добавляют 10% водный раствор гидрокарбоната калия (200 мл) и перемешивают в течение 15 мин при комнатной температуре. После отделения органического слоя водный слой экстрагируют CHCl3 (100 мл × 3). Органические слои объединяют, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сушат в вакууме. После сушки указанное в заголовке соединение получают в виде коричневого масла (11,0 г, количественный выход).
1H ЯМР (CDCl3) δ 2,51 (с, 3H), 3,91 (т, 2H, J=3,5 Гц), 5,21 (т, 2H, J=3,5 Гц).
Стадия 4: тиазолидин-4-илиденамин
Смесь 4-метилтио-2,5-дигидротиазола (10,7 г) и хлорида аммония (6,4 г) в сухом этаноле (400 мл) кипятят с возвращением флегмы в течение 27,5 ч. Реакционную смесь охлаждают до комнатной температуры и выпаривают при пониженном давлении. Остаток растворяют в хлороформе (300 мл) и 10% водном растворе гидрокарбоната калия (200 мл), затем перемешивают в течение 20 мин при комнатной температуре. После отделения органического слоя водный слой экстрагируют хлороформом (100 мл × 5). Органические слои объединяют, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сушат в вакууме, получая сырой тиазолидин-4-илиденамин (5,5 г) в виде коричневого твердого вещества, который содержится в продукте, которым является этокси-производное и 4-метилтио-2,5-дигидротиазол, который является исходным материалом. Соотношение указанных трех соединений определено 1Н-ЯМР как 61:34:5 соответственно.
1H ЯМР (CDCl3) δ 3,75 (т, 2H, J=2,8 Гц), 4,97 (т, 2H, J=2,9 Гц).
Стадия 5: 7H-имидазо[1,2-c]тиазол-2-карбальдегид
Раствор 2-бром-3-изопропоксипропеналя (6,9 г) в сухом ацетонитриле (326 мл) добавляют к раствору сырого тиазолидин-4-илиденамина (3,3 г) в сухом ацетонитриле (326 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 19,5 ч, добавляют триэтиламин (4,9 мл) и затем кипятят с возвращением флегмы в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и затем выпаривают при пониженном давлении. Остаток растворяют в дихлорметане (300 мл) и промывают 50% водным раствором гидрокарбоната калия (20 г). После фильтрования и разделения водный слой экстрагируют дихлорметаном (50 мл × 4). Органические слои объединяют, сушат (MgSO4) и фильтруют. Фильтрат выпаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и элюируют смесью CHCl3-MeOH (100:3), получая сырой 7H-имидазо[1,2-c]тиазол-2-карбальдегид в виде коричневого твердого вещества. Сырой продукт подвергают перекристаллизации дважды из смеси CHCl3-н-гексан (1-й раз: 30:5, 2-й: 30:60) при 0°C, получая требуемый альдегид в виде бледно-коричневых кристаллов (Выход: 1,84 г, 15%).
1H ЯМР (CDCl3) δ 4,09 (т, 2H, J=1,3 Гц), 5,08 (т, 2H, J=1,2 Гц), 7,63 (с, 1H), 9,81 (с, 1H).
Стадия 6: (5R)(6Z)-6-(7H-имидазо[1,2-c]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота, натриевая соль
7H-имидазо[1,2-c]тиазол-2-карбальдегид (841 мг) добавляют к раствору в сухом ацетонитриле (116 мл) безводного MgBr2 (2,93 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (116 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 99,7%) (2,51 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (2,20 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 4 ч при -20°C и обрабатывают уксусным ангидридом (1,26 мл) и DMAP (160 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении.
Остаток растворяют в THF (53 мл) и ацетонитриле (25 мл). Свежеактивированную Zn пыль (15,1 г) и 0,5 M фосфатный буфер (pH 6,5, 78 мл) добавляют к смеси. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (321 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-9/1). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (1,1 г, 51%, pH 7,5). Т.пл. 145°C (с разложением).
1H ЯМР (D2О) δ 3,85 (с, 2H), 4,88 (с, 2H), 6,32 (с, 1H), 6,78 (с, 1H), 6,85 (с, 1H), 7,27 (с, 1H).
Пример 14
Получение (5R,6Z)-7-оксо-6-[(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метилен]-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: диэтил 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбоксилат
К раствору диэтил-3,5-пиразолдикарбоксилата (2,17 г, 10 ммоль) в ацетонитриле (10 мл) в атмосфере азота добавляют карбонат калия (2,07 г, 15 ммоль) и 2-бромэтокси-трет-бутилдиметилсилан (2,90 г, 12 ммоль). Смесь перемешивают при кипячении с возвращением флегмы в течение 18 ч. Затем ее охлаждают до комнатной температуры, разбавляют этилацетатом (20 мл) и фильтруют через Magnesol. Фильтрующую прокладку элюируют 2 × 10 мл этилацетатом и объединенный фильтрат выпаривают. Остаток растворяют в гексанах и пропускают через колонку силикагеля (70 г). После элюирования гексанами (100 мл) колонку элюируют этилацетатом. Этилацетатный элюент выпаривают, получая 3,71 г бесцветного масла. МС m/e 371 (MH+).
Стадия 2: 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-диметанол
К раствору диэтил-1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбоксилата (0,74 г, 2 ммоль) в метиленхлориде (8 мл) в атмосфере азота добавляют 12 мл 1,0 M раствора гидрида диизобутилалюминия в метиленхлориде при 0°C. После перемешивания при 0°C в течение 0,5 ч смесь нагревают до комнатной температуры в течение 0,5 ч. Затем ее гасят 15 мл насыщенного раствора хлорида аммония и экстрагируют этилацетатом. Органический экстракт промывают насыщенным раствором соли, сушат над безводным сульфатом натрия и выпаривают, получая 0,44 г белого твердого вещества. Т.пл. 82-83°C; МС m/e 287 (MH+).
Стадия 3: 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбальдегид
К перемешиваемому раствору 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-диметанола (1,18 г, 4 ммоль) в метиленхлориде (20 мл) добавляют 4-метилморфолин-N-оксид (2,89 г, 24 ммоль) и молекулярное сито 4A (4 г). Реакционную смесь перемешивают при комнатной температуре в течение 10 мин и затем обрабатывают пиррутенатом тетрапропиламмония (0,15 г, 0,4 ммоль). Перемешивание продолжают в течение 2 ч. Раствор метиленхлорида концентрируют и разбавляют простым эфиром (40 мл). Смесь фильтруют через прокладку силикагеля (40 г) и фильтрующую прокладку элюируют 2 × 20 мл простым эфиром. Объединенный элюент промывают 1 н. HCl и насыщенным раствором соли, сушат над безводным сульфатом натрия и выпаривают, получая 0,79 г белого твердого вещества. Т.пл. 63-64°C, МС m/e 283 (MH*).
Стадия 4: 4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбальдегид
К раствору 1-(2-{[трет-бутил(диметил)силил]окси}этил)-1H-пиразол-3,5-дикарбальдегида (1,02 г, 6,07 ммоль) в THF (30 мл) добавляют 6,68 мл 1,0 M раствора фторида тетрабутиламмония в THF при 0°C. После перемешивания в течение 1 ч смесь обрабатывают 10 мл насыщенного раствора хлорида аммония и экстрагируют этилацетатом. Органический раствор промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют через Magnesol и выпаривают. Сырое клейкое вещество промывают гексанами, сушат в вакууме и затем растворяют в метиленхлориде (20 мл). К этому раствору добавляют 4-метилморфолин-N-оксид (2,89 г, 24 ммоль) и молекулярное сито 4A (6 г). Смесь перемешивают при комнатной температуре в течение 10 мин и затем обрабатывают пирутенатом тетрапропиламмония (0,11 г, 0,3 ммоль). Перемешивание продолжают в течение 2 ч. Раствор метиленхлорида концентрируют и разбавляют этилацетатом (40 мл). Смесь фильтруют через прокладку силикагеля (40 г) и фильтрующую прокладку элюируют 2 × 20 мл этилацетатом. Объединенный элюент промывают 1 н. HCl и насыщенным раствором соли, сушат над безводным сульфатом натрия и выпаривают, получая 0,30 г белого твердого вещества. Т.пл. 135-136°C, МС m/e 167 (MH+).
Стадия 5: 4-нитробензил-(5R)-6-[(ацетилокси)(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
К раствору MgBr2 (0,46 г, 2,52 ммоль) в ацетонитриле (13 мл) в атмосфере азота добавляют 4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбальдегид (0,14 г, 0,84 ммоль) при комнатной температуре при перемешивании. Затем добавляют раствор 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,32 г, 0,84 ммоль) в THF (13 мл) и смесь охлаждают до -20°C. Добавляют триэтиламин (0,35 мл, 2,52 ммоль) и смесь перемешивают при -20°C в темноте в течение 4 ч. Затем ее обрабатывают уксусным ангидридом (0,2 мл, 2,0 ммоль) и 4-N,N-диметиламинопиридином (12 мг, 0,1 ммоль) и выдерживают при 0°C в течение 18 ч. Смесь концентрируют и остаток растворяют в этилацетате. Этилацетатный раствор промывают 5% лимонной кислотой, насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушат над безводным сульфатом натрия и выпаривают. Сырой материал подвергают хроматографии на силикагеле (EtOAc-CH2Cl2/1:5), получая 0,27 г не совсем белого твердого вещества. Т.пл. 107-110°C, МС m/e 595 (MH+).
Стадия 6: (5R,6Z)-7-оксо-6-[(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
К раствору 4-нитробензил-(5R)-6-[(ацетилокси)(4-оксо-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилата (0,22 г, 0,37 ммоль) в THF (15 мл) в атмосфере азота добавляют 15 мл раствора фосфатного буфера (0,5 M, pH 6,5) и 80 мг 10% Pd/C. Смесь гидрогенизируют при 40-50 фунт/кв.дюйм в течение 3 ч и затем фильтруют через целит. Фильтрующую прокладку промывают THF и фильтрат экстрагируют этилацетатом. Органический экстракт сушат над безводным сульфатом магния и выпаривают. Остаток промывают простым эфиром, получая 0,07 г желтого твердого вещества. МС m/e 320 (MH+).
1H ЯМР (ДМСО-d6) δ 4,55-4,57 (м, 2H), 4,76-4,80 (м, 2H), 6,50 (с, 1H), 6,63 (с, 1H), 7,58 (с, 1H), 7,76 (с, 1H).
Пример 15
Получение 6-(6,7-дигидро-4H-тиено[3,2-c]пиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение 6,7-дигидро-4H-тиено[3,2-c]пиран-2-карбальдегида
POCl3 (3,83 мл, 50 ммоль) добавляют по каплям к охлаждаемому на ледяной бане DMF (3,85 мл, 50 ммоль) в течение 3 минут. Добавляют DCM (20 мл) и баню удаляют, когда реакционная среда становится вязкой. Реакцию поддерживают при 23°С в течение 2 ч. Затем снова охлаждают до 0°С. Затем 4H-пиран-4-он (5 г, 50 ммоль) в 10 мл DCM добавляют по каплям в течение 3 минут. Реакцию поддерживают при 0°С в течение 2 ч. Смесь выливают на раствор ацетата натрия со льдом и экстрагируют DCM (2 × 200). Объединенные органические слои сушат над сульфатом магния. Отфильтровывают осушающий агент и концентрируют, получая 5,0 г продукта. Соединение растворяют в DCM (200 мл) и добавляют 6,0 г этил 2-6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-карбальдегидацетата и 10 мл TEA. Смесь кипятят с возвращением флегмы в течение 18 ч. Затем ее промывают водой и сушат над сульфатом магния. Затем ее фильтруют, концентрируют и подвергают флэш-хроматогафии с 20% этилацетатом в гексане. Собранный материал растворяют в 100 мл THF и впрыскивают LAH (150 мл, 0,5 M в THF) и оставляют при 23°С на 10 минут. Затем его кипятят с возвращением флегмы в течение 18 ч. Гасят при 23°С добавлением воды и в конце 1 н. HCl, чтобы осветлить смесь. Экстрагируют этилацетатом (2 × 200 мл) и объединенные органические слои сушат над сульфатом магния. Фильтрование и концентрирование дают 2,3 г продукта. Сырой материал растворяют в DCM (300 мл) и добавляют диоксид марганца (15 г). Реакцию проводят при 23°С в течение 0,5 ч. Затем добавляют 2 × 15 г оксиданта через каждые полчаса. Материал затем фильтруют через прокладку целита и концентрируют. Колоночная флэш-хроматография дает 1,206 г (14% выход) маслянистого продукта.
H-ЯМР: δ 9,84 (с, 1H), 7,41 (с, 1H), 4,74 (с, 2H), 4,00 (т, 2H, J=5,6 Гц), 2,96 (т, 2H, J=5,6 Гц); МС: 169,1 (М+H).
Стадия 2: Получение 6-(6,7-дигидро-4H-тиено[3,2-c]пиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-4H-тиено[3,2-c]пиран-2-карбальдегид (336 мг, 2 ммоль) растворяют в 20 мл ацетонитрила и затем добавляют бромид магния (516 мг, 2 ммоль) в атмосфере N2. Смесь перемешивают при 23°С в течение получаса. Затем 4-нитробензиловый сложный эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (770 мг, 2 ммоль) в 20 мл THF впрыскивают весь сразу и смесь немедленно охлаждают до -20°С. Затем впрыскивают триэтиламин (1 мл) и смесь перемешивают при -20°С в течение трех часов. Затем впрыскивают уксусный ангидрид (0,4 мл) и смесь перемешивают при 0°С в течение 18 ч. Реакционную среду затем разбавляют 400 мл этилацетата и промывают 100 мл 5% лимонной кислоты, 100 мл насыщенного раствора бикарбоната натрия и 100 мл насыщенного раствора соли. Органический слой затем сушат над сульфатом магния, фильтруют и концентрируют. Колоночная флэш-хроматография с использованием 20% этилацетата в гексане дает 491 мг (41%) продукта. Этот продукт затем растворяют в 15 мл THF и 15 мл водного фосфатного буфера (pH 6,5). Смесь затем подвергают воздействию водорода при 45 фунт/кв.дюйм в течение одного часа с 0,5 г 10% палладия на углероде. Затем ее фильтруют через прокладку целита и концентрируют в вакууме, чтобы удалить большую часть THF. Раствор затем охлаждают до нуля градусов и подщелачивают до pH 8 1 н. гидроксидом натрия. Затем ее очищают посредством ВЭЖХ с обращенной фазой с использованием 2 литров воды и затем 5% ацетонитрила в воде. Воду затем удаляют концентрированием в вакууме и собирают 100 мг (38%) продукта. Т.пл.: >250°C.
H-ЯМР: δ 7,36 (с, 1H), 7,15 (с, 1H), 6,55 (с, 1H), 6,44 (с, 1H), 4,61 (с, 2H), 3,88 (м, 2H), 2,86 (м, 2H), 2,27 (м, 2H), 1,43 (т, 3H); МС: 320,3 (М-H).
Пример 16
Получение 6-(6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение 6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-карбальдегида
POCl3 (4,02 мл, 43 ммоль) добавляют по каплям к охлаждаемому на ледяной бане DMF (3,34 мл, 43 ммоль) в течение 3 минут. Добавляют DCM (20 мл) и баню удаляют, когда реакционная среда становится вязкой. Реакционную среду поддерживают при 23°С в течение 2 ч. Затем ее охлаждают до 0°С снова. Тетрагидротиопиран-4-он (5 г, 43 ммоль) в 10 мл DCM затем добавляют по каплям в течение 3 минут. Реакционную среду поддерживают при 0°С в течение 2 ч. Разбавляют DCM (250 мл) и потом промывают 200 мл охлажденного льдом насыщенного водного раствора ацетата натрия. Органический слой сушат над сульфатом натрия. Отфильтровывают осушающий агент, концентрирование и колоночная флэш-хроматография с использованием 10% этилацетата в гексане дают 1,3 г (8 ммоль) продукта. Соединение растворяют в DCM (100 мл) и добавляют 1,2 мл (11 ммоль) этил-2-меркаптоацетата и 1 мл TEA. Смесь кипятят с возвращением флегмы в течение 18 ч. Затем ее промывают водой и сушат над сульфатом магния. Фильтрование, концентрирование и флэш-хроматография с 20% этилацетатом в гексане дают 1,1 г (11% выход) продукта.
H-ЯМР: δ 6,68 (с, 1H), 4,73 (с, 2H), 3,68 (с, 2H), 3,04 (т, 2H, J=7,6 Гц), 2,91 (т, 2H, J=7,6 Гц),; МС (EI): 185,99 (М+).
1,1 г этилового сложного эфира (4,8 ммоль) 6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-карбоновой кислоты растворяют в 100 мл THF и впрыскивают LAH (40 мл, 0,5M в DMG) и реакционную смесь оставляют при 23°С на 10 минут. Затем ее кипятят с возвращением флегмы в течение 18 ч. Гасят при 23°С водой (10 мл). Органический слой декантируют и остальное промывают 20 мл DCM. Объединенные органические слои сушат над сульфатом натрия. Фильтрование, концентрирование и колоночная флэш-хроматография с 10-20% этилацетатом дают 940 мг сырого продукта. Этот сырой материал растворяют в DCM (40 мл) и добавляют диоксид марганца (2 грамма). Реакцию проводят при 23°С в течение получаса. Материал затем фильтруют через прокладку целита и концентрируют. Колоночная флэш-хроматография дает 320 мг (36%) продукта.
H-ЯМР: δ 9,82 (с, 1H), 7,46 (с, 1H), 3,56 (с, 2H), 3,15 (т, 2H, J=7,2 Гц), 2,95 (т, 2H, J=7,2 Гц),; МС (EI): 228,02 (М+).
Стадия 2: Получение 6-(6,7-дигидро-4H-тиено[3,2-c]тиопиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7-Дигидро-4H-тиено[3,2-c]тиопиран-2-карбальдегид (320 мг, 1,72 ммоль) растворяют в 17 мл ацетонитрила и затем добавляют эфират бромида магния (450 мг, 1,74 ммоль) в атмосфере N2. Смесь перемешивают при 23°С в течение получаса. Затем впрыскивают 4-нитробензиловый сложный эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (660 мг, 1,72 ммоль) в 17 мл THF весь сразу и смесь немедленно охлаждают до -20°С. После этого впрыскивают триэтиламин (1 мл) и смесь перемешивают при -20°С в течение трех часов. Затем впрыскивают уксусный ангидрид (0,4 мл) и смесь перемешивают при 0°С в течение 18 ч. Реакционную среду затем разбавляют 400 мл этилацетата и промывают 100 мл 5% лимонной кислоты, 100 мл насыщенного раствора бикарбоната натрия и 100 мл насыщенного раствора соли. Органический слой затем сушат над сульфатом магния, фильтруют и концентрируют. Колоночная флэш-хроматографии с использованием 20% этилацетата в гексане дает 461 мг (44%) продукта. Этот продукт затем растворяют в 20 мл THF и 20 мл водного фосфатного буфера (pH 6,5). Смесь затем подвергают воздействию водорода под давлением 40 фунт/кв.дюйм в течение полутора часов с 0,5 г 10% палладия на углероде. Затем фильтруют через прокладку целита и концентрируют в вакууме, чтобы удалить большую часть THF. Раствор затем охлаждают до нуля градусов и подщелачивают до pH 8 1 н. гидроксидом натрия. Затем его очищают посредством ВЭЖХ с обращенной фазой с использованием 2 литров воды и потом 5% ацетонитрила в воде. Воду затем удаляют концентрированием в вакууме и собирают 21 мг (8,6%) продукта. Т.пл.:>250°C
H-ЯМР: 7,34 (с, 1H), 7,18 (с, 1H), 6,59 (с, 1H), 6,44 (с, 1H), 3,71 (с, 2H), 2,93 (с, 2H), 2,50 (с, 2H); МС: 338,0 (М+H).
Пример 17
Получение 6-(5-Метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение (5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метанола
Диэтиловый сложный эфир 6,7-дигидро-4H-тиено[3,2-c]пиридин-2,5-дикарбоновой кислоты (46 грамм, 163 ммоль) растворяют в 200 мл THF. В раствор впрыскивают LAH (1M, THF) 300 мл при 23°С. Затем все это перемешивают при 23°С в течение 18 ч. Реакционную смесь гасят 10 мл воды и сушат непосредственно над сульфатом натрия. Фильтрование и концентрирование дают 29,3 грамма (160 ммоль, 98%) сырого продукта.
H-ЯМР: 6,55 (с, 1H), 4,70 (с, 2H), 3,41 (с, 2H), 2,86 (т, 2H, J=5,6 Гц), 2,73 (т, 2H, J=5,6 Гц), 2,38 (с, 3H); МС: 184,0 (М+H).
Стадия 2: Получение 5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбальдегида
ДМСО (1,7 мл, 24 ммоль) в 5 мл CH2Cl2 охлаждают до -50 - -60°С. Оксалилхлорид (1 мл, 11 ммоль) в 20 мл DCM затем добавляют в течение 5 минут при 50°С. Смесь выдерживают при -50°С в течение 5 минут и затем добавляют 1,67 г (9 ммоль) (5-метил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метанола в 20 мл DCM при 50°С и смесь перемешивают в течение других 15 минут при 50°С. Затем добавляют триэтиламин (7 мл) при -50°С и спустя 5 минут баню удаляют и смесь естественным образом нагревается вплоть до 23°С. Ее промывают 100 мл воды и экстрагируют 100 мл этилацетата. Объединенные органические слои сушат над сульфатом магния. Фильтрование, концентрирование и колоночная флэш-хроматография с использованием 0-15% метанола в этилацетате дают 736 мг (45% выход) продукта.
H-ЯМР: 9,81 (с, 1H), 7,42 (с, 1H), 3,56 (с, 2H), 3,00 (т, 2H, J=5,6 Гц), 2,91 (т, 2H, J=5,6 Гц), 2,51 (с, 3H); МС: 182,1 (М+H).
Стадия 3: Получение 6-(5-метил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Этиловый сложный эфир 2-формил-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновой кислоты (724 мг, 4 ммоль) растворяют в 40 мл ацетонитрила и затем добавляют эфират бромида магния (1,2 г, 4,65 ммоль) в атмосфере N2. Смесь перемешивают при 23°С в течение получаса. Затем впрыскивают 4-нитробензиловый сложный эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,54 г, 4 ммоль) в 40 мл THF весь сразу и смесь немедленно охлаждают до -20°С. Затем впрыскивают триэтиламин (2 мл) и смесь перемешивают при -20°С в течение 3 ч. Затем впрыскивают уксусный ангидрид (0,66 мл) и смесь перемешивают при 0°С в течение 48 ч. Реакционную среду затем разбавляют 500 мл этилацетата и промывают 50 мл 5% лимонной кислоты, 50 мл насыщенного раствора бикарбоната натрия и 50 мл насыщенного раствора соли. Другие 300 мл этилацетата используют для промывания каждого водного раствора. Объединенные органические слои затем сушат над сульфатом натрия. Фильтрование, концентрирование и флэш-хроматография с использованием 20% этилацетата в гексане дают 1,56 грамма (64% выход) продукта. Этот продукт затем растворяют в 20 мл THF и 20 мл водного фосфатного буфера (pH 6,5). Смесь затем подвергают воздействию водорода под давлением 40 фунт/кв.дюйм в течение двух часов с 0,5 г 10% палладия на углероде. Затем ее фильтруют через прокладку целита и концентрируют в вакууме, чтобы удалить большую часть THF. Раствор затем охлаждают до нуля градусов и подщелачивают до pH 8 1 н. гидроксидом натрия. Затем его очищают посредством ВЭЖХ с обращенной фазой, используя 2 литра воды, а потом 5% ацетонитрил в воде. Воду затем удаляют концентрированием в вакууме и собирают 112 мг (13%) продукта. Т.пл.: >250°С.
H-ЯМР: δ 7,48 (с, 1H), 7,37 (с, 1H), 7,21 (с, 1H), 7,10 (с, 1H), 3,41 (с, 2H), 2,88 (с, 2H), 2,68 (с, 2H), 2,37 (с, 3H); МС: 335,0 (М+H).
Пример 18
Получение этилового сложного эфира 2-(2-карбокси-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-илиденметил)-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновой кислоты
Этиловый сложный эфир 2-формил-6,7-дигидро-4H-тиено[3,2-c]пиридин-5-карбоновой кислоты (480 мг, 2 ммоль) растворяют в 20 мл ацетонитрила и затем добавляют эфират бромида магния (516 мг, 2 ммоль) в атмосфере N2. Смесь перемешивают при 23°С в течение получаса. Затем впрыскивают 4-нитробензиловый сложный эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (770 мг, 2 ммоль) в 20 мл THF весь сразу и смесь немедленно охлаждают до -20°С. Затем впрыскивают триэтиламин (1 мл) и смесь перемешивают при -20°С в течение 3 ч. Затем впрыскивают уксусный ангидрид (0,4 мл) и смесь перемешивают при 0°С в течение 48 ч. Реакционную среду затем разбавляют 200 мл этилацетата и промывают последовательно 50 мл 5% лимонной кислоты, 50 мл насыщенного раствора бикарбоната натрия и 50 мл насыщенного раствора соли. Органический слой затем сушат над сульфатом натрия. Фильтрование, концентрирование и флэш-хроматография с использованием 20% этилацетата в гексане дает 690 мг (50% выход) продукта. Фракцию этого продукта (456 мг, 0,69 ммоль) затем растворяют в 15 мл THF и 15 мл водного фосфатного буфера (pH 6,5). Смесь затем подвергают воздействию водорода под давлением 40 фунт/кв.дюйм в течение двух часов с 0,5 г 10% палладия на углероде. Затем ее фильтруют через прокладку целита и концентрируют в вакууме, чтобы удалить большую часть THF. Раствор затем охлаждают до нуля градусов и подщелачивают до pH 8 1 н. гидроксидом натрия. Затем его очищают посредством ВЭЖХ с обращенной фазой, используя 2 литра воды, а потом 5% ацетонитрил в воде. Воду затем удаляют концентрированием в вакууме и собирают 18 мг (5%) продукта. Т.пл.: >250°С.
H-ЯМР: 7,35 (с, 1H), 7,24 (с, 1H), 6,61 (с, 1H), 6,45 (с, 1H), 4,48 (с, 2H), 4,08 (кв, 2H, J=7,2 Гц), 3,68 (м, 2H), 2,87 (м, 2H), 1,20 (т, 3H, J=7,2 Гц); МС: 393,0 (М+H).
Пример 19
Получение 7-оксо-6-(6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение 6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-карбальдегида
Тиокапролактам (6,45 г, 50 ммоль) растворяют в 400 мл CH2Cl2 и потом добавляют метилиодид (16 мл, 5 экв.). Смесь перемешивают в атмосфере азота в течение 18 ч. Затем ее обрабатывают 100 мл карбоната калия (50%, водный). Органический слой затем сушат над сульфатом магния. После фильтрования и концентрирования получают 7,3 г материала. Этот материал растворяют в 300 мл этанола и добавляют 2,83 г хлорида аммония. Смесь кипятят с возвращением флегмы в течение 1 ч. Затем растворитель удаляют в вакууме. К половине материала добавляют 200 мл этанола с последующим добавлением 1,35 г (25 ммоль) метоксида натрия и 4,8 г (25 ммоль) 2-бром-3-изопропоксипропеналя и смесь перемешивают при 23°С в течение 2 ч. Затем растворитель удаляют и 200 мл хлороформа добавляют наряду с 10 мл триэтиламина. Смесь кипятят с возвращением флегмы в течение 2 ч и затем охлаждают до 23°С. Реакционную среду распределяют между 300 мл DCM и 2×150 карбоната калия (50%). Органический слой сушат над сульфатом магния. После фильтрования и концентрирования получают 2,1 г маслянистого продукта.
H-ЯМР: 9,62 (с, 1H), 7,60 (с, 1H), 6,61 (с, 1H), 6,45 (с, 1H), 4,58 (с, 2H), 2,96 (2м, H), 1,90 (м, 2H), 1,72 (м, 2H); МС: 164,9 (М+H).
Стадия 2: Получение 7-оксо-6-(6,7,8,9-тетрагидро-5H-имидазо[1,2-a]азепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
6,7,8,9-Тетрагидро-5H-имидазо[1,2-a]азепин-2-карбальдегид (1,312 г, 8 ммоль) растворяют в 80 мл ацетонитрила и затем добавляют эфират бромида магния (2,94 г, 8 ммоль) в атмосфере N2. Смесь перемешивают при 23°С в течение получаса. Затем впрыскивают 4-нитробензиловый сложный эфир 6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,155 г, 3 ммоль) в 60 мл THF весь сразу и смесь немедленно охлаждают до -20°С. Затем впрыскивают триэтиламин (4 мл) и смесь перемешивают при -20°С в течение 4 ч. Затем уксусный ангидрид (1 мл) впрыскивают и смесь перемешивают при 0°С в течение 20 ч. Реакционную среду затем разбавляют 500 мл этилацетата и промывают 100 мл 5% лимонной кислоты, 100 мл насыщенного раствора бикарбоната натрия и 100 мл насыщенного раствора соли. Органический слой затем сушат над сульфатом натрия. Фильтрование, концентрирование и флэш-хроматография с использованием 20% этилацетата в гексане дает 800 мг продукта. Этот продукт затем растворяют в 20 мл THF и 20 мл водного фосфатного буфера (pH 6,5). Смесь затем подвергают воздействию водорода под давлением 40 фунт/кв.дюйм в течение 1 ч с 0,5 г 10% палладия на углероде. Затем ее фильтруют через прокладку целита и концентрируют в вакууме, чтобы удалить большую часть THF. Раствор затем охлаждают до нуля градусов и подщелачивают до pH 8 1 н. гидроксидом натрия. Затем его очищают посредством ВЭЖХ с обращенной фазой, используя 2 литра воды, а потом 5% ацетонитрил в воде. Воду затем удаляют посредством концентрирования в вакууме и собирают 131 мг (31%) продукта. Т.пл.: >250°C.
H-ЯМР: δ 7,78 (с, 1H), 7,02 (с, 1H), 6,94 (с, 1H), 6,36 (с, 1H), 3,92 (м, 2H), 2,80 (м, 2H), 1,78 (м, 2H), 1,61 (м, 2H), 1,54 (м, 2H); МС: 318,2 (М+H).
Пример 20
Получение натриевой соли (5R),(6Z)-6-(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: этиловый сложный эфир 7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты
Et3N (6,27 мл), PhCHO (4,92 мл) добавляют последовательно к раствору в EtOH (81 мл) гидрохлорида этилового сложного эфира 5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (9,47 г) при комнатной температуре и перемешивают в течение 3 ч в атмосфере азота. Затем NaBH3CN (2,97 г) добавляют к реакционной смеси и перемешивают в течение 19 ч. Смесь фильтруют через прокладку целита и разбавляют CH2Cl2 и промывают 50% водным раствором K2CO3. Органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью CHCl3-ацетон (1/0˜9/1) и CHCl3-MeOH (19/1˜9/1). Указанное в заголовке соединение получают в виде бледно-желтых кристаллов (4,16 г, 36%).
1H ЯМР (CDCl3) δ 1,36 (т, 3H, J=7,1 Гц), 2,87 (т, 2H, J=5,2 Гц), 3,71 (с, 2H), 3,75 (с, 2H), 4,01 (м, 2H), 4,34 (кв, 2H, J=7,1 Гц), 7,25-7,34 (м, 5H), 7,51 (с, 1H).
Стадия 2: 7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид
1,01 M раствор DIBAL в толуоле (1 мл + 0,2 мл + 0,3 мл) добавляют к раствору в сухом CH2Cl2 (5 мл) этилового сложного эфира 7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбоновой кислоты (283 мг) в атмосфере азота при -78°C и перемешивают в течение 1,5 ч. Смесь гасят 1 M HCl (5 мл). Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают 50% водным раствором K2CO3 и водный слой экстрагируют CH2Cl2. Объединенный органический слой сушат (K2CO3) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью CHCl3-ацетон (9/1˜4/1) и CHCl3-MeOH (19/1). Указанное в заголовке соединение получают в виде бесцветных кристаллов (148 мг, 61%).
1H ЯМР (CDCl3) δ 2,90 (т, 2H, J=5,5 Гц), 3,74 (с, 2H), 3,76 (с, 2H), 4,06 (т, 2H, J=5,5 Гц), 7,28-7,35 (м, 5H), 7,53 (с, 1H), 9,80 (с, 1H).
Стадия 3: 4-нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (смесь диастереоизомеров)
7-Бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-карбальдегид (139 мг) добавляют к раствору в сухом ацетонитриле (8,7 мл) безводного MgBr2 (325 мг) в атмосфере азота при комнатной температуре. Раствор в сухом THF (8,7 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (223 мг) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (0,24 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 5 ч при -20°C и обрабатывают уксусным ангидридом (0,11 мл) и DMAP (7 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (3/1-1/1). Указанное в заголовке соединение получают в виде смеси двух диастереоизомеров (80/20, пурпурное аморфное твердое вещество, 233 мг, 61%).
1H ЯМР (CDCl3) δ 1,99 (с, 0,8×3H), 2,23 (с, 0,2×3H), 2,83˜2,89 (м, 2H), 3,68 (д, 2H, J=4,9 Гц), 3,71 (с, 2H), 3,94˜4,13 (м, 2H), 5,27 (д, 1H, J=13,6 Гц), 5,41 (д, 0,2×1H, J=13,6 Гц), 5,45 (д, 0,8×1H, J=13,6 Гц), 6,05 (с, 0,2×1H), 6,28 (с, 0,8×1H), 6,31 (с, 0,8×1H), 6,790 (с, 0,2×1H), 6,793 (с, 0,2x1H), 7,01 (с, 0,8×1H), 7,27˜7,36 (м, 5H), 7,42 (с, 0,2×1H), 7,46 (с, 0,8×1H), 7,61 (д, 2H, J=8,6 Гц), 8,22 (д, 2H, J=8,6 Гц).
Стадия 4: натриевая соль (5R),(6Z)-6-(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-a]пиразин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,27 г) растворяют в THF (55 мл) и ацетонитриле (25 мл). Свежеактивированную Zn пыль (5,08 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 80 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (79 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-4/1). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (390 мг, 49%, pH 7,7). Т.пл. 180°C (с разложением).
1H ЯМР (D2О) δ 2,84˜2,95 (м, 2H), 3,61 (д, 2H, J=7,2 Гц), 3,67 (с, 2H), 3,96 (т, 2H, J=5,7 Гц), 6,43 (с, 1H), 6,89 (с, 1H), 6,93 (с, 1H), 7,28˜7,37 (м, 6H).
Пример 21
Получение (5R,6Z)-7-оксо-6-{[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-илметилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 2-формил-[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено][3,2-с]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридина (1,05 г, 5,2 ммоль) в DMF (20 мл), добавляют гидрохлорид 3-пиколилхлорида (0,852 г, 5,2 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, хорошо промывают водой и сушат над безводным MgSO4. Ее фильтруют и концентрируют. Продукт очищают колоночной хроматографией на SiO2, элюируя этилацетатом. Бледно-желтое полутвердое вещество. Выход: 800 мг, 59%. M+H 259.
Стадия 2: 4-нитробензил-6-[(ацетилокси)[5-(пиридин-3-илметил]-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин (516 мг, 2,0 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (390 мг, 1,5 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 700 мг, 51%. M+H 685 и 687.
Стадия 3: (5R,6Z)-6-{[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетилокси)[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (686 мг, 1,0 ммоль) растворяют в THF (20 мл) и ацетонитриле (10 мл). Свежеактивированную Zn пыль (5,2 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C, получая желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой НР-21. Вначале колонку элюируют деионизированной водой (2 литра) и позднее смесью 10% CAN:вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном и фильтруют. Сушат. Выход: 50 мг, 12%, в виде желтых кристаллов. Т.пл. 134-136°C. (M+H)412.
1H ЯМР (ДМСО-d6) δ д 2,8 (м, 2H), 2,92 (уш.м, 2H), 3,6 (м, 2H), 3,86 (с, 2H), 6,3 (с, 1H), 6,41 (с, 1H), 7,17 (с, 1H), 7,29 (с, 1H), 7,35 (м, 1H), 7,7 (м, 1H), 8,48 (д, 1H), 8,54 (с, 1H).
Пример 22
Получение (5R,6Z)-7-оксо-6-{[5-(пиридин-3-карбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты:
Стадия 1: 2-формил-[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено][3,2-с]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)пиридина (606 мг, 3,0 ммоль) в DMF (20 мл), добавляют гидрохлорид никотиноилхлорида (531 мг, 3,0 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, хорошо промывают водой и сушат над безводным MgSO4. Ее фильтруют и концентрируют. Продукт очищают колоночной хроматографией на SiO2, элюируя этилацетатом. Бледно-желтое полутвердое вещество. Выход: 600 мг, 73%. M+H 273.
Стадия 2: 4-нитробензил-6-[(ацетилокси)[5-[пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)карбонил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин (400 мг, 1,4 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (772 мг, 2,0 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (619 мг, 2,4 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 300 мг, 30%. Т.пл. 71°C. M+H 701.
Стадия 3: натриевая соль (5R,6Z)-6-{[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензил-6-[(ацетилокси)[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (800 мг, 1,14 ммоль) растворяют в THF (20 мл) и ацетонитриле (10 мл). Свежеактивированную Zn пыль (5,2 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 28 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C, получая желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой НР-21. Вначале колонку элюируют деионизированной водой (2 литра) и позднее смесью 10% CAN:вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном и фильтруют. Сушат. Выход: 50 мг, 12%, в виде желтых кристаллов. Т.пл. 195°C. (M+H)426.
Пример 23
Получение (5R,6Z)-7-оксо-6-{[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 2-формил-[5-(фенилацетил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин
К перемешиваемому раствору 2-(формил)-6,7-дигидротиено[3,2-c]-5(4H)-пиридина (0,41 мг, 2 ммоль) в DMF (20 мл) добавляют фенилацетилхлорид (0,35 мг, 2,2 ммоль) и N,N-диизопропилэтиламин (10 мл, избыток) при комнатной температуре. Реакционную смесь перемешивают в течение 24 ч и гасят водой. Реакционную смесь экстрагируют хлороформом, хорошо промывают водой и сушат над безводным MgSO4. Ее фильтруют и концентрируют. Продукт очищают колоночной хроматографией на SiO2, элюируя этилацетатом. Белое твердое вещество. Выход: 510 мг, 89%. M+H 286.
Стадия 2: 4-нитробензил-6-[(ацетилокси)[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2-Формил-[5-(фенилацетил)-4,5,6,7-тетрагидротиено][3,2-c]пиридин (340 мг, 1,2 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (390 мг, 1,0 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (310 мг, 1,2 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 360 мг, 50%. M+H 713.
Стадия 3: (5R,6Z)-6-{[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
4-Нитробензил-6-[(ацетилокси)[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (300 мг, 0,4 ммоль) растворяют в THF (50 мл) и 0,5 M фосфатном буфере (pH 6,5, 28 мл). Проводят гидрогенизацию под давлением 40 фунт/кв.дюйм в присутствии 10% Pd/C (80 мг) в течение 2 ч, в конце реакционную смесь фильтруют через прокладку целита и концентрируют. Отделенное желтое твердое вещество растворяют в этилацетате и хорошо промывают водой. Органический слой сушат и концентрируют. Отделенное желтое твердое вещество растирают с диэтиловым простым эфиром и фильтруют. Желтое твердое вещество промывают тщательно диэтиловым простым эфиром и получают соединение 95% чистоты. Выход: 160 мг, 91%. Желтое твердое вещество, т.пл. 166-169°C. (M+H) 439.
Пример 24
Получение натриевой соли (5R),(6Z)-6-(5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-карбальдегид
м-Хлорпербензойную кислоту (содержание 69%) (6,36 г) добавляют к раствору в CH2Cl2 (111 мл) 6,7-дигидро-4H-пиразоло[5,1-c][1,4]тиазин-2-карбальдегида (1,86 г) при 0°C. Реакционную смесь перемешивают в течение 0,5 ч при той же температуре и перемешивают в течение 18 ч при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении. Остаток растирают с 10 мл THF и фильтруют, получая кристаллы. Фильтрат концентрируют при пониженном давлении. Остаток растирают с 5 мл THF и фильтруют, получая кристаллы. Объединенные кристаллы сушат при пониженном давлении, получая указанное в заголовке соединение в виде бесцветных кристаллов (1,96 г, 89%).
1H ЯМР (CDCl3) δ 3,60 (т, 2H, J=6,1 Гц), 4,47 (с, 2H), 4,87 (т, 2H, J=6,1 Гц), 6,71 (с, 1H), 9,94 (с, 1H).
Стадия 2: 4-нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
5,5-Диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-карбальдегид (1,95 г) добавляют к раствору в сухом ацетонитриле (112 мл) безводного MgBr2 (содержание 98%) (5,48 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (112 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 96,5%) (3,88 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (содержание 99%) (3,79 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают уксусным ангидридом (содержание 97%) (3,79 мл) и DMAP (содержание 99%) (120 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 16 ч при 0°C. К реакционной смеси добавляют уксусный ангидрид (содержание 97%) (0,95 мл) и DMAP (содержание 99%) (120 мг) одной порцией. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4), а потом концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (CHCl3: ацетон = 19:1-4:1), получая указанное в заголовке соединение в виде бледно-коричневого аморфного твердого вещества (диастереосмесь (8:2), 1,35 г, 22%).
1H ЯМР (CDCl3) δ 2,07 (с, 3H×0,2), 2,25 (с, 3H×0,8), 3,45-3,60 (м, 2H), 4,39 (д, 1H, J=17,0 Гц), 4,44 (д, 1H, J=17,0 Гц), 4,65-4,78 (м, 2H), 5,28 (д, 1H, J=13,5 Гц), 5,43 (д, 1H×0,8, J=13,5 Гц), 5,44 (д, 1H×0,2, J=13,5 Гц), 6,05 (с, 1H×0,8), 6,20 (с, 1H×0,8), 6,22 (с, 1H×0,2H), 6,38 (с, 1H×0,2), 6,39 (с, 1H×0,2), 6,79 (с, 1H×0,8), 7,42 (с, 1H×0,8), 7,44 (с, 1H×0,2), 7,60 (д, 2H, J=8,7 Гц), 8,24 (д, 2H, J=8,7 Гц).
Стадия 3: натриевая соль (5R),(6Z)-6-(5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,33 г) растворяют в THF (19 мл) и ацетонитриле (9 мл). Свежеактивированную Zn пыль (5,32 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 27 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Нерастворимый материал отфильтровывают и промывают H2O (27 мл). К фильтрату добавляют H2O (27 мл) и промывают этилацетатом (27 мл) и водный слой охлаждают до 3°C и добавляют 1 M HCl, чтобы довести рН до 2,5. Смесь перемешивают в течение 1 д при той же температуре и добавляют H2O (55 мл), затем перемешивают в течение 4 д при той же температуре. Смесь перемешивают в течение 10 ч при комнатной температуре. Полученную смесь охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8. Смесь концентрируют в высоком вакууме при 35°C. Концентрат обрабатывают колоночной хроматографией на смоле Diaion HP-21 (80 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-9/1). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (306 мг, 38%, pH 7,4). Т.пл. 180°C (с разложением).
1H ЯМР (D2О) δ 3,83 (т, 2H, J=6,1 Гц), 4,68 (с, 2H), 4,72 (т, 2H, J=6,1 Гц), 6,37 (с, 1H), 6,40 (с, 1H), 6,95 (с, 1H), 6,98 (с, 1H).
Пример 25
Получение натриевой соли (5R),(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Пиперазин-2-карбоновая кислота, дигидрохлорид:
Указанное в заголовке соединение получают таким же путем, как M. T. Wu и сотрудники (Bioorg. Med. Chem. Lett. 1993, 3, 2023-2028).
Стадия 1: 1-(4-нитробензиловый) сложный эфир пиперазин-1,3-дикарбоновой кислоты
CuCO3·Cu(OH)2·H2O (15,8 г) добавляют к раствору в H2O (275 мл) дигидрохлорида пиперазин-2-карбоновой кислоты (22,3 г), затем смесь кипятят с возвращением флегмы и перемешивают в течение 10 мин. Нерастворимый материал отфильтровывают и промывают горячей H2O (165 мл). Фильтрат охлаждают до комнатной температуры и NaHCO3 (9,2 г) и 1,4-диоксан (220 мл) добавляют к темносинему раствору. Смесь охлаждают до 0°C и NaHCO3 (18,5 г) и 50% раствор 4-нитробензил-хлороформиат в 1,4-диоксане (61,7 г) добавляют к смеси в течение 0,5 ч. После перемешивания в течение дополнительных 1,5 ч при 0°C осадок отфильтровывают и промывают холодной H2O (140 мл), EtOH (100 мл), ацетоном (200 мл) и Et2O (100 мл), затем ему позволяют сохнуть при пониженном давлении до получения бледно-голубых кристаллов. Кристаллы добавляют к раствору в 1 моль/л HCl (330 мл) EDTA-2Na (20,5 г) в течение 30 мин и перемешивают в течение 2 ч при комнатной температуре. Суспензию фильтруют и отфильтрованный материал разбавляют EtOH-H2O (7:3, 550 мл) и кипятят с возвращением флегмы в течение 10 мин. Реакционную смесь фильтруют, получая бесцветные кристаллы. Перекристаллизацию из фильтрата проводят 3 раза, получая дополнительные кристаллы. Объединенные кристаллы сушат при пониженном давлении, получая указанное в заголовке соединение (26,25 г, 77%) в виде бесцветных кристаллов.
1H ЯМР (D2О) δ 2,54-2,61 (м, 1H), 2,89 (дт, 2H, J=12,7, 3,4 Гц), 2,97 (уш., 1H), 3,13 (уш., 1H), 3,62-4,04 (м, 2H), 5,16 (с, 2H), 7,49 (д, 2H, J=8,6 Гц), 8,14 (д, 2H, J=8,6 Гц).
Стадия 2: 5-(4-нитробензилоксикарбонил)-3-оксо-3a,4,6,7-тетрагидро-3H-2-окса-1,5-диаза-7a-азониаинден-3a-ид
Раствор в H2O (300 мл) NaNO2 (содержание 98,5%) (6,66 г) добавляют к раствору в уксусной кислоте (864 мл) 1-(4-нитробензилового) сложного эфира пиперазин-1,3-дикарбоновой кислоты (26,72 г) в атмосфере азота при 0°C в течение 0,5 ч и перемешивают в течение 1 ч. Дополнительно раствор в H2O (132 мл) NaNO2 (содержание 98,5%) (2,41 г) добавляют к раствору при 0°C в течение 0,5 ч и перемешивают в течение 1 ч. Раствор концентрируют при пониженном давлении и к остатку добавляют H2O (500 мл). Раствор экстрагируют AcOEt (5 раз) и органический слой промывают насыщенным раствором соли. Смесь сушат над MgSO4, фильтруют и концентрируют при пониженном давлении, получая сырой 1-(4-нитробензиловый) сложный эфир 4-нитрозопиперазин-1,3-дикарбоновой кислоты в виде бледно-коричневого аморфного вещества (27,83 г (брутто), 25,77 г (нетто), 88,2%).
Раствор в THF (10 мл) трифторуксусного ангидрида (24,0 г) добавляют к раствору в THF (371 мл) сырого 1-(4-нитробензилового) сложного эфира 4-нитрозопиперазин-1,3-дикарбоновой кислоты в атмосфере азота при 0°C в течение 15 мин. Раствор перемешивают в течение 1,5 ч при 0°C и в течение 1 ч при комнатной температуре. Раствор в THF (5 мл) трифторуксусного ангидрида (8,0 г) добавляют к раствору в течение 5 мин и перемешивают в течение 20 ч при комнатной температуре. К раствору добавляют трифторуксусный ангидрид (8,0 г) в течение 5 мин и раствор перемешивают в течение 4 ч. Осадок фильтруют и промывают THF и Et2O. Фильтрат концентрируют при пониженном давлении. Остаток растирают с THF, фильтруют и промывают Et2O. Эти материалы объединяют и сушат при пониженном давлении, получая указанное в заголовке соединение в виде бесцветных кристаллов (22,3 г, 91%).
1H ЯМР (CDCl3) δ 4,06 (т, 2H, J=5,4 Гц), 4,37 (т, 2H, J=5,4 Гц), 4,63 (с, 2H), 5,30 (с, 2H), 7,54 (д, 2H, J=8,7 Гц), 8,25 (д, 2H, J=8,7 Гц).
Стадия 3: 5-(4-нитробензиловый) сложный эфир 2-этилового сложного эфира 6,7-дигидро-4H-пиразоло[1,5-a]пиразин-2,5-дикарбоновой кислоты
Этилпропиолат (содержание 99%) (8,28 г) добавляют к раствору в о-ксилоле (348 мл) 5-(4-нитробензилоксикарбонил)-3-оксо-3a,4,6,7-тетрагидро-3H-2-окса-1,5-диаза-7a-азониаинден-3a-ида (22,3 г) в атмосфере азота и кипятят с возвращением флегмы в течение 16 ч. Раствор концентрируют при пониженном давлении, затем подвергают колоночной хроматографии на силикагеле 3 раза (н-гексан/AcOEt = 2/1-1/3). Указанное в заголовке соединение получают в виде бледно-желтых кристаллов (16,78 г, 64%). Кроме этого, получают 5-(4-нитробензиловый) сложный эфир 3-этилового сложного эфира 6,7-дигидро-4H-пиразоло[1,5-a]пиразин-3,5-дикарбоновой кислоты в виде бледно-желтых кристаллов (6,18 г, 24%).
1H ЯМР (CDCl3) δ 1,39 (т, 3H, J=7,1 Гц), 4,01 (т, 2H, J=5,5 Гц), 4,31 (т, 2H, J=5,5 Гц), 4,40 (кв, 2H, J=7,1 Гц), 4,79 (с, 2H), 5,29 (с, 2H), 6,64 (с, 1H), 7,54 (д, 2H, J=8,6 Гц), 8,24 (д, 2H, J=8,6 Гц).
Стадия 4: 4-нитробензиловый сложный эфир 2-гидроксиметил-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты
LiBH4 (640 мг) и MeOH (1,2 мл) добавляют к раствору в THF (267 мл) 5-(4-нитробензилового) сложного эфира 2-этилового сложного эфира 6,7-дигидро-4H-пиразоло[1,5-a]пиразин-2,5-дикарбоновой кислоты (10 г) в атмосфере азота при комнатной температуре и перемешивают в течение 3 ч при 40°C. Дополнительный LiBH4 (523 мг) и MeOH (1,0 мл) добавляют к раствору и перемешивают в течение 1 ч при 40°C и 1 ч при 50°C. Смесь подкисляют 3 моль/л HCl до pH 2 и перемешивают в течение 1 ч при комнатной температуре, затем твердый K2CO3 добавляют к раствору, чтобы довести рН до 8. Нерастворимый материал отфильтровывают и фильтрат экстрагируют AcOEt. Органический слой сушат (K2CO3) и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (CHCl3/MeOH =49/1-19/1), получая указанное в заголовке соединение в виде бледно-желтых кристаллов (8,44 г, 95%).
1H ЯМР (CDCl3) δ 1,69 (уш., 1H), 3,98 (т, 2H, J=5,5 Гц), 4,19 (т, 2H, J=5,5 Гц), 4,65 (с, 2H), 4,75 (с, 2H), 5,28 (с, 2H), 6,08 (с, 1H), 7,53 (д, 2H, J=8,7 Гц), 8,24 (д, 2H, J=8,7 Гц).
Стадия 5: 4-нитробензиловый сложный эфир 2-формил-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты
MnO2 (активированный) (84,2 г) добавляют к раствору в CHCl3-MeOH (95:5, 253 мл) 4-нитробензилового сложного эфира 2-гидроксиметил-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты (8,42 г) и смесь кипятят с возвращением флегмы в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. К фильтрату добавляют силикагель (20 г) и растворитель удаляют при пониженном давлении, получая силикагель, покрытый сырым реагентом. Указанный силикагель подвергают адсорбции в процессе колоночной хроматографии на силикагеле и колонку элюируют CHCl3-MeOH (49/1 до 19/1). Указанное в заголовке соединение получают в виде желтых кристаллов (2,82 г, 34%).
1H ЯМР (CDCl3) δ 4,05 (т, 2H, J=5,5 Гц), 4,32 (т, 2H, J=5,5 Гц), 4,81 (с, 2H), 5,29 (с, 2H), 6,62 (с, 1H), 7,54 (д, 2H, J=8,7 Гц), 8,24 (д, 2H, J=8,7 Гц), 9,93 (с, 1H).
Стадия 6: 4-нитробензиловый сложный эфир 2-{(RS)-ацетокси[(5R,6RS)-6-бром-2-(4-нитробензилоксикарбонил)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-ил]метил}-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты
4-Нитробензиловый сложный эфир 2-формил-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты (2,71 г) добавляют к раствору в сухом ацетонитриле (164 мл) безводного MgBr2 (содержание 98%) (6,17 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (164 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 96,5%) (3,27 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (содержание 99%) (9,24 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 1,5 ч при -20°C и обрабатывают уксусным ангидридом (содержание 97%) (3,19 мл) и DMAP (содержание 99%) (203 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 1 ч при 0°C. Уксусный ангидрид (3,19 мл) добавляют к раствору и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли. Органический слой сушат (MgSO4) с последующим концентрированием при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле три раза (н-гексан - AcOEt (1/1 до 2/3), CHCl3-ацетон (29/1 до 19/1) и CHCl3-ацетон (29/1)). Указанное в заголовке соединение получают в виде желтого аморфного вещества (диастерео-смесь (64:36), 3,30 г, 53%).
1H ЯМР (CDCl3) δ 2,06 (с, 3H×0,36), 2,26 (с, 3H×0,64), 3,95-4,04 (м, 2H), 4,18 (с, 2H), 4,73 (д, 1H, J=18,2 Гц), 4,78 (д, 1H, J=18,2 Гц), 5,28 (д, 1H, J=13,5 Гц), 5,28 (с, 2H), 5,43 (д, 1H×0,64, J=13,5 Гц), 5,44 (д, 1H×0,36), 6,06 (с, 1H×0,64), 6,08 (с, 1H×0,64), 6,24 (с, 1H×0,36), 6,27 (с, 1H×0,36), 6,41 (с, 1H×0,36), 6,79 (с, 1H×0,64), 7,42 (с, 1H×0,64), 7,44 (с, 1H×0,36), 7,53 (д, 2H, J=8,6 Гц), 7,60 (д, 2H, J=8,8 Гц), 8,24 (д, 2H, J=8,8 Гц), 8,24 (д, 2H, J=8,6 Гц).
Стадия 7: натриевая соль (5R),(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
К раствору в THF (43 мл) и ацетонитриле (20 мл) 4-нитробензилового сложного эфира 2-{(RS)-ацетокси[(5R,6RS)-6-бром-2-(4-нитробензилоксикарбонил)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-ил]метил}-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты быстро добавляют Zn пыль (12,36 г) с 0,5 M фосфатным буфером (pH 6,5, 63 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Нерастворимый материал отфильтровывают и промывают H2O (63 мл). Фильтрат промывают этилацетатом (63 мл) и водный слой охлаждают до 3°C и добавляют 1 M HCl, чтобы довести рН до 2,5. Смесь перемешивают в течение 4 ч при той же температуре и добавляют H2O (63 мл) и 1 M HCl, чтобы довести рН до 2,5, затем перемешивают в течение 17 ч при той же температуре. К смеси добавляют 1 M NaOH, чтобы довести рН до 8. Смесь концентрируют в высоком вакууме при 35°C. Концентрат обрабатывают колоночной хроматографией на смоле Diaion HP-21 (124 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-95/5). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (288 мг, 22%, pH 8,8). Т.пл. 160°C (с разложением).
1H ЯМР (D2О) δ 2,94 (т, 2H, J=5,6 Гц), 3,67 (д, 1H, J=17,2 Гц), 3,70 (д, 1H, J=17,2 Гц), 3,82 (т, 2H, J=5,6 Гц), 5,84 (с, 1H), 6,03 (с, 1 H), 6,65 (с, 1H), 6,67 (с, 1H).
Пример 26
Получение производного 5,5-диметил-2-пиперидона натриевой соли (5R,6Z)-6-(5,5-диметил-4H-1,6a-диазапентален-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
5-5-Диметил-2-пиперадинон получают по методу Nagasawa (J. Med. Chem., 20, 1176 (1977)).
Стадия 1: 3,3-дихлор-5,5-диметил-2-пиперидон
К холодному (0°C) перемешиваемому раствору 5,5-диметил-2-пиперидона (30,2 г, 0,24 моль) в 475 мл CHCl3 добавляют PCl5 (57,1 г, 0,26 моль) с такой скоростью, что температура никогда не превышает 7°C. После завершения добавления перемешивание продолжают в течение 10 мин. Сульфурилхлорид (96,6 г, 0,72 моль) медленно добавляют и смесь нагревают с возвращением флегмы в течение 1 ч. Раствор концентрируют при пониженном давлении. Остаток охлаждают на ледяной бане и разбавляют 250 мл воды со льдом. Продукт затем экстрагируют CHCl3 (6 × 250 мл) и органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью CHCl3-MeOH (50:1). Указанное в заголовке соединение получают в виде белого твердого вещества (41,3 г, 88,8%). (J. Med. Chem., 20, 1176 (1977)).
1H ЯМР (CDCl3) δ 1,17 (с, 6H), 2,76 (с, 2H), 3,19 (д, 2H, J=3,0 Гц), 6,82 (уш.с, 1H).
Стадия 2: 3-хлор-5,5-диметил-2-пиперидон
К 40,8 г (0,21 моль) 3,3-дихлор-5,5-диметил-2-пиперидона, растворенного в 410 мл AcOH, добавляют 10% Pd/C (влажность 50%, 6,2 г) и NaOAc·3H2O (62,4 г, 0,46 моль) и смесь гидрогенизируют под давлением 300 кПа в течение 20 мин. Давление водорода доводят до 300 кПа каждые 5 мин. Катализатор удаляют фильтрованием и фильтрат концентрируют при пониженном давлении. К остатку добавляют CHCl3 (400 мл) и воду (300 мл) и водный слой нейтрализуют 4 моль/л NaOH. Смесь отделяют и водный слой экстрагируют CHCl3 (5 × 300 мл) и органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью гексан-AcOEt (1:1). Указанное в заголовке соединение получают в виде белого твердого вещества (20,4 г, 59,9%). (J. Med. Chem., 20, 1176 (1977)).
1H ЯМР (CDCl3) δ 1,10 (с, 3H), 1,12 (с, 3H), 2,02 (дд, 1H, J=10,8, 13,6 Гц), 2,20 (ддд, 1H, J=2,2, 6,7, 13,6 Гц), 2,97 (ддд, 1H, J=2,3, 3,9, 12,1 Гц), 3,22 (д, 1H, J=12,1 Гц), 4,44 (дд, 1H, J=6,8, 10,7 Гц), 6,66 (уш.с, 1H).
Стадия 3: 4,4-диметилпирролидин-2-карбоновая кислота
Суспензию 20,4 г (0,13 моль) 3-хлор-5,5-диметил-2-пиперидона и 45,2 г (0,14 моль) Ba(OH)3-8H2O в 252 мл воды нагревают в аппарате Parr при 150°C в течение 6 ч. Затем добавляют 18,6 г (0,14 моль) сульфата аммония. Осадок отфильтровывают и раствор концентрируют при пониженном давлении до сухости. Сырую 4,4-диметилпирролидин-2-карбоновую кислоту получают в виде белого твердого вещества (37,5 г). (J. Med. Chem., 20, 1176 (1977), EP 0447704 A1, стр. 17).
1H ЯМР (D2О) δ 1,10 (с, 3H), 1,11 (с, 3H), 1,88 (дд, 1H, J=7,8, 13,2 Гц), 2,21 (дд, 1H, J=9,2, 13,2 Гц), 3,12 (дд, 2H, J=11,5, 23,5 Гц), 4,22 (дд, 1H, J=8,1, 8,9 Гц).
Стадия 4: 5,5-диметил-3-оксо-3a,4-дигидро-3H,6H-2-окса-5-1-аза-6a-азонио-3a-пенталенид
К суспензии 37,5 г сырой 4,4-диметилпирролидин-2-карбоновой кислоты в 420 мл AcOH добавляют раствор 13,3 г (0,19 моль) NaNO2 в 210 мл воды в течение 15 мин при комнатной температуре и перемешивают в течение 3 ч. Раствор концентрируют при пониженном давлении. К остатку добавляют ацетон (250 мл) и осадок отфильтровывают и раствор концентрируют при пониженном давлении до сухости и получают сырую 4,4-диметил-1-нитрозопирролидин-2-карбоновую кислоту в виде коричневого масла.
К раствору сырой 4,4-диметил-1-нитрозопирролидин-2-карбоновой кислоты в 252 мл сухого THF добавляют трифторуксусный ангидрид (81,3 г, 0,39 моль) в атмосфере азота при 0°C и перемешивают в течение 6 ч при 0°C. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью н-гексан - AcOEt (2:1). Указанное в заголовке соединение получают в виде коричневого твердого вещества (12,0 г, 61,7%).
1H ЯМР (CDCl3) δ 1,38 (с, 6H), 2,71 (с, 2H), 4,12 (с, 2H).
Стадия 5: этиловый сложный эфир 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбоновой кислоты
Раствор 5,5-диметил-3-оксо-3a,4-дигидро-3H,6H-2-окса-5-1-аза-6a-азонио-3a-пенталенида (10,8 г, 0,07 моль) и этилпропиолата (10,8 мл, 0,11 моль) в o-ксилоле (350 мл) кипятят с возвращением флегмы в атмосфере азота в течение 16 ч. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью н-гексан - AcOEt (3:1). Указанное в заголовке соединение получают в виде бледно-коричневого твердого вещества (4,63 г, 31,7%) и этиловый сложный эфир 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-карбоновой кислоты получают в виде желтого твердого вещества (4,73 г, 32,4%).
Этиловый сложный эфир 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбоновой кислоты: 1H ЯМР (CDCl3) δ 1,29 (с, 6H), 1,40 (т, 3H, J=7,1 Гц), 2,71 (с, 2H), 3,93 (с, 2H), 4,39 (кв, 2H, J=7,1 Гц), 6,54 (с, 1H).
Этиловый сложный эфир 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-3-карбоновой кислоты: 1H ЯМР (CDCl3) δ 1,32 (с, 6H), 1,33 (т, 3H, J=7,1 Гц), 2,89 (с, 2H), 3,90 (с, 2H), 4,26 (кв, 2H, J=7,1 Гц), 7,90 (с, 1H).
Стадия 6: 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбальдегид
К 4,63 г (22,2 ммоль) этилового сложного эфира 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-карбоновой кислоты в 222 мл сухого THF добавляют LiAlH4 (0,85 г, 22,3 ммоль) в атмосфере азота при 0°C и затем перемешивают в течение 1 ч. Смесь гасят водой (5,0 мл) и осадок фильтруют через прокладку целита и прокладку промывают водой (50 мл) и THF (150 мл). Фильтрат концентрируют при пониженном давлении и затем добавляют воду (50 мл). Водный слой экстрагируют CHCl3 (5 × 100 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-ил)метанол получают в виде желтого твердого вещества (3,19 г).
К 3,19 г сырого (5,5-диметил-5,6-дигидро-4H-пирроло[1,2-b]пиразол-2-ил)метанола в 222 мл CHCl3 добавляют активированный MnO2 (18,5 г) в атмосфере азота при комнатной температуре и затем кипятят с возвращением флегмы в течение 1 ч. Смесь фильтруют через прокладку целита и фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью гексан-AcOEt (3:1). Указанное в заголовке соединение получают в виде коричневого твердого вещества (2,48 г, 68,0% от сложного эфира).
1H ЯМР (CDCl3) δ 1,32 (с, 6H), 2,73 (с, 2H), 3,95 (с, 2H), 6,52 (с, 1H), 9,90 (с, 1Н).
Стадия 7: натриевая соль (5R)(6Z)-6-(5,5-диметил-4H-1,6a-диазапентален-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в сухом ацетонитриле (16 мл) 5,5-диметил-5,6-дигидро-4H-пирроло[1,2-6]пиразол-2-карбальдегида (2,48 г, 15,1 ммоль) добавляют к раствору в сухом ацетонитриле (90 мл) MgBr2 (3,07 г, 16,4 ммоль) в атмосфере азота при комнатной температуре и затем смесь перемешивают в течение 15 мин. Раствор в сухом THF (106 мл) п-нитробензил-(5R,6S)-6-бромпенем-3-карбоксилата (5,30 г, 13,8 ммоль) добавляют и смесь охлаждают до -20°C и затем добавляют триэтиламин (4,6 мл, 33,0 ммоль) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4-диметиламинопиридином (172 мг, 1,4 ммоль) и уксусным ангидридом (2,6 мл, 27,6 ммоль) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 16 ч при 0°C. Этилацетат (420 мл) и водный раствор 1 моль/л лимонной кислоты (210 мл) добавляют к реакционной смеси и разделяют ее. Органический слой промывают насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(5,5-диметил-4H-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного вещества.
Свежеактивированную Zn пыль (32,0 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 167 мл) к раствору в THF (114 мл) и ацетонитриле (53 мл) сырого п-нитробензилового сложного эфира (5R)-6-[ацетокси(5,5-диметил-4H-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционный раствор охлаждают при 0°C и затем pH доводят до 8,0. Этилацетат (85 мл) добавляют к смеси и фильтруют через прокладку целита. Прокладку промывают водой (120 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 50 мл). Объединенные органические слои охлаждают при 0°C и затем pH доводят до 8,5. Смесь концентрируют до 325 г и затем подвергают колоночной хроматографии на смоле Diaion HP-21 (240 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой (480 мл) и затем водным раствором ацетонитрила (10%, 480 мл, 20%, 720 мл). Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (2,00 г, 42,8%, pH 7,16). Т.пл. 150°C (с разложением).
1H ЯМР (D2О) δ 1,19 (с, 6H), 2,67 (с, 2H), 3,85 (с, 2H), 6,15 (с, 1H), 6,45 (с, 1H), 6,96 (с, 1H), 7,03 (с, 1H); IR (KBr) 3422, 1752, 1683, 1598, 1557 см-1; λмакс. (Н2О) 296,198 нм.
Пример 27
Получение натриевой соли (5R),(6Z)-6-(5,6-дигидро-4H-циклопента[b]фуран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: метиловый сложный эфир 5,6-дигидро-4H-циклопента[b]фуран-2-карбоновой кислоты
Указанное в заголовке соединение получают согласно процедуре Tim Johnson и сотрудников (Synlett 2001, 5, 646-648).
Стадия 2: (5,6-дигидро-4H-циклопента[b]фуран-2-ил)метанол
Метиловый сложный эфир 5,6-дигидро-4H-циклопента[b]фуран-2-карбоновой кислоты (2,24 г) добавляют к раствору в THF (59 мл) LiAlH4 (511 мг) в атмосфере азота при 0°C и перемешивают в течение 1 ч при 0°C. Смесь гасят 10 мл воды и фильтруют. Фильтрат концентрируют при пониженном давлении и полученный водный раствор экстрагируют CHCl3. Органический слой промывают насыщенным раствором соли и сушат над MgSO4 и фильтруют. Фильтрат концентрируют, получая указанное в заголовке соединение в виде желтого масла (1,86 г, количественный выход).
1H ЯМР (CDCl3) δ 1,66 (т, 1H, J=5,9 Гц), 2,38-2,46 (м, 2H), 2,50-2,55 (м, 2H), 2,65-2,70 (м, 2H), 4,54 (д, 2H, J=5,9 Гц), 6,15 (с, 1H).
Стадия 3: 5,6-дигидро-4H-циклопента[b]фуран-2-карбальдегид
Активированный MnO2 (9,3 г) добавляют к раствору в CHCl3 (135 мл) (5,6-дигидро-4Н-циклопента[b]фуран-2-ил)метанола (1,86 г) и кипятят с возвращением флегмы в течение 1 ч в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (9/1-7/1). Указанное в заголовке соединение получают в виде желтых кристаллов (1,51 г, 77%).
1H ЯМР (CDCl3) δ 2,47-2,57 (м, 2H), 2,63 (т, 2H, J=6,8 Гц), 2,78 (т, 2H, J=7,3 Гц), 7,06 (с, 1H), 9,44 (с, 1H).
Стадия 4: 4-нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(5,6-дигидро-4H-циклопента[b]фуран-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в ацетонитриле (50 мл) 5,6-дигидро-4H-циклопента[b]фуран-2-карбальдегида (1,33 г) добавляют к раствору в сухом ацетонитриле (101 мл) безводного MgBr2 (содержание 98%) (5,52 г) в атмосфере азота при комнатной температуре. Раствор в сухом THF (151 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (содержание 96,5%) (3,91 г) добавляют к смеси, охлаждают до -20°C и добавляют Et3N (содержание 99%) (8,28 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (содержание 97%) (4,13 мл) и DMAP (содержание 99%) (121 мг) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 16 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4), затем фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (н-гексан:AcOEt = 4:1 - 3:1), получая указанное в заголовке соединение в виде коричневого аморфного твердого вещества (3,34 г, 61%).
1H ЯМР (CDCl3) δ 2,21 (с, 3H), 2,40-2,48 (м, 2H), 2,53 (т, 2H, J=7,0 Гц), 2,69 (т, 2H, J=7,0 Гц), 5,28 (д, 1H, J=13,5 Гц), 5,43 (д, 1H, J=13,5 Гц), 6,00 (с, 1H), 6,37 (с, 1H), 6,71 (с, 1H), 7,41 (с, 1H), 7,60 (д, 2H, J=8,1 Гц), 8,24 (д, 2H, J=8,1 Гц).
Стадия 5: натриевая соль (5R),(6Z)-6-(5,6-дигидро-4H-циклопента[b]фуран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензиловый сложный эфир (5R,6RS)-6-[(RS)-ацетокси(5,6-дигидро-4H-циклопента[b]фуран-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (3,28 г) растворяют в THF (46 мл) и ацетонитриле (21 мл). Свежеактивированную Zn пыль (13,12 г) быстро добавляют с 0,5 M фосфатным буфером (pH 6,5, 67 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,25 ч при комнатной температуре. Реакционную смесь фильтруют через прокладку целита. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой охлаждают до 3°C и добавляют 1 M NaOH, чтобы довести рН до 8,0. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (181 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-85/15). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанный в заголовке сырой продукт (288 мг). Его очищают колоночной хроматографией на смоле Diaion HP-21 (100 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют смесью H2O-MeCN (1/0-85/15). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (185 мг, 10%, pH 7,2). Т.пл. 170°C (с разложением).
1H ЯМР (D2О) δ 2,24-2,30 (м, 2H), 2,37 (т, 2H, J=6,5 Гц), 2,52-2,57 (т, 2H, J=7,1 Гц), 6,32 (с, 1H), 6,55 (с, 1H), 6,73 (с, 1H), 6,86 (с, 1H).
Пример 28
Получение натриевой соли (5R)(6Z}-6-(4,5-дигидро-6-тиа-1,7a-диазаинден-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: гидрохлорид DL-тетрагидро-1,3-тиазин-4-карбоновой кислоты
Гидрохлорид DL-тетрагидро-1,3-тиазин-4-карбоновой кислоты получают согласно методу Lewis (J. Med. Chem., 21, 1070 (1978)).
Стадия 2: 4,5-дигидро-3aH,7H-2-окса-3-оксо-6-тиа-1-аза-7a-азониоинден
К суспензии гидрохлорида DL-тетрагидро-1,3-тиазин-4-карбоновой кислоты (48,6 г, 0,26 моль) в 666 мл AcOH добавляют раствор 27,4 г (0,40 моль) NaNO2 в 333 мл воды в течение 16 мин при комнатной температуре и перемешивают в течение 3 ч. Раствор концентрируют при пониженном давлении. Ацетон (300 мл) добавляют к остатку и осадок отфильтровывают. Фильтрат концентрируют при пониженном давлении до сухости и сырую 3-нитрозо[1,3]тиазинан-4-карбоновую кислоту получают в виде коричневого аморфного твердого вещества.
К раствору сырой 3-нитрозо[1,3]тиазинан-4-карбоновой кислоты в 530 мл сухого THF добавляют трифторуксусный ангидрид (168,4 г, 0,80 моль) в течение 60 мин в атмосфере азота при 0°C и перемешивают в течение 5 ч при 0°C. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью н-гексан - AcOEt (1:2). Указанное в заголовке соединение получают в виде коричневого порошка (28,0 г, 67,0%).
1H ЯМР (CDCl3) δ 3,00 (т, 2H, J=5,7 Гц), 3,07 (т, 2H, J=5,7 Гц), 5,16 (с, 2H).
Стадия 3: этиловый сложный эфир 4,5-дигидро-6-тиа-1,7a-диазаинден-2-карбоновой кислоты
Раствор 4,5-дигидро-3aH,7H-2-окса-3-оксо-6-тиа-1-аза-7a-азониоиндена (28,0 г, 0,18 моль) и этилпропиолата (27,0 мл, 0,27 моль) в о-ксилоле(590 мл) кипятят с возвращением флегмы в атмосфере азота в течение 16 ч. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью н-гексан-AcOEt (3:1). Указанное в заголовке соединение получают в виде бледно-коричневых игольчатых кристаллов (22,1 г, 58,7%) и этиловый сложный эфир 4,5-дигидро-6-тиа-1,7a-диазаинден-3-карбоновой кислоты получают в виде бледно-коричневых кристаллов (12,7 г, 33,9%).
Этиловый сложный эфир 4,5-дигидро-6-тиа-1,7a-диазаинден-2-карбоновой кислоты: 1H ЯМР (CDCl3) δ 1,39 (т, 3H, J=7,1 Гц), 2,98 (т, 2H, J=6,1 Гц), 3,21 (т, 2H, J=6,1 Гц), 4,40 (кв, 2H, J=7,1 Гц), 5,17 (с, 2H), 6,60 (с, 1H).
Этиловый сложный эфир 4,5-дигидро-6-тиа-1,7a-диазаинден-3-карбоновой кислоты: 1H ЯМР (CDCl3) δ 1,34 (т, 3H, J=7,1 Гц), 2,99 (т, 2H, J=6,1 Гц), 3,45 (т, 2H, J=6,1 Гц), 4,28 (кв, 2H, J=7,1 Гц), 5,11 (с, 2H), 7,85 (с, 1H).
Стадия 4: 4,5-дигидро-6-тиа-1,7a-диазаинден-2-карбальдегид
К 22,1 грамма (0,10 моль) этилового сложного эфира 4,5-дигидро-6-тиа-1,7a-диазаинден-2-карбоновой кислоты в 520 мл сухого THF добавляют LiAlH4 (3,95 г, 0,10 моль) в атмосфере азота при 0°C и затем перемешивают в течение 45 мин. Смесь гасят водой (20 мл) и осадок фильтруют через прокладку целита и прокладку промывают водой (100 мл) и THF (250 мл). Фильтрат концентрируют при пониженном давлении и затем добавляют воду (300 мл). Водный слой экстрагируют CH2Cl2 (6 × 500 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой продукт получают в виде бледно-желтых кристаллов (17,2 г).
К 17,2 г сырого (4,5-дигидро-6-тиа-1,7a-диазаинден-2-ил)метанола в 520 мл CHCl3 добавляют активированный MnO2 (88,0 г) в атмосфере азота при комнатной температуре и затем кипятят с возвращением флегмы в течение 2 ч. Смесь фильтруют через прокладку целита и фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью гексан-AcOEt (2:1). Указанное в заголовке соединение получают в виде желтых кристаллов (13,0 г, 74,5%).
1H ЯМР (CDCl3) δ 3,00 (т, 2H, J=6,0 Гц), 3,23 (т, 2H, J=6,0 Гц), 5,18 (с, 2H), 6,58 (с, 1H), 9,92 (с, 1H).
Стадия 5: натриевая соль (5R)(6Z)-6-(4,5-дигидро-6-тиа-1,7a-диазаинден-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в сухом ацетонитриле (11 мл) 4,5-дигидро-6-тиа-1,7a-диазаинден-2-карбальдегида (1,70 г, 10,1 ммоль) добавляют к раствору в сухом ацетонитриле (60 мл) MgBr2 (2,03 г, 11,0 ммоль) в атмосфере азота при комнатной температуре и затем смесь перемешивают в течение 10 мин. Раствор в сухом THF (71 мл) п-нитробензил-(5R,6S)-6-бромпенем-3-карбоксилата (3,55 г, 9,2 ммоль) добавляют и смесь охлаждают до -20°C и затем добавляют триэтиламин (3,1 мл, 22,2 ммоль) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4-диметиламинопиридином (0,11 г, 0,9 ммоль) и уксусным ангидридом (1,8 мл, 18,6 ммоль) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Этилацетат (280 мл) и водный раствор 1 моль/л лимонной кислоты (140 мл) добавляют к реакционной смеси и разделяют ее. Органический слой промывают насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(4,5-дигидро-6-тиа-1,7a-диазаинден-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного твердого вещества.
Свежеактивированную Zn пыль (21,4 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 112 мл) к раствору в THF (76 мл) и ацетонитриле (36 мл) сырого п-нитробензилового сложного эфира (5R)-6-[ацетокси(4,5-дигидро-6-тиа-1,7a-диазаинден-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционный раствор охлаждают при 0°C и затем pH доводят до 8,0. Этилацетат (56 мл) добавляют к смеси и фильтруют через прокладку целита. Прокладку промывают водой (150 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 30 мл). Объединенные органические слои охлаждают при 0°C и затем pH доводят до 8,0. Смесь концентрируют до 236 г и затем подвергают колоночной хроматографии на смоле Diaion HP-21 (480 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой (960 мл) и затем водным раствором ацетонитрила (5%, 960 мл, 10%, 960 мл, 20%, 960 мл). Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (1,28 г, 40,5%, pH 7,45). Т.пл. 200°C (с разложением).
1H ЯМР (D2О) δ 2,95 (т, 2H, J=6,1 Гц), 3,12 (т, 2H, J=6,1 Гц), 5,08 (с, 2H), 6,23 (с, 1H), 6,46 (с, 1H), 6,97 (с, 1H), 7,01 (с, 1H); IR (KBr) 3382, 1752, 1684, 1597, 1554 см-1; λмакс. (H2O) 366, 292, 197 нм.
Пример 29
Получение натриевой соли (5R),(6Z)-6-(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиризин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение 5,5-диметил-2-пиперидона
5,5-Диметил-2-пиперадинон (1) получают по методу Nagasawa (J. Med. Chem., 23, 1176 (1977)).
Стадия 2: Получение 3,3-диметил-6-метокси-2,3,4,5-тетрагидропиридина
Тетрафторборат триметилоксония (97%, 11,9 г, 78 ммоль) добавляют к раствору в сухом дихлорметане (156 мл) 5,5-диметил-2-пиперидона (9,93 г, 78 ммоль) при комнатной температуре и перемешивают в течение 14 ч. Реакционную смесь нейтрализуют 10% водным раствором гидрокарбоната натрия и органический слой отделяют. Водный слой экстрагируют этилацетатом (3 × 120 мл), затем объединенный органический слой промывают 10% водным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и указанное в заголовке соединение получают в виде бледно-желтого масла (9,0 г, 82,0%).
1H ЯМР (CDCl3) δ 0,92 (с, 6H), 1,49 (т, 2H, J=7,0 Гц), 2,18 (т, 2H, J=7,0 Гц), 3,19 (с, 2H), 3,63 (с, 3H).
Стадия 3: моногидрохлорид 5,5-диметилпиперидин-2-илиденамина
Смесь 3,3-диметил-6-метокси-2,3,4,5-тетрагидропиридина (9,0 г, 64 ммоль) и хлорида аммония (3,4 г, 64 ммоль) в сухом этаноле (160 мл) нагревают до кипения с возвращением флегмы в течение 2 ч. Реакционную смесь затем концентрируют при пониженном давлении и указанное в заголовке соединение получают в виде белого твердого вещества (9,9 г, 94,6%).
1H ЯМР (ДМСО-d6) δ 0,95 (с, 6H), 1,52 (т, 2H, J=6,9 Гц), 2,55 (т, 2H, J=6,9 Гц), 2,99 (д, 2H, J=2,1 Гц).
Стадия 4: 6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-карбальдегид и 6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-3-карбальдегид
Смесь 2-бром-3-гидроксипропеналя (10,1 г, 67 ммоль), моногидрата п-толуолсульфоновой кислоты (0,13 г, 0,6 ммоль) и 2-пропанола (12,6 мл, 165 ммоль) в циклогексане (100 мл) подвергают азеотропной перегонке, пока температура пара не превысит 80°C. Реакционную смесь концентрируют при пониженном давлении. Остаток растворяют в сухом EtOH (200 мл). Раствор в сухом EtOH (350 мл) моногидрохлорида 5,5-диметилпиперидин-2-илиденамина (9,9 г, 61 ммоль) и раствор в сухом EtOH (50 мл) NaOMe (28%, 11,7 г, 61 ммоль) добавляют при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 2 ч и затем реакционный раствор удаляют в вакууме. Остаток растворяют в CHCl3 (300 мл) и добавляют триэтиламин (8,5 мл, 61 ммоль) и затем реакционную смесь нагревают до температуры кипения с возвращением флегмы в течение 2 ч. Реакционную смесь охлаждают до комнатной температуры и затем реакционный раствор удаляют в вакууме. Остаток растворяют в CH2Cl2 (600 мл) и промывают 50% водным растворои K2CO3 (2 × 200 мл). Объединенный водный раствор экстрагируют CH2Cl2 (2 × 200 мл). Объединенный органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируют смесью CHCl3-метанол (50:1) и получают указанное в заголовке соединение 6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-карбальдегид (коричневое твердое вещество, 4,4 г, 40,7%) и 6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-3-карбальдегид (оранжевое твердое вещество, 1,7 г, 15,8%).
6,6-Диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-карбальдегид: 1H ЯМР (CDCl3) δ 1,10 (с, 6H), 1,78 (т, 2H, J=6,9 Гц), 2,95 (т, 2H, J=6,9 Гц), 3,71 (с, 2H), 7,46 (с, 1H), 9,83 (с, 1H).
6,6-Диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-3-карбальдегид: 1H ЯМР (CDCl3) δ 1,09 (с, 6H), 1,74 (т, 2H, J=6,8 Гц), 2,97 (т, 2H, J=6,8 Гц), 4,05 (с, 2H), 7,74 (с, 1H), 9,64 (с, 1H).
Стадия 5: натриевая соль (5R),(6Z)-6-(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиризин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в сухом ацетонитриле (28 мл) 6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-карбальдегида (4,55 г, 26 ммоль) добавляют к раствору в сухом ацетонитриле (152 мл) MgBr2 (5,22 г, 28 ммоль) в атмосфере азота при комнатной температуре и затем смесь перемешивают в течение 10 мин. Раствор в сухом THF (180 мл) п-нитробензил-(5R,6S)-6-бромпенем-3-карбоксилата (8,94 г, 23 ммоль) добавляют и смесь охлаждают до -20°C и затем добавляют триэтиламин (7,8 мл, 56 ммоль) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4-диметиламинопиридином (0,29 г, 2,4 ммоль) и уксусным ангидридом (4,4 мл, 47 ммоль) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 16 ч при 0°C. Этилацетат (715 мл) добавляют к реакционной смеси и затем органический слой промывают 1 моль/л водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного твердого вещества.
Свежеактивированную Zn пыль (53,6 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 282 мл) к раствору в THF (192 мл) и ацетонитриле (90 мл) п-нитробензилового сложного эфира (5R)-6-[ацетокси(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 ч при комнатной температуре. Реакционную смесь охлаждают при 0°C и затем pH доводят до 7,6. Этилацетат (140 мл) добавляют к реакционной смеси и затем смесь фильтруют через прокладку целита и прокладку промывают водой (200 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 50 мл). pH объединенного органического слоя доводят до 8,1 и смесь концентрируют до 584 г. 1 моль/л NaOH добавляют, чтобы довести рН до 8,2 и подвергают колоночной хроматографии на смоле Diaion HP-21 (420 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют 2,5% (2 объема слоя), 5% (2 объема слоя), 10% (1 объем слоя) и 20% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая сырую натриевую соль (5R),(6Z)-6-(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиризин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде желтого аморфного твердого вещества (1,19 г).
Сырую натриевую соль (5R),(6Z)-6-(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-a]пиризин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты очищают препаративной ВЭЖХ (Mightysil RP-18 GP (5 ), Kanto Chemical Co. Inc., 35 × 250 мм, 0,05 моль/л фосфатный буфер (pH 7,2):CH3CN = 70:30, 20 мл/мин). Очищенный продукт обессоливают колоночной хроматографией на смоле Diaion HP-21 (50 мл) и указанное в заголовке соединение получают в количестве 230 мг (2,8%) в виде желтого аморфного твердого вещества. Т.пл. 210°C (с разложением).
1H ЯМР (D2О) δ 0,91 (с, 3H), 0,93 (с, 3H), 1,63 (т, 2H, J=6,8 Гц), 2,72 (т, 2H, J=6,8 Гц), 3,60 (с, 2H), 6,44 (с, 1H), 6,90 (с, 1H), 6,91 (с, 1H), 7,19 (с, 1H).
Пример 30
Получение (5R),(6Z)-6-(5,6-дигидро-8-H-имидазо[2,1-c][1,4]тиазин-3-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в сухом ацетонитриле (40 мл) 5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-3-карбальдегида (813 мг) добавляют к раствору в сухом ацетонитриле (40 мл) MgBr2 (2,2 г) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Добавляют раствор в сухом THF (80 мл) п-нитробензил-(5R, 6S)-6-бромпенем-3-карбоксилата (2,1 г), смесь охлаждают до -20°C, затем добавляют триэтиламин (1,7 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3,5 ч при -20°C и обрабатывают 4,4-диметиламинопиридином (64 мг) и уксусным ангидридом (0,9 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 14 ч при 0°C. К реакционной смеси добавляют 10% водный раствор лимонной кислоты (500 мл) и водный слой экстрагируют этилацетатом (3 × 200 мл). Органический слой промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и элюируют смесью CH2Cl2-ацетон (20:1), получая сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(5,6-дигидро-8H-имидазо[2,1-c][1,4]тиазин-3-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты в виде коричневого твердого вещества.
Твердое вещество, полученное указанной хроматографией, растворяют в THF (11 мл). Свежеактивированную Zn пыль (1,4 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 11 мл). Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 ч при комнатной температуре. Реакционный раствор фильтруют через прокладку целита и прокладку промывают водой (26 мл) и н-бутанолом (26 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 5 мл). Объединенный органический слой концентрируют до 18 г, добавляют 1 моль/л NaOH, чтобы довести рН до 7,3, и подвергают колоночной хроматографии на смоле Diaion HP-21 (20 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 5% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (81 мг). Т.пл. 145°C (с разложением).
1H ЯМР (D2О) δ 3,05-3,08 (м, 1H), 3,83 (с, 1H), 4,13-4,16 (м, 1H), 6,37 (с, 1H), 6,91 (с, 1H), 7,01 (с, 1H), 7,04 (с, 1H); IR (KBr) 3371, 1770, 1672, 1613 см-1; λмакс. (H2O) 314 нм.
Пример 31
Получение натриевой соли (5R)(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение 3-оксо-3a,4-дигидро-3H,6H-2-окса-5-тиа-1-аза-6a-азонио-3a-пенталенида
Концентрированную HCl (15 мл) и NaNO2 (16,6 г) добавляют к раствору в H2O (166 мл) L-тиопролина (24,3 г) в атмосфере азота при 0°C и перемешивают в течение 2 ч. Раствор экстрагируют CH2Cl2, органический слой сушат над MgSO4 и концентрируют при пониженном давлении, получая сырое N-нитрозо-соединение в виде желтого твердого вещества.
Трифторуксусный ангидрид (5,0 мл) добавляют к раствору в THF (350 мл) сырого N-нитрозо-тиопролина в атмосфере азота при 0°C и перемешивают в течение 5 ч при 0°C. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (1:1). Указанное в заголовке соединение получают в виде бледно-коричневого твердого вещества (4,0 г, 15,1%).
1H ЯМР (CDCl3) δ 4,04 (т, 2H, J=1,7 Гц), 5,40 (т, 2H, J=1,7 Гц).
Стадия 2: Получение этилового сложного эфира 4H-5-тиа-1,6a-диазапентален-2-карбоновой кислоты
Этилпропиолат (3,1 мл) добавляют к раствору в о-ксилоле (130 мл) 3-оксо-3a,4-дигидро-3H,6H-2-окса-5-тиа-1-аза-6a-азонио-3a-пенталенида (4,0 г) в атмосфере азота и кипятят с возвращением флегмы в течение 19 ч. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью н-гексан-AcOEt (4:1). Указанное в заголовке соединение получают в виде желтого твердого вещества (2,7 г, 49,3%) и этиловый сложный эфир 4H-5-тиа-1,6a-диазапентален-3-карбоновой кислоты получают в виде бледно-желтых кристаллов (1,2 г, 21,7%).
1H ЯМР (CDCl3) δ 1,40 (т, 3H, J=7,1 Гц), 4,11 (д, 2H, J=2,1 Гц), 4,40 (кв, 2H, J=7,1 Гц), 5,24 (т, 2H, J=1,6 Гц), 6,61 (с, 1H).
Стадия 3: Получение (4H-5-тиа-1,6a-диазапентален-2-ил)метанола
LiBH4 (содержание 90%) (459 мг) добавляют к раствору в простом эфире (126 мл) этилового сложного эфира 4H-5-тиа-1,6a-диазапентален-2-карбоновой кислоты (2,5 г) и MeOH (0,77 мл) в атмосфере азота при комнатной температуре, затем кипятят с возвращением флегмы в течение 1,5 ч. Смесь гасят 1 моль/л HCl (25 мл) и перемешивают в течение 1 ч при комнатной температуре. Смесь нейтрализуют насыщенным раствором гидрокарбоната натрия и разделяют. Водный слой экстрагируют дихлорметаном (10 × 25 мл). Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют AcOEt. Указанное в заголовке соединение получают в виде бледно-желтого твердого вещества (1,7 г, 87,9%).
1H ЯМР (CDCl3) δ 2,95 (т, 1H, J=5,6 Гц), 4,07 (с, 2H), 4,62 (д, 2H, J=5,1 Гц), 5,13 (т, 1H, J=1,6 Гц), 6,04 (с, 1H).
Стадия 4: Получение 4H-5-тиа-1,6a-диазапентален-2-карбальдегида
Раствор в сухом дихлорметане (8 мл) диметилсудьфоксида (2,2 мл) добавляют по каплям к раствору в сухом дихлорметане (110 мл) оксалилхлорида (2,0 мл) при -78°C. Реакционную смесь перемешивают в течение 15 мин при той же температуре. Раствор в сухом дихлорметане (40 мл) (4H-5-тиа-1,6a-диазапентален-2-ил)метанола (1,7 г) добавляют по каплям к реакционной смеси при -78°C и перемешивание продолжают в течение дополнительных 15 мин. Реакционной смеси позволяют нагреться до -45°C и перемешивают в течение 1 ч. Триэтиламин (11,3 мл) добавляют по каплям и реакционной смеси позволяют нагреться до 0°C. Через 20 мин добавляют насыщенный раствор хлорида аммония (50 мл) и воду (100 мл) и разделяют смесь. Водный слой экстрагируют AcOEt (3 × 150 мл). Объединенные органические слои промывают водой (200 мл) и насыщенным раствором соли (200 мл), сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью гексан-AcOEt (1:1). Указанное в заголовке соединение получают в виде желтого твердого вещества (1,7 г, количественный выход).
1H ЯМР (CDCl3) δ 4,13 (с, 2H), 5,26 (д, 2H, J=1,4 Гц), 6,59 (с, 1H), 9,90 (с, 1H).
Стадия 5: Получение натриевой соли (5R)(6Z)-7-оксо-6-(4H-5-тиа-1,6a-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в сухом ацетонитриле (92 мл) 4H-5-тиа-1,6a-диазапентален-2-карбальдегида (1,7 г) добавляют к раствору в сухом ацетонитриле (92 мл) MgBr2 (5,0 г) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 мин. Раствор в сухом THF (184 мл) п-нитробензил-(5R,6S)-6-бромпенем-3-карбоксилата (4,3 г) добавляют и смесь охлаждают до -20°C, затем добавляют триэтиламин (7,4 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 ч при -20°C и обрабатывают 4-диметиламинопиридином (138 мг) и уксусным ангидридом (2,1 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Водный раствор 1 моль/л лимонной кислоты (1000 мл) добавляют к реакционной смеси и водный слой экстрагируют этилацетатом (3 × 400 мл). Объединенные органические слои промывают водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(4H-5-тиа-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного вещества.
Свежеактивированную Zn пыль (19,3 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 100 мл) к раствору в THF (100 мл) сырого п-нитробензилового сложного эфира (5R)-6-[ацетокси(4H-5-тиа-1,6a-диазапентален-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2,5 ч при комнатной температуре. Реакционный раствор фильтруют через прокладку целита и прокладку промывают водой (200 мл) и н-бутанолом (200 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 50 мл). Объединенные органические слои концентрируют до 90 г, добавляют 1 моль/л NaOH, чтобы довести рН до 8,0, и подвергают колоночной хроматографии на смоле Diaion HP-21 (180 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 15% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (634 мг, 17,4%, pH 7,25). Т.пл. 150°C (с разложением).
1H ЯМР (D2О) δ 4,00 (с, 2H), 5,09 (с, 2H), 6,14 (с, 1H), 6,36 (с, 1H), 6,91 (с, 1H), 6,92 (с, 1H); IR (KBr) 3381, 1752, 1683, 1600, 1558 см-1; λмакс. (H2O) 292, 196 нм.
Пример 32
Получение натриевой соли (5R)(6Z)-6-(2,3-дигидропиразоло[5,1-b]тиазол-6-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение 3-оксо-3a,4-дигидро-3Н,6H-2-окса-4-тиа-1-аза-6a-азонио-3a-пенталенида
К суспензии тиазолидин-2-карбоновой кислоты (39,9 г, 0,30 моль) в 1000 мл уксусной кислоты добавляют раствор 31,0 г (0,45 моль) нитрита натрия в 500 мл воды в течение 13 минут при комнатной температуре и перемешивают в течение 5 часов. Реакционный раствор концентрируют при пониженном давлении. К остатку добавляют ацетон (500 мл) и осадок фильтруют через прокладку целита. Прокладку промывают ацетоном (500 мл). Фильтрат концентрируют при пониженном давлении до сухости и сырую 3-нитрозотиазолидин-2-карбоновую кислоту получают в виде желтого твердого вещества.
К раствору сырой 3-нитрозотиазолидин-2-карбоновой кислоты в 600 мл сухого тетрагидрофурана добавляют трифторуксусный ангидрид (189,6 г, 0,90 моль) в течение 20 минут в атмосфере азота при 0°C и перемешивают в течение 19 часов при 0°C. Раствор концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью н-гексан-этилацетат (1:1). Указанное в заголовке соединение получают в виде бледно-коричневых кристаллов (19,2 г, 44,5%).
1H ЯМР (CDCl3) δ 3,98 (т, 2H, J=7,7 Гц), 4,65 (т, 2H, J=7,7 Гц).
Стадия 2: Получение этилового сложного эфира 2,3-дигидропиразоло[5,1-b]тиазол-6-карбоновой кислоты и этилового сложного эфира 2,3-дигидропиразоло[5,1-b]тиазол-7-карбоновой кислоты
Этилпропиолат (20,3 мл, 0,20 моль) добавляют к раствору в o-ксилоле (600 мл) 3-оксо-3a,4-дигидро-3H,6H-2-окса-4-тиа-1-аза-6a-азонио-3a-пенталенида (19,2 г, 0,13 моль) в атмосфере азота и кипятят с возвращением флегмы в течение 21 часа. Раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью н-гексан-этилацетат (2:21 до 1:1). Смесь этилового сложного эфира 2,3-дигидропиразоло[5,1-b]тиазол-6-карбоновой кислоты и этилового сложного эфира 2,3-дигидропиразоло[5,1-b]тиазол-7-карбоновой кислоты получают в виде коричневого масла в соотношении 1:1,5 соответственно. (21,2 г, Выход: 80,0%).
Этиловый сложный эфир 2,3-дигидропиразоло[5,1-b]тиазол-6-карбоновой кислоты: 1H ЯМР (CDCl3) δ 1,39 (т, 3H, J=7,1 Гц), 3,82 (т, 2H, J=7,5 Гц), 4,39 (кв, 2H, J=7,1 Гц), 4,42 (т, 2H, J=7,5 Гц), 6,52 (с, 1H).
Этиловый сложный эфир 2,3-дигидропиразоло[5,1-b]тиазол-7-карбоновой кислоты: 1H ЯМР (CDCl3) δ 1,34 (т, 3H, J=7,1 Гц), 3,85 (т, 2H, J=7,8 Гц), 4,28 (кв, 2H, J=7,1 Гц), 4,39 (т, 2H, J=7,8 Гц), 7,87 (с, 1H).
Стадия 3: 2,3-дигидропиразоло[5,1-b]тиазол-6-карбальдегид и 2,3-дигидропиразоло[5,1-b]тиазол-7-карбальдегид
К смеси [21,2 г (0,11 моль)] этилового сложного эфира 2,3-дигидропиразоло[5,1-b]тиазол-6-карбоновой кислоты и этилового сложного эфира 2,3-дигидропиразоло[5,1-b]тиазол-7-карбоновой кислоты в 540 мл сухого тетрагидрофурана добавляют LiAlH4 (4,05 г, 0,11 моль) в атмосфере азота при 0°C и затем перемешивают в течение 2,5 часов при комнатной температуре. Смесь гасят водой (15 мл) и осадок фильтруют через прокладку целита. Прокладку промывают водой (100 мл) и тетрагидрофураном (500 мл). Фильтрат концентрируют при пониженном давлении и затем добавляют воду (150 мл). Водный слой экстрагируют дихлорметаном (15 × 250 мл). Объединенные органические слои сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и смесь (2,3-дигидропиразоло[5,1-b]тиазол-6-ил)метанола и (2,3-дигидропиразоло[5,1-b]тиазол-7-ил)метанола получают в виде бледно-коричневого масла (15,5 г).
К смеси [15,5 г (0,10 моль)] (2,3-дигидропиразоло[5,1-b]тиазол-6-ил)метанола и (2,3-дигидропиразоло[5,1-b]тиазол-7-ил)метанола в 500 мл хлороформа добавляют активированный MnO2 (77,7 г) в атмосфере азота при комнатной температуре и затем кипятят с возвращением флегмы в течение 3 часов. Смесь фильтруют через прокладку целита и фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле и затем колонку элюируют смесью гексан-этилацетат (2:1 до 1:1). Указанное в заголовке соединение 2,3-дигидропиразоло[5,1-b]тиазол-6-карбальдегид получают в виде желтых кристаллов (2,50 г, 15,2%) и 2,3-дигидропиразоло[5,1-b]тиазол-7-карбальдегид получают в виде бледно-коричневого твердого вещества (5,57 г, 33,8%).
2,3-Дигидропиразоло[5,1-b]тиазол-6-карбальдегид: 1H ЯМР (CDCl3) δ 3,86 (т, 2H, J=7,5 Гц), 4,45 (т, 2H, J=7,5 Гц), 6,50 (с, 1H), 9,83 (с, 1H).
2,3-Дигидропиразоло[5,1-b]тиазол-7-карбальдегид: 1H ЯМР (CDCl3) δ 3,92 (т, 2H, J=7,9 Гц), 4,40 (т, 2H, J=7,9 Гц), 7,91 (с, 1H), 9,76 (с, 1H).
Стадия 4: Получение натриевой соли (5R)(6Z)-6-(2,3-дигидропиразоло[5,1-b]тиазол-6-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Раствор в сухом ацетонитриле (19 мл) 2,3-дигидропиразоло[5,1-b]тиазол-6-карбальдегида (2,50 г, 16,2 ммоль) добавляют к раствору в сухом ацетонитриле (106 мл) MgBr2 (3,67 г, 19,9 ммоль) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 минут. Раствор в сухом тетрагидрофуране (125 мл) п-нитробензил-(5R, 6S)-6-бромпенем-3-карбоксилата (6,23 г, 16,2 ммоль) добавляют и смесь охлаждают до -20°C, затем добавляют триэтиламин (5,4 мл, 38,7 ммоль) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 3 часов при -20°C и обрабатывают 4-диметиламинопиридином (198 мг, 1,62 ммоль) и уксусным ангидридом (3,1 мл, 32,9 ммоль) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 16 часов при 0°C. Этилацетат (500 мл) добавляют к реакционной смеси и затем органический слой промывают водным раствором 1 моль/л лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении и сырой п-нитробензиловый сложный эфир (5R)-6-[ацетокси(2,3-дигидропиразоло[5,1-b]тиазол-6-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты получают в виде коричневого аморфного твердого вещества.
Свежеактивированную Zn пыль (37,4 г) быстро добавляют с 0,5 моль/л фосфатным буфером (pH 6,5, 196 мл) к раствору в тетрагидрофуране (134 мл) и ацетонитриле (62 мл) п-нитробензилового сложного эфира (5R)-6-[ацетокси(2,3-дигидропиразоло[5,1-b]тиазол-6-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 1,5 часов при комнатной температуре. Реакционную смесь охлаждают при 0°C и затем pH доводят до 8,0. Этилацетат (100 мл) добавляют к реакционной смеси. Смесь фильтруют через прокладку целита и прокладку промывают водой (300 мл). Водный слой отделяют и затем органический слой экстрагируют 0,5 моль/л фосфатным буфером (pH 6,5, 2 × 50 мл). pH объединенного органического слоя доводят до 8,0 и смесь концентрируют до 426 г. Концентрат доводят до pH 8,0 и подвергают колоночной хроматографии на смоле Diaion HP-21 (540 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой (1 объем слоя) и затем 5% (2 объема слоя), 10% (2 объема слоя) и 20% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде оранжевого аморфного твердого вещества (2,09 г, 39,2%, pH 7,10). Т.пл. 150°C (с разложением).
1H ЯМР (D2О) δ 3,75 (т, 2H, J=7,5 Гц), 4,27 (т, 2H, J=7,5 Гц), 6,00 (с, 1H), 6,34 (с, 1H), 6,85 (с, 1H), 6,94 (с, 1H); IR (KBr) 3392, 1755, 1596, 1554 см-1; λмакс. (H2O) 290, 223 нм.
Пример 33
Получение натриевой соли (5R)(6Z)-6-(2,3-дигидропиразоло[5,1-b]оксазол-6-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение этил-2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-карбоксилата
К перемешиваемой суспензии этил-5-гидрокси-1H-пиразол-3-карбоксилата (10,34 г, 0,66 моль) и 36,62 г карбоната калия в 500 мл ацетонитрила добавляют 13,68 г 1,2-дибромметана и кипятят с возвращением флегмы в течение 16 часов. Реакционной смеси позволяют остыть до комнатной температуры, затем фильтруют, твердое вещество промывают ацетонитрилом. Фильтрат концентрируют до масла. Остаток растворяют в этилацетате и экстрагируют водой. Органическую фазу сушат над MgSO4 и выпаривают до сухости. Получают 5,80 г желаемого продукта (48%).
Стадия 2: Получение 2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-метанола
К перемешиваемому раствору этил-2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-карбоксилата (5,47 г, 35 ммоль) в 100 мл THF добавляют 1,05 г боргидрида лития и 1,54 г метанола. Раствор нагревают при 40°С в течение 2,5 часов. Реакционную смесь гасят 1 н. HCl и доводят до pH 1,3 и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь доводят K2CO3 до рН 8. Реакционную смесь экстрагируют этилацетатом. Органический слой сушат над MgSO4 и концентрируют до масла и подвергают колоночной хроматографии, получая 2,68 г желательного продукта (65%).
Стадия 3: Получение 2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-карбальдегида
К перемешиваемому раствору 2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-метанола (2,60 г, 18,5 ммоль) в 60 мл CH3Cl добавляют 12,9 г MnO2. Суспензию кипятят с возвращением флегмы в течение 1,5 часов в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. Фильтрат концентрируют, получая желтое масло. Продукт очищают хроматографией. Получают 2,15 г продукта (84,3%).
Стадия 4: 4-нитробензил-(5R)-6-[(ацетилокси)(2,3-дигидропиразоло [5,1-b][1,3]оксазол-6-ил)-)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-карбальдегид (607 мг, 4,3 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (1,54 г, 4,6 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (2,21 г, 8,5 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 1,9 г, 81%. M+H 566.
H ЯМР (CDCl3) 8,24 (2H, д, J=6,6 Гц), 7,60 (2H, д, J=6,6 Гц), 7,44 (1H, с), 6,34 (1H, с), 6,23 (1H, с), 5,56 (1H, с), 5,44 (1H, д, J=10,2 Гц), 5,27 (1H, д, J=10,2 Гц), 5,04 (2H, м), 4,30 (2H, м), 2,10 (3H, с).
Анализ. Рассчитано для C21H17BrN4O8S: C 44,61, H 3,03, N 9,91
Обнаружено: C 45,00, H 3,14, N 9,53.
Стадия 5: натриевая соль (5R,6Z)-6-(2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
4-Нитробензил-6-[(ацетилокси)(2,3-дигидропиразоло[5,1-b][1,3]оксазол-6-ил)-)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (700 мг, 1,2 ммоль) растворяют в THF (20 мл), ацетонитриле (10 мл) и 0,5 M фосфатном буфере (pH 6,5, 28 мл) и гидрогенизируют над 10% Pd/C под давлением 40 фунт/кв. дюйм. Спустя 4 ч реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C, получая желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой НР21. Вначале колонку элюируют деионизированной водой (2 литра) и позднее смесью 10% ацетонитрил:вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном и фильтруют. Сушат. Выход: 276 мг, 73%, в виде желтого аморфного твердого вещества. (M+H+Na) 314.
1H ЯМР (D2О) δ 6,97 (1H, с), 6,95 (1H, с), 6,46 (1H, с), 5,56 (1H, с), 5,07 (2H, д, J=6,3 Гц), 4,30 (2H, т, J=6,3 Гц).
Пример 34
Получение (5R,6Z)-6-[(5-ацетил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен]оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (смесь E+Z изомеров, натриевая соль)
Стадия 1: 5-ацетил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-карбальдегид
К холодной (0°C) суспензии 1,5 г (7,4 ммоль) гидрохлорида 4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбальдегида в 50 мл метиленхлорида в атмосфере N2 в сухих условиях добавляют по каплям при перемешивании 2,6 мл (2,5 экв.) триэтиламина. РС перемешивают в течение 30 мин при 0°C и раствор 0,7 г (8,1 ммоль, 1,1 экв.) ацетилхлорида в 15 мл метиленхлорида добавляют по каплям, РС позволяют достичь КТ и перемешивают в течение 3 часов. Фильтруют через прокладку целита, фильтрат промывают 3 × 50 мл воды, сушат, выпаривают, получая 1,1 г (71,4%) указанного в заголовке соединения, вязкое масло, (M+H)+ 210,3.
Стадия 2: Получение 4-нитробензил-(5R)-6-[(ацетилокси)(5-ацетил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2карбоксилата
5-ацетил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-карбальдегид (540 мг, 2,57 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (950 мг, 2,5 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (2,21 г, 8,5 ммоль) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1).
Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 870 мг, 53%. Т.пл. 46-48°C. (M+H)+ 637,6.
1H ЯМР (CDCl3) δ 2,15 (т, 6H), 2,8-3,0 (м, 2H), 3,7-3,9 (м, 2H), 4,58-4,68 (м, 2H), 5,30-5,45 (дд, 2H), 5,85 (д, 1H), 6,71 (с, 1H), 6,95 (с, 1H), 7,35-7,45 (д, 1H), 7,60 (дд, 2H), 8,25 (дд, 2H).
Стадия 3: (5R,6Z)-6-[(5-ацетил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метилен]оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота (Смесь E+Z изомеров. Натриевая соль)
Раствор 0,77 г (1,21 ммоль) 4-нитробензил-(5R)-6-[(ацетилокси)(5-ацетил-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилата в 40 мл THF и 40 мл раствора фосфатного буфера (pH 6,36) гидрогенизируют под давлением 40 фунт/кв.дюйм в течение 3 часов в присутствии 0,4 г катализатора 10% палладия на углероде. Реакционную смесь фильтруют через прокладку целита, фильтрат доводят до pH 8,0, концентрируют в вакууме, остаток очищают на колонке с обращенной фазой (амберлит), используя смесь 5%-10% ACM/вода в качестве растворителя, получая 0,107 г (23%) указанного в заголовке соединения, красноватых кристаллов, т.пл. 362,4°C, (M+H)+ 409,5.
1H ЯМР: δ 2,08 (с, 3H), 2,80-2,95 (м, 1H), 3,74 (м, 2H), 3,9-4,06 (д, 2H), 6,32-6,42 (с, 1H), 6,50-6,60 (с, 1H), 6,98-7,20 (с, 1H), 7,30-7,40 (с, 1H).
Пример 35
Получение (5R,6Z)-6-(6,7-дигидро-4Н-пиразоло[5,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: 4-нитрозоморфолин-3-карбоновая кислота
К раствору морфолин-3-карбоновой кислоты (6,96 г, 52 ммоль) в воде (20 мл) при 0°C в атмосфере азота добавляют концентрированную хлористоводородную кислоту (4 мл), а затем нитрит натрия (5,0 г, 72 ммоль) маленькими порциями. Смесь перемешивают при 0°C в течение 1 ч и затем концентрируют в вакууме при 30-35°C. Остаток перемешивают с 200 мл ацетона и фильтруют. Фильтрат выпаривают и остаток обрабатывают 50 мл THF и концентрируют. Процесс повторяют с 2 × 50 мл THF, получая 11,87 г светло-желтой пены. MC (ESI) m/z 159,2 (M-H).
Стадия 2: 6,7-дигидро-4Н-[1,2,3]оксидиазоло[4,3-c][1,4]оксазин-8-ий-3-олат
Сырую 4-нитрозоморфолин-3-карбоновую кислоту (11,0 г) со стадии 1 растворяют в THF (250 мл) и охлаждают до 0°C. Раствор трифторуксусного ангидрида (7,4 мл, 52 ммоль) в THF (20 мл) добавляют при перемешивании в течение 10 мин. Полученную смесь перемешивают при 0°C в течение 5 ч и нагревают до комнатной температуры в течение 16 ч. Растворитель выпаривают и остаток разбавляют 250 мл этилацетата и перемешивают с 30 г безводного карбоната калия. Смесь фильтруют через прокладку силикагеля и фильтрат выпаривают. Остаток промывают смесью этилацетат-простой эфир, получая 3,80 г белого твердого вещества. Т.пл. 132-133°C, MC (ESI) m/z 143,1 (M+H).
Стадия 3: этил-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбоксилат
К частичному раствору 6,7-дигидро-4H-[1,2,3]оксидиазоло[4,3-c][1,4]оксазин-8-ий-3-олата (3,41 г, 24 ммоль) в o-ксилоле (80 мл) добавляют этилпропиолат (2,7 мл, 26 ммоль). Смесь перемешивают при 140°C в течение 3 ч. Затем добавляют дополнительные 2,0 мл (19 ммоль) этилпропиолата и смесь перемешивают при кипячении с возвращением флегмы в течение 18 ч. Конечный раствор выпаривают в вакууме и остаток растворяют в смеси метиленхлорида и гексанов (1:5). Раствор пропускают через прокладку силикагеля и фильтрующую прокладку элюируют смесью метиленхлорид-гексаны, а затем этилацетатом. Этилацетатный элюент выпаривают и остаток промывают гексанами, получая 4,10 г белого твердого вещества. Т.пл. 63°C; МС (ESI) m/z 197,1 (M+H).
Стадия 4: 6,7-дигидро-4Н-пиразоло[5,1-c][1,4]оксазин-2-илметанол
К раствору этил-6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбоксилата (1,57 г, 8,0 ммоль) в метиленхлориде (30 мл) добавляют 24 мл 1,0 M раствора гидрида диизобутилалюминия в метиленхлориде при 0°C в атмосфере азота. После перемешивания в течение 0,5 ч при 0°C смесь нагревают до комнатной температуры в течение 2 ч. Затем ее обрабатывают 30 мл насыщенного раствора хлорида аммония и экстрагируют этилацетатом. Органический раствор промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и выпаривают, получая 1,27 г бесцветного масла. МС (ESI) m/z 155,3 (M+H).
Стадия 5: 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбальдегид
К раствору 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-илметанола (1,08 г, 7,0 ммоль) в 1,2-дихлорэтане (30 мл) добавляют 5,4 г активированного диоксида марганца при комнатной температуре при перемешивании. Смесь нагревают до 60°C в течение 1 ч и затем перемешивают при комнатной температуре в течение 16 ч. Конечную смесь фильтруют через колонку силикагеля с находящимся на самом верху целитом. Фильтрующую прокладку элюируют метиленхлоридом, а затем этилацетатом. Этилацетатный элюент выпаривают и остаток растирают, получая 0,81 г белого твердого вещества. Т.пл. 91°C. МС (ESI) m/z 153,2 (M+H).
Стадия 6: 4-нитробензил-(5R)-6-[(ацетилокси)(6,7-дигидро-4Н-пиразоло[5,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
К раствору MgBr2 (0,94 г, 5,1 ммоль) в ацетонитриле (25 мл) в атмосфере азота добавляют 6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-карбальдегид (0,26 г, 1,7 ммоль) при комнатной температуре при перемешивании. Затем добавляют раствор 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,58 г, 1,5 ммоль) в THF (25 мл) и смесь охлаждают до -20°C. Вводят триэтиламин (0,71 мл, 5,1 ммоль) и смесь перемешивают при -20°C в темноте в течение 5 ч. Затем ее обрабатывают уксусным ангидридом (0,6 мл, 6,0 ммоль) и 4-N,N-диметиламинопиридином (24 мг, 0,2 ммоль) и выдерживают при 0°C в течение 18 ч. Смесь концентрируют и остаток растворяют в этилацетате. Этилацетатный раствор промывают 5% лимонной кислотой, насыщенным раствором бикарбоната натрия и насыщенным раствором соли, сушат над безводным сульфатом натрия и выпаривают. Сырой материал подвергают хроматографии на силикагеле (EtOAc-CH2Cl2/1:5), получая 0,77 г белой пены. МС (ESI) m/z 578,9 (M+H).
Стадия 7: (5R,6Z)-6-(6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновая кислота
К раствору 4-нитробензил-(5R)-6-[(ацетилокси)(6,7-дигидро-4H-пиразоло[5,1-c][1,4]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилата (0,35 г, 0,6 ммоль) в THF (20 мл) в атмосфере азота добавляют 20 мл раствора фосфатного буфера (0,5 M, pH 6,5) и 120 мг 10% Pd/C. Смесь гидрогенизируют под давлением 40-50 фунт/кв.дюйм в течение 3 ч и затем фильтруют через целит. Фильтрующую прокладку промывают THF и фильтрат экстрагируют этилацетатом. Органический экстракт сушат над безводным сульфатом магния и выпаривают. Остаток промывают простым эфиром, получая 0,09 г желтого твердого вещества. HRMS: рассчитано для C13H11N3O4S 305,0470; обнаружено (ESI+), 306,05434.
1H ЯМР (ДМСО-d6) δ 4,07-4,09 (т, 2H), 4,13-4,17 (т, 2H), 4,82 (с, 2H), 6,36 (с, 1H), 6,55 (с, 1H), 7,17 (с, 1H), 7,55 (с, 1H), 12,80 (уш.с, 1H).
Пример 36
Получение натриевой соли (5R)(6Z)-6-(6,7-5H-дигидропиразоло[5,1-b]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Стадия 1: Получение этил-6,7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбоксилата
К перемешиваемой суспензии этил-5-гидрокси-1H-пиразол-3-карбоксилата (10,34 г, 0,66 моль) и 36,62 г карбоната калия в 500 мл ацетонитрила добавляют 14,7 г 1,3-дибромпропана и кипятят с возвращением флегмы в течение 16 часов. Реакционной смеси позволяют остыть до комнатной температуры, затем фильтруют, твердое вещество промывают ацетонитрилом. Фильтрат концентрируют до масла. Остаток растворяют в этилацетате и экстрагируют водой. Органическую фазу сушат над MgSO4 и выпаривают до сухости. Получают 8,80 г желательного продукта (68%). Т.пл. 44-46°C. (M+H)+ 197,1.
Стадия 2: Получение 2,3-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-илметанола
К перемешиваемому раствору 6,7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбоксилата (4,0 г, 20 ммоль) в 100 мл THF добавляют 0,71 г боргидрида лития и 1,03 г метанола. Раствор нагревают при 40°С в течение 2,5 часов. Реакционную смесь гасят 1 н. HCl и доводят до pH 1,3 и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь доводят до рН 8 с помощью K2CO3. Реакционную смесь экстрагируют этилацетатом. Органический слой сушат над MgSO4 и концентрируют до масла и подвергают колоночной хроматографии, получая 2,08 г желательного продукта (67%). (M+H) 155.
Стадия 3: Получение 6,7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбальдегида
К перемешиваемому раствору 2,3-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-илметанола (2,08 г, 13,5 ммоль) в 60 мл CH3Cl добавляют 9,38 г MnO2. Суспензию кипятят с возвращением флегмы в течение 2 часов в атмосфере азота. Реакционную смесь фильтруют через прокладку целита. Фильтрат концентрируют, получая желтое масло. Продукт очищают хроматографией. Получают 2,15 г продукта (78%).
Стадия 4: 4-нитробензил-(5R)-6-[(ацетилокси)(6,7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат
6,7-Дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбальдегид (330 мг, 2 ммоль) и раствор в сухом THF (20 мл) 4-нитробензилового сложного эфира (5R,6S)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (0,794 г, 2,2 ммоль) добавляют последовательно к раствору в сухом ацетонитриле (15 мл) безводного MgBr2:O(Et)2 (1,2 г) в атмосфере аргона при комнатной температуре. После охлаждения до -20°C добавляют Et3N (2,0 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 2 ч при -20°C и обрабатывают уксусным ангидридом (1,04 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 15 ч при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют через прокладку целита. Прокладку промывают этилацетатом. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют смесью этилацетат:гексан (1:1). Собранные фракции концентрируют при пониженном давлении и смесь диастереоизомеров забирают на следующую стадию. Бледно-желтое аморфное твердое вещество. Выход: 0,76 г, 65%. M+H 579.
Стадия 5: натриевая соль (5R)(6Z)-6-(6,7-5H-дигидропиразоло[5,1-b]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0] гепт-2-ен-2-карбоновой кислоты
4-Нитробензил-(5R)-6-[(ацетилокси)(6,7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-ил)метил]-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилат (350 мг, 0,6 ммоль) растворяют в THF (20 мл), ацетонитриле (10 мл) и 0,5 M фосфатном буфере (pH 6,5, 28 мл) и гидрогенизируют над 10% Pd/C под давлением 40 фунт/кв.дюйм. Спустя 4 ч реакционную смесь фильтруют, охлаждают до 3°C и добавляют 0,1 M NaOH, чтобы довести рН до 8,5. Фильтрат промывают этилацетатом и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C, получая желтый осадок. Продукт очищают хроматографией с обращенной фазой на колонке со смолой НР21. Вначале колонку элюируют деионизированной водой (2 литра) и позднее смесью 10% ацетонитрил:вода. Фракции, содержащие продукт, собирают и концентрируют при пониженном давлении при комнатной температуре. Желтое твердое вещество промывают ацетоном и фильтруют. Сушат. Выход: 103 мг, 52%, в виде желтого аморфного твердого вещества. (M+H+Na) 327.
1H ЯМР (D2О) δ 6,97 (1H, с), 6,93 (1H, 8), 6,47 (1H, с), 5,65 (1H, с) 4,28 (2H, м), 4,10 (2H, м), 2,21 (2H, м).
Пример 37
Получение динатриевой соли (5R),(6Z)-6-[5-(3-карбоксипропионил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты
Указанное соединение получают процедурами, описанными в общих чертах в приведенных выше примерах. Исходя из натриевой соли (5R),(6Z)-6-{5-[3-(4-нитробензилоксикарбонил)пропионил]-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (467 мг) и гидрогенизируя ее над Pd/C (10%), изолируют 276 мг (74%) динатриевой соли (5R),(6Z)-6-[5-(3-карбоксипропионил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты в виде желтого аморфного твердого вещества. Т.пл. 180°C (с разложением).
1H ЯМР (D2О) δ 2,41 (т, 2H), 2,42 (т, 2H), 2,67 (т, 2H), 2,72 (т, 2H), 3,95-4,09 (м, 2H), 4,18 (т, 2H), 4,28 (т, 2H), 4,75 (с, 2H), 4,87 (с, 2H), 6,33 (с, 1H), 6,34 (с, 1H), 6,53 (с, 1H), 7,00 (с, 1H), 7,09 (с, 1H).
Пример 38
Получение натриевой соли (5R),(6Z)-6-[5-(2-метоксиацетил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты
Натриевая соль (5R),(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (Пример 25)
К раствору в THF (64 мл) и H2O (64 мл) натриевой соли (5R),(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (Пример 25) (638 мг) медленно добавляют 0,1 M водный NaOH, чтобы довести рН до 12,5, при 0°C. К смеси добавляют метоксиацетилхлорид (0,28 мл) в течение 5 мин. Смесь перемешивают в течение 0,5 ч при 0°C и метоксиацетилхлорид (0,09 мл) добавляют к смеси. После перемешивания смеси в течение 0,5 ч при той же температуре добавляют 0,1 M водн. NaOH, чтобы довести рН до 8,05. Смесь концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (78 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют H2O-MeCN (1:0 до 9:1). Объединенные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (509 мг, 65%, pH 7,58). Т.пл. 170°C (с разложением).
1H ЯМР (D2О) δ 3,28 (с, 3H×1/2), 3,29 (с, 3H×1/2), 3,78 (т, 2H×1/2, J=5,4 Гц), 3,89-3,93 (м, 2H×1/2), 4,09 (т, 2H×1/2, J=5,4 Гц), 4,14 (т, 2H×1/2, J=5,4 Гц), 4,20 (с, 2H×1/2), 4,25 (с, 2H×1/2), 4,61 (с, 2H×1/2), 4,66 (с, 2H×1/2), 6,19 (с, 1H×1/2), 6,22 (с, 1H×1/2), 6,37 (с, 1H×1/2), 6,372 (с, 1H×1/2), 6,87 (с, 1H), 6,93 (с, 1H).
Пример 39
Получение натриевой соли (5R),(6Z)-6-[5-(2-метоксиацетил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты
(4,5,6,7-Тетрагидропиразоло[1,5-a]пиразин-2-ил)метанол
Метанол (150 мл) добавляют к смеси 4-нитробензилового сложного эфира 2-гидроксиметил-6,7-дигидро-4H-пиразоло[1,5-a]пиразин-5-карбоновой кислоты (Пример 25) (2,38 г) и 10% Pd-C (50% влажность, 1,19 г). Реакционную смесь перемешивают в течение 2 часов в атмосфере водорода. Смесь фильтруют и концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют 50% метанолом в хлороформе. Указанное в заголовке соединение получают в виде белого твердого вещества (1,08 г, 98%).
1H ЯМР (400 МГц, CD3OD) δ 3,22-3,25 (м, 2H), 3,99 (с, 2H), 4,03-4,06 (м, 2H), 4,52 (с, 2H), 6,06 (с, 1H).
[5-(4,5-Дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметанол
Раствор хлорида водорода (2 моль/л) в простом диэтиловом эфире (0,7 мл) добавляют к раствору в метаноле (20 мл) (4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-ил)метанола (1,08 г) и 2-метилсульфанил-4,5-дигидротиазола (1,03 г). Реакционную смесь кипятят с возвращением флегмы в течение 4 дней. Смесь гасят небольшим количеством насыщенного раствора карбоната калия, сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем колонку элюируют 10% метанолом в хлороформе. Указанное в заголовке соединение получают в виде белого твердого вещества (1,49 г, 89%).
1H ЯМР (400 МГц, CDCl3) δ 2,04 (уш.с, 1H), 3,39 (т, 2H, J=7,5 Гц), 3,90 (т, 2H, J=5,3 Гц), 4,06 (т, 2H, J=7,5 Гц), 4,21 (т, 2H, J=5,3 Гц), 4,66 (с, 2H), 4,69 (с, 2H), 6,07 (с, 1H).
Натриевая соль (5R),(6Z)-6-[5-(4,5-дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Активированный оксид марганца (IV) (16,75 г) добавляют к смеси раствора в хлороформе (180 мл) [5-(4,5-дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-ил]метанола (3,35 г) при комнатной температуре. Реакционную смесь кипятят с возвращением флегмы в течение 1 ч. После кипячения с возвращением флегмы смесь фильтруют через прокладку целита и фильтрат концентрируют при пониженном давлении. Остаток сушат в вакууме и сырой 5-(4,5-дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-карбальдегид получают в виде бесцветного твердого вещества. Полученный таким образом сырой альдегид (2,56 г) добавляют к раствору в сухом ацетонитриле (200 мл) MgBr2 (7,36 г) в атмосфере азота при комнатной температуре, затем смесь перемешивают в течение 10 минут. Раствор в сухом THF (200 мл) WLJ 20,014 (4,16 г) добавляют и смесь охлаждают до -20°C. Затем добавляют триэтиламин (11,3 мл) одной порцией. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь перемешивают в течение 1,5 часов при -20°C и обрабатывают 4-диметиламинопиридином (132 мг) и уксусным ангидридом (4,2 мл) одной порцией. Реакционную смесь нагревают до 0°C и перемешивают в течение 20 часов при 0°C. Смесь разбавляют этилацетатом и промывают 5% водным раствором лимонной кислоты, насыщенным раствором гидрокарбоната натрия и насыщенным раствором соли. Органический слой сушат (MgSO4) и фильтруют. Фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, затем элюируют смесью н-гексан-AcOEt (1:2) и смесью хлороформ-метанол (9:1). Получают 4-нитробензиловый сложный эфир (5R,6RS)-6-{(RS)-ацетокси[5-(4,5-дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (5,41 г, 75,4%).
4-Нитробензиловый сложный эфир (5R,6RS)-6-{(RS)-ацетокси[5-(4,5-дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-ил]метил}-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (5,41 г) растворяют в THF (76 мл) и ацетонитриле (35 мл). Свежеактивированную Zn пыль (21,6 г) и 0,5 моль/л фосфатный буфер (pH 6,5, 111 мл) добавляют к смеси. Реакционный сосуд покрывают фольгой, чтобы исключить свет. Реакционную смесь энергично перемешивают в течение 2 часов при 30-35°C. Реакционную смесь охлаждают при 0°C и затем pH доводят до 7,6. Этилацетат добавляют к реакционной смеси и фильтруют через прокладку целита. Прокладку промывают водой и водный слой отделяют. Водный слой концентрируют в высоком вакууме при 35°C. Концентрат подвергают колоночной хроматографии на смоле Diaion HP-21 (170 мл, Mitsubishi Kasei Co. Ltd.). После адсорбирования колонку элюируют водой и затем 5%-15% водным раствором ацетонитрила. Объединенные активные фракции концентрируют в высоком вакууме при 35°C и лиофилизуют, получая указанное в заголовке соединение в виде сырого желтого аморфного твердого вещества (1,60 г).
Сырое соединение очищают препаративной ВЭЖХ (Mightysil RP-18GP, KANTO CHEMICAL CO., INC., 35 × 250 мм, 0,05 моль/л фосфатный буфер (pH 7,1):ацетонитрил = 80:20, 25 мл/мин) с последующим обессоливанием на смоле Diaion HP-21 (150 мл, Mitsubishi Kasei Co. Ltd.), получая указанное в заголовке соединение в виде желтого аморфного твердого вещества (1,06 г, 31,5%, pH 8,33). Т.пл. 100°C (с разложением).
1H ЯМР (D2О) δ 3,18 (т, 2H, J=7,6 Гц), 3,60 (т, 2H, J=5,3 Гц), 3,73 (т, 2H, J=7,6 Гц), 3,94 (т, 2H, J=5,3 Гц), 4,37 (с, 2H), 6,01 (с, 1H), 6,21 (с, 1H), 6,77 (с, 1H), 6,78 (с, 1H); IR (KBr) 3381, 1752, 1606 см-1; λмакс. (H2O) 369, 291, 208 нм.
Пример 40
Получение натриевой соли (5R),(6Z)-6-[5-(2-метоксиацетил)-4,5,6,7-тетрагидропиразоло[1,5-a]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты
Получение этил-2-[(ацетилокси)((5R)-6-бром-2-{[(4-нитробензил)окси]карбонил}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-ил)метил]-4,7-дигидрофуро[2,3-c]пиридин-6(5H)-карбоксилата
Указанное в заголовке соединение получают из 0,669 г метил-2-формил-4,7-дигидрофуро[2,3-c]пиридин-6(5H)-карбоксилата и 1,155 г 4-нитробензил-(5R)-6-бром-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоксилата с выходом 1,65 грамма продукта (84%), который используют непосредственно на следующей стадии. МС: 652,2 (M+H)
Получение (5R,6Z)-6-{[6-(этоксикарбонил)-4,5,6,7-тетрагидрофуро[2,3-c]пиридин-2-ил]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты
Указанное в заголовке соединение получают из 1,65 г этил-2-[(ацетилокси)((5R)-6-бром-2-{[(4-нитробензил)окси]карбонил}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-ил)метил]-4,7-дигидрофуро[2,3-c]пиридин-6(5H)карбоксилата с выходом 0,386 г продукта (41%). Т.пл.: разлагается при 175°C. МС: 375,0 (M-H).
1H ЯМР (D2О) δ 6,91 (с, 1H), 6,84 (с, 1H), 6,62 (с, 1H), 6,39 (с, 1H), 4,41 (уш., 2H), 4,04 (кв, 2H, J=5 Гц), 3,52 (уш., 2H), 2,42 (уш., 2H), 1,14 (т, 3H, J=5 Гц).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 6-АЛКИЛИДЕНПЕНЕМА | 2003 |
|
RU2317297C2 |
| 2-ТИОЗАМЕЩЕННЫЕ КАРБАПЕНЕМЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2130457C1 |
| ПРОИЗВОДНЫЕ 3β-АЛКЕНИЛПЕНАМА И ИХ ФАРМАЦЕВТИЧЕСКИ СОВМЕСТИМЫЕ СОЛИ | 1994 |
|
RU2139290C1 |
| КОНДЕНСИРОВАННОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ АМИНОПИРРОЛИДИНА | 2007 |
|
RU2443698C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНИЛЛАКТАМОВ ИЛИ ИХ СОЛЕЙ | 1992 |
|
RU2130938C1 |
| БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ГАММА-АМИНОКИСЛОТЫ | 2008 |
|
RU2446148C2 |
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2531588C2 |
| СРЕДСТВА, ИНДУЦИРУЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА, ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2568611C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДОНОВЫХ КАРБОНОВЫХ КИСЛОТ, СПОСОБ И ПРОМЕЖУТОЧНЫЕ ДЛЯ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ | 1993 |
|
RU2161154C2 |
Изобретение относится к соединениям формулы (I) и их фармацевтически приемлемьм солям в качестве ингибиторов β-лактамаз, способу их получения, фармацевтической композиции на их основе, а также к способу лечения с их использованием. Соединения могут найти применение для лечения бактериальных инфекций. В общей формуле (I)
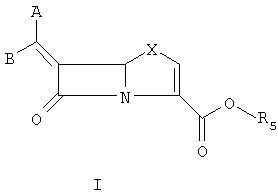
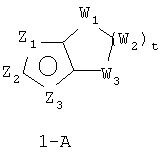
один из А и В означает водород, а другой означает необязательно замещенную конденсированную бициклическую гетероарилгруппу; при условии, что если ароматической кольцевой частью бициклической гетероарилгруппы является имидазол, неароматическая кольцевая часть не может содержать атом S, смежный с головным атомом углерода мостиковой группы; Х означает S; R5 означает Н, C1-С6-алкил или С5-С6-циклоалкил; или его фармацевтически приемлемая соль, где бициклической гетероарилгруппой является (1-А), где один из Z1, Z2 и Z3 независимо означают S, а другие CR2 или S, при условии, что один из Z1-Z3 означает углерод и связан с остальной частью молекулы; W1, W2 и W3 независимо означают CR4R4, S, О или N-R1, при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы; t=1-4; R1 означает Н, C1-С6-алкил, С5-С7-циклоалкил, -С=O-арил, -С=O(С1-С6)-алкил, -С=O(С5-С6)-циклоалкил, арил-C1-С6-алкил, необязательно замещенный C1-С6-алкокси; гетероалкил-С1-С6-алкил или С=O(гетероарил), где гетероарил представляет собой 6-членное кольцо, содержащее 1 атом азота, R2 означает водород, C1-С6-алкил, R4 означает Н, C1-С6-алкил. 8 н. и 21 з.п. ф-лы, 3 табл.
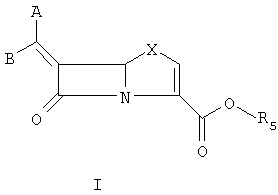
где один из А и В означает водород, а другой означает необязательно замещенную конденсированную бициклическую гетероарилгруппу; при условии, что если ароматической кольцевой частью бициклической гетероарилгруппы является имидазол, неароматическая кольцевая часть не может содержать атом S, смежный с головным атомом углерода мостиковой группы;
Х означает S;
R5 означает Н, C1-С6-алкил или С5-С6-циклоалкил;
или его фармацевтически приемлемая соль,
где бициклической гетероарилгруппой является
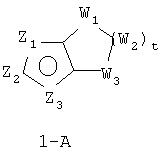
где один из Z1, Z2 и Z3 независимо означают S, а другие CR2 или S, при условии, что один из Z1-Z3 означает углерод и связан с остальной частью молекулы;
W1, W2 и W3 независимо означают CR4R4, S, О или N-R1, при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
t=1-4;
R1 означает Н, C1-С6-алкил, С5-С7-циклоалкил, -С=O-арил, -С=O(С1-С6)-алкил, -С=O(С5-С6)-циклоалкил, арил-С1-С6-алкил, необязательно замещенный C1-С6-алкокси; гетероалкил-C1-С6-алкил или С=O(гетероарил), где гетероарил представляет собой 6-членное кольцо, содержащее 1 атом азота,
R2 означает водород, C1-С6-алкил,
R4 означает Н, C1-С6-алкил.
где один из А и В означает водород, а другой означает необязательно замещенную конденсированную бициклическую гетероарилгруппу; при условии, что если ароматической кольцевой частью бициклической гетероарилгруппы является имидазол, неароматическая кольцевая часть не может содержать атом S, смежный с головным атомом углерода мостиковой группы;
Х означает S;
R5 означает Н, C1-С6-алкил или С5-С6-циклоалкил;
или его фармацевтически приемлемая соль,
где бициклической гетероарилгруппой является
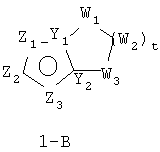
где Z1, Z2 и Z3 независимо означают CR2, N, О, S или NH при условии, что один из Z1-Z3 означает углерод и связан с остальной частью молекулы;
W1, W2 и W3 независимо означают CR4R4, S, О или N-R1, при условии, что не происходит образования ни S-S, ни O-O, ни S-O связи с образованием насыщенной кольцевой системы;
t=1-4;
Y1 и Y2 независимо означают N или С при условии, что если ароматической кольцевой частью бициклической гетероарилгруппы является имидазол, неароматическая кольцевая часть не может содержать атом S, смежный с головным атомом углерода мостиковой группы, и
R1 означает Н, C1-С6-алкил, С5-С7-циклоалкил, -С=O(С1-С6)-алкил, -С=O(С5-С6)-циклоалкил, -С(O)С1-С2-алкилкарбоксил, -С(=O)С1-С6-алкокси; или гетероарил, представляющий собой 5-членное кольцо, содержащее атом N и/или S;
R2 означает водород,
R4 означает Н или C1-С6-алкил.

где А, В и R5 имеют значения, определенные в п.1 или 2.
(5R,6Z)-6-[(5-бензил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метилен]-7-оксо-4-тиа-1-азабицикло [3.2.0] гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(7-метил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-7-оксо-6-(5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R,6Z)-6-{[5-(4-метоксибензил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(5,6-дигидро-8Н-имидазо[2,1-с][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(6,7-дигидро-5Н-пирроло[1,2-а]имидазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(5,6-дигидро-8Н-имидазо[2,1-с][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(5,6-дигидро-4Н-пирроло[1,2-b]пиразол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-а]пиридин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(7-метил-6-оксо-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-6-(6,7-дигидро-4Н-пиразоло[5,1-с][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-7-оксо-6-(4Н-5-тиа-1,6а-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-6-(7Н-имидазо[1,2-с]тиазол-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
(5R,6Z)-7-оксо-6-[(4-оксо-6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-ил)метилен]-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
6-(6,7-дигидро-4Н-тиено[3,2-с]пиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
6-(6,7-дигидро-4Н-тиено[3,2-с]тиопиран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
6-(5-метил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
этилового сложного эфира 2-(2-карбокси-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-6-илиденметил)-6,7-дигидро-4Н-тиено[3,2-с]пиридин-5-карбоновой кислоты;
7-оксо-6-(6,7,8,9-тетрагидро-5Н-имидазо[1,2-а]азепин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(7-бензил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
(5R,6Z)-7-оксо-6-{[5-(пиридин-3-илметил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
(5R,6Z)-7-оксо-6-{[5-(пиридин-3-илкарбонил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты, и
(5R,6Z)-7-оксо-6-{[5-(фенилацетил)-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(5,5-диоксо-4,5,6,7-тетрагидро-5λ6-пиразоло[5,1-c][1,4]тиазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-7-оксо-6-(4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
(5R)(6Z)-6-(5,5-диметил-4Н-1,6а-диазапентален-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты, натриевая соль 5,5-диметил-2-пиперидона;
натриевой соли (5R),(6Z)-6-(5,6-дигидро-4Н-циклопента[b]фуран-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-6-(4,5-дигидро-6-тиа-1,7а-диазаинден-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-(6,6-диметил-5,6,7,8-тетрагидроимидазо[1,2-а]пиризин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
(5R),(6Z)-6-(5,6-дигидро-8-Н-имидазо[2,1-с][1,4]тиазин-3-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-7-оксо-6-(4Н-5-тиа-1,6а-диазапентален-2-илметилен)-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-6-(2,3-дигидропиразоло[5,1-b]тиазол-6-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-6-(2,3-дигидропиразоло[5,1-b]оксазол-6-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
(5R,6Z)-6-[(5-ацетил-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ил)метилен]оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты (смесь E+Z изомеров, натриевой соли);
(5R,6Z)-6-(6,7-дигидро-4Н-пиразоло[5,1-с][1,4]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
натриевой соли (5R)(6Z)-6-(6,7-5Н-дигидропиразоло[5,1-b]оксазин-2-илметилен)-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты;
динатриевой соли (5R),(6Z)-6-[5-(3-карбоксипропионил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-[5-(2-метоксиацетил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-[5-(4,5дигидротиазол-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-илметилен]-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты;
натриевой соли (5R),(6Z)-6-{[6-(этоксикарбонил)-4,5,6,7-тетрагидрофуро[2,3-с]пиридин-2-ил]метилен}-7-оксо-4-тиа-1-азабицикло[3.2.0]гепт-2-ен-карбоновой кислоты.
определенного по любому одному из пп.1-16 или его фармацевтически приемлемой соли.
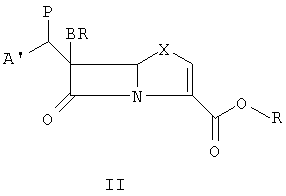
где А' означает А или В имеют значения, определенные в пп.1, 2, Х означает О или S, Р означает сложноэфирную уходящую группу, R означает удаляемую защитную группу, чтобы получить соединение формулы I
где А, В и Х имеют значения, определенные в п.1 или 2,
R5 означает водород; за время, достаточное для образования указанных соединений в условиях, эффективных для его образования или его фармацевтически приемлемой соли.
где А' означает А или В имеют значения, определенные в п.1 или 2, Х означает О или S, Р означает сложноэфирную уходящую группу, R означает удаляемую защитную группу, чтобы получить соединение формулы I
где А, В и X имеют значения, определенные в п.1 или 2,
R5 означает C1-С6-алкил или С5-С6-циклоалкил; за время, достаточное для образования соединений формулы I, в условиях, эффективных для его образования.
| WO 9410178 А, 11.05.1994 | |||
| BENNET I.S | |||
| et al | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| IV | |||
| Kidney stability, serum binding and additional biological evaluation of racemic derivatives | |||
| The J | |||
| of Antibiot | |||
| Циркуль-угломер | 1920 |
|
SU1991A1 |
| WO 8700525 A, 29.01.1987 | |||
| BROOM N.J.P | |||
| et al | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |

